Submitted:
30 January 2026
Posted:
02 February 2026
You are already at the latest version
Abstract
Rabbits’ genetic improvement has evolved from traditional phenotypic selection and crossbreeding to a more sophisticated genomic and gene-editing technologies. The comparative efficacy and effectiveness of the current genomic methodologies is crucial for sustainable animal agriculture. As global demand for animal protein continues to widen, efficient genetic im-provement strategies have become increasingly necessary to meet consumption needs. This review evaluates traditional and modern approaches to rabbit genetic improvement, comparing their efficacy, implementation challenges, and future prospects in sustainable rabbit production systems. The findings show that traditional breeding, while foundational, is of-ten constrained by the low heritability of key traits, long generation intervals, and environmental influences. The advent of molecular genetics has introduced more precise tools, such as microsatellites markers, SNPs markers and Whole genome sequencing techniques, which promise to accelerate genetic gain for economically important traits like growth rate, feed efficiency, litter size, disease resistance among others. The review also found that gene editing technologies including Zinc-Finger Nucleases, Transcription Activator-Like Endonucleases and Clustered Regularly Interspaced Short Palin-dromic Repeats-Cas9 play vital role in the improvement of rabbit genetics. However, implementation success depends heavily on population structure, economic resources, and the availability of technical infrastructures especially in developing countries. Integrating the strengths of all the technologies synergistically offers the greatest potential for sustainable rabbit genetic improvement.
Keywords:
gene editing
; quantitative trait
; marker-assisted selection
; microsatellite markers
; CRISPR-Cas9
; gene flow
1. Introduction
Rabbits (Oryctolagus cuniculus) production represents rapidly expanding sector of global livestock agriculture. According to [1] there is the need to increase food production in order to achieve food sufficiency while maintaining environmental sustainability including checking climate change, avoiding deforestation, conserving biodiversity as well as ensuring animal health and welfare [3,4,5,6]. Rabbit production serves as alternative source of protein that has the potential to support food supply. The advantages of rabbits as micro-livestock include; short generation intervals, high prolificacy, efficient feed conversion ratios, and high-quality protein which position it as an idea species for sustainable meat production. This has necessitated the need to improve the genetics of wild and domesticated rabbits especially those in developing countries to meet the food supply of the growing population. Traditional methods of rabbit genetic improvements have successfully been implemented and enhanced production traits through systematic phenotypic selection and breeding programs. However, these traditional methods faced multiple limitations in solving complex polygenic traits, maintaining genetic diversity, and responding to rapidly changing environmental challenges. Further improvement in traditional approaches of rabbit breeding involved the use of pedigree analysis. This is where ancestry family lineage was used to predict breeding values and select better potential species as parent of the next generation. The main challenge is that evaluating rabbits based on phenotypes and pedigree information of progeny is too expensive and time consuming. This is because some traits of interest are only recorded late in life (longevity), sex limited, required animals to be sacrificed (meat quality), or required animals to be exposed to conditions that would hamper the ability to market or export their germplasm (disease resistance). The discovery of the structure of DNA coupled with the development of molecular markers has gained popularity in rabbit genetic improvement. DNA based markers are now used in rabbit genetic improvement such as RAPD, AFLD, SSR among others. However, these markers also have some limitations; they are only able to detect monogenic traits and most importantly could not cover a larger proportion of the genome. The genomic era has introduced biotechnological tools that has overcome these traditional breeding limitations. Thus, high-throughput sequencing technologies by the use of single nucleotide polymorphism (SNP) arrays, and Whole genome sequencing have enabled precise identification of genetic variants affecting polygenic economically important traits. These developments have catalysed the transition from phenotype-based to genotype-based selection strategies, fundamentally transforming approaches to genetic improvement in rabbit populations worldwide. The use of molecular genetics technologies potentially offers a way to select breeding animals at an early age (embryos); to select for a wide range of traits and to enhance reliability in predicting the mature phenotype of the individual. Therefore, this review was to narrate the development of rabbit genetic improvement from traditional approaches to recent genomic methods assessing their technical feasibility, cost-effectiveness, and practical implementation.
2. Traditional Methods of Rabbit Genetic Improvement
Traditional methods of rabbit genetic improvement, developed through many years of empirical observation and practice, form the foundation upon which modern rabbit breeding programs are built [7]. These systems have enabled animal breeders and geneticist to enhance desirable traits leading to increased productivity, improved adaptation to local environments, and enhanced economic returns [8,9,10,11]. The method relies primarily on phenotypic selection, pedigree analysis, and progeny testing to identify superior breeding stock [15,16,17,18]. Mass selection is the simplest form of phenotypic selection which involves choosing breeding animals based on their individual performance or observable traits such as growth rate, reproductive performance, and meat quality. This system of rabbit genetic improvement has been effective for traits that are highly heritable such as growth rate. The primary advantage of traditional methods lies in their simplicity and the relatively low cost associated with implementation. However, traditional methods struggle with long generation interval and may not accurately reflect the underlying genetic potential of the rabbits. Additionally, traditional methods often do not account for the complex interactions between multiple traits, which can lead to unintended consequences, such as the exacerbation of undesirable traits. Furthermore, traditional breeding practices are increasingly challenged by the need for rapid genetic improvement in response to changing market demands and environmental conditions. Selection for traits with low heritability (fertility, disease resistance) or those expressed late in life remains particularly challenging. Historically, the selection of breeding stock has focused on specific traits deemed desirable in a given socio-economic context. For example, in some regions, reproductive traits such as litter size and maternal ability have been prioritized, while in others, growth rate and feed conversion efficiency have been emphasized. This trait-specific selection has led to notable improvements in production traits, although it has also raised concerns regarding inbreeding and the potential loss of genetic diversity. These limitations of phenotypic selection have prompted geneticist to explore more advanced methodologies that leverage genetic information to enhance breeding outcomes.
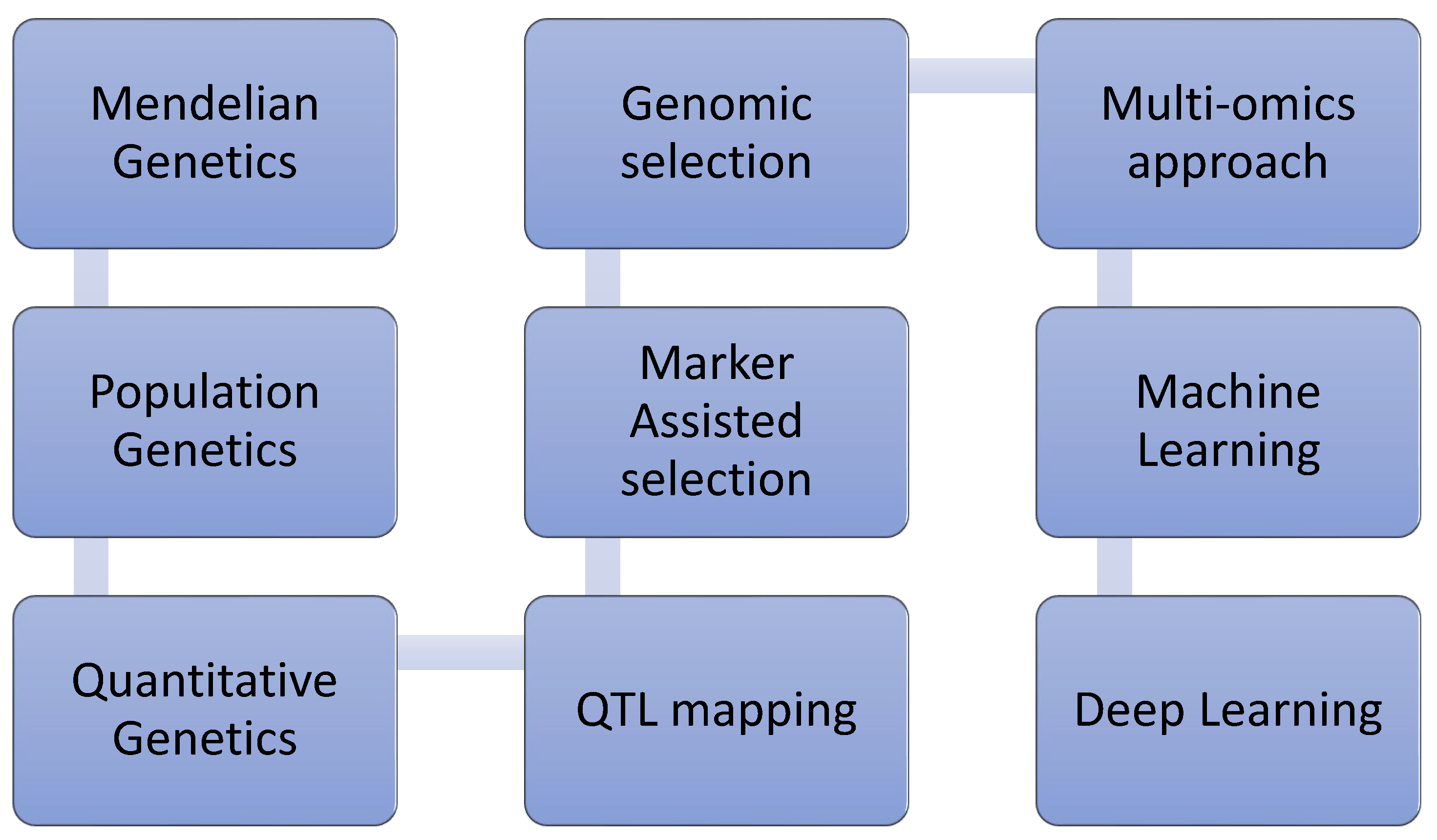
Figure 1.
Overview of the evolution of livestock genetic improvement from traditional to modern deep learning methods [19].
Figure 1.
Overview of the evolution of livestock genetic improvement from traditional to modern deep learning methods [19].
2.1. Quantitative Trait Loci Mapping and Its Application in Rabbits
Quantitative trait loci (QTL) mapping is a key genomic approach for identifying chromosomal regions associated with complex traits that exhibit continuous phenotypic variation and are controlled by multiple genes and environmental factors. In rabbits, QTL mapping has gained renewed importance over the past decade due to advances in genomic resources, including the availability of the rabbit reference genome and high-density SNP markers [20,21]. These developments have strengthened the use of rabbits both as a livestock species and as a model organism for studying the genetic basis of economically and biologically important traits. Most traits of interest in rabbit production systems such as growth rate, body weight, feed efficiency, carcass composition, reproductive performance, and disease resistance are quantitative in nature. Traditional selection methods based on phenotypic and pedigree information have contributed to genetic improvement in rabbits; however, their effectiveness is limited for quantitative traits [22,23,24]. QTL mapping provides a powerful complementary approach by linking phenotypic variation to molecular markers distributed across the genome, thereby improving understanding of trait architecture and enhancing selection efficiency.
Modern QTL mapping in rabbits is typically conducted using dense SNP data generated from SNP arrays or next-generation sequencing technologies. Compared with earlier microsatellite-based studies, SNP-based QTL mapping offers higher genomic resolution and greater statistical power to detect loci with small to moderate effects [25,26]. These studies often employ experimental crosses or well-characterized breeding populations and apply mixed linear models or genome-wide association study (GWAS) frameworks to identify genomic regions associated with target traits [27]. Recent studies have identified multiple QTL associated with body weight at different growth stages, daily gain, and feed efficiency, indicating that the genetic control of growth is dynamic and stage-specific [27,28]. Many of these QTL regions harbor genes involved in muscle development, energy metabolism, and endocrine regulation, reflecting the complex biological processes underlying growth performance. The identification of such regions provides valuable targets for genetic improvement and contributes to a better understanding of growth biology in rabbits.
Carcass and meat quality traits have also been investigated using QTL mapping and GWAS approaches. Traits such as carcass yield, muscle mass, fat deposition, and meat quality characteristics are critical for market value and consumer acceptance. Recent genomic studies in rabbits have reported QTL associated with carcass composition and fatness traits, often overlapping with genes involved in myogenesis and lipid metabolism [29,30]. These findings support the potential use of genomic information to improve meat quality while maintaining or enhancing growth performance. Reproductive traits represent another important application of QTL mapping in rabbits, although their genetic analysis is particularly challenging due to low heritability and strong environmental influence. Recent studies have identified genomic regions associated with litter size, fertility, and reproductive longevity, confirming the polygenic nature of these traits [9,22]. While individual QTL generally explain a small proportion of phenotypic variance, their combined effects can be exploited through genomic selection to improve reproductive efficiency in rabbit populations. QTL mapping has also contributed to research on health, robustness, and immune-related traits in rabbits. Disease susceptibility remains a major constraint in intensive rabbit production systems, and improving genetic resistance is a key objective for sustainable breeding programs. Recent genomic studies have reported associations between specific genomic regions and immune response traits, highlighting the role of host genetics in disease resistance [9]. Incorporating such information into breeding strategies may reduce reliance on antibiotics and improve animal welfare.
Beyond its direct application in breeding, QTL mapping serves as a foundation for functional genomics and candidate gene identification. Advances in genome annotation and comparative genomics have facilitated the identification of positional candidate genes within QTL regions, allowing biological interpretation of marker–trait associations (Carneiro et al., 2014; Zhang et al., 2019). This integrative approach strengthens the link between statistical associations and biological mechanisms and reinforces the value of rabbits as a model species for complex trait analysis. Despite recent progress, QTL mapping in rabbits still faces several challenges. Limited population sizes, population-specific effects, and genotype-by-environment interactions can reduce the reproducibility of detected QTL across studies and production systems [23] These limitations highlight the need for larger datasets, multi-population analyses, and the integration of QTL mapping with genomic selection and machine learning approaches. In recent years, genomic selection has become the dominant strategy in rabbit breeding, using genome-wide marker information to predict breeding values with higher accuracy than traditional methods. Nevertheless, QTL mapping remains highly relevant, as it provides critical insight into the genetic architecture of traits and supports the identification of biologically meaningful genomic regions that can be prioritized in selection programs [32]. The combined use of QTL mapping and genomic selection is particularly promising for traits influenced by loci with moderate effects.
2.2. Marker Assisted Selection (MAS) in Rabbit Breeding
Modern approaches such as MAS have gained traction due to their potential to accelerate genetic gains and improve the accuracy of selection. The purpose of rabbit breeding programs is to predict the genetic merit and thus allow targeted combinations of desired alleles to improve the performance of the next generation. Apparently, phenotypic traits does not show all the inherited traits according to Mendelian inheritance [33]. Therefore, genetic markers allow for tracing detailed information on the inherited genes of the genome other than such observed by the phenotype [34]. These markers, typically DNA sequences or genes linked to specific traits, allowed for identifying rabbits carrying favourable genetic variants without the need for costly and time-consuming phenotypic evaluations. MAS also involves the use of genetic markers to locate chromosomal regions that have proved to have a large influence on economic traits. This occurs by identifying genetic markers associated with candidate genes and analysing individual genetic makeup by genotyping to find the presence or absence of the markers [9,11]. The advance use of molecular genetic technologies prospectively presents the way to select the breeding animals at an early stage (embryo); also, to select for a superior variety of traits. Molecular markers are not considered normal genes because they usually serve no biological purpose [33,34]. They are recognizable DNA sequences inherited according to the normal rules of inheritance and can be discovered at particular sites across the genome. The types of molecular markers are hybridization-based DNA markers (RFLPS, Oligonucleotide fingerprinting), Polymerase Chain reaction-based DNA marker (RAPDs, microsatellite/SSRs, AFLPs) and DNA Chip and Sequencing-based DNA markers (SNPs,). Also, there are three generations of molecular marker tools which are; first generation (RAPD, AFLP, SSCP, SSR), second-generation (mtDNA, DNA microarray or DNA biochip, low and high-density of SNP chips, Whole Genome Sequencing WGS), and third-generation (single-molecule real-time-SMRT). The putting into practice of marker-based information for genetic improvement depends on the choice of an appropriate marker system for a given application. Selection of markers for different applications are influenced by certain factors; (1) the degree of polymorphism (PIC), (2) the automation of the analysis, (3) radioisotopes used, (4) reproducibility of the technique, and (4) the cost involved. Recently, microsatellites, SNPs and WGS are the most commonly used in rabbit genomic characterization studies. The advantage of using MAS is that the effect of genes on production is directly measured on the genetic makeup of the animal and not estimated from the phenotype. When MAS is used in a population, the frequency of the favourable QTL allele is quickly increased during the first generations compared to traditional selection based on BLUP. However, MAS is challenged with large number of Offspring required from each half-sib family in order to estimate unbiased effects. MAS is more profitable than traditional selection for sex-limited traits (milk yield, egg production), low heritability traits or traits that are a poor predictor of breeding value (litter size, fertility). Hence, MAS are used as a tool to reduce generation interval through early selection, even before maturity and to select those traits which are observed in only one sex. Increased cost involved in sample collection for genotyping and complete genotype information in MAS is a major limitation in a breeding scheme. MAS represents an intermediate step between traditional phenotypic selection and full genomic selection, offering practical advantages in situations where whole-genome prediction is not yet fully implemented or cost-effective. The effectiveness of MAS depends largely on the identification of reliable markers that are tightly linked to genes or QTL controlling target traits. Recently, most MAS-related studies in rabbits have relied on SNP markers identified through QTL mapping and genome-wide association studies (GWAS), supported by the availability of the rabbit reference genome [35]. SNP markers offer high genomic coverage, stability, and ease of automation, making them suitable for routine application in breeding programs. Mar
Growth and production traits are among the primary targets of MAS in rabbit breeding due to their direct economic importance. Studies conducted over the past decade have identified SNP markers and genomic regions associated with body weight at different ages, average daily gain, and feed efficiency [27]. Incorporating such markers into selection schemes enables breeders to identify superior animals early in life, reduce generation intervals, and increase selection intensity. This is particularly valuable in rabbit breeding, where rapid turnover and short generation intervals already provide a strong foundation for genetic improvement. MAS has also been explored for improving carcass composition and meat quality traits in rabbits. Traits such as muscle yield, fat deposition, and carcass quality are difficult or impossible to measure on selection candidates without slaughter. Marker information linked to these traits allows indirect selection while preserving breeding stock [36]. The use of MAS for carcass traits is therefore especially relevant in nucleus breeding programs, where accurate selection decisions have long-term impacts on commercial production.
Reproductive performance is another area where MAS offers significant potential benefits. Traits such as litter size, fertility, and reproductive longevity generally have low heritability and are strongly influenced by environmental factors, making genetic progress through conventional selection slow [37]. Marker-assisted approaches can improve selection accuracy by capturing genetic variation that is not easily detected through phenotypic records alone. Although individual markers typically explain a small proportion of phenotypic variance, their combined use can enhance reproductive performance when integrated into selection indices. MAS has also been investigated for improving health, robustness, and immune-related traits in rabbits. Disease susceptibility remains a major constraint in intensive rabbit production systems, particularly under tropical and suboptimal management conditions. Recent studies have identified genetic markers associated with immune response traits and disease resistance, suggesting that MAS could be used to improve robustness and reduce reliance on antibiotics [38]. This application aligns with global efforts to promote sustainable livestock production and antimicrobial stewardship.
One major challenge is that most economically important traits are controlled by many loci with small effects, reducing the impact of selection based on a limited number of markers [3]. Additionally, marker–trait associations may be population-specific, limiting their transferability across breeds, lines, or production environments. Genotype-by-environment interactions can further reduce the consistency of marker effects, particularly in diverse production systems [22]. These challenges underscore the importance of validating markers in the target breeding population before routine application.
In recent years, genomic selection has emerged as a more comprehensive alternative to MAS, using genome-wide marker information to predict breeding values. Nevertheless, MAS remains relevant in rabbit breeding, particularly in small or resource-limited programs where high-density genotyping of all selection candidates may not be feasible [40]. MAS can also complement genomic selection by prioritizing major-effect loci, improving biological understanding of traits, and supporting selection decisions for specific breeding objectives.
Furthermore, MAS plays an important role in the transition from QTL discovery to applied breeding. By integrating markers associated with well-characterized QTL into selection schemes, MAS provides a practical pathway for translating genomic research into measurable genetic gain [22,23,24]. This is especially relevant in rabbit breeding programs focused on niche markets, local adaptation, or conservation of genetic resources.
2.3. Genomic Selection in Rabbit Genetic Improvement
Genomic selection (GS) is an advanced breeding approach that uses genome-wide marker information to predict the genetic merit of individuals and accelerate genetic improvement for complex traits. Since its conceptual development and subsequent application in livestock, GS has gained increasing relevance in rabbit breeding due to the availability of high-density SNP data, improved statistical models, and expanding genomic resources for the rabbit genome [40]. Unlike MAS, which relies on a limited number of markers linked to major QTL, captures the combined effects of thousands of loci across the genome, making it particularly suitable for traits controlled by many genes with small effects.
Rabbit production and fitness traits, including growth rate, body weight, feed efficiency, carcass composition, reproductive performance, and disease resistance, are typically polygenic and influenced by environmental factors. Conventional selection based on phenotypic and pedigree information has contributed to genetic progress in rabbits; however, its efficiency is often constrained by low heritability, delayed trait expression, and the need for slaughter or sex-specific measurements [41]. GS overcomes many of these limitations by enabling early and more accurate prediction of breeding values using genomic estimated breeding values (GEBVs), thereby reducing generation intervals and increasing selection intensity. The implementation of GS in rabbits relies on the establishment of a reference population with both high-quality phenotypic records and dense genome-wide genotypic information. Statistical models such as genomic best linear unbiased prediction (GBLUP), single-step GBLUP (ssGBLUP), and Bayesian methods are commonly used to estimate GEBVs [7].
Growth and production traits have been among the first to benefit from GS in rabbits. Studies conducted using commercial and experimental rabbit lines have reported improved prediction accuracy for body weight, average daily gain, and feed efficiency when genomic information is included in the evaluation models [8,9].
GS has also shown strong potential for improving carcass and meat quality traits in rabbits. These traits are difficult or costly to measure on selection candidates, as they typically require slaughter. By using genomic data from relatives or reference populations, GS enables indirect selection for carcass yield, fatness, and meat quality characteristics without compromising breeding stock [2,5,9]. This approach enhances selection efficiency and supports balanced improvement of growth and carcass traits.
Reproductive traits represent one of the most challenging targets for genetic improvement in rabbits due to their low heritability and strong environmental influence. Recent studies have demonstrated that GS can increase prediction accuracy for traits such as litter size, fertility, and reproductive longevity compared to traditional models [41]. By capturing genome-wide genetic variation, GS improves the ability to select breeding animals with superior reproductive performance, contributing to enhanced productivity and sustainability of rabbit breeding systems. Health, robustness, and disease resistance traits have also become important targets of GS in rabbits, particularly in response to increasing concerns about animal welfare and antimicrobial use. Genomic approaches have been applied to immune-related traits and indicators of robustness, showing that GS can contribute to improved resilience under intensive and variable production conditions [6,7]. This aligns with global strategies aimed at promoting sustainable livestock production and reducing dependence on antibiotics.
Despite its advantages, the implementation of GS in rabbit breeding faces several challenges. The cost of genotyping, the need for large and well-phenotyped reference populations, and the management of genomic data can limit widespread adoption, particularly in small-scale or resource-limited breeding programs [20]. Additionally, prediction accuracy may decline when genomic models are applied across genetically distant populations or different production environments due to genotype-by-environment interactions. Continuous updating of reference populations and validation of models are therefore essential to maintain the effectiveness of GS. In recent years, GS has increasingly been integrated with other genomic tools, including QTL mapping, marker-assisted selection, and functional genomics. This integrative approach enhances biological understanding of complex traits and supports the identification of genomic regions with major effects that can be prioritized in selection programs [24,25]. Advances in statistical learning and machine learning methods are also being explored to further improve prediction accuracy and model robustness in rabbit GS.
The high marker density provided by SNP arrays enabled the implementation of GS in rabbit breeding programs. This is a paradigm shift from traditional pedigree-based and marker-assisted selection to a more sophisticated approach of genotyping. GS utilizes genome-wide SNP information to calculate genomic estimated breeding values (GEBVs), capturing the collective effects of numerous small-effect loci distributed across the genome [27]. Simulation and empirical studies have demonstrated the potential of GS in rabbits. [33] reported genomic prediction accuracies ranging from 0.30 to 0.55 for litter size traits using ~150,000 SNPs in a commercial rabbit line, representing substantial improvements over pedigree-based BLUP predictions. For growth and carcass traits, prediction accuracies reached 0.45-0.68, with the highest accuracies observed for traits with moderate to high heritability and adequate reference population sizes (>1,000 individuals) [1,2]. The implementation of GS offers several strategic advantages: reduced generation intervals through early selection before phenotypic evaluation, increased accuracy for low-heritability traits and sex-limited traits, enhanced genetic gain through improved selection intensity, and the ability to select for traits that are expensive or difficult to measure [2,3,4]. Thus, GS is a form of MAS in which genetic markers covering the whole genome are used so that all QTL are in linkage disequilibrium with at least one marker. In GS, SNP effects are estimated using genotype individuals that are phenotype for the characteristics of interest and the genomic estimated breeding values (GEBVs) can be predicted for any genotyped individuals by using only its SNP genotypes and estimated SNP effect [1]. GS has gained more popularity than traditional MAS because there was no need to unearth the QTL related to target traits and also no need of phenotyping the offspring. GS involves two types of populations which are training population (TP) that comprises both genotypic and phenotypic data and also breeding population (BP) which consist of candidate breeding lines with only genotypic data. Data from TP are used to train a statistical model to estimate the effect of each assayed marker and then calculate the estimated breeding values for each genotyped individual in BP to rank the lines without phenotyping. These reserved individuals can serve as parental lines that may be intimate with each other to pyramid favourable alleles for the next cycle of selection. In a chronological sequence, genomics became the cutting-edge technology today due to its evolution from genome sequencing, including approaches such as GWAS, whole-genome prediction (WGP), and genome-wide selection of complex traits. GS provides more accurate estimates for breeding value earlier in the life of breeding animals, giving more selection accuracy and allowing lower generation intervals. Reference populations for GS need to be large, with thousands of animals measured for phenotype and genotype. The smaller the effective size of the breeding population, the larger the DNA segments they potentially share and the more accurate genomic prediction will be. The relative contribution of information from relatives in the reference population will be larger if the baseline accuracy is low, but such information is limited to closely related individuals and does not last over generations.
2.4. Genome or Gene Editing (Gned) in Rabbit Genetic Improvement
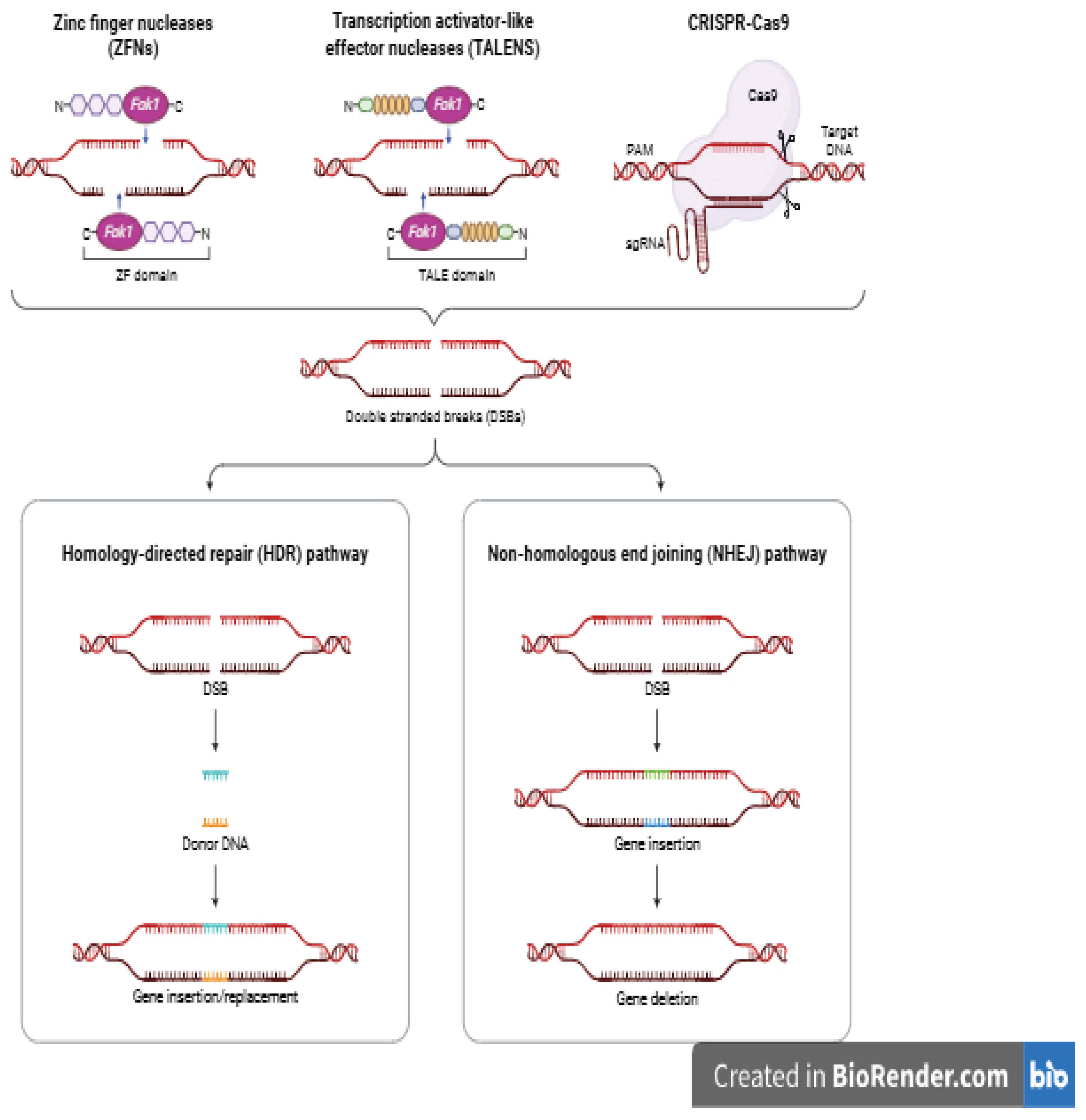
The traditional transfer of genetic material or DNA in a controlled and deliberate manner between animals was through induced mutagenesis and transgenesis. Transgenesis is a novel approach where a transgene or exogenous DNA is injected into an early-stage embryo of the host genome. The so-called transgenic animals were first produced by pronuclear zygote microinjection of foreign DNA, a random phenomenon that often resulted in unexpected, variable and inefficient transgene expression patterns leading to low efficiency and associated birth defects. Mutagenesis is also the intentional induction of genetic mutations in order to introduce desirable trait [37,38]. This occurs by exposure of the animals to chemicals, and radiation to induce genetic changes in the genome. Recently, biotechnological tools have been used precisely to change the DNA with the aim to induce traits of economic importance in order to maximise production efficiency, improve health and welfare. This involves using a site-directed nuclease (SDN) to target a specific DNA sequence to introduce a double-stranded break (DSBs) in the DNA at a targeted location in the genome [19]. When cells attempt to repair DSBs, the process can result in gene disruption or knockout. Alternatively, if a donor repair template is provided, the outcome can be the insertion or knock-in of an allele or gene from the same or different species. One method that cells use to repair DSBs is non-homologous end joining (NHEJ), where the two broken ends are brought back together and the phosphodiester bonds are reformed. This method is error-prone and often results in small insertions or deletions (indels) at the cleavage site due to mistakes in the repair process. These alter the SDNs target site and prevent further cleavage events. An alternative repair mechanism is homology-directed repair (HDR) using homologous DNA as a repair template. A DNA repair template can be added with desired modifications between regions of homology to either side of the DSB. This can be used to introduce a range of genome edits, from point mutations to whole-gene insertions. The path to repair largely depends on the stage in the cell cycle and the proteins that associate with the broken DNA ends. These applications include the use of SDNs such as mega nucleases (MNs), zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats/Cas9 system (CRISPR/Cas9). SDNs possess long recognition sites and are capable of breaking or cutting DNA double helix strands at a target site and then achieve various types of genetic modification through (NHEJ) and (HDR). These DNA nucleases mediate targeted genetic alterations by enhancing the DNA mutation rate via induction of DSB at a predetermined genomic site. In contrast to all other DNA nucleases that rely on protein--DNA binding, CRISPR/Cas9 uses RNA to establish a specific binding of its DNA nuclease. ZFNs and TALENs are artificial DNA cutting enzymes (nucleases) with a DNA--protein binding domain that directs the nucleases to a target sequence in the genome. However, CRISPR/Cas9 adopts the microinjection of the site-directed nucleases (in the form of plasmids, mRNAs, or proteins) into one-cell-stage animal embryos (zygotes) which can effectively generate genome-edited offspring. One setback of these endonucleases is that inappropriately designed could lead to off-target mutations that may result in a silent mutation or loss of specific function. However, GnEd offers several promising opportunities including; (1) used in functional genomics to elucidate gene function and identify causal variants underlying monogenic traits (2) used to precisely introduce useful genetic variation into structured rabbit breeding programs (3) used to accelerate the rate of genetic progress by enabling the change of the germ cell lineage of commercial breeding rabbits with cells derived from genetically elite lines [24,25].
Zinc-Finger Nucleases (ZFNs)
ZFNs are composed of two independent regions; a recognition domain of zinc-fingers and a non-specific nuclease called fokl which create the DSB. The DNA-binding domain of ZFNs consists of an assembly of zinc finger domains, each of which recognizes three DNA base pairs. At the C-terminus end of the DNA binding domain is a FokI cleavage domain, which dimerizes to form a non-specific nuclease. When two ZFNs bind to adjacent target sites on opposite DNA strands, the nuclease subunits combine to form a functional nuclease and create a targeted DSB. The capability to add the nuclease to the N′ end allows both ZFNs to bind the DNA in the same direction, increasing the possibility of finding a suitable ZFN-binding site on the target sequence. ZFNs are also small in size and are more susceptible to off-target effects and have proven to have detrimental effects on cell proliferation.
Transcription Activator-Like Effector Nucleases (TALENs)
TALENs are also made of two independent parts. The first part originates from the TALEs. The TALEs are naturally produced by a genus of Proteobacteria and a plant pathogen such as Xanthomonas, a gram-negative bacterium, that can infect a wide variety of plant species. TALEs bind to specific DNA sequences of the bacteria host, act as transcription factors and activate the expression of plant genes that aid bacterial infection. After binding, artificial TALEs are now designed to target desired DNA sequences. The TALEs are then fused with a FokI nuclease domain which again creates the DSB. TALENs contain a DNA-binding region consisting of a series of 33- to 35-aa-long units that each recognize a specific base pair as determined by two variable residues near the middle of each unit [9]. TALENs have increased specificity due to its straightforward design and longer recognition sites. A slight disadvantage is the increased size which makes delivery to cells more challenging compared to ZFNs [10].
CRISPR/Cas9 System
Clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 is a system of adaptive immunity found in many bacterial and archaeal species, where it functions to protect against invading viruses [11,12]. This system uses small noncoding RNAs to target a Cas nuclease to specific DNA sequences. The most widely used system at present is CRISPR/Cas9, which is based on the system of Streptococcus pyogenes and uses a short guide RNA (sgRNA) complexed with Cas9 nuclease. CRISPR/Cas9 has been found to be the most commonly used livestock genetic editing tool which uses short guide RNA (sgRNA) based in the system of streptococcus. There has been enormous application of CRISPR/Cas9 in livestock production which include the breeding of Angus cattle that carry heat-tolerant gene called ‘Slick’ [30,31,32] which help them to survive in a global heating condition. [40] has also produced polled cattle by removing horn buds through gene editing and could potentially replace the routine dehorning procedures. The Commonwealth Scientific and Industrial Research Organization (CSIRO) is currently undertaking proof-of-concept research to show that CRISPR-Cas9 could be used to produce chickens that express a fluorescent marker on the sex chromosomes. When the eggs are illuminated with a fluorescent light, the male embryos fluoresce, and are not put in an incubator. The design of CRISPR/Cas9 is straightforward and can be accomplished quickly as the only part that needs redesign is the guide RNA. The CRISPR/Cas9 system also allows multiplexing to generate organisms with multiple mutations or large chromosomal deletions [1,2]. In livestock breeding initiatives, the primary objective is achieving heritable genetic modifications that transfer through reproductive lineages to accomplish breed enhancement. This can be accomplished through two main approaches in mammalian farm animals: modifying body cells followed by reproductive cloning through somatic cell nuclear transfer (SCNT), or directly altering fertilized embryos. Gene deletions, and to a more limited extent gene additions, in farm mammals are commonly accomplished by working with cultured cells before applying SCNT techniques [20,21,22]. Conducting modifications within cell cultures enables more effective detection of genetic alterations, and allows examination of multiple cell populations from the same line, increasing the likelihood of identifying cells containing all desired changes. Direct embryo modification offers the benefit of creating genetically diverse breeding stock, since each embryo develops into a genetically unique individual, contrasting with animals produced from identical cell lines. However, using SCNT to generate embryos from modified cells significantly diminishes the procedure’s effectiveness due to poor survival rates of healthy offspring, especially pronounced in bovine species. Genetic modification tools can be introduced into recipient cells through mechanical techniques or carrier systems (either viral or non-viral vectors). Genetically modified farm mammals have primarily been created using mechanical approaches, which encompass electrical stimulation of body cells (commonly embryonic connective tissue cells) and direct injection, or alternatively electrical stimulation, of fertilized eggs. Genetic editing tools may be introduced as fully formed proteins (or RNA-protein combinations for CRISPR/Cas systems), or as genetic instructions contained within RNA or DNA molecules. Utilizing proteins (and somewhat less frequently RNA) provides enhanced precision over editor function, which proves crucial for minimizing unintended DNA cutting incidents. The necessity for a template sequence during homology-directed repair introduces an additional component (DNA) to the delivery combination, complicating the process. Individual repair templates need matching sequences that surround the intended modification site, typically requiring 50-1000 base pairs for each of the two surrounding regions. Unincorporated linear DNA molecules can harm fertilized eggs, necessitating careful limitation of administered amounts. Electroporation employs high-intensity electrical pulses to create temporary openings in cellular membranes. These openings permit genetic modification tools to move from the surrounding medium into the cell\’s interior. Nuclear-directing sequences can facilitate the movement of genome editing proteins through the nuclear envelope into the cell\’s control centre. When introduced into the cell interior, genetic instructions from circular DNA molecules are processed after the nuclear boundary breaks down during cell division or after passage through membrane channels. Nucleofection represents a specialized electroporation technique that simultaneously creates openings in both cellular and nuclear membranes, enabling faster introduction of genetic editing tools or DNA constructs directly into the nucleus. While electroporation has conventionally been applied to modify laboratory-grown cell populations, recent studies demonstrate its effectiveness on fertilized embryos [41]. Direct cytoplasmic microinjection (CPI) has traditionally served as the primary method for introducing genome modification tools into fertilized eggs of livestock species. More recently, electroporation has emerged as a promising alternative approach, demonstrating success in generating insertion-deletion mutations (via non-homologous end joining) in pig and cattle embryos. In contrast to CPI, which requires individual needle-based delivery of editing components to each zygote, electroporation enables simultaneous treatment of multiple embryos, thereby reducing both time investment and technical skill requirements. Direct zygotic delivery of genome editing tools circumvents the limitations associated with somatic cell nuclear transfer (SCNT), though it introduces the challenge of genetic mosaicism when editing occurs after the single-cell stage during early embryonic development, along with uncertain modification efficiency. Screening can be performed through genetic analysis of blastocyst-stage biopsies before implantation into surrogate mothers, this approach may compromise the survival potential of transferred embryos. When mosaic offspring are produced, breeding to subsequent generations becomes necessary to obtain uniform, homozygous animals lacking mosaicism. Direct introduction of genome editing components into fertilized eggs has proven effective for creating targeted gene knockouts in developing embryos, though mosaicism may impair the transmission of modifications to offspring. However, achieving precise targeted gene insertions remains challenging. The HDR mechanism functions predominantly in proliferating cells during the S and G2 phases of the cell cycle, reaching peak activity only near the completion of the initial DNA replication round in single-cell zygotes. Somatic cell electroporation is the standard method when HDR is necessary, though only limited cases have documented successful HDR through cytoplasmic injection of double-stranded DNA into livestock embryos [6,7]. In contrast, single-stranded oligodeoxynucleotides (ssODN) have been effectively employed by multiple research teams to generate precise nucleotide changes or small sequence insertions/deletions via HDR mechanisms [17,18,19]. An alternative strategy for incorporating complete genes into embryos was demonstrated in sheep engineered to produce two melatonin biosynthesis proteins in their milk using β-casein regulatory sequences [7]. Non-homologous linear double-stranded DNA was directly injected into embryonic cytoplasm alongside Cas9 nuclease and a guide RNA directed against the myostatin gene. These DNA constructs integrated through homology-independent repair mechanisms at the target location in approximately 35% of surviving offspring, with roughly 26% carrying both transgenes. Genetic modification in birds presents unique challenges due to the difficulty of accessing avian embryos. Consequently, most chicken genome editing research focuses on primordial germ cells (PGCs). These cells travel through the embryonic circulation between 48-60 hours of development while migrating toward the developing reproductive organs. Chicken PGCs can be easily maintained in laboratory culture where genetic modification tools can be introduced, commonly as DNA constructs delivered through lipofection techniques. Lipofection represents a non-viral delivery method that utilizes electrostatic interactions between lipids and genetic material to create lipid-DNA complexes (lipoplexes) that cells can internalize. Modified primordial germ cells (PGCs) can be evaluated and introduced into the circulatory system of chicken embryos that have undergone chemical or genetic germline depletion through an opening created in the eggshell. As an alternative approach, direct intravenous administration can target circulating PGCs using transfection compounds. The resulting avian specimens can subsequently reproduce to generate genetically modified progeny. It is crucial to acknowledge that numerous procedures involved in livestock genome modification are labour-intensive and currently demonstrate poor efficiency rates. Multiple sequential steps and unpredictable biological factors exist, including reproductive cell harvesting and development, delivery of modification components, embryo cloning and implantation into hormonally synchronized recipient females, each presenting distinct challenges and limitations. Embryonic microinjection to create chimeric animals, followed by selective breeding to obtain heterozygous edited descendants, represents a lengthy and costly process in large agricultural species. Numerous genome modification applications necessitate homozygous alterations to guarantee single-copy transmission in first-generation offspring, or to accommodate alleles exhibiting recessive inheritance patterns. The intricate nature and poor success rates associated with these methodologies render livestock genome editing far from standardized practice under current conditions. Recent gene-editing technologies, particularly CRISPR-Cas systems, represent the frontier of precision breeding approaches. While fewer commercial applications exist compared to GS, proof-of-concept studies demonstrate powerful potential. [8] successfully edited the myostatin gene in pigs to increase muscle development, while [9] produced hornless dairy cattle through precise genetic alterations mirroring natural polled mutations. Unlike earlier transgenic approaches that inserted foreign DNA, modern gene editing can create genetic changes indistinguishable from those occurring naturally, potentially addressing some regulatory and consumer acceptance concerns [16,17]. However, [20] highlighted persistent ethical questions regarding animal welfare, agrobiodiversity, and socioeconomic implications of such technologies.
Figure 2.
Modern gene editing technologies used in livestock genetic improvement.
Multi-Omics in Rabbit Genetic Improvement
The integration of multi-omics technologies has emerged as a transformative approach in animal breeding, enabling a comprehensive understanding of the biological mechanisms underlying complex traits. Since 2015, advances in high-throughput sequencing, mass spectrometry, and bioinformatics have facilitated the generation and integration of diverse omics datasets, including genomics, transcriptomics, proteomics, metabolomics, and epigenomics. In rabbit genetic improvement, multi-omics approaches are increasingly recognized for their potential to complement traditional quantitative genetics and GS by linking genetic variation to molecular function and phenotypic expression [10,11,12].
Genomics forms the foundation of multi-omics applications in rabbits, providing genome-wide information on genetic variation through SNP arrays and whole-genome sequencing. While GS captures the additive effects of genome-wide markers, it does not directly explain the biological pathways through which genetic variants influence traits. Integrating genomic data with other omics layers enhances the interpretation of marker–trait associations and improves understanding of trait architecture [39,40,41]. In rabbits, genomic resources developed over the past decade have created opportunities to expand beyond single-layer analyses toward integrative multi-omics frameworks.
Transcriptomics is a key component of multi-omics studies, providing insights into gene expression patterns across tissues, developmental stages, and environmental conditions. RNA sequencing (RNA-seq) has been used in rabbits to identify differentially expressed genes associated with growth, muscle development, reproduction, and immune response. Integrating transcriptomic data with genomic information allows the identification of expression quantitative trait loci (eQTL), which link genetic variants to gene expression levels and help prioritize candidate genes within QTL regions [29,30]. This approach improves biological interpretation and supports more informed selection decisions.
Proteomics and metabolomics further extend the functional resolution of multi-omics by characterizing downstream molecular phenotypes that are more closely related to observable traits. Proteomic analyses provide information on protein abundance and post-translational modifications, while metabolomics captures metabolic profiles reflecting physiological status and environmental interactions. Although applications in rabbits are still emerging, studies in livestock species demonstrate that integrating proteomic and metabolomic data with genomic information can reveal biomarkers for growth efficiency, meat quality, fertility, and health traits [20,21,22]. These approaches hold strong potential for improving selection accuracy in rabbit breeding programs.
Epigenomics represents another important omics layer, focusing on heritable changes in gene regulation that do not involve alterations in DNA sequence, such as DNA methylation and histone modifications. Epigenetic mechanisms play a crucial role in regulating gene expression in response to environmental factors, nutrition, and management practices. Since 2015, epigenomic studies in livestock have highlighted the importance of epigenetic variation in growth, reproduction, and immune function. In rabbits, integrating epigenomic data with genomic and transcriptomic information can improve understanding of genotype-by-environment interactions and developmental plasticity, which are particularly relevant under diverse production conditions [33,34]. One of the major applications of multi-omics in rabbit genetic improvement is the identification of biologically meaningful candidate genes and pathways underlying complex traits. By integrating genomic, transcriptomic, proteomic, and metabolomic data, researchers can move beyond statistical associations to establish causal links between genetic variation and phenotype. This systems-level understanding enhances the reliability of marker discovery and supports the development of more effective selection tools, including refined marker-assisted and GS strategies [12,13,14]. Multi-omics approaches also offer significant potential for improving the prediction of complex traits through integrative models. Combining omics datasets with advanced statistical and machine learning methods can improve the accuracy of breeding value predictions by incorporating functional information alongside genomic markers. Recent studies in livestock have shown that integrating multi-omics data can enhance prediction accuracy, particularly for traits with low heritability or strong environmental influence [1,2,3]. Such approaches are highly relevant for rabbit breeding programs aiming to improve reproduction, robustness, and disease resistance. Despite their promise, the application of multi-omics in rabbit genetic improvement faces several challenges. High costs, limited sample sizes, data integration complexity, and the need for specialized bioinformatics expertise can constrain widespread adoption. Additionally, most multi-omics studies have been conducted in experimental or research populations, highlighting the need for validation in commercial and diverse production systems. Addressing these challenges will require collaborative efforts, standardized protocols, and the development of cost-effective omics platforms tailored to rabbit breeding [20,21,22]. Table 1 show the comparative analysis o genetic improvement methods in rabbits production from traditional approaches to modern machine learning algorithms.

Table 2.
Gene Editing Technologies Applied to Livestock Genetic Improvement.
| Technology | Development Period | Key Components | Advantages | Disadvantages | Efficiency in Livestock | Applications in Livestock |
|---|---|---|---|---|---|---|
| Zinc-Finger Nucleases (ZFNs) | 1996-2003 | Zinc finger DNA-binding domain, FokI cleavage domain (18-36 bp recognition) | First-generation precision tool, smaller size aids delivery | Complex design, time-consuming, higher off-target rate | 1-5% in embryos | Limited use; proof-of-concept in model organisms |
| TALENs | 2009-2011 | TAL effector repeat arrays (33-35 aa units), FokI nuclease (30-40 bp recognition) | Straightforward design, higher specificity, longer recognition sites | Large size (challenging delivery), labor-intensive assembly | 5-15% in embryos | Early applications in pig and cattle; limited rabbit studies |
| CRISPR-Cas9 | 2012-2013 | Guide RNA (sgRNA), Cas9 protein, PAM sequence (20 bp guide + 3 bp PAM) | Simple design, rapid implementation, allows multiplexing, cost-effective | PAM sequence requirement limits targets, potential off-target effects | 20-80% in embryos | Heat tolerance (cattle), polled cattle, disease resistance, myostatin editing |
| Base Editors | 2016-2017 | Catalytically dead Cas9, cytidine or adenine deaminase, guide RNA | Precise single nucleotide changes (C→T or A→G), no DSB, reduced indels | Limited to specific base transitions, narrow editing window | 10-60% in embryos | Emerging in livestock; SNP correction, disease resistance alleles |
| Prime Editing | 2019 | Cas9 nickase (H840A), reverse transcriptase, pegRNA template | Versatile (insertions, deletions, all base changes), high precision, no DSB | Lower efficiency, complex pegRNA design, larger construct size | 5-40% in embryos | Proof-of-concept in mice and livestock cells; not widely applied |
DSB = Double-Strand Break; PAM = Protospacer Adjacent Motif; HDR = Homology-Directed Repair; NHEJ = Non-Homologous End Joining; GxE = Genotype-by-Environment interactions.
Application of Machine Learning in Rabbit’s Genetic Improvement
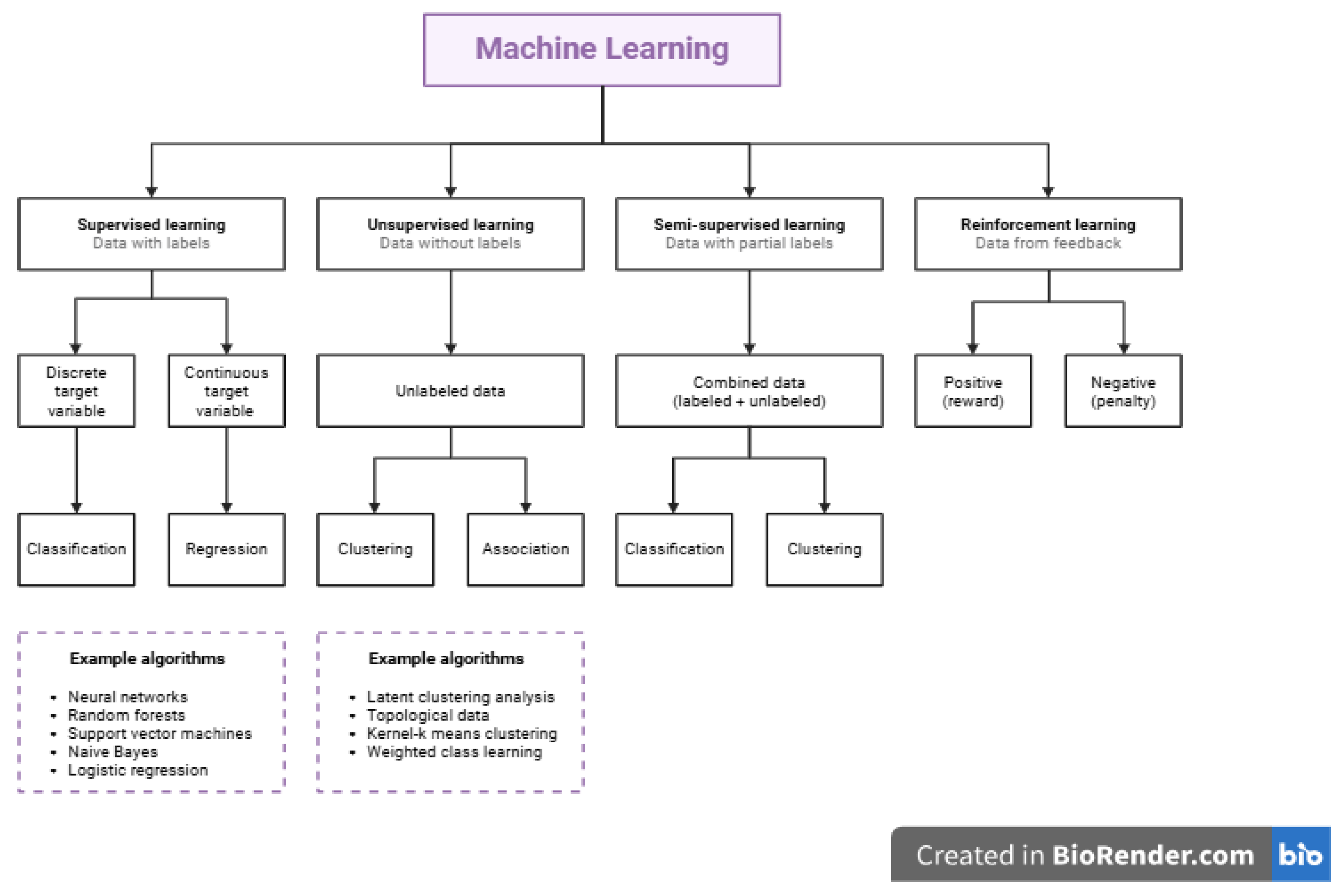
Machine learning (ML) has emerged as a powerful analytical framework for extracting complex patterns from large and high-dimensional datasets, offering new opportunities for genetic improvement in livestock species. Since 2015, the increasing availability of genomic, phenotypic, and environmental data, coupled with advances in computational power, has stimulated growing interest in the application of ML methods to animal breeding. In rabbit genetic improvement, ML approaches are increasingly being explored as complementary or alternative tools to traditional statistical models for predicting breeding values, identifying important genomic features, and improving selection decisions [20,21,23].
Rabbit breeding programs generate diverse datasets, including pedigree information, growth and reproduction records, carcass traits, health indicators, and genome-wide SNP data. Traditional linear models, such as best linear unbiased prediction (BLUP) and genomic BLUP (GBLUP), assume linear relationships between markers and phenotypes, which may not fully capture the complex, non-linear, and interactive nature of genomic data. Machine learning methods, by contrast, are well suited to modeling non-linear relationships, high-order interactions, and large numbers of predictors, making them attractive for genomic prediction and decision support in rabbit breeding [11,12,13]. One of the primary applications of ML in rabbit genetic improvement is genomic prediction of breeding values. Algorithms such as random forests, support vector machines, gradient boosting, artificial neural networks, and deep learning models have been applied to predict complex traits using genome-wide marker data. Recent studies in livestock, including rabbits and related species, have demonstrated that ML models can achieve prediction accuracies comparable to or, in some cases, higher than conventional GS models, particularly for traits influenced by non-additive genetic effects [40,41]. These approaches enable the estimation of genomic estimated breeding values without relying strictly on parametric assumptions.
Growth and production traits are among the most promising targets for ML-based prediction in rabbits due to the availability of large phenotypic datasets and their economic importance. Machine learning models have been shown to effectively integrate SNP data with performance records to predict body weight, growth rate, and feed efficiency. The ability of ML algorithms to capture complex interactions among loci and between genetic and environmental factors enhances prediction performance, especially in heterogeneous production systems [21,24]. This capability is particularly relevant for rabbit production in variable environments, where genotype-by-environment interactions play a significant role.
Reproductive traits, which are typically characterized by low heritability and strong environmental influence, represent another area where ML approaches offer substantial advantages. Traits such as litter size, fertility, and reproductive longevity are difficult to predict accurately using traditional models alone. Machine learning methods can incorporate a wide range of predictors, including genomic, physiological, and environmental variables, to improve prediction accuracy for reproductive performance [30,31]. Improved prediction of these traits can enhance selection decisions and contribute to greater reproductive efficiency in rabbit breeding programs. Machine learning has also been applied to carcass quality, health, and robustness traits in rabbits. Carcass traits often require slaughter for measurement, while health-related traits may be difficult to quantify precisely. ML models can leverage indirect indicators, genomic information, and correlated traits to predict these complex phenotypes. Recent livestock studies have shown that ML approaches are effective in identifying genomic regions and markers associated with disease resistance and immune response, supporting more resilient breeding strategies.
Figure 3.
A diagram showing the types of machine learning algorithms.
Conclusion
Rabbit breeders have a long history of changing the genetic composition to improve productivity and fulfil expanding human needs for food and other animal products. This has only recently become achievable through traditional breeding and selection, which is a laboriously slow process of gradually collecting genetic advantages over an extended length of time. The introduction of transgenic biotechnological approaches offered the ability for producing genetic modifications with higher impact and within a single generation. But at first, the technology was constrained by restrictions and technological issues that have since mostly been resolved by steady advancements. The advent of genome or editing technologies, combined with homologous recombination, has added a new level of efficiency and precision that holds much promise for rabbit genetic improvement based on increasing knowledge of the phenotypic impact of genetic sequence variants. The transition to genome-edited rabbit with precise sequence changes without the use of exogenous DNA could accelerate up the path to market, provided that agricultural applications of this new technology deliver compelling benefits for animals, consumers, and the environment in addition to incentives for producers.
Author Contributions
The manuscript was writing by Richard Asante Botwe and Bismark Yeboah while Samuel Ayeh Ofori served as the reviewer.
Funding
This research received funding from Green Africa Youth Organization a non-governmental organization that fo-cuses on environmental sustainability and community development.
Institutional Review Board Statement
This review did not require ethics approval as it involved the analysis of previously published studies and did not include primary data collection from human participants. All included studies had obtained appropriate ethical approvals from their respective institutional review boards or ethics committees as reported in the original publications.
Data Availability Statement
The datasets supporting the conclusions of this systematic review are available from the corresponding author upon reasonable request. All data extracted from the included studies are presented in the supplementary materials. The search strategies and full database search results are available in the appendices.
Conflicts of Interest
The authors declare that they have no competing interests or conflicts of interest that could have influenced the conduct or reporting of this systematic review.:
Abbreviations
| The following abbreviations are used in this manuscript | . |
| DNA | Deoxyribonucleic acid |
| ML | Machine Learning |
| GS | Genomic selection |
| SNP TALEN |
Single Nucleotide Polymorphism Transcription Activator-Like Endonucleases |
References
- Allain, D.; Bonnet, M.; Gilbert, H.; Ruesche, J. Genetic resistance to rabbit diseases: A review. World Rabbit Sci. 2015, 23, 123–137. [Google Scholar]
- Bishop, T.F.; Van Eenennaam, A.L. Genome editing approaches to livestock improvement. Nat. Rev. Genet. 2020, 21, 655–668. [Google Scholar]
- Carlson, D.F.; Lancto, C.A.; Zang, B.; Kim, E.S.; Walton, M.; Oldeschulte, D.; et al. Production of hornless dairy cattle from genome-edited cell lines. Nat. Biotechnol. 2016, 34, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.; Rubin, C.J.; Di Palma, F.; Albert, F.W.; Alföldi, J.; Martinez Barrio, A.; et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 2014, 345, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Cenariu, M.; Pall, E.; Cenariu, D. Embryo biopsy and preimplantation genetic diagnosis. J. Assist. Reprod. Genet. 2012, 29, 919–927. [Google Scholar]
- Chafai, M.; Moula, N.; Antoine-Moussiaux, N.; Farnir, F. Sustainable rabbit production systems under climate change. Animals 2023, 13, 251. [Google Scholar] [CrossRef]
- Clark, S.J.; Van der Werf, J.; Kinghorn, B. Breeding and genetics of livestock sustainability. Anim. Prod. Sci. 2013, 53, 832–839. [Google Scholar] [CrossRef]
- Eggen, A. The development and application of genomic selection as a new breeding paradigm. Anim. Front. 2012, 2, 10–15. [Google Scholar] [CrossRef]
- FAO. The Future of Food and Agriculture—Drivers and Triggers for Transformation; FAO: Rome, Italy, 2023. [Google Scholar] [CrossRef]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Scotti, E.; Martelli, P.L.; et al. A genome-wide association study for rabbit growth traits. J. Anim. Breed. Genet. 2015, 132, 195–204. [Google Scholar] [CrossRef]
- Fontanesi, L.; Bertolini, F.; Dall’Olio, S.; Russo, V. Multi-omics approaches in livestock genetics. Brief. Funct. Genomics 2018, 17, 421–432. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- García, M.L.; Baselga, M.; Piles, M. Genetic parameters and genomic regions for litter size in rabbits. Anim. Genet. 2017, 48, 273–280. [Google Scholar] [CrossRef]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Goddard, M.E.; Hayes, B.J.; Meuwissen, T.H. Genomic selection in livestock populations. Genet. Res. 2010, 92, 413–421. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Ibeagha-Awemu, E.M.; Peters, S.O.; Akwanji, K.A.; Imumorin, I.G.; Zhao, X. High-density genome-wide genotyping in livestock. Front. Genet. 2019, 10, 121. [Google Scholar] [CrossRef]
- Kramer, B.; Meijboom, F.L.B. Ethical issues in farm animal breeding. J. Agric. Environ. Ethics 2022, 35, 1–20. [Google Scholar]
- Kwon, S.G.; Lee, S.H.; Kim, T.W. Genomic tools and marker-assisted selection in livestock. Genes 2024, 15, 44. [Google Scholar] [CrossRef]
- Lillico, S.G.; Proudfoot, C.; Carlson, D.F.; Stverakova, D.; Neil, C.; Blain, C.; et al. Mammalian genome engineering using CRISPR systems. Transgenic Res. 2016, 25, 509–524. [Google Scholar]
- Ma, T.; Tao, J.; Yang, M.; He, C.; Tian, X.; Zhang, X.; et al. Sheep production of melatonin proteins via CRISPR/Cas9. Sci. Rep. 2017, 7, 12259. [Google Scholar]
- Menchaca, A.; Dos Santos-Neto, P.C.; Cuadro, F. CRISPR in livestock reproduction. Reprod. Fertil. Dev. 2020, 32, 202–215. [Google Scholar]
- Middelveld, R.; Macnaghten, P. Gene editing and livestock futures. Sci. Technol. Hum. Values 2021, 46, 693–720. [Google Scholar]
- Oliveira, H.R.; Brito, L.F.; Lourenco, D.A.L.; Silva, F.F.; Jamrozik, J.; Schenkel, F.S. Traditional and genomic breeding strategies. J. Anim. Sci. 2023, 101, skac403. [Google Scholar] [CrossRef]
- Park, K.E.; Park, C.H. Precision genome editing in livestock. BMB Rep. 2017, 50, 516–523. [Google Scholar]
- Piles, M.; Tusell, L.; Sánchez, J.P. Genomic selection in rabbits. J. Anim. Breed. Genet. 2017, 134, 485–497. [Google Scholar] [CrossRef]
- Piles, M.; García, M.L.; Tusell, L. Genomic tools for improving rabbit robustness. Animals 2021, 11, 713. [Google Scholar] [CrossRef]
- Proudfoot, C.; Carlson, D.F.; Huddart, R.; Long, C.R.; Pryor, J.H.; King, T.J.; et al. Genome edited pigs lacking myostatin. Sci. Rep. 2015, 5, 14337. [Google Scholar] [CrossRef]
- Rosell, J.M.; de la Fuente, L.F.; Parra, J.L. Disease resistance genetics in rabbits. World Rabbit Sci. 2019, 27, 189–203. [Google Scholar]
- Satué, K.; Tusell, L.; Piles, M. Genomic prediction of litter size in rabbits. Genet. Sel. Evol. 2019, 51, 66. [Google Scholar]
- Schultz-Bergin, M. Ethical issues in genome editing of animals. Philos. Technol. 2018, 31, 513–533. [Google Scholar]
- Stock, J.; Reents, R.; Simianer, H. Livestock breeding in the genomic era. Anim. Front. 2020, 10, 10–17. [Google Scholar] [CrossRef]
- Suravajhala, P.; Kogelman, L.J.A.; Kadarmideen, H.N. Multi-omics data integration in livestock. Brief. Bioinform. 2016, 17, 520–537. [Google Scholar] [CrossRef]
- Tan, W.; Proudfoot, C.; Lillico, S.G.; Whitelaw, C.B.A. Gene targeting in livestock. Trends Biotechnol. 2016, 34, 781–790. [Google Scholar] [CrossRef]
- Tanihara, F.; Hirata, M.; Otoi, T.; Yamanaka, K. Genome editing by electroporation in livestock embryos. Sci. Rep. 2018, 8, 493. [Google Scholar]
- Tusell, L.; García, M.L.; Piles, M. Genomic prediction in rabbit breeding programs. Animal 2019, 13, 2461–2468. [Google Scholar] [CrossRef]
- Van Eenennaam, A.L. Gene editing of livestock. Annu. Rev. Anim. Biosci. 2019, 7, 403–427. [Google Scholar]
- Velasco-Galilea, M.; Sánchez, J.P.; Piles, M. Genomic evaluation of growth traits in rabbits. Anim. Genet. 2021, 52, 471–482. [Google Scholar] [CrossRef]
- Wei, J.; Wagner, S.; Luttermann, C. Precise genome editing using ssODNs. Nucleic Acids Res. 2018, 46, 3736–3745. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Gong, H.; Cui, L.; Zhang, W.; Ma, J.; et al. Genome-wide association study for growth traits in rabbits. BMC Genet. 2019, 20, 40. [Google Scholar]
Table 1.
Comparative Analysis of Genetic Improvement Methods in Rabbits.
| Method | Time Period | Key Technologies | Advantages | Limitations | Prediction Accuracy | Generation Interval | Cost | Best Application |
|---|---|---|---|---|---|---|---|---|
| Traditional Selection | Pre-1990s | Phenotypic selection, pedigree analysis, progeny testing | Simple implementation, low cost, no specialized equipment | Long generation intervals, limited accuracy for low heritability traits | 0.20-0.40 | 6-12 months | Low | Highly heritable traits (growth rate) |
| QTL Mapping | 1990s-2000s | Microsatellite markers, linkage analysis | Identifies chromosomal regions, provides biological insights | Population-specific, limited genome coverage, labor-intensive | 0.25-0.45 | 6-12 months | Moderate | Candidate genes, monogenic traits |
| Marker-Assisted Selection | 2000s-2010s | SNP markers, candidate genes, GWAS | Early selection, useful for sex-limited traits, reduces phenotyping costs | Limited to major-effect loci, population-specific markers | 0.30-0.50 | 3-6 months | Moderate | Sex-limited traits, carcass quality, disease resistance |
| Genomic Selection | 2010s-present | High-density SNP arrays (150K-600K), GBLUP, ssGBLUP | Genome-wide effects, high accuracy, enables early selection | Requires large reference populations (>1000), expensive genotyping | 0.45-0.68 | 3-6 months | High | All polygenic traits, routine breeding programs |
| Gene Editing | 2015-present | CRISPR-Cas9, TALENs, ZFNs | Precise targeted changes, single generation modifications | Regulatory challenges, high costs, off-target risks, ethical concerns | N/A | Single gen. | Very High | Disease resistance, productivity genes, functional genomics |
| Machine Learning/AI | 2020-present | Random forests, neural networks, deep learning | Captures non-linear effects, handles complex interactions | Requires large datasets, ‘black box’ problem, computationally intensive | 0.50-0.70+ | 3-6 months | High | Complex traits with non-additive effects, GxE interactions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



