
Submitted:
08 January 2026
Posted:
09 January 2026
You are already at the latest version
Abstract
Background/Objectives: Inherited retinal diseases (IRDs) represent a genetically heterogeneous group of disorders caused by mutations in over 280 genes with more than 3,100 identified variants. While gene-specific replacement therapies have achieved landmark success with voretigene neparvovec (Luxturna) for RPE65-associated retinal dystrophy, developing individual therapies for each genetic subtype remains impractical. This review examines gene-agnostic gene therapy approaches with neuroprotection and immunomodulation that target common pathophysiological mechanisms shared across multiple IRD genotypes. Methods: We reviewed the literature on neuroprotective and immunomodulatory gene therapy strategies for IRDs, focusing on neurotrophic factors and complement system modulation. Results: Neuroprotective approaches delivering neurotrophic factors—including pigment epithelium-derived factor (PEDF), ciliary neurotrophic factor (CNTF), rod-derived cone viability factor (RdCVF), brain-derived neurotrophic factor (BDNF), fibroblast growth factors, glial cell line-derived neurotrophic factor (GDNF), and proinsulin—have demonstrated photoreceptor preservation across multiple preclinical IRD models regardless of the underlying genetic mutation. The recent FDA approval of CNTF cell-based gene therapy (Encelto) for macular telangiectasia type 2 validates this therapeutic paradigm. Complement system inhibition represents another gene-agnostic strategy, with intravitreal complement inhibitors approved for geographic atrophy secondary to age-related macular degeneration and gene therapy approaches targeting C3, C5, or delivering soluble complement regulators under investigation for IRDs. Combination strategies simultaneously addressing multiple pathogenic pathways may offer synergistic benefits. Conclusions: Gene-agnostic approaches targeting neuroprotection and immunomodulation offer a scalable therapeutic paradigm capable of benefiting patients across the spectrum of IRD genotypes, potentially transforming treatment for conditions where mutation-specific therapies remain unavailable.
Keywords:
inherited retinal disease
; gene therapy
; neuroprotection
; immunomodulation
; gene agnostic
1. Introduction
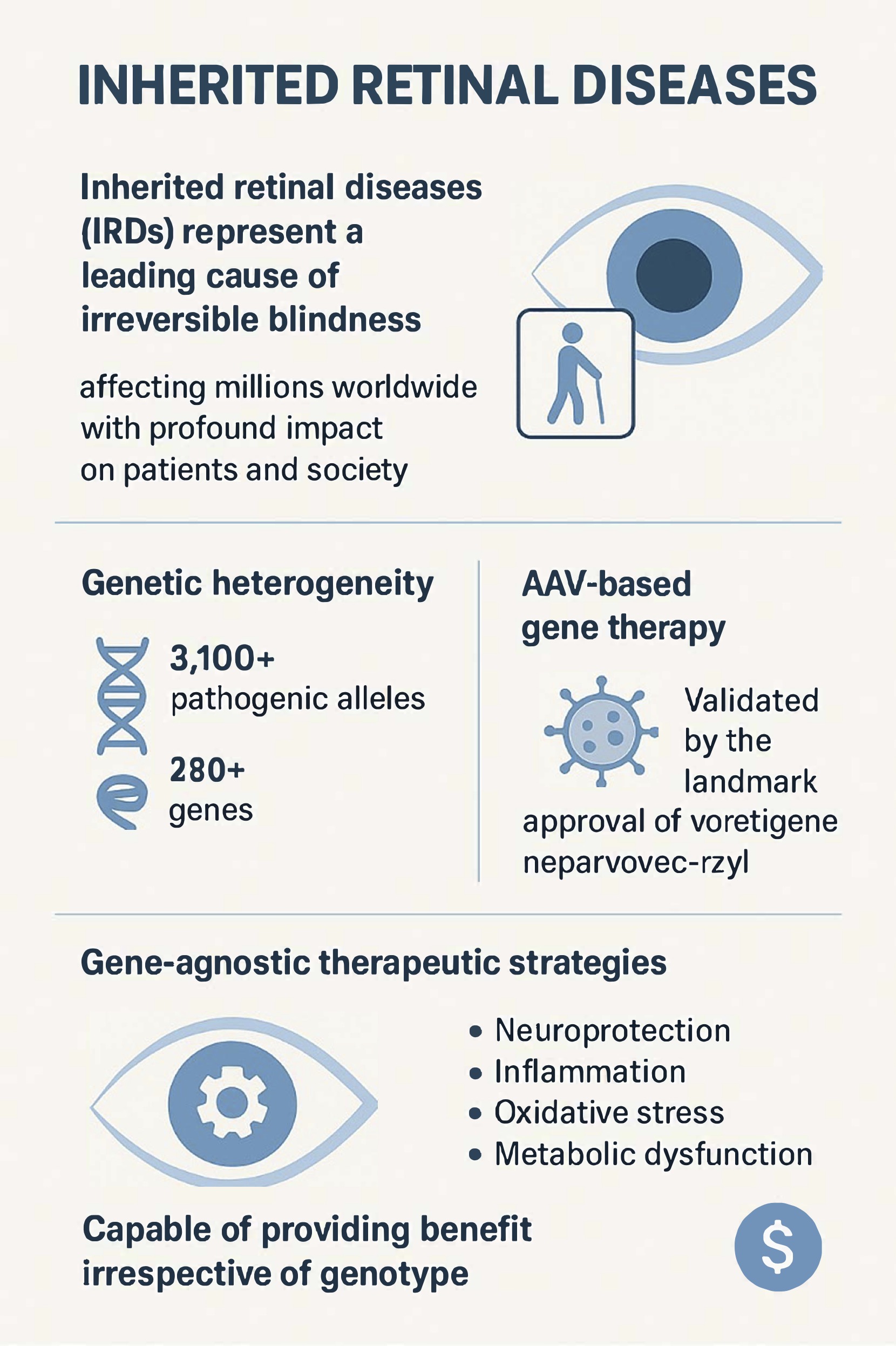
Inherited retinal diseases (IRDs) represent a leading cause of irreversible blindness in both children and the working-age population, collectively affecting millions worldwide with profound impact on patients and society [1]. In our clinical experience, we regularly encounter patients and families facing the devastating reality of progressive vision loss with no approved treatment options. These disorders are characterized by progressive degeneration of photoreceptors or the retinal pigment epithelium (RPE), leading to visual field loss and eventual blindness. Although the landmark approval of voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics, Philadelphia, PA, USA) for confirmed biallelic RPE65-mediated retinal dystrophy validated the feasibility of adeno-associated viral (AAV)-based gene replacement therapy, this success also underscored the limitations of gene-specific approaches [2].
A major barrier is the genetic heterogeneity underlying IRDs. More than 3,100 pathogenic alleles across over 280 genes have been identified as causal for IRDs [3,4,5]. Whilst it is becoming increasingly possible to develop gene therapies for rare diseases, including efforts to streamline and standardize manufacturing methods, there are still significant hurdles in identifying relevant genes, developing and delivering vector construct, performing preclinical validation, and completing clinical trials [6]. This presents a task that is scientifically and economically challenging given the rarity of individual mutations. Furthermore, many patients remain genetically undiagnosed, excluding them from gene-targeted treatments. We have witnessed this frustration firsthand in our own practices, among patients with clear phenotypic features of IRD who cannot access Luxturna or enroll in gene replacement trials simply because their causative mutation remains unidentified. Even in genetically confirmed cases, gene augmentation is limited by factors such as large gene size, dominant-negative mutations, or advanced photoreceptor loss that precludes meaningful recovery [7].
These constraints have catalyzed growing interest in gene-agnostic therapeutic strategies capable of providing benefit irrespective of genotype [8,9]. Such approaches aim to preserve or restore vision by targeting common mechanisms of degeneration including neuroprotection, inflammation, oxidative stress, and metabolic dysfunction. From an economic perspective, these interventions may also prove more scalable and cost-effective than individualized gene therapies [10]. Equally disheartening are those patients we see with confirmed genetic diagnoses for whom no gene replacement therapy exists or is under development, leaving us with few options beyond supportive care and watching disease progression unfold. Consequently, this review focuses on preservation-oriented strategies aimed at slowing or halting photoreceptor degeneration, focusing on neurotrophic factor delivery and complement modulation strategies (Table 1). Alternative gene-agnostic strategies include modifier gene therapy approaches such as OCU400 (AAV-hNR2E3, Ocugen, Malvern, PA, USA), which delivers the NR2E3 transcription factor via AAV5 to restore photoreceptor homeostasis; this approach is mechanistically distinct from the neurotrophic factor delivery and complement modulation strategies that are the focus of our review [11]. Similarly, optogenetic approaches, which confer light sensitivity to remaining retinal cells and target patients with end stage disease and profound vision loss, represent a complementary gene-agnostic paradigm reviewed elsewhere.
2. Neuroprotective Approaches
Neuroprotective interventions seek to preserve existing photoreceptors, thereby delaying or preventing irreversible visual loss. These therapies act broadly, independent of specific genetic mutations, by stabilizing cellular metabolism, reducing cell death, and modulating trophic signaling [12,13].
2.1. Growth Factor-Based Therapies
2.1.1. Pigment Epithelium-Derived Factor (PEDF)
PEDF is a 50-kDa glycoprotein secreted by the RPE with neurotrophic, anti-angiogenic, and anti-inflammatory properties [14]. PEDF levels in the eye decline with retinal degeneration [15,16], age-related macular degeneration (AMD) [17], and age [18]. Furthermore, loss of PEDF may cause age-related changes in the retina and RPE [19]. Its neuroprotective mechanisms include promotion of photoreceptor survival, reduction of oxidative stress, and regulation of apoptotic pathways [20,21]. Its neuroprotective actions are mediated largely through its receptor PEDF-R (PNPLA2) on photoreceptor inner segments [22]. Mechanistically, PEDF preserves calcium homeostasis via activation of the plasma membrane calcium pump (PMCA), preventing calcium-induced calpain/BAX/apoptosis-inducing factor (AIF)-mediated cell death, while also inhibiting AIF nuclear translocation and increasing Bcl-2 expression [23]. Additionally, PEDF promotes photoreceptor differentiation, rhodopsin polarization, and neurite outgrowth [24]. These pleiotropic mechanisms position PEDF as a broad neuroprotective agent targeting multiple IRD-associated pathways [25].
PEDF has demonstrated robust neuroprotective efficacy in multiple rodent models of IRD [26,27]. In rd10 mice, which carry a mutation in the PDE6B gene causing progressive rod photoreceptor death, PEDF protein levels in the RPE decline with age, coinciding with photoreceptor degeneration [16]. Furthermore, PEDF deficiency in SERPINF1 null mice crossed with rd10 mice resulted in accelerated retinal degeneration compared to rd10 mice alone, implying PEDF's protective role in IRD and demonstrating that PEDF deficiency increases susceptibility to retinal degeneration [28]. Administration of recombinant PEDF or PEDF-derived peptides containing the neurotrophic region significantly increased outer nuclear layer (ONL) thickness and reduced degenerating photoreceptors in rd10 retinal explants, in vivo models, and human retinal organoids [16,29] . In RhoP23H/+ mice and other models of retinitis pigmentosa, vector-mediated PEDF gene transfer prevented photoreceptor cell death, inhibited AIF nuclear translocation, and preserved retinal structure over extended periods [23]. Lastly, PEDF-derived peptides, 17-mer and H105A, delivered by topical eye drop not only successfully reached the retina, increased rhodopsin and opsin levels, reduced cell death markers (BAX/Bcl2 ratio), and promoted photoreceptor survival but also achieved improvement in retinal function, as measured by electroretinogram (ERG), in both rd10 and RhoP23H/+ mice [30]. Furthermore, retinal organoids exposed to oxidative stress via cigarette smoke extract demonstrated less photoreceptor death when treated with H105A [30]. These findings established PEDF as a potent, receptor-dependent neuroprotectant across diverse genetic models of IRDs.
Efforts to deliver PEDF via topical eye drops, intravitreal injection, protein microcarriers, and AAV-mediated gene therapy have all demonstrated sustained intraocular bioactivity [31,32,33]. PEDF-derived peptides (particularly 17-mer fragments containing the neurotrophic domain) have been formulated as eye drops and shown to penetrate ocular tissues, reaching the retina at physiologically relevant concentrations [29]. Topical administration of bioactive PEDF 17-mer peptide eye drops in microphthalmia (Mitf) mutant mice with RPE deficiency-associated retinal degeneration partially prevented photoreceptor loss, reduced apoptosis, and increased expression of rod and cone genes [29]. Topical delivery of PEDF-derived peptides, 17-mer and H105A, successfully reached the retina, promoted photoreceptor survival and improved retinal function in two retinitis pigmentosa mouse models [30]. Intravitreal injection of AAV vectors delivering H105A delayed photoreceptor degeneration for up to six months in RhoP23H/+ mice [30]. Intravitreal injection of PEDF, 17-mer, and H105A has demonstrated significant protection of cone photoreceptors from LED-induced phototoxic damage in rodent models, with this neuroprotective effect mediated through the 17-mer region's interaction with the PEDF receptor [27,34]. Intravitreal adenoviral gene transfer of PEDF in Lewis rats significantly protected photoreceptors from light-induced apoptotic cell death, preserving photoreceptor density and improving ERG function compared to control eyes [35]. Subretinal injection of a third-generation lentiviral vector based on simian immunodeficiency virus (SIV-hPEDF) has achieved safe and stable transgene expression of PEDF maintained for over five years in nonhuman primates and significant delay of retinal degeneration in rodent RP models [36,37,38,39].
Despite promising preclinical results, clinical translation remains limited for back of the eye pathologies, due to challenges in dosing and durability. For instance, the inverse scaling of vitreous volume to ocular surface area between mouse and human eye raises significant challenges in achieving therapeutic levels following topical application. Only a single phase 1 clinical trial has been conducted to date, evaluating PEDF for macular neovascularization, with limited published outcomes [40]. A phase 1/2a investigator-initiated clinical trial (jRCT2073180024) to evaluate the safety and efficacy in patients with RP is ongoing; however, efficacy and safety data are not yet available [41]. The relatively short half-life of recombinant PEDF protein necessitates frequent administrations, which is impractical for chronic retinal diseases [42,43,44]. While gene therapy offers sustained expression, questions remain regarding optimal vector design, route of administration, immune responses to viral vectors, and long-term safety [45,46]. Future optimization of delivery routes, vectors and sustained release systems will be essential to realize PEDF’s therapeutic potential in human IRDs.
2.1.2. Ciliary Neurotrophic Factor (CNTF)
CNTF promotes photoreceptor survival through the JAK/STAT pathway activation and modulation of Müller glial responses [47,48,49]. In multiple rodent and canine models, CNTF slowed photoreceptor loss and maintained ONL integrity by direct neuroprotective effects, modulation of glial responses, and metabolic support, with efficacy demonstrated across multiple delivery platforms [50,51,52]. Translation of these promising preclinical neuroprotective results in human retinal degenerative diseases has proved to vary by disease and outcome measure.
Revakinagene taroretcel-lwey (Encelto, Neurotech Pharmaceuticals, Cumberland, RI, USA) made history in March 2025 as the first therapy approved for use by the United States Food and Drug Administration (FDA) for the treatment of adults with idiopathic macular telangiectasia type 2 (MacTel) [53]. Revakinagene taroretcel-lwey is an allogeneic encapsulated cell-based gene therapy providing sustained delivery of recombinant CNTF (rhCNTF) to promote the survival and maintenance of photoreceptors. FDA approval was based on the results of two phase 3 trials which demonstrated that revakinagene taroretcel-lwey significantly slowed the loss of macular photoreceptors, as measured with optical coherence tomography (OCT), in MacTel patients over 24 months. Revakinagene taroretcel-lwey significantly slowed the rate of ellipsoid zone (EZ) area loss compared to sham in both NTMT-03-A (NCT03316300; 0.075 vs. 0.166 mm² over 24 months; difference: -0.091 mm², p < 0.0001) and NTMT-03-B (NCT03319849; 0.111 vs. 0.160 mm²; difference: -0.049 mm², p = 0.0186) [54].
In retinitis pigmentosa (RP), multicenter, sham-controlled trials (NCT01530659 NCT00447980, NCT00447993) found that CNTF delivered via intraocular encapsulated cell implants was safe and achieved long-term intraocular protein release [55,56]. However, these studies did not show significant benefit in primary visual function endpoints (visual acuity or field sensitivity). In fact, long-term follow-up showed greater visual field loss from baseline than sham eyes, which was reversable upon removing the implant [56]. Some secondary outcomes, such as increased retinal thickness and stabilization of vision in subgroups, were observed, but overall, efficacy was less robust than in animal models [55].
For geographic atrophy (GA) secondary to AMD, a phase 2 trial found that high-dose CNTF stabilized visual acuity and increased retinal thickness compared to sham, especially in patients with better baseline vision [57]. Further trials are being carried out for visual restoration in glaucoma (NCT04577300, NCT01408472, NCT02862938) and achromatopsia (NCT01648452) showing the potential breadth for a gene agnostic approach outside of a single disease. In the latter case, CNTF has been observed to increase outer segment growth and may have the potential to augment achromatopsia gene replacement therapies [58].
2.1.3. Rod-Derived Cone Viability Factor (RdCVF)
RdCVF is a thioredoxin-like protein secreted by rod photoreceptors that maintains cone viability by promoting glucose uptake through the basigin-1 (BSG1)/GLUT1 complex. RdCVF binds to BSG1 on cone photoreceptors, which interacts with GLUT1 to increase glucose entry into cones, thereby enhancing cone metabolic efficiency through stimulation of aerobic glycolysis and support of the high metabolic demands of cone outer segment renewal. This mechanism is central to the secondary cone degeneration observed in IRDs, where rod loss leads to a reduction in RdCVF secretion, depriving cones of the metabolic support necessary for survival [59].
In rd1 and rd10 mouse models, delivery of RdCVF as a recombinant protein, or via a gene therapy has markedly preserved cone structure and visual function. AAV-mediated expression of RdCVF in these models resulted in delayed cone loss, improved photopic ERG responses, and sustained visual acuity [60,61,62]. These effects are mutation-independent, as RdCVF acts on a metabolic pathway common to all cones, regardless of the underlying genetic defect.
Given its gene-independent mechanism and ability to rescue cones metabolically, RdCVF is considered one of the most promising broad-spectrum therapeutic candidates for IRDs. Preclinical safety and efficacy data have supported the initiation of first-in-human studies of RdCVF for IRD [60,61,63]. SPVN06 (SparingVision, Paris, France) is a subretinal AAV-based gene therapy delivering both RdCVF and Rod derived Cone Viability Factor Long form (RdCVFL), an enzyme which protects cones against oxidative stress. The ongoing Phase I/II PRODYGY trial (NCT05748873) evaluating SPVN06 in subjects with advanced rod-cone dystrophy (RCD) due to mutations in the RHO, PDE6A, or PDE6B gene has demonstrated a strong safety profile up to one year after injection, with no significant intraocular inflammation or immune response [64]. Specific peer-reviewed human efficacy data remains pending.
In summary, RdCVF’s role in promoting cone glucose uptake via the BSG1/GLUT1 complex provides a direct mechanistic link between rod loss and cone degeneration, and its mutation-independent rescue of cones positions it as a leading candidate for IRD therapy, with clinical translation underway.
2.2. Other Neuroprotective Strategies
Additional neurotrophic and metabolic factors including brain-derived neurotrophic factor (BDNF), fibroblast growth factors (FGF), glial-derived neurotrophic factor (GDNF), and metabolic modulators like proinsulin have shown protective effects across various retinal degeneration models. Although translation of these agents to clinical use remains preliminary, the breadth of preclinical evidence underscores the potential of multi-factor neuroprotection as a gene-agnostic therapeutic platform.
BDNF has been extensively studied for its neuroprotective and restorative effects in retinal diseases. Sustained BDNF expression in the retina delays photoreceptor cell death and preserves retinal function in models of IRD and oxidative damage, primarily through tropomyosin receptor kinase B (TrkB) receptor signaling and anti-apoptotic pathways [65,66,67]. Gene therapy approaches, such as AAV-mediated BDNF/TrkB delivery, have achieved sustained neuroprotection and long-term signaling enhancement in preclinical models, with no adverse effects on retinal structure or function [68,69]. These strategies are advancing toward clinical trials, with novel nanoparticle platforms enabling efficient and safe delivery to Müller cells and demonstrating synergistic effects when combined with metabolic modulators like oligomycin [70]. However, translation to routine clinical use is limited by challenges in maintaining therapeutic levels and receptor downregulation.
FGFs, particularly FGF2, have shown significant photoreceptor rescue in IRD models. Intravitreal FGF2 improves photoreceptor morphology and, when combined with agents like minocycline, yields additive neuroprotective effects by reducing microglial activation and enhancing cell survival [65,71]. While FGF2 and other FGFs are recognized for their neuroprotective potential, clinical translation is still preliminary, with most evidence derived from preclinical studies.
GDNF and related neurotrophic factors play interdependent roles in retinal neuroprotection, modulating neuronal survival, differentiation, and glial responses. GDNF has demonstrated efficacy in models of diabetic retinopathy and IRD, often acting synergistically with other trophic factors [67,72]. GDNF, alone or in combination with BDNF, has been safely delivered via sustained-release microspheres in animal models, supporting RGC survival and migration of RPE cells without toxicity or apoptosis [73]. These delivery systems are being developed for long-term intravitreal administration, with the potential for personalized dosing in future clinical applications.
Metabolic modulators such as proinsulin exert neuroprotective effects by attenuating oxidative stress, enhancing mitochondrial integrity, and supporting cell survival. Proinsulin activates the insulin receptor-A pathway in the retina, preserving synaptic connectivity and prolonging visual function in RP models without systemic metabolic effects [74]. These agents reduce neuroinflammation and promote metabolic efficiency, directly countering the metabolic deficits seen in degenerating retinas. These approaches are highlighted as promising gene-agnostic strategies, but human clinical data remain limited [75].
While multi-factor neuroprotective agents have demonstrated strong preclinical efficacy and safety, most are still progressing through proof-of-concept and safety studies. The major challenge is translating robust animal model results into meaningful clinical outcomes, with ongoing trials focused on refining delivery, patient selection, and endpoints [75,76].
3. Regulation of the Complement System
3.1. Complement Dysregulation in IRDs
The complement cascade, traditionally viewed as a defense mechanism, is increasingly recognized as a contributor to chronic retinal inflammation and photoreceptor loss in both acquired retinal degenerations and IRD. Evidence of complement dysregulation has been documented in Stargardt disease, RP, and Leber congenital amaurosis, where upregulation of C3 and C5 components parallels findings in AMD [77,78,79,80,81,82]. Overactivation of complement can trigger microglial recruitment, cytokine release, and formation of the membrane attack complex (MAC), which exacerbates cell death and accelerates retinal degeneration.
In Stargardt disease, both human donor eyes and mouse models show increased deposition of C3 fragments and MAC on the RPE, with reduced levels of complement regulatory proteins such as factor H. This dysregulation leads to chronic inflammation and accelerates photoreceptor degeneration. Gene therapy to increase complement regulation in the RPE (e.g., AAV-CRRY, AAV-RORA) reduces C3 activation, MAC deposition, and slows photoreceptor loss in mouse models, directly implicating complement activation in disease pathogenesis [77,78,83].
In RP, upregulation of complement component coincides with photoreceptor degeneration and microglial activation. C3 and its receptor CR3 on microglia mediate both protective clearance of apoptotic photoreceptors and, when dysregulated, contribute to neurotoxicity and chronic inflammation [79]. Overactivation of complement can trigger microglial recruitment, cytokine release, and MAC deposition, potentially accelerating death of stressed but potentially recoverable photoreceptors. Similar complement dysregulation is observed in Leber congenital amaurosis and other IRDs.
In summary, complement overactivation is a common pathogenic mechanism in AMD and IRDs that drives chronic inflammation and photoreceptor loss. However, the complement pathway also plays a critical role in retinal health. Therefore, therapies that restore complement activity to normal homeostatic levels – rather than fully suppressing it - offer a promising, gene-agnostic approach to reducing retinal inflammation and preserving vision in these disorders.
3.2. Complement Modulation as a Therapeutic Strategy
Current anti-complement strategies for IRDs include intravitreal pharmacologic agents targeting central complement components (C3 and C5 inhibitors) and gene therapy approaches designed to provide sustained intraocular complement regulation. Recent FDA approvals of the C3 inhibitor pegcetacoplan (Syfovre, Apellis Pharmaceuticals, Waltham, MA, USA) and the C5 inhibitor avacincaptad pegol (Izervay, Astellas Pharma, Tokyo, Japan) have advanced the management of GA secondary to dry AMD. These agents have not yet been approved or systematically studied in IRDs but reducing pathological complement activation offers a rational, inflammation-modulating approach to slow disease progression in IRDs [84].
There is strong and growing preclinical and clinical evidence that dampening complement activity within the retina can preserve retinal structure, reduce inflammation, and delay secondary degeneration in IRDs [84,85,86]. In mouse models of retinal degeneration, pharmacologic or genetic attenuation of C3 and C5 reduces complement activation, microglial recruitment, and formation of MAC, leading to decreased photoreceptor loss and preservation of retinal structure [87]. For example, downregulation of both classical and alternative pathway C3 and C5 convertases was required to reduce progressive rod and cone degeneration in models of retinal atrophy, highlighting the importance of targeting both arms of the cascade for maximal protection [87]. Building upon its approval in AMD, avacincaptad pegol had been evaluated in a phase 2b clinical trial for autosomal recessive Stargardt disease 1 (STGD1) (NCT03364153), with endpoints including safety, atrophy progression, and visual function, but clinical results are not yet available [88]. Safety data from AMD trials indicate a favorable profile, with most adverse events related to the injection procedure and an increased risk of macular neovascularization.
Gene therapy approaches using AAV vectors to deliver complement regulatory proteins such as CRRY or CR2-fH have shown robust efficacy in preclinical IRD models. In a Stargardt disease mouse model, subretinal AAV-CRRY increased local complement regulation, significantly reduced C3/C3b deposition, slowed photoreceptor degeneration, and improved visual chromophore levels, directly linking complement attack to disease progression and demonstrating structural and functional rescue [77]. Similarly, AAV-mediated delivery of CR2-fH to the RPE reduced excessive complement activation, improved RPE and Bruch’s membrane integrity, and preserved visual function in models of RPE damage [89]. Similar strategies are being explored for other IRDs and AMD, with the goal of achieving long-term complement control from a single treatment [90,91].
Overall, both pharmacologic and gene therapy-based complement modulators have shown the ability to reduce inflammation, preserve retinal structure, and delay secondary degeneration in preclinical IRD models. While human efficacy data in IRDs are still emerging, the mechanistic rationale, promsing preclinical results, and favorable safety profiles in related retinal diseases support complement dampening as a promising adjunctive or combination therapy for IRDs [84,86].
4. Combination Therapeutic Strategies
4.1. Rationale for Multi-Target Approaches
The majority of IRDs are characterized by complex, multifactorial pathophysiology involving interconnected pathogenic pathways, including oxidative stress, mitochondrial dysfunction, inflammation, excitotoxicity, and multiple forms of cell death. This complexity provides a strong rationale for multi-target therapeutic approaches that address complementary mechanisms simultaneously, potentially achieving synergistic neuroprotection that exceeds the efficacy of single-agent therapies.
The convergence of multiple pathogenic pathways in IRDs is well established. Photoreceptor cell death in IRDs is driven by calcium dyshomeostasis and excitotoxicity, oxidative stress and mitochondrial dysfunction, and neuroinflammation, mechanisms that are common to all neurodegenerative diseases [92,93,94]. These pathways are not independent but rather interact in positive feedback loops: for example, oxidative stress activates inflammatory responses, which in turn exacerbate mitochondrial dysfunction and cell death [95]. Similarly, photoreceptor degeneration triggers microglial activation and the release of pro-inflammatory cytokines such as TNFα, which further promotes cell death through multiple mechanisms [96].
Multi-target approaches offer the potential for additive effects by simultaneously interrupting multiple nodes in these interconnected pathways. The concept of combination therapy has been highly successful in treating chronic diseases such as cancer, AIDS, hypertension, glaucoma, and Parkinson's disease, where targeting complementary mechanisms produces superior outcomes compared to monotherapy [92]. In IRDs, preclinical studies have demonstrated that combined inhibition of three enzymes that act sequentially in photoreceptor degeneration can reduce cell death more effectively than single-agent treatment [97]. Similarly, modulation of multiple innate immune pathways has emerged as a promising strategy to prevent or delay vision loss in retinal degenerative diseases [90]. Furthermore, combinations of antioxidants can slow rod photoreceptor degeneration in rd1 mice more effectively than single agents, and simultaneous inhibition of both the Fas and autophagy pathways produces greater protective effects in models of autosomal dominant retinitis pigmentosa (adRP) [98,99].
Gene-agnostic, multi-target therapies are particularly attractive for IRDs given the extreme genetic heterogeneity of these disorders. With over 280 genes implicated in IRDs, developing individualized gene therapies for each mutation is impractical and costly [9]. In contrast, targeting common downstream pathways such as oxidative stress, inflammation, and metabolic failure offers potential clinical benefits to all IRD patients regardless of their specific genetic defect [100]. This approach is especially valuable for patients with advanced disease or unknown genetic mutations, where gene replacement is not feasible.
Finally, by targeting complementary mechanisms, combination therapies may achieve therapeutic effects at lower doses compared to monotherapies, thereby potentially improving safety profiles by reducing the risk of dose-dependent toxicity whilst maintaining or enhancing efficacy outcomes [92].
4.2. Promising Combinations
Multi-target therapeutic strategies combining neuroprotective, anti-inflammatory, metabolic, and immunomodulatory agents to achieve synergistic effects are under early development to treat IRDs. Combining neurotrophic factors with anti-inflammatory agents addresses two critical pathogenic mechanisms in IRDs. Neurotrophic factors such as CNTF, BDNF, and FGFs promote photoreceptor survival through activation of pro-survival signaling pathways, while anti-inflammatory agents reduce microglial activation and cytokine release [75,95]. For example, CNTF not only activates the JAK/STAT pathway in photoreceptors but also modulates Müller glial responses and reduces neuroinflammation, providing dual neuroprotective and anti-inflammatory effects. Similarly, BDNF has been shown to modulate neuroinflammation and support synaptic integrity in addition to its direct anti-apoptotic effects on photoreceptors.
PEDF is a multifunctional protein with neuroprotective, anti-angiogenic, and anti-inflammatory properties, while anti-complement treatments further targets the inflammatory cascade that contributes to photoreceptor loss [101,102]. Combining these approaches could provide potentiated benefit by simultaneously supporting photoreceptor survival and reducing complement-mediated inflammation and MAC formation [42,90]. This strategy is particularly relevant for IRDs such as Stargardt disease and RP, where complement dysregulation has been documented [103].
Combining other neurotrophic growth factors (e.g., CNTF, BDNF, GDNF) with complement pathway regulators addresses both the loss of trophic support and the chronic inflammatory environment in degenerating retinas. Growth factors promote photoreceptor survival and metabolic support, while pathological complement suppression reduces microglial recruitment, cytokine release, and MAC-mediated cell death. This combination may be particularly effective in advanced IRDs, where both photoreceptor loss and inflammation are prominent.
Oxidative stress is another central pathogenic mechanism in IRDs, and combining antioxidants with neuroprotective factors targets both the upstream cause (reactive oxygen species accumulation) and downstream consequences (cell death signaling) of retinal degeneration [13]. In a rabbit model, PEDF has demonstrated efficacy in blunting the damage to photoreceptors caused by paraquat, a known oxidant [104]. In addition to PEDF, N-acetylcysteine (NAC), a potent antioxidant, is currently in clinical trials for retinitis pigmentosa and has shown promise in reducing oxidative damage and slowing photoreceptor loss. In the phase 1 FIGHT RP trial (NCT03063021), oral NAC proved to be safe and well tolerated in patients with moderately advanced retinitis pigmentosa, with dose-dependent improvements in visual acuity and macular sensitivity over 24 weeks [105,106]. Based on these promising phase 1 results, NAC Attack, a phase 3 randomized, placebo-controlled trial (NCT05537220) is currently active but not recruiting. Evidence for oxidative stress being a key component of photoreceptor cell death in IRDs comes from the observations in end stage choroideremia patients. In these patients, the photoreceptors entirely degenerate but so too does the choroid. As a result, the foveal cones can survive for many years, indeed decades, despite advanced outer retinal loss in the periphery. The difference between choroideremia and most other end stage IRDs is that the choroid also degenerates due to the RP loss in choroideremia and this would naturally reduce the oxygen levels in the subretinal space [107,108].
Oral Tinlarebant, which is an inhibitor of vitamin A accumulation, has also shown Phase 3 success in the DRAGON Trial (NCT05244304) for Stargardt disease meeting its primary efficacy endpoint of a 36% reduction in the growth rate of retinal lesions, measured as definitely decreased autofluorescence (DDAF) by fundus autofluorescence imaging, compared with placebo. Combining NAC or Tinlarebant with neurotrophic factors such as PEDF, CNTF, or RdCVF could enhance mitochondrial integrity, reduce oxidative stress, and provide metabolic support to photoreceptors, potentially achieving synergistic neuroprotection [109].
A comprehensive, multi-target strategy using repositioned drugs that simultaneously address multiple pathogenic mechanisms shared across all retinal degenerations represents a rational and promising therapeutic approach [92]. For example, pharmacology-based drug repurposing and combination treatments have shown mutation-agnostic efficacy in retinopathy models, improving photoreceptor survival and function across diverse genetic backgrounds [110]. Other solutions such as combining a calcium channel blocker (to reduce excitotoxicity), an antioxidant (to mitigate oxidative stress), and an anti-inflammatory agent (to suppress microglial activation) could provide broad-spectrum neuroprotection across different IRDs. This strategy has shown promise in preclinical models and is being explored for translation to clinical trials [92].
In summary, multi-target therapeutic approaches for IRDs are grounded in the complex, multifactorial pathophysiology of these diseases and offer the potential for synergistic neuroprotection by simultaneously addressing oxidative stress, inflammation, metabolic failure, and cell death. The analogy would be glaucoma, where individual genetic mechanisms are not targeted but broader therapies aimed at several mechanisms that lead to neuronal cell loss are very successful at preventing disease progression. This includes, for instance, reducing aqueous production, increasing outflow through the trabecular meshwork and uveal tissues, as well as reducing intracranial pressure. A similar multifactorial approach is likely to benefit patients who have genetic predisposition to photoreceptor neuronal cell loss. Promising combination strategies include neuroprotection plus anti-inflammation, neurotrophins combined with anti-complement therapy, and antioxidants combined with growth factors, all of which are supported by robust preclinical evidence and are advancing toward clinical translation.
5. Conclusions and Future Directions
Gene-agnostic approaches offer broad therapeutic potential for IRDs due to the extreme genetic and phenotypic heterogeneity of these disorders. With over 280 causative genes and thousands of unique variants, gene-specific therapies cannot feasibly address the majority of IRD patients. Gene-agnostic strategies, such as neuroprotection, immune modulation, and metabolic support, target common downstream pathways or provide functional rescue independent of the underlying mutation. These approaches can benefit patients regardless of genotype, mild or moderate disease stage, or even when the causative gene is unknown, making them highly attractive for large-scale clinical application [8,9,111]. As clinicians who have counseled countless patients facing untreatable IRD, we view these gene-agnostic strategies as offering genuine hope to the majority of patients who currently have none.
Combination strategies may provide superior outcomes by simultaneously addressing multiple pathogenic mechanisms that converge in IRDs. Recent preclinical and translational studies demonstrate that multi-target therapies, including combinations of neuroprotective agents, anti-inflammatory drugs, and metabolic modulators, can achieve synergistic effects and broader efficacy than monotherapies. Combining neuroprotective and immunomodulatory mechanisms within a single gene therapy vector represents a compelling approach, as it could address two distinct but converging pathogenic pathways with a single intervention, potentially achieving synergistic benefit while simplifying clinical delivery. This paradigm is supported by the success of combination therapies in other chronic diseases and is increasingly reflected in IRD clinical trial pipelines. Important challenges remain, including establishing optimal dosing regimens, demonstrating long-term durability of therapeutic effect, and identifying which patients and disease stages are most likely to benefit. For instance, specific IRDs may respond to complement inhibition or neurotrophins better than others, and late stage disease could be refractory to therapy relative to early stage disease due to severe loss of photoreceptors or synapses. Rigorous clinical trial design and appropriate endpoint selection will be essential to address these questions. Fortunately, from a regulatory standpoint, a single vector expressing multiple transgenes does not trigger the increased burden that the FDA imposes on combination drug therapies [112]. However, it remains unclear if the FDA will allow an anatomic primary endpoint for IRD approval, as it has for MacTel and potentially for GA [113]. European agencies tend to only approve functional endpoints. Given the potential for a disconnect between retinal cell preservation and visual function, as for instance seen with the revakinagene taroretcel-lwey trials, a functional outcome may be required [56].
There is a critical need for continued investment in biomarker development and clinical trial infrastructure to accelerate the translation of these therapies. Validated anatomical, physiological, and functional biomarkers are essential for reliably measuring treatment effects, stratifying patients, and optimizing trial design. Emerging endpoints with advanced imaging, adaptive optics, and novel functional assays are being incorporated into IRD trials to better capture clinically meaningful changes [114,115]. Robust clinical trial networks and standardized assessment protocols will be vital for efficiently evaluating gene-agnostic and combination therapies across diverse patient populations. Initiatives such as the NCATS-led Platform Vector Gene Therapy (PaVe-GT) pilot project, which aims to streamline and standardize AAV manufacturing methods, may help reduce development costs and accelerate the path to clinical translation for gene-agnostic therapies targeting rare disease populations [116].
Looking ahead, combination therapies that integrate neuroprotection with anti-inflammatory and metabolic support may yield superior functional outcomes and longer-lasting vision preservation for IRD patients regardless of their genetic cause. By bypassing the need for individualized gene correction, gene-agnostic and combination strategies can be deployed more broadly, rapidly and affordably, addressing the unmet needs of patients we see daily who lack access to gene-specific therapies, whether due to unidentified mutations, rare genotypes without active development programs, or presentation at advanced disease stages. As the therapeutic landscape evolves, these modalities are poised to transform IRD care, offering hope for vision preservation and restoration to a much wider spectrum of patients.
Author Contributions
L.W.R., S.P.B., A.O., and K.B. performed the literature review and were major contributors to the writing, editing, and compilation of this manuscript. R.E.M., R.L.A., C.C.W., A.C.H., C.D.R., and D.E. contributed to the writing and editing of the manuscript. T.A.C. developed the initial concept for the review, performed the literature review, and was a major contributor to the writing, editing, and compilation of this manuscript. All authors have read and agreed to the published version of this manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
Ciulla would like to disclose that he is president and chief executive officer of Ikarovec Ltd in Norwich, United Kingdom, and chief medical advisor and chair of the scientific advisory board for Clearside Biomedical in Alpharetta, Georgia. Dr. Ciulla discloses financial relationships with Clearside Bio, Nanoscope, Viridian Therapeutics, and Ikarovec. Rowe reports no relevant disclosures.
References
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.C.; Fujinami-Yokokawa, Y.; Daich Varela, M.; Pontikos, N.; Kalitzeos, A.; Mahroo, O.A.; Webster, A.R.; et al. Phenotyping and Genotyping Inherited Retinal Diseases: Molecular Genetics, Clinical and Imaging Features, and Therapeutics of Macular Dystrophies, Cone and Cone-Rod Dystrophies, Rod-Cone Dystrophies, Leber Congenital Amaurosis, and Cone Dysfunction Syndromes. Prog Retin Eye Res 2024, 100, 101244. [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (AAV2-hRPE65v2) in Patients with RPE65-Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [CrossRef]
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited Retinal Diseases: Linking Genes, Disease-Causing Variants, and Relevant Therapeutic Modalities. Prog Retin Eye Res 2022, 89, 101029. [CrossRef]
- Abu Elasal, M.; Mousa, S.; Salameh, M.; Blumenfeld, A.; Khateb, S.; Banin, E.; Sharon, D. Genetic Analysis of 252 Index Cases with Inherited Retinal Diseases Using a Panel of 351 Retinal Genes. Genes (Basel) 2024, 15, 926. [CrossRef]
- Branham, K.; Samarakoon, L.; Audo, I.; Ayala, A.R.; Cheetham, J.K.; Daiger, S.P.; Dhooge, P.; Duncan, J.L.; Durham, T.A.; Fahim, A.T.; et al. Characterizing the Genetic Basis for Inherited Retinal Disease: Lessons Learned From the Foundation Fighting Blindness Clinical Consortium’s Gene Poll. Invest Ophthalmol Vis Sci 2025, 66, 12. [CrossRef]
- Rubanyi, G.M. The Future of Human Gene Therapy. Mol Aspects Med 2001, 22, 113–142. [CrossRef]
- Burnight, E.R.; Giacalone, J.C.; Cooke, J.A.; Thompson, J.R.; Bohrer, L.R.; Chirco, K.R.; Drack, A.V.; Fingert, J.H.; Worthington, K.S.; Wiley, L.A.; et al. CRISPR-Cas9 Genome Engineering: Treating Inherited Retinal Degeneration. Prog Retin Eye Res 2018, 65, 28–49. [CrossRef]
- Chew, L.A.; Iannaccone, A. Gene-Agnostic Approaches to Treating Inherited Retinal Degenerations. Front Cell Dev Biol 2023, 11, 1177838. [CrossRef]
- John, M.C.; Quinn, J.; Hu, M.L.; Cehajic-Kapetanovic, J.; Xue, K. Gene-Agnostic Therapeutic Approaches for Inherited Retinal Degenerations. Front Mol Neurosci 2022, 15, 1068185. [CrossRef]
- Orkin, S.H.; Reilly, P. MEDICINE. Paying for Future Success in Gene Therapy. Science 2016, 352, 1059–1061. [CrossRef]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a Common Non-Apoptotic Cell Death Mechanism in Hereditary Retinal Degeneration. PLoS One 2014, 9, e112142. [CrossRef]
- Tolone, A.; Sen, M.; Chen, Y.; Ueffing, M.; Arango-Gonzalez, B.; Paquet-Durand, F. Pathomechanisms of Inherited Retinal Degeneration and Perspectives for Neuroprotection. Cold Spring Harb Perspect Med 2023, 13, a041310. [CrossRef]
- Tombran-Tink, J.; Barnstable, C.J. PEDF: A Multifaceted Neurotrophic Factor. Nat Rev Neurosci 2003, 4, 628–636. [CrossRef]
- Wang, Y.; Subramanian, P.; Shen, D.; Tuo, J.; Becerra, S.P.; Chan, C.-C. Pigment Epithelium-Derived Factor Reduces Apoptosis and pro-Inflammatory Cytokine Gene Expression in a Murine Model of Focal Retinal Degeneration. ASN Neuro 2013, 5, e00126. [CrossRef]
- Hernández-Pinto, A.; Polato, F.; Subramanian, P.; Rocha-Muñoz, A. de la; Vitale, S.; de la Rosa, E.J.; Becerra, S.P. PEDF Peptides Promote Photoreceptor Survival in Rd10 Retina Models. Exp Eye Res 2019, 184, 24–29. [CrossRef]
- Holekamp, N.M.; Bouck, N.; Volpert, O. Pigment Epithelium-Derived Factor Is Deficient in the Vitreous of Patients with Choroidal Neovascularization Due to Age-Related Macular Degeneration. Am J Ophthalmol 2002, 134, 220–227. [CrossRef]
- Ogata, N.; Matsuoka, M.; Imaizumi, M.; Arichi, M.; Matsumura, M. Decrease of Pigment Epithelium-Derived Factor in Aqueous Humor with Increasing Age. Am J Ophthalmol 2004, 137, 935–936. [CrossRef]
- Rebustini, I.T.; Crawford, S.E.; Becerra, S.P. PEDF Deletion Induces Senescence and Defects in Phagocytosis in the RPE. Int J Mol Sci 2022, 23, 7745. [CrossRef]
- Jakobsen, T.S.; Adsersen, R.L.; Askou, A.L.; Corydon, T.J. Functional Roles of Pigment Epithelium-Derived Factor in Retinal Degenerative and Vascular Disorders: A Scoping Review. Invest Ophthalmol Vis Sci 2024, 65, 41. [CrossRef]
- Brook, N.; Brook, E.; Dharmarajan, A.; Chan, A.; Dass, C.R. Pigment Epithelium-Derived Factor Regulation of Neuronal and Stem Cell Fate. Exp Cell Res 2020, 389, 111891. [CrossRef]
- Comitato, A.; Subramanian, P.; Turchiano, G.; Montanari, M.; Becerra, S.P.; Marigo, V. Pigment Epithelium-Derived Factor Hinders Photoreceptor Cell Death by Reducing Intracellular Calcium in the Degenerating Retina. Cell Death Dis 2018, 9, 560. [CrossRef]
- Murakami, Y.; Ikeda, Y.; Yonemitsu, Y.; Onimaru, M.; Nakagawa, K.; Kohno, R.; Miyazaki, M.; Hisatomi, T.; Nakamura, M.; Yabe, T.; et al. Inhibition of Nuclear Translocation of Apoptosis-Inducing Factor Is an Essential Mechanism of the Neuroprotective Activity of Pigment Epithelium-Derived Factor in a Rat Model of Retinal Degeneration. Am J Pathol 2008, 173, 1326–1338. [CrossRef]
- Michelis, G.; German, O.L.; Villasmil, R.; Soto, T.; Rotstein, N.P.; Politi, L.; Becerra, S.P. Pigment Epithelium-Derived Factor (PEDF) and Derived Peptides Promote Survival and Differentiation of Photoreceptors and Induce Neurite-Outgrowth in Amacrine Neurons. J Neurochem 2021, 159, 840–856. [CrossRef]
- Pagan-Mercado, G.; Becerra, S.P. Signaling Mechanisms Involved in PEDF-Mediated Retinoprotection. Adv Exp Med Biol 2019, 1185, 445–449. [CrossRef]
- Polato, F.; Becerra, S.P. Pigment Epithelium-Derived Factor, a Protective Factor for Photoreceptors in Vivo. Adv Exp Med Biol 2016, 854, 699–706. [CrossRef]
- Ortín-Martínez, A.; Valiente-Soriano, F.J.; García-Ayuso, D.; Alarcón-Martínez, L.; Jiménez-López, M.; Bernal-Garro, J.M.; Nieto-López, L.; Nadal-Nicolás, F.M.; Villegas-Pérez, M.P.; Wheeler, L.A.; et al. A Novel in Vivo Model of Focal Light Emitting Diode-Induced Cone-Photoreceptor Phototoxicity: Neuroprotection Afforded by Brimonidine, BDNF, PEDF or bFGF. PLoS One 2014, 9, e113798. [CrossRef]
- Dixit, S.; Polato, F.; Samardzija, M.; Abu-Asab, M.; Grimm, C.; Crawford, S.E.; Becerra, S.P. PEDF Deficiency Increases the Susceptibility of Rd10 Mice to Retinal Degeneration. Experimental Eye Research 2020, 198, 108121. [CrossRef]
- Chen, Y.; Yang, J.; Geng, H.; Li, L.; Li, J.; Cheng, B.; Ma, X.; Li, H.; Hou, L. Photoreceptor Degeneration in Microphthalmia (Mitf) Mice: Partial Rescue by Pigment Epithelium-Derived Factor. Dis Model Mech 2019, 12, dmm035642. [CrossRef]
- Bernardo-Colón, A.; Bighinati, A.; Parween, S.; Debnath, S.; Piano, I.; Adani, E.; Corsi, F.; Gargini, C.; Vergara, N.; Marigo, V.; et al. H105A Peptide Eye Drops Promote Photoreceptor Survival in Murine and Human Models of Retinal Degeneration. Commun Med (Lond) 2025, 5, 81. [CrossRef]
- Vigneswara, V.; Esmaeili, M.; Deer, L.; Berry, M.; Logan, A.; Ahmed, Z. Eye Drop Delivery of Pigment Epithelium-Derived Factor-34 Promotes Retinal Ganglion Cell Neuroprotection and Axon Regeneration. Mol Cell Neurosci 2015, 68, 212–221. [CrossRef]
- Qu, Q.; Park, K.; Zhou, K.; Wassel, D.; Farjo, R.; Criswell, T.; Ma, J.-X.; Zhang, Y. Sustained Therapeutic Effect of an Anti-Inflammatory Peptide Encapsulated in Nanoparticles on Ocular Vascular Leakage in Diabetic Retinopathy. Front Cell Dev Biol 2022, 10, 1049678. [CrossRef]
- Bai, T.; Cui, B.; Xing, M.; Chen, S.; Zhu, Y.; Lin, D.; Guo, Y.; Du, M.; Wang, X.; Zhou, D.; et al. Stable Inhibition of Choroidal Neovascularization by Adeno-Associated Virus 2/8-Vectored Bispecific Molecules. Gene Ther 2024, 31, 511–523. [CrossRef]
- Valiente-Soriano, F.J.; Di Pierdomenico, J.; García-Ayuso, D.; Ortín-Martínez, A.; Miralles de Imperial-Ollero, J.A.; Gallego-Ortega, A.; Jiménez-López, M.; Villegas-Pérez, M.P.; Becerra, S.P.; Vidal-Sanz, M. Pigment Epithelium-Derived Factor (PEDF) Fragments Prevent Mouse Cone Photoreceptor Cell Loss Induced by Focal Phototoxicity In Vivo. Int J Mol Sci 2020, 21, 7242. [CrossRef]
- Imai, D.; Yoneya, S.; Gehlbach, P.L.; Wei, L.L.; Mori, K. Intraocular Gene Transfer of Pigment Epithelium-Derived Factor Rescues Photoreceptors from Light-Induced Cell Death. J Cell Physiol 2005, 202, 570–578. [CrossRef]
- Ikeda, Y.; Yonemitsu, Y.; Miyazaki, M.; Kohno, R.; Murakami, Y.; Murata, T.; Goto, Y.; Tabata, T.; Ueda, Y.; Ono, F.; et al. Acute Toxicity Study of a Simian Immunodeficiency Virus-Based Lentiviral Vector for Retinal Gene Transfer in Nonhuman Primates. Human Gene Therapy 2009, 20, 943–954. [CrossRef]
- Miyazaki, M.; Ikeda, Y.; Yonemitsu, Y.; Goto, Y.; Sakamoto, T.; Tabata, T.; Ueda, Y.; Hasegawa, M.; Tobimatsu, S.; Ishibashi, T.; et al. Simian Lentiviral Vector-Mediated Retinal Gene Transfer of Pigment Epithelium-Derived Factor Protects Retinal Degeneration and Electrical Defect in Royal College of Surgeons Rats. Gene Ther 2003, 10, 1503–1511. [CrossRef]
- Ikeda, Y.; Goto, Y.; Yonemitsu, Y.; Miyazaki, M.; Sakamoto, T.; Ishibashi, T.; Tabata, T.; Ueda, Y.; Hasegawa, M.; Tobimatsu, S.; et al. Simian Immunodeficiency Virus-Based Lentivirus Vector for Retinal Gene Transfer: A Preclinical Safety Study in Adult Rats. Gene Ther 2003, 10, 1161–1169. [CrossRef]
- Stable Retinal Gene Expression in Nonhuman Primates via Subretinal Injection of SIVagm-Based Lentiviral Vectors | Human Gene Therapy Available online: https://www.liebertpub.com/doi/10.1089/hum.2009.009 (accessed on 28 November 2025).
- Rasmussen, H.; Chu, K.W.; Campochiaro, P.; Gehlbach, P.L.; Haller, J.A.; Handa, J.T.; Nguyen, Q.D.; Sung, J.U. Clinical Protocol. An Open-Label, Phase I, Single Administration, Dose-Escalation Study of ADGVPEDF.11D (ADPEDF) in Neovascular Age-Related Macular Degeneration (AMD). Hum Gene Ther 2001, 12, 2029–2032.
- Hisai, T.; Murakami, Y.; Kusano, K.; Kobayakawa, Y.; Ikeda, Y. Phase 1/2a Clinical Trial Protocol for Lentiviral Vector-Based Retinal Gene Therapy to Slow the Progression of Retinitis Pigmentosa. Methods Mol Biol 2026, 2974, 239–248. [CrossRef]
- Warner, E.F.; Vaux, L.; Boyd, K.; Widdowson, P.S.; Binley, K.M.; Osborne, A. Ocular Delivery of Pigment Epithelium-Derived Factor (PEDF) as a Neuroprotectant for Geographic Atrophy. Aging Dis 2024, 15, 2003–2007. [CrossRef]
- Cayouette, M.; Smith, S.B.; Becerra, S.P.; Gravel, C. Pigment Epithelium-Derived Factor Delays the Death of Photoreceptors in Mouse Models of Inherited Retinal Degenerations. Neurobiol Dis 1999, 6, 523–532. [CrossRef]
- Amaral, J.; Fariss, R.N.; Campos, M.M.; Robison, W.G.; Kim, H.; Lutz, R.; Becerra, S.P. Transscleral-RPE Permeability of PEDF and Ovalbumin Proteins: Implications for Subconjunctival Protein Delivery. Invest Ophthalmol Vis Sci 2005, 46, 4383–4392. [CrossRef]
- Wang, J.-H.; Zhan, W.; Gallagher, T.L.; Gao, G. Recombinant Adeno-Associated Virus as a Delivery Platform for Ocular Gene Therapy: A Comprehensive Review. Mol Ther 2024, 32, 4185–4207. [CrossRef]
- Duarte, F.; Arsenijevic, Y. Precision Gene Therapy: Tailoring rAAV-Mediated Gene Therapies for Inherited Retinal Dystrophies (IRDs). Mol Aspects Med 2025, 106, 101424. [CrossRef]
- Rhee, K.D.; Nusinowitz, S.; Chao, K.; Yu, F.; Bok, D.; Yang, X.-J. CNTF-Mediated Protection of Photoreceptors Requires Initial Activation of the Cytokine Receptor Gp130 in Müller Glial Cells. Proc Natl Acad Sci U S A 2013, 110, E4520-4529. [CrossRef]
- Peterson, W.M.; Wang, Q.; Tzekova, R.; Wiegand, S.J. Ciliary Neurotrophic Factor and Stress Stimuli Activate the Jak-STAT Pathway in Retinal Neurons and Glia. J Neurosci 2000, 20, 4081–4090. [CrossRef]
- Xue, W.; Cojocaru, R.I.; Dudley, V.J.; Brooks, M.; Swaroop, A.; Sarthy, V.P. Ciliary Neurotrophic Factor Induces Genes Associated with Inflammation and Gliosis in the Retina: A Gene Profiling Study of Flow-Sorted, Müller Cells. PLoS One 2011, 6, e20326. [CrossRef]
- Cayouette, M.; Behn, D.; Sendtner, M.; Lachapelle, P.; Gravel, C. Intraocular Gene Transfer of Ciliary Neurotrophic Factor Prevents Death and Increases Responsiveness of Rod Photoreceptors in the Retinal Degeneration Slow Mouse. J Neurosci 1998, 18, 9282–9293. [CrossRef]
- Cayouette, M.; Gravel, C. Adenovirus-Mediated Gene Transfer of Ciliary Neurotrophic Factor Can Prevent Photoreceptor Degeneration in the Retinal Degeneration (Rd) Mouse. Hum Gene Ther 1997, 8, 423–430. [CrossRef]
- Tao, W.; Wen, R.; Goddard, M.B.; Sherman, S.D.; O’Rourke, P.J.; Stabila, P.F.; Bell, W.J.; Dean, B.J.; Kauper, K.A.; Budz, V.A.; et al. Encapsulated Cell-Based Delivery of CNTF Reduces Photoreceptor Degeneration in Animal Models of Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 2002, 43, 3292–3298.
- l.ferguson@neurotechusa.com Neurotech’s ENCELTOTM (Revakinagene Taroretcel-Lwey) Approved by the FDA for the Treatment of Adults with Idiopathic Macular Telangiectasia Type 2 (MacTel) Available online: https://www.neurotechpharmaceuticals.com/neurotechs-enceltotm-revakinagene-taroretcel-lwey-approved-by-the-fda-for-the-treatment-of-macular-telangiectasia-type-2-mactel/ (accessed on 13 April 2025).
- Chew, E.Y.; Gillies, M.; Jaffe, G.J.; Gaudric, A.; Egan, C.; Constable, I.; Clemons, T.; Aaberg, T.; Manning, D.C.; Hohman, T.C.; et al. Cell-Based Ciliary Neurotrophic Factor Therapy for Macular Telangiectasia Type 2. NEJM Evidence 2025, 4, EVIDoa2400481. [CrossRef]
- Birch, D.G.; Weleber, R.G.; Duncan, J.L.; Jaffe, G.J.; Tao, W.; Ciliary Neurotrophic Factor Retinitis Pigmentosa Study Groups Randomized Trial of Ciliary Neurotrophic Factor Delivered by Encapsulated Cell Intraocular Implants for Retinitis Pigmentosa. Am J Ophthalmol 2013, 156, 283-292.e1. [CrossRef]
- Birch, D.G.; Bennett, L.D.; Duncan, J.L.; Weleber, R.G.; Pennesi, M.E. Long-Term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am J Ophthalmol 2016, 170, 10–14. [CrossRef]
- Zhang, K.; Hopkins, J.J.; Heier, J.S.; Birch, D.G.; Halperin, L.S.; Albini, T.A.; Brown, D.M.; Jaffe, G.J.; Tao, W.; Williams, G.A. Ciliary Neurotrophic Factor Delivered by Encapsulated Cell Intraocular Implants for Treatment of Geographic Atrophy in Age-Related Macular Degeneration. Proc Natl Acad Sci U S A 2011, 108, 6241–6245. [CrossRef]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [CrossRef]
- Byrne, L.C.; Dalkara, D.; Luna, G.; Fisher, S.K.; Clérin, E.; Sahel, J.-A.; Léveillard, T.; Flannery, J.G. Viral-Mediated RdCVF and RdCVFL Expression Protects Cone and Rod Photoreceptors in Retinal Degeneration. J Clin Invest 2015, 125, 105–116. [CrossRef]
- Marie, M.; Churet, L.; Gautron, A.-S.; Farjo, R.; Mizuyoshi, K.; Stevenson, V.; Khabou, H.; Léveillard, T.; Sahel, J.-A.; Lorget, F. Preclinical Safety and Biodistribution of SPVN06, a Novel Gene- and Mutation-Independent Gene Therapy for Rod-Cone Dystrophies. Gene Ther 2025. [CrossRef]
- Mei, X.; Chaffiol, A.; Kole, C.; Yang, Y.; Millet-Puel, G.; Clérin, E.; Aït-Ali, N.; Bennett, J.; Dalkara, D.; Sahel, J.-A.; et al. The Thioredoxin Encoded by the Rod-Derived Cone Viability Factor Gene Protects Cone Photoreceptors Against Oxidative Stress. Antioxid Redox Signal 2016, 24, 909–923. [CrossRef]
- Noel, J.; Jalligampala, A.; Marussig, M.; Vinot, P.-A.; Marie, M.; Butler, M.; Lorget, F.; Boissel, S.; Leveillard, T.D.; Sahel, J.A.; et al. SPVN06, a Novel Mutation-Independent AAV-Based Gene Therapy, Protects Cone Degeneration in a Pig Model of Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci. 2021, 62, 1189–1189.
- Audo, I.; Barale, P.-O.; Everett, L.A.; Lauer, A.K.; Martel, J.N.; Blouin, L.; Gautron, A.-S.; Messeca, N.; Loustalot, F.; Meur, A.L.; et al. PRODYGY: A First-in-Human Trial of Rod-Derived Cone Viability Factor (RdCVF) Gene Therapy in Subjects with Rod-Cone Dystrophy. Invest. Ophthalmol. Vis. Sci. 2025, 66, 1442–1442.
- Abed, E.; Corbo, G.; Falsini, B. Neurotrophin Family Members as Neuroprotectants in Retinal Degenerations. BioDrugs 2015, 29, 1–13. [CrossRef]
- Okoye, G.; Zimmer, J.; Sung, J.; Gehlbach, P.; Deering, T.; Nambu, H.; Hackett, S.; Melia, M.; Esumi, N.; Zack, D.J.; et al. Increased Expression of Brain-Derived Neurotrophic Factor Preserves Retinal Function and Slows Cell Death from Rhodopsin Mutation or Oxidative Damage. J Neurosci 2003, 23, 4164–4172. [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, Growth Factors and BDNF-TrkB Signalling in Retinal Degeneration. Int J Mol Sci 2016, 17, 1584. [CrossRef]
- Osborne, A.; Khatib, T.Z.; Songra, L.; Barber, A.C.; Hall, K.; Kong, G.Y.X.; Widdowson, P.S.; Martin, K.R. Neuroprotection of Retinal Ganglion Cells by a Novel Gene Therapy Construct That Achieves Sustained Enhancement of Brain-Derived Neurotrophic Factor/Tropomyosin-Related Kinase Receptor-B Signaling. Cell Death Dis 2018, 9, 1007. [CrossRef]
- Osborne, A.; Khatib, T.Z.; Whitehead, M.; Mensah, T.; Yazdouni, S.; Nieuwenhuis, B.; Ali, Z.; Ching, J.; Watt, R.; Kishi, N.; et al. Dose-Ranging and Further Therapeutic Evaluation of a Bicistronic Humanized TrkB-BDNF Gene Therapy for Glaucoma in Rodents. Mol Neurodegener Adv 2025, 1, 3. [CrossRef]
- Cao, Y.; Yin, X.; Wu, L.; Huang, D.; Wang, Z.; Wu, F.; Jiang, J.; Chen, G.; Wang, Q. High-Efficiency Ocular Delivery of Brain-Derived Neurotrophic Factor and Oligomycin for Neuroprotection in Glaucoma. Adv Mater 2025, 37, e2500623. [CrossRef]
- Di Pierdomenico, J.; Scholz, R.; Valiente-Soriano, F.J.; Sánchez-Migallón, M.C.; Vidal-Sanz, M.; Langmann, T.; Agudo-Barriuso, M.; García-Ayuso, D.; Villegas-Pérez, M.P. Neuroprotective Effects of FGF2 and Minocycline in Two Animal Models of Inherited Retinal Degeneration. Invest Ophthalmol Vis Sci 2018, 59, 4392–4403. [CrossRef]
- Rana, D.; Dhankhar, S.; Chauhan, R.; Saini, M.; Singh, R.; Kumar, P.; Singh, T.G.; Chauhan, S.; Devi, S. Targeting Neurotrophic Dysregulation in Diabetic Retinopathy: A Novel Therapeutic Avenue. Mol Biol Rep 2025, 52, 570. [CrossRef]
- Arranz-Romera, A.; Hernandez, M.; Checa-Casalengua, P.; Garcia-Layana, A.; Molina-Martinez, I.T.; Recalde, S.; Young, M.J.; Tucker, B.A.; Herrero-Vanrell, R.; Fernandez-Robredo, P.; et al. A Safe GDNF and GDNF/BDNF Controlled Delivery System Improves Migration in Human Retinal Pigment Epithelial Cells and Survival in Retinal Ganglion Cells: Potential Usefulness in Degenerative Retinal Pathologies. Pharmaceuticals (Basel) 2021, 14, 50. [CrossRef]
- Sánchez-Cruz, A.; Hernández-Pinto, A.; Lillo, C.; Isiegas, C.; Marchena, M.; Lizasoain, I.; Bosch, F.; de la Villa, P.; Hernández-Sánchez, C.; de la Rosa, E.J. Insulin Receptor Activation by Proinsulin Preserves Synapses and Vision in Retinitis Pigmentosa. Cell Death Dis 2022, 13, 383. [CrossRef]
- Pan, W.W.; Wubben, T.J.; Zacks, D.N. Promising Therapeutic Targets for Neuroprotection in Retinal Disease. Curr Opin Ophthalmol 2025, 36, 247–252. [CrossRef]
- Hill, D.; Compagnoni, C.; Cordeiro, M.F. Investigational Neuroprotective Compounds in Clinical Trials for Retinal Disease. Expert Opin Investig Drugs 2021, 30, 571–577. [CrossRef]
- Lenis, T.L.; Sarfare, S.; Jiang, Z.; Lloyd, M.B.; Bok, D.; Radu, R.A. Complement Modulation in the Retinal Pigment Epithelium Rescues Photoreceptor Degeneration in a Mouse Model of Stargardt Disease. Proc Natl Acad Sci U S A 2017, 114, 3987–3992. [CrossRef]
- Hu, J.; Pauer, G.J.; Hagstrom, S.A.; Bok, D.; DeBenedictis, M.J.; Bonilha, V.L.; Hollyfield, J.G.; Radu, R.A. Evidence of Complement Dysregulation in Outer Retina of Stargardt Disease Donor Eyes. Redox Biol 2020, 37, 101787. [CrossRef]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-Dependent Microglial Clearance Protects Photoreceptors in Retinitis Pigmentosa. J Exp Med 2019, 216, 1925–1943. [CrossRef]
- Kim, B.J.; Mastellos, D.C.; Li, Y.; Dunaief, J.L.; Lambris, J.D. Targeting Complement Components C3 and C5 for the Retina: Key Concepts and Lingering Questions. Prog Retin Eye Res 2021, 83, 100936. [CrossRef]
- de Jong, S.; Tang, J.; Clark, S.J. Age-Related Macular Degeneration: A Disease of Extracellular Complement Amplification. Immunol Rev 2023, 313, 279–297. [CrossRef]
- Armento, A.; Ueffing, M.; Clark, S.J. The Complement System in Age-Related Macular Degeneration. Cell. Mol. Life Sci. 2021, 78, 4487–4505. [CrossRef]
- Akula, M.; McNamee, S.M.; Love, Z.; Nasraty, N.; Chan, N.P.M.; Whalen, M.; Avola, M.O.; Olivares, A.M.; Leehy, B.D.; Jelcick, A.S.; et al. Retinoic Acid Related Orphan Receptor α Is a Genetic Modifier That Rescues Retinal Degeneration in a Mouse Model of Stargardt Disease and Dry AMD. Gene Ther 2024, 31, 413–421. [CrossRef]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement Inhibition as a Therapeutic Strategy in Retinal Disorders. Expert Opin Biol Ther 2019, 19, 335–342. [CrossRef]
- Hussain, R.M.; Ciulla, T.A.; Berrocal, A.M.; Gregori, N.Z.; Flynn, H.W.; Lam, B.L. Stargardt Macular Dystrophy and Evolving Therapies. Expert Opin Biol Ther 2018, 18, 1049–1059. [CrossRef]
- West, E.E.; Woodruff, T.; Fremeaux-Bacchi, V.; Kemper, C. Complement in Human Disease: Approved and up-and-Coming Therapeutics. Lancet 2024, 403, 392–405. [CrossRef]
- Katschke, K.J.; Xi, H.; Cox, C.; Truong, T.; Malato, Y.; Lee, W.P.; McKenzie, B.; Arceo, R.; Tao, J.; Rangell, L.; et al. Classical and Alternative Complement Activation on Photoreceptor Outer Segments Drives Monocyte-Dependent Retinal Atrophy. Sci Rep 2018, 8, 7348. [CrossRef]
- Astellas Pharma Global Development, Inc. A Phase 2b Randomized, Double-Masked, Controlled Trial to Establish the Safety and Efficacy of ZimuraTM (Complement C5 Inhibitor) Compared to Sham in Subjects With Autosomal Recessive Stargardt Disease; clinicaltrials.gov, 2025;
- Annamalai, B.; Parsons, N.; Nicholson, C.; Obert, E.; Jones, B.; Rohrer, B. Subretinal Rather Than Intravitreal Adeno-Associated Virus-Mediated Delivery of a Complement Alternative Pathway Inhibitor Is Effective in a Mouse Model of RPE Damage. Invest Ophthalmol Vis Sci 2021, 62, 11. [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of Three Key Innate Immune Pathways for the Most Common Retinal Degenerative Diseases. EMBO Mol Med 2018, 10, e8259. [CrossRef]
- Guymer, R.H.; Campbell, T.G. Age-Related Macular Degeneration. Lancet 2023, 401, 1459–1472. [CrossRef]
- Maneu, V.; Lax, P.; De Diego, A.M.G.; Cuenca, N.; García, A.G. Combined Drug Triads for Synergic Neuroprotection in Retinal Degeneration. Biomed Pharmacother 2022, 149, 112911. [CrossRef]
- Comitato, A.; Schiroli, D.; La Marca, C.; Marigo, V. Differential Contribution of Calcium-Activated Proteases and ER-Stress in Three Mouse Models of Retinitis Pigmentosa Expressing P23H Mutant RHO. Adv Exp Med Biol 2019, 1185, 311–316. [CrossRef]
- Kutluer, M.; Huang, L.; Marigo, V. Targeting Molecular Pathways for the Treatment of Inherited Retinal Degeneration. Neural Regen Res 2020, 15, 1784–1791. [CrossRef]
- Pinilla, I.; Maneu, V.; Campello, L.; Fernández-Sánchez, L.; Martínez-Gil, N.; Kutsyr, O.; Sánchez-Sáez, X.; Sánchez-Castillo, C.; Lax, P.; Cuenca, N. Inherited Retinal Dystrophies: Role of Oxidative Stress and Inflammation in Their Physiopathology and Therapeutic Implications. Antioxidants (Basel) 2022, 11, 1086. [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int J Mol Sci 2021, 22, 2096. [CrossRef]
- Dong, Y.; Yan, J.; Yang, M.; Xu, W.; Hu, Z.; Paquet-Durand, F.; Jiao, K. Inherited Retinal Degeneration: Towards the Development of a Combination Therapy Targeting Histone Deacetylase, Poly (ADP-Ribose) Polymerase, and Calpain. Biomolecules 2023, 13, 581. [CrossRef]
- Sanz, M.M.; Johnson, L.E.; Ahuja, S.; Ekström, P. a. R.; Romero, J.; van Veen, T. Significant Photoreceptor Rescue by Treatment with a Combination of Antioxidants in an Animal Model for Retinal Degeneration. Neuroscience 2007, 145, 1120–1129. [CrossRef]
- Yang, M.; Yao, J.; Jia, L.; Kocab, A.J.; Zacks, D.N. Neuroprotection of Photoreceptors by Combined Inhibition of Both Fas and Autophagy Pathways in P23H Mice. Cell Death Dis 2025, 16, 469. [CrossRef]
- Martinez Velazquez, L.A.; Ballios, B.G. The Next Generation of Molecular and Cellular Therapeutics for Inherited Retinal Disease. Int J Mol Sci 2021, 22, 11542. [CrossRef]
- Zhang, S.X.; Wang, J.J.; Gao, G.; Shao, C.; Mott, R.; Ma, J. Pigment Epithelium-Derived Factor (PEDF) Is an Endogenous Antiinflammatory Factor. FASEB J 2006, 20, 323–325. [CrossRef]
- Park, K.; Jin, J.; Hu, Y.; Zhou, K.; Ma, J. Overexpression of Pigment Epithelium-Derived Factor Inhibits Retinal Inflammation and Neovascularization. Am J Pathol 2011, 178, 688–698. [CrossRef]
- Kaur, G.; Singh, N.K. The Role of Inflammation in Retinal Neurodegeneration and Degenerative Diseases. Int J Mol Sci 2021, 23, 386. [CrossRef]
- Kanan, Y.; Zhang, J.; Bernardo-Colón, A.; Debnath, S.; Khan, M.; Becerra, S.P.; Campochiaro, P.A. Rabbit Model of Oxidative Stress-Induced Retinal Degeneration. Free Radic Biol Med 2025, 231, 48–56. [CrossRef]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-Acetylcysteine Improves Cone Function in Retinitis Pigmentosa Patients in Phase I Trial. J Clin Invest 2020, 130, 1527–1541. [CrossRef]
- Kong, X.; Hafiz, G.; Wehling, D.; Akhlaq, A.; Campochiaro, P.A. Locus-Level Changes in Macular Sensitivity in Patients with Retinitis Pigmentosa Treated with Oral N-Acetylcysteine. Am J Ophthalmol 2021, 221, 105–114. [CrossRef]
- Khan, I.; Ramzan, F.; Tayyab, H.; Damji, K.F. Rekindling Vision: Innovative Strategies for Treating Retinal Degeneration. Int J Mol Sci 2025, 26, 4078. [CrossRef]
- Leinonen, H.; Zhang, J.; Occelli, L.M.; Seemab, U.; Choi, E.H.; L P Marinho, L.F.; Querubin, J.; Kolesnikov, A.V.; Galinska, A.; Kordecka, K.; et al. A Combination Treatment Based on Drug Repurposing Demonstrates Mutation-Agnostic Efficacy in Pre-Clinical Retinopathy Models. Nat Commun 2024, 15, 5943. [CrossRef]
- Zuzic, M.; Striebel, J.; Pawlick, J.S.; Sharma, K.; Holz, F.G.; Busskamp, V. Gene-Independent Therapeutic Interventions to Maintain and Restore Light Sensitivity in Degenerating Photoreceptors. Prog Retin Eye Res 2022, 90, 101065. [CrossRef]
- Thirunavukarasu, A.J.; Raji, S.; Cehajic Kapetanovic, J. Visualising Treatment Effects in Low-Vision Settings: Proven and Potential Endpoints for Clinical Trials of Inherited Retinal Disease Therapies. Gene Ther 2025. [CrossRef]
- Guidelines on Clinical Assessment of Patients with Inherited Retinal Degenerations - 2022 Available online: https://www.aao.org/education/clinical-statement/guidelines-on-clinical-assessment-of-patients-with (accessed on 28 November 2025).
- PaVe-GT Available online: https://pave-gt.ncats.nih.gov (accessed on 27 December 2025).

Table 1.
Gene-Agnostic Therapeutic Approaches for Inherited Retinal Diseases.
| Therapeutic Factor | Category | Key Mechanism of Action | Development Status |
|---|---|---|---|
| PEDF | Neuroprotection | Neurotrophic, anti-angiogenic, and anti-inflammatory properties; protects photoreceptors via suppression of apoptotic pathways | Preclinical |
| CNTF | Neuroprotection | Activates neuroprotective signaling via STAT3 pathway; promotes photoreceptor survival | FDA Approved (Encelto for MacTel Type 2) |
| RdCVF | Neuroprotection | Rod-secreted factor promoting cone survival; stimulates aerobic glycolysis in cones | Clinical trials |
| BDNF | Neuroprotection | Promotes neuronal survival via TrkB receptor signaling; supports photoreceptor viability | Preclinical |
| FGF | Neuroprotection | Multiple growth factors supporting retinal neuron survival and development | Preclinical |
| GDNF | Neuroprotection | Glial-derived factor promoting photoreceptor survival | Preclinical |
| Proinsulin | Neuroprotection | Activates survival pathways; reduces oxidative stress in photoreceptors | Preclinical |
| Complement C3 inhibitors | Immunomodulation | Blocks complement cascade at C3 level; reduces inflammation and cell damage | Clinical trials (GA approved) |
| Complement C5 inhibitors | Immunomodulation | Inhibits terminal complement pathway; prevents membrane attack complex formation | Clinical trials (GA approved) |
| Soluble complement regulators | Immunomodulation | Gene therapy delivery of endogenous complement regulatory proteins | Preclinical/Early clinical |
Abbreviations: PEDF, pigment epithelium-derived factor; CNTF, ciliary neurotrophic factor; RdCVF, rod-derived cone viability factor; BDNF, brain-derived neurotrophic factor; FGF, fibroblast growth factor; GDNF, glial cell line-derived neurotrophic factor; GA, geo-graphic atrophy; MacTel, macular telangiectasia.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



