
Submitted:
02 December 2025
Posted:
04 December 2025
You are already at the latest version
Abstract
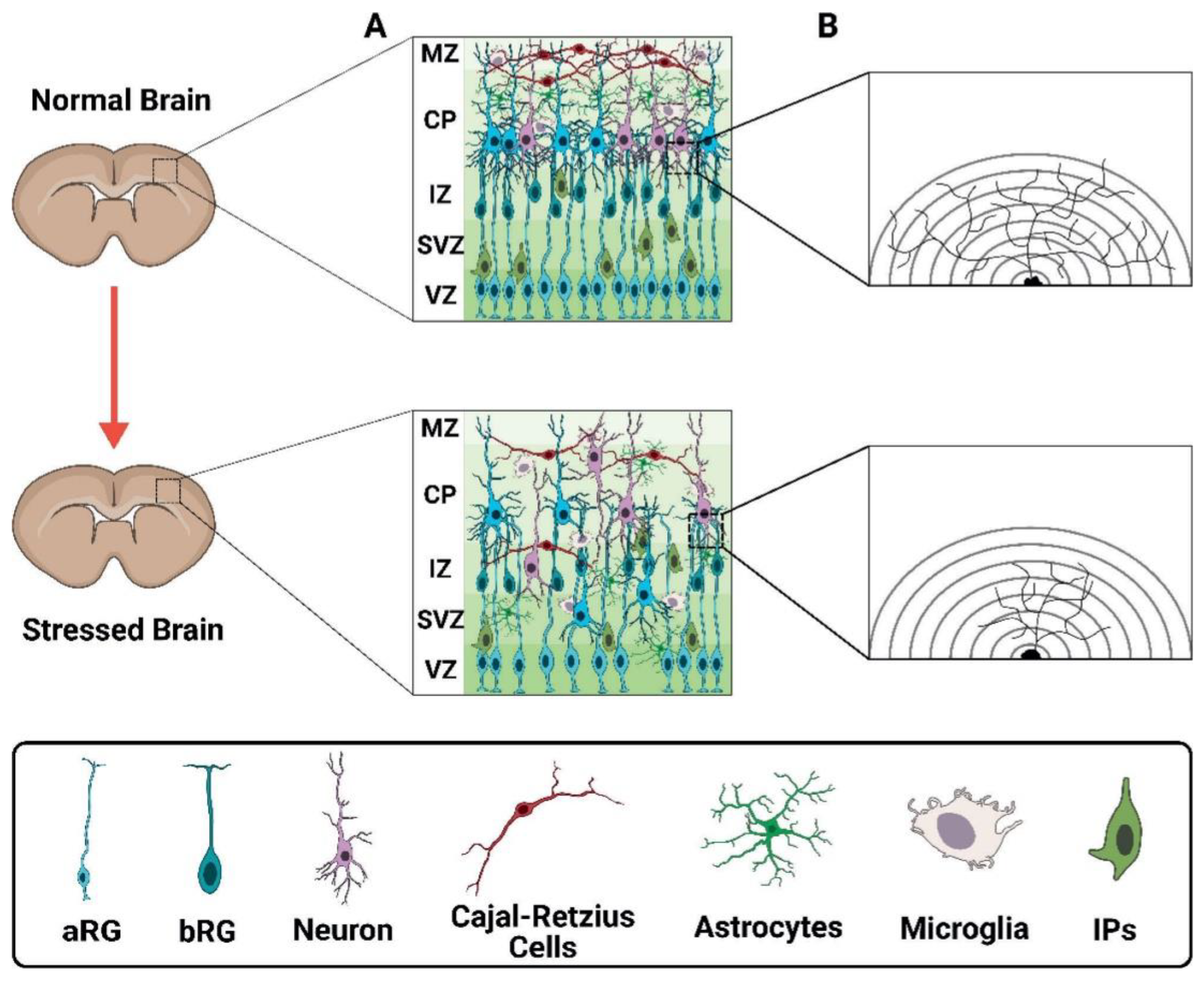
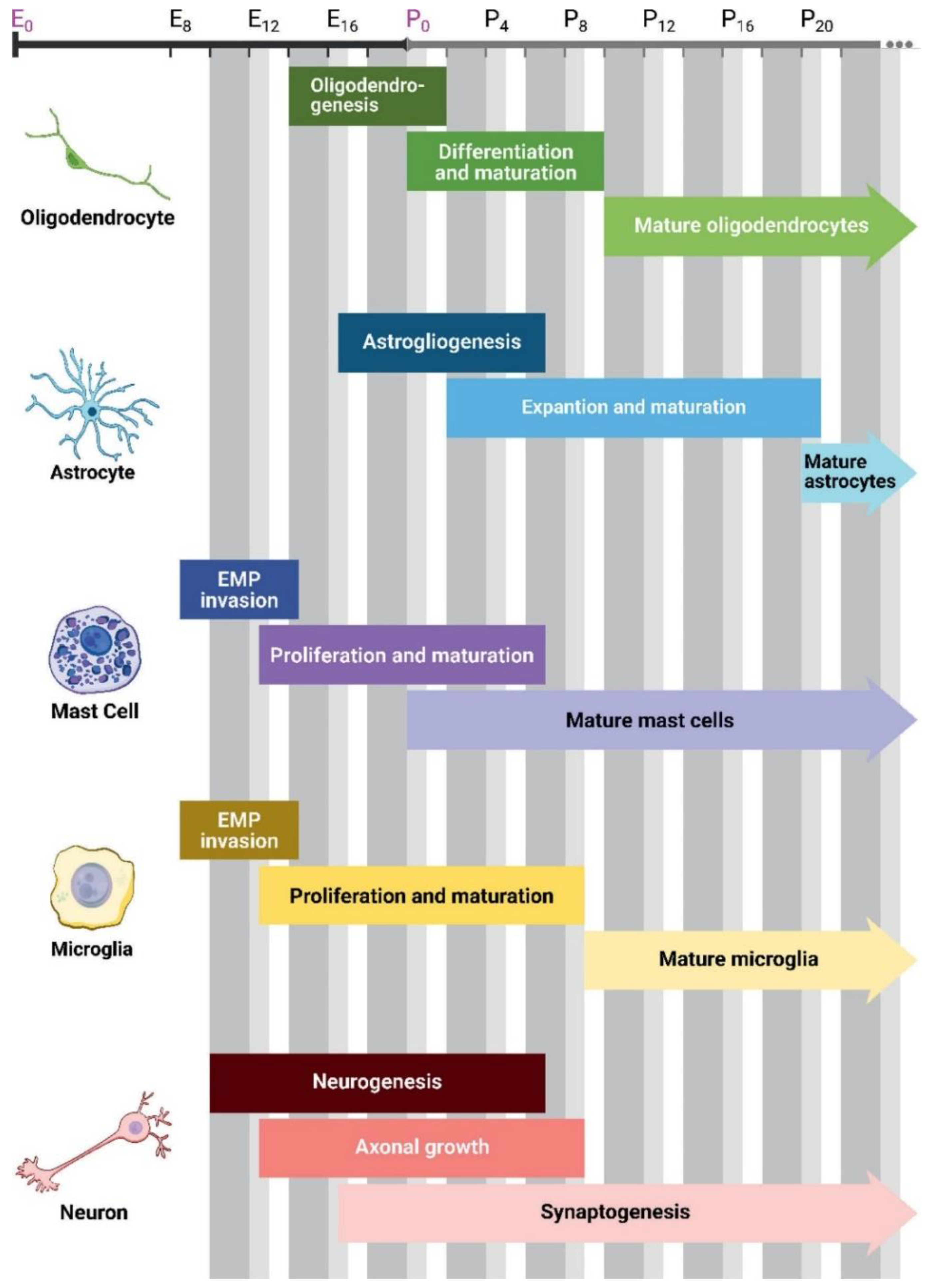
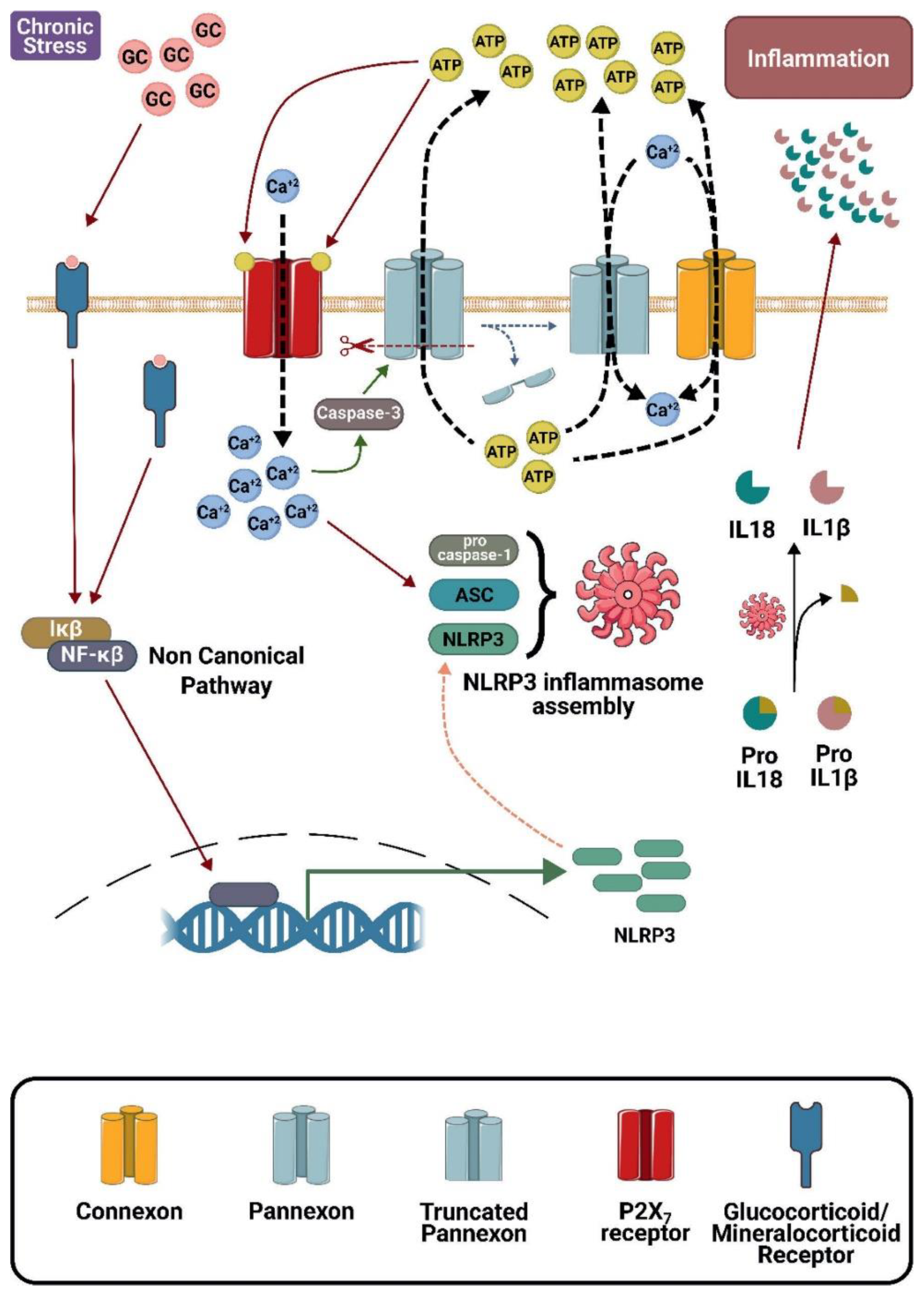
In pathological conditions, elevated activity of connexin and pannexin hemichannels facilitates Adenosine triphosphate (ATP) efflux and Ca2+ influx, activating metabolic pathways of neuroinflammation. While a small insult could result in a protective inflammatory response, more intense and/or prolonged insults induce cell death, causing tissue dysfunction. In the brain, different stressors elevate glucocorticoid (GC) levels that are sensed by mast cells and microglia, and this response persists for a long time, causing continuous inflammasome activation and release of IL-1β and IL-18. These proinflammatory cytokines, together with those released by mast cells, activate astrocytes and oligodendrocytes, which in turn release glutamate and ATP, and altogether reduce neuronal functionality and survival. The extent of neuroinflammation also depends on host features that result in different degrees of alterations during brain ontogeny, consequently changing the brain cytoarchitecture and leading to spectrums of behavioral diseases. Selective hemichannel blockers have been recently discovered and shown to reduce neuroinflammation, as well as neuronal suffering and symptoms linked to adult models of depression and epilepsy. These blockers can serve as tools to dissect the role of neuroinflammation in behavioral diseases. Early treatment during brain ontogeny could reduce detrimental impacts on the brain cytoarchitecture, inducing behavioral alterations elicited in adulthood.
Keywords:
1. Introduction
2. Molecular Aspects of Neuroinflammation and Anti-Inflammatory Agents
3. Stress and Inflammation
4. Mast Cells, Microglia, and Inflammation
5. Connexons, Pannexons, and Neuroinflammation
6. Epigenetic Changes Induced by Stress
7. Concluding Remarks
Acknowledgements
Conflicts of Interest
References
- Robinson R, Lähdepuro A, Tuovinen S, et al. Maternal Hypertensive Pregnancy Disorders and Mental and Behavioral Disorders in the Offspring: a Review. Curr Hypertens Rep 2021;23:30. [CrossRef]
- Kelly SJ, Day N, Streissguth AP. Effects of prenatal alcohol exposure on social behavior in humans and other species. Neurotoxicol Teratol 2000;22:143–9. [CrossRef]
- Cattane N, Richetto J, Cattaneo A. Prenatal exposure to environmental insults and enhanced risk of developing Schizophrenia and Autism Spectrum Disorder: focus on biological pathways and epigenetic mechanisms. Neurosci Biobehav Rev 2020;117:253–78. [CrossRef]
- Shin SH, Kim YK. Early Life Stress, Neuroinflammation, and Psychiatric Illness of Adulthood. Adv Exp Med Biol 2023;1411:105–34. [CrossRef]
- Penisson M, Ladewig J, Belvindrah R, et al. Genes and Mechanisms Involved in the Generation and Amplification of Basal Radial Glial Cells. Front Cell Neurosci 2019;13:1–21. [CrossRef]
- Zhang J, Tabassum S, Jiang J, et al. Optimized Golgi-Cox Staining Validated in the Hippocampus of Spared Nerve Injury Mouse Model. Front Neuroanat 2020;14:1–10. [CrossRef]
- Ryan Kissinger. Macrophage. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/310 (accessed May 11, 2025).
- Ryan Kissinger. Astrocyte. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/40 (accessed May 11, 2025).
- Ryan Kissinger. Immature Organoid Brush. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/251 (accessed May 11, 2025).
- Reemst K, Noctor SC, Lucassen PJ, et al. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front Hum Neurosci 2016;10:1–28. [CrossRef]
- Nishiyama A, Shimizu T, Sherafat A, et al. Life-long oligodendrocyte development and plasticity. Semin Cell Dev Biol 2021;116:25–37. [CrossRef]
- Li X, Liu G, Yang L, et al. Decoding Cortical Glial Cell Development. Neurosci Bull 2021;37:440–60. [CrossRef]
- Vivi E, Di Benedetto B. Brain stars take the lead during critical periods of early postnatal brain development: relevance of astrocytes in health and mental disorders. Mol Psychiatry 2024;29:2821–33. [CrossRef]
- Farhy-Tselnicker I, Allen NJ. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev 2018;13:7–19. [CrossRef]
- Nayak D, Roth TL, McGavern DB. Microglia Development and Function. Annu Rev Immunol 2014;32:367–402. [CrossRef]
- St. John AL, Rathore APS, Ginhoux F. New perspectives on the origins and heterogeneity of mast cells. Nat Rev Immunol 2023;23:55–68. [CrossRef]
- Stagni F, Giacomini A, Guidi S, et al. Timing of therapies for Down syndrome: the sooner, the better. Front Behav Neurosci 2015;9:1–18. [CrossRef]
- John Chilton. Mast Cell. SciDrawIo 2024. [CrossRef]
- Francis He. Oligodendrocyte progenitor cell. SciDrawIo 2023. [CrossRef]
- John Chilton. Astrocyte. SciDrawIo 2020. [CrossRef]
- John Chilton. Microglia reactive. SciDrawIo 2020. [CrossRef]
- John Chilton. Neuron. SciDrawIo 2020. [CrossRef]
- Oronsky B, Caroen S, Reid T. What Exactly Is Inflammation (and What Is It Not?). Int J Mol Sci 2022;23:14905. [CrossRef]
- Rea K, Dinan TG, Cryan JF. The microbiome: A key regulator of stress and neuroinflammation. Neurobiol Stress 2016;4:23–33. [CrossRef]
- Maras PM, Molet J, Chen Y, et al. Preferential loss of dorsal-hippocampus synapses underlies memory impairments provoked by short, multimodal stress. Mol Psychiatry 2014;19:811–22. [CrossRef]
- Chenani A, Weston G, Ulivi AF, et al. Repeated stress exposure leads to structural synaptic instability prior to disorganization of hippocampal coding and impairments in learning. Transl Psychiatry 2022;12:381. [CrossRef]
- Aguirre A, Maturana CJ, Harcha PA, et al. Possible Involvement of TLRs and Hemichannels in Stress-Induced CNS Dysfunction via Mastocytes, and Glia Activation. Mediators Inflamm 2013;2013:1–17. [CrossRef]
- Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol 2017;17:545–58. [CrossRef]
- Ryan Kissinger. Inflammasome. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/255 (accessed May 11, 2025).
- Ryan Kissinger. Receptor. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/429 (accessed May 11, 2025).
- Ryan Kissinger. DNA. NIAID Visual & Medical Arts 2024. https://bioart.niaid.nih.gov/bioart/123 (accessed May 11, 2025).
- Servier Medical Art. Membrane (Yellow-Orange). Servier Medical Art 2025. https://bioicons.com/?query=membrane-2d-yelloworange%20 (accessed May 11, 2025).
- Servier Medical Art. Channel (Yellow). Servier Medical Art 2025. https://smart.servier.com/smart_image/smart-yellow-channel-tubes-overview/ (accessed May 11, 2025).
- Servier Medical Art. Channel (Red). Servier Medical Art 2025. https://smart.servier.com/smart_image/smart-red-channel-overview/ (accessed May 11, 2025).
- Servier Medical Art. Channel (Blue Sky). Servier Medical Art 2025. https://smart.servier.com/smart_image/blue-sky-channels/ (accessed May 11, 2025).
- Madrigal J. The Increase in TNF-α Levels Is Implicated in NF-κB Activation and Inducible Nitric Oxide Synthase Expression in Brain Cortex after Immobilization Stress. Neuropsychopharmacology 2002;26:155–63. [CrossRef]
- Sorrells SF, Caso JR, Munhoz CD, et al. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009;64:33–9. [CrossRef]
- Franchi L, Eigenbrod T, Núñez G. Cutting Edge: TNF-α Mediates Sensitization to ATP and Silica via the NLRP3 Inflammasome in the Absence of Microbial Stimulation. The Journal of Immunology 2009;183:792–6. [CrossRef]
- Petrilli V, Papin S, Tschopp J. The inflammasome. Current Biology 2005;15:R581. [CrossRef]
- Sandhu JK, Kulka M. Decoding mast cell-microglia communication in neurodegenerative diseases. Int J Mol Sci 2021;22:1–22. [CrossRef]
- Maturana CJ, Aguirre A, Sáez JC. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev Neurobiol 2017;77:625–42. [CrossRef]
- Chen R, Yin C, Fang J, et al. The NLRP3 inflammasome: an emerging therapeutic target for chronic pain. J Neuroinflammation 2021;18:84. [CrossRef]
- Grol MW, Zelner I, Dixon SJ. P2X 7-mediated calcium influx triggers a sustained, PI3K-dependent increase in metabolic acid production by osteoblast-like cells. Am J Physiol Endocrinol Metab 2012;302:561–75. [CrossRef]
- Liang X, Samways DSK, Wolf K, et al. Quantifying Ca2+ current and permeability in ATP-gated P2X7 receptors. Journal of Biological Chemistry 2015;290:7930–42. [CrossRef]
- Cotrina ML, Lin JH-C, Alves-Rodrigues A, et al. Connexins regulate calcium signaling by controlling ATP release. Proceedings of the National Academy of Sciences 1998;95:15735–40. [CrossRef]
- Arcuino G, Lin JH-C, Takano T, et al. Intercellular calcium signaling mediated by point-source burst release of ATP. Proceedings of the National Academy of Sciences 2002;99:9840–5. [CrossRef]
- Gustin A, Kirchmeyer M, Koncina E, et al. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS One 2015;10:e0130624. [CrossRef]
- Albalawi F, Lu W, Beckel JM, et al. The P2X7 Receptor Primes IL-1β and the NLRP3 Inflammasome in Astrocytes Exposed to Mechanical Strain. Front Cell Neurosci 2017;11:1–14. [CrossRef]
- Zhang X, Wang R, Hu D, et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv 2020;6:eabb8680. [CrossRef]
- Li D-X, Wang C-N, Wang Y, et al. NLRP3 inflammasome-dependent pyroptosis and apoptosis in hippocampus neurons mediates depressive-like behavior in diabetic mice. Behavioural Brain Research 2020;391:112684. [CrossRef]
- Mychasiuk R, Gibb R, Kolb B. Prenatal stress alters dendritic morphology and synaptic connectivity in the prefrontal cortex and hippocampus of developing offspring. Synapse 2012;66:308–14. [CrossRef]
- Radley JJ, Rocher AB, Miller M, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cerebral Cortex 2006;16:313–20. [CrossRef]
- Liston C, Miller MM, Goldwater DS, et al. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. Journal of Neuroscience 2006;26:7870–4. [CrossRef]
- Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol 2017;17:233–47. [CrossRef]
- Habib KE, Gold PW, Chrousos GP. Neuroendocrinology of stress. Endocrinol Metab Clin North Am 2001;30:695–728. [CrossRef]
- Kemeny ME. The Psychobiology of Stress. Curr Dir Psychol Sci 2003;12:124–9. [CrossRef]
- Chan S, Debono M. Replication of cortisol circadian rhythm: new advances in hydrocortisone replacement therapy. Ther Adv Endocrinol Metab 2010;1:129–38. [CrossRef]
- Dickmeis T. Glucocorticoids and the circadian clock. Journal of Endocrinology 2009;200:3–22. [CrossRef]
- Liddle GW. Analysis of circadian rhythms in human adrenocortical secretory activity. Arch Intern Med 1966;117:739–43.
- Smith LK, Cidlowski JA. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog Brain Res 2010;182:1–30. [CrossRef]
- Adcock IM, Mumby S. Glucocorticoids. Handb Exp Pharmacol, vol. 237, Springer New York LLC; 2016, p. 171–96. [CrossRef]
- Galon J, Franchimont D, Hiroi N, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. The FASEB Journal 2002;16:61–71. [CrossRef]
- Seckl JR. 11β-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol 2004;4:597–602. [CrossRef]
- Mairesse J, Lesage J, Breton C, et al. Maternal stress alters endocrine function of the feto-placental unit in rats. American Journal of Physiology-Endocrinology and Metabolism 2007;292:E1526–33. [CrossRef]
- Jensen Peña C, Monk C, Champagne FA. Epigenetic Effects of Prenatal Stress on 11β-Hydroxysteroid Dehydrogenase-2 in the Placenta and Fetal Brain. PLoS One 2012;7:e39791. [CrossRef]
- Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc 2004;1:239–46. [CrossRef]
- Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun 2007;21:259–72. [CrossRef]
- Căruntu C, Boda D, Musat S, et al. Stress-Induced Mast Cell Activation in Glabrous and Hairy Skin. Mediators Inflamm 2014;2014:1–9. [CrossRef]
- Orellana JA, Moraga-Amaro R, Díaz-Galarce R, et al. Restraint stress increases hemichannel activity in hippocampal glial cells and neurons. Front Cell Neurosci 2015;9:1–12. [CrossRef]
- Noorlander CW, Tijsseling D, Hessel EVS, et al. Antenatal Glucocorticoid Treatment Affects Hippocampal Development in Mice. PLoS One 2014;9:e85671. [CrossRef]
- Esposito P, Chandler N, Kandere K, et al. Corticotropin-Releasing Hormone and Brain Mast Cells Regulate Blood-Brain-Barrier Permeability Induced by Acute Stress. J Pharmacol Exp Ther 2002;303:1061–6. [CrossRef]
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 2005;4:141–94. [CrossRef]
- Ph.D. J. Glucocorticoid Hormones and Early Brain Development in Schizophrenia. Neuropsychopharmacology 2002;27:309–18. [CrossRef]
- Panettieri RA, Schaafsma D, Amrani Y, et al. Non-genomic Effects of Glucocorticoids: An Updated View. Trends Pharmacol Sci 2019;40:38–49. [CrossRef]
- Vianna P, Bauer ME, Dornfeld D, et al. Distress conditions during pregnancy may lead to pre-eclampsia by increasing cortisol levels and altering lymphocyte sensitivity to glucocorticoids. Med Hypotheses 2011;77:188–91. [CrossRef]
- Kline SA, Mega MS. Stress-Induced Neurodegeneration: The Potential for Coping as Neuroprotective Therapy. Am J Alzheimers Dis Other Demen 2020;35:1–13. [CrossRef]
- Kay VR, Rätsep MT, Figueiró-Filho EA, et al. Preeclampsia may influence offspring neuroanatomy and cognitive function: A role for placental growth factor†. Biol Reprod 2019;101:271–83. [CrossRef]
- Rose’Meyer R. A review of the serotonin transporter and prenatal cortisol in the development of autism spectrum disorders. Mol Autism 2013;4:37. [CrossRef]
- Welberg LAM, Seckl JR. Prenatal Stress, Glucocorticoids and the Programming of the Brain. J Neuroendocrinol 2001;13:113–28. [CrossRef]
- Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav 2011;59:279–89. [CrossRef]
- Oberlander TF, Weinberg J, Papsdorf M, et al. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008;3:97–106. [CrossRef]
- Noorlander CW, Visser GHA, Ramakers GMJ, et al. Prenatal corticosteroid exposure affects hippocampal plasticity and reduces lifespan. Dev Neurobiol 2008;68:237–46. [CrossRef]
- Vázquez DM, Neal CR, Patel PD, et al. Regulation of corticoid and serotonin receptor brain system following early life exposure of glucocorticoids: Long term implications for the neurobiology of mood. Psychoneuroendocrinology 2012;37:421–37. [CrossRef]
- Głombik K, Kukla-Bartoszek M, Curzytek K, et al. The Effects of Prenatal Dexamethasone Exposure on Brain Metabolic Homeostasis in Adulthood: Implications for Depression. Int J Mol Sci 2023;24:1156. [CrossRef]
- Hiroi R, Carbone DL, Zuloaga DG, et al. Sex-dependent programming effects of prenatal glucocorticoid treatment on the developing serotonin system and stress-related behaviors in adulthood. Neuroscience 2016;320:43–56. [CrossRef]
- Huang C-C, Lin H-R, Liang Y-C, et al. Effects of Neonatal Corticosteroid Treatment on Hippocampal Synaptic Function. Pediatr Res 2007;62:267–70. [CrossRef]
- Huang W. Effect of corticosteroids on brain growth in fetal sheep. Obstetrics & Gynecology 1999;94:213–8. [CrossRef]
- Huang WL, Harper CG, Evans SF, et al. Repeated prenatal corticosteroid administration delays astrocyte and capillary tight junction maturation in fetal sheep. International Journal of Developmental Neuroscience 2001;19:487–93. [CrossRef]
- Uno H, Lohmiller L, Thieme C, et al. Brain damage induced by prenatal exposure to dexamethasone in fetal rhesus macaques. I. Hippocampus. Developmental Brain Research 1990;53:157–67. [CrossRef]
- Tauber SC, Schlumbohm C, Schilg L, et al. Intrauterine Exposure to Dexamethasone Impairs Proliferation But Not Neuronal Differentiation in the Dentate Gyrus of Newborn Common Marmoset Monkeys. Brain Pathology 2006;16:209–17. [CrossRef]
- Kolb B, Gibb R. Plasticity in the prefrontal cortex of adult rats. Front Cell Neurosci 2015;9:1–11. [CrossRef]
- Arnsten AFT. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 2009;10:410–22. [CrossRef]
- Tauchi M, Zhang R, D’Alessio DA, et al. Role of central glucagon-like peptide-1 in hypothalamo-pituitary-adrenocortical facilitation following chronic stress. Exp Neurol 2008;210:458–66. [CrossRef]
- Frank MG, Hershman SA, Weber MD, et al. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014;40:191–200. [CrossRef]
- Bharti V, Tan H, Zhou H, et al. Txnip mediates glucocorticoid-activated NLRP3 inflammatory signaling in mouse microglia. Neurochem Int 2019;131:104564. [CrossRef]
- Yue N, Huang H, Zhu X, et al. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J Neuroinflammation 2017;14:102. [CrossRef]
- Zhang B, Zhang Y, Xu T, et al. Chronic dexamethasone treatment results in hippocampal neurons injury due to activate NLRP1 inflammasome in vitro. Int Immunopharmacol 2017;49:222–30. [CrossRef]
- Cory-Slechta DA, Sobolewski M, Varma G, et al. Developmental lead and/or prenatal stress exposures followed by different types of behavioral experience result in the divergence of brain epigenetic profiles in a sex, brain region, and time-dependent manner: Implications for neurotoxicology. Curr Opin Toxicol 2017;6:60–70. [CrossRef]
- Zhu X, Wu Y, Pan J, et al. Neuroinflammation Induction and Alteration of Hippocampal Neurogenesis in Mice following Developmental Exposure to Gossypol. International Journal of Neuropsychopharmacology 2021;24:419–33. [CrossRef]
- Harcha PA, Garcés P, Arredondo C, et al. Mast cell and astrocyte hemichannels and their role in alzheimer’s disease, als, and harmful stress conditions. Int J Mol Sci 2021;22:1–19. [CrossRef]
- Dong H, Zhang X, Qian Y. Mast cells and neuroinflammation. Med Sci Monit Basic Res 2014;20:200–6. [CrossRef]
- Grootens J, Ungerstedt JS, Nilsson G, et al. Deciphering the differentiation trajectory from hematopoietic stem cells to mast cells. Blood Adv 2018;2:2273–81. [CrossRef]
- Galli SJ, Kalesnikoff J, Grimbaldeston MA, et al. Mast cells as “tunable” effector and immunoregulatory cells: Recent advances. Annu Rev Immunol 2005;23:749–86. [CrossRef]
- Wang L, Sikora J, Hu L, et al. ATP Release from Mast Cells by Physical Stimulation: A Putative Early Step in Activation of Acupuncture Points. Evidence-Based Complementary and Alternative Medicine 2013;2013:1–7. [CrossRef]
- Huang X, Lan Z, Hu Z. Role and mechanisms of mast cells in brain disorders. Front Immunol 2024;15:1445867. [CrossRef]
- McGrath KE, Frame JM, Palis J. Early hematopoiesis and macrophage development. Semin Immunol 2015;27:379–87. [CrossRef]
- Bolós M, Perea JR, Avila J. Alzheimer’s disease as an inflammatory disease. Biomol Concepts 2017;8:37–43. [CrossRef]
- Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022;110:3458–83. [CrossRef]
- Frick LR, Williams K, Pittenger C. Microglial Dysregulation in Psychiatric Disease. Clin Dev Immunol 2013;2013:1–10. [CrossRef]
- Duarte Y, Quintana-Donoso D, Moraga-Amaro R, et al. The role of astrocytes in depression, its prevention, and treatment by targeting astroglial gliotransmitter release. Proceedings of the National Academy of Sciences 2024;121:1–12. [CrossRef]
- Sáez JC, Brañes MC, Corvalán LA, et al. Gap junctions in cells of the immune system: structure, regulation and possible functional roles. Brazilian Journal of Medical and Biological Research 2000;33:447–55. [CrossRef]
- Schalper KA, Sánchez HA, Lee SC, et al. Connexin 43 hemichannels mediate the Ca 2+ influx induced by extracellular alkalinization. American Journal of Physiology-Cell Physiology 2010;299:C1504–15. [CrossRef]
- Abudara V, Retamal MA, Del Rio R, et al. Synaptic Functions of Hemichannels and Pannexons: A Double-Edged Sword. Front Mol Neurosci 2018;11:1–24. [CrossRef]
- Adamson SE, Leitinger N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett, vol. 588, Elsevier; 2014, p. 1416–22. [CrossRef]
- Yi WJ, Kim TS. Melatonin protects mice against stress-induced inflammation through enhancement of M2 macrophage polarization. Int Immunopharmacol 2017;48:146–58. [CrossRef]
- Salgado M, Márquez-Miranda V, Ferrada L, et al. Ca 2+ permeation through C-terminal cleaved, but not full-length human Pannexin1 hemichannels, mediates cell death. Proceedings of the National Academy of Sciences 2024;121:1–10. [CrossRef]
- Palacios-Prado N, Soto PA, López X, et al. Endogenous pannexin1 channels form functional intercellular cell–cell channels with characteristic voltage-dependent properties. Proceedings of the National Academy of Sciences 2022;119:1–11. [CrossRef]
- Ślusarczyk J, Trojan E, Głombik K, et al. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neurosci 2015;9:1–14. [CrossRef]
- Wang Y, Su Y, Yu G, et al. Reduced Oligodendrocyte Precursor Cell Impairs Astrocytic Development in Early Life Stress. Advanced Science 2021;8:2101181. [CrossRef]
- Çalışkan G, Müller A, Albrecht A. Long-term impact of early-life stress on hippocampal plasticity: Spotlight on astrocytes. Int J Mol Sci 2020;21:1–19. [CrossRef]
- Bravo-Tobar ID, Fernández P, Sáez JC, et al. Long-term effects of stress resilience: Hippocampal neuroinflammation and behavioral approach in male rats. J Neurosci Res 2021;99:2493–510. [CrossRef]
- Yang Y, Xing M-J, Li Y, et al. Reduced NLRP3 inflammasome expression in the brain is associated with stress resilience. Psychoneuroendocrinology 2021;128:105211. [CrossRef]
- Siebert AP, Ma Z, Grevet JD, et al. Structural and functional similarities of calcium homeostasis modulator 1 (CALHM1) ion channel with connexins, pannexins, and innexins. Journal of Biological Chemistry 2013;288:6140–53. [CrossRef]
- Hernández-Salinas R, Vielma AZ, Arismendi MN, et al. Boldine Prevents Renal Alterations in Diabetic Rats. J Diabetes Res 2013;2013:1–12. [CrossRef]
- Toro CA, Johnson K, Hansen J, et al. Boldine modulates glial transcription and functional recovery in a murine model of contusion spinal cord injury. Front Cell Neurosci 2023;17:1–16. [CrossRef]
- Li H, Guo A, Salgado M, et al. The connexin hemichannel inhibitor D4 produces rapid antidepressant-like effects in mice. J Neuroinflammation 2023;20:191. [CrossRef]
- Cisterna BA, Vargas AA, Puebla C, et al. Active acetylcholine receptors prevent the atrophy of skeletal muscles and favor reinnervation. Nat Commun 2020;11:1073. [CrossRef]
- Guo A, Zhang H, Li H, et al. Inhibition of connexin hemichannels alleviates neuroinflammation and hyperexcitability in temporal lobe epilepsy. Proceedings of the National Academy of Sciences 2022;119:1–12. [CrossRef]
- Walrave L, Pierre A, Albertini G, et al. Inhibition of astroglial connexin43 hemichannels with TAT-Gap19 exerts anticonvulsant effects in rodents. Glia 2018;66:1788–804. [CrossRef]
- Chen B, Yang L, Chen J, et al. Inhibition of Connexin43 hemichannels with Gap19 protects cerebral ischemia/reperfusion injury via the JAK2/STAT3 pathway in mice. Brain Res Bull 2019;146:124–35. [CrossRef]
- Chen Y, Wang L, Zhang L, et al. Inhibition of Connexin 43 Hemichannels Alleviates Cerebral Ischemia/Reperfusion Injury via the TLR4 Signaling Pathway. Front Cell Neurosci 2018;12:1–15. [CrossRef]
- Dierks A, Vanucci-Bacqué C, Schäfer A-M, et al. The Bioactive Phenolic Agents Diaryl Ether CVB2-61 and Diarylheptanoid CVB4-57 as Connexin Hemichannel Blockers. Pharmaceuticals 2022;15:1173. [CrossRef]
- Yang HW, Kho AR, Lee SH, et al. A phosphodiesterase 4 (PDE4) inhibitor, amlexanox, reduces neuroinflammation and neuronal death after pilocarpine-induced seizure. Neurotherapeutics 2024;21:e00357. [CrossRef]
- Agarwal S, Vyas P, Nirwan N, et al. Effect of lacosamide on neuroinflammation-mediated seizures comorbid with depression in C57BL/6 mice– Role of kynurenine pathway. Epilepsy & Behavior 2021;123:108262. [CrossRef]
- Mat Nor MN, Rupenthal ID, Green CR, et al. Connexin Hemichannel Block Using Orally Delivered Tonabersat Improves Outcomes in Animal Models of Retinal Disease. Neurotherapeutics 2020;17:371–87. [CrossRef]
- Kim Y, Griffin JM, Nor MNM, et al. Tonabersat Prevents Inflammatory Damage in the Central Nervous System by Blocking Connexin43 Hemichannels. Neurotherapeutics 2017;14:1148–65. [CrossRef]
- Lyon H, Shome A, Rupenthal ID, et al. Tonabersat inhibits connexin43 hemichannel opening and inflammasome activation in an in vitro retinal epithelial cell model of diabetic retinopathy. Int J Mol Sci 2021;22:1–12. [CrossRef]
- NIH National Library of Medicine. Tonabersat Clinical Trials. ClinicalTrialsGov 2025. https://www.clinicaltrials.gov/search?intr=Tonabersat (accessed May 13, 2025).
- Yi C, Ezan P, Fernández P, et al. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017;65:1607–25. [CrossRef]
- Cipriani A, Zhou X, Del Giovane C, et al. Comparative efficacy and tolerability of antidepressants for major depressive disorder in children and adolescents: a network meta-analysis. The Lancet 2016;388:881–90. [CrossRef]
- Toth M. Epigenetic Neuropharmacology: Drugs Affecting the Epigenome in the Brain. Annual Review of Pharmacology and Toxicology Annu Rev Pharmacol Toxicol 2021 2025;25:6.
- Trerotola M, Relli V, Simeone P, et al. Epigenetic inheritance and the missing heritability. Hum Genomics 2015;9:17. [CrossRef]
- Yang R, Gautam A, Getnet D, et al. Epigenetic biotypes of post-traumatic stress disorder in war-zone exposed veteran and active duty males. Mol Psychiatry 2021;26:4300–14. [CrossRef]
- Yuan M, Yang B, Rothschild G, et al. Epigenetic regulation in major depression and other stress-related disorders: molecular mechanisms, clinical relevance and therapeutic potential. Signal Transduct Target Ther 2023;8:309. [CrossRef]
- Schiele MA, Domschke K. Epigenetics at the crossroads between genes, environment and resilience in anxiety disorders. Genes Brain Behav 2018;17:1–15. [CrossRef]
- Mychasiuk R, Muhammad A, Kolb B. Chronic stress induces persistent changes in global DNA methylation and gene expression in the medial prefrontal cortex, orbitofrontal cortex, and hippocampus. Neuroscience 2016;322:489–99. [CrossRef]
- Perisic T, Zimmermann N, Kirmeier T, et al. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 2010;35:792–805. [CrossRef]
- Zimmermann N, Zschocke J, Perisic T, et al. Antidepressants inhibit DNA methyltransferase 1 through reducing G9a levels. Biochemical Journal 2012;448:93–102. [CrossRef]
- Rapanelli M, Williams JB, Ma K, et al. Targeting histone demethylase LSD1 for treatment of deficits in autism mouse models. Molecular Psychiatry 2022 27:8 2022;27:3355–66. [CrossRef]
- Ookubo M, Kanai H, Aoki H, et al. Antidepressants and mood stabilizers effects on histone deacetylase expression in C57BL/6 mice: Brain region specific changes. J Psychiatr Res 2013;47:1204–14. [CrossRef]
- Belujon P, Grace AA. Dopamine system dysregulation in major depressive disorders. International Journal of Neuropsychopharmacology 2017;20:1036–46. [CrossRef]
- Su Y, Li H, Zhang W, et al. Connexin43 hemichannel blockade turns microglia neuroprotective and mitigates cognitive deficits in a mouse model of amyloidosis. Nat Commun 2025;16:5621. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



