
Submitted:
06 January 2026
Posted:
07 January 2026
You are already at the latest version
Abstract
FAM3A, FAM3B, FAM3C and FAM3D are members of “family with sequence similarity 3” (FAM3) gene family, an emerging class of cytokine-like proteins with a unique structural globular beta-beta-alpha fold and distinct biological functions. With widespread expression in tissue, organs and in many cell types, their specific roles in human diseases have been the focus of much research. FAM3A acts as a positive regulator of metabolic health, typically activating canonical pro-survival and metabolic pathways. FAM3B, also called PANDER (PANcreatic DERived Factor) exerts critical physiological functions in the regulation of glycemic levels via promotion of hepatic glucose production and pancreatic beta-cell insulin secretion. FAM3C, also named ILEI (Interleukin-like EMT inducer), is involved as inducer of epithelial-mesenchymal transition (EMT) and cancer metastasis, as well as osteoblast differentiation and bone mineralization. FAM3D is a gut secreted protein and potential regulator of gastrointestinal homeostasis and microbiota-induced inflammation. Here we provide an overview of previous studies supporting that FAM3 proteins can binding to putative membrane receptors and co-partners, including Fibroblast Growth Factor Receptor (FGFR), Leukemia Inhibitory Factor Receptor (LIFR), Formyl Peptide Receptor (FPR1/2), to activate diverse downstream signaling pathways on different cellular contexts. Basic and clinical studies suggest that FAM3 family influence both obesity, diabetes, and other metabolic disorders, thus its expression may have diagnostic potential. The differential and often cancer-specific expression patterns make members of the FAM3 family promising candidates for biomarkers and therapeutic targets of some types of neoplasia.
Keywords:
FAM3 family
; diabetes
; cancer
; metabolism reprogramming
; EMT
; receptors
1. Introduction
Aerobic glycolysis, or Warburg effect, is a metabolic process where glucose is converted to lactate in the presence of oxygen, a phenomenon first observed by Otto Warburg around 100 year ago [1]. The Warburg effect is now recognized as a specific metabolic reprogramming in which tumor cells achieve a fine-tuned balance between ATP (Adenosine Triphosphate) and mitochondrial radical oxygen species (ROS) production [2]. This balance maintains an optimal biosynthesis of nucleotides, lipids, and proteins to meet the needs for cell proliferation and survival ad aeternum. The metabolic reprogramming observed in cancer cells has a significant and striking overlap with systemic metabolic perturbations in the patients suffering chronic and metabolic diseases such as diabetes, cardiovascular disease, and obesity [3,4,5,6]. Epidemiological studies demonstrated that patients with Type 2 diabetes mellitus (T2DM) have an increased risk of mortality from many different types of cancer, including liver, colon, pancreas, bladder and female breast cancers [6,7]. The possible biological link T2DM and carcinogenesis is hyperinsulinemia, which is caused by either resistance to endogenous insulin or exogenous insulin levels, hyperglycemia, and fat-induced chronic inflammation [6,7,8]. The clinical epidemiological studies have also confirmed the link between obesity and high risk of developing several types of cancers, including colorectal, endometrial, kidney, esophageal, breast, liver, bladder, pancreatic, thyroid, and liver cancers [5,6,7,8]. The β-cell dysfunction in obesity and T2DM cause chronic hyperglycemia and insulin resistance, which fuel multifactorial and multistep pathways to cancer cell growth, as tumor cells prefer glucose and do not become insulin resistant like normal cells [9].
Many factors and nutrients produced by malignant cells and non-cancer cells within an acidic microenvironment can shift cancer cell morphology to enhance their potential to spread [10,11]. To do this cancer cells can adopt cancer stem cell (CSC)-like properties which involve a process named epithelial-mesenchymal transition (EMT) or MET, depending on the context and stimuli [12]. This process is regulated by the transcription factors, including Zinc finger protein SNAI1 (Snail), SNAI2 (Slug), Twist-related protein (Twist), and Zinc finger E-box-binding homeobox 1 (ZEB1/2) in response to transforming growth factor beta (TGF-β) [13,14]. During this morphological hybrid process, cancer cells lose E-cadherin (epithelial feature) and gain N-cadherin (mesenchymal feature) expression while vimentin is overexpressed [13,14]. EMT is stimulated by TGF-β via a canonical pathway mediated by SMAD4 phosphorylation or via non-canonical pathway mediated by the phosphatidylinositol 3-kinase (PI3K)/AKT (protein kinase B, PKB)/mechanistic target of rapamycin (mTOR). However, studies have demonstrated that many other growth factors as well as metabolic conditions, including those conditions that drive metabolic reprogramming, can induce or revert EMT to promote proliferation, metastasis and drug resistance in cancer cells [10,11,13,14].
In 2002, Zhu and collaborators developed a computational algorithm named Ostensible Folding Recognizer (ORF) to search for predicted four-helical bundle protein sequences that form the secondary structure common to the cytokine family [15]. Surprisingly a new family of “cytokine-like” proteins designated the Family with sequence similarity 3 or FAM3 was characterized. These “cytokine-like” proteins with homology to FAM3A mRNA were named FAM3B, FAM3C, and FAM3D [15]. The crystal structure X-ray studies of murine recombinant FAM3B/PANDER protein revealed a unique globular β-β-α fold, which comprises two anti-parallel β sheets lined by three short helices arranged to form a highly conserved water-filled cavity [16]. This fold does not resemble the four-helix bundle of the classical cytokine family, such as the TNF family, or the cysteine-knot growth factor family [16]. FAM3 family member transcripts have a wide tissue distribution and their products play pleiotropic roles in mitochondrial ATP production, regulation of glucose and lipid metabolism [17]. The deregulation of these mechanisms leads to various pathologies, including cancer [17,18]. In this review, we update on the FAM3 cytokine-like proteins, their putative receptors and signaling pathways that support their canonical and non-canonical molecular functions in the regulation of both physiological processes in chronic metabolic diseases and EMT in cancers.
2. FAM3A
FAM3A is a mitochondrial protein ubiquitously expressed among all cells of tissues and organs of humans and rodents [19,20]. FAM3A participates in mitochondrial ATP production increasing oxidative phosphorylation efficiency and mitochondrial biogenesis and respiratory chain activity [19,20]. Human FAM3A gene is located at chromosome X and is translated in a protein that contains 230 amino acid residues. The expression of this gene may be regulated by peroxisome proliferator-activated receptor gamma (PPAR-γ). FAM3A suppresses hepatic gluconeogenesis and lipogenesis by activating protein kinase B (Akt) and adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) pathways [21]. A reduced hepatic FAM3A expression causes fasting hyperglycemia and hepatic lipid deposition associated with nonalcoholic liver disease [22,23]. Beyond liver, FAM3A also regulates the functions of vascular smooth muscle cells (VSMCs) and adipocytes by modulating ATP-Akt pathways [24,25,26,27]. C/EBPβ is a transcriptional activator of FAM3A and its overexpression significantly inhibited the efficiency of preadipocytes to differentiate into adipocytes [23]. FAM3A protects neuronal cells against apoptosis triggered by oxidative and endoplasmic reticulum (ER) stress via PI3K/Akt pathways [28,29]. The FAM3A knockout mice model displayed a reduced ATP production in hepatocytes, smooth muscle cells, and endothelial cells [22]. The FAM3A gene deficiency increased macrophage and neutrophil cell infiltration, and facilitated tubular cell pyroptosis after kidney and liver ischemia/reperfusion injury [30]. FAM3B
FAM3B is a secreted protein that contains 235 amino acids, a secretion signal peptide (1-29) and its internal sequence two disulfide bonds between C63-C69 and C91-C229 are responsible for the formation of intermolecular disulfide bridges and protein folding [15]. The purified protein displays two molecules of 20 and 21 kDa [15]. The murine FAM3B solved to 2.3A° resolution revealed a ββα fold, which is strikingly different from the classical four-helix bundle of the cytokine family members [16]. FAM3B has a marked expression in pancreatic tissue, and is also highly expressed in organs such as prostate, testis, small and large intestines, stomach, kidney, heart and brain [31]. FAM3B is expressed in β-cells of islets and co-localizes within insulin granules and is co-secreted with insulin and because of it, the protein was first named PANDER (PANcreatic DERived Factor) [17,32,33,34]. FAM3B full-length RNA is sensitively regulated by glucose. Upon glucose stimulation, the cleaved protein precursor is increased in both insulinoma cells and primary islets [17,35,36]. Glucose induces FAM3B gene expression via multiple signaling pathways, which include Ca2+ protein kinase A (PKA), Ca2+ protein kinase C (PKC), extracellular signal-regulated kinase 1 (ERK1/2), and cAMP-responsive element-binding protein (CREB). The FAM3B expression is also stimulated via phosphoinositide 3-kinase (PI3K) and radical oxygen species (ROS)-related pathways [35,36,37]. The first studies about FAM3B/PANDER metabolic activities found that PANDER protein induces apoptosis in insulin-secreting beta-cells via a caspase-3-dependent mechanism [33,35,38]. This result is controversial since this effect could be attributed to glucose toxicity (glycotoxicity). Moreover, one study showed that siRNA targeting human FAM3B message decreases cell viability and triggers apoptosis in various types of cancer cell lines [39]. FAM3B gene knockout mice are viable and fertile and display normal endocrine pancreatic morphology [40,41,42]. However, they display a fairly paradoxical metabolic phenotype depending on genetic background. The initial PANDER knockout transgenic mice (PANKO) on a mixed genetic background displayed a defective insulin secretion in response to glucose and arginine [40,41,42]. A pure-bred PANDER knockout made on a C57BL/6 mouse background (PANKO-C57) showed enhanced glucose tolerance compared to wild-type mice [43]. The animal liver seems more sensitive to insulin (at least under fasting), but also accumulates more fat/glycogen when stimulated, indicating a reprogramming of hepatic insulin signaling. In addition, males and females had increased weight gain. In resume, targeted disruption PANDER/FAM3B gene demonstrated that protein contributes to maintain normal pancreatic β-cell function regulating hepatic gluconeogenic gene expression [40,41,42,43]. Further investigations in these mice models should reveal additional and novel findings on the FAM3B physiological activities.
3. FAM3C
FAM3C gene is located on chromosome 7 and is composed of 10 exons and 9 introns [15]. The resolved crystallized structure of FAM3C protein shows that this molecule forms a new ββα fold, which is unique and conserved within the family members [16]. The FAM3C protein contains 227 amino acids and can exist as a monomer or dimer in certain types of tumor cells, but the formation of the active dimer appears to be essential for its function in tumor growth and metastasis [44,45]. FAM3C has four cysteine residues that form two disulfide bridges (Cys 58-86, and Cys 64-221). In addition to these four conserved cysteines, FAM3C carries an additional cysteine (Cys 185), which is responsible for its dimerization [44,45]. A protein-protein docking study with a refined 1.8Å structure containing four α-helices, nine β-sheets, two disulfide bridges, allowed the identification of four potential FAM3C inhibitors: N-hydroxy-L-arginine, normetanephrine, deazaneplanocin and PR-619 [46].
The FAM3C protein is expressed in specialized epithelial cells in the pancreas, breast, duodenum, and salivary gland [47]. Several types of cancer also express FAM3C, most notably breast, lung, stomach, and esophageal cancers. There are two forms of FAM3C: secreted and intracellular. The intracellular form of FAM3C contains 24-amino acid signal peptide that directs it to the endoplasmic reticulum and Golgi complex. This peptide is later cleaved by plasmin, which positively regulates the secretion of FAM3C after the urokinase-type plasminogen activator receptor (uPAR) signaling in cell membrane [48]. The endogenous form of FAM3C can be localized in granules/vesicles or in the cytoplasm itself. In normal tissues, granular expression is common. Because of its key role in epithelial-mesenchymal transition (EMT) and metastasis, FAM3C was named Interleukin-like EMT inducer (ILEI) [49]. EMT is a process activated by transcription factors, such as the SNAIL, TWIST and ZEB families, in response to TGF-β receptor activation [14]. E-cadherin, vimentin and N-cadherin increases are major biomarkers of EMT. FAM3C/ILEI binds to leukemia inhibitory factor receptor (LIFR) and activate STAT3 signaling pathway to induce metastasis in both human and murine cancer cells [50]. The analysis of fam3c −/− transgenic mice reveal a cortical bone phenotype indicating that FAM3C protein participates in the regulation of mesenchymal stromal cells and osteoblast differentiation and mineralization [51]. Additionally, in vitro experiments revealed that FAM3C may exert its action via the down-regulation of transcription factor Runx2 in osteoblasts [52]. (FAM3C also regulate bone morphology, bone volume, and osteogenic differentiation. The initial characterization of a Tet-ON inducible FAM3C/ILEI transgenic mouse strain confirmed that overexpression of fam3c gene leads to a shortened lifespan, reduced body weight and microcytic hypochromic anemia [53]. FAM3C overexpression caused liver cirrhosis and marked fibrosis, a marker of EMT, but did not cause spontaneous tumor formation in any organs of the transgenic mice [53].
4. FAM3D
FAM3D, also known as oncoprotein-induced transcript 1 (Oit1), is a secreted protein that exerts an important role in the maintenance of gut microbiome and nutritional homeostasis [54]. The protein contains 224 amino acids with homology of 53% to FAM3A, 28% to FAM3B and 50% to FAM3C [15,16]. FAM3D has constitutively high expression in intestinal Goblet and Paneth cells of digestive system and also a moderate level of expression in lung and pancreas [54]. FAM3D is upregulated after intake of a high-fat diet both in mice and in humans. Higher plasma levels are observed after the postprandial period and more pronounced levels at high fat diets, which support its role in gastrointestinal nutritional homeostasis [54]. FAM3D may act as a new chemokine-like factor activating the G protein coupled receptors Formyl Peptide Receptor (FPR)1 and FPR2 during inflammation [55,56]. FAM3D expression is increased after dextran sulfate sodium (DSS)-induced colitis, supporting its role as a human ulcerative colitis as well as inflammation-induced carcinogenesis [55]. According, transgenic mice with FAM3D deficiency showed reduced colon length, body weight and survival after chronic colitis induced by lower DSS doses. The same pathological features were observed after azoxymethane (AOM)-induced tumorigenesis in mice whose displayed increased number of larger tumors. Confirming FAM3D protective properties, infusion of Adv-FAM3D protein restored colitis in fam3d -/- transgenic as compared to wild-type mice [55]. The studies also confirmed that fam3d -/- transgenic mice had an impaired microbiota in which was identified an increase number of the microbial species Lactobacillus/Lactococcus, Peptostreptococcus and Firmicutes phyla, and a decreased number of segmented filamentous bacterium species Eubacterium rectale, Enterococcus faecalis and Enterobacteria phyla [57]. Another study showed that fam3d −/− mice exhibited decreased neutrophil infiltration during abdominal aortic aneurysm of the aorta [56]. Therefore, current evidence supports that FAM3D has an important role in the physiological homeostasis of gut mucosal barrier, acting as a chemokine in recruiting neutrophils to inflammatory reaction, and in this way, protecting from dysbiosis and carcinogenesis. The Table 1 displays the molecular characteristics, chromosome localization, tissue distribution and major impact of overexpression or deficiency of each member of the FAM3 family in metabolism, body development and diseases.
5. Roles of FAM3 Proteins in Type 2 Diabetes (T2DM)
FAM3 cytokine-like proteins have prominent roles in body physiology by regulating pancreatic β-cell function and insulin effects in peripheral tissues [17,57]. Human and animal model studies have shown evidence to suggest that the expression and secretion of FAM3 cytokine-like proteins may serve as biomarkers for the early detection of T2DM and some metabolic disorders [11,57].
5.1. FAM3 Proteins Affect Insulin Secretion and β-Cell Function
Because its high expression in the endocrine pancreas, the first studies about FAM3B functions were dedicated to elucidate its role in pancreatic β-cells physiology [17,33,35]. Confocal microscopy studies revealed that FAM3B antibody staining is highly expressed in the islets of Langerhans and is co-located with insulin and glucagon. FAM3B protein is detected in both pancreatic β- and α- pancreatic cells. FAM3B was first associated with apoptosis of β-cells and pathogenesis of type I diabetes and after the gene is transiently expressed in the mouse insulinoma cell line β-TC3 [33,35]. It seems that FAM3B signaling cascade to promote apoptosis is distinct from that normally activated by well-known cytotoxic cytokines such as IL-1βIFN-γ and TNF-α [32,33,35,58]. These cytokines promote the activation of transcription factors NF-κB and STAT1 to induce β-cell demise, which does not appear to be activated by FAM3B. However, IFN-γ induces expression and regulation of the FAM3B in islets and insulin-secreting β-TC3 cells, suggesting that deleterious effects of IF-γ on β cells may be at least partially achieved through FAM3B activation [58]. A gene network study demonstrated that FAM3B protein induces cell death in β-cells via increase of caspase-3 expression and activation and cell cycle blockade by regulatory protein p21 [32,33].
Glucose stimulates FAM3B expression and secretion together with insulin via multiple interactive signaling pathways [35]. Pancreatic duodenal homeobox 1 (PDX1), a pancreatic β-cell–specific transcriptional factor binds to FAM3B promoter to activate its transcription [59]. The reciprocal participation of the phosphatidylinositol 3-kinase (PI3K) pathway and reactive oxygen species (ROS) have also been considered in the glucose-induced FAM3B gene expression in pancreatic β-cells [35]. To determine effects on glucose homeostasis, insulin sensitivity, and lipid metabolism, a transgenic mouse exclusively overexpressing FAM3B under the pancreatic-duodenal homeobox-1 (PDX1) promoter was generated [40,41]. Despite the overexpression, PANTG mice exhibited normal islet and pancreatic morphology, with no signs of lymphocytic infiltration, and the insulin and glucagon expression within islets was comparable to WT mice. The PANTG transgenic mice exhibited fasting hyperglycemia, impaired glucose tolerance, and hepatic insulin resistance, which are features of hepatic insulin resistance (SHIR)-like phenotype. These data support a potential role of FAM3B in the development of SHIR and T2D [40,41]. A recent investigation demonstrated that FAM3B can bind to fibroblast growth factor receptors (FGFRs) and activate downstream ERK signaling to promote posterior development of Xenopus embryous [60]. Overexpression of Xenopus FAM3B mRNA inhibited the formation of Xenopus cephalic (head) structure and induced an ectopic tail-like structure. Conversely, depletion of fam3b gene message with antisense morpholino oligonucleotides (MOs) resulted in macrocephaly (enlarged head), further confirming its role in promoting embryos posterior development [60]. Moreover, FAM3B protein could bind to the ectodomain of FGFR1 in solution, suggesting that FGFRs are putative receptors or binding partners in some contexts [60]. Any embryonic malformation was observed in gene knockout mice models [40,41]. Specifically, further studies are needed to establish if FGFR is responsible for mediating the effects of FAM3B in the induction of insulin secretion by pancreas and particularly apoptosis in pancreatic β-cells, in mammalian models and its potential role in pancreatic diseases.
5.2. FAM3 Proteins Regulate Hepatic Glucose Production and Lipid Metabolism
Binding of FAM3B protein to hepatic cell membranes has suggested a possible existence of a specific membrane receptor for FAM3B in liver [61]. Effects of recombinant FAM3B to this putative receptor in HepG2 hepatoma cells and primary murine hepatocytes caused inhibition of insulin intracellular signaling that included decreased activation of insulin receptor, insulin receptor substrate-1 (IRS-1), PI3K/Akt pathway, which were accompanied with increased cAMP levels and activation of cAMP-responsive element binding protein (CREB) signaling, leading to increased production of glucose [61,62].
The analysis of transgenic mice overexpressing FAM3B (PANTG mice) under food deprivation revealed significant metabolic alterations, including hyperglycemia, compensatory hyperinsulinemia, decreased liver triglycerides, and elevated corticosterone levels [40,41,63]. In vitro studies with primary hepatocytes showed high levels of glucose production and expression of the gluconeogenic genes after treatment with FAM3B recombinant protein. In addition to the extracellular actions, it was reported that intracellular FAM3B promotes lipogenesis, lipid accumulation and compromises insulin signaling in the liver by increasing FOXO1 activity. Conversely, overexpression of FOXO1 increased hepatic FAM3B levels in steatotic livers of diabetic db/db mice as well as in mice fed with high fat diet [31]. FAM3B is co-stored and co-secreted with insulin, and high FAM3B levels in fat mice may be due to a free-fatty acids (FFA)-induced stimulation of insulin secretory vesicles [63]. Thus, FFAs would increase FAM3B levels and long-term HFD feeding in mice. FAM3B protein is abundantly present in the nucleus of hepatocytes, where it functions as a novel coactivator of FOXO1 [31]. Under obese conditions, an increase in nuclear FAM3B-FOXO1 interaction retains FOXO1 in the nucleus, leading to increased gluconeogenesis in hepatocytes [24]. Compared to FAM3B mixed (PANTG) mice, FAM3B knockout C57BL/6 (PANKO-C57) mice had exhibited high glucose resistance, decreased fasting insulin and C-peptide levels, increased fasting leptin levels and the weight body of male and female mice [40,41,43,62,63]. Overall, the data confirmed that FAM3B regulates hepatic lipogenesis via suppression of P3K and Akt pathway, and this abolishes the activation of sensors like sterol regulatory element binding protein (SREBP) transcription factor.
FAM3A and FAM3C have opposite effects to FAM3B in glucose and lipid metabolism. FAM3A plays important role in regulation of hepatic and glucose metabolism via ATP-binding to P2 receptors (P2X and P2Y subtypes) which, in turn, trigger the PI3K/Akt signaling by way of a Ca2+ /calmodulin (CaM)-dependent mechanism and independent of insulin [22]. Blockade of P2 receptors or inhibition of phospholipase C (PLC) and IP3R, attenuatedFAM3A-mediated PI3K/Akt activation, resulting in hyperglycemia [22]. FAM3C overexpression reduced gluconeogenic and lipogenic genes in diabetic mouse livers with the suppression of gluconeogenesis and lipid deposition [64]. In culture hepatocytes, FAM3C increased the expression of heat shock factor 1 (HSF1), calmodulin (CaM) and phosphorylated Akt (pAkt), and these events occurred with nuclear exclusion and inactivation of FOXO1 [64]. FAM3D also activates calmodulin-Akt pathway to suppress gluconeogenesis in hepatocytes [65]. FAM3D bind and activate FPR1/FPR1 and this upregulate the expression of heterogeneous nuclear ribonucleoprotein U (hnRNP U), which recruits the glucocorticoid receptor (GR) to the promoter region of the short-chain acyl-CoA dehydrogenase (SCAD) gene and stearoyl-CoA desaturase 1 (SCD1) [55,56,65]. These enzymes are involved in the lipid oxidation in hepatocytes [65]. Therefore, the FAM3 proteins can engage multiple pathways to regulate liver lipid metabolism and glucose production.
5.3. Roles of FAM3 Proteins in T2DM and Metabolic Syndrome: Clinical Studies
Hyperglycemia increases FAM3B circulating levels, and this response may be associated with impaired β-cell function and death [35,66]. Thus, several cross-sectional and cohort analyses were performed to investigate the potential clinical association of FAM3B with T2DM and metabolic syndrome. Serum FAM3B levels were significantly elevated in 24 long-standing diabetics patients when compared to 39 recently diagnosed diabetics and 16 controls [67]. The β-cell dysfunction in diabetic patients was investigated using various biochemical parameters. The study found that elevated levels of FAM3B in long-standing diabetics was positively associated to proinsulin (PI), PI/Insulin and PI/C-peptide ratios and negatively associated with HOMA2-%β [67]. A cross-sectional study investigated the association of serum FAM3B levels with HbA1c and fasting blood glucose (FBG) in individuals with T2DM [68]. This study found that FAM3B concentration was pourer associated with insulin, HOMA-β, HOMA-IR and adiponectin [68]. A recent bioinformatics study using gene expression omnibus (GEO) datasets has shown that FAM3B is one of three most relevant and differentially expressed genes in the placenta tissue from mothers with gestational diabetes mellitus (GDM) and mothers with non-GDM and normal glucose metabolism [69]. Therefore, further studies are required to prove the association between FAM3B secretion and pancreatic β-cell dysfunction in T2D pathogenesis.
A cross-sectional study with 212 Chinese individuals, aging between 40 and 65 years, showed that elevated levels of FAM3B protein in the plasma correlated significantly with various components of metabolic syndrome (MetS) and with metabolic score (r = 0. 529, P < 0.001) [70]. Circulating levels of FAM3B were also associated with an increased risk ratio of impaired glucose tolerance or diabetes mellitus [70]. In agreement with this finding, a 5-year prospective study with a cohort 212 individuals showed that serum FAM3B level is closely associated with long-term MetS risk and islet β-cells dysfunction in the population without MetS [71]. In both studies, high circulating levels of FAM3B were positively correlated with fasting plasma glucose, fasting plasma insulin, LDL lipoprotein, triglycerides, blood pressure and HOMA-IR [70,71]. Therefore, elevated circulating levels of FAM3B might be a predictor of newly-onset MetS in those individuals presenting impaired glucose tolerance (IGT), diabetes mellitus, overweight or obesity. Four independent cohort studies investigated the associations of FAM3C genetic variants among Chinese children with overweight, obesity and quantitative lipid traits [72]. The analyses showed evidence that some FAM3C gene variants increase the risk for overweight and obesity, a trait associated with gene-by-sex interaction [72]. Therefore, more studies with larger sample size and diverse population need to validate these findings.
5.4. Role of FAM3 Proteins in Non-Alcoholic Fatty Liver Disease (NAFLD)
Non-alcoholic fatty liver disease (NAFLD) is a pathophysiological process covering steatosis (simple fat accumulation), non-alcoholic steatohepatitis (NASH) and cirrhosis in the liver [73,74]. NAFLD is a strong risk factor for many diseases including hyperlipidemia, insulin resistance and T2DM. FAM3A, FAM3B, FAM3C and FAM3D are potential novel regulators of hepatic glucose and lipid metabolism via the modulation of gluconeogenic and lipogenic biochemical processes [73,74]. In the liver, elevated FAM3B represses the activities of the Akt and AMPK pathways, reducing hepatic lipogenesis and gluconeogenesis [31,57]. Physical exercise significantly reduced FAM3B expression with an increase in FAM3A expression in the livers of db/db mice [31,57,73,74]. Liver-specific FAM3A overexpression reduced triglyceride accumulation and prevented diet-induced steatosis in mice [22]. Accordingly, FAM3A knockdown caused increased lipid deposition, inflammation, and insulin resistance [30]. FAM3C overexpression in the liver does not drive steatosis directly, but contributes to NASH progression and fibrosis [53]. As demonstrated in mice models, injection of FAM3D recombinant protein or FAM3D gene overexpression significantly suppressed glucose production, promoted nuclear exclusion of FOXO1, and reduced lipid deposition in the presence of FFAs [65]. Accordingly, FAM3D-deficient mice exhibited exaggerated hyperglycemia and steatosis after high-fat diet (HFD) feeding [65]. Then, an interesting question raised is how this imbalance happens. As discussed above, FAM3B expression is activated by FFAs and require FOXO1 activity, while FAM3A expression is induced by PPARγ activation, but it is repressed by high levels of FFAs and insulin [75,76]. Thus, one possibility is that FAM3A and FAM3C may repress FAM3B expression by Akt-mediated inhibition of FOXO1 activity. On the other hand, FAM3B may repress hepatic FAM3A and FAM3C expression by inducing FFAs accumulation in hepatocytes. Moreover, FAM3B may also upregulate FAM3A expression via the activation of PPARγ in hepatocytes [31]. FAM3D is produced in an autocrine/paracrine manner in liver and its expression is increased in steatosis in human and mouse livers [65]. FAM3D expression seems to act as compensatory mechanism against this metabolic dysregulation since it enhances fatty acid oxidation, which reduces lipid deposition in hepatocytes and ameliorates hepatic steatosis [65]. Further studies are needed to understand how the imbalance among FAM3A, FAM3B, FAM3C and FAM3D expression and their signaling networks contributes to steatosis and other fatty liver diseases. The Table 2 displays the primary molecular functions of each member of the FAM3 family, and the key molecular mechanisms for their participation in diabetes and metabolic syndrome.
6. FAM3 Proteins in Cancer Progression
Tumor progression depends on a complex interaction among extracellular matrix components (EMC), non-immune and immune normal cells and cancer cells within a tumor environment (TMA) [77,78]. Tumor tissue is composed of highly heterogeneous cancer cell clones arising from random events of mutation, deletion or amplification at genome level, which foster tumor evolution [79]. Various processes are essentials for cancer progression, including angiogenesis, EMT, migration and adhesion which depend on the production of many growth factors, cytokines, nutrients and metabolites produced from local cells and at distant sites [77,78]. FAM3 like-cytokines exert pleiotropic functions that connect intracellular signaling pathways to tumor metabolism, cell survival and progression. FAM3 family genes are emerging biomarkers for the diagnostic and prognostic in 33 types of human cancers as revealed in one study using the Cancer Genome Atlas (TCGA) and the Clinical Proteomic Tumor Analysis Consortium (CPTAC) databases [80]. FAM3C expression was negatively correlated with FAM3A, FAM3B, and FAM3D expression, while FAM3A expression was positively correlated with FAM3D, and FAM3B expression in the stemness scores based on RNA message expression and DNA methylation [80]. FAM3C was the first member of the FAM3 cytokine family to be implicated in the epithelial-mesenchymal transition (EMT) and cancer cell metastasis [49]. A potential role in induction of EMT and cancer metastasis is also attributed to FAM3B, including human colon cancer, esophageal squamous cell carcinoma and prostate cancer [81,82]. FAM3B was recently identified as one of senescence-associated genes with a potential role in carcinogenesis [83]. Expression of FAM3B was associated positively with sensitivity of tumors to many cytotoxic drugs (78 of 89 drugs), implying that patients with high expression of FAM3B may be better responders from chemotherapy regimen [80]. Overall, investigations using cancer-derived cell lines and multiomic bioinformatics analyses in cancer patients are helping to explore the feasibility of using FAM3 family members as biomarker and prognostic factor to some types of cancer, as it will be described below.
6.1. Breast Cancer
The roles of FAM3B and FAM3C in breast tumor progression have been investigated in vivo in mouse models and in culture using human breast carcinoma cell lines. The normal murine mammary gland epithelial (NMuMG) cells and mammary epithelial (EpH4) cells transformed with oncogenic Ras (EpRas) have been used as model for TGF-β-induced EMT [49,84]. In the first study, it was observed that EpH4-derivated cells with a stable FAM3C expression were able to increase cell migration and cell plasticity [49]. FAM3C expressing cells had a significant increase in expression of vimentin, a mesenchymal marker, with no change in E-cadherin expression. In agreement, mouse xenograft assays revealed lung metastasis formation and tumor growth upon subcutaneous inoculation of EpH4-derivated cells with a stable FAM3C expression. FAM3C promotes EMT independently of TGF-β signaling, suggesting that other candidates are regulated by EMT gene expression [49]. A clinical investigation with tissue breast human cancer array (TMA) made of samples of 56 breast cancer patients revealed that 43 samples were FAM3C positive. The expression correlated with high risk of metastasis development and shortened survival. Few tumors retained granular FAM3C staining as well, but it was the cytoplasmatic staining which was linked with a major decrease in metastasis-free and overall survival [49].
TGF-β coordinates translational activation of specific transcripts required for EMT program [13,14]. One investigation showed that FAM3C transcripts increases upon TGF-β stimulation [84]. To do this, TGF-β promoted the activation of Akt2, that in turn, caused phosphorylation of heterogeneous nuclear ribonucleoprotein E1(hnRNP E1) and this caused the release and activation of a 33-nucleotide TGF-β-activated translation element (BAT). The BAT element is a specific structural RNA element found in the 3’ untranslated region (3’-UTR) of many target mRNAs, including Dab2 (disabled-2) and FAM3C/ILEI [84]. This regulatory mechanism involving FAM3C and TGF-β signaling axis and EMT program was explored in MDA-MB-231 breast tumor cell line [84]. FAM3C overexpression caused upregulation of the Ying-Yang 1 (YY1), a transcription factor that directly activate heat shock factor 1(HSF) gene transcription. Thus, FAM3C overexpression promoted the proliferation and migration of MDA-MB-231 cells with YY1 and HSF1 up-regulation [85].
FAM3C cleavage is necessary for its activity, since noncleavable FAM3B form resulted failed activity [48]. The uPAR (uroquinase plasminogen activator receptor) gene knockdown in two breast cancer cells line demonstrated the requirement of plasmin enzymatic activity for FAM3C release from the secretory vesicles. This studies suggest that plasminogen-urokinase plasminogen activator receptor system regulates FAM3B biological activity in breast cancer progression [48].
We have explored the role of FAM3B in MDA-MB-231, a triple negative breast cancer (TNBC) tumor cell line and MCF-7, the estrogen hormone-responsive cell line [86]. We derived subclone cell lines that overexpress FAM3B gene and protein. FAM3B overexpression protected MDA-MB-231 cells from cell death induced by TNF-α and staurosporine. The protective effects were associated with increased expression of Bcl-2 and Bcl-xL anti-apoptotic genes, concomitantly with a reduced caspase-3 activity. FAM3B overexpression increased cell migration and wound healing activities in MDA-MB-231 cells, a EMT phenotype. In fact, FAM3B expression caused upregulation of EMT-related genes Slug, Snail, TGFBR2, vimentin, N-cadherin, MMP-2, MMP-9, and MMP-14 in MDA-MB-231 subclone cell line [86]. The in silico analysis of gene expression data obtained from RNA sequencing from The Cancer Genome Atlas (TCGA) revealed that FAM3B is highly expressed in a group of patients diagnosed with TNBC subtypes, poor prognosis and reduced overall survival [86]. The TNBC are now classified using gene expression profile in six major molecular subtypes, namely basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR) [87,88]. Future studies should help to confirm FAM3B utility to classify breast cancer patients particularly TNBC subtypes M and MSL with enriched gene expression associated with epithelial-mesenchymal-transition and stemness. The presence of FAM3B may also serve as a predictive biomarker to those patients whose will have better response to chemotherapy, as indicated in other study [80].
6.2. Colorectal Cancer
FAM3B and FAM3C have been involved in intestine tumor progression by different mechanisms. Increased FAM3B expression in colon cancer cell lines was able to promote cancer cell survival, migration and invasion [89]. Similarly, FAM3C expression in colorectal cancer was also implicated in increased cancer cell migration and lower survival rates. Samples of colorectal cancer from patients with high FAM3C expression also had lower survival rates [48,90]. On the other hand, unlike its relatives, FAM3D seems to act on intestine homeostasis, ensuring protection against carcinogenesis.
The activity of FAM3B in colon and intestine carcinoma has been demonstrated using colon human cell lines HCT116, HT29 and HCT8 cell lines, and microglial murine N9, myoblast C2C12, pancreatic β-cell Min6 murine cell lines and human liver cancer SMM-7721 and lung cancer A549 cell lines [39]. FAM3B silencing by RNA interference in human colon carcinoma cell line HCT8 resulted in upregulated Fas membrane (CD95) and cleavage of caspase-8 and caspase-9, which were accompanied by a decrease of BCL-2 antiapoptotic protein and an increase of pro-apoptotic protein Bax. Annexin V membrane exposure and PARP cleavage were observed in murine cell lines N9, C2C12, Min6 and in human cell lines HCT116 and A549. Apoptotic cell death induced by FAM3B silencing was mediated by a p53-dependent mechanism as demonstrated by p53 phosphorylation and degradation of Mdm2 protein [39]. Therefore, FAM3B protein may function by increasing expression of the genes of survival pathways in cancer cells. FAM3A regulates mitochondrial energy metabolism and therefore intracellular ROS generation. Accordingly, overexpression of FAM3A significantly increased cellular resistance of neuronal HT22 cells line from injuries caused by hydrogen peroxide (H2O2). [28]. FAM3A treatment also reduced mitochondrial cytochrome c release and decreased activation of caspase-3, two biomarkers of apoptosis, and these effects were associated with increased the activation of PI3K/Akt signaling pathway.
FAM3B mRNA multiple alternative variants were found in human colorectal adenocarcinoma [81]. These splicing alternatives variants translated to a non-secretory form named FAM3B-258 isoform [81]. Overexpression FAM3B-258 transcripts in colorectal cell lines induced a spindle-like morphology and promoted an increase of cell migration and invasion. These events accompanied the expression of cdc42, an important small GTPase of Rho family involved in the reorganization of actin cytoskeleton and modulation of cell-matrix interaction. Moreover, these dramatic changes in cell morphology and motility induced by FAM3B-258 were associated with expression of Slug and Snail, which act as transcriptional repressor of E-cadherin in HCT116 overexpressing cells [81]. The authors observed that after silencing Slug expression in HCT116 cells overexpressing FAM3B-258 gene, as expected, there was an increase in mRNA of adhesion molecules JAM and E-cadherin, an event accompanied by a decrease of Cdc42 protein expression. Moreover, in vivo assays using NUDE mice intravenously injected with HCT116 cells overexpressing FAM3B-258 protein demonstrated that the isoform increases lung metastasis [81]. Additionally, the analyses of tissue samples from patients by immune-histochemical stain and specific antibody showed enhanced cytoplasmic FAM3B expression in colorectal tumor tissue, compared to adjacent non-tumor samples. The histological analyses of tumors with FAM3C upregulation also showed increased vimentin and reduced E-cadherin expression [81].
6.3. Prostate Cancer
The role of FAM3C in the regulation of prostate cancer progression has been investigated in vitro and in vivo using mouse and human cell lines PC3 and DU145, both with lowest expression, LNCaP, with detectable expression, and PCS-440-010, a normal cell prostate cell line PCS-440-010 [91]. This study demonstrated that E3 ligase UBE4A targets FAM3C protein for proteasome degradation [91]. When this UBE4A E ligase was overexpressed in PC3 and DU145 cell lines or the proteasome mediated degradation was inhibited by MG-132, a FAM3C active isoform could be released through proteolytic activity or inhibited, respectively [91]. This possible post-translational control mechanism of FAM3C controlled by UBE4A ligase in normal tissues and tumor cells need to be further investigated.
The deletion or loss FAM3B gene has been found to attenuate androgen receptor signaling pathways, which is associated in prostate cancer hormone resistance and disease progression [92]. FAM3B gene is among four interstitial genes that are downregulated in TMPRSS2-ERG fusion-positive metastatic castration-resistant prostate cancers compared with primary prostate cancers [92]. Patients with TMPRSS2-ERG fusions and FAM3B downregulation frequently have pelvic lymph node metastases, and poorer disease specific and overall survival. Thus, genes in this region have been considered to function as tumor-suppressor genes. The tumor suppression effects of FAM3B was investigate in in vitro cell culture and in vivo with xenografts of LNCaP, C4-2, and LNCaP95 prostate cancer cells with or without constitutive or doxycycline-induced FAM3B expression [92]. LNCaP95 and C4-2 are castration and hormone resistant and LNCaP derivatives are androgen sensitive and TMPRSS2-ERG fusion negative. FAM3B expression is low in the LNCaP, C4-2, and LNCaP95 cell lines. This study demonstrated that FAM3B is an androgen receptor transcriptional target. An additional study investigated the tumor growth in a transgenic model by crossing Fam3b -/- mice with Hi-Myc mice, which express high Myc transcription factor levels specifically in the prostate. Overall these studies demonstrated that the loss of FAM3B induces rapid epithelial proliferation, invasive adenocarcinoma, angiogenesis, and reduces apoptosis [92].
We investigated the role of FAM3B in a DU145 cell line which does not express the androgen receptor, thus it is not stimulated by androgens [93]. We selected s DU145 cell subclone lines from lentivirus-mediated FAM3B overexpression and examined rates of cell proliferation and colony formation in in vitro in cell culture and tumor growth in vivo studies in NUDE mice [93]. We observed that cells became resistance to cell death induced by the cytokine TNF-α and this anti-apoptotic effect was related to up-regulation of anti-apoptotic proteins B-cell lymphoma 2 (Bcl-2), Bcl-xL and no change in pro-apoptotic Bcl-2 associated protein X (Bax) expression. The overexpression of FAM3B in DU145 cells also increased their ability to tumor growth in NUDE mice. FAM3B could not inhibit cell death types, like autophagy and necrosis, induced by the cytotoxic drugs including staurosporine, camptothecin and etoposide [93]. For instance, pan-cancer investigation showed that expression of FAM3B in various tumor types influence positively the sensitivity to many types of drugs (78 of 89 drugs), implying that patients with high FAM3B expression have a reduced DNA damage response and better response to chemotherapy [80]. Together, these studies providence evident that FAM3B can induce protection to cell death and its deletion promote prostate tumor growth through androgen receptor signaling.
6.4. Melanoma
FAM3C has been implicated in a process named phenotypic switching in which high microphtalmia-related transcription factor (MITF) expressing (MITF-high) proliferative cells switch to a low expressing (MITF-low) invasive state in melanoma cells [94,95]. MITF-low invasive melanoma cells have high FAM3C expression. Results from in vitro assays showed that knockdown of FAM3C with stably transduced shRNA attenuated invasive low MITF expressing cell potential but not MITF expression or chemoresistance of melanoma cells [94,95]. FAM3B expression was higher in a metastatic cell line 1205Lu as compared to WM983B, a less aggressive melanoma cell line. Gene expression analysis showed that FAM3B regulates the MITF-low invasive phenotype via expression of JARID1B, HIF-2α, and BDNF, but EMT transcription factors like ZEB1 and ZEB2 were not regulated. According to the authors comments, the regulation of FAM3C in the melanoma cell line model seems to be independent of TGF-β/AKT2/hnRNP-E1, degradation by the ubiquitin/proteasome system, or autophagy. They suggest an alternative mechanism in which paracrine production of FAM3B by tumor cells could activate the immune system. This mechanism may involve the participation of multiple molecular factors and cell types [94,95].
6.5. Esophageal Cancers
Two recent studies support the role of FAM3B in head and neck cancer, in special esophageal carcinomas [96,97]. FAM3C mRNA expression was remarkably upregulated in a small group of patients with esophageal squamous cell carcinoma (ESCC) compared with their non-tumor counterpart [96]. Kaplan-Meier analysis to estimate the 7-year overall survival rate among groups with low and high expression of FAM3C revealed that those with high expression had a poorer prognosis shorter overall survival rate [96]. A following study confirmed that FAM3B gene expression was up-regulated in esophageal tumor surgical samples from patients with esophageal squamous cell carcinoma and also ESCC cell lines (ECA109, KYSE150, KYSE9706 and TE-1) as compared to matched adjacent normal tissues and normal esophageal epithelial cell line, HET-1A [97]. Established overexpression of FAM3B inhibited cell death of ECA109 cells and increased tumor growth in xenografted nude mice, as well as promoted ESCC cell migration and invasion. On contrary, siRNA-mediated FAM3B gene knockdown in ECA109 cells decreased the expression of the proteins p-AKT, MDM2, Snail and N-cadherin protein expression while increased p53 and E-cadherin expression [97]. Complementary experiment demonstrated that the rates of apoptosis measured by DNA fragmentation by flow cytometry, were reduced by up-regulated expression of FAM3B but increased by down-regulated expression (knockdown) of FAM3B mRNA. The authors concluded that FAM3B may be suppressing the AKT-MDM2-P53 pathway to induce tumor progression [97]. Another study also confirmed that knockdown of FAM3B in colon cancer cell lines triggers apoptosis through a p53-dependent pathway [39]. The p53-MDM2 pathway plays a crucial role in regulating apoptosis, a mechanism mediated by its ubiquitination by MDM2 and degradation by the ubiquitin-proteasome system. It is interesting that wild p53 facilitates Slug degradation through upregulation of MDM2, repressing the expression of this key pro-EMT transcription factor [98]. Therefore, this complex feedback regulatory network in control of both apoptosis and EMT by FAM3B requires further investigation.
6.6. Gastric Cancers
A previous screening study for cisplatin sensitive and resistant cell lines, manifested by the ability to survive under toxic production of reactive oxygen species (ROS), identified high levels of FAM3B in human gastric cancer cell line (AGS) cisplatin-resistant [82]. FAM3B overexpression could induce cisplatin resistance of gastric cancer cells by reducing cisplatin-induced apoptosis [99]. The authors of these studies also demonstrated AGS cells overexpressing FAM3B had upregulated Snail. When Snail was inhibited, EMT and cisplatin resistance triggered by FAM3B were reversed [82,99]. HGC27, a gastric tumor cell line with a low FAM3B expression treated with exogenous FAM3B had an increase in cell migration and phosphorylation of Akt, PI3K and GSK3β protein and drastic reduction in E-cadherin expression [100]. Moreover, FAM3B knockdown increased E-cadherin levels and reduced vimentin levels. These events occur with a decreased phosphorylated Akt, P13K, GSK3β and downregulation of the transcription factors Snail and Slug [100]. Histological investigations in tumor tissue samples from patients found a cytoplasmic distribution FAM3B protein, whereas more stable granular distribution was seen in normal mucosa [100]. A similar finding was reported in another study based on IHC of 150 gastric cancer tissues cases [99]. The results showed cytoplasmic FAM3B protein expression occurred in 70% tumor tissues with a positive correlation with vimentin and downregulation of E-cadherin. Patients with higher FAM3B tissue expression had shortened overall survival rates, compared to cases with lower protein expression [99].
6.7. Hepatocarcinoma
The role of FAM3C in hepatocarcinoma (HCC) was investigated employing a model of EMT based on immortalized p19ARF null hepatocytes (MIM), which display tumor growth upon expression of oncogenic Ras and undergo EMT through the synergism of Ras and transforming growth factor (TGF)-β [101]. FAM3 could promote EMT after upregulation of platelet derived growth factor (PDGF-C) and PDGFR-β in murine mammary hepatocellular cell lines transformed with Ras oncogene [101]. In vitro and in vivo studies with V12-C40-Ras in MIM1-4 cell lines with overexpression FAM3B plus oncogenic Ras showed significant increases in tumor growth, tumorigenicity, cytoplasmatic E-cadherin, nuclear β-catenin expression with lower levels of membrane β-catenin. FAM3C seems to signaling through PDGF isoforms that induce nuclear increase of β-catenin and pY-Stat3 [101]. An important finding communicated in Lahsning study regarding EMT induction by Ras/FAM3C versus Ras/TGF-β hepatocytes and mammary cells was the confirmation that FAM3C-induced EMT did not involve or required autocrine TGF-β secretion. This study also investigated of FAM3C expression and localization in relation to HCC patient prognosis. A tissue array from 69 human hepatocarcinoma samples revealed that the majority cases (66%) with poorly differentiated tumors samples with a poor prognosis were positive for cytoplasmatic FAM3C staining [101]. Therefore, cytoplasmic localization of FAM3C protein may predict poor differentiation and prognosis of human HCC patients.
6.8. Lung Cancer
Aberrant expression of FAM3C protein was found in A549 lung tumor cell line [102]. This transcriptional enhancement in A549 lung tumor cells could be inhibited by expression of short hairpin to RNA to Poly r(C)-binding protein 1 (PCBP1). Activation of TGF-β promoted PCBP1 phosphorylation at serine-43 through protein kinase Bβ/Akt2, and this promote PCBP-1 to induce FAM3B transcripts and its translation subsequently caused EMT [101]. FAM3C could induce and maintain CD24low/CD44high expression in the subpopulation of A549 cells treated with TGF-β [102]. A recent study investigated the role of FAM3C in the release of extracellular vesicles (EVs) [102]. EVs FAM3C positive was observed in the lung cancer cell lines H1650, H226, A549, H1299, SKMES-1, Calu-1, H2172, ChaGo-K, H520 and H217, but it was not observed in normal lung fibroblast cell line. A549 cells engineered to overexpress FAM3C showed stronger invasion and metastatic potential when injected in mice via tail. FAM3C-tumor-derived EVs also increased lung metastasis in xenografts models. Accordingly, higher FAM3C concentrations were detected in EVs extracted from plasma samples of non-small cell lung (NSCLC) patients compared to those of healthy subjects [103]. Biochemical studies suggested that FAM3C may promote migration and invasiveness through association with Ras-related protein RalA, a Ras GTPase, and the activation of the kinases Src and just another kinase (JNK), as well as activation of nuclear-factor-κB (NF-κB) transcription factor, resulting in the expression of cyclin D and Aurora A [103]. FAM3C expression was elevated in adjacent tumor tissues and associated with worse prognosis. It remains to be investigated whether FAM3C act as an oncogene that transform subpopulation of progenitor cells into lung cancers.
6.9. Oral Cancer
A tissue tumor microarray (TMA) immunohistochemical analysis and differential gene expression investigation demonstrated that FAM3B mRNA and protein are downregulated in oral squamous metastatic cell carcinoma (OSCC) [104]. This reduced FAM3B expression in OSCC was correlated with increased tumorigenesis and apoptotic cell death inhibition. In addition, this study demonstrated that lower FAM3B expression may be regulated by miRNAs, possibly miR-181a, miR-181b, miR-200b, and miR-200c, as revealed using a bioinformatics algorithm for targeted gene prediction [104].
The expression FAM3C was also investigated in OSCC in another study [105]. It was found FAM3C was highly expressed in oral cavity carcinoma, oropharyngeal carcinoma, and floor of the mouth carcinoma, as compared with normal oral mucosa and epithelial dysplasia [105]. (Wu et al., 2019). Increased FAM3C levels correlated with the negative immune checkpoint protein: programmed death ligand-1 (PD-L1), VISTA, B7-H4 as well as Slug, SRY-Box Transcription Factor 2 (SOX2) and aldehyde dehydrogenase-1 (ALDH1), as revealed by immunohistochemical staining [105]. The results suggest that overexpression of PD-L1 on tumor cells correlate with its EMT status. There was no remarkable difference in the expression of FAM3C among pathological grades, metastatic lymph nodes, but it correlates positivity with free survival [105]. In according the results of Shibata and coworkers, FAM3B is frequently down-regulated in primary OSCC samples [104]. This occur with increased expression of FAM3C [1054]. Despite the high amino acid sequence homology between FAM3B and FAM3C, FAM3C carries an additional cysteine (Cys 185), which is responsible for its dimerization [44,45]. Thus, they may have different binding affinity to their own receptors and candidate partners resulting in opposite effects in certain pathological conditions.
7. Conclusions and Perspectives
Recent studies are beginning to provide more evidence that FAM3 family – composed of FAM3A, FAM3B, FAM3C, and FAM3D, are cytokine-like proteins with pleiotropic functions, orchestrating physiological and pathological cellular processes in context dependent manner. They are particularly involved in glucose and lipid metabolism (FAM3A, FAM3B), embryogenesis (FAM3B), osteogenic differentiation (FAM3C), adipocyte differentiation (FAM3A) and gastrointestinal homeostasis (FAM3D). In addition, these proteins may have intriguingly roles in carcinogenesis and metastasis, acting as oncogene (FAM3C), tumor suppressor gene (FAM3B, FAM3D), or either conferring resistance to cell death (FAM3A, FAM3B) or susceptibility to chemotherapeutic agents (FAM3B), or promoting of EMT (FAM3B, FAM3C) or immune cell recruitment to inflammatory sites (FAM3D).
The knockout animal model studies confirmed that these cytokine-like proteins are particularly involved in glucose and lipid metabolism via not yet fully understood mechanisms. Studies have described the opposing effects of FAM3A, FAM3C and FAM3D to FAM3B effects in the metabolism in a cell- and tissue-specific manner. While FAM3B induces signal pathways to promote hepatic lipogenesis and gluconeogenesis, FAM3A, FAM3C and FAM3D drive downstream signals to suppress gluconeogenesis and lipogenesis and insulin resistance. The potential cytotoxicity of FAM3B in pancreatic β-cells and dysfunction associated with T2D remains to be further investigated at clinical setting.
The results of the bioinformatics analyses in large public cancer genome databases showed that FAM3C expression was negatively correlated with FAM3A, FAM3B, and FAM3D expression in various types of cancers [80]. Malignancies with up-regulation of FAM3C gene expression were mainly enriched in genes of p53 signaling pathway. Expression of FAM3A was negatively correlated with stromal score, immune score and estimate score, and these scores correlated with a poor prognosis. Expression of FAM3B was associated with the stronger drug sensitivity of cancer cells as examined by a patients’ response to standard chemotherapies [80]. Expression of FAM3B and FAM3C increased either the risk or decreased the survival of patients with many types of cancers [80]. Protein p53 has pivotal roles in triggering apoptosis and limiting glycolysis (Warburg effect). These effects stem from p53’s transcriptional activation of hundreds of genes for cell cycle, DNA repair, survival, apoptosis or cellular metabolism [106]. However, a variety of mutations in coding sequence provoke abnormal function or inactivation of p53 protein signaling pathway to limit tumor development or anti-tumour immune responses [107]. It is likely that p53 may act as regulator FAM3 proteins in one side favoring and, in other side, opposing their effects on tumor growth, drug resistance and induction EMT. FAM3 effects may occur along cancer cell progress, in which cells produce stemness factors enabling them to increasing their capacity for drug resistance, invasion and metastasis. FAM3 proteins may bind to their owner receptor (not yet identified) or to partner growth factor and cytokine receptors to execute some specific activities. Figure 1 shows the signaling pathways of the potential candidate receptors of four FAM3 proteins and TGF-β receptors and crosstalk of downstream signaling cascades to EMT program and other induced-cellular activities.
TGF-β can induce either cell cycle arrest, apoptosis, resistance to cell death by TNF-α or EMT, depending on a complex network of intracellular signaling pathways [13,14,108]. TGF-β drives EMT by activating both the primary Smad-dependent pathway (Smad2/3/4 complex) and non-canonical pathways (MAPKs, PI3K/AKT) [13,14]. TGF-β induced EMT program may be upregulated by FAM3C and FAM3B since they also increase the expression of transcription factors such as Snail, Slug and other genes, in many types of cancer cells. There is biochemical evidence that FAM3C binds to LIFR receptor, a receptor shared with the IL-6 cytokine family members. LIF binds to LIFR/gp130 complex to induce EMT through the activation of STAT3 [109]. It is also demonstrated that FAM3B can bind to FGFRs [60]. FGF family members bind to FGFRs triggering the activation of associated tyrosine kinases and various branches of signaling pathways, like JAK-STAT, MAPK and PI3-kinase pathways. These signaling pathways can promote proliferation, inhibition of apoptosis, drug resistance, angiogenesis, EMT and many other cellular outcomes [110]. FGFRs can directly interact and form complexes with platelet-derived growth factors (PDGFR) and epithelial growth factor receptors (EGFR) in cancer cells with persistent oncogenic signaling [110]. FGFRs can also interact with a large number of extracellular matrix components to initiate non-canonical signaling pathways [110]. Therefore, FAM3 proteins may hijack raft-localized receptors to dysregulate normal or cancer-related pathways. FAM3B protein may act as monomers while FAM3C as dimers. The gathering and clustering of FAM3 proteins and oncogenic growth factor receptors (examples, FGFR, EGFR, PDGFR) in the membrane lipid rafts should dramatically enhances the efficiency, sensitivity, and specificity of signal transduction pathways in cancer cells. It remains to be determined which degree of expression and signaling activation will lead to either stimulation or inhibition of cell proliferation, differentiation, growth arrest, resistance to apoptosis or EMT. Advanced 3D cell system models (such as organoids and spheroids) may be appropriated to test these hypotheses. Therefore, future studies aiming at the identification of the FAM3 own receptors, co-receptors and adaptors and their canonical and non-canonical signaling pathways will be instrumental for better characterization of their functions on metabolic diseases and cancer.
Author Contributions
Conceptualization, HMGM, JEB; article search and selection, critical reading and discussion, JMO, IDSC, HMGM, JEB; preparation of tables and figures, writing of original draft manuscript, HMGM, JEB, IDSC, BM, JMO; final review and editing, JEB, BRB, and HMGM. All authors have read and agreed to the published version of the manuscript.
Funding
The research of the authors was supported by Brazilian Foundation of Research (FAPESP 2007/04513-1, 2017/17062-0, 2018/08540-8) and the National Council for Scientific and Technological Development (486048/2011-0, 0312206/2016-0).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data used in this study are included in the main text.
Acknowledgments
We would like to express our sincere gratitude to all our students and lab technicians for commitment, dedicated work and assistance in the conduction of animal, cell culture and molecular biology studies. Also, EACH-USP colleagues who provided lab infrastructure and reagents to complete the experiments.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
EMT, the epithelial-mesenchymal transition, TGFR, transforming growth factor β receptor; FPR, formyl peptide receptor; LIFR, leukemia inhibitory factor receptor; FGFR, fibroblast growth factor receptor; RAS, Rat sarcoma virus; RAF, proto-oncogene serine/threonine-protein kinase; MEK, mitogen-activated protein kinase kinase; MEK, dual specificity mitogen-activated protein kinase kinase; G, guanine nucleotide-binding proteins; YY1, multifunctional zinc-finger transcription factor; HSF1, Heat shock factor 1; USF1 Upstream Transcription Factor 1; hnRNP E1, heterogeneous nuclear ribonucleoprotein E1; JAK, Janus kinase; STAT; signal transducer and activator of transcription 3; ERK, extracellular signal-regulated kinases; p38, p38 mitogen-activated protein kinase; JNK, Jun N-terminal kinase; PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase B; PLC, phospholipase C, PIP3, phosphatidylinositol (3,4,5)-trisphosphate; GSK3, glycogen synthase kinase-3; MDM2, murine double minute 2; STAT3, signal transducer and activator of transcription 3; TORC2, mammalian target of rapamycin complex 2; SMAD, Mothers Against Decapentaplegic; CDC42, cell division cycle 42; SNAIL, Zinc finger protein SNAI1; SLUG, Zinc finger protein SNAI2; Twist, Twist-related protein (Twist); ZEB, Zinc finger E-box-binding homeobox; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated protein X; MMP, matrix metallopeptidase; P53, tumor protein; Cyt c, cytochrome C, ATP, adenosine triphosphate; ROS, reactive oxygen species.
References
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der carcinomzelle. Biochem. Z. 1924, 152, 319. [Google Scholar]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: still emerging. Cell Metab. 2022, 34(3), 355–377. [Google Scholar] [CrossRef]
- Cannata, D.; Fierz, Y.; Vijayakumar, A.; LeRoith, D. Type 2 diabetes and cancer: what is the connection? Mt Sinai J Med. 2010, 77(2), 197–213. [Google Scholar]
- Wojciechowska, J.; Krajewski, W.; Bolanowski, M.; Kręcicki, T.; Zatoński, T. Diabetes and cancer: a review of current knowledge. Exp. Clin Endocrinol Diabetes 2016, 124(5), 263–75. [Google Scholar] [CrossRef]
- Le, A.; Udupa, S.; Zhang, C. The metabolic interplay between cancer and other diseases. Trends Cancer 2019, 5(12), 809–21. [Google Scholar] [CrossRef]
- Scully, T.; Ettela, A.; LeRoith, D.; Gallagher, E.J. Obesity, type 2 diabetes, and cancer risk. Front. Oncol. 2021, 10, 615375. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: the increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95(3), 727–48. [Google Scholar] [CrossRef]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and abnormal glucose and lipid metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: what do metabolic outliers teach us? Cell 2012, 148(6), 1132–44. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133(4), 704–15. [Google Scholar] [CrossRef] [PubMed]
- Farahzadi, R.; Valipour, B.; Fathi, E.; Pirmoradi, S.; Molavi, O.; Montazersaheb, S.; Sanaat, Z. Oxidative stress regulation and related metabolic pathways in epithelial-mesenchymal transition of breast cancer stem cells. Stem Cell Res. Ther. 2023, 14(1), 342. [Google Scholar] [CrossRef]
- Nwokolo, G.C.; Ganesan, T.S.; Pors, K.; Falconer, R.A.; Smarakan, S. Cancer stem cells in focus: Deciphering the dynamic functional landscape of stemness in cancer. Biochim Biophys Acta Rev Cancer 2025, 1880(5), 189440. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell. 2019, 49(3), 361–374. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Baker, D.; Ten; Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20(11), 2767. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, G.; Patel, A.; McLaughlin, M.M.; Silverman, C.; Knecht, K.; et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics 2002, 80(2), 144–50. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, G.; Patel, A.; McLaughlin, M.M.; Silverman, C.; Knecht, K.; et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics 2002, 80(2), 144–50. [Google Scholar] [CrossRef]
- Wilson, C.G.; Robert-Cooperman, C.E.; Burkhardt, B.R. PANcreatic-DERived factor: novel hormone PANDERing to glucose regulation. FEBS Lett. 2011, 585(14), 2137–43. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22(2), 142–158. [Google Scholar] [CrossRef]
- Zhang, Y.; Wan, J.; Liu, S.; Hua, T.; Sun, Q. Exercise induced improvements in insulin sensitivity are concurrent with reduced NFE2/miR-432-5p and increased FAM3A. Life Sci. 2018, 207, 23–9. [Google Scholar] [CrossRef]
- Yang, W.; Wang, J.; Chen, Z.; Chen, J.; Meng, Y.; Chen, L.; et al. NFE2 induces miR-423-5p to promote gluconeogenesis and hyperglycemia by repressing the hepatic FAM3A-ATP-Akt pathway. Diabetes. 2017, 66(7), 1819–32. [Google Scholar] [CrossRef]
- Song, Q.; Gou, W.L.; Zou, Y.L. FAM3A protects against glutamate-induced toxicity by preserving calcium homeostasis in differentiated PC12 cells. Cell Physiol. Biochem. 2017, 44(5), 2029–41. [Google Scholar] [CrossRef]
- Wang, C.; Chi, Y.; Li, J.; Miao, Y.; Li, S.; Su, W.; et al. FAM3A activates PI3K p110α/Akt signaling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology 2014, 59(5), 1779–90. [Google Scholar] [CrossRef]
- Kang, T.; Peng, D.; Bu, G.; Gu, H.; Zhang, F.; Zhang, R.; Zhou, Y.; Xiong, Y.; Lei, M. Transcriptional regulation analysis of FAM3A gene and its effect on adipocyte differentiation. Gene 2016, 20(595(1)), 92–98. [Google Scholar] [CrossRef]
- Chi, Y.; Meng, Y.; Wang, J.; Yang, W.; Wu, Z.; Li, M.; et al. FAM3B (PANDER) functions as a co-activator of FOXO1 to promote gluconeogenesis in hepatocytes. J. Cell. Mol. Med. 2019, 23(3), 1746–58. [Google Scholar] [CrossRef]
- Jia, S.; Chen, Z.; Li, J.; Chi, Y.; Wang, J.; Li, S.; et al. FAM3A promotes vascular smooth muscle cell proliferation and migration and exacerbates neointima formation in rat artery after balloon injury. J. Mol Cell Cardiol. 2014, 74, 173–82. [Google Scholar] [CrossRef]
- Xiang, R.; Chen, J.; Li, S.; Yan, H.; Meng, Y.; Cai, J.; et al. VSMC-specific deletion of FAM3A attenuated Ang II-promoted hypertension and cardiovascular hypertrophy. Circ. Res. 2020, 126(12), 1746–59. [Google Scholar] [CrossRef]
- Su, Y.; Cui, X.; Li, M.; Jiang, D.; Chen, R.; Zhou, Y. FAM3A: a novel mitochondrial protein for the treatment of ischemic diseases. Cell Cycle 2025, 24(5-8), 71–85. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Gou, W.L.; Zhang, R. FAM3A Protects HT22 cells against hydrogen peroxide-induced oxidative stress through activation of PI3K/Akt but not MEK/ERK pathway. Cell Physiol. Biochem. 2015, 37(4), 1431–41. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Gao, Q.; Chen, T.; Wen, T.; Wu, P.; Luo, X.; Chen, Q.Y. FAM3A ameliorates brain impairment induced by hypoxia-ischemia in neonatal rat. Cell Mol. Neurobiol. 2023, 43(1), 251–264. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yuan, F.; Xiong, Y.; Tang, Y.; Li, Z.; Ai, J.; Miao, J.; Ye, W.; Zhou, S.; Wu, Q.; Wang, X.; et al. FAM3A plays a key role in protecting against tubular cell pyroptosis and acute kidney injury. Redox Biol. 2024, 74, 103225. [Google Scholar] [CrossRef]
- Li, J.; Chi, Y.; Wang, C.; Wu, J.; Yang, H.; Zhang, D.; Zhu, Y.; Wang, N.; Yang, J.; Guan, Y. Pancreatic-derived factor promotes lipogenesis in the mouse liver: role of the Forkhead box 1 signaling pathway. Hepatology 2011, 53(6), 1906–16. [Google Scholar] [CrossRef]
- Burkhardt, B.R.; Greene, S.R.; White, P.; Wong, R.K.; Brestelli, J.E.; Yang, J.; et al. PANDER-induced cell-death genetic networks in islets reveal central role for caspase-3 and cyclin-dependent kinase inhibitor 1A (p21). Gene 2006, 369, 134–41. [Google Scholar] [CrossRef]
- Cao, X.; Gao, Z.; Robert, C.E.; Greene, S.; Xu, G.; Xu, W.; et al. Pancreatic-derived factor (FAM3B), a novel islet cytokine, induces apoptosis of insulin-secreting beta-cells. Diabetes 2003, 52(9), 2296–303. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, J.R.; Robert-Cooperman, C.E.; Wu, J.; Young, R.A.; Wolf, B.A.; Burkhardt, B.R. Characterization of the expression, localization, and secretion of PANDER in alpha-cells. Mol. Cell. Endocrinol. 2010, 325, 36–45. [Google Scholar] [CrossRef]
- Yang, J.; Robert, C.E.; Burkhardt, B.R.; Young, R.A.; Wu, J.; Gao, Z.; et al. Mechanisms of glucose-induced secretion of pancreatic-derived factor (PANDER or FAM3B) in pancreatic beta-cells. Diabetes. 2005, 54(11), 3217–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, O.; Cai, K.; Pang, S.; Wang, T.; Qi, D.; Zhu, Q.; et al. Mechanisms of glucose-induced expression of pancreatic-derived factor in pancreatic beta-cells. Endocrinology 2008, 149(2), 672–80. [Google Scholar] [CrossRef]
- Burkhardt, B.R.; Yang, M.C.; Robert, C.E.; Greene, S.R.; McFadden, K.K.; Yang, J.; et al. Tissue-specific and glucose-responsive expression of the pancreatic derived factor (PANDER) promoter. Biochim. Biophys Acta 2005, 1730(3), 215–25. [Google Scholar] [CrossRef]
- Cao, X.; Yang, J.; Burkhardt, B.R.; Gao, Z.; Wong, R.K.; Greene, S.R.; et al. Effects of overexpression of pancreatic derived factor (FAM3B) in isolated mouse islets and insulin-secreting betaTC3 cells. Am. J. Physiol. Endocrinol. Metabol. 2005, 289, E543-50. [Google Scholar] [CrossRef]
- Mou, H.; Li, Z.; Yao, P.; Zhuo, S.; Luan, W.; Deng, B.; Qian, L.; Yang, M.; Mei, H.; Le, Y. Knockdown of FAM3B triggers cell apoptosis through p53-dependent pathway. Int. J. Biochem. Cell Biol. 2013, 45(3), 684–91. [Google Scholar] [CrossRef]
- Robert-Cooperman, C.E.; Carnegie, J.R.; Wilson, C.G.; Yang, J.; Cook, J.R.; Wu, J.; et al. Targeted disruption of pancreatic-derived factor (PANDER, FAM3B) impairs pancreatic beta-cell function. Diabetes 2010, 59(9), 2209–18. [Google Scholar] [CrossRef] [PubMed]
- Robert-Cooperman, C.E.; Dougan, G.C.; Moak, S.L.; Athanason, M.G.; Kuehl, M.N.; Bell-Temin, H.; et al. PANDER transgenic mice display fasting hyperglycemia and hepatic insulin resistance. J. Endocrinol. 2014, 220(3), 219–31. [Google Scholar] [CrossRef] [PubMed]
- Athanason, M.G.; Ratliff, W.A.; Chaput, D.; MarElia, C.B.; Kuehl, M.N.; Stevens, S. M., Jr.; Burkhardt, B.R. Quantitative proteomic profiling reveals hepatic lipogenesis and liver X receptor activation in the PANDER transgenic model. Mol. Cell. Endocrinol. 2016, 436, 41–49. [Google Scholar] [CrossRef]
- Moak, S.L.; Dougan, G.C.; MarElia, C.B.; Danse, W.A.; Fernandez, A.M.; Kuehl, M.N.; et al. Enhanced glucose tolerance in pancreatic-derived factor (PANDER) knockout C57BL/6 mice. Dis. Model. Mech. 2014, 7(11), 1307–15. [Google Scholar] [CrossRef]
- Jansson, A.M.; Csiszar, A.; Maier, J.; Nyström, A.C.; Ax, E.; Johansson, P.; Schiavone, L.H. The interleukin-like epithelial-mesenchymal transition inducer ILEI exhibits a non-interleukin-like fold and is active as a domain-swapped dimer. J. Biol. Chem. 2017, 292(37), 15501–15511. [Google Scholar] [CrossRef]
- Kral, M.; Klimek, C.; Kutay, B.; Timelthaler, G.; Lendl, T.; Neuditschko, B.; Gerner, C.; Sibilia, M.; Csiszar, A. Covalent dimerization of interleukin-like epithelial-to-mesenchymal transition (EMT) inducer (ILEI) facilitates EMT, invasion, and late aspects of metastasis. FEBS J. 2017, 284(20), 3484–3505. [Google Scholar] [CrossRef]
- Flores, R.M.A.; Pantaleão, S.Q.; Araujo, S.C.; Malpartida, H.M.G.; Honorio, K.M. Structural analysis of factors related to FAM3C/ILEI dimerization and identification of inhibitor candidates targeting cancer treatment. Comput. Biol. Chem. 2023, 104, 107869. [Google Scholar] [CrossRef] [PubMed]
- Katahira, T.; Nakagiri, S.; Terada, K.; Furukawa, T. Secreted factor FAM3C (ILEI) is involved in retinal laminar formation. Biochem. Biophys. Res. Commun. 2010, 392, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Kutay, B.; Wirth, S.; Schmidt, U.; Macho-Maschler, S.; Schreiber, M.; et al. Interleukin-like epithelial-to-mesenchymal transition inducer activity is controlled by proteolytic processing and plasminogen-urokinase plasminogen activator receptor system-regulated secretion during breast cancer progression. Breast Cancer Res. 2014, 16(5), 433. [Google Scholar] [CrossRef]
- Waerner, T.; Alacakaptan, M.; Tamir, I.; Oberauer, R.; Gal, A.; Brabletz, T.; et al. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell 2006, 10(3), 227–39. [Google Scholar] [CrossRef]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019, 38(20), 3794–3811. [Google Scholar] [CrossRef]
- Määttä, J.A.; Bendre, A.; Laanti, M.; Büki, K.G.; Rantakari, P.; Tervola, P.; Saarimäki, J.; Poutanen, M.; Härkönen, P.; Väänänen, K. FAM3C modulates osteogenic cell differentiation and affects bone volume and cortical bone mineral density. Bonekey Rep. 2016, 5, 787. [Google Scholar] [CrossRef]
- Bendre, A.; Büki, K.G.; Määttä, J.A. Fam3c modulates osteogenic differentiation by down-regulating Runx2. Differentiation 2017, 93, 50–57. [Google Scholar] [CrossRef]
- Schmidt, U.; Uluca, B.; Vokic, I.; Malik, B.; Kolbe, T.; Lassnig, C.; Holcmann, M.; et al. Inducible overexpression of a FAM3C/ILEI transgene has pleiotropic effects with shortened life span, liver fibrosis and anemia in mice. PLoS One 2023, 18(9), e0286256. [Google Scholar] [CrossRef]
- de Wit, N.J.; IJosterink, E.; Keshtkar, S.; Hooiveld, G.J.; Mensink, R.P.; et al. Oit1/Fam3D, a gut-secreted protein displaying nutritional status-dependent regulation. J. Nutr. Biochem. 2012, 23(11), 1425–33. [Google Scholar] [CrossRef]
- Peng, X.; Xu, E.; Liang, W.; Pei, X.; Chen, D.; Zheng, D.; et al. Identification of FAM3D as a new endogenous chemotaxis agonist for the formyl peptide receptors. J. Cell Sci. 2016, 129(9), 1831–42. [Google Scholar] [CrossRef]
- He, L.; Fu, Y.; Deng, J.; Shen, Y.; Wang, Y.; Yu, F.; Xie, N.; Chen, Z.; Hong, T.P.; et al. Deficiency of FAM3D (Family With Sequence Similarity 3, Member D), a novel chemokine, attenuates neutrophil recruitment and ameliorates abdominal aortic aneurysm development. Arterioscler. Thromb. Vasc. Biol. 2018, 38(7), 1616–1631. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Peng, X.; Li, Q.; Wang, P.; Lv, P.; Song, Q.; et al. FAM3D is essential for colon homeostasis and host defense against inflammation associated carcinogenesis. Nat. Commun. 2020, 11(1), 5912. [Google Scholar] [CrossRef]
- Xu, W.; Gao, Z.; Wu, J.; Wolf, B.A. Interferon-gamma-induced regulation of the pancreatic derived cytokine FAM3B in islets and insulin-secreting betaTC3 cells. Mol. Cell Endocrinol. 2005, 240(1-2), 74–81. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, B.R.; Cook, J.R.; Young, R.A.; Wolf, B.A. PDX-1 interaction and regulation of the Pancreatic Derived Factor (PANDER, FAM3B) promoter. Biochim. Biophys. Acta 2008, 1779(10), 645–51. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, X.; Wang, P.; He, Q.; Huang, H.; Zheng, T.; Li, Y.; Jia, H.; Xu, L.; Zhao, H.; et al. The cytokine FAM3B/PANDER is an FGFR ligand that promotes posterior development in Xenopus. Proc. Natl. Acad. Sci. U S A 2021, 118(20), e2100342118. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, Z.; Robert, C.E.; Burkhardt, B.R.; Gaweska, H.; Wagner, A.; et al. Structure-function studies of PANDER, an islet specific cytokine inducing cell death of insulin-secreting beta cells. Biochemistry 2005, 44(34), 11342–52. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.G.; Schupp, M.; Burkhardt, B.R.; Wu, J.; Young, R.A.; Wolf, B.A. Liver-specific overexpression of pancreatic-derived factor (PANDER) induces fasting hyperglycemia in mice. Endocrinology 2010, 151(11), 5174–84. [Google Scholar] [CrossRef] [PubMed]
- Robert-Cooperman, C.E.; Wilson, C.G.; Burkhardt, B.R. PANDER KO mice on high-fat diet are glucose intolerant yet resistant to fasting hyperglycemia and hyperinsulinemia. FEBS Lett. 2011, 585(9), 1345–9. [Google Scholar] [CrossRef]
- Chen, Z.; Ding, L.; Yang, W.; Wang, J.; Chen, L.; Chang, Y.; et al. Hepatic activation of the FAM3C-HSF1-CaM pathway attenuates hyperglycemia of obese diabetic mice. Diabetes 2017, 66(5), 1185–97. [Google Scholar] [CrossRef]
- Hu, Y.; Li, J.; Li, X.; Wang, D.; Xiang, R.; Liu, W.; Hou, S.; Zhao, Q.; Yu, X.; Xu, M.; Zhao, D.; Li, T.; Chi, Y.; Yang, J. Hepatocyte-secreted FAM3D ameliorates hepatic steatosis by activating FPR1-hnRNP U-GR-SCAD pathway to enhance lipid oxidation. Metabolism 2023, 146, 155661. [Google Scholar] [CrossRef]
- Yang, J.; Gao, Z.; Robert, C.E.; Burkhardt, B.R.; Gaweska, H.; Wagner, A.; et al. Structure-function studies of PANDER, an islet specific cytokine inducing cell death of insulin-secreting beta cells. Biochemistry 2005, 44(34), 11342–52. [Google Scholar] [CrossRef]
- Shehata, M.M.; Kamal, M.M.; El-Hefnawy, M.H.; El-Mesallamy, H.O. Association of serum pancreatic derived factor (PANDER) with beta-cell dysfunction in type 2 diabetes mellitus. J. Diabetes Complications 2017, 31(4), 748–52. [Google Scholar] [CrossRef]
- MarElia, C.B.; Kuehl, M.N.; Shemwell, T.A.; Alman, A.C.; Burkhardt, B.R. Circulating PANDER concentration is associated with increased HbA1c and fasting blood glucose in Type 2 diabetic subjects. J Clin. Transl. Endocrinol. 2018, 11, 26–30. [Google Scholar] [CrossRef]
- Chen, M.; Cao, X.; Huang, S.; Yang, J.; Bao, J.; Su, M. Evaluation of biomarkers and immune microenvironment of gestational diabetes mellitus evidence from omics data and machine learning. Sci Rep. 2025, 15(1), 35531. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Yang, C.; Lai, F.; Hong, Z.; Lin, H.; Liu, J.; et al. Elevated circulating level of a cytokine, pancreatic-derived factor, is associated with metabolic syndrome components in a Chinese population. J Diabetes Investig. 2016, 7(4), 581–6. [Google Scholar] [CrossRef]
- Wang, H.; Yu, F.; Zhang, Z.; Hou, Y.; Teng, W.; Shan, Z.; et al. Effects of circulating member B of the family with sequence similarity 3 on the risk of developing metabolic syndrome and its components: A 5-year prospective study. J. Diabetes Investig. 2018, 9(4), 782–8. [Google Scholar] [CrossRef]
- Song, Q.; Song, J.; Li, C.; Wang, Y.; Qi, L.; Wang, H. Genetic variants in the FAM3C gene are associated with lipid traits in Chinese children. Pediatr. Res. 2021, 89(3), 673–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, W.; Wang, J.; Meng, Y.; Guan, Y.; Yang, J. FAM3 gene family: a promising therapeutical target for NAFLD and type 2 diabetes. Metabolism 2018, 81, 71–82. [Google Scholar] [CrossRef]
- Yang, J.; Guan, Y. Family with sequence similarity 3 gene family and nonalcoholic fatty liver disease. J Gastroenterol. Hepatol. 2013, 28 Suppl 1, 105–11. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Luo, Y.; Wang, J.; Meng, Y.; Sun, L.; et al. repurposing doxepin to ameliorate steatosis and hyperglycemia by activating FAM3A signaling pathway. Diabetes 2020, 69(6), 1126–39. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Li, J.; Li, N.; Chen, Z.; Ma, L.; Peng, W.; et al. FAM3A enhances adipogenesis of 3T3-L1 preadipocytes via activation of ATP-P2 receptor-Akt signaling pathway. Oncotarget 2017, 8(28), 45862–73. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–74. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Sanchez-Veja, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173(2), 321–337. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.T.; Ma, D.D.; Gong, Q.; Lin, Z.Y.; Li, Z.H.; Ye, J.X.; Qin, C.H.; Jin, W.D.; Zhang, J.X.; Zhang, Z.Y. FAM3 family genes are associated with prognostic value of human cancer: a pan-cancer analysis. Sci. Rep. 2023, 13(1), 15144. [Google Scholar] [CrossRef]
- Li, Z.; Mou, H.; Wang, T.; Xue, J.; Deng, B.; Qian, L.; et al. A non-secretory form of FAM3B promotes invasion and metastasis of human colon cancer cells by upregulating slug expression. Cancer Lett. 2013, 328(2), 278–84. [Google Scholar] [CrossRef]
- Huang, R.; Wang, H.; Hong, J.; Wang, Z.; Wu, J.; Huang, O.; He, J.; Chen, W.; Li, Y.; Chen, X.; Shen, K. A senescence-associated signature refines the classification of different modification patterns and characterization of tumor immune microenvironment infiltration in triple-negative breast cancer. Front. Pharmacol. 2023, 14, 1191910. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Duan, C. Upregulation of FAM3B promotes cisplatin resistance in gastric cancer by inducing epithelial-mesenchymal transition. Med. Sci. Monit. 2020, 26, e921002. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12(3), 286–93. [Google Scholar] [CrossRef]
- Yang, W.; Feng, B.; Meng, Y.; Wang, J.; Geng, B.; Cui, Q.; et al. FAM3C-YY1 axis is essential for TGF-beta-promoted proliferation and migration of human breast cancer MDA-MB-231 cells via the activation of HSF1. J. Cell. Mol. Med. 2019, 23(5), 3464–75. [Google Scholar] [CrossRef]
- Caldeira, I.D.S.; Giovanini, G.; Adorno, L.F.; Fernandes, D.; Ramos, C.R.; Cruz-Visalaya, S.R.; et al. Antiapoptotic and prometastatic roles of cytokine FAM3B in triple-negative breast cancer. Clin. Breast Cancer 2024, 24(7), e633–e644.e2. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, D.; Fu, Q.; Wang, M.; Cheng, Z.; Cao, Y.; Wang, L.; Liu, J.; Zhao, Y. DNA methylation-driven gene FAM3D promotes colorectal cancer growth via the ATF4-SESN2-mTORC1 pathway. Aging (Albany NY) 2024, 16(19), 12866–12892. [Google Scholar] [CrossRef]
- Gao, Z.H.; Lu, C.; Wang, Z.N.; Song, Y.X.; Zhu, J.L; Gao, P.; et al. ILEI: a novel marker for epithelial-mesenchymal transition and poor prognosis in colorectal cancer. Histopathology 2014, 65(4), 527–38. [Google Scholar] [CrossRef]
- Sun, Y.; Jia, X.; Gao, Q.; Liu, X.; Hou, L. The ubiquitin ligase UBE4A inhibits prostate cancer progression by targeting interleukin-like EMT inducer (ILEI). IUBMB Life 2017, 69(1), 16–21. [Google Scholar]
- Ma, T.; Jin, L.; Bai, S.; Liu, Z.; Wang, S.; Shen, B.; Cho, Y.; Cao, S.; Sun, M.J.S.; Fazli, L.; et al. Loss of feedback regulation between FAM3B and androgen receptor driving prostate cancer progression. J. Natl. Cancer Inst. 2024, 116(3), 421–433. [Google Scholar]
- Maciel-Silva, P.; Caldeira, I.; de Assis Santos, I.; Carreira, A. C. O.; Siqueira, F. R.; Antonioli, E.; Goldberg, A. C.; Belizário, J. E.; Garay-Malpartida, H. M. FAM3B/PANDER inhibits cell death and increases prostate tumor growth by modulating the expression of Bcl-2 and Bcl-XL cell survival genes. BMC Cancer 2018, 18(1), 90. [Google Scholar]
- Noguchi, K.; Dalton, A.C.; Howley, B.V.; McCall, B.J.; Yoshida, A.; Diehl, J.A.; et al. Interleukin-like EMT inducer regulates partial phenotype switching in MITF-low melanoma cell lines. PLoS One 2017, 12(5), e0177830. [Google Scholar] [CrossRef]
- Noguchi, K.; Dincman, T.A.; Dalton, A.C.; Howley, B.V.; McCall, B.J.; Mohanty, B.K.; et al. Interleukin-like EMT inducer (ILEI) promotes melanoma invasiveness and is transcriptionally up-regulated by upstream stimulatory factor-1 (USF-1). J. Biol. Chem. 2018, 293(29), 11401–14. [Google Scholar] [PubMed]
- Zhu, Y.H.; Zhang, B.; Li, M.; Huang, P.; Sun, J.; Fu, J.; et al. Prognostic significance of FAM3C in esophageal squamous cell carcinoma. Diagn. Pathol. 2015, 10, 192. [Google Scholar] [CrossRef] [PubMed]
- He, S.L.; Wang, W.P.; Yang, Y.S.; Li, E.M.; Xu, L.Y.; Chen, LQ. FAM3B promotes progression of oesophageal carcinoma via regulating the AKT-MDM2-p53 signalling axis and the epithelial-mesenchymal transition. J. Cell. Mol. Med. 2019, 23(2), 1375–85. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell. Biol. 2011, 13(3), 317–23. [Google Scholar] [CrossRef]
- Yin, S.; Chen, F.; Ye, P.; Yang, G. Overexpression of FAM3C protein as a novel biomarker for epithelial-mesenchymal transition and poor outcome in gastric cancer. Int. J. Clin. Exp. Pathol. 2018, 11(8), 4247–56. [Google Scholar]
- Shi, M.; Duan, G.; Nie, S.; Shen, S.; Zou, X. Elevated FAM3C promotes cell epithelial- mesenchymal transition and cell migration in gastric cancer. Onco Targets Ther. 2018, 11, 8491–505. [Google Scholar]
- Lahsnig., C.; Mikula, M.; Petz, M.; Zulehner, G.; Schneller, D.; van Zijl, F.; et al. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene 2009, 28(5), 638–50. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Sheng, W.; Zhang, X.; Jiao, S.; Li, F. ILEI drives epithelial to mesenchymal transition and metastatic progression in the lung cancer cell line A549. Tumour Biol. 2014, 35(2), 1377–82. [Google Scholar]
- Thuya, W.L.; Kong, L.R.; Syn, N.L.; Ding, L.W.; Cheow, E.S.H.; Wong, R.T.X.; et al. FAM3C in circulating tumor-derived extracellular vesicles promotes non-small cell lung cancer growth in secondary sites. Theranostics 2023, 13(2), 621–638. [Google Scholar]
- Shiiba, M.; Ishige, S.; Saito, Y.; Shimizu, T.; Minakawa, Y.; Kasamatsu, A.; et al. Down-regulated expression of family with sequence similarity 3, member B (FAM3B), in oral squamous cell carcinoma. Oral Sci. Internal 2012, 9(1), 9–16. [Google Scholar]
- Wu, C.C.; Xiao, Y.; Li, H.; Mao, L.; Deng, W.W.; Yu, G.T.; et al. Overexpression of FAM3C is associated with poor prognosis in oral squamous cell carcinoma. Pathol. Res. Pract. 2019, 215(4), 772–8. [Google Scholar] [CrossRef]
- Kastenhuber, E. R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170(6), 1062–1078. [Google Scholar] [CrossRef]
- Blagih, J.; Buck, M. D.; Vousden, K. H. p53, cancer and the immune response. J. Cell Science 2020, 133(5), jcs237453. [Google Scholar]
- Belizario, J.E.; Dinarello, C.A. Interleukin 1, interleukin 6, tumor necrosis factor, and transforming growth factor beta increase cell resistance to tumor necrosis factor cytotoxicity by growth arrest in the G1 phase of the cell cycle. Cancer Res. 1991, 51(9), 2379–85. [Google Scholar] [PubMed]
- Yue, X.; Zhao, Y.; Zhang, C.; Li, J.; Liu, Z.; Liu, J.; Hu, W. Leukemia inhibitory factor promotes EMT through STAT3-dependent miR-21 induction. Oncotarget 2016, 7(4), 3777–90. [Google Scholar]
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast growth factor receptors (FGFRs) and noncanonical partners in cancer signaling. Cells 2021, 10(5), 1201. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
EMT program stimulated by canonical and non-canonical pathways induced by TGF-β and crosstalk with FAM3 protein pathways in cancer cells. After binding to a tetrameric complex of type I and type II receptors (TβRI and TβRII), TGF-β induced signals activate SMAD2, which then combine with SMAD4, move to the nucleus and activate Slug and Snail transcription factors. Similar to TGFβ, FAM3B and FAM3C proteins, after binding their putative membrane receptors, LIFR and FGFR, respectively, also activate these transcription factors, to promote the expression of gene programs for EMT. The expression MMP-2, MMP-9, N-cadherin and vimentin promote the morphological changes associated with EMT, migration and metastasis. FAM3D targets mitochondrial respiratory pathway, increases ATP production, and inhibits ROS effects and cell death pathway by apoptosis. FAM3D binds to FPR1/2 receptors and activates other signaling molecules and regulatory cascades involved in neutrophil chemotaxis.
Figure 1.
EMT program stimulated by canonical and non-canonical pathways induced by TGF-β and crosstalk with FAM3 protein pathways in cancer cells. After binding to a tetrameric complex of type I and type II receptors (TβRI and TβRII), TGF-β induced signals activate SMAD2, which then combine with SMAD4, move to the nucleus and activate Slug and Snail transcription factors. Similar to TGFβ, FAM3B and FAM3C proteins, after binding their putative membrane receptors, LIFR and FGFR, respectively, also activate these transcription factors, to promote the expression of gene programs for EMT. The expression MMP-2, MMP-9, N-cadherin and vimentin promote the morphological changes associated with EMT, migration and metastasis. FAM3D targets mitochondrial respiratory pathway, increases ATP production, and inhibits ROS effects and cell death pathway by apoptosis. FAM3D binds to FPR1/2 receptors and activates other signaling molecules and regulatory cascades involved in neutrophil chemotaxis.
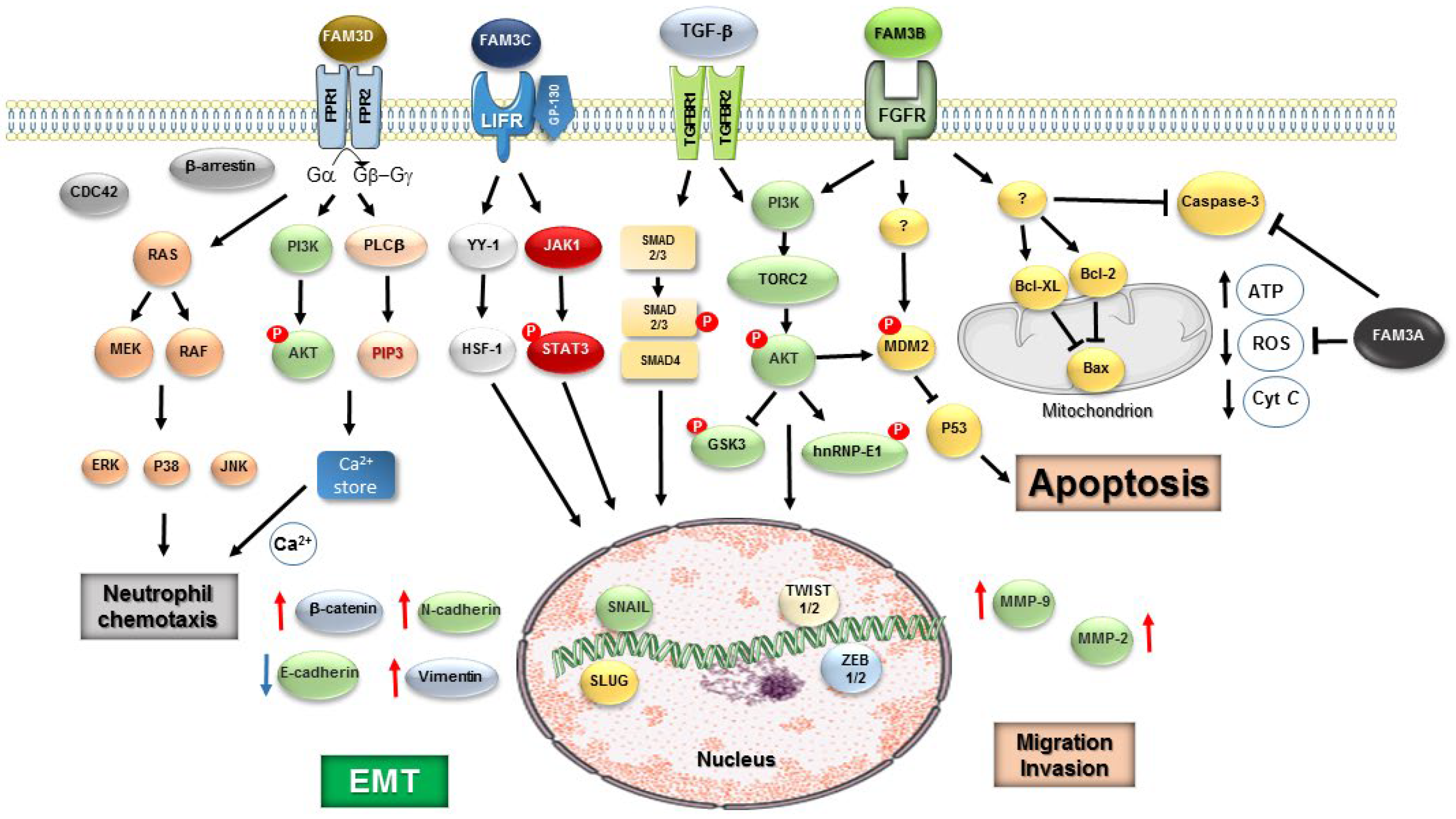

Table 1.
FAM3 Family molecular and functional feature.
| GENE / CHROMOSSOMAL LOCALIZATION | PROTEN FUNCTIONS | TISSUE DISTRIBUTION | IMPACT OF DEFICIENCY / KNOCKOUT PHENOTYPE |
| FAM3A Human chr Xq28 |
Mitochondrial-associated cytokine-like protein; interacts with F1-ATP synthase, enhances oxidative phosphorylation and ATP synthesis; regulates glucose and lipid metabolism | Highly expressed in heart, skeletal muscle, liver, brain, adipose tissue | Reduced mitochondrial ATP production, insulin resistance, impaired glucose tolerance, increased inflammation, worsened outcomes after an acute kidney and liver injury by ischemia / reperfusion |
| FAM3B / PANDER Human chr 21q22.3 |
Secreted cytokine-like protein; binds to receptors on pancreatic α- and β-cells; induces ER stress and apoptosis, and disrupts insulin secretion | Pancreatic islets (β-cells and α-cells); lower levels in liver and adipose tissue | Resistance to diet-induced glucose intolerance, altered glucagon secretion, increased body weight and adiposity; impaired hepatic glucose regulation; protection against β-cell apoptosis |
| FAM3C / ILEI Human chr 7q31.31 |
Secreted signaling protein; regulates epithelial–mesenchymal transition (EMT); involved in neurodevelopment and cancer metastasis | Broad: brain, liver, lung, pancreas, placenta; high in neurons and epithelial tissues | Accelerated osteogenic differentiation and mineralization, developmental defects in neural cells and memory-based behaviors; impaired EMT; reduced tumor invasion and metastasis in cancer models |
| FAM3D / Oit1 Human chr 3p14.2 |
Chemokine-like secreted protein; binds to formyl peptide receptors FPR1/FPR2; modulates neutrophil and macrophage recruitment; regulates gut inflammation | Gastrointestinal tract, immune cells, liver, adipose tissue | Disrupted gut immune homeostasis, increased susceptibility to colitis, impaired neutrophil chemotaxis; increase the development of abdominal aortic aneurysm |
Table 2.
FAM3 Family in Diabetes & Metabolic Syndrome.
| FAM3 MEMBER | PRIMARY METABOLIC FUNCTION | EFFECTS IN DIABETES | EFFECTS IN METABOLIC SYNDROME | KEY MECHANISMS INVOLVED |
| FAM3A | Mitochondrial energy regulator; insulin sensitizer | Pathogenic: induces β-cell apoptosis, ER stress, decreases insulin secretion; increases hepatic insulin resistance |
Promotes central metabolic syndrome, ↑ fasting glucose, ↑ hepatic TG, ↑ inflammation) |
Impaired insulin signaling, ↑ gluconeogenesis, ↑ lipogenesis, β-cell apoptosis via ER stress |
| FAM3B / PANDER | Endocrine factor from pancreatic α/β-cells; insulin resistance promoter |
Pathogenic: induces β-cell apoptosis, ER stress, and decreases insulin secretion; increases hepatic insulin resistance | Promotes central metabolic syndrome, ↑ fasting glucose, ↑ hepatic TG, ↑ inflammation) |
Impaired insulin signaling, ↑ gluconeogenesis, ↑ lipogenesis, β-cell apoptosis via ER stress |
| FAM3C / ILEI | Secreted cytokine-like protein; regulator of adipose inflammation, fibrosis and EMT | Protective: regulated glucose/lipid metabolism, restore CaM-Akt pathway, improve insulin resistance | Promotes overweight and obesity associated with genetic variants | Activate mTOR-SREBP1-FAS lipogenic pathway; adipose tissue remodeling |
| FAM3D / Oit1 | Secreted cytokine-like protein; regulator of Gut–liver axis, inhibits glucagon secretion via activation of prohormone convertase 2 (PC2) | Protective: reduces inflammation and improves glucose tolerance | Reduces obesity-associated inflammation and protects against fatty liver deposition | Increase lipid oxidation, reduce lipid deposition via FPR1/hnRNP/short-chain Acyl-CoA dehydrogenase (SCAD) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



