
Submitted:
27 January 2026
Posted:
29 January 2026
You are already at the latest version
Abstract
Computational biology has completely changed the paradigm of drug development, moving it from random screening to a logical, predictive science. Three fundamental computational approaches Structure-Based Drug Design (SBDD), Ligand-Based Drug Design (LBDD), and Network Pharmacology are integrated in this review's potent, synergistic framework. In order to uncover important treatment targets, we show how these approaches function together as a coherent pipeline, with Network Pharmacology offering a systems-level blueprint of disease mechanisms. This realization immediately drives LBDD for intelligent screening utilizing pharmacophore and QSAR models in the absence of structural data, and SBDD for atomic-level rational design in the presence of it. Importantly, we stress that the foundation of this integrated strategy is early and iterative in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling, which guarantees the development of molecules with the best possible safety and drug-likeness. A new era of effective, multifaceted pharmaceutical development is ushered in by this technique, which de-risks the discovery process and speeds up the time from target identification to viable lead candidate by combining different disciplines into a single workflow.
Keywords:
molecular docking
; molecular dynamic simulation
; network pharmacology
; structure-based drug design
; ligand-based drug design
1. Introduction
The search for novel medicinal molecules is a difficult, expensive, and time-consuming process that has historically relied on high-throughput screening and chance discovery. However, the development of advanced algorithms and computing power has sparked a paradigm change and ushered in a period of logical drug design. Nowadays, computer-aided drug design, or CADD, is a crucial component of pharmaceutical research since it makes it possible to quickly identify and optimize lead compounds with increased accuracy and efficiency. These techniques serve as a potent filter by simulating molecular interactions in silico, identifying the most promising candidates for experimental validation and lowering the risk associated with the early phases of drug development [1,2]. .
Modern drug development uses a diverse computational toolkit that includes a number of complementary approaches. The foundational idea of Paul Ehrlich’s “corpora non agunt nisi fixata” (substances do not act unless bound) is operationalized via Structure-Based Drug Design (SBDD) [3]. As it has been shown previously, it uses the three-dimensional structure of a biological target to predict how tiny molecules can bind to its active site, leading the logical design of powerful inhibitors [4,5,6]. Ligand-Based Drug Design (LBDD) provides an alternate route in the absence of structural information by using pharmacophore modeling and Quantitative Structure-Activity Relationship (QSAR) analysis to infer the key characteristics of bioactivity from known active molecules. A more comprehensive, systems-level viewpoint has been provided more recently with the emergence of network pharmacology. Understanding the polypharmacology of natural products is made easier using this method, which goes beyond the “one drug, one target” concept to map the intricate network of interactions between multi-target medications and disease-associated pathways [7,8].
Although these approaches are frequently addressed separately, their integration is where their real potential resides. A more robust and thorough discovery pipeline is produced via a synergistic strategy in which network pharmacology, LBDD, and SBDD confirm and inform one another. Additionally, the pipeline’s ultimate objective is to produce a viable medication rather than just a strong binder, therefore early use of in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling is essential for evaluating pharmacokinetics and drug-likeness.
This review offers a thorough examination of these fundamental computational techniques. From target preparation and molecular docking to dynamic validation using Molecular Dynamics (MD) simulations and LBDD, we will go into detail about the core ideas and procedures of SBDD. The network pharmacology concept and its use in deconvoluting intricate pharmacological effects will be discussed. Their significance in lead optimization will be emphasized by a rigorous examination of ADMET prediction systems. In conclusion, we will summarize how the combination of these fields, enhanced by AI and machine learning, is creating a new, more effective, and predictive future for drug discovery.
2. Structure-Based Drug Design
Structure-based drug design, or SBDD, is philosophically predicated on this claim. By evaluating their three-dimensional structures and determining binding affinity at the active site, SBDD forecasts and maximizes the interaction between a ligand and its target protein [9]. Computational techniques that are combined to evaluate binding energetics, conformational flexibility, and ligand-target interactions include molecular docking, fragment-based screening, and molecular dynamics (MD) simulations [10], as it is shown in Figure 1. The first FDA-approved HIV-1 protease inhibitors, Saquinavir and Amprenavir, which revolutionized HIV treatment in the 1990s, are among the many therapeutic advances that have resulted from this approach [4,5,6]. Moreover, SBDD has played a crucial role in identifying potent Pim-1 kinase inhibitors, offering promising therapeutic avenues for cancer treatment [11].
2.1. Target Preparation
The Protein Data Bank (PDB), a valuable resource for structure-based drug design (SBDD), has grown significantly as a result of advancements in structural biology. However, many therapeutic targets’ structures are still unknown due to experimental restrictions [12,13]. Computational techniques are used to forecast three-dimensional protein structures based on amino acid sequences. Tools like MODELLER and SwissModel are used in homology modeling, which is based on a template from a homologous protein (usually having a sequence similarity of >40%) [14,15]. When no template exists, ab initio methods such as I-TASSER and QUARK predict structures by identifying the most stable conformations [16,17]. The field has been revolutionized by AI-based tools like AlphaFold, which achieves near-experimental accuracy [18]. All predicted structures must subsequently be validated for quality using techniques including Ramachandran plots [19,20]. Consequently, obtaining and validating a precise 3D structure, whether determined experimentally or computationally, remains the essential foundation for successful SBDD.
2.2. Binding Site Prediction
The binding site of a protein is a small cavity or pocket where a ligand binds to activate, inhibit, or modify the protein [21]. Although co-crystallized protein–ligand complexes provide valuable structural information for SBDD, in silico methods can be utilized to predict potential binding sites in the absence of such information [22]. Several factors influence the accuracy of these predictions, such as template similarity and pocket size [23]. Van der Waals interaction energies between the protein and small probes are calculated by energy-based approaches such as Q-SiteFinder to identify and rank energetically favorable clusters as potential binding sites [24] (See Table 1). Alternatively, blind docking can be employed to search the entire protein surface for potential ligand-binding pockets in the absence of any prior knowledge. Blind docking can also uncover surprising binding mechanisms and interactions, despite its lack of precision [25,26]. The prediction of binding sites is supported by a number of computational methods, such as DeepSite, DoGSiteScorer, POCASA, Fpocket, RaptorX-Binding Site, COACH, and PocketDepth [27,28,29,30,31,32,33].
2.3. Virtual Screening
Virtual screening (VS) is a powerful computer method used in medicinal chemistry to find potential lead compounds [38]. Using molecular docking, which ranks ligands according to binding affinity, it entails screening sizable databases of drug-like compounds against target proteins with known 3D structures [39]. In vitro tests are then used to validate the top-performing compounds, or “hits” [38]. There are two types of VS: structure-based (SBVS) and ligand-based (LBVS). LBVS uses molecular similarity or pharmacophore modeling to find active scaffolds and uses biological information to differentiate between active and inactive compounds [40]. On the other hand, SBVS relies on thorough structural data of the target protein to predict and assess ligand–protein interactions using docking algorithms with scoring functions. As shown in Table 2, compound libraries such as ZINC, PubChem, DrugBank, and ChEMBL are commonly used.
2.4. Molecular Docking and Scoring Functions
Molecular docking is a basic computational technique in structure-based drug design (SBDD) that forecasts the affinity and binding conformation (pose) of a small molecule (ligand) to a target protein [41]. This method is crucial for virtual screening (VS), which enables the efficient ranking of compounds from large libraries based on their predicted interactions with biological targets [42]. The most common approach, semi-flexible docking, permits ligand flexibility while preserving the protein’s rigidity; rigid docking treats both molecules as static, offering computational speed for high-throughput tasks; and flexible docking takes into account both the ligand and the protein’s conformational changes, offering the most realistic but computationally demanding simulation [43,44].
Docking programs use complex search techniques to navigate the large conformational space of a flexible ligand. Systematic approaches employ pre-generated conformational libraries or investigate rotational bonds in predetermined increments [45,46]. Monte Carlo simulations and genetic algorithms are examples of stochastic methods that incorporate random modifications that are either accepted or rejected depending on probability distributions [46]. Molecular Dynamics (MD) and other modeling approaches are used to achieve complete protein flexibility [43].
Scoring functions, which are mathematical models used to score postures and predict binding affinities, are essential to the precision of docking. They include functions that are empirical, knowledge-based, physics-based, and increasingly machine learning-based [47]. AutoDock Vina, Glide, and GOLD are well-known examples of conventional docking software (Table 3). Deep learning techniques are currently revolutionizing the field. For pose prediction and virtual screening, tools such as EquiBind, which makes direct “blind” predictions, DiffDock, a diffusion model that gives confidence estimates, and GNINA, which scores using convolutional neural networks, greatly outperform conventional techniques in terms of speed and accuracy [48,49,50].
2.5. Molecular Dynamics Simulation
In the 1970s, a computer technique known as molecular dynamics (MD) simulation was developed to model the intrinsic flexibility of proteins, which is crucial for processes such as ligand binding [73]. By resolving Newton’s equations of motion and forecasting the temporal evolution of a molecular system, MD simulations provide atomic-level insights into protein dynamics, conformational changes, and the stability of protein-ligand complexes [74]. As a result, MD is particularly helpful in drug development for docking posture validation, allosteric binding site identification, and virtual screening [75].
A typical process starts with setting up the system topology with tools like CharmGUI or AMBER, and then uses force fields like AMBER, CHARMM, or GROMOS to simulate atomic movements [76,77,78]. The produced trajectories are analyzed using software like Xmgrace to calculate stability metrics including radius of gyration, hydrogen bonding, Root Mean Square Deviation (RMSD), and Root Mean Square Fluctuation (RMSF) (Turner, 2005). The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) methods are commonly used for a more accurate determination of binding affinity because they more closely reproduce experimental results than conventional docking scores [80,81].
To get over computational constraints, the field is still developing. Longer, microsecond-scale simulations are now made possible by the use of Graphics Processing Units (GPUs) [82]. Additionally, by applying MM to the surrounding environment and QM to the reactive core, hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) techniques offer a balanced approach to investigating chemical reactions [83]. GROMACS, NAMD, AMBER, and CHARMM are well-known software programs that facilitate these sophisticated simulations [77,78,84,85].
Drug design has benefited greatly from the use of molecular dynamics (MD) simulations, which help to make sense of intricate biological processes. They are valuable because they can provide time-resolved, atomic-level information that is often unavailable through experimental methods. This ability is demonstrated in research on the effects of anticancer drugs, such as how cannabinoids inhibit glucose-6-phosphate dehydrogenase (G6PD) and how chemicals from Withania somnifera alter the KAT6A enzyme [86,87]. MD simulations provide dynamic information that is frequently unavailable to experimental approaches, making them a valuable tool for comprehending drug-target interactions and creating more potent treatments.
3. Ligand-Based Drug Design
A method called ligand-based drug design, or LBDD, is used when the active chemicals that inhibit a protein are known but its three-dimensional structure is unknown. This method is predicated on the idea that molecules with a high degree of physical and structural similarity are likely to exhibit similar biological activities [88,89]. Important LBDD methods that direct the creation of improved analog compounds and assist in identifying the characteristics that affect biological activity include pharmacophore modeling and quantitative structure-activity relationship (QSAR) modeling [90]. Compound libraries are usually screened to identify new candidates with similar features in order to identify known active compounds for the first time [12,91].
3.1. Similarity Searches
The basis of ligand-based drug development is the molecular similarity notion, which maintains that molecules with similar structures are likely to have comparable biological functions. Using a known active component as a template, this technique uses similarity searches to identify new chemical entities with comparable characteristics [92,93]. This technique is crucial in situations where the target protein’s structure is unavailable because it only needs ligand information. In order to find new bioactive ligands and improve their pharmacokinetic and efficacious characteristics, it is frequently employed for in silico screening [94].
3.2. Pharmacophore Modeling
Pharmacophore modeling is one computer method for identifying the essential steric and electronic properties a drug need to bind a biological target [95]. When the three-dimensional structure of a protein is not available, this method which is based on the analysis of known active ligands is essential. The procedure entails creating a list of active substances and determining their common chemical characteristics, such as hydrophobic areas and donors/acceptors of hydrogen bonds that are essential for biological activity [96,97]. These attributes are then used to create a 3D query model based on these properties. Pharmacophore modeling uses specialized software such as like LigandScout, HipHop, HypoGen, and PHASE to generate 3D queries. These models can then virtually screen large compound libraries to identify novel chemical entities that share the essential pharmacophore features and are thus predicted to exhibit similar biological activity [98,99,100].
A major limitation of classical models is their static nature, which simplifies dynamic binding interactions. So, sophisticated methods like the “dynophore” have been devised to circumvent this. Molecular Dynamics (MD) simulations, which capture the dynamic aspect of binding, are included in the dynophore approach to offer data on interaction frequencies and a more accurate representation of the ligand-target complex [101].
3.3. Quantitative Structure-Activity Relationship
The ligand-based Quantitative Structure-Activity Relationship (QSAR) technique creates mathematical models that link chemical structure to experimental activity by using molecular descriptors (MDs) or fingerprints (FPs) to predict biological activity. Support Vector Machines (SVM), Random Forests (RF), Polynomial Regression, Multiple Linear Regression (MLR), and Artificial Neural Networks (ANN) are a few of the machine learning (ML) and deep learning (DL) approaches commonly used in QSAR development [102]. In contrast to pharmacophore models, QSAR detects positive or negative effects associated to structure and quantitatively predicts activity. In drug design, QSAR is widely used for lead compound optimization, analogue activity prediction, and scaffold discovery. Through mathematical equations, classical 2D-QSAR links structure to activity using steric, electronic, and hydrophobic characteristics [103]. Force field-based models are used in advanced 3D-QSAR techniques, such as CoMFA and CoMSIA [104,105], which provide 3D contour maps for interpretation.
In pharmaceutical research and development, QSAR is an essential computational method [105,106]. While QSAR uses statistical modelling to connect physicochemical characteristics (such as molecular weight, lipophilicity, and electronic distribution) with bioactivity, pharmacophore mapping finds the structural elements that are responsible for biological activity (such as hydrogen bond donors/acceptors, hydrophobic regions, and aromatic rings) [107]. Model building and validation are made easier by contemporary tools such as MOE, Schrodinger, SYBYL-X, Open3DQSAR, Discovery Studio, and MOE (Table 4). By improving pharmacokinetics, potency, and selectivity, these models help optimize leads and cut down on time and expense associated with drug development [108]. One well-known example is the modelling of HDAC inhibitors, which are essential in cancer treatments, by combining QSAR with structure-based pharmacophores utilizing libraries of natural compounds [109]. Through ROC analysis and virtual screening, new HDAC-inhibiting scaffolds were discovered as a result of these efforts. The work showed how QSAR models anticipate analogue activity and direct hit-to-lead optimization [110].
4. Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) Properties
Pharmacology, the study of how drugs interact with biological systems, is the field that essentially directs drug discovery. Pharmacodynamics (PD), which studies how a drug affects the body, and pharmacokinetics (PK), which studies how the body affects a drug, specifically its Absorption, Distribution, Metabolism, and Excretion (ADME), are the two primary subfields of this science [2]. When the critical aspect of Toxicity is included, this full profile is known as ADMET. For a molecule to be considered a viable drug candidate, it must not only be active and specific in vitro but also possess favorable ADMET properties in vivo. A compound is unlikely to become a drug unless it is efficiently absorbed, adequately distributed, metabolically stable, and safely eliminated [111]. Therefore, optimizing ADMET profiles without sacrificing biological efficacy is a primary objective following lead compound identification. In the past, the main framework for forecasting drug-likeness was Lipinski’s Rule of Five, which defined it by four important parameters: molecular weight (<500 Da), lipophilicity (cLogP <5), hydrogen bond donors (<5), and hydrogen bond acceptors (<10) [112]. These days, more advanced computational technologies frequently complement this early rule-based filtering. Nowadays, it’s common practice to estimate thorough pharmacokinetic profiles early in the discovery phase using predictive platforms like SwissADME, PreADMET, ALOGPS, and DrugMint (Table 5).
5. Network Pharmacology
Network pharmacology provides a methodical framework for elucidating the complex mechanisms of bioactive compounds, particularly those included in herbal remedies, by means of a multi-step process that blends computer predictions with experimental confirmation [7]. Finding active compounds and their canonical SMILES structures from medicinal plants is the first step in the process, which involves collecting disease-associated genes from public sources and applying data mining. Then, transcriptome data analysis and computational target prediction are employed to find potential compound-target and disease-target interactions [128]. Venn diagrams are utilized to find common targets between chemicals and disorders as potential therapeutic targets, and statistical cutoffs are employed to find significantly connected genes [129]. Protein-protein interaction (PPI) networks constructed from these shared targets help identify hub genes and key active components that regulate disease-relevant biological pathways [130]. Subsequent network analysis, including topological assessment and functional enrichment analysis through KEGG and GO, reveals crucial subnetworks and pathways regulated by these targets [131]
Both experimental techniques and high-throughput in silico technologies are used to validate computational predictions. By forecasting binding affinities, molecular docking is essential for confirming ligand-target interactions and successfully bridging the gap between conventional and contemporary pharmacological techniques [132]. In addition, network pharmacology, fueled by the rise of omics technologies (genomics, proteomics, metabolomics), presents a transformative approach to cancer drug discovery by overcoming the cost and time limitations of traditional methods [133]. This has been demonstrated in studies of Prunella vulgaris for breast cancer, which showed strong binding affinities of its active components to disease target. Similar results were observed with Artemisia argi against hepatocellular carcinoma and Nauclea latifolia against breast cancer [8,134,135]. Gene expression microarray analysis, supported by datasets from repositories like GEO, provides additional validation by assessing transcriptional changes across hundreds of genes under various experimental conditions [136].
6. Structure Framework of Computational Drug Discovery
Contemporary drug discovery is increasingly driven by integrated computational frameworks that unify multiple in silico strategies into a single, predictive pipeline (Figure 1). In this framework, network pharmacology provides a systems-level perspective by elucidating disease-associated molecular networks, identifying key regulatory targets, and capturing the complexity of polygenic and pathway-driven pathologies. This holistic understanding establishes a rational foundation for target prioritization and therapeutic hypothesis generation.
Building upon these insights, structure-based drug design (SBDD) is employed when high-resolution target structures are available, enabling rational ligand design through binding-site identification, molecular docking, and dynamic stability assessment. In parallel, ligand-based drug design (LBDD) complements this approach in scenarios where structural information is limited, leveraging known bioactive compounds to perform similarity searches, pharmacophore modeling, and quantitative structure–activity relationship (QSAR) analyses. The bidirectional integration of SBDD and LBDD ensures iterative refinement and cross-validation of candidate molecules.
Critically, early and iterative ADMET profiling is embedded throughout the pipeline to evaluate pharmacokinetic behavior, drug-likeness, and safety liabilities, thereby filtering out suboptimal candidates at the earliest possible stage. The synergistic integration of network pharmacology, SBDD, and LBDD substantially surpasses the predictive power of any single methodology, reducing attrition risk and accelerating the translation of molecular insights into high-confidence therapeutic candidates.
7. Critical Challenges
Despite tremendous progress, computational drug discovery still faces a number of important obstacles. The ability to accurately anticipate binding affinities is a major drawback. The scoring systems used to quantify binding free energy frequently lack the precision necessary to consistently rank ligands, resulting in false positives and negatives in virtual screens, despite the fact that molecular docking is quite good for posture prediction [47]. Furthermore, a significant obstacle is the dynamic nature of biomolecules by nature. The majority of docking procedures fail to capture the conformational changes and allosteric effects that are essential for binding and function because they regard the protein receptor as rigid or semi-flexible [137]. It is still computationally intensive and challenging to generalize to account for solvation effects and the function of important water molecules in binding sites.
In LBDD, the quality and quantity of training data have a direct effect on the prediction performance of QSAR and pharmacophore models. Because of its extrapolation challenges, these models often fail to accurately forecast the activity of compounds with novel scaffolds beyond the chemical space of the training set [106]. It is necessary to integrate and validate the enormous volumes of heterogeneous data from omics technologies in order to build physiologically meaningful and predictive network models in network pharmacology [133]. Lastly, although though ADMET prediction methods are very useful, their precision depends on the quality of the underlying data, and it is still a challenging task to predict complex in vivo phenomena from simple chemical structures, such as organ-specific toxicity and drug-drug interactions [124].
8. Conclusion and Perspectives
This study highlights the strategic value of integrating network pharmacology with ligand-based and structure-based drug design approaches to rationalize and accelerate early-stage drug discovery. By combining systems-level target identification with molecular-level interaction analysis, this integrated computational framework enables a more comprehensive understanding of compound–target–pathway relationships than any single methodology alone. Network pharmacology provides a holistic view of disease-associated biological networks and polypharmacological effects, LBDD facilitates efficient identification and prioritization of chemically relevant candidates based on known bioactivity patterns, and SBDD offers atomistic insight into binding modes, interaction stability, and target specificity. Together, these complementary strategies form a coherent and rational pipeline for lead identification and optimization in complex diseases.
Despite the strengths of this integrated approach, it is essential to recognize its inherent limitations. All predictions presented herein are derived from in silico analyses and should be interpreted as hypothesis-generating rather than definitive evidence of biological efficacy. Molecular docking scores and molecular dynamics stability metrics, while informative, do not directly translate into pharmacological activity or clinical benefit. Nevertheless, by narrowing the chemical and biological search space, this computational framework substantially improves efficiency and cost-effectiveness, thereby enabling more focused and rational experimental validation in subsequent in vitro and in vivo studies.
Looking ahead, the continued convergence of computational drug discovery with artificial intelligence and machine learning is expected to further enhance the predictive power and scalability of integrated pipelines such as the one presented in this work. Advances in deep learning–based docking algorithms, protein structure prediction platforms, and generative chemistry models are rapidly transforming virtual screening and lead optimization. The incorporation of dynamic interaction descriptors, such as MD-informed pharmacophores, along with multi-omics and single-cell data within network pharmacology frameworks, will enable increasingly precise characterization of disease mechanisms and therapeutic responses. Ultimately, the development of unified, end-to-end in silico platforms capable of jointly predicting target engagement, pharmacokinetics, safety, and efficacy holds the promise of reducing attrition rates and accelerating the translation of computational discoveries into clinically relevant therapeutics. In this context, the integrative strategy presented in this study provides a solid conceptual and methodological foundation for future computational and experimental drug discovery efforts
Authorship Contributions: Cromwel Tepap Zemnou: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Supervision, Resources, Writing – original draft, Writing – review & editing. Gabriel Tchuente KAMSU: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Ramelle Ngakam: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. Etienne Junior Tcheumeni: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflicts of Interests
The authors report there are no competing interests to declare.
Funding
This work was not supported by any funding or grant.
Data Availability Statement
The authors confirm that the data supporting the article are available within the article.
Acknowledgments
None.
References
- Segall, M.D.; Barber, C. Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov. Today 2014, 19, 688–693. [Google Scholar] [CrossRef]
- Zemnou, C.T. In silico Comparative Study of the Anti-Cancer Potential of Inhibitors of Glucose-6-Phosphate Dehydrogenase Enzyme Using ADMET Analysis, Molecular Docking, and Molecular Dynamic Simulation. Adv. Theory Simulations 2025, 8, 2400757. [Google Scholar] [CrossRef]
- Bosch, F.; Rosich, L. The Contributions of Paul Ehrlich to Pharmacology: A Tribute on the Occasion of the Centenary of His Nobel Prize. Pharmacology 2008, 82, 171–179. [Google Scholar] [CrossRef]
- Craig, J.; Duncan, I.; Hockley, D.; Grief, C.; Roberts, N.; Mills, J. Antiviral properties of Ro 31-8959, an inhibitor of human immunodeficiency virus (HIV) proteinase. Antivir. Res. 1991, 16, 295–305. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.E.; Baker, C.T.; Dwyer, M.D.; Murcko, M.A.; Rao, B.G.; Tung, R.D.; Navia, M.A. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 1995, 117, 1181–1182. [Google Scholar] [CrossRef]
- Niu, W.-H.; Wu, F.; Cao, W.-Y.; Wu, Z.-G.; Chao, Y.-C.; Peng, F.; Liang, C. Network pharmacology for the identification of phytochemicals in traditional Chinese medicine for COVID-19 that may regulate interleukin-6. Biosci. Rep. 2021, 41, BSR20202583. [Google Scholar] [CrossRef]
- Zemnou, C.T. Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Revealed the Molecular Targets and Potential Mechanism of Nauclea Latifolia in the Treatment of Breast Cancer. Chem. Biodivers. 2024, 22, e202402423. [Google Scholar] [CrossRef] [PubMed]
- Rognan, D. Structure-Based Approaches to Target Fishing and Ligand Profiling. Mol. Informatics 2010, 29, 176–187. [Google Scholar] [CrossRef]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef]
- Ren, J.-X.; Li, L.-L.; Zheng, R.-L.; Xie, H.-Z.; Cao, Z.-X.; Feng, S.; Pan, Y.-L.; Chen, X.; Wei, Y.-Q.; Yang, S.-Y. Discovery of Novel Pim-1 Kinase Inhibitors by a Hierarchical Multistage Virtual Screening Approach Based on SVM Model, Pharmacophore, and Molecular Docking. J. Chem. Inf. Model. 2011, 51, 1364–1375. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19. BioMed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Proteomics: High-Throughput Methods; Kobe, B., Guss, M., Huber, d.T., Eds.; Humana Press: Totowa, NJ, USA, 2008; pp. 145–159. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins: Struct. Funct. Bioinform. 2011, 79, 147–160. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77 (Suppl. S9), 114–122. [Google Scholar] [CrossRef]
- Wang, L.; Song, Y.; Wang, H.; Zhang, X.; Wang, M.; He, J.; Li, S.; Zhang, L.; Li, K.; Cao, L. Advances of Artificial Intelligence in Anti-Cancer Drug Design: A Review of the Past Decade. Pharmaceuticals 2023, 16, 253. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Bentham Science Publisher. Methods for the Prediction of Protein-Ligand Binding Sites for Structure-Based Drug Design and Virtual Ligand Screening. Curr. Protein Pept. Sci. 2006, 7, 395–406. [Google Scholar] [CrossRef]
- Chen, K.; Mizianty, M.J.; Gao, J.; Kurgan, L. A Critical Comparative Assessment of Predictions of Protein-Binding Sites for Biologically Relevant Organic Compounds. Structure 2011, 19, 613–621. [Google Scholar] [CrossRef]
- Laurie, A.T.R.; Jackson, R.M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Grimm, M.; Dai, W.-T.; Hou, M.-C.; Xiao, Z.-X.; Cao, Y. CB-Dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, W.; Bell, E.W.; Yin, M.; Zhang, Y. EDock: blind protein–ligand docking by replica-exchange monte carlo simulation. J. Chemin- 2020, 12, 37. [Google Scholar] [CrossRef]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef]
- Kalidas, Y.; Chandra, N. PocketDepth: A new depth based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol. 2008, 161, 31–42. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Shiwang, L.; Li, W.; Liu, S.; Xu, J. RaptorX-Property: A Web Server for Protein Structure Property Prediction. Nucleic Acids Res. 2016, 44, W430–W435. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2009, 26, 46–52. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J.E. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, W469–W473. [Google Scholar] [CrossRef]
- Kalidas, Y.; Chandra, N. PocketDepth: A new depth based algorithm for identification of ligand binding sites in proteins. J. Struct. Biol. 2008, 161, 31–42. [Google Scholar] [CrossRef]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Phatak, S.S.; Stephan, C.C.; Cavasotto, C.N. High-throughput andin silicoscreenings in drug discovery. Expert Opin. Drug Discov. 2009, 4, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Rescoring and Linearly Combining: A Highly Effective Consensus Strategy for Virtual Screening Campaigns. Int. J. Mol. Sci. 2019, 20, 2060. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. Advances and Challenges in Protein-Ligand Docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins: Struct. Funct. Bioinform. 1992, 13, 206–222. [Google Scholar] [CrossRef]
- Oshiro, C.M.; Kuntz, I.D.; Dixon, J.S. Flexible ligand docking using a genetic algorithm. J. Comput. Mol. Des. 1995, 9, 113–130. [Google Scholar] [CrossRef]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein–ligand docking: Current status and future challenges. Proteins: Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Corso, G.; Stärk, H.; Jing, B.; Barzilay, R.; Jaakkola, T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking. arXiv 2023, arXiv:2210.01776. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wang, S.; Tang, X. EQUIBIND: A geometric deep learning-based protein-ligand binding prediction method. Drug Discov. Ther. 2023, 17, 363–364. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: molecular docking with deep learning. J. Chemin- 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Desai, P.V.; Patny, A.; Sabnis, Y.; Tekwani, B.; Gut, J.; Rosenthal, P.; Srivastava, A.; Avery, M. Identification of Novel Parasitic Cysteine Protease Inhibitors Using Virtual Screening. 1. The ChemBridge Database. J. Med. Chem. 2004, 47, 6609–6615. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Vasilevich, N.I.; Aksenova, E.A.; Kazyulkin, D.N.; Afanasyev, I.I. General Ser/Thr Kinases Pharmacophore Approach for Selective Kinase Inhibitors Search as Exemplified by Design of Potent and Selective Aurora A Inhibitors. Chem. Biol. Drug Des. 2016, 88, 54–65. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Joy, S.; Nair, P.S.; Hariharan, R.; Pillai, M.R. Detailed Comparison of the Protein-Ligand Docking Efficiencies of GOLD, a Commercial Package and ArgusLab, a Licensable Freeware. Silico Biol. 2006, 6, 601–605. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Englebienne, P.; Moitessier, N. Docking Ligands into Flexible and Solvated Macromolecules. 1. Development and Validation of FITTED 1.0. J. Chem. Inf. Model. 2007, 47, 435–449. [Google Scholar] [CrossRef]
- Zhao, Y.; Sanner, M.F. FLIPDock: Docking flexible ligands into flexible receptors. Proteins: Struct. Funct. Bioinform. 2007, 68, 726–737. [Google Scholar] [CrossRef]
- Venkatachalam, C.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Cheng, T.M.; Blundell, T.L.; Fernandez-Recio, J. pyDock: Electrostatics and desolvation for effective scoring of rigid-body protein–protein docking. Proteins: Struct. Funct. Bioinform. 2007, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; A Porter, K.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins: Struct. Funct. Bioinform. 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Studio: Discovery studio - Google Scholar. Available online: https://scholar.google.com/scholar_lookup?title=Discovery%20Studio%2C%20Accelrys%20%5B2.1%5D&author=D.%20Studio&publication_year=2008 (accessed on 23 October 2025).
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef]
- Yang, J.; Chen, C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins: Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Karim, M.; Islam, M. N.; Azad Jewel, G. M. N. Silico identification of potential drug targets by subtractive genome analysis of Enterococcus faecium DO, Feb. 15; Bioinformatics, 2020. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef]
- Nair, P.C.; Malde, A.K.; Drinkwater, N.; Mark, A.E. Missing Fragments: Detecting Cooperative Binding in Fragment-Based Drug Design. ACS Med. Chem. Lett. 2012, 3, 322–326. [Google Scholar] [CrossRef]
- Christen, M.; Hünenberger, P.H.; Bakowies, D.; Baron, R.; Bürgi, R.; Geerke, D.P.; Heinz, T.N.; Kastenholz, M.A.; Kräutler, V.; Oostenbrink, C.; et al. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Turner: XMGRACE, Version 5.1. 19 - Google Scholar. Available online: https://scholar.google.com/scholar_lookup?title=XMGRACE%2C%20Version%205.1.%2019&author=P.%20Turner&publication_year=2005 (accessed on 16 October 2025).
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, Y.; Chen, Y. GPU accelerated molecular dynamics simulation of thermal conductivities. J. Comput. Phys. 2007, 221, 799–804. [Google Scholar] [CrossRef]
- Walker, R.C.; Crowley, M.F.; Case, D.A. The implementation of a fast and accurate QM/MM potential method in Amber. J. Comput. Chem. 2007, 29, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: a Parallel, Object-Oriented Molecular Dynamics Program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Tepap, C.Z.; Anissi, J.; Bounou, S.; Zanchi, F.B. In Silico Approach for Assessment of the Anti-Tumor Potential of Cannabinoid Compounds by Targeting Glucose-6-Phosphate Dehydrogenase Enzyme. Chem. Biodivers. 2024, 21, e202401338. [Google Scholar] [CrossRef]
- Zemnou, C.T.; Karim, E.M.; Chtita, S.; Zanchi, F.B. Impact of mutations on KAT6A enzyme and inhibitory potential of compounds from Withania somnifera using computational approaches. Comput. Biol. Med. 2025, 190, 110041. [Google Scholar] [CrossRef]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Prathipati, P.; Dixit, A.; Saxena, A.K. Computer-Aided Drug Design: Integration of Structure-Based and Ligand-Based Approaches in Drug Design. Curr. Comput. Aided-Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Huerta-Yépez, S.; Moreno-Vargas, L.M. Application of computational methods for anticancer drug discovery, design, and optimization. Boletin Medico Del Hosp. Infant. De Mex. 2016, 73, 411–423. [Google Scholar] [CrossRef]
- Prathipati, P.; Dixit, A.; Saxena, A.K. Computer-Aided Drug Design: Integration of Structure-Based and Ligand-Based Approaches in Drug Design. Curr. Comput. Aided-Drug Des. 2007, 3, 133–148. [Google Scholar] [CrossRef]
- Bender, A.; Jenkins, J.L.; Scheiber, J.; Sukuru, S.C.K.; Glick, M.; Davies, J.W. How Similar Are Similarity Searching Methods? A Principal Component Analysis of Molecular Descriptor Space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef]
- Johnson, M. A.; Maggiora, G. M.; A. C., S. Meeting, Concepts and applications of molecular similarity. Wiley, 1990. Available online: https://cir.nii.ac.jp/crid/1970304959821169570 (accessed on 16 October 2025).
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Van Drie, J.H. Generation of three-dimensional pharmacophore models. WIREs Comput. Mol. Sci. 2013, 3, 449–464. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- F. Güner, Pharmacophore Perception, Development, and Use in Drug Design. In Internat’l University Line; 2000.
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Rakers, C. “Dynophores: Novel Dynamic Pharmacophores-Implementation of Pharmacophore Generation Based on Molecular Dynamics Trajectories and Their Graphical Representation”. Available online: https://www.semanticscholar.org/paper/Dynophores:-Novel-Dynamic-of-Pharmacophore-Based-on-Rakers/a7fec7f5d233ec81274b2b4c77c6d2d57e461083 (accessed on 16 October 2025).
- Mendenhall, J.; Meiler, J. Improving quantitative structure–activity relationship models using Artificial Neural Networks trained with dropout. J. Comput. Mol. Des. 2016, 30, 177–189. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. p-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular Similarity Indices in a Comparative Analysis (CoMSIA) of Drug Molecules to Correlate and Predict Their Biological Activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2013, 57, 4977–5010. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational Methods in Drug Discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef]
- Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Pai, P.; Kumar, A.; Shetty, M.G.; Kini, S.G.; Krishna, M.B.; Satyamoorthy, K.; Babitha, K.S. Identification of potent HDAC 2 inhibitors using E-pharmacophore modelling, structure-based virtual screening and molecular dynamic simulation. J. Mol. Model. 2022, 28, 119. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Hou, Z.; Zhang, Z. Pharmacophore and QSAR Studies to Design Novel Histone Deacetylase 2 Inhibitors. Chem. Biol. Drug Des. 2012, 79, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J. ADMET—turning chemicals into drugs. Nat. Biotechnol. 2001, 19, 722–726. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Dimitrov, S.D.; Diderich, R.; Sobanski, T.; Pavlov, T.S.; Chankov, G.V.; Chapkanov, A.S.; Karakolev, Y.H.; Temelkov, S.G.; Vasilev, R.A.; Gerova, K.D.; et al. QSAR Toolbox – workflow and major functionalities. SAR QSAR Environ. Res. 2016, 27, 203–219. [Google Scholar] [CrossRef]
- Jing, P.; Zhao, S.; Ruan, S.; Sui, Z.; Chen, L.; Jiang, L.; Qian, B. Quantitative studies on structure–ORAC relationships of anthocyanins from eggplant and radish using 3D-QSAR. Food Chem. 2014, 145, 365–371. [Google Scholar] [CrossRef]
- Tosco, P.; Balle, T. Open3DQSAR: a new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef]
- Ambure, P.; Halder, A.K.; Díaz, H.G.; Cordeiro, M.N.D.D.S. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef]
- Vainio, M.J.; Johnson, M.S. McQSAR: A Multiconformational Quantitative Structure−Activity Relationship Engine Driven by Genetic Algorithms. J. Chem. Inf. Model. 2005, 45, 1953–1961. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Dong, J.; Zhu, G.; Ma, L.; Qian, Y.; Zhu, X.; Chen, Y. Experimental characterization of a SESAM mode-locked Yb:YAG thin disk laser. In High-Power Lasers and Applications IX; SPIE, 2018; pp. 105–111. [Google Scholar] [CrossRef]
- Nunes, A.M.V.; Andrade, F.d.C.P.d.; Filgueiras, L.A.; Maia, O.A.d.C.; Cunha, R.L.; Rodezno, S.V.; Filho, A.L.M.M.; Carvalho, F.A.d.A.; Braz, D.C.; Mendes, A.N. preADMET analysis and clinical aspects of dogs treated with the Organotellurium compound RF07: A possible control for canine visceral leishmaniasis? Environ. Toxicol. Pharmacol. 2020, 80, 103470. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Tanchuk, V.Y. Application of Associative Neural Networks for Prediction of Lipophilicity in ALOGPS 2.1 Program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Singla, D.; Mondal, A.K.; Raghava, G.P. DrugMint: a webserver for predicting and designing of drug-like molecules. Biol. Direct 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Shaker, B.; Yu, M.-S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.-S.; Na, D. LightBBB: computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef]
- Banerjee, P.; O Eckert, A.; Schrey, A.K.; Preissner, R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Yu, M.-S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.-H.; Bae, M.-A.; Lee, B.H.; Shin, D.-S.; Oh, K.-S.; Ceong, H.; et al. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform. 2019, 20. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- (PDF) ToxiPred: A Server for Prediction of Aqueous Toxicity of Small Chemical Molecules in T. Pyriformis. ResearchGate 2025. [CrossRef]
- Seo, E.-J.; Klauck, S.M.; Efferth, T.; Panossian, A. Adaptogens in chemobrain (Part III): Antitoxic effects of plant extracts towards cancer chemotherapy-induced toxicity - transcriptome-wide microarray analysis of neuroglia cells. Phytomedicine 2019, 56, 246–260. [Google Scholar] [CrossRef]
- Zhao, F.; Guochun, L.; Yang, Y.; Shi, L.; Xu, L.; Yin, L. A network pharmacology approach to determine active ingredients and rationality of herb combinations of Modified-Simiaowan for treatment of gout. J. Ethnopharmacol. 2015, 168, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, R.; Lu, J.; Jiang, Y.; Huang, T.; Cai, Y. Protein-protein interaction networks as miners of biological discovery. Proteomics 2022, 22, e2100190. [Google Scholar] [CrossRef]
- Du, H.; Zhao, X.; Zhang, A. Identifying potential therapeutic targets of a natural product Jujuboside B for insomnia through network pharmacology. Plant Sci. Today 2014, 1, 69–79. [Google Scholar] [CrossRef]
- Tao, Q.; Du, J.; Li, X.; Zeng, J.; Tan, B.; Xu, J.; Lin, W.; Chen, X.-L. Network pharmacology and molecular docking analysis on molecular targets and mechanisms of Huashi Baidu formula in the treatment of COVID-19. Drug Dev. Ind. Pharm. 2020, 46, 1345–1353. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Liu, K.; Hu, X.; Gu, X. Advances in drug discovery based on network pharmacology and omics technology. Curr. Pharm. Anal. 2024, 21, 33–43. [Google Scholar] [CrossRef]
- Tepap, C.Z.; Ibrahim, R.H. Network Pharmacology-based Approach to Investigate the Mechanism of Folium Artemisiae Argyi in the Treatment of Hepatocellular Carcinoma. Int. J. Adv. Life Sci. Res. 2025, 08, 74–94. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, T.; Zhou, X.; Tang, X.; Gao, R.; Xu, L.; Wang, L.; Zhou, Z.; Lin, J.; Hu, Y. Network pharmacology based virtual screening of active constituents of Prunella vulgaris L. and the molecular mechanism against breast cancer. Sci. Rep. 2020, 10, 15730. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. In Statistical Genomics; Springer: New York, NY, USA, 2016; pp. 93–110. [Google Scholar]
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins: Struct. Funct. Bioinform. 1992, 13, 206–222. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Synergistic computational workflow integrating ligand-based drug design (LBDD), structure-based drug design (SBDD), and network pharmacology (NP) for rational drug discovery. LBDD enables identification and optimization of lead compounds based on chemical similarity, pharmacophore features, and QSAR models. SBDD validates ligand–target interactions through molecular docking, binding mode analysis, and molecular dynamics simulations. NP provides a systems-level perspective by identifying disease-relevant targets and pathways.
Figure 1.
Synergistic computational workflow integrating ligand-based drug design (LBDD), structure-based drug design (SBDD), and network pharmacology (NP) for rational drug discovery. LBDD enables identification and optimization of lead compounds based on chemical similarity, pharmacophore features, and QSAR models. SBDD validates ligand–target interactions through molecular docking, binding mode analysis, and molecular dynamics simulations. NP provides a systems-level perspective by identifying disease-relevant targets and pathways.
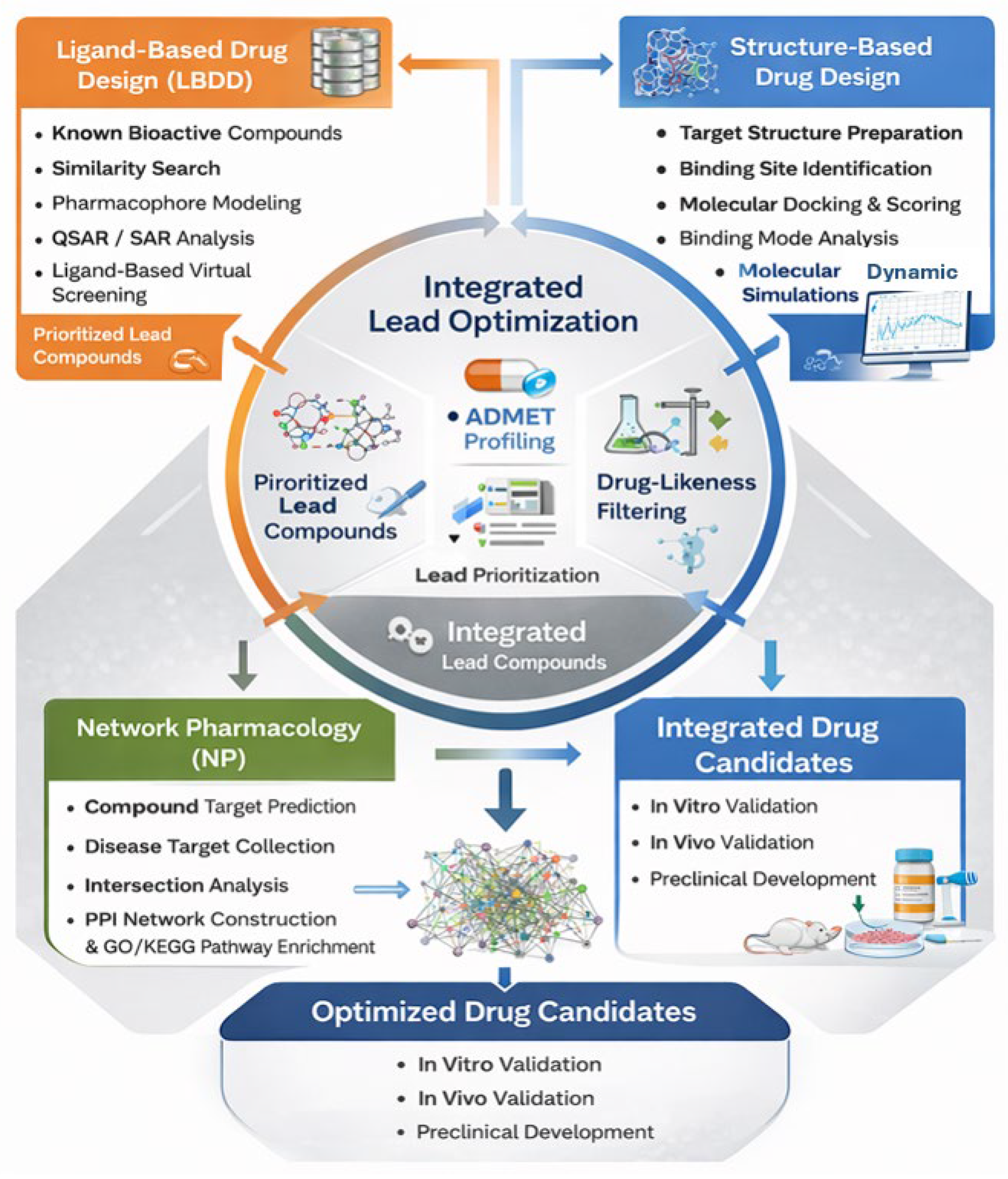

Table 1.
List of ligand binding site prediction tools.
| Name | Description | URL | Ref. |
|---|---|---|---|
| CASTp 3.0 | Identifies and provides detailed geometric measurements of all potential surface pockets and interior cavities in a protein structure. | http://sts.bioe.uic.edu/castp/ | [34] |
| SiteMap | Employs a grid-based method to locate and rank putative binding sites according to a proprietary score (SiteScore) that evaluates their ligand-binding suitability. | https://www.schrodinger.com/products/sitemap | [35] |
| Fpocket | An open-source, fast geometry-based pocket detection algorithm designed for large-scale binding site prediction. | https://github.com/DisorderedProteins/fpocket | [29] |
| 3DLigandSite | Utilizes homology modeling; it predicts a 3D structure for an input sequence and infers binding sites by mapping ligands from homologous protein-ligand complexes. | http://www.sbg.bio.ic.ac.uk/~3dligandsite/ | [36] |
| PocketDepth | A geometry-based method that identifies ligand binding pockets by evaluating the depth and shape of protein surface cavities. | http://proline.physics.iisc.ernet.in/pocketdepth/ | [37] |
| DeepSite | A deep learning-based method that uses a 3D convolutional neural network to predict protein binding pockets. | https://www.playmolecule.com/deepsite/ | [27] |
| DoGSiteScorer | An automated pocket detection and analysis tool that calculates physicochemical properties and druggability of predicted binding pockets. | https://proteins.plus/ | [30] |
| POCASA | Predicts ligand binding sites by scanning the protein surface for regions that can accommodate a spherical probe. | http://altair.sci.hokudai.ac.jp/g6/Research/POCASA_e.html | [33] |
| Q-SiteFinder | Identifies binding sites by computing the van der Waals interaction energies between the protein and a methyl probe. | http://www.modelling.leeds.ac.uk/qsitefinder/ | [24] |
| RaptorX-Binding Site | Predicts ligand binding sites using a combination of template-based and ab initio methods, leveraging deep learning for quality assessment. | http://raptorx2.uchicago.edu/ | [31] |
| COACH | A meta-server approach that combines predictions from multiple methods (e.g., TM-SITE, S-SITE) to generate consensus ligand binding site predictions. | https://yanglab.nankai.edu.cn/COACH/ | [32] |
Table 2.
Overview of Publicly Accessible Chemical Libraries for In Silico Screening.
| Database | Description | URL | Ref. |
|---|---|---|---|
| Zinc15 | A comprehensive repository of over 230 million commercially available, “ready-to-dock” small molecules in 3D formats. | https://zinc15.docking.org/ | [51] |
| PubChem | A vast public repository containing information on approximately 11 million unique chemical structures, alongside bioactivity data from high-throughput screening assays. | https://pubchem.ncbi.nlm.nih.gov/ | [52] |
| ChemBridge | Provides a diverse collection of over 1.3 million small molecules, including both structurally diverse libraries and target-focused sets for screening. | https://www.chembridge.com/ | [53] |
| BindingDB | A specialized database focusing on measured binding affinities, containing data for over 1.2 million interactions between 520,000+ compounds and 5,500+ protein targets. | http://bindingdb.org | [54] |
| Asinex | Offers a curated library of more than 90,000 lead-like compounds, designed for efficient virtual screening and hit identification. | http://www.asinex.com/ | [55] |
Table 3.
Molecular Docking and Virtual screening Platforms.
| Tool | Brief Description | URL | Ref. |
|---|---|---|---|
| AutoDock Vina | Known for its speed and accuracy; utilizes a hybrid global optimization algorithm and accommodates flexibility in receptor side chains. | http://vina.scripps.edu/ | [56] |
| Glide | Employs hierarchical filters for rapid docking with multiple precision modes (SP, XP, HTVS) for scoring and ranking ligand poses. | https://www.schrodinger.com/glide | [57] |
| GOLD | Uses a genetic algorithm for conformational sampling and evaluates poses with functions like GoldScore and ChemScore. | https://www.ch.cam.ac.uk/computing/software/gold-suite | [58] |
| DOCK | A comprehensive suite that factors in desolvation, conformational entropy, and solvation, while supporting receptor flexibility. | http://dock.compbio.ucsf.edu/ |
[59] |
| MOE | An integrated software platform featuring robust tools for molecular modeling, virtual screening, and structure-based design, including docking. | https://www.chemcomp.com/Products.htm | [60] |
| HADDOCK | A highly adaptable approach for modeling diverse complexes, including protein-ligand, protein-protein, and protein-nucleic acid interactions. | https://wenmr.science.uu.nl/haddock2.4/ | [61] |
| FITTED | A genetic algorithm-based program designed to handle macromolecular flexibility and the role of key water molecules in binding. | http://mgltools.scripps.edu/documentation/links/fitted | [62] |
| FlipDock | Docks a flexible ligand into a fully flexible receptor binding site to address full conformational flexibility. | http://flipdock.scripps.edu/ | [63] |
| LigandFit | Combines cavity detection with a Monte Carlo conformational search to generate and score ligand poses within active sites. | https://www.phenix-online.org/documentation/reference/ligandfit.html | [64] |
| pyDOCK | A fast, efficient web server for rigid-body docking using an advanced scoring function. | https://life.bsc.es/pid/pydockweb/ | [65] |
| ClusPro | An FFT-based server optimized for the rapid and accurate prediction of peptide-protein interactions. | https://cluspro.bu.edu/publications.php | [66] |
| ZDock | Predicts protein-protein interactions using an FFT-based algorithm to explore rotational and translational space. | http://zdock.umassmed.edu | [67] |
| PatchDock | Uses a geometry-based algorithm to predict structures of protein-protein and protein-small molecule complexes. | https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php | [68] |
| FlexX | Utilizes a fragment-based method, placing core fragments in the binding site and reconstructing the complete ligand for scoring. | https://www.biosolveit.de/SeeSAR/#FlexX | [69] |
| Discovery Studio | A comprehensive modeling environment integrating docking with molecular dynamics, QM/MM, pharmacophore modeling, and QSAR. | https://www.discoverystudio.com/discovery.studio | [70] |
| Surflex-Dock | A platform within the BioPharma suite providing tools for structure preparation, virtual screening, and ligand modeling. | https://www.biopharmics.com/ | [71] |
| GEMDOCK | Uses a generic evolutionary method and a proprietary empirical scoring function for predicting ligand binding. | http://gemdock.life.nctu.edu.tw/dock/ | [72] |
| EquiBind | A deep learning model that performs direct, “blind” prediction of ligand binding poses without the need for a traditional search procedure. | https://github.com/HannesStark/EquiBind | [49] |
| DiffDock | A diffusion-based generative model for molecular docking that provides confidence estimates for its predicted ligand poses. | https://github.com/gcorso/DiffDock | [48] |
| GNINA | Utilizes convolutional neural networks (CNNs) for both pose prediction and scoring, offering high accuracy in structure-based virtual screening. | https://github.com/gnina/gnina | [50] |
Table 4.
Software Tools for Quantitative Structure-Activity Relationship (QSAR) Modeling.
| Tool | Description | URL | Ref. |
|---|---|---|---|
| QSAR ToolBox | An integrated platform that facilitates chemical grouping and read-across by combining experimental data, computational tools, and theoretical knowledge to identify structurally similar compounds. | https://qsartoolbox.org/ | [113] |
| SYBYL-X | A comprehensive molecular modeling suite for small and macromolecular design, supporting lead identification and optimization through various computational methods. | https://chemweb.ir/downloads/sybyl-x-suite/ | [114] |
| Open3DQSAR | A specialized tool for generating 3D-QSAR models through pharmacophore mapping and partial least squares (PLS) regression analysis. | http://open3dqsar.sourceforge.net/ | [115] |
| QSAR-Co | Enables the construction of multi-target classification QSAR models using machine learning algorithms such as Random Forest and Linear Discriminant Analysis. | https://sites.google.com/view/qsar-co | [116] |
| McQSAR | Utilizes a Monte Carlo-based genetic algorithm for the automated generation and optimization of QSAR models. | http://users.abo.fi/mivainio/mcqsar/index.php | [117] |
Table 5.
Web-servers for the prediction of the ADMET properties of compounds.
| Name | Brief Description | URL | Ref. |
|---|---|---|---|
| SwissADME | Computes physicochemical properties and predicts pharmacokinetic (ADME) parameters. | http://www.swissadme.ch/ | [118] |
| ADMETlab | A comprehensive platform for the systematic evaluation of ADMET properties. | http://admet.scbdd.com/ | [119] |
| PreADMET 2.0 | Predicts absorption, distribution, metabolism, excretion, and toxicity parameters. | https://preadmet.bmdrc.kr/preadmet-pc-version-2-0/ | [120] |
| ALOGPS 2.1 | Predicts key physicochemical properties like lipophilicity (LogP) and water solubility. | http://www.vcclab.org/lab/alogps/ | [121] |
| DrugMint | Assesses the drug-likeness of small molecules using various screening rules. | https://webs.iiitd.edu.in/oscadd/drugmint/ | [122] |
| LightBBB | Predicts blood-brain barrier (BBB) permeability for chemical compounds. | http://bioanalysis.cau.ac.kr:7030/ | [123] |
| ProTox-II | A virtual lab for predicting the toxicity of small molecules, including organ toxicity. | http://tox.charite.de/protox_II/ | [124] |
| CardPred | Estimates cardiotoxicity risk by predicting hERG channel blockade liability. | http://bioanalysis.cau.ac.kr:7050/ | [125] |
| ToxinPred2 | Predicts and assists in the design of toxic versus non-toxic peptides. | https://webs.iiitd.edu.in/raghava/toxinpred2/index.html | [126] |
| ToxiPred | Predicts compound toxicity using quantitative structure-activity relationship (QSAR) models. | http://crdd.osdd.net/oscadd/toxipred/ | [127] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



