1. Introduction
The ability of plants to sense and respond to biotic stressors, such as fungal, bacterial, and viral pathogens, is crucial for survival. Mosses share innate immune systems with vascular plants and evolved defense mechanisms that further enable pathogen resistance. For example, during the attack of a fungal predator, moss plants can detect a pathogen-associated molecular pattern (PAMP) at surface-localized pattern-recognition receptors (PRRs) and initiate a PAMP-triggered immune response. Once activated, cells exhibit a simultaneous rapid influx of cytosolic calcium (Ca
2+) and activation of the mitogen-activated protein kinase (MAPK) phosphorylation cascade. The resulting defense response propagates plant-wide and includes production of reactive oxygen species (ROS), defense gene activation, and hormone synthesis leading to programmed cell death, growth inhibition, and cell wall reinforcement [
1,
2,
3,
4]. Many processes modulate pathogen resistance including circadian rhythms; for example,
Arabidopsis susceptibility to
B. cineria fungus is decreased at dawn when biotrophic predators primarily release spores [
5].
Calcium signaling plays a vital role in these responses. Stimulation with chitin oligosaccharides, a component of the fungal cell wall, caused plant-wide cytoplasmic Ca
2+ oscillations and induction of defense gene expression [
3]. Calcium influx alone by ionomycin treatment was sufficient to induce the acute phase of defense gene expression, suggesting a direct role in the process, but not the sustained, elevated level following chitin exposure. The details by which calcium levels and oscillation frequency affect gene expression and other pathogen responses, and how calcium and other signals propagate within and between cells, remain largely undetermined [
6].
This study addresses these questions using new experimental methods for the measurement and analysis of pathogen-triggered Ca
2+ responses at plant-wide, single-cell, and subcellular levels. The moss
P. patens is an advantageous model for studying plant-pathogen interactions due to its conserved defense mechanisms, genomic toolset, rapid growth, and small size [
7]. In particular, the juvenile protonemal stage of
P. patens has a filamentous morphology composed of linear and branching chains of chloronemal (CH) and caulonemal (CA) cells. Chloronemata contain an abundance of chloroplasts for photosynthetic energy production. Caulonemata are longer, thinner, and faster growing than chloronemata [
8,
9], attributes that are key for building plant mass and for nutrient acquisition. These cell types exhibit different calcium responses related to their position within the filament and growth status. For example, the apical regions of growing caulonema tip cells exhibit Ca
2+ oscillations that promote the dissipation of actin filaments as required for growth [
3,
10]. Prior studies utilizing protonemata have focused primarily on these growing tip cells, or on average calcium levels across the entire plant which obscures cell-type variation and signal propagation [
6].
To explore individual cell responses across entire plants, we used wide-field fluorescent imaging of plants expressing a genetically-encoded calcium indicator [
11,
12,
13]. Recently, microfluidic devices were fabricated to grow and image plants while keeping them physically within a narrow imaging focal plane [
9,
14,
15]. In addition, microfluidics enables the study of biological phenomena in precise chemical and mechanical microenvironments [
16,
17,
18]. Specifically, laminar fluid flow permits precise and predictable spatiotemporal stimulus patterns, including the ability to rapidly introduce and remove chemicals [
19]. Here, we present microfluidic devices capable of dynamically stimulating multiple plants at once during time-lapse calcium imaging. Similar to prior devices, we demonstrated that protonemata housed in microfluidics triggered intracellular calcium waves comparable to studies that stimulated plants on coverslips. Further, we show for the first time how calcium dynamics react to stimulus removal and to repeated stimulation for different durations. We additionally examined the effects of adaptation time within the microfluidic environment, timing of stimulation relative to circadian cycles, and variations among cell types.
In functional imaging studies, data analysis can be a substantial bottleneck. Each timelapse image stack may require manual annotation and corrections to address sample movement or illumination artifacts. To increase experimental throughput, reduce bias, and increase reproducibility, we developed and validated a semi-automatic, unbiased clustering algorithm to locate contiguous regions within the plants exhibiting distinct calcium dynamics. These regions included whole individual cells as well as subcellular regions. We report the spontaneous and chitin-elicited responses across cell types and subcellular regions to long and short chitin exposures, and highlight differences in response due to time of day, adaptation in the device. Together, these microscopy, microfluidic, and data analysis methods enable the broad study of plant responses to dynamic, reversible stimuli and underlying calcium signaling.
3. Discussion
This study expands upon prior investigations of calcium oscillations in
P. patens moss protonemal cells, combining epifluorescent calcium imaging of plants expressing the GCaMP indicator with microfluidic devices for rapid and reversible chemical stimulation. We demonstrated tip-localized calcium oscillations indicative of tip-growth and plant-wide calcium oscillations upon introduction of chitin oligosaccharides, and measured calcium dynamics that resembled prior studies on agar and in microfluidic growth chambers [
1,
3]. We further illustrate, for the first time: (1) calcium response variations among different cell types and subcellular regions; (2) response modulation by adaptation time and by experimental time of day relative to circadian rhythms; (3) responses to the sudden
removal of chitin stimulus; (4) responses to repeated pulses and response adaptation over repeated stimulation; and (5) the ability to drive plant-wide calcium oscillations at a prescribed frequency. We characterized calcium wave dynamics for each experimental condition across several parameters, including rates, durations, intervals, probabilities, and variabilities, as a baseline for comparison and to inform future experimental designs. We validated an unbiased, pixel-based k-means clustering approach to automatically extract ROIs exhibiting calcium wave dynamics which differ in time of initiation, direction of propagation, or oscillation frequency, and developed data analysis software to extract, summarize, and combine results from multiple individual plants.
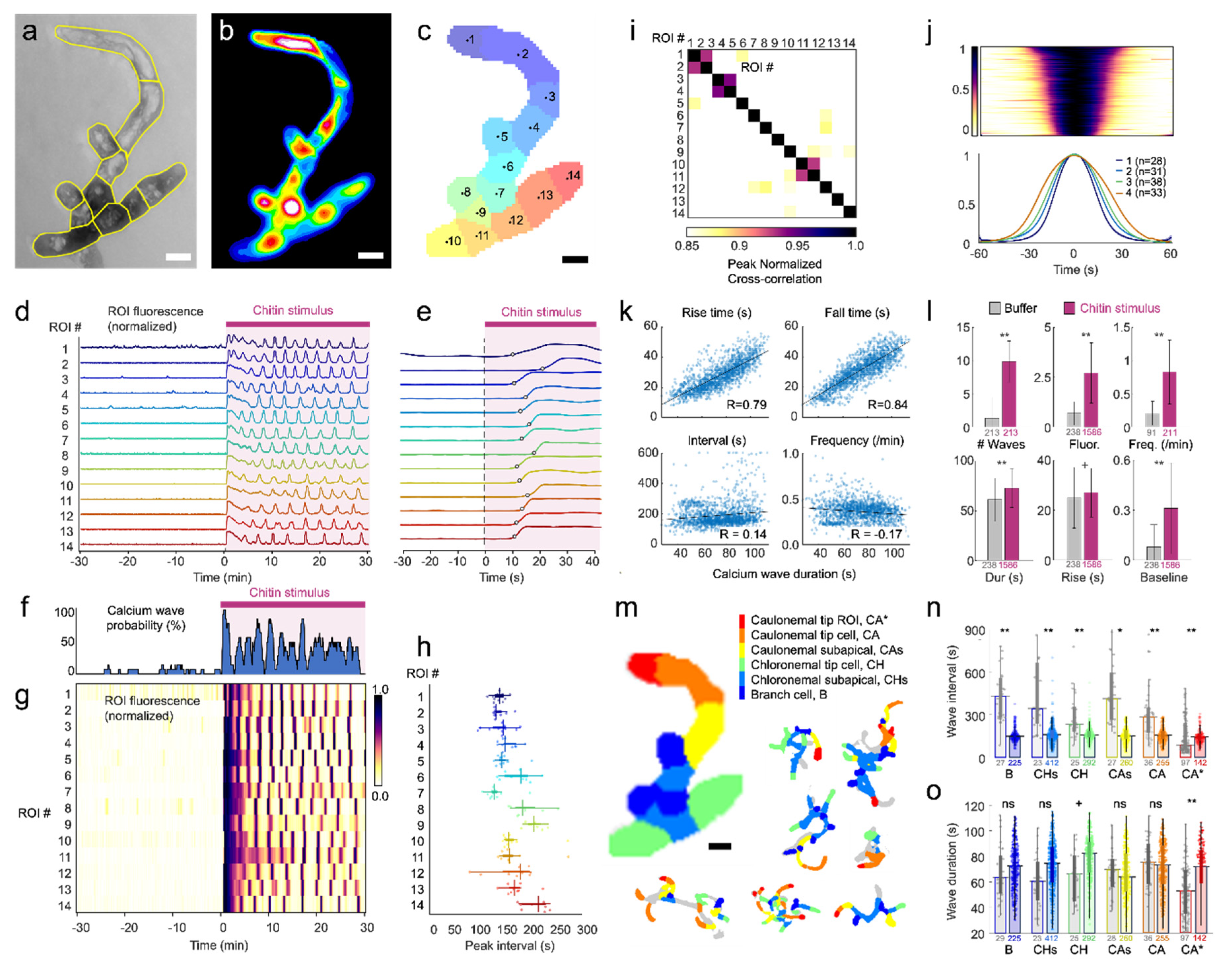
The automatic segmentation algorithm reduced data analysis time 10-fold, from upwards of an hour (for typical sized week-old protonemal plants) to ~5 min, removed bias implicit to manual segmentation, and identified physiologically-relevant subcellular regions that exhibited unique calcium dynamics, such as the apical CA* region versus the rest of the CA tip cell. These regions (for example, ROI #1 in
Figure 2), exhibited basal calcium fluctuations prior to chitin addition characteristic of tip growth.
This study validated the use of microfluidic devices for studying chitin-induced Ca
2+ waves at the plant, cell, and subcellular levels. The prolonged 30 min stimulation experiment most closely matched prior studies exposing moss protonemal tissues on agar pads to a sudden increase in chitin. Here, moss housed in microfluidic arenas demonstrated a similar rapid increase in cytosolic Ca
2+ and comparable plant-wide oscillation interval (2 – 3 min interquartile range, vs. over 2 min interval on agar [
3]). Before stimulation, growing apical regions of the caulonemal cells (CA* region) oscillated at a rate comparable to agar (2.4 – 2.6 per min vs. ~2 per min on agar [
3]). We further characterized cell-type differences and found that while the frequency of chitin-induced waves was relatively consistent across all cell types, the waves in chloronemal and branch cells were slower and longer than caulonemal cells. Chloronemal tip cells exhibited Ca
2+ waves and chitin-induced changes uniformly across the cell, with similar dynamics to those seen in the apical subcellular region of caulonemal tip cells, reflective of growth occurring in both tip cell types but more apically localized in the polarized caulonemal cells [
8].
Chitin stimulation induced stereotyped, high amplitude calcium waves with a symmetrical rise and fall that scaled with wave duration, which varied four-fold across all conditions from about 30 to 120 s (mean 77 s; median 79 s; 65 – 91 s interquartile range). These wave dynamics did not correlate with wave interval, suggesting that wave initiation occurred by signaling processes separate from calcium levels. Oscillation intervals were consistent within each cell and ROI, at least over the time scale of an hour. Chitin-induced waves were initiated shortly after stimulus presentation, with an onset delay that varied several-fold (mean 32 s; median 24 s; 14 – 40 s interquartile range). These delays align with the timing of CERK1 activation by chitin and downstream MAPK phosphorylation [
1]. Interestingly, initiation delays were longer after recent calcium activity, and shortest after quiescent periods, suggesting a “refractory period” in which components involved in calcium wave initiation accumulate or become sequestered during calcium activity and slowly recover responsiveness. Initiated waves followed their prescribed rise and fall and could not be terminated by removal of the stimulus, again suggesting a disconnect between the initiation (by chitin stimulation) and the progression of each calcium wave.
Circadian rhythms enable plants to manage daily cycles, such as the optimization of photosynthesis, stomatal opening, and metabolic processes to align with diurnal light and dark cycles, boost growth, and enhance fitness. The responsiveness of cells to external stimuli and internal signals is regulated by circadian rhythms though differential gene expression. In
P. patens, cryptochrome CRY1 encodes when blue light affects gene expression by translocation into the nucleus, and clock genes such as CCA1 determine how strongly blue light is perceived by modulating blue-light receptors PHOT1/2, which implement phototropic responses at the cellular level [
23]. Moss exhibit Ca
2+ transients shortly after blue light [
24], and we found that blue light excitation of the calcium sensor could directly trigger calcium responses under specific conditions.
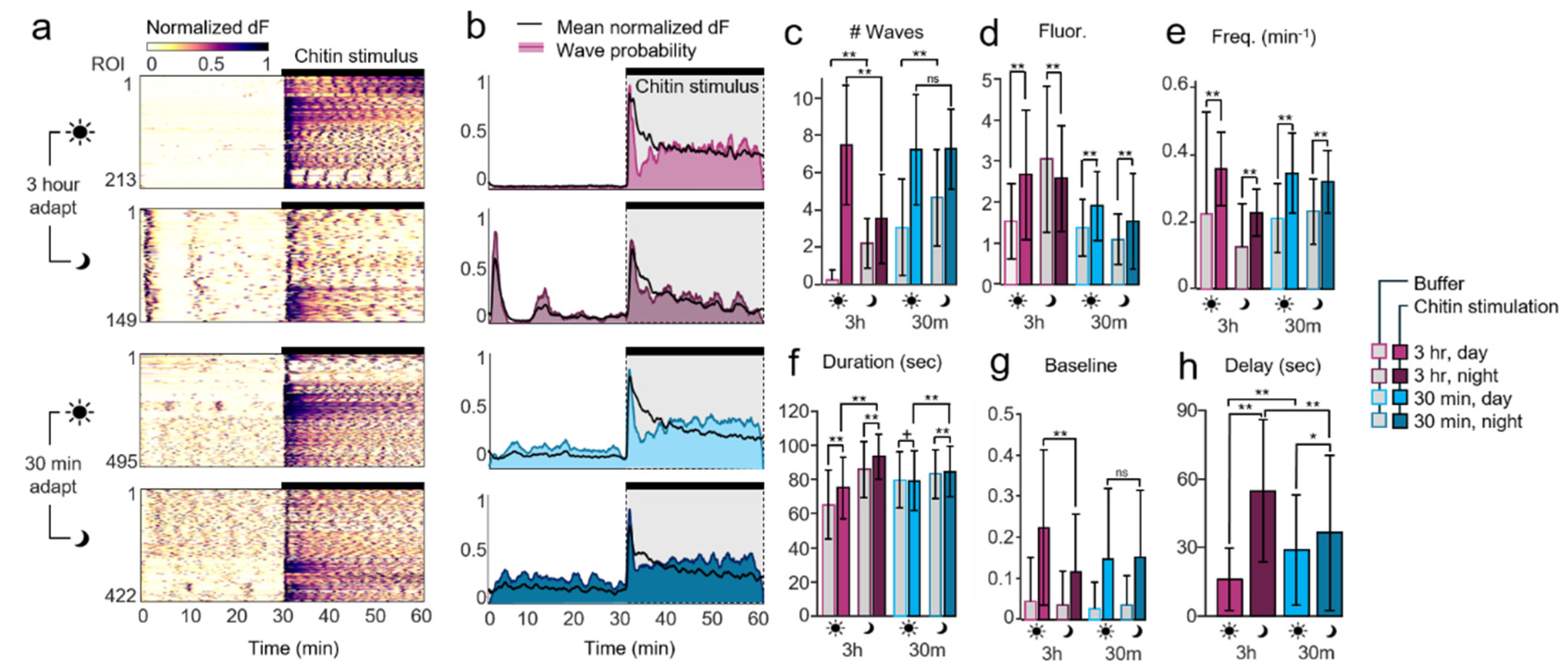
Light sensitivity motivated examination of different adaptation times in darkness prior to calcium recordings, as well as testing at various times during the day and night. The time of day affected calcium dynamics prior to chitin stimulation in long-adapted plants: in daytime, only caulonemal apical tip regions exhibited fluctuations in Ca
2+ indicative of tip growth, while the rest of the cells remained silent. In contrast, plants stimulated at night exhibited an initial wave of Ca
2+ during the first minute of imaging, reflecting sensitivity to blue light excitation. Following a brief quiescent period, spontaneous calcium waves propagated within and between adjacent cells, suggesting intercellular communication through plasmodesmata or apoplastic mechanisms [
25]. Unlike long-adapted plants, short-adapted plants responded similarly at different times of day, suggesting that adaptation in the microfluidic device is required to observe sensitivity to circadian rhythms. Short-adapted plants also exhibited far more spontaneous Ca
2+ activity prior to chitin stimulation, regardless of time of day.
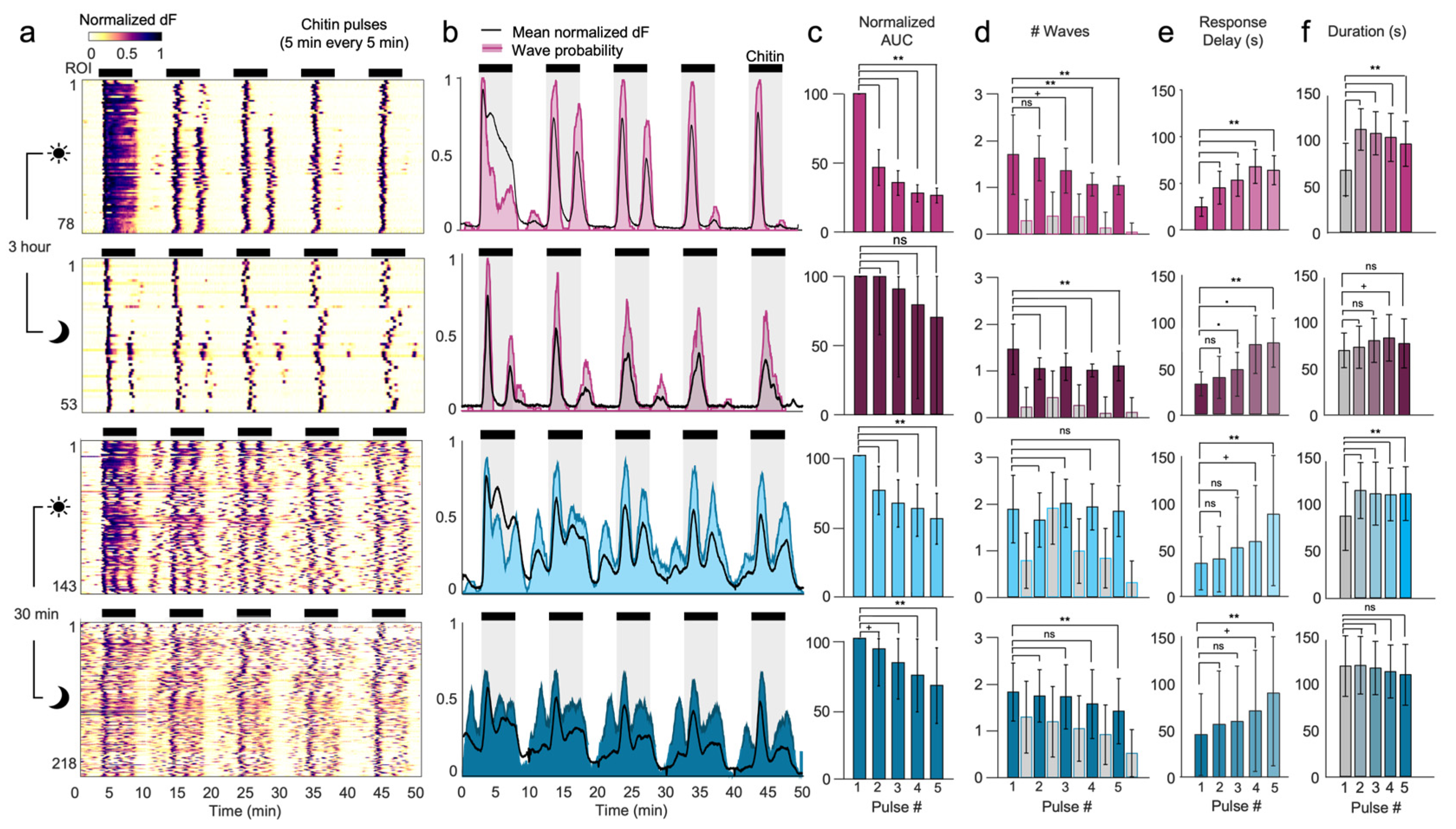
Repeated stimulation of living systems often elicits diminishing responses over time, such as adaptation of internal signals and habituation of organism behavior. A related memory phenomenon, priming, refers to the exogenous application of a substance to plants to enhance their natural defense responses and their tolerance to future stress presentations [
26]. Because the microfluidic system can reversibly apply and remove chemical stimuli, we assessed for the first time whether calcium responses would adapt or change over multiple chitin stimulations. Indeed, stimulation with five cycles of 5 min chitin and 5 min of rest revealed a gradual response adaptation, with progressively lower calcium response levels, fewer calcium waves, longer wave intervals, longer onset delays, and decreased wave synchronicity. It is not yet clear how these history- and memory-dependent changes occur.
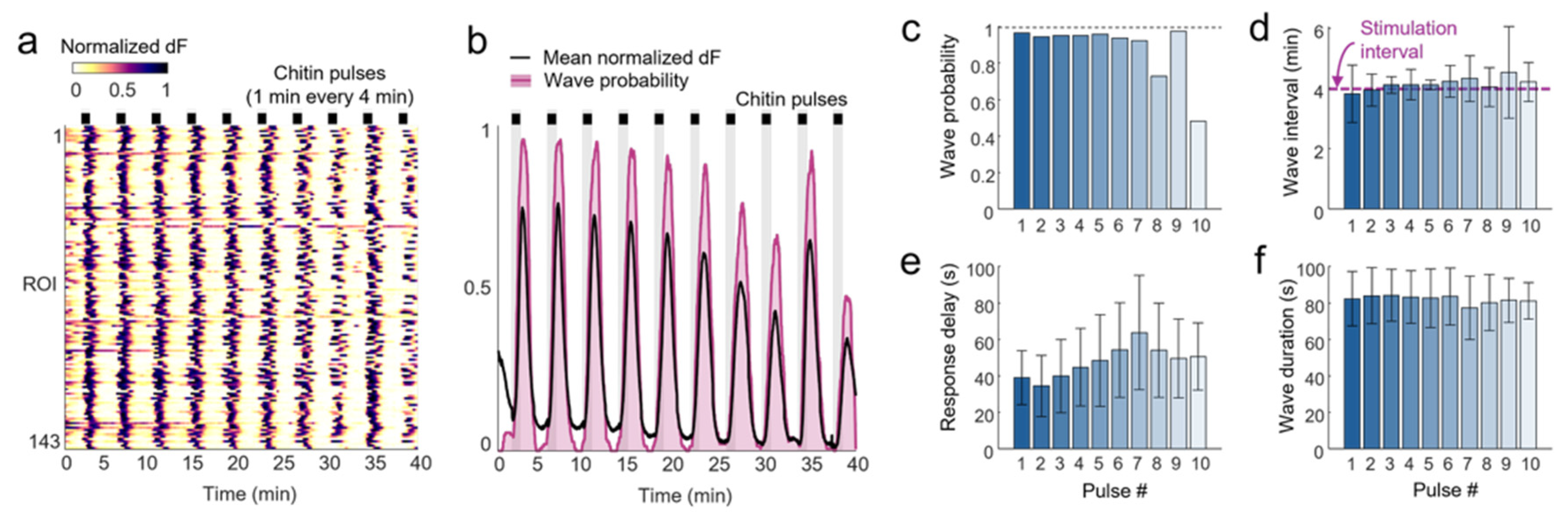
We also demonstrated a novel ability to entrain calcium oscillations to the prescribed frequency of chitin stimulation. As the sudden increase in chitin pulses elicited calcium waves in nearly all ROIs within 1 min, we investigated whether 1 min pulses of chitin could induce singular, synchronized calcium waves across plants. At a stimulation interval of 4 min, we found that nearly all ROIs exhibited single oscillations entrained at the same frequency as chitin stimulation. This external control of calcium wave frequency can be used to decode how wave frequency modulates gene expression.
In this study, all cells of the plant were stimulated equally and simultaneously by a uniform, high concentration of chitin oligosaccharides [
3], representing a strong fungal attack. Beyond microfluidic control of chemical
timing, devices with multiple inlets can control
spatial chemical patterns by exploiting laminar flow physics [
28,
29]. Future experiments could stimulate a single cell within the plant by directing a narrow, micron-scale stream of chitin stimulus, and then trace signal propagation from the target cell across the plant. It will be interesting to observe the relation of calcium activity in adjacent cells when they are not exposed to the same external stimulus, and whether calcium waves propagate across cell boundaries under these conditions. This experimental setup may better resemble the initial encounter of a pathogen at a local point on the plant.
The characteristics of calcium waves, including symmetry and consistency, onset delays following stimulus addition, and inability to halt waves in progress by stimulus removal, all provide hints to the underlying signaling mechanisms. The lack of direct synchronization or calcium propagation across adjacent cells suggests signaling by other molecules. Dynamic mathematical models that produce simulated responses consistent with experimental data will be useful to investigate and identify these signals and to design perturbation experiments to further interrogate calcium signaling [
27]. Together, this toolset should aid in the further study of calcium signaling in plants, including mechanisms of signal propagation, regulation of gene expression, and protection against external challenge.
4. Materials and Methods
4.1. Plant Material and Growth Conditions
All plant lines used in this study were derived from the moss Physcomitrium patens (Gransden strain). Moss lines were cultured in a Percival growth chamber at 25 °C under white light (Fluorescent Light bulbs cool white GE F17-T8 at ~100 µmol/sec/m2) and a 16 h/8 h light-dark cycle for 6-8 days before being used in experiments. Sub-culturing was done with an Omni Tissue Homogenizer (TH) set at half power and using 7 mm hard tissue Omni tip plastic probes. Plants were grown on cellophane disks (AA Packaging Limited, UK) placed on top of 1% agar WPi plant medium (described below) on 10 cm Petri dishes. One plate of one-week-old tissue was ground in 4 ml of water for ~10 sec. Two new plates were inoculated with 1 ml each, and the other two milliliters were sieved with a Falcon disposable cell strainer (70 µm pore size) to select for very small plants. The flow-through was placed onto two plates (1 ml each).
4.2. Solutions
The WPi plant medium was modified from {Ashton and Cove, 1977}: 1 mM MgSO4· MgSO4·7H2O, 1 mM Ca(NO3)2·4H2O, 4 mM KNO3, 89 µM Fe-EDTA, 1.84 mM KH2PO4, 9.9 µM H3BO3, 220 nM CuSO4·5H2O, 1.97 µM MnCl2·4H2O, 230 nM CoCl2·6H2O, 190 nM ZnSO4·7H2O, 168 nM KI, 100 nM NaMoO4·2H2O, pH 5.5. A 10X stock solution of 22 mg/ml chitin was prepared in water and stored at -20 degrees Celsius. The chitin stock was thawed and diluted in the media described above to a concentration of 2.2 mg/ml on the day of each experiment. The buffer solution was filtered through a 0.45 μm PES syringe filter (Genessee Scientific, USA; Cat#: 25-245) when creating the stimulus solution and passively before flowing into the microfluidic device to reduce the likelihood of aggregates entering the imaging area. The likelihood of aggregates increases with the length of time the stimulus solution is kept static. To allow the visualization and quantification of the fluid flow dynamics within the chamber and to verify the timing of stimulation, fluorescein was added to either the stimulus or buffer solutions for all experiments at a concentration of 0.1-1 μg/ml.
4.3. Microfluidic Device Design and Fabrication
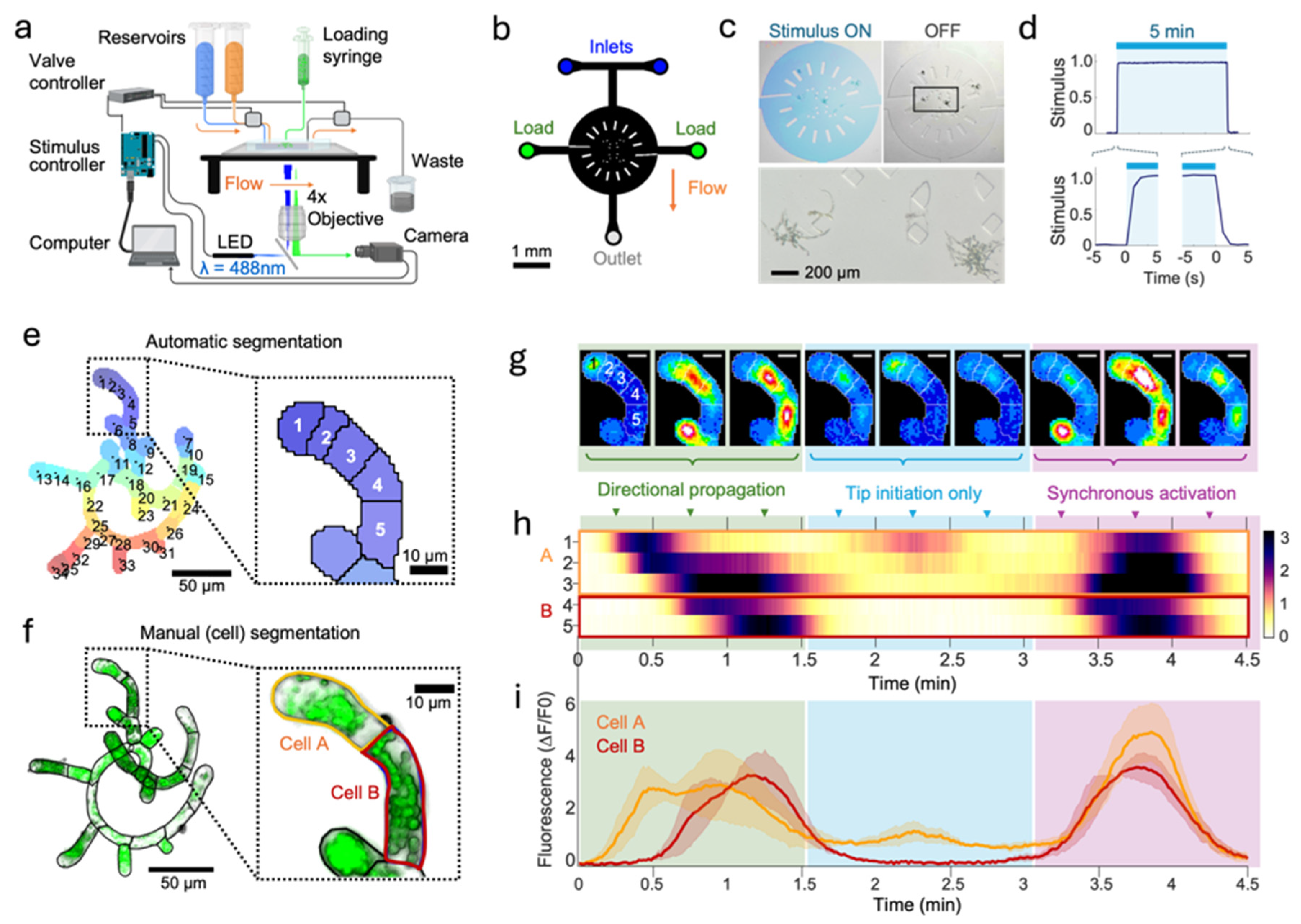
To investigate dynamic plant-fungal interactions, we created a monolayer microfluidic device design capable of rapid, reversible switching between chemical stimuli. A chamber height of 35 or 50 μm was chosen to keep small moss plants withing the 50 μm depth of field of the fluorescent imaging setup. The design features two fluid inlet ports that connect to the imaging area via a T-shaped channel (
Figure 1b) to enable the addition, removal, and spatial control of stimulus solution from the receptive field of the plants. Devices also feature two U-shaped traps inspired by (Sakai et al., 2019) aligned with each lateral loading port to locate plants centrally in the imaging area and provide support. The 3 mm diameter imaging area matched the microscope field of view and the surrounding support posts (100 x 200 μm) prevented collapse when plasma bonding. The device master molds were created by photolithography and soft lithography following methods previously described (Lagoy et. al., 2022). Briefly, a 35 μm or 50 μm layer of SU8 2035 photoresist (Microchem) was applied to a 4-inch wafer (University Wafer) and exposed through a 25,000-dpi photomask (CAD/Art Services) to generate a master mold. Polydimethylsiloxane (PDMS, Sylgard 184, mixed 1:10 by weight) was cast to create ~5mm thick microfluidic devices. Inflow, loading, and outflow ports were punched using a dermal punch (Miltex, 1 mm), then devices were cleaned and plasma bonded (Harrick PDC-32G, 18W, 45 sec) to a 25 x 75 mm glass slide. Assembled arenas were degassed in a vacuum desiccator for at least an hour before loading plant medium through the outlet port. This step ensures removal of air bubbles within the microfluidic channels prior to loading plants.
4.4. Experiment Setup
All solutions were filtered using 0.22 um PES syringe filters on the day of the experiment. Plant medium and chitin stimulus solutions were placed into 60 mL syringe reservoirs and covered with Parafilm. Microfluidic experiments were prepared by assembling fluid reservoirs and tubing as previously described (White et al., 2024; Lagoy et al., 2021). Inline 0.22 um Luer filters were connected below each reservoir and the 3-way Luer stopcock valve. Reservoirs were connected to the microfluidic device using tubing 0.020” Tygon microbore tubing connected to a blunt 23-gauge Luer stub adapter on one end and a ½” long 19-gauge stainless steel tube on the other end. Bubbles were removed from each fluid line before connecting to the degassed microfluidic device and filling it with plant medium (White et al., 2024). For each experiment, plants were randomly selected from the colony and placed in a dish of buffer solution. Plants were drawn into loading tubing with a 3-mL syringe, then gently injected into the arena using the loading ports and allowed to adapt to the microfluidic environment for 30 min to 3 h with continuous medium flow before imaging.
4.5. Automated Timelapse Imaging and Stimulation
Images were acquired using an inverted widefield epifluorescence microscope (Applied Scientific Instruments RAMM) with a 4x/0.28 NA objective (Olympus). Frames were acquired at 1 fps using a Hamamatsu Orca Flash 4.0 sCMOS camera mounted with a 1.0x c-Mount adapter. Binning was set to 2 to enhance the signal-to-noise ratio (SNR) of acquired signals for a spatial resolution of 0.345 pixels/μm (2.9 μm/pixel). A blue LED (Mightex GCS-470-50) excited GCaMP for 10 ms each acquisition frame. A 3-way solenoid valve (Bio-Chem Fluidics, PN: 075P2-S642) actuated by a ValveLink 8.2 (Automate Scientific) directed a stimulus or buffer solution into the microfluidic arena. Image capture, fluorescence illumination, and stimulus delivery were synchronized via digital trigger signals coordinated by an open-source system previously described in (White, et. al., 2023; Lawler, et. al, 2021). This system uses Micro-manager microscopy software (version 1.4.24) in streaming mode to acquire timelapse images and communicate experiment parameters by serial commands to an Arduino Uno microcontroller which coordinates digital trigger pulses (White, 2023).
Timelapse recordings of up to 60 minutes were conducted at room temperature (20-22 °C), beginning with plant medium flow. Chitin stimulation was introduced after 30 mins and persisted for 30 mins (
Figure 2 and
Figure 3), or chitin pulses were repeatedly introduced and removed for different durations and intervals (
Figure 4 and
Figure 5).
4.6. Structural Image Acquisition
After time-lapse recordings, microfluidic devices were moved to a Zeiss inverted microscope with an Axiocam 503 camera to identify individual cell borders. Plants were stained by flowing 0.1 µg/mL Direct Yellow 96 (Solophenyl Flavine 7GF5E) through the device for 30 min to label cell wall components. Brightfield and epifluorescence images were acquired at higher magnification (10x/0.3 NA) in Z-stacks with a 1.3 µm step size. Cell borders were manually outlined in FIJI (Schindelin, 2012) with the segmented line tool and validated between brightfield and stained images. These cell outlines provided the gold standard to evaluate the performance of the cluster generation algorithm and the initial number of ROIs per cell in the clustering algorithm, described below.
4.7. Image Processing Pipeline for Automated ROI Segmentation
4.7.1. Foreground-Background Detection
Regions of distinct calcium signal dynamics were automatically identified using an image processing pipeline depicted in
Figure 6. First, raw TIFF stacks generated from the timelapse acquisition were cropped and processed with a FIJI macro (
binaryMaskGeneration.ijm). The raw image stack was flattened by maximum z-projection and background subtraction was applied using radius values between 25 – 90 pixels depending on plant size. The resulting image was median filtered (radius = 2.5 pixels) to reduce noise and maintain contrast around the edges. To identify plant regions, an automatic threshold using the “
Triangle” method was applied. Non-plant noise was removed by selecting pixel areas at least 200 pixels
2 and circularity larger than 0.9. Each binary mask was validated by manual inspection.
Figure 6.
Automated plant ROI segmentation pipeline and data reduction. (top row) A binary mask is generated indicating plant pixels versus background pixels. The raw image stack (3D; m x n x t; in this example, 114 pixels wide x 158 pixels tall x 3600 frames) is reduced to an m x n binary mask image. (middle row) Calcium fluorescence image stacks are normalized to scale-independent Z-scores for each pixel, which equalize all pixels within a cell that respond with similar temporal dynamics. (bottom row) Data is flattened to a 2D matrix of each plant pixel Z-score over time (here, 5996 plant pixels x 3600 time points). K-means clustering grouped all pixels within 39 predicted clusters based on plant size (~150 pixel2 per ROI). Clusters were reorganized to the plant geometry and filtered for size and connectedness. Filtered ROI numbers typically increase over initial cluster number. For example, a divided cluster is shown separated into four ROIs with noise pixels removed. Fluorescence intensity from the background-subtracted stack is integrated across each of the resulting 45 ROIs in this example, normalized to minimum intensity, yielding a reduced 2D dataset (45 ROIs x 3600 timepoints). .
Figure 6.
Automated plant ROI segmentation pipeline and data reduction. (top row) A binary mask is generated indicating plant pixels versus background pixels. The raw image stack (3D; m x n x t; in this example, 114 pixels wide x 158 pixels tall x 3600 frames) is reduced to an m x n binary mask image. (middle row) Calcium fluorescence image stacks are normalized to scale-independent Z-scores for each pixel, which equalize all pixels within a cell that respond with similar temporal dynamics. (bottom row) Data is flattened to a 2D matrix of each plant pixel Z-score over time (here, 5996 plant pixels x 3600 time points). K-means clustering grouped all pixels within 39 predicted clusters based on plant size (~150 pixel2 per ROI). Clusters were reorganized to the plant geometry and filtered for size and connectedness. Filtered ROI numbers typically increase over initial cluster number. For example, a divided cluster is shown separated into four ROIs with noise pixels removed. Fluorescence intensity from the background-subtracted stack is integrated across each of the resulting 45 ROIs in this example, normalized to minimum intensity, yielding a reduced 2D dataset (45 ROIs x 3600 timepoints). .
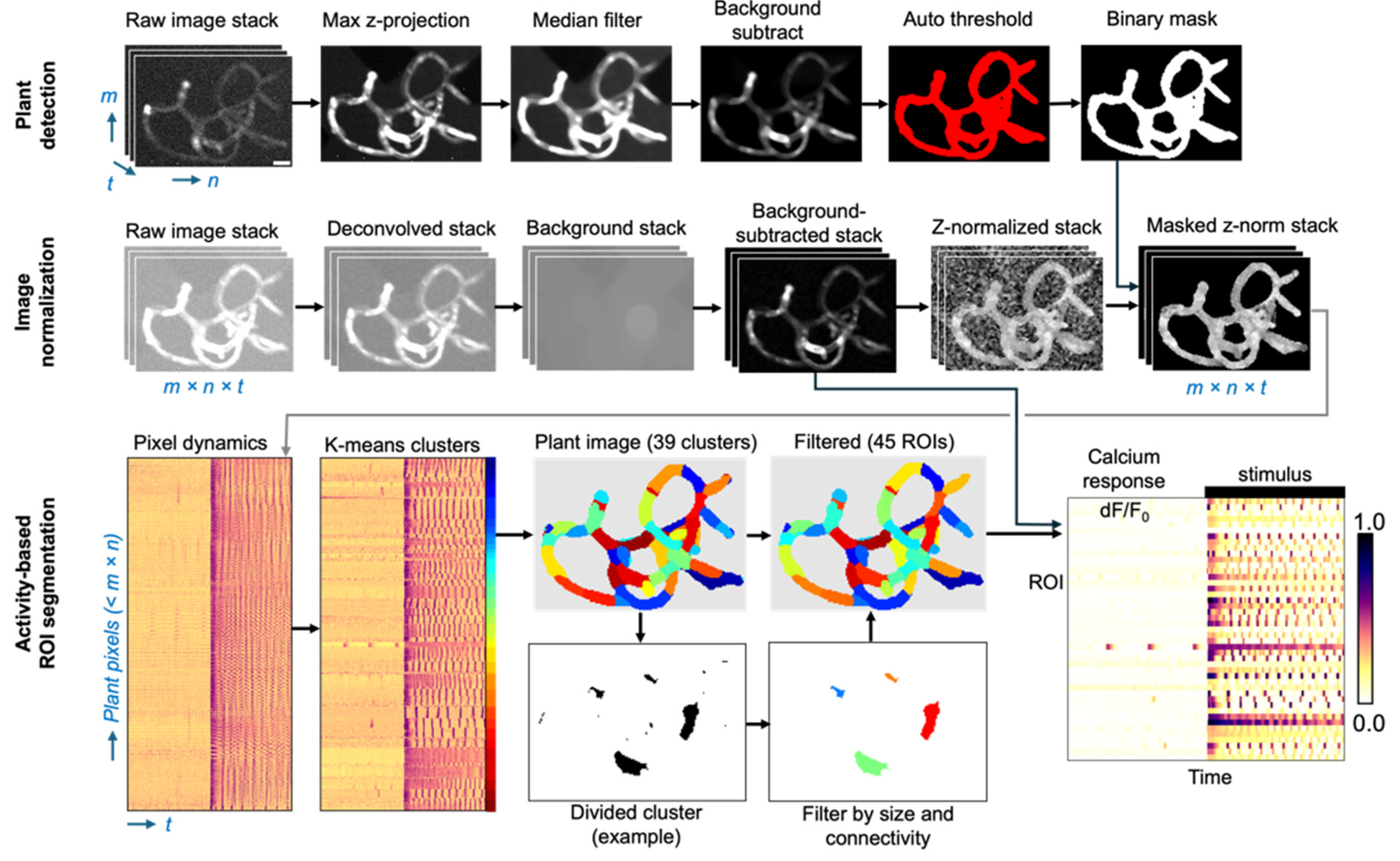
4.7.2. Fluorescence Image Correction and Normalization
To improve signal focus and reduce background fluorescence, each widefield image stack was deconvolved using a theoretical point spread function (PSF) assuming Abbe resolution limits (0.61 lambda / NA = 1.1 µm) using the ‘deconvolucy’ MATLAB command and ‘edgetaper’ to reduce ringing artifacts generated from iterative methods. We set the number of iterations to 10, which consistently generated smoother images and reduced the impact of out-of-focus blur without amplifying noise or introducing artifacts in the images (Ronneberger, 2008). This deconvolution step improved the outcome of the clustering algorithm, better matching ROIs to plant geometry.
GCaMP fluorescence (Fpixel) was obtained by subtracting background fluorescence from each pixel as follows. The previously generated binary masks were morphologically opened (MATLAB “imopen” function, with ‘disk’ structuring element and radius 15 pixels) to remove any structures smaller than 10 μm in diameter from the background images. The background images were then subtracted from the deconvolved image stack before quantification.
Fluorescence intensity is greatest in the center of each cell and lowest at the cell borders. Therefore, data were normalized to Z-scores per pixel before ROI detection, to group pixels by their dynamics irrespective of their time-averaged intensity. For each pixel, the mean intensity over time (
µ) was subtracted from each time point (
x) and divided by the standard deviation over time (
σ), i.e.
. Z-normalized pixels are more uniform across each cell (
Figure 6, middle row left). Next, background pixels were removed using the binary plant mask.
4.7.3. Data Reduction
To automatically identify contiguous pixel “regions of interest” (ROIs) with similar temporal dynamics, we first performed K-means clustering on a 2D matrix representation of the Z-normalized image stack. Each row of the 2D matrix represents a single pixel value over time (
Figure 6, lower left). The clustering algorithm groups pixels into a pre-specified number of clusters given by the initial cell number estimated from manual observation. Where plants overlapped, the cluster number was increased to account for potential summation of fluorescence from both cells. Clustering used the distance metric “cosine” to maximize the similarity of pixels within the same cluster and the difference between separate clusters. We used the ‘
OnlinePhase’ setting and 10 replicates with random initializations to guarantee a global minimization of distance metrics. The time course for each ROI was further reduced into 120 s windows surrounding the peak of ca2+ waves. Peaks were identified using the
‘findpeaks’ MATLAB function with a prominence threshold of 0.35 times the Median Absolute Deviation, chosen to capture both the spontaneous and chitin-driven oscillations. In contrast, standard deviation-based thresholding missed some spontaneous peaks or included false positive detections.
4.8. Statistical Analysis
Data are represented as the mean ± standard deviation (SD) unless otherwise noted, typically median and interquartile range (25 to 75 percentile) for non-normally distributed distributions. Non-responsive ROIs were excluded from categorization and quantification if the maximum increase in fluorescence (ΔF) within 5 minutes of stimulation was less than the baseline (F0) fluorescence, typically lacked waves or oscillations to quantify. In comparison, the typical chitin-induced responses generated initial peaks around 250% ΔF/F0. Sample sizes for each analysis (waves, ROIs, or plants) are reported in the figure legends. Statistics were performed using either a one-way ANOVA with Bonferroni’s correction for multiple comparisons, or two-tailed t-test using the Statistics and Machine Learning Toolbox in MATLAB (v. 2023a).