
Submitted:
29 January 2026
Posted:
30 January 2026
You are already at the latest version
Abstract
Constructed wetlands (CW) are a low-cost alternative for wastewater treatment, where microbial communities play a key role in the biotransformation of pollutants, including sulfur compounds. This study reports the identification of cultivable bacteria involved in the sulfur cycle (SC) in a subsurface-flow CW located in Tetipac, Mexico. Water, sediment, and rhizosphere samples were collected during four sampling events and plated on a mineral medium with thiosulfate. Colony-forming units were quantified, and 15 isolates were genetically identified by partial 16S rRNA gene sequencing. Bacterial abundance was higher in the rhizosphere, and the cultivable fraction was dominated by Pseudomonadota, particularly Gammaproteobacteria, accompanied by Bacteroidota and several previously uncultured lineages; genera such as Achromobacter, Chitinophaga, Enterobacter, Pseudomonas, Raoultella and Stenotrophomonas were recovered. Biochemical assays revealed heterogeneous metabolic profiles, with several isolates showing activities comparable to canonical sulfur-oxidizing bacteria (SOB). Most isolates oxidized thiosulfate and a substantial proportion oxidized elemental sulfur, with higher metabolic performance in rhizosphere isolates and a positive association with BOD₅ removal. Overall, these results indicate that the Tetipac wetland harbors a cultivable community of largely non-canonical SOB whose mixotrophic versatility and spatial differentiation suggest a key contribution to the SC and organic matter degradation in CW.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling
2.2. Physicochemical Characterization of the Wetland System
2.3. Culture-Based Isolation and Colony-Forming Units Quantification
2.4. Genomic DNA Extraction and PCR Amplification of the 16S rRNA Gene
2.5. Phylogenetic Analysis
2.6. Metabolic Screening of Isolates Using Sulfur and Carbon Substrates
2.7. Determination of Catalase and Oxidase Enzymatic Activities
3. Results
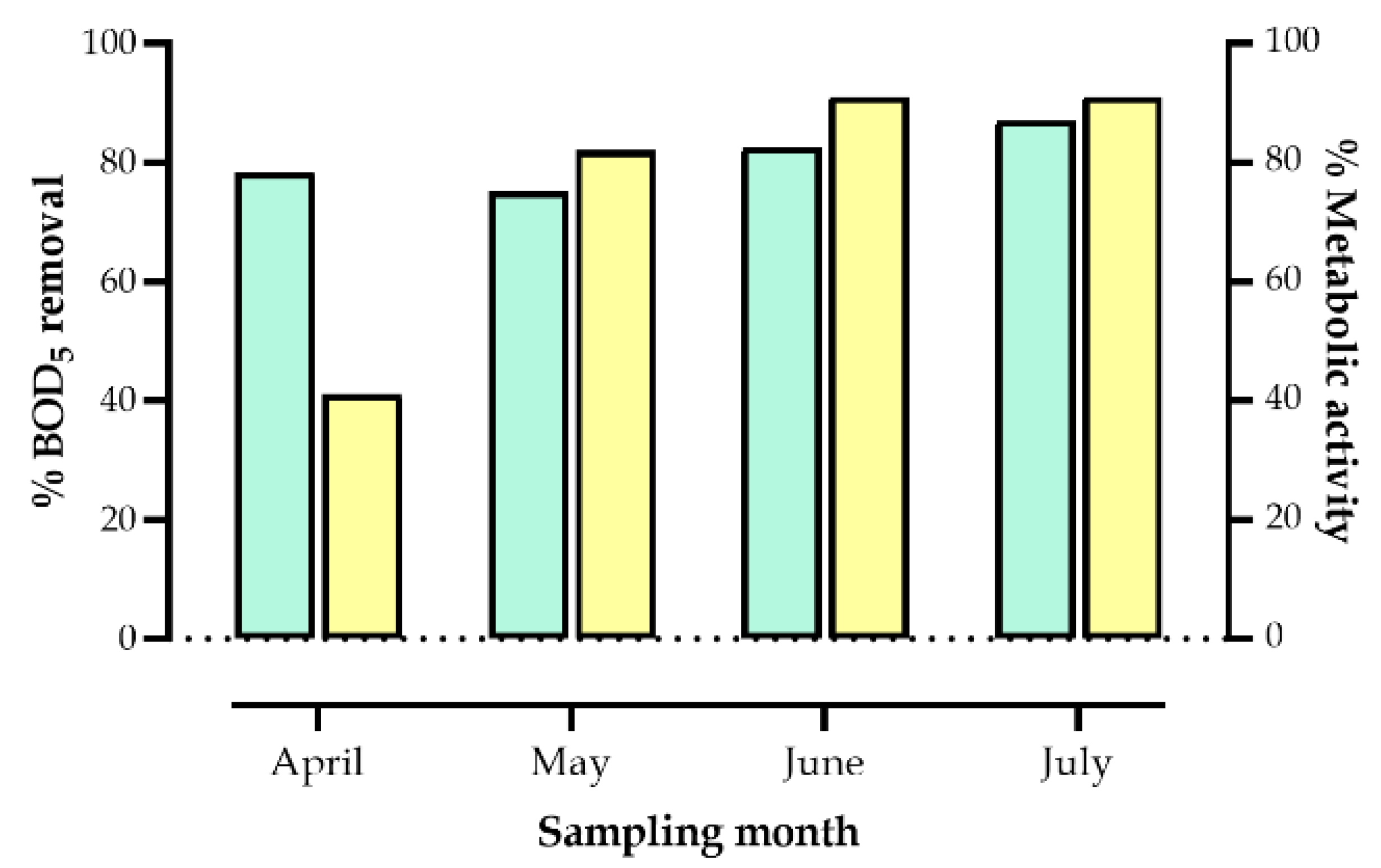
3.1. Physicochemical Parameters of the Constructed Wetland During the Sampling Period
3.2. Cultivable Bacterial Abundance Among Wetland Compartments
3.3. Taxonomic Composition of Culturable Bacterial Isolates
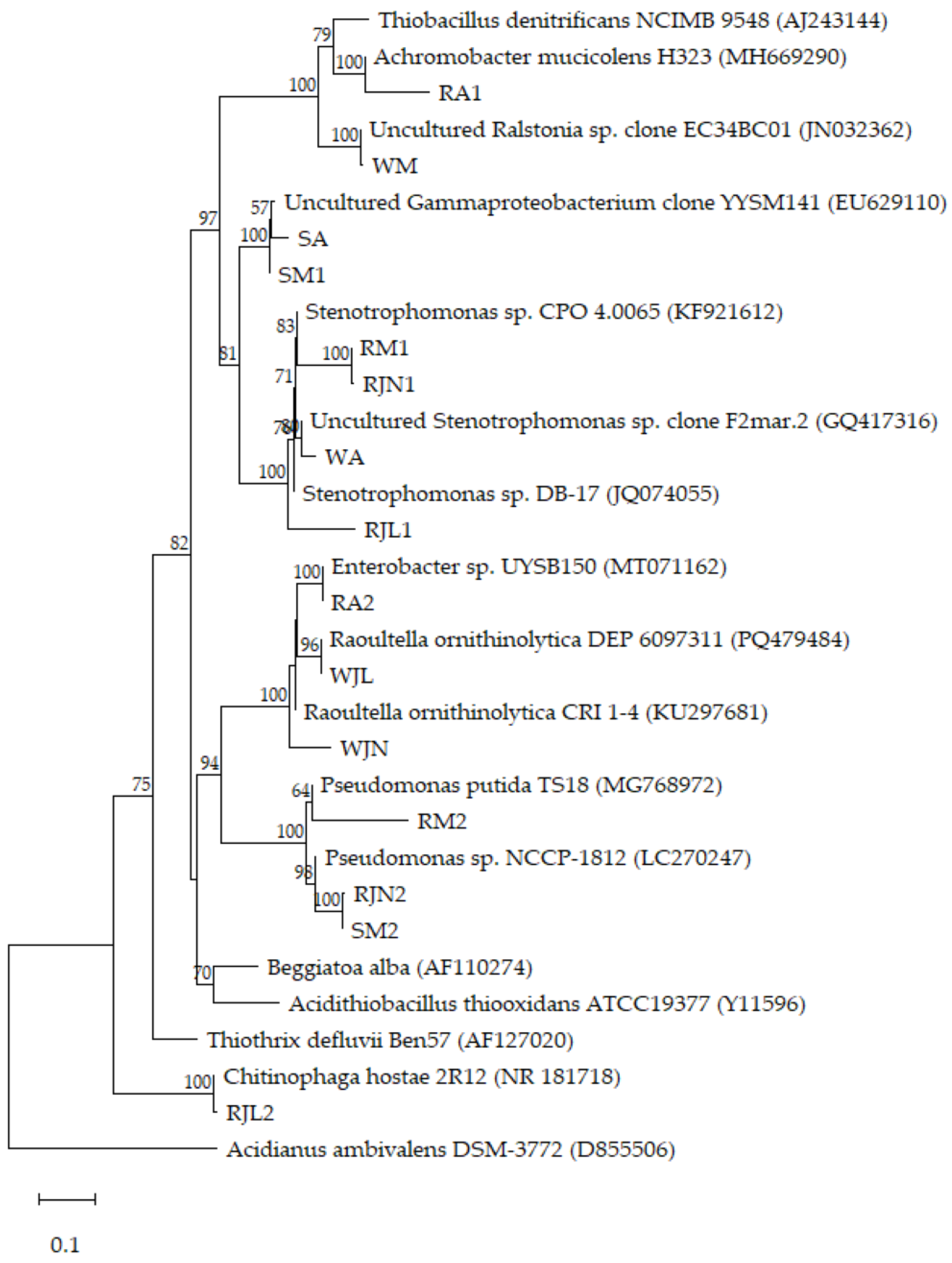
3.4. Phylogenetic Affiliation of Bacterial Isolates and Links to Environmental Origin
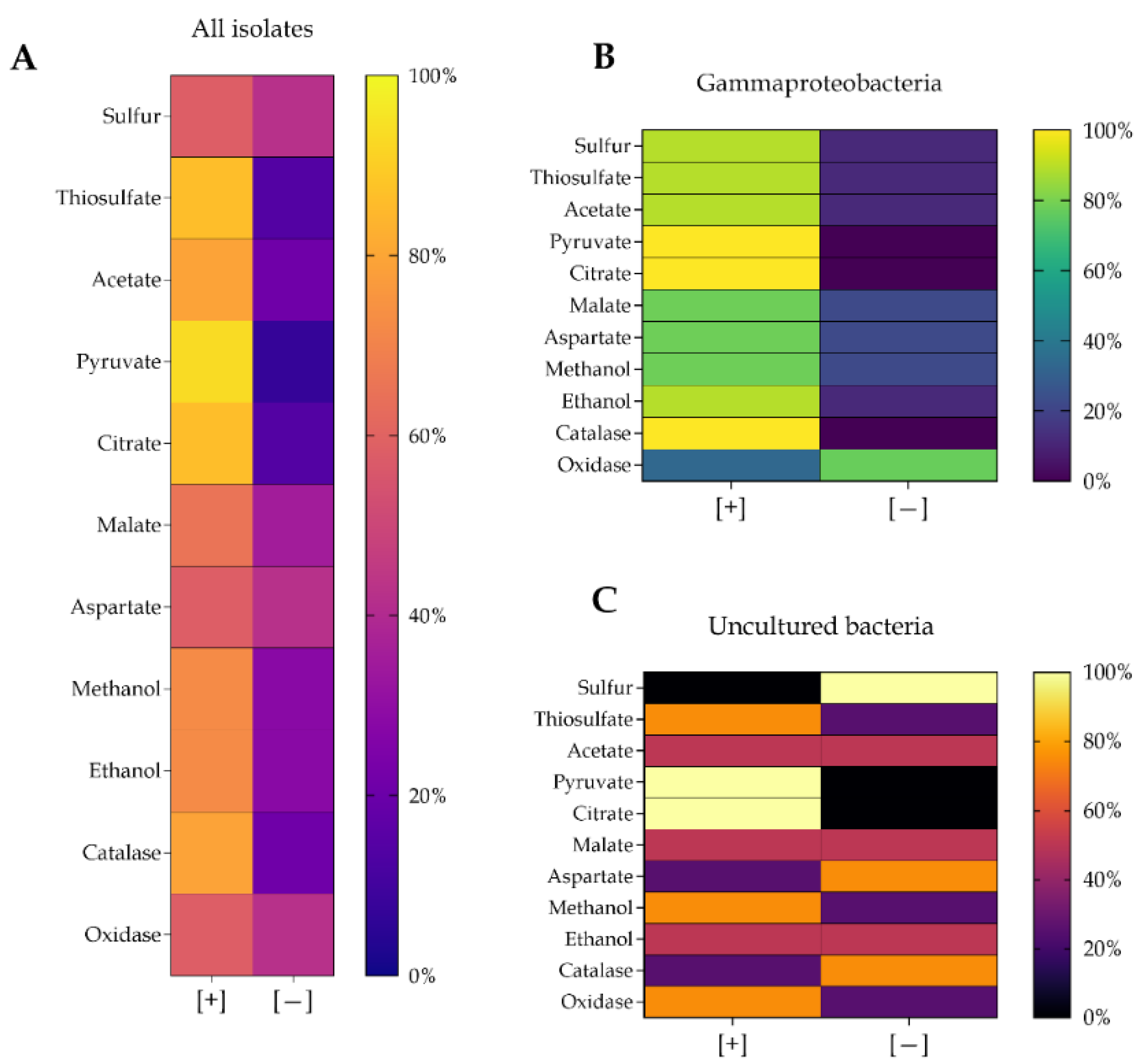
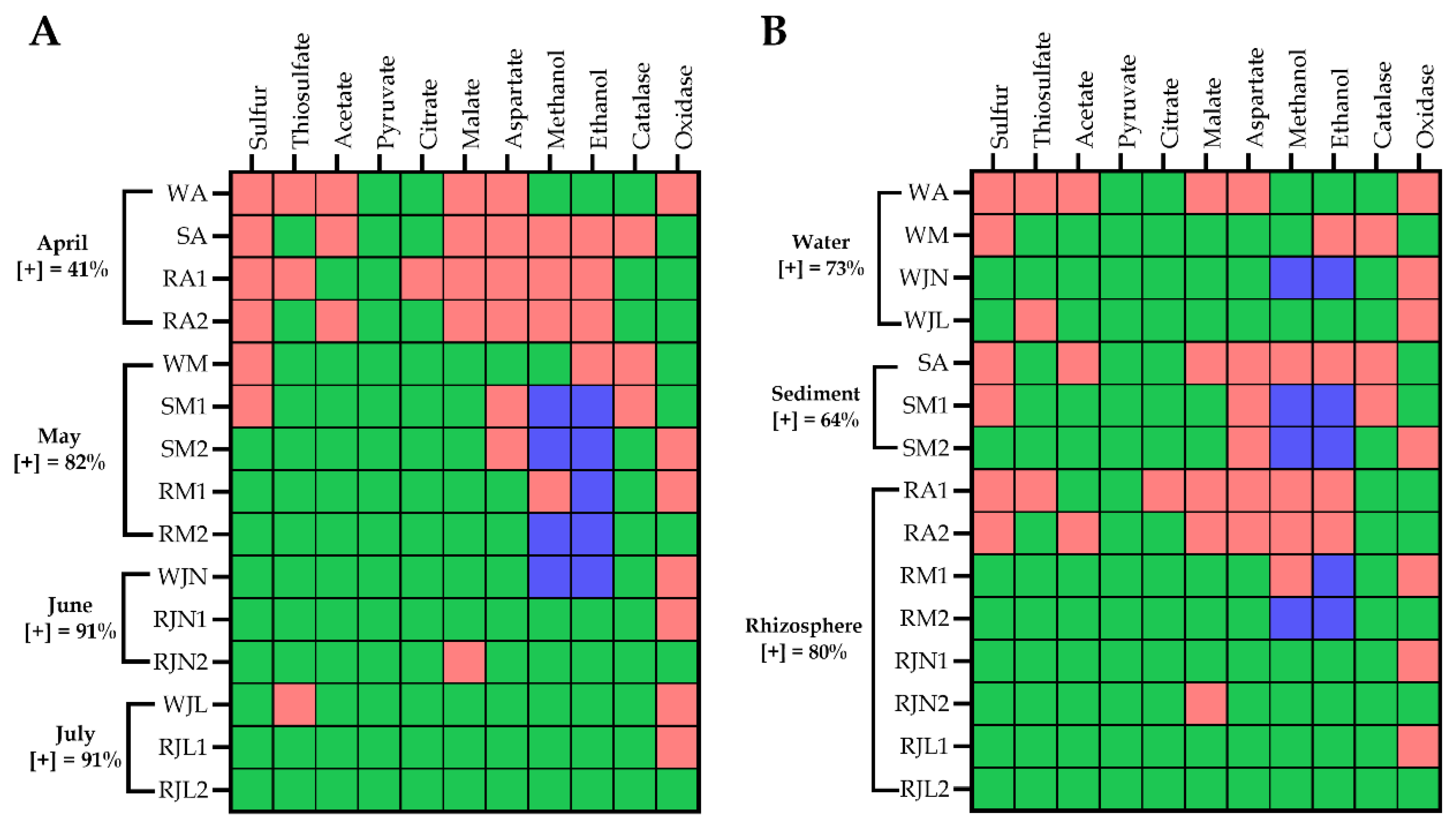
3.5. Metabolic Diversity and Functional Links Between Bacterial Isolates and Wetland Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CW | Constructed wetland |
| SC | Sulfur cycle |
| SOB | Sulfur-oxidizing bacteria |
| DO | Dissolved oxygen |
| BOD | Biochemical oxygen demand |
| CFU | Colony-forming units |
References
- Vymazal, J. Constructed wetlands for wastewater treatment. Water 2010, 2, 530–549. [Google Scholar] [CrossRef]
- Kurzbaum, E.; Kirzhner, F.; Armon, R. Improvement of water quality using constructed wetland systems. Rev. Environ. Health 2012, 27, 59–64. [Google Scholar] [CrossRef]
- Bodelier, P.L.E.; Dedysh, S.N. Microbiology of wetlands. Front. Microbiol. 2013, 4, 79. [Google Scholar] [CrossRef]
- Wu, S.; Kuschk, P.; Wiessner, A.; Müller, J.; Saad, R.A.; Dong, R. Sulphur transformations in constructed wetlands for wastewater treatment: A review. Ecol. Eng. 2013, 52, 278–289. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, Y.; Zhou, Q.; Huang, J.; Vymazal, J.; Kuschk, P. Sulfate removal and sulfur transformation in constructed wetlands: The roles of filling material and plant biomass. Water Res. 2016, 102, 572–581. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Arslan, M.; Kappelmeyer, U.; Musezahl, I.; Wiessner, A.; Müller, J.A. Spatial characterization of microbial sulfur cycling in horizontal-flow constructed wetland models. Chemosphere 2022, 309, 136605. [Google Scholar] [CrossRef]
- Knossow, N.; Blonder, B.; Eckert, W.; Turchyn, A.V.; Antler, G.; Kamyshny, A., Jr. Annual sulfur cycle in a warm monomictic lake with sub-millimolar sulfate concentrations. Geochem. Trans. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y. Geochemical cycle and environmental effects of sulfur in lakes. IOP Conf. Ser. Mater. Sci. Eng. 2018, 394, 052039. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Zurita, F.; Vera-Puerto, I.L.; Maine, A.; Arias, C.; Herazo, L.C.S. Use of constructed wetlands in Latin America: Assessment of current status, opportunities and challenges in Mexico, Chile and Argentina. J. Environ. Manag. 2025, 395, 127900. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.G.; Bardischewsky, F.; Rother, D.; Quentmeier, A.; Fischer, J. Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 2005, 8, 253–259. [Google Scholar] [CrossRef]
- Ghosh, W.; Dam, B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 2009, 33, 999–1043. [Google Scholar] [CrossRef]
- Klier, K.M.; Martin, C.; Langwig, M.V.; Anantharaman, K. Evolutionary history and origins of Dsr-mediated sulfur oxidation. ISME J. 2024, 18, wrae167. [Google Scholar] [CrossRef]
- Twible, L.E.; Whaley-Martin, K.; Chen, L.X.; Colenbrander Nelson, T.; Arrey, J.L.S.; Jarolimek, C.V.; King, J.J.; Ramilo, L.; Sonnenberg, H.; Banfield, J.F.; Apte, S.C.; Warren, L.A. pH and thiosulfate dependent microbial sulfur oxidation strategies across diverse environments. Front. Microbiol. 2024, 15, 1426584. [Google Scholar] [CrossRef]
- Rana, K.; Rana, N.; Singh, B. Applications of sulfur oxidizing bacteria. In Microbial Biotechnology: Applications in Agriculture and Environment; Salwan, R., Sharma, V., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 131–136. [Google Scholar] [CrossRef]
- Kumari, A.; Raj, S.; Singh, S.K.; Verma, K.K.; Mishra, P.K. Ecological functions of microbes in constructed wetlands for natural water purification. Water 2025, 17, 2947. [Google Scholar] [CrossRef]
- Lin, S.; Mackey, H.R.; Hao, T.; Guo, G.; Van Loosdrecht, M.C.; Chen, G. Biological sulfur oxidation in wastewater treatment: A review of emerging opportunities. Water Res. 2018, 143, 399–415. [Google Scholar] [CrossRef]
- Sánchez, O. Constructed wetlands revisited: Microbial diversity in the -omics era. Microb. Ecol. 2016, 73, 722–733. [Google Scholar] [CrossRef]
- Ercole, E.; Adamo, M.; Lumini, E.; Fusconi, A; Mucciarelli, M. Alpine constructed wetlands: A metagenomic analysis reveals microbial complementary structure. Sci. Total Environ. 2022, 822, 153640. [Google Scholar] [CrossRef] [PubMed]
- Seth, N.; Vats, S.; Lakhanpaul, S.; Arafat, Y.; Mazumdar-Leighton, S.; Bansal, M.; Babu, C.R. Microbial community diversity of an integrated constructed wetland used for treatment of sewage. Front. Microbiol. 2024, 15, 1355718. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Long, Y.; Yu, G.; Wang, G.; Zhou, Z.; Li, P.; Zhang, Y.; Yang, K.; Wang, S. A review on microorganisms in constructed wetlands for typical pollutant removal: Species, function and diversity. Front. Microbiol. 2022, 13, 845725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Bera, T.; Roy, S.; Vuong, P.; Jana, C.; Sarkar, D.J.; Devi, M.S.; Jana, A.K.; Rout, A.K.; Kaur, P.; Das, B.K.; Behera, B.K. Investigating bio-remediation capabilities of a constructed wetland through spatial successional study of the sediment microbiome. NPJ Clean Water 2023, 6, 1. [Google Scholar] [CrossRef]
- Jagetiya, S.P.B. Isolation and characterization of indigenous bacterial strains from constructed wetland with biofertilizer potential. J. Chem. Health Risks 2024, 14, 6638. [Google Scholar]
- Lipps, W.C.; Braun-Howland, E.B.; Baxter, T.E. (Eds.) Standard Methods for the Examination of Water and Wastewater, 24th ed.; American Public Health Association; American Water Works Association; Water Environment Federation; APHA Press: Washington, DC, USA, 2023. [Google Scholar]
- Starosvetsky, J.; Zukerman, U.; Armon, R.H. A simple medium modification for isolation, growth and enumeration of Acidithiobacillus thiooxidans syn. Thiobacillus thiooxidans from water samples. J. Microbiol. Methods 2013, 92, 178–182. [Google Scholar] [CrossRef]
- Breakwell, D.; Woolverton, C.; MacDonald, B.; Smith, K.; Robison, R. Colony morphology protocol. ASM Microbiol. 2007. Available online: https://asm.org/protocols/colony-morphology-protocol (accessed on 10 March 2025).
- Hassan, A.A.; Akineden, O.; Kress, C.; Estuningsih, S.; Schneider, E.; Usleber, E. Characterization of the gene encoding the 16S rRNA of Enterobacter sakazakii and development of a species-specific PCR method. Int. J. Food Microbiol. 2007, 116, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, Chapter 2, Unit 2.3. [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetics Analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Reiner, K. Catalase test protocol. ASM Microbiol. 2010. Available online: https://asm.org/Protocols/Catalase-Test-Protocol (accessed on 10 March 2025).
- Shields, P.; Cathcart, L. Oxidase test. ASM Microbiol. 2010. Available online: https://asm.org/Image-Gallery/Oxidase-Test (accessed on 10 March 2025).
- Rani, A.; Chauhan, M.; Kumar Sharma, P.; Kumari, M.; Mitra, D.; Joshi, S. Microbiological dimensions and functions in constructed wetlands: A review. Curr. Res. Microb. Sci. 2024, 7, 100311. [Google Scholar] [CrossRef] [PubMed]
- Abata, O.E.; Adesorioye, K.C.; Babaniyi, E.E.; Adebomi, J.I. Constructed wetlands for wastewater treatment and role of wetlands in water quality improvement and filtration. In Wetlands: Ecology, Conservation and Management; Babaniyi, B.R., Aransiola, S.A., Babaniyi, E.E., Maddela, N.R., Eds.; Springer: Cham, Switzerland, 2025; pp. 297–323. [Google Scholar] [CrossRef]
- Grinberga, L.; Celms, A.; Dzicio, J.; Guc̆howski, A.; Grabua, D.; Gr̄īnfeld̄e, I.; Lauva, D.; Sas, W. Analysis of the removal of BOD5, COD and suspended solids in subsurface flow constructed wetland in Latvia. Acta Sci. Pol. Archit. 2022, 20, 21–28. [Google Scholar] [CrossRef]
- Shi, B.; Cheng, X.; Jiang, S.; Pan, J.; Zhu, D.; Lu, Z.; Jiang, Y.; Liu, C.; Guo, H.; Xie, J. Unveiling the power of CODN on constructed wetlands in a short-term experiment: Exploring microbiota co-occurrence patterns and assembly dynamics. Sci. Total Environ. 2024, 912, 169568. [Google Scholar] [CrossRef]
- Castañer, C.M.; Bellver-Domingo, À.; Hernández-Sancho, F. Environmental and economic approach to assess a horizontal subsurface flow wetland in developing area. Water Resour. Manag. 2020, 34, 3761–3778. [Google Scholar] [CrossRef]
- Saeed, T.; Alam, M.K.; Miah, M.J.; Majed, N. Removal of heavy metals in subsurface flow constructed wetlands: Application of effluent recirculation. Environ. Sustain. Indic. 2021, 12, 100146. [Google Scholar] [CrossRef]
- Wu, S.; Kuschk, P.; Brix, H.; Vymazal, J.; Dong, R. Development of constructed wetlands in performance intensifications for wastewater treatment: A nitrogen and organic matter targeted review. Water Res. 2014, 57, 40–55. [Google Scholar] [CrossRef]
- Singh, K.K.; Vaishya, R.C. Municipal wastewater treatment uses vertical flow followed by horizontal flow in a two-stage hybrid constructed wetland planted with Calibanus hookeri and Canna indica (Cannaceae). Water Air Soil Pollut. 2022, 233, 510. [Google Scholar] [CrossRef] [PubMed]
- Amponsah, S.K.; Frimpong, F.; Danquah, E.O.; Amankwa-Yeboah, P.; Amengor, N.E.; Dzomeku, J.B.; Agyemang, S.M.; Adu, J.K.; Frimpong, T.; Azumah, D.D. Performance of a horizontal subsurface flow constructed wetland in treating aquaculture wastewater. J. Ecol. Eng. 2024, 25, 53–61. [Google Scholar] [CrossRef]
- Wang, T.; Li, X.; Wang, H.; Xue, G.; Zhou, M.; Ran, X.; Wang, Y. Sulfur autotrophic denitrification as an efficient nitrogen removal method for wastewater treatment towards lower organic requirement: A review. Water Res. 2023, 245, 120569. [Google Scholar] [CrossRef]
- Payan-Villalva, M.G.; Ramírez-Pereda, B.; Mendívil-García, K.; Ortiz-Marín, A.D.; Ro-Sosa, A.; Amabilis-Sosa, L.E. Performance of a horizontal subsurface flow constructed wetland for the efficient reduction of pollution due to shrimp farm wastewater. J. Environ. Sci. Health Part A 2025, 60, 283–292. [Google Scholar] [CrossRef]
- Zhai, J.; Zou, J.; He, Q.; Ning, K.; Xiao, H. Variation of dissolved oxygen and redox potential and their correlation with microbial population along a novel horizontal subsurface flow wetland. Environ. Technol. 2012, 33, 1999–2006. [Google Scholar] [CrossRef]
- Ozengin, N. Application of artificial neural network in horizontal subsurface flow constructed wetland for nutrient removal prediction. Appl. Ecol. Environ. Res. 2016, 14, 305–324. [Google Scholar] [CrossRef]
- Liu, G.; He, T.; Liu, Y.; Chen, Z.; Li, L.; Huang, Q.; Xie, Z.; Xie, Y; Wu, L.; Liu, J. Study on the purification effect of aeration-enhanced horizontal subsurface-flow constructed wetland on polluted urban river water. Environ. Sci. Pollut. Res. 2019, 26, 12867–12880. [Google Scholar] [CrossRef]
- Aregu, M.B.; Asfaw, S.L.; Khan, M.M. Developing horizontal subsurface flow constructed wetland using pumice and Chrysopogon zizanioides for tannery wastewater treatment. Environ. Syst. Res. 2021, 10, 1. [Google Scholar] [CrossRef]
- Pérez, Y.; Vargas, E.; García-Cortés, D.; Hernández, W.; Checo, H.; Juregui-Haza, U. Efficiency and effectiveness of systems for the treatment of domestic wastewater based on subsurface flow constructed wetlands in Jarabacoa, Dominican Republic. Water Sci. Eng. 2023, 17, 118–128. [Google Scholar] [CrossRef]
- Conti, F.; Rada, E.C.; Viotti, P.; Raboni, M. Removal and survival of fecal indicators in a constructed wetland after UASB pre-treatment. Sustainability 2021, 13, 9302. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Xiong, J.; Li, L.; Zhao, B.; Sohail, I.; He, Z. A constructed wetland system with aquatic macrophytes for cleaning contaminated runoff storm water from urban area in Florida. J. Environ. Manag. 2021, 280, 111794. [Google Scholar] [CrossRef]
- Montero-Martínez, M.J.; Castañeda-Chávez, M.D.R.; Lango-Reynoso, F.; Navarrete-Rodríguez, G.; Martínez-Cárdenas, L. Removal of pathogenic bacteria in a horizontally fed subsurface constructed wetland hybrid system. J 2023, 6, 492–507. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Mavrodi, O.; Bhowmik, N.; Weller, D.; Thomashow, L.; Mavrodi, D. Bacterial biofilms as an essential component of rhizosphere plant–microbe interactions. Methods Microbiol. 2023, 53, 3–48. [Google Scholar] [CrossRef]
- Mishra, T.; Tiwari, P.B.; Kanchan, S.; Kesheri, M. Advances in microbial bioremediation for effective wastewater treatment. Water 2025, 17, 3196. [Google Scholar] [CrossRef]
- Kochi, L.Y.; Freitas, P.L.; Maranho, L.T.; Juneau, P.; Gomes, M.P. Aquatic macrophytes in constructed wetlands: A fight against water pollution. Sustainability 2020, 12, 9202. [Google Scholar] [CrossRef]
- Singh, P.; Singh, G.; Singh, A.; Mishra, V.K.; Shukla, R. Macrophytes for utilization in constructed wetland as efficient species for phytoremediation of emerging contaminants from wastewater. Wetlands 2024, 44, 2. [Google Scholar] [CrossRef]
- Huang, Y.; An, W.; Ning, T.; Ma, Z.; Li, Y.; Liu, K.; Ji, L.; Liu, H.; Hui, D.; Ren, H. Functionality of bacterial communities in constructed wetlands used for water purification: Influence of root components and seasonality. Front. Plant Sci. 2025, 16, 1480099. [Google Scholar] [CrossRef] [PubMed]
- Cakin, I.; Morrissey, B.; Marcello, L.; Gaffney, P.P.J.; Pap, S.; Taggart, M.A. A comparison between constructed wetland substrates: Impacts on microbial community and wastewater treatment. Chemosphere 2024, 364, 143179. [Google Scholar] [CrossRef]
- Grüterich, L.; Wilson, M.; Jensen, K.; Streit, W.R.; Mueller, P. Transcriptomic response of wetland microbes to root influence. iScience 2024, 27, 110890. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liang, J.; Liu, R.; Hu, C.; Qu, J. Metagenomic analysis reveals microbial diversity and function in the rhizosphere soil of a constructed wetland. Environ. Technol. 2014, 35, 2521–2527. [Google Scholar] [CrossRef]
- Alavi, A.F.; Malik, S.; Ahmed, S.; Ahmed, B.; Salman, M.; Farooq, A.; Naz, I. Mitigation of antibiotic resistance: The efficiency of a hybrid subsurface flow constructed wetland in the removal of resistant bacteria in wastewater. Int. J. Phytoremediat. 2025, 27, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Salih, L.I.F.; Rasheed, R.O.; Muhammed, S.M. Raoultella ornithinolytica as a potential candidate for bioremediation of heavy metal from contaminated environments. J. Microbiol. Biotechnol. 2023, 33, 895–908. [Google Scholar] [CrossRef]
- Jia, Q.; Jin, H.; Jin, X.; Zhu, X.; Cui, C. Emergence of tigecycline-resistant Raoultella ornithinolytica with tetX-carrying plasmid from swine wastewater in China. Front. Microbiol. 2025, 16, 1642708. [Google Scholar] [CrossRef]
- Xu, S.; Xing, Y.; Liu, S.; Huang, Q.; Chen, W. Role of novel bacterial Raoultella sp. strain X13 in plant growth promotion and cadmium bioremediation in soil. Appl. Microbiol. Biotechnol. 2019, 103, 3887–3897. [Google Scholar] [CrossRef]
- Fu, W.; Zhao, Y.; Wang, Q.; Yu, X.; Song, Z.; Duan, P.; Xu, M; Zhang, X.; Rao, Z. Characterization of simultaneous removal of nitrogen and phosphorus by novel Raoultella ornithinolytica strain YX-4 and application in real farm wastewater treatment. Bioresour. Technol. 2023, 129922. [Google Scholar] [CrossRef] [PubMed]
- Thijs, S.; Van Hamme, J.; Gkorezis, P.; Rineau, F.; Weyens, N.; Vangronsveld, J. Draft genome sequence of Raoultella ornithinolytica TNT, a trinitrotoluene-denitrating and plant growth-promoting strain isolated from explosive-contaminated soil. Genome Announc. 2014, 2, e00491-14. [Google Scholar] [CrossRef]
- Isler, B.; Kidd, T.J.; Stewart, A.G.; Harris, P.; Paterson, D.L. Achromobacter infections and treatment options. Antimicrob. Agents Chemother. 2020, 64, e01025-20. [Google Scholar] [CrossRef]
- Jadhav, K.; Jadhav, I. Sulfur oxidation by Achromobacter xylosoxidans strain WSP05 reveals ecological widening over which thiotrophs are distributed. World J. Microbiol. Biotechnol. 2017, 33, 192. [Google Scholar] [CrossRef] [PubMed]
- Nyamath, S.; Subburamu, K.; Kalyanasundaram, G.T.; Balachandar, D.; Suresh, M.; Anandham, R. Multifarious characteristics of sulfur-oxidizing bacteria residing in rice rhizosphere. Folia Microbiol. 2024, 69, 395–405. [Google Scholar] [CrossRef]
- Zhong, X.; Teng, X.; Tang, B.; Lin, X.; Chen, J.; Zhao, R.; Wu, Q.; Huang, X. Constructed wetland treating mine drainage wastewater: Performance and microbial mechanisms. J. Water Process Eng. 2024, 60, 105244. [Google Scholar] [CrossRef]
- Xie, E.; Ding, A.; Zheng, L.; Lu, C.; Wang, J.; Huang, B.; Xiu, H. Seasonal variation in populations of nitrogen-transforming bacteria and correlation with nitrogen removal in a full-scale horizontal flow constructed wetland treating polluted river water. Geomicrobiol. J. 2016, 33, 338–346. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; He, X. Effects of selected root exudate components on nitrogen removal and development of denitrifying bacteria in constructed wetlands. Water 2017, 9, 430. [Google Scholar] [CrossRef]
- Sauvêtre, A.; Schröder, P. Uptake of carbamazepine by rhizomes and endophytic bacteria of Phragmites australis. Front. Plant Sci. 2015, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Roy, S.; Mester, J.C. A review of metal and antibiotic resistance of microbial strains and their bioremediation ability. In Urban Watershed Microbiology; Tiquia-Arashiro, S.M., Medema, G., Ubomba-Jaswa, E., Urakawa, H., Ella, V., Libardi, N., Eds.; Springer: Cham, Switzerland, 2025; Volume 1. [Google Scholar] [CrossRef]
- Jiao, Y.; Yuan, Q.; Wang, W.; Yan, L.; Mu, X.; Li, H.; Zhang, S. Vallisneria natans tolerance and response of microbial community in wetlands to excess nutrients loading. Ecol. Indic. 2021, 131, 108179. [Google Scholar] [CrossRef]
- He, S.W.; Ma, R.; Zhao, Y.Y.; An, L.; Huang, J.H.; Zhang, Q.; Han, J.G.; Zhang, X.X. Chitinophaga hostae sp. nov., isolated from the rhizosphere soil of Hosta plantaginea. Int. J. Syst. Evol. Microbiol. 2022, 72, 005335. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, D.; He, F.; Zhang, Y.; Wu, Z. Application of constructed wetland for water pollution control in China during 1990–2010. Ecol. Eng. 2012, 47, 189–197. [Google Scholar] [CrossRef]
- Feng, H.; An, J.; Wang, H.; Miao, X.; Yang, G.; Feng, H.; Wu, Y.; Ma, X. The ecological healthcare benefits and influences of plant communities in urban wetland parks. Forests 2023, 14, 2257. [Google Scholar] [CrossRef]
- Zha, Y.; Chong, H.; Yang, P.; Ning, K. Microbial dark matter: From discovery to applications. Genom. Proteom. Bioinform. 2022, 20, 867–881. [Google Scholar] [CrossRef]
- Li, W.J.; Rekadwad, B.N.; Jiao, J.Y.; Salam, N. Exploring microbial dark matter and the status of bacterial and archaeal taxonomy: Challenges and opportunities in the future. In Modern Taxonomy of Bacteria and Archaea; Li, W.J., Jiao, J.Y., Salam, N., Rao, M.P.N., Eds.; Springer: Singapore, 2024. [Google Scholar] [CrossRef]
- Osburn, E.D.; McBride, S.G.; Strickland, M.S. Microbial dark matter could add uncertainties to metagenomic trait estimations. Nat. Microbiol. 2024, 9, 1427–1430. [Google Scholar] [CrossRef]
- Abdallah, R.Z.; Elbehery, A.H.A.; Ouf, A.; Siam, R. Microbial dark matter spearheading the biogeochemical cycle in the Solar Lake of Taba, Egypt. Curr. Res. Microb. Sci. 2025, 9, 100433. [Google Scholar] [CrossRef]
- Kumar, R.; Mishra, A. Functional gene diversity and metabolic potential of uncultured bacteria. In Microbial Diversity in Ecosystem Sustainability and Biotechnological Applications; Das, S., Dash, H.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2024; pp. 481–491. [Google Scholar] [CrossRef]
- Fulke, A.B.; Sharma, N.; Nadekar, J. Darkness to discovery: A comprehensive mini-review on culturable and non-culturable microbial diversity from deep sea. Microb. Ecol. 2025, 88, 77. [Google Scholar] [CrossRef]
- Ji, M.; Ma, B.; Dong, J.; Liu, S.; Shi, Y.; Bu, M.; Wang, L.; Liu, L. Mining microbial dark matter: Advanced cultivation techniques for bioactive compound discovery. Pharmaceuticals 2025, 18, 1583. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wen, L.; He, C.; Chen, X.; Si, L.; Li, H.; Liang, Y.; Zheng, W.; Guo, F. Sequencing-guided re-estimation and promotion of cultivability for environmental bacteria. Nat. Commun. 2024, 15, 9051. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Begum, F.; Ullah, I.; Jalal, N.; Shaw, P. Peeling off the layers from microbial dark matter (MDM): Recent advances, future challenges, and opportunities. Crit. Rev. Microbiol. 2025, 51, 1–21. [Google Scholar] [CrossRef]
- Iasur-Kruh, L.; Hadar, Y.; Milstein, D.; Gasith, A.; Minz, D. Microbial population and activity in wetland microcosms constructed for improving treated municipal wastewater. Microb. Ecol. 2010, 59, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Bouali, M.; Zrafi, I.; Bakhrouf, A.; Chaussonnerie, S.; Sghir, A. Bacterial structure and spatiotemporal distribution in a horizontal subsurface flow constructed wetland. Appl. Microbiol. Biotechnol. 2013, 98, 3191–3203. [Google Scholar] [CrossRef]
- He, G.; Yi, F.; Zhou, S.; Lin, J. Microbial activity and community structure in two terrace-type wetlands constructed for the treatment of domestic wastewater. Ecol. Eng. 2014, 67, 198–205. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhu, J.N.; Liu, Q.F.; Liu, Y.; Liu, M.; Liu, L.; Zhang, Q. Comparison of the diversity of root-associated bacteria in Phragmites australis and Typha angustifolia L. in artificial wetlands. World J. Microbiol. Biotechnol. 2013, 29, 1499–1508. [Google Scholar] [CrossRef]
- Kumar, V.; Ameen, F.; Verma, P. Unraveling the shift in bacterial communities profile grown in sediments co-contaminated with chlorolignin waste of pulp–paper mill by metagenomics approach. Front. Microbiol. 2024, 15, 1350164. [Google Scholar] [CrossRef]
- Vetrovský, T.; Baldrian, P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE 2013, 8, e57923. [Google Scholar] [CrossRef]
- Ramasamy, D.; Mishra, A.K.; Lagier, J.C.; Padhmanabhan, R.; Rossi, M.; Sentausa, E.; Raoult, D.; Fournier, P.E. A polyphasic strategy incorporating genomic data for the taxonomic description of novel bacterial species. Int. J. Syst. Evol. Microbiol. 2014, 64, 384–391. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Castro, J.C.; Kyrpides, N.C.; Cole, J.R.; Tiedje, J.M.; Konstantinidis, K.T. How much do rRNA gene surveys underestimate extant bacterial diversity? Appl. Environ. Microbiol. 2018, 84, e00014-18. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; Sodergren, E.; Weinstock, G.M. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Kämpfer, P.; Glaeser, S.P. Prokaryotic taxonomy in the sequencing era—the polyphasic approach revisited. Environ. Microbiol. 2012, 14, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B.M. Taxonomic note: A place for DNA–DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Rossi-Tamisier, M.; Benamar, S.; Raoult, D.; Fournier, P.E. Cautionary tale of using 16S rRNA gene sequence similarity values in identification of human-associated bacterial species. Int. J. Syst. Evol. Microbiol. 2015, 65, 1929–1934. [Google Scholar] [CrossRef]
- Christensen, H.; Korczak, B.M.; Bojesen, A.M.; Kuhnert, P.; Frederiksen, W.; Bisgaard, M. Classification of organisms previously reported as the SP and Stewart–Letscher groups, with descriptions of Necropsobacter gen. nov. and of Necropsobacter rosorum sp. nov. for organisms of the SP group. Int. J. Syst. Evol. Microbiol. 2011, 61, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Tanaka, Y.; Matsuzawa, H.; Muramatsu, M.; Meng, X.Y.; Hanada, S.; Mori, K.; Kamagata, Y. Armatimonas rosea gen. nov., sp. nov., of a novel bacterial phylum, Armatimonadetes phyl. nov., formally called the candidate phylum OP10. Int. J. Syst. Evol. Microbiol. 2011, 61, 1442–1450. [Google Scholar] [CrossRef]
- Stride, M.C.; Polkinghorne, A.; Miller, T.L.; Groff, J.M.; Lapatra, S.E.; Nowak, B.F. Molecular characterization of “Candidatus Parilichlamydia carangidicola”, a novel Chlamydia-like epitheliocystis agent in yellowtail kingfish, Seriola lalandi (Valenciennes), and the proposal of a new family, “Candidatus Parilichlamydiaceae” fam. nov. (order Chlamydiales). Appl. Environ. Microbiol. 2013, 79, 1590–1597. [Google Scholar] [CrossRef]
- Dar, R.; Bandh, S.A.; Shafi, S.; Shameem, N. Bacterial diversity of the rock–water interface in freshwater ecosystem. In Freshwater Microbiology; Bandh, S.A., Shafi, S., Shameem, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 73–104. [Google Scholar] [CrossRef]
- da Rocha, U.N.; van Overbeek, L.; van Elsas, J.D. Exploration of hitherto-uncultured bacteria from the rhizosphere. FEMS Microbiol. Ecol. 2009, 69, 313–328. [Google Scholar] [CrossRef]
- Youseif, S.H.; El-Megeed, F.H.A.; Humm, E.A.; Maymon, M.; Mohamed, A.H.; Saleh, S.A.; Hirsch, A.M. Comparative analysis of the cultured and total bacterial community in the wheat rhizosphere microbiome using culture-dependent and culture-independent approaches. Microbiol. Spectr. 2021, 9, e00678-21. [Google Scholar] [CrossRef]
- Kato, S.; Yamagishi, A.; Daimon, S.; Kawasaki, K.; Tamaki, H.; Kitagawa, W.; Abe, A.; Tanaka, M.; Sone, T.; Asano, K.; Kamagata, Y. Isolation of previously uncultured slow-growing bacteria by using a simple modification in the preparation of agar media. Appl. Environ. Microbiol. 2018, 84, e00807-18. [Google Scholar] [CrossRef]
- Kato, S.; Terashima, M.; Yama, A.; Sato, M.; Kitagawa, W.; Kawasaki, K.; Kamagata, Y. Improved isolation of uncultured anaerobic bacteria using medium prepared with separate sterilization of agar and phosphate. Microbes Environ. 2020, 35, ME19060. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, D.K.; Khulan, A.; Kim, J. Development of a novel cultivation technique for uncultured soil bacteria. Sci. Rep. 2019, 9, 6666. [Google Scholar] [CrossRef]
- Chang, J.; Yoon, I.; Lee, J.; Kim, K.; An, J.; Kim, K. Arsenic detoxification potential of aox genes in arsenite-oxidizing bacteria isolated from natural and constructed wetlands in the Republic of Korea. Environ. Geochem. Health 2010, 32, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kouki, S.; Saidi, N.; Mhiri, F.; Nasr, H.; Cherif, H.; Ouzari, H.; Hassen, A. Isolation and characterization of facultative mixotrophic ammonia-oxidizing bacteria from constructed wetlands. J. Environ. Sci. 2011, 23, 1699–1708. [Google Scholar] [CrossRef]
- Dong, X.; Reddy, G.B. Ammonia-oxidizing bacterial community and nitrification rates in constructed wetlands treating swine wastewater. Ecol. Eng. 2012, 40, 189–197. [Google Scholar] [CrossRef]
- Lee, Y.J.; Romanek, C.S.; Mills, G.L.; Davis, R.C.; Whitman, W.B.; Wiegel, J. Gracilibacter thermotolerans gen. nov., sp. nov., an anaerobic, thermotolerant bacterium from a constructed wetland receiving acid sulfate water. Int. J. Syst. Evol. Microbiol. 2006, 56, 2089–2093. [Google Scholar] [CrossRef]
- Guo, S.Z.; Wu, T.; Zhu, H.Z.; Yan, L.; Liu, Z.P.; Li, D.F.; Jiang, C.Y.; Liu, S.J.; Shen, X.H. Niabella beijingensis sp. nov. and Thermomonas beijingensis sp. nov., two bacteria from constructed wetland. Int. J. Syst. Evol. Microbiol. 2022, 72, 005280. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.Y.; Nam, I.H.; Kim, S.J. Undibacter mobilis gen. nov., sp. nov. isolated from an artificial wetland in Okcheon, Korea. Int. J. Syst. Evol. Microbiol. 2022, 72, 005369. [Google Scholar] [CrossRef]
- Gillis, M.; Vandamme, P.; De Vos, P.; Swings, J.; Kersters, K. Polyphasic taxonomy. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–10. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Chuvochina, M.; Oren, A.; Parks, D.H.; Soo, R.M. Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J. 2021, 15, 1879–1892. [Google Scholar] [CrossRef]
- Salcedo Sánchez, E.R.; Martínez, J.M.E.; Morales, M.M.; Talavera Mendoza, O.; Alberich, M.V.E. Ecological and health risk assessment of potential toxic elements from a mining area water and sediments: The San Juan–Taxco river system, Guerrero, Mexico. Water 2022, 14, 518. [Google Scholar] [CrossRef]
- Pu, Y.; Li, Y.; Zhu, L.; Cheng, Y.; Nuamah, L.A.; Zhang, H.; Chen, H.; Du, G.; Wang, L.; Song, C. Long-term assessment on performance and seasonal optimal operation of a full-scale integrated multiple constructed wetland–pond system. Sci. Total Environ. 2023, 862, 161219. [Google Scholar] [CrossRef] [PubMed]
- Tlili, H.; Bali, M.; Boukchina, R. Seasonal evaluation of pollutant removal in a vertical constructed wetland in Tunisia’s arid climate. Int. J. Phytoremediat. 2025, 27, 1602–1617. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.M.; Kareem, S.L. Evaluating the role of hydraulic retention time (HRT) in pollutant removal efficiency using Arundo donax in vertical subsurface flow constructed wetlands. Bioremediat. J. 2024, 28, 1–15. [Google Scholar] [CrossRef]
- Hao, K.; Zhao, Y.; Wei, T.; Wen, D.; Liu, A.; Huang, Y.; Chen, Z. Recent advances of constructed wetlands utilization under cold environment: Strategies and measures. Process Saf. Environ. Prot. 2025, 201, 107530. [Google Scholar] [CrossRef]
- Murray, L.; Fullerton, H.; Moyer, C.L. Microbial metabolic potential of hydrothermal vent chimneys along the submarine ring of fire. Front. Microbiol. 2024, 15, 1399422. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, S.; Jiang, S.; Zhang, D.; Sha, Z. Diversity and potential metabolic characteristics of culturable copiotrophic bacteria that can grow on low-nutrient medium in Zhenbei seamount in the South China Sea. Microb. Ecol. 2024, 87, 157. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, W.; Liu, Y.; Cai, M.; Luo, Z.; Li, M. Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap Trench. Front. Microbiol. 2018, 9, 2402. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Z.; Lu, T.; Yu, Y.; Peñuelas, J.; Zhu, Y.; Qian, H. Gammaproteobacteria, a core taxon in the guts of soil fauna, are potential responders to environmental concentrations of soil pollutants. Microbiome 2021, 9, 196. [Google Scholar] [CrossRef]
- Cai, X.; Li, J.; Guan, F.; Luo, X.; Yuan, Y. Unveiling metabolic characteristics of an uncultured Gammaproteobacterium responsible for in situ PAH biodegradation in petroleum polluted soil. Environ. Microbiol. 2021, 23, 7093–7104. [Google Scholar] [CrossRef]
- Mellado, M.; Vera, J. Microorganisms that participate in biochemical cycles in wetlands. Can. J. Microbiol. 2021, 67, 771–788. [Google Scholar] [CrossRef]
- Bhargawa, P.K.; Kumar, A.; Singh, A.; Kumar, R. A metagenomics profiling of rhizospheric microbes in constructed wetlands for Azo dye degradation: Managing abiotic stress for sustainable practices. Total Environ. Microbiol. 2025, 14, 100045. [Google Scholar] [CrossRef]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 2020, 19, 225–240. [Google Scholar] [CrossRef]
- Salam, N.; Xian, W.; Asem, M.D.; Xiao, M.; Li, W. From ecophysiology to cultivation methodology: Filling the knowledge gap between uncultured and cultured microbes. Mar. Life Sci. Technol. 2020, 2, 132–147. [Google Scholar] [CrossRef]
- Liu, S.; Moon, C.D.; Zheng, N.; Huws, S.; Zhao, S.; Wang, J. Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation. Microbiome 2022, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Rossell-Móra, R.; Amann, R. Past and future species definitions for Bacteria and Archaea. Syst. Appl. Microbiol. 2015, 38, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hatzenpichler, R.; Krukenberg, V.; Spietz, R.L.; Jay, Z.J. Next-generation physiology approaches to study microbiome function at single cell level. Nat. Rev. Microbiol. 2020, 18, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Forchielli, E.; Sher, D.; Segrè, D. Metabolic phenotyping of marine heterotrophs on refactored media reveals diverse metabolic adaptations and lifestyle strategies. mSystems 2022, 7, e00070-22. [Google Scholar] [CrossRef]
- Liu, Y.; Li, T.; Yang, C.; Deng, H. Bacterial energy metabolism. In Reference Module in Life Sciences; Tang, Y.W., Hindiyeh, M.Y., Liu, D., Sails, A., Spearman, P., Zhang, J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2024; pp. 177–200. [Google Scholar] [CrossRef]
- Dewachter, L.; Fauvart, M.; Michiels, J. Bacterial heterogeneity and antibiotic survival: Understanding and combatting persistence and heteroresistance. Mol. Cell 2019, 76, 255–267. [Google Scholar] [CrossRef]
- Nitschke, W.; Farr, O.; Gaudu, N.; Truong, C.; Guyot, F.; Russell, M.J.; Duval, S. The winding road from origin to emergence of life. Life 2024, 14, 607. [Google Scholar] [CrossRef]
- Palleroni, N.J. Stenotrophomonas . In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–20. [Google Scholar] [CrossRef]
- Ouattara, A.S.; Le Mer, J.; Joseph, M.; Macarie, H. Transfer of Pseudomonas pictorum Gray and Thornton 1928 to genus Stenotrophomonas as Stenotrophomonas pictorum comb. nov., and emended description of the genus Stenotrophomonas. Int. J. Syst. Evol. Microbiol. 2017, 67, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Malviya, D.; Varma, A.; Singh, U.B.; Singh, S.; Saxena, A.K. Unraveling the mechanism of sulfur nutrition in pigeonpea inoculated with sulfur-oxidizing bacteria. Front. Microbiol. 2022, 13, 927702. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, W.; Liu, L.; Guo, L.; Li, X.; Liu, Y. Anti-Stenotrophomonas maltophilia mechanism of rose essential oil: A metabolomic study. Food Bioproc. Technol. 2023, 17, 2693–2705. [Google Scholar] [CrossRef]
- Li, Z.; Nandakumar, R.; Madayiputhiya, N.; Li, X. Proteomic analysis of 17-estradiol degradation by Stenotrophomonas maltophilia. Environ. Sci. Technol. 2012, 46, 5947–5955. [Google Scholar] [CrossRef]
- Zhang, L.; Song, C.; Xu, Y.; Shi, Y.; Liu, X. Isolation, characterization and S2− oxidation metabolic pathway of a sulfur-oxidizing strain from a black-odor river in Beijing. Water Sci. Technol. Water Supply 2022, 22, 3729–3743. [Google Scholar] [CrossRef]
- Sun, Q.; Fang, Y.; Liu, W.; Xie, N.; Dong, H.; Guadie, A.; Liu, Y.; Cheng, H.; Wang, A. Synergistic between autotrophic and heterotrophic microorganisms for denitrification using bio-S as electron donor. Environ. Res. 2023, 231, 116047. [Google Scholar] [CrossRef]
- Jochum, L.M.; Schreiber, L.; Marshall, I.P.G.; Jørgensen, B.B.; Schramm, A.; Kjeldsen, K.U. Single-cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans-related Deltaproteobacteria widely distributed in marine sediment. Front. Microbiol. 2018, 9, 2038. [Google Scholar] [CrossRef]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.M.; Huntemann, M.; et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2021, 39, 499–509. [Google Scholar] [CrossRef]
- Pacheco-Valenciana, A.; Tausch, A.; Veseli, I.; Dharamshi, J.E.; Bergland, F.; Delgado, L.F.; Rodríguez-Gijón, A.; Andersson, A.F.; Garcia, S.L. Microbial model communities exhibit widespread metabolic interdependencies. Commun. Biol. 2025, 8, 1752. [Google Scholar] [CrossRef]
- Dong, X.; Greening, C.; Rattray, J.E.; Chakraborty, A.; Chuvochina, M.; Mayumi, D.; Dolfing, J.; Li, C.; Brooks, J.M.; Bernard, B.B.; et al. Metabolic potential of uncultured bacteria and archaea associated with petroleum seepage in deep-sea sediments. Nat. Commun. 2019, 10, 1816. [Google Scholar] [CrossRef]
- Srinivas, P.; Peterson, S.B.; Gallagher, L.A.; Wang, Y.; Mougous, J.D. Beyond genomics in Patescibacteria: A trove of unexplored biology packed into ultrasmall bacteria. Proc. Natl. Acad. Sci. USA 2024, 121, e2419369121. [Google Scholar] [CrossRef]
- Tong, X.; Luo, D.; Leung, M.H.Y.; Lee, J.Y.Y.; Shen, Z.; Jiang, W.; Mason, C.E.; Lee, P.K.H. Diverse and specialized metabolic capabilities of microbes in oligotrophic built environments. Microbiome 2024, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Busse, H.; Auling, G. Achromobacter . In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–14. [Google Scholar] [CrossRef]
- Jie, S.; Han, J.; Jia, S.; Tan, N.; Jin, W. Achromobacter sp. JWJ-09 based on methanol co-metabolism efficiently enhanced the degradation of PAHs in coking wastewater and domestic sewage using activated sludge. J. Water Process Eng. 2025, 76, 108152. [Google Scholar] [CrossRef]
- D’Souza, G.; Kost, C. Experimental evolution of metabolic dependency in bacteria. PLoS Genet. 2016, 12, e1006364. [Google Scholar] [CrossRef]
- Baumgartner, M.; Roffler, S.; Wicker, T.; Pernthaler, J. Letting go: Bacterial genome reduction solves the dilemma of adapting to predation mortality in a substrate-restricted environment. ISME J. 2017, 11, 2258–2266. [Google Scholar] [CrossRef]
- Ekkers, D.M.; Tusso, S.; Moreno-Gámez, S.; Rillo, M.C.; Kuipers, O.P.; van Doorn, G.S. Trade-offs predicted by metabolic network structure give rise to evolutionary specialization and phenotypic diversification. Mol. Biol. Evol. 2022, 39, msac124. [Google Scholar] [CrossRef] [PubMed]
- Serra Moncadas, L.; Hofer, C.; Bulzu, P.A.; Pernthaler, J.; Andrei, A.S. Freshwater genome-reduced bacteria exhibit pervasive episodes of adaptive stasis. Nat. Commun. 2024, 15, 3421. [Google Scholar] [CrossRef]
- LeBlanc, N.; Charles, T.C. Bacterial genome reductions: Tools, applications, and challenges. Front. Genome Ed. 2022, 4, 957289. [Google Scholar] [CrossRef]
- Zhu, M.; Dai, X. Shaping of microbial phenotypes by trade-offs. Nat. Commun. 2024, 15, 4238. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Kawamura, Y.; Ezaki, T. Ralstonia . In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–21. [Google Scholar] [CrossRef]
- Swanner, E.D.; Nell, R.M.; Templeton, A.S. Ralstonia species mediate Fe-oxidation in circumneutral, metal-rich subsurface fluids of Henderson mine, CO. Chem. Geol. 2011, 284, 339–350. [Google Scholar] [CrossRef]
- Li, G.; Li, H.; Xiao, K.; Bao, P. Thiosulfate reduction coupled with anaerobic ammonium oxidation by Ralstonia sp. GX3-BWBA. ACS Earth Space Chem. 2020, 4, 2426–2434. [Google Scholar] [CrossRef]
- Lee, H.; Jeon, B.; Oh, M. Microbial production of ethanol from acetate by engineered Ralstonia eutropha. Biotechnol. Bioprocess Eng. 2016, 21, 402–407. [Google Scholar] [CrossRef]
- Imazaki, I.; Nakaho, K. Pyruvate-amended modified SMSA medium: Improved sensitivity for detection of Ralstonia solanacearum. J. Gen. Plant Pathol. 2009, 76, 52–61. [Google Scholar] [CrossRef]
- Yuan, C.; An, T.; Li, X.; Zou, J.; Lin, Z.; Gu, J.; Hu, R.; Fang, Z. Genomic analysis of Ralstonia pickettii reveals the genetic features for potential pathogenicity and adaptive evolution in drinking water. Front. Microbiol. 2024, 14, 1272636. [Google Scholar] [CrossRef]
- Thakur, T.K.; Barya, M.P.; Dutta, J.; Mukherjee, P.; Thakur, A.; Swamy, S.L.; Anderson, J.T. Integrated phytobial remediation of dissolved pollutants from domestic wastewater through constructed wetlands: An interactive macrophyte–microbe-based green and low-cost decontamination technology with prospective resource recovery. Water 2023, 15, 3877. [Google Scholar] [CrossRef]
- Grimont, P.A.; Grimont, F. Enterobacter . In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; Wiley: Hoboken, NJ, USA, 2015; pp. 1–17. [Google Scholar] [CrossRef]
- Manter, D.K.; Hunter, W.J.; Vivanco, J.M. Enterobacter soli sp. nov., a lignin-degrading γ-proteobacteria isolated from soil. Curr. Microbiol. 2011, 62, 1044–1049. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Nazir, R.; Sessitsch, A.; Elhottová, D.; Korenblum, E.; van Overbeek, L.S.; van Elsas, J.D. The new species Enterobacter oryziphilus sp. nov. and Enterobacter oryzendophyticus sp. nov. are key inhabitants of the endosphere of rice. BMC Microbiol. 2013, 13, 164. [Google Scholar] [CrossRef]
- Akita, H.; Matsushika, A.; Kimura, Z.I. Enterobacter oligotrophica sp. nov., a novel oligotroph isolated from leaf soil. MicrobiologyOpen 2019, 8, e00843. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, I.H.; Lee, W.J.; Lee, J. Characterization of thiosulfate-oxidizing Enterobacter hormaechei JH isolated from barnyard manure. Korean J. Chem. Eng. 2008, 25, 1131–1135. [Google Scholar] [CrossRef]
- Andrs-Barrao, C.; Alzubaidy, H.; Jalal, R.; Mariappan, K.G.; de Zelicourt, A.; Bokhari, A.; Artyukh, O.; Alwutayd, K.; Rawat, A.; Shekhawat, K.; et al. Coordinated bacterial and plant sulfur metabolism in Enterobacter sp. SA187-induced plant salt stress tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2107417118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, X.; Zhang, Z.; Chen, G.; Jiang, F. Elemental sulfur as an electron acceptor for organic matter removal in a new high-rate anaerobic biological wastewater treatment process. Chem. Eng. J. 2017, 331, 16–22. [Google Scholar] [CrossRef]
- Zapata-Morales, A.L.; La Torre, M.C.A.; Hernández-Morales, A.; La Cruz, R.F.G. Isolation of cultivable bacteria associated with the root of Typha latifolia in a constructed wetland for the removal of diclofenac or naproxen. Water Air Soil Pollut. 2020, 231, 8. [Google Scholar] [CrossRef]
- La Rosa, R.; Behrends, V.; Williams, H.D.; Bundy, J.G.; Rojo, F. Influence of the Crc regulator on the hierarchical use of carbon sources from a complete medium in Pseudomonas. Environ. Microbiol. 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Sievert, S.M.; Kiene, R.P.; Schulz-Vogt, H.N. The sulfur cycle. Oceanography 2007, 20, 117–123. Available online: https://www.jstor.org/stable/24860050. [CrossRef]
- Lens, P. Sulfur cycle. In Encyclopedia of Ecology; Schaechter, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 361–369. [Google Scholar] [CrossRef]
- Pokorna, D.; Zabranska, J. Sulfur-oxidizing bacteria in environmental technology. Biotechnol. Adv. 2015, 33, 1246–1259. [Google Scholar] [CrossRef]
- Sturman, P.J.; Stein, O.R.; Vymazal, J.; Kröpfelová, L. Sulfur cycling in constructed wetlands. In Wastewater Treatment, Plant Dynamics and Management in Constructed and Natural Wetlands; Vymazal, J., Ed.; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef]
- Mateos, K.; Chappell, G.; Klos, A.; Le, B.; Boden, J.; Steken, E.; Anderson, R. The evolution and spread of sulfur cycling enzymes reflect the redox state of the early Earth. Sci. Adv. 2023, 9, eade4847. [Google Scholar] [CrossRef]
- Tanabe, T.S.; Bach, E.; D’Ermo, G.; Mohr, M.G.; Hager, N.; Pfeiffer, N.; Guiral, M.; Dahl, C. A cascade of sulfur transferases delivers sulfur to the sulfur-oxidizing heterodisulfide reductase-like complex. Protein Sci. 2024, 33, e5014. [Google Scholar] [CrossRef] [PubMed]
- Berben, T.; Overmars, L.; Sorokin, D.Y.; Muyzer, G. Diversity and distribution of sulfur oxidation-related genes in Thioalkalivibrio, a genus of chemolithoautotrophic and haloalkaliphilic sulfur-oxidizing bacteria. Front. Microbiol. 2019, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Maile-Moskowitz, A.; Brown, C.L.; Rumi, M.A.; Moumi, N.A.; Majeed, H.; Finkielstein, C.V.; Ceci, A.; Gonzalez, R.; Xia, K.; McDaniel, L.; et al. Relating antimicrobial use to wastewater resistance gene patterns via metagenomic analysis of two neighboring treatment plants circa the COVID-19 pandemic. NPJ Antimicrob. Resist. 2025, 3, 82. [Google Scholar] [CrossRef] [PubMed]

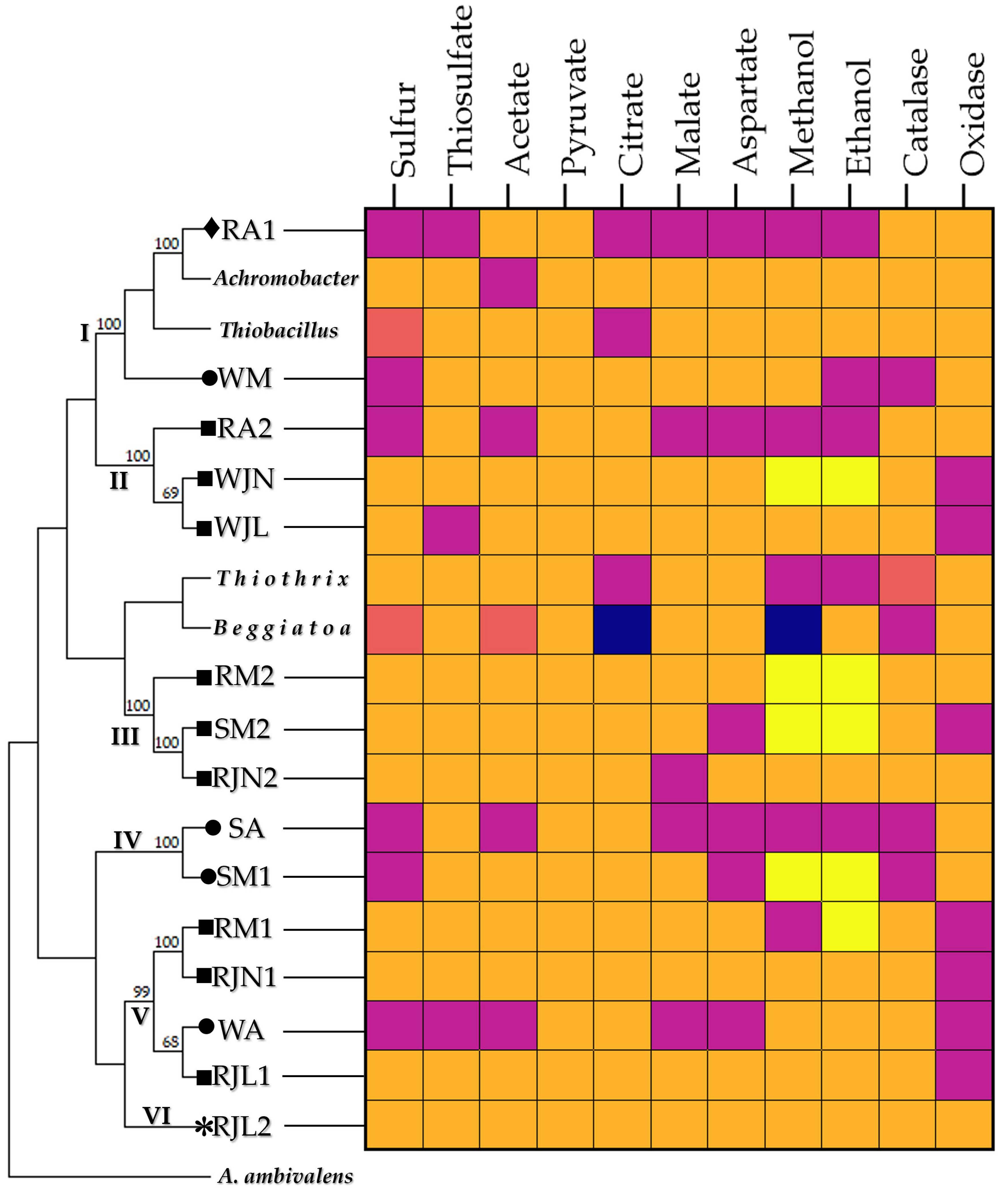

| pH | Temperature (°C) | Dissolved oxygen | BOD₅ | BOD₅ removal | |||||
|---|---|---|---|---|---|---|---|---|---|
| Sampling month | Inlet | Outlet | Inlet | Outlet | Inlet | Outlet | Inlet | Outlet | |
| April | 6.82 | 7.02 | 24.3 | 24.5 | 1.21 | 1.82 | 765.69 | 166.44 | 78.3 |
| May | 6.96 | 7.09 | 23.3 | 23.3 | 2.02 | 1.41 | 359.7 | 89.7 | 75.1 |
| June | 7.09 | 6.96 | 22.01 | 20.5 | 0.51 | 0.46 | 363.95 | 63.71 | 82.5 |
| July | 6.8 | 6.4 | 21 | 23 | 1.01 | 0.6 | 300 | 39.8 | 87.0 |
| Mean ± SD | 6.92 ± 0.12 |
6.87 ± 0.27 |
22.65 ± 1.25 |
22.82 ± 1.45 |
1.19 ± 0.54 |
1.07 ± 0.56 |
447.33 ± 185.53 |
89.91 ± 47.57 |
80.6 ± 4.4 |
| Sampling month |
Water (CFU/mL) | Sediment (CFU/g) | Rhizosphere (CFU/g) | Number of morphotypes |
|---|---|---|---|---|
| April | 184 | 4990 | 5,650 | 5 |
| May | 233 | 42,090 | 33,200 | 8 |
| June | 492 | NG | 23,370 | 3 |
| July | 221 | NG | 27,150 | 6 |
| Mean ± SD | 282±122 | 11,770±17,623 | 22,342±10,255 |
| Isolate code | Class | Closest match (NCBI BLAST) | Habitat/Function | % identity |
|---|---|---|---|---|
| WA |
Uncultured bacterium |
Uncultured Stenotrophomonas sp. clone F2mar.2 (GQ417316) | Biological degreasing system | 97.83 |
| SA | Uncultured Gammaproteobacterium clone YYSM141 (EU629110) | Forest soil | 99.24 | |
| RA1 | Betaproteobacteria |
Achromobacter mucicolens H323 (MH669290) |
Endophytic bacteria of pine tree | 93.87 |
| RA2 | Gammaproteobacteria |
Enterobacter sp. UYSB150 (MT071134) |
Plant growth-promoting bacteria | 100 |
| WM |
Uncultured bacterium |
Uncultured Ralstonia sp. clone EC34BC01 (JN032362) | Cave; urine-degrading bacteria | 99.43 |
| SM1 | Uncultured Gammaproteobacterium clone YYSM141 (EU629110) | Forest soil | 99.71 | |
| SM2 | Gammaproteobacteria |
Pseudomonas sp. NCCP-1812 (LC270247) |
Environmental samples; antibiotic resistance testing | 94.19 |
| RM1 | Stenotrophomonas sp. CPO 4.0065 (KF921612) | Rhizosphere; hydrocarbon-degrading bacteria | 91.12 | |
| RM2 | Pseudomonas putida TS18 (MG768972) | Plant | 85.17 | |
| WJN | Raoultella ornithinolytica CRI 1-4 (KU297681) | Rhizosphere; constructed wetland | 92.61 | |
| RJN1 | Stenotrophomonas sp. CPO 4.0065 (KF921612) | Rhizosphere; hydrocarbon-degrading bacteria | 90.83 | |
| RJN2 |
Pseudomonas sp. NCCP-1812 (LC270247) |
Environmental samples; antibiotic resistance testing | 93.78 | |
| WJL | Raoultella ornithinolytica DEP_6097311 (PQ479484) | Wastewater treatment plant | 99.71 | |
| RJL1 | Stenotrophomonas sp. DB-17 (JQ074055) | Metal rich soil | 89.15 | |
| RJL2 | Chitinophagia | Chitinophaga hostae 2R12 (NR_181718) | Rhizosphere | 99.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



