Submitted:
11 January 2026
Posted:
13 January 2026
You are already at the latest version
Abstract
Enzyme performance parameters, including the turnover number and specificity constant, exhibit remarkable diversity due to biological evolution and natural selection. In some bacterial and human enzymes, catalytic efficiencies approach fundamental physical limits, underscoring the importance of physical constraints on enzymatic function. A deeper understanding of these constraints, particularly in far-from-equilibrium irreversible processes, is therefore essential for rational enzyme engineering. Such constraints are most naturally addressed within the frameworks of nanothermodynamics and stochastic thermodynamics, which remain relatively unfamiliar to much of the molecular biology community. Recent theoretical and experimental advances indicate that classical enzyme kinetic parameters are not independent, but are systematically linked to energetic dissipation. In particular, enzymes appear to occupy a characteristic dissipation plane defined by entropy production, reflecting the coupled influence of thermodynamic principles and evolutionary selection. In this review, we synthesize evidence across diverse enzyme families demonstrating correlated increases in housekeeping dissipation, evolutionary divergence, and enzymatic performance. Together, these findings support dissipation as a physically grounded parameter that connects enzyme kinetics, biological evolution, and nonequilibrium thermodynamics.
Keywords:
evolution
; enzyme efficiency
; turnover number
; evolutionary distances
; partial entropy production
; thermodynamic constraints
; scale‐invariant dissipation plane
1. Introduction
Although there is no universal scientific consensus on origin and a precise definition of life [1,2], modern biology converges on measurable hallmarks such as metabolism, growth, and energy dissipation. Among these, metabolic activity provides a quantifiable physical signature of living systems. Vigorously reproducing bacterial or yeast cells acts as a measurable heat source. Exported dissipation is also associated with the evolutionary maturation of complex heterotrophs, endotherms, and ecological systems [3,4,5]. Thus, the additional heat dissipated into the environment indicates the presence of metabolism in living cells [6].
Interest in the relationship between life and entropy production due to metabolism has increased markedly over the past decade. That viewpoint from physics is well on its way to enter the mainstream thinking about the origin and evolutionary divergence of molecular biology [7,8,9,10,11,12,13]. However, we should keep in mind that there would be no metabolism and molecular biology without highly evolved enzymes that speed up chemical reactions by extraordinary amounts [14,15]. At the molecular level, enzymes act as the primary mediators through which biological systems harvest and channel available free energy into directed, dissipative processes. Understanding how enzymes function in catalysis conventionally emphasizes the roles of structure and electrostatics [16]. Still, dynamics enter through the mobility of functionally important residues, transitions among conformational substates, hydride transfer, and changes in loop movements [17,18,19]. These factors collectively determine enzyme kinetic and thermodynamic parameters. Predicting kinetic parameters is one recent research avenue [20,21,22,23,24]. Finding statistically meaningful connections between enzyme kinetic parameters and the dissipation caused by evolved or designed enzymes is a less well-explored research avenue. This review focuses on the relationship between enzyme kinetic parameters and entropy production during catalysis. We examine theoretical frameworks, experimental evidence, and computational approaches that connect enzymatic rate enhancement with thermodynamic dissipation, with particular emphasis on how evolutionary pressures may have shaped these properties.
High dissipation characterizes far-from-equilibrium processes. While such processes are not unique to living systems, their sustained and regulated manifestation through metabolism is a defining feature of life [25]. When associated with inanimate events, driven irreversible processes direct the physical evolution of the system. The same must hold for living systems. Biological systems therefore operate under universal thermodynamic constraints, with evolutionary selection acting on molecular mechanisms—particularly enzymes—that exploit these constraints. Consequently, physical and biological evolution are intimately linked in shaping enzyme function [26]. Such a line of thinking leaves us with the question about the specific way in which interconnected physical and biological evolution left its imprint on enzymes. In this review, we analyze whether commonly measured enzyme kinetic parameters—such as turnover numbers and catalytic efficiencies—exhibit systematic relationships with entropy production during catalysis. By integrating perspectives from enzymology, nonequilibrium thermodynamics, and evolutionary biology, we aim to assess whether the dissipation landscape represents an emergent design principle of evolved enzymes.
2. Tools Choice from Irreversible Thermodynamics
Let us use the symbol σ for local entropy production. For isothermal conditions, the dissipation function is proportional to σ: where T is the absolute temperature. Entropy production measures the irreversibility of a process, while the dissipation T∙σ measures the energy cost of that irreversibility, i.e., how much free energy is lost as heat. In living cells or organisms, enzymes operate in an open, energy-driven system far from thermodynamic equilibrium. The system can exchange energy (heat, light, chemical work), matter (nutrients, products, ions), and information (signaling, regulation) with its environment. Irreversible thermodynamics is an appropriate tool to study living systems. When we focus on enzymes, the question becomes: should we use modern stochastic thermodynamics for microscopic irreversible processes [7,27] or the older Prigogine-like description [28,29], which is suitable for driven open systems? However, the definition of entropy production differs between the modern and coarse-grained approaches.
Stochastic thermodynamics treats the system and the heat bath as a closed universe. It then defines the entropy production as the sum of the system term and bath entropy change [27]. Initial requirements of no matter flow across the system boundary [7,30] were subsequently relaxed [31,32,33,34,35]. The constant-temperature requirement was also relaxed [32,36,37]. However, the assumption of a closed, energy-conserving “universe” needed to derive stochastic thermodynamics from the microdynamics of a closed total system remained the best way to consistently define entropy production microscopically. Some ingenious exceptions were found on how to go around that obstacle, too. For instance, one can add an information reservoir to the system and a heat bath to get a closed total system [38,39]. Also, for active matter and self-driven systems, the coarse-grained effective baths can be added to restore the ability to calculate the entropy production rates [40,41,42,43]. The “environment” is then effectively open due to the postulated existence of a large thermostat that is not explicitly included.
Over the last two decades, stochastic thermodynamics has been applied to model biomolecules in vitro, such as a motor protein in a well-defined chemical bath (ATP/ADP + buffer) [44] or a single attached enzyme [45,46]. Nevertheless, it’s not the simplest or best framework for modeling a living cell or enzymes coupled to multiple reservoirs (chemical, mechanical, electrical, heat, radiation), which can exchange energy and matter with their surroundings. Those systems are open and far from equilibrium, where energy fluxes through the system maintain order and drive metabolism — exactly the domain of Prigogine’s approach.
The central square of Figure 1 represents an open system. Given multiple inputs and outputs (arrows), one can choose the conditions for the system of interest. For short time periods, we can approximate the homeostasis of the living system as a steady-state condition, in which there is no change in entropy, temperature, or pressure. The radiation field can also be excluded when the proteins of interest do not include light detectors, light converters, or photosystem complexes. That simplifies the measurement of the entropy produced within the system (due to irreversible processes), which is completely exported to maintain the steady-state zero entropy change for the system. Microcalorimetric techniques [47,48,49] are used to achieve that goal.
The decomposition of entropy production σ into the sum of forces Xi and corresponding flows Ji (as depicted in equation (1) and the central square of Figure 1) is the essential relation of irreversible thermodynamics. The bilinear form ΣJiXi is a consequence of the second law of thermodynamics and the local equilibrium assumption [50]. At fixed thermodynamic forces and positive σ(Ji) = ΣJiXi, only a maximum can be the extremum of σ (maximum entropy production principle) [50]. The principle enables the calculation of actual (optimal) thermodynamic flows. In 2017, Martyushev and Celezneff [51] showed that replacing the assumption of local equilibrium with the postulate of scale invariance σ = σ(J) allows one to construct nonequilibrium thermodynamics that not only includes all the results of classical linear nonequilibrium thermodynamics, but also allows one to describe nonlinear systems that are extremely far from equilibrium. Very few exceptions have been claimed regarding the ΣJiXi bilinearity in non-equilibrium thermodynamics [52]. Other authors re-examined these exceptions (see Chapter 4).
Forces and flows are generalized quantities. Enzyme biochemistry considers chemical affinities as forces and corresponding conversions of substrates into products as flows. Equation (1) is the theoretical way to calculate entropy production when we can measure or calculate all flow-force couples relevant to the biochemical pathway of interest. We do not have to worry about how entropy is defined, whether we are close or arbitrarily far from thermodynamic equilibrium, nor whether the relationship between flows and forces is linear or nonlinear. For enzyme catalyzed reactions , where is the affinity for each reaction i at the temperature T. While the dissipation is due to internal irreversible processes (flows) according to Prigogine’s definition, forces can be either internal or external. External forces are treated as boundary conditions.
When averaged, the microscopic definition of entropy production from stochastic thermodynamics also reduces to the equation (1) definition [53,54]. Nevertheless, Prigogine’s definition has been applied to small-system nanothermodynamics in biochemistry and stochastic thermodynamics by Terrel L. Hill and other researchers [32,55,56,57,58,59,60,61,62]. Tanford (1982) [63] offered the critique of Hill’s bookkeeping for free-energy transduction, but modern developments tend to support Hill’s viewpoint [64,65,66].
In Hill’s approach, one can start from measured and estimated microscopic rate constants to calculate the probability of enzyme states as the enzyme cycles among conformations and excited states during the transformation of substrates into products. Our attention is then focused on the functionally essential conformational states of the enzyme of interest, rather than on small organic molecules serving as substrates or products. From steady-state probabilities, all fluxes and forces can be easily calculated using the diagram method (appropriate for simpler kinetic schemes) or computational tools (for more complex schemes). Terrel L. Hill used toy models to illustrate how such calculations can determine the efficiency of free-energy transduction in the steady state. He explicitly stated that light-driven systems are not amenable to such an approach.
3. Entropy Production can Be Decomposed into Productive and Waste Parts
The idea that entropy production can also be decomposed into “productive” (useful, flux-carrying, ligand-transforming, or energy-transducing) and “futile” (waste, thermal, or dissipative slip) parts merits short elaboration because readers may not be familiar with it. Also, some renowned scientists who have made fundamental contributions to nonequilibrium thermodynamics are strongly opposed to focusing on entropy production or dissipation. To cite Professor R. Dean Astumian [67]: “The focus on dissipation is misguided. Dissipation is waste and drives nothing other than heating.” However, classical network thermodynamics and stochastic thermodynamics often examine cycle decomposition of entropy production. The theory can be applied to enzyme-controlled processes. Enzyme catalysis is necessarily cyclical in nature because substrates are altered, but the enzyme itself always returns to its original state. For instance, productive and futile pathways for cycle-associated free-energy transduction and entropy production contributions were studied in detail by Terrel L. Hill [55,56,68,69] and further developed by Jürgen Schnakenberg [70] and other researchers [71,72,73,74,75].
Interestingly, both productive and futile cycles are subject to regulation and feedback [76,77,78,79,80,81,82]. For instance, the productive ATP synthesis/hydrolysis cycle is actively switched, inhibited, or modulated, which directly regulates both output and dissipation [83,84,85]. The rate and dissipation of the productive ATP-generating cycle are controlled by energy demand (ADP, ATP, NADH levels, membrane potential, and pH gradient across membranes) [86,87,88]. The productive pumping cycle of Na⁺/K⁺-ATPase (which moves Na⁺ and K⁺ across the membrane via ATP hydrolysis) is regulated by accessory proteins and phosphorylation, thereby controlling both flux and energetic cost [89,90]. For regulated dissipation in the productive cycle of molecular motor proteins, kinesin and myosin are good examples of how load and gating alter cycle kinetics, partitioning ATP energy between productive motion and futile/regulatory internal transitions [44].
4. Extensions of Terrell Hill’s Theoretical Approach
Meszéna and Westerhoff examined whether photosynthesis can be described as any other biochemical process [91]. They found expressions for the non-zero photon chemical potential and the net rate of absorption. By showing that photosynthetic and photochemical processes can be expressed using a generalized free energy for photons, they enabled a thermodynamically rigorous description of light-driven biochemical cycles. Their expressions for fluxes and forces associated with the radiation field can be incorporated into the bilinear form (1) to calculate the entropy production rate (EPR). When thermal dissipation of the excited state is included, we have the light-driving cycle that enables photosynthesis [92]. Juretić and Županović extended the framework to irreversible thermodynamics with radiation fields for the case of bacterial photosynthesis, treating photons analogously to reactants with an effective chemical potential/affinity [93,94]. As a result, the entropy production retains a bilinear form even when the radiation fields are far from equilibrium. Other authors also increased the scope of nonequilibrium thermodynamics of chemical reaction networks to include incoherent light as a source of free energy by defining a chemical potential of photons under local-equilibrium assumptions, thereby allowing photon fluxes and affinities to be introduced [95]. Landi and Paternostro provided a modern theoretical background for how entropy production expressions can be generalized in quantum and classical systems (including radiation) beyond the classical flux–force linear regime [96].
Our other extension of Hill’s theoretical approach is the possibility to find maximal partial entropy production (MPEP) for a chosen i→j transition [2,93,97,98]. The MPEP conform with the general formulation of the maximum entropy production principle (MEPP) [99,100] that can be formulated as “at each level of description, with preset external constraints, the local relationship between the cause and the response of a complex nonequilibrium system is established in order to maximize the entropy production.” The maximum in a chosen transition arises from a simple trade-off between thermodynamic flux and force when we vary only the forward microscopic rate constant kij. There were no restrictions on network complexity, the nonlinearity of the force-flux response, or the distance from the equilibrium state. Considering the transitions between macromolecular steady states provides a more detailed decomposition of overall entropy production than the cycle-associated decomposition we considered in Chapter 3. Intriguing results are facilitated rate-limiting transitions when optimal kij values are close to the observed values [2,93,97,98,101,102]. Such transitions in productive, free-energy transducing cycles are, as a rule, the latest and slowest recovery steps, involving obligatory proton shuffling or transport between conformations or compartments. The examples we studied in the 2003-2021 period extended physical applications of MEPP [50,103,104,105,106,107,108,109] to enzyme kinetics.
In applying these advances, we always strived to compare theoretical predictions with experimental observations. Our goal was to determine whether the observed kinetic parameters are close to the predicted optimal kinetic parameters for specific biochemical systems. Emerging insights allowed us to postulate the evolution-coupling hypothesis – biological evolution does not act independently from the physical (thermodynamic) evolution [26,110]. Evolution in biology is a broader concept than classical Darwinian evolution [111,112,113,114], but the recognition of universal physical evolution (thermodynamic evolution) is the essential achievement in physics [115] that gained only a sparse foothold in modern biology [116,117]. There is an astonishing opinion difference about the thermodynamic evolution concept among experts from different research fields.
Although the term thermodynamic evolution is not widely used within enzyme evolution research per se, thermodynamic principles play a central role in many physical and natural systems across disciplines. In non-equilibrium thermodynamics, the entropy production rate is a fundamental measure of how systems evolve over time under gradients of temperature, concentration, or chemical potentials, governing processes from chemical reactions to fluid flows and plasma dynamics. For instance, in materials science and crystallization, the rate of entropy production per unit area can govern morphological pattern formation as a system moves away from equilibrium, influencing dendritic growth and surface instabilities [118]. In thermodynamic modeling of complex systems and machine performance, changes in entropy production are directly linked to energy dissipation and conversion efficiency, such as in heat engines or reactive flows studied in fluid mechanics and process engineering [119,120]. Similarly, in astrophysics, the thermodynamic properties of large-scale phenomena such as coronal mass ejections (CMEs) evolve as a function of density, temperature, and pressure as the system propagates outwards from the sun, reflecting changes in internal entropy and energy distribution during expansion [121]. Moreover, recent interdisciplinary work has begun to apply “thermodynamic evolution” concepts to biological systems, proposing that higher entropy production is associated with greater dissipation and may correlate with improved efficiency or complexity in biological catalysts such as enzymes. In these kinetic models, more specialized enzymes tend to dissipate more free energy, suggesting a thermodynamically driven component to their evolutionary trajectory [26]. Thus, while researchers focused on enzyme evolution or molecular biology may not routinely frame their work in terms of thermodynamic evolution, non-equilibrium thermodynamics and entropy production provide a unifying language that connects physical pattern formation, energy dissipation, and the behavior of complex systems across scales — from crystals and plasmas to evolving biological molecules. There is a firm feedback between biological and thermodynamic evolution. Despite being constrained by physical laws, biological evolution can accelerate or slow down thermodynamic evolution [2,26].
5. Examples of Appplications Combining Nanothermodynamis and Entropy Production Principles in Bioenergetics
Firstly, we examined simplified photosynthetic models of bacterial photosynthesis and bacteriorhodopsin regarding the initial irreversible photosynthetic steps [93]. In both models, free energy transduction in the productive cycle converts photon-free energy into the proton-motive force. Experimentally determined kinetic constants were published by van Rotterdam in his PhD thesis [122] for the initial photosynthetic steps of the purple photosynthetic Eubacterium Rhodobacter sphaeroides. For our work, we simplified his kinetic scheme to the three-state and five-state model by retaining only the essential kinetic steps of the an-oxygenic chlorophyll-based photosynthetic cycle. In an iterative self-consistent procedure, we simultaneously sought the maximum partial entropy production in two final transitions from the productive cycle leading to active proton transport and the recovery of the bacteriochlorophyll ground state [2,93,110].
At that time, we did not have proof that the maximum in the partial edge-wise entropy production can always be found. Nevertheless, we claimed that we applied the maximum entropy production principle (MEPP). Despite the model’s simplicity and theoretical shortcomings, several interesting results followed. The dominant contribution to total entropy production of 80 to 90% is the sum of partial entropy productions in the productive (non-slip) pathway. For different choices of light intensity and the radiationless rate constant to the ground state, we obtained optimal quantum yields ranging from 0.93 to 0.98, in accord with experiments. The optimized model reproduced the backpressure regulation of energy transduction, namely that proton pumping slows with increasing transmembrane potential [123]. The microscopic recovery rate constant is rate-limiting. Its predicted optimal value, krecovery = 10.1 s-1, was in good agreement with the observed value, krecovery = 8.6 s-1 [122]. An optimal value of free-energy transduction efficiency in the range of 17-19% and a close-to-80% loss of photon input power as heat also agrees with experimental observations [2].
The other example we studied is the bacteriorhodopsin from Halobacterium salinarium. It is the simplest and smallest light-activated proton pump that nature designed [124]. The MPEP application yielded an optimal recovery kij that was similar to the observed ground-state restoration rate constant. Also, the predicted hierarchy of characteristic time values was in rough agreement with the observed values. Interestingly, these results were robust with respect to the kinetic scheme we used, the number of functional states, and the presence or absence of a slip transition [2,93,94,125]. Relatively low thermodynamic efficiency in converting photon-free energy into the electrochemical proton gradient (10-16%) begs the question of why nature disregards the majority of available energy. We can assume that fundamental physical and functional (biological) characteristics of energy conversion have become tightly regulated and optimized during biological evolution. If this is so, we expect that MPEP optimization (with all constraints taken into account) cannot change the already optimal values of these parameters inferred from measurements by much. Computational experiments confirmed these expectations [98,102,125]. Biological evolution used almost all available options to accelerate enzyme evolution toward more efficient dissipation of free energy gradients, often with a coincidental increase in catalytic performance as a positive side effect. Indeed, only small increases in entropy production, net proton flux, and energy conversion efficiency can be achieved after MPEP optimization in the last (recovery) step of the bacteriorhodopsin photocycle [2,98].
The distribution of microscopic kinetic constant values over many orders of magnitude is the well-known and frequently observed feature of light-activated biological cycles. It starts with initial picosecond transitions and proceeds to the final (recovery) millisecond step [126,127,128]. That result follows naturally from our optimization method for maximal partial entropy productions [2,98], but, as far as we can tell, it has not been reproduced using the same or different extremal principles. Nothing similar could be obtained after the application of Prigogine’s minimum entropy production theorem [28].
Dhanuka et al. [129] developed an ambitious framework of ecosystems as adaptive living circuits. Local dissipation along the chosen circuit edge can be tuned to reach a near-maximal value when subjected to sufficient drive. Their “local dissipation” definition needs semantic clarification, as other authors have used different names for the same mathematical entity. We settled on the phrase “partial entropy production” [2,26,97,130] for the equivalent definition from the thermodynamic network theory, but not before using other phrases, such as “entropy production in each transition between neighboring states” [93], “free-energy dissipation associated with corresponding transition” [110], and “transitional entropy production” [98,125]. The mathematical equivalence is apparent when the ij edge energy dissipation [129]:
is compared to the partial dissipation for the net mean transition flux i→j [55,98]:
where kB is the Boltzmann constant, T is temperature, pi are state probabilities, kij are microscopic kinetic constants, and the number of moles n is taken to be one. The physical and biological context is very different. In our case, we do not deal with living species as edges of the circuits, but with macromolecules (enzymes), their functionally important conformational states, and their interstate transitions, which may involve smaller molecules (substrates, products) with which they interact. The decomposition of total entropy production (or dissipation) into positive partial contributions is a generally accepted procedure in irreversible thermodynamics and bioenergetics [2].
Still, two clarifications would help with the phrase „partial entropy production”. Firstly, the term “partial entropy production” is used in recent literature to indicate incomplete knowledge [131] rather than a desired positive additive partition of a fully known total. Secondly, recent decomposition proposals of entropy production for general nonlinear dynamics distinguish the housekeeping cycling mode (due to external driving) and the excess part originating from the relaxation mode [132]. The housekeeping positive contribution is the entropy production part which can be considered as productive, as we discussed in Chapter 3, but with different mathematical and physical meanings from those we used in our biochemical network models. However, Yoshimura et al. [132] also provided the mathematical decomposition of housekeeping contribution into the sum of positive JeXe products, where Xe is the thermodynamic force on edge e, while Je is the current on edge e. In a steady-state situation, when each node in a biochemical kinetic scheme represents the conformation of an enzyme or an enzyme complexed with small molecules, and each edge represents a bidirectional transition between conformations, this is equivalent to the EPR decomposition used by Terrel L. Hill [55] (see his equation 4.28) and us (our publications [2,26,93,97,98,101,102,125,130] in the 2003-2025 period). Thus, the essential caveat about our usage of the “partial entropy production” phrase is its connection to the edge-wise or transition-wise contribution to the known total EPR.
For instance, when the transition-state parameter κ (defining the angular position for the transition state) is varied in modeling F1-ATPase, a single joint maximum is found for the information entropy and the entropy production in the transition responsible for ATP synthesis or hydrolysis [97]. In the intact F0F1-ATP-synthase, ATP synthesis or hydrolysis is coupled to transmembrane proton translocation. After MPEP application, the optimal κoptimal = 0.602 for chloroplast’s ATPase turned out to be very close to observed κobserved = 0.598 [133]. The transition state theory and our modeling reproduced the sigmoidal shape of the ATP synthesis flux observed by Pänke and Rumberg [133]. Sigmoidal enzyme kinetics is well known as a sign of cooperativity and metabolic regulation [134,135,136,137]. The inflection point of a sigmoidal reaction rate curve is the point of maximal sensitivity to variable chemical potential when the ATP synthesis rate is maximally sensitive to the electrochemical proton gradient. We found that the inflection point is also a) the point of maximal information entropy, b) the point of maximal partial entropy production, and c) the point of the best far-from-equilibrium linearity in the force-flux relationship. Thus, we claimed that the optimal metabolic control is achieved for maximal partial entropy production [97]. Subsequently, metabolic regulation was linked to various versions of the maximal entropy production principle [94,101,138,139,140,141,142,143,144,145,146,147].
In conclusion, we addressed in this chapter the fundamental question linking bioenergetics and nonequilibrium physics: can the evolved dissipative steps that facilitate biomolecular function be identified by their contribution to the housekeeping entropy production? We have found that dominant entropy-production transitions are coupled to nonequilibrium conformational switches that enable ATP synthesis or the active transport of protons. Such localized or edge-wise transitions can be optimized by using the MPEP theorem [98]. Optimized parameters are often close to the observed values in accord with MEPP [100].
We have also recently examined simpler one-cycle enzymes that maintain nonequilibrium concentrations of biologically important small molecules without free-energy transduction [26,130,148]. The question we asked was whether there is a statistically significant relationship between biochemical (kinetic constants) and physical performance parameters (housekeeping entropy production). It turned out that enzyme kinetic parameters, such as the turnover number kcat and the Michaelis-Menten constant KM, are not independent of each other or of the total entropy produced. The specificity constant, as the ratio of these two parameters (kcat/KM), is proportional to the dissipated energy in the log-log frame. It is worthwhile to note that determining kcat and KM is often regarded as a complete characterization of any enzyme-catalyzed reaction [49]. However, the connection between biochemical and physical parameters is less well known. It survived the eons of biological evolution and exhibits a clear trend: more evolved members of the same enzyme family are associated with greater specificity toward their substrate and higher entropy production [2,130]. By examining a large enough set of well-known enzyme families, we discovered yet another general rule – specialized enzymes are considerably better dissipators from bi-functional and generalist enzymes, thus confirming Jensen’s hypothesis (that specialized enzymes emerged from generalized enzymes during biological evolution) [149,150] in an unexpected way [130].
6. The Dissipation-Scaling Plane
A sufficiently large dataset of enzymes with known microscopic rate constants enables not only the calculation of kcat, KM, and overall entropy production for individual enzymes, but also statistically meaningful analyses of the interdependence between biochemical and physical parameters. In two recent studies [26,130], we restricted the dataset to enzymes operating through a single productive catalytic cycle, characterized by a single steady-state flux passing through two, three, or four enzyme conformational states, with or without bound ligands. All analyses were performed under steady-state conditions.
Figure 2 presents a three-dimensional log–log–log representation of these results for 75 enzymes. Although this visualization does not introduce new data, it provides an alternative perspective on our recent two-dimensional analyses [130]. The 3D plot highlights the existence of a dissipation-scaling plane that accommodates enzymes spanning the full spectrum of catalytic performance, from sluggish enzymes (yellow and green points) to those approaching catalytic perfection (dark pink points). Notably, 20 of the 75 reactions are catalyzed by artificially mutated enzymes. Their inclusion enables partial disentanglement of purely physical constraints imposed by protein structure from biologically evolved structure–function relationships. Remarkably, engineered enzymes populate the same dissipation plane as their naturally evolved counterparts, with nearly identical goodness-of-fit values and statistically indistinguishable scaling slopes [130]. This observation demonstrates that the dissipation–specificity scaling relationship persists even in the absence of evolutionary history, underscoring its physical origin and generality. Figure 2 further reveals that enzymes with near-optimal catalytic performance are associated with exceptionally high dissipation. Among these are three bacterial β-lactamases with corrected Ambler sequences [98,151,152,153]: Staphylococcus aureus PC1, Escherichia coli RTEM, and Bacillus cereus Lac1. The red connecting lines illustrate their evolutionary relationship, which is accompanied by concomitant increases in both the catalytic constant kcat and overall dissipation.
While the log–log–log representation is effective for identifying scaling trends, it compresses relative distances and obscures evolutionary relationships within enzyme families. To address this limitation, Figure 3a plots evolutionary distances on the y-axis in a conventional three-dimensional space, with kcat on the x-axis and dissipation on the z-axis. For β-lactamases, the ranking PC1 < RTEM < Lac1 reproduces previous results [98], not only in terms of evolutionary distance but also for kcat and dissipation. Thus, more evolutionarily derived β-lactamases exhibit improved catalytic performance at the cost of increased housekeeping dissipation. A similar trend is observed for human cyclophilins, where the evolutionary ranking CypA < CypB < CypC coincides with increasing kcat values and dissipation, in agreement with our recent findings (Figure 6 in [130]).
Figure 3b further clarifies these relationships by replacing kcat with enzyme efficiency kcat/KM on the x-axis. Together, Figure 3a and Figure 3b corroborate our phylogenetic analyses, showing that increasing evolutionary distance from the putative ancestral sequence is accompanied by systematic increases in dissipation as well as in the kinetic parameters kcat and kcat/KM [130]. Figure 3b also illustrates a rare but informative case in which engineered glutamate racemase variants—single-site (K29A, R214A, P99A, Y221A, K106A, R25A, Q86A) and double-site (R214A/K106A) mutants—exhibit higher turnover numbers and greater dissipation than the wild-type enzyme [26,130,154]. In the log–log–log representation of Figure 2, all glutamate racemase variants collapse onto an effectively single point on the dissipation-scaling plane. Figure 2 and Figure 3 together emphasize the dominance of physical constraints over evolutionary fine-tuning in determining scaling behavior.
Across all enzymes examined, encompassing four of the seven major EC classes and all three life domains (Bacteria, Archaea, and Eukarya, with 18 from Homo sapiens), dissipation exhibits a highly statistically significant correlation with both kcat and kcat/KM [130]. Generalist and specialized enzymes occupy the same dissipation plane, despite specialized enzymes displaying a median dissipation approximately 350-fold higher (Figure 4 in [130]). Even enzymes differing by nearly ten orders of magnitude in net forward flux—from carbonic anhydrase II and ketosteroid isomerase to glucose isomerase and 2-hydroxyisobutyryl-CoA mutase—remain constrained to this same plane. These observations demonstrate a synergy between physical (thermodynamic) constraints and biological evolution, which together define a dissipation landscape along which enzymes can evolve toward higher or lower performance. While biological context determines the evolutionary trajectory along the plane, physical laws restrict all enzymes—including engineered variants—to remain within it.
When expressed as a power law in the log–log representation of dissipation versus enzyme efficiency, the observed slope of approximately 0.72 implies that dissipation (or entropy production under isothermal conditions) scales as (kcat/KM)0.72. Intriguingly, Kleiber’s law [155] falls within the confidence interval of this slope, raising the question of whether sublinear scaling in enzyme catalysis may share a common physical origin with metabolic scaling at the organismal level, as discussed in [130]. Within the same confidence interval, kcat scales nearly linearly with dissipation (∼(kcat)0.97 ), as expected if the generalized flux–force product JX approximates the turnover number. However, this approximation holds only when backward fluxes are negligible. For the majority of the 75 enzymes analyzed, backward fluxes are significant, with ratios of kcat/(J = J(+) - J(-)) exceeding 2.0 for 43 enzymes and reaching values above 100 for certain mutants, such as the D-psicose 3-epimerase R215K variant.
7. Conclusions, Limitations, and Future Directions
Dissipation constraints are shared between physics, which sets the upper and lower bounds, and biology, which regulates dissipation within these bounds. The total dissipation spans many orders of magnitude across different enzymes, as do turnover numbers and catalytic activities. In this review, we presented evidence that physical and kinetic enzyme parameters are not independent. All are required for a meaningful characterization of enzyme functionality. This interdependence becomes apparent only when a complete set of microscopic kinetic constants is collected for a large number of enzymes, allowing the dissipation landscape—manifested as a dissipation-scaling plane—to emerge.
The biological context determines how enzymes from the same family evolve across this dissipation plane, whereas physical laws restrict all enzymes, including engineered variants, to remain within it. Physical (thermodynamic) evolution, long recognized in physics, is therefore not absent from biology. On the contrary, physical and biological evolution appear to be tightly coupled and often act synergistically. A defining capability of living systems is their ability to accelerate or decelerate physical evolution, as reflected by increases or decreases in enzyme dissipation along the dissipation plane. For example, evolutionary pressure toward more efficient dissipation of free-energy gradients can, as a positive side effect, enhance catalytic performance.
Several limitations currently restrict the universality of the proposed scale-invariant performance–dissipation relationships. Integral membrane proteins essential for bioenergetics [2] and oxidoreductases from EC class 1, the most populous enzyme class, are not yet represented in the enzyme datasets used to construct the dissipation-scaling plane. Complex kinetic schemes, the difficulty of obtaining microscopic kinetic constants, and a historical lack of interest in comprehensive kinetic characterization further limit dataset completeness. These challenges represent clear targets for future work aimed at expanding the bioinformatic foundation required for similar analyses. Moreover, defining appropriate in vivo substrate and product concentrations under homeostatic steady-state conditions remains non-trivial. It may be fundamentally incompatible with the requirement that all reactions proceed in the forward direction under near-physiological driving forces.
A more comprehensive dataset, combined with deeper integration of stochastic thermodynamics, will be required to assess the full generality of these findings. Notably, an alternative method for calculating total entropy production for a single enzyme (β-galactosidase) [161] yielded results consistent with both the present approach and our recent publication [148]. That study reached the same conclusion: increased enzyme efficiency is associated with higher total dissipation. Whether the correlated increases in evolutionary distance, catalytic performance, and housekeeping dissipation observed here extend broadly across enzyme families and throughout the history of life remains an open question. Figure 2 and Figure 3 provide only two representative examples—bacterial β-lactamases and human cyclophilins.
Nevertheless, recent advances in far-from-equilibrium thermodynamics applied to biological catalysis suggest that the dissipation-scaling plane may represent a generic organizing principle for enzyme evolution and design. Future research may either falsify or confirm this principle and, critically, provide mechanistic explanations for the apparent emergence of order from diversity in enzyme kinetic and thermodynamic properties. One plausible mechanistic basis for dissipation–performance relationships is the directional motion of particles within the enzyme nanoworld triggered by substrate capture. Although thermodynamics alone cannot identify the nature of these particles, concerted proton transfer or proton shuttling through hydrogen-bonded networks frequently provides a compelling explanation for directional flux. Optimization of proton-transfer rate-limiting steps using the maximum entropy production principle (MPEP) is one example of the many extremal principles proposed in biology, described in this review in the context of enzyme kinetics. To our knowledge, no authoritative comparative evaluation of such tenets exists in enzymology or, more broadly, in the life sciences—a gap that remains open for future investigation.
In physics, order can emerge when light interacts with chaotic Brownian systems [162]. Similarly, the conversion of ionic gradients into electrochemical energy through selective transport of Na⁺, K⁺, H⁺, and Cl⁻ may have contributed to the formation of the first cells [163,164], representing another example of complex order arising from external forcing. Research at the interface of nonequilibrium physics and biology is therefore entering a particularly promising phase, with important implications for enzyme engineering, bioenergetics, evolutionary theory, and fundamental biophysics [165,166,167,168,169,170].
Author Contributions
D.J. wrote a manuscript and generated figures. B.B.M. verified the ranking order reproducibility of evolutionary distances for β-lactamses and participated in the review and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
New evolutionary distances for β-lactamases and the methods used to derive them are briefly described in the legend to Figure 3a. These calculations mainly served to verify our older results. Otherwise, no new data were created or analyzed in this study. Data sharing does not apply to this article.
Acknowledgments
One of us (D.J.) is pleased to acknowledge valuable insights from letters exchanged with the Professor Leonid Martyushev. During the preparation of this manuscript, the authors used ChatGPT (OpenAI) for language refinement and for assistance in the initial drafting of figures. All content was critically reviewed, edited, and validated by the authors, who take full responsibility for the final version of the manuscript.
References
- England: J. Every life is on fire: How Thermodynamics explains the origins of living things. Hachette UK, 2020.
- Juretić, D. Bioenergetics: A Bridge Across Life and Universe; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2021. ISBN: 978-0-8153-8838-8. [CrossRef]
- 3. von Stockar, U.; Marison, I.; Janssen, M.; Patiño, R. Biothermodynamics of live cells: A tool for biotechnology and biochemical engineering. J. Non-Equilib. Thermodyn. 2010, 35, 415–475. [CrossRef]
- Nielsen, S.N.; Müller, F.; Marques, J.C.; Bastianoni, S.; Jørgensen, S.E. Thermodynamics in Ecology-An Introductory Review. Entropy (Basel) 2020, 22(8), 820. [CrossRef]
- Glazier, D.S. Power and Efficiency in Living Systems. Sci. 2024, 6(2), 28; [CrossRef]
- Ulanowicz, R.E.; Hannon, B.M. Life and the production of entropy. Proc. R. Soc. Lond. B. 1987, 232, 181–192. [CrossRef]
- Seifert, U. Stochastic thermodynamics, fluctuation theorems and molecular machines. Rep. Prog. Phys. 2012, 75(12), 126001. [CrossRef]
- England, J.L. Statistical physics of self-replication. J. Chem. Phys. 2013, 139, 121923. [CrossRef]
- England, J.L. Dissipative adaptation in driven self-assembly. Nat. Nanotechnol. 2015, 10, 919–923. [CrossRef]
- Riedel, C.; Gabizon, R.; Wilson, C.A.M.; Hamadani, K.; Tsekouras, K.; Marqusee, S.; Pressé, S.; Bustamante, C. The heat releasedduring catalytic turnover enhances the diffusion of an enzyme. Nature 2015, 517, 227–230. [CrossRef]
- Gnesotto, F.S.; Mura, F.; Gladrow, J.; Broedersz, C.P. Broken detailed balance and non-equilibrium dynamics in living systems: a review. Rep. Prog. Phys. 2018, 81(6), 066601. [CrossRef]
- Wagoner, J.A.; Dill, K.A. Opposing pressures of speed and efficiency guide the evolution of molecular machines. Mol. Biol. Evol. 2019, 36, 2813–2822. [CrossRef]
- Lineweaver, C.H. Beyond the Second Law: Darwinian Evolution as a Tendency for Entropy Production to Increase. Entropy (Basel) 2025, 27(8), 850. [CrossRef]
- Wolfenden, R. Benchmark Reaction Rates, the Stability of Biological Molecules in Water, and the Evolution of Catalytic Power in Enzymes. Annu. Rev. Biochem. 2011, 80, 645–667. 10.1146/annurev-biochem-060409-093051.
- Edwards, D.R.; Lohman, D.C.; Wolfenden, R. Catalytic proficiency: the extreme case of S-O cleaving sulfatases. J. Am. Chem. Soc. 2012, 134(1), 525-531. [CrossRef]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [CrossRef]
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116(11), 6503-6515. [CrossRef]
- Agarwal, P.K.; Bernard, D.N.; Bafna, K.; Doucet, D. Enzyme dynamics: Looking beyond a single structure. ChemCatChem 2020, 12(19), 4704-4720. [CrossRef]
- Klinman, J.P.; Kohen, A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 2013, 82, 471-496. [CrossRef]
- Kroll, A.; Engqvist, M.K.M.; Heckmann, D.; Lercher, M.J. Deep learning allows genome-scale prediction of Michaelis constants from structural features. PLoS Biol. 2021,19, e3001402. [CrossRef]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep Learning-Based kcat Prediction Enables Improved Enzyme-Constrained Model Reconstruction. Nat. Catal. 2022, 5(8), 662−672. [CrossRef]
- Kroll, A.; Rousset, Y.; Hu, X.-P.; Liebrand, N.A.; Lercher, M.J. Turnover number predictions for kinetically uncharacterized enzymes using machine and deep learning. Nat. Commun. 2023,14, 4139. [CrossRef]
- Yu, H.; Deng, H.; He, J.; Keasling, J.D.; Luo, X. UniKP: a unified framework for the prediction of enzyme kinetic parameters. Nat. Commun. 2023,14, 8211. [CrossRef]
- Boorla, V.S.; Maranas, C.D. CatPred: a comprehensive framework for deep learning in vitro enzyme kinetic parameters. Nat. Commun. 2025, 16(1), 2072. [CrossRef]
- Annila, A.; Baverstock, K. Genes without prominence: a reappraisal of the foundations of biology. J. R. Soc. Interface, 2014, 11, 20131017. [CrossRef]
- Juretić, D. Exploring the evolution-coupling hypothesis: do enzymes’ performance gains correlate with increased dissipation? Entropy 2025, 27, 365. [CrossRef]
- Crooks, G.E. Entropy production fluctuation theorem and the nonequilibrium work relation for free energy differences. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics 1999, 60(3), 2721-2726. [CrossRef]
- Prigogine, I. Introduction to Thermodynamics of Irreversible Processes; Wiley: New York, NY, USA, 1967.
- Kondepudi, D.; Prigogine, I. Modern Thermodynamics: From Heat Engines to Dissipative Structures; Johh Willey & Sons 2015, 2nd ed.: Chichester, UK. ISBN 978-1-118-371-81-7.
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997, 78, 2690-2693. 10.1103/PhysRevLett.78.2690.
- Seifert, U. Entropy production along a stochastic trajectory and an integral fluctuation theorem. Phys. Rev. Lett. 2005, 95(4), 040602. [CrossRef]
- Esposito, M.; Van den Broeck, C. Three detailed fluctuation theorems. Phys. Rev. Lett. 2010, 104(9), 090601. [CrossRef]
- Polettini, M.; Bulnes-Cuetara, G.; Esposito, M. Conservation laws and symmetries in stochastic thermodynamics. Phys. Rev. E, 2016, 94(5), 052117. [CrossRef]
- Horowitz, J.M.; Esposito, M. Work producing reservoirs: Stochastic thermodynamics with generalized Gibbs ensembles. Phys. Rev. E, 2016, 94, 02 0102. [CrossRef]
- Horowitz, J.M.; Gingrich, T.R. Thermodynamic uncertainty relations constrain non-equilibrium fluctuations. Nat. Phys., 2020, 16(1), 15–20. [CrossRef]
- Celani, A.; Bo, S.; Eichhorn, R.; Aurell, E. Anomalous thermodynamics at the microscale. Phys. Rev. Lett. 2012, 109(26), 260603. [CrossRef]
- Falasco, G.; Esposito, M. Local detailed balance across scales: from diffusions to jump processes and beyond. Phys. Rev. E, 2017, 95(5), 052142. [CrossRef]
- Sagawa, T.; Ueda, M. Generalized Jarzynski Equality under Nonequilibrium Feedback Control. Phys. Rev. Lett., 2010, 104 (9), 090602. [CrossRef]
- Horowitz, J.M.; Esposito, M. Thermodynamics with Continuous Information Flow. Physical Review X, 2014, 4, 031015. [CrossRef]
- Fodor, É.; Nardini, C.; Cates, M.E.; Tailleur, J.; Visco, P.; van Wijland, F. How far from equilibrium is active matter? Phys. Rev. Lett. 2016, 117(3), 038103. [CrossRef]
- Speck, T. Stochastic thermodynamics for active matter. EPL (Europhysics Letters), 2016, 114(3), 30006. [CrossRef]
- Mandal, D.; Klymko, K.; DeWeese, M.R. Entropy Production and Fluctuation Theorems for Active Matter. Phys. Rev. Lett. 2017, PRL 119, 258001. [CrossRef]
- Seifert, U. From stochastic thermodynamics to thermodynamic inference. Annu. Rev. Condens. Matter Phys., 2019, 10, 171–192. [CrossRef]
- Takaki, R.; Mugnai, M.L.; Thirumalai, D. Information flow, gating, and energetics in dimeric molecular motors. Proc. Natl. Acad. Sci. USA 2022, 119(46), e2208083119. [CrossRef]
- English, B.P.; Min, W.; van Oijen, A.M.; Lee, K.T.; Luo, G.; Sun, H.; Cherayil, B.J.; Kou, S.C.; Xie, X.S. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat. Chem. Biol. 2006, 2(2), 87-94. [CrossRef]
- Singh, D.; Punia, B.; Chaudhury S. Theoretical Tools to Quantify Stochastic Fluctuations in Single-Molecule Catalysis by Enzymes and Nanoparticles. ACS Omega 2022, 7(51), 47587-47600. [CrossRef]
- Wang Y.; Wang G.; Moitessier N.; Mittermaier, A.K. Enzyme Kinetics by Isothermal Titration Calorimetry: Allostery, Inhibition, and Dynamics. Front. Mol. Biosci. 2020, 7, 583826. [CrossRef]
- Falconer, R.J.; Schuur, B.; Mittermaier, A.K. Applications of isothermal titration calorimetry in pure and applied research from 2016 to 2020. J. Mol. Recognit. 2021, 34(10), e2901. [CrossRef]
- Mazzei, L.; Ranieri, S.; Silvestri, D.; Greene-Cramer, R.; Cioffi, C.; Montelione, G.T.; Ciurli, S. An isothermal calorimetry assay for determining steady state kinetic and Ensitrelvir inhibition parameters for SARS-CoV-2 3CL-protease. Sci. Rep. 2024, 14(1), 32175. [CrossRef]
- Martyushev, L.M.; Seleznev, V.D. Maximum entropy production principle in physics, chemistry and biology. Phys. Rep. 2006, 426, 1–45. [CrossRef]
- Martyushev, L.M.; Celezneff, V. Nonequilibrium Thermodynamics and Scale Invariance. Entropy 2017, 19, 126;. [CrossRef]
- Essex, C. Radiation and the violation of bilinearity in the thermodynamics. Planet. Space. Sci. 1984, 32, 1035–1043. [CrossRef]
- Tomé, T.; de Oliveira, M.J. Stochastic thermodynamics and entropy production of chemical reaction systems. J. Chem. Phys. 2018, 148, 224104 .
- Tomé, T.; de Oliveira, M.J. Irreversible thermodynamics and Glansdorff-Prigogine principle derived from stochastic thermodynamics. J. Stat. Mech. 2025, 2025(6), 063202.. [CrossRef]
- Hill, T.L. Free Energy Transduction in Biology: The Steady State Kinetic and Thermodynamic Formalism. Academic Press: New York, NY, USA, 1977. [CrossRef]
- Hill, T.L.; Eisenberg E. Can free energy transduction be localized at some crucial part of the enzymatic cycle? Q. Rev. Biophys. 1981, 14(4), 463-511. [CrossRef]
- Hill, T.L. Thermodynamics of Small Systems; Dover: New York, 1994.
- Hill, T.L. Perspective: Nanothermodynamics. Nano Letters, 2001a, 1(3), 111-112.
- Hill, T.L. A Different Approach to Nanothermodynamics. Nano Letters, 2001b, 1(5), 273-275.
- Hill, T.L.; Chamberlin, R.V. Fluctuations in Energy in Completely Open Small Systems. Nano Letters, 2002, 2(6), 609-613.
- Andrieux, D.; Gaspard, P. A fluctuation theorem for currents and nonlinear response coefficients. J. Stat. Mech. 2007, P02006. [CrossRef]
- Qian, H. Hill’s small systems nanothermodynamics: a simple macromolecular partition problem with a statistical perspective. J. Biol. Phys. 2012, 38(2), 201-207. [CrossRef]
- Tanford, C. Mechanism of active transport: free energy dissipation and free energy transduction. Proc. Natl. Acad. Sci. USA 1982, 79(21), 6527-31. [CrossRef]
- Brown, A.I.; Sivak, D.A. Theory of Nonequilibrium Free Energy Transduction by Molecular Machines. Chemical Reviews, 2020, 120(1), 434–459. [CrossRef]
- Wachtel, A.; Rao, R.; Esposito, M. Free-energy transduction in chemical reaction networks: From enzymes to metabolism. J. Chem. Phys. 2022, 157(2), 024109. [CrossRef]
- Leighton, M.P.; Sivak, D.A. Flow of Energy and Information in Molecular Machines. Annu. Rev. Phys. Chem. 2025, 76(1), 379-403. [CrossRef]
- Astumian, R.D. Trajectory and Cycle-Based Thermodynamics and Kinetics of Molecular Machines: The Importance of Microscopic Reversibility. Acc. Chem. Res. 2018, 51, 2653–2661. [CrossRef]
- Hill, T.L.; Simmons, R.M. Free energy levels and entropy production associated with biochemical kinetic diagrams. Proc. Natl. Acad. Sci. USA, 1976, 73(1), 95-99. [CrossRef]
- Hill, T.L. Steady-state kinetic formalism applied to multienzyme complexes, oxidative phosphorylation, and interacting enzymes. Proc. Natl. Acad. Sci. USA, 1976, 73(12), 4432-4436. [CrossRef]
- Schnakenberg, J. Network theory of microscopic and macroscopic behavior of master equation systems. Rev. Mod. Phys. 1976, 48, 571-585. [CrossRef]
- Qian, H. Cycle kinetics, steady state thermodynamics and motors-a paradigm for living matter physics. J. Phys. Condens. Matter 2005, 17(47), S3783-94. [CrossRef]
- Lipowsky, R.; Liepelt, S. Chemomechanical Coupling of Molecular Motors: Thermodynamics, Network Representations, and Balance Conditions. J. Stat. Phys. 2008, 130, 39–67. [CrossRef]
- Altaner, B.; Grosskinsky, S.; Herminghaus, S.; Katthän, L.; Timme, M.; Vollmer, J. Network representations of nonequilibrium steady states: Cycle decompositions, symmetries, and dominant paths. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012, 85(4 Pt 1), 041133. [CrossRef]
- Biddle, J.W.; Gunawardena, J. Reversal symmetries for cyclic paths away from thermodynamic equilibrium. Phys. Rev. E, 2020, 101(6-1), 062125. [CrossRef]
- Mugnai, M.L.; Hyeon, C.; Hinczewski, M., Thirumalai, D. Theoretical perspectives on biological machines. Rev. Mod. Phys., 2020, 92(2), 025001. [CrossRef]
- Samoilov, M.; Plyasunov, S.; Arkin, A.P. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc. Natl. Acad. Sci. USA 2005, 102(7), 2310-2315.. [CrossRef]
- Qian, H.; Beard, D.A. Metabolic futile cycles and their functions: a systems analysis of energy and control. Syst. Biol. (Stevenage) 2006, 153(4), 192-200. [CrossRef]
- Boehr, D.D. Editorial: Allosteric functions and inhibitions: structural insights. Front. Mol. Biosci. 2024, 11, 1363100. [CrossRef]
- Rivoire, O. A role for conformational changes in enzyme catalysis. Biophys. J. 2024, 123(12), 1563-1578. [CrossRef]
- McCullagh, M.; Zeczycki, T.N.; Kariyawasam, C.S.; Durie, C.L.; Halkidis, K.; Fitzkee, N.C.; Holt, J.M.; Fenton, A.W. What is allosteric regulation? Exploring the exceptions that prove the rule! J. Biol. Chem. 2024, 300(3), 105672. [CrossRef]
- Ito, S.; Kobayashi, C.; Yagi, K.; Sugita, Y. Toward understanding whole enzymatic reaction cycles using multi-scale molecular simulations. Curr. Opin. Struct. Biol. 2025, 95, 103153. [CrossRef]
- Aykac Fas, B.; Akbas Buz, Z.E.; Haliloglu, T. Global dynamics behind enzyme catalysis, evolution, and design. Curr. Opin. Struct. Biol. 2025, 94, 103131. [CrossRef]
- Miranda-Astudillo, H.; Zarco-Zavala, M.; García-Trejo, J.J.; González-Halphen, D. Regulation of bacterial ATP synthase activity: A gear-shifting or a pawl–ratchet mechanism? FEBS J. 2021, 288(10), 3159-3163. [CrossRef]
- Liu, Y.; Yu, J.; Wang, M.; Zeng. Q.; Fu, X.; Chang, Z. A high-throughput genetically directed protein crosslinking analysis reveals the physiological relevance of the ATP synthase “inserted” state. FEBS J. 2021, 288(9), 2989-3009. [CrossRef]
- Brunetta, H.S.; Jung, A.S.; Valdivieso-Rivera, F.; de Campos Zani, S.C.; Guerra, J.; Furino, V.O.; Francisco, A.; Berçot, M.; Moraes-Vieira, P.M.; Keipert, S. et al. IF1 is a cold-regulated switch of ATP synthase hydrolytic activity to support thermogenesis in brown fat. EMBO J. 2024, 43(21), 4870–4891. [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038.. [CrossRef]
- Hahn, A.; Vonck, J.; Mills, D.J.; Meier ,T.; Kühlbrandt, W. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 2018, 360(6389), eaat4318. [CrossRef]
- Pham, L.; Arroum, T.; Wan, J.; Pavelich, L.; Bell, J.; Morse, P.T.; Lee, I.; Grossman, L.I.; Sanderson, T.H.; Malek, M.H.; Hüttemann, M. Regulation of mitochondrial oxidative phosphorylation through tight control of cytochrome c oxidase in health and disease - Implications for ischemia/reperfusion injury, inflammatory diseases, diabetes, and cancer. Redox Biol. 2024, 78, 103426. [CrossRef]
- Mishra, N.K.; Habeck, M.; Kirchner, C.; Haviv , H.; Peleg , Y.; Eisenstein, M.; Apell, H.J.; Karlish, S.J.D. Molecular Mechanisms and Kinetic Effects of FXYD1 and Phosphomimetic Mutants on Purified Human Na,K-ATPase. J. Biol. Chem. 2015, 290(48), 28746-28759. [CrossRef]
- Cordeiro, B.M.; Leite Fontes, C.F.; Meyer-Fernandes, J.R. Molecular Basis of Na, K-ATPase Regulation of Diseases: Hormone and FXYD2 Interactions. Int. J. Mol. Sci. 2024, 25(24), 13398. [CrossRef]
- Meszéna, G.; Westerhoff, H.V. Non-equilibrium thermodynamics of light absorption. J. Phys. A.: Math. Gen. 1999, 32, 301–311. [CrossRef]
- Juretić, D.; Westerhoff, H.V. Variation of efficiency with free-energy dissipation in models of biological energy transduction. Biophys. Chem. 1987, 28, 21–34. [CrossRef]
- Juretić, D.; Županović, P. Photosynthetic models with maximum entropy production in irreversible charge transfer steps. J. Comp. Biol. Chem. 2003, 27, 541–553. [CrossRef]
- Dobovišek, A.; Županović, P.; Brumen, M.; Juretić, D. Maximum entropy production and maximum Shannon entropy as germane principles for the evolution of enzyme kinetics, in: Dewar, R.C.; Lineweaver, C.H.; Niven, R.K.; Regenauer-Lieb. K. (Eds.), Beyond the Second Law, Springer-Verlag, Berlin, Heidelberg, Germany, 2014, pp. 361–382. [CrossRef]
- Penocchio, E.; Rao, R.; Esposito, M., Nonequilibrium Thermodynamics of Light-Induced Reactions J. Chem. Phys. 2021, 155, 114101. [CrossRef]
- Landi, G.T.; Paternostro, M. Irreversible Entropy Production: From Classical to Quantum Rev. Mod. Phys. 2021, 93, 035008 . [CrossRef]
- Dewar, R.; Juretić, D.; Županović, P. The functional design of the rotary enzyme ATP synthase is consistent with maximum entropy production. Chem. Phys. Lett. 2006, 430, 177–182. [CrossRef]
- Juretić, D.; Simunić, J.; Bonačić Lošić, Ž. Maximum entropy production theorem for transitions between enzyme functional states and its application. Entropy 2019, 21, 743. [CrossRef]
- Martyushev, L.M.; Konovalov, M.S. Thermodynamic model of nonequilibrium phase transitions. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2011, 84(1 Pt 1), 011113. [CrossRef]
- Martyushev, L.M. Maximum entropy production principle: History and current status. Phys.-Usp. 2021, 64, 558-583. [CrossRef]
- Dobovišek, A.; Županović, P.; Brumen, M.; Bonačić-Lošić, Ž.; Kuić, D.; Juretić, D. Enzyme kinetics and the maximum entropy production principle. Biophys. Chem. 2011, 154(2-3), 49-55. [CrossRef]
- Bonačić Lošić, Ž.; Donđivić, T., Juretić, D. Is the catalytic activity of triosephosphate isomerase fully optimized? An investigation based on maximization of entropy production. J. Biol. Phys. 2017, 43, 69–86. [CrossRef]
- Ziegler, H. Some extreme principles in irreversible thermodynamics with application to continuum mechanics in: Sneddon, I.N.; Hill, R. (Eds.), Progress in Solid Mechanics, vol. 4, pp. 93–193, North-Holland, Amsterdam, 1963.
- Paltridge, G.W. Climate and thermodynamic systems of maximum dissipation, Nature, 1979, 279, 630–631. [CrossRef]
- Dewar, R.C. Information theory explanation of the fluctuation theorem, maximum entropy production and self-organized criticality in non-equilibrium stationary states. J. Phys. A Math. Gen. 2003, 36, 631–641. [CrossRef]
- Dewar, R.C. Maximum entropy production and the fluctuation theorem. J. Phys. A Math. Gen. 2005, 38, L371–L381. [CrossRef]
- Dewar, R.C.; Maritan, A.A. Theoretical Basis for Maximum Entropy Production. In Beyond the Second Law; Dewar, R.C., Lineweaver, C.H., Niven, R.K., Regenauer-Lieb, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 49–71.
- Martyushev, L.; Seleznev, V. Maximum entropy production: application to crystal growth and chemical kinetics. Curr. Opin. Chem. Eng. 2015, 7, 23–31. [CrossRef]
- Kleidon, A. Working at the limit: a review of thermodynamics and optimality of the Earth system. Earth Syst. Dynam. 2023, 14, 861–896. [CrossRef]
- Juretić, D.; Županović, P. The free-energy transduction and entropy production in initial photosynthetic reactions, In Non-equilibrium Thermodynamics and the Production of Entropy Kleidon, A.; Lorenz, R.D., Eds.; Springer-Verlag, Berlin, Germany, 2005, Chapter 13, pp. 161-171. [CrossRef]
- Laland, K.N.; Uller, T.; Feldman, M.W.; Sterelny, K.; Müller, G.B.; Moczek, A.; Jablonka, E.; Odling-Smee, J. The extended evolutionary synthesis: its structure, assumptions and predictions. Proc. R. Soc. B 2015, 282: 20151019. [CrossRef]
- Müller, G.B. Why an extended evolutionary synthesis is necessary. Interface Focus 2017, 7, 20170015. [CrossRef]
- Demetrius, L.A. Directionality theory and the origin of life. R. Soc. Open Sci. 2024, 11, 230623. [CrossRef]
- Mueller, S.A.; Merondun, J.; Lečić, S.; Wolf, J.B.W. Epigenetic variation in light of population genetic practice. Nat. Commun. 2025, 16(1), 1028. [CrossRef]
- Arto, A.; Stanley, S. Physical foundations of evolutionary theory. J. Non-Equilib. Thermodyn. 2010, 35, 301–321. [CrossRef]
- Goldenfeld, N.; Woese, C. Life is Physics: Evolution as a Collective Phenomenon Far From Equilibrium. Annu. Rev. Condens. Matter Phys. 2011, 2, 375-399. [CrossRef]
- Zanetti-Polzi, L.; Daidone, I.; Iacobucci, C.; Amadei, A. Thermodynamic Evolution of a Metamorphic Protein: A Theoretical-Computational Study of Human Lymphotactin. Protein J. 2023, 42, 219–228. [CrossRef]
- Hill, A. Entropy production as the selection rule between different growth morphologies. Nature 1990, 348, 426–428. [CrossRef]
- Pal, Rajinder. On the Gouy–Stodola theorem of thermodynamics for open systems. International Journal of Mechanical Engineering Education 2017, 45(2), 194–206. [CrossRef]
- Gonzalez-Ayala, J.; Santillan, M.; Santos, M.J.; Calvo Hernandez, A.; Mateos Roco, J.M. Optimization and Stability of Heat Engines: The Role of Entropy Evolution. Entropy 2018, 20, 865. [CrossRef]
- Sheoran J.; Pant, V.; Patel, R.; Banerjee D. Evolution of the thermodynamic properties of a coronal mass ejection in the inner Corona. Frontiers in Astronomy and Space Sciences, 2023,10, 27. [CrossRef]
- 122. Van Rotterdam, B. Control of Light-Induced Electron Transfer in Bacterial Photosynthesis, PhD thesis: University of Amsterdam, Amsterdam, The Netherlands, 1998.
- Van Rotterdam, B.J.; Westerhoff, H.V.; Visschers, R.W.; Bloch, D.A.; Hellingwerf, K.J.; Jones, M.R.; Crielaard, W. Pumping capacity of bacterial reaction centers and backpressure regulation of energy transduction. Eur. J. Biochem. 2001, 268, 958–970. [CrossRef]
- Wickstrand, C.; Dods, R.; Royant, A.; Neutze, R. Bacteriorhodopsin: Would the real intermediates please stand up? Biochim. Biophys. Acta 2015, 1850, 536–553. [CrossRef]
- Juretić, D.; Bonačić Lošić, Ž.; Kuić, D.; Simunić, J.; Dobovišek, A., The maximum entropy production requirement for proton transfers enhances catalytic efficiency for β-lactamases. Biophys. Chem. 2019, 244, 11–21. [CrossRef]
- Perrino, A.P.; Miyagi, A.; Scheuring, S. Single molecule kinetics of bacteriorhodopsin by HS-AFM. Nat. Commun. 2021, 12(1), 7225. [CrossRef]
- Petrovszki, D.; Krekic, S.; Valkai, S.; Heiner, Z.; Dér, A. All-Optical Switching Demonstrated with Photoactive Yellow Protein Films. Biosensors (Basel) 2021, 11(11), 432. [CrossRef]
- Jacobson, D.R.; Perkins, T.T. Quantifying a light-induced energetic change in bacteriorhodopsin by force spectroscopy. Proc. Natl. Acad. Sci. USA 2024, 121(7), e2313818121. [CrossRef]
- Dhanuka, A.; Flamholz, A.I.; Murugan, A.; Goyal, A. Ecosystems as adaptive living circuits. bioRxiv [Preprint]. 2025, Jun 29, 06.27.661910. [CrossRef]
- Juretić, D.; Bruvo Mađarić, B. Scale-invariant dissipation underlies enzyme catalytic performance. Biosystems 2025, 258, 105568. [CrossRef]
- Bisker, G.; Polettini, M.; Gingrich, T.R.; Horowitz, J.M. Hierarchical bounds on entropy production inferred from partial information. J. Stat. Mech. 2017, 093210. [CrossRef]
- Yoshimura, K.; Kolchinsky, A.; Dechant, A.; Ito, S. Housekeeping and excess entropy production for general nonlinear dynamics. Phys. Rev. Research 2023, 5, 013017. [CrossRef]
- Pänke, O.; Rumberg, B. Kinetic modeling of rotary CF0F1-ATP synthase: storage of elastic energy during energy transduction. Biochim. Biophys. Acta 1999, 1412(2), 118-128. [CrossRef]
- Whittington, A.C.; Larion, M.; Bowler, J.M.; Ramsey, K.M.; Brüschweiler, R.; Miller, B.G. Dual allosteric activation mechanisms in monomeric human glucokinase. Proc. Natl. Acad. Sci. USA 2015, 112(37), 11553-11558. [CrossRef]
- Dixon, R.E.; Navedo, M.F.; Binder, M.D.; Santana, L.F. Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol. Rev. 2021, 102(3), 1159–1210. [CrossRef]
- Liao, J.; Shahul Hameed, U.F.; Hoffmann, T.D.; Kurze, E.; Sun, G.; Steinchen, W.; Nicoli, A.; Di Pizio, A.; Kuttler, C.; Song C. et al. β-Carotene alleviates substrate inhibition caused by asymmetric cooperativity. Nat. Commun. 2025, 16(1), 3065. [CrossRef]
- Punekar, N.S. Regulation of Enzyme Activity. In: ENZYMES: Catalysis, Kinetics and Mechanisms. Springer, Singapore, 2025, pp 527-561. [CrossRef]
- Bordel, S.; Nielsen, J. Identification of flux control in metabolic networks using non-equilibrium thermodynamics. Metab. Eng. 2010, 12(4), 369-377. [CrossRef]
- Vallino, J.J. Ecosystem biogeochemistry considered as a distributed metabolic network ordered by maximum entropy production. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365(1545), 1417-1427. [CrossRef]
- Unrean, P.; Srienc, F. Metabolic networks evolve towards states of maximum entropy production. Metab. Eng. 2011, 13(6), 666-673. [CrossRef]
- Lan, G.; Pablo Sartori, P.; Neumann, S.; Sourjik, V.; Tu, Y. The energy-speed-accuracy tradeoff in sensory adaptation. Nat. Phys. 2012, 8, 422–428. [CrossRef]
- Weber, J.K.; Shukla, D.; Pande, V.S. Heat dissipation guides activation in signaling proteins. Proc. Natl. Acad. Sci. USA 2015, 112(33), 10377-10382. [CrossRef]
- Britton, S.; Alber, M.; Cannon, W.R. Enzyme activities predicted by metabolite concentrations and solvent capacity in the cell. J. R. Soc. Interface 2020, 17, 20200656. [CrossRef]
- King E.; Holzer J.; North J.A.; Cannon, W.R. An approach to learn regulation to maximize growth and entropy production rates in metabolism. Front. Syst. Biol. 2023, 3, 981866. [CrossRef]
- Dobovišek, A.; Blaževič, T.; Kralj, S.; Fajmut, A. Enzyme cascade to enzyme complex phase-transition-like transformation studied by the maximum entropy production principle. Cell Reports Physical Science, 2025, 6(2), 102400. [CrossRef]
- Sawada, Y.; Daigaku, Y.; Toma, K. Maximum Entropy Production Principle of Thermodynamics for the Birth and Evolution of Life. Entropy 2025, 27, 449. [CrossRef]
- Srienc, F.; Barrett, J. Predicting the Rate Structure of an Evolved Metabolic Network. Metabolites 2025, 15(3), 200. [CrossRef]
- Juretić, D.; Bonačić Lošić, Ž. Theoretical improvements in enzyme efficiency associated with noisy rate constants and increased dissipation. Entropy 2024, 26, 151. [CrossRef]
- Jensen, R.A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409-25. [CrossRef]
- Copley, S.D. Toward a systems biology perspective on enzyme evolution. J. Biol. Chem. 2012, 287(1), 3-10. [CrossRef]
- Ambler, R.P. The Amino Acid Sequence of Staphylococcus aureus Penicillinase. Biochem. J. 1975, 151, 197–218. [CrossRef]
- Thatcher, D.R. The Partial Amino Acid Sequence of the Extracellular P-Lactamase I of Bacillus cereus 569/H. Biochem. J. 1975, 147, 313–326. [CrossRef]
- Christensen, H.; Martin, M.T.;Waley, G. beta-lactamases as fully efficient enzymes. Determination of all the rate constants in the acyl-enzyme mechanism. Biochem. J. 1990, 266, 853–861.
- Mehboob, S.; Guo, L.; Fu, W.; Mittal, A.; Yau, T.; Truong, K.; Johlfs, M.; Long, F.; Fung, L. W.-M.; Johnson, M.E. Glutamate racemase dimerization inhibits dynamic conformational flexibility and reduces catalytic rates. Biochemistry 2009, 48, 7045–7055. [CrossRef]
- Kleiber, M. Body size and metabolic rate. Physiol. Rev. 1947, 27, 511–541. https://doi. org/10.1152/physrev.1947.27.4.511.
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Briefings in Bioinformatics, 2019, 20 (4), 1160–1166. [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution, 2015, 32 (1), 268–274. [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods, 2017, 14 (6), 587–589. [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Molecular Biology and Evolution, 2018, 35 (2), 518–522. [CrossRef]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic Biology, 2006, 55 (4), 539–552. [CrossRef]
- Banerjee, K.; Bhattacharyya, K. States with identical steady dissipation rate in reaction networks: A non-equilibrium thermodynamic insight in enzyme efficiency. Chem. Phys. 2014, 438, 1–6. [CrossRef]
- Zhang, X.; Chen, P.; Li, M.; Shi, Y.; Hasman, E.; Wang, B.; Chen, X. Brownian spin-locking effect Nat. Mater 2025, Nov 18. Online ahead of print. [CrossRef]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [CrossRef]
- Lee, H.-E.; Okumura, T.; Ooka, H; Adachi, K.; Hikima, T.; Hirata , K.; Kawano, Y.; Matsuura, H.; Yamamoto Masaki, Yamamoto, Masahiro et al. Osmotic energy conversion in serpentinite-hosted deep-sea hydrothermal vents. Nat. Commun. 2024, 15(1), 8193. [CrossRef]
- Fang, X.; Oriola, D.; Grill, S.W. Nonequilibrium physics in biology. Reviews of Modern Physics, 2019, 91(4), 045004. [CrossRef]
- Niebel, B.; Leupold, S.; Heinemann, M. An upper limit on Gibbs energy dissipation governs cellular metabolism. Nat. Metab. 2019, 1, 125–132 . [CrossRef]
- Wortel, M.T.; Peters, H.; Hulshof, J.; Teusink, B.; Bruggeman, F.J. Metabolic enzyme cost explains variable trade-offs between microbial growth rate and yield. PLoS Comput. Biol. 2021, 17, e1008656. [CrossRef]
- Mehdi Molaei, M.; Redford, S.A.; Chou, W.-H.; Scheff, D.; de Pablo,J.J.; Oakes, P.W.; Gardel, M.L. Measuring response functions of active materials from data. Proc. Natl. Acad. Sci. USA 2023, 120(42), e2305283120. [CrossRef]
- Bo, S.; Celani, A.; Eichhorn, R.; Aurell, E. Stochastic Thermodynamics of Enzyme Catalysis: Efficiency, Speed, and Dissipation. Phys. Rev. Lett. 2023, 130, 098401. [CrossRef]
- Umeda, K.; Nishizawa, K.; Nagao, W.; Inokuchi, S.; Sugino, Y.; Ebata, H.; Mizuno, D. Activity-dependent glassy cell mechanics II: Nonthermal fluctuations under metabolic activity. Biophys. J. 2023, 122(22), 4395-4413. [CrossRef]
Figure 1.
A general Prigogine-like scheme for an open steady-state system performing irreversible free-energy transduction [28,29]. All generalized forces Xi and corresponding flows Ji (radiation, heat, matter) contribute to the entropy production density σ expressed as the sum of JiXi products. In particular, enzymes or living systems convert high free-energy μ and low-entropy S substrates into low free-energy and high-entropy products.
Figure 1.
A general Prigogine-like scheme for an open steady-state system performing irreversible free-energy transduction [28,29]. All generalized forces Xi and corresponding flows Ji (radiation, heat, matter) contribute to the entropy production density σ expressed as the sum of JiXi products. In particular, enzymes or living systems convert high free-energy μ and low-entropy S substrates into low free-energy and high-entropy products.
Figure 2.
The dissipation plane viewpoint of our recent results [130]. Seventy-five points for 75 enzyme-catalyzed reactions are mostly contained within the brown plane in such a 3D log-log-log representation that the x-axis is log10(kcat), the y-axis is log10(KM), and the z-axis is log10(Dissiption/(RT)). Points are colored by the specificity constant (kcat/KM), also named enzyme efficiency. The vertical projections to the (x,y) plane (dotted lines) highlight five reactions associated with an increased overall dissipation, ranked as CAII (carbonic anhydrase II) > KSI (ketosteroid isomerase) > Lac1 > RTEM > PC1. The last three enzymes are evolutionarily related β-lactamases from bacteria, likely descended from the same ancestor. That is why we highlighted them with black arrows and red connecting lines.
Figure 2.
The dissipation plane viewpoint of our recent results [130]. Seventy-five points for 75 enzyme-catalyzed reactions are mostly contained within the brown plane in such a 3D log-log-log representation that the x-axis is log10(kcat), the y-axis is log10(KM), and the z-axis is log10(Dissiption/(RT)). Points are colored by the specificity constant (kcat/KM), also named enzyme efficiency. The vertical projections to the (x,y) plane (dotted lines) highlight five reactions associated with an increased overall dissipation, ranked as CAII (carbonic anhydrase II) > KSI (ketosteroid isomerase) > Lac1 > RTEM > PC1. The last three enzymes are evolutionarily related β-lactamases from bacteria, likely descended from the same ancestor. That is why we highlighted them with black arrows and red connecting lines.
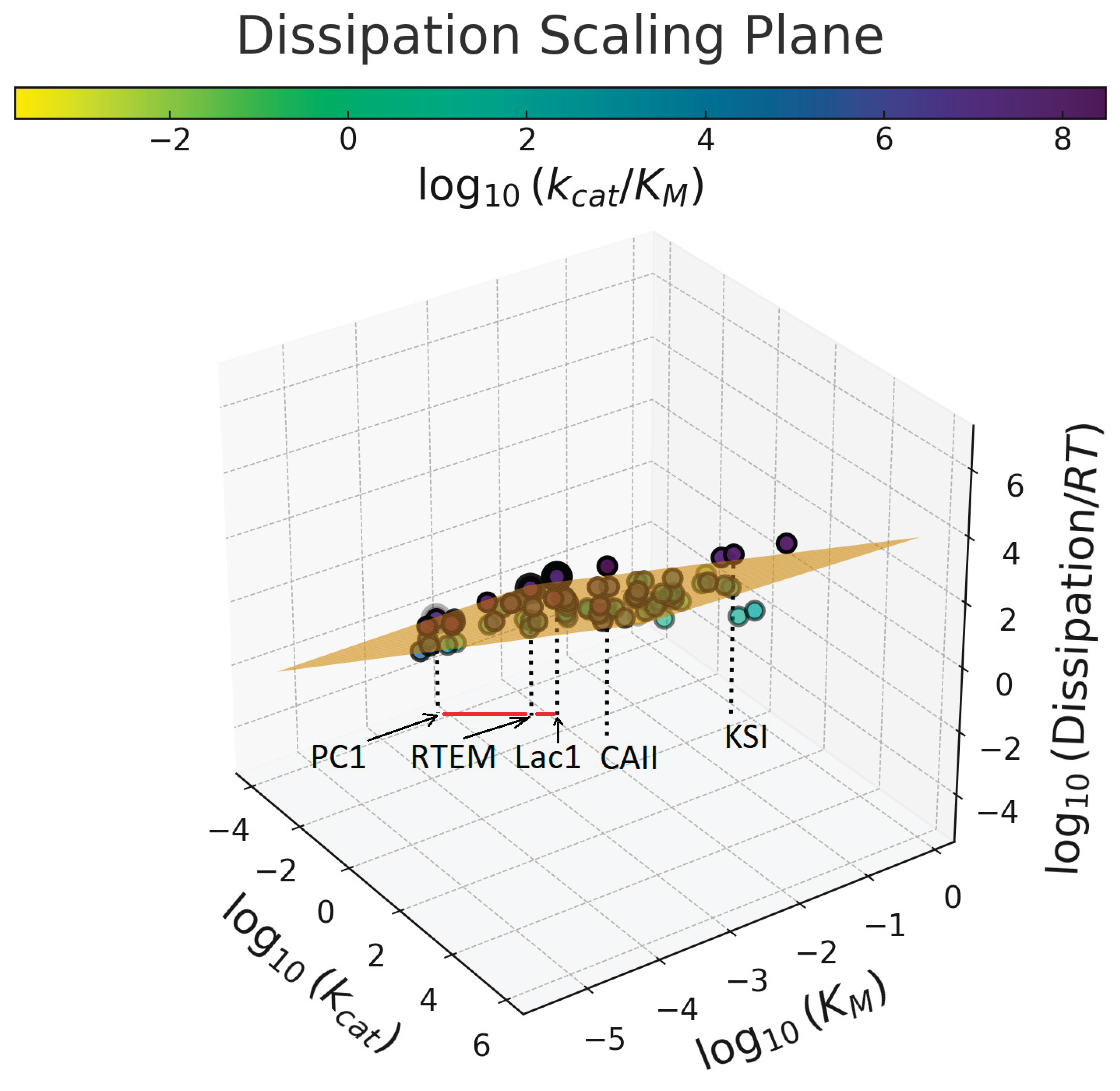
Figure 3.
(a). The connection of biochemical, evolutionary, and physical parameters for cyclophilins (blue points and connecting lines) and β-lactamases (red points and connecting lines). The methods already described [130] were applied to find evolutionary distances. There was no change in cyclophilin distances, and we reproduced the ranking of evolutionary distances for bacterial β-lactamases: PC1 = 0.8119 < RTEM = 1.1456 < Lac1 = 1.1797. In brief, β-lactamase sequences used in [125,130] were aligned in the MAFFT program under the E-Ins-I algorithm [156]. Evolutionary distances (in substitutions per site) were read from the branch lengths of a maximum likelihood tree constructed in the IQTree program [157] under the LG+G+I model selected by ModelFinder [158]; ultrafast bootstrap (UFBoot) with 1000 iterations and SH-aLRT branch test were used for branch support assessment, as implemented in IQTree [159,160]. There was no change in other parameter values we recently used [130]. Orange rectangles for cyclophilins do not illustrate very well the association between increased evolutionary distances (CypA = 0.451 < CypB = 0.632 < CypC = 0.783) and higher overall dissipation, but Figure 3b resolves that issue. (b). The CypA < CypB < CypC ranking is clear here for the catalytic constant kcat and Dissipation/(RT) (both parameters are expressed in inverse seconds)[130], but enzyme efficiency (kcat/KM) ranking is different. All engineered glutamase racemase mutants [154] are regularly clustered together (red points) in the dissipation plane due to physical constraints. They exhibit higher dissipation and kcat values than the wild-type enzyme (green point).
Figure 3.
(a). The connection of biochemical, evolutionary, and physical parameters for cyclophilins (blue points and connecting lines) and β-lactamases (red points and connecting lines). The methods already described [130] were applied to find evolutionary distances. There was no change in cyclophilin distances, and we reproduced the ranking of evolutionary distances for bacterial β-lactamases: PC1 = 0.8119 < RTEM = 1.1456 < Lac1 = 1.1797. In brief, β-lactamase sequences used in [125,130] were aligned in the MAFFT program under the E-Ins-I algorithm [156]. Evolutionary distances (in substitutions per site) were read from the branch lengths of a maximum likelihood tree constructed in the IQTree program [157] under the LG+G+I model selected by ModelFinder [158]; ultrafast bootstrap (UFBoot) with 1000 iterations and SH-aLRT branch test were used for branch support assessment, as implemented in IQTree [159,160]. There was no change in other parameter values we recently used [130]. Orange rectangles for cyclophilins do not illustrate very well the association between increased evolutionary distances (CypA = 0.451 < CypB = 0.632 < CypC = 0.783) and higher overall dissipation, but Figure 3b resolves that issue. (b). The CypA < CypB < CypC ranking is clear here for the catalytic constant kcat and Dissipation/(RT) (both parameters are expressed in inverse seconds)[130], but enzyme efficiency (kcat/KM) ranking is different. All engineered glutamase racemase mutants [154] are regularly clustered together (red points) in the dissipation plane due to physical constraints. They exhibit higher dissipation and kcat values than the wild-type enzyme (green point).
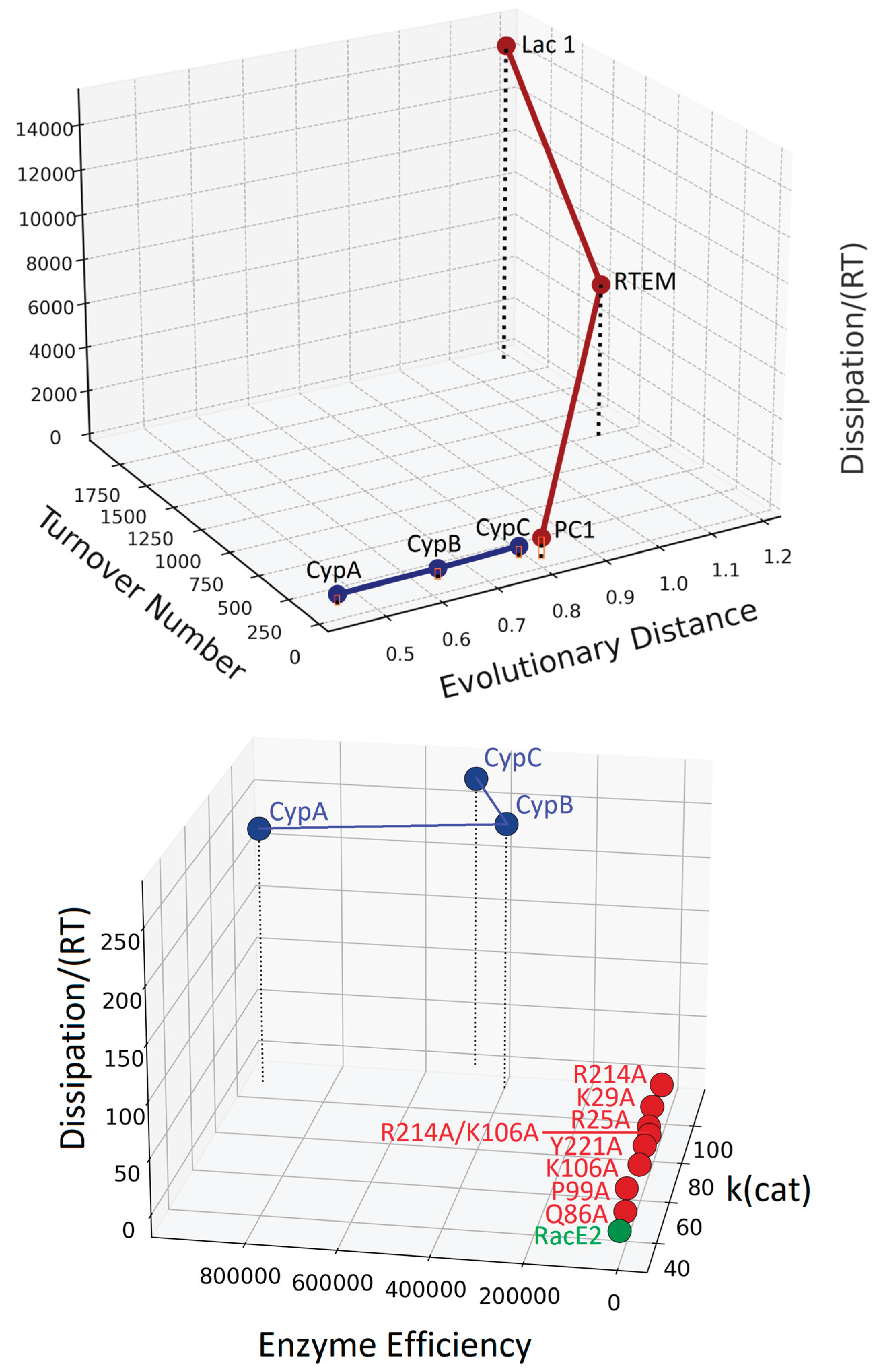
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



