Submitted:
01 December 2025
Posted:
03 December 2025
You are already at the latest version
Abstract
Distribution water-extractable particulate (colloid) organic matter (WEOM) in narrow (nano- to micrometer) size fractions of chernozem soil by sequential filtration on track-etched membranes were studied. Multimodal (IR and fluorescence) two-dimensional correlation (2D-COS) spectroscopy was used. Protocols for attenuated total reflectance (ATR) FTIR of WEOM are proposed. ATR-FTIR 2D-COS provides larger volume of information on characteristic bands compared to traditional FTIR, especially in C–H ranges (3000–2800 and 1450–1300 cm–1). Fluorescence excitation-emission-matrix 2D-COS showed that the indexes and ratios of humic- to protein-like compounds are reproducible, exhibit significant variation among size fractions, with maximum amounts of saturated humic-like compounds in the largest (2–10 μm) and finest factions (0.01–0.03 μm) while medium fractions (0.05–1 μm) are dominated by fulvic acids and fresh organic matter. Heterospectral fluorescence–IR 2D-COS enhanced the accuracy of identification and assessment of WEOM group composition and showed that C–H IR band intensities correlate with tyrosine-like EEM bands and biogenic fluorescence in-dexes, while carboxylic components, with humate-like bands and humification fluo-rescence indexes. Element profiles in WEOM fractions correlate with fluorescence in-dexes; humification indexes, with P, S, Cr, Mg, Ca, Cu, and Zn; biogenic, with Mg, P, Cr, Cd, K, S, and Ca.
Keywords:
two-dimensional correlation spectroscopy
; IR spectroscopy
; fluorescence spectroscopy
; dissolved organic matter
; water-extractable particulate soil organic matter
; soil colloids
; chernozem
; size fractionation
; ultrafiltration
; track-etched membranes
1. Introduction
Dissolved organic matter (DOM) is found in varying amounts in all types of natural waters, including soil solutions, surface and groundwater of various types, water from swamps, rivers, lakes, seas, and oceans [1]. DOM is mostly made up of organic compounds of various, often unknown composition, due to complexity, especially of humic nature, which are assessed as specific compounds, the result of the transformation of plant substances in the process of soil formation. Such compounds make up to 50% of the organic matter of natural waters [2]. The roles of DOM in nature are diverse: the substances that make it up are not only a source of energy and nutrients for soil microflora and terrestrial plants, but also carry out the transport of substances of various nature within the soil profile, determine the redistribution of matter in the landscape, and participate in the cycles of carbon and nitrogen at the global, planetary level [3,4].
In soils, DOM composition and properties are influenced by the nature and intensity of agricultural cultivations, which change the composition of soil organic matter (SOM) as a whole and the soil structure [5,6]. A change in DOM composition indicates a change in SOM as a whole [7]; the material composition of DOM and the ratio of its constituent chemical phases are specific for different sources and may serve an indicator of the origin of organic matter of naturally existing substances [3] as well as anthropogenically altered areas, e.g., storm sewage [8]. DOM as a carrier of exogenous chemicals affects their mobility, ability to transform, thus changing their bioavailability or toxicity [1,9].
The criteria by which DOM is determined and distinguished are now mainly abridged to separation from the initial solution, whether natural water or an aqueous extract from a soil sample, fractions with a particle size of less than 0.45 μm. It is obvious that this fraction is quite heterogeneous and includes particles not only of different nature (including inorganic), but also of various sizes. During the movement of the water flows, the dimensional heterogeneity of DOM determines the gradient of concentrations of certain compounds that are part of it, which in turn leads to the redistribution of matter both in the soil profile or landscape, and in the catchment areas of water bodies, river estuaries and coastal waters of seas and oceans [4,10]. At the same time, changes in the composition of soil organic matter during active agricultural use also affect the material, and hence fractional, composition of DOM.
In addition, SOM is often divided into free, existing as aggregated solid organic substrates, and as organic matter, bound to the mineral matrix into organo-clay particles, including colloidal sizes (less than 2 μm or even less than 1 μm) [11]. The latter part of SOM is referred to as soil colloids or water-extractable particulate soil organic matter (WEOM) [12]. Studies of two size fractions of 53–2000 μm and less than 53 μm of aggregated particulate organic matter and non-aggregated organo-clay WEOM after chemical dispersion and obtained data on the differences in their material composition associated with biogeochemical features of soil formation [13]. However, there are no more detailed studies of SOM based on a separate analysis of the size fractions isolated from the colloidal, the most mobile component of the soil phase [11].
In this regard, to be able to predict the spatial redistribution of matter, it is important to consider the features of the composition of smaller fractions that make up DOM, to develop methods for isolating and analyzing these fractions. As shown previously [14], track-etched membranes can be used to isolate and separate WEOM.
The rapid development of membrane separation and purification technologies has led to the new generation of membranes obtained by high-energy ion beam bombardment (track-etched membranes), which are characterized by straight through channels with the same width and narrow distribution by size (deviations from the nominal amount are units of percent) like on sieves [15,16]. However, there are also certain drawbacks, primarily related to the effects of the interaction of the surface charge and the charges of the separated particles, which also affect the separation and particles with a smaller size do not pass through the channels due to the occurrence of electrostatic repulsion. Nevertheless, this effect can be neutralized by selecting separation conditions (e.g., by recharging the surface of membranes or particles with neutral salt buffers) [17,18]. The choice of filter material is also important. Membranes made of organic and inorganic materials are now available. Among the organic materials, polycarbonate and PET seem to be optimum as they are transparent and colorless in the UV/vis range, thus providing direct absorption-spectra and optical microscopic measurements directly on membranes without sample removal. The absence of own fluorescence of these materials makes it possible to carry out spectrofluorometric measurements on membranes as well.
Among the methods of WEOM measurements, fluorescence spectrometry is currently most often used for its qualitative composition [19,20,21,22,23,24,25,26,27]. This method provides high sensitivity and allows for non-destructive analysis [26]. Various variants of spectrofluorimetry are used, e.g., gathering emission spectra at a fixed excitation wavelength or synchronous fluorescence spectra [27]. However, over the last 25 years, one of the most used variants of fluorescence spectrometry for DOM analysis is fluorescence excitation-emission matrix (EEM or total synchronous fluorescence) techniques [28,29]. Usually, EEM is accompanied by their analysis using various chemometric methods of multivariate calibration; one of these generally accepted models is parallel factor analysis (PARAFAC) [30]. This method separates a given set of EEM into the data on fluorescing components of different types that correspond to certain groups of constituent compounds [31].
IR spectroscopy also has a special position in studying the molecular composition of soils, and especially the structural and group composition of SOM [32,33,34,35,36,37,38,39,40,41,42]. On the one hand, it is possible to obtain structural-group information about the entire molecular composition of the soil, including SOM [32,34,35,36,38,39,40,41,42,43,44,45,46,47]. On the other hand, IR spectroscopy also works with solutions obtained after various types of extraction, which makes it possible to compare these data with NMR spectroscopy or mass spectrometry data [48]. FTIR shows the possibilities of studying differences in narrow soil fractions [49,50].
Elemental analysis is one of the most common methods for analyzing fractions of natural organic matter [51,52]. ICP-AES analysis combined with direct injection of suspensions developed for nanomaterials [53] provides the information on the bulk contents of elements to determine the total mineral composition of the studied samples without preliminary decomposition (in the case of membrane washouts) [14,37,54]. Recently, combined analysis of complex samples using both molecular and atomic analytical techniques (multimodal spectroscopy) have shown the possibility to reveal a large volume of new information on such samples including soils [55,56,57]. Here, the main approach is to use correlation analysis [58], including more complex techniques like generalized two-dimensional correlation spectroscopy (2D-COS).
The technique of 2D-COS is an enhancement on traditional spectroscopic methods, which turns one-dimensional spectra into two-dimensional maps (matrices) to identify correlations between individual bands in the spectrum [59,60]. The mathematical basis of 2D-COS makes it possible to find the relationship between individual bands in a single spectrum (homospectral 2D-COS) or in two spectra: in different regions for the same test object or for the spectra of the same object by different methods (both are heterospectral 2D-COS). Apart from revealing the correlations between spectral features due to various mechanisms of interaction, 2D-COS simplifies complex spectra with many overlapping bands and may increase spectral resolution by separating overlapping bands along the second axis of the correlation map. The spectral sets in 2D-COS are built by affecting the test sample by various factors, which are called external perturbations (sample-changing factors). Usually, they are heating [61,62,63,64,65,66], pH [67] or selective chemical reactions [68,69]. However, any physical or chemical fractionation, separating the sample into characteristic size fractions, is also a very efficient external perturbation for 2D-COS [49]. In fluorescence spectroscopy, 2D-COS is used for studying fluorescence quenching factors and changes, mainly conformational, in biomolecules [70]. Fluorescence 2D-COS can resolve overlapping spectral information and reveal the predominant types of soil organic matter [71] and aids in finding changes in humic substances due to pollutant binding and environmental factors [72].
In IR spectroscopy, 2D-COS aids in identifying overtones and Raman bands, as well correlations between the bands of inorganic and organic constituents [73,74,75,76]. 2D-COS is used in IR spectroscopy to study polymers or living objects [77,78]. In soil analysis, IR 2D-COS expands the volume of data on SOM for similar samples without additional sample preparation. In particular, , IR 2D-COS reveals the bands of functional groups on soil particle surface, as well as both aromatic and aliphatic components of SOM, including in the long-wavelength region (1000 cm–1 and below), with predominant mineral component contribution [79,80] and can aid in identifying the land use type [49]. Heterospectral 2D COS of soils 2D-COS based on FTIR and fluorescence techniques is not widespread and is used for obtaining increased information volume of whole SOM structure, interactions, and spatial distribution especially for metal binding or biofilms [81,82]. There is almost no data on the use of multimodal 2D-COS on soil particles, especially narrow and fine fractions of WEOM.
Thus, the aim of this study was to demonstrate the possibilities of the multimodal spectroscopy of fluorescence and FTIR, along with atomic-emission spectroscopies combined with two-dimensional correlation analysis on narrow soil WEOM fractions of nano- to micrometer size range and to characterize the distribution patterns of organic matter and microelements in the wet-sieved narrow fractions of chernozem soil.
2. Materials and Methods
2.1. Samples and Chemicals
Samples of Kursk chernozem (Haplic chernozem) were taken from the territory Federal State Budgetary Institution “Kursk Federal Agrarian Scientific Center” were used to prepare water-extractable particulate organic matter [49,83].
All aqueous solutions were prepared on deionized water (Type I, 18.2 MΩ cm at 25 °C; a Milli-Q Academic system, Merck Millipore, Darmstadt, Germany). Sodium azide to preserve the samples was from Molekula Ltd. (Darlington, UK (purity > 99.5%). To acidify, samples were treated with nitric acid (69%, PA-ACS-ISO grade) from Panreac (Barcelona, Spain).
2.2. Fractionation
At the first stage, soil samples were sieved using a Retsch AS 200 sieving machine (Haan, Germany) equipped with sieves with diameters of 40, 20, 10, and 5 μm (Precision Eforming LLC, Cortland, USA). After the first round (an amplitude, 0.85 mm; time, 5 min), the whole set of sieves was cleansed with 50 mL of deionized water, and the sample was sieved another round under the same conditions.
The details for the second stage of sequential membrane fractionation are given elsewhere [14]. Sets of track-etched membranes made of polycarbonate (GVS Filter Technology, Bologna, Italy; pore size, 5.0, 2.0, 1.0, 0.8, 0.4, 0.2, 0.1, 0.05, 0.03, and 0.01 µm) and made of PET (REATRAK-Filter, Obninsk, Russia; 5.0, 4.0, 3.0, 2.0, 0.4, and 0.2 µm) were used throughout. Fractionation was carried out on a Mark 410 oil-free vacuum pump (Rocker Scientific Co., Ltd., Kaohsiung, Taiwan) and a home-made setup containing two 1-L vacuum filtration flasks. One of the flasks is equipped with a stainless-steel frit and funnel (Rocker Scientific Co., Ltd., Kaohsiung, Taiwan), the other flask, with a porous glass filter, glass funnel, and an aluminum clamp (Borosil Ltd., Mumbai, India).
2.3. ATR-FTIR Measurements and Data Processing
A Vertex 70 (Bruker Optik GmbH, Ettlingen, Germany) with a KBr beam splitter, a DLATGS detector, and a diamond ATR crystal with a temperature controller (GladiATR™, Pike Technologies, Madison, WI, USA) was used for ATR-FTIR of aqueous samples as films produced by heating the ATR crystal to a preset temperature. FTIR spectra were processed using OPUS 8.5 software. Conditions are given in Table 1. The procedure for the FTIR measurements is also given in detail in Appendix A.
2.4. Fluorescence Measurements
A Fluorolog FL3-22-Tau fluorescence spectrometer (Horiba Jobin Yvon, Montpellier France) equipped with a 450 W xenon lamp, and two double-emission was used. To analyze the fluorescent organic constituents, EEMs were obtained. Conditions of fluorescence measurements are summarized in Table S2, Supplementary materials. Various variants of FS are used in the works, i.e., the emission spectrum at a fixed excitation wavelength, synchronous monochromators, and one double-excitation monochromator. To eliminate the influence of the internal filter effect and self-absorption, the samples were diluted with deionized water so that the absorbance of the solutions did not exceed 0.1 at a wavelength of 254 nm.
Regions of the excitation-emission matrix corresponding to certain groups of compounds—humic-like (A and C), tyrosine and protein-like (B), and tryptophan-, phenol-, and protein-like—were selected according to [84,85], Figure 1 (green frames; their values are summed up in Table S3, Supplementary information) and Table S3 (supplementary information. Also, to test and increase selectivity and sensitivity, broader regions were selected, Figure 1 (light magenta frames), they are summed up in Table 2.
It was assumed that the integration of the selected regions (fluorescence regional integration, FRI) represents cumulative response of the WEOM components [27]. Thus, the volume under the surface of the i-th region of an EEM can be calculated using eq. (1). However, the resulting excitation–emission matrices are discrete, so eq. (2) is correct in this case. The fraction for each area according to (3) [27]:
Here, I(λEx, λEm) is the fluorescence intensity at the excitation wavelength λEx and the emission wavelength λEm; ∆λEx is the excitation wavelength step; ∆λEm is the emission wavelength step; and Pi is the partial sum fraction by volume for the i-th area of the excitation matrix-emissions, using which it is possible to estimate the fluorescence components ratio.
Fluorescence indexes according to [25]—humification index (HIXEM), freshness index (BIX) (β/α), fluorescence index (FI), and T/C peak ratio—were calculated as protocoled in [84,86,87,88,89,90,91,92,93]; Table S4, Supplementary information. Also, the ratios of total intensities of regions corresponding to humic-like substances (A + C or A* + C*) to biologically active compounds (regions B + T or B* + T*) were calculated. Also, the approach of two regions IV and V that reflect the integral fluorescence signals (signatures) of humic-like (Region IV, em = 405–450 nm at ex = 290–310 nm, also aromatic structures) and protein-like (Region V, em = 400–410 nm at ex 275–300 nm, fresh organic sources) materials [27].
2.5. Other Equipment and Measurements
For element analysis, an axial ICP–OES 720-ES instrument with an SPS3 autosampler (Agilent Technologies, Santa Clara, CA, USA) was used throughout. The details and conditions of measurements were as in [14] and are summarized in the Supplementary materials. To study the leaching of the plasticizer from polycarbonate analytical track membranes during membrane filtration, the absorption spectra of aqueous filtrates and flushing water in the visible and ultraviolet regions were recorded using a Cary 4000 spectrophotometer (Agilent Technologies, Santa Clara, CA, USA) and a quartz spectrophotometric cell (optical path length, 1 cm).
2.6. Correlation and 2D-COS Analysis
Simple correlation analysis was made using Origin Pro software (OriginLab, Northampton, MA, USA). Building 2D-COS maps was implemented using OriginLab Origin Pro 2021b using 2D Correlation Spectroscopy Analysis app (OriginLab Technical Support, https://www.originlab.com/fileExchange/details.aspx?fid=497).
Matrix spectra (assemblies) for 2D-COS were assembled with the sieved fraction size as a perturbation variable. The spectral assemblies were built by the size increase from fine to coarse fractions. In all cases in this study, the spectra were averaged taking into account the average numerical size of fractions and the Pareto function was used to reduce the dominance of large peaks).
For the purposes of 2D-COS data processing, the numerical values of the average fraction sizes (0.01, 0.02, 0.075, 0.15, 0.3, 0.6, 0.9, 1.5, 3.5, 7.5, and 10 µm were used as the values for the perturbation variable in 2D-COS calculations.
Homospectral synchronous and asynchronous maps were built for each size fraction for the whole measured ranges of FTIR and fluorescence measurements. Synchronous maps were not normalized; asynchronous maps were normalized to the average value of all the correlation.
Heterospectral FTIR synchronous and asynchronous maps were built for each size fraction for the whole studied range and for characteristic ranges 700–650 cm–1, 900–800 cm–1, 1800–1200 cm–1, and 3000–2800 cm–1. The normalization was the same as for homospectral 2D-COS.
Heterospectral FTIR–fluorescence synchronous and asynchronous maps were built for each size fraction for the characteristic range 280–510 nm of the fluorescence spectrum (excitation, 265 nm) and for characteristic IR ranges 700–650 cm–1, 900–800 cm–1, 1800–1200 cm–1, and 3000–2800 cm–1. The normalization was the same as for homospectral 2D-COS.
2.7. Experimental Procedures
Membrane plasticizer leaching was used exactly as described previously [14] and given in the Supplementary information (Procedure S1). An experiment was also carried out, which consisted in sequentially passing 50 mL of deionized water (a total of 250 mL) through a membrane with a pore size of 0.05 μm to check the leaching of the plasticizer in dynamics.
Extraction of organic matter was made for cold extracts. A 40-g sample of chernozem, previously ground in a jasper mortar, was placed in a 700-mL Erlenmeyer flask. Then 400 mL of deionized water at room temperature (1 g soil per 10 mL of deionized water) was added [94]. The resulting suspension was shaken on a laboratory shaker for 1 h [95] and then defended for 30 min.
WEOM particle size fractionation [14] by sequential membrane filtration was used exactly as described previously [14] and given in the Supplementary information (Procedure S3). Before each step of filtration, the system was rinsed with 100 mL of deionized water to remove the remaining plasticizer from membranes and wash the vessels. Thus, the following fractions were obtained: 2-5 µm, 1–2 µm, 0.8–1 µm, 0.4–0.8 µm, 0.2–0.4 µm, 0.1–0.2 µm, 0.05–0.1 µm, 0.03–0.05 µm, 0.01–0.03 µm, and < 0.01 µm. Washing particles from membranes was used exactly as described previously [14] and given in the Supplementary information (Procedure S4).
3. Results and Discussion
3.1. Degree of Plasticizer Extraction from Membranes During Membrane Filtration
Since in this work, membranes are used to separate organic substances (albeit in a sorbed state) with subsequent analysis by sensitive methods, the first stage was to study the effect of the membrane material on the resulting fractions, since in addition to the main component (in this case, polycarbonate), membranes also contain a plasticizer. For this purpose, spectrophotometry was used to assess the total content of leachable organic compounds (and fluorescence spectrometry we analyzed flushing water and filtrates obtained during blank experiments. From the spectra in the UV and visible region (Figure 2) for polycarbonate membranes of absorption shows that the highest absorbance is observed for a flush water sample obtained for a membrane with a pore size of 0.01 μm (0.055 at λ = 200 nm), the lowest for a membrane filtrate of 1 μm (0.02 at λ = 200 nm). This difference can be explained by the duration of filtration: for membranes with smaller pore sizes, the process took a longer time, thus a larger amount of the plasticizer passed into solution.
During the leaching test of the plasticizer from a membrane with a pore size of 0.05 μm over time (Figure 2, inset) found that the highest absorbance is observed when the membrane is washed with the first 50 mL of water (0.1 at λ = 200 nm). For subsequent filtrates, the optical density was already less than 0.02, therefore, it can be assumed that most of the plasticizer is washed out during the first two stages of their washing.
Spectrofluorimetric analysis (Figure S1, Supplementary information) showed the presence of organic matter, including humic-like compounds, in the washes and filtrate for membranes with a pore size of 1 and 0.01 μm, which is indicated by the characteristic outlines of the obtained fluorescent matrices. The latter may be associated with the vital activity of microorganisms in the obtained samples. The obtained intensities for these fluorescent EEMs are significantly lower than for that characteristic to fractions of SOM. Based on the data obtained, it was decided that the membranes should be washed with 100 mL of fresh deionized water before use, to wash out the plasticizer.
Thus, we confirmed the initial hypothesis that the plasticizer can be extracted from the membranes during the fractionation process and affect the determination of organic substances. However, such an impact can be estimated as insignificant on the methods of analysis that are used in the work. However, a procedure for washing 100 mL of deionized water has been proposed to reduce this effect, primarily on fluorescence spectra.
3.2. FTIR Analysis
To separate inorganic and SOM constituents, the entire examined mid-IR and far IR range (4000–150 cm–1), was separated into four regions. These zones are the hydrogen-bond and CH-region (4000–2800 cm–1), the SOM region (1700–1170 cm–1), the Matrix I region (quartz overtone region, 1170–800 cm–1), and the Matrix II region (quartz lattice region, 800–150 cm–1) [36,47,49,50,96,97]. These areas include the predominant SOM or matrix bands and may be easily and consistently divided into distinct groups for all comparable samples. However, certain provisional borders, such as those at 1170 and 1700 cm–1 are acceptable [47]. The ranges of 4000−3775 and 2560−2000 cm−1 containing bands of atmospheric water, carbon dioxide and artifacts associated with the ATR−FTIR diamond crystal absorption are excluded from the analysis. The bands are summed up in Table 3.
3.2.1. Hydrogen-Bond and CH-Regions (4000–2800 cm–1)
This range of SOM IR spectra is largely related to the vibrations of OH groups in clay minerals, absorbed water as well as aliphatic carbon chain stretching. So, on the shoulder of a wide band of OH-groups (Figure 3), for all fractions with a particle size greater than 1 μm, bands of hydroxyl groups associated with metals in clay minerals at 3696 and 3620 cm–1 are visible. In turn, a narrow band at 3390 cm–1 is characteristic of sodium azide, which was used as a preservative. It should be noted that this band was not observed for all fractions, which may be due to problems in depositing the sample in the form of a film on the ATR diamond crystal (see below). The relatively narrow bands at 2926 and 2854 cm–1 are also of great interest. In the literature [97], these bands are often associated with C–H stretching in aliphatic chains. It must be mentioned that unlike other bands, bands assigned to the C–H stretching are pronounceable both for larger (> 1 μm) (Figure 3, a) and smaller (< 1 μm) (Figure 3, b) size fractions.
In the CH range (Figure 4, a) in general, a greater number of methylene groups in medium and small fractions are found, while in large fractions, methyl group is manifested as dominating. It is noteworthy that in the middle fractions, the band at 2800 cm–1, which is usually correspond to ‘aldehyde’ neighboring C–H is manifested (which may be the reflection if sugar contents).
3.2.2. SOM Region (1700–1170 cm–1)
In the spectral range of 2500–1250 cm–1 (Table 3 and Figure 4, b), only two distinct bands were observed for all fractions with a particle size greater than 1 µm. The first of them, the composite band at 1630 cm–1, is associated in the existing literature data with stretching vibrations –C=C– in aromatic compounds that are part of soil organic matter [50]. At the same time, this band can be a manifestation of vibrations in soil water and SiO2 (Table 3). Also, bands in the range of 1650–1600 cm–1 can be associated with asymmetric C–O vibrations in carboxylates, which may indicate the presence of lignin and/or various aromatic and/or aliphatic carboxylates in the fractions [142]. This band will also be discussed below. The band at 1410 cm–1, in turn, can be attributed to vibrations of the hydroxyl group associated with magnesium in minerals such as brucite [109].
In large fractions, the band of water absorption is visible, which practically disappears in medium and small fractions (Figure 4, b). Carboxylic species manifest themselves in all the fractions (Figure 4, b). While in the smallest fractions they appear as carboxylate bands, in the rest of fractions they are bands of undissociated carboxylic acids. Figure 6 shows that in small fractions, the bands corresponding to CH groups at 1500 and 1300 cm–1 are more pronounced.
3.2.3. Matrix II and I regions (1170–800 and 800–150 cm–1)
In the range of 1250–100 cm–1 (Figure 5) of the obtained IR spectra, bands related to the inorganic soil matrix predominate[97]. Most of the intense bands in the spectra are associated with Si-O vibrations in quartz or silicates (Table 3). For example, the band centered at ca. 1020 cm–1 is attributed to Si-O stretching vibrations in quartz and/or clay minerals. Also, in this range there are bands related to vibrations of the Mg(Al)–OH group (915, 780, 750, and 430 cm–1) in such clay minerals as kaolinite and smectite, and iron oxides (655 cm–1). At the same time, the organic component also contributes to many of these bands. In several studies [143,144], these bands are also associated with the presence of cellulose and lignin (1090 and 1030 cm–1) in SOM, with C–H vibrations in aromatic and aliphatic compounds (800 and 605 cm–1).
In general, in the long-wavelength range of the spectrum (less than 800 cm–1), vibration bands of carbonates, quartz, and iron oxides predominate. However, at the same time, C–H vibration bands in aromatic compounds and/or N–H vibrations (out-of-plane vibrations in primary amines and wagging vibrations in secondary amines) can be present in this range [99].
The principal limit of fraction size is 1 μm, before which alpha-quartz dominates in the spectra (Figure 3). For smaller fractions, quartz bands are almost invisible, and no new bands appear (Figure 3). All fractions are available in four ranges: 3000–2750 (CH), 1750–1200 (SOM), 900–800, and 700–650 cm–1 (both are silicate, possibly SOM); Figure 4 and Figure 5.
It is confirmed that quartz dominates in large fractions (Figure 5, in medium fractions bands of carbon dioxide are visible). Silicate dominance is seen in large fractions (Figure 5, b), while in finer fractions, the higher intensities of bands corresponding to aluminosilicate and alumina are found. The data correlate well with elemental analysis of these fractions [14]. In medium and small fractions, Al–O bands are manifested (satisfactorily correlated with elemental analysis, [14]), Figure 5, b. Carbonate bands appear everywhere (pronounced in large areas, shoulder bands in small and medium-sized ones), Figure 5b.
3.3. IR Correlations
To test the attribution hypotheses, homospectral 2D-COS maps were constructed, Figure 6. In the silicate range 700–650 cm–1 (Figure 6, a) as expected, the quartz band correlates with itself, a very weak correlation with probable iron oxide band at 550–590 cm–1. From the asynchronous map (Figure 6, b), one can see that the band changes differently than the background around it. In total, 2D-COS information fully confirms the analysis of direct ATR FTIR data for this range.
In the range 900–800 cm–1 (Figure 6, c), the synchronous map shows that the band at 880 cm–1 should be assigned not as a carbonate band as it is well correlated with other SiO2 bands. Asynchronous map (Figure 6, d) also says that the band of 880 cm–1 is consistent with the bands of Al–O and is not the band of quartz. Overall, the bands show the correlation of Al and Si, which is consistent with [14]. In the range 1800–1200 cm–1 (Figure 6, e), the water band dominates; there is a weak correlation of carboxylates with it (the synchronous map). The asynchronous map (Figure 6, f) shows that carboxylate bands change in the way opposite to C–H. In the range of 3000–2800 cm–1 (Figure 6, g), synchronous maps show an obvious correlation of methylene, while asynchronous reveal different behavior of the region of sugars and the region of other C–H. Nevertheless, this information fully confirms the data obtained from the traditional spectra giving no new information. Thus, IR homospectral correlations did not provide any new valuable information and mainly confirm the attribution of the IR spectra themselves.
For additional verification, synchronous heterospectral correlations of regions are constructed (Figure 7), asynchronous maps do not provide any reliable information and probably need even larger numbers of fractions. The comparison of ‘inorganic’, low-wavenumber ranges of 700–650 cm–1 and 900–800 cm–1 related to quartz and aluminosilicates (Figure 7, a) show that the questionable band of iron oxide (see above) is most likely a silicate as it correlates with most silicate bands found by IR spectra and homospectral IR correlations. Comparison of SOM dominating ranges, 1800–1200 cm–1 and 3000–2800 cm–1 (Figure 7, b), shows excellent correlation of the ranges of bending and stretch vibrations of –C–H. This fact confirms that the ranges 1500–1390 cm–1, which cannot be elucidated from direct FTIR measurements, is C–H. Interregional heterospectral correlation of regions of 1800–1200 cm–1 and 900–800 cm–1 (Figure 7, c) shows that the adsorbed water band at 1630–1610 cm–1 correlates with silicate (bands at 900 and 700 cm–1).
Thus, most of the conclusions on the band assignment made by direct IR spectra are confirmed except for carbonate and iron oxide. However, heterospectral IR–IR correlations of the same spectra show much larger volume of information a provide a way to extend the information on the bands, based on the C–H range, confirming other hydrocarbon groups and all allow excluding unreliable information like of iron oxide, which proves to be silicate matrix band.
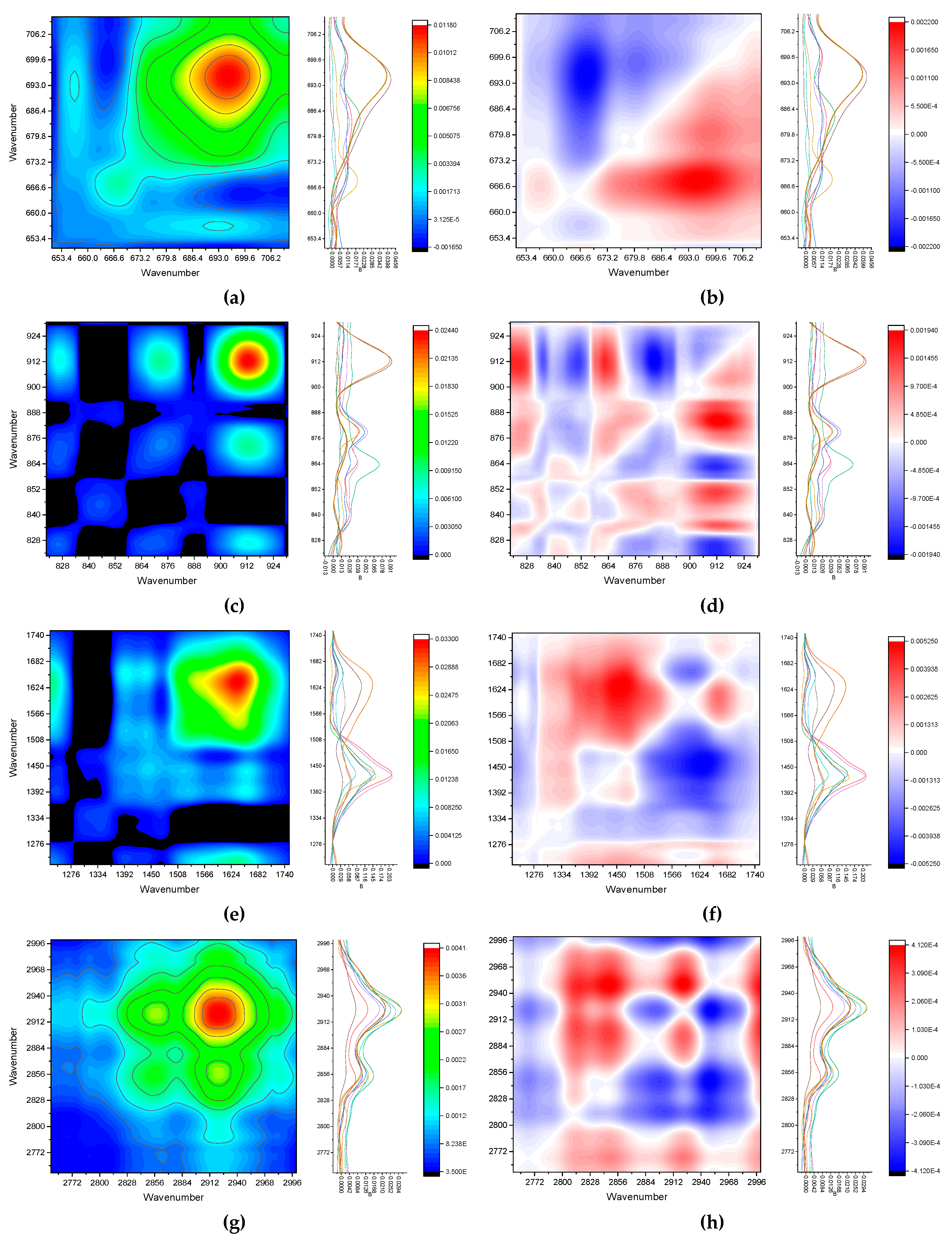
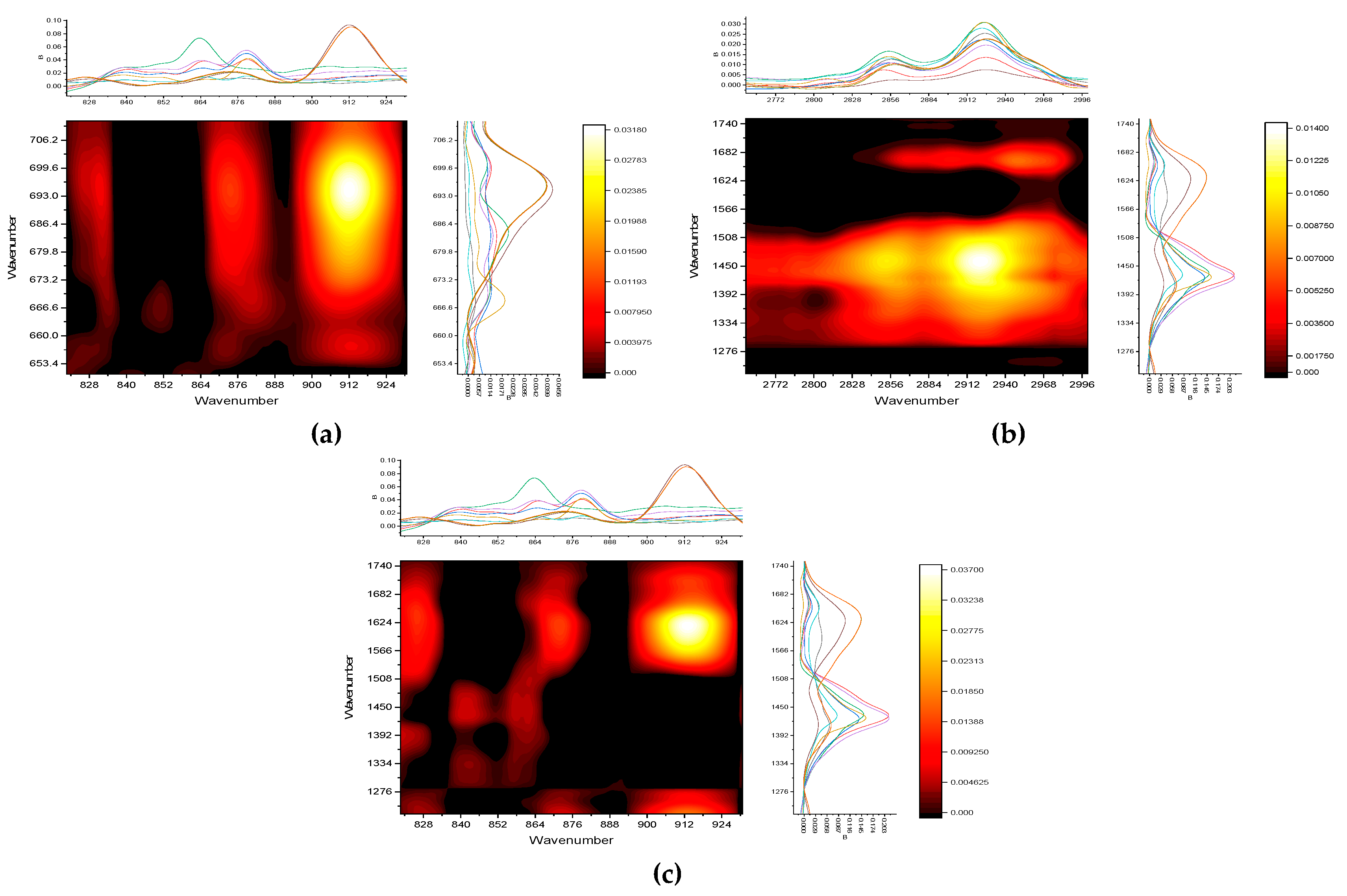
Figure 6.
Synchronous (a, c, e, and g) and asynchronous (b, d, f, and h) homospectral ATR FTIR 2D-COS maps of particulate SOM for the ranges (a and b) 700–650 cm–1; (c and d) 900–800 cm–1; (e and f) 1800–1200 cm–1; and (g and h) 3000–2800 cm–1.
Figure 6.
Synchronous (a, c, e, and g) and asynchronous (b, d, f, and h) homospectral ATR FTIR 2D-COS maps of particulate SOM for the ranges (a and b) 700–650 cm–1; (c and d) 900–800 cm–1; (e and f) 1800–1200 cm–1; and (g and h) 3000–2800 cm–1.
Figure 7.
Synchronous heterospectral ATR FTIR 2D-COS maps of particulate SOM for the ranges (a) 700–650 cm–1 and 900–800 cm–1; (b) 1800–1200 cm–1 and 3000–2800 cm–1; and (c) 1800–1200 cm–1 and 900–800 cm–1.
Figure 7.
Synchronous heterospectral ATR FTIR 2D-COS maps of particulate SOM for the ranges (a) 700–650 cm–1 and 900–800 cm–1; (b) 1800–1200 cm–1 and 3000–2800 cm–1; and (c) 1800–1200 cm–1 and 900–800 cm–1.
3.4. Fluorescence Measurements
Correspondence of various regions of EEM to certain groups of compounds was first made in 1996 [85], in which, however, not regions, but individual bands corresponding to these regions were marked. So, the authors write that the B band of tyrosine- and protein-like substances can be detected at a pair of wavelengths (ex, em) = (275, 310 nm), the T band (tryptophan- and protein-like compounds) at a pair (275, 340 nm). Two bands are identified in the work, which the author correlates with humic-like substances (bands A and C) can be found at (ex, em) = (260, 380–460 nm) and (ex, em) = (350, 420-480 nm, respectively. The existence of a marine humic-like compounds region should also be noted. However, this does not mean that it cannot be present in such samples, since this band was found in a non-marine environment [25]. In this study, marine-origin humic-like substances were not considered as the preliminary studies showed that the corresponding range show very weak and irreproducible intensities in EEMs.
In [84], the intervals were somewhat expanded: the bands became regions. The authors of this work also introduce slightly different notations for these areas (Table S3, Supplementary information). In later works, the ranges were also slightly expanded [25,145], and new groups of compounds were added in addition to those already mentioned. Thus, in [145], such groups of compounds as hydrophobic fulvic acids or soluble waste products of microbes can be distinguished, while not dividing the area of humic acids into two areas, as in previous works. The EEM fluorescence spectra for different size fractions are shown, with the fluorescence intensity being normalized on xenon lamp emission intensity (Figure 8).
In general, the shape of the matrices was comparable for all the obtained fractions and expectedly, marine based peak of humic substances was negligible in all the fractions, and it was excluded from the consideration (Table S3, supplementary information). However, integral intensities and the positions of fluorescence-intensity maxima of the resulting peaks (A, C, B, and T) and their ratios varied significantly throughout the size profile. For fractions above 10 µm, 5–10 µm, 2–5 µm, 0.05–0.1 µm (less intense that other fractions in this list), and also below 0.01 µm, peaks at (ex, em) = (260–270; 450–490 nm) (A) and (ex, em) = (315–330; 420–460 nm) (C) designated to humic-like substances are the most pronounceable (Figure 8). According to [146], A and C peak positions depend on their origin, fulvic or humic acids. In all the fractions the above major peak positions show that they are contributions from mainly fulvic components of SOM. However, a secondary, red-shifted peaks in regions A and C at (ex, em) = (260–270; 510–540 nm) for A and (ex, em) = (350–365; 510–540 nm) for C are rather clearly seen for all the above fractions and also for the fraction 0.2–0.4 µm, which is quite different from all other fractions. This is the evidence of the contribution of humic acids [146], which seem to dominate the fractions of 2–5 µm and 0.2–0.4 µm.
The other group consists of fractions of 1–2 µm, 0.8–1 µm, 0.4–0.8 µm, 0.1–0.2 µm, 0.03–0.05 µm, and 0.01–0.03 µm. For these samples, the tyrosine-like substances peak B at (ex, em) = (270–280; 300–320 nm) is more distinguishable than humic- or tryptophan-like bands. The rest of the samples (2–5 µm, 0.2–0.4 µm, and 0.1–0.2 µm) contained bands corresponding both with humic-like, and tyrosine-like substances. The most intense EEMs were for fractions above 10 µm, 5–10 µm, and below 0.01 µm when the fluorescence intensities of other fractions were much less in their values. It is worth mentioning that the largest and finest fractions show similar EEMs, quite different from medium fractions.
Obtained fractions significantly differ from each other in terms of the fluorescent components content of organic matter. Figure 8 shows that the initial and final fractions are identical to each other in composition (in this case, a change in the content of less than 5 units was not considered significant). However, the intermediate fractions differ very significantly. Thus, from fractions with a larger particle size to fractions with a smaller one, there is a tendency for a decrease in the total content of humic-like compounds and an increase in the content of tyrosine-like substances with a maximum value of 50.3% for a fraction of 0.03–0.05 μm. At the same time, the content of tryptophan-like compounds for all fractions is <1%, which may be due to insufficient coverage of this EEM region during the study. In many studies, for example, in [27], the main band of tryptophan-like compounds is at λex = 200–250 nm, but in the present work, the excitation wavelength range λex = 240–550 nm was analyzed.
The analysis of main fluorescence parameters show that the approaches to select humic- and non-humic compounds by A, C, B, and T ranges [84,85] and Regions IV and V [27] show the same picture(Figure 9, a); both decrease for fine fractions below 1 µm and then start to increase for fractions below 0.1 µm, the finest fractions show the values close to large fractions. The difference of data for regions selected in the previous studies (Table S3 [84,85]) and the regions expanded from the experimental values (Table 2) is negligible, but the reproducibility of values obtained for expanded ranges is better because the excitation ranges for the studied samples are redshifted compared to previous studies, and the regions are broader, which is accounted for in the expanded ranges A*, C*, B*, and T*.
Figure S2 shows that the contribution of peak A to the total value of humic-like compounds is approximately the same, and the main difference in the amounts of humic-like compounds is peak C, mainly to contribution of the secondary band at 525 nm of more condensed humic substances in the largest and the finest fractions (Figure 8). This is well correlated with the increase in methylene groups in IR spectra at 2925 and 2860 cm–1 (Figure 4, a).
Figure 9, a inset shows the profile of the sums of humic-like and non-humic components along the profile (a detailed picture for all the expanded ranges A*, C*, B*, and T* is given in the supplementary information, Figure S2). It shows the similarity of the largest fraction of 5–10 μm and the finest fraction of below 0.01 μm. Similarly, the next large fraction of 2–5 μm and the second finest fraction of 0.01–0.03 μm also show the same values. For all these for fraction, the humic-like compounds dominate (80%). For the middle range of fractions 0.03–2 μm, the ratio of humic to protein-like compounds decreases (Figure S3, Supplementary information 3-fold compared to the largest and finest fractions, and the percentage of humic-like compounds is around 60%.
As for main fluorescence indicators, their values along the profile of size fractions id different (Figure 9, b). FI changes rather significantly, while there are no certain trends the values along the size profile. FI values for the majority of fractions are ca. 1.2, which evidences main terrestrially derived (allochthonous, plant-produced) DOM, while for fractions 0.03–0.05, 0.05–0.1, 0.4–0.8, and above 1 μm the values are ca. 1.5, which shows in increase in the contribution from autochthonous (microbially derived, recently produced) sources.
BIX index also changes little; the values are around 1 meaning a considerable share of newly produced DOM [147]. The changes in BIX correlate well (reciprocally) with the changes in the ratio of humic and non-humic compounds (Figure 9, a and b): the lower BIX values of fractions of 5–10 μm and 2–5 μm indicate higher humic-like substance amounts.
T/C index changes to the maximum extent Figure S4, a, medium fractions are the largest, the values for fractions 2–0.03 μm show values above 1, in the range 0.8–2, which indicates recently appeared organic constituents, probably of fresh (microbial) origin [148]. For the largest and the finest fractions T/C values are significantly below 0.5, which shows the dominance of condensed organic compounds. This indicator is quite correlated with the data from IR spectra: the medium fractions show methylene groups in IR spectra at 2925 and 2860 cm–1 (Figure 4, a) as well as much higher intensities of bands, corresponding to CH at C–O bands at 2800 cm–1 (Figure 4, a) and carboxylic acids (Figure 4, b).
HIXEM humification index values also differ significantly (Figure S4, b, Supplementary information) and they correlate quite well with the total intensities of the bands and regions (Figure 9, a). HIXEM decrease rather significantly in medium fractions compared to two largest and two finest, from the values of 0.9–1 to 0.7. This is the indication of less condensed constituents in medium fractions [87], which correlates with dominating fulvic components indicated by the BIX and FI and the redshifted secondary bands in bands A and C (Figure 8). Along with BIX values for these fractions of 0.8–1.0 (Figure 9, b) and HIX of 3–5 (Figure S4, b) probably indicating weak humic character of SOM in these fractions with strong contribution from autochthonous components [149]. Much higher values of HIXEM (above 10) for the largest and finest fractions, according to [87] result from lower H/C ratios in SOM (more aromatic, less hydrogen-saturated), i.e., from more condensed, terrestrially derived, well-humified WEOM leading to redshifted fluorescence emission. This conclusion supports the conclusion on BIX and F/I indexes. In total, HIXEM is well correlated with intensities of A and C peaks, and their sum (Figure 10).
Correlations of other indexes BIX, T/C peak ratio, and FI are expectedly correlated with intensities of B and T peaks and their sum (Figure 10). As B peak has the minimum intensity of all the regions, its contribution to total correlations is lower than for peak T. In total, indexes are well correlated with intensities of whole areas (Figure 9, a, either as sum of corresponding bands A+C and B+T (Table 3 and Table S3, Supplementary information) or Regions IV and V according to [27]. Among all the fluorescence indexes, the lowest correlation with other indexes is for FI index. Also, HIXEM and T/C values are reversely correlated; other indexes are not significantly correlated. Thus, Figure 10 demonstrates a clear separation of fluorescent markers associated with humified organic matter (A, C) and that of microbial and root plant origin (T, B). It should be noted that the BIX index, which is actually an indicator of fresh organic matter, weakly positively correlates with B (tyrosine-like structures), T (tryptophan-like), A + T structures, and weakly negatively with humified (A, C, A + C), indicating a generally low biological activity associated with the isolated fractions of microbiota. A similar conclusion was made in the previous study of water-soluble SOM of similar soil as a whole [7].
Thus, fluorescence data, both excitation–emission matrices and the main fluorescence indicator show a noncontradictory picture of most condensed non-oxidized WEOM in largest particles above 5 μm and below 0.03 μm, and higher amounts of less-condesed and more oxidized SOM in the middle fractions, with the domination of fulvic acids and a much higher fractions of fresh (tryptophan-like) organic matter in these fractions. These data agree well with the data from IR spectra.
3.5. IR–Fluorometry Heterospectral Correlations
A synchronous correlation of all four bands A, C, B, and T with the main range (280–510 nm) of the fluorescence spectrum at an excitation of 265 nm (Figure S5, Supplementary information) with characteristic bands of IR spectra was carried out (Figure 11).
Comparison of fluorescence spectra with the IR region of 700–650 cm–1 (Figure 11, a) shows that quartz silicate correlates with the region of tryptophan-like compounds (320–400 nm) and does not correlate with tyrosine-like (up to 300 nm) and humic-like (400 nm and above). This fully agrees with the heterospectral correlation with the IR region of 900–800cm–1 (Figure 11, b).
On the contrary, comparison of fluorescence spectra with the IR region 1700–1200 cm–1 shows that tyrosine-like bands (300–320 nm) mainly correlate with C–H IR bands at 1450–1300 cm–1, while humic-like fluorescence bands at 380–500 nm correlate well with carboxylates and carboxylic acids (1690–1650, 1570–1450, and 1390–1270 cm–1), and nothing correlates with tryptophan-like ones (Figure 11, c). This agrees well the conclusion on the fulvic-acid amounts as a difference in the medium and the largest and finest fractions (see the previous section. Adsorbed water IR bands do not correlate with anything.
The comparison of fluorescence spectra with IR region 3000–2800 cm–1 (Figure 11, d) fully confirms the 2D-COS for region 1700–1200 cm–1: the entire range 2950–2810 cm–1 correlates with tyrosine and humate-like fluorescence bands, there are no correlations of IR bands in this range with tryptophan-like substances (band T) in the fluorescence spectra.
Thus, heterospectral fluorescence–FTIR 2D-COS not only makes the direct correlations between the types of organic matter based on certain functional groups in IR spectra, but it provides a more detailed confirmation of fluorescence EEM regions and indexes. In the case studied, it fully confirms that contribution of low-molecular (fulvic) acids in the medium fractions of WEOM and the higher contribution of fresh (microbial-based) organic matter in these fractions.
3.6. Correlations of Fluorescence Indexes with ICP-AES
To extend the possibilities of 2D-COS and to connect the multimodal molecular spectroscopy studies, we connected the fluorescence and FTIR data obtained in this study with the elemental composition of these fractions previously found by ICP–AES [14]. In this case, fraction size was used as a 2D-COS perturbation variable, and the element profile found for each fraction was used as an element spectrum, 2D-COS of FTIR and element analysis showed no reliable information on most trace elements. Thus only simple correlation analysis was used (band intensity vs. concentrations found by ICP–AES, and showed good correlation of Si contents with all major quartz and silicate bands (which agrees well with the previous data on humic substances [37]) and a moderate correlation of Ca and carbonate bands in IR spectra (Figure 4, b and Figure 5,a), the coefficient of correlation is above 0.7. However, the data obtained in this study show that some extra handing of IR spectra, especially of such complex samples as concentrated narrow-size particle fractions to exclude the artefact correlations with some background or noise features; hence, it should be the subject of a separate study.
The same is true for fluorescence spectra or EEM; direct 2DCOS correlations are not possible without extra protocols (or algorithms) on data handling to exclude high false positive 2D-COS correlation bands with background signals. On the contrary, fluorescence indexes and other integrated parameters of EEM or fluorescence spectra are easy to correlate with similar discrete element profiles (Figure 12).
It was found that there are distinct correlations between fluorescent indexes of organic matter composition and gross element fraction composition. It is obvious that the indicators associated with humification state (peaks A and C, as well as the HIXEM humification and Fl indexes) are weakly positively correlated with the elements associated with the mineral silicate component (Si and K). This probably indicates (and confirms) the predominant role of aggregates with sizes of 2–10 μm characterized mainly by silicate, namely silicon-silicate mineralogical composition [150] in humification processes [151].
The only element that has a strong positive correlation of intensities of A and C peaks as well as with the HIXEM humification index, is Mn (Figure 12). Manganese actively forms complex compounds with organic ligands [152] and usually found in humic substances in relatively large amounts [37]. In same time, in concordance with the data of fluorescence and IR and multimodal 2D-COS, Mn shows strong negative correlations with peaks B and T related to fresh organic matter and corresponding indexes T/C peak ratio as well as BIX.
The same is true for biogenic elements. The correlation of humification indicators with P, S, Cr, Mg, Ca, Cu, and Zn is either strong negative or absent, which indicates a significant degree of transformation of the original organic matter, which agrees with the humate-related bands of carboxylic acids in IR spectra and HIXEM values above 5 for largest and finest fractions.
Indexes associated with predominantly microbial (peak T) and root (peak B) tryptophan- and tyrosine-like constituents, on the contrary, show direct, and rather strong, correlations with the biogenic elements Mg, P, Cr, Cd, K, S, and Ca (Figure 12). It is worth mentioning that BIX index shows a considerably high coefficient of correlation of ca. 0.6 with Ca and S, which agrees with the existing data on BIX index correlation with bioavailable nutrient elements as well as microbial activity, which in turn often is associates with increased contents of Ca and S in both soils and aquatic systems [153]. These biogenic indexes show a negative correlation with Si, Al, and Fe, and Ti, which is due to the origin of these substances, their low degree of transformation and preservation in the composition of associates.
4. Conclusions
As a result of this study the possibilities of multimodal heterospectral two-dimensional correlation analysis of narrow soil fractions were demonstrated, and both modalities—fluorescence and FTIR spectroscopies—have showed their contribution in a larger volume of information of 2D-COS analysis. 2D-COS modality in FTIR increased the reliability of band assignment, especially in the range 1800–1100 cm–1, where bands from inorganic, carboxylic, and hydrocarbon components are significantly overlapped. The heterospectral IR–fluorescence 2D-COS was even more relevant as it provided the cross-confirmation of IR bands in various regions and showed the correlations of various bands in EEM for fresh (biogenic, non-humic) and humic components. Certainly, within the frames of this study, it was not possible to make the compete characterization of the analytical possibilities of these techniques, and some drawbacks–like possible correlations with background values were found, which require some further work with algorithmics of data handling prior 2D-COS or some post-operations. Nevertheless, the capabilities of the technique for such samples as narrow soil fractions are shown. We also believe that inclusion of atomic-emission spectroscopy to two-dimensional correlation analysis on soil fractions is also a very promising technique and the further work should also be focused on full-scale two-dimensional correlation analysis of ICP–AES with molecular-spectroscopy techniques. Also. Some work is to be done with sample preparation, e.g., using microwave autoclave decomposition to analyze the membranes themselves to control the quantity of flushing and more correct element analysis; as well as implementation of nondestructive/destructive sample preparation for two-step molecular and atomic multimodal spectrochemical analysis of WEOM or similar samples.
From the viewpoint of the samples—narrow soil fractions of WEOM of nano- to micrometer size range of chernozem soil—the distribution patterns of organic matter and microelements showed a rather distinct distribution and significantly different fraction, which can be divided into two parts. The first part is two largest (2–10 μm) and two finest factions (0.01–0.03 μm) characterized by less carboxylated and saturated humic substances according to both fluorescence and FTIR, which dominates over the fresh (biogenic) organic matter. The other group is medium fractions (0.05–1 μm), which are characterized by higher contents of fulvic acids and higher percentage of fresh organic matter. The elucidation of this data requires more in-depth studies of various types of soils but this study shows the possibility to obtain characteristic data on nano- to micrometer size soil particles that can be used for soil characterization, classification and, potentially remediation and recultivations based on the detailed analysis of wide-size range soil particle composition.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Conditions of ICP–AES measurements; Table S2: Conditions of EEM fluorescence measurements; Table S3: Regions of the excitation (Ex)-emission (Em) matrix corresponding to certain groups of compounds; Table S4: Fluorescence indexes calculated for WEOM fractions; Figure S1: Excitation–emission matrices for (a) a filtrate after passing water through 1 μm; (b) a 1 μm membrane flush; and (c) a 0.01 μm membrane flush during blank experiments using polycarbonate analytical track-etched membranes; Figure S2: Relative abundances of humic- and non-humic components according to individual fluorescence integrated intensities regions A and C (humic-like compounds) and B and T (non-humic-like compounds); Figure S3: Ratio of the proportions of humic and biochemical compounds to the average particle size of fractions; Figure S4: Distribution of integral indicators, (a) T/C peak ratio and (b) humification index (HIXEM), between narrow fractions; Figure S5: Smoothed fluorescence spectra at 265 nm excitation, normalized for total fluorescence for each spectrum.
Author Contributions
Conceptualization, M.P., D.V., and O.R.; methodology, D.V., S.O., and O.R.; formal analysis, M.P.; investigation, S.O. and O.R.; resources, D.V..; data curation, S.O. and M.P.; writing—original draft preparation, M.P.; writing—review and editing, M.P., D.V., and O.R.; visualization, M.P.; supervision, M.P.; project administration, M.P.; funding acquisition, M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation, grant no. 25-13-00088.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 2D-COS | two-dimensional correlation spectroscopy |
| ATR | attenuated total reflection |
| DOM | dissolved organic matter |
| EEM | excitation–emission matrix |
| FTIR | Fourier-transform infrared (spectroscopy) |
| HS | humic substances |
| ICP-AES | atomic-emission spectroscopy with inductively coupled plasma |
| SOM | soil organic matter |
| WEOM | water-extractable organic matter |
Appendix A: Methodological Features of the Analysis of Size Fractions of Soil Water-Soluble Matter Using ATR IR Spectroscopy
We chose ATR-FTIR spectroscopy to study the obtained size fractions since it is possible to obtain and analyze aqueous samples in the form of a film by heating the diamond crystal of the ATR attachment to a given temperature. In addition, ATR IR provides more opportunities for analyzing spectra in the inorganic soil matrix region containing many bands associated with silicates and clay minerals [47]. Based on the existing literature data, the analysis of aqueous size fractions using this method has not been previously carried out. However, the structural analysis of soil fractions with a particle size of 20 µm to 5 mm obtained by dry sieve fractionation using ATR FTIR was done, which allows the study of aqueous fractions with smaller size particles.
In terms of the shape of the spectrum and the intensity ratio of the bands, the reproducibility of the obtained fractions spectra using ATR FTIR is quite high. However, some bands (638 and 174 cm-1 are assigned to sodium azide) may be absent. This discrepancy between the spectra of the same fraction can be explained by how the sample was deposited on the ATR diamond crystal. The obtained spectrum is influenced both by the volume of the sample and, accordingly, its crystallization on the surface of the diamond crystal of the ATR unit of the IR spectrometer (Figure A1). This issue is especially important for the quantitative comparison of spectra, but this requires separate research and was beyond the scope of this work.
Figure A1.
Film of sodium azide (4 μL, 2.5 g/L) on a diamond crystal of an ATR FTIR spectrometer.

Thus, this method can in principle be used to obtain spectra of narrow fractions using the technique of drying a drop on the ATR crystal surface. This technique allows microvolumes of fraction. If necessary, the signal-to-noise ratio can be increased by repeated drying. However, a quantitative comparison of the obtained spectra is significantly complicated by the effect of solution crystallization. Nevertheless, we believe that this can be overcome in the future by choosing the drying conditions (drying rate and salt composition) and by applying a drop onto a crystal.
References
- Zhang, H.; Zheng, Y.; Wang, X.C.; Wang, Y.; Dzakpasu, M. Characterization and biogeochemical implications of dissolved organic matter in aquatic environments. J Environ Manage 2021, 294, 113041. [Google Scholar] [CrossRef]
- Karavanova, E.I. Dissolved organic matter: Fractional composition and sorbability by the soil solid phase (Review of literature). Eurasian Soil Sci. 2013, 46, 833–844. [Google Scholar] [CrossRef]
- Ding, Y.; Shi, Z.; Ye, Q.; Liang, Y.; Liu, M.; Dang, Z.; Wang, Y.; Liu, C. Chemodiversity of Soil Dissolved Organic Matter. Environ Sci Technol 2020, 54, 6174–6184. [Google Scholar] [CrossRef]
- Xenopoulos, M.A.; Barnes, R.T.; Boodoo, K.S.; Butman, D.; Catalán, N.; D’Amario, S.C.; Fasching, C.; Kothawala, D.N.; Pisani, O.; Solomon, C.T.; et al. How humans alter dissolved organic matter composition in freshwater: Relevance for the Earth’s biogeochemistry. Biogeochemistry 2021, 154, 323–348. [Google Scholar] [CrossRef]
- Bu, R.; Ren, T.; Lei, M.; Liu, B.; Li, X.; Cong, R.; Zhang, Y.; Lu, J. Tillage and straw-returning practices effect on soil dissolved organic matter, aggregate fraction and bacteria community under rice-rice-rapeseed rotation system. Agriculture, Ecosystems & Environment 2020, 287, 106681. [Google Scholar] [CrossRef]
- Mielnik, L.; Hewelke, E.; Weber, J.; Oktaba, L.; Jonczak, J.; Podlasiński, M. Changes in the soil hydrophobicity and structure of humic substances in sandy soil taken out of cultivation. Agriculture, Ecosystems & Environment 2021, 319, 107554. [Google Scholar] [CrossRef]
- Kulikova, N.A.; Kholodov, V.A.; Farkhadov, Y.R.; Ziganshina, A.R.; Zavarzina, A.G.; Karpukhin, M.M. Dissolved Organic Matter of Chernozems of Different Use: The Relationship of Structural Features and Mineral Composition. Mosc. Univ. Soil Sci. Bull. 2024, 79, 19–27. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, H.; Jia, Q.; Liu, H.; Ye, J. Interactions of anthropogenic and terrestrial sources drive the varying trends in molecular chemodiversity profiles of DOM in urban storm runoff, compared to land use patterns. Sci. Total Environ. 2022, 817, 152990. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, M.; Zhang, G.; Li, F.; Chen, H.; Leng, Y. The nature of dissolved organic matter determines the biosorption capacity of Cu by algae. Chemosphere 2020, 252, 126465. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Seo, H.; Han, H.; Seo, J.; Kim, T.; Kim, G. Conservative behavior of terrestrial trace elements associated with humic substances in the coastal ocean. Geochim. Cosmochim. Acta 2021, 308, 373–383. [Google Scholar] [CrossRef]
- Yu, W.; Huang, W.; Weintraub-Leff, S.R.; Hall, S.J. Where and why do particulate organic matter (POM) and mineral-associated organic matter (MAOM) differ among diverse soils? Soil Biol. Biochem. 2022, 172, 108756. [Google Scholar] [CrossRef]
- Corvasce, M.; Zsolnay, A.; D’Orazio, V.; Lopez, R.; Miano, T.M. Characterization of water extractable organic matter in a deep soil profile. Chemosphere 2006, 62, 1583–1590. [Google Scholar] [CrossRef]
- Olayemi, O.P.; Kallenbach, C.M.; Wallenstein, M.D. Distribution of soil organic matter fractions are altered with soil priming. Soil Biol. Biochem. 2022, 164, 108494. [Google Scholar] [CrossRef]
- Volkov, D.S.; Rogova, O.B.; Ovseenko, S.T.; Odelskii, A.; Proskurnin, M.A. Element Composition of Fractionated Water-Extractable Soil Colloidal Particles Separated by Track-Etched Membranes. Agrochemicals 2023, 2, 561–580. [Google Scholar] [CrossRef]
- Mashentseva, A.A.; Sutekin, D.S.; Rakisheva, S.R.; Barsbay, M. Composite Track-Etched Membranes: Synthesis and Multifaced Applications. Polymers (Basel) 2024, 16, 2616. [Google Scholar] [CrossRef]
- Kaya, D.; Keçeci, K. Review—Track-Etched Nanoporous Polymer Membranes as Sensors: A Review. J. Electrochem. Soc. 2020, 167, 037543. [Google Scholar] [CrossRef]
- Armstrong, J.A.; Bernal, E.E.; Yaroshchuk, A.; Bruening, M.L. Separation of ions using polyelectrolyte-modified nanoporous track-etched membranes. Langmuir 2013, 29, 10287–10296. [Google Scholar] [CrossRef]
- de Grooth, J.; Oborný, R.; Potreck, J.; Nijmeijer, K.; de Vos, W.M. The role of ionic strength and odd–even effects on the properties of polyelectrolyte multilayer nanofiltration membranes. J. Membr. Sci. 2015, 475, 311–319. [Google Scholar] [CrossRef]
- Huguet, A.; Vacher, L.; Saubusse, S.; Etcheber, H.; Abril, G.; Relexans, S.; Ibalot, F.; Parlanti, E. New insights into the size distribution of fluorescent dissolved organic matter in estuarine waters. Org. Geochem. 2010, 41, 595–610. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, S.; Ruan, L.; Yang, M.; Shi, W.; Zhang, H.; Li, W. Evaluating Soil Dissolved Organic Matter Extraction Using Three-Dimensional Excitation-Emission Matrix Fluorescence Spectroscopy. Pedosphere 2017, 27, 968–973. [Google Scholar] [CrossRef]
- Ly, Q.V.; Hur, J. Further insight into the roles of the chemical composition of dissolved organic matter (DOM) on ultrafiltration membranes as revealed by multiple advanced DOM characterization tools. Chemosphere 2018, 201, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Shen, Y.; Sun, J.; Liang, S.; Fan, H.; Tan, J.; Wang, X.; Huang, X.; Waite, T.D. Correlating fluorescence spectral properties with DOM molecular weight and size distribution in wastewater treatment systems. Environ. Sci. Water Res. Technol. 2018, 4, 1933–1943. [Google Scholar] [CrossRef]
- Bao, T.; Wang, P.; Hu, B.; Shi, Y. Investigation on the effects of sediment resuspension on the binding of colloidal organic matter to copper using fluorescence techniques. Chemosphere 2019, 236, 124312. [Google Scholar] [CrossRef]
- Shi, M.S.; Huang, W.S.; Hsu, L.F.; Yeh, Y.L.; Chen, T.C. Fluorescence of Size-Fractioned Humic Substance Extracted from Sediment and Its Effect on the Sorption of Phenanthrene. Int. J. Environ. Res. Public Health 2019, 16, 11. [Google Scholar] [CrossRef]
- Gabor, R.S.; Baker, A.; McKnight, D.M.; Miller, M.P. Fluorescence Indices and Their Interpretation. In Aquatic Organic Matter Fluorescence, Coble, P.G., Lead, J., Baker, A., Reynolds, D.M., Spencer, R.G.M., Eds.; Cambridge Environmental Chemistry Series; Cambridge Univ Press: Cambridge, 2014; pp. 303–338. [Google Scholar]
- Zsolnay, Á. Dissolved organic matter: Artefacts, definitions, and functions. Geoderma 2003, 113, 187–209. [Google Scholar] [CrossRef]
- Chen, W.; Westerhoff, P.; Leenheer, J.A.; Booksh, K. Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter. Environ Sci Technol 2003, 37, 5701–5710. [Google Scholar] [CrossRef]
- Matilainen, A.; Gjessing, E.T.; Lahtinen, T.; Hed, L.; Bhatnagar, A.; Sillanpaa, M. An overview of the methods used in the characterisation of natural organic matter (NOM) in relation to drinking water treatment. Chemosphere 2011, 83, 1431–1442. [Google Scholar] [CrossRef]
- Sillanpää, M.; Matilainen, A.; Lahtinen, T. Characterization of NOM. In Natural Organic Matter in Water, Sillanpää, M., Ed.; Butterworth-Heinemann: 2015; pp. 17–53.
- Bro, R. Multivariate calibration. Anal. Chim. Acta 2003, 500, 185–194. [Google Scholar] [CrossRef]
- He, W.; Hur, J. Conservative behavior of fluorescence EEM-PARAFAC components in resin fractionation processes and its applicability for characterizing dissolved organic matter. Water Res. 2015, 83, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Linker, R.; Shaviv, A. Characterization of soils using photoacoustic mid-infrared spectroscopy. Appl. Spectrosc. 2007, 61, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Linker, R.; Shaviv, A. Identification of agricultural Mediterranean soils using mid-infrared photoacoustic spectroscopy. Geoderma 2008, 143, 85–90. [Google Scholar] [CrossRef]
- Du, C.; Zhou, J.; Wang, H.; Chen, X.; Zhu, A.; Zhang, J. Determination of soil properties using Fourier transform mid-infrared photoacoustic spectroscopy. Vib. Spectrosc 2009, 49, 32–37. [Google Scholar] [CrossRef]
- Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. Photoacoustic and photothermal methods in spectroscopy and characterization of soils and soil organic matter. Photoacoustics 2020, 17, 100151. [Google Scholar] [CrossRef]
- Volkov, D.; Rogova, O.; Proskurnin, M. Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy 2021, 11, 1822. [Google Scholar] [CrossRef]
- Karpukhina, E.; Mikheev, I.; Perminova, I.; Volkov, D.; Proskurnin, M. Rapid quantification of humic components in concentrated humate fertilizer solutions by FTIR spectroscopy. J. Soils Sed. 2018, 19, 2729–2739. [Google Scholar] [CrossRef]
- Boguta, P.; Sokolowska, Z.; Skic, K. Use of thermal analysis coupled with differential scanning calorimetry, quadrupole mass spectrometry and infrared spectroscopy (TG-DSC-QMS-FTIR) to monitor chemical properties and thermal stability of fulvic and humic acids. PLoS ONE 2017, 12, e0189653. [Google Scholar] [CrossRef] [PubMed]
- Morra, M.J.; Marshall, D.B.; Lee, C.M. FT--IR analysis of aldrich humic acid in water using cylindrical internal reflectance. Commun. Soil Sci. Plant Anal. 2008, 20, 851–867. [Google Scholar] [CrossRef]
- Tatzber, M.; Stemmer, M.; Spiegel, H.; Katzlberger, C.; Haberhauer, G.; Mentler, A.; Gerzabek, M.H. FTIR--spectroscopic characterization of humic acids and humin fractions obtained by advanced NaOH, Na4P2O7, and Na2CO3 extraction procedures. J. Plant Nutr. Soil Sci. 2007, 170, 522–529. [Google Scholar] [CrossRef]
- Tanykova, N.; Petrova, Y.; Kostina, J.; Kozlova, E.; Leushina, E.; Spasennykh, M. Study of Organic Matter of Unconventional Reservoirs by IR Spectroscopy and IR Microscopy. Geosciences 2021, 11, 277. [Google Scholar] [CrossRef]
- Dudek, M.; Kabała, C.; Łabaz, B.; Mituła, P.; Bednik, M.; Medyńska-Juraszek, A. Mid-Infrared Spectroscopy Supports Identification of the Origin of Organic Matter in Soils. Land 2021, 10, 215. [Google Scholar] [CrossRef]
- Woolverton, P.; Dragila, M.I. Characterization of hydrophobic soils: A novel approach using mid-infrared photoacoustic spectroscopy. Appl. Spectrosc. 2014, 68, 1407–1410. [Google Scholar] [CrossRef]
- Yuan, Y.; Cai, X.; Tan, B.; Zhou, S.; Xing, B. Molecular insights into reversible redox sites in solid-phase humic substances as examined by electrochemical in situ FTIR and two-dimensional correlation spectroscopy. Chem. Geol. 2018, 494, 136–143. [Google Scholar] [CrossRef]
- Slaný, M.; Jankovič, Ľ.; Madejová, J. Structural characterization of organo-montmorillonites prepared from a series of primary alkylamines salts: Mid-IR and near-IR study. Applied Clay Science 2019, 176, 11–20. [Google Scholar] [CrossRef]
- Madejová, J.; Sekeráková, Ľ.; Bizovská, V.; Slaný, M.; Jankovič, Ľ. Near-infrared spectroscopy as an effective tool for monitoring the conformation of alkylammonium surfactants in montmorillonite interlayers. Vib. Spectrosc 2016, 84, 44–52. [Google Scholar] [CrossRef]
- Volkov, D.; Rogova, O.; Proskurnin, M. Organic Matter and Mineral Composition of Silicate Soils: FTIR Comparison Study by Photoacoustic, Diffuse Reflectance, and Attenuated Total Reflection Modalities. Agronomy 2021, 11, 1879. [Google Scholar] [CrossRef]
- Machado, W.; Franchini, J.C.; de Fatima Guimaraes, M.; Filho, J.T. Spectroscopic characterization of humic and fulvic acids in soil aggregates, Brazil. Heliyon 2020, 6, e04078. [Google Scholar] [CrossRef]
- Proskurnin, M.A.; Volkov, D.S.; Rogova, O.B. Two-Dimensional Correlation IR Spectroscopy of Humic Substances of Chernozem Size Fractions of Different Land Use. Agronomy 2023, 13, 1696. [Google Scholar] [CrossRef]
- Krivoshein, P.K.; Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. FTIR Photoacoustic and ATR Spectroscopies of Soils with Aggregate Size Fractionation by Dry Sieving. ACS Omega 2022, 7, 2177–2197. [Google Scholar] [CrossRef]
- Krasner, S.W.; Croué, J.P.; Buffle, J.; Perdue, E.M. Three approaches for characterizing NOM. Journal AWWA 1996, 88, 66–79. [Google Scholar] [CrossRef]
- Abbtbraun, G. Basic characterization of Norwegian NOM samples ? Similarities and differences. Environ. Int. 1999, 25, 161–180. [Google Scholar] [CrossRef]
- Volkov, D.S.; Proskurnin, M.A.; Korobov, M.V. Elemental analysis of nanodiamonds by inductively-coupled plasma atomic emission spectroscopy. Carbon 2014, 74, 1–13. [Google Scholar] [CrossRef]
- Karpukhina, E.A.; Vlasova, E.A.; Volkov, D.S.; Proskurnin, M.A. Comparative Study of Sample-Preparation Techniques for Quantitative Analysis of the Mineral Composition of Humic Substances by Inductively Coupled Plasma Atomic Emission Spectroscopy. Agronomy 2021, 11, 2453. [Google Scholar] [CrossRef]
- Lu, B.; Wang, X.; Hu, C.; Xing, J.; Zhu, S.; Li, X. On-site rapid quantitative assessment of wet soil nutrients based on laser-induced breakdown spectroscopy fingerprint spectral lines corrected by near-infrared spectroscopy. Comput. Electron. Agric. 2025, 237, 110741. [Google Scholar] [CrossRef]
- Lu, B.; Wang, X.; Hu, C.; Zhu, S.; Li, X. Comparing atomic spectroscopy, molecular spectroscopy and multi-source spectroscopy synergetic fusion for quantitation of total potassium in culture substrates. J. Anal. At. Spectrom. 2025, 40, 1536–1551. [Google Scholar] [CrossRef]
- Newland, T.G.; Pitts, K.; Lewis, S.W. Multimodal spectroscopy with chemometrics for the forensic analysis of Western Australian sandy soils. Forensic Chemistry 2022, 28, 100412. [Google Scholar] [CrossRef]
- Hayes, E.; Greene, D.; O’Donnell, C.; O’Shea, N.; Fenelon, M.A. Spectroscopic technologies and data fusion: Applications for the dairy industry. Front Nutr 2022, 9, 1074688. [Google Scholar] [CrossRef] [PubMed]
- Noda, I. Generalized Two-Dimensional Correlation Method Applicable to Infrared, Raman, and other Types of Spectroscopy. Appl. Spectrosc. 1993, 47, 1329–1336. [Google Scholar] [CrossRef]
- Czarnecki, M.A. Interpretation of Two-Dimensional Correlation Spectra: Science or Art? Appl. Spectrosc. 1998, 52, 1583–1590. [Google Scholar] [CrossRef]
- Müller, M.; Buchet, R.; Fringeli, U.P. 2D-FTIR ATR Spectroscopy of Thermo-Induced Periodic Secondary Structural Changes of Poly-(L)-lysine: A Cross-Correlation Analysis of Phase-Resolved Temperature Modulation Spectra; 1996; Volume 100, pp. 1 0810–10825.
- Noda, I.; Liu, Y.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy Study of Temperature-Dependent Spectral Variations of N -Methylacetamide in the Pure Liquid State. 1. Two-Dimensional Infrared Analysis; 1996; Volume 100, pp. 8674–8680.
- Shin, H.; Jung, Y.M.; Lee, J.; Chang, T.; Ozaki, Y.; Bin Kim, S. Structural Comparison of Langmuir−Blodgett and Spin-Coated Films of Poly(tert-butyl methacrylate) by External Reflection FTIR Spectroscopy and Two-Dimensional Correlation Analysis; 2002; Volume 18.
- Zhang, J.; Tsuji, H.; Noda, I.; Ozaki, Y. Weak Intermolecular Interactions during the Melt Crystallization of Poly(l-lactide) Investigated by Two-Dimensional Infrared Correlation Spectroscopy; 2004; Volume 108.
- Yang, I.-S.; Jung, Y.M.; Bin Kim, S.; V. Klein, M. Two-Dimensional Correlation Analysis of Superconducting YNi2B2C Raman Spectra; 2005; Volume 19, pp. 281–284. 19.
- Jeong Kim, H.; Bin Kim, S.; Kim, J.; Jung, Y.M.; Yeol Ryu, D.; A Lavery, K.; P. Russell, T. Phase Behavior of a Weakly Interacting Block Copolymer by Temperature-Dependent FTIR Spectroscopy; 2006; Volume 39.
- Liu, Y.; Chen, Y.R.; Ozaki, Y. Two-dimensional visible/near-infrared correlation spectroscopy study of thermal treatment of chicken meats. J Agric Food Chem 2000, 48, 901–908. [Google Scholar] [CrossRef]
- Nakano, T.; Shimada, S.; Saitoh, R.; Noda, I. Transient 2D IR Correlation Spectroscopy of the Photopolymerization of Acrylic and Epoxy Monomers. Appl. Spectrosc. 1993, 47, 1337–1342. [Google Scholar] [CrossRef]
- Wang, Y.; Murayama, K.; Myojo, Y.; Tsenkova, R.; Hayashi, N.; Ozaki, Y. Two-Dimensional Fourier Transform Near-Infrared Spectroscopy Study of Heat Denaturation of Ovalbumin in Aqueous Solutions. J. Phys. Chem. B 1998, 102, 6655–6662. [Google Scholar] [CrossRef]
- Ghosh, A.; Enderlein, J. Advanced fluorescence correlation spectroscopy for studying biomolecular conformation. Curr. Opin. Struct. Biol. 2021, 70, 123–131. [Google Scholar] [CrossRef]
- Liu, D.; Hao, Y.; Gao, H.; Yu, H.; Li, Q. Applying synchronous fluorescence spectra with Gaussian band fitting and two-dimensional correlation to characterize structural composition of DOM from soils in an aquatic-terrestrial ecotone. Sci. Total Environ. 2023, 859, 160081. [Google Scholar] [CrossRef]
- Xia, M.-M.; Dong, G.-M.; Yang, R.-J.; Li, X.-C.; Chen, Q. Study on fluorescence interaction between humic acid and PAHs based on two-dimensional correlation spectroscopy. J. Mol. Struct. 2020, 1217, 128428. [Google Scholar] [CrossRef]
- Noda, I.; Dowrey, A.E.; Marcott, C. Recent Developments in Two-Dimensional Infrared (2D IR) Correlation Spectroscopy. Appl. Spectrosc. 1993, 47, 1317–1323. [Google Scholar] [CrossRef]
- Noda, I.; Dowrey, A.E.; Marcott, C.; Story, G.M.; Ozaki, Y. Generalized Two-Dimensional Correlation Spectroscopy. Appl. Spectrosc. 2000, 54, 236A–248A. [Google Scholar] [CrossRef]
- Noda, I.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy—Applications in Vibrational and Optical Spectroscopy. 2004.
- Noda, I. Advances in two-dimensional correlation spectroscopy. Vib. Spectrosc 2004, 36, 143–165. [Google Scholar] [CrossRef]
- Buchet, R.; Wu, Y.; Lachenal, G.; Raimbault, C.; Ozaki, Y. Selecting Two-Dimensional Cross-Correlation Functions to Enhance Interpretation of Near-Infrared Spectra of Proteins. Appl. Spectrosc. 2001, 55, 155–162. [Google Scholar] [CrossRef]
- Noda, I. Progress in two-dimensional (2D) correlation spectroscopy. J. Mol. Struct. 2006, 799, 2–15. [Google Scholar] [CrossRef]
- Yu, H.; Qu, F.; Zhang, X.; Shao, S.; Rong, H.; Liang, H.; Bai, L.; Ma, J. Development of correlation spectroscopy (COS) method for analyzing fluorescence excitation emission matrix (EEM): A case study of effluent organic matter (EfOM) ozonation. Chemosphere 2019, 228, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, X.; Zheng, D.; Song, H.; Han, P.; Yuen, P. Prediction of the Soil Organic Matter (SOM) Content from Moist Soil Using Synchronous Two-Dimensional Correlation Spectroscopy (2D-COS) Analysis. Sensors 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Habibul, N.; Liu, X.Y.; Sheng, G.P.; Yu, H.Q. FTIR and synchronous fluorescence heterospectral two-dimensional correlation analyses on the binding characteristics of copper onto dissolved organic matter. Environ Sci Technol 2015, 49, 2052–2058. [Google Scholar] [CrossRef]
- Yu, G.H.; Tang, Z.; Xu, Y.C.; Shen, Q.R. Multiple fluorescence labeling and two dimensional FTIR-13C NMR heterospectral correlation spectroscopy to characterize extracellular polymeric substances in biofilms produced during composting. Environ Sci Technol 2011, 45, 9224–9231. [Google Scholar] [CrossRef] [PubMed]
- Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A.; Farkhodov, Y.R.; Markeeva, L.B. Thermal stability of organic matter of typical chernozems under different land uses. Soil Tillage Res. 2020, 197, 104500. [Google Scholar] [CrossRef]
- Parlanti, E.; Wörz, K.; Geoffroy, L.; Lamotte, M. Dissolved organic matter fluorescence spectroscopy as a tool to estimate biological activity in a coastal zone submitted to anthropogenic inputs. Org. Geochem. 2000, 31, 1765–1781. [Google Scholar] [CrossRef]
- Coble, P.G. Characterization of marine and terrestrial DOM in seawater using excitation-emission matrix spectroscopy. Mar. Chem. 1996, 51, 325–346. [Google Scholar] [CrossRef]
- Zsolnay, A.; Baigar, E.; Jimenez, M.; Steinweg, B.; Saccomandi, F. Differentiating with fluorescence spectroscopy the sources of dissolved organic matter in soils subjected to drying. Chemosphere 1999, 38, 45–50. [Google Scholar] [CrossRef]
- Ohno, T. Fluorescence inner-filtering correction for determining the humification index of dissolved organic matter. Environ Sci Technol 2002, 36, 742–746. [Google Scholar] [CrossRef]
- Huguet, A.; Vacher, L.; Relexans, S.; Saubusse, S.; Froidefond, J.M.; Parlanti, E. Properties of fluorescent dissolved organic matter in the Gironde Estuary. Org. Geochem. 2009, 40, 706–719. [Google Scholar] [CrossRef]
- Wilson, H.F.; Xenopoulos, M.A. Effects of agricultural land use on the composition of fluvial dissolved organic matter. Nature Geoscience 2008, 2, 37–41. [Google Scholar] [CrossRef]
- McKnight, D.M.; Boyer, E.W.; Westerhoff, P.K.; Doran, P.T.; Kulbe, T.; Andersen, D.T. Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity. Limnol. Oceanogr. 2001, 46, 38–48. [Google Scholar] [CrossRef]
- Cory, R.M.; McKnight, D.M. Fluorescence spectroscopy reveals ubiquitous presence of oxidized and reduced quinones in dissolved organic matter. Environ Sci Technol 2005, 39, 8142–8149. [Google Scholar] [CrossRef]
- Baker, A. Fluorescence excitation-emission matrix characterization of some sewage-impacted rivers. Environ Sci Technol 2001, 35, 948–953. [Google Scholar] [CrossRef]
- Miller, M.P.; McKnight, D.M.; Cory, R.M.; Williams, M.W.; Runkel, R.L. Hyporheic exchange and fulvic acid redox reactions in an Alpine stream/wetland ecosystem, Colorado Front Range. Environ Sci Technol 2006, 40, 5943–5949. [Google Scholar] [CrossRef] [PubMed]
- Kalbitz, K.; Angers, D.; Kaiser, K.; Chantigny, M. Extraction and Characterization of Dissolved Organic Matter. In Soil Sampling and Methods of Analysis, Second Edition, Carter, M.R., Gregorich, E.G., Eds.; CRC Press: Boca Raton, 2007. [Google Scholar]
- Chantigny, M.H.; Harrison-Kirk, T.; Curtin, D.; Beare, M. Temperature and duration of extraction affect the biochemical composition of soil water-extractable organic matter. Soil Biol. Biochem. 2014, 75, 161–166. [Google Scholar] [CrossRef]
- Proskurnin, M.A.; Volkov, D.S.; Rogova, O.B. Temperature Dependences of IR Spectral Bands of Humic Substances of Silicate-Based Soils. Agronomy 2023, 13, 1740. [Google Scholar] [CrossRef]
- Krivoshein, P.K.; Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. FTIR photoacoustic spectroscopy for identification and assessment of soil components: Chernozems and their size fractions. Photoacoustics 2020, 18, 100162. [Google Scholar] [CrossRef] [PubMed]
- Saleem, S.; Ashiq, M.N.; Manzoor, S.; Ali, U.; Liaqat, R.; Algahtani, A.; Mujtaba, S.; Tirth, V.; Alsuhaibani, A.M.; Refat, M.S.; et al. Analysis and characterization of opto-electronic properties of iron oxide (Fe2O3) with transition metals (Co, Ni) for the use in the photodetector application. Journal of Materials Research and Technology 2023, 25, 6150–6166. [Google Scholar] [CrossRef]
- Peltre, C.; Bruun, S.; Du, C.; Thomsen, I.K.; Jensen, L.S. Assessing soil constituents and labile soil organic carbon by mid-infrared photoacoustic spectroscopy. Soil Biol. Biochem. 2014, 77, 41–50. [Google Scholar] [CrossRef]
- Bryant, J.I. Vibrational Spectrum of Sodium Azide Single Crystals. The Journal of Chemical Physics 1964, 40, 3195–3203. [Google Scholar] [CrossRef]
- Attenuated Total Reflectance Infrared (ATR-IR) Spectrum of Sodium azide. Available online: https://spectrabase.com/spectrum/9i9JWsCo4j4 (accessed on 18.05).
- Koike, C.; Noguchi, R.; Chihara, H.; Suto, H.; Ohtaka, O.; Imai, Y.; Matsumoto, T.; Tsuchiyama, A. Infrared Spectra of Silica Polymorphs and the Conditions of Their Formation. Astrophys. J. 2013, 778, 60. [Google Scholar] [CrossRef]
- Yu, H.-G.; Nyman, G. The Infrared and Uv-Visible Spectra of Polycyclic Aromatic Hydrocarbons Containing (5, 7)-Member Ring Defects: A Theoretical Study. Astrophys. J. 2012, 751, 3. [Google Scholar] [CrossRef]
- Changwen, D.; Jianmin, Z.; Goyne, K.W. Organic and inorganic carbon in paddy soil as evaluated by mid-infrared photoacoustic spectroscopy. PLoS ONE 2012, 7, e43368. [Google Scholar] [CrossRef]
- Lucas, S.; Tognonvi, M.T.; Gelet, J.L.; Soro, J.; Rossignol, S. Interactions between silica sand and sodium silicate solution during consolidation process. Journal of Non-Crystalline Solids 2011, 357, 1310–1318. [Google Scholar] [CrossRef]
- Changwen, D.; Jing, D.; Jianmin, Z.; Huoyan, W.; Xiaoqin, C. Characterization of Greenhouse Soil Properties Using Mid-infrared Photoacoustic Spectroscopy. Spectrosc. Lett. 2011, 44, 359–368. [Google Scholar] [CrossRef]
- Calderón, F.J.; Reeves, J.B.; Collins, H.P.; Paul, E.A. Chemical Differences in Soil Organic Matter Fractions Determined by Diffuse--Reflectance Mid--Infrared Spectroscopy. Soil Sci. Soc. Am. J. 2011, 75, 568–579. [Google Scholar] [CrossRef]
- Calderón, F.J.; Mikha, M.M.; Vigil, M.F.; Nielsen, D.C.; Benjamin, J.G.; Reeves, J.B. Diffuse-Reflectance Mid-infrared Spectral Properties of Soils under Alternative Crop Rotations in a Semi-arid Climate. Commun. Soil Sci. Plant Anal. 2011, 42, 2143–2159. [Google Scholar] [CrossRef]
- Hofmeister, A.M.; Bowey, J.E. Quantitative Infrared Spectra of Hydrosilicates and Related Minerals. Mon. Not. R. Astron. Soc. 2006, 367, 577–591. [Google Scholar] [CrossRef]
- Tan, C.Z. Optical interference in overtones and combination bands in α-quartz. Journal of Physics and Chemistry of Solids 2003, 64, 121–125. [Google Scholar] [CrossRef]
- Madejová, J.; Komadel, P. Baseline Studies of the Clay Minerals Society Source Clays: Infrared Methods. Clays Clay Miner. 2024, 49, 410–432. [Google Scholar] [CrossRef]
- Russell, J.D.; Fraser, A.R. Infrared methods. In Clay Mineralogy: Spectroscopic and Chemical Determinative Methods, Wilson, M.J., Ed.; Springer Netherlands: Dordrecht, 1994; pp. 11–67.
- Kronenberg, A.K. Chapter 4. Hydrogen Speciation and Chemical Weakening of Quartz. In Silica; De Gruyter: Berlin, Germany, 1994; pp. 123–176. [Google Scholar]
- Nguyen, T.T.; Janik, L.J.; Raupach, M. Diffuse reflectance infrared fourier transform (DRIFT) spectroscopy in soil studies. Soil Res. 1991, 29, 49–67. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Elsevier Science: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Colthup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy; Elsevier Science: 1990.
- Asselin, M.; Sandorfy, C. Anharmonicity and Hydrogen Bonding. The in-plane OH Bending and its Combination with the OH Stretching Vibration. Canadian Journal of Chemistry 1971, 49, 1539–1544. [Google Scholar] [CrossRef]
- Bock, J.A.N.; Su, G.J. Interpretation of the Infrared Spectra of Fused Silica. J. Am. Ceram. Soc. 1970, 53, 69–73. [Google Scholar] [CrossRef]
- Spitzer, W.G.; Kleinman, D.A. Infrared Lattice Bands of Quartz. Phys. Rev. 1961, 121, 1324–1335. [Google Scholar] [CrossRef]
- So, R.T.; Blair, N.E.; Masterson, A.L. Carbonate mineral identification and quantification in sediment matrices using diffuse reflectance infrared Fourier transform spectroscopy. Environ. Chem. Lett. 2020, 18, 1725–1730. [Google Scholar] [CrossRef]
- Schütz, M.K.; Lopes, N.F.; Cenci, A.; Ketzer, J.M.M.; Einloft, S.; Dullius, J.; Ligabue, R. Influence of Alkaline Additives and Buffers on Mineral Trapping of CO2 under Mild Conditions. Chemical Engineering & Technology 2018, 41, 573–579. [Google Scholar] [CrossRef]
- Max, J.J.; Chapados, C. Isotope effects in liquid water by infrared spectroscopy. III. H2O and D2O spectra from 6000 to 0 cm(-1). J. Chem. Phys. 2009, 131, 184505. [Google Scholar] [CrossRef]
- Invernizzi, C.; Rovetta, T.; Licchelli, M.; Malagodi, M. Mid and Near-Infrared Reflection Spectral Database of Natural Organic Materials in the Cultural Heritage Field. Int. J. Anal. Chem. 2018, 2018, 7823248. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Anbalagan, G.; Pandi, S. Raman and infrared spectra of carbonates of calcite structure. J. Raman Spectrosc. 2006, 37, 892–899. [Google Scholar] [CrossRef]
- Myszka, B.; Schussler, M.; Hurle, K.; Demmert, B.; Detsch, R.; Boccaccini, A.R.; Wolf, S.E. Phase-specific bioactivity and altered Ostwald ripening pathways of calcium carbonate polymorphs in simulated body fluid. RSC Adv 2019, 9, 18232–18244. [Google Scholar] [CrossRef]
- Rogovska, N.; Laird, D.A.; Chiou, C.-P.; Bond, L.J. Development of field mobile soil nitrate sensor technology to facilitate precision fertilizer management. Precision Agriculture 2018, 20, 40–55. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry, Meyers, R.A., McKelvy, M.L., Eds.; Wiley: 2000.
- Traore, M.; Kaal, J.; Martinez Cortizas, A. Differentiation between pine woods according to species and growing location using FTIR-ATR. Wood Sci Technol 2018, 52, 487–504. [Google Scholar] [CrossRef]
- Bishop, J.L.; Lane, M.D.; Dyar, M.D.; King, S.J.; Brown, A.J.; Swayze, G.A. Spectral properties of Ca-sulfates: Gypsum, bassanite, and anhydrite. Am. Mineral. 2014, 99, 2105–2115. [Google Scholar] [CrossRef]
- Kunimatsu, K.; Samant, M.G.; Seki, H. In-situ FT-IR spectroscopic study of bisulfate and sulfate adsorption on platinum electrodes. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1989, 258, 163–177. [Google Scholar] [CrossRef]
- Ellerbrock, R.; Stein, M.; Schaller, J. Comparing amorphous silica, short-range-ordered silicates and silicic acid species by FTIR. Sci. Rep. 2022, 12, 11708. [Google Scholar] [CrossRef] [PubMed]
- Ellerbrock, R.H.; Stein, M.; Schaller, J. Comparing silicon mineral species of different crystallinity using Fourier transform infrared spectroscopy. Front. Environ. Chem. 2024, 5. [Google Scholar] [CrossRef]
- Launer, P.J.; Arkles, B. Infrared Analysis of Organosilicon Compounds. In Silicon Compounds. In Silicon Compounds: Silanes & Silicones, 3rd ed.; Gelest Inc.: Morrisville, PA, 2013. [Google Scholar]
- Hofmeister, A.M.; Keppel, E.; Speck, A.K. Absorption and reflection infrared spectra of MgO and other diatomic compounds. Mon. Not. R. Astron. Soc. 2003, 345, 16–38. [Google Scholar] [CrossRef]
- Taylor, W.R. Application of infrared spectroscopy to studies of silicate glass structure: Examples from the melilite glasses and the systems Na2O-SiO2 and Na2O-Al2O3-SiO2. Journal of Earth System Science 1990, 99, 99–117. [Google Scholar] [CrossRef]
- Brusentsova, T.N.; Peale, R.E.; Maukonen, D.; Harlow, G.E.; Boesenberg, J.S.; Ebel, D. Far infrared spectroscopy of carbonate minerals. Am. Mineral. 2010, 95, 1515–1522. [Google Scholar] [CrossRef]
- Rodionov, A.N. Vibration Spectra and Structures of the Simplest Aromatic Derivatives of Group I–VI Elements. Russian Chemical Reviews 1973, 42, 998–1010. [Google Scholar] [CrossRef]
- Parolo, M.E.; Savini, M.C.; Loewy, R.M. Characterization of soil organic matter by FT-IR spectroscopy and its relationship with chlorpyrifos sorption. J Environ Manage 2017, 196, 316–322. [Google Scholar] [CrossRef]
- Chang, Z.; Yue, Y.; Qu, Y.; Li, S.; Wu, F.; Liu, H. Properties of Aluminosilicate Glasses Prepared by Red Mud with Various [Al2O3]/[CaO] Mass Ratios. Journal of Wuhan University of Technology-Mater. Sci. Ed. 2018, 33, 363–367. [Google Scholar] [CrossRef]
- Rana, D.; Upterworth, A.L.; Winghart, M.O.; Sebastiani, D.; Nibbering, E.T.J. Azide Anion Interactions with Imidazole and 1-Methylimidazole in Dimethyl Sulfoxide. J Phys Chem B 2025, 129, 8192–8200. [Google Scholar] [CrossRef]
- Richter, T.A.; Malhotra, M.L.; Möller, K.D. Far infrared absorption of lattice modes of solid sodium azide. Materials Research Bulletin 1970, 5, 203–206. [Google Scholar] [CrossRef]
- Artz, R.R.E.; Chapman, S.J.; Jean Robertson, A.H.; Potts, J.M.; Laggoun-Défarge, F.; Gogo, S.; Comont, L.; Disnar, J.-R.; Francez, A.-J. FTIR spectroscopy can be used as a screening tool for organic matter quality in regenerating cutover peatlands. Soil Biol. Biochem. 2008, 40, 515–527. [Google Scholar] [CrossRef]
- Latham, K.G.; Matsakas, L.; Figueira, J.; Rova, U.; Christakopoulos, P.; Jansson, S. Examination of how variations in lignin properties from Kraft and organosolv extraction influence the physicochemical characteristics of hydrothermal carbon. Journal of Analytical and Applied Pyrolysis 2021, 155, 105095. [Google Scholar] [CrossRef]
- Javier-Astete, R.; Jimenez-Davalos, J.; Zolla, G. Determination of hemicellulose, cellulose, holocellulose and lignin content using FTIR in Calycophyllum spruceanum (Benth.) K. Schum. and Guazuma crinita Lam. PLoS ONE 2021, 16, e0256559. [Google Scholar] [CrossRef]
- Qiao, W.; Wang, X.; Liu, X.; Zhen, X.; Guo, J.; Wang, S.; Yang, F.; Chen, G.; Zhang, B. Characterization of Dissolved Organic Matter in Deep Geothermal Water from Different Burial Depths Based on Three-Dimensional Fluorescence Spectra. Water 2017, 9, 266. [Google Scholar] [CrossRef]
- Sierra, M.M.; Giovanela, M.; Parlanti, E.; Soriano-Sierra, E.J. Fluorescence fingerprint of fulvic and humic acids from varied origins as viewed by single-scan and excitation/emission matrix techniques. Chemosphere 2005, 58, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Moore, O.C.; Holt, A.D.; Richards, L.A.; McKenna, A.M.; Spencer, R.G.M.; Lapworth, D.J.; Polya, D.A.; Lloyd, J.R.; van Dongen, B.E. Characterisation of dissolved organic matter in two contrasting arsenic-prone sites in Kandal Province, Cambodia. Org. Geochem. 2024, 198, 104886. [Google Scholar] [CrossRef]
- He, J.; Wu, X.; Zhi, G.; Yang, Y.; Wu, L.; Zhang, Y.; Zheng, B.; Qadeer, A.; Zheng, J.; Deng, W.; et al. Fluorescence characteristics of DOM and its influence on water quality of rivers and lakes in the Dianchi Lake basin. Ecol. Indic. 2022, 142, 109088. [Google Scholar] [CrossRef]
- Wang, X.; Tong, Y.; Chang, Q.; Lu, J.; Ma, T.; Zhou, F.; Li, J. Source identification and characteristics of dissolved organic matter and disinfection by-product formation potential using EEM-PARAFAC in the Manas River, China. RSC Adv 2021, 11, 28476–28487. [Google Scholar] [CrossRef]
- Petrofanov, V.L. Role of the soil particle-size fractions in the sorption and desorption of potassium. Eurasian Soil Sci. 2012, 45, 598–611. [Google Scholar] [CrossRef]
- Semenov, V.M.; Tulina, A.S.; Semenova, N.A.; Ivannikova, L.A. Humification and nonhumification pathways of the organic matter stabilization in soil: A review. Eurasian Soil Sci. 2013, 46, 355–368. [Google Scholar] [CrossRef]
- Kabata-Pendias, A. Trace Elements in Soils and Plants; CRC Press: 2010.
- Liu, Y.; Sun, J.; Wang, X.; Liu, X.; Wu, X.; Chen, Z.; Gu, T.; Wang, W.; Yu, L.; Guo, Y.; et al. Fluorescence Characteristics of Chromophoric Dissolved Organic Matter in the Eastern Indian Ocean: A Case Study of Three Subregions. Front. Mar. Sci. 2021, 8, 742595. [Google Scholar] [CrossRef]
Figure 1.
Regions of the excitation–emission matrix that correspond to certain groups of compounds. Green frames correspond to previously used boundaries [84,85] and light magenta frames, to broader boundaries (marked with asterisks) also used in this study; fraction of 0.01–0.03 µm.
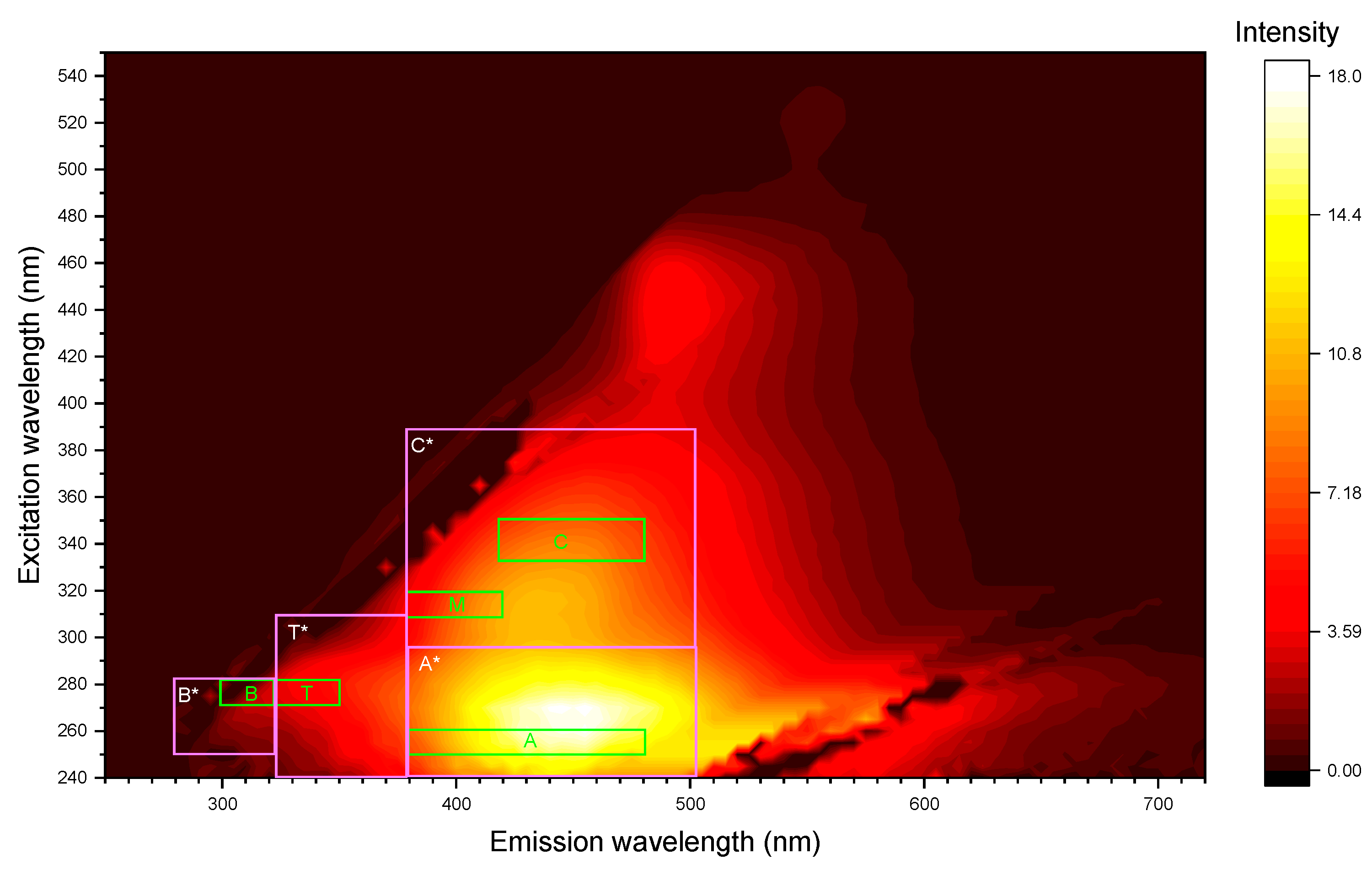
Figure 2.
Absorption spectra of flushing water and filtrates obtained during blank experiments using polycarbonate analytical track membranes with different pore sizes and (inset) absorption spectra (with a logarithmic Y axis) of a series of filtrates obtained sequentially (violet, 1; blue, 2; green, 3; yellow, 4; and red, 5) in blank experiments using polycarbonate analytical track membranes with a pore size of 0.05 μm.
Figure 2.
Absorption spectra of flushing water and filtrates obtained during blank experiments using polycarbonate analytical track membranes with different pore sizes and (inset) absorption spectra (with a logarithmic Y axis) of a series of filtrates obtained sequentially (violet, 1; blue, 2; green, 3; yellow, 4; and red, 5) in blank experiments using polycarbonate analytical track membranes with a pore size of 0.05 μm.
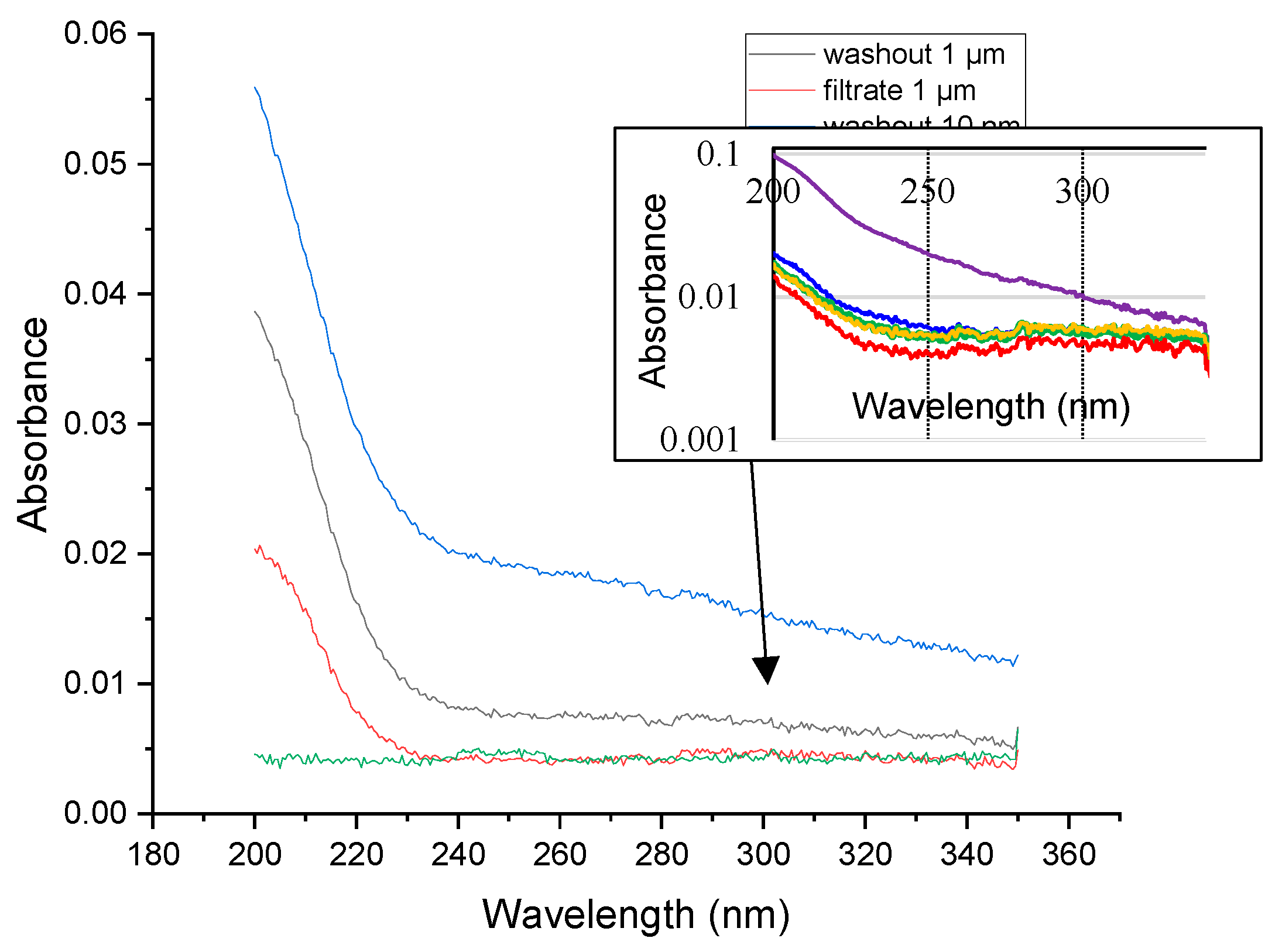
Figure 3.
Full smoothed normalized IR spectra (4000–100 cm–1) for (a) four large fractions with a particle size of 1 to 10 µm, azide bands removed. For clarity, the abscissa axis is represented on a logarithmic scale, and the spectra are shifted (orange rectangles indicate areas where most absorption bands of narrow fine fractions appear) and (b) fractions with a particle size of less than 1 μm.
Figure 3.
Full smoothed normalized IR spectra (4000–100 cm–1) for (a) four large fractions with a particle size of 1 to 10 µm, azide bands removed. For clarity, the abscissa axis is represented on a logarithmic scale, and the spectra are shifted (orange rectangles indicate areas where most absorption bands of narrow fine fractions appear) and (b) fractions with a particle size of less than 1 μm.
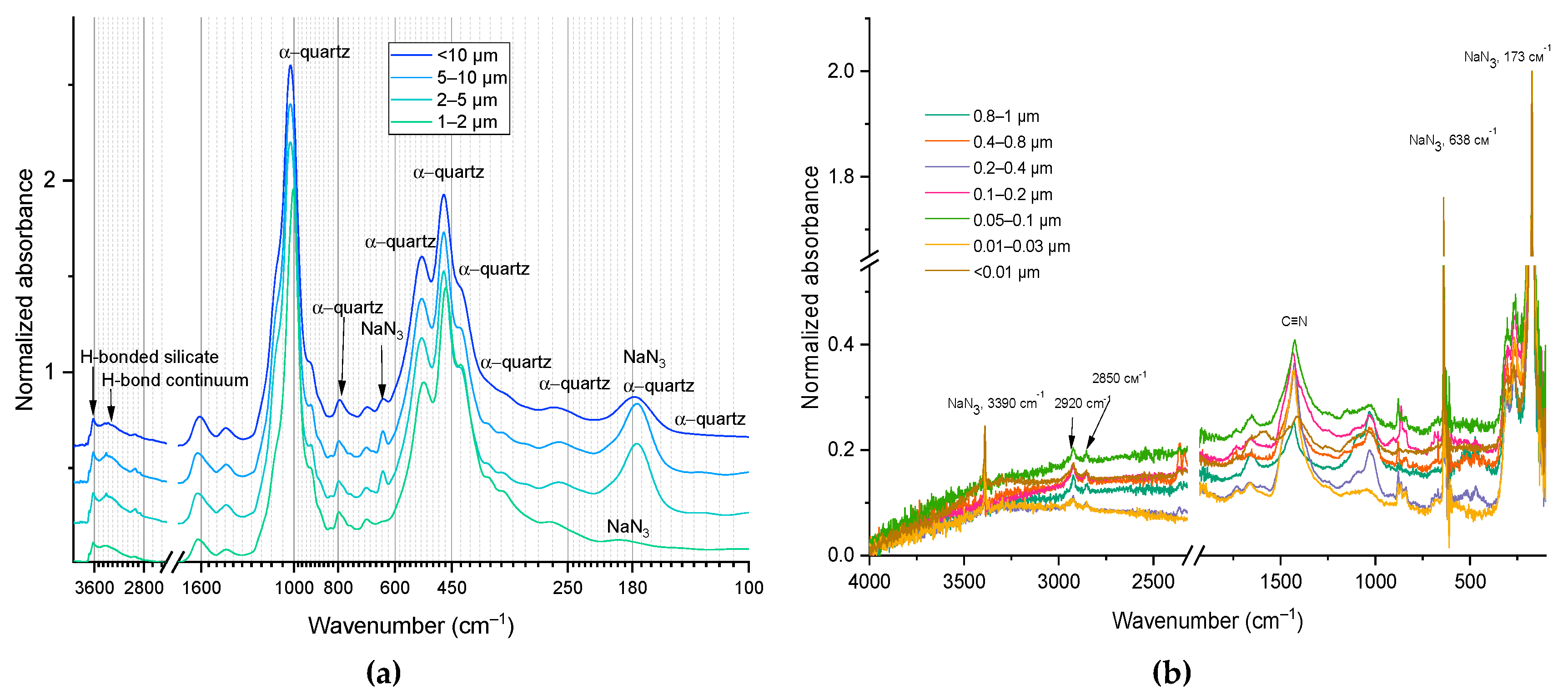
Figure 4.
Smoothed IR spectra with baseline correction for the selected range of wavenumbers: (a) 3100–2700 cm–1 and (b) 1800–1200 cm–1. For the purposes of 2D-COS data processing, the numerical values of the average fraction size are shown.
Figure 4.
Smoothed IR spectra with baseline correction for the selected range of wavenumbers: (a) 3100–2700 cm–1 and (b) 1800–1200 cm–1. For the purposes of 2D-COS data processing, the numerical values of the average fraction size are shown.
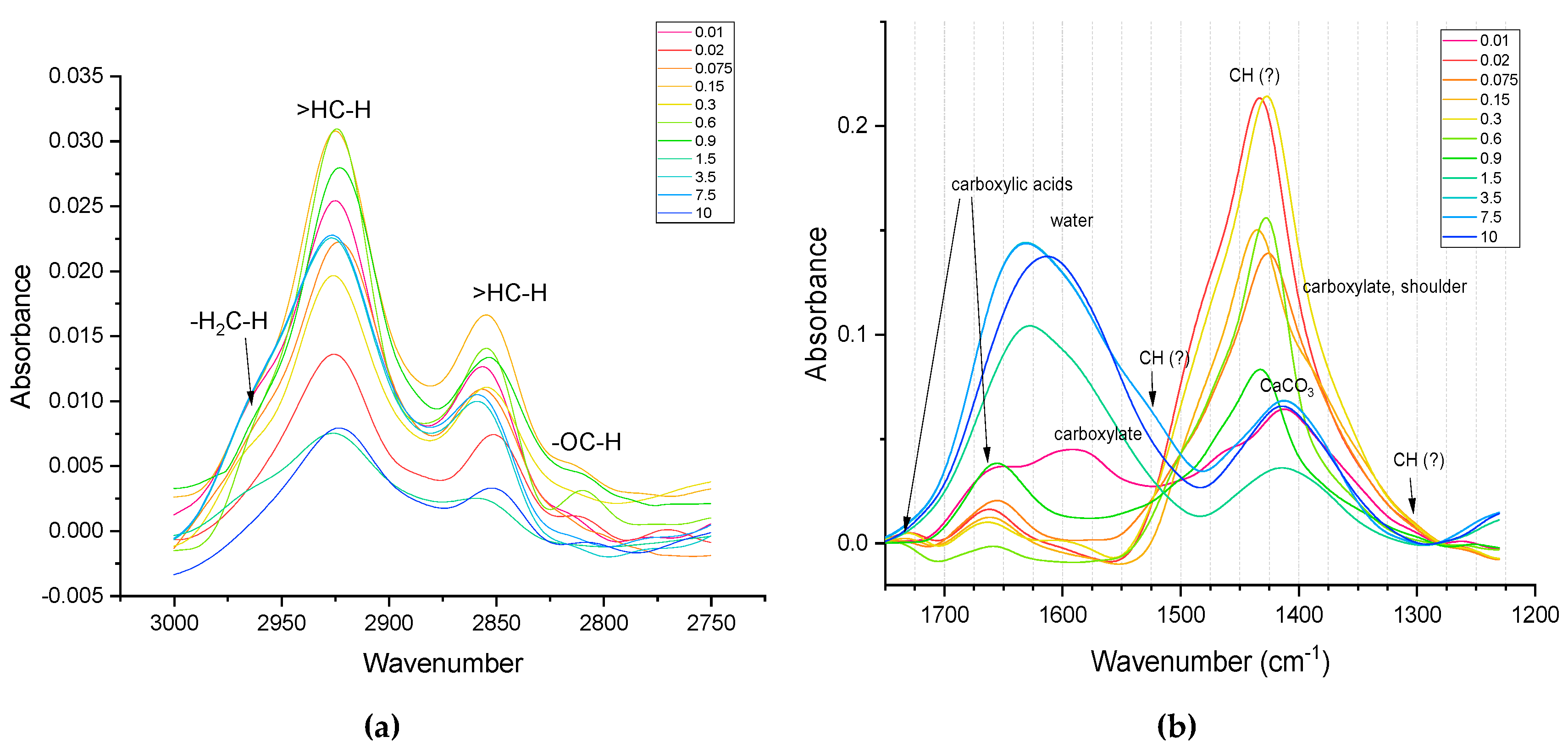
Figure 5.
Smoothed spectra with baseline correction for the selected range of wavenumbers; (a) 930–810 and (b) 710–650 cm–1. For the purposes of 2D-COS data processing, the numerical values of the average fraction size are shown.
Figure 5.
Smoothed spectra with baseline correction for the selected range of wavenumbers; (a) 930–810 and (b) 710–650 cm–1. For the purposes of 2D-COS data processing, the numerical values of the average fraction size are shown.
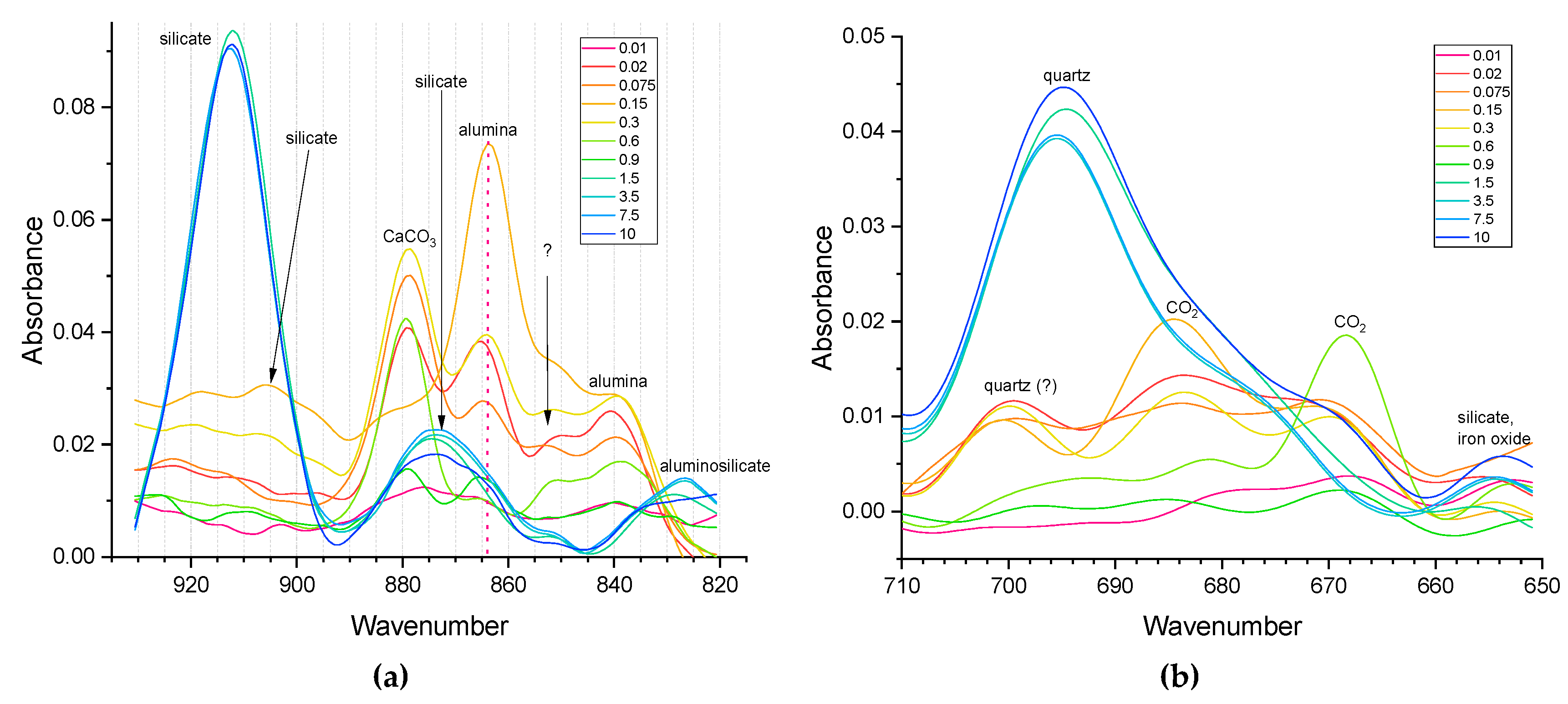
Figure 8.
Excitation–emission matrices for obtained narrow size fractions of chernozem soils.
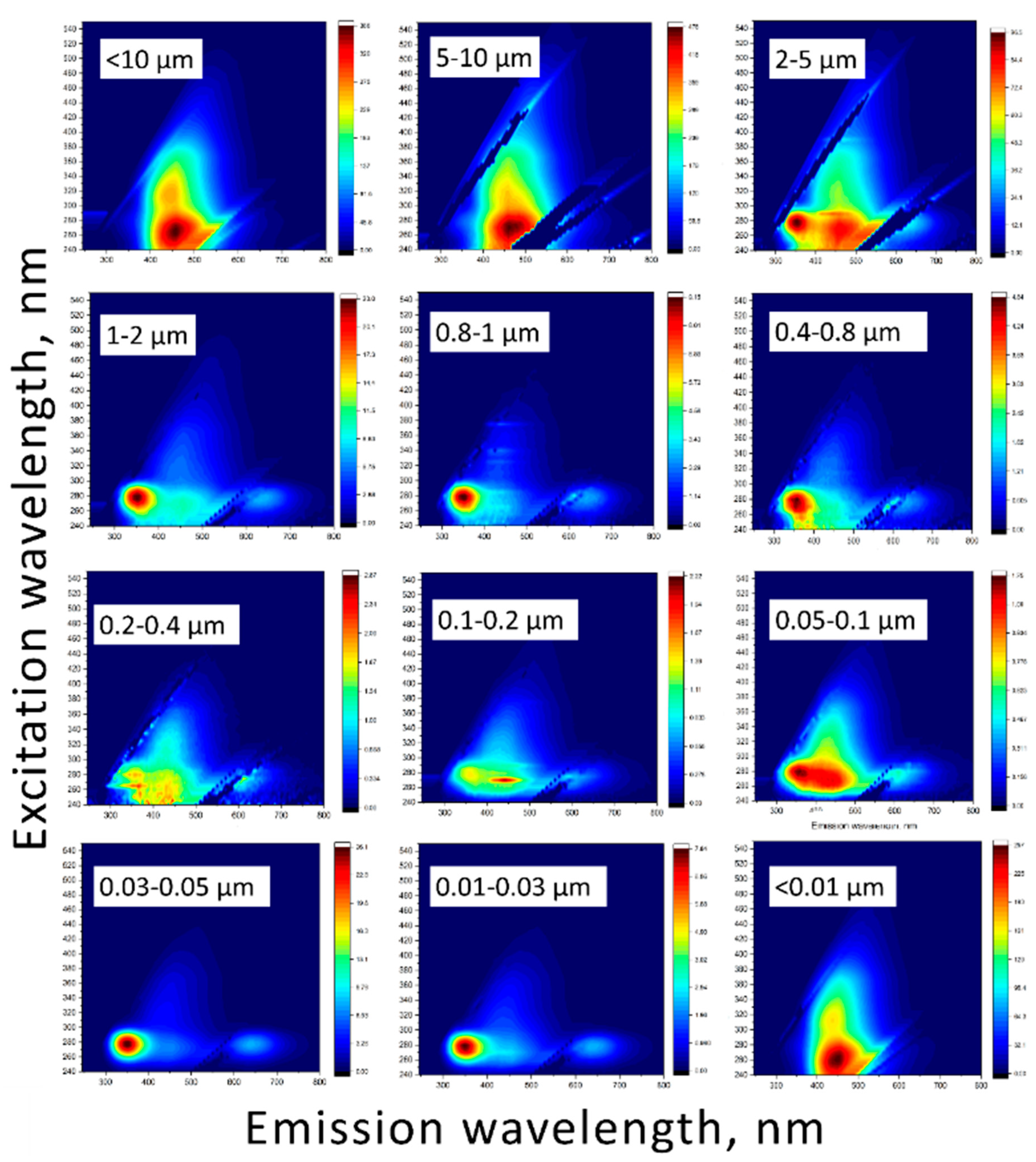
Figure 9.
Distribution of fluorescent organic matter components, (a) region and peak area ratios and (b) integral indicators (freshness index (BIX) (β/α) and fluorescence index (FI)) between narrow fractions. Inset in (a) are relative abundances of humic- (HS) and non-humic (BE) components according to sums of AC–BT intensities; red line in (b) marks the value of 1.
Figure 9.
Distribution of fluorescent organic matter components, (a) region and peak area ratios and (b) integral indicators (freshness index (BIX) (β/α) and fluorescence index (FI)) between narrow fractions. Inset in (a) are relative abundances of humic- (HS) and non-humic (BE) components according to sums of AC–BT intensities; red line in (b) marks the value of 1.
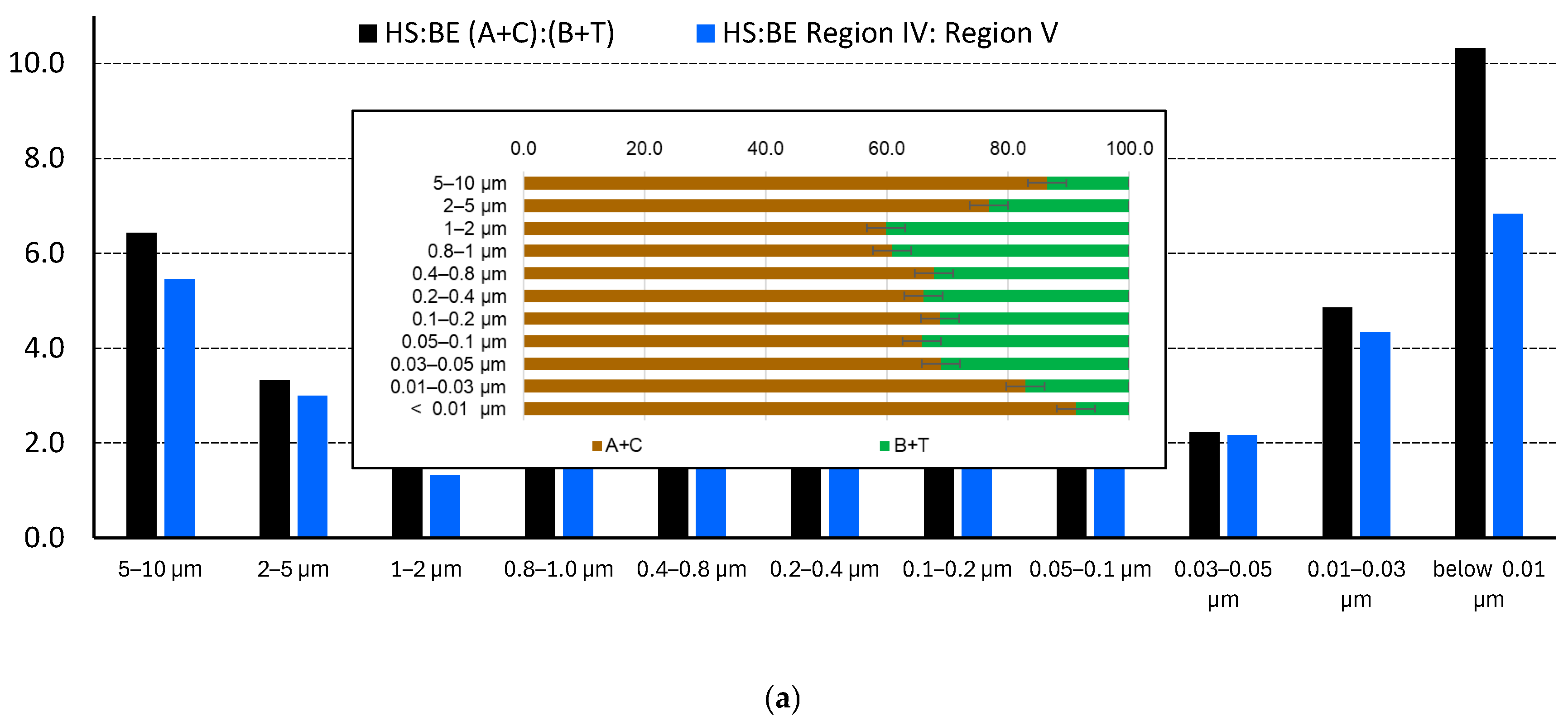
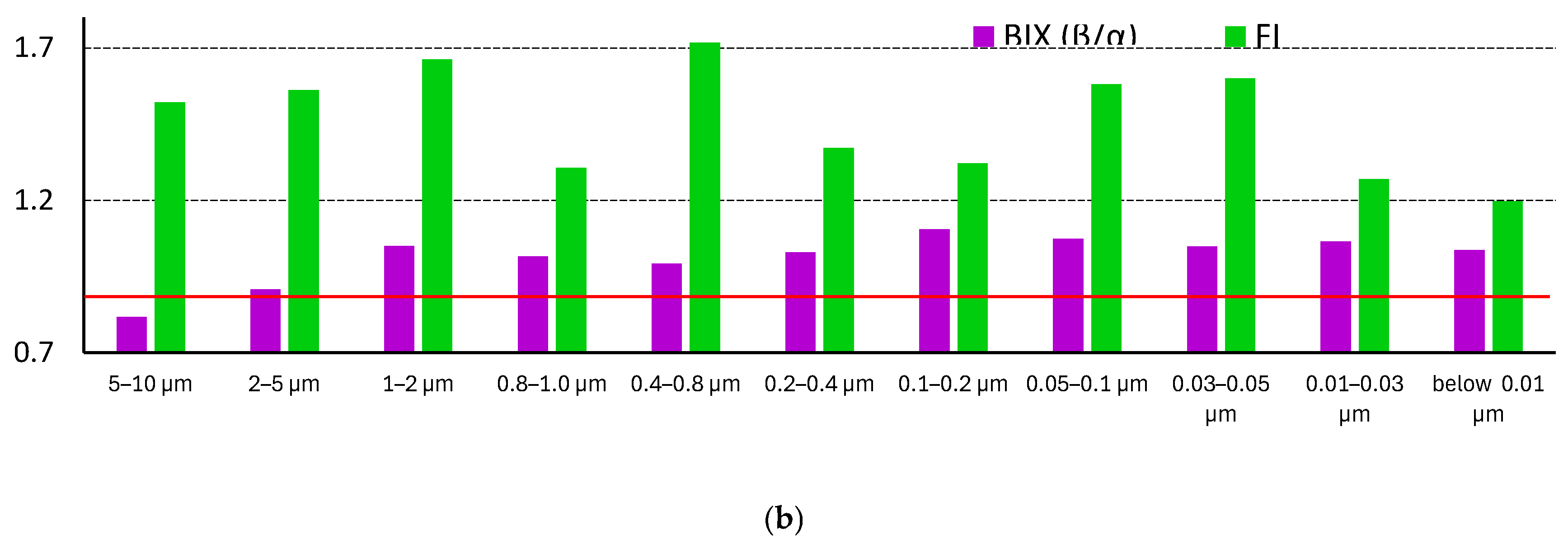
Figure 10.
Correlations of area intensities and fluorescence indicators (humification index (HIXEM), freshness index (BIX) (β/α), fluorescence index (FI), and T/ C peak ratio), divided into mainly highly humified (brown background) and freshly created SOM (green background).
Figure 10.
Correlations of area intensities and fluorescence indicators (humification index (HIXEM), freshness index (BIX) (β/α), fluorescence index (FI), and T/ C peak ratio), divided into mainly highly humified (brown background) and freshly created SOM (green background).
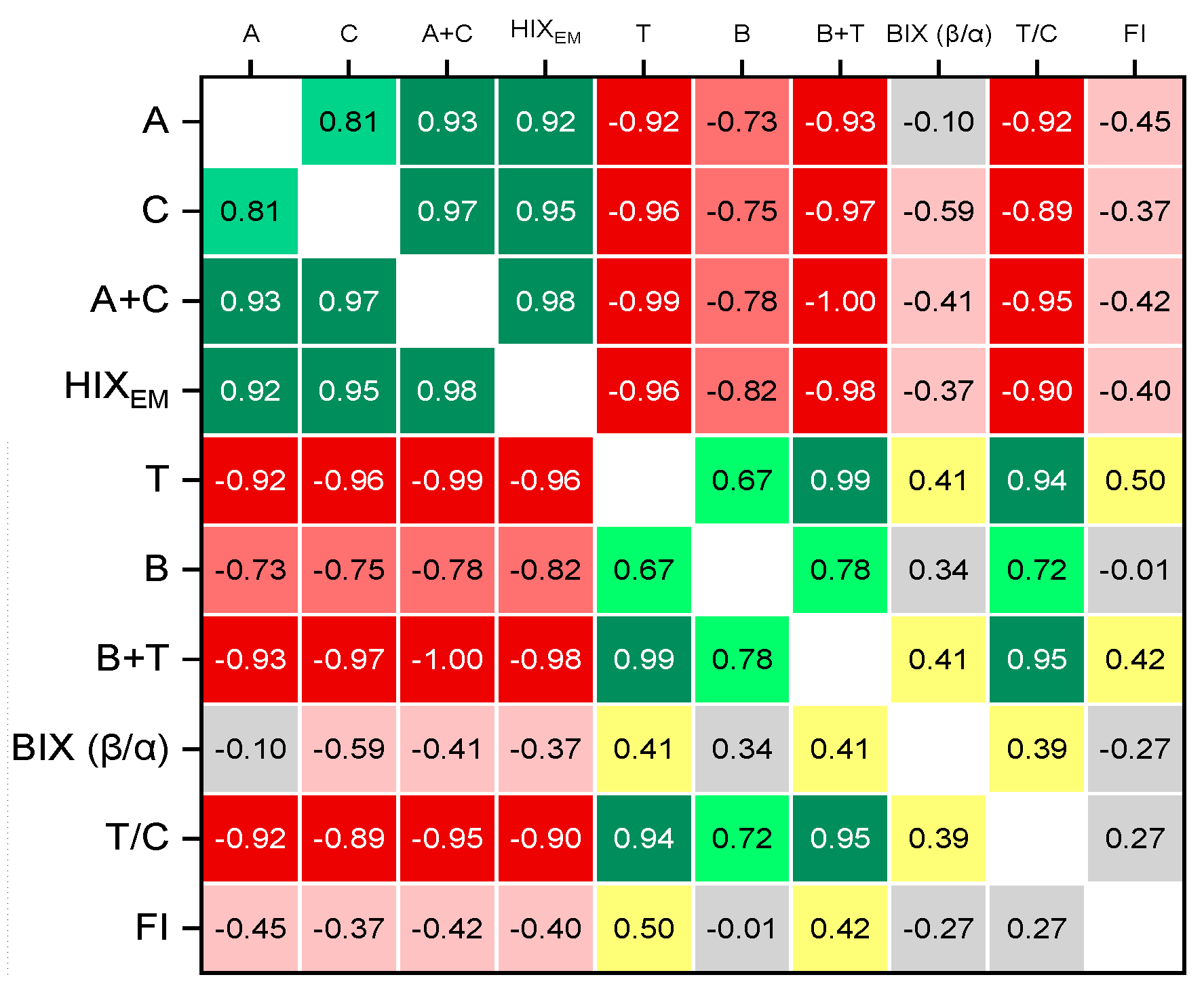
Figure 11.
Synchronous heterospectral IR–fluorescence 2D-COS maps of particulate SOM for the ranges (a) 300–500 nm (fluorescence) and 650–750 cm–1 (FTIR); (b) 300–500 nm (fluorescence) and 800–950 cm–1 (FTIR); (c) 300–500 nm (fluorescence) and 1800–1200 cm–1 (FTIR); and (d) 300–500 nm (fluorescence) and 3000–2700 cm–1 (FTIR).
Figure 11.
Synchronous heterospectral IR–fluorescence 2D-COS maps of particulate SOM for the ranges (a) 300–500 nm (fluorescence) and 650–750 cm–1 (FTIR); (b) 300–500 nm (fluorescence) and 800–950 cm–1 (FTIR); (c) 300–500 nm (fluorescence) and 1800–1200 cm–1 (FTIR); and (d) 300–500 nm (fluorescence) and 3000–2700 cm–1 (FTIR).
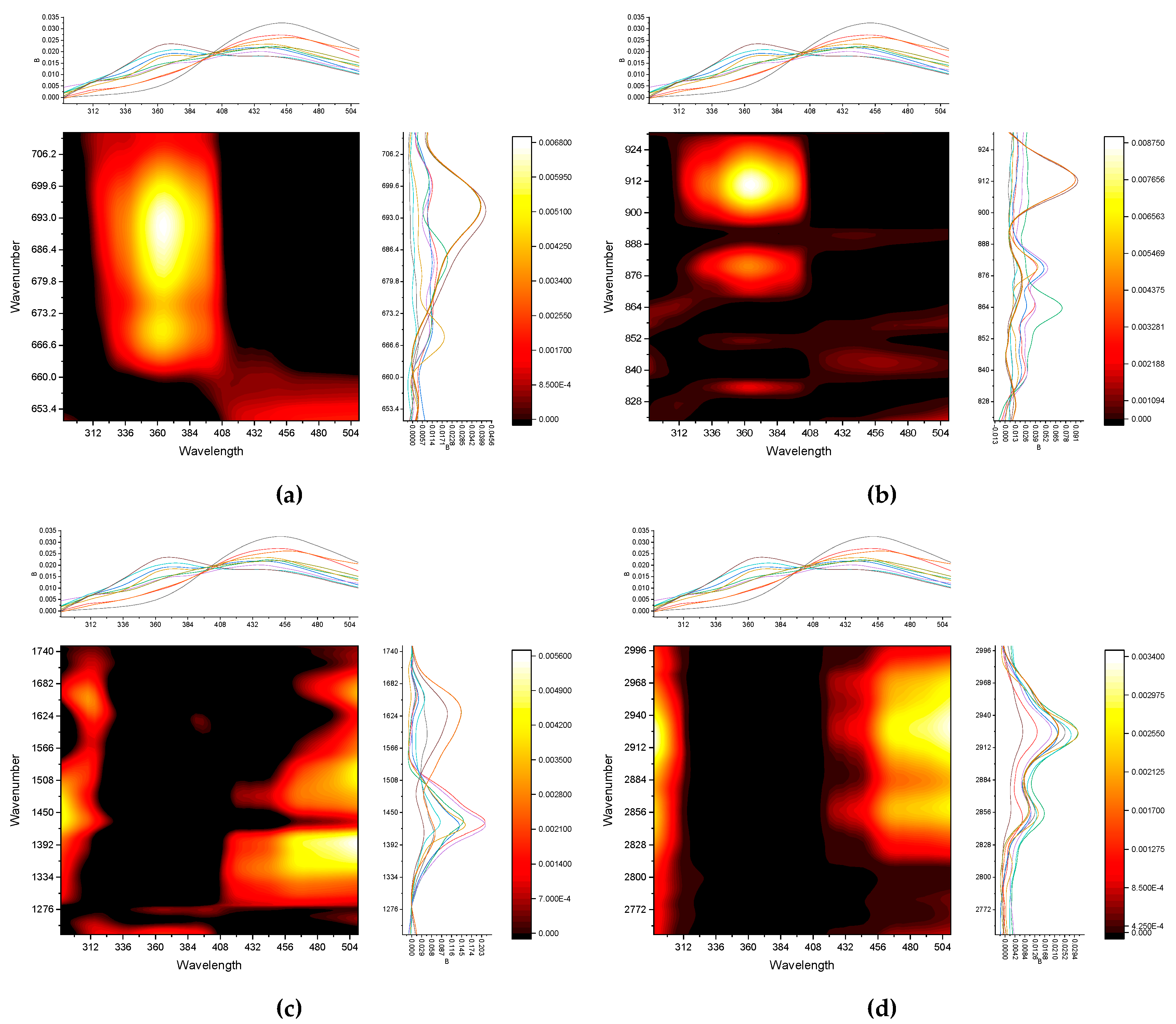
Figure 12.
Correlations of fluorescence indexes, molar fractions of elements in fractions with ICP-AES except for the fraction of 5–10 μm due to high amounts of all the elements in this fraction that distort overall correlations.
Figure 12.
Correlations of fluorescence indexes, molar fractions of elements in fractions with ICP-AES except for the fraction of 5–10 μm due to high amounts of all the elements in this fraction that distort overall correlations.
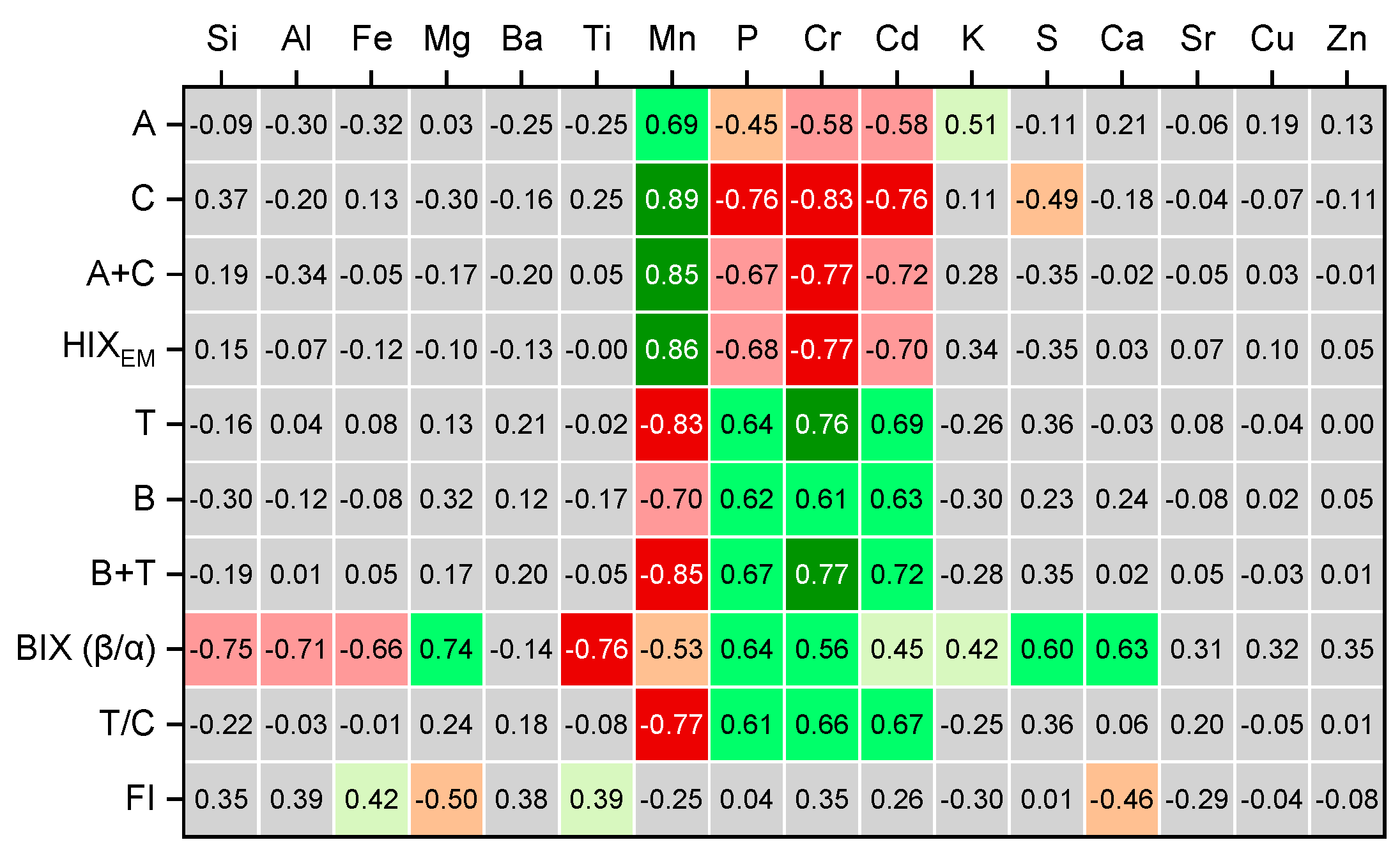

Table 1.
Conditions for registration of ATR-FTIR spectra.
| Parameter | Value |
| Measuring range, cm–1 | 4000–100 |
| Resolution, cm–1 | 2 |
| Number of background scans | 128 |
| Number of sample scans | 128 |
| Phase resolution | 16 |
| Phase Correction | Mertz |
| Apodization Function | Blackman–Harris, 3-term |
| Zero fill factor | 2 |
| Aperture, mm | 8 |
| Source | mid-IR |
| Beam Splitter | wide mid-IR–far IR (Si) |
Table 2.
Regions of expanded (marked with the asterisk) excitation (Ex)-emission (Em) matrix corresponding to certain groups of compounds [84,85].
| Band name | Ex wavelength range, nm | Em wavelength range, nm | Groups of substances |
| А* | 240–300 | 380–505 | humic-like |
| В* | 250–285 | 280–320 | tyrosine-, protein-like |
| С* | 300–390 | 380–505 | humic-like |
| Т* | 250–285 | 320–400 | tryptophan-, phenol-, protein-like |
Table 3.
Band assignments for WEOM size fractions by ATR−FTIR [35,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138].
| Wavenumber, cm–1 | Inorganic (matrix) constituents | Organic constituents |
| 3695–3690 | unbonded SiO–H stretch, tilted (kaolinite, clay) [113] | — |
| 3625–3620 | hydrogen-bonded SiO–H…H2O stretch (amorphous species) [111,112] | — |
| 3390 | combination band NaN3 ≅ 1350+2043 [100,101] | — |
| 3390 and 3270 | antisymmetric and symmetric hydrogen-bond ensembles | — |
| 2960 | — | antisymmetric stretch of (alkene) methylene groups [104,106,107,108] |
| 2940–2920 and 2855–2850 |
— | antisymmetric and symmetric stretch of methylene groups [104,106,107,108] |
| 2810 | — | (?) C–H stretching adjacent to carbonyls |
| 2770 | — | — |
| 2670 | — | hydrogen-bonded O-H stretching vibrations in carboxylic acids [50] |
| 2380 | ambient CO2 | — |
| 2340 | ambient CO2 | — |
| 2040 | NaN3 | — |
| 1740–1730 | — | Carbonyl, conjugated or esters |
| 1680–1670 | — | Carboxyl, antisymmetric stretch, or Amide I alkene –C=C– stretch, (?) substituted aromatics |
| 1645–1630 | bend (v2) of the covalent bonds of liquid absorbed water [122] and OH groups, O–H stretch | — |
| 1620–1615 | hydrogen-bonded sioh…h2o, ho–h stretch (amorphous) [113] | — |
| 1580 | — | Carboxylate, antisymmetric stretch |
| 1520 | — | Aromatic C=C stretch Amide II band (primarily –N–H bending and C–N stretching) SiO2 combination band [110] |
| 1500 | — | aromatic C=C stretch |
| 1480–1460 | — | Scissoring C–H bend (deformation) antisymmetric bending in –CH3 [123] (?) C=C stretching and ring breathing vibrations in aromatic compounds |
| 1450–1440 | carbonate, antisymmetric stretch [114], dolomite [124] | — |
| 1415–1405 | carbonate, antisymmetric stretch calcite [125], clay or carbonate minerals [109] | — |
| 1390–1380 | — | Carboxylate, symmetric stretch nitrate from nitrogen fertilizers [126] symmetric bend in –CH3 [127] |
| 1310–1300 | — | –C–H bend (deformation) vibrations, including amorphous and crystalline cellulose [128] |
| 1280 | — | Carboxyl, antisymmetric stretch or SiO2 combination band |
| 1120–1100 | and O–Si–O stretch in crystalline/amorphous SiO2 species | — |
| 1090 | Carbonate, symmetric stretch [125] | Cellulose |
| 1070–1050 | SiO2, (kaolinite, illite) O–Si–O lattice antisymmetric stretch [119,131,132] |
— |
| 1035 | quartz lattice O–Si–O stretch | Carbohydrates + PO43 stretching (1100–1000 cm–1) [138] |
| 1010 | Si–O–Si stretch [131,132] | Carbohydrates + PO43 stretching (1100–1000 cm–1) [138] |
| 975 | amorphous silica, Si–OH including biogenic [131,132] | — |
| 930 | Silicate, aluminosilicate, overtone [118] | — |
| 912 | –Si–O– [105]; overtone SiO2 ≅2×450 in aluminosilicates and silicates + Al–OH bending [99] | — |
| 900 | Si–O stretching in silicates | — |
| 890 | Si–O–Si stretch in quartz | — |
| 880–875 | Carbonate, out-of-plane bend [125] | — |
| 865 | Si–O–Si stretching (sheet silicates, some aluminosilicates [131,139] | — |
| 850 | Si–O (quartz/silicate, aluminosilicate) | — |
| 840 | –Si–O– [105] | — |
| 825 | Si–O stretch in feldspars and aluminosilicate [131,139] | — |
| 810–805 | symmetric stretching vibration Si–O–Si, silica, amorphous [102] | — |
| 797 | O–Si–O stretch | C–H bending (non-aromatic) |
| 715 | Carbonate, in-plane bend [125] | — |
| 697 | Si–O–Si bend (including aluminosilicates) [131,139] | C–H aromatic compounds, bending |
| 685 | Si–O–Si (crystalline forms, aluminosilicates) [131] | — |
| 668 | CO2 | — |
| 655 | Al–O–Si in aluminosilicates | — |
| 638 | NaN3 out-of-plane bending [140] | — |
| 525–520 | silicate O–Si–O bend [119], including bending or deformation modes of silicate frameworks or associated alumina environments in complex silicates [135] | — |
| 510 | O–Si–O or Si–O–Si bending in both crystalline and amorphous silica species | — |
| 460–450 | O–Si–O bending of bridging oxygens | — |
| 430 | O–Si–O bending of bridging oxygens Mg–OH, Al–OH (clay minerals) | С–С in-phase vibrations |
| 375 | R(SiO4) [109] | — |
| 347 | SiO2 | — |
| 308 | crystalline matrix (clay or carbonate minerals) [136] | — |
| 296 | lattice vibrations | — |
| 263 | α-quartz [102] | — |
| 225 | lattice vibrational modes in minerals and crystalline materials (involving collective movement of atoms or ions in the crystal lattice) [137] | — |
| 200–190 | crystalline matrix (clay or carbonate minerals) [136] | — |
| 174 | NaN3 lattice [141] | — |
| 130 | crystalline matrix (clay or carbonate minerals) | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



