Submitted:
29 November 2025
Posted:
03 December 2025
You are already at the latest version
Abstract
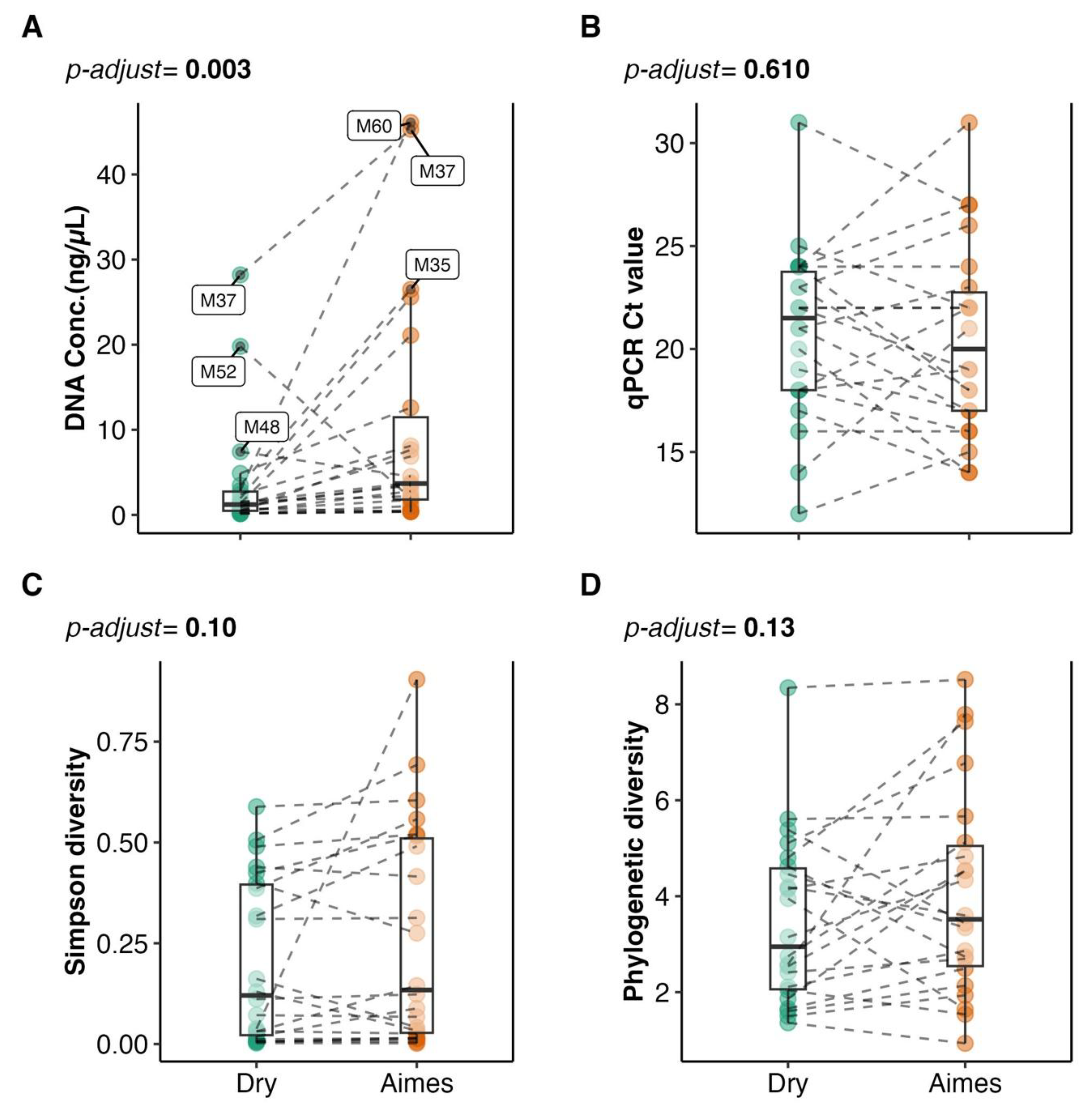
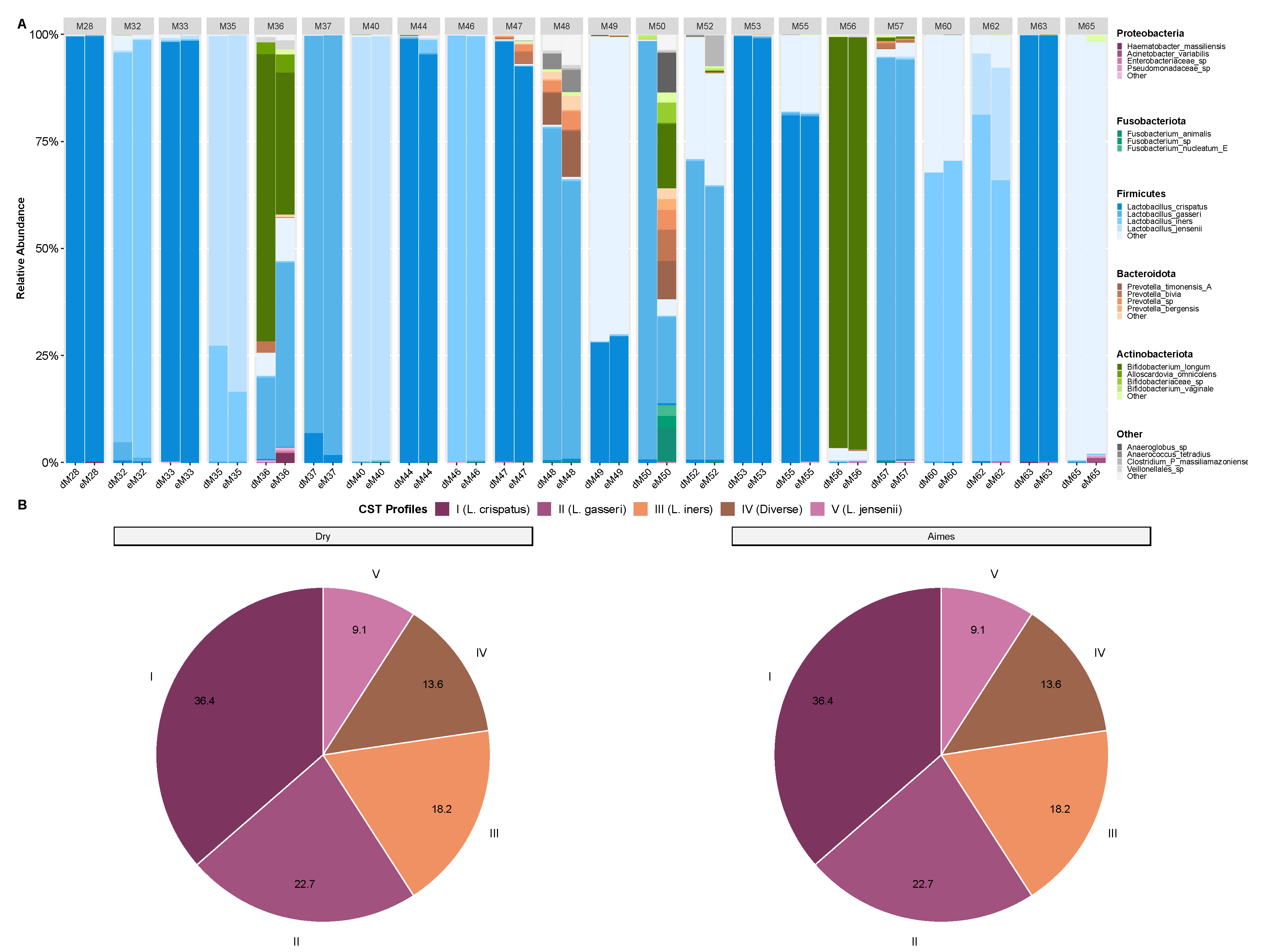
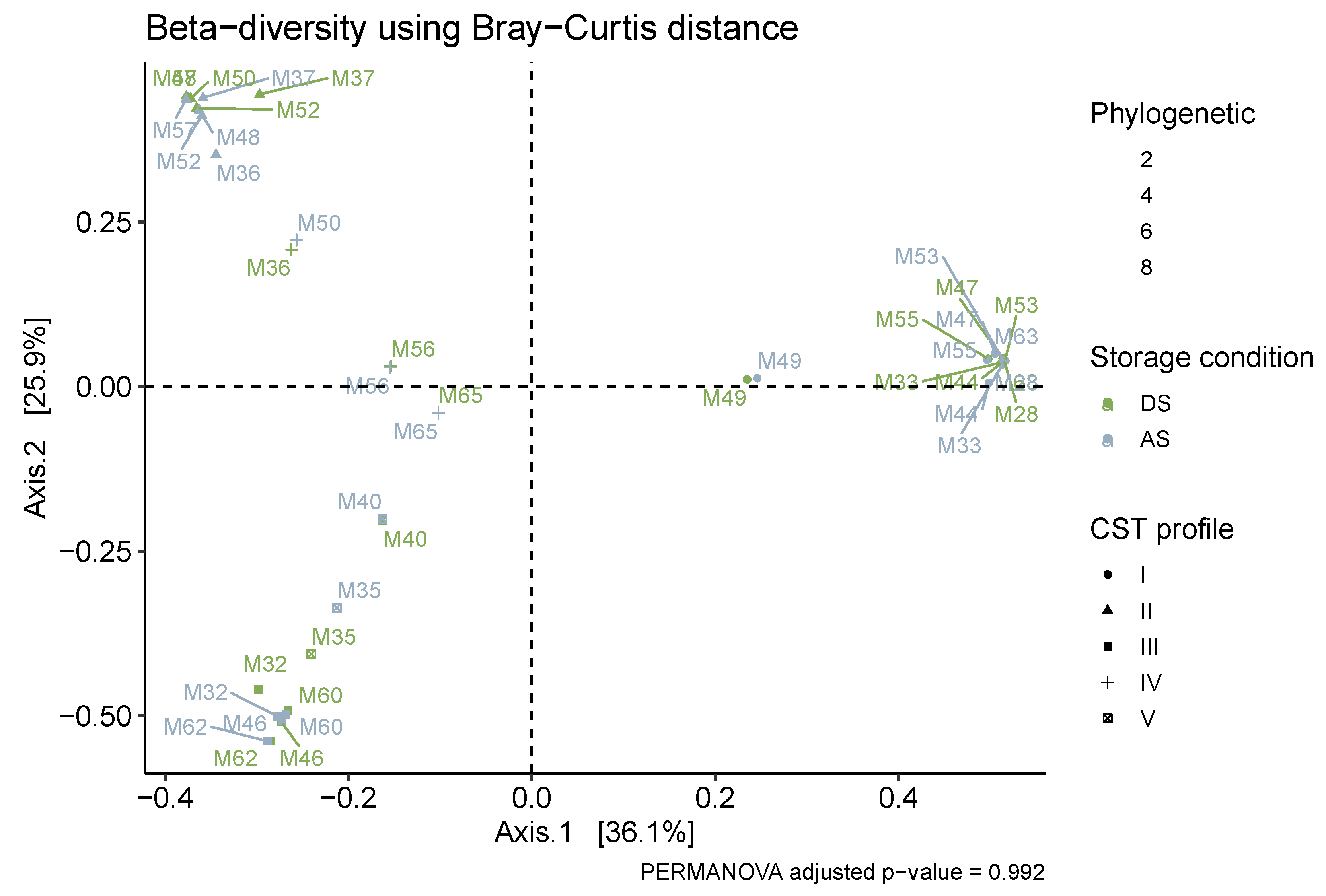
Vaginal microbiome composition has been linked to risk of preterm birth (PTB), a persistent global health challenge. 16S rRNA microbial profiling has identified specific vaginal community state types (CSTs) that have been associated with PTB risk. Diagnostic profiling requires standardised pre-analytical protocols. We evaluated two storage methods and validated a curated, vagina-specific 16S rRNA gene database (VagDB) to enhance annotation. Paired Copan FLOQ swabs from 22 women at high PTB risk were processed for either (a) dry/immediate freezing or (b) Amies-stabilisation/refrigeration. Amplicon sequence variants were generated via 16S rRNA gene (V4) PCR and Illumina sequencing. We assessed diversity, composition, and community state type (CST) allocation. Amies-stabilised samples yielded significantly higher DNA (p = 0.003), but this did not alter species richness, evenness, or community structure. VagDB enhanced species-level resolution. PCoA showed robust clustering by participant and CST (p < 0.001), irrespective of storage; CST concordance exceeded 90%. Routinely collected vaginal swabs in stabilisation medium with an 8–72-hour refrigeration window yields reliable data, supporting the integration of vaginal microbiome profiling into clinical PTB risk assessment.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Sample Collection
2.3. DNA Extraction
2.5. Amplicon Generation and Sequencing
2.6. Bioinformatics
2.7. Statistical Analysis
3. Results
3.1. Impact of Storage Condition on DNA Yield and PCR Amplification
3.2. Sample Read Counts and Alpha Diversity Comparisons
3.3. Microbiota Composition and CST Accuracy
4. Discussion
4.1. Strengths and Limitations
4.2. Addressing the Taxonomic Database Issue
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Belizário, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef]
- Diop, K.; Dufour, J.C.; Levasseur, A.; Fenollar, F. Exhaustive repertoire of human vaginal microbiota. Hum. Microbiome J. 2019, 11, 100051. [Google Scholar] [CrossRef]
- Haque, M.M.; Merchant, M.; Kumar, C.N.S.; Jayaram, A.; Jeyaseelan, L.; K, J. First-trimester vaginal microbiome diversity: A potential indicator of preterm delivery risk. Sci. Rep. 2017, 7, 16145–16155. [Google Scholar] [CrossRef]
- Parker, J.; MacIntyre, D.A.; Bennett, P.R.; Kyrgiou, M.; Terzidou, V. Cervicovaginal microbiome and metabolite profiles are altered remote from term in women who subsequently deliver preterm. BJOG 2016, 123, 45. [Google Scholar]
- Miller, E.; Beasley, D.E.; Dunn, R.R.; Archie, E.A. Lactobacilli Dominance and Vaginal pH: Why is the Human Vaginal Microbiome Unique? Front. Microbiol. 2016, 7, 1936. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Bieda, J.; Chaemsaithong, P.; Miranda, J.; Chaiworapongsa, T.; Ravel, J. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2014, 2, 18. [Google Scholar] [CrossRef]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Bieda, J.; Chaemsaithong, P.; Miranda, J.; Chaiworapongsa, T.; Ravel, J. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef]
- Ravel, J.; Brotman, R.M.; Gajer, P.; Fadrosh, D.; Zhou, X.; McCulle, S.L.; Abdo, Z.; Forney, L.J. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 2013, 1, 29. [Google Scholar] [CrossRef]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef] [PubMed]
- Fettweis, J.M.; Brooks, J.P.; Serrano, M.G.; Sheth, N.U.; Girerd, P.H.; Edwards, D.J.; Strauss, J.F., 3rd; Jefferson, K.K.; Buck, G.A. Species-level classification of the vaginal microbiome. BMC Genomics 2012, 13, S17. [Google Scholar] [CrossRef] [PubMed]
- Lamont, R.F.; Sobel, J.D.; Akins, R.A.; Hassan, S.S.; Chaiworapongsa, T.; Kusanovic, J.P.; Romero, R. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG 2011, 118, 533–549. [Google Scholar] [CrossRef]
- Ma, B.; France, M.T.; Crabtree, J.; Holm, J.B.; Humphrys, M.S.; Brotman, R.M.; Ravel, J. A comprehensive non-redundant gene catalog reveals extensive within-community intraspecies diversity in the human vagina. Nat. Commun. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; DiGiulio, D.B.; Goltsman, D.S.A.; Sun, C.L.; Costello, E.K.; Jeganathan, P.; Biggio, J.R.; Wong, R.J.; Druzin, M.L.; Shaw, G.M.; et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. USA 2017, 114, 9966–9971. [Google Scholar] [CrossRef]
- Kindinger, L.M.; Bennet, P.R.; Lee, Y.S.; MacIntyre, D.A.; Marchesi, J.R.; Smith, A.; Cacciatore, S.; Terzidou, V. The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome 2017, 5, 6. [Google Scholar] [CrossRef]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal microbiome: rethinking health and disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef]
- Dunn, A.B.; Jordan, S.; Baker, B.J.; Carlson, S.E. The Microbiome and Complement Activation: A Mechanistic Model for Preterm Birth. Biol. Res. Nurs. 2017, 19, 295–307. [Google Scholar] [CrossRef]
- Nadeau, H.C.; Subramaniam, A.; Andrews, W.W. Infection and preterm birth. Semin. Fetal Neonatal Med. 2016, 21, 100–105. [Google Scholar] [CrossRef]
- Keelan, J.A.; Payne, M.S. Vaginal microbiota during pregnancy: Pathways of risk of preterm delivery in the absence of intrauterine infection? Proc. Natl. Acad. Sci. USA 2015, 112, E6414. [Google Scholar] [CrossRef]
- Petricevic, L.; Domig, K.J.; Nierscher, F.J.; Sandhofer, M.J.; Fidesser, M.; Krondorfer, I.; Husslein, P.; Kneifel, W.; Kiss, H. Characterisation of the vaginal Lactobacillus microbiota associated with preterm delivery. Sci. Rep. 2014, 4, 5712. [Google Scholar] [CrossRef]
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.B.; Narwal, R.; Adler, A.; Vera Garcia, C.; Rohde, S.; Say, L.; et al. Born Too Soon: The global epidemiology of 15 million preterm births. Reprod. Health 2013, 10, S2. [Google Scholar] [CrossRef]
- Tabatabaei, N.; Eren, A.M.; Barreiro, L.B.; Yotova, V.; Dumaine, A.; Allard, C.; Fraser, W.D. Vaginal microbiome in early pregnancy and subsequent risk of spontaneous preterm birth: a case–control study. BJOG 2019, 126, 349–358. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.P.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef]
- Freitas, A.C.; Chaban, B.; Bocking, A.; Rocco, M.; Yang, S.; Hill, J.E.; Money, D.M. Increased richness and diversity of the vaginal microbiota and spontaneous preterm birth. Microbiome 2018, 6, 117. [Google Scholar] [CrossRef]
- Jean, S.; Brochu, V.; M, D. Multi-omic Microbiome Profiles in the Female Reproductive Tract in Early Pregnancy. Infect. Microbes Dis. 2019, 1, 49–60. [Google Scholar] [CrossRef]
- Donders, G.G.; Van Calsteren, K.; Bellen, G.; Reybrouck, R.; Van den Bosch, T.; Riphagen, I.; Van Lierde, S. Predictive value for preterm birth of abnormal vaginal flora, bacterial vaginosis and aerobic vaginitis during the first trimester of pregnancy. BJOG 2009, 116, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.G.; Rutherford, A.J.; Wilson, J.D. Influence of bacterial vaginosis on conception and miscarriage in the first trimester: cohort study. BMJ 1999, 319, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D., Jr.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, R.D.; Raaby, M.; Funch, P.; Puetz, A. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Sci. Rep. 2019, 9, 17287. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Andersson, A.F. Analysing Microbial Community Composition through Amplicon Sequencing: From Sampling to Hypothesis Testing. Front. Microbiol. 2017, 8, 1561. [Google Scholar] [CrossRef]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; McLaren, M.; Callahan, B. Understanding and interpreting community sequencing measurements of the vaginal microbiome. BJOG 2020, 127, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Marotz, C.; Cavagnero, K.J.; Song, S.J.; McDonald, D.; Wandro, S.; Humphrey, G.; Knight, R. Evaluation of the Effect of Storage Methods on Fecal, Saliva, and Skin Microbiome Composition. mSystems 2021, 6, e01329-20. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, K.G.; Audette, C.D.; Sebeck, D.; Tucker, K.A. Comparison of 3 swab transport systems for direct release and recovery of aerobic and anaerobic bacteria. Diagn. Microbiol. Infect. Dis. 2008, 62, 471–473. [Google Scholar] [CrossRef]
- Mattei, V.; Vici, F.; Sisti, D.; Rocchi, M.; Bulli, L.; Citterio, B. Evaluation of Methods for the Extraction of Microbial DNA From Vaginal Swabs Used for Microbiome Studies. Front. Cell. Infect. Microbiol. 2019, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Bassis, C.M.; Erb-Downward, J.R.; Young, V.B.; Huffnagle, G.B. Comparison of stool versus rectal swab samples and storage conditions on bacterial community profiles. BMC Microbiol. 2017, 17, 78. [Google Scholar] [CrossRef]
- Bai, G.; Gajer, P.; Nandy, M.; Ma, B.; Yang, H.; Sakamoto, J.; Blanchard, M.H.; Ravel, J.; Brotman, R.M. Comparison of storage conditions for human vaginal microbiome studies. PLoS ONE 2012, 7, e36934. [Google Scholar] [CrossRef]
- Edgar, R. Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ 2018, 6, e5030. [Google Scholar] [CrossRef]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT — how do these taxonomies compare? BMC Genomics 2017, 18, 114. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and "All-species Living Tree Project (LTP)" taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Edgar, R.C. Accuracy of microbial community diversity estimated by closed- and open-reference OTUs. PeerJ 2017, 5, e3889. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009-15. [Google Scholar] [CrossRef]
- Huang, L.; et al. A multi-kingdom collection of 33,804 reference genomes for the human vaginal microbiome. Nat. Microbiol. 2024, 9, 2185–2200. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.K.; et al. Application of ecosystem-specific reference databases for increased taxonomic resolution in soil microbial profiling. Front. Microbiol. 2022, 13, 969460. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, B.D.; Bokulich, N.A.; McDonald, D.; Knight, R.; Caporaso, J.G.; Huttley, G.A. Species abundance information improves sequence taxonomy classification accuracy. Nat. Commun. 2019, 10, 4643. [Google Scholar] [CrossRef]
- Ali, A.; Christophersen, C.T.; Keelan, J.A. Vaginal microbial profiling in a preterm birth high-risk cohort using shallow shotgun metagenomics. Microbiol. Aust. 2021, 42, 69–74. [Google Scholar] [CrossRef]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gligor, K.; Holt, J.M.; Hansen, A.J.; Knights, D. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018, 3, e00069-18. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; et al. Metagenomic Information Recovery from Human Stool Samples Is Influenced by Sequencing Depth and Profiling Method. Genes 2020, 11, 1380. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghalith, G.A.; Montassier, E.; Ward, H.N.; Knights, D. SHI7 Is a Self-Learning Pipeline for Multipurpose Short-Read DNA Quality Control. mSystems 2018, 3, e00202-17. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Lopera, J.; et al. Development and Evaluation of Whole Cell-and Genomic DNA-based Microbiome Reference Standards. Preprint 2021.
- France, M.T.; Ma, B.; Gajer, P.; Brown, S.; Humphrys, M.S.; Holm, J.B.; Waetjen, L.E.; Brotman, R.M.; Ravel, J. VALENCIA: a nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 2020, 8, 166. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- Patil, I. Visualizations with statistical details: The 'ggstatsplot' approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Lewis, F.M.T.; Bernstein, K.T.; Aral, S.O. Vaginal Microbiome and Its Relationship to Behavior, Sexual Health, and Sexually Transmitted Diseases. Obstet. Gynecol. 2017, 129, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; MacIntyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Le-Re, T.; Kote-Jarai, Z.; Kyrgiou, M. Comparison of vaginal microbiota sampling techniques: cytobrush versus swab. Sci. Rep. 2017, 7, 9802. [Google Scholar] [CrossRef]
- Tomaiuolo, R.; Veneri, C.; Nucci, F.; D'Alessandro, A.; Pesapane, F.; Sarno, L.; De Guida, M. Microbiota and Human Reproduction: The Case of Female Infertility. High-Throughput 2020, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Bayar, E.; Bennett, P.R.; Chan, D.; Sykes, L.; MacIntyre, D.A. The pregnancy microbiome and preterm birth. Semin. Immunopathol. 2020, 42, 487–499. [Google Scholar] [CrossRef]
- Payne, M.S.; et al. A specific bacterial DNA signature in the vagina of Australian women in midpregnancy predicts high risk of spontaneous preterm birth (the Predict1000 study). Am. J. Obstet. Gynecol. 2021, 224, 206.e1–206.e23. [Google Scholar] [CrossRef] [PubMed]
- Hočevar, K.; Maver, A.; Vidmar, R.; Hodžić, A.; Kušar, D.; Verdenik, I. Vaginal microbiome signature is associated with spontaneous preterm delivery. Front. Med. 2019, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Casals-Pascual, C.; et al. Microbial Diversity in Clinical Microbiome Studies: Sample Size and Statistical Power Considerations. Gastroenterology 2020, 158, 1524–1528. [Google Scholar] [CrossRef]
- Li, T.; et al. Evaluation of the vaginal microbiome in clinical diagnosis and management of vaginal infectious diseases. Chin. Med. J. 2019, 132, 1100–1103. [Google Scholar] [CrossRef]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef]
- Virtanen, S.; et al. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS ONE 2017, 12, e0181477. [Google Scholar] [CrossRef]
- France, M.; et al. VALENCIA: A nearest centroid classification method for vaginal microbial communities based on composition. Research Square 2020.
- Qian, X.-B.; et al. A guide to human microbiome research: study design, sample collection, and bioinformatics analysis. Chin. Med. J. 2020, 133, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



