
Submitted:
18 December 2025
Posted:
19 December 2025
You are already at the latest version
Abstract
High-grade serous ovarian cancer (HGSOC) is the most prevalent and aggressive form of epithelial ovarian cancer, characterized by high recurrence rates and poor clinical outcomes. In this study, we identify molecular signatures associated with recurrence by conducting integrative transcriptomic and proteomic analyses on paired primary and recurrent HGSOC tissues from 34 patients. RNA sequencing and proteomic profiling revealed 185 differentially expressed genes (DEGs) and 36 differentially expressed proteins (DEPs) linked to recurrence. Pathway Enrichment and Ingenuity Pathway Analysis highlighted the involvement of immune cell trafficking, cell signaling, and MAPK pathway activation in recurrent tumors. Survival analysis identified seven DEGs significantly correlated with recurrence-free survival; among these, IL7R, IRF8, and PTPRC were upregulated in recurrent tumors and associated with poor prognosis, while NSG1 was downregulated and linked to favorable outcomes. Immunohistochemistry validated the differential expression of these markers at the protein level. Proteomic analysis demonstrated that recurrent tumor-specific DEGs are functionally linked to MAPK signaling. Co-expression analyses revealed dynamic regulatory interactions between DEGs and DEPs, suggesting context-dependent molecular shifts during recurrence. This integrative multi-omics approach reveals key molecular alterations underlying HGSOC recurrence and identifies IL7R, IRF8, PTPRC, and NSG1 as potential prognostic biomarkers and therapeutic targets. Our findings provide a foundation for targeted strategies to improve outcomes for patients with recurrent HGSOC.
Keywords:
high-grade serous ovarian cancer
; recurrence
; transcriptomics
; proteomics
1. Introduction
Epithelial ovarian cancer (EOC) has the highest mortality rate among all gynecological cancers [1]. This cancer type comprises five distinct histological subtypes, the most common of which is high-grade serous ovarian cancer (HGSOC), making up 70% of invasive EOCs. The other subtypes are low-grade serous (<5%), endometrioid (10%), clear cell (10%), and mucinous (3%) [2,3]. The standard treatment for HGSOC, the most aggressive subtype of EOC, consists of optimal debulking surgery followed by adjuvant chemotherapy with a platinum-based cytotoxic agent and paclitaxel [4,5]. However, 70–80% of patients with advanced HGSOC experience recurrence within 14–24 months after first-line treatment, primarily due to cancer metastasis and the development of drug resistance [6,7,8,9,10,11,12]. Discovering effective therapeutic targets continues to be a key unresolved objective in the field [13,14,15].
Considerable research efforts have been directed toward the molecular and genetic characteristics of HGSOC to facilitate the development of more effective and personalized treatment strategies. Despite major advancements in therapeutic approaches over the past two decades, including traditional cytotoxic drugs and emerging agents with diverse mechanisms, such as VEGF inhibitors, PARP inhibitors, and anti-PD/PD-L1 inhibitors, treatment of HGSOC remains highly challenging [16,17,18]. Therefore, gaining a deeper understanding of the underlying biology of the disease is essential for the development of innovative and more effective therapeutic strategies [19].
In recent years, there has been a notable increase in studies utilizing whole-genome sequencing, whole-transcriptome sequencing, and single-cell analysis to analyze public datasets [20,21,22,23]. However, limited research has focused on comparing RNA sequencing results between recurrent and primary ovarian cancer using actual tissue samples from the same patients. This is primarily because obtaining recurrent tumor samples longitudinally from the same patient is challenging in real-world clinical practice due to the lack of established consensus on performing secondary debulking surgery.
In this study, we conducted a comparative transcriptomic analysis using formalin-fixed paraffin-embedded (FFPE) samples obtained from paired primary and recurrent HGSOC tissues collected longitudinally from the same patients in real-world clinical settings. To further elucidate downstream effects, we performed complementary proteomic analysis on fresh-frozen tissues, enabling more detailed insight into molecular mechanisms associated with recurrence. Our objective was to identify clinically relevant differentially expressed genes (DEGs) associated with disease recurrence in HGSOC.
2. Results
2.1. Patient characteristics
The clinicopathological characteristics of the 34 enrolled patients are summarized in Table 1. The median age at diagnosis was 53 years (range 41–69), and the median serum CA125 level at diagnosis was 639.5 U/mL (range 50.0–20388.1). The majority of patients was diagnosed at FIGO stage III (82.4%), with high-grade tumors (76.5%, G3), consistent with the typical presentation of HGSOC. BRCA status was available for 27 patients, with BRCA1 and BRCA2 mutations identified in 11.8% and 5.9% of cases, respectively. Expert pathologists reviewed all tissue samples to confirm histological subtype and tumor content. Samples with inadequate tumor proportion or poor RNA/protein quality were excluded prior to analysis.
2.2. Gene expression alterations between matched primary and recurrent HGSOC
To identify potential biomarkers of HGSOC recurrence, we analyzed differences in gene expression of protein-coding genes between matched primary and recurrent tumor samples. Using the criteria of FDR < 0.01 and |log2FC| > 1, we identified 185 DEGs with 151 (81.6%) genes upregulated and 34 (18.4%) downregulated in recurrent HGSOC (Figure 1B, Supplementary Table 1). The gene expression patterns of the two groups were visualized using a heatmap (Figure 1C). These findings highlight a significant difference in gene expression patterns between primary and recurrent HGSOC.
2.3. Ingenuity pathway analysis reveals key molecular networks in recurrent HGSOC
IPA was conducted to explore the molecular networks and biological functions associated with 185 DEGs, revealing 11 significant networks with network scores higher than 20 (Supplementary Table 2). The highest-scoring network (score = 34) was associated with cell morphology, immunological diseases, and organismal injury. In this network, the algorithm focused on activating the BCR complex and several transcriptional regulators, including IRF8, BATF, and IKZF3. Based on this observation, the activation of multiple complexes, including various immunoglobulin complexes and the STAT5A/B complex, was predicted to be higher in recurrent HGSOC compared to primary HGSOC (Figure 1D). The second network (score = 34) was enriched for cell-to-cell signaling, hematological system development, and immune cell trafficking, with increased expression of genes CCR7, ITGAL, CD28, PTPRC, CD5, GZMB, and GZMH. This pattern indicated the activation of RAS, mTORC1, p85-PIK3R, and NGF alongside inhibition of the MHC II complex (Figure 1E). The third network (score = 32) involved 17 focus molecules linked to endocrine, gastrointestinal, and metabolic disorders. This network predicted activation of complexes such as MAPK, NOTCH, VEGF, and the NF-κB family based on the observed expression of genes, including TNFSF11, S100B, TLR10, and TLR8 (Figure 1F). Taken together, these findings imply that functions of DEGs were closely associated with the molecular characteristics of cancer, highlighting their potential functions in cancer recurrence.
2.4. Pathway enrichment analysis identifies molecular features underlying recurrence in HGSOC
Canonical pathway analysis with IPA revealed 27 significant pathways associated with DEGs (FDR < 0.01). Significant pathways mainly consisted of cell signaling pathways and pathways implicated in cancer mechanisms, such as “Molecular Mechanisms of Cancer” and “Breast Cancer Regulation by Stathmin 1” (Figure 2A). ORA was performed using the KEGG database and GO: BP gene sets to further validate these findings. In the KEGG analysis, 10 significantly enriched pathways (p < 0.05) were identified including those involving cell adhesion molecules, natural killer cell-mediated cytotoxicity, and the PD-1/PD-L1 pathway in cancer (Figure 2B). GO: BP analysis highlighted similar processes, which were related to immune cell activation, cell adhesion, and cell signaling (Figure 2C). In summary, we identified significant gene expression differences between primary and recurrent HGSOC tissues, with DEGs enriched in pathways related to cell signaling and cancer mechanisms, providing potential insights into the molecular drivers of HGSOC recurrence.
2.5. Survival analysis identifies prognostic DEGs in recurrent HGSOC
Survival analysis was performed using clinical data from patients to identify DEGs significantly associated with HGSOC recurrence. This analysis revealed 11 DEGs exhibiting significant differences in recurrence-free survival between high and low expression groups in primary tumors (p < 0.01; Supplementary Table 3). Among these, we prioritized seven candidate DEGs based on their tumor-specific expression profiles and known or predicted prognostic relevance. We generated a heatmap using RNA sequencing data from matched samples to illustrate the expression differences of the seven selected DEGs between primary and recurrent HGSOC tissues (Figure 3A). GPR68, IL7R, IRF8, PTPRC, and WDFY4 showed higher expression in recurrent tumors, whereas C15orf39 and NSG1 were more highly expressed in primary tumors. Survival analysis revealed that high expression of GPR68, IL7R, IRF8, PTPRC, and WDFY4 in primary tumors was associated with poorer prognosis, with the high-expression group showing worse outcomes than the low-expression group. In contrast, C15orf39 and NSG1 were linked to more favorable outcomes (Figure 3B). As C15orf39 is currently categorized as an uncharacterized protein, the remaining six DEGs were selected for subsequent IHC validation.
2.6. Experimental validation of DEGs between primary and recurrent HGSOC
To validate the RNA sequencing results at the protein level, we performed IHC using a TMA. The TMA consisted of paired primary and recurrent tumor tissues from 34 HGSOC patients, with patient information organized based on the primary tumor diagnosis. All included tissues were of the serous subtype, and 91% of patients were classified as stage 3 or 4. During the IHC process, some tissue cores were lost, leading to variation in the number of patients analyzed for each protein, as summarized in Supplementary Table 4. Protein expression was quantified using QuPath for IL7R (n = 32), IRF8 (n = 33), GPR68 (n = 32), PTPRC (n = 32), WDFY4 (n = 30), and NSG1 (n = 31). The protein expression levels of IL7R, PTPRC, and NSG1 were consistent with the RNA sequencing results. Specifically, IL7R and PTPRC were significantly upregulated in recurrent HGSOC tissues, whereas NSG1 showed reduced expression. These differences were statistically significant based on the paired t-test. Although IRF8 did not reach statistical significance, it exhibited a trend of increased expression in recurrent tissues, similar to the RNA sequencing results (Figure 4). In conclusion, the expression patterns of four of six proteins (IL7R, PTPRC, NSG1, and IRF8) were consistent with the RNA sequencing findings, supporting their potential role in HGSOC recurrence.
2.7. Co-expression of DEGs and their regulatory interactions with DEPs in primary and recurrent HGSOC
We first examined the co-expression relationships among the four DEGs to investigate potential regulatory interactions. The expression levels of IL7R, IRF8, and PTPRC were significantly upregulated in recurrent tumors compared to primary tumors and were strongly correlated across both tumor types (p < 0.05; Figure 5A, Supplementary Table 5). Consistent with the survival analysis results, these three DEGs exhibited similar co-expression patterns with one other, whereas the primary tumor-specific DEG NSG1, which was downregulated in recurrent tumors, displayed an opposite expression trend. These findings suggest that IL7R, IRF8, and PTPRC are functionally linked or involved in a shared molecular pathway distinct from that of NSG1.
We performed a proteomic assay to further explore the biological significance of these candidate genes at the protein level. Among the 360 proteins that met quality control criteria, 36 differentially expressed proteins (DEPs) were identified between primary and recurrent HGSOC (p < 0.05), with 17 upregulated and 19 downregulated in recurrent tumors. To gain insight into potential regulatory interactions and downstream effects, we next examined the co-expression relationships between the DEGs and DEPs using data from the seven patients with both RNA sequencing and proteomic profiles (Figure 5B, Supplementary Table 6). Unlike the consistent co-expression patterns observed among DEGs across both tumor types, DEG-DEP correlations exhibited divergent patterns between primary and recurrent HGSOC.
In primary HGSOC, the recurrent tumor-specific DEG IL7R exhibited a positive correlation with AMBP and HDGF, while showing a negative correlation with FEN1, LRRC25, RIGI, and TBL1X (p < 0.05). Additionally, AMBP was positively correlated with IRF8. Conversely, the primary tumor-specific DEG NSG1 did not exhibit significant associations with protein expression in primary tumors.
In recurrent HGSOC, the recurrent tumor-specific DEGs IL7R, IRF8, and PTPRC displayed significant correlations with seven proteins. Positive correlations were observed with CHAC2, ERBIN, and LTA4H, while negative correlations were identified with HAVCR1 and NPY. Co-expression between DEGs and two DEPs (RIGI and TBL1X) was also detected. Notably, RIGI and TBL1X, both of which were downregulated in recurrent tumors, showed positive correlations with DEGs in recurrent tumors, whereas they were negatively correlated in primary tumors. This reversal in correlation direction was unique to RIGI and TBL1X, as the other five proteins maintained consistent patterns with the DEGs across tumor types (p < 0.05), suggesting a context-dependent shift in regulatory relationships during tumor progression. Similar to its behavior in primary tumors, NSG1 did not exhibit significant associations with protein expression in recurrent tumors. These findings highlight the dynamic nature of DEG–DEP interactions across disease stages and suggest that the functional roles of recurrent tumor-specific DEGs are shaped by distinct molecular contexts in primary versus recurrent HGSOC.
To determine whether the DEPs were associated with molecular pathways predicted to be activated in our network analysis, we categorized them into functional gain and functional loss groups based on a comprehensive literature review. The functional gain group, which included CHAC2, ERBIN, LTA4H, HAVCR1, and NPY, was primarily involved in the RAS–RAF–MAPK signaling pathway. In contrast, proteins in the functional loss group were associated with a broader range of pathways, including the PI3K–AKT–mTOR pathway, the MAPK pathway, base excision repair, and NF-κB signaling (Supplementary Table 7). Collectively, our proteomic analysis suggests that recurrent tumor-specific DEGs are predominantly linked to the MAPK pathway through multiple mechanisms, underscoring their potential roles in HGSOC recurrence.
3. Discussion
In this study, we performed integrative transcriptomic and proteomic analyses of matched primary and recurrent HGSOC tissues from the same patient and identified distinct molecular alterations—highlighting key genes, proteins, and regulatory networks as potential biomarkers and therapeutic targets in ovarian cancer recurrence. RNA sequencing analysis identified 185 DEGs between primary and recurrent tumors, with 151 genes upregulated and 34 downregulated in recurrent HGSOC. Pathway enrichment analysis using IPA revealed molecular networks involved in immune cell trafficking and cell morphology regulation, which are critical for immune evasion and metastasis. Survival analysis incorporating patient clinical data further narrowed these to seven key candidate genes associated with recurrence: IL7R, IRF8, PTPRC, NSG1, GPR68, WDFY4, and C15orf39. IHC analysis to validate the protein-level expression of the selected genes confirmed significant upregulation of IL7R, IRF8, and PTPRC in recurrent tumors, whereas NSG1 exhibited a decreasing trend. Furthermore, lower expression levels of IL7R, IRF8, and PTPRC, along with higher NSG1 expression, were associated with improved prognosis, highlighting their potential as prognostic biomarkers. Functional validation further demonstrated that IL7R, IRF8, and PTPRC contribute to tumor progression by promoting immune evasion, whereas NSG1 might exert tumor-suppressive effects.
Previous studies analyzing matched primary and recurrent HGSOC tissues highlighted the dynamic nature of the tumor microenvironment (TME), revealing profound alterations in immune composition, stromal interactions, and vascular remodeling that contribute to immune evasion and therapeutic resistance [24,25,26,27,28,29,30]. Recurrent tumors frequently exhibit enhanced immune evasion mechanisms, including upregulation of immune checkpoint molecules (e.g., PD-1/PD-L1, CD80/CTLA-4) and dysregulated immune cell trafficking, reflecting complex immunoregulatory processes [31,32]. Among immune cells in the TME, tumor-associated macrophages (TAMs) and regulatory T cells (Tregs) are pivotal in establishing an immunosuppressive niche [26]. Additional contributors to treatment resistance include cancer stem cells (CSCs) and autophagy, which are implicated in chemotherapy failure and disease relapse [30,33]. Furthermore, stromal and vascular components actively shape the TME; cancer-associated fibroblasts facilitate tumor proliferation, invasion, and immune evasion, while endothelial cells drive angiogenesis through VEGF signaling [34]. Although angiogenesis remains essential for tumor growth, recent evidence suggests that microvessel density and VEGF expression act more as supportive factors than direct drivers of disease progression — emphasizing the need for combination therapies targeting multiple aspects of the TME [29]. Advances in multi-omic approaches have identified therapeutic strategies targeting the TME, such as anti-angiogenic agents (e.g., bevacizumab), TAM-targeted therapies, and immune checkpoint inhibitors (e.g., PD-1/PD-L1 blockade), which show clinical potential [24,35]. Nevertheless, resistance to immune-based therapies remains a challenge, particularly in recurrent tumors with high PD-L1 expression. Continued investigation is needed to refine these strategies, enhance patient stratification, and develop novel combinatorial therapies that effectively modulate the TME and improve clinical outcomes in ovarian cancer.
Our study highlights the importance of analyzing matched primary and recurrent HGSOC tissues to gain deeper insights into the molecular alterations driving tumor recurrence. By characterizing these changes, we emphasize the need for a more comprehensive understanding of the mechanisms underlying recurrence, which is crucial for developing targeted therapeutic strategies to disrupt these processes and improve patient outcomes. Among the genes identified, IL7R, IRF8, PTPRC, and NSG1, which have not been thoroughly explored or recognized for their relevance in ovarian cancer recurrence in previous studies, were notably associated with tumor recurrence and clinical prognosis, warranting further investigation into their potential roles in HGSOC biology.
Although IL-7 plays a critical role in enhancing anti-tumor immunity through its immunomodulatory effects, increasing evidence suggests that activation of its receptor, IL-7R, paradoxically might promote tumor progression by supporting tumor cell proliferation and facilitating immune evasion via modulation of the TME [36]. Due to its dual role in cancer biology, IL-7R has emerged as a potential therapeutic target, prompting efforts to exploit its signaling axis to improve the efficacy and safety of cancer immunotherapies [37]. IL-7 restores CD8+ T cell function by reducing exhaustion markers such as PD-1, while simultaneously promoting the expansion of Tregs, which can suppress anti-tumor immunity [36,38,39]. This dual effect suggests that IL-7R upregulation in recurrent HGSOC facilitates immune escape by disrupting the balance between cytotoxic and regulatory immune cell populations within the TME. Mechanistically, IL-7R signaling activates several key oncogenic pathways—including JAK/STAT5, PI3K/AKT/mTOR, Ras/ERK, and MAPK—which regulate genes involved in cell proliferation, apoptosis resistance, and immune modulation [36]. Inhibition of IL-7R signaling has been investigated across various malignancies, particularly hematologic cancers such as T-cell and B-cell acute lymphoblastic leukemia (T-ALL and B-ALL), with multiple therapeutic strategies under development [40,41,42,43,44]. These include monoclonal antibodies targeting IL-7Rα (CD127) to block IL-7-mediated signaling and JAK inhibitors like ruxolitinib that suppress the downstream STAT5 pathway [40,44,45,46]. More recently, chimeric antigen receptor T cells were engineered to interfere with IL-7R signaling, offering another promising avenue [47,48,49,50]. In addition to hematologic malignancies, IL-7/IL-7R signaling is implicated in the progression of various solid tumors. In prostate cancer, high IL-7 expression correlates with poor prognosis, with IL-7R activation promoting invasion and migration via the AKT/NF-κB pathway and matrix metalloproteinase (MMP) regulation [51,52]. Similarly, IL-7 enhances bladder cancer invasion through NF-κB–mediated MMP-9 expression and promotes lung cancer proliferation by upregulating cyclin D1 via the c-FOS/c-Jun pathway [53].
IRF8, a member of the interferon regulatory factor (IRF) family, plays essential roles in hematopoiesis and type I interferon signaling [54]. Although traditionally considered a tumor suppressor [20,55,56], recent studies suggest a pro-oncogenic role for IRF8, particularly in acute myeloid leukemia (AML), where elevated IRF8 expression correlates with poor prognosis [57,58,59]. Mechanistically, IRF8 promotes leukemic cell proliferation by regulating STAT3, a key component of the JAK/STAT signaling pathway. Reduced IRF8 expression decreases STAT3 and pSTAT3 levels, suppressing AML growth through downregulation of Cyclin A and Cyclin B1, which induces S-phase arrest [57]. In addition to its role in AML, elevated IRF8 expression is associated with poor survival in lung adenocarcinoma based on TCGA data [60]. In clear cell renal cell carcinoma, TAMs express high levels of IRF8, contributing to CD8+ T cell exhaustion [61]. High IRF8-TAM gene signatures correlate with increased T cell dysfunction, particularly in tumors with abundant CD8+ T cell infiltration [62]. IRF8-expressing TAMs contribute to tumor-reactive cytotoxic T lymphocyte (CTL) exhaustion by presenting tumor antigens and inducing PD-1 expression on CD8+ T cells. In the absence of IRF8, TAMs fail to drive T cell exhaustion, highlighting the importance of IRF8-mediated antigen presentation. High IRF8 expression in TAMs correlates with poor prognosis in CD8+ T cell-rich tumors, suggesting that IRF8-dependent TAM functions sustain CTL exhaustion [61]. Although IRF8 is linked to favorable outcomes in some cancers, our findings suggest that the IRF8-TAM gene signature predicts immune dysfunction in ovarian cancer, highlighting the complex and context-dependent role of IRF8 within the TME.
The leukocyte common antigen PTPRC (CD45) is a transmembrane glycoprotein broadly expressed on hematopoietic cells, except mature erythrocytes, and plays a pivotal role in immune cell activation and signaling [63]. While PTPRC has traditionally served as a pan-leukocyte marker, emerging evidence suggests it also has prognostic and functional significance in several malignancies [64,65,66,67]. In colorectal cancer, a subset of epithelial tumor cells aberrantly expresses PTPRC, and elevated expression in these cells is linked to poor tumor regression and shorter recurrence-free survival following chemoradiotherapy [64]. Mechanistically, PTPRC enhances Wnt/β-catenin signaling by reducing β-catenin tyrosine phosphorylation, preventing its degradation and promoting nuclear accumulation. It, in turn, increases transcription of Wnt target genes, supporting CSC survival, self-renewal, and metastatic potential—highlighting a tumor-intrinsic role for PTPRC in promoting chemoresistance and progression. Similarly, in hematologic malignancies such as AML, PTPRC promotes tumor growth by localizing to lipid rafts, where it enhances GM-CSF signaling through activation of Src family kinases [67]. These findings underscore the context-dependent and multifaceted functions of PTPRC in cancer. Our study extends this knowledge to HGSOC, where we found that high PTPRC expression is significantly associated with poor prognosis. As in colorectal cancer, PTPRC drives HGSOC progression by promoting therapy resistance and CSC-related properties.
NSG1 is involved in endocytosis, apoptosis regulation, and intracellular trafficking of membrane receptors such as AMPA-type glutamate receptors, transferrin receptors, and neurotensin receptors (NTSR1/2) [68]. Through this regulation, NSG1 modulates intracellular signaling pathways that influence tumor cell proliferation, migration, and progression. NSG1 is also a transcriptional target of the tumor suppressor p53. Under genotoxic stress, p53 induces NSG1 expression, which in turn promotes tumor suppression by facilitating endocytic degradation of oncogenic receptors like EGFR and integrins, inhibiting pro-tumorigenic signaling [69,70,71]. Consistent with this mechanism, low NSG1 expression—as observed in our study—is associated with impaired tumor suppressor function and poor prognosis. Overexpression of NSG1 reduces tumor cell viability, potentially by inducing endoplasmic reticulum stress. In contrast, reduced NSG1 expression leads to decreased CHOP activation and weakened apoptotic responses to DNA damage, promoting tumor survival [69]. Beyond its tumor-intrinsic role, NSG1 also influences the TME. Low NSG1 expression correlates with reduced infiltration of CD8+ T cells and M1 macrophages, alongside increased activation of M2 macrophages, contributing to immune evasion and tumor progression [68].
Furthermore, integrative correlation analysis revealed that specific DEGs, such as IL7R, IRF8, and PTPRC, were positively correlated with DEPs like CHAC2, ERBIN, and LTA4H and negatively correlated with HAVCR1 and NPY. These results suggest complex gene–protein interactions in recurrent HGSOC, many of which converge on key oncogenic pathways such as MAPK, NOTCH, VEGF, and NF-κB signaling. Pathway enrichment analysis further highlighted the involvement of these signaling axes in recurrence, tumor progression, and treatment resistance.
Importantly, our multi-omics approach offers a comprehensive view of the molecular drivers underlying recurrence and might inform the development of precision medicine strategies. Integrating genomic and proteomic profiling is essential for identifying novel druggable targets, particularly in treatment-resistant ovarian cancer. Further studies should validate these candidate biomarkers in larger patient cohorts and investigate their roles in immune checkpoints and personalized treatment approaches.
However, several limitations of this study must be acknowledged. First, the relatively small sample sizes in the primary/recurrent tumor and validation cohorts increase the risk of overfitting. Second, discrepancies between mRNA-based Kaplan–Meier survival data and some protein-level findings highlight the need for additional investigation. Third, although this study provides functional insights into IL7R, IRF8, PTPRC, and NSG1, further mechanistic studies are required to elucidate their interactions and downstream signaling pathways. Despite these limitations, our findings offer novel insights into the mechanisms of HGSOC recurrence through the characterization of key gene and protein expression changes and their interaction networks. These results have important implications for prognosis prediction and the development of individualized therapeutic strategies.
4. Materials and Methods
4.1. Sample collection and preparation
Sixty-eight matched primary and first recurrent HGSOC tissue samples were obtained from 34 patients who were initially diagnosed and continuously treated at Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea, between 2009 and 2019. Clinicopathological data, including time to first recurrence or death from ovarian cancer, were retrieved from electronic medical records. The study was approved by the Institutional Review Board of Gangnam Severance Hospital (IRB No. 3-2021-0380), and informed consent was obtained from all patients. FFPE blocks and fresh-frozen tissues were obtained from the Human Tissue Bank and the Korea Gynecologic Cancer Bank at Gangnam Severance Hospital (Resource No. HTB-P2022-3). All research procedures were conducted in accordance with all relevant ethical regulations including the principles of the Declaration of Helsinki. All samples included in the study met the following criteria: availability of matched primary and first recurrent HGSOC samples, no history of neoadjuvant chemotherapy prior to initial surgery, and sufficient histological quality of the primary and recurrent tumor samples. All specimens were reviewed by a pathologist specializing in gynecologic oncology, and only those with tumor purity greater than 80% were included in the analysis. RNA sequencing was performed on FFPE samples from 24 patients, using paired primary and recurrent tumor tissues. Concurrently, proteomic analysis was conducted on matched fresh-frozen tissue samples from 19 patients. Nine patients provided both FFPE and frozen tissue samples, enabling integrated transcriptomic and proteomic analyses. The remaining 25 patients contributed only one type of sample each, precluding integrated transcriptomic and proteomic analyses (Figure 1A). Immunohistochemistry (IHC) analysis using tissue microarray (TMA) samples was conducted on a distinct set of paired primary and recurrent tumor tissues from 34 patients.
4.2. RNA sequencing
Total RNA concentration was measured using the Quant-IT RiboGreen assay (Invitrogen). Samples were analyzed using the TapeStation RNA ScreenTape system (Agilent Technologies) to determine the DV200 value (percentage of RNA fragments >200 bp). Following the manufacturer’s protocol, 100 ng of total RNA was used to construct sequencing libraries using the Agilent SureSelect RNA Direct kit. Total RNA was first fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments were then reverse-transcribed into first-strand cDNA using random primers, followed by second-strand cDNA synthesis. The resulting cDNA fragments underwent end repair, addition of a single ‘A’ base, and adapter ligation. The products were subsequently purified and enriched via PCR to generate the cDNA library. The Agilent SureSelect XT Human All Exon V6+UTRs Kit was used following the standard Agilent SureSelect Target Enrichment protocol. To capture human exonic regions, 250 ng of the cDNA library was combined with hybridization buffers, blocking mixes, RNase block, and 5 µL of the SureSelect All Exon capture library. Hybridization to the capture baits was performed at 65 °C using a thermal cycler with a heated lid set at 105 °C for 24 h. The captured library was then washed and subjected to a second round of PCR amplification.
The final purified product was quantified using the KAPA Library Quantification Kit for Illumina sequencing platforms, following the qPCR Quantification Protocol Guide (KAPA BIOSYSTEMS, #KK4854). Its quality was assessed using the TapeStation D1000 ScreenTape system (Agilent Technologies, #5067-5582). Indexed libraries were submitted for sequencing on an Illumina NovaSeq platform (Illumina, Inc., San Diego, CA, USA), and paired-end (2×100 bp) sequencing was performed by Macrogen, Inc.
4.3. Data preprocessing and differential expression analysis of RNA sequencing data
Low-quality and adapter sequences were trimmed using TrimGalore with a quality score cutoff of 20 [72]. Subsequently, the STAR 2-pass method was utilized to align raw data to the human reference genome (GRCh38.p14) using STAR aligner, and the gencode v47 annotation file was utilized for gene annotation [73]. Differential expression analysis was performed with R package DESeq2 to extract the magnitude (log2 ratio of fold change (log2FC)) and significance (p-value) of the gene expression value between groups [74]. The p-value was adjusted using the Benjamini-Hochberg procedure, and genes with false discovery rate (FDR) < 0.01 and an absolute log2 fold change > 1 were defined as DEGs. Gene expression was visualized using a volcano plot generated with the R package ‘ggrepel’ and a heatmap produced with the R package ‘pheatmap.’
4.4. Ingenuity pathway analysis
The complete list of DEGs was uploaded to the Ingenuity Pathway Analysis (IPA) platform for candidate molecule analysis. For network analysis, focus molecules were identified based on their documented interactions with other molecules in the QIAGEN Knowledge Base. Networks were constructed by maximizing the interconnectedness of focus molecules, and additional molecules from the QIAGEN Knowledge Base were incorporated to merge smaller networks into larger ones. Each network was restricted to a maximum of 35 molecules to ensure clarity. Network significance was assessed using Fisher’s Exact Test, with scores calculated as the negative base-10 logarithm of the p-value, where higher scores denoted greater statistical significance of the observed relationships.
4.5. Pathway enrichment analysis
For canonical pathway analysis using IPA, the ratio was determined using an in-house script based on the formula
Ratio = (N of detected genes in pathway)/(N of all genes in pathway)
R package ‘clusterProfiler’ was employed for the over-representation analysis (ORA) [75]. Reference gene sets were sourced from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Gene Ontology Biological Process (GO: BP) gene sets [76,77]. Multiple testing correction was performed using the Benjamini-Hochberg method, with gene sets considered statistically significant if the FDR was less than 0.05. For these gene sets, the ratio was calculated in the same way: as the proportion of DEGs that overlapped with the gene set, relative to the total number of genes in that gene set.
4.6. Survival analysis
Survival differences between the two groups were analyzed using the survival and survminer packages in R [78,79]. The optimal cutpoint for gene expression was determined using the ‘surv_cutpoint’ and ‘surv_categorize’ functions. Then, Kaplan–Meier survival curves were generated using the ‘survfit’ and ‘ggsurvplot’ functions to evaluate survival probabilities in patient groups stratified by high and low expression. Genes with log-rank p-values less than 0.01 were considered statistically significant.
4.7. Tissue microarray and immunohistochemistry
TMA blocks were constructed using FFPE donor blocks from paired primary and recurrent tumors of 34 ovarian cancer patients. Each tissue core had a diameter of 1.5 mm, and the TMA blocks were sectioned into 5-µm-thick slices using a rotary microtome. The TMA sections were incubated at 65 °C for 20 min; deparaffinized in xylene for 15 min; and transferred to 100% ethanol, 90% ethanol, and 70% ethanol for 5 min each. The sections were then rinsed with deionized water. For antigen retrieval, the sections were subjected to heat-mediated pretreatment with 10 mM citrate buffer (pH 6.0 or 9.0) for 10 min in a microwave. The sections were then immersed in methanol containing 3% hydrogen peroxide for 10 min, followed by incubation with primary antibodies for 120 min. The primary antibodies used were IL7R (Abcam, #ab259806), IRF8 (Cell Signaling, #83413), GPR68 (Abcam, #ab61420), PTPRC (Cell Signaling, #13917), WDFY4 (Abcam, #ab122661), and NSG1 (Invitrogen, #PA5-36497). The secondary antibody, EnVision™ Rabbit/Mouse reagent (DAKO, #K5007), was applied for 60 min. Sections were visualized with 3,3-diaminobenzidine tetrachloride (DAB; DAKO, #K3468) and counterstained with hematoxylin (DAKO, #S3309) for 5 min. All procedures were carried out at room temperature (25 °C).
4.8. Evaluation of immunohistochemistry staining
Stained TMA sections were scanned at 400× optical magnification using an Axioscan 7 Microscope Slide Scanner (ZEISS). The scanned images were analyzed using QuPath 0.4.4, a quantitative pathology and bioimage analysis software. Representative images were captured at 2.5× and 50× digital magnification within QuPath. Each TMA core was designated as a region of interest for analysis, and automated cell segmentation was performed using the default algorithm in QuPath, with parameter adjustments. The algorithm classified cells into stained and unstained populations, and the percentage of stained cells relative to the total cell count was quantified on a scale of 0 to 100.
4.9. Proteomic assay data generation
The fresh-frozen tissues were transferred to bead tubes on ice and lysed using T-PER™ buffer with protease inhibitors. Homogenization was performed using a FastPrep-24™ classic homogenizer, followed by multiple cycles of centrifugation and supernatant collection. Protein concentration was measured using bovine serum albumin, and all samples were adjusted to a uniform concentration. The prepared samples were analyzed using the Olink Explore Oncology II panel (Uppsala, Sweden), comprising 386 oncology-related antibodies. This technology relies on the precise binding of target proteins to antibody probes, which are tagged with dual oligonucleotides. The subsequent DNA sequences are quantitatively detected through microfluidic RT-qPCR amplification of the oligonucleotides. Expression values were normalized using experimental controls, and the results were presented as normalized protein expression values, log2-transformed. This experiment was conducted by DNA Link (Seoul, Korea).
4.10. Data preprocessing and analysis of proteomics data
NPX values were used for differential expression analysis of proteins that passed quality control. The comparison was performed using empirical Bayes (eBayes) moderated t-tests implemented through the limma R package [80]. Proteins with a p-value less than 0.05 were considered differentially expressed proteins (DEPs). Co-expression was analyzed using Spearman’s rank correlation coefficients, and a correlation with a p-value less than 0.05 was considered significant.
5. Conclusions
Our integrative multi-omics analysis reveals novel molecular mechanisms driving HGSOC recurrence, identifying IL7R, IRF8, PTPRC, and NSG1 as potential biomarkers and therapeutic targets. By contributing to immune evasion, tumor progression, and treatment resistance, these genes illuminate critical pathways in HGSOC recurrence, underscoring their potential as both prognostic markers and therapeutic targets. Future research should validate these findings in larger cohorts and further explore their roles in modulating the TME and immune response. Additionally, combination therapies targeting these molecules, alongside immune checkpoint inhibitors or chemotherapy, might improve clinical outcomes for recurrent HGSOC patients.
Author Contributions
Conceptualization, M.-A.K., J.N., M.-R.H., and J.-H.K.; methodology, J.Y.K. and H.-Y.S.; validation, J.Y.K. and H.-Y.S.; formal analysis, J.N.; investigation, M.-A.K., A.J., and H.C.; resources, M.-R.H., H.C, and J.-H.K.; data curation, J.N., M.-R.H., and J.-H.K.; writing—original draft preparation, M.-A.K. and J.N.; writing—review and editing, M.-R.H., and J.-H.K.; visualization, J.N. and M.-A.K.; supervision, M.-R.H., and J.-H.K.; project administration, M.-R.H. and J.-H.K.; funding acquisition, M.-R.H. and J.-H.K. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
The study was approved by the Institutional Review Board of Gangnam Severance Hospital (IRB No. 3-2021-0380), and informed consent was obtained from all patients. Approval date: 5 November 2021).
Informed Consent Statement
Written informed consent has been obtained from all subjects involved in the study.
Data Availability Statement
The transcriptomic data produced in this study are publicly available through the Gene Expression Omnibus (GEO) database with accession number GSE295041.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Romero, I.; Leskela, S.; Mies, B.P.; Velasco, A.P.; Palacios, J. Morphological and molecular heterogeneity of epithelial ovarian cancer: Therapeutic implications. EJC Suppl 2020, 15, 1–15. [CrossRef]
- Punzon-Jimenez, P.; Lago, V.; Domingo, S.; Simon, C.; Mas, A. Molecular management of high-grade serous ovarian carcinoma. Int J Mol Sci 2022, 23.
- Lalwani, N.; Prasad, S.R.; Vikram, R.; Shanbhogue, A.K.; Huettner, P.C.; Fasih, N. Histologic, molecular, and cytogenetic features of ovarian cancers: Implications for diagnosis and treatment. Radiographics 2011, 31, 625–646.
- Network, N.C.C. Nccn clinical practice guidelines in oncology: Ovarian cancer including fallopian tube cancer and primary peritoneal cancer (version 2.2025). Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1453 (accessed on May 30, 2025.
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L. et al. Association between brca1 and brca2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390.
- Fu, R.; Hu, R.; Li, W.; Lv, X.; Zhao, H.; Li, F. Unveiling drug resistance pathways in high-grade serous ovarian cancer(hgsoc): Recent advances and future perspectives. Front Immunol 2025, 16, 1556377.
- Adzibolosu, N.; Alvero, A.B.; Ali-Fehmi, R.; Gogoi, R.; Corey, L.; Tedja, R.; Chehade, H.; Gogoi, V.; Morris, R.; Anderson, M. et al. Immunological modifications following chemotherapy are associated with delayed recurrence of ovarian cancer. Front Immunol 2023, 14, 1204148.
- Nero, C.; Vizzielli, G.; Lorusso, D.; Cesari, E.; Daniele, G.; Loverro, M.; Scambia, G.; Sette, C. Patient-derived organoids and high grade serous ovarian cancer: From disease modeling to personalized medicine. J Exp Clin Cancer Res 2021, 40, 116. [CrossRef]
- Ushijima, K. Treatment for recurrent ovarian cancer-at first relapse. J Oncol 2010, 2010, 497429.
- Mahmood, R.D.; Morgan, R.D.; Edmondson, R.J.; Clamp, A.R.; Jayson, G.C. First-line management of advanced high-grade serous ovarian cancer. Curr Oncol Rep 2020, 22, 64.
- 11. Lim, H.J.a.L., W. Targeted therapy in ovarian cancer. Womens Health 2016, 12, 363–378.
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382.
- Wang, G.; Yang, H.; Wang, Y.; Qin, J. Ovarian cancer targeted therapy: Current landscape and future challenges. Front Oncol 2025, 15, 1535235.
- Moufarrij, S.; O'Cearbhaill, R.E. Novel therapeutics in ovarian cancer: Expanding the toolbox. Curr Oncol 2023, 31, 97–114.
- Dinkins, K.; Barton, W.; Wheeler, L.; Smith, H.J.; Mythreye, K.; Arend, R.C. Targeted therapy in high grade serous ovarian cancer: A literature review. Gynecol Oncol Rep 2024, 54, 101450.
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol 2018, 81, 17–38.
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat Rev Dis Primers 2016, 2, 16061.
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S. et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018, 379, 2495–2505.
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253.
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J. et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494.
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell rna-seq. PLoS One 2018, 13, e0206785.
- Harbin, L.M.; Gallion, H.H.; Allison, D.B.; Kolesar, J.M. Next generation sequencing and molecular biomarkers in ovarian cancer-an opportunity for targeted therapy. Diagnostics (Basel) 2022, 12.
- Li, H.; Zheng, X.; Gao, J.; Leung, K.S.; Wong, M.H.; Yang, S.; Liu, Y.; Dong, M.; Bai, H.; Ye, X. et al. Whole transcriptome analysis reveals non-coding rna's competing endogenous gene pairs as novel form of motifs in serous ovarian cancer. Comput Biol Med 2022, 148, 105881.
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor microenvironment in ovarian cancer: Function and therapeutic strategy. Front Cell Dev Biol 2020, 8, 758.
- Westergaard, M.C.W.; Milne, K.; Pedersen, M.; Hasselager, T.; Olsen, L.R.; Anglesio, M.S.; Borch, T.H.; Kennedy, M.; Briggs, G.; Ledoux, S. et al. Changes in the tumor immune microenvironment during disease progression in patients with ovarian cancer. Cancers (Basel) 2020, 12. [CrossRef]
- Ojalvo, L.S.; Thompson, E.D.; Wang, T.L.; Meeker, A.K.; Shih, I.M.; Fader, A.N.; Cimino-Mathews, A.; Emens, L.A. Tumor-associated macrophages and the tumor immune microenvironment of primary and recurrent epithelial ovarian cancer. Hum Pathol 2018, 74, 135–147. [CrossRef]
- Stanske, M.; Wienert, S.; Castillo-Tong, D.C.; Kreuzinger, C.; Vergote, I.; Lambrechts, S.; Gabra, H.; Gourley, C.; Ganapathi, R.N.; Kolaschinski, I. et al. Dynamics of the intratumoral immune response during progression of high-grade serous ovarian cancer. Neoplasia 2018, 20, 280–288. [CrossRef]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of pd-l1 on tumor cells is associated with reduced mhc i expression and pd-l1 expression increases in recurrent serous ovarian cancer. Sci Rep 2017, 7, 42929.
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Glajzer, J.; Kulbe, H.; Trillsch, F.; Mustea, A. et al. Characterisation of tumour microvessel density during progression of high-grade serous ovarian cancer: Clinico-pathological impact (an octips consortium study). Br J Cancer 2018, 119, 330–338.
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Ganapathi, R.N.; Glajzer, J.; Kulbe, H.; Trillsch, F. et al. Exploring the clonal evolution of cd133/aldehyde-dehydrogenase-1 (aldh1)-positive cancer stem-like cells from primary to recurrent high-grade serous ovarian cancer (hgsoc). A study of the ovarian cancer therapy-innovative models prolong survival (octips) consortium. Eur J Cancer 2017, 79, 214–225.
- Li, Y.R.; Halladay, T.; Yang, L. Immune evasion in cell-based immunotherapy: Unraveling challenges and novel strategies. J Biomed Sci 2024, 31, 5.
- Kreuzinger, C.; Geroldinger, A.; Smeets, D.; Braicu, E.I.; Sehouli, J.; Koller, J.; Wolf, A.; Darb-Esfahani, S.; Joehrens, K.; Vergote, I. et al. A complex network of tumor microenvironment in human high-grade serous ovarian cancer. Clin Cancer Res 2017, 23, 7621–7632. [CrossRef]
- Wang, J.; Garbutt, C.; Ma, H.; Gao, P.; Hornicek, F.J.; Kan, Q.; Shi, H.; Duan, Z. Expression and role of autophagy-associated p62 (sqstm1) in multidrug resistant ovarian cancer. Gynecol Oncol 2018, 150, 143–150.
- Liu, H.; Zhou, L.; Cheng, H.; Wang, S.; Luan, W.; Cai, E.; Ye, X.; Zhu, H.; Cui, H.; Li, Y. et al. Characterization of candidate factors associated with the metastasis and progression of high-grade serous ovarian cancer. Chin Med J (Engl) 2023, 136, 2974–2982.
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging therapeutic strategies to overcome drug resistance in cancer cells. Cancers (Basel) 2024, 16.
- Wang, C.; Kong, L.; Kim, S.; Lee, S.; Oh, S.; Jo, S.; Jang, I.; Kim, T.D. The role of il-7 and il-7r in cancer pathophysiology and immunotherapy. Int J Mol Sci 2022, 23.
- Liang, J.; Zhu, L.; Li, J.; Wu, K.; Zhang, M.; Ma, S.; Chen, X.; Xia, B. Comprehensive analysis to identify il7r as a immunotherapy biomarker from pan-cancer analysis to in vitro validation. Discov Oncol 2024, 15, 509.
- Pellegrini, M.; Calzascia, T.; Elford, A.R.; Shahinian, A.; Lin, A.E.; Dissanayake, D.; Dhanji, S.; Nguyen, L.T.; Gronski, M.A.; Morre, M. et al. Adjuvant il-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat Med 2009, 15, 528–536.
- Gao, J.; Zhao, L.; Wan, Y.Y.; Zhu, B. Mechanism of action of il-7 and its potential applications and limitations in cancer immunotherapy. Int J Mol Sci 2015, 16, 10267–10280.
- Courtois, L.; Cabannes-Hamy, A.; Kim, R.; Delecourt, M.; Pinton, A.; Charbonnier, G.; Feroul, M.; Smith, C.; Tueur, G.; Pivert, C. et al. Il-7 receptor expression is frequent in t-cell acute lymphoblastic leukemia and predicts sensitivity to jak inhibition. Blood 2023, 142, 158–171.
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R. et al. Targeted sequencing identifies associations between il7r-jak mutations and epigenetic modulators in t-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 1301–1310. [CrossRef]
- Oliveira, M.L.; Akkapeddi, P.; Ribeiro, D.; Melao, A.; Barata, J.T. Il-7r-mediated signaling in t-cell acute lymphoblastic leukemia: An update. Adv Biol Regul 2019, 71, 88–96.
- Almeida, A.R.M.; Neto, J.L.; Cachucho, A.; Euzebio, M.; Meng, X.; Kim, R.; Fernandes, M.B.; Raposo, B.; Oliveira, M.L.; Ribeiro, D. et al. Interleukin-7 receptor alpha mutational activation can initiate precursor b-cell acute lymphoblastic leukemia. Nat Commun 2021, 12, 7268. [CrossRef]
- Cramer, S.D.; Aplan, P.D.; Durum, S.K. Therapeutic targeting of il-7ralpha signaling pathways in all treatment. Blood 2016, 128, 473–478.
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K. et al. A fully human anti-il-7ralpha antibody promotes antitumor activity against t-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168.
- Lodewijckx, I.; Cools, J. Deregulation of the interleukin-7 signaling pathway in lymphoid malignancies. Pharmaceuticals (Basel) 2021, 14.
- Sakunrangsit, N.; Khuisangeam, N.; Inthanachai, T.; Yodsurang, V.; Taechawattananant, P.; Suppipat, K.; Tawinwung, S. Incorporating il7 receptor alpha signaling in the endodomain of b7h3-targeting chimeric antigen receptor t cells mediates antitumor activity in glioblastoma. Cancer Immunol Immunother 2024, 73, 98.
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R. et al. Constitutive signaling from an engineered il7 receptor promotes durable tumor elimination by tumor-redirected t cells. Cancer Discov 2017, 7, 1238–1247.
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered il-7 receptor enhances the therapeutic effect of axl-car-t cells on triple-negative breast cancer. Biomed Res Int 2020, 2020, 4795171.
- Vorri, S.C.; Holl, N.J.; Leeming, M.; Apostolova, P.; Marple, A.; Ravich, J.W.; Canbaz, A.; Rahnama, R.; Choe, J.; Modi, A. et al. Activation of cell-intrinsic signaling in car-t cells via a chimeric il7r domain. Cancer Res Commun 2024, 4, 2359–2373.
- Seol, M.A.; Kim, J.H.; Oh, K.; Kim, G.; Seo, M.W.; Shin, Y.K.; Sim, J.H.; Shin, H.M.; Seo, B.Y.; Lee, D.S. et al. Interleukin-7 contributes to the invasiveness of prostate cancer cells by promoting epithelial-mesenchymal transition. Sci Rep 2019, 9, 6917. [CrossRef]
- Qu, H.; Zou, Z.; Pan, Z.; Zhang, T.; Deng, N.; Chen, G.; Wang, Z. Il-7/il-7 receptor axis stimulates prostate cancer cell invasion and migration via akt/nf-kappab pathway. Int Immunopharmacol 2016, 40, 203–210.
- Ming, J.; Jiang, G.; Zhang, Q.; Qiu, X.; Wang, E. Interleukin-7 up-regulates cyclin d1 via activator protein-1 to promote proliferation of cell in lung cancer. Cancer Immunol Immunother 2012, 61, 79–88.
- Honda, K.; Takaoka, A.; Taniguchi, T. Type i interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360.
- Luo, X.; Xiong, X.; Shao, Q.; Xiang, T.; Li, L.; Yin, X.; Li, X.; Tao, Q.; Ren, G. The tumor suppressor interferon regulatory factor 8 inhibits beta-catenin signaling in breast cancers, but is frequently silenced by promoter methylation. Oncotarget 2017, 8, 48875–48888.
- Liang, J.; Lu, F.; Li, B.; Liu, L.; Zeng, G.; Zhou, Q.; Chen, L. Irf8 induces senescence of lung cancer cells to exert its tumor suppressive function. Cell Cycle 2019, 18, 3300–3312.
- Zhuang, H.; Li, F.; Xu, Y.; Pei, R.; Chen, D.; Liu, X.; Li, S.; Ye, P.; Yuan, J.; Lian, J. et al. Loss of irf8 inhibits the growth of acute myeloid leukemia cells. Ann Hematol 2023, 102, 1063–1072.
- Liss, F.; Frech, M.; Wang, Y.; Giel, G.; Fischer, S.; Simon, C.; Weber, L.M.; Nist, A.; Stiewe, T.; Neubauer, A. et al. Irf8 is an aml-specific susceptibility factor that regulates signaling pathways and proliferation of aml cells. Cancers (Basel) 2021, 13.
- Pogosova-Agadjanyan, E.L.; Kopecky, K.J.; Ostronoff, F.; Appelbaum, F.R.; Godwin, J.; Lee, H.; List, A.F.; May, J.J.; Oehler, V.G.; Petersdorf, S. et al. The prognostic significance of irf8 transcripts in adult patients with acute myeloid leukemia. PLoS One 2013, 8, e70812.
- Li, L.; Peng, M.; Xue, W.; Fan, Z.; Wang, T.; Lian, J.; Zhai, Y.; Lian, W.; Qin, D.; Zhao, J. Integrated analysis of dysregulated long non-coding rnas/micrornas/mrnas in metastasis of lung adenocarcinoma. J Transl Med 2018, 16, 372.
- Nixon, B.G.; Kuo, F.; Ji, L.; Liu, M.; Capistrano, K.; Do, M.; Franklin, R.A.; Wu, X.; Kansler, E.R.; Srivastava, R.M. et al. Tumor-associated macrophages expressing the transcription factor irf8 promote t cell exhaustion in cancer. Immunity 2022, 55, 2044–2058 e2045.
- Muhitch, J.B.; Hoffend, N.C.; Azabdaftari, G.; Miller, A.; Bshara, W.; Morrison, C.D.; Schwaab, T.; Abrams, S.I. Tumor-associated macrophage expression of interferon regulatory factor-8 (irf8) is a predictor of progression and patient survival in renal cell carcinoma. J Immunother Cancer 2019, 7, 155.
- Al Barashdi, M.A.-O.; Ali, A.; McMullin, M.A.-O.; Mills, K. Protein tyrosine phosphatase receptor type c (ptprc or cd45). J Clin Pathol 2021, 74, 548–552.
- Park, S.Y.; Kim, J.Y.; Jang, G.B.; Choi, J.H.; Kim, J.H.; Lee, C.J.; Lee, S.; Baek, J.H.; Park, K.K.; Kim, J.M. et al. Aberrant activation of the cd45-wnt signaling axis promotes stemness and therapy resistance in colorectal cancer cells. Theranostics 2021, 11, 8755–8770.
- Lv, Z.a.W., Tianming and Cao, Xin and Sun, Mengting and Qu, Yuan. The role of receptor-type protein tyrosine phosphatases in cancer. Precision Medical Sciences 2023, 12, 48–57.
- An, J.; Chen, P.; Li, X.; Li, X.; Peng, F. Identification of potential hub genes and biological mechanism in rheumatoid arthritis and non-small cell lung cancer via integrated bioinformatics analysis. Transl Oncol 2024, 45, 101964. [CrossRef]
- Saint-Paul, L.; Nguyen, C.H.; Buffiere, A.; Pais de Barros, J.P.; Hammann, A.; Landras-Guetta, C.; Filomenko, R.; Chretien, M.L.; Johnson, P.; Bastie, J.N. et al. Cd45 phosphatase is crucial for human and murine acute myeloid leukemia maintenance through its localization in lipid rafts. Oncotarget 2016, 7, 64785–64797. [CrossRef]
- Zhang, H.; Li, Y.; Wang, R.; Hu, X.; Wang, Z. Neuron-specific gene family member 1 is a potential new therapeutic target associated with immune cell infiltration for breast cancer. Breast Cancer (Dove Med Press) 2024, 16, 769–783.
- Kudoh, T.; Kimura, J.; Lu, Z.G.; Miki, Y.; Yoshida, K. D4s234e, a novel p53-responsive gene, induces apoptosis in response to DNA damage. Exp Cell Res 2010, 316, 2849–2858.
- Ohnishi, S.; Futamura, M.; Kamino, H.; Nakamura, Y.; Kitamura, N.; Miyamoto, Y.; Miyamoto, T.; Shinogi, D.; Goda, O.; Arakawa, H. Identification of neep21, encoding neuron-enriched endosomal protein of 21 kda, as a transcriptional target of tumor suppressor p53. Int J Oncol 2010, 37, 1133–1141. [CrossRef]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P. et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341.
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12.
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal rna-seq aligner. Bioinformatics 2013, 29, 15–21.
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol 2014, 15, 550.
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. et al. Clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021, 2, 100141.
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Matsuura, Y.; Ishiguro-Watanabe, M. Kegg: Biological systems database as a model of the real world. Nucleic Acids Res 2025, 53, D672–D677.
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat Genet 2000, 25, 25–29.
- Therneau, T.M.a.G., Patricia M. Modeling survival data: Extending the cox model. Springer: New York, 2000.
- Kassambara, A.a.K., Marcin and Biecek, Przemyslaw. Survminer: Drawing survival curves using 'ggplot2'. R package version 0.5.0: 2024.
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res 2015, 43, e47.
Figure 1.
Transcriptomic alterations and molecular interaction networks in matched primary and recurrent high-grade serous ovarian cancer. (A) Schematic representation of the patient cohort included in this study. A total of 34 patients with matched primary and recurrent high-grade serous ovarian cancer samples were analyzed. Green segments indicate patients subjected to RNA sequencing, while blue segments indicate those analyzed by proteomics. (B) Volcano plot illustrating differentially expressed genes (DEGs) between primary and recurrent tumors. Genes with a false discovery rate (FDR) < 0.05 and absolute log2 fold change > 1 are considered significant. Upregulated genes in recurrent tumors are shown in orange, downregulated genes in purple, and non-significant genes in grey. (C) Heatmap displaying the expression profiles of DEGs across all samples. Columns represent individual tumor samples (primary: yellow; recurrent: green), and rows represent DEGs. Color intensity corresponds to expression levels, ranging from blue (low expression) to red (high expression). (D–F) Gene regulatory networks generated using Ingenuity Pathway Analysis (IPA) based on the DEGs identified between primary and recurrent tumors. Nodes represent molecules, with red and green indicating observed upregulation and downregulation, respectively. Orange and blue nodes denote molecules predicted by IPA to be activated or inhibited. Solid and dashed lines represent direct and indirect molecular interactions, respectively, based on curated literature and experimental evidence.
Figure 1.
Transcriptomic alterations and molecular interaction networks in matched primary and recurrent high-grade serous ovarian cancer. (A) Schematic representation of the patient cohort included in this study. A total of 34 patients with matched primary and recurrent high-grade serous ovarian cancer samples were analyzed. Green segments indicate patients subjected to RNA sequencing, while blue segments indicate those analyzed by proteomics. (B) Volcano plot illustrating differentially expressed genes (DEGs) between primary and recurrent tumors. Genes with a false discovery rate (FDR) < 0.05 and absolute log2 fold change > 1 are considered significant. Upregulated genes in recurrent tumors are shown in orange, downregulated genes in purple, and non-significant genes in grey. (C) Heatmap displaying the expression profiles of DEGs across all samples. Columns represent individual tumor samples (primary: yellow; recurrent: green), and rows represent DEGs. Color intensity corresponds to expression levels, ranging from blue (low expression) to red (high expression). (D–F) Gene regulatory networks generated using Ingenuity Pathway Analysis (IPA) based on the DEGs identified between primary and recurrent tumors. Nodes represent molecules, with red and green indicating observed upregulation and downregulation, respectively. Orange and blue nodes denote molecules predicted by IPA to be activated or inhibited. Solid and dashed lines represent direct and indirect molecular interactions, respectively, based on curated literature and experimental evidence.
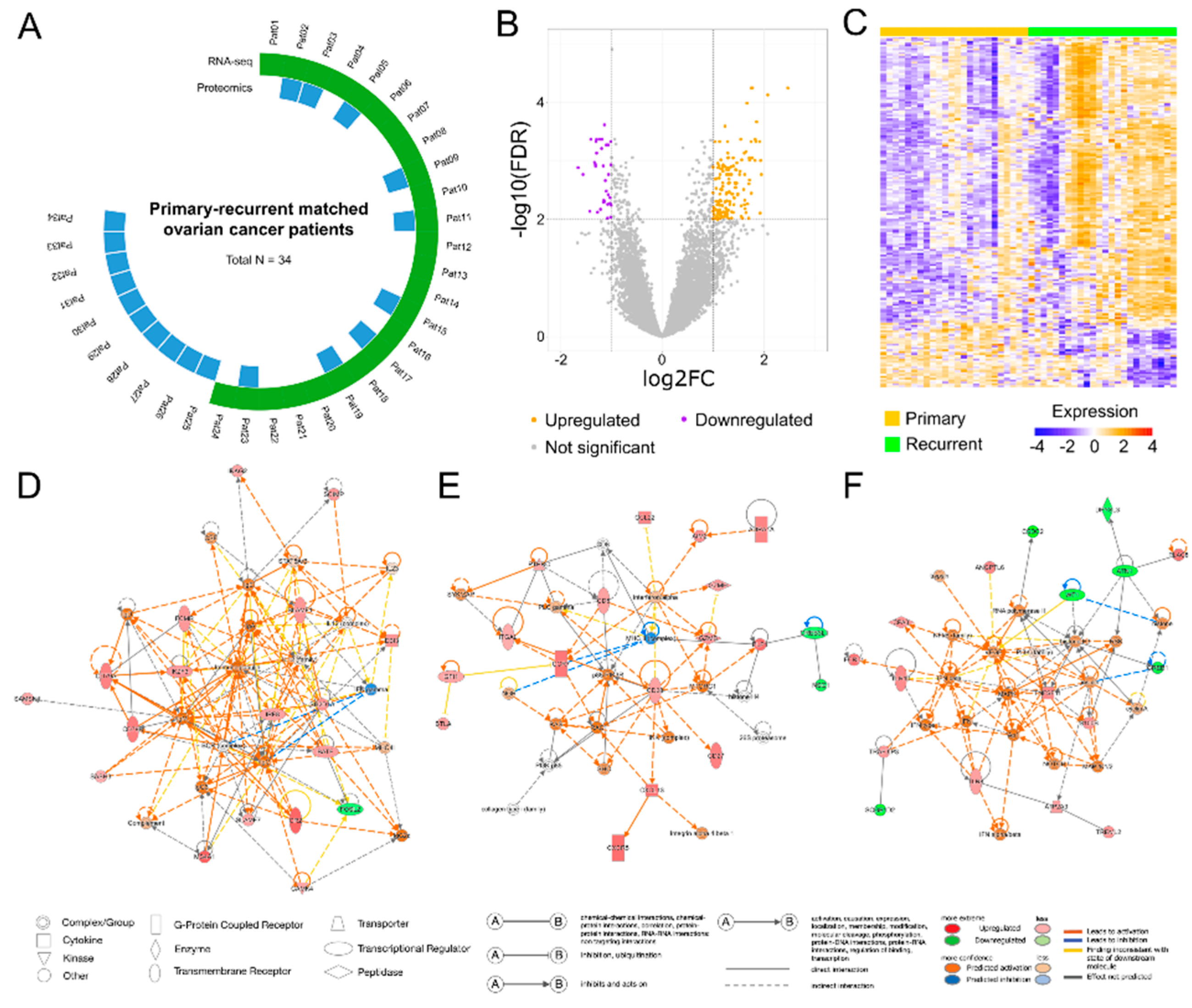
Figure 2.
Pathway enrichment analysis of differentially expressed genes between matched primary and recurrent high-grade serous ovarian cancer samples. (A) Canonical pathway analysis based on the QIAGEN Knowledge Base. The top 20 significantly enriched pathways are displayed. Dot color indicates predicted pathway activity: red for activated pathways, blue for inhibited pathways, and grey for pathways with undetermined directionality. (B) Over-representation analysis (ORA) of KEGG pathways. The top 10 significantly enriched pathways are shown based on differentially expressed genes overlap. (C) ORA of Gene Ontology Biological Process (GO: BP) terms. The top 20 enriched biological processes are displayed. Gene ratios on the x-axis indicate the proportion of input DEGs associated with each pathway or process.
Figure 2.
Pathway enrichment analysis of differentially expressed genes between matched primary and recurrent high-grade serous ovarian cancer samples. (A) Canonical pathway analysis based on the QIAGEN Knowledge Base. The top 20 significantly enriched pathways are displayed. Dot color indicates predicted pathway activity: red for activated pathways, blue for inhibited pathways, and grey for pathways with undetermined directionality. (B) Over-representation analysis (ORA) of KEGG pathways. The top 10 significantly enriched pathways are shown based on differentially expressed genes overlap. (C) ORA of Gene Ontology Biological Process (GO: BP) terms. The top 20 enriched biological processes are displayed. Gene ratios on the x-axis indicate the proportion of input DEGs associated with each pathway or process.
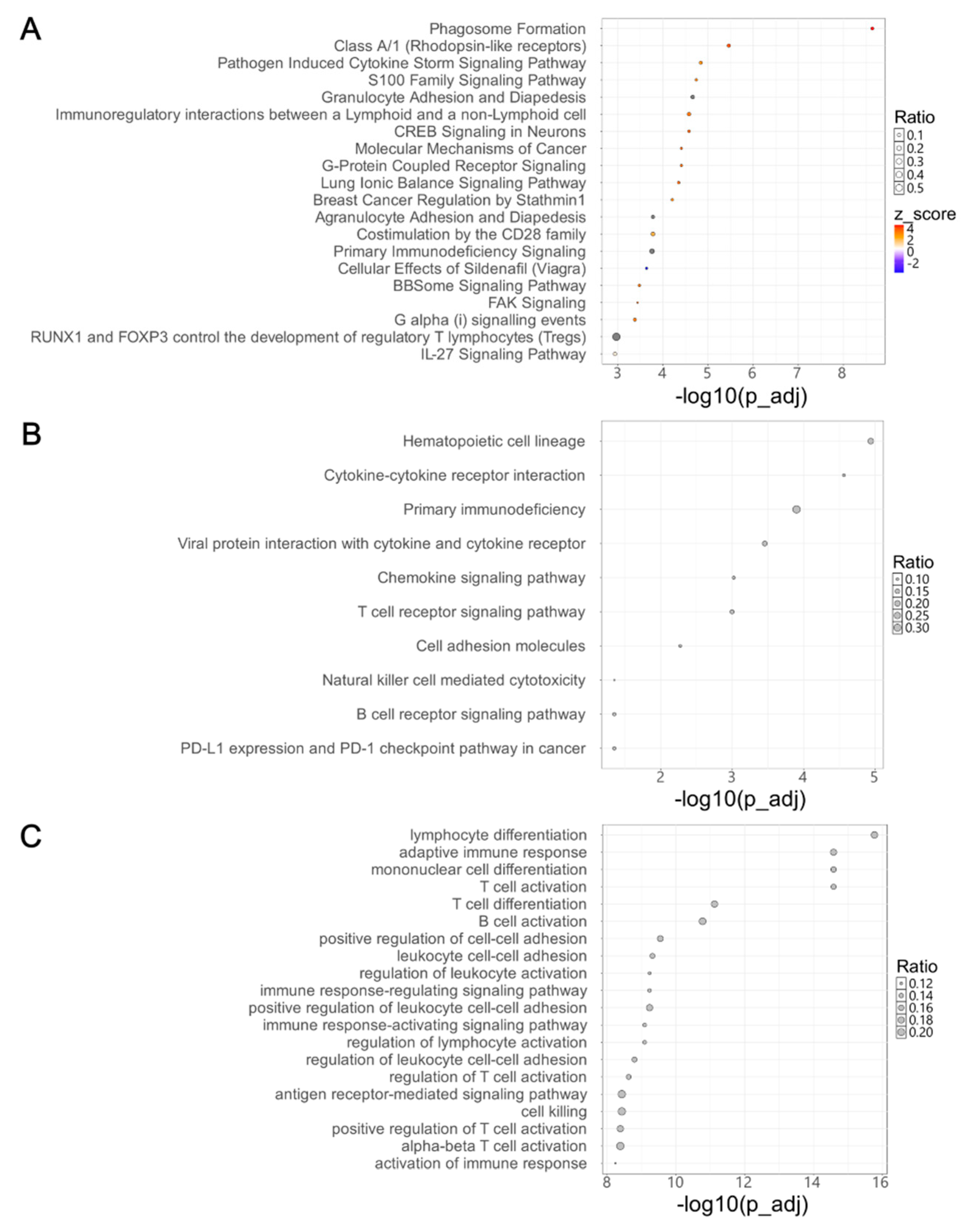
Figure 3.
Differential expression and prognostic significance of candidate differentially expressed genes in primary and recurrent high-grade serous ovarian cancer. (A) Heatmap showing the expression levels of selected differentially expressed genes (DEGs) in primary and recurrent tumor samples. Columns represent individual tumor samples (primary: yellow; recurrent: green), and rows represent DEGs. Color intensity corresponds to expression levels, ranging from blue (low expression) to red (high expression). (B) Kaplan-Meier curves illustrate time to recurrence for patients stratified into high- and low-expression groups for each indicated DEG (GPR68, IL7R, IRF8, PTPRC, WDFY4, C15orf39, NSG1). Statistical significance was assessed using the log-rank test, with p-values shown on each plot.
Figure 3.
Differential expression and prognostic significance of candidate differentially expressed genes in primary and recurrent high-grade serous ovarian cancer. (A) Heatmap showing the expression levels of selected differentially expressed genes (DEGs) in primary and recurrent tumor samples. Columns represent individual tumor samples (primary: yellow; recurrent: green), and rows represent DEGs. Color intensity corresponds to expression levels, ranging from blue (low expression) to red (high expression). (B) Kaplan-Meier curves illustrate time to recurrence for patients stratified into high- and low-expression groups for each indicated DEG (GPR68, IL7R, IRF8, PTPRC, WDFY4, C15orf39, NSG1). Statistical significance was assessed using the log-rank test, with p-values shown on each plot.
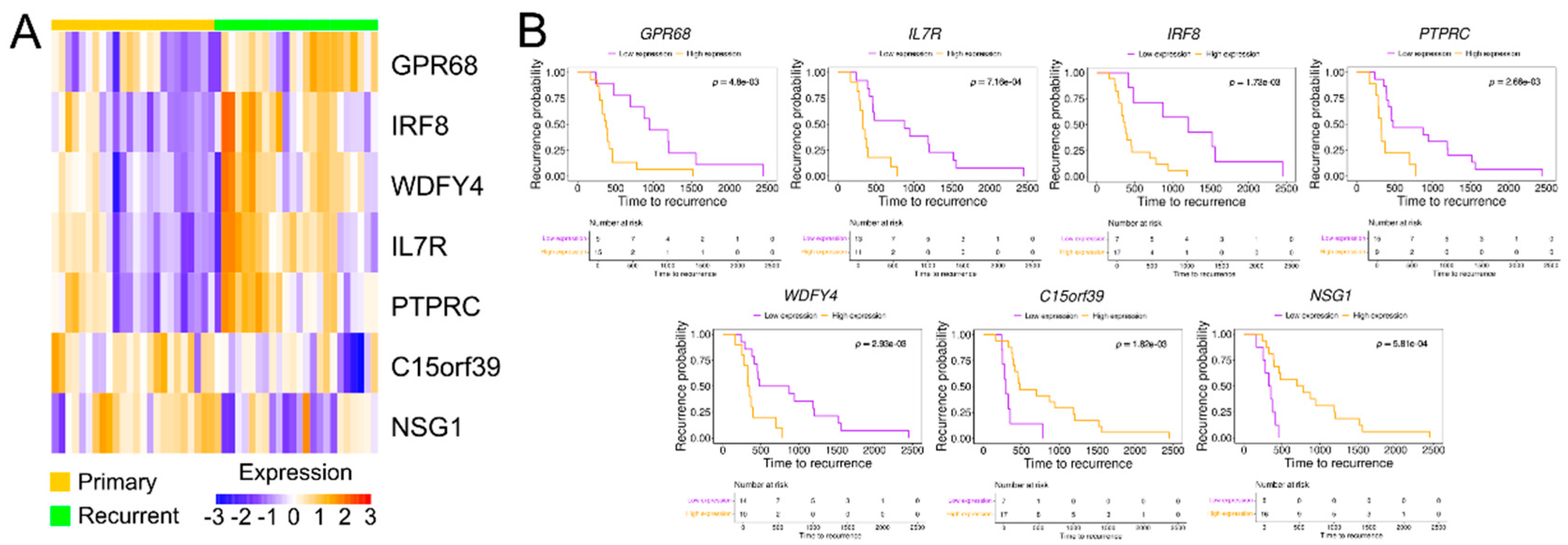
Figure 4.
Differential protein expression of six candidate genes in matched primary and recurrent high-grade serous ovarian cancer tissues by immunohistochemistry. (A) Representative immunohistochemical staining of IL7R, IRF8, GPR68, PTPRC, WDFY4, and NSG1 in tissue microarrays derived from paired primary and recurrent tumor samples. Scale bars represent 50 or 100 µm. Notable differential expression is observed for IL7R, PTPRC, and NSG1 between matched tumor pairs. (B) Paired dot plots depicting immunohistochemical staining scores for each of the six proteins. Statistical significance of expression differences between primary and recurrent tumor samples was assessed using paired t-tests.
Figure 4.
Differential protein expression of six candidate genes in matched primary and recurrent high-grade serous ovarian cancer tissues by immunohistochemistry. (A) Representative immunohistochemical staining of IL7R, IRF8, GPR68, PTPRC, WDFY4, and NSG1 in tissue microarrays derived from paired primary and recurrent tumor samples. Scale bars represent 50 or 100 µm. Notable differential expression is observed for IL7R, PTPRC, and NSG1 between matched tumor pairs. (B) Paired dot plots depicting immunohistochemical staining scores for each of the six proteins. Statistical significance of expression differences between primary and recurrent tumor samples was assessed using paired t-tests.
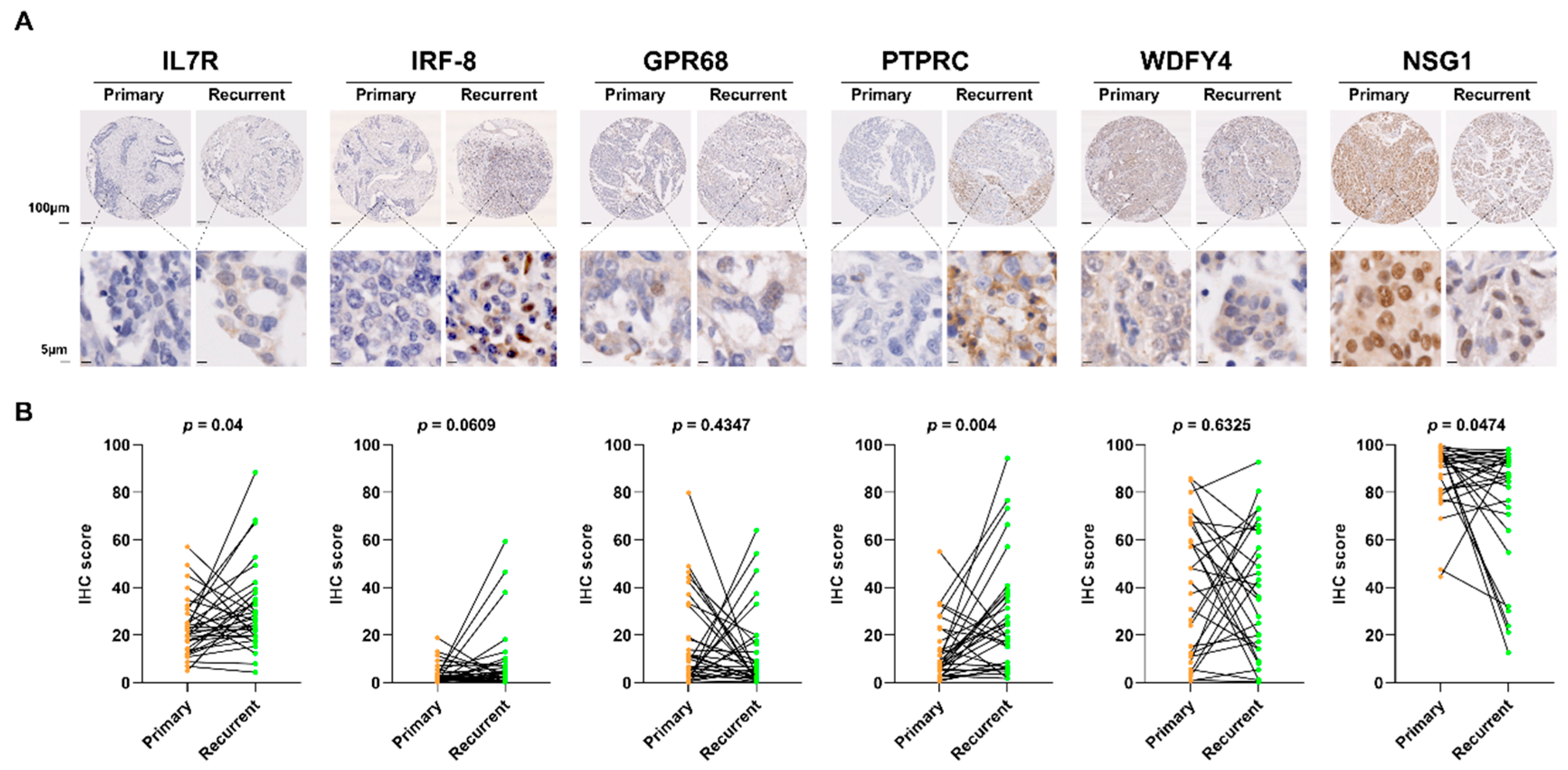
Figure 5.
Co-expression relationships between differentially expressed genes and proteins in primary and recurrent high-grade serous ovarian cancer. (A) Dot plot representing co-expression patterns among four differentially expressed genes (DEGs) in primary and recurrent tumor samples. (B) Dot plot showing co-expression relationships between DEGs and differentially expressed proteins (DEPs) in primary and recurrent tumor samples.
Figure 5.
Co-expression relationships between differentially expressed genes and proteins in primary and recurrent high-grade serous ovarian cancer. (A) Dot plot representing co-expression patterns among four differentially expressed genes (DEGs) in primary and recurrent tumor samples. (B) Dot plot showing co-expression relationships between DEGs and differentially expressed proteins (DEPs) in primary and recurrent tumor samples.
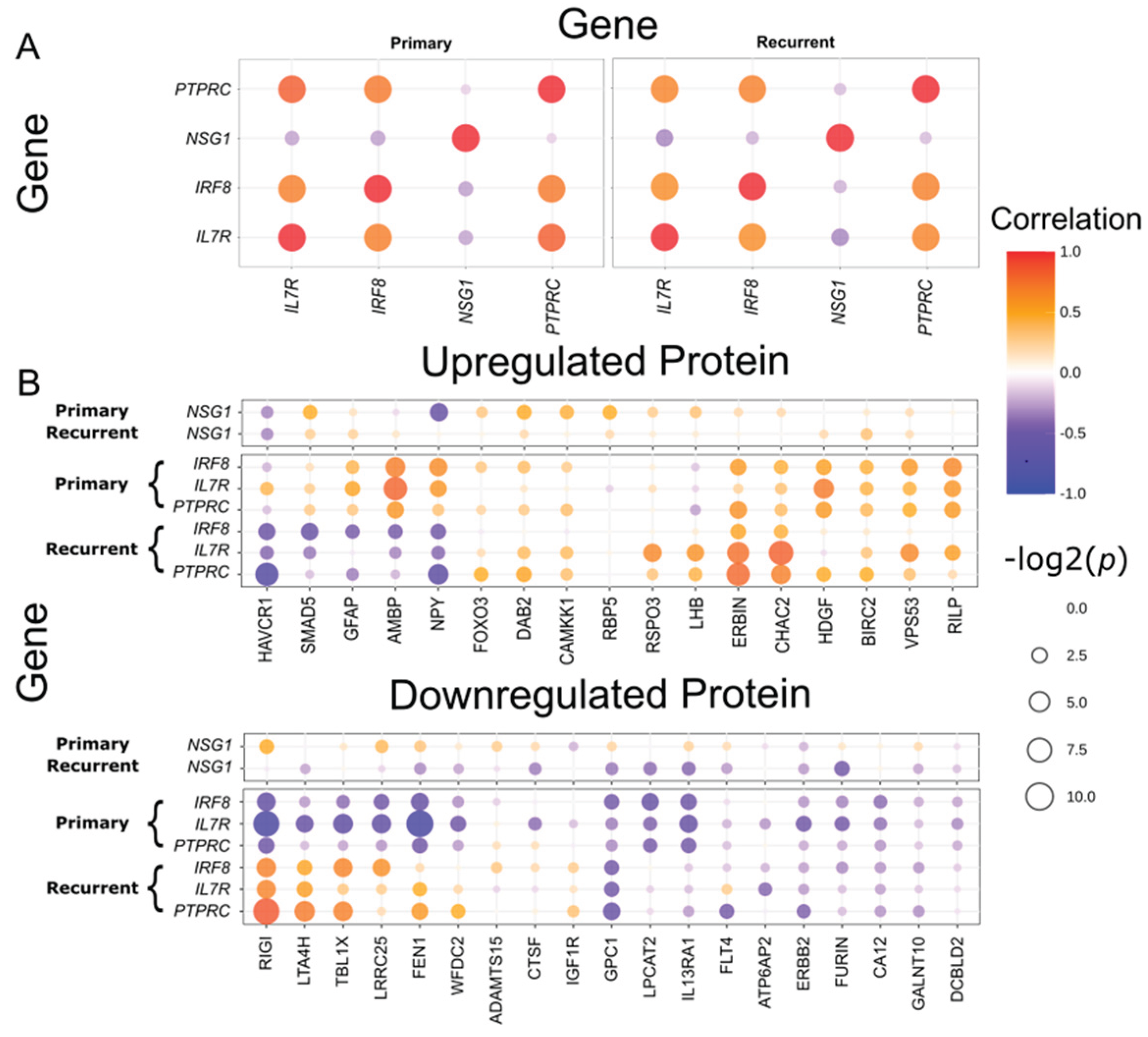

Table 1.
Clinicopathological characteristics of enrolled 34 patients.
| Characteristics | N (Median) | (%) |
|---|---|---|
| All cases | 34 | 100 |
| Age (years) | (53) range 41 - 69 | |
| CA125 at diagnosis (U/mL) | (639.5) range 50.0-20388.1 | |
| FIGO staging | ||
| I, II | 1 | 2.9 |
| III | 28 | 82.4 |
| IV | 5 | 14.7 |
| Histological grade | ||
| G2 | 6 | 17.6 |
| G3 | 26 | 76.5 |
| unknown | 2 | 5.9 |
| BRCA status | ||
| BRCA wild type | 21 | 61.8 |
| BRCA1 mutation | 4 | 11.8 |
| BRCA2 mutation | 2 | 5.9 |
| unknown | 7 | 20.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



