
Submitted:
04 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Gut microbiota are integral to host health and ecological adaptation, yet their interactions with environmental microbial communities remain understudied in migratory waterbirds. Using high-throughput 16S rRNA gene sequencing, we compared gut microbiota of captive and wild Siberian cranes (Leucogeranus leucogeranus) and their associations with soil microbiota in the Poyang Lake wetland. Alpha diversity was significantly higher in soil than in gut microbiota, with captive cranes exhibiting greater microbial richness and evenness than wild individuals. Beta diversity analysis revealed distinct gut and soil microbiomes, with partial overlap between captive and wild crane gut microbiota. Firmicutes dominated gut communities, with Ligilactobacillus and Romboutsia enriched in captive cranes, whereas Acidobacteria were predominant in soil. Potential pathogens (e.g., Escherichia-Shigella) were more abundant in wild cranes and soil. LEfSe analysis identified 34 differentially enriched taxa, and microbial network analysis indicated stronger gut–soil microbial associations than those between captive and wild hosts, suggesting that environmental microbiota may serve as reservoirs for host colonization. These findings highlight the ecological dynamics shaping gut microbiota in response to captivity and environmental exposure, providing insights into microbial contributions to conservation strategies for Siberian cranes.
Keywords:
1. Introduction
2. Materials and Methods
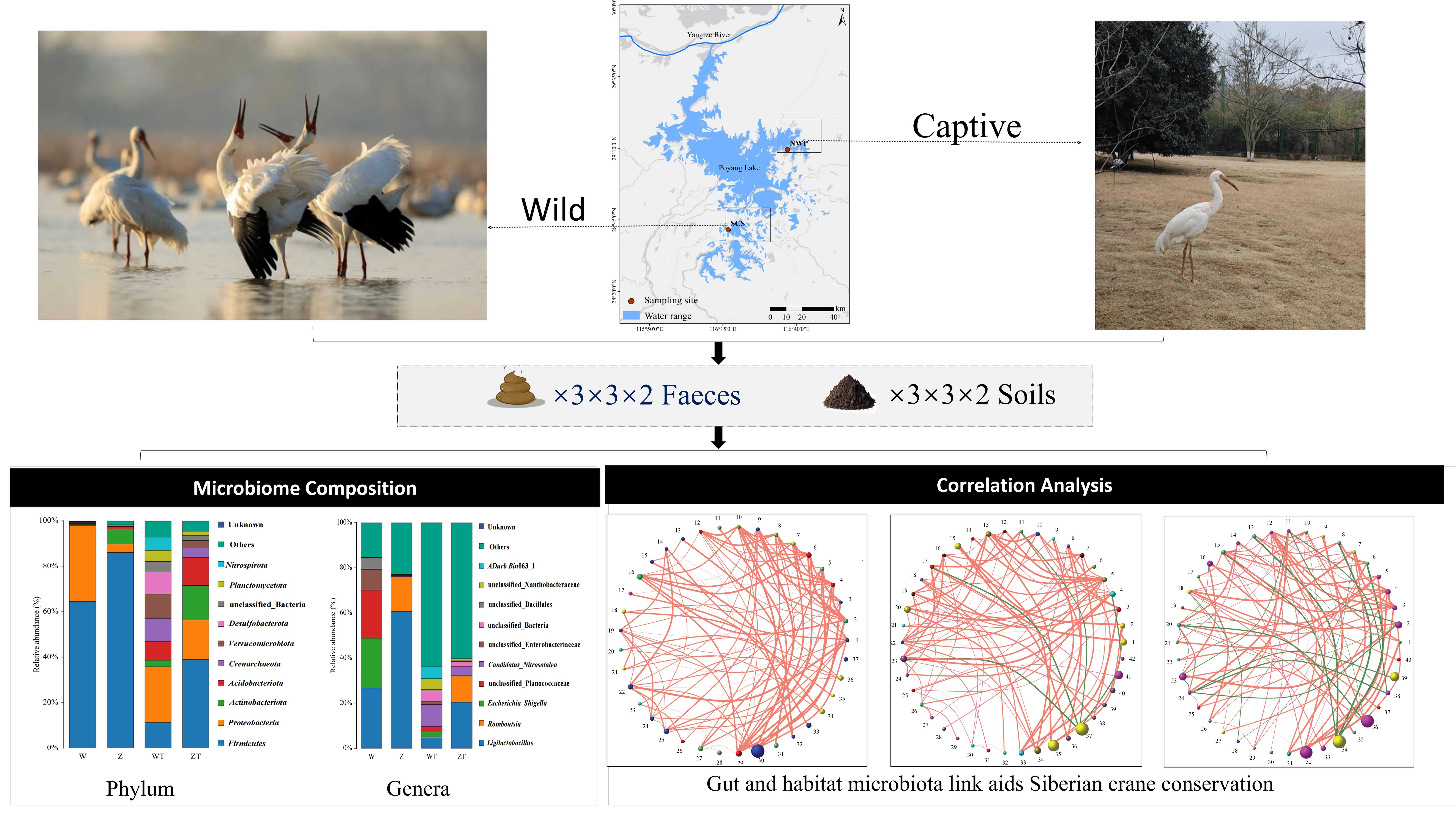
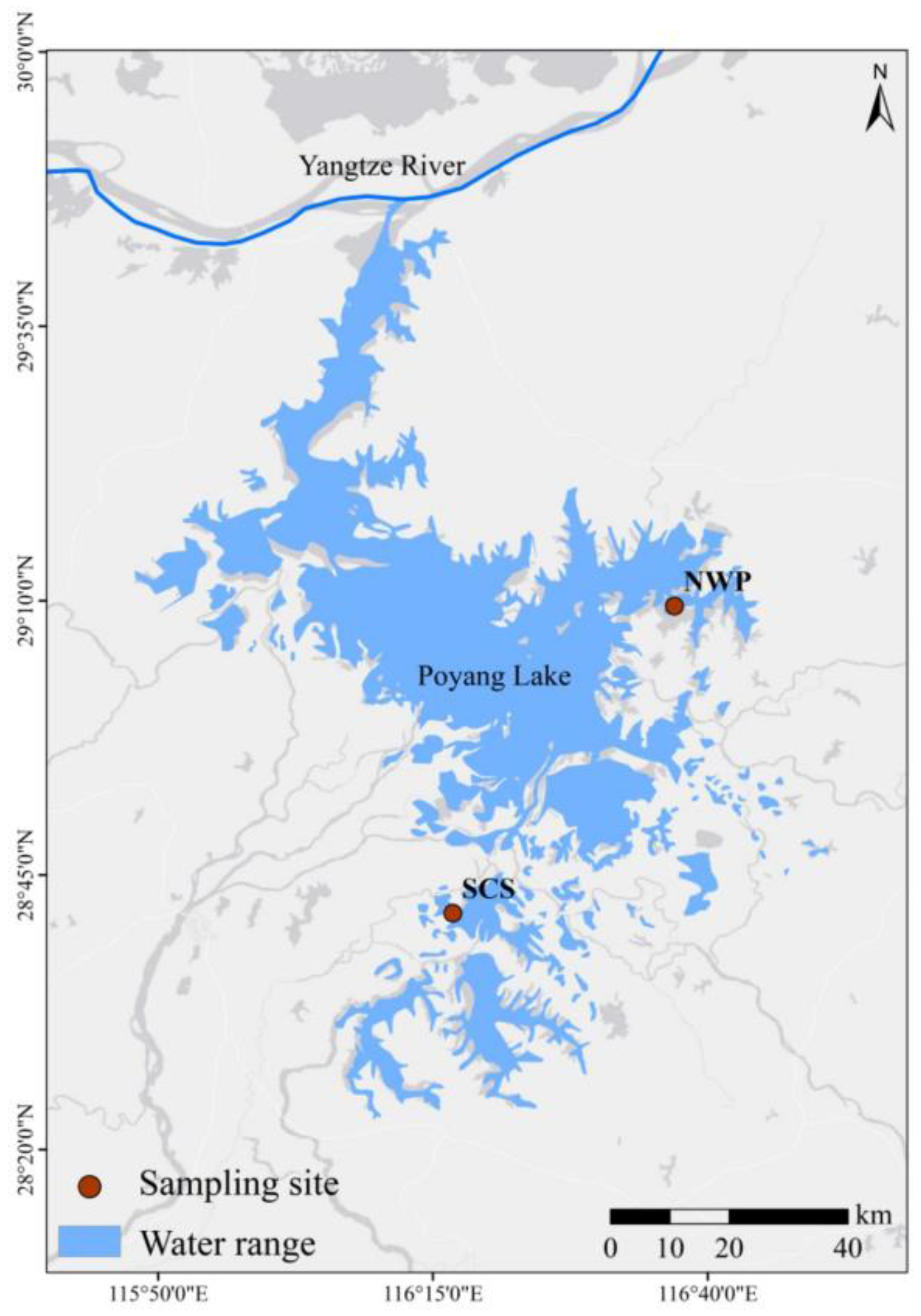
2.1. Study Area and Samples Collection
2.2. DNA Extraction, Amplification and High-Throughput Sequencing
2.3. Bioinformatics Processing and Ecological Statistical Analysis
- Raw data quality control: Raw sequencing reads were quality-filtered using Trimmomatic-v0.33 (SLIDINGWINDOW:4:20, MINLEN:100). Adapter sequences were removed using cutadapt-v1.91.
- Amplicon Sequence Variant (ASV) Analysis: The high-resolution ASV analysis was performed to characterize microbial community composition with single-nucleotide precision. Cleaned sequencing reads were processed using the DADA2 pipeline (v1.16.0) implemented in QIIME2 (v2020.6) to generate exact biological sequence variants (ASVs) [24].
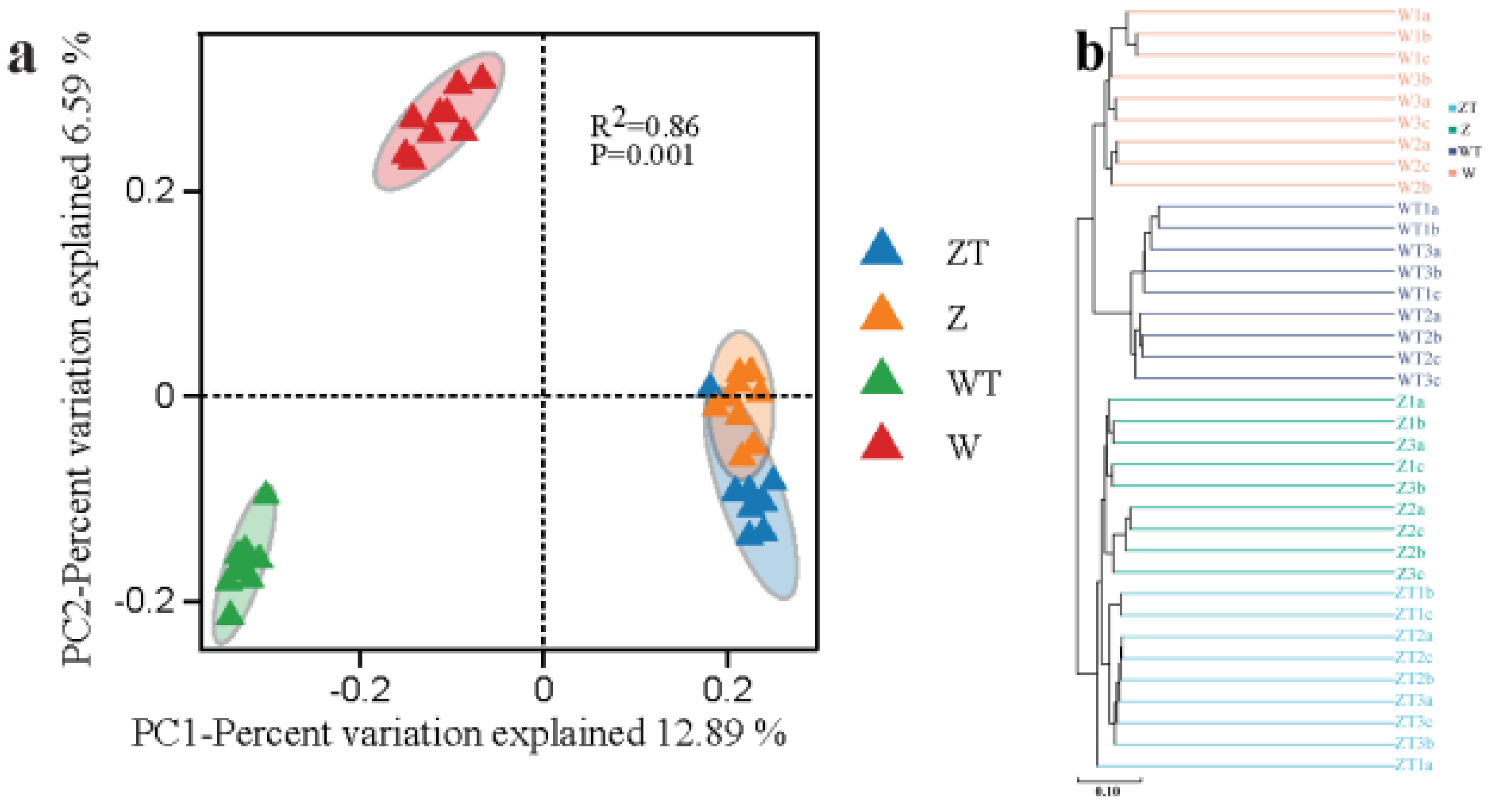
- Diversity Analysis: Alpha diversity was assessed using QIIME2-v2020.06. Beta diversity was assessed using principal coordinates analysis (weighted UniFrac PCoA), with significance testing via PERMANOVA (999 permutations) [25]. Hierarchical clustering of samples was analyzed using Bray-Curtis distance.
- Taxonomic Annotation and Biomarker Analysis: Taxonomic classification of amplicon sequences was performed using the SILVA database (version 138) with a confidence threshold of 85% [26]. Sequences were assigned to taxonomic ranks using the naïve Bayesian classifier implemented in QIIME2 [25]. To minimize potential misclassifications, only sequences with a confidence score above the predefined threshold were retained for downstream analyses. The community composition of each sample was statistically analyzed at various levels (phylum, class, order, family, genus, species). QIIME2 was used to generate abundance tables at different classification levels, and R was used to visualize the community structure of each sample at various taxonomic levels. Furthermore, biomarker taxa were identified using LEfSe (Kruskal-Wallis test, LDA score > 4.5, and FDR-adjusted p-values for multiple testing correction) [27].
- Cross-Domain Correlation and Network Analysis: Inter-domain microbial correlations were assessed using SparCC methods (Sparse Correlations for Compositional Data) with thresholds of |r| > 0.6 and p < 0.01. Network visualization was performed in Gephi, employing the Fruchterman-Reingold force-directed layout [28].
3. Results
3.1. Distinct Microbial Diversity, Composition, and Host-Environment Overlap Between Gut and Soil Microbiota
3.2. Phylum and Genus-Level Divergence in Gut and Soil Microbiota of Captive and Wild Siberian Cranes
3.3. Differential Microbial Abundance in Gut and Soil Microbiomes Revealed by LEfSe Analysis
3.4. Network-Based Insights into Gut and Soil Microbiota Interactions in Siberian Cranes

4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Liao, C.; Wu, L.; Tang, J.; Chen, J.; Lei, C.; Zheng, L.; Zhang, C.; Liu, Y.; Xavier, J.; et al. Ecological dynamics of the gut microbiome in response to dietary fiber. ISME J. 2022, 16, 2040–2055. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, E.C.; Metcalfe, N.B.; Llewellyn, M.S. The potential role of the gut microbiota in shaping host energetics and metabolic rate. J. Anim. Ecol. 2020, 89, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Chen, J.; Liu, K.; Tang, M.; Yang, Y. The avian gut microbiota: diversity, influencing factors, and future directions. Front. Microbiol. 2022, 13, 934272. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.; Gong, M.; Zheng, C.; Zhang, C.; Li, H.; Wen, W.; Wang, Y.; Liu, G. Diet-induced microbiome shifts of sympatric overwintering birds. Appl. Microbiol. Biotechnol. 2021, 105, 5993–6005. [Google Scholar] [CrossRef]
- Li, L.; Ye, J.; Yu, M.; Jiang, J.; Guo, X.; Yu, W.; Rong, K. Dynamic changes in the avian gut microbiome in response to diverse lifestyles. Appl. Microbiol. Biotechnol. 2025, 167, 331–344. [Google Scholar] [CrossRef]
- Hill, S.C.; François, S.; Thézé, J.; Smith, A.L.; Simmonds, P.; Perrins, C.M.; van der Hoek, L.; Pybus, O.G. Impact of host age on viral and bacterial communities in a waterbird population. ISME J. 2023, 17, 215–226. [Google Scholar] [CrossRef]
- Eliades, S.J.; Brown, J.C.; Colston, T.J.; Fisher, R.N.; Niukula, J.B.; Gray, K.; Siler, C.D. Gut microbial ecology of the critically endangered Fijian crested iguana (Brachylophus vitiensis): Effects of captivity status and host reintroduction on endogenous microbiomes. Ecol. Evol. 2021, 11, 4731–4743. [Google Scholar] [CrossRef]
- Tang, S.; Li, Y.; Huang, C.; Yan, S.; Li, Y.; Chen, Z.; Wu, Z. Comparison of gut microbiota diversity between captive and wild tokay gecko (Gekko gecko). Front. Microbiol. 2022, 13, 897923. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Chen, Q.; Ding, H. Effects of diet shift on the gut microbiota of the critically endangered Siberian crane. Avian Res. 2023, 14, 100108. [Google Scholar] [CrossRef]
- Heijtz, R.D.; Wang, S.G.; Anura, F.; Q., Y.; Bjorkholm, B.; Samuelsson, A.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.C.; Bordenstein, S. Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 2016, 26, 1175–1189. [Google Scholar] [CrossRef]
- Zheng, Z.X. Birds of the World, 2nd ed.; Science Press: Beijing, China, 2002. [Google Scholar]
- Hou, J.; Li, L.; Wang, Y.; Wang, W.; Zhan, H.; Dai, N.; Lu, P. Influences of submerged plant collapse on diet composition, breadth, and overlap among four crane species at Poyang Lake, China. Front. Zool 2021, 18, 24. [Google Scholar] [CrossRef]
- Shao, M.; Guo, H.; Jiang, J. Population sizes and group characteristics of Siberian Crane (Leucogeranus leucogeranus) and Hooded Crane (Grus monacha) in Poyang Lake Wetland. Zool. Res. 2014, 35, 373–379. [Google Scholar] [CrossRef]
- Wang, Y.; Long, Z.; Zhang, Y.; Li, X.; Zhang, X.; Su, H. Host genetic background rather than diet-induced gut microbiota shifts of sympatric black-necked crane, common crane and bar-headed goose. Front. Microbiol. 2023, 12, 1270716. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Sheng, Y.; Xiao, L.; Yang, H.; Yang, W.; Jian, M. Gut microbiota structure and function of Siberian crane (Grus leucogeranus) overwintering in Poyang Lake. Acta Microbiol. Sin. 2023, 63, 4302–4314. [Google Scholar] [CrossRef]
- Fierer, N.; Wood, S.; Mesquita, C.P.B.D. How microbes can, and cannot, be used to assess soil health. Soil Biol. Biochem. 2021, 153, 108111. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-Gonzalez, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.t.; Singh, B.K.; Fiere, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Zhou, D.; Bai, Z.; Zhang, H.; Li, N.; Bai, Z.; Cheng, F.; Lu, Z. Soil is a key factor influencing gut microbiota and its effect is comparable to that exerted by diet for mice. F1000Res 2018, 7, 1588. [Google Scholar] [CrossRef]
- Shao, M.Q. Bird Diversity and Ecological Habits of Poyang Lake and Five River Systems; Science Press: Beijing, China, 2021. [Google Scholar]
- Dennis, K.L.; Wang, Y.W.; Blatner, N.R.; Wang, S.Y.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A.; et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013, 73, 5905–5913. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 2018, 6, 140. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xia, P.; Wang, P.; Yu, H.; Giesy, J.P.; Zhang, Y.; Zhang, X. Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 2016, 6, 33350. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righin, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Van, T.B.; Van, M.V.; et al. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 10376–10381. [Google Scholar] [CrossRef]
- McKenzie, V.J.; Song, S.J.; Delsuc, F.; Prest, T.L.; Oliverio, A.M.; Korpita, T.M.; et al. The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 2017, 57, 690–704. [Google Scholar] [CrossRef]
- Chen, J.; Xu, Y.; Liu, Y.; Liu, K.; Wu, Y.; Zhang, Y.; Yang, Y. Gut microbiota analysis and gene function prediction among young and adult Larus saundersi with habitat soil in the Yellow River Delta. Bioresour. Technol. Rep. 2022, 19, 100960. [Google Scholar] [CrossRef]
- Justel-Díez, M.; Delgadillo-Nuño, E.; Gutiérrez-Barral, A.; García-Otero, P.; Alonso-Barciela, I.; Pereira-Villanueva, P.; Álvarez-Salgado, X.A.; Velando, A.; Teira, E.; Fernández, E. Inputs of seabird guano alter microbial growth, community composition and the phytoplankton–bacterial interactions in a coastal system. Environ. Microbiol. 2023, 25, 1155–1173. [Google Scholar] [CrossRef]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe. 2021, 29, 1366–1377. [Google Scholar] [CrossRef]
- Wu, H.; Wu, N.; Liu, X.; Zhang, L.; Zhao, D. Diet drives gut bacterial diversity of wild and semi-captive common cranes (Grus grus). Animals 2024, 14, 1566. [Google Scholar] [CrossRef]
- Ai, C.; Zhang, S.; Zhang, X.; Guo, D.; Zhou, W.; Huang, S. Distinct responses of soil bacterial and fungal communities to changes in fertilization regime and crop rotation. Geoderma 2018, 319, 156–166. [Google Scholar] [CrossRef]
- Zhang, N.; Zhou, L.; Yang, Z.; Gu, J. Effects of food changes on intestinal bacterial diversity of wintering hooded cranes (Grus monacha). Animals 2021, 11, 433. [Google Scholar] [CrossRef]
- Zhao, X.; Ye, W.; Xu, W.; Xu, N.; Zheng, J.; Chen, R.; Liu, H. Changes in the diversity and composition of gut microbiota of Red-Crowned Cranes (Grus japonensis) after avian influenza vaccine and anthelmintic treatment. Animals 2022, 12, 1183. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef]
- Diaz, J.; Reese, A.T. Possibilities and limits for using the gut microbiome to improve captive animal health. Anim. Microbiome 2021, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.V.D.M.; Coelho, B.D.O.; Júnior, A.I.M.; Thomaz-Soccol, V.; Soccol, C.R. How to select a probiotic? A review and update of methods and criteria. Biotechnol. Adv. 2018, 36, 2060–2076. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. bioRxiv 2019, 845511. [Google Scholar] [CrossRef]
- Haidar-Ahmad, N.; Manigat, F.O.; Silue, N.; Pontier, S.M.; Campbell-Valois, F.X. A tale about Shigella: Evolution, plasmid, and virulence. Microorganisms 2023, 11, 1709. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.L.; Wiley, N.; Cooke, A.C.; Johnson, C.N.; Fouhy, F.; Reichert, M.S.; de la Hera, I.; Crane, J.M.S.; Kulahci, I.G.; Ross, R.P.; et al. Diet induces parallel changes to the gut microbiota and problem solving performance in a wild bird. Sci. Rep. 2020, 10, 20783. [Google Scholar] [CrossRef] [PubMed]
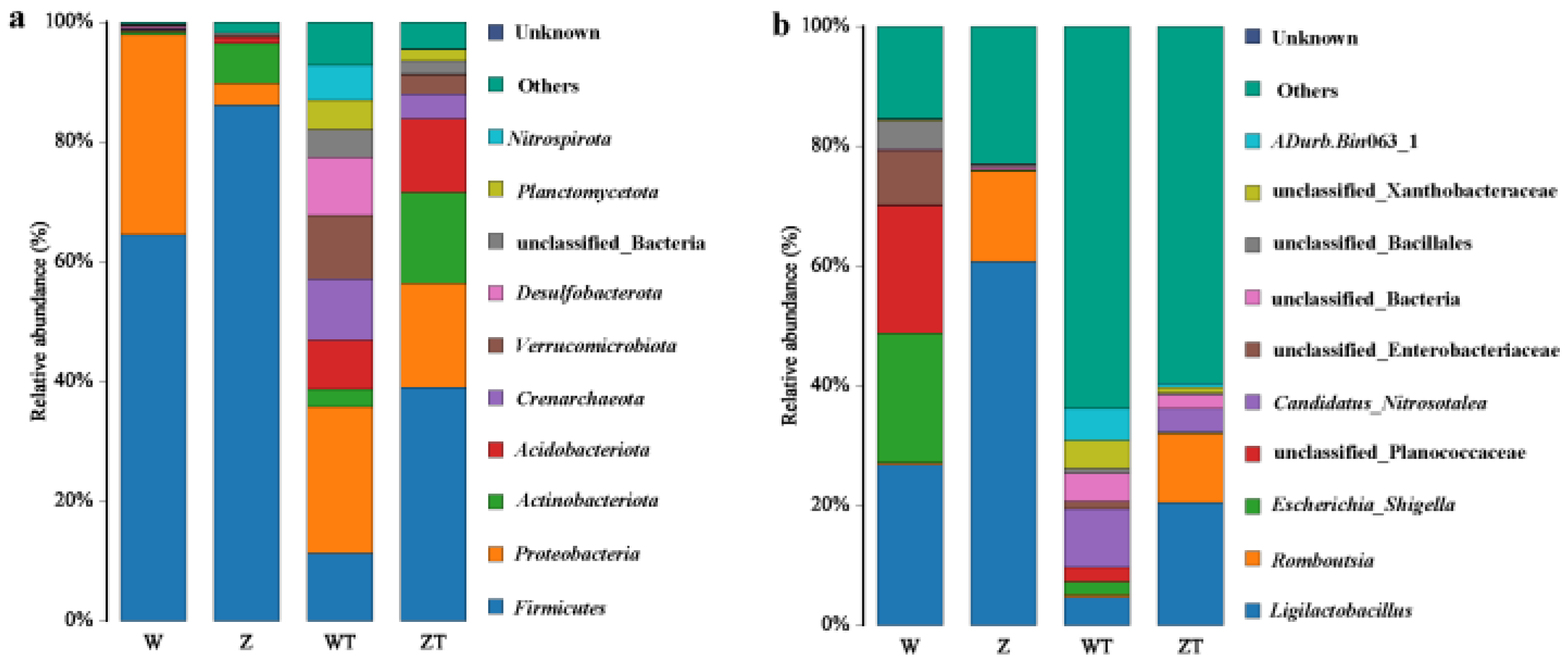
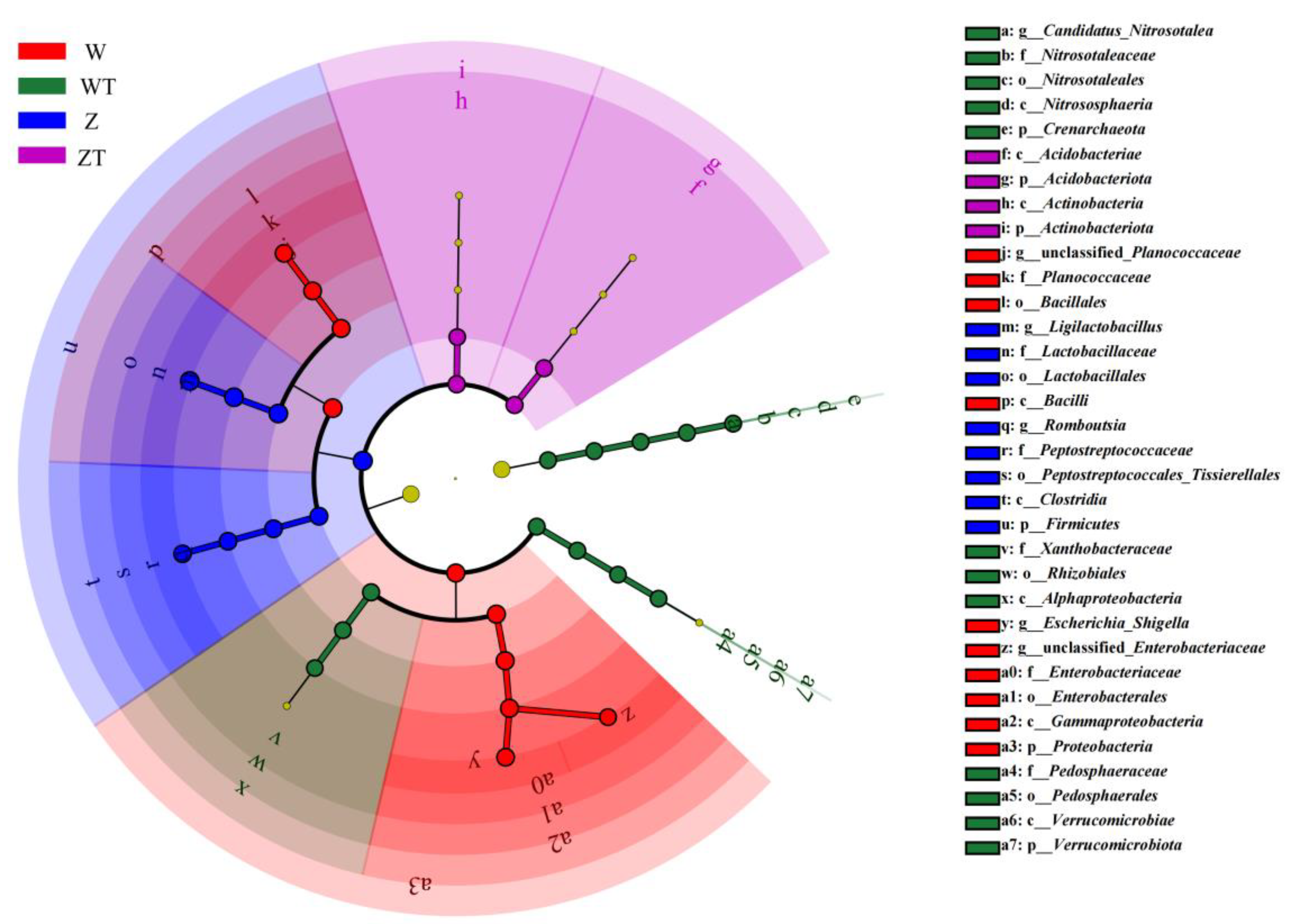
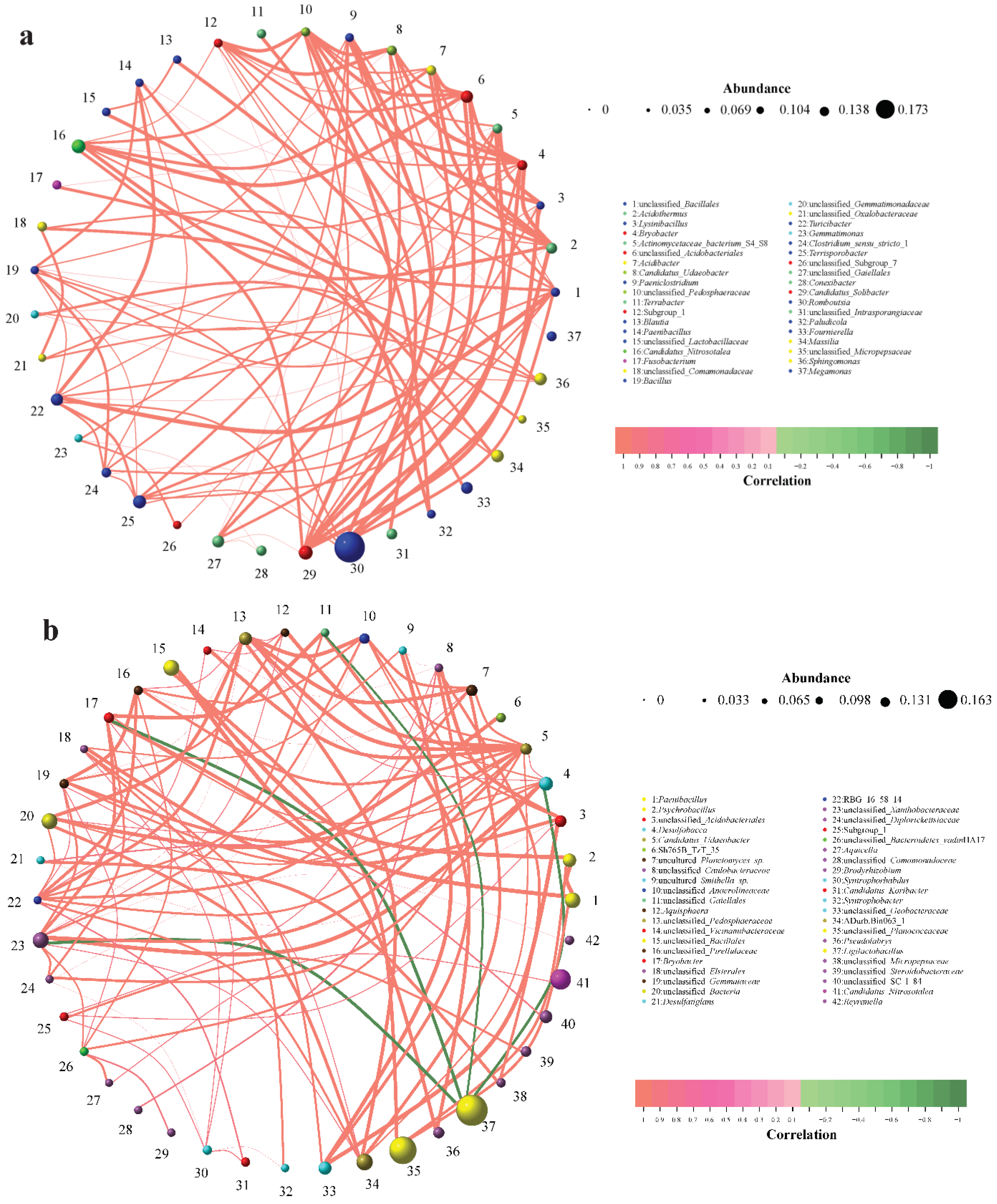
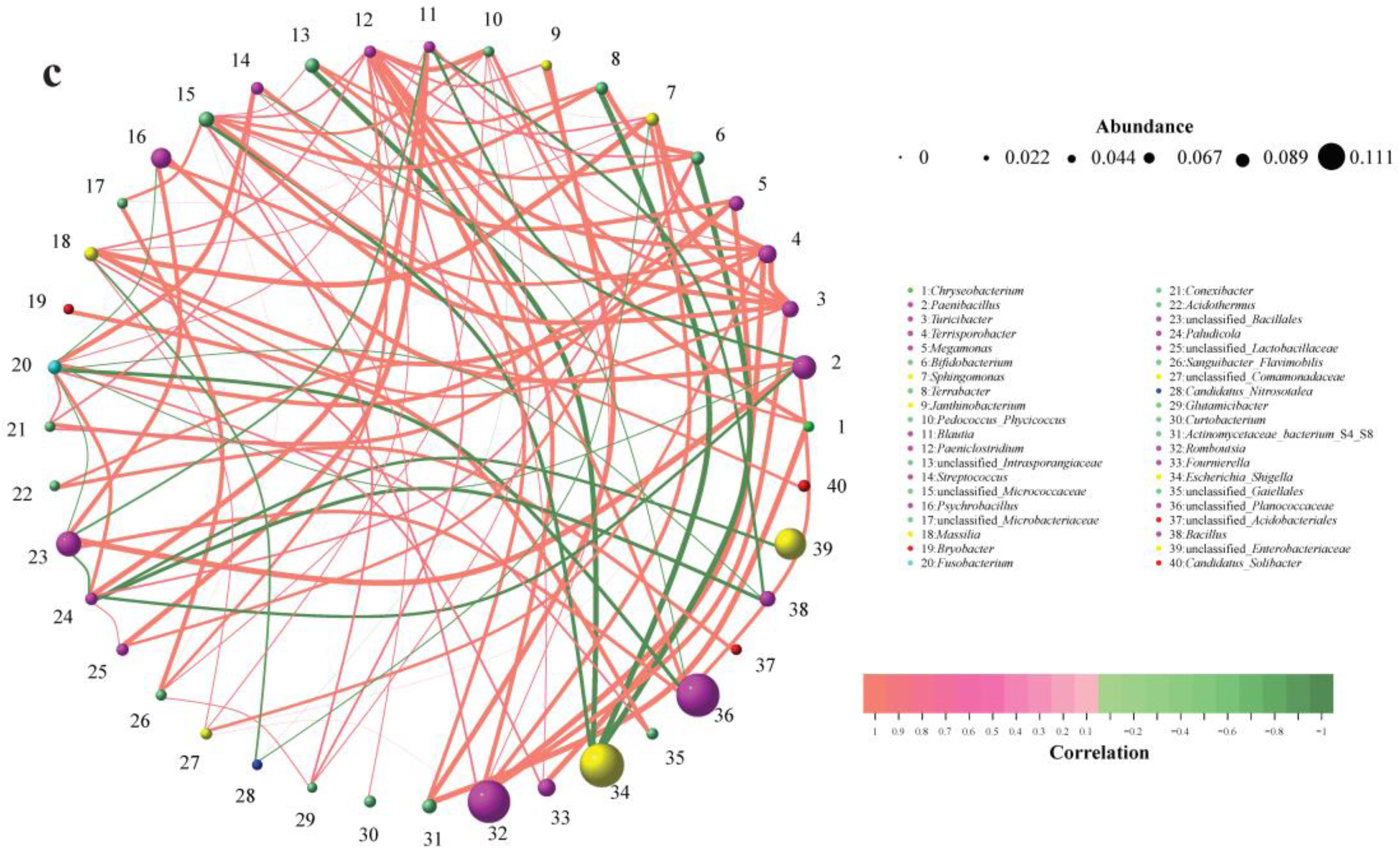

| Sample | ASV | ACE | Chao1 | Simpson | Shannon |
| W | 220±38c | 229±13c | 239±13c | 0.794±0.010c | 3.213±0.111d |
| Z | 415±93b | 418±31b | 421±30b | 0.821±0.012c | 3.965±0.091c |
| WT | 734±134a | 739±45a | 743±44a | 0.989±0.002a | 8.027±0.075a |
| ZT | 676±241a | 678±80a | 681±80a | 0.949±0.015b | 6.916±0.278b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



