
Submitted:
06 December 2025
Posted:
08 December 2025
You are already at the latest version
Abstract
Mitogen-activated protein kinase (MAPK) signaling cascade is fundamental in regulating cellular proliferation and differentiation, cell survival as well as cell death via apoptosis. Disruption in MAPK signaling cascade at any point can lead to evasion of apoptosis and unchecked cell growth and proliferation leading to oncogenesis. This narrative review describes MAPK pathway dysregulation, their therapeutic targets, and resistance mechanisms. The therapeutic targeting of MAPK pathway is complex owing to dual context dependent roles of several kinases in the signaling cascade. Despite therapeutic effectiveness of MAPK inhibitors, cancers cells develop chemoresistance that needs to be targeted via bypassing several molecular signaling points, pairing of MAPK inhibitors with multiple immune agents and targeting MAPK pathway downstream of ERK to prevent its reactivation mechanisms using combination therapies, downstream signaling regulators and PROTACs. Additionally, MAPK mediated regulation of ferroptosis is a novel oncological therapeutic targeting strategy for controlling tumor progression. The emerging therapies targeting MAPK pathway should be designed considering these crosstalks, compensatory signaling mechanism activation and the impact of tumor microenvironment.
Keywords:
cell survival
; tumor microenvironment
; mitogen-activated protein kinases
; carcinogenesis
; cell proliferation
Introduction
Cancer is a leading cause of morbidity and mortality all around the world, accounting for almost 14.5% of total annual global mortalities and 8.8% of Disability-adjusted life years [1]. The aberrant molecular signaling in cancer cells is one of the essential component in development of cancer [2]. Of all the molecular pathways regulating cell signaling and growth, Mitogen-activated protein kinase (MAPK) signaling cascade is fundamental in regulating cellular proliferation and differentiation, cell survival as well as cell death via apoptosis [3]. Moreover, this cascade is cornerstone signaling pathway for mediating cellular response to stress [4]. Under physiological conditions, MAPK signaling cascade is tightly regulated by certain extracellular stimuli like cytokines, receptor tyrosine kinases and growth factors. These stimuli activates RAS which leads to translocation of Extracellular signal-regulated kinase (ERK), Raf and MAPK/ERK kinase (MEK), hence modulating genetic expression, progression of cell cycle and their downstream effects [5].
Disruption in MAPK signaling cascade at any point can lead to evasion of apoptosis and unchecked cell growth and proliferation leading to oncogenesis [6]. The dysregulation of MAPK pathway and recurrent mutations are observed frequently across a wide-spectrum of malignancies in humans [7]. According to Maik-Rachline, Ras mutations accounts for approximately 30% of mutations in all tumors [8]. In a large genomic study by Sinkala et al., MAPK mutations accounted for almost 58% of mutations in all cancer types, with some of the cancer cells expressing exceptionally higher frequencies ([9]. Literature have identified the occurrence of MAPK signaling dysregulation including thyroid cancers, melanomas, colorectal cancers and several others, which makes it an attractive therapeutic target for treatment of oncological diseases [7]. Targeting MAPK pathway, an essential mediators of cell cycle, results in impaired cell growth and apoptosis of tumor cells [10]. Several treatment strategies for cancer have been developed to target MAPK pathway at various stages of clinical trials with variable results [10].
However, the complexity of MAPK signaling in the form of complicated feedback loops, crosstalks with other signaling pathways and resistance to targeted therapy via activation of bypass signaling pathways might affect the therapeutic success of targeted therapies [11]. Overall, the key role of MAPK pathway in tumorigenesis necessitates the need for studying the role of MAPK as an essential therapeutic target in cancer. This narrative review aimed to describe molecular underpinnings of MAPK pathway dysregulation, identify currently available therapeutic targets, determine resistance mechanisms, and underscore the future directions required for improving patient outcomes.
Overview of the MAPK Pathway in Cancer
MAPK signaling cascade comprises of sequential three to five layers of kinases with variable representation in different type of cells [12]. It includes basic core unit of first three layers available in all types of cells including MAPK kinase kinase kinase (MAP4K), MAPK kinase kinase (MAP3K), MAPK kinase (MAPKK), MAPK and MAPK-activated protein kinases (MAPKAPK) and the last two layers comprising of MAPK and MAPK-activated protein kinases (MAPKAPK) [12]. The MAPK signaling cascade are broadly classified into four categories on the basis of structural layers including ERK1/2, c-Jun N-terminal kinase (JNK), p38 MAPK and ERK5.[12] Extracellular stimulus triggers the activation of MAPKKK by phosphorylation which in turn phosphorylates MAPKKs [13]. Resultantly, the tripeptide motif (Thr-X-Tyr) in the activation loop of MAP kinase is activated, which mediates downstream signaling. In some cells, this three-tier MAPK cascade is supplemented with additional two-tiers of transactivation domain which contains a nucleus localization sequence at the carboxyl terminal [13].
Canonical MAPK/ERK Cascade
Of all the discussed MAPK signaling cascades, MAPK/ERK signaling is most frequently discussed in cancer, owing to its effective role in cell proliferation and differentiation [10]. The phosphorylation cascade begins with RAS, an essential upstream protein in RAF/MEK/ERK pathway. The bindings of ligands with receptor tyrosine kinases (RTKs) results in loading of RAS with GTP and recruitment of adapter proteins like GRB2/SOS.[14] This results in activation of RAF kinases (ARAF, BRAF, CRAF), which leads to phosphorylation and activation of dual-specificity kinases MEK1/2, followed by phosphorylation of ERK1/2 [10]. The phosphorylated ERK1/2 is translocated into the nucleus where several transcription factors are activated including ELK1, c-Fos, and c-Myc. These factors are pivotal in regulating cell cycle, cell metabolism and cell survival [10,15,16]. The specificity of outcomes in whole signaling cascade is largely affected by amplitude of stimulatory signal, the duration of stimulation, type of subcellular localization, and the kind of scaffold proteins activated in pathway [17].
Figure 1.
Canonical MAPK cascade.

Oncogenic Mutations in MAPK/ERK Cascade
The well-controlled MAPK/ERK cascade undergoes unchecked over activation in various cancers owing to activating mutations in components of MAPK cascade which drives constitutive signaling, rendering it as always-on-pathway. This drives uncontrolled proliferation of cells, with angiogenesis, evasion of apoptosis and metastasis [7]. Ras and Raf mutations are encountered most frequently in various cancers with ras mutations accounting for almost 30% of all cancer types [18]. Among Ras mutations, most frequently found oncogenic drive is KRAS mutations observed in almost 97.7% of pancreatic ductal adenocarcinoma, 44.7% of colorectal carcinoma, and 30.9% of lung carcinomas [19]. NRAS mutations are common in melanomas and hematological malignancies whereas HRAS mutations are frequently observed in head and neck squamous cancers (4.7%) as well as urothelial carcinomas (5.9%) [19]. This variation in distribution of various oncogenic mutations in MAPK signaling in different cancers makes it an attractive therapeutic target, yet some of these mutations are challenging due to undruggable structure [20]. The diversity in mutations highlights the need for precision therapeutic approaches tailored according to mutation type and tumor context.

Table 1.
MAPK mutations in various cancer types.
| Cancer type | MAPK mutation |
| Pancreatic ductal adenocarcinoma[19,21] | KRAS , BRAF |
| Colorectal carcinoma [22,23] | KRAS (G12D,G12C,G12S, G13D, Q61R, Q61H, Q61L) NRAS (G13A and Q61H), BRAF (V600) |
| Breast carcinoma [24,25] | KRAS , NRAS, MKP1,MKP2 |
| Lung carcinoma[26,27] | BRAF |
| Biliary carcinoma | MKP1, MKP2, JNK activity, MKK4 |
Dysregulation Beyond Mutations
JNK (c-Jun N-Terminal Kinase) Pathway — A Double-Edged Sword and Therapeutic Opportunities
Apart from MAPK/RAS/RAF/ERK pathway, c-Jun N-terminal kinase (JNK) pathway is another type of MAPK signaling cascade comprising of serine/threonine kinases, which are activated by stress, reactive oxygen species, cytokine storm and genotoxic stress [28]. The resultant phosphorylation of transcription factors like c-Jun, ATF2, p53, c-Myc and apoptotic proteins like BCL-2 and cytoskeletal regulators, hence serving as a mediator of cellular stress response and cell-fate decisions [28]. Unlike MPK/ERK cascade which undergo point mutations, JNK signaling is dysregulated by isoform imbalance, shifting of scaffold proteins and disturbance in upstream stimulus. This renders JNK a functional therapeutic target in various cancers [29].
Dual Roles: Apoptosis Promoter vs Tumor Facilitator
The biology of JNK in cancers is context-dependent, hence playing a dual role in cancers. In some cancers, the activation of JNK promotes apoptosis while in some other conditions, it leads to increased tumor cell proliferation, increased tumor invasion, survival and therapy resistance [30]. The outcomes of JNK activation largely depends upon the amplitude and duration of JNK activation, cross-talk of JNK with other molecular cascades (PI3K and p38/ERK) and relative activity of JNK isoforms (JNK1 vs JNK2 vs JNK3) [30]. This tissue and stimulus dependent effect of JNK enables it as an essential therapeutic target in oncology (Table 2).
p38 MAPK Pathway in Oncogenesis
The p38, also known as stress associated protein kinases (SAPK), are usually activated by genotoxic or environmental stress [38]. There are four types of p38 MAPKs, including p38α, p38β (MAPK11); p38γ (MAPK12); and p38δ (MAPK13) coded by MAPK14, MAPK11, MAPK12 and MAPK13 respectively [39,40]. The activation of p38 signaling occurs by dual phosphorylation of Thr and Tyr on the Thr‒Gly‒Tyr motif, located on kinase subdomain VIII [41]. The activation of p38 MAPK pathway in environmental stress and toxins is mediated through phosphorylation of Threonine 180 and 182 residues via MAP3Ks and MKK3/6 [38,42,43,44,45]. Apart from this, several non-canonical pathways might also results in activation of p38 signaling in T-lymphocytes, and myocytes [46]. This pathway regulates cellular proliferation, differentiation and survival, apoptosis and stress response [40,46].
Dual Role of p38 Signaling Cascade in Oncogenesis
The role of p38 pathway in cancers is complex with literature variable in stating it as a tumor suppressor pathway on one hand and tumor promoter pathway on the other side. p38α ablation results in modulation of Epo expression which increases proliferation of hematopoietic progenitor cells [47]. Similarly, p38α deficient mice were found susceptible to develop lung cancer and liver cancers [48,49]. The downregulation of cell cycle proteins mediated by p38-phosphorylated RB also inhibits cell proliferation [50]. Contrary to this, deletion of p38α gene in breast cancer model resulted in decreased tumor volume [51]. The dual role of p38 signaling in oncogenesis is provided in Table 3.
Oncological Targeting of MAPK Pathway
Many researchers are struggling to develop therapeutic targets of JNK pathway to halt the progress of cancer cell growth. In pancreatic cancer, several JNK inhibitors have been developed till now with variable isoform selectivity. SP600125, a reversible AP-competitive pan JNK-inhibitor, has revealed great effectiveness in treatment of pancreatic cancer [31,59]. SP600125, when combined with radiotherapy, has resulted in decreased resistance of metastatic tetraploid cells by inhibition of JNK pathway [60]. Bentamapimod (AS602801), ATP-competitive inhibitor pan-JNK inhibitor, is effective in reducing cell survival and tumorigenesis in pancreatic cancers, glioblastomas and ovarian cancers [61,62,63]. In endometrial cancers, Bentamapimod induced regression of endometrial lesions by inhibition of JNK signaling which resulted in inhibition of cytokine secretion and inhibiting progesterone resistance [64]. Lichocholane, JNK1-specific inhibitor, inhibits cancer cell survival by competing with JIP1 scaffolding protein in binding with JNK1 [65]. JNK-inhibitor-IX, a JNK2 specific ATP competitive inhibitor, has shown superior effectiveness in halting the progress of PANC-1, most commonly cell line in pancreatic cancers which is resistant to JNK inhibition [66,67]. Several BRAF and MEK inhibitors like Vemurafinib, Dabrafinib, Gefitinib are used in various cancers like melanomas, thyroid cancers and lung cancers and many others [68]. Sorafenib also targets RAF/MEK/ERK pathway to limit the progression of hepatocellular carcinoma[69]. Ulixertinib, an ERK inhibitor, have been used in non-small cell lung carcinoma, colorectal carcinoma and melanoma [70]. It has also shown great effectiveness in pediatric low-grade glioma models when used as monotherapy as well as an adjunct to BH3-mimetics, chemotherapy or MEK inhibitors [71]. The combination of Dabrafenib and trametinib, the BRAF and MEK inhibitors respectively, has shown improved progression free survival and overall survival in several trials of patients with BRAF-mutant solid tumors [72]. Sotorasib, KRASG12C irreversible inhibitor, revealed a significantly greater median progression free survival in patients with advanced non-small cell lung cancer (NSCLC) when compared with docetaxel i.e., 5.6 versus 4.7 months, p<0.001) [73]. Similarly another KRASG12C irreversible inhibitor, Adagrasib, is proven to be effective in treatment of KRASG12C mutated NSCLC in patients refractory to platinum-based chemotherapy and anti-programmed death ligand 1 therapy [74]. Selumitinib, a MEK1/2 inhibitor, has shown clinical and radiological improvement in patients suffering from inoperable symptomatic plexiform neurofibroma with effective safety profile [75]. Tunlametinib, another MEK inhibitor, resulted in significant antitumor activity in patients with inoperable, stage III/4 NRAS-mutant melanomas with prior exposure to immunotherapy [76]. Additionally, several combinational-sequential strategies have been proposed in literature to improve clinical efficacy of MAPK targeting, while overcoming drug resistance [77]. Wang et al. proposed that sequencing only two doses of anti-PDL-1 or anti CTLA-4 agents before initiating MAPK inhibitors in melanoma results in improves their antitumor activity and efficacy of clinical therapy [77].
MAPK Targeting and Drug-Resistances
Despite therapeutic effectiveness of agents targeting MAPK signaling cascades in cancers, it is evident from literature that several resistance mechanisms have been emerged that limits the durability of therapeutic effects of these drugs. Sturm et al. proposed negative feedback amplification as resistance mechanism against MEK inhibitors which keeps ERK signaling intact [78]. Combining MEK inhibitors with RAF inhibitors overcome the negative feedback amplification of MEK hence is now used as standard therapy in malignant melanoma [79,80]. Unfortunately, several other escape mechanisms have also been developed against RAF inhibitors which leads to reactivation of ERK pathways including RAF protein dimerization, RAF amplification and splicing mutations [81]. Additionally activation of alternative pathways have also resulted in resistance against combination therapies of MEK inhibitors with RAF inhibitors [82]. According to one in vitro study, targeting p38 pathway via inhibitors results in increased proliferation of pancreatic cancer cells by activation of JNK pathway [83]. Zhong et al. proposed inhibition of pancreatic cancer cell growth via p38 MAPK activation which inhibits JNK pathway [83]. Ning et al. also proposed the phosphorylation activation of JNK p38 pathway by inhibition of p38 pathway and vice versa [84]. Morphine, on low dose, can promote pancreatic cancer progression by activation of JNK pathway secondary to p38 inhibition, whereas in high doses, it activates p38 leading to suppression of JNK pathway and hence suppression of cancer growth [84]. This highlights the compensatory activation of parallel pathways in cancers may limit the therapeutic success of isolated inhibitors. Additionally feedback reactivation of receptor tyrosine kinases [85], epigenetic modulation [86] and tumor microenvironment [87] also played an important role in mediating therapeutic resistance as shown in Figure 2.
Targeting Drug Resistance in MAPK Tumor Therapy
The clinical effectiveness of MAPK-targeted tumor therapy is limited by several resistance mechanisms identified above that needs to be considered while initiating MAPK targeted antitumor therapy in patients suffering from oncological diseases. Studies from recent literature are basically focused on three mechanistic approaches including combination therapies aimed at bypassing several molecular signaling points, pairing of MAPK inhibitors with multiple immune agents and targeting MAPK pathway downstream of ERK to prevent its reactivation [88,89].
The combination therapy of axitinib with pembrolizumab and avelumab ahs shown an improved overall survival, progression-free survival and response rate in renal cell carcinoma [90,91]. Similarly, in EGFR-mutant metastatic NSLC, the use of erlotinib with ramucirumab, resulted in improved progression free survival in RELAY Phase III trial [92]. In hepatocellular carcinoma, the use of combination immunotherapy with MAPK inhibitors i.e., atezolizumab and bevacizumab with sorafenib, resulted in improved overall survival and progression-free survival when compared to monotherapy [93]. In phase III clinical trial of advanced renal cell carcinoma, CheckMate-9ER, overall response and survival rate was higher in patients who received nivolumab plus cabozantinib in comparison to sunitinib alone [94]. However, not all combination therapies were effective as Kelley et al. observed no significant difference in overall survival rate of melanomas when treated with sorafenib alone or combination therapy with cabozantinib and atezolizumab [95]. This controversy highlights the need for future clinical trials. Nelfinavir, HIV-1 protease inhibitor, is an essential salvage therapy for treatment of non-mutational drug tolerance developed in BRAF and NRAS mutant melanomas during therapy phase owing to PAX-3 mediated increased expression of MITF gene [96]. In ovarian carcinomas, an alpha1-antiagonist, Nafttopidil, has shown greater efficacy in overcoming resistance to MEK inhibitors via activation of JNK signaling pathway and stimulation of BH3-only protein expression [97].
Kinases downstream of ERK, including p90 ribosomal S6 kinase (RSK) involved in transcription and translation mediated by Y-box binding protein 1, also results in improved tumor cell survival despite treatment with chemotherapy [98]. Targeting YB-1 by RSK inhibitors re-sensitises vemurafinib-resistant melanoma cells to BRAF inhibitors [99]. A novel RSK inhibitor, PMD-026, has shown high specificity in triple negative breast carcinoma [100]. In several phase 1 clinical trials of metastatic triple negative breast carcinoma, PMD-026 has shown effective safety profile when compared with inhibitors with MAPK pathway [100,101]. RSK inhibitors are also tested in prostate carcinoma in in-vitro and in-vivo studies [102]. RSK inhibitors are known to improve the response of endogenous and adaptive T cells directed against melanocytic differentiation antigens, hence improving responsiveness of melanoma cells towards immunotherapy [103]. The combination of CDK4/6 inhibitors along with MAPK inhibitors is another therapeutic option to target cancers resistant to MAPK inhibitors alone. This combinatorial therapy works by modulation of cell cycle and altered transcriptional expression of CDK6 as well as AP-1 transcription factors [104]. The combined therapy of RAF inhibitors along with abemaciclib, a CDK4/6 inhibitor, resulted in regression of tumors in KRAS, NRAS and BRAF mutant xenograft model [105]. It results in induction of apoptosis in BRAF inhibitor resistant cells and stimulates cell cycle arrest in non-resistant cells [106]. Similarly, combination therapy comprising of trametinib and palbociclib resulted in NRAS-mutant melanoma models [107]. In neuroblastoma models, combination therapy of binimetinib and ribociclib has resulted in impaired tumor growth [108].
Recent advancement in targeting the resistance to anti-cancer therapy has resulted in development of proteolysis-targeting chimeras (PROTACs). In BRAF-mutant cancers, BRAF-PROTACs were developed by Cullgen as compound 12 and 23 by linking BI882370, a pan-RAF inhibitor, and vemurafinib to CRBN thalidomide ligand [109]. Similarly, SJF-0628, results in degradation of BRAF mutant cancers in a dose-dependent manner within four hours with no ERK phosphorylation for upto 72 hours and only 30% recovery of BRAF activity after washout within 24 hours, depicting the extended catalytic activity of BRAF-PROTAC treatment [110]. P4b, another CRBN recruiting PROTAC developed by dabrafenib and BI882370, resulted in reduced cellular proliferation in vemurafinib-resistant cells [111]. In KRASG12D mutant pancreatic cancer, a CRBN-based PROTAC, ZJK-807, has shown its effectiveness in targeting chemoresistance towards MRTX1133 by uniquely modulating signaling mechanism of TNF and eukaryotic ribosomal biogenesis, hence suppressing the growth of chemoresistant cells [112].
Role of MAPK Targeting with Ferroptosis Regulation in Oncology
Ferroptosis, a form of programmed cell death which occurs due to increased levels of iron and lipid peroxidation of fatty acids in cell membranes, is highly modulated by MAPK signaling cascade [113]. Increased cellular levels of reactive oxygen species induces ferroptosis via MAPK signaling cascade [114]. Dixon et al. also proposed the role of MAPK signaling cascade in modulating the sensitivity of ferroptosis by ERK/MEK activity [113]. Increased activation of erastin-mediated stimulation of ferroptosis have been observed in RAS mutated cancer cell lines [115]. The molecular mechanism through which Ferroptosis is modulated by MAPK signaling includes regulation of iron-ion homeostasis, regulation of lipid and amino acid metabolism as well and altered expression levels of various factors including nuclear factor erythrocyte-related factor 2 (Nrf2) and voltage-dependent anion channels (VDAC) that are present between mitochondria and the cytoplasm [116,117,118]. Riaz et al. also found MAPK1 as an essential ferroptosis-related gene in triple negative breast cancers via its role in cellular proliferation and susceptibility to ferroptosis. Conversely, the inhibition of MAPK pathway results in improved ferroptosis by disrupting cellular signaling especially in cancers with NF2 or RAS mutation [119].The summary of molecular mechanism through which MAPK signaling cascade modulates ferroptosis is provided below in Figure 3:
MAPK Modulation of Ferroptosis- Therapeutic Role in Oncology
The RAS synthetic lethal screen have described the active role of RAS oncogene in modulating ferroptosis via erastin and RSL3 [120,121]. The inhibition of RAF/MAPK pathway have shown to reverse the cytoxicity mediated by erastin or RSL3 in RAS-mutant tumor cells via modulation of iron-metabolism genes expression [118]. Any mutation in epidermal growth factor receptor (EGFR), results in increased ferroptosis sensitivity in mammary epithelial cells and non-small cell lung cancers [122]. The evasion of ferroptosis in RAS-mutant cells have established its vulnerability in lung-cancer [123].
Several studies have described the use of regulating Ferroptosis in cancer cells to limit their growth [116,124,125]. In KRAS mutated colorectal carcionoma, Cetuximab, an anti-EGFR antibody, leads to depletion of Glutathione (GSH) when combined with β-Elemene by inducing iron-dependent accumulationof reactive oxygen species [125]. Similarly, in breast cancer, therapeutic induction of ferroptosis is possible by blocking the transport of iron via using Siramesine alongwith Lapatinib, inhibitor of EGFR and HER2 [126,127]. In adenocarcinoma of lung with EGFR-mutant strain, induction of ferroptosis halt progression of cancer by Vorinostat, a kinase inhibitor which mediates the decreased expression of SLC7A11 [124].
JNK activators have also shown a positive sensitization effect of cancer cells towards ferroptosis. SP600125, a JNK-IN-8 activator, promotes nuclear translocation of RB1CC1 (RB1-inducible coiled-coil 1) and stimulates ferroptosis sensitivity. RB1CC1, an essential factor for development of autophagososme, modulates cellular proliferation via positive regulation of lipid ROS [128,129]. The Ser537 phosphorylation-dependent nuclear translocation of RB1CC1, initiates the transcriptional reprogramming which improves ferroptosis sensitization. The nuclear RB1CC1 increases the amount of histone modifications (H4K12Ac) at ferroptosis-linked enhancers via recruitment of elongator acetyltransferase complex subunit 3 (ELP3) through forkhead box (FOX)-binding motifs, hence stimulating the ferroptosis associated genes like CHCHD3 to improve mitochondrial functioning, as seen in lung cancer models[130]. Despite this positive role of JNK activators in sensitizing cancer cells towards ferroptosis, the dual role of JNK activators described in JNK activators need to be considered while prescribing therapy as it has shown its proinflammatory effects in healthy tissues [131].
Anisomycin, a p38MAPK activator, promotes phosphorylation of H3S10, which activates NCOA4 gene. NCOA4, a ferroptosis stimulator gene, is involved in recruitment of FTH1 to autophagosomes, hence driving its lysosomal degradation. This results in increased iron pool mediated by ferritinophagy, which stimulates cell death in hepatic cancer cells owing to altered iron levels which stimulates lipid peroxidation via Fenton reaction. Simultaneously, anisomycin –negatively regulates the solute carrier family 7 member 11 (SLC7A11) via p38-MAPK pathway via non-GPX4 inactivation [132].
In KRAS-mutant cancers with G12Ci resistant cells, there is downregulation of SOX2, SLC7A11 and SLC40A1. Treatment of KRAS-mutant cancers with G12Ci results in feedback activation of MAK pathway, which modulates ferroptosis in cancer cells, hence proposing MAPK-SOX2 axis as an essential target for modulation of ferroptosis in G12Ci resistant tumors via regulation of intracellular GSH synthesis [133].
Conclusion
ERK, JNK and p38 pathways are essential molecular signaling cascades that regulate the oncogenic transformation of cells throughout the body. Any dysregulation in these signaling cascades as a result of genetic mutations, aberrant kinase activity or disturbed feedback activation results in tumorigenesis and malignancies. Although isolated targeting of each pathway till point of clinical translation have been developed, the therapeutic targeting of MAPK pathway is complex owing to dual context dependent roles of several kinases in the signaling cascade. Despite therapeutic effectiveness of MAPK inhibitors, cancers cells develop chemoresistance that needs to be targeted via bypassing several molecular signaling points, pairing of MAPK inhibitors with multiple immune agents and targeting MAPK pathway downstream of ERK to prevent its reactivation mechanisms using combination therapies, downstream signaling regulators and PROTACs. Additionally, MAPK mediated regulation of ferroptosis is a novel oncological therapeutic targeting strategy for controlling tumor progression. The emerging therapies targeting MAPK pathway should be designed considering these interpathway interactions, compensatory signaling mechanism activation and the impact of tumor microenvironment.
References
- Wu, Z.; Xia, F.; Lin, R. Global burden of cancer and associated risk factors in 204 countries and territories, 1980–2021: a systematic analysis for the GBD 2021. J. Hematol. Oncol. 2024, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gong, L.; Ye, J. The Role of Aberrant Metabolism in Cancer: Insights Into the Interplay Between Cell Metabolic Reprogramming, Metabolic Syndrome, and Cancer. Front. Oncol. 2020, 10, 942. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Guo, Y. MAPK/ERK Signaling in Tumorigenesis: mechanisms of growth, invasion, and angiogenesis. EXCLI J. 2025, 24, 854–79. [Google Scholar]
- Burotto, M.; Chiou, V.L.; Lee, J.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef]
- Sinkala, M.; Nkhoma, P.; Mulder, N.; Martin, D.P. Integrated molecular characterisation of the MAPK pathways in human cancers reveals pharmacologically vulnerable mutations and gene dependencies. Commun. Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct. Target. Ther. 2023, 8, 1–38. [Google Scholar] [CrossRef]
- Li, Y.; Dong, Q.; Cui, Y. Synergistic inhibition of MEK and reciprocal feedback networks for targeted intervention in malignancy. Cancer Biol. Med. 2019, 16, 415–434. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. MAP Kinase Signal. Protoc. 2010, 661, 3–38. [Google Scholar] [CrossRef]
- Ng, G.Y.Q.; Loh, Z.W.-L.; Fann, D.Y.; Mallilankaraman, K.; Arumugam, T.V.; Hande, M.P. Role of Mitogen-Activated Protein (MAP) Kinase Pathways in Metabolic Diseases. Genome Integr. 2024, 15, 20230003. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras activation revisited: role of GEF and GAP systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef]
- Schulze, A.; Nicke, B.; Warne, P.H.; Tomlinson, S.; Downward, J. The Transcriptional Response to Raf Activation Is Almost Completely Dependent on Mitogen-activated Protein Kinase Kinase Activity and Shows a Major Autocrine Component. Mol. Biol. Cell 2004, 15, 3450–3463. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zi, X.; Koontz, Z.; Kim, A.; Xie, J.; Gorlick, R.; Holcombe, R.F.; Hoang, B.H. Blocking Wnt/LRP5 signaling by a soluble receptor modulates the epithelial to mesenchymal transition and suppresses met and metalloproteinases in osteosarcoma Saos-2 cells. J. Orthop. Res. 2007, 25, 964–971. [Google Scholar] [CrossRef]
- Ma, Y.; Nicolet, J. Specificity models in MAPK cascade signaling. FEBS Open Bio 2023, 13, 1177–1192. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef]
- Saiki, Y.; Jiang, C.; Ohmuraya, M.; Furukawa, T. Genetic Mutations of Pancreatic Cancer and Genetically Engineered Mouse Models. Cancers 2021, 14, 71. [Google Scholar] [CrossRef]
- Hassan, S.; Khatoon, A.; Bukhari, U.; Mirza, T. ANALYSIS OF COMMON SOMATIC MUTATIONS IN COLORECTAL CARCINOMA AND ASSOCIATED DYSREGULATED PATHWAYS. J. Ayub Med Coll. Abbottabad 2023, 35, 137–143–137–143. [Google Scholar] [CrossRef]
- Benmokhtar, S.; Laraqui, A.; El Boukhrissi, F.; Hilali, F.; Bajjou, T.; Jafari, M.; El Zaitouni, S.; Baba, W.; El Mchichi, B.; Elannaz, H.; et al. Clinical Significance of Somatic Mutations in RAS/RAF/MAPK Signaling Pathway in Moroccan and North African Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. 2022, 23, 3725–3733. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Gonzalez, C.; Ferrell, M.; Giza, R.; Syed, M.P.; Magge, T.; Bao, R.; Singhi, A.D.; Saeed, A.; Sahin, I.H. Identification of MAPK and mTOR pathway alterations in HER2-amplified colorectal cancer. J. Clin. Oncol. 2024, 42, 185–185. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Cheng, Z.; Malbon, C.C. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett. 2003, 191, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, Z.; Guo, C.; Wang, Y.; Li, B.; Wang, S.; Bai, N.; Chen, H.; Xue, J.; Wang, D.; et al. Super multiple primary lung cancers harbor high-frequency BRAF and low-frequency EGFR mutations in the MAPK pathway. npj Precis. Oncol. 2024, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- bo, Shen H; Li, J; shan, Yao Y; hua, Yang Z; jie, Zhou Y; Chen, W; et al. Impact of Somatic Mutations in Non-Small-Cell Lung Cancer: A Retrospective Study of a Chinese Cohort. Cancer Manag Res. 2020, 12, 7427–37. [Google Scholar]
- Barr, R.K.; Bogoyevitch, M.A. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 2001, 33, 1047–1063. [Google Scholar] [CrossRef]
- Yan, H.; He, L.; Lv, D.; Yang, J.; Yuan, Z. The Role of the Dysregulated JNK Signaling Pathway in the Pathogenesis of Human Diseases and Its Potential Therapeutic Strategies: A Comprehensive Review. Biomolecules 2024, 14, 243. [Google Scholar] [CrossRef]
- Signal integration by JNK and p38 MAPK pathways in... - Google Scholar [Internet]. 11 Oct 2025. Available online: https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Signal+integration+by+JNK+and+p38+MAPK+pathways+in+cancer+development+-+PubMed+%5BInternet%5D.+%5Bcited+2025+Sept+21%5D.+Available+from%3A+https%3A%2F%2Fpubmed.ncbi.nlm.nih.gov%2F19629069%2F&btnG=.
- Tian, X.; Traub, B.; Shi, J.; Huber, N.; Schreiner, S.; Chen, G.; Zhou, S.; Henne-Bruns, D.; Knippschild, U.; Kornmann, M. c-Jun N-terminal kinase 2 suppresses pancreatic cancer growth and invasion and is opposed by c-Jun N-terminal kinase 1. Cancer Gene Ther. 2021, 29, 73–86. [Google Scholar] [CrossRef]
- Cui, C; Zhang, H; Yang, C; Yin, M; Teng, X; Yang, M; et al. Inhibition of JNK Signaling Overcomes Cancer-Associated Fibroblast-Mediated Immunosuppression and Enhances the Efficacy of Immunotherapy in Bladder Cancer. Cancer Res. 2024, 84(24), 4199–213. [Google Scholar] [CrossRef]
- Wu, X.; Chen, X.; Liu, X.; Jin, B.; Zhang, Y.; Wang, Y.; Xu, H.; Wan, X.; Zheng, Y.; Xu, L.; et al. LINC02257 regulates colorectal cancer liver metastases through JNK pathway. Heliyon 2024, 10, e30841. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, B.; Cheng, S.; Fan, H.; Liu, S.; Zhou, B.; Liu, W.; Liang, R.; Tang, Y.; Zhang, Y. KDELR2 knockdown synergizes with temozolomide to induce glioma cell apoptosis through the CHOP and JNK/p38 pathways. Transl. Cancer Res. 2021, 10, 3491–3506. [Google Scholar] [CrossRef]
- Hu, C.; Zuo, K.; Li, K.; Gao, Y.; Chen, M.; Hu, R.; Liu, Y.; Chi, H.; Wang, H.; Qin, Y.; et al. p38/JNK Is Required for the Proliferation and Phenotype Changes of Vascular Smooth Muscle Cells Induced by L3MBTL4 in Essential Hypertension. Int. J. Hypertens. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Yue, W.Y.; Clark, J.J.; Fernando, A.; Domann, F.; Hansen, M.R. Contribution of persistent C-Jun N-terminal kinase activity to the survival of human vestibular schwannoma cells by suppression of accumulation of mitochondrial superoxides. Neuro-Oncology 2011, 13, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Garufi, A.; Trivedi, P.; Frati, L.; D'Orazi, G.; Faggioni, A.; et al. JNK and Macroautophagy Activation by Bortezomib Has a Pro-Survival Effect in Primary Effusion Lymphoma Cells. PLOS ONE 2013, 8, e75965. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- p38 MAP-kinases pathway regulation, function and role in human diseases - PubMed [Internet]. 9 Oct 2025. Available online: https://pubmed.ncbi.nlm.nih.gov/17481747/.
- A, C; Jj, SE. p38γ and p38δ: From Spectators to Key Physiological Players. Trends Biochem Sci [Internet]. 2017 June [cited 2025 Oct 9], 42. Available online: https://pubmed.ncbi.nlm.nih.gov/28473179/.
- Yn D, A C, Gm T, P C, Ar N. Activation of the MAP kinase homologue RK requires the phosphorylation of Thr-180 and Tyr-182 and both residues are phosphorylated in chemically stressed KB cells. FEBS Lett [Internet]. 8 May 1995, 364. Available online: https://pubmed.ncbi.nlm.nih.gov/7750576/.
- Dérijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.-H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent Human MAP-Kinase Signal Transduction Pathways Defined by MEK and MKK Isoforms. Science 1995, 267, 682–685. [Google Scholar] [CrossRef]
- Enslen, H.; Raingeaud, J.; Davis, R.J. Selective Activation of p38 Mitogen-activated Protein (MAP) Kinase Isoforms by the MAP Kinase Kinases MKK3 and MKK6. J. Biol. Chem. 1998, 273, 1741–1748. [Google Scholar] [CrossRef]
- Alonso, G.; Ambrosino, C.; Jones, M.; Nebreda, A.R. Differential Activation of p38 Mitogen-activated Protein Kinase Isoforms Depending on Signal Strength. J. Biol. Chem. 2000, 275, 40641–40648. [Google Scholar] [CrossRef]
- Remy, G.; Risco, A.M.; Iñesta-Vaquera, F.A.; González-Terán, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell. Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Bonney, E.A. Mapping out p38MAPK. Am. J. Reprod. Immunol. 2017, 77. [Google Scholar] [CrossRef]
- Tamura, K; Sudo, T; Senftleben, U; Dadak, AM; Johnson, R; Karin, M. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell 2000, 102(2), 221–31. [Google Scholar] [CrossRef]
- Ventura, J.J.; Tenbaum, S.; Perdiguero, E.; Huth, M.; Guerra, C.; Barbacid, M.; Pasparakis, M.; Nebreda, A.R. p38α MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 2007, 39, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.-Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1α Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Gubern, A.; Joaquin, M.; Marquès, M.; Maseres, P.; Garcia-Garcia, J.; Amat, R.; González-Nuñez, D.; Oliva, B.; Real, F.X.; de Nadal, E.; et al. The N-Terminal Phosphorylation of RB by p38 Bypasses Its Inactivation by CDKs and Prevents Proliferation in Cancer Cells. Mol. Cell 2016, 64, 25–36. [Google Scholar] [CrossRef]
- Wada, M.; Canals, D.; Adada, M.; Coant, N.; Salama, M.F.; Helke, K.L.; Arthur, J.S.; Shroyer, K.R.; Kitatani, K.; Obeid, L.M.; et al. P38 delta MAPK promotes breast cancer progression and lung metastasis by enhancing cell proliferation and cell detachment. Oncogene 2017, 36, 6649–6657. [Google Scholar] [CrossRef]
- Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells - PubMed [Internet]. 9 Oct 2025. Available online: https://pubmed.ncbi.nlm.nih.gov/29805078/.
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.-A.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 Activation in Human Non–Small Cell Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef]
- A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis - PubMed [Internet]. 9 Oct 2025. Available online: https://pubmed.ncbi.nlm.nih.gov/24216180/.
- P38 kinase in gastrointestinal cancers - PMC [Internet]. 9 Oct 2025. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC10501902/.
- Gupta, J.; Del Barco Barrantes, I.; Igea, A.; Sakellariou, S.; Pateras, I.S.; Gorgoulis, V.G.; Nebreda, A.R. Dual Function of p38α MAPK in Colon Cancer: Suppression of Colitis-Associated Tumor Initiation but Requirement for Cancer Cell Survival. Cancer Cell 2014, 25, 484–500. [Google Scholar] [CrossRef]
- Tomás-Loba, A.; Manieri, E.; González-Terán, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M.; Romero-Becerra, R.; Rodríguez, E.; Pintor-Chocano, A.; Feixas, F.; et al. p38γ is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560. [Google Scholar] [CrossRef]
- Kumar, B; Koul, S; Petersen, J; Khandrika, L; Hwa, JS; Meacham, RB; et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res. 2010, 70(2), 832–41. [Google Scholar] [CrossRef]
- Konno, T.; Ninomiya, T.; Kohno, T.; Kikuchi, S.; Sawada, N.; Kojima, T. c-Jun N-terminal kinase inhibitor SP600125 enhances barrier function and elongation of human pancreatic cancer cell line HPAC in a Ca-switch model. Histochem. 2014, 143, 471–479. [Google Scholar] [CrossRef]
- Jemaà, M.; Boubaker, N.S.; Kerkeni, N.; Huber, S.M. JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis. Int. J. Mol. Sci. 2025, 26, 1209. [Google Scholar] [CrossRef]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.-I.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed]
- Seino, M; Okada, M; Shibuya, K; Seino, S; Suzuki, S; Ohta, T; et al. Requirement of JNK signaling for self-renewal and tumor-initiating capacity of ovarian cancer stem cells. Anticancer Res. 2014, 34(9), 4723–31. [Google Scholar] [PubMed]
- Bentamapimod (JNK Inhibitor AS602801) Induces Regression of Endometriotic Lesions in Animal Models | Request PDF. ResearchGate [Internet]. 9 Aug 2025. Available online: https://www.researchgate.net/publication/281515285_Bentamapimod_JNK_Inhibitor_AS602801_Induces_Regression_of_Endometriotic_Lesions_in_Animal_Models.
- Yao, K; Chen, H; Lee, MH; Li, H; Ma, W; Peng, C; et al. Licochalcone A, a natural inhibitor of c-Jun N-terminal kinase 1. Cancer Prev Res Phila Pa 2014, 7(1), 139–49. [Google Scholar] [CrossRef]
- Shi, J.; Yang, X.; Kang, Q.; Lu, J.; Denzinger, M.; Kornmann, M.; Traub, B. JNK inhibitor IX restrains pancreatic cancer through p53 and p21. Front. Oncol. 2022, 12, 1006131. [Google Scholar] [CrossRef]
- JNK pathway inhibition selectively primes pancreatic cancer stem cells to TRAIL-induced apoptosis without affecting the physiology of normal tissue resident stem cells - PubMed [Internet]. 9 Oct 2025. Available online: https://pubmed.ncbi.nlm.nih.gov/26840266/.
- Luke, JJ; Hodi, FS. Ipilimumab, Vemurafenib, Dabrafenib, and Trametinib: Synergistic Competitors in the Clinical Management of BRAF Mutant Malignant Melanoma. The Oncologist 2013, 18(6), 717–25. [Google Scholar] [CrossRef]
- Jing, W.; Shuo, L.; Yingru, X.; Min, M.; Runpeng, Z.; Jun, X.; Dong, H. Artesunate promotes sensitivity to sorafenib in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2019, 519, 41–45. [Google Scholar] [CrossRef]
- Sullivan, RJ; Infante, JR; Janku, F; Wong, DJL; Sosman, JA; Keedy, V; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8(2), 184–95. [Google Scholar] [CrossRef]
- Sigaud, R.; Rösch, L.; Gatzweiler, C.; Benzel, J.; von Soosten, L.; Peterziel, H.; Selt, F.; Najafi, S.; Ayhan, S.; Gerloff, X.F.; et al. The first-in-class ERK inhibitor ulixertinib shows promising activity in mitogen-activated protein kinase (MAPK)-driven pediatric low-grade glioma models. Neuro-Oncology 2022, 25, 566–579. [Google Scholar] [CrossRef]
- Jhanji, V.; Duncan, J.; Gardner, T.; Hughes, G.K.; McIntire, R.; Peña, A.M.; Ladd, C.; Gardner, B.; Moore, T.; Garrett, E.; et al. Assessing Patient Risk, Benefit, and Outcomes in Drug Development: A Decade of Dabrafenib and Trametinib Clinical Trials. Mol. Cancer Ther. 2025, 24, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- de Langen, A.J.; Johnson, M.L.; Mazieres, J.; Dingemans, A.-M.C.; Mountzios, G.; Pless, M.; Wolf, J.; Schuler, M.; Lena, H.; Skoulidis, F.; et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRASG12C mutation: a randomised, open-label, phase 3 trial. Lancet 2023, 401, 733–746. [Google Scholar] [CrossRef]
- Jänne, PA; Riely, GJ; Gadgeel, SM; Heist, RS; Ou, SHI; Pacheco, JM; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N Engl J Med 2022, 387(2), 120–31. [Google Scholar] [CrossRef]
- Santo, V.E.; Passos, J.; Nzwalo, H.; Carvalho, I.; Santos, F.; Martins, C.; Salgado, L.; e Silva, C.; Vinhais, S.; Vilares, M.; et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: a single-institution experience. J. Neuro-Oncology 2020, 147, 459–463. [Google Scholar] [CrossRef]
- Wei, X.; Zou, Z.; Zhang, W.; Fang, M.; Zhang, X.; Luo, Z.; Chen, J.; Huang, G.; Zhang, P.; Cheng, Y.; et al. A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-mutant melanoma. Eur. J. Cancer 2024, 202, 114008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, S.; Yang, Z.; Algazi, A.P.; Lomeli, S.H.; Wang, Y.; Othus, M.; Hong, A.; Wang, X.; Randolph, C.E.; et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell 2021, 39, 1375–1387.e6. [Google Scholar] [CrossRef]
- Sturm, O.E.; Orton, R.; Grindlay, J.; Birtwistle, M.; Vyshemirsky, V.; Gilbert, D.; Calder, M.; Pitt, A.; Kholodenko, B.; Kolch, W. The Mammalian MAPK/ERK Pathway Exhibits Properties of a Negative Feedback Amplifier. Sci. Signal. 2010, 3, ra90–ra90. [Google Scholar] [CrossRef]
- Faghfuri, E.; Nikfar, S.; Niaz, K.; Faramarzi, M.A.; Abdollahi, M. Mitogen-activated protein kinase (MEK) inhibitors to treat melanoma alone or in combination with other kinase inhibitors. Expert Opin. Drug Metab. Toxicol. 2018, 14, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Wahid, M.; Jawed, A.; Mandal, R.K.; Dar, S.A.; Akhter, N.; Somvanshi, P.; Khan, F.; Lohani, M.; Areeshi, M.Y.; Haque, S. Recent developments and obstacles in the treatment of melanoma with BRAF and MEK inhibitors. Crit. Rev. Oncol. 2018, 125, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed]
- Lu, H; Liu, S; Zhang, G; Wu, Bin; Zhu, Y; Frederick, DT; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550(7674), 133–6. [Google Scholar] [CrossRef]
- Zhong, Y; Naito, Y; Cope, L; Naranjo-Suarez, S; Saunders, T; Hong, SM; et al. Functional p38 MAPK identified by biomarker profiling of pancreatic cancer restrains growth through JNK inhibition and correlates with improved survival. Clin Cancer Res Off J Am Assoc Cancer Res 2014, 20(23), 6200–11. [Google Scholar] [CrossRef]
- Ning, J.; Chen, X.; Li, Q.; Yang, D.; Xie, C.; Qin, S.; Jiang, H. Bidirectional effects of morphine on pancreatic cancer progression via the p38/JNK pathway. Sci. Rep. 2024, 14, 1–11. [Google Scholar] [CrossRef]
- Massarweh, S; Osborne, CK; Creighton, CJ; Qin, L; Tsimelzon, A; Huang, S; et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68(3), 826–33. [Google Scholar] [CrossRef]
- Gupta, R.; Bugide, S.; Wang, B.; Green, M.R.; Johnson, D.B.; Wajapeyee, N. Loss of BOP1 confers resistance to BRAF kinase inhibitors in melanoma by activating MAP kinase pathway. Proc. Natl. Acad. Sci. 2019, 116, 4583–4591. [Google Scholar] [CrossRef]
- Xie, H; Li, C; Dang, Q; Chang, LS; Li, L. Infiltrating mast cells increase prostate cancer chemotherapy and radiotherapy resistances via modulation of p38/p53/p21 and ATM signals. Oncotarget 2016, 7(2), 1341–53. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Huang, M.; Lin, Q.; Fang, Q.; Liu, J.; Chen, X.; Liu, L.; Zhan, X.; Shan, H.; et al. The multi-molecular mechanisms of tumor-targeted drug resistance in precision medicine. Biomed. Pharmacother. 2022, 150, 113064. [Google Scholar] [CrossRef]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martinez-Cardus, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed]
- Rini: Pembrolizumab plus axitinib versus sunitinib... - Google Scholar [Internet]. Available from. https://scholar.google.com/scholar_lookup?title=Pembrolizumab%20plus%20axitinib%20versus%20sunitinib%20for%20advanced%20renal-cell%20carcinoma&publication_year=2019&author=B.I.%20Rini&author=E.R.%20Plimack&author=V.%20Stus&author=R.%20Gafanov&author=R.%20Hawkins&author=D.%20Nosov&author=F.%20Pouliot&author=B.%20Alekseev&author=D.%20Soulieres&author=B.%20Melichar&author=I.%20Vynnychenko&author=A.%20Kryzhanivska&author=I.%20Bondarenko&author=S.J.%20Azevedo&author=D.%20Borchiellini&author=C.%20Szczylik&author=M.%20Markus&author=R.S.%20McDermott&author=J.%20Bedke&author=S.%20Tartas&author=Y.H.%20Chang&author=S.%20Tamada&author=Q.%20Shou&author=R.F.%20Perini&author=M.%20Chen&author=M.B.%20Atkins&author=T.%20Powles&author=K.-.%20Investigators, 24 Nov 2025.
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial - The Lancet Oncology [Internet]. 24 Nov 2025. Available online: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(19)30634-5/abstract.
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Cella: Patient-reported outcomes with first-line... - Google Scholar [Internet]. 24 Nov 2025. Available online: https://scholar.google.com/scholar_lookup?title=Patient-reported%20outcomes%20with%20first-line%20nivolumab%20plus%20cabozantinib%20versus%20sunitinib%20in%20patients%20with%20advanced%20renal%20cell%20carcinoma%20treated%20in%20CheckMate%209ER%3A%20an%20open-label%2C%20randomised%2C%20phase%203%20trial&publication_year=2022&author=D.%20Cella&author=R.J.%20Motzer&author=C.%20Suarez&author=S.I.%20Blum&author=F.%20Ejzykowicz&author=M.%20Hamilton&author=J.F.%20Wallace&author=B.%20Simsek&author=J.%20Zhang&author=C.%20Ivanescu&author=A.B.%20Apolo&author=T.K.%20Choueiri.
- Kelley. VP10-2021: cabozantinib (C) plus atezolizumab... - Google Scholar [Internet]. Available from. https://scholar.google.com/scholar_lookup?title=VP10-2021%3A%20Cabozantinib%20%20plus%20atezolizumab%20%20versus%20sorafenib%20%20as%20first-line%20systemic%20treatment%20for%20advanced%20hepatocellular%20carcinoma%20%3A%20Results%20from%20the%20randomized%20phase%20III%20COSMIC-312%20trial&publication_year=2022&author=R.K.%20Kelley&author=T.%20Yau&author=A.L.%20Cheng&author=A.%20Kaseb&author=S.%20Qin&author=A.X.%20Zhu&author=S.%20Chan&author=W.%20Sukeepaisarnjaroen&author=V.%20Breder&author=G.%20Verset&author=E.%20Gane&author=I.%20Borbath&author=J.D.%20Gomez%20Rangel&author=P.%20Merle&author=F.M.%20Benzaghou&author=K.%20Banerjee&author=S.%20Hazra&author=J.%20Fawcett&author=L.%20Rimassa, 24 Nov 2025.
- Smith, MP; Brunton, H; Rowling, EJ; Ferguson, J; Arozarena, I; Miskolczi, Z; et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer Cell 2016, 29(3), 270–84. [Google Scholar] [CrossRef] [PubMed]
- Bim, Puma. Noxa upregulation by Naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor Trametinib - PubMed [Internet]. 24 Nov 2025. Available online: https://pubmed.ncbi.nlm.nih.gov/32424251/.
- Stratford, A.L.; Fry, C.J.; Desilets, C.; Davies, A.H.; Cho, Y.Y.; Li, Y.; Dong, Z.; Berquin, I.M.; Roux, P.P.; E Dunn, S. Y-box binding protein-1 serine 102 is a downstream target of p90 ribosomal S6 kinase in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, 1–12. [Google Scholar] [CrossRef]
- Kosnopfel, C.; Sinnberg, T.; Sauer, B.; Niessner, H.; Schmitt, A.; Makino, E.; Forschner, A.; Hailfinger, S.; Garbe, C.; Schittek, B. Human melanoma cells resistant to MAPK inhibitors can be effectively targeted by inhibition of the p90 ribosomal S6 kinase. Oncotarget 2017, 8, 35761–35775. [Google Scholar] [CrossRef]
- Jayanthan, A.; Yue, L.; Huynh, M.-M.; Los, G.; Dunn, S.E. Abstract 5378: PMD-026, a first in class oral RSK inhibitor, demonstrates activity against hormone receptor positive breast cancer with acquired CDK4/6 inhibitor resistance. Cancer Res. 2022, 82. [Google Scholar] [CrossRef]
- Beeram, M.; Chalasani, P.; Wang, J.S.; Mina, L.A.; Shatsky, R.A.; Trivedi, M.S.; Wesolowski, R.; Hurvitz, S.A.; Han, H.S.; Patnaik, A.; et al. First-in-human phase 1/1b expansion of PMD-026, an oral RSK inhibitor, in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2021, 39, e13043–e13043. [Google Scholar] [CrossRef]
- Ushijima, M; Shiota, M; Matsumoto, T; Kashiwagi, E; Inokuchi, J; Eto, M. An oral first-in-class small molecule RSK inhibitor suppresses AR variants and tumor growth in prostate cancer. Cancer Sci 2022, 113(5), 1731–8. [Google Scholar] [CrossRef]
- Kosnopfel, C.; Wendlinger, S.; Niessner, H.; Siewert, J.; Sinnberg, T.; Hofmann, A.; Wohlfarth, J.; Schrama, D.; Berthold, M.; Siedel, C.; et al. Inhibition of p90 ribosomal S6 kinases disrupts melanoma cell growth and immune evasion. J. Exp. Clin. Cancer Res. 2023, 42, 1–14. [Google Scholar] [CrossRef]
- Scheiblecker, L.; Kollmann, K.; Sexl, V. CDK4/6 and MAPK—Crosstalk as Opportunity for Cancer Treatment. Pharmaceuticals 2020, 13, 418. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Gong, X.; Zhang, Y.; Van Horn, R.D.; Yin, T.; Huber, L.; Burke, T.F.; Manro, J.; Iversen, P.W.; Wu, W.; et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 2017, 37, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V; Burke, TF; Huber, L; Van Horn, RD; Zhang, Y; Buchanan, SG; et al. The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther. 2014, 13(10), 2253–63. [Google Scholar] [CrossRef] [PubMed]
- Kwong, LN; Costello, JC; Liu, H; Jiang, S; Helms, TL; Langsdorf, AE; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012, 18(10), 1503–10. [Google Scholar] [CrossRef]
- Hart, LS; Rader, J; Raman, P; Batra, V; Russell, MR; Tsang, M; et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin Cancer Res. 2017, 23(7), 1785–96. [Google Scholar] [CrossRef]
- Han, X.-R.; Chen, L.; Wei, Y.; Yu, W.; Chen, Y.; Zhang, C.; Jiao, B.; Shi, T.; Sun, L.; Zhang, C.; et al. Discovery of Selective Small Molecule Degraders of BRAF-V600E. J. Med. Chem. 2020, 63, 4069–4080. [Google Scholar] [CrossRef]
- Alabi, S.; Jaime-Figueroa, S.; Yao, Z.; Gao, Y.; Hines, J.; Samarasinghe, K.T.G.; Vogt, L.; Rosen, N.; Crews, C.M. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Posternak, G.; Tang, X.; Maisonneuve, P.; Jin, T.; Lavoie, H.; Daou, S.; Orlicky, S.; de Rugy, T.G.; Caldwell, L.; Chan, K.; et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol. 2020, 16, 1170–1178. [Google Scholar] [CrossRef]
- Liu, Z.; Zheng, H.; Tian, Y.; Li, Z.; Zhang, S.; Zhang, S.; Ma, S.; Wang, X.; Qin, C. ZJK-807: A Selective PROTAC Degrader of KRASG12D Overcoming Resistance in Pancreatic Cancer. J. Med. Chem. 2025, 68, 20103–20129. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Son, Y.; Kim, S.; Chung, H.-T.; Pae.
- Ye, F; Chai, W; Xie, M; Yang, M; Yu, Y; Cao, L; et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am J Cancer Res. 2019, 9(4), 730–9. [Google Scholar]
- Wang, X.; Tan, X.; Zhang, J.; Wu, J.; Shi, H. The emerging roles of MAPK-AMPK in ferroptosis regulatory network. Cell Commun. Signal. 2023, 21, 1–17. [Google Scholar] [CrossRef]
- Ko, W.-C.; Shieh, J.-M.; Wu, W.-B. P38 MAPK and Nrf2 Activation Mediated Naked Gold Nanoparticle Induced Heme Oxygenase-1 Expression in Rat Aortic Vascular Smooth Muscle Cells. Arch. Med Res. 2020, 51, 388–396. [Google Scholar] [CrossRef]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef]
- Riaz, T.; Saleem, M.A.; Khan, M.U.M.; Rashid, M.A.R.; Zubair, M. Ferroptosis as a Therapeutic Avenue in Triple-Negative Breast Cancer: Mechanistic Insights and Prognostic Potential. Biomedicines 2025, 13, 2037. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Poursaitidis, I.; Wang, X.; Crighton, T.; Labuschagne, C.; Mason, D.; Cramer, S.L.; Triplett, K.; Roy, R.; Pardo, O.E.; Seckl, M.J.; et al. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Rep. 2017, 18, 2547–2556. [Google Scholar] [CrossRef]
- Bartolacci, C.; Andreani, C.; Vale, G.; Berto, S.; Melegari, M.; Crouch, A.C.; Baluya, D.L.; Kemble, G.; Hodges, K.; Starrett, J.; et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 2022, 13, 1–19. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, B.; Zhong, C.; Xu, K.; Wang, Z.; Hofman, P.; Nagano, T.; Legras, A.; Breadner, D.; Ricciuti, B.; et al. Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. [CrossRef] [PubMed]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.; Mazière, J.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Wood, ER; Truesdale, AT; McDonald, OB; Yuan, D; Hassell, A; Dickerson, SH; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64(18), 6652–9. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307–e2307. [Google Scholar] [CrossRef]
- Behrends: Network organization of the human autophagy system - Google Scholar [Internet]. 1 Dec 2025. Available online: https://scholar.google.com/scholar_lookup?title=Network%20organization%20of%20the%20human%20autophagy%20system&publication_year=2010&author=B.%20Christian.
- Chano, T.; Ikebuchi, K.; Ochi, Y.; Tameno, H.; Tomita, Y.; Jin, Y.; Inaji, H.; Ishitobi, M.; Teramoto, K.; Nishimura, I.; et al. RB1CC1 Activates RB1 Pathway and Inhibits Proliferation and Cologenic Survival in Human Cancer. PLOS ONE 2010, 5, e11404–e11404. [Google Scholar] [CrossRef]
- Xue: Tumour cells are sensitised to ferroptosis via... - Google Scholar [Internet]. 1 Dec 2025. Available online: https://scholar.google.com/scholar_lookup?title=Tumour%20cells%20are%20sensitised%20to%20ferroptosis%20via%20RB1CC1-mediated%20transcriptional%20reprogramming&publication_year=2022&author=X.%20Xiangfei.
- Dai, Z.; Liu, J.; Zeng, L.; Shi, K.; Peng, X.; Jin, Z.; Zheng, R.; Zeng, C. Targeting ferroptosis in cancer therapy: Mechanisms, strategies, and clinical applications. Cell Investig. 2025, 1. [Google Scholar] [CrossRef]
- Chen, W.; Yang, W.; Zhang, C.; Liu, T.; Zhu, J.; Wang, H.; Li, T.; Jin, A.; Ding, L.; Xian, J.; et al. Modulation of the p38 MAPK Pathway by Anisomycin Promotes Ferroptosis of Hepatocellular Carcinoma through Phosphorylation of H3S10. Oxidative Med. Cell. Longev. 2022, 2022, 6986445. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, X.; Fan, Y.; Zhou, L.; Duan, Y.; Li, S.; Sun, Z.; Zhang, C.; Yang, H.; Yuan, W.; et al. Reactivation of MAPK-SOX2 pathway confers ferroptosis sensitivity in KRASG12C inhibitor resistant tumors. Redox Biol. 2024, 78, 103419. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Drug resistance mechanism in MAPK signaling cascade.
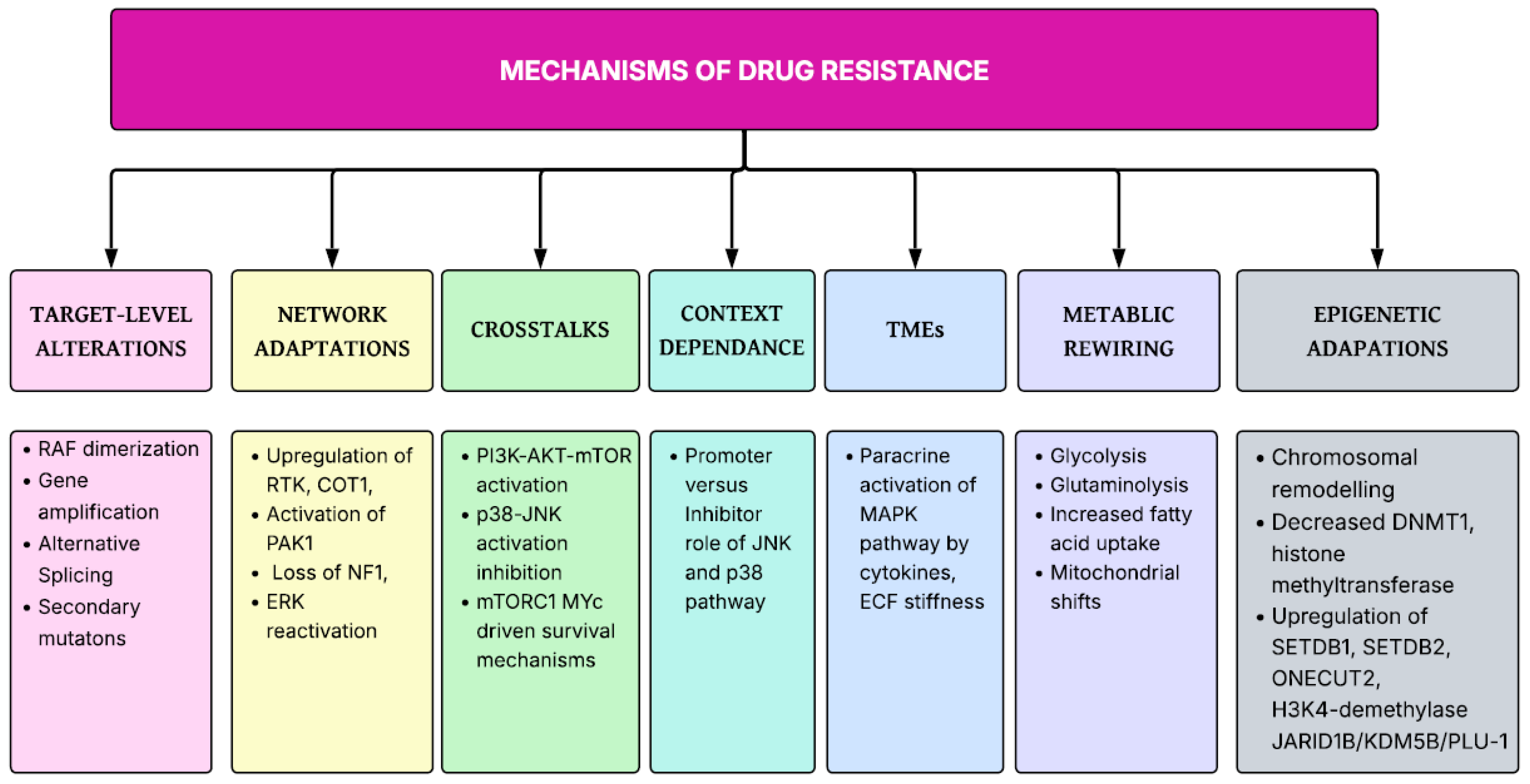
Figure 3.
MAPK modulation of Ferroptosis.
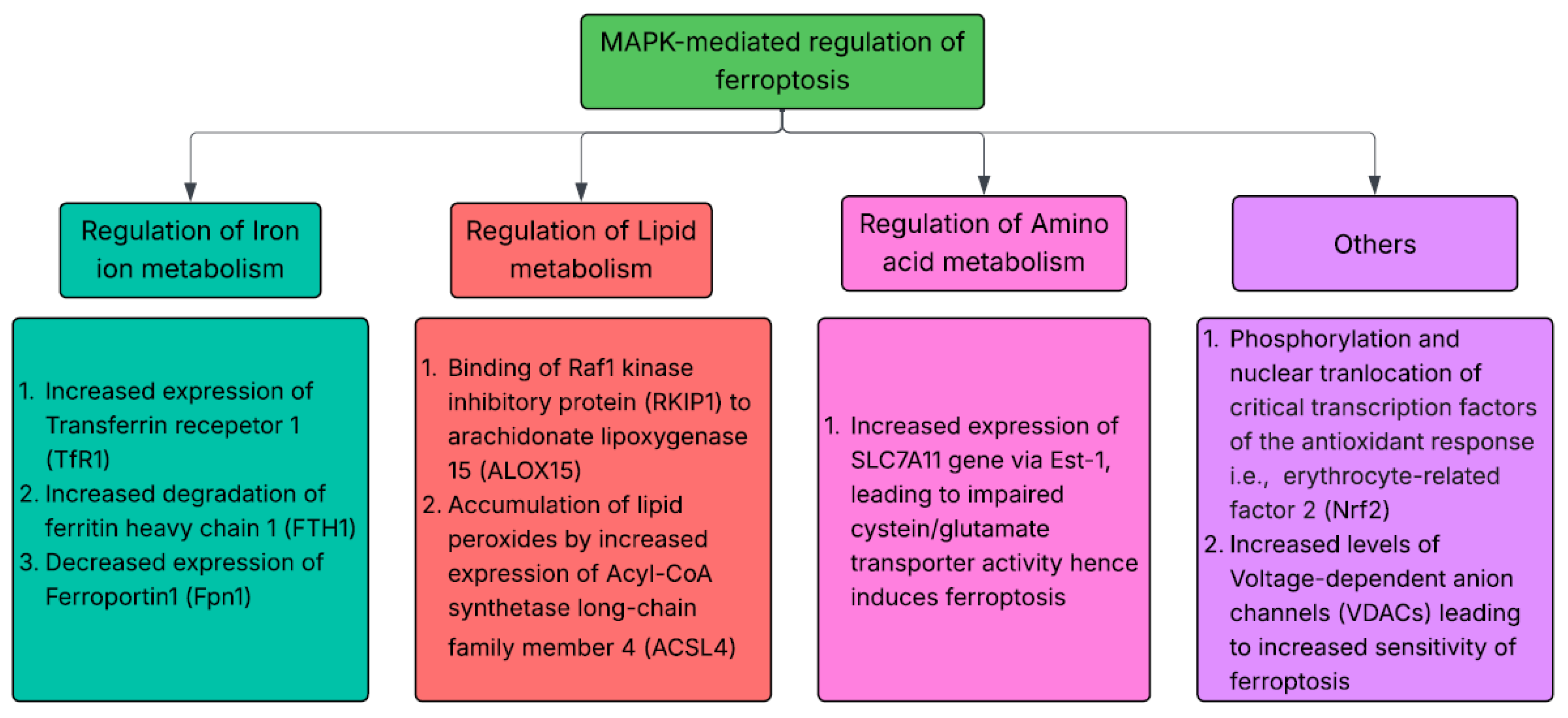
Table 2.
Dual role of JNK cascade in oncogenesis.
| Cancer type | Mechanism and Therapeutic implications | Dual role | Ref |
| Pancreatic cancer | ● JNK2 inhibition increases invasion ● JNK1 inhibition leads to tumor growth suppression Need for Isoform selective therapeutic targeting strategies |
Pro-apoptotic and Pro-tumorigenic | [31] |
| Bladder cancer | ● JNK inhibition decreases cancer-associated fibroblasts mediated expression of thymic stromal lymphopoietin (TSLP) required for creating immunosuppressive microenvironment JNK inhibition with Anti- PD-1 treatment is effective against bladder cancer |
Pro-tumorigenic | [32] |
| Colorectal carcinoma | ● LINC02257/JNK axis leads to colorectal liver metastasis | Pro-tumorigenic | [33] |
| Glioma | ● Pro-apoptotic in glioma cells, pro-proliferative in vascular smooth muscle cells | Pro-apoptotic and Pro-tumorigenic | [34,35] |
| Vestibular Schwannoma | ● Inhibits apoptosis of cancer cells by limiting ROS accumulation | Pro-tumorigenic | [36] |
| Lymphoma | ● Inhibits apoptosis of cancer cells by limiting ROS accumulation Combination therapy of bortezomib with JNK inhibitors required |
Pro-tumorigenic | [37] |
Table 3.
Dual role of p38 signaling cascade in oncogenesis.
| Type of cancer | p38 isoform | Evidence | Reference |
| Breast cancer | p38α | Deletion leads to altered DNA damage response after | [52] |
| p38δ | Deletion leads to decreased tumor volume | [51] | |
| Lung cancers | - | Increased p38 kinase activation | [53] |
| Head and Neck cancers | - | Hyperactivated p38 in tissue samples | [54] |
| Colon cancers | p38γ | Increased expression leading to increased proliferation | [55] |
| p38α | Increased expression leading to increased proliferation | [56] | |
| Liver cancers | p38γ | deletion or inhibition reduces formation of liver tumors induced by chemicals | [57] |
| Bladder carcinoma | p38α | Inhibition leads to reduced invasion of cancer cells by diminishing MMP-2/9 activities | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



