1. Introduction
Parasitic plants represent one of the most intriguing ecological guilds, having evolved at least 12 times independently throughout the evolutionary history of flowering plants (Nickrent 2020). The transition to parasitism entails profound morphological and physiological modifications, the most critical being the development of the haustorium. This specialized organ not only functions as a physiological bridge for water and nutrient extraction but also establishes an intimate, cell-to-cell physical connection between the parasite and its host (Teixeira-Costa 2021).
This physical intimacy has unexpected genomic consequences: the haustorium facilitates the exchange of macromolecules, including RNA and DNA, promoting Horizontal Gene Transfer (HGT) at rates significantly higher than in free-living plants (Mower et al., 2010; Xi et al. 2013; Sanchez-Puerta et al. 2017; Davis and Xi 2015; Góralski et al. 2022). Although HGT is typically considered a rare phenomenon in eukaryotic evolution, this haustorial connection may drive an accumulation of foreign genetic material in parasitic plants. This accumulation is particularly striking in non-photosynthetic holoparasites, which depend entirely on their host for survival (Yang et al. 2016; 2019). HGT is especially prevalent in the mitochondrial genome (mtDNA) of parasitic plants. In certain lineages (e.g., Rafflesiaceae, Orobanchaceae, Cynomorium, Lophophytum), the mtDNA can incorporate genes from multiple historical and current hosts (Xi et al. 2013; Cusimano and Renner 2019; Feng and Wicke 2025; Sanchez-Puerta et al. 2017).
The dynamics of these horizontal acquisitions and the evolutionary fate vary considerably among holoparasitic plants. While mtDNAs in some lineages retain large, often functional foreign fragments (Sanchez-Puerta et al. 2023; Cai et al. 2021; Garcia et al. 2021), close relatives may exhibit virtually no mitochondrial HGT (Yu et al. 2022). In addition, evidence from the holoparasitic Cuscuta and Orobanchaceae reveals that significant nuclear HGT can occur independently of mitochondrial dynamics. In these lineages, the nuclear genome has acquired multiple functional nuclear genes, despite the absence of major mitochondrial HGT (Yang et al. 2016; Vogel et al. 2018). A comprehensive understanding of the full extent and diversity of HGT in holoparasites requires the exploration of remaining key lineages, including the holoparasitic Hydnoraceae (Piperales), which comprise the genera Hydnora and Prosopanche. Despite the loss of photosynthetic capacity and the reduction of their plastome (Yu et al. 2022), little is known about the full extent of horizontal gene flow that impacts their mitochondrial and nuclear genomes, as well as the timing of these evolutionary events.
The genus Prosopanche (comprising seven species) exhibits a documented host range spanning 11 diverse angiosperm families, including Solanales, Malvales, Fabales, and Malpighiales, among others (Hatt et al. 2025). However, the host range diversity is not uniformly distributed across species of Prosopanche. While P. bonacinae is a generalist associated with all 11 reported host families, the remaining species are specialists, each restricted to a single host family. This is the case for P. americana and P. panguanensis, which have been exclusively documented parasitizing Fabaceae (Hatt et al. 2022; Sequeira et al. 2018; Martel et al. 2018; Cocucci 1975; 1965; 1976). The draft mtDNA of P. americana exhibits limited signals of HGT in the coding regions, namely short patches within the genes cox1 and atp8, (Yu et al. 2023) (Yu et al. 2023). However, the intergenic regions, which comprise ~90% of the mtDNA, remain unexplored, and the nuclear genome has likewise not been examined.
In this study, we present a comprehensive analysis of nuclear and mitochondrial HGT in Prosopanche americana. Using a comparative phylogenomic approach that integrates mitochondrial genomes and nuclear transcriptomes, we investigate the origin and timing of foreign DNA acquisitions. We leverage available data from sister species, P. panguanensis and P. bonacinae, to distinguish between recent and ancestral events. Our results uncover a multi-layered landscape wherein P. americana possesses genetic material from donors belonging to at least five distinct orders (including Solanales, Malvales, and Fabales), providing new insights into the extensive history of host interactions that have shaped this genomic architecture over time.
2. Materials and Methods
2.1. Mitochondrial Genome Assemblies
To detect and characterize mitochondrial HGT in
Prosopanche americana, we analyzed the published draft mitochondrial DNA (mtDNA) sequence (
Table S1). This genome was assembled into 43 contigs totaling 426,953 bp and contains 39 native protein-coding genes, two of which show evidence of chimerism due to partial recombination with a foreign homolog (Yu et al. 2023).
Draft mitochondrial genomes from P. panguanensis and P. bonacinae were assembled de novo from paired-end reads downloaded in SRA, NCBI (accessions SRR31419645 and SRR31419646, respectively) using GetOrganelle v.1.7.5 (Jin et al. 2020) with the embryophyte mitochondrial database (-F embplant_mt). To optimize graph resolution, we utilized specific k-mer lengths of 21, 85, and 105, with a maximum of 2 extension rounds. Mitochondrial contigs and were identified via BLASTn searches in Bandage with default parameters (Wick et al. 2015) using the P. americana mtDNA as query. This process recovered 168 mitochondrial contigs for P. bonacinae (total length: 132 kb) and 357 contigs for P. panguanensis (total length: 444 kb). Using the same BLASTn-based approach, protein-coding regions were identified in the assembled contigs to include in the phylogenetic analyses.
2.2. Identification of High-Confidence Mitochondrial HGT Candidate Intergenic Regions
The phylogenetic origin of the intergenic regions in the draft mtDNAs of
P. americana and
P. panguanensis was initially assessed by BLASTN searches against a suite of custom databases designed around the host spectrum of
Prosopanche (Hatt et al. 2025). To account for ancestral HGT events from donors other than the actual hosts, these databases were constructed at the Order level, including: (i) reported hosts, incorporating NCBI sequences from Solanales, Fabales, Rosales, Aquifoliales, Apiales, Sapindales, Malvales, Caryophyllales, Malpighiales, Poales; (ii) the native Order, Piperales; and (iii) the rest of the angiosperms (excluding parasitic species to avoid confounding HGT signals). The database sequence retrieval was automated using the rentrez R package. All available records up to August 2025 were downloaded, filtering for mitochondrial sequences with a length greater than 4,000 bp (
Table S2). BLASTN hits were plotted using the SUSHI R package v.1.20.0 (Phanstiel et al. 2014).
To further evaluate the phylogenetic origin of the intergenic mitochondrial regions in the draft mtDNAs of
P. americana and
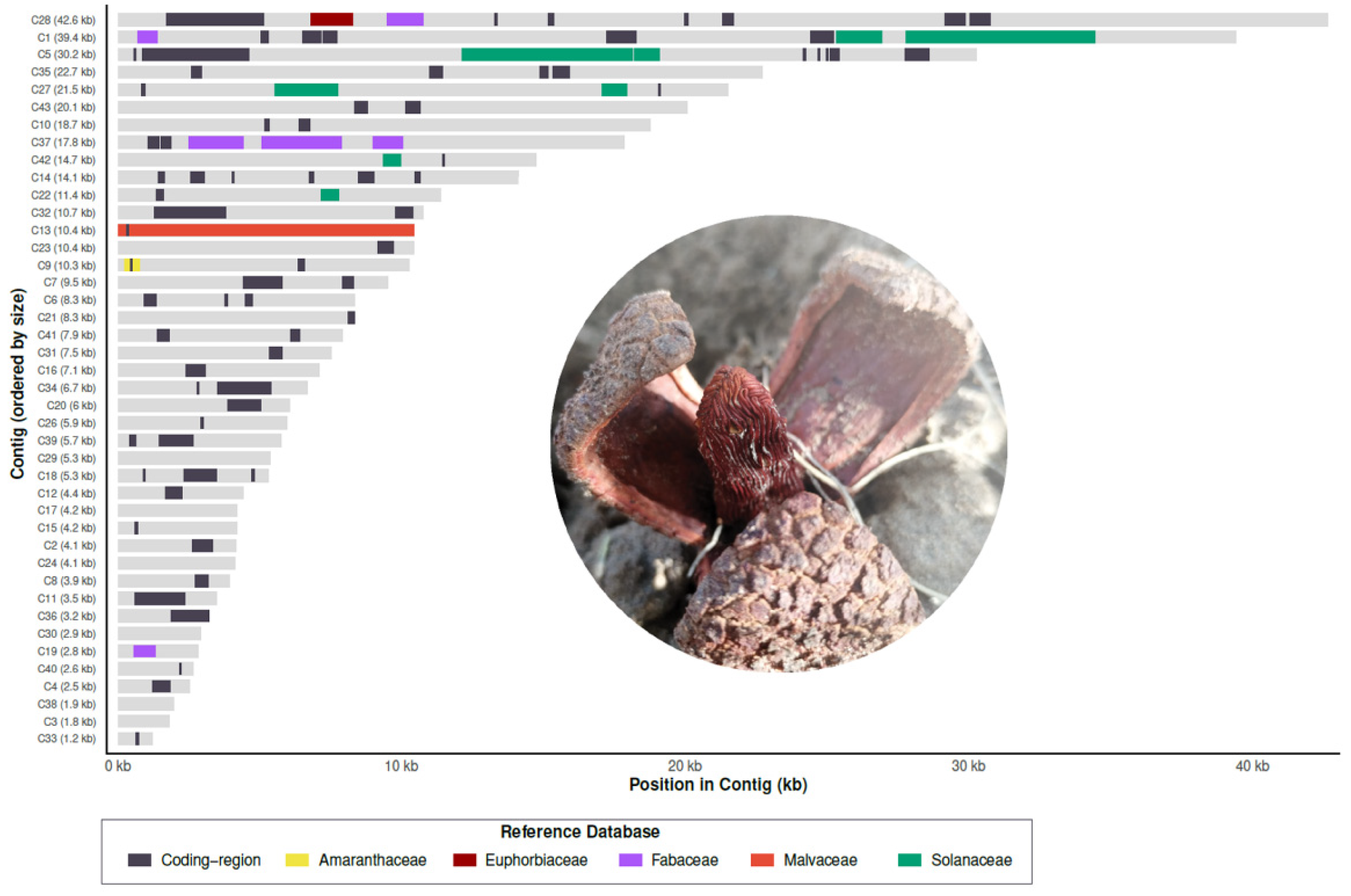
P. panguanensis, we implemented a pipeline integrating BLAST filtering, interval collapsing, and taxon-aware conflict resolution. Initial BLAST alignments were filtered to retain only high-confidence HGT hits (hits from reported hosts with percent identity ≥ 90%, alignment length ≥ 200 bp), and overlapping matches derived from the same donor lineage were subsequently merged into contiguous blocks. To resolve phylogenetic assignment conflicts where blocks from different lineages overlapped, we prioritized the taxon exhibiting the highest percent identity, utilizing bit score and alignment length as secondary tie-breakers. Finally, a greedy interval-selection algorithm was applied to generate a non-overlapping genomic tiling (“no-gap” strategy), ensuring that each genomic position was assigned to the strongest available evidence of foreign origin. This was visualized as a distribution of best organellar BLAST hits across the mitochondrial contigs (
Figure 1), where colors other than grey correspond to foreign mitochondrial DNA tracts from different donors (e.g., Solanaceae, Fabaceae). To confirm their foreign origin, phylogenetic trees were reconstructed for regions with BLASTn hits in at least two different lineages, provided they met a > 60% coverage threshold. Homologous DNA sequences were aligned using AliViewv1.30 (Larsson 2014), and trees were reconstructed using RAxMLv8 with 1,000 bootstrap replicates under the GTRGAMMA model. The resulting topologies were visualized in iTOL (Letunic and Bork 2024).
To assess the timing of the HGT events, the foreign regions identified in P. americana were BLASTn-searched against the mitochondrial contigs of P. bonacinae and P. panguanensis. This allowed us to evaluate the conservation of HGT events across the genus.
2.3. Sequencing and Assembly of the Prosopanche americana Transcriptome
A flower from an individual of Prosopanche americana (Hydnoraceae) parasitizing Neltuma flexuosa (Fabaceae) was collected in January 2023 in Phillips, Junín Department, Mendoza Province, Argentina (33° 12 ′ 28.008 ′′ S 68° 21 ′ 47.916 ′′ W). Total RNA was extracted using a protocol suitable for highly viscous samples rich in polysaccharides (Zeng and Yang 2002) and purified with the RNAqueous kit (Invitrogen, Waltham, MA), which employs columns with glass fiber filters. The rRNA-depleted total RNA was sequenced on the Illumina Novaseq S6000 platform at Shanghai Neo-Biotechnology Co., Ltd., Shanghai, China. The obtained RNA-seq data were analyzed with FastQC v0.12.1. The FastQC report showed that all parameters assessed were acceptable except for the ‘per base sequence content’ parameter, which displayed a deviation in the first 12 nucleotides of the reads. These nucleotides were trimmed using Trimmomatic v0.40. The RNA was extracted from flowers of the holoparasitic plants for several reasons: (i) no leaves are present in these plants, (ii) flower tissues are not contaminated with host tissue, and (iii) success with inflorescences has been reported previously (Sanchez-Puerta et al. 2017; Roulet et al. 2020; Garcia et al. 2021).
Reads quality-filtered with Trimmomatic were assembled
de novo using Trinity v.2.8.4 (Grabherr et al. 2011), with the following parameters SS_lib_type RF –min_contig_length 100 on the SARTOI computational cluster at the IBAM UNCuyo-CONICET. The transcriptome integrity was assessed using BUSCO (Waterhouse et al. 2018), which revealed complete gene recovery for approximately 91%, 52%, and 47% of single-copy orthologs representing conserved gene families within Eukaryota (n=303), Embryophyta (n=1,440), and Eudicots (n=2,121), respectively (
Table S3). These recovery rates are consistent with expectations for the relatively complete transcriptome of a holoparasitic plant, given the likely widespread evolutionary loss of genes associated with photosynthetic functions and vegetative structures (Sun et al. 2018; Cai et al. 2021; Chen et al. 2023; Svetlikova et al. 2025). Open Reading Frames (ORFs) were extracted from the transcriptome assembly using TransDecoder v.5.7.0 (
https://github.com/TransDecoder/TransDecoder), which yielded an initial set of 120,164 ORFs exceeding 100 bp in length.
2.4. Orthogroup Inference and Identification of Foreign Nuclear Genes
A custom multi-stage bioinformatics pipeline was designed and implemented to identify genes acquired from its potential hosts into the
P. americana nuclear genome. The workflow (
Figure S1) encompassed: (1) data preparation and filtering, (2) orthogroup identification, (3) candidate selection and phylogenetic inference, and (4) validation and curation of HGT events.
The initial set of ORFs underwent a rigorous sequential filtering process. First, 8,893 sequences corresponding to Transposable Elements (TEs) were identified and removed using HMMER v.3.3.2 (Potter et al. 2018) with profiles from the Dfam database (-value < 0.05). Subsequently, to eliminate biological contaminants (e.g., fungi, metazoans), the remaining 111,271 sequences were taxonomically annotated with eggNOG-mapper v.5.0 (Cantalapiedra et al. 2021). All sequences without an assignment within Viridiplantae (n = 15,812) were discarded, resulting in a high-confidence final transcriptome for
Prosopanche composed of 95,459 sequences. Gene family identification was performed using OrthoFinder v.2.5.4 (Emms and Kelly 2019). The analysis included the filtered
Prosopanche translated ORFs along with the proteomes from 45 reference species, encompassing host lineages from the orders Solanales, Fabales, and Malvales, which were identified as donors in the
Prosopanche mitochondrial genome, as well as pertinent outgroups (see
Table S4 for the complete species list). This analysis generated 101,329 orthogroups (OGs). To restrict the phylogenetic analysis to high-probability HGT candidates, a two-step filtering strategy was applied:
Similarity Filter (BLASTP): We selected OGs where at least one Prosopanche sequence had its best hit (BLASTP, e-value < 1e-5) against a species from the host families.
Phylogenetic Informativeness: OGs with a minimum of three taxa were retained to ensure robust topological inference.
2.5. Phylogenetic Reconstruction of Nuclear Foreign Candidate Genes
The amino acid sequences for each candidate OG were aligned using MAFFT v.7 (Katoh et al. 2013) (L-INS-i algorithm). To optimize the signal-to-noise ratio, the alignments were processed with ClipKIT v.2 (Steenwyk et al. 2020) in gappy mode (-m gappy). Phylogenetic reconstruction was conducted using Maximum Likelihood (ML) in IQ-TREE v.2.2 (Nguyen et al. 2015), employing the PlantQ substitution model and 1000 replicates of Ultrafast Bootstrap (UFBoot).
2.6. Nuclear HGT Event Detection and Curation
HGT identification in phylogenetic analyses was automated via a custom R script (using the ape package). HGT events were considered when: (i) the Prosopanche sequence was nested within a host family clade, or positioned as a sister to the host family, and (ii) the basal node of the clade presented robust support (UFBoot > 95%).
The resulting orthogroup containing foreign transcripts (n = 142) underwent exhaustive cleaning. To eliminate redundancy due to isoforms, CD-HIT (-c 0.95) was applied, followed by a manual selection of the longest isoform. Functional annotation (Mercator, InterProScan, eggNOG) enabled us to rule out cryptic contaminants that were undetected in previous steps. Finally, to distinguish functional genes from potential transposons, sequences with domains or descriptions associated with TE machinery (e.g., reverse transcriptase, transposase) were excluded. The final high-confidence dataset consisted of 305 horizontally transferred transcripts.
2.7. Functional Annotation of Foreign Nuclear Genes
Gene Ontology (GO) enrichment analysis was performed using the PANTHER Classification System (v.19.0, release 2024-08-07;
http://www.pantherdb.org). We utilized the Statistical Overrepresentation Test to compare the input gene list against the
Arabidopsis thaliana reference genome. Significance was determined using Fisher’s Exact Test with a False Discovery Rate (FDR) correction for multiple testing. Terms with an adjusted P-value (FDR) < 0.05 were considered significantly enriched.
3. Results
3.1. Characterization and Origin of Mitochondrial HGTs in Prosopanche americana
BLASTn analysis against custom databases, including the host orders of the genus
Prosopanche (Hatt et al. 2025)(Hatt et al., 2025), the order Piperales (to which
P. americana belongs), and all other angiosperms (updated as of August 2025), revealed that 55% of the
Prosopanche americana mtDNA is covered by hits from other angiosperm mtDNAs (
Figure S2). A comprehensive inspection of the 43 contigs in the draft mtDNA of
P. americana identified 18 distinct foreign regions exceeding 500 bp in length. The origin of these foreign regions was confirmed by phylogenetic analyses whenever possible; in other cases, donor assignment relied on the exclusive presence of the fragment in specific host lineages (
Figure S3). The most heavily represented host orders as potential donors were Solanales (20.2 kb), Malvales (10.4 kb), and Fabales (8.5 kb) (
Figure 1). Additionally, smaller fragments attributed to Euphorbiaceae (1,460 bp) and Caryophyllales (500 bp) were detected (
Figure 1;
Figure S2;
Table S4).
To assess the timing of these acquisitions, we screened for these foreign regions in the draft mtDNAs of
P. panguanensis and
P. bonacinae. Thirteen of the 18 identified regions were present in
P. panguanensis, while none were detected in
P. bonacinae. It should be noted that the apparent absence of foreign sequences in the
P. bonacinae draft mitochondrial assembly may be an artifact due to missing data. In all phylogenetic trees where
P. panguanensis sequences were available,
P. americana and
P. panguanensis formed a monophyletic group, suggesting that these HGT events occurred in their common ancestor (
Figure S3). These ancestral HGT events were identified as foreign DNA donated by all five host orders.
To further explore the evolutionary dynamics of HGT in the genus, we inspected the
P. panguanensis draft mtDNA (198 contigs) and identified 58 distinct foreign regions exceeding 500 bp, distributed across 44 contigs (
Figure S4). Interestingly, while
P. americana retains Solanales as the predominant donor; the HGT landscape in
P. panguanensis is dominated by Fabales (44 kb), followed by Solanales (13 kb), Malpighiales (6 kb), Malvales (4.5 kb), and Caryophyllales (4 kb). Additionally, smaller fragments attributed to Poales (800 bp) and Sapindales (700 bp) were detected (
Figure S4), revealing a distinct HGT profile characterized by a marked enrichment of sequences from the current host lineage compared to
P. americana.
3.2. HGT Impact on Mitochondrial Coding Regions
Phylogenetic analyses of protein-coding genes in the three species of
Prosopanche revealed a predominant native origin of all genes, in agreement with Yu et al. (2023) (
Figure S5). The exceptions were
cox1 and
atp8, where the chimerism previously reported for
P. americana was also identified in
P. panguanensis and
P. bonacinae.
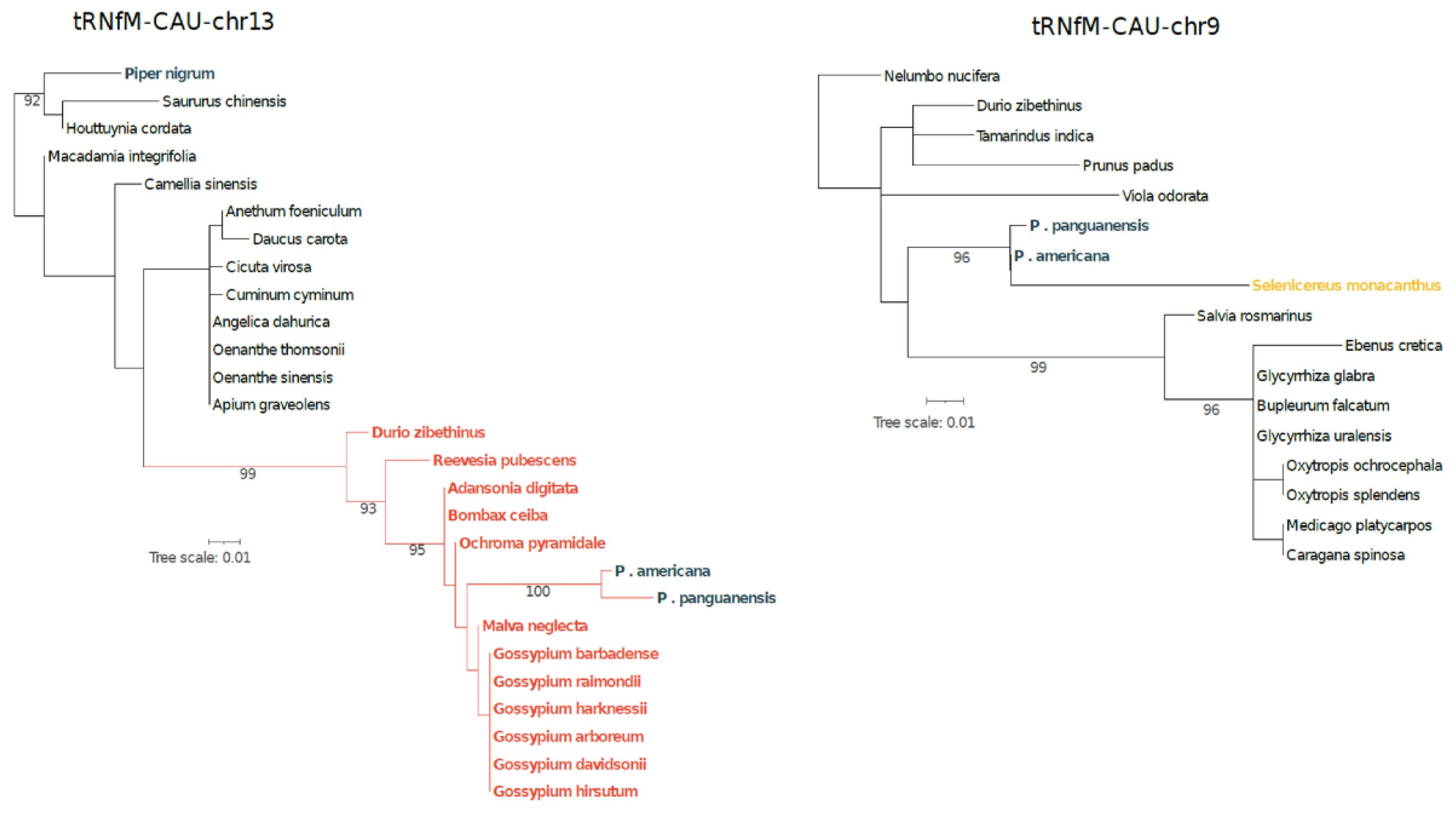
Gene mapping within the HGT blocks revealed the presence of two foreign
trnfM. One copy is located on chromosome 9 embedded within a genomic block derived from Caryophyllales, while the other resides on chromosome 13 within a Malvaceae-derived region. Phylogenetic analyses of the trnfM and 100 bp flanking regions confirmed these affinities (
Figure 2).
3.3. HGT from Different Hosts in the Nuclear Genome of Prosopanche americana
To identify potential HGT candidates, the curated
Prosopanche transcriptome was analyzed in a comparative framework alongside 45
Viridiplantae species (
Table S4), leading to the inference of 101,329 orthogroups (OGs). We identified candidate OGs in which
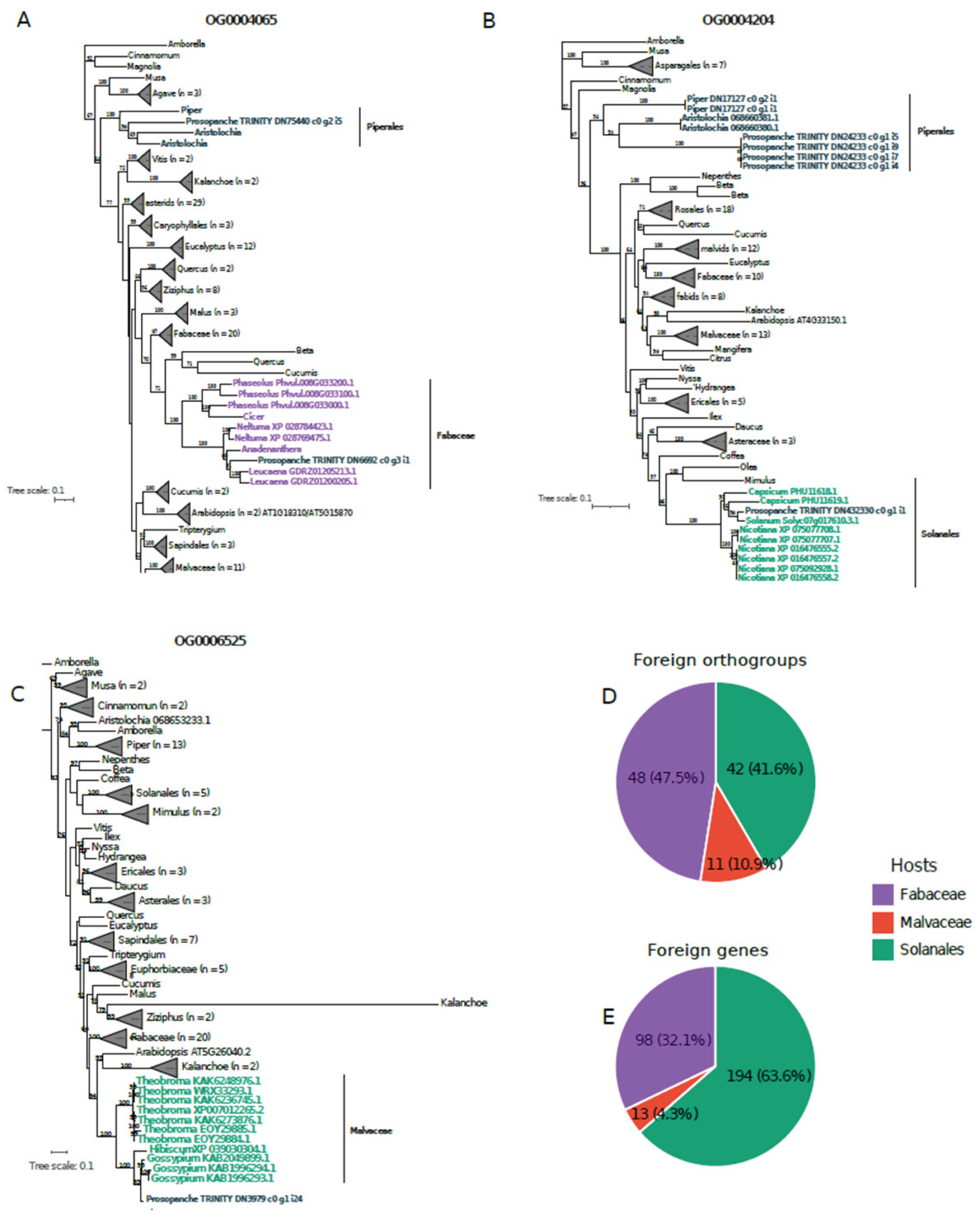
Prosopanche sequences exhibited a BLAST ‘best-hit’ to species within Fabaceae (651 OGs), Malvaceae (676 OGs), or Solanaceae (370 OGs). These groups defined the primary search space for subsequent phylogenetic validation. Following rigorous phylogenetic analyses (see Materials and Methods), we ultimately identified 101 Orthogroups exhibiting robust evidence of HGT from the three analyzed donor lineages, comprising a total of 305 horizontally acquired transcripts with bootstrap support higher than 95% (
Table S6). Fabaceae was the most represented donor lineage in terms of orthogroups (47 OGs), followed by Solanaceae (42 OGs) and Malvaceae (11 OGs) (
Figure 3). However, when accounting for the total number of individual transcripts, Solanaceae exhibited a marked increase in relative abundance. This indicates that Solanaceae-derived OGs tend to contain a higher number of transcripts with affinity to Solanaceae in
Prosopanche compared to those from other donors (
Figure 3).
3.4. Functional Analyses of Foreign Nuclear Transcripts
Of the 305 foreign genes detected in the
P. americana transcriptome, 113 were successfully annotated using one of the four methods employed, namely String, EggNOG, Mercator, or the
Arabidopsis TAIR code (
Table S6). Within the annotated fraction, Fisher’s exact test identified significantly overrepresented Gene Ontology (GO) terms (FDR < 0.05) across various categories (
Table S7). Regarding Biological Processes (BP), the highest fold enrichment values (>40) were observed for terms related to specific metabolic pathways, including “L-serine metabolic process” and “one-carbon metabolic process.” Terms associated with “photosynthesis, light harvesting,” and “organic acid biosynthetic process” were also significantly overrepresented.
In terms of Cellular Components (CC), the analysis showed a significant enrichment for categories associated with intercellular connections, specifically “plasmodesma” and “cell-cell junction.” Additionally, terms related to plastid structure, such as “plastid envelope,” were identified as significantly enriched in the HGT gene set.
Functional classification via Mercator revealed a striking correlation between the function of transferred genes and their phylogenetic origin (
Figure S6). We observed a distinct segregation in the “Photosynthesis” category (Mercator Bin 1). Remarkably, all transcripts associated with photosynthetic light reactions (e.g., Photosystems, LHCs) were exclusively derived from Solanales, an ancestral host lineage (
Figure S6, Bin 1). Conversely, the current host lineage (Fabales) contributed no genes to this category. The phylogenetic affinity of photosynthesis-related transcripts (Mercator Bin 1) with Solanales—an ancestral host lineage— and not with Neltuma (Fabaceae)-actual host- provides strong evidence against host tissue contamination during RNA extraction. The complete absence of Fabales-derived photosynthetic transcripts, in contrast to the robust presence of Solanales-derived sequences, confirms that these are endogenous genomic insertions acquired before the lineage’s host shift.
4. Discussion
4.1. Molecular Fossils of an Ancestral Generalist in the mtDNA and in the Nucleus of Prosopanche
The HGT landscape of P. americana presents a paradox: while it is currently a strict specialist of Fabaceae (Hatt et al. 2025), its genome acts as a reservoir of historical ecological interactions. We identified a complex mosaic of foreign sequences from Solanales, Malvales, Malpighiales, and Caryophyllales—lineages absent from its modern host range. The presence of 13 shared mitochondrial regions between P. americana and P. panguanensis, recovered as sister lineages in our phylogenies, confirms that these are not recent events or artifacts. Instead, they represent ancestral HGT events that predated their speciation.
Our results suggest that the Prosopanche ancestor was a generalist, similar to the extant P. bonacinae, and that many of those hosts donated both mitochondrial and nuclear DNA. The use of HGT to identify previously unknown host relationships has been documented in other holoparasites (Roulet et al. 2020; Cusimano and Renner 2019; Davis and Wurdack 2004). In the mtDNA of the holoparasite Ombrophytum subterraneum (Balanophoraceae, Santalales), HGT analysis unveiled historical interactions with hosts (e.g., Lamiaceae, Apocynaceae) not currently parasitized by the species (Roulet et al. 2020). Interestingly, the largest foreign inserts in our study originate from Solanales and Malvales, suggesting that these now-lost interactions were once central to the parasite’s biology, before the lineage narrowed its host range toward Fabaceae.
The foreign DNA was captured before the speciation event and the subsequent ecological shift toward host specialization in the P. americana lineage. Current interactions with Fabaceae have left surprisingly small mitochondrial genomic footprints (~8.5 kb in P. americana) compared to the larger tracts retained from ancestral hosts. The presence of foreign regions from different donors and the minimal impact of HGT from the current Fabaceae donor in P. americana suggest 2 scenarios. First, the transition from a generalist to a specialist (the “ecological shift”) might be a relatively recent event in the Prosopanche radiation. Alternatively, the lineage may have experienced a peak of genomic plasticity followed by a cessation of horizontal acquisitions after the speciation event and host specialization.
4.2. Comparative HGT Dynamics and Functional Landscape
Our results reinforce the idea that the transition to holoparasitism does not uniformly entail the massive acquisition of foreign DNA into the mtDNA. While some Balanophoraceae, like Lophophytum, act as a reservoir of host genes (Sanchez-Puerta et al. 2017; 2019; Gatica-Soria et al. 2025), its relative Rhopalocnemis exhibits virtually no organellar HGT (Yu et al. 2022). In Prosopanche, we observe a distinctive scenario. Despite its ancient Cretaceous origin—which theoretically offered ample time for accumulation—the mitochondrial genome exhibits remarkably low levels of HGT. This disparity suggests that lineage-specific intrinsic factors differ even among closely related Prosopanche species. While P. americana shows a restricted uptake or retention of mitochondrial DNA from its current host (Fabales, ~8.5 kb), its sister species P. panguanensis displays a comparatively higher retention of Fabales-derived sequences (~44 kb).
4.3. Functional Bias in Nuclear Retained Sequences
Regarding the functional role of these foreign genes, although functional characterization was limited by annotation availability (~40% of transferred genes), the retained foreign genes do not reflect metabolic adaptation. The enrichment in plastid-related terms (e.g., thylakoids, photosystems) is intriguing in light of the extensive convergent loss of photosynthetic genes documented in other holoparasitic plants (Cai et al. 2021; Cai 2023; Chen et al. 2020). In a non-photosynthetic parasite, this does not imply a functional “rescue” of metabolic activity. In contrast to findings in other holoparasitic lineages where HGT has been linked to the acquisition of adaptive traits (Zou et al. 2026; Yang et al. 2016) our results likely reflect the donor’s genomic architecture. Since these genes are abundant and often found in high-copy numbers in the donor’s organellar or nuclear genomes, they may simply be more likely to be captured. The retention of these sequences might be a byproduct of neutral genomic integration rather than an adaptive requirement for the holoparasitic lifestyle (Richardson and Palmer 2006).
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org; Table S1. Genbank accession numbers of the Prosopanche spp. data sets. Table S2. List of NCBI mitochondrial sequences (>4 kb) included in the custom reference databases for BLASTn searches. Table S3. BUSCO assessment of the transcriptome of Prosopanche americana. TableS4-Inventory of mitochondrial foreign sequences in Prosopanche americana and assessment of their presence in the sister species P. panguanensis. Table S5. Species included in the OrthoFinder analyses. Table S6. Functional annotation and classification of Prosopanche transcripts associated with different host plant families Table S7. Gene Ontology (GO) enrichment analysis of horizontally transferred transcripts identified in the Prosopanche americana transcriptome. Figure S1. Bioinformatic workflow for the identification and validation of Horizontal Gene Transfer (HGT) events in the nuclear transcriptome of Prosopanche americana. The pipeline follows four sequential phases. Phase 1 (Quality Control): The predicted proteome was filtered to remove transposable elements (TEs) using HMMER and non-Viridiplantae contaminants via eggNOG taxonomic annotation. Phase 2 (Orthology): Orthogroups were inferred using OrthoFinder, including Prosopanche, potential host lineages (Fabaceae, Solanaceae, Malvaceae), and outgroups. Preliminary candidates were selected based on BLASTP best-hit criteria against host families. Phase 3 (Phylogenomics): Candidates underwent rigorous phylogenetic validation. Sequences were aligned (MAFFT) and trimmed (ClipKIT) before Maximum Likelihood tree reconstruction (IQ-TREE). Trees were topologically filtered to retain only those supporting a robust Parasite-Host clustering. Phase 4 (Curation): The final set of HGT candidates was curated by removing redundancy (CD-HIT) and discarding remaining sequences functionally annotated as TE machinery. Numbers in parentheses indicate the count of sequences or candidates retained at key steps. Figure S2. Visualization of BLASTn homology searches for the Prosopanche americana mitochondrial contigs. BLASTn hits between Prosopanche and a custom angiosperm mitochondrial database are organized by taxonomic order, as described in Hatt et al. (2025). The Y-axis represents different angiosperm lineages, including potential hosts (e.g., Solanales, Fabales, Malvales) and other reference groups. Colors indicate percentage identity (pident), ranging from blue (lower identity) to red (high identity). Gray arrows below the tracks indicate the position and transcriptional direction of annotated mitochondrial genes. Note that vertical “stacks” of hits across multiple orders typically represent conserved coding regions (e.g., atp1, ccmB), while high-identity hits restricted to specific lineages suggest Horizontal Gene Transfer (HGT) events. Figure S3. Maximum likelihood (ML) phylogenetic analyses of foreign regions shared by taxa beyond the parasite and the primary host. Numbers at nodes indicate bootstrap support values. Clades corresponding to Malvales, Solanales, and Caryophyllales are highlighted in orange, green, and yellow, respectively. Figure S4. Horizontal Gene Transfer (HGT) landscape in the Prosopanche panguanensis mtDNA. Each horizontal bar represents a single contig, with the X-axis showing the position in kilobases (kb). The BLAST analysis was conducted against custom databases including host orders, the order Piperales, and all other angiosperms. Colored segments represent regions identified as high-confidence HGT candidates, where the P. panguanensis sequence showed its best BLAST hit against a specific host family (see legend). These hits met the filtering criteria: p-identity > 90 and length > 200 bp. The color key below the graph indicates the specific host family that provided the best hit for the HGT region. The light gray background of the contigs represents regions that did not meet the HGT criteria. This includes sequences that did not yield a significant BLAST hit, hits broadly against all angiosperms (e.g., “Other Angiosperms” in the full database), and hits only against the native order Piperales. Note that only contigs containing identified HGT regions are displayed; entirely native contigs are excluded from the figure. Figure S5: Phylogenetic trees of mitochondrial genes identified in the draft mitochondrial assemblies of P. bonacinae and P. panguanensis. Maximum Likelihood (ML) phylogenies were inferred using RAxML with 1,000 bootstrap replicates. For genes where only partial sequences were recovered, this status is indicated following the taxon name. These trees illustrate the phylogenetic placement of mitochondrial loci identified within the genomic assemblies of both holoparasitic species. Figure S6. Functional landscape of horizontally transferred genes in the Prosopanche americana nuclear genome. HGT candidates were annotated using the Mercator pipeline and grouped by biological pathway (Y-axis) across the three main donor lineages (X-axis). The size of each circle is proportional to the number of transcripts assigned to each category. Note the specific enrichment of photosynthesis-related genes (e.g., Photosystem subunits, Light harvesting) derived exclusively from Solanaceae, in contrast to their absence in the current host lineage (Fabaceae).
Author Contributions
Conceptualization, LG and MVSP; Methodology, LG and MVSP; Formal Analysis, LG & MER & LAG; Writing – Original Draft Preparation, LG; Writing – Review & Editing, MVS.; Funding Acquisition, MVSP & LG.
Funding
This work was supported by grants from Fondo para la Investigación Científica y Tecnológica (grant # PICT2019-03067) and Universidad Nacional de Cuyo (grant # M048-T1) to LEG.
Data Availability Statement
The genomic data generated for this study have been deposited in NCBI: Illumina DNAseq (SRRxxxxxxx).
Conflicts of Interest
“The authors declare no conflict of interest.”.
References
- Cai, Liming. Rethinking Convergence in Plant Parasitism through the Lens of Molecular and Population Genetic Processes. American Journal of Botany 2023, 110(5), e16174. [Google Scholar] [CrossRef] [PubMed]
- Cai, Liming; Arnold, Brian J.; Xi, Zhenxiang; et al. Deeply Altered Genome Architecture in the Endoparasitic Flowering Plant Sapria Himalayana Griff. (Rafflesiaceae). Current Biology 2021, 31(5), 1002–1011.e9. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, Carlos P; Hernández-Plaza, Ana; Letunic, Ivica; Bork, Peer; Huerta-Cepas, Jaime. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Molecular Biology and Evolution 2021, 38(12), 5825–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, Xiaoli; Fang, Dongming; Wu, Chenyu; et al. Comparative Plastome Analysis of Root- and Stem-Feeding Parasites of Santalales Untangle the Footprints of Feeding Mode and Lifestyle Transitions. Genome Biology and Evolution 2020, 12(1), 3663–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, Xiaoli; Fang, Dongming; Xu, Yuxing; et al. Balanophora Genomes Display Massively Convergent Evolution with Other Extreme Holoparasites and Provide Novel Insights into Parasite–Host Interactions. Nature Plants 2023, 9(10), 1627–42. [Google Scholar] [CrossRef]
- Cocucci, Alfredo. Estudios en el genero Prosopanche (Hydnoraceae); Sec. 2. (Cordoba), 1965. [Google Scholar]
- Cocucci, ALfredo.
Estudios en el genero Prosopanche II
. Organización de la Flor 1975, 8 (December), 7–15. [Google Scholar]
- Cocucci, Alfredo. Estudios En El Genero Prosopanche III, Embriologia; Sec. 9. (Cordoba), 1976. [Google Scholar]
- Cusimano, Natalie; Renner, Susanne S. Sequential Horizontal Gene Transfers from Different Hosts in a Widespread Eurasian Parasitic Plant, Cynomorium Coccineum. American Journal of Botany 2019, 106(5), 679–89. [Google Scholar] [CrossRef]
- Davis, Charles C.; Wurdack, Kenneth J. Host-to-Parasite Gene Transfer in Flowering Plants: Phylogenetic Evidence from Malpighiales. Science 2004, 305(5684), 676–78. [Google Scholar] [CrossRef]
- Davis, Charles C; Xi, Zhenxiang. Horizontal Gene Transfer in Parasitic Plants. Current Opinion in Plant Biology 2015, 26 (August), 14–19. [Google Scholar] [CrossRef]
- Emms, David M.; Kelly, Steven. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biology 2019, 20(1), 238. [Google Scholar] [CrossRef]
- Feng, Yanlei; Wicke, Susann. Systemic Organellar Genome Reconfiguration along the Parasitic Continuum in the Broomrape Family (Orobanchaceae). Plant and Cell Physiology 2025, pcaf131. [Google Scholar] [CrossRef]
- Garcia, Laura E.; Edera, Alejandro A.; Palmer, Jeffrey D.; Sato, Hector; Sanchez-Puerta, M. Virginia. Horizontal Gene Transfers Dominate the Functional Mitochondrial Gene Space of a Holoparasitic Plant. New Phytologist 2021, 229(3), 1701–14. [Google Scholar] [CrossRef] [PubMed]
- Gatica-Soria, Leonardo Martin; Roulet, M. Emilia; Tulle, Walter D.; Sato, Hector A.; Barrandeguy, M. Eugenia; Sanchez-Puerta, M. Virginia. Highly Variable Mitochondrial Chromosome Content in a Holoparasitic Plant Due to Recurrent Gains of Foreign Circular DNA. Physiologia Plantarum 2025, 177(2). [Google Scholar] [CrossRef] [PubMed]
- Góralski, Grzegorz; Denysenko-Bennett, Magdalena; Burda, Anna; Staszecka-Moskal, Natalia; Kwolek, Dagmara. Achievements in Horizontal Gene Transfer Studies in Parasitic Plants. Acta Biologica Cracoviensia s. Botanica 2022, 17–28. [Google Scholar] [CrossRef]
- Grabherr, Manfred G; Haas, Brian J; Yassour, Moran; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nature Biotechnology 2011, 29(7), 644–52. [Google Scholar] [CrossRef]
- Hatt, Sebastian A; Grace, Olwen M; Zuntini, Alexandre R; Cameron, Duncan D; Thorogood, Chris J. Parasitic Plants Show Striking Convergence in Host Preference across Angiosperm Lineages. Annals of Botany 2025, 135(6), 1135–46. [Google Scholar] [CrossRef]
- Hatt, Sebastian A; Thorogood, Chris J; Bolin, Jay F; Musselman, Lytton J; Cameron, Duncan D; Grace, Olwen M. A Taxonomic Revision of the Genus Hydnora (Hydnoraceae) 2022.
- Jin, Jian-Jun; Yu, Wen-Bin; Yang, Jun-Bo; et al. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biology 2020, 21(1), 241. [Google Scholar] [CrossRef]
- Larsson, Anders. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30(22), 3276–78. [Google Scholar] [CrossRef]
- Letunic, Ivica; Bork, Peer. Interactive Tree of Life (iTOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Research 2024, 52(W1), W78–82. [Google Scholar] [CrossRef]
- Martel, Carlos; Fernandez-Hilario, Robin; TELLO, JUAN; Arteaga, Robert; Gerlach, Günter. Prosopanche Panguanensis (Aristolochiaceae), a New Species from Central Peru. Phytotaxa 2018, 364 (August), 241. [Google Scholar] [CrossRef]
- Mower, Jeffrey P; Stefanović, Saša; Hao, Weilong; et al. Horizontal Acquisition of Multiple Mitochondrial Genes from a Parasitic Plant Followed by Gene Conversion with Host Mitochondrial Genes. BMC Biology 2010, 8(1), 150. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Lam-Tung; Schmidt, Heiko A.; Von Haeseler, Arndt; Minh, Bui Quang. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 2015, 32(1), 268–74. [Google Scholar] [CrossRef] [PubMed]
- Nickrent, Daniel L. Parasitic Angiosperms: How Often and How Many? TAXON 2020, 69(1), 5–27. [Google Scholar] [CrossRef]
- Phanstiel, Douglas H.; Boyle, Alan P.; Araya, Carlos L.; Snyder, Michael P. Sushi.R: Flexible, Quantitative and Integrative Genomic Visualizations for Publication-Quality Multi-Panel Figures. Bioinformatics 2014, 30(19), 2808–10. [Google Scholar] [CrossRef]
- Potter, Simon C; Luciani, Aurélien; Eddy, Sean R; Park, Youngmi; Lopez, Rodrigo; Finn, Robert D. HMMER Web Server: 2018 Update. Nucleic Acids Research 2018, 46(W1), W200–204. [Google Scholar] [CrossRef]
- Richardson, A. O.; Palmer, J. D. Horizontal Gene Transfer in Plants. Journal of Experimental Botany 2006, 58(1), 1–9. [Google Scholar] [CrossRef]
- Roulet, M. Emilia; Garcia, Laura E.; Gandini, Carolina L.; Sato, Hector; Ponce, Gabriela; Sanchez-Puerta, M. Virginia. Multichromosomal Structure and Foreign Tracts in the Ombrophytum Subterraneum (Balanophoraceae) Mitochondrial Genome. Plant Molecular Biology 2020, 103(6), 623–38. [Google Scholar] [CrossRef]
- Sanchez-Puerta, M Virginia; Ceriotti, Luis F; Gatica-Soria, Leonardo M; Roulet, M Emilia; Garcia, Laura E; Sato, Hector A. Invited Review Beyond Parasitic Convergence: Unravelling the Evolution of the Organellar Genomes in Holoparasites. Annals of Botany 2023, 132(5), 909–28. [Google Scholar] [CrossRef]
- Sanchez-Puerta, M. Virginia; Edera, Alejandro; Gandini, Carolina L.; et al. Genome-Scale Transfer of Mitochondrial DNA from Legume Hosts to the Holoparasite Lophophytum Mirabile (Balanophoraceae). Molecular Phylogenetics and Evolution 2019, 132 (March), 243–50. [Google Scholar] [CrossRef]
- Sanchez-Puerta, M. Virginia; García, Laura E.; Wohlfeiler, Josefina; Ceriotti, Luis F. Unparalleled Replacement of Native Mitochondrial Genes by Foreign Homologs in a Holoparasitic Plant. The New Phytologist (England) 2017, 214(1), 376–87. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, Andrea; Rocamundi, Nicolás; Ferrer, M.; Baranzelli, Matias; Marvaldi, Adriana. Unveiling the History of a Peculiar Weevil-Plant Interaction in South America: A Phylogeographic Approach to Hydnorobius Hydnorae (Belidae) Associated with Prosopanche Americana (Aristolochiaceae). Diversity 2018, 10(2), 33. [Google Scholar] [CrossRef]
- Steenwyk, Jacob L.; Buida, Thomas J.; Li, Yuanning; Shen, Xing-Xing; Rokas, Antonis. ClipKIT: A Multiple Sequence Alignment Trimming Software for Accurate Phylogenomic Inference. PLOS Biology 2020, 18(12), e3001007. [Google Scholar] [CrossRef] [PubMed]
- Sun, Guiling; Xu, Yuxing; Liu, Hui; et al. Large-Scale Gene Losses Underlie the Genome Evolution of Parasitic Plant Cuscuta Australis. Nature Communications 2018, 9(1), 2683. [Google Scholar] [CrossRef]
- Svetlikova, Petra; Su, Huei-Jiun; Suetsugu, Kenji; Husnik, Filip. Phylogenomics Clarifies Balanophora Evolution, Metabolic Retention in Reduced Plastids, and the Origins of Obligate Agamospermy. New Phytologist 2025, nph.70761. [Google Scholar] [CrossRef]
- Teixeira-Costa, Luiza. A Living Bridge between Two Enemies: Haustorium Structure and Evolution across Parasitic Flowering Plants. Brazilian Journal of Botany 2021, 44(1), 165–78. [Google Scholar] [CrossRef]
- Vogel, Alexander; Schwacke, Rainer; Denton, Alisandra K.; et al. Footprints of Parasitism in the Genome of the Parasitic Flowering Plant Cuscuta Campestris. Nature Communications 2018, 9(1), 2515. [Google Scholar] [CrossRef]
- Waterhouse, Robert M.; Seppey, Mathieu; Simao, Felipe A.; et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Molecular Biology and Evolution 2018, 35(3), 543–48. [Google Scholar] [CrossRef]
- Wick, Ryan R.; Schultz, Mark B.; Zobel, Justin; Holt, Kathryn E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31(20), 3350–52. [Google Scholar] [CrossRef]
- Xi, Zhenxiang; Wang, Yuguo; Bradley, Robert K.; et al. Massive Mitochondrial Gene Transfer in a Parasitic Flowering Plant Clade. PLoS Genetics 2013, 9(2), e1003265. [Google Scholar] [CrossRef]
- Yang, Zhenzhen; Wafula, Eric K.; Kim, Gunjune; et al. Convergent Horizontal Gene Transfer and Cross-Talk of Mobile Nucleic Acids in Parasitic Plants. Nature Plants 2019, 5(9), 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Yang, Zhenzhen; Zhang, Yeting; Wafula, Eric K.; et al. Horizontal Gene Transfer Is More Frequent with Increased Heterotrophy and Contributes to Parasite Adaptation. Proceedings of the National Academy of Sciences 2016, 113(45). [Google Scholar] [CrossRef]
- Yu, Runxian; Chen, Xudong; Long, Lingjie; et al. De Novo Assembly and Comparative Analyses of Mitochondrial Genomes in Piperales. Genome Biology and Evolution 2023, 15(3), evad041. [Google Scholar] [CrossRef]
- Yu, Runxian; Sun, Chenyu; Zhong, Yan; et al. The Minicircular and Extremely Heteroplasmic Mitogenome of the Holoparasitic Plant Rhopalocnemis Phalloides. Current Biology 2022, 32(2), 470–479.e5. [Google Scholar] [CrossRef]
- Zeng, Ying; Yang, Tao. RNA Isolation from Highly Viscous Samples Rich in Polyphenols and Polysaccharides. Plant Molecular Biology Reporter 2002, 20(4), 417–417. [Google Scholar] [CrossRef]
- Zou, Rong; Huang, Jian; Xie, Hong; et al. A Chromosome-Level Genome Assembly of Cistanche Deserticola Provides Insights into Its Evolution and Molecular Mechanisms of Parasitism. Plant Communications 2026, 7(1), 101581. [Google Scholar] [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |