
Submitted:
20 January 2025
Posted:
20 January 2025
You are already at the latest version
Abstract
Background: Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D) is a rare metabolic disorder that impairs the body's ability to produce ketone bodies and regulate energy metabolism. Diagnosing HMGCS2D is challenging because patients typically remain asymptomatic unless experiencing fasting or illness. Due to the absence of reliable biochemical markers, genetic testing has become the definitive method for diagnosing HMGCS2D. Methods: This study included 19 patients from 14 unrelated families diagnosed with HMGCS2D in our department between October 2018 and October 2024. The clinical presentations, biochemical findings, molecular characteristics, and management strategies were systematically summarized and analyzed. Results: Of the 19 cases studied, 16 were symptomatic and three were asymptomatic. The onset of the first acute episode occurred between 10 days and 28 months of age. Triggers for the initial crisis in the symptomatic cases included poor feeding (93.8%), vomiting (56.3%), diarrhea (25.0%), and fever (18.8%). Clinical manifestations during the first episode were lethargy/coma (81.3%), rapid breathing (68.8%), hepatomegaly (56.3%), shock (37.5%), and seizures (18.8%). Biochemical abnormalities observed included elevated plasma transaminases (100%), metabolic acidosis (75%), hypoglycemia (56.3%), and elevated plasma ammonia levels (31.3%). Additionally, low free carnitine levels were found in seven cases, elevated C2 levels in one case, dicarboxylic aciduria in two cases, and ketonuria in two cases. Abnormal brain MRI findings were detected in three patients. Genetic analysis revealed seven HMGCS2 gene variants across the 19 cases. Notably, a novel variant, c.407A>T (p.D136V), was identified and has not been reported in any existing databases. Two common variants, c.559+1G>A and c.1090T>A (p.F364I), were present in 11 out of 19 cases (57.9%) and 10 out of 19 cases (55.5%), respectively. Implementation of high glucose infusion and proactive management strategies—such as preventing prolonged fasting and providing enteral carbohydrate/glucose infusion during illness—effectively reduced the rate of acute relapses following accurate diagnosis. Currently, all 19 patients are alive, with ages ranging from five months to 14 years, and exhibit normal physical development. Conclusions: To the best of our knowledge, this study represents the first reported cases of HMGCS2D in Vietnamese patients. Our findings contribute to a broader understanding of the clinical phenotype and expand the known spectrum of HMGCS2 gene variants, enhancing current knowledge of this rare metabolic disorder.
Keywords:
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency
; HMGCS2 variant
; Vietnamese
; p.D136V
; c.559+1G>A
; p.F364I
; asymptomatic
1. Introduction
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D, OMIM #605911) is a rare metabolic disorder that impairs the body’s ability to produce ketone bodies and regulate energy metabolism, particularly during fasting or illness [1]. This condition is caused by a deficiency of the HMG-CoA synthase enzyme, which plays a key role in the ketogenesis pathway by catalyzing the formation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) from acetyl-CoA and acetoacetyl-CoA [2]. This step is essential for the synthesis of acetoacetate and its derivatives, 3-hydroxybutyric acid and acetone, in the liver. Ketone bodies act as an alternative energy source for the brain and muscles when glucose is scarce, such as during fasting or prolonged physical activit [3]. In the absence of the HMG-CoA enzyme, the body cannot effectively produce ketone bodies and becomes entirely dependent on glucose for energy [4]. During fasting or illness, when glucose stores are depleted, the lack of alternative en-ergy sources leads to symptoms like hypoglycemia, vomiting, dehydration, lethargy, poor feeding, developmental delays in infants, seizures, and other neurological issues [5,6,7]. Persistent and intractable metabolic acidosis has also been observed [8,9]. Several cases have even resulted in early death (before two years of age) [9,10,11]; however, other cases have remained asymptomatic [10,12].
The diagnosis of HMGCS2D primarily relies on an HMG-CoA synthase enzyme assay; however, distinguishing between mitochondrial HMG-CoA synthase and cytosolic HMG-CoA synthase in liver homogenates is challenging, limiting the use of this assay [13]. Additionally, other biochemical indicators such as hypoketosis, elevated free fatty acids, normal acylcarnitines, ketonuria, and dicarboxyluria are not specific to HMGCS2D alone [7]. Recently, biomarkers like 4-hydrox-6-methyl-2-pyrone have been identified for HMGCS2D [14]; but this is not part of routine laboratory testing. These factors highlight the diagnostic challenges of HMGCS2D, especially in asymptomatic patients during non-stress con-ditions, due to clinical overlap with other metabolic disorders and the absence of reliable biochemical markers [7,15]. As a result, genetic testing has become the definitive diagnostic tool for HMG-CoA synthase deficiency, utilizing methods such as Sanger sequencing, targeted next- generation sequencing, and exome sequencing [11,13,15].
The HMG-CoA enzyme is encoded by the HMGCS2 gene (OMIM #600234), located on chromosome 1p12, consisting of 10 exons with distinct domains and features [11]. Pathogenic variants in the HMGCS2 gene lead to partial or complete loss of HMG-CoA enzyme function, resulting in HMGCS2D. Since the first reported case in 1997, more than 60 cases with around 40 different HMGCS2 pathogenic variants have been documented [11]. The majority of pathogenic variants are found in exons 2 and 4 [11,14,16]. The c.1201G>T (p.E401*) variant is the most common mutation identified in Chinese patients [11], while the c.634G>A (p.G212R) variant is more prevalent among individuals of European descent [14,17,18]. A genotype-phenotype correlation has been observed in HMGCS2D, where patients with biallelic truncation mutations tend to present with more severe clinical symptoms and exhibit a higher mortality rate [11].
In this study, we describe clinical, biochemical, molecular characteristics, along with the management strategies, of 19 cases from 14 unrelated families diagnosed with HMGCS2D.
2. Results
2.1. Clinical and Biochemical Characteristics
Nineteen cases of HMGCS2D from 14 unrelated families were diagnosed between October 2018 and October 2024 at the Center of Endocrinology, Metabolism, Genetic/Genomics, and Molecular Therapy, Vietnam National Children’s Hospital. Five families had siblings affected by HMGCS2D: P1 and P2, P3 and P4, P10 and P11, P13 and P14, and P16 and P17. Among the cases, 16 were symptomatic, while three (P4, P11, and P17) were asymptomatic, as shown in Table 1. The age of onset for the first acute episode ranged from 10 days to 28 months. Triggers for the initial crisis in the sixteen symptomatic cases included poor feeding (15/16, 93.8%), vomiting (9/16, 56.3%), diarrhea (4/16, 25.0%), and fever (3/16, 18.8%). The primary clinical features during the first episode were lethargy or coma (13/16, 81.3%), rapid breathing (11/16, 68.8%), hepatomegaly (9/16, 56.3%), shock (6/16, 37.5%), and seizures (3/16, 18.8%).

Table 1.
Clinical symptoms of 19 patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency.
Table 1.
Clinical symptoms of 19 patients with mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency.
| Pt | Age of onset | Gender | Family history | Days of onset | Fever | Vomiting | Diarrhea | Poor feeding | Rapid breathing | Seizure | Hepatomegaly | Lethargy/coma | Others |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 5m | M | + | 3.0 | - | - | - | + | + | - | + | + | Shock |
| P2 | 7m | F | + | 1.0 | + | + | - | + | + | - | - | + | Shock |
| P3 | 14m | M | + | 1.0 | - | + | + | + | + | - | + | + | Shock |
| P4* | - | M | + | - | - | - | - | - | - | - | - | - | - |
| P5 | 18m | M | - | 2.0 | + | - | - | + | + | - | + | + | Shock |
| P6 | 8m | F | - | 1.0 | - | + | - | + | + | - | + | + | Shock |
| P7 | 10d | F | - | 1.0 | - | - | - | + | + | - | NA | + | Jaundice |
| P8 | 28m | M | - | 5.0 | - | + | + | + | + | + | + | + | Pallor |
| P9 | 13m | M | - | 4.0 | - | + | - | + | + | + | + | + | - |
| P10 | 8m | F | + | 1.0 | - | - | - | + | - | - | + | + | Shock |
| P11* | - | M | + | - | - | - | - | - | - | - | - | - | - |
| P12 | 7m | F | - | 3.0 | - | + | + | + | + | - | + | + | - |
| P13 | 18m | M | + | 1.0 | - | - | + | + | - | - | + | - | - |
| P14 | 5m | F | + | 0.5 | - | + | - | + | - | - | - | - | - |
| P15 | 6m | F | - | 2.0 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| P16 | 5m | F | + | 2.0 | - | - | - | + | - | + | NA | + | - |
| P17* | - | M | + | - | - | - | - | - | - | - | - | - | - |
| P18 | 10m | F | - | 2.0 | - | + | - | + | + | - | NA | + | - |
| P19 | 6m | F | - | 2.0 | + | + | - | + | + | - | - | + | Loss weight |
M, male; F: female; NA, not analyzed; +, present; -, not present; m, month; y, year; d, day. P1 and P2 are siblings; P3 and P4 are siblings; P10 and P11 are siblings; P13 and P14 are siblings; P16 and P17 are siblings. Three cases, P4, P11, and P17 were asymptomatic and diagnosed through family screening.
Hypoglycemia was observed in nine cases, with a median blood glucose level of 2.2 mmol/L (ranging from 0.1 to 2.48 mmol/L) (Table 2). Notably, patient P10 exhibited the highest level of hyperglycemia, which required insulin infusion. Metabolic acidosis was present in 12 of the 16 symptomatic cases, with a median pH of 7.18 (Table 2). Four cases—P8, P13, P14, and P16—did not exhibit metabolic acidosis. Elevated plasma ammonia levels were detected in five cases (25%), specifically in P6, P7, P10, P16, and L18. Severe hyperammonemia was recorded in P7, with a plasma ammonium concen-tration of 983 µmol/L. All 16 symptomatic cases had elevated plasma transaminase levels. Plasma creatine kinase was assessed in seven cases, and elevated levels (ranging from 297 to 3,267 UI/L) were found in four patients: P3, P8, P14, and P16. Additionally, seven cases showed low free carnitine (plasma C0) levels, while only one case exhibited a high plasma C2 level. Dicarboxylic aciduria was identified in two cases, and keto-nuria was detected in five cases.
Brain magnetic resonance imaging (MRI) was performed in 11 cases, and three of these cases exhibited brain abnormalities (Figure 1). Case P3 presented with symmetric hyperintensities in both the head of the caudate nuclei and the putamina (Figure 1a). Case P9 showed bilateral hyperintensities in the periventricular deep white matter and the left parietal cortex, along with enhancement in the subcortical regions of the left frontal and parietal lobes (Figure 1b). Case P12 demonstrated bilateral ventricular dila-tion and widening of the temporal subarachnoid space, predominantly on the left side (Figure 1c).
2.2. Molecular Analyses
A total of seven deleterious HMGCS2 gene variants were identified in 19 patients with HMGCS2D, consisting of one nonsense variant, two splicing variants, and four missense variants (Table 3 and Figure 2). Homozygous variants were found in seven patients: P1, P2, P5, P7, P9, P16, and P17 (Table 3). The remaining 12 patients carried compound heterogeneous variants (Table 3). Among these, a novel variant, c.407A>T (p.D136V), has not been reported in any database. Five other variants—c.334C>T (p.R112W), c.559+1G>A, c.682C>T (p.R228*), c.1090T>A (p.F364I), and c.1502G>C (p.R501P)—have been classified as likely pathogenic or pathogenic in the ClinVar database. In contrast, the c.407A>T (p.D136V) and c.850+1G>A variants were not listed in ClinVar. According to the ACMG classification, c.407A>T (p.D136V) was categorized as a likely pathogenic variant, while c.850+1G>A was identified as a pathogenic variant (Table 3). Two common variants, c.559+1G>A and c.1090T>A (p.F364I), were detected in 11 out of 19 cases (57.9%) and 10 out of 19 cases (55.5%), respectively (Table 3 and Figure 2). Sanger sequencing in three families confirmed that patients P18, P10, P11, P13, and P14 inherited the mutant alleles from their parents (Figure 3).
2.3. Outcomes
Management of the first episode for the 16 symptomatic cases involved various interventions. Mechanical ventilation was required in 11 cases for two to six days, ac-id-base correction was administered in 13 cases for three to 72 hours, and glucose in-fusion was provided in 15 cases for one to seven days. Continuous veno-venous hemofiltration (CVVH) was performed in seven cases for 19 to 82 hours. Antibiotic therapy was given to 14 cases for two to 28 days, and L-carnitine supplementation was administered in 13 cases. Additionally, patient P10 experienced hyperglycemia that required insulin infusion (0.05 UI/kg/hour) to maintain normal blood glucose levels for four hours. Patient P7, who developed severe hyperammonemia (983 µmol/L), was treated with sodium benzoate supplementation. All patients recovered from the first episode, with normalization of metabolic acidosis, transaminase, and CK levels. Only patient P3 experienced neurological sequelae, while the remaining patients showed normal mental and motor development. After discharge, 15 patients continued L-carnitine supplementation, and two of them also received arginine therapy.
Table 4.
Management and outcome at the first episode.
| Pt | Initial diagnosis | Management in first crisis | Outcome of first crisis | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ventilation (days) | Acidotic correction (hours) | Glucose infusion (days) | CVVH (hours) | Antibiotics (days) | Others | Recovered MA (hours) | Alive | Neurological | ||
| P1 | Organic aciduria | 3 | 34 | 4 | 34 | 13 | L-carnitine, biotin, B12 | 13 | Yes | No |
| P2 | HMGCS2D | 2 | 48 | 2 | 19 | 10 | L-carnitine | 48 | Yes | No |
| P3 | Glutaric acidemia II | 5 | 26 | 3 | 20 | 20 | L-carnitine, B2, coenzyme Q10 | 11 | Yes | Yes |
| P5 | MMA/GA II | 3 | 33 | 6 | 0 | 16 | L-carnitine, B2, B12, biotin | 33 | Yes | No |
| P6 | Myocarditis, # IEM | 4 | 72 | 6 | 82 | 28 | L-carnitine | 72 | Yes | No |
| P7 | Glutaric acidemia II | 2 | 48 | 7 | 0 | 19 | L-carnitine | 48 | Yes | No |
| P8 | Hypoglycemia/IEMs | 0 | 24 | 4 | 0 | 0 | L-carnitine | 24 | Yes | No |
| P9 | IEMs | 6 | 48 | 4 | 0 | 21 | 0 | 48 | Yes | No |
| P10 | Glycogen storage | 2 | 25 | 2 | 32 | 15 | L-carnitine, biotin, B12 | 14 | Yes | No |
| P12 | CUD | 0 | 18 | 2 | 0 | 12 | L-carnitine | 18 | Yes | No |
| P13 | Elevated transaminase | 0 | 0 | 0 | 0 | 7 | 0 | 0 | Yes | No |
| P14 | HMGCS2D | 0 | 0 | 1 | 0 | 0 | L-carnitine | 0 | Yes | No |
| P15 | IEM | 3 | 0 | 3 | 48 | 3 | L-carnitine, | 72 | Yes | No |
| P16 | FAOD | 3 | 3 | 1 | 0 | 2 | L-carnitine, biotin, B12 | 48 | Yes | No |
| P18 | IEM | 3 | 3 | 3 | 72 | 7 | L-carnitine, B12, arginine, biotin, | 72 | Yes | No |
| P19 | Glutaric aciduria II | 0 | 10 | 5 | 0 | 9 | L-carnitine, B2, coenzyme Q10 | 48 | Yes | No |
| Sum | 11/16 | 13/16 | 15/16 | 7/16 | 14/16 | 14/16 | ||||
CVVH, continuous veno-venous hemofiltration; HMGCS2D, mitochondrial HMG-CoA synthase deficiency; MMA/GA II, methylmalonic acidemia/glutaric acidemia II; IEM, inborn errors of metabolism; CUD, carnitine uptake defect; FAOD, fatty acid oxidation disorders.
It took between 10 days and 16 months to achieve an accurate diagnosis through molecular analysis. During this waiting period, seven patients experienced a total of eleven recurrent acute episodes between their first crisis and the confirmed diagnosis. Following genetic confirmation, all 19 patients were advised to avoid prolonged fasting. Sixteen sympto-matic patients received management with L-carnitine and maltodextrin. Post-diagnosis care for all patients involved fasting avoidance, sup-plementation with L-carnitine and maltodextrin, glucose infusions, and carbohydrate-rich fluids during illnesses, effectively preventing further acute episodes. Only two recurrent acute attacks occurred in two pa-tients after receiving accurate diagnosis and appropriate management. Currently, all 19 patients are alive, with ages ranging from five months to 14 years. All patients have normal physical development, and 18 out of 19 patients show normal psychomotor development without any complications.
Table 5.
Accurated diagnosis and follow–up.
| Pt | Age of AD | Time to achieve AD | Management before AD | Crisis numbers before AD | Current age | Crisis numbers after AD | Height (SDS) | Weight (SDS) | DQ |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 21m | 16m | L-carnitine, Biotin, B12 | 4 (2 times: CVVH) | 7y2m | 1 | -0.2 | -0.4 | Normal |
| P2 | 7m | 10d | L-carnitine | 1 | 1y11m | 0 | -0.2 | 0.2 | Normal |
| P3 | 17m | 3m | L-carnitine | 1 | 4y | 0 | NA | -2.1 | Neurological sequelae |
| P4 | 6y | - | - | 0 | 9y8m | 0 | 0.1 | 0.7 | Normal |
| P5 | 26m | 8m | L-carnitine | 0 | 7y8m | 0 | 1.2 | 1.7 | Normal |
| P6 | 9m | 1m | L-carnitine | 1 | 4y3m | 0 | 1.1 | 0.8 | Normal |
| P7 | 1m | 1m | L-carnitine | 0 | 2y5m | 0 | 0.8 | 1.2 | Normal |
| P8 | 3y3m | 11m | L-carnitine | 2 | 7y3m | 0 | -0.8 | -1.1 | Normal |
| P9 | 17m | 4m | L-carnitine | 0 | 3y | 0 | -0.5 | -1.6 | Normal |
| P10 | 10m | 2m | L-carnitine | 0 | 2y5m | 0 | -1.0 | -0.4 | Normal |
| P11 | 12y | - | - | 14y1m | 0 | 0.2 | -1.2 | Normal | |
| P12 | 8m | 1m | L-carnitine | 0 | 13m | 0 | 1.4 | 0.7 | Normal |
| P13 | 19m | 1m | Arginine, vitamin E, L-carnitine | 0 | 3y4m | 0 | 1.7 | 0.7 | Normal |
| P14 | 5m | - | - | 0 | 5m | 1 | 3.1 | -0.9 | Normal |
| P15 | 10m | 4m | Arginine, L-carnitine | 1 (CVVH) | 18m | 0 | -0.8 | -0.8 | Normal |
| P16 | 8m | 3m | L-carnitine, Biotin, vitamin B12 | 1 | 14m | 0 | 0 | 0 | Normal |
| P17 | 4y6m | - | - | - | 5y | 0 | 0.3 | -0.2 | Normal |
| P18 | 12m | 2m | L-carnitine | 0 | 17m | 0 | -0.9 | -0.2 | Normal |
| P19 | 9m | 3m | L-carnitine, CoQ10, vitamin B2 | 0 | 19m | 0 | 0.1 | -0.8 | Normal |
M, month; y, year; AD, accurate diagnosis; CVVH, continuous veno-venous hemofiltration diagnosis; DQ, development quotient.
3. Discussion
Diagnosing HMGCS2D based on biochemical profiles presents important challenges. In our department, both the HMG-CoA synthase enzyme assay and specific urinary bio-chemical markers such as 4-hydroxy-6-methyl-2-pyrone (4-HMP) are unavailable. Wu et al. suggested that a high C2 to C0 ratio, combined with urinary dicarboxylic acids during episodes of acute hypoglycemia and metabolic acidosis, could serve as an additional biochemical marker for HMGCS2D [11]. However, none of our patients fully met these criteria. For example, patient P16 ex-hibited a high C2/C0 ratio (7.5), urinary dicarboxylic acids, and hypoglycemia but did not present with metabolic acidosis. Patient P19 had a high C2/C0 ratio (5.9), urinary dicarboxylic acids, and metabolic acidosis but maintained normal glucose levels. Notably, patient P13 only showed elevated transaminase levels. This variability in clinical and biochemical presentations during the first acute episode made it extremely difficult to promptly and accurately diagnose HMGCS2D in our patients. Initially, the 14 probands were misdiagnosed with various conditions, including organic acidurias, fatty acid ox-idation disorders, and glycogen storage diseases. Our findings emphasize the essential role of genetic testing in accurately diagnosing HMGCS2D [7,10,11,12,19]. All patients in this study were confirmed to have biallelic mutations in the HMGCS2 gene. Additionally, five siblings were diagnosed with HMGCS2D through family genetic screening. Among these five, three asymptomatic patients (P4, P11, and P17) displayed no clinical symptoms and maintained normal blood test results up to their current ages of five to 14 years. In contrast, their younger siblings experienced typical clinical manifestations between five and 14 months of age. Previous studies have reported only three asymptomatic cases [10,12]. The identification of our three asymptomatic patients further expands the database of asymptomatic individuals with HMGCS2D.
The median age of the first acute episodes in our study was seven months (ranging from 10 days to 28 months), which is slightly younger than the median age of nine months reported in other studies [11]. Notably, one patient (P7) experienced an acute episode at just 10 days old, despite having no prior digestive issues or infections. However, the patient's mother had a history of polyhydramnios and gestational diabetes starting at 24 weeks of gestation, which required insulin treatment, and the infant's birth weight was 3,900 grams. These factors—gestational diabetes, polyhydramnios, and high birth weight—may have contributed to neonatal hypoglycemia, potentially triggering the acute episode. Additionally, the initial acute episodes in our patients often followed digestive symptoms such as poor feeding (15 out of 16 cases), vomiting (9 out of 16), and diarrhea (four out of 16), which is consistent with findings from other studies [10,11]. The onset of HMGCS2D typically occurs in infancy, often after digestive disturbances that impair nutrient absorption and increase catabolic activity, leading to enhanced gluconeogenesis and reliance on ketone bodies for energy. The clinical symptoms ob-served in the 16 symptomatic patients during acute episodes included poor appetite, vomiting, diarrhea, fever, rapid breathing, lethargy or coma, convulsions, and shock. These symptoms closely resemble those of sepsis or acute brain syndrome, making diagnosis particularly challenging [20,21].
Hypoglycemia is considered one of the diagnostic indicators for HMGCS2D; however, it is not a defining feature during metabolic crises, as supported by previous studies [17,22] and confirmed by our study findings. In our study, four out of 16 symptomatic patients maintained normal glucose levels during episodes of metabolic decompensation. Additionally, patient P10 presented with considerable hyperglycemia (24.5 mmol/L) accompanied by metabolic acidosis, ketonuria, and elevated transaminase levels on the first day of the acute episode, closely resembling diabetic ketoacidosis. Similar instances of hyperglycemia in HMGCS2D have been reported in a patient from Thailand [16] and another from Turkey [10]. The etiology for this dysglycemia in HMGCS2D remains unclear.
Additionally, brain damage detected through MRI during acute episodes was observed in three out of 11 examined patients. The brain lesions were located in the bilateral putamen and caudate nuclei, as well as in the white matter of the cerebral cortex, along with dilated lateral ventricles. These findings are consistent with brain abnormalities reported in patients with HMGCS2D in previous studies [7,11,17].
In this study, the variants c.559+1G>A and c.1090T>A (p.F364I) were identified as the two most common mutations in Vietnamese patients with HMGCS2D, each occurring in more than 50% of cases. The c.559+1G>A variant affects the 5’ splice site of intron 2 in the HMGCS2 gene and is predicted to disrupt the donor splice site, supported by a high Δ score of 0.97 in SpliceAI. This disruption likely interferes with the normal splicing process of the HMGCS2 gene. This variant has been reported in the population database dbSNP155 (rs587603096) and classified as pathogenic in the ClinVar database (ID: 859738). Similar splice site disruptions have been observed in other individuals with HMGCS2D [11,12]. The second most common variant, c.1090T>A, results in the substitution of phenylala-nine with isoleucine at position 364 (p.F364I) of the HMGCS2 protein. Phenylalanine contains an aromatic side chain, whereas isoleucine has a branched hydrocarbon side chain. This amino acid change may alter the protein's side chain structure, potentially affecting the conformation and function of the HMGCS2 protein. Additionally, the c.1502G>C (p.R501P) variant was found in three patients and has been previously reported as a common pathogenic variant in Thai patients with HMGCS2D [16]. Arginine 501 (R501) has been predicted to be a critical amino acid for the proper function of the HMGCS2 protein, and the R501P substitution leads to the loss of a salt bridge between arginine 501 and aspartic acid 101 [23]. This structural disruption considerably impacts the protein’s stability and function. Furthermore, functional assays of the R501P mutation demonstrated a complete loss of HMGCS2 enzymatic activity [23]. The fourth identified variant, c.334C>T (p.R112W), was previously reported in a patient of Romanian origin [15]. Although the mutated p.R112W protein showed moderate expression levels (22.4%) compared to the wildtype protein, enzyme activity assays revealed a complete absence of enzymatic function in the mutant protein [15]. The fifth variant, a nonsense mutation c.682C>T (p.R228*), has not been previously reported in individuals with HMGCS2-related disorders. However, this variant is classified as pathogenic in ClinVar database. The c.682C>T mutation introduces a premature termination codon at residue 228 (p.Arg228*) of the HMGCS2 protein, resulting in an partly deletion or absence of HMGCS2 protein product.
Our study, to the best of our knowledge, is the first to report the association of the c.407A>T (p.D136V) and c.850+1G>A variants with HMGCS2D. The sixth variant, c.407A>T, results in the substitution of aspartic acid at position 136 with valine. This D136V mutation disrupts three hydrogen bonds between D136 and the neighboring amino acids K137, S138, and K139 (Figure 4), leading to an unstable HMGCS2 protein structure. The seventh variant, c.850+1G>A, is located at the 5’ splice site of intron 5 in the HMGCS2 gene. This mutation is predicted to disrupt the canonical donor splice site, supported by a high Δ score of 0.99, indicating an important impact on proper splicing. Although this variant is listed in the dbSNP database (rs112412189), it has not previously been reported in individuals affected by HMGCS2-related disorders.
According to a previous review publication [11], six out of 49 patients with HMGCS2D died due to hypoglycemic crises and severe met-abolic acidosis during acute episodes. HMGCS2D impairs the body's ability to produce ketone bodies and regulate energy metabolism, particularly during periods of fasting or illness. Therefore, high glucose infusion and proactive management—such as preventing prolonged fasting and providing enteral carbohydrates or glucose infusion during ill-ness—are essential for effective treatment. This management approach considerably reduced the acute relapse rate in our patients, decreasing from 11 to two recurrent crises after an accurate diagnosis. . Symptomatic patients were treated with L-carnitine sup-plementation, frequent meals, and maltodextrin. To date, none of the patients in our study have died, and only one patient has experienced neurological sequelae.
4. Materials and Methods
4.1. Individuals
This study included both retrospective and prospective analyses of 19 Vietnamese children from 14 unrelated families diagnosed with HMGCS2D at the Center of Endo-crinology, Metabolism, Genetic/Genomics, and Molecular Therapy, Vietnam National Children’s Hospital.
4.2. Clinical Characteristics
Clinical symptoms such as fever, vomiting, diarrhea, poor feeding, breathing dif-ficulty, seizures, hepatomegaly, and lethargy were closely monitored and documented. Urinary organic acid analysis was conducted using Gas Chromatography-Mass Spec-trometry (GC/MS; Shimadzu model QP 5000, Japan) following the method described in a previous study [24]. Acylcarnitine analysis was performed using Guthrie blotting paper [25,26]. Dried blood samples were automatically punched into 3.2 mm diameter circles with an automatic puncher (WallacAutoPuncher™; PerkinElmer, Waltham, MA). Three 3.2 mm diameter circles were placed in an Amicon Ultra 0.5 (10K) tube containing 100 µL of distilled water and incubated at 370C for 2 hours with occasional shaking. The mixture was then centrifuged at 12,500 rpm for 15 minutes. The resulting extracted solution was transferred to a new tube, and 110 μL of stable-isotope internal standards in methanol was added for further analysis. The isotope standards used in the analysis included 2 nmol of glycine-2H2; 1.5 nmol each of alanine-2H3, valine, and leucine-2H10; 0.5 nmol each of methionine-2H3, phenylalanine-2H5, and arginine-13C6; 0.8 nmol of tyrosine-2H2; 0.2 nmol of citrulline-2H3; 3 nmol of glutamine-2H5; 100 pmol of carnitine-2H3; 100/3 pmol of acetyl-carnitine-2H3; 50/3 pmol each of propionyL-carnitine-2H3 and glutaryL-carnitine-2H9; 10 pmol of butyryl-carnitine-2H3; and 20/3 pmol each of octanoyL-carnitine-2H3 and palmitoyl-carnitine-2H9. The mass spectrometry system used was a triple-stage mass spectrometer, Model TSQ7000 (Thermo-Quest, Tokyo, Japan), equipped with a Model LC10 HPLC system and a Model SIL-10ADVP autoinjector (Shimadzu, Kyoto, Japan). The autoinjector introduced the derivatized sample (13 μL) at a flow rate of 20 μL/s using 50% acetonitrile in water at 1.9-minute intervals. The resolution of the mass spectra was automatically adjusted, and data were collected in the receiving channels for 1.2 minutes per sample. Images for amino acid and acylcarnitine analysis were captured, and peak data were automatically processed using Analyst 1.5.1 software (AB SCIEX, Foster City, CA).
4.3. Genetic Analysis
Genomic DNA was extracted from whole blood samples using the QIAamp DNA Blood Kit (Qiagen, Hilden, Germany). Genetic screening was conducted at Invitae (Invitae Corporation, San Francisco, CA, USA) and Center for Gene and Protein Research, Hanoi Medical University using a comprehensive glycogen storage disease and fatty acid oxidation defects panel that included 46 genes comprising the following genes: ACADM (NM_000016.5), ACADS (NM_000017.3), ACADSB (NM_001609.3), ACADVL (NM_000018.3), AGL (NM_000642.2), ALDOA (NM_000034.3), CPT1A (NM_001876.3), CPT2 (NM_000098.2), ENO3 (NM_053013.3), ETFA (NM_000126.3), ETFB (NM_001985.2), ETFDH (NM_004453.3), FBP1 (NM_000507.3), G6PC (NM_000151.3), GAA (NM_000152.3), GBE1 (NM_000158.3), GYG1 (NM_004130.3), GYS1 (NM_002103.4), GYS2 (NM_021957.3), HADH (NM_005327.4), HADHA (NM_000182.4), HADHB (NM_000183.2), HMGCL (NM_000191.2), HMGCS2 (NM_005518.3), LAMP2 (NM_002294.2), LDHA (NM_005566.3), MLYCD (NM_012213.2), NADK2 (NM_001085411.2), PFKM (NM_000289.5), PGAM2 (NM_000290.3), PGM1 (NM_002633.2), PHKA1 (NM_002637.3), PHKA2 (NM_000292.2), PHKB (NM_000293.2), PHKG2 (NM_000294.2), POLG (NM_002693.2), PYGL (NM_002863.4), PYGM (NM_005609.3), RBCK1 (NM_031229.3), SLC22A5 (NM_003060.3), SLC25A20 (NM_000387.5), SLC2A2 (NM_000340.1), SLC37A4 (NM_001164277.1), SLC52A1 (NM_017986.3), SLC52A2 (NM_024531.4), and SLC52A3 (NM_033409.3).
The pathogenicity of the identified variants was predicted using Mutation Taster [27] and according to the ClinVar database and the guidelines provided by American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines [28]. The splicing variants were further in silico analyzed using the SpliceAI tool [29]. To confirm the identified variants, exons 2, 3, 4, 6, and 9 of the HMGCS2 gene were amplified using specifically designed oligonucleotide primers, which are available upon request. The PCR products were sequenced using the 3500 Genetic Analyzer capillary electrophoresis system (Life Technologies, Foster City, CA, USA). The reference sequence used for HMGCS2 was NM_005518.3. Additionally, the impact of the variants on the three-dimensional structure of the human mitochondrial 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2, PDB ID: 2WYA) was modeled using Swiss-PdbViewer version 4.1.0[30].
4.4. Management
The patients were initially admitted to the hospital with misdiagnoses of other inborn errors of metabolism or unknown elevated transaminase levels. Acute management included the administration of a 10% dextrose infusion with a glucose delivery rate of 8–10 mg/kg/min, prompt initiation of oral feeding when possible, correction of acidosis using sodium bicarbonate or continuous veno-venous hemofiltration (CVVH), supplementation with L-carnitine, and treatment of triggering factors, such as antibiotics for infections and antipyretic drugs for fever. Proactive management strategies focused on preventing fasting. For infants aged 0–4 months, feeding was scheduled every three hours, and for infants older than four months up to 12 months, fasting was limited to no more than eight hours. Beyond infancy, regular feeding schedules were maintained. During illness, patients were aggressively managed with oral or enteral carbohydrate-rich fluids every 3–4 hours, along with 10% dextrose infusion at 1.5 times the maintenance rate during periods of illness, poor oral intake, or preoperative fasting. L-carnitine supplementation was also continued to support energy metabolism.
5. Conclusions
We have described 16 symptomatic and three asymptomatic Vietnamese patients diagnosed with HMG-CoA synthase deficiency. Seven pathogenic or likely pathogenic HMGCS2 variants were identified, including two common variants, c.559+1G>A and c.1090T>A, as well as a novel variant, c.407A>T. Following an accurate diagnosis, pro-active management was implemented, resulting in a reduction in the acute relapse rate. All patients are currently alive and exhibit normal physical development.
Author Contributions
Conceptualization, K.N.N. and C.D.V.; methodology, K.N.N., T.M.D., N.H.T. and C.D.V.; software, K.N.N., N.L.N., T.T.Q.T. and P.L.N.; validation, T.M.D., B.P.T., V.K.T., T.H.T., N.H.T. and C.D.V.; formal analysis, K.N.N., B.P.T., T.K.G.D., N.L.N., T.T.N. and T.H.T.; investigation, K.N.N., T.B.N.C., T.K.G.D., T.T.N., T.T.Q.T., L.T.P. and P.L.N.; data curation, T.M.D., T.B.N.C., B.P.T., V.K.T. and N.H.T.; writing—original draft preparation, K.N.N. and N.L.N.; writing—review and editing, K.N.N., T.B.N.C., N.L.N., V.K.T., T.T.N., L.T.P., P.L.N., T.H.T., N.H.T. and C.D.V.; visualization, T.M.D., T.B.N.C., B.P.T., T.K.G.D., N.L.N., V.K.T., T.T.N., T.T.Q.T., L.T.P. and C.D.V.; supervision, C.D.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Vietnam Ministry of Science and Technology under grant number ĐTĐL.CN-133/21.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Hanoi Medical University Institutional Ethical Review Board (Approval No: 940/GCN-HĐĐĐNCYSH-ĐHYHN on 30 June 2023).
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bouchard, L.; Robert, M.-F.; Vinarov, D.; Stanley, C.A.; Thompson, G.N.; Morris, A.; Leonard, J.V.; Quant, P.; Hsu, B.Y.L.; Boneh, A.; et al. Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Clinical Course and Description of Causal Mutations in Two Patients. Pediatr Res 2001, 49, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Hegardt, F.G. Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase: A Control Enzyme in Ketogenesis. Biochemical Journal 1999, 338, 569–582. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of Ketone Bodies during Exercise and Training: Physiological Basis for Exogenous Supplementation. J Physiol 2017, 595, 2857–2871. [Google Scholar] [CrossRef]
- Shao, X.; Tang, Y.; Long, H.; Gu, H.; Zhang, J.; Deng, P.; Zhao, Y.; Cen, X. HMG-CoA Synthase 2 Drives Brain Metabolic Reprogramming in Cocaine Exposure. Neuropharmacology 2019, 148, 377–393. [Google Scholar] [CrossRef] [PubMed]
- Sait, H.; Srivastava, S.; Kumar, S.; Varughese, B.; Pandey, M.; Venkatramaiah, M.; Chaudhary, P.; Moirangthem, A.; Mandal, K.; Kapoor, S. Inborn Errors of Ketogenesis: Novel Variants, Clinical Presentation, and Follow-Up in a Series of Four Patients. J Pediatr Genet 2024, 13, 22–28. [Google Scholar] [CrossRef]
- Sass, J.O.; Fukao, T.; Mitchell, G.A. Inborn Errors of Ketone Body Metabolism and Transport: An Update for the Clinic and for Clinical Laboratories. J Inborn Errors Metab Screen 2018, 6, 2326409818771101. [Google Scholar] [CrossRef]
- Conboy, E.; Vairo, F.; Schultz, M.; Agre, K.; Ridsdale, R.; Deyle, D.; Oglesbee, D.; Gavrilov, D.; Klee, E.W.; Lanpher, B. Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Unique Presenting Laboratory Values and a Review of Biochemical and Clinical Features. JIMD Rep 2018, 40, 63–69. [Google Scholar] [CrossRef]
- Ma, D.; Yu, D. [Mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase deficiency: a case report and literature review]. Zhongguo Dang Dai Er Ke Za Zhi 2018, 20, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Miao, J.-K.; Yu, C.-W.; Wan, K.-X.; Zhang, J.; Yuan, Z.-J.; Yang, J.; Wang, D.-J.; Zeng, Y.; Zou, L. Severe Clinical Manifestation of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency Associated with Two Novel Mutations: A Case Report. BMC Pediatr 2019, 19, 344. [Google Scholar] [CrossRef]
- Kılıç, M.; Dorum, S.; Topak, A.; Yazıcı, M.U.; Ezgu, F.S.; Coskun, T. Expanding the Clinical Spectrum of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency with Turkish Cases Harboring Novel HMGCS2 Gene Mutations and Literature Review. Am J Med Genet A 2020, 182, 1608–1614. [Google Scholar] [CrossRef]
- Wu, S.; Shen, L.; Chen, Q.; Gong, C.; Yang, Y.; Wei, H.; Cao, B.; Chen, Y. Clinical, Biochemical, Molecular, and Outcome Features of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency in 10 Chinese Patients. Front Genet 2021, 12, 816779. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, Y.-L.; Liu, M.; Chen, J.-J.; Li, X.-Q.; Cao, B.-Y.; Gong, C.-X. Clinical, Biochemical, Molecular and Therapeutic Characteristics of Four New Patients of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency. Clin Chim Acta 2020, 509, 83–90. [Google Scholar] [CrossRef]
- Arnedo, M.; Ramos, M.; Puisac, B.; Gil-Rodríguez, M.C.; Teresa, E.; Pié, Á.; Bueno, G.; Ramos, F.J.; Gómez-Puertas, P.; Pie, J.; et al. Mitochondrial HMG–CoA Synthase Deficiency. In Advances in the Study of Genetic Disorders; IntechOpen, 2011; pp. 189–204, ISBN 978-953-307-305-7.
- Pitt, J.J.; Peters, H.; Boneh, A.; Yaplito-Lee, J.; Wieser, S.; Hinderhofer, K.; Johnson, D.; Zschocke, J. Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Urinary Organic Acid Profiles and Expanded Spectrum of Mutations. J Inherit Metab Dis 2015, 38, 459–466. [Google Scholar] [CrossRef]
- Puisac, B.; Marcos-Alcalde, I.; Hernández-Marcos, M.; Tobajas Morlana, P.; Levtova, A.; Schwahn, B.C.; DeLaet, C.; Lace, B.; Gómez-Puertas, P.; Pié, J. Human Mitochondrial HMG-CoA Synthase Deficiency: Role of Enzyme Dimerization Surface and Characterization of Three New Patients. Int J Mol Sci 2018, 19, 1010. [Google Scholar] [CrossRef] [PubMed]
- Rojnueangnit, K.; Maneechai, P.; Thaweekul, P.; Piriyanon, P.; Khositseth, S.; Ittiwut, C.; Chetruengchai, W.; Kamolvisit, W.; Theerapanon, T.; Suphapeetiporn, K.; et al. Expanding Phenotypic and Mutational Spectra of Mitochondrial HMG-CoA Synthase Deficiency. Eur J Med Genet 2020, 63, 104086. [Google Scholar] [CrossRef]
- Conlon, T.A.; Fitzsimons, P.E.; Borovickova, I.; Kirby, F.; Murphy, S.; Knerr, I.; Crushell, E. Hypoglycemia Is Not a Defining Feature of Metabolic Crisis in Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency: Further Evidence of Specific Biochemical Markers Which May Aid Diagnosis. JIMD Rep 2020, 55, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Penzien, J.M.; Bielen, R.; Casals, N.; Aledo, R.; Pié, J.; Hoffmann, G.F.; Hegardt, F.G.; Mayatepek, E. The Diagnosis of Mitochondrial HMG-CoA Synthase Deficiency. J Pediatr 2002, 140, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, A.D.; Cooper, H.; Lee, C.U. Mitochondrial HMG-CoA Synthase Deficiency: A Cyclic Vomiting Mimic Without Reliable Biochemical Markers. J Investig Med High Impact Case Rep 2024, 12, 23247096241267154. [Google Scholar] [CrossRef]
- Czempik, P.F.; Pluta, M.P.; Krzych, Ł.J. Sepsis-Associated Brain Dysfunction: A Review of Current Literature. Int J Environ Res Public Health 2020, 17, 5852. [Google Scholar] [CrossRef] [PubMed]
- Purdie, F.R.; Honigman, B.; Rosen, P. Acute Organic Brain Syndrome: A Review of 100 Cases. Ann Emerg Med 1981, 10, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Takami, Y.; Yamada, K.; Kobayashi, H.; Hasegawa, Y.; Sasai, H.; Otsuka, H.; Takeshima, Y.; Fukao, T. A Japanese Case of Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Synthase Deficiency Who Presented with Severe Metabolic Acidosis and Fatty Liver without Hypoglycemia. JIMD Rep 2019, 48, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Fam, S.; Chen, H.; Wilson, S.; Ayers, K.; Hughes, J.; Sloan-Bena, F.; Calvel, P.; Robevska, G.; Puisac, B.; Kusz-Zamelczyk, K.; et al. The Gene Encoding the Ketogenic Enzyme HMGCS2 Displays a Unique Expression during Gonad Development in Mice. PLoS One 2020, 15, e0227411. [Google Scholar] [CrossRef]
- Nguyen, K.N.; Tran, V.K.; Nguyen, N.L.; Can, T.B.N.; Dang, T.K.G.; Nguyen, T.H.; Do, T.T.M.; Phuong, L.T.; Tran, T.H.; Ta, T.V.; et al. Hyperornithinemia–Hyperammonemia–Homocitrullinuria Syndrome in Vietnamese Patients. Medicina 2024, 60, 1877. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Kikawa, Y.; Mayumi, M.; Tanaka, Y.; Sudo, M.; Kado, N. Modifications in Electrospray Tandem Mass Spectrometry for a Neonatal-Screening Pilot Study in Japan. J Chromatogr B Biomed Appl 1999, 731, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hirano, S.; Hata, I.; Tanaka, Y.; Sudo, M.; Sakura, N.; Tajima, T.; Yamaguchi, S. Newborn Mass Screening and Selective Screening Using Electrospray Tandem Mass Spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci 2002, 776, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Magnetic resonance imaging (MRI) of three Vietnamese cases with Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D). (a) P3: Coronal FLAIR, axial T1-weighted, and axial T2-weighted images showed symmetric hyperintensities involving both head of caudate nuclei and putamina. (b) P9: Coronal FLAIR image showed bilateral hyperintensities of periventricular deep white matters and left parietal cortex; axial T1-weighted post-gadolinium image showed enhancement of subcortical area in the left frontal and parietal lobe; axial T2-weighted image showed bilateral symmetric hyperintensities involving putamen. (c) P12: Axial T2-weighted MR image showed bilateral ventricular dilatation and widening of temporal subarachnoid space predominant in the left side.
Figure 1.
Magnetic resonance imaging (MRI) of three Vietnamese cases with Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency (HMGCS2D). (a) P3: Coronal FLAIR, axial T1-weighted, and axial T2-weighted images showed symmetric hyperintensities involving both head of caudate nuclei and putamina. (b) P9: Coronal FLAIR image showed bilateral hyperintensities of periventricular deep white matters and left parietal cortex; axial T1-weighted post-gadolinium image showed enhancement of subcortical area in the left frontal and parietal lobe; axial T2-weighted image showed bilateral symmetric hyperintensities involving putamen. (c) P12: Axial T2-weighted MR image showed bilateral ventricular dilatation and widening of temporal subarachnoid space predominant in the left side.
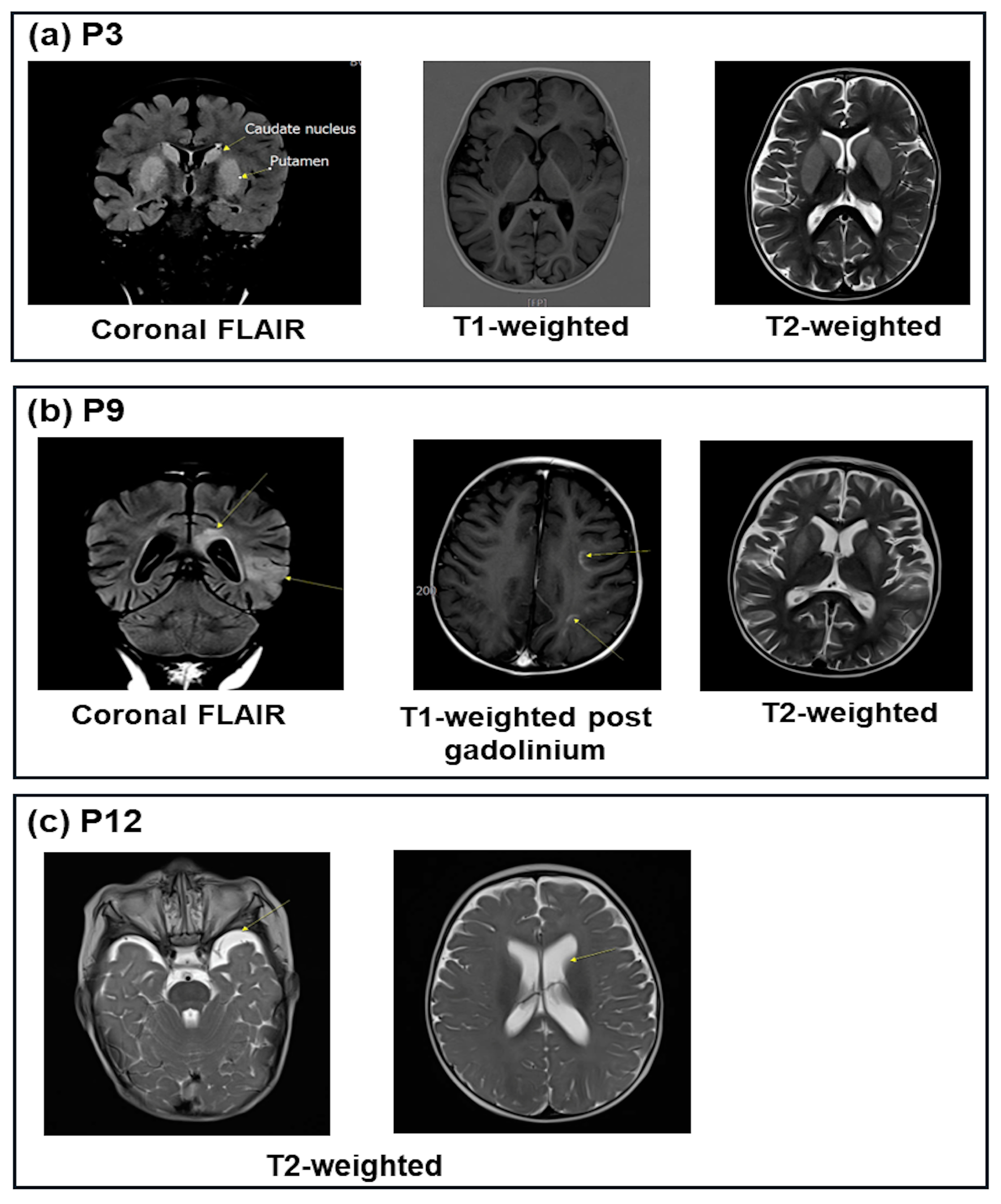
Figure 2.
Scheme of the distribution of HMGCS2 variants in 19 Vietnamese patients with mitochondrial HMG-CoA synthase deficiency.
Figure 2.
Scheme of the distribution of HMGCS2 variants in 19 Vietnamese patients with mitochondrial HMG-CoA synthase deficiency.
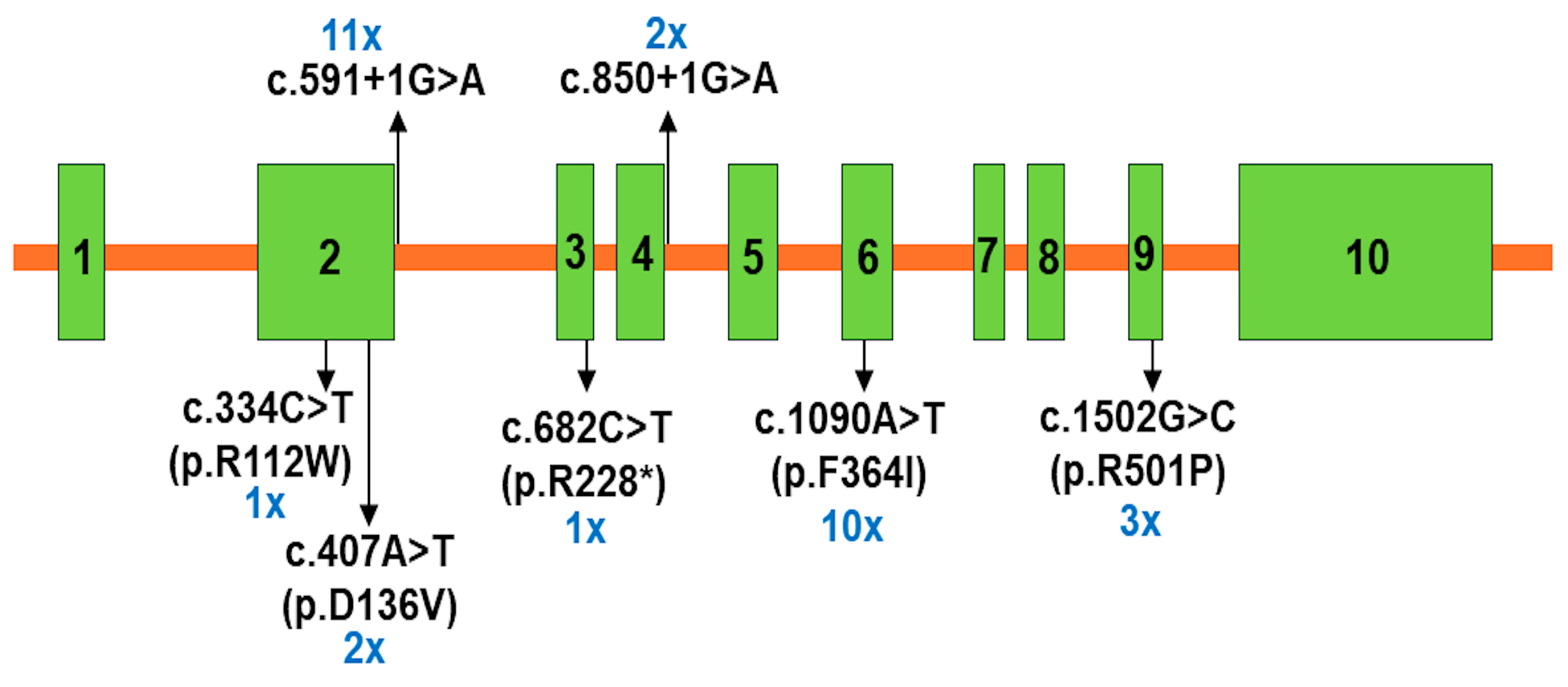
Figure 3.
Pedigree and Sanger sequencing chromatograms of HMGCS2 variants in the three families of the study. Rectangles and circles represent males and females; “-/” represents for normal allele; arrows indicate the probands; partially filled symbols indicate carrier parents; filled symbol represent affected individuals; Het, heterozygous; WT, wild-type. Patient P8 inherited c.334C>T (p.R112W) from her father and c.591+1G>A from her mother. Patient P10 and P11 inherited c.407A>T (p.D136V) from their father and c.850+1G>A from their mother. Patient P13 and P14 inherited c.1090T>A (p.F364I) from their father and c.1502G>C (p.R501P) from their mother.
Figure 3.
Pedigree and Sanger sequencing chromatograms of HMGCS2 variants in the three families of the study. Rectangles and circles represent males and females; “-/” represents for normal allele; arrows indicate the probands; partially filled symbols indicate carrier parents; filled symbol represent affected individuals; Het, heterozygous; WT, wild-type. Patient P8 inherited c.334C>T (p.R112W) from her father and c.591+1G>A from her mother. Patient P10 and P11 inherited c.407A>T (p.D136V) from their father and c.850+1G>A from their mother. Patient P13 and P14 inherited c.1090T>A (p.F364I) from their father and c.1502G>C (p.R501P) from their mother.
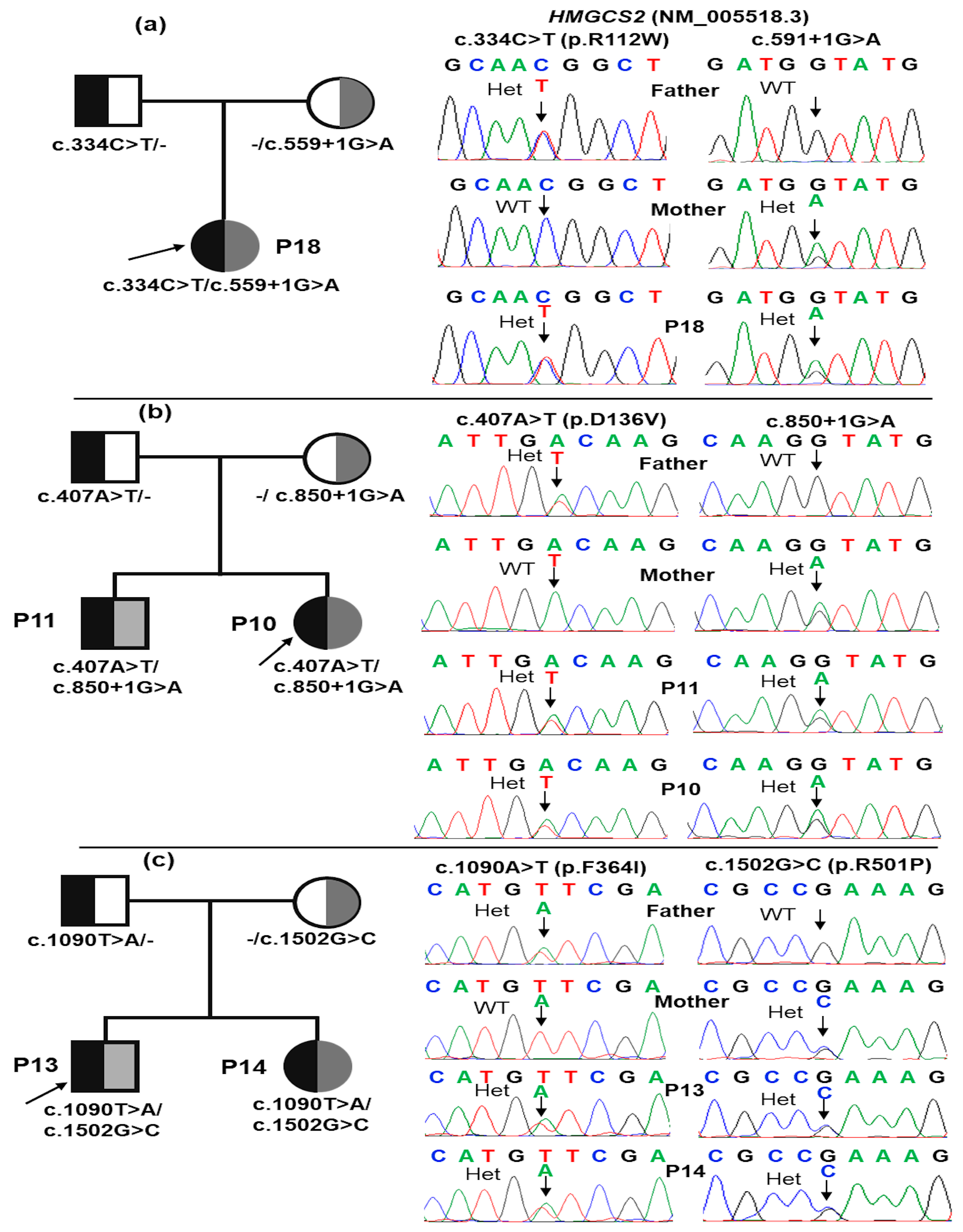
Figure 4.
The D136V change in three-dimensional structure of human mitochondrial 3-hydroxy-3-methylglutaryl- coenzyme a synthase 2 (PDB ID: 2WYA). Wildtype is D136 and mutant is V136. The D136V change causes losses of H-bond between D136 and K137, S138, and K139.
Figure 4.
The D136V change in three-dimensional structure of human mitochondrial 3-hydroxy-3-methylglutaryl- coenzyme a synthase 2 (PDB ID: 2WYA). Wildtype is D136 and mutant is V136. The D136V change causes losses of H-bond between D136 and K137, S138, and K139.
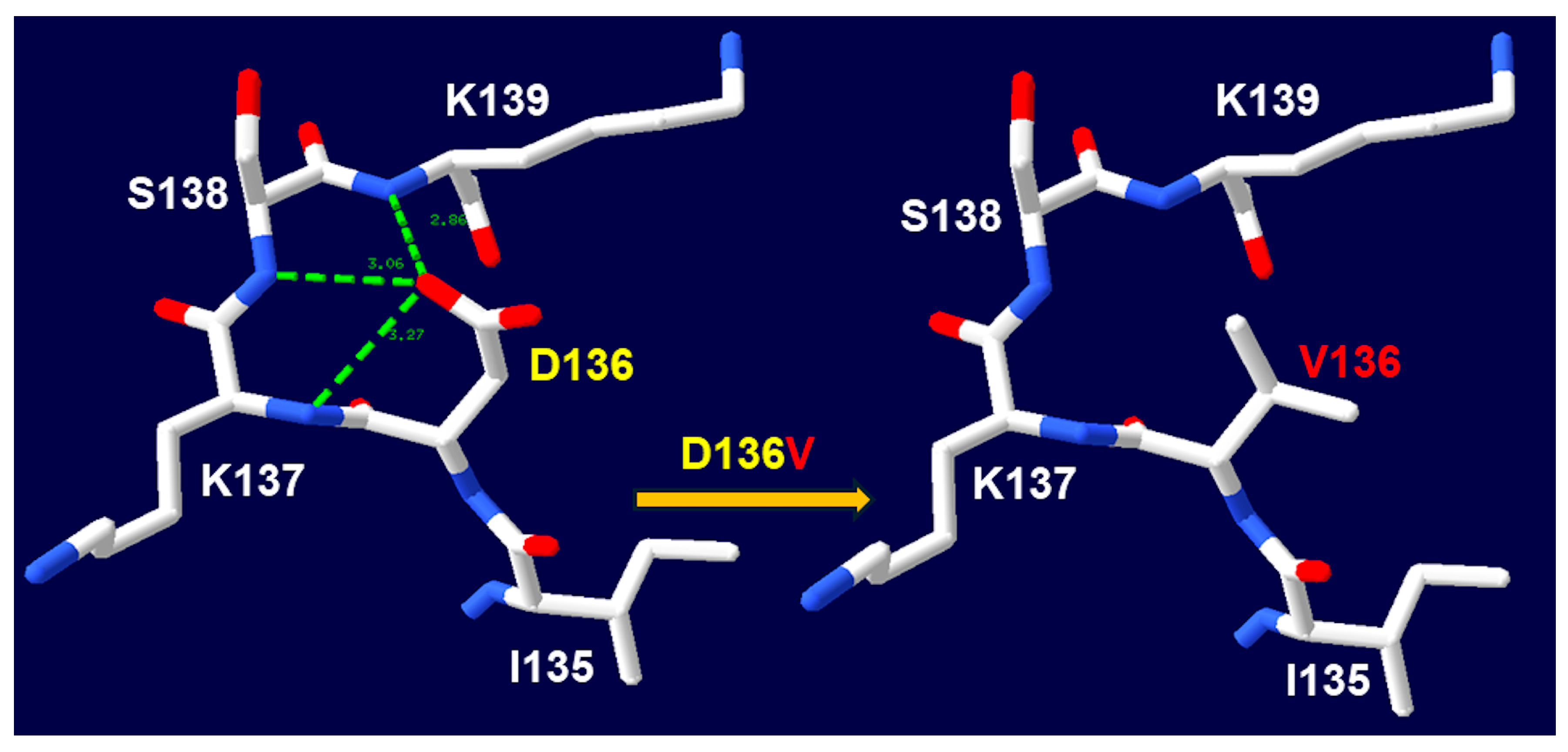
Table 2.
Biochemical features of 16 symptomatic cases at the first episode.
| Pt | Blood glucose mmol/L | metabolic acidosis | Plasma ammoniac μmol/L | ALT UI/L | AST UI/L | CK IU/L | Plasma C0 µmol/L | Plasma C2 µmol/L | Urinary dicarboxylic acid | Ketonuria | Brain MRI | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH | HCO3 mmol/L | BE | |||||||||||
| P1 | ↓2.0 | 6.96 | 4.2 | -27.0 | 98.4 | ↑88 | ↑138 | NA | 13.2 | 8.5 | - | 1+ | Normal |
| P2 | ↓1.1 | 7.25 | 5.3 | -22.0 | 64.2 | ↑62 | ↑112 | 126.8 | 14.9 | 9.5 | - | Neg | Normal |
| P3 | ↓0.8 | 7.12 | 9.6 | -21.0 | 51.6 | ↑108 | ↑209 | ↑2287 | ↓4.4 | 27.0 | - | 1+ | Lesions of the bilateral putamen caudate |
| P5 | 4.8 | 7.30 | 5.9 | -20.0 | 38.4 | ↑212 | ↑159 | NA | 9.1 | 9.5 | - | 3+ | Normal |
| P6 | ↓1.8 | 6.99 | 4.6 | -27.0 | ↑109.2 | ↑83 | ↑290 | NA | ↓5.0 | 4.6 | - | Trace | NA |
| P7 | 3.9 | 6.86 | 2.7 | -29.0 | ↑983.0 | ↑56 | ↑139 | NA | ↓5.0 | 4.7 | - | Neg | NA |
| P8 | ↓0.1 | 7.47 | 25.2 | 0.5 | 37.2 | ↑133 | ↑148 | ↑297 | ↓6.0 | 20.4 | - | Neg | Normal |
| P9 | 3.82 | 7.00 | 8.6 | -20.0 | 49.2 | ↑126 | ↑143 | NA | 16.7 | 8.8 | - | Neg | Bilateral cerebrum lesions |
| P10 | ↑25.45 | 6.89 | 1.5 | -31.0 | ↑156.6 | ↑57 | ↑222 | NA | 6.9 | 27.3 | - | Trace | Normal |
| P12 | ↓2.48 | 7.37 | 7.5 | -15.6 | 36.4 | ↑132 | ↑270 | NA | ↓3.8 | 19.7 | - | 1+ | Slight ventilation dilation |
| P13 | 4.5 | 7.40 | 21.0 | -4.0 | 31.8 | ↑465 | ↑691 | ↑1198 | 10.1 | 22.5 | - | Neg | NA |
| P14 | 4.46 | 7.39 | 21.5 | -3.0 | 52.4 | ↑56 | ↑97 | 67 | 26.2 | 11.1 | - | Neg | NA |
| P15 | ↓2.4 | 7.05 | 3.2 | -24.0 | 71.0 | ↑105 | ↑280 | NA | ↓3.3 | 18.5 | - | Neg | NA |
| P16 | ↓0.9 | 7.41 | 23.0 | NA | ↑244.0 | ↑56 | ↑90 | ↑3267 | 8.4 | ↑62.8 | + | Neg | Normal |
| P18 | ↓2.08 | 7.10 | 3.0 | -21.0 | ↑226.0 | 32 | ↑84 | NA | 12.2 | 14.7 | - | Neg | Normal |
| P19 | 3.65 | 7.27 | 4.5 | -21.0 | 43.8 | ↑131 | ↑121 | 59 | ↓3.8 | 22.3 | + | Neg | Normal |
BE, base excess; AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase; ↓, low level; ↑, elevated level, -, none; +, yes; Neg, negative; 1+, positive with ketone level < 1.5 mmol/L; 3+, positive with 3.9 mmol/L ≤ ketone level < 7.8 mmol/L; NA, not analyzed; MRI, magnetic resonance imaging. In the metabolic acidosis columns, the bold font indicates that metabolic acidosis.
Table 3.
Molecular findings in 19 Vietnamese patients with HMGCS2D.
| Patients | Genotype | AA change | Effect |
Mutation Taster | dbSNP | ClinVar | AMCG classification | |
|---|---|---|---|---|---|---|---|---|
| 06 (P1, P2, P7, P9, P16, P17) |
Hom | c.559+1G>A | - | Splicing | Deleterious | rs587603096 | Pathogenic 859738 |
Pathogenic (PVS1, PM1, PM2, PM3, PP1, PP3, PP4, and PP5) |
| 05 (P3, P4, P6, P8, P12) |
CH | c.559+1G>A | - | Splicing | Deleterious | rs587603096 | Pathogenic 859738 |
Pathogenic (PVS1, PM1, PM2, PM3, PP1, PP3, PP4, and PP5) |
| c.1090T>A | F364I | Missense | Deleterious | rs1652807016 | Pathogenic 859739 | Likely pathogenic (PM1, PM2, PM3, PP1, PP3, PP4, and PP5) | ||
| 01 (P5) | Hom | c.1090T>A | F364I | Missense | Deleterious | rs1652807016 | Pathogenic 859739 | Likely pathogenic (PM1, PM2, PM3, PP1, PP3, PP4, and PP5) |
| 02 (P10, P11) |
CH | c.407A>T | D136V | Missense | Deleterious | - | - | Likely pathogenic (PM1, PM2, PM5, PP1, PP3, and PP4) |
| c.850+1G>A | - | Splicing | Deleterious | rs112412189 | - | Pathogenic (PVS1, PM2, PM3, PP1, PP3, and PP4) | ||
| 03 (P13, P14, P15) |
CH | c.1090T>A | F364I | Missense | Deleterious | rs1652807016 | Pathogenic 859739 | Likely pathogenic (PM1, PM2, PM3, PP1, PP3, PP4, and PP5) |
| c.1502G>C | R501P | Missense | Deleterious | rs372079931 | Pathogenic 452101 |
Pathogenic (PS3, PM1, PM2, PM3, PP1, PP3, PP4, and PP5) | ||
| 01 (P18) |
CH | c.682C>T | R228* | Nonsense | Deleterious | rs763531478 | Pathogenic 2080601 |
Pathogenic (PVS1, PM2, PM3, PP3, and PP4) |
| c.1090T>A | F364I | Missense | Deleterious | rs1652807016 | Pathogenic 859739 | Likely pathogenic (PM1, PM2, PM3, PP1, PP3, PP4, and PP5) | ||
| 01 (P19) |
CH | c.334C>T | R112W | Missense | Deleterious | rs768707273 | Likely pathogenic SCV003828997.2 |
Likely pathogenic (PM1, PM2, PM3, PP3, and PP4) |
| c.559+1G>A | - | Splicing | Deleterious | rs587603096 | Pathogenic 859738 |
Pathogenic (PVS1, PM1, PM2, PM3, PP1, PP3, PP4, and PP5) | ||
AA: amino acid; Hom, homozygous; CH, compound heterozygous; ACMG, American College of Medical Genetics and Genomics; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



