
Submitted:
30 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Equine Infectious Anemia (EIA) is a retroviral disease of equids, for which there is no vac-cine particularly adapted to American viral strains. In this work we searched for possible epitope regions for the surface proteins gp90 and gp45, rationally employing the latest available bioinformatics tools that constitute the state of the art in the field. We selected eight regions that contain numerous overlapping epitopes that have a high coverage amongst American viral strains and designed a chimeric envelope protein with those proteins fused in tandem as a novel vaccine candidate. In silico predictors were used to analyze chimeric protein physicochemical and immunogenic properties, as well as its al-lergenicity and toxicity. Protein structure was predicted and validated, and its ability to trigger cytotoxic immune responses was predicted by molecular docking to ELA alleles. The proposed sequence is predicted to be highly immunogenic and sets the base for a novel EAIV vaccine that could be used to protect against several American field strains.
Keywords:
bioinformatics
; equine infectious anemia virus
; env-glycoproteins
; B and T-cell epitopes
1. Introduction
Equine infectious anemia virus (EIAV) causes a persistent infection in horses that typically shows a variable disease course with acute, chronic, and inapparent stages [1]. Acute and chronic stages are defined by waves of viraemia, with fever, anemia, thrombocytopenia and various wasting signs. One-year post-infection, animals progress to lifelong inapparent carriers with low levels of viral replication in monocyte-rich tissue reservoirs, due to a strict immunologic control over virus replication [2]. Meanwhile the EIAV quasi-species coexist and the horse’s immune system is exposed to high levels of antigenic heterogeneity, especially in Env glycoproteins, gp90 and gp45, which are the first and main immunogens exposed [3,4].
EIAV gp90 has a complex of discontinuous sequences in its C-terminal two-thirds, that contain the binding domains responsible for attachment to equine lentivirus receptor-1 (ELR-1) [5,6]. The EIAV exhibits great genetic variability for efficient evasion from host immunity [7]. Conserved and variable regions exist principally within gp90 protein, which has the maximum exposure to the immune system.
In contrast, gp45 glycoprotein is more shielded from the immune system and plays the role of fusing by its highly conserved fusion peptide to the host cell during viral entry [8].
In order to design a vaccine, most studies have been limited to the identification of specific B- and T-cell epitopes in EIAV glycoproteins, following experimental infection [9,10,11,12,13,14]. We have previously identified four peptides representing conserved regions of these glycoproteins, recognized by antibodies and peripheral blood mononuclear cells from naturally infected animals [15,16,17,18].
The protective efficacy of these vaccines can range from preventing disease to causing death due to EIAV replication activity [19]. The only attenuated vaccine that has been used to provide 85% protection against different natural EIAV strains, despite showing a 32% envelope protein divergency, was developed in China and applied up to 1990 [7,20,21]. Chinese studies have suggested that its effectiveness could be related to the diversity of this strain, mainly associated with the envelope proteins, mimicking the diversity of the viral quasi-species during the inapparent stage [7,22]. Despite being largely protective, vaccination has been discontinued because it did not allow the differentiation between infected and vaccinated animals, so there are currently no vaccines available.
The introduction of Reverse Vaccinology [23] in vaccine development together with the growing knowledge of the host immune response led to new insights in vaccine design using in silico epitope predictions. Immunoinformatics tools use several approaches such as creation and management of databases, definition of both structural and functional signatures and the design and application of predictive tools. These tools allow us to identify multiple epitopes useful to the design of vaccines targeting conserved epitopes in rapidly mutating pathogens, such as EIAV [24,25,26].
The purpose of this study is to design a novel vaccine candidate by predicting B- and T-cell epitopes on EIAV envelope proteins using bioinformatics and immunoinformatics tools. This will allow us to set the foundations for the rational design and development of an EIAV vaccine that could offer broad coverage against American strains of the virus.
2. Materials and Methods
The step-by-step procedure of in silico analysis is depicted in Figure 1.
2.1. Retrieval of the Target Sequences and Multiple Sequence Alignment (MSA)
The target sequences of EIAV ENV polyprotein were retrieved from the UNIPROT database [27]. Given that our goal is to design a vaccine primarily targeting American viral strains, only sequences belonging to this continent were selected and the ENV polyprotein sequences having less than 99% identical residues were included. To analyze the sequence similarity and obtain a consensus sequence among all the selected ones, the MAFFT [28] program was used with its default values, from the Jalview [29] interface. A consensus sequence was obtained from the MSA by placing the most frequent residue in each column.
2.2. Prediction of Protein Boundaries
PROCLEAVE software [30], which predicts potential enzymatic cleavage sites using both sequence and structural information, was run on the consensus sequence to establish the boundaries of each domain. Whenever possible, the sites defined in previous publications were included.
2.3. Epitope Prediction
2.3.1. CD8+ Epitope Prediction (ELA I)
It was performed using the MHCflurry-2.0 [31] program, which allows the prediction of pan-allelic peptides presented by ELA I (Equine Lymphocyte Antigen, equivalent to MHC-I). It employs convolutional neural networks. The program was run locally on all of the sequences selected. Among the epitopes found (nine residues), we kept those with a Presentation Percentile of less than 0.5%, which represents the acceptance of up to this value of false positives.
2.3.2. CD4+ Epitope Prediction (ELA-II)
MHC class II predictions were carried out to identify 15-mer epitopes using the NetMHCpan-4.2 [32] program installed locally. The alleles for ELA-II (Equine Lymphocyte Antigen, equivalent to MHC-II) predictions were selected and downloaded from the Immuno Polymorphism Database (IPD) [33]. The program was run locally on all of the sequences selected. Peptides with score_EL > 0.5 cut off were used to select most likely binding epitopes.
2.3.3. IFN-γ inducing MHC class II binding peptides
To identify peptides able to induce IFN-γ production by Th-cells, we used the "scan" program described by Dhanda et al [34], the IFNepitope web server (http://crdd.osdd.net/raghava/ifneepitope), which is able to predict IFN-γ induction with a maximum accuracy of 82.10% and to classify the epitopes between those that generate IFN-γ and those that do not.
2.3.4. Linear B-Cell Epitope Prediction
Linear B-cell epitope prediction was performed using the BEPIPRED-2.0 [35] program on the consensus sequence. Peptides with score EL > 0.6 cut off were used to select most likely binding epitopes.
2.4. Criteria for Selection of Immunogenic Sequences
Taking into account the predicted epitopes in the previous steps, a number of regions containing overlapping epitopes recognized by ELA-I and/or ELA-II and/or BCR of B-cells were defined, mostly within conserved domains. Each region was conformed with at least two types of predicted epitopes. Furthermore, those epitopes within the Principal Neutralizing Domain of gp90 that are associated with binding to the ELR-1 were also included.
In order to ensure optimal safety, the allergenic properties of each region were analyzed by a combination of three programs, AllerTOP v.2; AlgPred and AllergenFP, https://www.ddg-pharmfac.net/AllerTOP/; https://webs.iiitd.edu.in/raghava/algpred2; https://ddg-pharmfac.net/AllergenFP/, respectively. After eliminating allergenic epitopes, we checked their toxicity by ToxinPred [36] (https://http://crdd.osdd.net/raghava/toxinpred/).
Vaccinal constructs containing overlapping epitope regions were designed using different amino acid linker sequences and tested in terms of their allergenicity and toxicity (AllerTOP v.2, AlgPred and AllergenFP), antigenicity (Vaxijen3.0, https://www.ddg-pharmfac.net/vaxijen3/home/; ANTIGENpro, https://scratch.proteomics.ics.uci.edu/explanation.html#ANTIGENpro) and solubility and stability (GRAVY, https://web.expasy.org/protparam/; CamSol, https://www-vendruscolo.ch.cam.ac.uk/camsolmethod.html and SOLUPROT, https://loschmidt.chemi.muni.cz/soluprot/).
2.5. Vaccinal Construct Structure Prediction
Secondary structure was predicted with PSIPRED 4.0 (http://bioinf.cs.ucl.ac.uk/psipred/), and tertiary structure with AlphaFold 3.0 (https://alphafoldserver.com/). Structure was refined with galaxyWEB (https://galaxy.seoklab.org/) and validated with Swissmodel Structure Assessment tools (https://swissmodel.expasy.org/assess).
2.6. Vaccinal Candidate Cellular Immunogenicity Assessment Via Molecular Docking
Molecular docking experiments were used to validate that the selected epitopes, based on sequence alone, could bind to their respective major histocompatibility complexes, considering possible molecular receptor-epitope interactions.
Comparative molecular docking analyses were run for the four highest-scoring cytotoxic T lymphocyte (CTL) epitopes with their respective major histocompatibility complex type I (ELA-I). Each specific ELA-I 3D model was obtained by homology modeling using the crystal structure of Equine MHC I (PDB code: 4zuv) as a template. Five models were generated for each of the ELA-I variants, using Modeller38 9.21 software. The model with the highest DOPE39 score was selected to perform each of the docking trials. Docking assays were performed using GalaxyPepDock40 software. Complexes (ELA-I - peptide) with the highest GalaxyPepDock score were relaxed using the Relax41 application, and then the binding energy was estimated using the Interface Analyzer42 implemented in the Rosetta package.
3. Results
3.1. In Silico Design and Validation Workflow
The strategy followed in this work to design a high coverage ENV based EIAV vaccine can be divided in three different sections: the first one involved the identification of conserved regions containing overlapping epitopes among different strains (Figure 1, A), the second one involved the assembly of the conserved regions with different linkers and the selection of the most appropriate one in terms of immunogenicity, stability, allergenicity and toxicity (Figure 1, B), and the final section aimed to predict the antigen’s structure and validate the generated model (Figure 1, C). Finally, the last step also included a docking study of the most important CD8(+) epitopes included in the antigen with ELA I molecules, as a predictor of the ability of the vaccinal construct to induce cellular immune responses.
3.2. MSA and Definition of Consensus Sequence
We identified 12 ENV polyprotein representative sequences of EIAV American strains and then used them to generate a multiple sequence alignment (MSA) (Figure 2), in order to visualize the conserved and variable regions between the sequences and to obtain a consensus sequence that would later allow us to design the high coverage vaccinal construct.
The variable regions matched the regions in gp90 protein (Figure 2, green squares), with the exception of two regions (AA 40-80 and 370-400). In contrast, gp45 protein is more conserved as it has been previously reported [2,7,21,22]. Nevertheless, region AA 760-800 in gp45 exhibits a certain degree of variability.
Protease cleavage sites were predicted using Procleave software and identified on the MSA. The furin cleavage sequence RHKR†DFGI (Figure 2, C1) was found to be among the 20 best scoring sequences predicted by the program (Table1-SI). An additional cleavage site was found on gp45 and included in the MSA (Figure 2, C2).
3.3. Epitope Prediction
As the objective of this work was to design a high coverage vaccine for EIAV American strains, those epitopes that had a high representation across the different strains of interest were the main target to include in the design. With this objective in mind, all the 12 sequences were analyzed through different T and B epitope predictors. This resulted in a set of epitopes for each sequence which were then compared with those obtained for the other sequences and represented with a frequency number in Figure 3.
3.3.1. CD8+ T-Cell Epitopes
MHC-Flurry2 was used with all the sequences described to identify ELA-I epitopes (Table-2-SI). Three regions with a high frequency of ELA-I epitopes were identified. The first one in gp90, between residues 100 and190, the second one in the interface of gp90 and gp45 (residues 480-580) and the third one between residues 620 and 700, spanning a fragment of gp45 ectodomain and transmembrane region.
3.3.2. CD4+ T-Cell Epitopes
NetMHCpan4.1 was used with all the sequences described to identify ELA-II epitopes (Table-3-SI). Five preliminary regions with a high frequency of occurrence of epitopes can be noted (Figure 3 B). The first sequence is located into the gp90 protein between residues 140 and 160, and the others, in the gp45 protein in fragments 490-530, 620-660, 710-740, and 760-820.
3.3.3. IFN-γ Inducing MHC Class II binding Peptides
IFN-γ high-frequency epitope regions were found at residues 140-160, 490-530, 620-660, 710-740 and 760-820. These regions coincide with CD4+ high-frequency epitope regions (Figure 3 C).
3.3.4. B-Cell Epitope Prediction
For B-cell epitopes we could define six regions in gp90 protein with high coverage: 20-30, 260-280, 300-310, 380-390, 435-455, and 475-490 (Figure 3 D). For gp45 protein, two regions localized into the intracytoplasmic domain have remarkably high scores between residues 690 and 750, and 780 and 810. Furthermore, three B-cell epitopes conserved among four different strains were found in the PND region (AA 197-235), which have the potential of eliciting neutralizing antibodies.
3.4. Selection of Polypeptide Sequences with Overlapping Epitopes
In order to optimize the size of the vaccinal construct, those regions with high frequency of overlapping epitopes as well as a variety of epitope types were selected to be included in the primary sequence. Eight regions were defined based on the presence of ELA-I and ELA-II recognized epitopes, including those associated with the secretion of IFN-γ and B-cell epitopes (Figure 3, red boxes). Region 1 is an exception, as it only contains high frequency of B-cell epitopes. In addition, region number 3 was included as it contains ELR-1 binding motif (AA 197-235), which is the domain associated with the first step of viral entry into cells and the principal neutralizing domain of the protein (Fig 3 and 4). As a general rule, gp90 variable residues were avoided (Figure 3, green rectangles), although some were included due to the high frequency of the epitopes observed among different strains in those regions (regions 1 and 2).
Despite the presence of numerous overlapping epitopes with high frequency among strains in the residues corresponding to the fusion peptide (AA 490-505), this region was not included due to its hydrophobic nature, which may lead to protein aggregation and instability of the immunogen if included.
A summary of the selected regions and types of epitopes included can be seen on Figure 4, where a preliminary sequence is depicted from the N to C-terminal end of the antigen sequence. Regions 1, 3 and 5 are mostly B-epitopes enriched regions with a high coverage among the different strains. The other five regions have a higher number of T-cell epitopes, with a variety of ELA-I, ELA-II and IFN-γ triggering sequences, as well as some B-epitopes as well. Taking into account the complete sequence, there is a total of 18 ELA-I epitopes, 9 ELA-II epitopes, 25 Th1 epitopes and 32 LB epitopes.
3.5. Vaccinal Antigen Design and in Silico Characterization
3.5.1. Linker Optimization
In order to join the high coverage overlapping epitope regions described in Figure 4, three different linkers were analyzed in term of their contribution for protein stability, solubility, toxicity, allergenicity and immunogenicity. Three different proteins were designed by joining each of the high coverage overlapping epitope regions to another one though one copy of the tested linker (Table-4-SI), and then by analyzing the complete primary sequences through different predictors (table 1). The first construct, which was designed with AAY linker, is predicted to be instable (instability index lower than 40), with low probability of adequate solubility (CamSol) and high probability of being an allergen (AllerTOPv.2). The second one, containing the flexible GGGGS linker, is also predicted to be instable (instability index lower than 40) and a probable allergen (AllergenFP). In contrast, the third construct containing the linker EAAAK, had acceptable values in all of the used predictors and thus was selected as the final antigenic sequence and named high coverage ENV (hcENV).
3.5.2. Prediction of the Vaccinal Candidate Structure
The secondary structure of hcENV obtained with PSIPRED (Figure 5 A) revealed that it is composed predominantly of random coils (46,5%), following by α-helices (37,3%) and β-strands (15.8%). This suggests a moderately helical protein with a substantial proportion of disordered or flexible regions. A model of the predicted tertiary structure of the protein was obtained with AlphaFOLD 3.0 and refined with galaxyWEB (Figure 5 B). A general inspection of the obtained model revealed higher percentages of α-helices and coil secondary structures (43.8% and 54.6% respectively) in comparison with ones obtained using PSIPRED, and a small percentage of residues forming β-strands (1.7%).
The short B epitope containing Region 1 (red) acquired an α-helix conformation, while Region 2 (orange) is composed of a larger α-helix, a small α-helix and a β-strand connected by coils. The Region 3 (yellow) is composed by a short α-helix that is a continuation of the previous EAAAK linker, and the rest adopts a coil structure that continues into Region 4 (green), which contains a β-strand that stablishes a β-sheet conformation with the Region 2 β-strand, and two small α-helix regions following by a long coil structure. This long coil encompasses the final residues of Region 4 and almost the totality of Region 5 (blue), with the exception of a small α-helix and an intra-region β-sheet. Region 6 (purple) and Region 7 (light pink) form a long α-helix that contains the linker in the middle as part of the structure. The rest of the protein contains two moderately long α-helices that seem to interact with each other, and a final long α-helix, all separated by short coil regions (the last two α-helices are part of Region 8, bright pink). A relatively long coil marks the C-terminal end of Region 8.
hcENV predicted structure was analyzed with SwissModel, and the local quality of the model was assessed (Figure 6 A). α-helices I to IV are the regions with the higher b-factor values, while helices V and VI have intermediate values, as well as β-sheet 1 and 2. Coil structures are the least reliable structures of the model. Nevertheless, the stereo-chemical quality of the model is very good, as 99.7% of the residues are located on the favoured regions of the Ramachandran plot (Figure 6 B). Furthermore, the normalized QMEAN value obtained for the model is similar to experimentally solved structures of proteins with a similar size to hcENV (Figure 6 C), indicating that the model has an acceptable quality.

Table 1.
Comparison of constructs designed with three different linkers. Sequences were compared in terms of their physicochemical properties (residue number, Molecular Weight (MW), Isoelectrical Point (IP)); stability (Instability Index); solubility (GRAVY, CamSol, SoluProt); immunogenicity (Vaxijen 3.0, ANTIGENpro); allergenicity (AllerTOPv.2, AllergenFP) and toxicity (Toxinpred2).
Table 1.
Comparison of constructs designed with three different linkers. Sequences were compared in terms of their physicochemical properties (residue number, Molecular Weight (MW), Isoelectrical Point (IP)); stability (Instability Index); solubility (GRAVY, CamSol, SoluProt); immunogenicity (Vaxijen 3.0, ANTIGENpro); allergenicity (AllerTOPv.2, AllergenFP) and toxicity (Toxinpred2).
| Linker | Residue number | MW | IP | Instability Index | Aliphatic Index | GRAVY | CamSol | SoluProt | Vaxijen 3.0 |
ANTIGEN pro |
Aller TOPv.2 |
AllergenFP | Toxinpred2 |
| Criteria for acceptance | - | - | - | < 40 | > 60 | < 0 | > 0,80 | > 0,50 | > 90% | > 0,90 | Non allergen | Non allergen | (-) |
| AAY | 347 | 39351 | 8.60 | 42.42 | 70.09 | -0.51 | 0,19 | 0,60 | 100% | 0.93 | Probable allergen | Non Allergen | (-) |
| GGGGS | 361 | 39421 | 8.65 | 47.57 | 63.49 | -0.58 | 0,83 | 0,61 | 100% | 0.97 | Non Allergen | Probable allergen | (-) |
| EAAAK | 361 | 40507 | 8.60 | 39.40 | 69.31 | -0.57 | 0,91 | 0,59 | 100% | 0.95 | Non Allergen | Non Allergen | (-) |
3.6. Docking of CD8+ T-Cell Epitopes on ELA-I
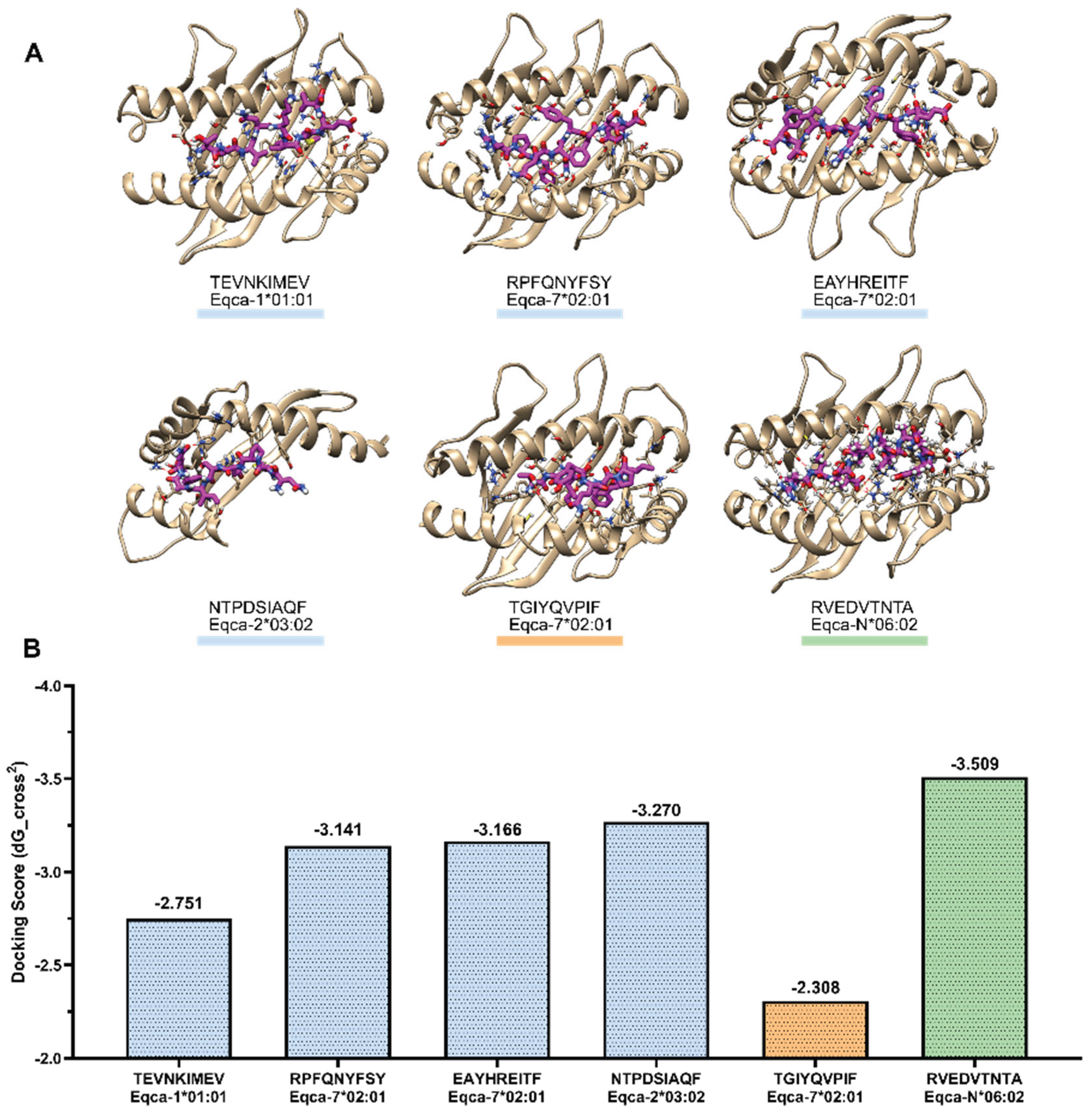
Due to the importance of cytotoxic T Lymphocytes responses to the resolution of early episodes of viraemia and clinical disease in EIAV-infected horses [9,11,12,14,37,38,39], hcENV representative ELA-I epitopes were tested in docking experiments with ELA-I alleles (Figure 7 A, Table-5-SI).
All the studied ELA-I epitopes showed a negative stabilizing score (Figure 7 B), which was normalized to the interface area of each complex. To validate the method, a peptide with low percentile affinity [TGIYQVPIF](356-405), expected to show the least favorable (least negative) score value and a crystallized solved ELA-epitope complex (PDB:4ZUV, RVEDVTNTA) [40], which should give a very favorable (most negative) docking value, were included.
Low percentile affinity peptide, [TGIYQVPIF](356-405) evidenced a high docking score despite the fact that MHC-Flurry predicted a low score value. The three epitopes of gp90 predicted by MHC-Flurry: [TEVNKIMEV](518-526), [RPFQNYFSY](142-150), [EAYHREITF](177-185) and one of gp45: [NTPDSIAQF](640-648) showed docking score values higher than the lowest value found for the low scoring reference, but lower than the highest value shown for the experimental complex. [RPFQNYFSY](142-150) and [EAYHREITF](177-185) interactions showed high score (ddG) values, and are stabilized by numerous hydrogen bonds, including several specific ones with the side chains. [TEVNKIMEV](518-526) epitope of gp90 showed the lowest docking value.
It is interesting to note that the complex of [NTPDSIAQF] (640-648) with Eqca-2*03:02 showed a good score despite the low solvent accessible surface of the complex interface (Figure 7 A), as Eqca-2*03:02 presents an N-terminal helix shorter than those of the other ELA-I molecules.
4. Discussion
In this work, we rationally applied well-founded principles of Reverse Vaccinology and Structural Vaccinology [25] in order to design a multiepitope EIAV vaccine against American strains, based on those with fully sequenced Env genes. The approach included the use of epitope prediction tools as well as solubility, antigenicity, allergenicity and toxicity predictors.
Epitope analysis was performed on envelope proteins because they were found to be a primary determinant of the efficacy of a lentiviral vaccine, despite their diversity [41,42,43]. An initial multiple sequence alignment was performed to obtain a consensus sequence and to identify conserved and variable regions among the different strains.
The variable regions matched the regions in gp90 protein, while gp45 protein is more conserved as it has been previously reported [2,7,21,22]. Principal Neutralizing Domain (PND), which is located on V3 of gp90, shows low sequence similarity between strains as previously reported [44,45]. On the other hand, the Fusion Peptide (FP) and the transmembrane protein are highly conserved.
Additionally, protein cleavage was predicted to confirm the localization of the gp90-gp45 boundary. The cleavage boundary by enzyme furin, between gp90 and gp45 predicted, is consistent with that described by other authors [46]. Our prediction indicates another possible cleavage site by cathepsin B and a matrix metallopeptidase-2 at the HLAG-VTGG site, within the cytoplasmic tail of gp45. Both the enzymes furin and cathepsin B have been reported but nobody described the matrix metallopeptidase-2 in horses until now [47]. This potential cleavage site is located in the same region than an experimentally found gp45 cleavage site, that was attributed to a viral protease [48]. The effect of this proteolytic cleavage on gp45 function is unknown, although the truncated product could play a role in protein trafficking, processing and virion egress. It has been proven that gp45 C-terminal end inhibits protein localization to plasma membrane, as well as gp90/gp45 furin cleavage and is directly linked to a decrease in virion production [49].
Epitope analysis focused on ELA-I, ELA-II, IFN-γ and B-cell epitopes due their importance in anti-viral immunity. gp90 and gp45 were extensively analyzed to identify epitopes that are present on several strains [11,38,39,41,50].
In this work, we predicted five ELA-I epitopes within residues 129-185, recognized by 15 ELA-I alleles. Indeed, we were able to demonstrate that the CTL epitope [RPFQNYFSY](142-150) had the best affinity score with the Eqca-7*02:01 allele. In previous experimental studies, we demonstrated that the synthetic peptide gp90-B [NAIECWGSFPGCRPFQNYFSYETNRSMHMDNNTATLLEAYHR](130-171) was recognized by ELA-I molecules in 4 of 5 non-apparent infected horses [50].
At the C-terminal end of this first region (129-185) we found two CTL epitopes (142-RPFQNYFSY-150 and 177-EAYHREITF-185) which are, to our knowledge, the first report of CTL epitopes in this area [11,38,39,41]
One of the earliest described immunodominant CD8+ T-cell epitopes in gp90 protein was Env-RW12 [RVEDVTNTAEYW], localized in the V3 region [9,38,39]. In the region 204-228 we detected a CTL epitope [RVEDVTNTTEY](217-227), recognized by four ELA-I alleles, that is quite similar to the epitope crystallized with the ELA receptor (4ZUV.pdb). However, the substitution of two amino acid residues was sufficient to decrease its potential binding capacity to the receptor in our docking studies (Table 5-SI). Nevertheless, Tagmyer et al [38] found a protective synthetic peptide [SNPV-RVEDVMNTTEY-WGFKW](213-232) associated with Env-specific CTL responses [11,39,43]. Given this experimental evidence, we decided to include [RVEDVTNTTEY](217-227) despite its predicted diminished binding capacity in comparison with Env-RW12 CD8+ T-cell epitope.
Within region 271-300 we predicted three CD8+ T-cell epitopes recognized by eight ELA-I alleles. Their sequences were included in the peptides [KRCPIDILYGIHPIRLCVQPDCTL](273-296) and [IHPIRLCVQPPFFLVQEKGI](283-302), associated with protective CTL immune response [11,50].
In the region (417-487) of the gp90 glycoprotein, we reported two other ELA-I epitopes. One of them (417-425) is within the synthetic peptide [NNYNCVVQSF-GVIGQAHLEL](416-435), associated with a protective CTL response [11,39]. The other, (472-480) is included in the gp90-A [ETWKLVKTSGI-TPLPISSEA-NTGLIRHKR](461-489) synthetic peptide, which was recognized by ELA-I and ELA-II molecules in earlier studies [50].
In the region 490-538 of gp45 protein, we found six ELA-I epitopes; the first two overlapped (504-512) and could be assembled by fifteen allotypes. The other four at the C-terminal end of this region were recognized by all alleles, except for the Eqca 2*01:01. This is the first report of potential ELA-I epitopes in this region so far [11,38,39,41,51].
In the selected region 620-663 we predicted two ELA-I epitopes. One of them, [NTPDSIAQF](640-648), was recognized by 12 allotypes with a high affinity percentile and a high score in molecular docking analysis, mainly with Eqca-2*03:02 alleles. The other one, [HIANWIPGL](655-663) was recognized by eight allotypes. A similar sequence [IGNWIPGL-GASSIKYIMFL](656-674), was found to induce CTL response in only two of the twelve horses immunized with EIAV D9 vaccine, so authors did not consider it as a protective CTL epitope [52].
The last CTL epitope (722-730) is within the sequence of the gp45-B(696-715) synthetic peptide and presented by ELA-I molecules, as previously described [50,52].
Many other synthetic peptides have been described in both env EIAV proteins to give positive results in CTL assays, but only eight were associated with a protective response [11,39,50,51]
Many other synthetic peptides have been described in both env EIAV proteins to give positive results in CTL assays, but only eight were associated with the protective response, four of them have very similar or even identical sequences to those five predictive ELA-I epitopes by our bioinformatic analysis in the present work
It was demonstrated the protective effect of IFN-γ production by CTL and Th cells induced by the attenuated vaccine EIAV DLV121 [7,10,38,52,53].Within the sequences analyzed we predicted 33 helper T-cell epitopes broadly reactive on most DQA/DQB2 and DQA/DRB1 haplotypes and 25 of them potentially associated with the secretion of IFN- γ.
Among the candidate regions defined, the region 129-185 presents eight overlapping Th-cell epitopes recognized by 34 pairs of ELA-II alleles, in agreement with previous experimental results obtained with the peptide [LNGSGQS-NAIECWGSFPGCR](123-142) and the gp90-B(130-171) peptide [13].
The second region rich in Th-cell epitopes spans residues 490-526. These epitopes are associated with IFN- γ production, and potentially presented by 42 pairs of ELA-II alleles. So far, experimental studies on this region are scarce, so its inclusion in a vaccine merits consideration.
Another interesting region is that comprising residues 620-663, which contains six Th1 overlapping epitopes broadly recognized by 32 pairs of alleles, principally with DQA alleles. In agreement, Tagmyer et al. described two peptides, 60 [MITFNTPDSIAQFGKDLWS](636-654) and 61 [AQFGKDLWSHIGNWIPGLGA](646-665), susceptible to recognition by DQA alleles and associated with protection against the experimental infection with homologous or heterologous strains [11,38].
Sterile immunity to the lentivirus challenge demands not only the CTL response, but also broadly neutralizing antibodies (bNAbs). One of the major goals of an HIV vaccine is to elicit bNAbs that can neutralize a broad spectrum of HIV-1 strains [54]. Although difficult to predict, a vaccine containing multiple epitopes including conserved sequences and those related to the interaction of the virion with its receptor, could be an attractive strategy [55,56]. The selected regions in this work would be consistent with this strategy.
We analyzed linear B-cell epitopes within both env proteins. We found 30 B-cell linear epitopes that match ELA-I and/or ELA-II epitopes. Indeed, many of them are consistent with previously published findings [3,4,15,16,18,57].
We found two B-cell epitopes overlapping T-cell epitopes (129-185). In agreement, antibodies from infected horses were shown to recognize these peptides in the same region, [WGSFPGC-RPFQNYFSYET](135-152) ; [SYETNRSMHMDN-NTATLLEAYHREITF](149-185), and [NAIECWGSFPGC-RPFQNYFSYETNRSMHMDN-NTATLLEAYHR](130-171) [3].
The region 207-227, displays interesting features as it contains the Principal Neutralizing Domain (PND, 197-235). Here we found B epitopes within the sequence related to the interaction with ELR-1 (212-223) [4]. Some potential disadvantages of this region are its variability and the four potentially glycosylable asparagines that could mask neutralizing antibody recognition [45]. Nevertheless, we decided to include this region and propose that it merits further experimental study as a vaccine candidate.
Most of the predicted B-cell epitopes were in the sequence 417-487, accordingly to experimental results that showed high reactivity in the C-terminal residues of gp90 molecules [18,57].
There are no predetermined variable domains in the gp45 protein, but it is recognized that the cytoplasmic tail (CT) (680-820) is more conserved than the ectodomain. The CT decreases the viraemia by suppressing the expression of Env thus the virus escapes immune recognition [49]. The first region of the CT is included in the region spanning residues 688-754, where we found four overlapping B-cell epitopes, in concordance with other reports [4]. It is not known if these antibodies could block its function but the strains of EIAV attenuated Chinese vaccine have a truncated CT downstream of the amino acid residue 710, reducing viral replication and pathogenicity in vivo [49].
The strategy used in this work led to the identification of T- and B-cell epitopes that were clustered to form eight candidate regions, five in gp90 and three in gp45, including 84 epitopes. Among the LB epitopes present in the different regions, three are localized in the PND with the potential ability of neutralizing 4 of the 12 strains included in the analysis, which is remarkable due to the high variability of that domain.
A set of proteins including each of the regions fused in tandem was designed, containing different linkers between each of the regions. The first linker, AAY, is the proteasome cleavage site sequence in mammalian cells and aims to achieve an efficient internal processing of the protein into their individual regions once it is internalized by the immune cells [58]. The second one, GGGGS, is a standard highly flexible sequence that has been widely used as linker in multiepitope vaccines [59,60,61,62]. The last one, EAAAK, is a rigid alfa-helix forming sequence that aims to maintain a fixed distance between the joined sequences [63,64,65]. The resulting theoretical sequences were tested by different web servers to predict their solubility, antigenicity, toxicity and allergenicity.
Based on the predictors, EAAAK linker was selected and the complete sequence of the proposed vaccinal candidate (hcENV) was analyzed to predict its secondary and tertiary structure. hcENV was predicted to adopt a globular structure with rigid α-helices separated by flexible regions. Therefore, the regions that are predicted to be rigid and solvent exposed would trigger B-cell responses efficiently, while the flexible regions would allow for effective processing and presentation of T-cell epitopes, having an overall balanced predicted immunogenicity [66].
Furthermore, hcENV potential ability to induce ELA-I triggered responses was assessed in docking experiments, where four peptides included in the vaccinal construct were tested against ELA-I molecules. Adequate binding of the peptides to their respective ELA-I molecules was predicted based on the high docking scores obtained for each complex, strengthening even more the potential immunogenicity of our vaccinal candidate. In fact, docking of peptides to human leukocyte antigens (HLA) is a strategy that has been previously applied for the design of vaccinal antigens [67,68,69].
5. Conclusions
In this work we present the in silico design of a high coverage EIAV vaccine that includes a diversity of conserved epitopes associated with a large number of American field strain variants. This vaccinal candidate contains peptides that can be recognized by several ELA-I and/or ELA-II alleles and potentially induce the secretion of IFN-γ by CD4+ T-cells. Furthermore, four ELA-I epitopes included in the final construct were tested against their ELA alleles in a docking study, were they showed adequate binding. In addition, some of the B-cell epitopes could potentially elicit neutralizing antibodies, contributing to broad even more the immune response triggered by this vaccinal candidate. The proposed sequence is predicted to be highly immunogenic and sets the base for a novel EAIV vaccine that could be used to protect against several American field strains.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.S.G., CV and A.S..; methodology, E.G. and A.S.G.; validation A.S.G., CV and A.S.; formal analysis, E.G. and A.S.G.; investigation, E.G., A.S.G. and AS.; writing—original draft preparation, E.G., A.S.G. and AS.; writing—review and editing, C.V. and A. S.; supervision, A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest
Abbreviations
The following abbreviations are used in this manuscript:
| AA | aminoacid |
| BCR | B-cell Receptor |
| CTL | Cytotoxic T-Lymphocyte |
| EIAV | Equine Infectious Anemia Virus |
| ELA | Equine Lymphocyte Antigen |
| ELR-1 | Equine Lentivirus Receptor-1 |
| ENV | Envelope |
| gp45 | Glycoprotein 45 |
| gp90 | Glycoprotein 90 |
| hcENV | High coverage ENV based chimera |
| IFN--γ | Gamma interferon |
| MHC | Major Histocompatibility Complex |
| MSA | Multiple Sequence Alignment |
References
- Sellon, D.C.; Fuller, F.J.; McGuire, T.C. The immunopathogenesis of equine infectious anemia virus. Virus Res. 1994, 32, 111–138. [CrossRef]
- Montelaro, R.C.; Ball, J.M.; Rushlow, K.E. Equine Retroviruses. The Retroviridae [Internet]. 1993 [cited 2025 Nov 30];257–360. Available from: https://link.springer.com/chapter/10.1007/978-1-4899-1627-3_5.
- Hussain, K.A.; Issel, C.J.; Schnorr, K.L.; Rwambo, P.M.; West, M.; Montelaro, R.C. Antigenic mapping of the envelope proteins of equine infectious anemia virus: identification of a neutralization domain and a conserved region on glycoprotein 90. Arch. Virol. 1988, 98, 213–224. [CrossRef]
- Chong, Y.H.; Ball, J.M.; Issel, C.J.; Montelaro, R.C.; E Rushlow, K. Analysis of equine humoral immune responses to the transmembrane envelope glycoprotein (gp45) of equine infectious anemia virus. J. Virol. 1991, 65, 1013–1018. [CrossRef]
- Zhang, B.; Jin, S.; Jin, J.; Li, F.; Montelaro, R.C. A tumor necrosis factor receptor family protein serves as a cellular receptor for the macrophage-tropic equine lentivirus. Proc. Natl. Acad. Sci. 2005, 102, 9918–9923. [CrossRef]
- Sun, C.; Zhang, B.; Jin, J.; Montelaro, R.C. Binding of equine infectious anemia virus to the equine lentivirus receptor-1 is mediated by complex discontinuous sequences in the viral envelope gp90 protein. J. Gen. Virol. 2008, 89, 2011–2019. [CrossRef]
- Lin, Y.; Wang, X.-F.; Wang, Y.; Du, C.; Ren, H.; Liu, C.; Zhu, D.; Chen, J.; Na, L.; Liu, D.; et al. Env diversity-dependent protection of the attenuated equine infectious anaemia virus vaccine. Emerg. Microbes Infect. 2020, 9, 1309–1320. [CrossRef]
- Henzy, J.E.; Johnson, W.E. Pushing the endogenous envelope. Philos. Trans. R. Soc. B: Biol. Sci. 2013, 368, 20120506. [CrossRef]
- Mealey RH, Zhang B, Leib SR, Littke MH, McGuire TC. Epitope specificity is critical for high and moderate avidity cytotoxic T lymphocytes associated with control of viral load and clinical disease in horses with equine infectious anemia virus. Virology [Internet]. 2003 [cited 2025 Nov 30];313:537–52. Available from: https://www.sciencedirect.com/science/article/pii/S0042682203003441?via%3Dihub.
- Fraser, D.G.; Oaks, J.L.; Brown, W.C.; McGuire, T.C. Identification of broadly recognized, T helper 1 lymphocyte epitopes in an equine lentivirus. Immunology 2002, 105, 295–305. [CrossRef]
- Tagmyer, T.L.; Craigo, J.K.; Cook, S.J.; Even, D.L.; Issel, C.J.; Montelaro, R.C. Envelope Determinants of Equine Infectious Anemia Virus Vaccine Protection and the Effects of Sequence Variation on Immune Recognition. J. Virol. 2008, 82, 4052–4063. [CrossRef]
- McGuire, T.C.; Leib, S.R.; Lonning, S.M.; Zhang, W.; Byrne, K.M.; Mealey, R.H. Equine infectious anaemia virus proteins with epitopes most frequently recognized by cytotoxic T lymphocytes from infected horses. J. Gen. Virol. 2000, 81, 2735–2739. [CrossRef]
- Liu, C.; Cook, F.R.; Cook, S.J.; Craigo, J.K.; Even, D.L.; Issel, C.J.; Montelaro, R.C.; Horohov, D.W. The determination of in vivo envelope-specific cell-mediated immune responses in equine infectious anemia virus-infected ponies. Veter- Immunol. Immunopathol. 2012, 148, 302–310. [CrossRef]
- McGuire, T.C.; Leib, S.R.; Mealey, R.H.; Fraser, D.G.; Prieur, D.J. Presentation and Binding Affinity of Equine Infectious Anemia Virus CTL Envelope and Matrix Protein Epitopes by an Expressed Equine Classical MHC Class I Molecule. J. Immunol. 2003, 171, 1984–1993. [CrossRef]
- Soutullo A, Verwimp V, Riveros M, Pauli R, Tonarelli G. Design and validation of an ELISA for equine infectious anemia (EIA) diagnosis using synthetic peptides. Vet Microbiol [Internet]. 2001 [cited 2025 Nov 30];79:111–21. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0378113500003527?via%3Dihub.
- Soutullo, A.; García, M.; Bailat, A.; Racca, A.; Tonarelli, G.; Borel, I.M. Antibodies and PMBC from EIAV infected carrier horses recognize gp45 and p26 synthetic peptides. Veter- Immunol. Immunopathol. 2005, 108, 335–343. [CrossRef]
- Bailat, A.S.; Soutullo, A.R.; García, M.I.; Veaute, C.M.; Garcia, L.; Racca, A.L.; Borel, I.S.M. Effect of two synthetic peptides mimicking conserved regions of equine infectious anemia virus proteins gp90 and gp45 upon cytokine mRNA expression. Arch. Virol. 2008, 153, 1909–1915. [CrossRef]
- Russi, R.C.; Garcia, L.; Cámara, M.S.; Soutullo, A.R. Validation of an indirect in-house ELISA using synthetic peptides to detect antibodies anti-gp90 and gp45 of the equine infectious anaemia virus. Equine Veter- J. 2022, 55, 111–121. [CrossRef]
- Meng, Q.; Lin, Y.; Ma, J.; Ma, Y.; Zhao, L.; Li, S.; Yang, K.; Zhou, J.; Shen, R.; Zhang, X.; et al. A Pilot Study Comparing the Development of EIAV Env-Specific Antibodies Induced by DNA/Recombinant Vaccinia-Vectored Vaccines and an Attenuated Chinese EIAV Vaccine. Viral Immunol. 2012, 25, 477–484. [CrossRef]
- Lin, Y.-Z.; Shen, R.-X.; Zhu, Z.-Y.; Deng, X.-L.; Cao, X.-Z.; Wang, X.-F.; Ma, J.; Jiang, C.-G.; Zhao, L.-P.; Lv, X.-L.; et al. An attenuated EIAV vaccine strain induces significantly different immune responses from its pathogenic parental strain although with similar in vivo replication pattern. Antivir. Res. 2011, 92, 292–304. [CrossRef]
- Wang, X.; Wang, S.; Lin, Y.; Jiang, C.; Ma, J.; Zhao, L.; Lv, X.; Wang, F.; Shen, R.; Kong, X.; et al. Genomic comparison between attenuated Chinese equine infectious anemia virus vaccine strains and their parental virulent strains. Arch. Virol. 2010, 156, 353–357. [CrossRef]
- Liu, C.; Wang, X.-F.; Wang, Y.; Chen, J.; Zhong, Z.; Lin, Y.; Wang, X. Characterization of EIAV env Quasispecies during Long-Term Passage In Vitro: Gradual Loss of Pathogenicity. Viruses 2019, 11, 380. [CrossRef]
- Korber, B.; Hraber, P.; Wagh, K.; Hahn, B.H. Polyvalent vaccine approaches to combat HIV-1 diversity. Immunol. Rev. 2017, 275, 230–244. [CrossRef]
- Hurwitz, J.L.; Bonsignori, M. Multi-Envelope HIV-1 Vaccine Development: Two Targeted Immune Pathways, One Desired Protective Outcome. Viral Immunol. 2018, 31, 124–132. [CrossRef]
- Rappuoli, R.; De Gregorio, E.; Del Giudice, G.; Phogat, S.; Pecetta, S.; Pizza, M.; Hanon, E. Vaccinology in the post−COVID-19 era. Proc. Natl. Acad. Sci. USA 2021, 118, e2020368118. [CrossRef]
- Oli AN, Obialor WO, Ositadimma M, Ifeanyichukwu, Odimegwu DC, Okoyeh JN, et al. Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther [Internet]. 2020 [cited 2025 Nov 30];9:13–30. Available from: https://www.dovepress.com/immunoinformatics-and-vaccine-development-an-overview-peer-reviewed-fulltext-article-ITT.
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [CrossRef]
- Li, F.; Leier, A.; Liu, Q.; Wang, Y.; Xiang, D.; Akutsu, T.; Webb, G.I.; Smith, A.I.; Marquez-Lago, T.; Li, J.; et al. Procleave: Predicting Protease-Specific Substrate Cleavage Sites by Combining Sequence and Structural Information. Genom. Proteom. Bioinform. 2020, 18, 52–64. [CrossRef]
- O’Donnell TJ, Rubinsteyn A, Laserson U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst [Internet]. 2020 [cited 2025 Nov 30];11:42-48.e7. Available from: https://www.cell.com/action/showFullText?pii=S2405471220302398.
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30–30. [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, 9. [CrossRef]
- Liu, C.; Cook, S.J.; Craigo, J.K.; Cook, F.R.; Issel, C.J.; Montelaro, R.C.; Horohov, D.W. Epitope shifting of gp90-specific cellular immune responses in EIAV-infected ponies. Veter- Immunol. Immunopathol. 2014, 161, 161–169. [CrossRef]
- Tagmyer, T.L.; Craigo, J.K.; Cook, S.J.; Issel, C.J.; Montelaro, R.C. Envelope-specific T-helper and cytotoxic T-lymphocyte responses associated with protective immunity to equine infectious anemia virus. J. Gen. Virol. 2007, 88, 1324–1336. [CrossRef]
- McGuire, T.C.; Fraser, D.G.; Mealey, R.H. Cytotoxic T Lymphocytes and Neutralizing Antibody in the Control of Equine Infectious Anemia Virus. Viral Immunol. 2002, 15, 521–531. [CrossRef]
- Yao, S.; Liu, J.; Qi, J.; Chen, R.; Zhang, N.; Liu, Y.; Wang, J.; Wu, Y.; Gao, G.F.; Xia, C. Structural Illumination of Equine MHC Class I Molecules Highlights Unconventional Epitope Presentation Manner That Is Evolved in Equine Leukocyte Antigen Alleles. J. Immunol. 2016, 196, 1943–1954. [CrossRef]
- Craigo, J.K.; Ezzelarab, C.; Cook, S.J.; Chong, L.; Horohov, D.; Issel, C.J.; Montelaro, R.C. Envelope Determinants of Equine Lentiviral Vaccine Protection. PLOS ONE 2013, 8, e66093. [CrossRef]
- Burton DR. What Are the Most Powerful Immunogen Design Vaccine Strategies? Cold Spring Harb Perspect Biol [Internet]. 2017 [cited 2025 Nov 30];9:a030262. Available from: http://cshperspectives.cshlp.org/content/9/11/a030262.full.
- Craigo, J.K.; Durkin, S.; Sturgeon, T.J.; Tagmyer, T.; Cook, S.J.; Issel, C.J.; Montelaro, R.C. Immune suppression of challenged vaccinates as a rigorous assessment of sterile protection by lentiviral vaccines. Vaccine 2007, 25, 834–845. [CrossRef]
- Wu, W.; Blythe, D.C.; Loyd, H.; Mealey, R.H.; Tallmadge, R.L.; Dorman, K.S.; Carpenter, S. Decreased Infectivity of a Neutralization-Resistant Equine Infectious Anemia Virus Variant Can Be Overcome by Efficient Cell-to-Cell Spread. J. Virol. 2011, 85, 10421–10424. [CrossRef]
- Sponseller, B.A.; Sparks, W.O.; Wannemuehler, Y.; Li, Y.; Antons, A.K.; Oaks, J.L.; Carpenter, S. Immune selection of equine infectious anemia virus env variants during the long-term inapparent stage of disease. Virology 2007, 363, 156–165. [CrossRef]
- Ball, J.M.; Payne, S.L.; Issel, C.J.; Montelaro, R.C. EIAV genomic organization: Further characterization by sequencing of purified glycoproteins and cDNA. Virology 1988, 165, 601–605. [CrossRef]
- Bastian, F.B.; Roux, J.; Niknejad, A.; Comte, A.; Costa, S.S.F.; de Farias, T.M.; Moretti, S.; Parmentier, G.; de Laval, V.R.; Rosikiewicz, M.; et al. The Bgee suite: integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Res. 2020, 49, D831–D847. [CrossRef]
- Rice, N.R.; E Henderson, L.; Sowder, R.C.; Copeland, T.D.; Oroszlan, S.; Edwards, J.F. Synthesis and processing of the transmembrane envelope protein of equine infectious anemia virus. J. Virol. 1990, 64, 3770–3778. [CrossRef]
- Wang, X.-F.; Wang, Y.-H.; Bai, B.; Zhang, M.; Chen, J.; Zhang, X.; Gao, M.; Wang, X. Truncation of the Cytoplasmic Tail of Equine Infectious Anemia Virus Increases Virion Production by Improving Env Cleavage and Plasma Membrane Localization. J. Virol. 2021, 95, e0108721. [CrossRef]
- Ricotti S, Barcarolo M, Garcia M, Soutullo A. Differences of immune Env-specific responses between newly EIAV infected and inapparent carrier horses. Front Immunol. 2015;6.
- Craigo, J.K.; Zhang, B.; Barnes, S.; Tagmyer, T.L.; Cook, S.J.; Issel, C.J.; Montelaro, R.C. Envelope variation as a primary determinant of lentiviral vaccine efficacy. Proc. Natl. Acad. Sci. 2007, 104, 15105–15110. [CrossRef]
- Zhang, X.; Wang, Y.; Liang, H.; Wei, L.; Xiang, W.; Shen, R.; Shao, Y. Correlation between the induction of Th1 cytokines by an attenuated equine infectious anemia virus vaccine and protection against disease progression. J. Gen. Virol. 2007, 88, 998–1004. [CrossRef]
- Wagner B, Goodman LB, Babasyan S, Freer H, Torsteinsdóttir S, Svansson V, et al. Antibody and cellular immune responses of naïve mares to repeated vaccination with an inactivated equine herpesvirus vaccine. Vaccine [Internet]. 2015 [cited 2025 Nov 30];33:5588–97. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0264410X15012530?via%3Dihub.
- Berg, F.T.v.D.; Makoah, N.A.; Ali, S.A.; Scott, T.A.; Mapengo, R.E.; Mutsvunguma, L.Z.; Mkhize, N.N.; Lambson, B.E.; Kgagudi, P.D.; Crowther, C.; et al. AAV-Mediated Expression of Broadly Neutralizing and Vaccine-like Antibodies Targeting the HIV-1 Envelope V2 Region. Mol. Ther. - Methods Clin. Dev. 2019, 14, 100–112. [CrossRef]
- Liu, H.; Su, X.; Si, L.; Lu, L.; Jiang, S. The development of HIV vaccines targeting gp41 membrane-proximal external region (MPER): challenges and prospects. Protein Cell 2018, 9, 596–615. [CrossRef]
- Yang X, Zhang X, Zhao X, Yuan M, Zhang K, Dai J, et al. Antibody-Dependent Enhancement: ″Evil″ Antibodies Favorable for Viral Infections. Viruses 2022, Vol 14, Page 1739 [Internet]. 2022 [cited 2025 Nov 30];14:1739. Available from: https://www.mdpi.com/1999-4915/14/8/1739/htm.
- Ball, J.M.; E Rushlow, K.; Issel, C.J.; Montelaro, R.C. Detailed mapping of the antigenicity of the surface unit glycoprotein of equine infectious anemia virus by using synthetic peptide strategies. J. Virol. 1992, 66, 732–742. [CrossRef]
- Ayyagari, V.S.; C., V.T.; K., A.P.; Srirama, K. Design of a multi-epitope-based vaccine targeting M-protein of SARS-CoV2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 40, 2963–2977. [CrossRef]
- Habib, A.; Liang, Y.; Xu, X.; Zhu, N.; Xie, J. Immunoinformatic Identification of Multiple Epitopes of gp120 Protein of HIV-1 to Enhance the Immune Response against HIV-1 Infection. Int. J. Mol. Sci. 2024, 25, 2432. [CrossRef]
- Huang, Q.; Lang, Q.; Li, Y.; Wang, F.; Han, X.; Luo, L.; Duan, X.; Cao, X.; Wang, Y.; Bai, Y.; et al. Highly scalable prefusion-stabilized RSV F vaccine with enhanced immunogenicity and robust protection. Nat. Commun. 2025, 16, 1–15. [CrossRef]
- Sun, C.; Kang, Y.-F.; Fang, X.-Y.; Liu, Y.-N.; Bu, G.-L.; Wang, A.-J.; Li, Y.; Zhu, Q.-Y.; Zhang, H.; Xie, C.; et al. A gB nanoparticle vaccine elicits a protective neutralizing antibody response against EBV. Cell Host Microbe 2023, 31, 1882–1897.e10. [CrossRef]
- Zhang, G.; Liu, W.; Gao, Z.; Chang, Y.; Yang, S.; Peng, Q.; Ge, S.; Kang, B.; Shao, J.; Chang, H. Antigenic and immunogenic properties of recombinant proteins consisting of two immunodominant African swine fever virus proteins fused with bacterial lipoprotein OprI. Virol. J. 2022, 19, 1–16. [CrossRef]
- Pang, F.; Long, Q.; Liang, S. Designing a multi-epitope subunit vaccine against Orf virus using molecular docking and molecular dynamics. Virulence 2024, 15, 2398171. [CrossRef]
- Tarrahimofrad, H.; Rahimnahal, S.; Zamani, J.; Jahangirian, E.; Aminzadeh, S. Designing a multi-epitope vaccine to provoke the robust immune response against influenza A H7N9. Sci. Rep. 2021, 11, 1–22. [CrossRef]
- Ismail, S.; Abbasi, S.W.; Yousaf, M.; Ahmad, S.; Muhammad, K.; Waheed, Y. Design of a Multi-Epitopes Vaccine against Hantaviruses: An Immunoinformatics and Molecular Modelling Approach. Vaccines 2022, 10, 378. [CrossRef]
- Scheiblhofer, S.; Laimer, J.; Machado, Y.; Weiss, R.; Thalhamer, J. Influence of protein fold stability on immunogenicity and its implications for vaccine design. Expert Rev. Vaccines 2017, 16, 479–489. [CrossRef]
- Mahdavi, M.; Moreau, V. In silico designing breast cancer peptide vaccine for binding to MHC class I and II: A molecular docking study. Comput. Biol. Chem. 2016, 65, 110–116. [CrossRef]
- I Abdelmageed, M.; Abdelmoneim, A.H.; I Mustafa, M.; Elfadol, N.M.; Murshed, N.S.; Shantier, S.W.; Makhawi, A.M. Design of a Multiepitope-Based Peptide Vaccine against the E Protein of Human COVID-19: An Immunoinformatics Approach. BioMed Res. Int. 2020, 2020, 2683286. [CrossRef]
- Zhang, X.W. A combination of epitope prediction and molecular docking allows for good identification of MHC class I restricted T-cell epitopes. Comput. Biol. Chem. 2013, 45, 30–35. [CrossRef]
Figure 1.
Flow chart showing the strategy followed to design a high coverage ENV based EIAV vaccine. A. Epitope identification across different strains. ELA-I, ELA-II, IFN-γ and linear B epitopes were identified on 12 EIAV ENV American strain sequences, while allergenic and toxic peptides were discarded. Those regions with the higher amount of overlapping epitopes and with the higher strain coverage were identified on a consensus sequence. B. Antigen design. Three antigens containing the regions identified in A linked by different linker sequences were designed and analyzed through different predictors to select the one with the highest immunogenicity, solubility and stability, as well as the lowest allergenicity and toxicity. C. Antigen validation. The secondary and tertiary structure of the final vaccinal construct antigen was predicted and validated, and its cellular CD8(+) T cell immunogenicity was predicted through docking of the higher scoring epitopes with ELA-I molecules.
Figure 1.
Flow chart showing the strategy followed to design a high coverage ENV based EIAV vaccine. A. Epitope identification across different strains. ELA-I, ELA-II, IFN-γ and linear B epitopes were identified on 12 EIAV ENV American strain sequences, while allergenic and toxic peptides were discarded. Those regions with the higher amount of overlapping epitopes and with the higher strain coverage were identified on a consensus sequence. B. Antigen design. Three antigens containing the regions identified in A linked by different linker sequences were designed and analyzed through different predictors to select the one with the highest immunogenicity, solubility and stability, as well as the lowest allergenicity and toxicity. C. Antigen validation. The secondary and tertiary structure of the final vaccinal construct antigen was predicted and validated, and its cellular CD8(+) T cell immunogenicity was predicted through docking of the higher scoring epitopes with ELA-I molecules.
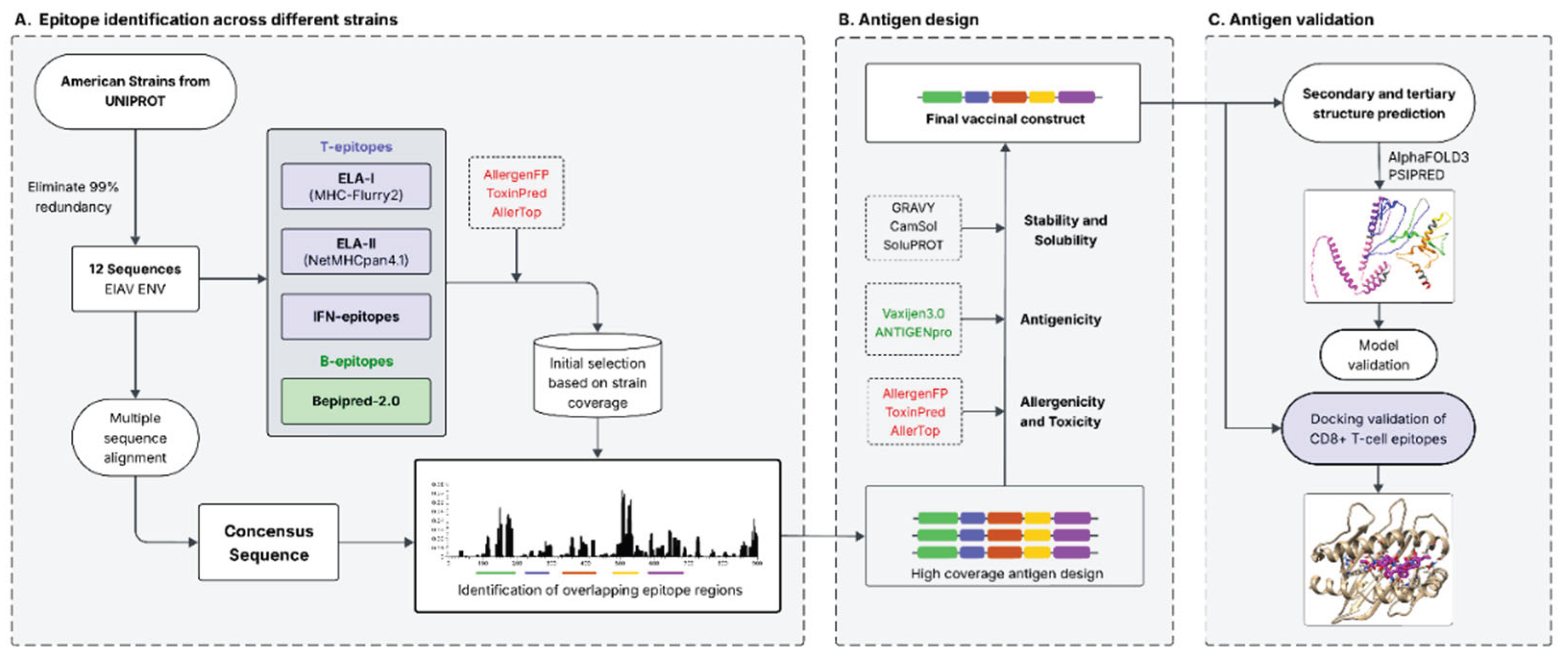
Figure 2.
Overview of ENV protein sequences. A. Schematic of ENV gp90 and gp45. The main regions of immature gp90 (purple, AA 1-489) and gp45 (pink, AA 490-904) are shown. Principal Neutralizing Domain (PND, AA 197-235) and the Fusion Peptide (FP, AA 490-505) are depicted in dark purple and vertical striped rectangles, respectively. Transmembrane domain (TM) in gp45 is shown as a dark pink rectangle (AA 663-683). C1 and C2 indicate predicted protein cleavage sites between residues 489-490 (Furin cleavage) and 732-733 (cathepsin B and a matrix Metalloprotease-2 cleavage, Table-1-SI) respectively. B. Multiple Sequence Alignment (MSA) of the ENV polyprotein of the American EIAV strains used in this work. The columns are colored in shades of blue according to their level of conservation. gp90 variable regions (V1-V8) are depicted as transparent green rectangles over the alignment. The features described in A are shown on the consensus sequence (PND, FP, TM, C1 and C2).
Figure 2.
Overview of ENV protein sequences. A. Schematic of ENV gp90 and gp45. The main regions of immature gp90 (purple, AA 1-489) and gp45 (pink, AA 490-904) are shown. Principal Neutralizing Domain (PND, AA 197-235) and the Fusion Peptide (FP, AA 490-505) are depicted in dark purple and vertical striped rectangles, respectively. Transmembrane domain (TM) in gp45 is shown as a dark pink rectangle (AA 663-683). C1 and C2 indicate predicted protein cleavage sites between residues 489-490 (Furin cleavage) and 732-733 (cathepsin B and a matrix Metalloprotease-2 cleavage, Table-1-SI) respectively. B. Multiple Sequence Alignment (MSA) of the ENV polyprotein of the American EIAV strains used in this work. The columns are colored in shades of blue according to their level of conservation. gp90 variable regions (V1-V8) are depicted as transparent green rectangles over the alignment. The features described in A are shown on the consensus sequence (PND, FP, TM, C1 and C2).
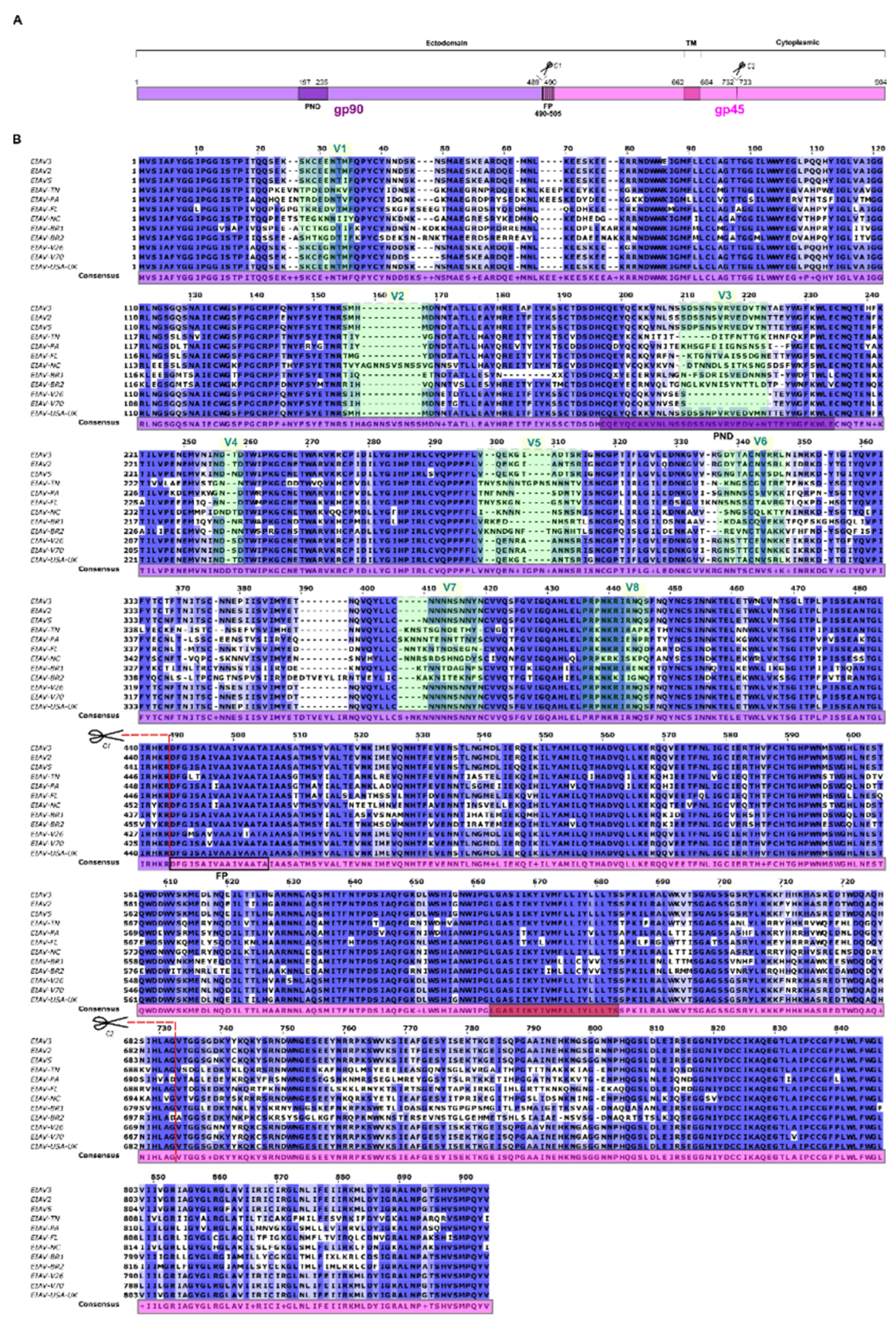
Figure 3.
Epitopes predicted on the EIAV envelope protein. Bars represent the frequencies of each residue predicted as an epitope. To simplify each graph, only residues that exceeded the cutoff value for each prediction method were used for the statistics. Long green stripes represent the variable zones of gp90 and red ones indicate the high coverage overlapping epitopes regions found. A) ELA-I epitopes predicted with MHCflurry-2.0 B) ELA-II epitopes predicted by NetMHCpan-4.2. C) IFN-gamma inducing MHC class II binding peptides D) Linear B-cell epitopes predicted by Bepipred.
Figure 3.
Epitopes predicted on the EIAV envelope protein. Bars represent the frequencies of each residue predicted as an epitope. To simplify each graph, only residues that exceeded the cutoff value for each prediction method were used for the statistics. Long green stripes represent the variable zones of gp90 and red ones indicate the high coverage overlapping epitopes regions found. A) ELA-I epitopes predicted with MHCflurry-2.0 B) ELA-II epitopes predicted by NetMHCpan-4.2. C) IFN-gamma inducing MHC class II binding peptides D) Linear B-cell epitopes predicted by Bepipred.
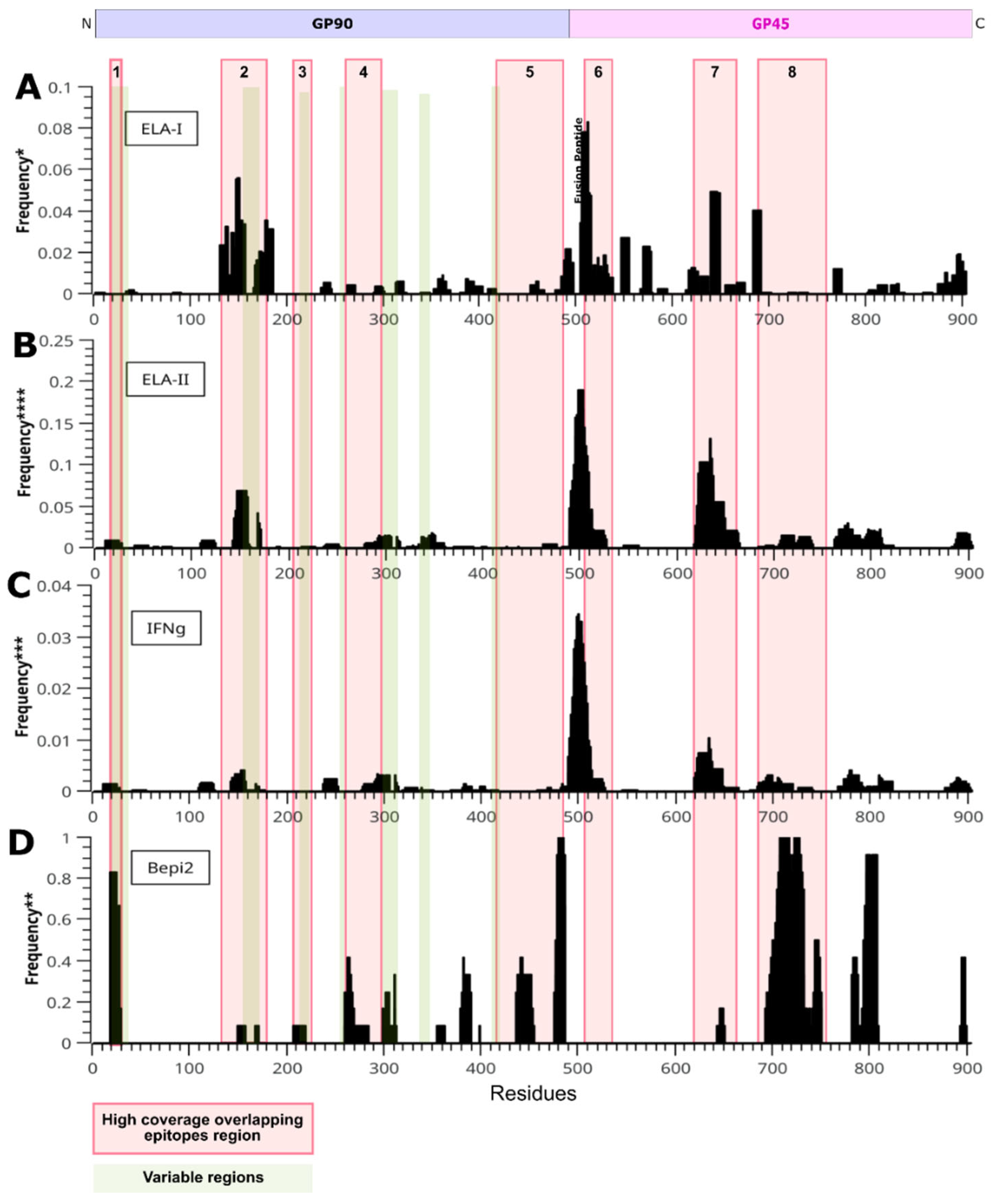
Figure 4.
Preliminary immunogenicity summary of the high coverage overlapping epitope regions. Regions are identified with numbers from 1 to 8 (right side), and their residues numbered according to the immature EIAV ENV consensus sequence. The number of Th1 epitopes is shown between brackets.
Figure 4.
Preliminary immunogenicity summary of the high coverage overlapping epitope regions. Regions are identified with numbers from 1 to 8 (right side), and their residues numbered according to the immature EIAV ENV consensus sequence. The number of Th1 epitopes is shown between brackets.
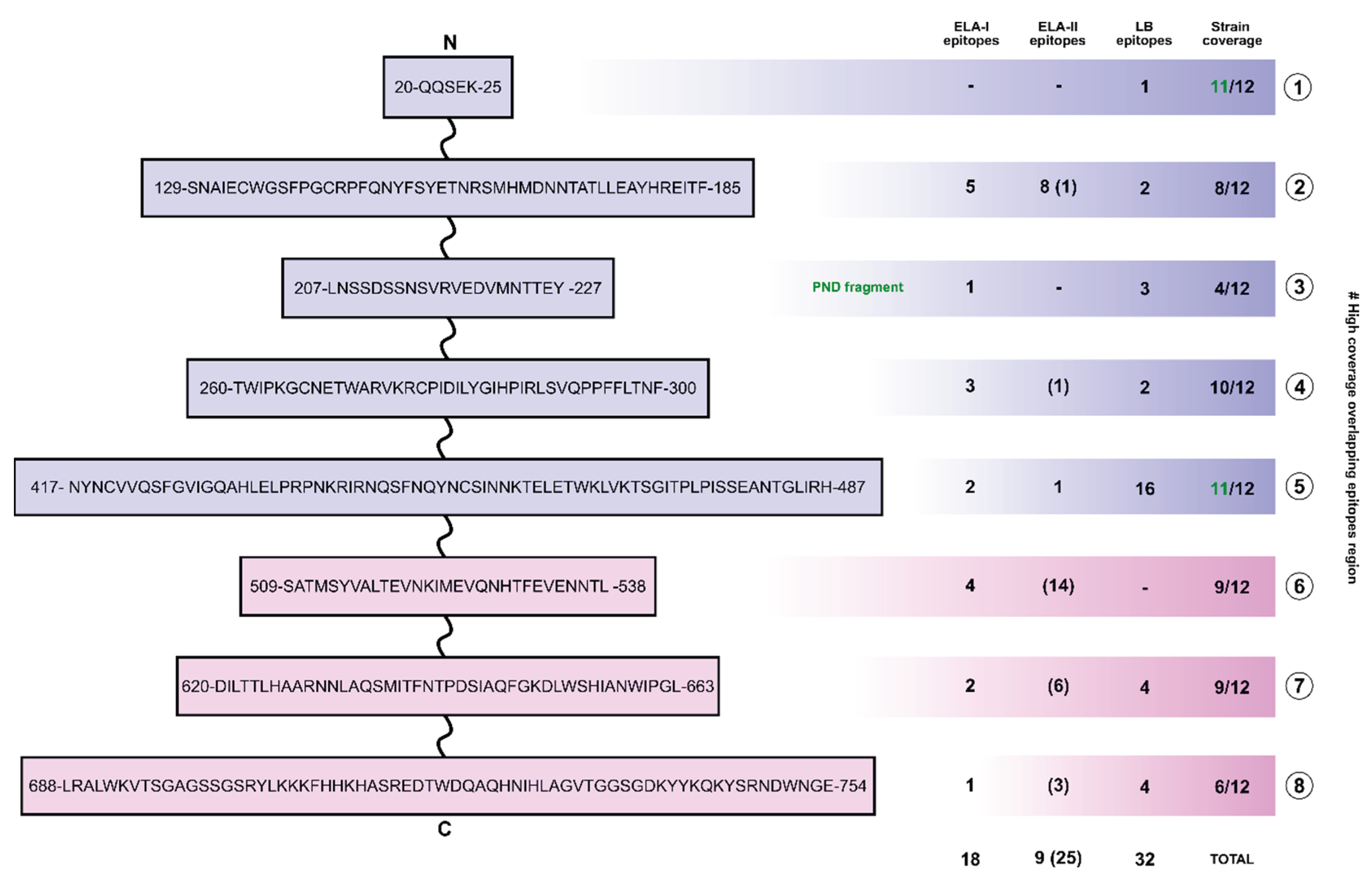
Figure 5.
Predicted secondary and tertiary structures of hcENV construct. A. Secondary structures as predicted by PSIPRED. High coverage regions are depicted in different colours on top of the sequence, while EAAAK linkers are shown in grey. Each residue is coloured in term of its predicted secondary structure: yellow for β-strand, pink for α-helix and grey for coils. B. Predicted tertiary structure of hcENV. A 3D model was generated with AlphaFOLD 3.0, and the structure was refined with GalaxyWEB suite. High coverage regions are depicted in different colours, while EAAAK linkers are shown in grey.
Figure 5.
Predicted secondary and tertiary structures of hcENV construct. A. Secondary structures as predicted by PSIPRED. High coverage regions are depicted in different colours on top of the sequence, while EAAAK linkers are shown in grey. Each residue is coloured in term of its predicted secondary structure: yellow for β-strand, pink for α-helix and grey for coils. B. Predicted tertiary structure of hcENV. A 3D model was generated with AlphaFOLD 3.0, and the structure was refined with GalaxyWEB suite. High coverage regions are depicted in different colours, while EAAAK linkers are shown in grey.
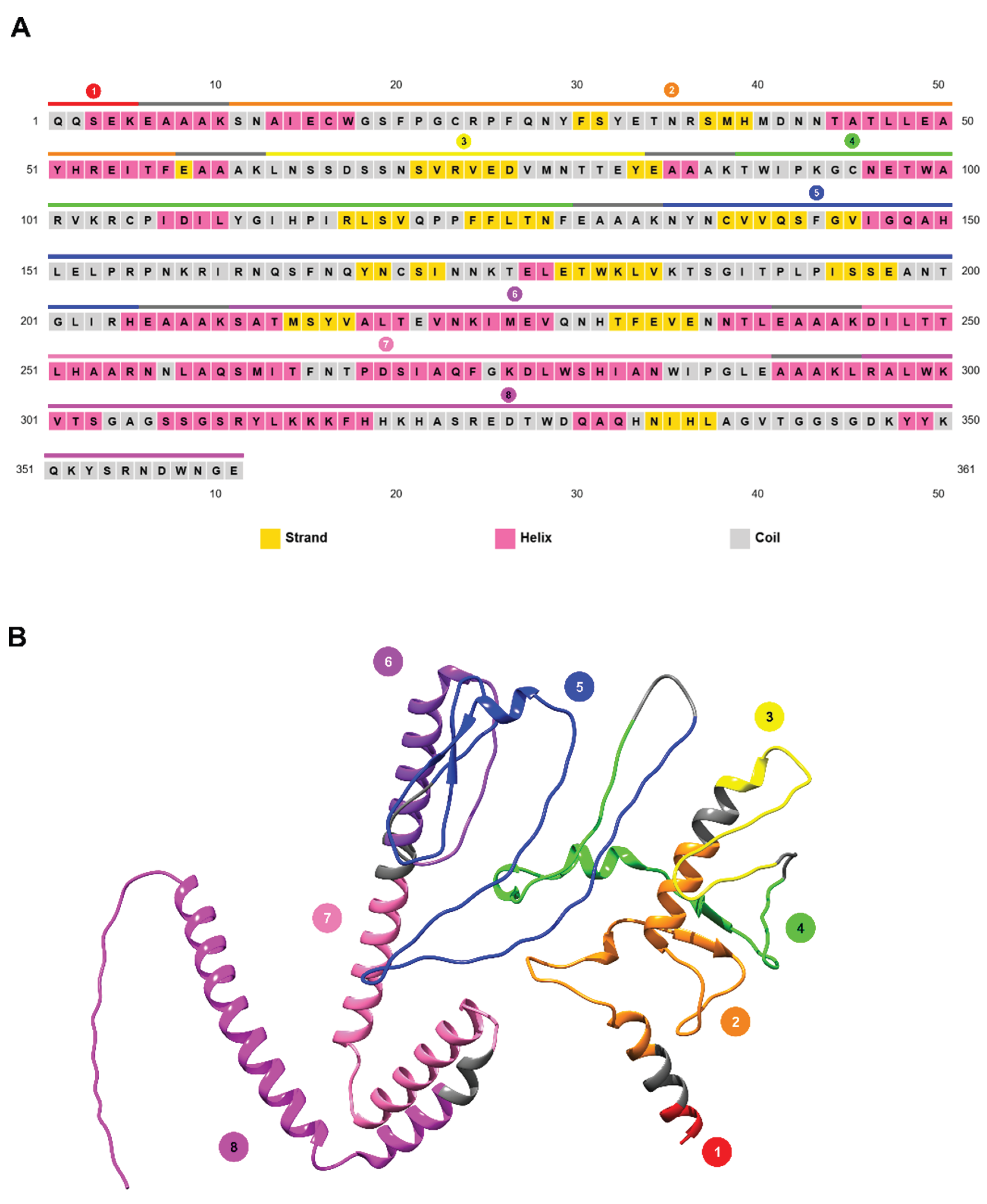
Figure 6.
Assessment of the quality of the model. A. Local quality estimate. hcENV 3D model was coloured according to the QMEAN local score of each residue. Residues with lower QMEAN values are depicted in red (lower quality), while the ones with higher values are coloured in blue (higher quality). Color scale ranges from 0,14 to 0,46 QMEAN local score. B. Ramachandran plot. C. Normalized QMEAN value of hcENV model. The value obtained for the model is compared to a set of non-redundant Protein Data Bank structures.
Figure 6.
Assessment of the quality of the model. A. Local quality estimate. hcENV 3D model was coloured according to the QMEAN local score of each residue. Residues with lower QMEAN values are depicted in red (lower quality), while the ones with higher values are coloured in blue (higher quality). Color scale ranges from 0,14 to 0,46 QMEAN local score. B. Ramachandran plot. C. Normalized QMEAN value of hcENV model. The value obtained for the model is compared to a set of non-redundant Protein Data Bank structures.
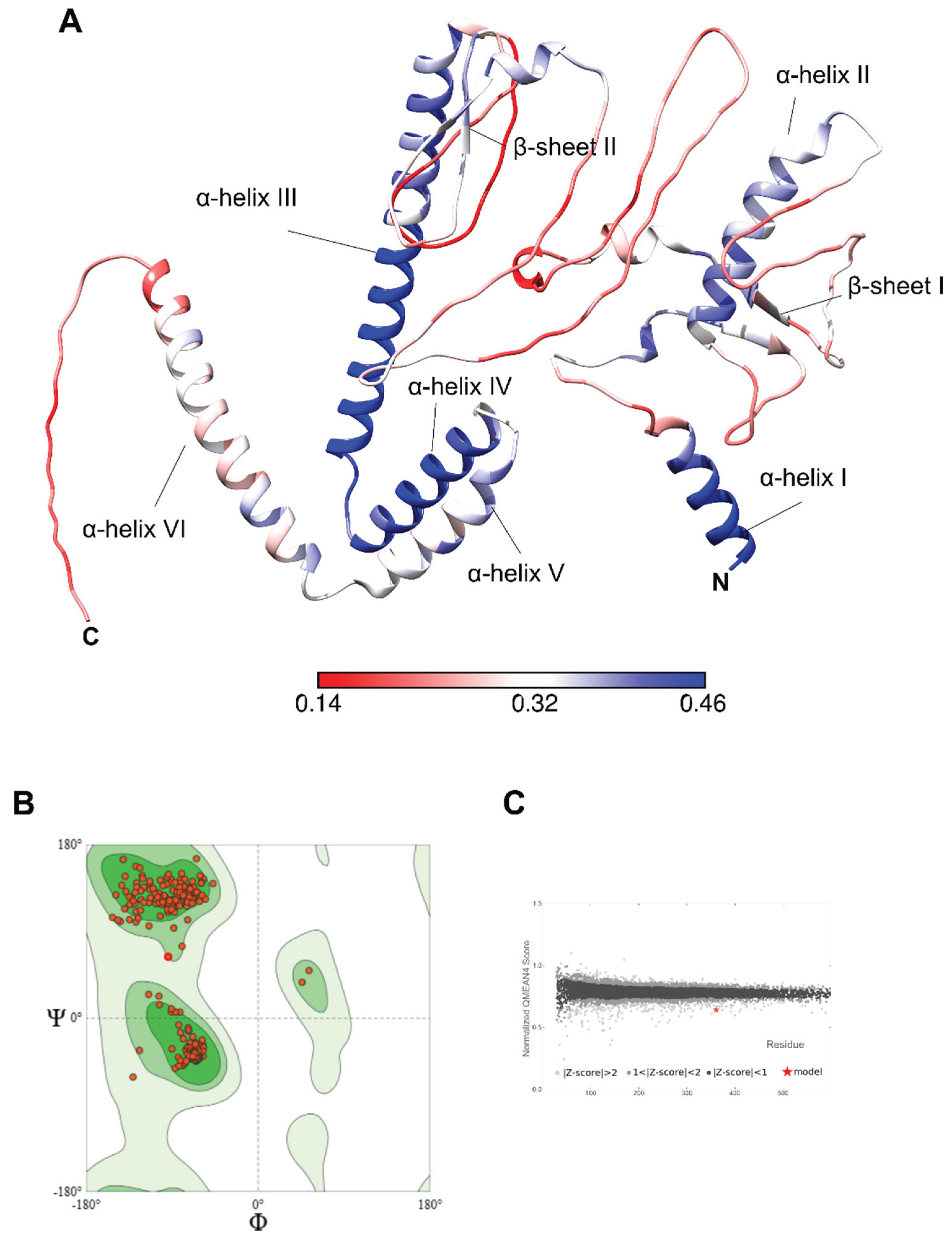
Figure 7.
Docking of epitopes with ELA-I molecules. A. Best structural results obtained for each docking experiment. Backbones of the ELA receptors are rendered as brown ribbons, with the side chains that interact with the epitope represented as sticks. The peptide epitopes are displayed as stick representation, with the carbon atoms colored in pink. All the other atoms are colored with CPK coloring scheme. With red dot lines are shown the hydrogen bonds formed between the receptor and the epitope. B. Docking score (DG_cross) obtained for each peptide-ELA complex. Docking assays were performed using GalaxyPepDock40 software. Complexes (ELA-I - peptide) with the highest GalaxyPepDock score were relaxed using the Relax41 application, and then the binding energy was estimated using the Interface Analyzer42 implemented in the Rosetta package. As controls, a peptide with MHC-Flurry low percentile affinity score value (TGIYQVPIF, orange) and a crystallized solved ELA-epitope complex (PDB:4ZUV, RVEDVTNTA green) were included in the analysis.
Figure 7.
Docking of epitopes with ELA-I molecules. A. Best structural results obtained for each docking experiment. Backbones of the ELA receptors are rendered as brown ribbons, with the side chains that interact with the epitope represented as sticks. The peptide epitopes are displayed as stick representation, with the carbon atoms colored in pink. All the other atoms are colored with CPK coloring scheme. With red dot lines are shown the hydrogen bonds formed between the receptor and the epitope. B. Docking score (DG_cross) obtained for each peptide-ELA complex. Docking assays were performed using GalaxyPepDock40 software. Complexes (ELA-I - peptide) with the highest GalaxyPepDock score were relaxed using the Relax41 application, and then the binding energy was estimated using the Interface Analyzer42 implemented in the Rosetta package. As controls, a peptide with MHC-Flurry low percentile affinity score value (TGIYQVPIF, orange) and a crystallized solved ELA-epitope complex (PDB:4ZUV, RVEDVTNTA green) were included in the analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



