Submitted:
21 January 2026
Posted:
21 January 2026
You are already at the latest version
Abstract
The cytoplasmic accumulation of TDP-43 aggregates remains a persistent pathological hallmark of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and limbic predominant age-related TDP-43 encephalopathy. The cell’s natural clearance mechanisms, the Ubiquitin-Proteasome System (UPS) and the autophagy-lysosome pathway (ALP), frequently fail due to a ‘vicious cycle’ created by the sequestration of essential downstream components by aberrant TDP-43, which interrupts autophagic flux. Classical autophagic activators (e.g., rapamycin) often initiate the pathway but cannot address downstream bottlenecks due to flux failure. This review revisits classical strategies and discusses newer approaches to modulate TDP-43 clearance, including TFEB activators, PROTACs (proteolysis-targeting chimeras), and antisense oligonucleotides (ASOs). We propose that adopting multi-targeting strategies and developing better biomarkers are vital for clinical success.
Keywords:
TDP-43 proteinopathy
; autophagy-lysosome pathway
; proteostasis
; neurodegeneration
; autophagic flux
; PROTACs
; frontotemporal dementia
; amyotrophic lateral sclerosis
1. Introduction
The formation of protein aggregates within neurons is a typical clinical hallmark of neurodegenerative diseases [1,2,3,4]. Neurons are particularly vulnerable to this aberrant accumulation due to their unique biology, increased metabolic demand, and post-mitotic status. Due to their limited division, they are unable to dilute the aggregates, thus subjecting them to cumulative damage [5,6]. Particularly, the accumulation of trans-active response DNA-binding protein (43 kDa) (TDP-43) in amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and recently, limbic predominant age-related TDP-43 encephalopathy (LATE; a form of dementia strikingly similar to Alzheimer’s disease (AD), but affects the limbic areas and is generally characterized by TDP-43 cytoplasmic aggregation) [7] has long intrigued experts and complicated the neurodegenerative disease landscape, underscoring its multifaceted nature [8,9,10].
Approximately 97% of ALS cases and 45% of FTD (FTD-TDP) cases exhibited TDP-43 aggregation [11,12], and a significant secondary pathology was also observed in AD and LATE with neuropathological changes (LATE-NC) cases [13,14,15]. The consequences of the accumulation of these aggregates are still highly debated, with questions remaining about whether the toxicity is plaque-intrinsic (gain-of-function) or more akin to sinks that sequester and inactivate functional TDP-43, leading to dysregulation of essential cellular functions (loss-of-function). However, earlier studies have indicated that TDP-43 pathogenicity is potentially a combination of both mechanisms [16,17,18]. Nevertheless, one pathological explanation of this aberrant accumulation posits that it is primarily driven by the impairment of cellular systems that clear these aggregates: the ubiquitin-proteasome system (UPS) for misfolded soluble monomers, and the autophagy-lysosome pathway (ALP) for larger aggregates [19,20].
This clearance deficit hypothesis suggests that TDP-43 proteinopathy is driven at least as much by the breakdown of degradation systems as by production or intrinsic aggregation; thus, restoring defective proteostasis and clearance systems becomes a viable therapeutic target. The UPS primarily processes soluble, misfolded TDP-43 monomers in the nucleus, while the ALP processes the larger cytoplasmic aggregates. These two systems are restricted to their target protein species, meaning that the failure of one results in the accumulation of its target species [19]. Studies have demonstrated that autophagy disruption plays a significant role in the pathogenesis of ALS and FTD, and that aberrant TDP-43 could interfere with lysosomal fusion and function, further contributing to its own uncontrolled cytoplasmic deposition [21,22]. It is therefore logical that stimulating these pathways could enhance proteostatic mechanisms, thereby promoting the clearance of TDP-43 aggregates. In this review, we will discuss the strategies based on this rationale as potential therapeutic avenues for TDP-43 pathologies.
2. Regulation of TDP-43 Monomer Accumulation via UPS Activation
The pathological cascade leading to TDP-43 aggregation begins with increased accumulation of its monomeric form. Apart from its natural tendency to aggregate (which can be further enhanced by mutations) [10], TDP-43 in postmortem brains and spinal cords of patients with sporadic ALS was reported to be unable to form multimers or dimers [23]. Furthermore, N-terminal dimerization-deficient TDP-43 was found to comprise pathological inclusion bodies in ALS motor neurons [23]. Several factors, such as post-translational modifications (e.g., acetylation, citrullination, C-terminal phosphorylation), contribute to the instability of RNA binding in the RNA recognition motifs (RRM) domains, exposing regions that cause the monomer to misfold and undergo liquid-liquid phase separation (LLPS) [24,25,26,27,28]. Misfolding of the monomers can bury lysines and linear degrons that E3 ligases and UPS shuttles recognize, while exposing other residues that favor self-association and prevent proteasome processing. This kinetically diverts the monomers towards phase separation instead of being tagged for UPS degradation [4,19,29].
2.1. The Role of Ubiquitination
Ubiquitination is an essential PTM that tags aberrant proteins for degradation by the UPS. Various ligases and factors drive this process (e.g., UBE2E3, parkin, VHL/CUL2, Znf129, etc.) [30]. However, this system can be disrupted by increased formation of TDP-43 aggregates, which can sequester these components or deplete the ubiquitin pool, thereby preventing the UPS cascade from being executed [30]. In other words, once TDP-43 oligomers begin to seed due to mutations and aberrant PTMs, they interact with and sequester essential UPS components (e.g., E2/E3 enzymes and ubiquitin), effectively disabling the very machinery that facilitates their own degradation. Therefore, fine-tuning the ubiquitination architecture rather than global depletion of aggregated TDP-43 was proposed as a viable therapeutic strategy to restore proteolytic flux and mitigate disease progression [30].
Early findings by Hans et al. on Drosophila models have identified UBE2E and ubiquitin isopeptidase Y (UBPY) as modulators of TDP-43 ubiquitination. They recognized that the UBE2E class of enzymes promotes ubiquitination, while UBPY reduces it [31]. However, UBE2E overexpression failed to reduce TDP-43 steady-state levels within the observation window as one would predict for proteasome-targeting ubiquitination. Moreover, forced ubiquitination by UBE2E3 shifts TDP-43 into insoluble fractions, and the authors attributed this to modifications in TDP-43’s tertiary or quaternary structure [31]. However, RNAi knockdown of DUB UBPY in Drosophila led to the accumulation of insoluble ubiquitinated TDP-43, thereby enhancing neurotoxicity. Logically, activation of UBPY could reduce insoluble ubiquitinated TDP-43 species; however, this parameter was not explicitly tested in the study. Nevertheless, the authors concluded that UBPY is a disease-modifying factor that could potentially suppress TDP-43 neurotoxicity [31].
More recently, Byrd et al. employed an unbiased yeast genome-wide screen using high-throughput dot blots and identified ESCRT complex factors (which induce membrane invagination) and K63-linked ubiquitination as key facilitators of TDP-43 endolysosomal clearance. Moreover, NEDD4, a HECT-type E3 ubiquitin ligase, was found to be involved in TDP-43 ubiquitination [32]. Additional transfection experiments that overexpressed NEDD4 rescued the reduction in cell viability that resulted from the overexpression of TDP-43-GFP and TDP-35-GFP [32]. Taken together, the findings suggest the therapeutic potential of NEDD4, although further studies are needed to confirm its clinical efficacy.
2.2. Other Strategies Involving the UPS
Several strategies have been developed to enhance proteasomal activity. In one early study, affinity probes based on pyrazolones, five-membered aromatic heterocyclic compounds [33], were synthesized and used to screen for high-affinity binding partners. This class of compounds was previously identified through a high-throughput screening of a >50,000-compound library in a G93A-SOD1 cell model [34]. Pyrazolones were confirmed to be neuroprotective in PC12-SOD1G93A cells [35]. Using proteomics, the regulatory subunits of the proteasome: PSMC1, PSMC3, and TCP-1, were characterized as putative targets of the probes [35]. Subsequent proteasome activation by the pyrazolones was demonstrated in the absence of exogenous proteasome inhibitors, as well as through the restoration of degradative function (using a fluorogenic substrate) in the cell model [33], making the probes among the first candidates with a promising ability to enhance proteasomal function.
In another study, the expression of HDAC6, a cytosolic deacetylase enzyme essential in the regulation of protein quality control at the interface between UPS impairment and compensatory autophagy, was modulated using plasmid transfection (overexpression) and siRNA (knockdown) [36]. Here, the authors demonstrated that HDAC6 overexpression decreased insoluble and cytosolic TDP-43 levels in a TDP-43-overexpressing cell model. Conversely, knockdown of Hdac6 increased total TDP-43 levels [36]. LC3-I/II levels (ubiquitin-like adaptor proteins on autophagosome membranes that facilitate phagophore expansion and protein recruitment) were also monitored and found to be significantly increased in TDP-43-overexpressing cells [36]. Their concentrations increased further when HDAC6 expression was enhanced, whereas Hdac6 knockdown completely abolished LC3-I/II levels. These findings suggest that HDAC6 regulates TDP-43-induced UPS impairment through the autophagy-lysosome pathway. The authors confirmed this hypothesis through Bafilomycin A1 (Baf), an autophagy inhibitor, and found no changes in TDP-43 levels despite HDAC6 overexpression [36].
Another study showed that the IκB kinase (IKK) complex, which typically phosphorylates IκB proteins for UPS degradation [37]could directly phosphorylate TDP-43 for UPS degradation [38]. IKKβ, together with IKKα and the scaffold subunit NEMO, is required for efficient phosphorylation of aberrant TDP-43 and its subsequent targeting to the UPS. Moreover, phosphorylation of the N-terminal residues Thr8 and Ser92 was identified as critical for the IKK-dependent reduction of cytoplasmic TDP-43 levels [38]. Most importantly, IKKβ was also shown to reduce only cytoplasmic aggregation-prone TDP-43 and had no considerable effect on wt-TDP-43 concentrations [38], suggesting that modulating IKKβ activity could represent a strategy for preferentially targeting pathological cytoplasmic TDP-43 while sparing physiological nuclear TDP-43.
Much more recently, the knockdown of RAD23A (via an inducible mutant TDP-43 HEK-293 cell line), a gene coding for a member of the Rad23 family of DNA repair/ubiquitin-proteasome shuttle proteins, reduced insoluble TDP-43 levels in the cell model as well as primary rat cortical neurons expressing A315T mTDP-43 [39]. Using a proteomic screen, USP13, a deubiquitinase, was found to be related to this cascade and a modifier of TDP-43-induced aggregation and cytotoxicity [39]. Knockdown of this protein also reduced the sarkosyl-insoluble mTDP-43 in both cell models, and reduced cell death of the rat motor neurons and improved locomotor deficits in C. elegans ALS models, making RAD23A and USP13 possible therapeutic targets for TDP-43 clearance [39].
Taken together, several routes or components could be harnessed to enhance the efficiency of the UPS in serving as a critical defense against the accumulation of pathological TDP-43 monomers. Nevertheless, the recurring evidence of UPS component sequestration and the reliance on compensatory mechanisms, such as HDAC6, suggest that the UPS alone may be inadequate when the load of accumulated monomers exceeds the critical substrate threshold or transitions into insoluble oligomers. This limitation highlights the need for a secondary line of defense capable of clearing larger aggregates. Therefore, the development of therapeutic strategies must extend beyond the UPS to the bulk degradation machinery of the cell: the autophagy-lysosome pathway.
3. Molecular Mechanisms of TDP-43 Clearance via Autophagy
3.1. Macroautophagy Pathway
The autophagic system is a conserved intracellular system for degrading long-lived proteins and organelles in lysosomes [40,41,42]. Three main types of autophagy have been described in the current literature: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [41,42]. Among the three types described, macroautophagy and CMA were reported to be associated with the degradation of TDP-43 aggregates [43,44]. Macroautophagy involves the initial sequestration of the aggregates within autophagosomes, which, upon maturation, shuttle their cargo to fuse with the hydrolase-containing lysosome for proteolysis [42]. Interestingly, studies have shown the dual properties of TDP-43, where its presence regulates autophagy while also acting as the substrate of the process.
For instance, a study by Leibiger et al. demonstrated that the endosomal vacuolar pathway and the vacuolar fusion machinery were critical for TDP-43 clearance and cell survival, whereas autophagy had a more complex and context-dependent contribution [22]. Interestingly, the study showed that the presence of TDP-43 interfered with lysosomal pathways and its own clearance [22]. Thus, it was suggested that in the absence of endolysosomal activity, autophagy facilitates TDP-43 clearance but simultaneously enhances TDP-43-triggered cell death, indicating that autophagy can be cytotoxic in this context despite its degradative role. These findings suggest that therapeutic strategies for TDP-43 proteinopathies should initially focus on restoring endolysosomal flux and vacuolar/lysosomal fusion, while carefully regulating autophagy to promote clearance, thereby avoiding lethal, TDP-43-dependent self-toxicity.
3.2. Chaperone-Mediated Autophagy (CMA)
CMA is a more selective process that operates via a cytosolic chaperone that recognizes a lysosomal targeting motif [42,45] and shuttles proteins to the lysosomal surface. The substrate proteins then interact with a membrane receptor, where lysosomal chaperones facilitate their entry into the lumen [42]. The selectivity of this pathway is beneficial under conditions that require minimal oxidative stress-induced damage or exposure to various toxic compounds, as only damaged proteins are removed without affecting intact ones [42,46,47]. In a study by Ormeño et al., the CMA was shown to be involved in the degradation of recombinant TDP-43 (rTDP-43). The authors demonstrated that CMA-positive lysosomes in rat liver specifically degraded rTDP-43 and contained endogenous TDP-43, providing evidence that TDP-43 could be a CMA substrate in vivo [44]. Previous studies have shown that CMA degradation occurs through the interaction of a specific targeting sequence (KFERQ) within the target protein with the heat shock cognate 70 kDa (Hsc70) chaperone [45,48,49,50]. The formed complex then interacts with the lysosomal-associated membrane protein isoform 2A (Lamp2A), which subsequently facilitates the translocation of the target protein into the lysosomal lumen [44]. The authors further investigated the compatibility of the CMA pathway for TDP-43 degradation through a competition assay using glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an established CMA substrate [51]. A constant concentration of rTDP-43 was incubated with increasing concentrations of GAPDH, revealing complete TDP-43 degradation in the absence of GAPDH. In contrast, no TDP-43 degradation was observed in samples pre-incubated with GAPDH [44]. Moreover, an upregulation in CMA components (Hsc70 and Lamp2A) was observed in response to the overexpression of the aggregation-prone form of TDP-43. GAPDH concentrations were also compared in TDP-43-aggregate expressing cells at 24 and 72 hours to check the possible influence of TDP-43 aggregates on CMA activity. Reduced concentrations were observed in the aggregate-expressing cells, with the decrease more evident at 24 hours [44]. These findings indicate that the CMA pathway is also involved in TDP-43 degradation.
3.3. TFEB-Mediated Lysosomal Biogenesis
A parallel study by Xia et al. (2016) explored the consequences of TDP-43 nuclear loss by knocking down TDP-43 expression in HeLa (non-neuronal) and SH-SY5Y (neuronal) cells. TDP-43 knockdown led to the nuclear translocation of transcription factor EB (TFEB), which is the master regulator of the ALP through the expression of autophagic gene products (e.g., ATG5, Beclin-1, and ATG9B) and lysosomal gene products (e.g., LAMP1, cathepsins, and subunits of ATPases) [52,53,54]. The translocation of TFEB was associated with the loss of raptor mRNA, leading to the disruption of mTORC1 activity and, consequently, a reduction in the efficiency of TFEB phosphorylation [52]. Since phosphorylation is required for TFEB to be sequestered in the cytosol, it translocates to the nucleus. It accumulates there, resulting in enhanced global gene expression involved in ALP and increased biogenesis of autophagosomes and lysosomes [52]. However, TDP-43 loss also impaired autophagosome-lysosomal fusion by reducing dynactin 1 expression, leading to the accumulation of immature autophagic vesicles that could not be processed [52]. This presents a scenario in which the significant upregulation of ALP components from an overactive TFEB (due to chronic mTORC1 inhibition) further drives the formation of autophagosomes that have nowhere to go, as they are unable to fuse with the lysosome due to a lack of dynactin 1, thereby overwhelming the ALP [52]. Therefore, restoring mTORC1 activity and physiological levels of dynactin 1 are potential strategies to mitigate the ALP’s failure to clear TDP-43 aggregates. Figure 1 summarizes the vicious cycle that results from the accumulation of these aggregates.
4. Pharmacological Autophagy Enhancers: Preclinical Evidence
Current and emerging interventions for driving the degradation of aberrant TDP-43 can be categorized by their specificity and their point of attack within the proteostatic network. These range from broad-spectrum mTOR-dependent inducers to newer high-precision molecular tools that target the transcript or the protein directly (Figure 2).
4.1. mTOR-Dependent Autophagy Inducers
The mTORC1 (mechanistic target of rapamycin complex 1) is a principal suppressor of autophagy that remains active when sufficient energy and amino acids are present in the cell (under favorable conditions) [55]. It actively suppresses the pathway by phosphorylating the ULK1-ATG13-FIP200 complex, the main kinase module that initiates autophagy [55]. The phosphorylated ULK1 remains inactive, preventing autophagosome formation and limiting the lysosomal recycling of cytoplasmic components. However, pharmacological or genetic interventions can inhibit mTORC1 activity, thereby activating the ULK1 complex to initiate autophagosome formation and enhance lysosomal degradation of misfolded proteins and damaged cellular components. In TDP-43 proteinopathies, where the UPS and ALP are overwhelmed by the uncontrolled accumulation of TDP-43 aggregates, the induction of autophagy through the inhibition of mTOR has been explored as a strategy to enhance cellular clearance capacity and alleviate the resulting neurotoxicity [55,56].
4.1.1. Rapamycin and Rapalogs
Previous attempts have been made to develop therapeutic strategies for neurodegenerative diseases by enhancing autophagy through rapamycin treatment. The results were promising and demonstrated that this approach was practical in facilitating the clearance of mutant huntingtin protein, α-synuclein, β-amyloid, and prions [57,58,59,60]. However, other studies have revealed the limitations of this approach, as the phenotype of an ALS mouse model (mutant SOD1G93A) could not be rescued [61]. The same adverse effects of this approach were also demonstrated in other AD animal models [62,63,64], thus showing that autophagic enhancement via rapamycin is not generally applicable to all neurodegenerative disease cases.
Nevertheless, an early study showed that mTOR inhibition by rapamycin could rescue learning and memory and improve motor neuron function in TDP-43 transgenic mice that presented FTLD-U (frontotemporal lobar degeneration with ubiquitin-positive inclusions) pathological phenotypes, and also slow its pathological progression [65]. The study also tested tamoxifen (a known mTOR inhibitor) and spermidine and carbamazepine, both of which activate autophagy through mTOR-independent pathways. The application of these analogs improved motor performance, attributed to autophagy activation, which lowered p62 and increased LCII/LC3-1, as well as NeuN+ (Neuronal Nuclei, RBFOX3; a pan-neuronal marker) neurons [65].
Unlike rapamycin and tamoxifen, which activate autophagy through the inhibition of mTORC1, spermidine, a natural polyamine in citrus and soybean, activates autophagy through a deacetylase called SIRT1 [66]. Earlier studies demonstrated the longevity-promoting effect of spermidine in yeast, nematodes, and flies via autophagy activation [67,68]. This involves coordinated (de)acetylation of multiple proteins across the acetylproteome, which, in combination, reconfigures the autophagic network into an autophagic state [66]. In contrast, carbamazepine, an anticonvulsant and mood-stabilizing drug, activates autophagy by depleting myo-inositol levels in the phosphoinositol pathway [69]. This was based on the increased autophagic clearance in macrophages and neurons without measurably inhibiting AKT or mTOR [69].
4.1.2. Monepantel
Monepantel is a commonly used veterinary anthelmintic in livestock [70,71,72]. It demonstrates off-target inhibition in mTOR signaling, which activates autophagy and triggers apoptosis [70,73]. In the Phase 1 MEND trial, the mean ALSFRS-R (ALS Functional Rating Scale – Revised) slope during monepantel treatment decreased relative to historical progression rates, with some analyses suggesting up to 40–60% attenuation of functional decline, but these estimates should be interpreted cautiously pending larger, randomized trials [70,74,75]. Moreover, the active metabolite monepantel sulfone was found in the CSF of ALS patients, which indicates that the drug and/or its metabolite can penetrate the BBB (blood-brain barrier), although detailed CNS pharmacokinetic parameters are yet to be reported [70,71,75]. Nevertheless, early trials reported that monepantel is generally well tolerated with no dose-limiting toxicities in small cohorts, and its prior veterinary and oncological applications were often reported to have a comparatively favorable safety profile; however, robust long-term human safety data in neurodegenerative populations are still limited [70,71,76].
4.2. mTOR-Independent Autophagy Inducers
Unlike rapamycin and rapalogs, which activate autophagy by directly inhibiting mTORC1, mTOR-independent inducers trigger autophagy without measurably suppressing mTOR activity, often preserving anabolic signaling while still boosting autophagic clearance. This distinction is particularly relevant in chronic neurodegenerative settings, where long-term mTORC1 inhibition may compromise neuronal growth, metabolism, and synaptic function, making pathway-selective strategies a favorable approach for therapeutic development [56,77,78].
4.2.1. Trehalose
Trehalose is a non-reducing disaccharide that is found in many organisms, including bacteria, yeast, fungi, insects, and invertebrates [79]. It is known as the hemolymph sugar in invertebrates and primarily maintains cellular integrity by protecting cells against external stresses, such as changes in temperature and humidity, and against oxidation, thereby preventing protein denaturation [79,80]. Earlier studies sought to uncover the mechanism by which the compound confers its protective effects, and some of these effects may be due to its influence on protein folding through direct trehalose-protein interactions [81]. Trehalose was later shown to prevent the aggregation and reduce the cytotoxicity of amyloid-β 40 and 42 (in vitro), which are associated with AD [81], and to inhibit the aggregation of pathologic proteins with polyglutamine repeats in Huntington’s disease mouse models [82].
In a groundbreaking study, Sarkar et al. (2006) revealed the crucial role of trehalose as an mTOR-independent autophagy activator that facilitated the clearance of mutant huntingtin and A30P and A53T mutants of α-synuclein in vitro [79]. Guided by these findings, Wang et al. (2018) tested the effect of the same compound on TDP-43 clearance in a cell culture model, revealing a reduction in TDP-43 accumulation through the autophagic degradation pathway [83]. The authors also showed that autophagy enhancement was driven by TFEB activation through immunofluorescence, which also correlated with increased LC3-I to LC3-II conversion [83]. The TFEB-associated activation was similarly demonstrated in a separate study using an ARpolyQ (androgen receptor with an elongated polyglutamine tract) cell model [84]. Additionally, trehalose was shown to activate the TFEB pathway through the action of calcineurin, a Ca2+-dependent serine/threonine phosphatase PPP3 that removes the inhibitory phosphate groups on TFEB, allowing its nuclear translocation and transcriptional activation [84,85]. The downregulation of Ppp3cb reduced the trehalose-induced nuclear translocation of TFEB, and treatment with cyclosporin A, a PPP3CB inhibitor, similarly reduced TFEB nuclear translocation and counteracted its dephosphorylation when TFEB was overexpressed [84]. These findings strongly suggest that PPP3CB activation is an essential mediator of TFEB translocation.
The effects of trehalose treatment on misfolded protein toxicity in neurodegenerative disease mouse models are promising. Yet, one significant limitation of trehalose is its susceptibility to intestinal TREH (trehalase [brush-border membrane glycoprotein]) when taken orally [86]. Thus, trehalose can be administered only intravenously if approved for human use. Given this limitation, TREH-resistant analogs, such as melibiose and lactulose, promoted TFEB nuclear translocation and ARpolyQ clearance. This effect was reversed upon TFEB downregulation, indicating that these analogs exhibit TFEB dependency, as does trehalose [84].
Another limitation is that most studies have reported increased LC3-II levels following trehalose treatment; however, this parameter alone is only indicative of autophagosome formation, and not necessarily complete autophagic flux. Using mRFP-GFP tandem fluorescent-tagged LC3 (tfLC3) assays that distinguish between autophagosomes and autolysosomes, subsequent studies revealed that trehalose treatment only increased autophagosome but not autolysosomal formation, suggesting an inhibition in autophagic flux [87]. Another study using the same approach in H4 glioma cells showed that trehalose treatment resulted in an inefficient delivery of LC3 from autophagosomes to autolysosomes [88].
Thus, while trehalose has been traditionally viewed as an mTOR-independent inducer of autophagy, prior studies have shown that its effects on the autophagy pathway are more complex than initially thought. Increased autophagosomes reflect not only enhanced initiation, but also impaired autophagic flux, possibly stemming from lysosomal dysfunction. These results emphasize the need for careful interpretation of trehalose-mediated autophagy markers and highlight that its therapeutic potential and limitations should be evaluated in the context of its full impact on the autophagy-lysosomal system.
4.2.2. Ibudilast
Ibudilast is a small-molecule phosphodiesterase (PDE) inhibitor primarily used for the treatment of pulmonary airway diseases such as asthma and chronic obstructive pulmonary disease (COPD) [89]. PDEs typically degrade cyclic nucleotides (e.g., cAMP, cGMP), which serve as second messengers that regulate muscle tone, inflammatory cell activation, and epithelial barrier function. The inhibition of specific PDE isoforms increases the levels of cAMP or cGMP, resulting in bronchodilation, anti-inflammatory effects, reduced vascular resistance, and improved alveolar-capillary barrier integrity [90,91]. Based on these properties, the drug was applied in clinical trials for the treatment of multiple sclerosis (MS, ongoing Phase III) and ALS (active Phase IIb). It showed neuroprotective effects in progressive MS, particularly reductions in brain atrophy, retinal degeneration, and lesion expansion [92]. In contrast, ALS data showed the drug as a mechanically compatible candidate, but the general efficacy data remain inconclusive pending the full trial results [93].
A study by Chen et al. (2020) showed that ibudilast enhanced the clearance of TDP-43 and SOD-1 aggregates in mutant transfected cellular (HEK) models [89]. Though primarily a PDE inhibitor, the authors showed that the drug drives autophagy activity through the increased nuclear translocation of TFEB, and through the inhibition of mTORC1 [89]. Despite not being strictly considered a classical or direct mTOR inhibitor and often categorized among mTOR-independent autophagy enhancers, the pro-autophagic/lysosomal action of ibudilast in ALS models appears to be associated with the modulation of the mTORC1-TFEB axis [89]. In this context, the drug exerts its effects upstream of mTORC1, rather than directly binding to the kinase; yet, this still requires decreased mTORC1 activity to accomplish full aggregate clearance through autophagy. Most importantly, the intermediate steps connecting PDE inhibition to mTORC1 remain incompletely defined.
4.3. Small Molecule TFEB Activators
Emerging studies have uncovered the potential role of other small molecules in enhancing autophagy via TFEB activation. An alternative pathway, specifically the MEK/ERK branch of the MAPK signaling cascade, was targeted in one study to suppress TFEB. The authors utilized trametinib, a MEK1/2 inhibitor, as the principal agent [94]. Their findings showed rescue of impaired neural structures, cognitive function, and hippocampal long-term potentiation in 5xFAD mice, as well as reduced amyloid-β deposition through activated autophagic-lysosomal processes, as demonstrated by the upregulation of related genes following trametinib administration [94]. More recently, clomiphene citrate, an FDA-approved selective estrogen receptor modulator, has been shown to activate TFEB and the autophagic-lysosomal pathway in neuronal cells, promoting the clearance of amyloid-β plaques and improving cognitive function in the APP/PS1 mouse model of Alzheimer’s disease (AD) [95].
Recently, a multiparameter high-throughput screen evaluated the nuclear translocation of endogenous TFEB and TFE3 (a related transcription factor that also drives the expression of autophagy-lysosome and stress-response genes) together with lysosomal function and cytotoxicity, allowing TFEB activation hits to be distinguished from generic lysosomal stressors [96]. Using the Published Kinase Inhibitor Set 2 (PKIS2) library, the approach yielded 74 hits, which included AKT-targeting 4-aryl-7-azaindoles that enhanced lysosome activity and some 2-aryl-4-anilino(pyridine-4-yl)-quinazoline series (e.g., NK140, NK176, NK177) that promoted the nuclear translocation of TFEB and TFE3, upregulated CLEAR genes, and drove the clearance of mutant HTT aggregates [96]. The parallel application of the DQ Red BSA assay, which assesses lysosomal function, enabled the further selection of compounds that preserve or enhance lysosomal function. Overall, the above findings imply that TFEB/TFE3 activation and enhanced CLEAR gene expression are achievable without the lysosomal dysfunction typically observed with chloroquine and related lysosomotropic compounds.
Finally, levacetylleucine (NALL, trade name Aqneursa™), an FDA-approved drug for the treatment of Niemann-Pick disease type C (NPC) — a genetic disorder that impairs lipid-trafficking and causes abnormal lipid accumulation in late endosomes and lysosomes — was recently shown in a preprint to rapidly induce nuclear translocation of TFEB and increase the expression of LAMP1, a lysosomal membrane protein essential for maintaining lysosomal integrity, function, and pH [97]. The authors further demonstrated that TFEB activation depends on the N-acetyl moiety rather than the L-leucine backbone, as L-leucine alone did not affect TFEB activation [97]. In contrast, the D-enantiomer (N-acetyl-D-leucine) was inactive, and the racemic mixture of the two forms (N-acetyl-DL-leucine) produced only a modest response, supporting the hypothesis that the D-enantiomer may antagonize the activity of the L-enantiomer [97]. Overall, the divergent activities of the L-, D-, and DL-enantiomers highlight the impact of stereochemistry in regulating lysosomal signaling.
4.4. Kinase Inhibitors with Autophagy-Enhancing Properties
Some kinase inhibitors that enhance autophagy are considered non-canonical mTORC1 inhibitors, as they typically modulate upstream or parallel signaling nodes (e.g., ERK, PDGFR-Akt, AMPK) and exert their effect on mTOR-ULK1-TFEB only secondarily, or even bypass mTOR. In contrast, mTOR-dependent agents primarily function by directly reducing mTORC1 kinase activity at the autophagy initiation complex. As a result, the former has the advantage of preserving more of the mTOR’s anabolic functions while increasing autophagic flux or lysosomal biogenesis, whereas the latter broadly activates mTORC1 targets (e.g., ULK1, ATG proteins, TFEB) with correspondingly wider metabolic consequences [56,98,99]. Below are some of these kinase inhibitors and their emerging applications in TDP-43 pathologies.
4.4.1. Bosutinib
Bosutinib, a drug used for the treatment of chronic myelogenous leukemia (CML), was among the 14 hits funneled from 27 primary hits in a phenotypic drug screen that utilized iPSC-derived motor neurons expressing the SOD1 mutation [100]. The 14 hits targeted the Src/c-Abl signaling axis, which controls cell survival, cytoskeletal dynamics, and the stress response. Inhibitors of this pathway have been reported to increase the survival rate of ALS motor neurons, and knockdown experiments have rescued these same neurons [100]. Bosutinib also improved autophagy, reduced misfolded SOD1 protein, and attenuated altered mitochondria-relevant gene expression, as well as increased the survival of ALS iPSC-derived motor neurons with mutations in TDP-43 or repeat expansions in C9orf72 in vitro [100]. Applying the drug in an ALS mouse model with a SOD1 mutation also modestly extended its survival, suggesting that the Src/c-Abl axis is a potentially valuable target for ALS drug development [100].
These pre-clinical findings gave way to a small-scale, placebo-controlled Phase I clinical trial, which demonstrated it to be safe and well-tolerated, with a subset of patients presenting decreased levels of plasma neurofilament light chain (NfL) and slowed rates of pathological progression [101]. Adverse events (AEs) reported were gastrointestinal AEs, liver function-related AEs, and rash, but the safety profile was consistent with that known for CML, and no ALS-specific AEs were observed [101]. Nevertheless, larger clinical trials to verify this efficacy and decide if bosutinib should proceed further in ALS development were still needed [101,102]. A Phase II observational study is currently evaluating safety and effectiveness in a small cohort of patients. It is ongoing in Japan and was designed as a real-world, observational effectiveness study using routinely collected clinical data, rather than a randomized controlled trial, to evaluate its safety and effectiveness [103,104].
4.4.2. Withaferin-A and Analogs
Withaferin-A (WFA) is an active withanolide derived from the medicinal herb Withania somnifera, and was shown to induce autophagy, reduce TDP-43 proteinopathy (RIPA-insoluble fraction), and improve cognitive function in transgenic FTLD mice expressing mutant TDP-43G348C [105]. WFA also demonstrated anti-inflammatory effects by reducing NF-κB activity and neuroinflammation in the mouse brain, and by increasing LC3BII [105]. However, the compound failed to modulate other autophagic markers, including Beclin-1, p62, and Atg-5 [105]. The reason remains unclear and contradicts another study, which found increased LC3BII, Atg-5, and Beclin-1 expression when the IκB-super-repressor transgene—mutant IκBα that blocks canonical NF-κB activation—was expressed in TDP-43G348C mice [106]. Further investigation is required to resolve these inconsistencies, particularly given that LC3BII expression reflects only increased autophagosome formation, which could stem from impaired degradation rather than enhanced autophagy. To accurately distinguish between enhanced autophagic flux and simple accumulation of autophagosomes in the above results, flux assays (e.g., lysosomal inhibition with chloroquine/bafilomycin, LC3 turnover assays, or LC3 immunostaining to assess autolysosomes) may be required in future studies employing WFA.
Another study explored the effects of IMS-088, a novel WFA analog and an antagonist of nuclear factor-κB essential modulator (NEMO), on vascular dementia [107]. The authors examined chronic cerebral hypoperfusion (CCH), which refers to a long-term reduction in blood flow to the brain caused by vascular disorders linked to conditions such as hypertension, diabetes, and atherosclerosis [108]. CCH is recognized as a major contributor to vascular dementia in older adults. It was induced experimentally in mice through unilateral occlusion of the common carotid artery, leading to chronic cerebral hypoxia and metabolic stress without causing immediate extensive infarction. This led to cytoplasmic mislocalization of TDP-43 in neurons, insoluble phospho-TDP-43 aggregates, chronic microglial activation, and the development of cognitive deficits and motor impairments [107]. Orally administering IMS-088 in CCH mice mitigated TDP-43 pathology, increased autophagy, and improved mental and motor deficits [107]. Moreover, LC3B1 and 2, as well as Beclin-1 levels, were upregulated following IMS-088 treatment, indicating increased autophagy, which likely promoted the clearance of phospho-TDP-43 aggregates [107].
5. Natural Compounds and Polyphenols
Naturally derived compounds and polyphenols are being recognized as viable candidates for enhancing TDP-43 clearance. They have been shown to influence aggregation and toxicity, while promoting neuronal resilience, and exhibit milder toxicity profiles compared to other synthetic molecules [109]. In contrast to more target-specific synthetic small molecules, natural compounds are usually characterized as multi-target, pleiotropic modulators of proteostasis and autophagy, often acting simultaneously at several nodes (e.g., redox balance, inflammatory signaling, mitochondrial function, and nutrient-sensing pathways) [110]. Their pro-autophagic effects typically result from indirect modulation of multiple pathways rather than direct binding to a single autophagy protein. Since multiple stress pathways contribute to TDP-43 pathology in chronic diseases, a therapeutic strategy that simultaneously enhances autophagy and provides antioxidant, anti-inflammatory, and mitochondrial protection may offer synergistic advantages [110,111]. These compounds also exploit overlapping mechanisms with synthetic drugs, but do so through upstream signaling rather than by directly inhibiting mTORC1 or kinases. This multi-pathway engagement may produce wider cytoprotective effects, but could also result in a more complex pharmacological profile and ambiguous effective central nervous system (CNS) exposures [112,113,114].
5.1. Curcumin
Curcumin is a natural compound derived from the powdered rhizome of Curcuma longa, more commonly known as turmeric [111,115]. It is a popular and widely studied polyphenol due to its broad-ranging benefits, including antioxidant, anti-inflammatory, antiproliferative, antitumor, analgesic, and anti-amyloid properties, among others [111]. Some studies further suggest its safety as a bioactive compound, even when administered in high doses [116], which adds to its broad appeal. In terms of autophagy activation, curcumin has been documented to stimulate the PI3K/Akt/mTOR, AMPK, MAPK/ERK1/2, Bcl-2, and Rab GTPase pathways [117].
Several studies have investigated the therapeutic potential of different curcuminoids in the treatment of ALS. One demonstrated the capability of bisdemethoxycurcumin (BDC) and its analogs to clear misfolded/aggregated Aβ/SOD1 in innate immune cells (U-937) by potentiating the expression of MGAT3, VDR, and TLR [118]. A distinct subset of anti-inflammatory curcuminoids was also described that potently inhibited COX-2 and leukotriene B4, as well as IL-17A, IL-6, IL-10, and TNF-α in SOD1-stimulated peripheral blood mononuclear cells from patients with sporadic ALS [118]. However, the study did not disclose the categorization of individual curcuminoids in each set; thus, potential overlap between SOD1/Aβ1-clearance-enhancing and anti-inflammatory analogs cannot be determined from the published data [118]. A later study by Song et al. (2016) revealed a new curcumin analog (C1) that activates TFEB by binding to its N-terminus, promoting its nuclear translocation without inhibiting MTOR. As a result, both autophagy and lysosomal pathways are enhanced in vitro and in vivo [119]. This study also confirmed increased autophagy flux by measuring the degradation of sequestosome 1 (SQSTM1), a selective autophagy substrate. The cells were treated with cycloheximide to prevent unintended upregulation of SQSTM1 caused by the stimulated TFEB [119]. Recently, solid lipid curcumin particles (SLCP) were administered orally to female Prp-TDP-43A315T mice, resulting in a significant reduction in pathological insoluble phosphorylated TDP-43 species, attenuation of disease progression, and improved survival and weight loss by regulating estradiol levels through CYP19A1 upregulation and CYP3A4 downregulation [120]. This implicated a CYP450-estrogen pathway rather than autophagy activation as the primary mechanism of action.
Curcumin is typically known as an autophagy modulator, but its influence on autophagy flux is highly context-dependent [111,113]. Many neurodegenerative and injury models (AD cells, SCI, aging cardiomyocytes) demonstrated that curcumin promotes autophagic flux or at least induces autophagy, usually through the inhibition of PI3K/Akt/mTOR or activation of AMPK/SIRT1 [121,122,123]. This correlates with the improved clearance of toxic proteins or damaged organelles; thus, autophagy is inferred to be protective in these scenarios [121,122,124]. In contrast, curcumin acts as a neuroprotective agent by inhibiting excessive or abnormal autophagy in more severe and sustained stress conditions, such as H2O2-induced oxidative stress in neural stem or progenitor cells, or ischemia-reperfusion models [122,125]. This regulation helps to normalize LC3-II/LC3-I, Beclin1, p-ERK, and p62 expression, thereby restoring proper autophagic flux [126,127]. Thus, autophagy-associated cell death is prevented, rather than driving further degradation. Considering these findings together, it seems that curcumin acts as an autophagic “buffer,” favoring autophagy when aggregate clearance is insufficient (e.g., increased accumulation, impaired axonal transport), but begins to dampen its effects when autophagic overstimulation contributes to increased cell death or maladaptive responses [121,122,127]. This likely reflects curcumin’s action at multiple upstream nodes (PI3K/Akt/mTOR, AMPK, SIRT1, ERK, ROS/p62-Keap-Nrf2), so the overall effect will depend on the cell’s current signaling state and the nature of the insult [111,126].
An early study by Duan et al. (2014) showed in mutant TDP-43Q331K and TDP-25 neuronal cells (NSC-34) that treatment with various curcumin derivatives reduced levels of TDP-43 fragments. Notably, monocarbonyl dimethoxycurcumin C (Compound C) significantly reduced the expression levels and aggregates of TDP-25-transfected cells [128]. Reduced lactate dehydrogenase (LDH) and malondialdehyde (MDA) levels in the NSC-34, following Compound C treatment, demonstrated the compound’s role in reducing oxidative stress [128]. The authors also showed that the compound significantly induced heme oxygenase-1 (HO-1), an antioxidant enzyme, compared with the other candidates. They previously showed a correlation between reduced HO-1 expression and mt-TDP-43 expression, which could contribute to increased oxidative damage [129]. Further investigation suggested that Compound C-induced HO-1 was only partially involved in TDP-43 fragment degradation [128].
5.2. EGCG
Epigallocatechin-3-gallate (EGCG), a polyphenol derived from green tea, has been reported to bind to TDP-43 with micromolar affinity and significantly prevent nucleation, thereby hindering TDP-43 aggregation in vitro and redirecting the protein to form less toxic oligomeric species [135,136]. EGCG is also a characterized modulator of autophagy in other cell types through AMPK/mTOR/ULK1 and CaMKKβ-dependent pathways, but a direct association of EGCG in the promotion of autophagic clearance of TDP-43 inclusions has not been reported, and current evidence mainly supports an anti-aggregation mechanism rather than cargo-specific engagement of the autophagy-lysosome system [137,138].
5.3. Metformin
Metformin is a drug commonly used in type 2 diabetes that activates the AMPK, subsequently triggering a p53-dependent metabolic stress response that enables p53-proficient cells to adapt but renders p53-dependent tumors selectively susceptible to apoptosis under nutrient limitation [139]. The drug is widely described as an indirect activator of AMPK that suppresses mTORC1 signaling, although some reports demonstrated it to inhibit mTOR via AMPK-independent pathways [140,141,142]. Since AMPK is an autophagy activator, its stimulation by metformin should be beneficial to reduce aggregates. For instance, metformin treatment decreased Aβ pathology in an in vivo model of AD (APP/PS1 mice) [143]. Observational studies done on diabetic patients also showed an association with a lower risk of AD [144,145]. However, these findings remain controversial since other studies found no associated lowered AD risk with metformin treatment in diabetic patients [146], while others showed an increase in AD risk following the same treatment [147,148]. However, no other reports explicitly describe its use for TDP-43 clearance.
Nevertheless, metformin is better positioned as a background adjunct instead of a primary TDP-43-clearing drug. In multiple preclinical models, metformin can activate AMPK and subsequently inhibit mTORC1 [149]. Through these and AMPK-independent pathways, it has been demonstrated to modulate autophagy, support mitochondrial function, and promote cellular homeostasis, albeit in a context-dependent manner [114,149]. Given its long safety record, metformin could be likened to a low-toxicity “metabolic scaffold” on which more potent TDP-43-targeted therapies can be layered in future combination regimens. However, this concept remains to be validated in ALS/FTD [110,114,149].
5.4. Resveratrol
Resveratrol (RSV) is a plant-derived polyphenol found in fruits and nuts that acts as a defense compound against environmental stress and pathogens [150,151]. Its antioxidative and anti-inflammatory properties allow it to scavenge reactive oxygen species (ROS) and influence redox-sensitive pathways [152]. Its suggested benefits (e.g., cardioprotective, neuroprotective, antitumor, anti-aging, and other metabolic effects) have made it widely available as a dietary supplement [151,153]. Earlier studies have demonstrated the SIRT-1-activating role of RSV, with an important nuance that could potentially lead to assay artifacts, as it has been shown that RSV can induce conformational changes in SIRT-1, making it preferentially bind fluorophore-labeled substrates that do not necessarily resemble its physiological targets [154]. Nevertheless, a subsequent study has shown that RSV treatment reversed the decreased SIRT1 and FOXO3a expression in human monocytic cells (THP-1) under hyperglycemic conditions [155]. Building on the knowledge that SIRT-1 influences the nuclear translocation of TFEB, resulting in the activation of the autophagy-lysosomal pathway, a study by Bao et al. (2016) used RSV as a SIRT-1 activator to stimulate microglial expression of TFEB in BV2 murine microglia. Here, they demonstrated that the resulting upregulated TFEB facilitated the degradation of fibrillar Aβ and reduced plaque formation through increased lysosomal biogenesis and activity [156]. Crucially, they also revealed a key mechanistic detail, wherein SIRT-1 activation of TFEB occurs through the deacetylation of the latter at Lys-116 [156]. More recently, another study uncovered a TFEB-related pathway, which is an endoplasmic reticulum (ER)-Ca2+ signaling cascade, as well as another cofactor, protein phosphatase 2A (PP2A), which dephosphorylates TFEB [150]. Based on these findings, RSV appears to be a TFEB activator that integrates SIRT-1, ER-Ca2+ signaling, and PP2A-mediated dephosphorylation. However, whether such TFEB activation is enough to drive TDP-43 clearance in neurodegenerative disease models remains unclear and requires further investigation.
6. Advanced Therapeutic Modalities
The results of mitigating TDP-43 aggregation and other neurodegenerative proteins through the modulation of clearance pathways are promising; however, relying solely on these direct approaches is insufficient to address the complexities of proteinopathies. Classical mTOR-dependent autophagy activators, such as rapamycin and related agents, have been shown to reduce inclusions and improve behavioral deficits in mouse models. However, they act broadly on multiple targets, have intrinsic toxicities, and could be dampened in settings where TDP-43-associated lysosomal and autophagic flux deficiencies become dominant [22,40,52,65].
To date, there are no disease-modifying therapies that directly correct TDP-43 mislocalization, aggregation, or loss of nuclear function in neurodegenerative diseases, highlighting both the incomplete control of TDP-43 clearance and the fact that enhancing proteostasis does not eliminate toxic species without unwanted effects [22,157]. Since TDP-43 can also interfere with the very systems that clear it up, classical approaches are vulnerable to self-amplifying loops of proteostatic failure; hence, the need for precision tools that directly recognize TDP-43 species for removal or restoration of specific functions.
6.1. Proteolysis-Targeting Chimeras (PROTACs)
PROTACs have been gaining attention due to their demonstrated potential to target proteins associated with neurodegenerative diseases, specifically [158,159]. PROTACs are heterobifunctional molecules that typically consist of an E3-ligase-recruiting moiety and a ligand for the targeted protein, linked by a linker. Upon forming a ternary complex, the molecular scaffold induces proximity that facilitates the transfer of ubiquitin to the target protein through the ubiquitin-proteasome system (UPS) [160]. Since these molecular scaffolds can be “reused” in a sense, they do not require the full stoichiometric concentrations required in traditional compounds [161]. A small amount of PROTAC is sufficient to tag and eliminate all levels of the target protein, thereby reducing the risk of side effects associated with larger doses [162].
Early proof-of-concept studies by Buckley et al. (2015) utilized HaloTag fusion proteins to demonstrate that small-molecule PROTACs could successfully recruit VHL E3 ligase to degrade specific targets [163]. While this validates the mechanism, the requirement for foreign fusion tags limits clinical application, especially when vulnerable areas, such as the CNS, are targeted, raising concerns about safety and immunogenicity.
In a more recent study, Tseng et al. (2023) designed and characterized four kinds of PROTACs with varying PEG linker lengths (2-5 ethylene glycol units) and showed via the filter trap assay that PROTAC-2 was the only candidate that facilitated the significant degradation of C-terminal TDP-43 aggregates (C-TDP-43) and alleviated C-TDP-43-induced cytotoxicity in Neuro-2a cells without impacting endogenous TDP-43 [164]. The C-TDP-43-binding end was a benzothiazole-aniline (BTA) derivative modified with a 6-O-PEG chain that connected to the linker. In contrast, the E3-ligase-recruiting end was a pomalidomide-based cereblon (CRBN) ligand, which was the same in all other PROTAC constructs [164]. PROTAC-2, with a linker length of 3 ethylene glycol units, was reported as the optimal candidate due to the most favorable spatial positioning between the BTA C-TDP-43 binder and CRBN recruiter, resulting in a more efficient and selective UPS-mediated degradation of toxic proteins [164].
Nevertheless, several challenges remain, including poor permeability through the BBB and target specificity. This is due to several factors, including the possible addition of new protein interfaces in the ternary complex that are absent in the binary complex, E3 ligase promiscuity, and dependence on structural epitope recognition rather than on the specific mutation [165,166,167]. Moreover, although abundant, E3 ligases in the brain exhibit limited diversity. More studies are needed to characterize their chemical properties, dynamic regulation, target specificity, and their specific role in the pathophysiology of neurodegeneration [168].
6.2. Antisense Oligonucleotides (ASOs)
The ASO-mediated modulation of transcripts associated with TDP-43 turnover has been explored in a limited but growing number of studies, suggesting a viable approach to regulating their levels and pathological accumulation. A prior study by Mann et al. (2019) featured an optogenetic expression construct that enabled selective induction of TDP-43 proteinopathy in HEK293 cells. The observed TDP-43 aggregates resulted from the aberrant interactions within the low-complexity (LCD) domains of the proteins [169]. Here, the addition of ASOs acted as molecular “scaffolds” that helped stabilize the LCD through steric or allosteric restraints, thereby preventing pathological phase transitions [169]. These findings underscore the importance of maintaining the RRM domains of TDP-43 bound to RNA to preserve its molecular stability and reduce the likelihood of oligomerization and aggregation.
In a subsequent study, the application of ASOs was extended to the fused in sarcoma (FUS) protein, an RNA-binding protein related to TDP-43 that is also predominantly nuclear and modulates RNA metabolism under physiological conditions [170,171]. Here, a series of FUS knock-in mouse lines expressing FUS mutations found in ALS (FUSP525L and FUSΔEX14), which presented progressive, age-dependent motor neuropathy. A non-allele-specific FUS ASO (ION363) was demonstrated to efficiently silence Fus and reduce postnatal FUS levels in the brain and spinal cord [171]. The authors also showed that repeated intrathecal injections of ION363 in an ALS patient with FUSP525L mutation lowered both wild-type and mutant FUS levels and markedly reduced FUS aggregates [171].
More recently, gapmer-type ASOs targeting TDP-43 have been developed using 2’-O,4’-C-ethylene nucleic acids (ENAs), which are more stable than traditional nucleic acids. Using a mouse model of ALS/FTD that expressed mutant human TDP-43, the authors demonstrated that the intracerebrovascular delivery of ENA-modified ASOs significantly reduced TDP-43 expression without any toxic consequences [172]. Notably, a single injection of ENA-modified ASOs resulted in the sustained improvement of behavioral abnormalities (e.g., anxiety-like behavior, hyperactivity) and the inhibition of cytoplasmic TDP-43 aggregation despite the restoration of TDP-43 initial levels [172].
Since TDP-43 dysfunction has multiple downstream consequences and the pathology itself is complex, targeting multiple downstream pathways could lead to better outcomes. A dual-modality strategy is being developed that can modulate faulty genes that influence the severity or progression of TDP-43 pathology and potentially correct specific TDP-43 loss-of-function phenotypes [173]. This approach could represent a significant shift in the field, given emerging mechanistic evidence that TDP-43 influences its own pathology by failing to modulate essential proteins, such as Stathmin-2 (STMN2), a microtubule-associated protein specifically expressed in neurons. This structural protein is required for axon outgrowth and maintenance, as well as for axonal regeneration following injury in vitro [174]. STMN2 loss in mice was reported to result in ALS-relevant pathology characterized by progressive neuropathy and significant loss of neuromuscular junctions [175]. Baughn et al. (2023) demonstrated that TDP-43 acts as a “steric blocker” that prevents the inclusion of a cryptic exon 2a into STMN2 pre-mRNA, which otherwise (due to loss of nuclear TDP-43) introduces an in-frame stop-codon and a premature polyadenylation signal that truncates the mature STMN2 mRNA, resulting in the loss of functional STMN2 [176]. The authors then developed ASO constructs that bind to exon 2a to prevent its inclusion, thereby rescuing regular STMN2 expression [176].
In developing ASOs for therapeutic targeting, emerging evidence has highlighted the need for alignment with specific repeat RNA species that most directly disrupt TDP-43 function, rather than simply reducing the number of pathological repeats. Notably, Rothstein et al. (2023) showed that ASOs targeting the antisense G2C4 repeat RNA of C9orf72 mitigated deficits in TDP-43 function in C9orf72 ALS/FTD patient-derived induced pluripotent stem cell (iPSC)- derived neurons (IPSNs) [177]. Previous clinical trials utilizing ASOs targeting the sense G4C2 repeat in C9orf72 ALS patients were terminated due to a lack of clinical efficacy [178,179]. The results in the Rothstein study showed that knocking down G4C2 does not correct the downstream consequences of C9orf72 ALS neurons, despite earlier data demonstrating robust reduction of sense dipeptide repeats (DPRs), whereas G2C4 knockdown (in a 15 and 20-day period) restored regular expression and splicing of TDP-43 mRNA targets [177]. They have also demonstrated that G2C4 antisense RNA alone is sufficient to induce TDP-43 loss-of-function, leading to altered expression of numerous proteins that contribute to disease pathology [177]. These findings underscore that depending solely on histological examination (i.e., decreased RNA foci, aggregates) is not enough to infer the restoration of downstream TDP-43 function, as G4C2-sense-targeting ASOs could reduce repeat RNA/DPR pathology and improve neuronal survival, but fail to normalize TDP-43-controlled gene expression and cryptic exon inclusion in C9orf72 iPSNs.
6.3. Gene Therapy Approaches
As for gene therapy approaches targeting TDP-43 accumulation or enhancing its clearance, recent studies have employed two main strategies: direct TDP-43 silencing and indirect approaches that reduce its pathology. A study by Russo et al. (2024) developed novel polymeric nanovectors for delivering TDP-43 small interfering RNAs (siRNAs) in neuronal cells [180]. These nanovectors are typically composed of cationic polymers that bind negatively charged siRNAs via electrostatic interactions, forming more stable complexes. This condenses siRNA into particles that can efficiently penetrate cells and promote endosomal escape, thereby increasing cytoplasmic delivery of the target TDP-43. Their nanovector formulations effectively reduced TDP-43 mRNA and protein levels to comparable levels observed in traditional lipid-based systems [180]. This provided the first evidence that polymeric nanovectors are a viable strategy for treating TDP-43 proteinopathies by directly silencing TDP-43.
Another study revealed that Rho guanine nucleotide exchange factor (RGNEF), an RNA-binding protein, was colocalized in TDP-43 aggregates under pathological stress. Extending this observation, the authors observed that HEK293T cells transfected with RGNEF had a significant survival benefit following treatment with arsenite or sorbitol [181]. This led to the suggestion that RGNEF protects cells under pathological stress. Drawing on this evidence, the same group mapped out the interactions. It was shown that the N-terminal fragment of RGNEF (NF242) directly interacts with the RRM domains of TDP-43, and that the IPT/TIG domain of NF242 is primarily responsible for this interaction [182]. Moreover, expression of NF242 in a fruit fly ALS model that overexpressed TDP-43 mitigated the neuropathological phenotype, resulting in increased lifespan, the abolition of motor deficits, and reversal of neurodegeneration [182]. These findings revealed a protective role for RGNEF against TDP-43 under cellular stress, which could be therapeutically harnessed to regulate TDP-43 pathology in ALS.
A recent attempt to exploit the TFEB pathway through its overexpression was also made to stimulate autophagy-lysosomal activity, albeit in a GRN-knockout (GRN-KO) cell model. The authors showed that GRN-KO cells exhibited increased nuclear localization of TFEB and increased expression of lysosomal transcripts, but impaired autophagy. [183]. Overexpression of TFEB increased lysosomal transcripts and partially restored autophagy. Upon injection of an adeno-associated virus (AAV) expressing mouse Tfeb into the thalamus of Grn-/-mice, lysosomal transcripts were increased, and levels of SCMAS, an indicator of lysosomal storage material, were reduced, indicating decreased lysosomal storage burden rather than loss of lysosomes [183]. Collectively, these results demonstrate that neuronal TFEB activation can pharmacologically rescue lysosomal dysfunction induced by GRN deficiency, a key upstream contributor to FTLD-TDP pathology. TFEB overexpression can likely provide mechanistic support for TDP-43 clearance strategies that exploit the autophagy-lysosomal pathway, both in GRN-related or broader TDP-43 pathologies.
Due to ongoing challenges in safely and efficiently delivering siRNAs to the CNS, one study modified endogenous small RNA processing in liver cells to generate siRNA-encapsulating small extracellular vesicles (SEVs) [184]. Initial treatment of a TDP-43 pathology (TDP-43M337V) mouse model using the siRNAs (IVSA-siR-TDP-43) effectively reduced TDP-43 aggregates and improved motor function and neuropathology. This was then expanded into an AAV-delivery system that contained IVSA-siR-TDP-43, resulting in sustained therapeutic effects in TDP-43-associated neurodegeneration [184]. By harnessing the patient’s own tissues as a long-term source of siRNA and leveraging endogenous RNA processing and natural vesicular trafficking across the BBB, this approach is minimally invasive, sustainable, and CNS-directed, without the delivery and safety challenges associated with traditional techniques.
6.4. Kinase Modulators and Signal Transduction
A growing repertoire of kinase-targeting approaches is designed to drive TDP-43 clearance by regulating upstream effectors of proteostatic pathways, rather than TDP-43 itself. IκB kinase (IKK), glycogen synthase kinase-3 beta (GSK3β), and Nemo-like kinase (NLK) are stress- and inflammation-response kinases that reconfigure post-translational modifications (PTMs), phase behavior, and trafficking, thereby influencing aberrant TDP-43 targeting by the UPS cascade or ALP pathways [38,185,186,187]. In particular, GSK3β was documented to have dual roles in promoting autophagic clearance and suppression, depending on the pathological context [186]. For instance, under stress conditions (e.g., ischemia, energy shortage), GSK3β was shown to favor autophagic clearance of damaged proteins to promote neuronal survival [188]. Conversely, GSK3β can suppress mTOR-mediated autophagy by interacting with its upstream pathways during energy-rich or inflammatory conditions [189].
Acting in parallel are the AMPK-mTOR-TFEB, ERK, and HDAC6-linked aggresome signaling pathways, which drive autophagic flux, lysosomal biogenesis, and microtubule transport, thereby facilitating the clearance of misfolded TDP-43 and its aggregates without altering TARDBP expression [190,191,192,193]. These kinase- and pathway-level interventions function as intermediaries between broader proteostatic enhancers (such as mTOR and ERK) and highly selective TDP-43-directed modalities. By modulating related pathways, the proteostatic network can be redirected toward more efficient processing of aberrant TDP-43 species without directly engaging the protein itself.
7. Challenges and Considerations
Several hurdles impede the clinical application of TDP-43 clearance strategies. First, the same pathways that clear TDP-43 are critical for normal proteostasis. Long-term modulation of UPS, ALP, or related kinases could lead to off-target effects, particularly in long-lived neurons [194]. Secondly, TDP-43 proteinopathy is spatiotemporally heterogeneous, meaning that nuclear loss of function, cytoplasmic aggregation, mitochondrial dysregulation, and lysosomal disruption do not emerge concurrently and critically imply that the therapeutic window and optimal target (aggregation vs. splicing vs. organelle quality control) could vary depending on the stage of the disease and brain region [8,195,196]. Third, the literature shows that several of the targeted pathways result in outcomes that are context-dependent, where the same intervention can be protective in one context but harmful in another [43,197]. This requires careful consideration of dosing, timing, and combination approaches rather than relying on the simple assumption that better outcomes arise from exhaustive clearance of aggregates [71,198]. Finally, TDP-43-specific biomarkers that report target engagement and burden are either not yet readily accessible or remain under development, making it difficult to confirm that these approaches are normalizing TDP-43 biology rather than merely reflecting nonspecific cytoprotective effects [199,200,201].
8. Conclusions
The contributory role of TDP-43 aggregates to neurodegeneration stems either from their own toxicities due to cytoplasmic crowding or from their sequestration of biologically active proteins, rendering them functionally incapacitated. The growing literature suggests a combination of these two roles, supporting the ‘clearance deficit hypothesis,’ which posits that worsening neuropathology results not only from the overexpression of aberrant proteins but also from their failure to be cleared by the UPS and ALP systems.
While UPS mainly serves as the first line of defense by processing soluble misfolded monomers, its capacity can be easily overwhelmed in pathological states. This leads to the accumulation of ubiquitinated proteins and activates stress-response signaling, which upregulates ALP. Although the ALP’s primary role is processing larger oligomers and aggregates, it can also degrade misfolded monomers. However, it could still collapse eventually without the UPS, resulting in an uncontrolled cascade of aggregation. Most critically, a recurring theme across the studies mentioned is the ‘vicious cycle’ in which TDP-43 actively compromises the very endolysosomal and autophagic systems responsible for its clearance. This leads to a therapeutic paradox: stimulating a compromised system still results in failed clearance if downstream ALP components and flux remain undermined.
Emerging therapeutic strategies, such as TFEB activators, PROTACs, and ASOs, are more target-specific than their broad-acting predecessors (e.g., rapamycin and rapalogs). Relying solely on activating the autophagic pathway via mTORC1 inhibition can be complicated, as mTORC1 sits at an upstream node shared with other controllers of protein synthesis (S6K, 4E-BP1), lipid and nucleotide synthesis, and cell growth [55]. Consequently, mTOR-dependent inducers may exert pleiotropic effects far beyond the scope of aggregate clearance.
Furthermore, reports showed mixed outcomes of rapamycin treatment, with clearance of related neurodegenerative diseases demonstrated in some but failed phenotypic rescue in mouse models in others [61,62,63,64]. These inconsistent results could be best explained by the vicious cycle described earlier, in which pathological TDP-43 can sequester and inactivate essential components of the lysosomal pathway, effectively dampening its effects. Since mTOR is an upstream activator that initiates ALP (by inhibiting it), complete clearance cannot proceed if autophagic flux is compromised due to the loss of components (e.g., E2/E3 ligases, ubiquitin, dynactin), leading to the accumulation of autophagosomes with nowhere to go. In contrast, TFEB activation with small molecules specifically modulates lysosomal biogenesis, thereby ensuring that the cell catches up with TDP-43-driven sequestration by replacing the lost components (Figure 3). Even more precise approaches include ASOs and PROTACs, which directly interact with TDP-43 to regulate its expression and prevent the sequestration of elements involved in cellular clearance pathways.
The viability of prospective therapeutic strategies could be improved if these go beyond single ALP interventions. Due to the spatiotemporal heterogeneity of TDP-43 pathology, a multi-target strategy, such as administering metformin to stabilize cellular metabolism and autophagic tone while delivering more precise ASOs and PROTACs to clear aggregates, may improve current outcomes. The goal is to ensure functional recovery and preserve autophagic flux rather than relying solely on ALP upregulation, which could lead to autophagosome accumulation without degradation. This can be achieved by developing reliable biomarkers that reflect successful target engagement and lysosomal function in vivo and could be a significant step forward in the transition from bench to clinical applications.
Author
Conceptualization, investigation, writing—original draft preparation, writing—review and editing.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing does not apply to this article, as no new data were created or analyzed in this study, and all information is derived from previously published sources as cited.
Acknowledgments
The author would like to thank Professors John P. Hulme and Seong Soo A. An for their expertise in drafting this review.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| AAV | Adeno-associated virus |
| ALP | Autophagy–lysosome pathway |
| ALS | Amyotrophic lateral sclerosis |
| ALSFRS-R | ALS Functional Rating Scale–Revised |
| AMPK | AMP-activated protein kinase |
| APP/PS1 | Amyloid precursor protein / presenilin-1 double-transgenic AD mouse model |
| ARpolyQ | Androgen receptor with an elongated polyglutamine tract |
| ASO | Antisense oligonucleotide |
| ATG | Autophagy-related gene/protein (e.g., ATG5, ATG9B) |
| BTA | Benzothiazole–aniline (TDP-43-binding scaffold in PROTACs) |
| CCH | Chronic cerebral hypoperfusion |
| CLEAR | Coordinated lysosomal expression and regulation |
| CML | Chronic myelogenous leukemia |
| CMA | Chaperone-mediated autophagy |
| COPD | Chronic obstructive pulmonary disease |
| CRBN | Cereblon |
| CSF | Cerebrospinal fluid |
| DPR | Dipeptide repeat |
| DUB | Deubiquitinase |
| ENAs | 2’-O,4’-C-ethylene nucleic acids |
| ERK | Extracellular signal-regulated kinase |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| ESCRT | Endosomal sorting complexes required for transport |
| FTD | Frontotemporal dementia |
| FTLD | Frontotemporal lobar degeneration |
| FTLD-U | Frontotemporal lobar degeneration with ubiquitin-positive inclusions |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GSK3β | Glycogen synthase kinase-3 beta |
| HDAC6 | Histone deacetylase 6 |
| HO-1 | Heme oxygenase-1 |
| Hsc70 | Heat shock cognate 70 kDa protein |
| HTT | Huntingtin protein |
| IKK | IκB kinase |
| IKKβ | IκB kinase beta |
| IVSA | In vivo self-assembled (siRNA platform, context-specific) |
| LAMP1 | Lysosome-associated membrane protein 1 |
| Lamp2A | Lysosome-associated membrane protein 2A |
| LATE | Limbic-predominant age-related TDP-43 encephalopathy |
| LATE-NC | LATE neuropathologic change |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| LDH | Lactate dehydrogenase |
| MAPK | Mitogen-activated protein kinase |
| MDA | Malondialdehyde |
| MS | Multiple sclerosis |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| NALL | N-acetyl-L-leucine |
| NEMO | NF-κB essential modulator |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NfL | Neurofilament light chain |
| NLK | Nemo-like kinase |
| NPC | Niemann–Pick disease type C |
| NeuN | Neuronal nuclei (RBFOX3; pan-neuronal marker) |
| PDE | Phosphodiesterase |
| PI3K | Phosphoinositide 3-kinase |
| PP2A | Protein phosphatase 2A |
| PROTAC | Proteolysis-targeting chimera |
| RGNEF | Rho guanine nucleotide exchange factor |
| RRM | RNA recognition motif |
| ROS | Reactive oxygen species |
| SCMAS | Subunit C of mitochondrial ATP synthase |
| SEV | Small extracellular vesicle |
| SIRT1 | Sirtuin 1 |
| SLCP | Solid lipid curcumin particles |
| SOD1 | Superoxide dismutase 1 |
| STMN2 | Stathmin-2 |
| TARDBP | Gene encoding TDP-43 |
| TDP-43 | Transactive response DNA-binding protein of 43 kDa |
| TFE3 | Transcription factor E3 |
| TFEB | Transcription factor EB |
| TREH | Trehalase |
| ULK1 | Unc-51-like kinase 1 |
| UPS | Ubiquitin–proteasome system |
References
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat Med 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dai, L.; Zhang, Z. Protein Aggregation in Neurodegenerative Diseases. Chinese Medical Journal 2025, 138, 2753–2768. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, J.; Chen, T.; Cai, J.; Ren, R. Protein Aggregation and Its Affecting Mechanisms in Neurodegenerative Diseases. Neurochemistry International 2024, 180, 105880. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Frontiers in Molecular Neuroscience 2019, 12. [Google Scholar] [CrossRef]
- Li, Q.; Pan, W.; Zhou, J.; Yu, H.; Xie, S. Targeting Protein Aggregation for the Treatment of Neurodegenerative Diseases. Medicine Plus 2024, 1, 100005. [Google Scholar] [CrossRef]
- Gorman, A.M. Neuronal Cell Death in Neurodegenerative Diseases: Recurring Themes around Protein Handling. J Cell Mol Med 2008, 12, 2263–2280. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE): Consensus Working Group Report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The Role of TDP-43 Propagation in Neurodegenerative Diseases: Integrating Insights from Clinical and Experimental Studies. Exp Mol Med 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Dhakal, S.; Wyant, C.E.; George, H.E.; Morgan, S.E.; Rangachari, V. Prion-like C-Terminal Domain of TDP-43 and α-Synuclein Interact Synergistically to Generate Neurotoxic Hybrid Fibrils. Journal of Molecular Biology 2021, 433, 166953. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J Biol Chem 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental Models and Reality. Acta Neuropathol 2017, 133, 177–196. [Google Scholar] [CrossRef]
- Waldron, F.M.; Spence, H.; Taso, O.S.; Read, F.L.; Sinha, I.R.; Irwin, K.E.; Wong, P.C.; Ling, J.P.; Gregory, J.M. Brain Iron as a Surrogate Biomarker of Pathological TDP-43 Identifies Brain Region-Specific Signatures in Ageing, Alzheimer’s Disease and Amyotrophic Lateral Sclerosis. 2025. [Google Scholar] [CrossRef]
- Wang, J.; Schneider, J.A.; Bennett, D.A.; Seyfried, N.T.; Young-Pearse, T.L.; Yang, H.-S. Plasma TDP-43 Is a Potential Biomarker for Advanced Limbic-Predominant Age-Related TDP-43 Encephalopathy Neuropathologic Change. Mol Neurodegener 2025, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 Is a Key Player in the Clinical Features Associated with Alzheimer’s Disease. Acta Neuropathol 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Fani, G.; Capitini, C.; Rusmini, P.; Poletti, A.; Cecchi, C.; Chiti, F. Quantitative Assessment of the Degradation of Aggregated TDP-43 Mediated by the Ubiquitin Proteasome System and Macroautophagy. The Faseb Journal 2017, 31, 5609–5624. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; Bigio, E.H.; Cairns, N.J.; Neumann, M.; Mackenzie, I.R.A.; Mann, D.M.A. Mechanisms of Disease in Frontotemporal Lobar Degeneration: Gain of Function versus Loss of Function Effects. Acta Neuropathol 2012, 124, 373–382. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Gains or Losses: Molecular Mechanisms of TDP43-Mediated Neurodegeneration. Nat Rev Neurosci 2012, 13, 38–50. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Budini, M.; Romano, V.; Quadri, Z.; Buratti, E.; Baralle, F.E. TDP-43 Loss of Cellular Function through Aggregation Requires Additional Structural Determinants beyond Its C-Terminal Q/N Prion-like Domain. Hum Mol Genet 2015, 24, 9–20. [Google Scholar] [CrossRef]
- Beckers, J.; Van Damme, P. The Role of Autophagy in the Pathogenesis and Treatment of Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Autophagy Reports 2025, 4, 2474796. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Büttner, S.; Braun, R.J. TDP-43 Controls Lysosomal Pathways Thereby Determining Its Own Clearance and Cytotoxicity. Human Molecular Genetics 2018, 27, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Oiwa, K.; Watanabe, S.; Onodera, K.; Iguchi, Y.; Kinoshita, Y.; Komine, O.; Sobue, A.; Okada, Y.; Katsuno, M.; Yamanaka, K. Monomerization of TDP-43 Is a Key Determinant for Inducing TDP-43 Pathology in Amyotrophic Lateral Sclerosis. Sci Adv 2023, 9, eadf6895. [Google Scholar] [CrossRef]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.-K.; Surewicz, W.K. The Role of Liquid-Liquid Phase Separation in Aggregation of the TDP-43 Low-Complexity Domain. J Biol Chem 2019, 294, 6306–6317. [Google Scholar] [CrossRef]
- Haider, R.; Shipley, B.; Surewicz, K.; Hinczewski, M.; Surewicz, W.K. Pathological C-Terminal Phosphomimetic Substitutions Alter the Mechanism of Liquid-Liquid Phase Separation of TDP-43 Low Complexity Domain. bioRxiv 2024, 2024.03.21.586202. [Google Scholar] [CrossRef] [PubMed]
- Rabdano, S.O.; Izmailov, S.A.; Luzik, D.A.; Groves, A.; Podkorytov, I.S.; Skrynnikov, N.R. Onset of Disorder and Protein Aggregation Due to Oxidation-Induced Intermolecular Disulfide Bonds: Case Study of RRM2 Domain from TDP-43. Sci Rep 2017, 7, 11161. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A.; et al. RRM Adjacent TARDBP Mutations Disrupt RNA Binding and Enhance TDP-43 Proteinopathy. Brain 2019, 142, 3753–3770. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chiang, M.; Toh, E.K.-W.; Chang, C.-F.; Huang, T. Molecular Mechanism of Oxidation-Induced TDP-43 RRM1 Aggregation and Loss of Function. FEBS Lett 2013, 587, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Tsekrekou, M.; Giannakou, M.; Papanikolopoulou, K.; Skretas, G. Protein Aggregation and Therapeutic Strategies in SOD1- and TDP-43- Linked ALS. Front Mol Biosci 2024, 11, 1383453. [Google Scholar] [CrossRef]
- Tran, N.-N.; Lee, B.-H. Functional Implication of Ubiquitinating and Deubiquitinating Mechanisms in TDP-43 Proteinopathies. Front. Cell Dev. Biol. 2022, 10, 931968. [Google Scholar] [CrossRef]
- Hans, F.; Fiesel, F.C.; Strong, J.C.; Jäckel, S.; Rasse, T.M.; Geisler, S.; Springer, W.; Schulz, J.B.; Voigt, A.; Kahle, P.J. UBE2E Ubiquitin-Conjugating Enzymes and Ubiquitin Isopeptidase Y Regulate TDP-43 Protein Ubiquitination. Journal of Biological Chemistry 2014, 289, 19164–19179. [Google Scholar] [CrossRef]
- Byrd, A.; Marmorale, L.; Addison, V.; Marcinowski, S.; Buchan, J.R. Rsp5/NEDD4 and ESCRT Regulate TDP-43 Toxicity and Turnover via an Endolysosomal Clearance Mechanism 2022.
- Zhao, Z.; Dai, X.; Li, C.; Wang, X.; Tian, J.; Feng, Y.; Xie, J.; Ma, C.; Nie, Z.; Fan, P.; et al. Pyrazolone Structural Motif in Medicinal Chemistry: Retrospect and Prospect. Eur J Med Chem 2020, 186, 111893. [Google Scholar] [CrossRef] [PubMed]
- Benmohamed, R.; Arvanites, A.C.; Kim, J.; Ferrante, R.J.; Silverman, R.B.; Morimoto, R.I.; Kirsch, D.R. Identification of Compounds Protective against G93A-SOD1 Toxicity for the Treatment of Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler 2011, 12, 87–96. [Google Scholar] [CrossRef]
- Trippier, P.C.; Zhao, K.T.; Fox, S.G.; Schiefer, I.T.; Benmohamed, R.; Moran, J.; Kirsch, D.R.; Morimoto, R.I.; Silverman, R.B. Proteasome Activation Is a Mechanism for Pyrazolone Small Molecules Displaying Therapeutic Potential in Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2014, 5, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kwon, Y.; Kim, S.; Jo, M.; Jeon, Y.-M.; Cheon, M.; Lee, S.; Kim, S.R.; Kim, K.; Kim, H.-J. The Role of HDAC6 in TDP-43-Induced Neurotoxicity and UPS Impairment. Front Cell Dev Biol 2020, 8, 581942. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Hu, F. IκB Kinase Thwarts Aggregation: Phosphorylating TDP-43 for Degradation. The Journal of Cell Biology 2024, 223. [Google Scholar] [CrossRef]
- Iguchi, Y.; Takahashi, Y.; Li, J.; Araki, K.; Amakusa, Y.; Kawakami, Y.; Kobayashi, K.; Yokoi, S.; Katsuno, M. IκB Kinase Phosphorylates Cytoplasmic TDP-43 and Promotes Its Proteasome Degradation. J Cell Biol 2024, 223, e202302048. [Google Scholar] [CrossRef]
- Dalton, C.; Mojsilovic-Petrovic, J.; Safren, N.; Snoznik, C.; Gebis, K.K.; Wang, Y.-Z.; Lamitina, T.; Savas, J.N.; Kalb, R.G. Ubiquitin Proteasome System Components, RAD23A and USP13, Modulate TDP-43 Solubility and Neuronal Toxicity 2025.
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin Rescues TDP-43 Mislocalization and the Associated Low Molecular Mass Neurofilament Instability. J Biol Chem 2009, 284, 27416–27424. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and Neurodegeneration: When the Cleaning Crew Goes on Strike. The Lancet Neurology 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy Induction Enhances TDP43 Turnover and Survival in Neuronal ALS Models. Nat Chem Biol 2014, 10, 677–685. [Google Scholar] [CrossRef]
- Ormeño, F.; Hormazabal, J.; Moreno, J.; Riquelme, F.; Rios, J.; Criollo, A.; Albornoz, A.; Alfaro, I.E.; Budini, M. Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front. Mol. Neurosci. 2020, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F. Peptide Sequences That Target Cytosolic Proteins for Lysosomal Proteolysis. Trends Biochem Sci 1990, 15, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-Mediated Autophagy during Oxidative Stress. Mol Biol Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Hildebrand, H.; Bomhard, E.M.; Dice, J.F. Direct Lysosomal Uptake of Alpha 2-Microglobulin Contributes to Chemically Induced Nephropathy. Kidney Int 1999, 55, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Isenman, L.D.; Dice, J.F. Secretion of Intact Proteins and Peptide Fragments by Lysosomal Pathways of Protein Degradation. J Biol Chem 1989, 264, 21591–21596. [Google Scholar] [CrossRef]
- Wing, S.S.; Chiang, H.L.; Goldberg, A.L.; Dice, J.F. Proteins Containing Peptide Sequences Related to Lys-Phe-Glu-Arg-Gln Are Selectively Depleted in Liver and Heart, but Not Skeletal Muscle, of Fasted Rats. Biochemical Journal 1991, 275, 165–169. [Google Scholar] [CrossRef]
- Huang, C.-C.; Bose, J.K.; Majumder, P.; Lee, K.-H.; Huang, J.-T.J.; Huang, J.K.; Shen, C.-K.J. Metabolism and Mis-Metabolism of the Neuropathological Signature Protein TDP-43. J Cell Sci 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Terlecky, S.R.; Dice, J.F.; Knecht, E. Selective Binding and Uptake of Ribonuclease A and Glyceraldehyde-3-Phosphate Dehydrogenase by Isolated Rat Liver Lysosomes. J Biol Chem 1994, 269, 26374–26380. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 Loss of Function Increases TFEB Activity and Blocks Autophagosome-Lysosome Fusion. EMBO J 2016, 35, 121–142. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR Regulation of Autophagy. FEBS Lett 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A Pharmacologic Target for Autophagy Regulation. J Clin Invest 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat Genet 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein Is Degraded by Both Autophagy and the Proteasome. J Biol Chem 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by Rapamycin Abolishes Cognitive Deficits and Reduces Amyloid-Beta Levels in a Mouse Model of Alzheimer’s Disease. PLoS One 2010, 5, e9979. [Google Scholar] [CrossRef]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schätzl, H.M. Lithium Induces Clearance of Protease Resistant Prion Protein in Prion-Infected Cells by Induction of Autophagy. J Neurochem 2009, 109, 25–34. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin Treatment Augments Motor Neuron Degeneration in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef]
- Bové, J.; Martínez-Vicente, M.; Vila, M. Fighting Neurodegeneration with Rapamycin: Mechanistic Insights. Nat Rev Neurosci 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Song, H.-J.; Garza, D.; Neufeld, T.P.; Salvaterra, P.M. Abeta42-Induced Neurodegeneration via an Age-Dependent Autophagic-Lysosomal Injury in Drosophila. PLoS ONE 2009, 4, e4201. [Google Scholar] [CrossRef]
- Zhang, S.; Salemi, J.; Hou, H.; Zhu, Y.; Mori, T.; Giunta, B.; Obregon, D.; Tan, J. Rapamycin Promotes Beta-Amyloid Production via ADAM-10 Inhibition. Biochem Biophys Res Commun 2010, 398, 337–341. [Google Scholar] [CrossRef]
- Wang, I.-F.; Guo, B.-S.; Liu, Y.-C.; Wu, C.-C.; Yang, C.-H.; Tsai, K.-J.; Shen, C.-K.J. Autophagy Activators Rescue and Alleviate Pathogenesis of a Mouse Model with Proteinopathies of the TAR DNA-Binding Protein 43. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Mariño, G.; Bennetzen, M.V.; Eisenberg, T.; Megalou, E.; Schroeder, S.; Cabrera, S.; Bénit, P.; Rustin, P.; Criollo, A.; et al. Spermidine and Resveratrol Induce Autophagy by Distinct Pathways Converging on the Acetylproteome. Journal of Cell Biology 2011, 192, 615–629. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat Cell Biol 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Büttner, S.; Eisenberg, T.; Carmona-Gutierrez, D.; Ruli, D.; Knauer, H.; Ruckenstuhl, C.; Sigrist, C.; Wissing, S.; Kollroser, M.; Fröhlich, K.-U.; et al. Endonuclease G Regulates Budding Yeast Life and Death. Molecular Cell 2007, 25, 233–246. [Google Scholar] [CrossRef]
- Vasconcelos-Ferreira, A.; Carmo-Silva, S.; Codêsso, J.M.; Silva, P.; Martinez, A.R.M.; França, M.C., Jr.; Nóbrega, C.; Pereira De Almeida, L. The Autophagy-enhancing Drug Carbamazepine Improves Neuropathology and Motor Impairment in Mouse Models of Machado–Joseph Disease. Neuropathology Appl Neurobio 2022, 48, e12763. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Tolerability, Safety, Pharmacokinetics and Preliminary Efficacy Study of Oral Monepantel in Individuals With Motor Neurone Disease 2021.
- PhD, A.L. Vet Drug Monepantel Slows ALS, MND Disease Progression in Trial. Available online: https://alsnewstoday.com/news/vet-drug-monepantel-slows-als-mnd-disease-progression-trial/ (accessed on 12 December 2025).
- Limited, P. PharmAust Announces Positive Phase 1 MEND Study Top-Line Results in MND / ALS. Available online: https://www.prnewswire.com/news-releases/pharmaust-announces-positive-phase-1-mend-study-top-line-results-in-mnd--als-302074155.html (accessed on 31 December 2025).
- Bahrami, F.; Pourgholami, M.H.; Mekkawy, A.H.; Rufener, L.; Morris, D.L. Monepantel Induces Autophagy in Human Ovarian Cancer Cells through Disruption of the mTOR/p70S6K Signalling Pathway. Am J Cancer Res 2014, 4, 558–571. [Google Scholar] [PubMed]
- Cotton, I. PharmAust’s Monepantel Study Shows 58% Slowdown in MND Progression. Available online: https://smallcaps.com.au/article/pharmaust-monepantel-study-shows-slowdown-mnd-progression (accessed on 31 December 2025).
- Meglio, M. PharmaAust’s ALS Agent Monepantel Meets Primary End Point in Phase 1 Study | NeurologyLive - Clinical Neurology News and Neurology Expert Insights. Available online: https://www.neurologylive.com/view/pharmaaust-als-agent-monepantel-meets-primary-end-point-phase-1 (accessed on 31 December 2025).
- Neurizon Therapeutics Limited An Open Label Extension Study to Investigate the Long Term Safety, Tolerability And Efficacy of Oral Monepantel in Individuals With Motor Neurone Disease Who Previously Completed Study MON-2021-001; clinicaltrials.gov, 2025.
- Valenzuela, V.; Nassif, M.; Hetz, C. Unraveling the Role of Motoneuron Autophagy in ALS. Autophagy 2018, 14, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Jeyabalan, N.; Liton, P.B. MTOR-Independent Induction of Autophagy in Trabecular Meshwork Cells Subjected to Biaxial Stretch. Biochim Biophys Acta 2014, 1843, 1054–1062. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a Novel mTOR-Independent Autophagy Enhancer, Accelerates the Clearance of Mutant Huntingtin and α-Synuclein *. Journal of Biological Chemistry 2007, 282, 5641–5652. [Google Scholar] [CrossRef]
- Chen, Q.; Haddad, G.G. Role of Trehalose Phosphate Synthase and Trehalose during Hypoxia: From Flies to Mammals. Journal of Experimental Biology 2004, 207, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Brown, C.R. Influence of Molecular and Chemical Chaperones on Protein Folding. Cell Stress Chaperones 1996, 1, 109–115. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose Alleviates Polyglutamine-Mediated Pathology in a Mouse Model of Huntington Disease. Nat Med 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, F.-T.; Wang, Y.-X.; Guan, R.-Y.; Chen, C.; Li, D.-K.; Bu, L.-L.; Song, J.; Yang, Y.-J.; Dong, Y.; et al. Autophagic Modulation by Trehalose Reduces Accumulation of TDP-43 in a Cell Model of Amyotrophic Lateral Sclerosis via TFEB Activation. Neurotox Res 2018, 34, 109–120. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose Induces Autophagy via Lysosomal-Mediated TFEB Activation in Models of Motoneuron Degeneration. Autophagy 2018, 15, 631–651. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal Calcium Signalling Regulates Autophagy through Calcineurin and TFEB. Nat Cell Biol 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Gudmand-Høyer, E.; Fenger, H.J.; Skovbjerg, H.; Kern-Hansen, P.; Madsen, P.R. Trehalase Deficiency in Greenland. Scand J Gastroenterol 1988, 23, 775–778. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Cho, E.-D.; Jung Ahn, W.; Won Lee, K.; Lee, S.-J.; Lee, H.-J. Is Trehalose an Autophagic Inducer? Unraveling the Roles of Non-Reducing Disaccharides on Autophagic Flux and Alpha-Synuclein Aggregation. Cell Death Dis 2017, 8, e3091–e3091. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.T.; Karaca, I.; Tamboli, I.Y.; Walter, J. Trehalose Alters Subcellular Trafficking and the Metabolism of the Alzheimer-Associated Amyloid Precursor Protein. J Biol Chem 2016, 291, 10528–10540. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast Enhances the Clearance of SOD1 and TDP-43 Aggregates through TFEB-Mediated Autophagy and Lysosomal Biogenesis: The New Molecular Mechanism of Ibudilast and Its Implication for Neuroprotective Therapy. Biochemical and Biophysical Research Communications 2020, 526, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Mokra, D.; Mokry, J. Phosphodiesterase Inhibitors in Acute Lung Injury: What Are the Perspectives? Int J Mol Sci 2021, 22, 1929. [Google Scholar] [CrossRef] [PubMed]
- Kawamatawong, T. Phosphodiesterase-4 Inhibitors for Non-COPD Respiratory Diseases. Front. Pharmacol. 2021, 12, 518345. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Varshosaz, J.; Darash, S.; Ebne-Ali-Heydari, Y. A Comprehensive Systematic Review of Ibudilast as a Neuroprotective Therapy for Progressive Multiple Sclerosis. Multiple Sclerosis and Related Disorders 2025, 104, 106807. [Google Scholar] [CrossRef]
- MS, M.W. MN-166 (Ibudilast) for ALS | ALS News Today.
- Chun, Y.S.; Kim, M.-Y.; Lee, S.-Y.; Kim, M.J.; Hong, T.-J.; Jeon, J.K.; Ganbat, D.; Kim, H.T.; Kim, S.S.; Kam, T.-I.; et al. MEK1/2 Inhibition Rescues Neurodegeneration by TFEB-Mediated Activation of Autophagic Lysosomal Function in a Model of Alzheimer’s Disease. Mol Psychiatry 2022, 27, 4770–4780. [Google Scholar] [CrossRef]
- Lin, J.; Yuan, Y.; Huang, C.; Zi, J.; Li, L.; Liu, J.; Wu, X.; Li, W.; Zhao, Q.; Li, Y.; et al. TFEB Agonist Clomiphene Citrate Activates the Autophagy-Lysosomal Pathway and Ameliorates Alzheimer’s Disease Symptoms in Mice. Journal of Biological Chemistry 2024, 300, 107929. [Google Scholar] [CrossRef]
- Carling, P.J.; Ryan, B.J.; McGuinness, W.; Kataria, S.; Humble, S.W.; Milde, S.; Duce, J.A.; Kapadia, N.; Zuercher, W.J.; Davis, J.B.; et al. Multiparameter Phenotypic Screening for Endogenous TFEB and TFE3 Translocation Identifies Novel Chemical Series Modulating Lysosome Function. Autophagy 2023, 19, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.C.; Brain, R.; Churchill, G.; Factor, M.; Fields, T.; Platt, F.; Patterson, M.; Strupp, M.; Galione, A. Stereospecific Rapid Activation of Transcription Factor EB (TFEB) by Levacetylleucine (NALL) 2025.
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.-G. The Association of AMPK with ULK1 Regulates Autophagy. PLOS ONE 2010, 5, e15394. [Google Scholar] [CrossRef] [PubMed]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR–Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl Pathway Is a Potential Therapeutic Target in Amyotrophic Lateral Sclerosis. Science Translational Medicine 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; Hanajima, R.; Egawa, N.; Ayaki, T.; Oki, R.; Fujita, K.; et al. Safety and Tolerability of Bosutinib in Patients with Amyotrophic Lateral Sclerosis (iDReAM Study): A Multicentre, Open-Label, Dose-Escalation Phase 1 Trial. eClinicalMedicine 2022, 53, 101707. [Google Scholar] [CrossRef]
- Carroll, E.; Scaber, J.; Huber, K.V.M.; Brennan, P.E.; Thompson, A.G.; Turner, M.R.; Talbot, K. Drug Repurposing in Amyotrophic Lateral Sclerosis (ALS). Expert Opinion on Drug Discovery 2025, 20, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Egawa, N.; Ayaki, T.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; Murakami, T.; Hanajima, R.; Kataoka, H.; et al. Protocol for a Phase 2 Study of Bosutinib for Amyotrophic Lateral Sclerosis Using Real-World Data: Induced Pluripotent Stem Cell-Based Drug Repurposing for Amyotrophic Lateral Sclerosis Medicine (iDReAM) Study. BMJ Open 2024, 14, e082142. [Google Scholar] [CrossRef] [PubMed]
- Center (APRC), A. and P.R. Leukemia Drug Decelerates Amyotrophic Lateral Sclerosis (ALS) Progression. Available online: https://sj.jst.go.jp/news/202408/n0801-01k.html (accessed on 12 December 2025).
- Kumar, S.; Phaneuf, D.; Julien, J.-P. Withaferin-A Treatment Alleviates TAR DNA-Binding Protein-43 Pathology and Improves Cognitive Function in a Mouse Model of FTLD. Neurotherapeutics 2021, 18, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Dutta, K.; Thammisetty, S.S.; Boutej, H.; Bareil, C.; Julien, J.-P. Mitigation of ALS Pathology by Neuron-Specific Inhibition of Nuclear Factor Kappa B Signaling. J Neurosci 2020, 40, 5137–5154. [Google Scholar] [CrossRef]
- Thammisetty, S.S.; Renaud, L.; Picher-Martel, V.; Weng, Y.C.; Calon, F.; Saikali, S.; Julien, J.-P.; Kriz, J. Targeting TDP-43 Pathology Alleviates Cognitive and Motor Deficits Caused by Chronic Cerebral Hypoperfusion. Neurotherapeutics 2021, 18, 1095–1112. [Google Scholar] [CrossRef] [PubMed]
- Valério Romanini, C.; Dias Fiuza Ferreira, E.; Correia Bacarin, C.; Verussa, M.H.; Weffort de Oliveira, R.M.; Milani, H. Neurohistological and Behavioral Changes Following the Four-Vessel Occlusion/Internal Carotid Artery Model of Chronic Cerebral Hypoperfusion: Comparison between Normotensive and Spontaneously Hypertensive Rats. Behav Brain Res 2013, 252, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Novak, V.; Rogelj, B.; Župunski, V. Therapeutic Potential of Polyphenols in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Antioxidants (Basel) 2021, 10, 1328. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the Balance of TDP-43, Mitochondria, and Autophagy: A Promising Therapeutic Strategy for Neurodegenerative Diseases. Transl Neurodegener 2020, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, X.; Pang, X.; Ma, K.; Li, L.; Song, Y.; Hou, D.; Wang, X. Pharmacological Effects, Molecular Mechanisms and Strategies to Improve Bioavailability of Curcumin in the Treatment of Neurodegenerative Diseases. Front. Pharmacol. 2025, 16. [Google Scholar] [CrossRef] [PubMed]
- Forouzanfar, F.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Neuroprotective Effects of Curcumin through Autophagy Modulation. IUBMB Life 2020, 72, 652–664. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and Neurodegeneration. J Clin Invest 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From Kitchen to Clinic. Biochem Pharmacol 2008, 75, 787–809. [Google Scholar] [CrossRef]
- Soleimani, V.; Sahebkar, A.; Hosseinzadeh, H. Turmeric (Curcuma Longa) and Its Major Constituent (Curcumin) as Nontoxic and Safe Substances: Review. Phytotherapy Research 2018, 32, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, A.; Cicero, A.F.G.; Panahi, Y.; Mohajeri, M.; Sahebkar, A. Curcumin: A Naturally Occurring Autophagy Modulator. Journal of Cellular Physiology 2019, 234, 5643–5654. [Google Scholar] [CrossRef]
- Cashman, J.R.; Gagliardi, S.; Lanier, M.; Ghirmai, S.; Abel, K.J.; Fiala, M. Curcumins Promote Monocytic Gene Expression Related to β-Amyloid and Superoxide Dismutase Clearance. Neurodegener Dis 2012, 10, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-X.; Sun, Y.-R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.-H.; Xu, Z.; Wang, M.-Z.; Liu, L.-F.; Huang, Y.-Y.; et al. A Novel Curcumin Analog Binds to and Activates TFEB in Vitro and in Vivo Independent of MTOR Inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.; Hsu, T.-I.; Hu, C.-J.; Huang, J.K.; Lee, Y.-C.; Hsieh, Y.-C.; Ahsan, A.; Huang, C.-C. Potential Role of Solid Lipid Curcumin Particle (SLCP) as Estrogen Replacement Therapy in Mitigating TDP-43-Related Neuropathy in the Mouse Model of ALS Disease. Experimental Neurology 2025, 383, 114999. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhou, F.; Xiong, X.; Zhang, X.; Li, S.; Li, X.; Gao, M.; Li, Y. Enhancing the Retrograde Axonal Transport by Curcumin Promotes Autophagic Flux in N2a/APP695swe Cells. Aging (Albany NY) 2019, 11, 7036–7050. [Google Scholar] [CrossRef]
- Li, W.; Yao, S.; Li, H.; Meng, Z.; Sun, X. Curcumin Promotes Functional Recovery and Inhibits Neuronal Apoptosis after Spinal Cord Injury through the Modulation of Autophagy. J Spinal Cord Med 44 37–45. [CrossRef] [PubMed]
- Yang, L.; Shi, J.; Wang, X.; Zhang, R. Curcumin Alleviates D-Galactose-Induced Cardiomyocyte Senescence by Promoting Autophagy via the SIRT1/AMPK/mTOR Pathway. Evidence-Based Complementary and Alternative Medicine 2022, 2022, 2990843. [Google Scholar] [CrossRef] [PubMed]
- He, H.-J.; Xiong, X.; Zhou, S.; Zhang, X.-R.; Zhao, X.; Chen, L.; Xie, C.-L. Neuroprotective Effects of Curcumin via Autophagy Induction in 6-Hydroxydopamine Parkinson’s Models. Neurochemistry International 2022, 155, 105297. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, J.; Feng, J. The Neuroprotective Effects of Curcumin Are Associated with the Regulation of the Reciprocal Function between Autophagy and HIF-1α in Cerebral Ischemia-Reperfusion Injury. Drug Des Devel Ther 2019, 13, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- LI, X.; SUN, P.; ZHANG, D.; YAN, L. Curcumin in Vitro Neuroprotective Effects Are Mediated by P62/Keap-1/Nrf2 and PI3K/AKT Signaling Pathway and Autophagy Inhibition. Physiol Res 2023, 72, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Luo, H.; Tang, J.; Ji, M.; Yu, D.; Yu, Q.; Cao, Z.; Mai, Y.; Zhang, B.; Chen, Y.; et al. Curcumin Inhibits Oxidative Stress and Autophagy in C17.2 Neural Stem Cell through ERK1/2 Signaling Pathways. Aging Med (Milton) 2024, 7, 559–570. [Google Scholar] [CrossRef]
- Duan, W.; Guo, Y.; Xiao, J.; Chen, X.; Li, Z.; Han, H.; Li, C. Neuroprotection by Monocarbonyl Dimethoxycurcumin C: Ameliorating the Toxicity of Mutant TDP-43 via HO-1. Mol Neurobiol 2014, 49, 368–379. [Google Scholar] [CrossRef]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-Binding Protein-43 Induces Oxidative Injury in Motor Neuron-like Cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Vareed, S.K.; Kakarala, M.; Ruffin, M.T.; Crowell, J.A.; Normolle, D.P.; Djuric, Z.; Brenner, D.E. Pharmacokinetics of Curcumin Conjugate Metabolites in Healthy Human Subjects. Cancer Epidemiol Biomarkers Prev 2008, 17, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Sabet, S.; Rashidinejad, A.; Melton, L.D.; McGillivray, D.J. Recent Advances to Improve Curcumin Oral Bioavailability. Trends in Food Science & Technology 2021, 110, 253–266. [Google Scholar] [CrossRef]
- Curcumin Uptake and Metabolism - Metzler - 2013 - BioFactors - Wiley Online Library. Available online: https://iubmb.onlinelibrary.wiley.com/doi/10.1002/biof.1042 (accessed on 7 January 2026).
- Olmos-Juste, R.; Alonso-Lerma, B.; Pérez-Jiménez, R.; Gabilondo, N.; Eceiza, A. 3D Printed Alginate-Cellulose Nanofibers Based Patches for Local Curcumin Administration. Carbohydrate Polymers 2021, 264, 118026. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ma, Q.; Qiu, C.; Wang, J.; Jin, Z.; Hu, Y. Encapsulation and Controlled Delivery of Curcumin by Self-Assembled Cyclodextrin Succinate/Chitosan Nanoparticles. Food Hydrocolloids 2024, 157, 110465. [Google Scholar] [CrossRef]
- Morando, M.A.; D’Alessandro, V.; Spinello, A.; Sollazzo, M.; Monaca, E.; Sabbatella, R.; Volpe, M.C.; Gervaso, F.; Polini, A.; Mizielinska, S.; et al. Epigallocatechin-3-Gallate Binds Tandem RNA Recognition Motifs of TDP-43 and Inhibits Its Aggregation. Sci Rep 2025, 15, 17879. [Google Scholar] [CrossRef]
- Meshram, V.D.; Balaji, R.; Saravanan, P.; Subbamanda, Y.; Deeksha, W.; Bajpai, A.; Joshi, H.; Bhargava, A.; Patel, B.K. Computational Insights Into the Mechanism of EGCG’s Binding and Inhibition of the TDP-43 Aggregation. Chem Biol Drug Des 2024, 104, e14640. [Google Scholar] [CrossRef]
- Holczer, M.; Besze, B.; Zámbó, V.; Csala, M.; Bánhegyi, G.; Kapuy, O. Epigallocatechin-3-Gallate (EGCG) Promotes Autophagy-Dependent Survival via Influencing the Balance of mTOR-AMPK Pathways upon Endoplasmic Reticulum Stress. Oxidative Medicine and Cellular Longevity 2018, 2018, 6721530. [Google Scholar] [CrossRef]
- Kim, H.-S.; Montana, V.; Jang, H.-J.; Parpura, V.; Kim, J. Epigallocatechin Gallate (EGCG) Stimulates Autophagy in Vascular Endothelial Cells. J Biol Chem 2013, 288, 22693–22705. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic Treatment with the Antidiabetic Drug Metformin Selectively Impairs P53-Deficient Tumor Cell Growth. Cancer Res 2007, 67, 6745–6752. [Google Scholar] [CrossRef]
- Amin, S.; Lux, A.; O’Callaghan, F. The Journey of Metformin from Glycaemic Control to mTOR Inhibition and the Suppression of Tumour Growth. British Journal of Clinical Pharmacology 2019, 85, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; Schmitz, G. Metformin: An Inhibitor of mTORC1 Signaling. Journal of Endocrinology, Diabetes and Obesity 2014, 2, 1–15. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.-F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, Independent of AMPK, Induces mTOR Inhibition and Cell-Cycle Arrest through REDD1. Cancer Res 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin Treatment Prevents Amyloid Plaque Deposition and Memory Impairment in APP/PS1 Mice. Brain, Behavior, and Immunity 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-Term Metformin Usage and Cognitive Function among Older Adults with Diabetes. Journal of Alzheimer’s Disease 2014, 41, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chin-Hsiao, T. Metformin and the Risk of Dementia in Type 2 Diabetes Patients. Aging and disease 2019, 10, 37–48. [Google Scholar] [CrossRef]
- Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.-M.; Hartikainen, S. Metformin and Risk of Alzheimer’s Disease Among Community-Dwelling People With Diabetes: A National Case-Control Study. J Clin Endocrinol Metab 2020, 105, e963–e972. [Google Scholar] [CrossRef] [PubMed]
- Imfeld, P.; Bodmer, M.; Jick, S.S.; Meier, C.R. Metformin, Other Antidiabetic Drugs, and Risk of Alzheimer’s Disease: A Population-Based Case–Control Study. Journal of the American Geriatrics Society 2012, 60, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Choi, D.-W.; Kim, K.J.; Cho, S.Y.; Kim, H.; Kim, K.Y.; Koh, Y.; Nam, C.M.; Kim, E. Association of Metformin Use with Alzheimer’s Disease in Patients with Newly Diagnosed Type 2 Diabetes: A Population-Based Nested Case–Control Study. Sci Rep 2021, 11, 24069. [Google Scholar] [CrossRef]
- Ondaro, J.; Hernandez-Eguiazu, H.; Garciandia-Arcelus, M.; Loera-Valencia, R.; Rodriguez-Gómez, L.; Jiménez-Zúñiga, A.; Goikolea, J.; Rodriguez-Rodriguez, P.; Ruiz-Martinez, J.; Moreno, F.; et al. Defects of Nutrient Signaling and Autophagy in Neurodegeneration. Front. Cell Dev. Biol. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Shi, J.; Du, K.; Wang, N.; Cai, W.; Liu, S.; Ding, Z.; Wang, Y.; Li, D. Resveratrol Promotes Lysosomal Function via ER Calcium-Dependent TFEB Activation to Ameliorate Lipid Accumulation. Biochem J 2021, 478, 1159–1173. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol Scavenges Reactive Oxygen Species and Effects Radical-Induced Cellular Responses. Biochem Biophys Res Commun 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- What Is Resveratrol? What Are the Benefits of Resveratrol? Available online: https://www.batigoz.com/en/health-guide/what-is-resveratrol (accessed on 7 January 2026).
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J Biol Chem 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.-M.; Chien, A.; Jialal, I.; Devaraj, S. Resveratrol Upregulates SIRT1 and Inhibits Cellular Oxidative Stress in the Diabetic Milieu: Mechanistic Insights. J Nutr Biochem 2012, 23, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zheng, L.; Zhang, Q.; Li, X.; Zhang, X.; Li, Z.; Bai, X.; Zhang, Z.; Huo, W.; Zhao, X.; et al. Deacetylation of TFEB Promotes Fibrillar Aβ Degradation by Upregulating Lysosomal Biogenesis in Microglia. Protein Cell 2016, 7, 417–433. [Google Scholar] [CrossRef]
- Liu, R.; Yang, G.; Nonaka, T.; Arai, T.; Jia, W.; Cynader, M.S. Reducing TDP-43 Aggregation Does Not Prevent Its Cytotoxicity. Acta Neuropathologica Communications 2013, 1. [Google Scholar] [CrossRef]
- Tomoshige, S.; Nomura, S.; Ohgane, K.; Hashimoto, Y.; Ishikawa, M. Discovery of Small Molecules That Induce the Degradation of Huntingtin. Angewandte Chemie International Edition 2017, 56, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Churcher, I. Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J Med Chem 2018, 61, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Kuemper, S.; Cairns, A.G.; Birchall, K.; Yao, Z.; Large, J.M. Targeted Protein Degradation in CNS Disorders: A Promising Route to Novel Therapeutics? Front. Mol. Neurosci. 2024, 17. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat Rev Drug Discov 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Pan, M.; Fu, Z.; Hou, H.; Yang, C.; Li, J. Proteolysis-Targeting Chimera (PROTAC): A Revolutionary Tool for Chemical Biology Research. Small Methods 2025, 9, 2500402. [Google Scholar] [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-L.; Lu, P.-C.; Lee, C.-C.; He, R.-Y.; Huang, Y.-A.; Tseng, Y.-C.; Cheng, T.-J.R.; Huang, J.J.-T.; Fang, J.-M. Degradation of Neurodegenerative Disease-Associated TDP-43 Aggregates and Oligomers via a Proteolysis-Targeting Chimera. J Biomed Sci 2023, 30, 27. [Google Scholar] [CrossRef]
- He, S.; Dong, G.; Sheng, C. Strategies for Precise Modulation of Protein Degradation. Acc. Chem. Res. 2025, 58, 1236–1248. [Google Scholar] [CrossRef]
- Ibrahim, S.; Umer Khan, M.; Khurram, I.; Rehman, R.; Rauf, A.; Ahmad, Z.; Aljohani, A.S.M.; Al Abdulmonem, W.; Quradha, M.M. Navigating PROTACs in Cancer Therapy: Advancements, Challenges, and Future Horizons. Food Sci Nutr 2025, 13, e70011. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, M.; Yang, Y.; Du, C.; Zhou, H.; Liu, C.; Chen, Y.; Fan, L.; Ma, H.; Gong, Y.; et al. An Overview of PROTACs: A Promising Drug Discovery Paradigm. Mol Biomed 2022, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Thakur, V.; Sardana, S.; Tiwari, V.; Sharma, D.; Kumar, A. Contemporary Trends in Targeted Protein Degradation for Neurodegenerative Diseases. European Journal of Medicinal Chemistry 2025, 300, 118110. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [PubMed]
- Codron, P.; Cassereau, J.; Vourc’h, P. InFUSing Antisense Oligonucleotides for Treating ALS. Trends in Molecular Medicine 2022, 28, 253–254. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense Oligonucleotide Silencing of FUS Expression as a Therapeutic Approach in Amyotrophic Lateral Sclerosis. Nat Med 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Takeuchi, T.; Maeta, K.; Ding, X.; Oe, Y.; Takeda, A.; Inoue, M.; Nagano, S.; Fujihara, T.; Matsuda, S.; Ishigaki, S.; et al. Sustained Therapeutic Benefits by Transient Reduction of TDP-43 Using ENA-Modified Antisense Oligonucleotides in ALS/FTD Mice. Mol Ther Nucleic Acids 2023, 31, 353–366. [Google Scholar] [CrossRef]
- Moorthy, G. Target ALS Grants and Core Resources Dedicated to TDP-43-Targeted Therapeutics. Target ALS 2025. [Google Scholar]
- Menge, S.; Decker, L.; Freischmidt, A. Restoring Expression of Stathmin-2: A Novel Strategy to Treat TDP-43 Proteinopathies. Sig Transduct Target Ther 2023, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Krus, K.L.; Strickland, A.; Yamada, Y.; Devault, L.; Schmidt, R.E.; Bloom, A.J.; Milbrandt, J.; DiAntonio, A. Loss of Stathmin-2, a Hallmark of TDP-43-Associated ALS, Causes Motor Neuropathy. Cell Reports 2022, 39, 111001. [Google Scholar] [CrossRef] [PubMed]
- Baughn, M.W.; Melamed, Z.; López-Erauskin, J.; Beccari, M.S.; Ling, K.; Zuberi, A.; Presa, M.; Gonzalo-Gil, E.; Maimon, R.; Vazquez-Sanchez, S.; et al. Mechanism of STMN2 Cryptic Splice-Polyadenylation and Its Correction for TDP-43 Proteinopathies. Science 2023, 379, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Baskerville, V.; Rapuri, S.; Mehlhop, E.; Jafar-Nejad, P.; Rigo, F.; Bennett, F.; Mizielinska, S.; Isaacs, A.; Coyne, A.N. G2C4 Targeting Antisense Oligonucleotides Potently Mitigate TDP-43 Dysfunction in Human C9orf72 ALS/FTD Induced Pluripotent Stem Cell Derived Neurons. Acta Neuropathol 2023, 147, 1. [Google Scholar] [CrossRef] [PubMed]
- Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis | Biogen. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results (accessed on 9 January 2026).
- Wave Life Sciences Terminates WVE-004 Program. Available online: https://www.thepharmaletter.com/biotech-news/wave-life-sciences-ends-wve-004-program (accessed on 9 January 2026).
- Russo, A.; Maiorano, G.; Cortese, B.; D’Amone, S.; Invidia, A.; Quattrini, A.; Romano, A.; Gigli, G.; Palamà, I.E. Optimizing TDP-43 Silencing with siRNA-Loaded Polymeric Nanovectors in Neuronal Cells for Therapeutic Applications: Balancing Knockdown and Function. Nanoscale 2024, 16, 22337–22349. [Google Scholar] [CrossRef]
- Cheung, K.; Droppelmann, C.A.; MacLellan, A.; Cameron, I.; Withers, B.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Rho Guanine Nucleotide Exchange Factor (RGNEF) Is a Prosurvival Factor under Stress Conditions. Molecular and Cellular Neuroscience 2017, 82, 88–95. [Google Scholar] [CrossRef]
- Droppelmann, C.A.; Campos-Melo, D.; Noches, V.; McLellan, C.; Szabla, R.; Lyons, T.A.; Amzil, H.; Withers, B.; Kaplanis, B.; Sonkar, K.S.; et al. Mitigation of TDP-43 Toxic Phenotype by an RGNEF Fragment in Amyotrophic Lateral Sclerosis Models. Brain 2024, 147, 2053–2068. [Google Scholar] [CrossRef]
- Nader, W.O.; Brown, K.S.; Boyle, N.R.; Kaplelach, A.K.; Abdelaziz, S.M.; Davis, S.E.; Aljabi, Q.; Hakim, A.R.; Davidson, A.G.; Vollmer, G.A.; et al. TFEB Overexpression Alleviates Autophagy-Lysosomal Deficits Caused by Progranulin Insufficiency. Sci Rep 2025, 15, 26217. [Google Scholar] [CrossRef]
- Wu, J.; Guo, J.; Wu, J.; Song, J.; Xu, J.; Lin, Y.; Huang, C.; Shi, C.; Li, J.; Li, C.; et al. In Vivo Self-Assembled siRNAs Ameliorate Neurological Pathology in TDP-43-Associated Neurodegenerative Disease. Brain 2025, awaf330. [Google Scholar] [CrossRef]
- Kellett, E.A.; Bademosi, A.T.; Walker, A.K. Molecular Mechanisms and Consequences of TDP-43 Phosphorylation in Neurodegeneration. Mol Neurodegener 2025, 20, 53. [Google Scholar] [CrossRef]
- Alhassan, H.H.; Janiyani, K.; Surti, M.; Adnan, M.; Patel, M. The Dual Role of Glycogen Synthase Kinase-3 Beta (GSK3β) in Neurodegenerative Pathologies: Interplay between Autophagy and Disease Progression. Front. Pharmacol. 2025, 16, 1693805. [Google Scholar] [CrossRef]
- Tejwani, L.; Jung, Y.; Kokubu, H.; Sowmithra, S.; Ni, L.; Lee, C.; Sanders, B.; Lee, P.J.; Xiang, Y.; Luttik, K.; et al. Reduction of Nemo-like Kinase Increases Lysosome Biogenesis and Ameliorates TDP-43–Related Neurodegeneration. J Clin Invest 2023, 133. [Google Scholar] [CrossRef]
- Gao, J.; Long, L.; Xu, F.; Feng, L.; Liu, Y.; Shi, J.; Gong, Q. Icariside II, a Phosphodiesterase 5 Inhibitor, Attenuates Cerebral Ischaemia/Reperfusion Injury by Inhibiting Glycogen Synthase Kinase-3β-mediated Activation of Autophagy. British J Pharmacology 2020, 177, 1434–1452. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hua, X.; Qin, T.; Zhang, J.; He, K.; Xia, Q. Inhibition of Glycogen Synthase Kinase 3β Protects Liver against Ischemia/Reperfusion Injury by Activating 5′ Adenosine Monophosphate-activated Protein Kinase-mediated Autophagy. Hepatology Research 2019, 49, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gong, S.; Zhang, H.; Chen, Y.; Liu, Y.; Hao, J.; Liu, H.; Li, X. From the Regulatory Mechanism of TFEB to Its Therapeutic Implications. Cell Death Discov. 2024, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, H.; Yuan, M.; Fan, H.; Cai, Z. Role of AMPK in Autophagy. Front. Physiol. 2022, 13, 1015500. [Google Scholar] [CrossRef]
- Palmieri, M.; Pal, R.; Sardiello, M. AKT Modulates the Autophagy-Lysosome Pathway via TFEB. Cell Cycle 2017, 16, 1237–1238. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Park, K.; Lee, M.-S. Current Status of Autophagy Enhancers in Metabolic Disorders and Other Diseases. Front Cell Dev Biol 2022, 10, 811701. [Google Scholar] [CrossRef]
- Kril, J. Neuropathology of Neurodegenerative Disorders. TBLSC 2021, 2021, e1005587. [Google Scholar] [CrossRef]
- Nasir, A.R.; Delpirou Nouh, C. TDP-43-Proteinopathy at the Crossroads of Tauopathy: On Copathology and Current and Prospective Biomarkers. Front. Cell. Neurosci. 2025, 19, 1671419. [Google Scholar] [CrossRef]
- Asakawa, K.; Tomita, T.; Shioya, S.; Handa, H.; Saeki, Y.; Kawakami, K. Intrinsically Accelerated Cellular Degradation Is Amplified by TDP-43 Loss in ALS-Vulnerable Motor Neurons in a Zebrafish Model. Nat Commun 2025, 16, 9213. [Google Scholar] [CrossRef]
- Chennampally, P.; Sayed-Zahid, A.; Soundararajan, P.; Sharp, J.; Cox, G.A.; Collins, S.D.; Smith, R.L. A Microfluidic Approach to Rescue ALS Motor Neuron Degeneration Using Rapamycin. Sci Rep 2021, 11, 18168. [Google Scholar] [CrossRef] [PubMed]
- San Gil, R.; Walker, A.K. Unlocking Disease-Modifying Treatments for TDP-43-Mediated Neurodegeneration. BioEssays 2025, 47, e202400257. [Google Scholar] [CrossRef] [PubMed]
- Hayes, L.R.; Kalab, P. Emerging Therapies and Novel Targets for TDP-43 Proteinopathy in ALS/FTD. Neurotherapeutics 2022, 19, 1061–1084. [Google Scholar] [CrossRef]
- Moorthy, G. Biomarkers: Pioneering Non-Invasive Tools for TDP-43 Detection. Target ALS; 2025. [Google Scholar]
Figure 1.
The vicious cycle of TDP-43-mediated proteostatic collapse. TDP-43 aggregates actively contribute to pathology rather than merely serving as passive metabolic waste. They sequester essential components of the UPS, such as ubiquitin and E2/E3 ligases, and disrupt the ALP by depleting related components (e.g., dynactin-1), which are essential for autophagosome-lysosomal fusion. This leads to a self-perpetuating “vicious cycle” in which TDP-43 disrupts the very complexes responsible for its clearance, thereby impairing autophagic flux. Created in https://BioRender.com.
Figure 1.
The vicious cycle of TDP-43-mediated proteostatic collapse. TDP-43 aggregates actively contribute to pathology rather than merely serving as passive metabolic waste. They sequester essential components of the UPS, such as ubiquitin and E2/E3 ligases, and disrupt the ALP by depleting related components (e.g., dynactin-1), which are essential for autophagosome-lysosomal fusion. This leads to a self-perpetuating “vicious cycle” in which TDP-43 disrupts the very complexes responsible for its clearance, thereby impairing autophagic flux. Created in https://BioRender.com.
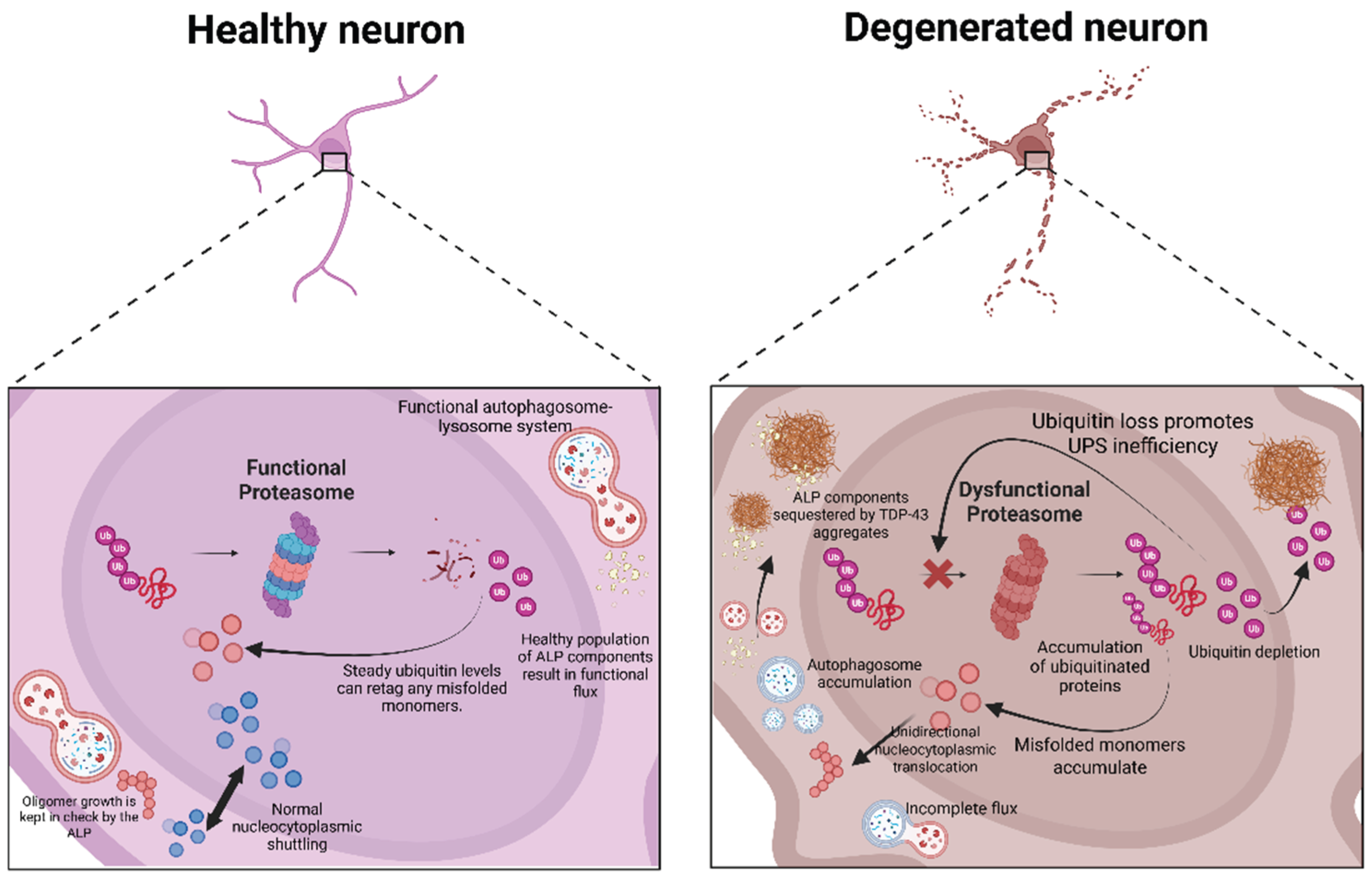
Figure 2.
Nested metabolic hierarchy of therapeutic interventions of TDP-43 proteinopathy. The schematic illustrates the transition from broad metabolic regulation to more precise targeting. (a) Systemic modulation (top panel) involves upstream inhibition of mTORC1 by rapamycin, which activates autophagy but also exerts pleiotropic effects on lipogenesis and metabolism. (b) TFEB-mediated pathway modulation (middle panel) is more downstream, focusing on lysosomal biogenesis and restoring ALP fusion to bypass the bottlenecks created by TDP-43 sequestration. (c) Molecular targeting (bottom panel) uses precision tools such as antisense oligonucleotides (ASOs) and proteolysis-targeting chimeras (PROTACs), which provide the highest specificity by targeting TARDBP mRNA or TDP-43 directly, thereby preventing the initiation of the vicious cycle. Created in https://BioRender.com.
Figure 2.
Nested metabolic hierarchy of therapeutic interventions of TDP-43 proteinopathy. The schematic illustrates the transition from broad metabolic regulation to more precise targeting. (a) Systemic modulation (top panel) involves upstream inhibition of mTORC1 by rapamycin, which activates autophagy but also exerts pleiotropic effects on lipogenesis and metabolism. (b) TFEB-mediated pathway modulation (middle panel) is more downstream, focusing on lysosomal biogenesis and restoring ALP fusion to bypass the bottlenecks created by TDP-43 sequestration. (c) Molecular targeting (bottom panel) uses precision tools such as antisense oligonucleotides (ASOs) and proteolysis-targeting chimeras (PROTACs), which provide the highest specificity by targeting TARDBP mRNA or TDP-43 directly, thereby preventing the initiation of the vicious cycle. Created in https://BioRender.com.
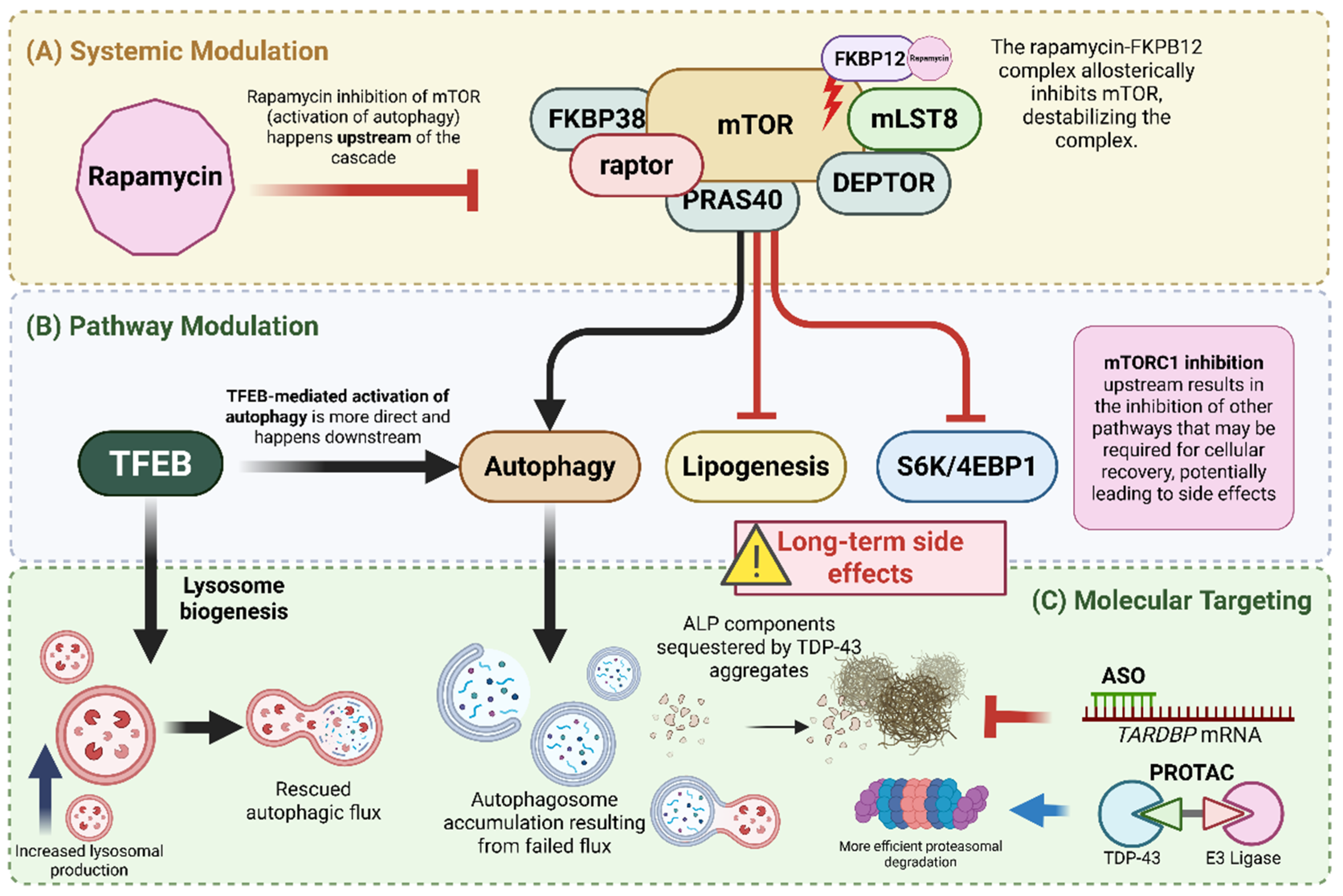
Figure 3.
Shifting the paradigm from autophagy induction to flux restoration. Traditional therapeutic approaches often emphasize initiating autophagy, which can lead to a bottleneck of accumulated, unprocessed autophagosomes if lysosomal function is impaired. Future approaches must focus on preserving autophagic flux to ensure completion of aggregate processing. To achieve this functional recovery, it will probably be necessary to use multi-target strategies that include both metabolic stabilization and precise degradation. Created in https://BioRender.com.
Figure 3.
Shifting the paradigm from autophagy induction to flux restoration. Traditional therapeutic approaches often emphasize initiating autophagy, which can lead to a bottleneck of accumulated, unprocessed autophagosomes if lysosomal function is impaired. Future approaches must focus on preserving autophagic flux to ensure completion of aggregate processing. To achieve this functional recovery, it will probably be necessary to use multi-target strategies that include both metabolic stabilization and precise degradation. Created in https://BioRender.com.
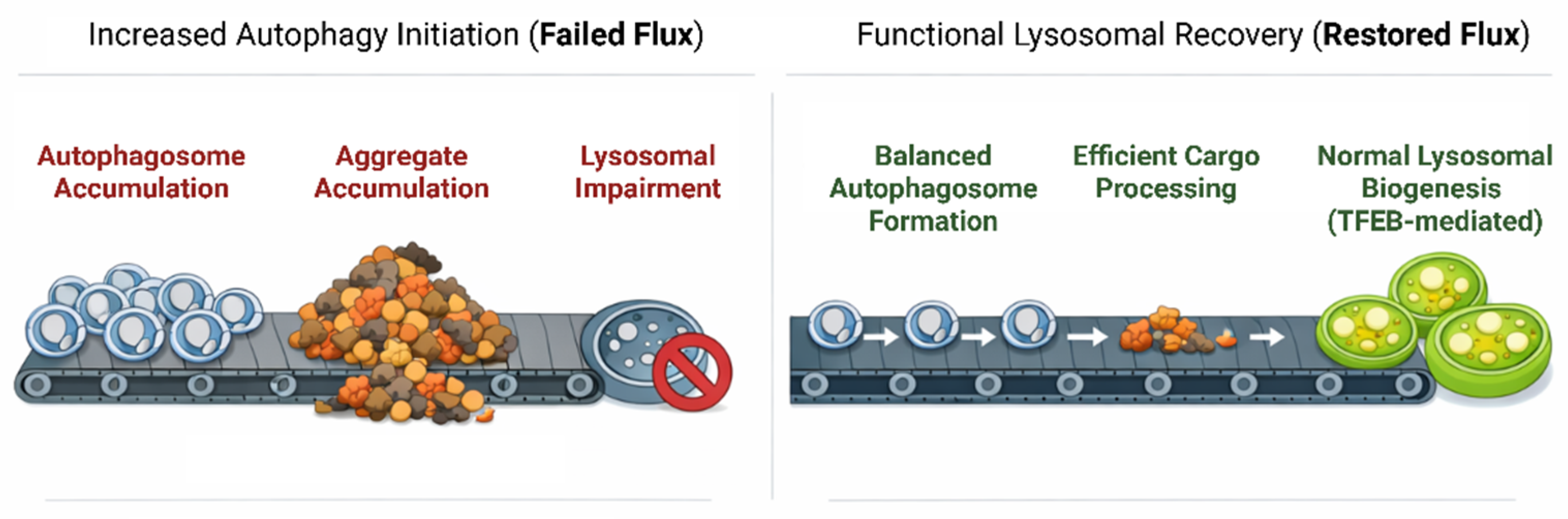
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



