
Submitted:
08 February 2026
Posted:
10 February 2026
You are already at the latest version
Abstract
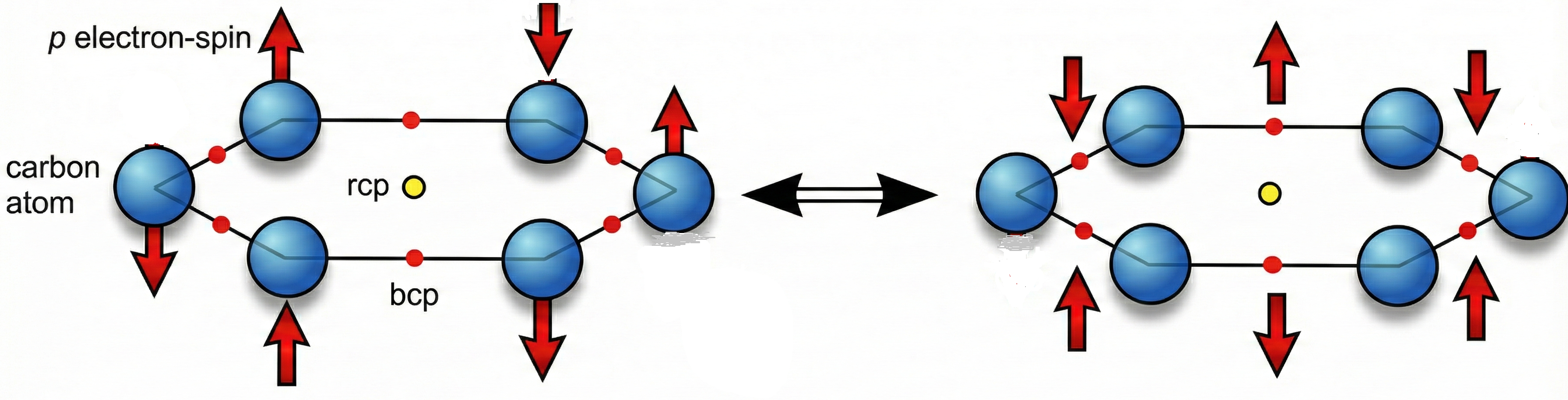
We present D2BIA_discrete, a zero-cost computational framework for predicting local aromaticity in linear acenes without quantum chemical calculations. The model partitions π-electron density using a 61/39 localized/delocalized ratio derived from spin-coupled valence bond and QTAIM analyses, combined with Slater-type orbital decay to estimate ring-center electron density. Three progressive refinements introduce molecular topology weighting, tunable bond-sharing (α), and depth-dependent attenuation (γ) parameters. Leave-one-n-out cross-validation on benzene through undecacene yields optimal parameters α=1.00 and γ=0.05, achieving ring-by-ring predictions with R²=0.456 and RMSE=2.394×10⁻³ against fluorescence aromaticity index (FLU). Model 2 provides excellent molecular-averaged correlations (R²>0.93 for FLU, PDI, HOMA) and reliable ring-level predictions for terminal and first internal rings. Local LONOCV D2BIA_discrete has excellent metrics with six-center index (R² = 0.922, R = 0.960, RMSE = 0.219, MAE = 0.139, MRE = 4.83%), showing importance of zero-cost D2BIA_discrete to obtain local aromaticity of linear acenes in agreement with SCI gradients for the same group. This is our third work based on the so-called discrete geometry chemistry.
Keywords:
D2BIA
; six-center index
; aromaticity index
; linear acenes
; discrete geometry chemistry
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



