Submitted:
10 February 2026
Posted:
11 February 2026
You are already at the latest version
Abstract
After following up of more than 9 years, we report three affected male siblings from a consanguineous family (PKSOK) in rural Baluchistan, Pakistan, presenting with a novel, levodopa/carbidopa-responsive Nocturnal Akinesia Syndrome (NAS). Clinical features include nocturnal hypotonia, developmental delay, mild intellectual disability, speech impairment, and psychomotor delay without classical metabolic crises or spasticity. Exome sequencing identified a predicted pathogenic homozygous missense variant [c.313G>A; p.(Gly105Ser)] in the Isovaleryl-CoA dehydrogenase (IVD) gene, traditionally associated with Isovaleric Acidemia (IVA). However, the clinical phenotype of affected individuals of family PKSOK deviates from classical IVA, exhibiting unique levodopa-responsive motor symptoms rather than metabolic decompensation. Protein-protein interaction analyses predicted IVD interactions with dopamine receptors DRD1 and DRD4, suggesting a functional link between metabolic enzymes and dopaminergic neurotransmission and circadian rhythm. The potential involvement of GABAergic pathways may further explain the motor phenotype and therapeutic responsiveness. This study expands the phenotypic spectrum of IVD variants, highlighting new mechanisms underlying movement disorders and offering important implications for diagnosis and targeted treatment in similar rare neurometabolic syndromes.
Keywords:
Nocturnal Akinesia Syndrome
; IVD
; Isovaleric Acidemia
; Isovaleryl-CoA dehydrogenase
; Levo Dopa
1. Introduction
Genetic diseases continue to challenge the human mankind by imparting distress and death in many especially in the developing world[1]. The research in genetic diseases however continues to be scarce as compared to the various communicable and non-communicable diseases. Similarly, the awareness and management of genetic disease remain dismal and even among the healthcare providers the knowledge remains rudimentary[2].
In 2015, a family from remote rural area of the geographically largest Baluchistan province of Pakistan was reported in the local media with peculiar clinical phenotype. Two affected members of the family reportedly were suffering from a flaccid paralysis only during the night while the sufferers were functional during the daytime. At that time, the third sibling was younger than one year and had not been diagnosed with the disease. A medical board evaluated the patients and found that the disease does not fit into any already known syndrome listed in Online Mendelian Inheritance in Man (OMIM) database. Due to their unique symptoms and daytime normalcy, the siblings were named the “SOLAR KIDS” in the social media.
To characterize this apparently new rare clinical entity, after institutional review board (IRB) approval, we conducted comprehensive clinical and genetic investigations, including clinical, biochemical, radiological studies, and family were followed up for more than 9 years. Results are these studies are presented here, which we expect will help in providing an insight into the clinical phenotype, as well as molecular basis of this rare syndrome involving Isovaleryl-CoA dehydrogenase (IVD) variant.
2. Material and Methods
2.1. Family Enrolment & Clinical Investigation
After institutional board review and approval, family PKSOK segregating nocturnal paralysis and mild intellectual disability (ID) was ascertained from the rural area of Baluchistan province of Pakistan. The clinical diagnosis was made through a battery of neurological, morphological, behavioral, ophthalmological, auditory, skeletal, and dermatological assessments. Front and side-pose facial photographs were taken for different morbidities. Peripheral blood samples were collected from the affected individuals, parents, normal siblings and all available relatives. Written informed consent was obtained from each participant, including the guardian of affected individuals/minors.
2.2. Exome Sequencing (ES)
ES was performed at the University of Maryland, Baltimore, USA as previously described[3]. Briefly, genomic DNA samples from two affected individuals were used to perform high-throughput sequencing and exome enrichment. Exome enrichment was performed by using Nimblegen SeqCap EX Exome v2.0 Library (Roche Diagnostics, San Francisco, CA), followed by generating approximately 30 million paired end reads with one hundred base pair Length. The Illumina HiSeq 4000 platform (Illumina, Inc. San Diego, CA) was used to perform massive parallel sequencing. Raw Data was analyzed by using GATK (the Broad Institute’s Genome Analysis Toolkit, Broad Institute, Cambridge, MA, USA) and the Illumina Chastity Filter with the Burrows Wheeler Aligner (BWA) (Broad Institute) was used for the alignment of reads. GATK was used to call the variant sites and the variant quality score recalibration method was used to filter the single nucleotide variant (SNV) calls. To prioritize the most relevant candidate variant from ES data, an in-house customized pipeline was used[4,5]. Sanger sequencing was performed to confirm the variant segregation among all the affected and non-affected individuals of the family.
2.3. Segregation Analysis
The online primer designing tool “Primer3 (v. 0.4.0)” (https://bioinfo.ut.ee/primer3-0.4.0/) was used to design the primers. For the amplification of identified variants, 2X Econotaq DNA Polymerase (Bioresearch Technologies, Radnor, PA, USA) was used. After PCR amplification of candidate genes, 3µl of PCR product is loaded on the 2% agarose gel containing EtBr. The size of amplified bands was determined by running 100bp DNA Ladder (Gene Ruler, Thermo Scientific INC., USA) in parallel well. ExoSap was performed followed by Sanger sequencing PCR and precipitation to process and analyze samples using ABI sequencer. Chromatograms generated were analyzed using SeqMan Pro by DNASTAR.
2.4. In-Silico Analysis
Several online available pathogenicity prediction tools and webservers, including MutationTaster (https://www.mutationtaster.org/), Varsome (https://varsome.com/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), PhyloP (http://compgen.cshl.edu/phastweb/), SIFT (https://sift.bii.a-star.edu.sg/), Marrvel (https://marrvel.org/), FATHMM (https://fathmm.biocompute.org.uk/), and GERP++ (https://github.com/tvkent/GERPplusplus), were used to assess the pathogenicity scores of IVD identified variant. Clustal Omega (http://www.clustal.org/omega/) was used for evolutionary conversation of mutated residue, while MetaDome (https://stuart.radboudumc.nl/metadome/) interface helped to assess the intolerance score. Furthermore, we performed string analysis and used gene-mania to identify the interacting partners of IVD protein. We also used Wiki Pathways (https://www.wikipathways.org/) to identify the IVD protein participating pathways. PyMol (TM) v 2.3.4 and HOPE (https://www3.cmbi.umcn.nl/hope/), were used to perform the 3D modelling of wild type and mutated residue to compare the effect of amino acid change. Further, to study the impact on interaction with other proteins ClusPro (https://cluspro.org/) and PyMOL (TM) v 2.3.4 were used.
3. Results
3.1. Clinical Assessment
As part of our ongoing studies on neurodevelopmental disorders, we came across an unusual form of neuromuscular weakness in three affected individuals of Pakistani kindred (Figure 1). A medical board comprising of physicians, neurologists, clinical and molecular geneticists thoroughly reviewed these patients, their clinical scenario has been summarized in Table 1. Three male children of a consanguineous family, aged 9 to 21 years, presented with nocturnal Akinesis and rigidity of all the limbs and truncal muscles (Figure 1B-C). Examination of the cranial nerves revealed partial loss of motor function. They remained well-oriented, had a normal sensory system, and tendon reflexes, during the ordeal as well as during the daytime. Muscle strength, limb movements and muscle tone reverted to being normal during the day or after oral Levodopa/Carbidopa even at night. The ailment starts during the first year of life characterized by a state of listlessness in the evening. The patients could not walk until after 2 years of age. The weakness involved axial and limb muscles; speech would get slow, but mentation remained normal. Axial weakness was more pronounced, and patients were unable to sit or hold their necks. The power of limb muscles was globally reduced to 3/5. The tone was normal, deep tendon reflexes were 2+ and plantar responses were down going bilaterally.
These three siblings are intellectually challenged. There is no history of diplopia, difficulty in swallowing, involuntary movements or birth asphyxia. At night, kids were dependent on the care takers for feeding and defecation. There is no history of seizures, ataxia and fecal or urinary incontinence. The weakness has been progressively worsening over the years, and the elderly is the most affected. There was no evidence of ptosis, extraocular ophthalmoplegia or palatal paralysis. Kids were afebrile; however, extremities were warm and sweaty. There was no evidence of respiratory distress and abdominal and cardiovascular examination was unremarkable. A pre- and post-exercise electromyography (EMG) conducted in the morning was normal and showed no decremental response on repetitive nerve stimulation. General physical examination was normal, and no apparent abnormalities of other organs were noted. Other investigations including blood count, serum biochemistry, nerve conduction studies (NCS), computed tomography (CT), and magnetic resonance imaging (MRI) were normal. Anticholinesterase antibodies, and antinuclear antibodies (ANA) tests were negative, while Tensilon test also did not show any improvement. However, administration of low dose of Levo Dopa resulted in the improvement of symptoms. The differential diagnosis included Hereditary dystonia (MIM#128100), Myasthenia gravis (MIM#254210), and Periodic paralysis (MIM#170400) including hypokalemic periodic paralysis. A thorough review of the literature, and OMIM database revealed that the spectrum of the phenotype observed in family PKSOK affected individuals did not fit into any defined clinical entity, therefore based on the clinical manifestations, we named this rare disorder as Nocturnal Akinesis Syndrome (NAS).
3.2. A Novel Biallelic Missense Variant in IVD Gene Segregates with NAS Phenotype
Next, we aimed to identify the causal genetic variant that underlies NAS. Through a systematic approach, ES sequencing followed by segregation analysis via Sanger revealed homozygous c.304G>A; p.(Gly105Ser) variant in exon 4 of IVD gene (NM_002225.5) in the three affected males, while both parents were obligated carrier, and normal siblings as well as other family members were either heterozygous or had homozygous wild type allele (Figure 1A). IVD gene has 13 alternatively spliced exons (Figure 2A), resulting in seven different transcripts. The c.304G>A variant identified in family PKSOK is located in the canonical exon of IVD. We did not find the same variant in the 200 ethnically matched control samples, and only six carriers are listed in gnomAD with allele frequency of 3.18x10-05.
Variants in IVD gene are primarily associated with the autosomal recessive disorder isovaleric acidemia (IVA)[6,7]. IVA is a metabolic disorder caused by a deficiency of the enzyme isovaleryl-CoA dehydrogenase, which leads to the accumulation of isovaleric acid, resulting in toxic effects such as metabolic acidosis, vomiting, neurological damage, and, if untreated, severe health complications. This disorder typically presents in the neonatal period or later with episodic symptoms and requires early diagnosis and management to prevent serious outcomes. However, none of our diseased individuals showed these symptoms, making this c.304G>A change even more unique for further analysis.
The c.304G>A (p.Gly105Ser) variant in IVD has been predicted to be deleterious by various in-silico tools (Table 2). Meta Dome intolerance scale analysis shows that p.(Gly105Ser) is located in the highly intolerant region of IVD. Missense Tolerance Ratio (MTR), a measure of regional intolerance to missense variation, analysis calculated a score of 0.854 for the identified variant, which was consistent with location of evolutionary conserved p.Gly105 residue within highly intolerant region of acetyl-CoA dehydrogenase domain of IVD (Figure 2B-D). The amino-terminal acetyl-CoA dehydrogenase domain is crucial for the enzymatic activity, while the neighboring acyl-CoA dehydrogenase middle domain, known to be involved in substrate binding[8]. Using GeneMANIA (https://genemania.org/) and STRING (https://string-db.org/) analyses, we predicted that the IVD gene interacts with several genes implicated in neurological and musculoskeletal conditions. These include PCCB, associated with neurological disorders; DPT, linked to osteoarthritis and muscular dystrophy; and ECHS1, involved in neurodegenerative diseases. Additionally, IVD shows connectivity with dopamine receptor genes DRD1 and DRD4, which have been associated with personality traits and ADHD (Figure 2E). Pathway analysis further indicated a potential role of IVD in GABA uptake (Figure 2F), prompting us to focus more closely on its interactions with DRD1 and DRD4.
3.3. The p.Gly105Ser Variant Is Predicted to Alter the Secondary Structure and Interactions of IVD with Dopamine Receptors
To gain further insights on the potential impact of identified variant on protein structure, we performed 3-dimensional (3D) protein modelling using available human IVD protein structure (PDB:1ivh), Pymol and HOPE. The p.Gly105 residue is predicted to form two hydrogen bonds, one with p.Glu108 (bond length 2.9Å) and the other with external protein (2.7Å), respectively. Substitution of glycine with serine at position 105 alters this pattern, resulting in three predicted hydrogen bonds, all with Glu108 (2.6 Å, 2.9 Å, and 3.5 Å, Figure 3A). The wild-type glycine residue, the most flexible of all residues, is located on the surface interface of the encoded protein. In contrast, the p.Gly105Ser substitution is predicted to impact the folding due to the larger size and interactions with other molecules.
Next, we analyzed protein-protein interactions of IVD with DRD1 and DRD4 (Figure 3B-C), known dopamine receptor binding partners involved in signaling pathways and circadian rhythm[9,10]. These interactions suggest potential functional crosstalk between IVD and dopaminergic signaling[11]. A total of 34 polar contacts, including hydrogen bonds and ionic contacts, were identified manually using PyMOL between IVD and DRD1 (Figure 3B). While only 5 polar contacts were identified between IVD and DRD4 (Figure 3C). The bond distances and amino acid residues that make up the docking interface are shown in the supplementary Table 1. All the bond distances were in the acceptable range and are representative of strong binding interaction. In view of the increased number of polar contacts and acceptable bond distances, the protein-protein complex was deemed stable. There could be a lot of hydrophobic interaction among residues of both proteins making the interaction between proteins more stable and stronger. Moreover, there was no unfavorable binding interaction within the docking interface which also contributes to the overall stability of the complex. However, when glycine is replaced by serine (p.Gly105Ser) only 12 polar contacts were identified between IVD and DRD1, and 22 interactions were lost. In contrast, the mutant IVD-DRD4 complex has a greater number of polar contacts, with 14 interactions observed, with overall less favorable energy scores, which makes it less stable despite more polar connections. Overall, the in-silico analysis suggests that the p.Gly105Ser variant, segregating with NAS in family PKSOK, likely has damaging impact on the protein secondary structure and affects the stability as well as interaction of IVD with other proteins.
4. Discussion
Recently, significant progress has been made in the identification of genetics of neuromuscular disorders. Yet many neurogenetic disorders remain uncharacterized. Here, we present a novel, levodopa/carbidopa-responsive Nocturnal Akinesia Syndrome (NAS), with salient clinical features include nocturnal hypotonia, developmental delay, mild intellectual disability, speech impairment, and psychomotor delay without classical metabolic crises or spasticity. A list of differentials included congenital myasthenia syndromes (OMIM# 601462, 605809, 616324, 615120, 610542, 614750, 616720, 616224, 618198), hereditary dystonia’s (OMIM# 128101, 224500, 128101, 607488, 612067), and periodic paralysis (OMIM# 170400, 613345, 170500, 170390) syndromes. Congenital myasthenia syndromes are a group of neuromuscular disorders characterized by fatiguability. Muscle weakness improves with rest and worsens with activity. Major findings in the neonatal period may include feeding difficulties, poor suck and cry, choking spells, eyelid ptosis, and facial, bulbar, and generalized weakness. Individuals with onset later in childhood show abnormal muscle fatigability, with difficulty in running or climbing stairs[12]. Almost 50% of cases of congenital myasthenia result from mutation in a single gene CHRNE[17]. Overall, mutations in 12 genes have been implicated in the causation of congenital myasthenia and all except one have autosomal recessive inheritance pattern[12]. In our patients, fatigable weakness suggests congenital myasthenia. However, dramatic diurnal variation, sparing of ocular and facial muscles and repeatedly normal EMG negate the possibility of congenital myasthenia.
Some hereditary dystonia’s also show marked diurnal variations. Segawa syndrome, for example, is characterized by dystonia’s that in some cases are pronounced in the evening and almost nonexistent in the morning. This disease is characterized by extrapyramidal symptoms. Tone of muscles in increased and can be of cog wheel type. A detailed examination of our patients showed no abnormality of muscle tone and therefore the diagnosis of hereditary dystonia is not likely. Juvenile paralysis agitans is another disorder that presents with early onset of extrapyramidal symptoms and shows improvement with L-dopa[13]. Symptoms usually begin in teens or earlier, however Parkinsons like features are predominant and the degree of diurnal variation seen in our patients is not reported in these cases. The illness of affected kids of this family from remote Baluchistan does not fall into any defined syndrome and poses a diagnostic as well as scientific challenge.
Based on the genetic IVD variant identified in family PKSOK, the differential also includes IVA (OMIM: 243500), a rare autosomal recessive disorder, with symptoms including feeding intolerance, developmental delay, lethargy, hypotonia, seizures, metabolic acidosis, pancytopenia and a characteristic “sweaty feet” odor. In contrast, the three cases reported here exhibit a novel NAS syndrome distinguished by levodopa-responsive nocturnal akinesia without classical intoxication crises or metabolic signs usually seen in IVA. Unlike typical IVA, the clinical presentation includes developmental and speech delays with mild intellectual disability but no seizures or acute metabolic symptoms. This indicates a phenotypic expansion of IVD-related disorders beyond metabolic dysfunction.
Previously, several population specific missense variants of IVS have been functional evaluated. Based on the severity, the evaluated variants were classified into three categories. For instance, the p.Ala314Val, p.Ser281Gly, p.Phe382Val, and p.Glu411Lys led to a severe loss of function, with enzyme activity remaining below 20%, while the p.Arg53Pro and p.Arg53Cys exhibited reduced enzyme activity to a lesser extent but had lower expression levels[14]. The p.Ala300Val variant harboring protein displayed enzyme activity and expression levels closest to the WT, with relatively mild symptoms in cases, including vomiting and tachypnea[14]. Based on our in silico analysis, we anticipate that the p.(Gly105Ser) variant associated with NAS may impair enzymatic activity differently or affect new functional domains, as demonstrated by protein-protein interaction analysis of IVD protein with DRD1 and DRD4 (dopamine receptors) through multiple polar contacts. Our in silico analysis provides a plausible mechanistic link between the metabolic enzyme and dopaminergic signaling pathways, potentially explaining the positive response to dopaminergic therapy in these patients. Further functional studies are warranted to elucidate the exact impact of the p.(Gly105Ser) variant on enzyme activity and dopaminergic receptor interactions.
Additionally, we assume GABAergic neurotransmission may also play an important role in the pathophysiology of NAS through valproic acid pathway (Figure 2E). GABA, the primary inhibitory neurotransmitter in the basal ganglia, is known to modulate motor control and is implicated in levodopa-responsive movement disorders[15,16]. Elevated GABA levels have been associated with gait disturbances and akinetic symptoms, while altered GABA receptor function contributes to motor dysfunction in Parkinsonian syndromes and related disorders[17,18].The interplay between dopaminergic and GABAergic systems and involvement of circadian rhythm genes could explain the nocturnal akinesis and its responsiveness to levodopa therapy observed in these patients. This aligns with emerging evidence suggesting that non-dopaminergic pathways, including GABAergic circuits, significantly influence motor symptoms and treatment outcomes in movement disorders[19].
Clinically, the nocturnal predominance of akinetic symptoms and responsiveness to Levodopa/Carbidopa therapy distinguishes NAS from other pediatric movement disorders and dystonia’s. The absence of classical signs like spasticity, epilepsy, or cerebellar ataxia further supports this uniqueness. The mild cognitive impairment and behavioral problems likely represent downstream consequences or comorbidities of the primary dysfunction. Our findings underscore the importance of comprehensive genetic and biochemical workup in unexplained movement disorders, especially in consanguineous populations where rare autosomal recessive conditions are more prevalent. The discovery of the IVD variant expands the genotype-phenotype correlations of this gene and advocates for consideration of dopaminergic therapy in similar cases. Longitudinal follow-up of these patients will provide insights into disease progression and long-term treatment efficacy. Broader screening of other patients with nocturnal akinesis or levodopa-responsive syndromes may identify additional cases and facilitate therapeutic strategies.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org.
References
- Verma, I.C.; Puri, R.D. Global burden of genetic disease and the role of genetic screening. In Seminars in fetal and neonatal medicine; Elsevier, 2015; pp. 354–363. [Google Scholar]
- Krakow, M.; Ratcliff, C.L.; Hesse, B.W.; Greenberg-Worisek, A.J. Assessing genetic literacy awareness and knowledge gaps in the US population: Results from the Health Information National Trends Survey. Public Health Genomics 2018, 20, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Usmani, M.A.; Ahmed, Z.M.; Magini, P.; Pienkowski, V.M.; Rasmussen, K.J.; Hernan, R.; et al. De novo and bi-allelic variants in AP1G1 cause neurodevelopmental disorder with developmental delay, intellectual disability, and epilepsy. The American Journal of Human Genetics 2021, 108, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Ghaffar, A.; Rasheed, F.; Rashid, M.; van Bokhoven, H.; Ahmed, Z.M.; Riazuddin, S.; et al. Biallelic in-frame deletion of SOX4 is associated with developmental delay, hypotonia and intellectual disability. European Journal of Human Genetics 2022, 30, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.; Vockley, J.; Willard, J.M.; Huey, J.C.; Sass, J.O.; Edland, S.D.; et al. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. The American Journal of Human Genetics 2004, 75, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Parimoo, B.; Tanaka, K. Molecular characterization of four different classes of mutations in the isovaleryl-CoA dehydrogenase gene responsible for isovaleric acidemia. Am J Hum Genet. 1991, 49, 147. [Google Scholar]
- Swigoňová, Z.; Mohsen, A.W.; Vockley, J. Acyl-CoA dehydrogenases: Dynamic history of protein family evolution. J Mol Evol. 2009, 69, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.R.; Chaurasia, S.S.; Hwang, C.K.; Michael Iuvone, P. Dopamine D4 receptor activation controls circadian timing of the adenylyl cyclase 1/cyclic AMP signaling system in mouse retina. European Journal of Neuroscience 2011, 34, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Grippo, R.M.; Purohit, A.M.; Zhang, Q.; Zweifel, L.S.; Güler, A.D. Direct midbrain dopamine input to the suprachiasmatic nucleus accelerates circadian entrainment. Curr. Biol. 2017, 27, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Cruz, A.; Salinas-Jazmín, N.; Valdés-Rives, A.; Velasco-Velázquez, M.A. DRD1 and DRD4 are differentially expressed in breast tumors and breast cancer stem cells: Pharmacological implications. Transl Cancer Res. 2022, 11, 3941. [Google Scholar] [CrossRef] [PubMed]
- Abicht, A.; Müller, J.S.; Lochmüller, H. Congenital myasthenic syndromes overview. 2021. [Google Scholar]
- Abicht, A.; Stucka, R.; Schmidt, C.; Briguet, A.; HoÈpfner, S.; Song, I.; et al. A newly identified chromosomal microdeletion and an N-box mutation of the AChRϵ gene cause a congenital myasthenic syndrome. Brain 2002, 125, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.; Knopp, W. Hereditary Parkinsonism-dystonia with sustained control by L-DOPA and anticholinergic medication. Adv Neurol. 1976, 14, 201–213. [Google Scholar] [PubMed]
- Ju, K.; Bai, F.; Xu, Y.; Li, Q.; Su, G.; Jin, Y.; et al. Structural Insights into Isovaleryl-Coenzyme A Dehydrogenase: Mechanisms of Substrate Specificity and Implications of Isovaleric Acidemia-Associated Mutations. Research 2025, 8, 0661. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Chinea, P.; Bezard, E. Basal ganglia circuits underlying the pathophysiology of levodopa-induced dyskinesia. Front Neuroanat. 2010, 4, 131. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.; Sivam, S.P. Dopamine and GABA Interaction in Basal Ganglia: GABA-A or GABA-B Receptor Stimulation Attenuates L-DOPA-Induced Striatal and Nigral ERK1/2 Signaling in a Rat Model of Parkinson’s Disease. J Behav Brain Sci. 2013, 3, 479–488. [Google Scholar] [CrossRef]
- Alharbi, B.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Elekhnawy, E.; Alharbi, H.; Alexiou, A.; et al. Role of GABA pathway in motor and non-motor symptoms in Parkinson’s disease: A bidirectional circuit. Eur J Med Res. 2024, 29, 205. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman Tuura, R.L.; Baumann, C.R.; Baumann-Vogel, H. Beyond dopamine: GABA, glutamate, and the axial symptoms of Parkinson disease. Front Neurol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A Novel biallelic missense variant in IVD gene segregates in the family. (A) Family structure of rural family. Double horizontal lines represent the consanguineous marriages, affected are shown by filled symbols. Genotypes for the identified IVD variant are given for the participating individuals. (B) Pictures of two affected male siblings at their first diagnosis in 2016. (C) Pictures of two affected male siblings along with their 3rd sibling in recent follow up visit.
Figure 1.
A Novel biallelic missense variant in IVD gene segregates in the family. (A) Family structure of rural family. Double horizontal lines represent the consanguineous marriages, affected are shown by filled symbols. Genotypes for the identified IVD variant are given for the participating individuals. (B) Pictures of two affected male siblings at their first diagnosis in 2016. (C) Pictures of two affected male siblings along with their 3rd sibling in recent follow up visit.
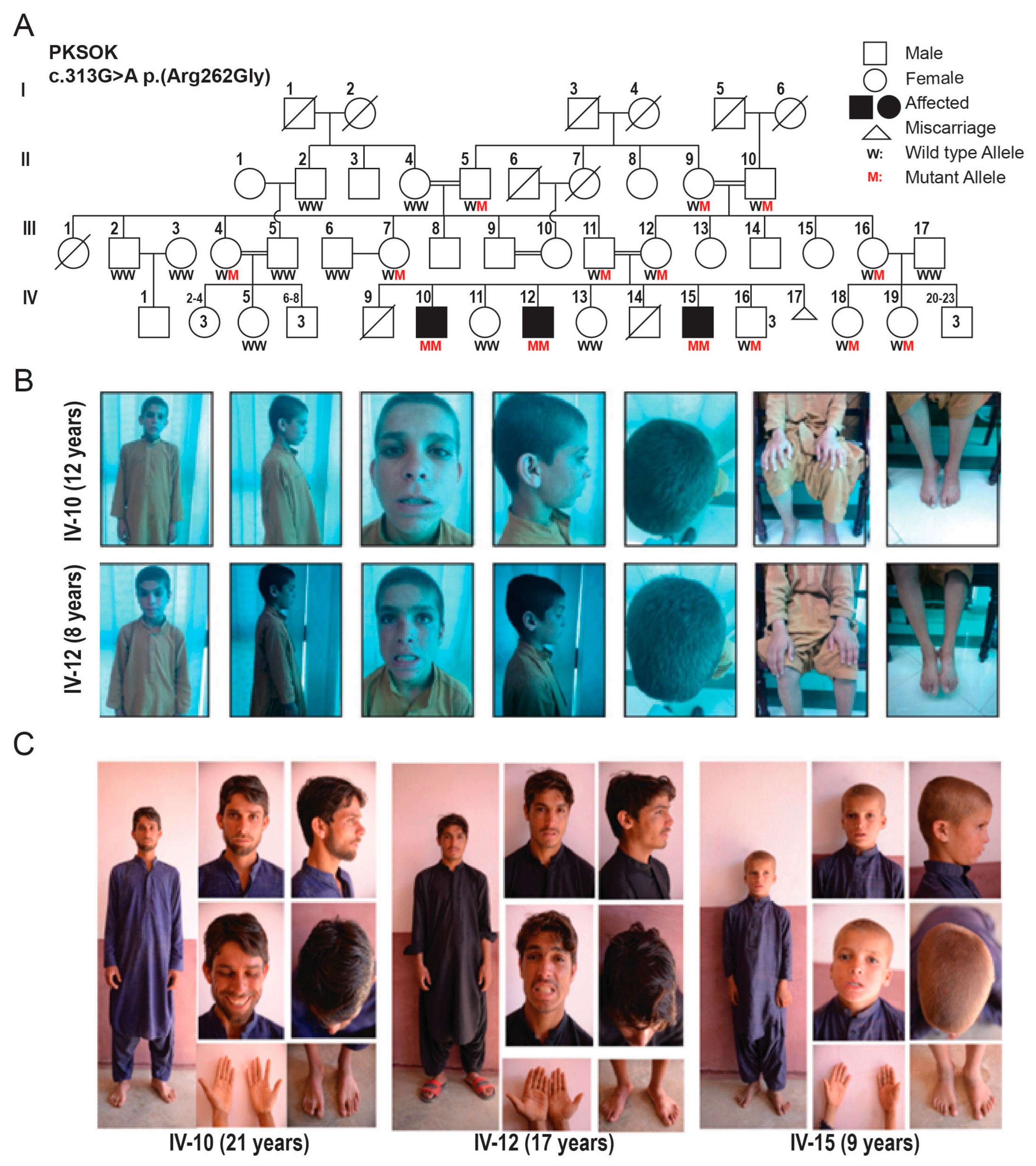
Figure 2.
Identified Biallelic variant is highly intolerant and conserved. (A) Structure of IVD gene, representing the identified variant position. (B) Missense Tolerance Ratio (MTR), a measure of regional intolerance to missense variation, analysis calculated a score of 0.854 for the IVD variant, which was consistent with location of p.Gly105 residue within highly intolerant region of IVD protein predicted by Meta Dome intolerance scale analysis. (C) Representation of IVD protein containing three acyl-CoA dehydrogenase domains. (D) Representation of valproic acid metabolic pathway and gamma-aminobutyric acid (GABA) synthesis. (E) Phylogenetic analysis showed that the p.Gly105 residue of IVD, mutated in rural family, is evolutionarily conserved in different species. (F) Interaction predicition reveal interaction of IVD with many known genes involved in neurological disorders and transporter proteins including DRD1 and DRD4 which are dopamine receptors.
Figure 2.
Identified Biallelic variant is highly intolerant and conserved. (A) Structure of IVD gene, representing the identified variant position. (B) Missense Tolerance Ratio (MTR), a measure of regional intolerance to missense variation, analysis calculated a score of 0.854 for the IVD variant, which was consistent with location of p.Gly105 residue within highly intolerant region of IVD protein predicted by Meta Dome intolerance scale analysis. (C) Representation of IVD protein containing three acyl-CoA dehydrogenase domains. (D) Representation of valproic acid metabolic pathway and gamma-aminobutyric acid (GABA) synthesis. (E) Phylogenetic analysis showed that the p.Gly105 residue of IVD, mutated in rural family, is evolutionarily conserved in different species. (F) Interaction predicition reveal interaction of IVD with many known genes involved in neurological disorders and transporter proteins including DRD1 and DRD4 which are dopamine receptors.
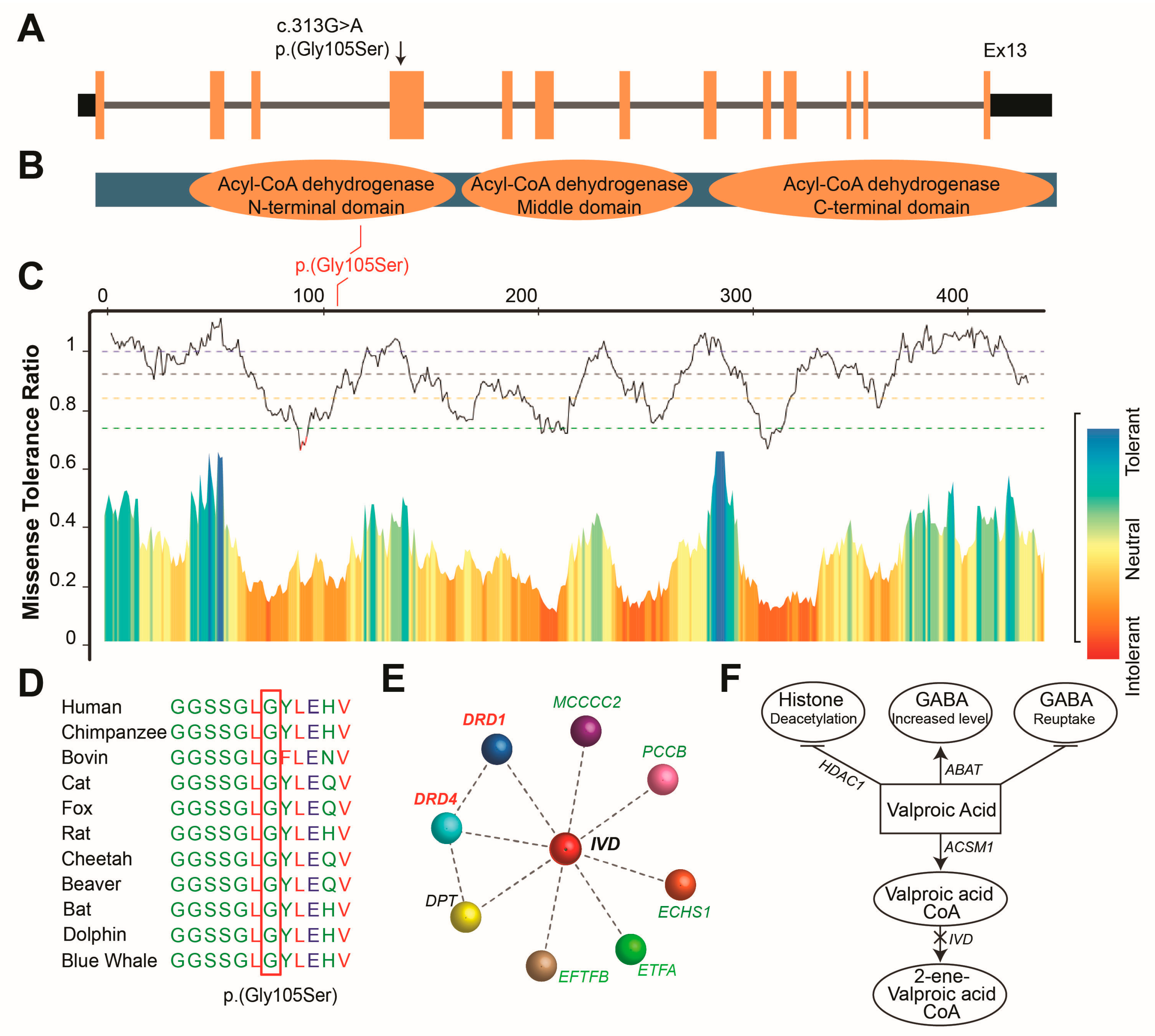
Figure 3.
Three-dimensional (3D)-Protein modeling, depict pathogenic nature of identified variant in IVD. (A) 3D protein structure of IVD. Magnified position of wild-type and p.Gly105Ser variant harbouring proteins. Hydrogen bonding between the residues is shown with dotted lines along with the distances in Å. Substitution of p.Gly105 with p.Ser105 is predicted to affect the activity of protein resulting in incorrect function of the protein and/or disruption in interaction with external domains, which might affect the signal transduction between the domains. (B) Depiction of Protein-Protein complex and Docking Interface. The receptor protein IVD (Shown in green) and the ligand protein DRD1 (shown in blue). The polar contacts are shown in figure depict both hydrogen bonds and ionic contacts. A total of 34 polar contacts were identified when annotated with wildtype IVD and DRD1 and only 12 polar contacts were identified when glycine is replaced by serine, affecting the docking between the IVD and DRD1. (C) Depiction of Protein-Protein complex and Docking Interface. The receptor protein IVD (Shown in green) and the ligand protein DRD4 (shown in blue). The polar contacts are shown in figure depict both hydrogen bonds and ionic contacts. A total of 5 polar contacts were identified when docking was performed between wildtype IVD and DRD1. In contrast, with IVD (Mut), DRD4 has a greater number of polar contacts, with 14 interactions observed, however, has less favorable energy scores, which makes it less stable even though it has more polar connections.
Figure 3.
Three-dimensional (3D)-Protein modeling, depict pathogenic nature of identified variant in IVD. (A) 3D protein structure of IVD. Magnified position of wild-type and p.Gly105Ser variant harbouring proteins. Hydrogen bonding between the residues is shown with dotted lines along with the distances in Å. Substitution of p.Gly105 with p.Ser105 is predicted to affect the activity of protein resulting in incorrect function of the protein and/or disruption in interaction with external domains, which might affect the signal transduction between the domains. (B) Depiction of Protein-Protein complex and Docking Interface. The receptor protein IVD (Shown in green) and the ligand protein DRD1 (shown in blue). The polar contacts are shown in figure depict both hydrogen bonds and ionic contacts. A total of 34 polar contacts were identified when annotated with wildtype IVD and DRD1 and only 12 polar contacts were identified when glycine is replaced by serine, affecting the docking between the IVD and DRD1. (C) Depiction of Protein-Protein complex and Docking Interface. The receptor protein IVD (Shown in green) and the ligand protein DRD4 (shown in blue). The polar contacts are shown in figure depict both hydrogen bonds and ionic contacts. A total of 5 polar contacts were identified when docking was performed between wildtype IVD and DRD1. In contrast, with IVD (Mut), DRD4 has a greater number of polar contacts, with 14 interactions observed, however, has less favorable energy scores, which makes it less stable even though it has more polar connections.
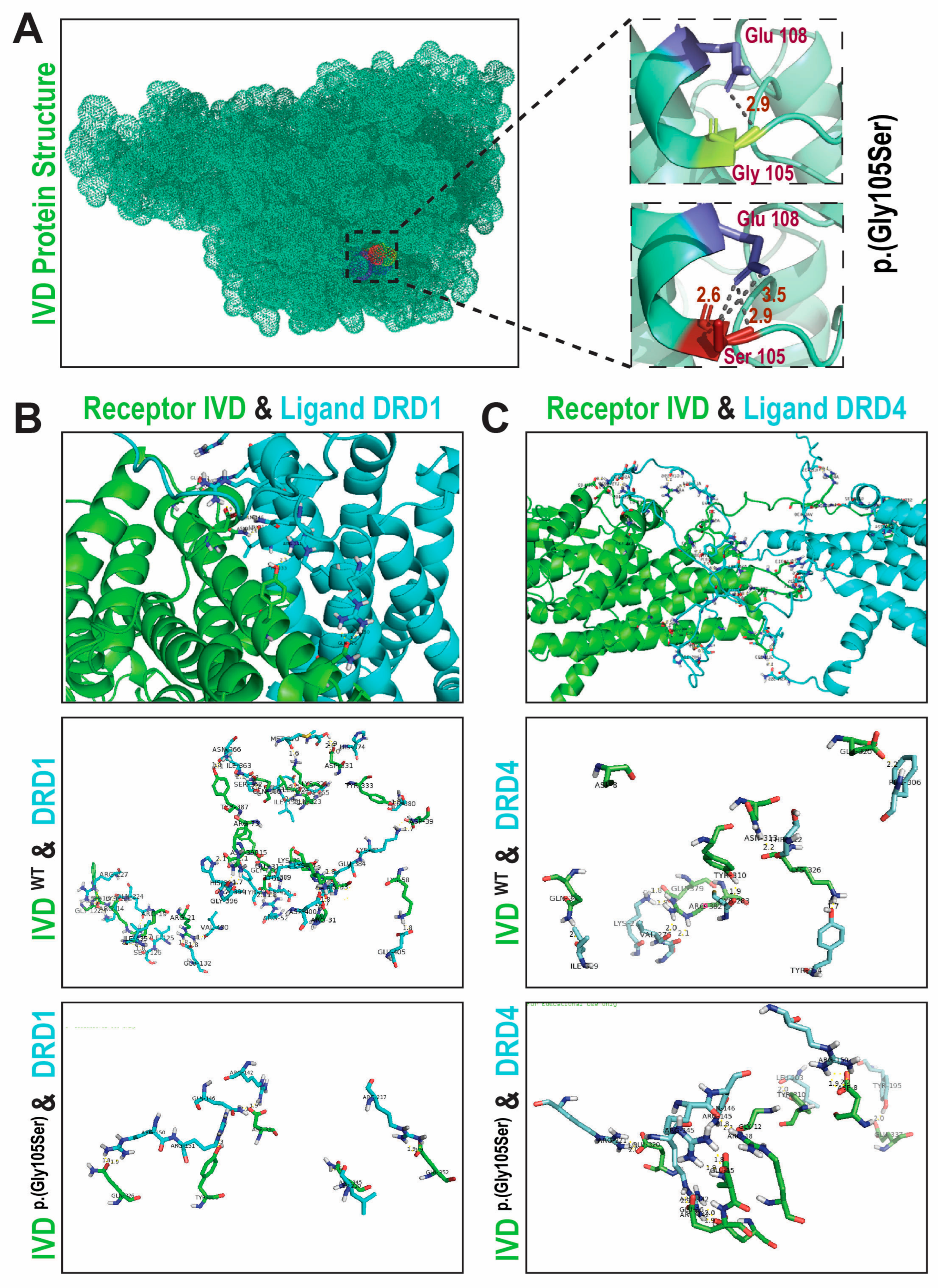

Table 1.
Clinical scenario of three affected Balochi male brothers.
| Ethnicity | Pakistani | ||
| Individual | IV:10 | IV:12 | IV:15 |
| Sex | Male | Male | Male |
| Age (years) | 21 years | 17 years | 9 years |
| Epilepsy | No | No | No |
| Intellectual disability | Mild | Mild | Mild |
| Weight | 49.8 Kg | 44.9 Kg | 19.7 Kg |
| Height | 163.5 cm | 153 cm | 113cm |
| Head Circumference | 54 cm | 54 cm | 49 cm |
| Head Shape | Normal | Normal | Normal |
| Hypotonia | Nocturnal | Nocturnal | Nocturnal |
| Speech Delay | Yes | Yes | Yes |
| Psychomotor delays | Yes | Yes | Yes |
| Ataxia | No | No | No |
| Spasticity | No | No | No |
| Behavioral Problem | Yes | Yes | Yes |
| Perinatal History | Delayed childhood milestones | Delayed childhood milestones | Delayed childhood milestones |
Table 2.
In silico analysis of identified pathogenic variants in IVD.
| Family | Balochi Family |
| Segregation | Homozygous |
| hg19 coordinates | 15:40702844 G>A |
| Nucleotide variant | c.313G>A |
| Amino acid substitution | p.(Gly105Ser) |
| ACMG Classification | Pathogenic Strong |
| ACMG Criteria | PP3b |
| gnomAD Frequencies | 0.0000318 (Not reported as homo) |
| CADD | 25.3 |
| REVEL | 0.831 |
| M-CAP | Damaging |
| Polyphen-2 HumDiv | Possibly Damaging |
| Polyphen-2 HumVar | Possibly Damaging |
| GERP++ | 5.07 |
| phyloP 100way Vertebrate | 6.674 |
| Mutation Taster | Disease Causing |
| FATHMM | Pathogenic |
| SIFT | Uncertain |
bPP3: Pathogenic computational prediction by 10 algorithms.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



