
Submitted:
12 November 2025
Posted:
13 November 2025
You are already at the latest version
Abstract
A series of azido- and cyclooctyne-functionalized N-hydroxysuccinimidyl esters (NHS esters) and benzotriazolides were prepared and used as N-acylation reagents to obtain azido- (BSA-1) and cyclooctyne-functionalized bovine serum albumin proteins (BSA-2), fluorescein derivatives 5 and 6, and homobifunctional linkers 3 and 4. Strain-promoted azide-alkyne cycloaddition (SPAAC) and copper-catalyzed azide-alkyne cycloaddition (CuAAC) of azido- functionalized fluorescent probe 5 and alkyne- functionalized fluorescent probe 6 with complementary functionalized proteins BSA-2 and BSA-1 yielded fluorescent cycloadducts BSA-2-5 and BSA-1-6. These cycloadducts were used to determine the loading of BSA-1 and BSA-2 with the respective azido and cyclooctyne groups based on their molar absorbances and fluorescence intensities. Dimerization through covalent cross-linking of BSA was then performed by SPAAC between azido-functionalized BSA-1 and cyclooctyne-functionalized BSA-2, and by treating BSA-1 and BSA-2 with 0.5 equiv. of com-plementary bis-cyclooctyne linker 4 and bis-azide linker 3. Although the formation of covalent dimers BSA-1-2-BSA, BSA-1-6-1-BSA, and BSA-2-5-2-BSA was detected by SDS-PAGE analysis, this was a minor process, and most of the functionalized BSA did not form covalent dimers.
Keywords:
protein cross-linking
; azide–alkyne cycloaddition
; fluorescein dyes
; amidation
; benzotriazolides
1. Introduction
Chemical modification of proteins is a key technique in bioconjugation that enables various synthetic molecules to be covalently attached to proteins. The resulting derivatized proteins serve as tools for investigating biological processes. An important modification method is cross-linking proteins by forming strong covalent bonds between specific amino acid residues within or between protein molecules. Cross-linking plays significant roles in stabilizing protein structures, maintaining tissue integrity and strength, altering protein functions, inducing pathological conditions, and facilitating interactions between proteins and between proteins and other molecules. Therefore, understanding and manipulating protein cross-linking processes are essential for elucidating biological mechanisms, developing therapeutic interventions, and engineering biomaterials with tailored properties [1,2,3].
Since its definition in 2001 [4], "click" chemistry has become an important method in modern synthetic organic chemistry encompassing various highly efficient reactions, such as nucleophilic opening of spring-loaded rings, non-aldol carbonyl chemistry, additions to C−C multiple bonds and cycloadditions. A typical “click” reaction should also be bioorthogonal, meaning it should not interact with biological systems. Among the various "click" reactions, the Cu-catalyzed azide−alkyne cycloaddition (CuAAC) [5,6] emerged as the first and is now the best-known example of a "click" reaction [7,8,9,10]. In biological systems, however, CuAAC is often not truly bioorthogonal due to possible interactions of copper ions with biomolecules [1]. This limitation was soon addressed by the introduction of a catalyst-free strain-promoted azide–alkyne cycloaddition (SPAAC) in 2004 [11,12,13,14]. Since then, CuAAC and SPAAC have found widespread application in connecting various types of small molecules and macromolecular entities and are now standard ligation tools in combinatorial synthesis [15,16,17] bioconjugation [18,19,20,21], and materials science [22,23].
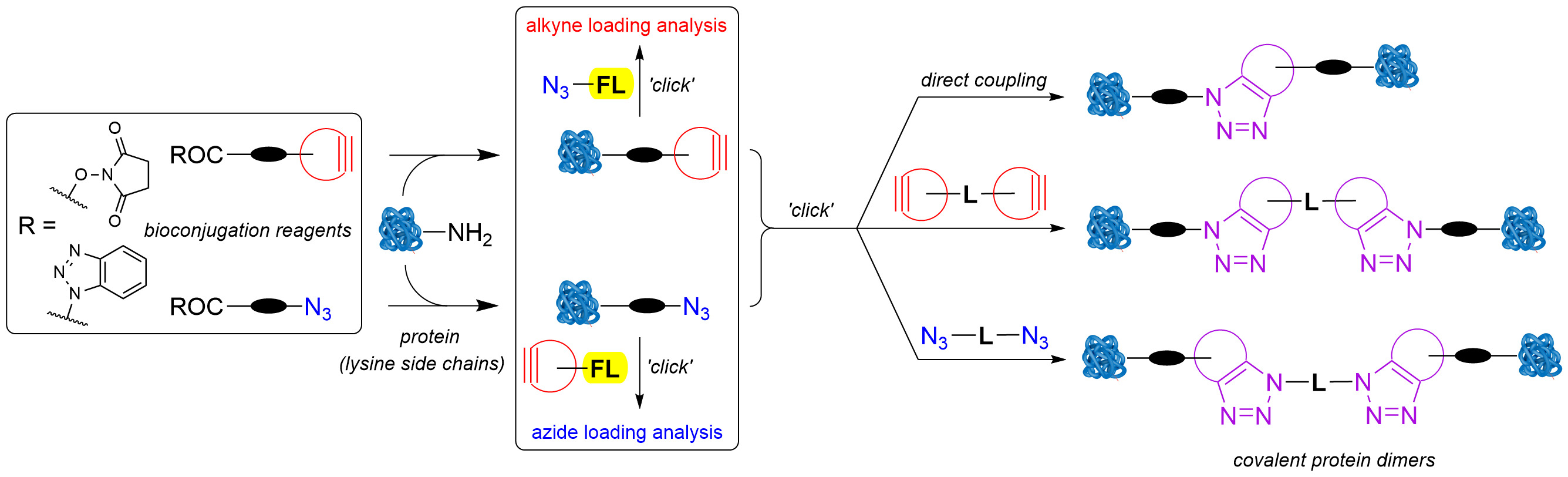
Recently, we focused on applied studies aimed at labeling and covalently cross-linking native proteins using novel synthetic organic molecules as bioconjugation reagents [24,25]. To minimize troubleshooting events and optimize time, we kept all aspects of this study as simple as possible. Accordingly, SPAAC and CuAAC were selected as the most suitable “click” reactions to begin with. For the same reason, fluorescein derivatives were chosen as fluorescent probes, due to their availability and well-known optical properties. This approach required the use of various readily available azide-, cyclooctyne-, and alkyne-functionalized reagents to achieve controlled covalent linking of two proteins. To avoid excessive cross-linking and protein oligomerization, each protein molecule should be tagged with only one binding functional group. The concept of linking native proteins is presented in Figure 1. Following known protocols [1,2,3], the lysine residues of a protein would be treated in parallel with excess N-acylation reagents 1 and 2 to obtain the azido- (P-1) and cycloalkyne-functionalized proteins (P-2). Linking these two would be achieved by SPAAC, either directly or via bifunctional spacers 3 and 4. The loading of each functional group would be determined spectrophotometrically from absorbances and/or emission intensities of fluorescently labeled proteins P-1-6 and P-2-5, obtained by SPAAC or CuAAC reactions between azide- and alkyne- functionalized proteins P-1 and P-2 and their complementary functionalized fluorescein derivatives 5 and 6. With optimal reaction conditions to achieve the desired functional group loading FG/P ~1, the proteins P-1 and P-2 could then be used in “click” cross-linking experiments (Figure 1).
Contrary to our expectations, synthesizing both novel and known reagents proved challenging despite extensive literature coverage on the subject. Therefore, we report the results of the first part of our ongoing study, the synthesis of a series of azide- and alkyne-functionalized bioconjugation reagents, fluorescent probes, and bifunctional linkers, as well as their application in the covalent cross-linking of bovine serum albumin (BSA).
2. Results and Discussion
2.1. Bioconjugation Reagents and Linkers Used in This Study
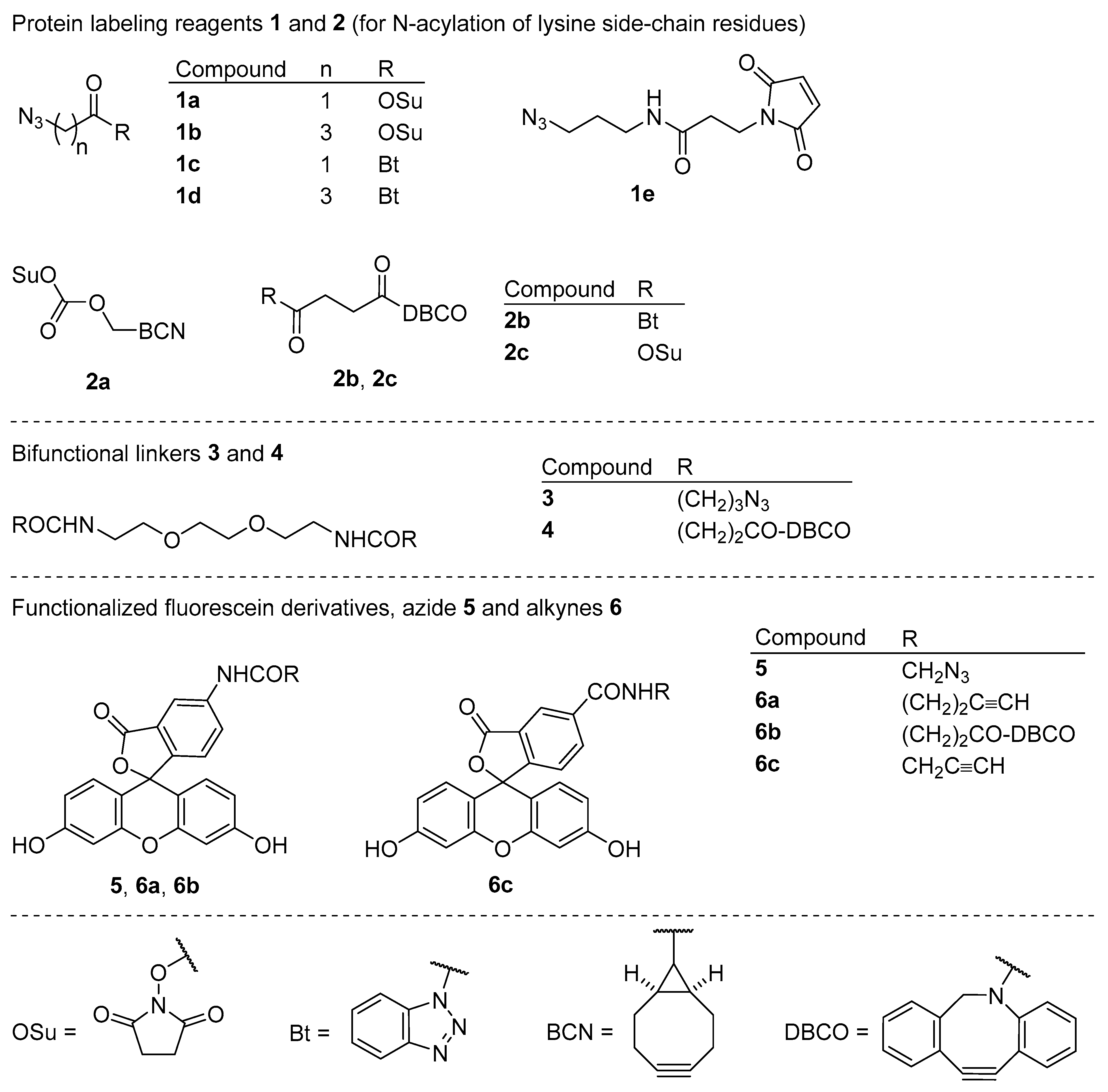
Bioconjugation reagents for functionalizing proteins with the azido group 1a [26], 1b [27], 1c [28], 1d, and 1e [29] and with the cyclooctyne group 2a [30,31], 2b, and 2c [32], homobifunctional “click” linkers 3 and 4, and “click” fluorescent probes 5 [33,34], 6a [35], 6b, and 6c [36,37], which were selected for use in this study, are shown in Figure 2. Compounds 1c, 1d, 2b, 3, 4, and 6b are novel and have not been previously reported in the literature. Bioconjugation reagents 1a, 1b, 1e, 2a, 2c, and fluorescent probe 6c are commercially available. Other reagents and linkers were synthesized from commercial precursors, and their syntheses are described in the following sections of this article.
2.2. Synthesis of Azido- and Cyclooctyne-Functionalized Conjugation Reagents 1 and 2 for N-Acylation of Lysine Side-Chain Residues
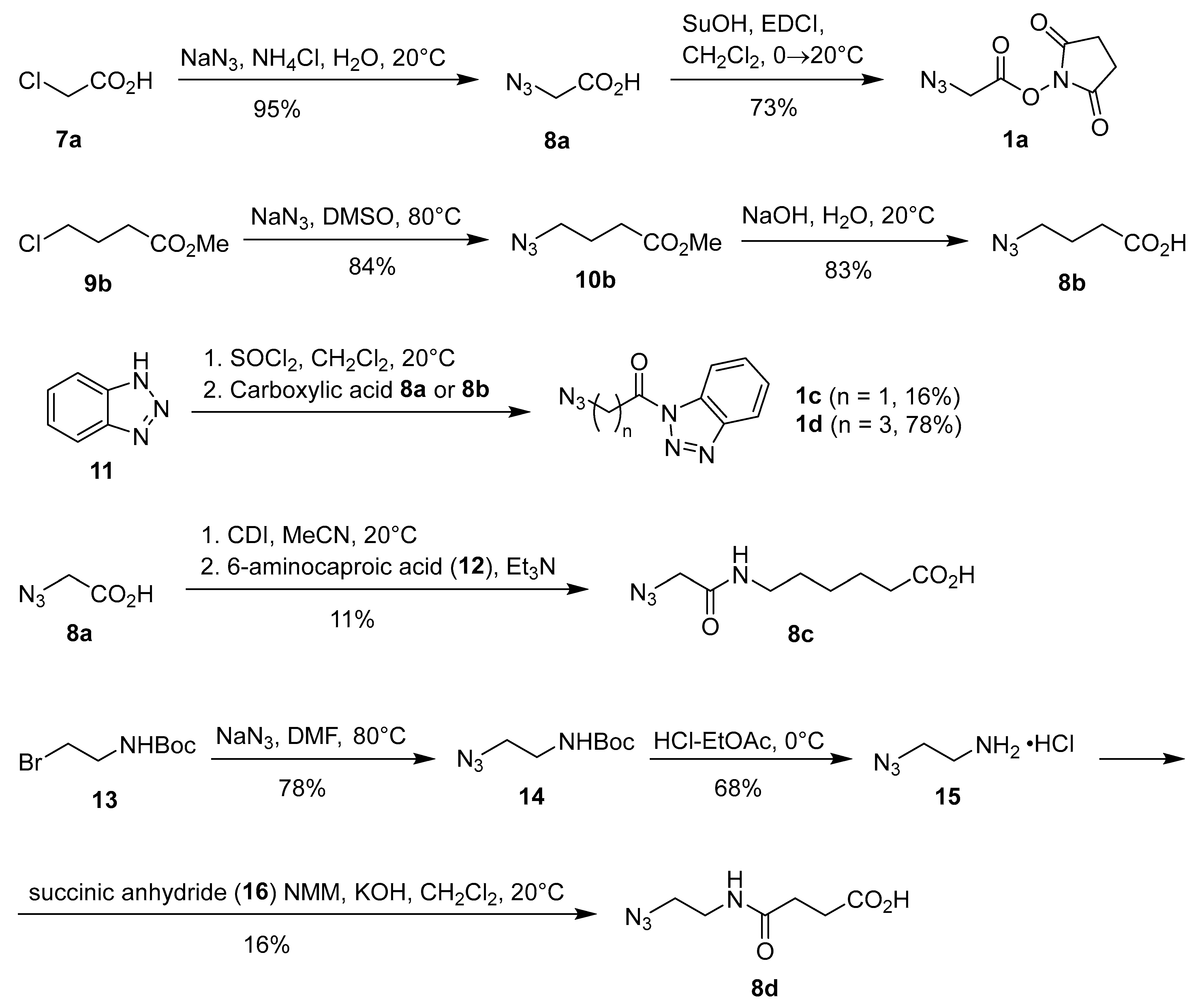
Due to high cost (typically >1000 €/g) of commercial reagents for covalent binding to the lysine residues of proteins, we decided to prepare at least some reagents following the literature procedures. The synthesis of azido-functionalized reagents 1, 8, 10, 13, and 14 is shown in Scheme 1. First, chloroacetic acid (7a) was treated with sodium azide in aqueous ammonium chloride at room temperature for three days, followed by acidification and extraction workup to give azidoacetic acid (8a) in almost quantitative yield [38]. Treatment of acid 8a with N-hydroxysuccinimide and N-ethyl-N’-[3-(dimethylamino)propyl]carbodiimide hydrochloride (EDCI) followed by extraction workup, then gave the desired N-succinimidyl azidoacetate (1a) in 73% yield [26]. Next, we synthesized 4-azidobutanoic acid (8b) in two steps from methyl 4-chlorobutanoate (9b) by treatment with NaN3 in DMSO at 80 °C and subsequent hydrolysis of methyl 4-azidobutanoate (10b) in 70% yield over two steps [39]. Since NHS esters are prone to hydrolysis with a typical half-life of 4–5 h at 0 °C and pH 7 [1,40], only freshly prepared stock solutions of NHS esters should be used in bioconjugation to minimize undesired hydrolysis. We also prepared more robust and stable benzotriazolide analogues 1c and 1d in 16% and 78% yield, respectively, by acylation of 1H-benzo[d][1,2,3]triazole (11) with azidoacetic acid (8a) and 4-azidobutyric acid (8b), following a general literature procedure for N-acylation of 1H-benzo[d][1,2,3]triazole (11) [41]. We also prepared the ‘extended’ azido acids 8c and 8d. Activation of azidoacetic acid (8a) with CDI, followed by treatment with a slight excess of 6-aminocaproic acid (12), gave the corresponding carboxamide 8c in 11% yield. Reaction of 2-(tert-butoxycarbonylamino)-1-bromoethane (13) with sodium azide in DMF gave the corresponding azide 14 [42,43], which was deprotected with HCl–EtOAc to furnish 2-azidoethylamine hydrochloride (15) [44,45]. Subsequent treatment of 15 with succinic anhydride (16) in the presence of excess base gave 4-[(2-azidoethyl)amino]-4-oxobutanoic acid (8d) in 16% yield (Scheme 1) [46].
The synthesis of cyclooctyne-functionalized acylation reagent 2b, compounds 20, 21, 23–26, and the attempted syntheses of (1R*,8S*)-bicyclo[6.1.0]non-4-yne-9-carboxylic acid (22) and (1R*,8S*)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (27) shown in Scheme 2. Acylation of benzotriazole 11 with 4-(11,12-didehydrodibenzo[b,f]azocin-5(6H)-yl)-4-oxobutanoic acid (DBCO-acid) (17), following a general literature procedure for the synthesis of N-acylbenzotriazoles [41] gave N-[4-(11,12-didehydrodibenzo[b,f]azocin-5(6H)-yl)-4-oxobutanyl]-1H-benzo[d][1,2,3]triazole (2b) in 82% yield. Next, we turned our attention to the preparation of exo/endo-(1R*,8S*)-bicyclo[6.1.0]non-4-yne-9-carboxylic acid (22). Rh-catalyzed reaction of ethyl diazoacetate (19) with a large excess of 1Z,5Z-1,5-cyclooctadiene (18) gave ethyl (1R*,8S*)-bicyclo[6.1.0]non-4-yne-9-carboxylate (20) as an endo/exo-mixture of isomers in 83% yield [47]. Bromination of 20 in dichloromethane gave the expected trans-adduct 21 [48] in 90% yield, however, attempts to obtain the target cyclooctyne 22 by base-induced elimination of two HBr molecules from 21 failed. We then tried to prepare 22 via hydrolysis of ester 20 [49,50], bromination of carboxylic acid 23 to obtain dibromo-acid 24 [51], and subsequent base-induced elimination of HBr from 24. However, the final step - base-induced elimination of HBr - failed again. The unsuccessful elimination of HBr from dibromo compounds 21 and 24 was surprising, since this step was not reported as problematic in the literature syntheses of bicyclo[6.1.0]non-4-yne derivatives [30,47,48,49,50,51,52]. In contrast to previous report [51], we did not encounter problems with the bromination and ester hydrolysis steps. Finally, we tried to synthesize exo/endo-[(1R*,8S*)-bicyclo[6.1.0]non-4-yn-9-yl]methanol (27) from ester 20 following the literature procedure [47,52]. In this case as well, the elimination step failed. Treatment of dibromo adduct 26 with t-BuOK in THF at 0 °C → 65 °C → 20 °C resulted in incomplete conversion, which did not improve after further treatment with LDA at –63 °C → 20 °C. Consequently, we were not able to isolate pure cyclooctyne 27 (Scheme 2). At this point, all further attempts to synthesize pure compounds 22 and 27 were abandoned, and we decided to use commercial [(1R*,8S*,9s)-bicyclo[6.1.0]non-4-yn-9-yl]methyl (2,5-dioxopyrrolidin-1-yl) carbonate (2a, see Figure 2) for cross-linking studies.
2.3. Synthesis of Azido- and Alkynyl-Functionalized Fluorecent Probes 5 and 6
The synthesis of fluorescent probes 5 and 6 is shown in Scheme 3. Treatment of 6-aminofluorescein (28) with NHS ester 1a gave the corresponding 2-azidoacetylfluorescein derivative 5 [33,34]. Similarly, acylation of 28 with 4-pentynoic acid (29) and DBCO-acid (17) in the presence of EDCI gave the alkynylated derivatives 6a [35] and 6b. As expected due to lower nucleophilicity of the anilino group, the conversions in both N-acylation reactions were incomplete and the yields of the corresponding anilides 5 (24%), 6a (24%), and 6b (8%) were low. Therefore, fluorescent probe 6c was also prepared by amidation of 6-carboxyfluorescein (30) following the literature procedure for the preparation of closely related fluorescein-6-carboxamides [53]. Compound 30 was first activated with di(succinimid-1-yl)carbonate (DSC) and the intermediate NHS ester was treated with excess propargylamine (31) to furnish the corresponding fluoresceine derivative 6c [36,37] albeit in only 6% yield (Scheme 3).
2.4. Synthesis of Bis Azido- (3) and Bis-Alkyne-Linkers 4
The synthesis of homobifunctional linkers is shown in Scheme 4. Compounds 3 and 4 were prepared by treatment of 2,2′-(ethylenedioxy)bis(ethylamine) (32) with 2 equiv. of benzotriazolides 1d and 2b, respectively. Next, we attempted to prepare the ‘extended’ diamines by reacting adipoyl chloride (33) and butane-1,4-diisothiocyanate (35) with two equivalents of diamine 32. Amidation of adipoyl chloride (33) with two equivalents of 32 following the literature procedure [54], gave macrocyclic 1,4-dioxa-7,14-diazacyclohexadecane-8,13-dione (34) as the only product in 95% yield. Notably, the reaction did not give the ‘extended’ diamine as reported in the literature [54]. At first glance, formation of macrocyclic compound 34 instead of the acyclic extended diamine was surprising. However, further literature search revealed that 34 is a known compound, which had been obtained previously by reacting 32 with dimethyl adipate [55,56]. On the other hand, addition of two equivalents of diamine 32 to diisothiocyanate 35 gave the expected product 36 in quantitative yield (Scheme 4).
2.5. Covalent Binding of Compounds 1 and 2 to BSA Protein
Finally, compounds 1–6 were tested as bioconjugation reagents. Bovine serum albumin (BSA) was selected as the model protein. N-Acylation reagents 1 and 2 were used for attachment of azido and alkyne functionality to BSA, fluorescent probes 5 and 6 as “click” analytical reagents for determination of the amount of functional groups attached to azido and cycloalkyne modified BSA, and “click” bifunctional linkers 3 and 4 as linkers for connecting two modified BSA.
The covalent binding of compounds 1 and 2 to BSA and the determination of the amount of functional group attached to BSA (i.e. functional group loading, FG/BSA) are presented in Scheme 5. Commercial HOSu esters 1a, 1b, 2a, and 2c were chosen as reagents for covalent binding to BSA by N-acylation of accessible primary amino groups in the side chains of lysine residues. Additionally, the maleimide coupling reagent 1e was selected, as at pH ~ 7 it can react with both lysine and cysteine residues of BSA through different binding modes (condensation and 1,4-addition) [1]. 6-Aminofluoresceine derivatives 5 and 6a were chosen as fluorescent probes for determining functional group loading (FG/BSA) in azide- (BSA-1) and alkyne-functionalized proteins (BSA-2). First, azide-functionalized proteins BSA-1a, BSA-1b, BSA-1e and cyclooctyne-functionalized proteins BSA-2a, and BSA-2c were prepared by treating BSA with 10 equiv. of NHS esters 1a, 1b, 1e, 2a, and 2c in PBS buffer at pH 7.4 and 20 °C for 4 h. To minimize competitive hydrolysis of NHS esters 1 and 2 [1,40], BSA solution was added to a 10-fold excess of solid reagents 1 and 2. Next, aliquots of the crude functionalized proteins BSA-1 and BSA-2 were taken, and excess reagents and small molecular byproducts were removed by desalting. The purified BSA-1 and BSA-2 were then analyzed for functional group loading (FG/BSA). SPAAC reactions of cyclooctyne-conjugates BSA-2a and BSA-2c with azido-functionalized fluorescent probe 5 and CuAAC reactions of azido-conjugates BSA-1a, BSA-1b, and BSA-1e with alkyne-functionalized fluorescent probe 6a were performed first, followed by purification by SEC-FPLC to yield fluorescently labeled proteins BSA-1a-6a, BSA-1b-6a, BSA-1e-6a, BSA-2a-5, and BSA-2c-5. Fractions containing the labeled proteins were collected, their absorbances were measured at 280 nm and 498 nm, and the corresponding FG/BSA values were then determined for each fraction based on the known molar absorbances of BSA at 280 nm and fluorescein derivatives 5 and 6 at 498 nm (Scheme 5, Method A). Molar loadings of azide-conjugates BSA-1a, BSA-1b, and BSA-1e were surprisingly low (~0.15), while molar loadings of cycloalkyne-functionalized conjugates BSA-2a and BSA-2c were 5.0 and 1.4, respectively (Scheme 5, Method A).
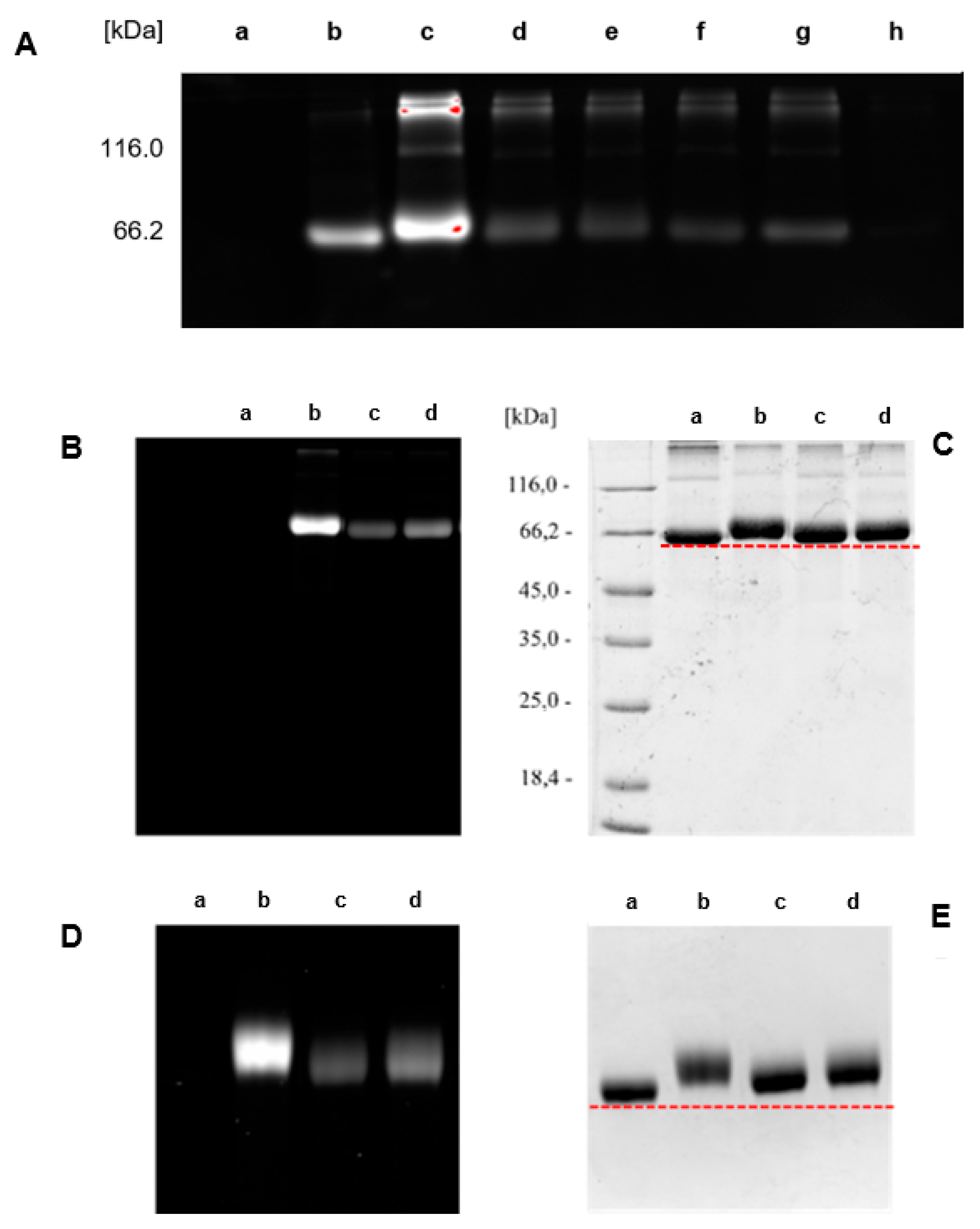
Since determination of FG/BSA following the above protocol was very time-consuming and laborious, it was not optimal for rapid FG/BSA analysis. Therefore, we decided to use a simplified procedure. First, a reference solution of fluorescently labeled conjugate BSA-2a-6a was prepared, FG/BSA = 2.2 was determined by Metod A and the conjugate was stored at 4 °C as a reference standard. Next, fluorescent conjugates BSA-1a-5, BSA-1b-5, BSA-1d-5, BSA-1e-5, BSA-2b-6a, and BSA-2c-6a were prepared and, together with the reference standard BSA-2a-6a, analyzed by SDS-PAGE. FG/BSA for conjugates BSA-1a-5 (0.5), BSA-1b-5 (4.6), BSA-1d-5 (0.4), BSA-1e-5 (0.9), BSA-2b-6a (0.0), and BSA-2c-6a (0.6) was determined based on their fluorescence intensities relative to the reference standard BSA-2a-6a (FG/BSA = 2.2) (Scheme 5, Method B). The obtained molar loading values were 0.4–0.9 in most cases. FG/BSA of BSA-1b-5 was significantly higher (4.6), while coupling with benzotriazolide 2b failed (FG/BSA = 0) (Scheme 5, Method B, Figure 3A). Low molar loadings (FG/BSA < 1) obtained after treating BSA with a large excess (10 equiv.) of coupling reagents 1 and 2 can be explained by incomplete conversion of solid reagents 1 and 2 due to their insolubility in aqueous media and concomitant partial hydrolysis, which competes with the N-acylation reaction. This explanation is supported by the observation that reagents 1 and 2 were not completely dissolved after stirring with BSA solution for 4 h. According to the FG/BSA values obtained, NHS esters 1a, 1b, 2a, and 2c were clearly superior to N-acylbenzotriazoles 1d and 2b as N-acylating reagents. Surprisingly, benzotriazolide 2b was completely ineffective (FG/BSA ~ 0) under the coupling conditions employed (Scheme 5, Figure 3A, entry h). Successful functionalization of BSA should result in a slight (~1%) increase of the molecular weight of BSA (66.5 kDa) by 0.5–0.7 kDa due to the attached linker (coupling reagent) and fluorescent probe. As indicated with red dashed lines in Figure 3C and Figure 3E, this increase of molecular weight was detectable on the SDS-PAGE gel, where spots of labeled BSA are shifted toward higher molecular weights. In addition, spots (b), (c), and (d) of labeled BSA are smeared or show dragging. This indicates that not all BSA molecules were functionalized with the same number of markers, resulting in a distribution of different molecular weights of products within a single spot (Figure 3D and Figure 3E) [57].
2.6. Attempted Covalent Cross-Linking of Azide- and Cyclooctyne-Functionalized Proteins BSA-1 and BSA-2
We were interested only in controlled cross-linking of BSA that would preferentially yield the “covalent dimers” of BSA. We did not want any kind of uncontrolled oligomerization or cross-linking of BSA to be the major process. Accordingly, covalent cross-linking of azide- and cyclooctyne-functionalized molecules BSA-1 and BSA-2 was performed in two ways: by direct coupling of BSA-1 and BSA-2 to obtain BSA-1-2-BSA (Scheme 6, Method C), and by coupling azide-functionalized BSA-1 with 0.5 equiv. of bis-cyclooctyne linker 4 and cyclooctyne-functionalized BSA-2 with 0.5 equiv. of bis-azide linker 3 to obtain BSA-1-4-1-BSA and BSA-2-3-2-BSA, respectively (Method D, Scheme 6). First, Method C was tested. Cyclooctyne-functionalized proteins BSA-2a and BSA-2c (FG/BSA = 5.0 and 1.4, respectively) were treated with azide-functionalized proteins BSA-1a, BSA-1b, and BSA-1e (FG/BSA = 0.2, 0.1, and 0.1, respectively) in a volume ratio BSA-2: BSA-1 = 1:1 and 1:5 at 20 °C for 4 h, and the products were analyzed by SDS-PAGE. In all cases, weak spots with Mw ~ 120 kDa, corresponding to BSA dimers were observed. However, since the same spot with Mw ~ 120 kDa was also observed with the BSA control, we concluded that covalent dimerization of BSA-1 and BSA-2 via the SPAAC reaction did not take place, or occurred only to a small extent (Figure 4A and Figure 4B). We reasoned that unsuccessful direct dimerization might be due to the size of the protein molecule, which prevents the functional groups from getting close enough to react (Scheme 6, Method C). Therefore, we tested Method D, hoping that this difficulty could be overcome by using longer bifunctional linkers 3 and 4, where each bifunctional reagent would react with two functionalized BSA molecules. Cyclooctyne-functionalized BSA-2a was treated with 0.5 equiv. of diazide 3 and azide-functionalized BSA-1b was treated with bis-cyclooctyne 4. The reactions were carried out at room temperature for 24 hours and then analyzed by SDS-PAGE. After comparing the spots of the non-functionalized BSA control (a) with those from the dimerization experiments (b and c), we concluded that most BSA molecules did not form dimers. Nevertheless, smeared spots with a molecular weight approximately 120 kDa indicate that some dimerized molecules were probably formed (Figure 5C).
3. Conclusions
NHS esters and benzotriazolides of azido- and cyclooctyne-functionalized carboxylic acids 1 and 2 were prepared and used as reagents for functionalization of primary amino groups of lysine’s’ side chains of BSA protein, 6-aminofluorescein (28), and 2,2'-[ethane-1,2-diylbis(oxy)]bis(ethan-1-amine) (32). It turned out in the course of this study that performing the syntheses of cyclooctyne and fluorescein derivatives to obtain bioconjugation reagents and fluorescent probes may sometimes be challenging. Acquiring specific skills is necessary for successful reproduction of known synthetic procedures. On the other hand, the biochemistry part of this study was relatively predictable. Treatment of BSA with excess reagents 1 and 2 gave the desired azido and cyclooctyne groups in functionalized proteins BSA-1 and BSA-2. The amount of azide and cyclooctyne groups attached was determined from absorbances and fluorescence intensities of cycloadducts BSA-1-6 and BSA-2-6 obtained by SPAAC and CuAAC reactions with complementary functionalized fluorescent probes 5 and 6. Covalent binding of functionalized BSA was performed by direct SPAAC between BSA-1 and BSA-2 and by binding BSA-1 or BSA-2 through SPAAC with 0.5 equiv. of complementary bis-azide and bis-cyclooctyne linkers 4 and 3. SDS-PAGE analysis showed weak spots with Mw ~ 120 kDa corresponding to dimers of BSA in all dimerization attempts. However, further optimization is required to obtain covalent dimers as the major products.
4. Experimental
4.1. General Methods
Melting points were determined on a Kofler micro hot stage and on a Mettler Toledo MP30 automated melting point system (Mettler Toledo, Columbus, OH, USA). The NMR spectra were recorded in CDCl3 and DMSO-d6 using Me4Si as the internal standard on a Bruker Avance III Ultrashield 500 and Bruker Avance Neo 600 instruments (Bruker, Billerica, MA, USA) at 500 and 600 MHz for 1H and at 125 and 150 MHz for 13C nucleus, respectively. Chemical shifts (δ) are given in ppm relative to Me4Si as internal standard (δ = 0 ppm) and vicinal coupling constants (J) are given in hertz (Hz). HRMS spectra were recorded on an Agilent 6224 time-of-flight (TOF) mass spectrometer equipped with a double orthogonal electrospray source under atmospheric pressure ionization (ESI) coupled to an Agilent 1260 high-performance liquid chromatograph (HPLC) (Agilent Technologies, Santa Clara, CA, USA). UV-vis spectra were recorded in MeOH using a Varian Cary Bio50 UV-Visible Spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Emission spectra were recorded on a PerkinElmer LS 50 B Luminescence spectrophotometer (PerkinElmer, Waltham, MA, USA). Fourier-transform infrared (FT-IR) spectra were obtained on a Bruker FTIR Alpha Platinum spectrophotometer (Bruker, Billerica, MA, USA) using attenuated total reflection (ATR) sampling technique. Microanalyses for C, H, and N were obtained on a Perkin-Elmer CHNS/O Analyzer 2400 Series II (PerkinElmer, Waltham, MA, USA). Column chromatography (CC) was performed on silica gel (Silica gel 60, particle size: 0.035–0.070 mm (Sigma-Aldrich, St. Louis, MO, USA). Acquisition and analysis of gels obtained after SDS-PAGE analysis and subsequent staining were performed on a Bio-Rad ChemiDoc MO Imaging System using BioRad Image Lab 6.1 Software for Windows (Bio-Rad, Hercules, CA, USA).
Unless otherwise stated, solutions in PBS buffer (10 mM Na2HPO4, 10 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) were used. Prior to use, the commercial BSA was purified by loading the sample onto a HiLoad Superdex 200 pg preparative SEC column (Cytiva), connected to an ÄKTA FPLC system. The column was equilibrated in PBS buffer, pH 7.4, and a flow rate of 1 mL·min−1 was used to separate proteins. Proteins eluting at volumes corresponding to monomeric BSA were collected and stored at −80 °C until further use.
Ascorbic acid, bis(2,5-dioxopyrrolidin-1-yl) carbonate (DSC), bromine, t-BuOK, t-BuONa, 1,1’-carbonyldiimidazole (CDI), copper(II) sulfate, N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDCI), 4-(dimethylamino)pyridine (DMAP), N-hydroxysuccinimide, LiAlH4, lithium diisopropylamide (LDA), N-methylmorpholine (NMM), sodium azide, Rh(OAc)2, thionyl chloride, tris[(1-benzyl-4-triazolyl)methyl]amine (TBTA), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), bovine serum albumin (BSA), 2,5-dioxopyrrolidin-1-yl 2-azidoacetate (1a), 2,5-dioxopyrrolidin-1-yl 4-azidobutanoate (1b), N-(3-azidopropyl)-3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanamide (1e), [(bicyclo[6.1.0]non-4-yn-9-yl)methyl] (2,5-dioxopyrrolidin-1-yl) carbonate (2a), 2,5-dioxopyrrolidin-1-yl 4-(11,12-didehydrodibenzo[b,f]azocin-5(6H)-yl)-4-oxobutanoate (2c), chloroacetic acid (7a), azidoacetic acid (8a), methyl 4-chlorobutyrate (9b), 1H-benzo[d][1,2,3]triazole (11), 6-aminocaproic acid (12), tert-butyl (2-bromoethyl)carbamate (13), succinic anhydride (16), 4-(11,12-didehydrodibenzo[b,f]azocin-5(6H)-yl)-4-oxobutanoic acid (17), Z,Z-1,5-cyclooctadiene (18), ethyl diazoacetate (19), 6-aminofluorescein (28), 4-pentynoic acid (29), 6-carboxyfluorescein (30), propargylamine (31), 2,2'-[ethane-1,2-diylbis(oxy)]bis(ethan-1-amine) (32), adipoyl chloride (33), and 1,4-diisothiocyanatobutane (35) are commercially available.
2,5-Dioxopyrrolidin-1-yl 2-azidoacetate (1a) [26], 3',6'-dihydroxy-3-oxo-N-(prop-2-yn-1-yl)-3H-spiro[isobenzofuran-1,9'-xanthene]-6-carboxamide (6c) [36,37], azidoacetic acid (8a) [38], 4-azidobutanoic acid (8b), methyl 4-azidobutanoate (10b) [39], and tert-butyl (2-azidoethyl)carbamate (14) [42,43], and were prepared following the literature procedures (see experimental procedures for the references).
Unless otherwise stated, solutions in PBS buffer (10 mM Na2HPO4, 10 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4) were used.
4.2. 2-Azidoacetic Acid (8a) [38]
This compound was prepared following slightly modified general literature procedure for the synthesis of alkyl azides [38]. NaN3 (672 mg, 10.5 mmol) and ammonium chloride (1.06 g, 20 mmol) were added to a solution of chloroacetic acid (7a) (945 mg, 10 mmol) in water (7 mL) and the mixture was stirred at room temperature for 72 h. The reaction mixture was acidified with aq. HCl to pH 2 and the product was extracted with Et2O (3×20 mL). The combined organic phases were dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 8a. Yield: 957 g (95%) of colorless oil. 1H NMR (500 MHz, CDCl3) δ 9.09 (br s, 1H), 4.14 (s, 2H). Spectral data are in agreement with the literature data [26,38].
4.3. 2,5-Dioxopyrrolidin-1-yl 2-Azidoacetate (1a) [26]
This compound was prepared following literature procedure [26]. The reaction was carried out under argon. A mixture of azidoacetic acid (8) (508 g, 5 mmol), N-hydroxysuccinimide (862 g, 7.5 mmol), and anh. CH2Cl2 (15 mL) was stirred at 0 °C for 10 min. Then, N-[3-(dimethylamino)propyl]-N′-ethylcarbodiimide hydrochloride (EDCI) (1.438 g, 7.5 mmol) was added and stirring was continued for 10 min. at 0 °C and then at room temperature for 24 h. The organic phase was washed with water (2×5 mL) and brine (5 mL), then dried over anh. Na2SO4, filtered, and the filtrate was evaporated in vacuo to give 1a. Yield: 729 mg (73%) of white solid. 1H NMR (500 MHz, CDCl3) δ 4.24 (s, 2H), 2.87 (s, 4H). Spectral data are in agreement with the literature data [26].
4.4. Synthesis of Methyl 4-Azidobutanoate (10b) [39]
This compound was prepared following slightly modified literature procedure [39]. Methyl 4-chlorobutanoate (9b) (609 μL, 682 mg, 5 mmol) was dissolved in DMSO (5 mL), NaN3 (975 mg, 15 mmol) was added, and the mixture was stirred at 80 °C for 12 h. Reaction mixture was cooled to room temperature, diluted with Et2O (20 mL), and washed with water (3×10 mL). The organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 10b. Yield: 603 mg (84%) of yellowish oil. 1H NMR (500 MHz, CDCl3) δ 3.69 (s, 3H), 3.35 (t, J= 6.7 Hz, 2H), 2.42 (t, J= 7.3 Hz, 2H), 1.91 (p, J= 7.0 Hz, 2H). νmax (ATR) 2093 (N3), 1732 (C=O), 1437, 1164, 1080, 897 cm–1. Spectral data are in agreement with the literature data [39].
4.5. Synthesis of 4-Azidobutanoic Acid (8b) [39]
This compound was prepared following modified literature procedure [39]. Methyl 4-azidobutanoate (10b) (603 mg, 4.2 mmol) was dissolved in MeOH (3 mL), 2 M aq. NaOH (4.2 mL, 8.4 mmol) was added, and the mixture was stirred at 20 °C for 1 h. Reaction mixture was acidified with 1 M aq. HCl to pH 1, and the product was extracted with Et2O (3×20 mL). The combined organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 8b. Yield: 460 mg (83%) of yellowish oil. 1H NMR (500 MHz, CDCl3) δ 11.13 (br s, 1H), 3.38 (t, J= 6.7 Hz, 2H), 2.48 (t, J= 7.3 Hz, 2H), 1.92 (p, J= 7.0 Hz, 2H). Spectral data are in agreement with the literature data [39].
4.6. General Procedure for the Synthesis of N-Acylbenzotriazoles 1c, 1d, and 2b
Compounds 1c, 1d, and 2b were prepared following the general literature procedure for the preparation of N-acylbenzotriazoles [41]. Under Ar, SOCl2 (150 µL, 2 mmol) was slowly added via syringe to a stirred solution of 1H-benzotriazole (1.00 g, 8 mmol) in anh. CH2Cl2 (50 mL) at r.t. and the mixture was stirred at r.t. for 30 min. Then, carboxylic acid 8a, 8b, or 17 (2 mmol) was added and a white precipitate that was formed within a few seconds was collected by filtration and washed with CH2Cl2 (2×10 mL). The combined filtrate was washed with 2 M aq. NaOH (30 mL), dried over anh. Na2SO4, filtered, and the filtrate was evaporated in vacuo to give 1c, 1d, and 2c.
4.6.1. 2-Azido-1-(1H-benzo[d][1,2,3]triazol-1-yl)ethan-1-one (1c) [28]
From 1H-benzo[d][1,2,3]triazole (11) (4.617 g, 38.8 mmol), SOCl2 (688 μL, 9.7 mmol), azidoacetic acid (8a) (980 mg, 9.7 mmol), the precipitate was washed with CH2Cl2 (2×50 mL), and the combined filtrate with 2 M aq. NaOH (3×60 mL). Yield: 322 mg (16%) of yellow solid, m.p. 56–64 °C. 1H NMR (500 MHz, CDCl3): δ 8.30 (ddt, J = 8.4, 4.2, 1.0 Hz, 1H), 8.17 (dt, J = 8.4, 0.9 Hz, 1H), 7.73 (ddt, J = 8.3, 7.0, 1.3 Hz, 1H), 7.57 (ddt, J = 8.1, 7.4, 0.9 Hz, 1H), 5.20 and 5.02 (2s, 1:2, 2H). 13C NMR (126 MHz, CDCl3): δ 172.3, 138.5, 126.7, 115.0, 50.4. m/z (HRMS) Found: 120.0509 [MH–N3CH2CO]+. C6H6N3 requires m/z = 120.0556. νmax (ATR) 2953, 2108, 1723 (C=O), 1413, 1278, 1202, 1012, 778, 737, 688 cm–1.
4.6.2. 4-Azido-1-(1H-benzo[d][1,2,3]triazol-1-yl)butan-1-one (1d)
From 1H-benzo[d][1,2,3]triazole (11) (1.67 g, 14 mmol), SOCl2 (247 μL, 3.5 mmol), 4-azidobutanoic acid (8b) (452 mg, 3.5 mmol), the precipitate was washed with CH2Cl2 (2×15 mL), and the combined filtrate with 2M aq. NaOH (3×20 mL). Yield: 625 mg (78%) of yellow solid, m.p. 48–50 °C. 1H NMR (500 MHz, CDCl3): δ 8.28 (br d, J = 8.3 Hz, 1H), 8.13 (br d, J = 8.3 Hz, 1H), 7.67 (ddd, J = 8.1, 7.1, 0.9 Hz, 1H), 7.52 (ddd, J = 8.1, 7.0, 0.9 Hz, 1H), 3.55 (t, J = 7.2 Hz, 2H), 3.53 (t, J = 6.6 Hz, 2H), 2.20 (p, J = 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 171.6, 146.3, 131.1, 130.6, 126.4, 120.3, 114.4, 50.6, 32.7, 23.7. m/z (HRMS) Found: 253.0813 [M+Na]+. C10H10N6NaO requires m/z = 253.0808. Anal. Calcd. for C10H10N6O: C, 52.17; H, 4.38; N, 36.50%. Found: C, 52.20; H, 4.12; N, 36.26%. νmax (ATR) 2099, 1742 (C=O), 1485, 1445, 1365, 1287, 1260, 1165, 1065, 1003, 966, 854, 771, 755, 630 cm–1.
4.6.3. N-[4-(11,12-Didehydro-5,6-dihydrodibenzo[b,f]azocin-5-yl)-4-oxobutanoyl]-1H-benzo[d][1,2,3]triazole (2b)
From 1H-benzo[d][1,2,3]triazole (11) (714 mg, 6 mmol), SOCl2 (106 μL, 1.5 mmol)), DBCO-acid (17) (457 mg, 1.5 mmol), the precipitate was washed with CH2Cl2 (2×5 mL), and the combined filtrate with 2M aq. NaOH (3×6 mL). Yield: 500 mg (82%) of pink solid, m.p. 160–163 °C. 1H NMR (500 MHz, CDCl3) δ 8.20 (d, J = 8.2 Hz, 1H), 8.08 (dd, J = 8.3, 0.9 Hz, 1H), 7.72 – 7.57 (m, 3H), 7.51 – 7.40 (m, 4H), 7.37 – 7.24 (m, 3H), 5.20 (d, J = 13.9 Hz, 1H), 3.80 (ddd, J = 18.4, 9.0, 5.1 Hz, 1H), 3.73 (d, J = 13.9 Hz, 1H), 3.38 (ddd, J = 18.4, 6.0, 5.0 Hz, 1H), 3.06 (ddd, J = 16.9, 9.0, 5.1 Hz, 1H), 2.20 (dt, J = 17.0, 5.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 171.6, 171.2, 151.4, 147.9, 146.0, 132.1, 131.0, 130.2, 129.3, 128.6, 128.3, 127.8, 127.2, 126.0, 125.5, 123.1, 122.8, 120.1, 115.1, 114.3, 107.6, 55.6, 53.5, 30.9, 28.6. m/z (HRMS) Found: 407.1491 [M+H]+. C25H19N4O2 requires m/z = 407.1491. νmax (ATR) 1727, 1652, 1479, 1448, 1387, 1350, 1307, 1226, 1165, 1063, 1005, 960, 802, 780, 765, 751, 649, 634 cm–1.
4.7. Synthesis of 6-(2-Azidoacetamido)hexanoic Acid (8c)
Under argon, CDI (2.00 g, 12.3 mmol) was added to a stirred solution of azidoacetic acid (8a) (1.22 g, 12.1 mmol) in anh. MeCN (30 mL) and the mixture was stirred at room temperature for 1 h. Then, 6-aminocaproic acid (12) (1.75 g, 13.3 mmol) was added and stirring under argon was continued for 2 h at room temperature and then for 24 h at 40 °C. Volatile components were evaporated in vacuo, the residue was dissolved in water (15 mL), acidified with 1 M aq. HCl to pH 1, and the product was extracted with EtOAc (3×70 mL). The combined organic phase was dried over anh. Na2SO4, filtered, and the filtrate was evaporated in vacuo to give 8c. Yield: 290 mg (11%) of yellow oil. 1H NMR (500 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.19, 8.08, and 7.72 (3br t, 3:1:1, J = 5.5 Hz, 1H), 4.27, 4.02, 3.99, and 3.78 (4s, 3:6:1:2, 2H), 3.06 and 3.00 (2q, 4:1, J = 6.8 Hz, 2H), 2.19 and 2.02 (2t, 5:1, J = 7.4 Hz, 2H), 1.52 – 1.45 (m, 2H), 1.44 – 1.34 (m, 2H), 1.29 – 1.17 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 174.9, 172.5, 172.3, 170.6, 169.1, 167.5, 166.2, 51.2, 49.9, 43.1, 42.0, 35.8, 34.0, 29.4, 29.1, 29.0, 26.4, 26.3, 25.5, 24.6, 21.5. Multiple signals for all nuclei are due to the presence of isomers (rotamers). m/z (HRMS) Found: 213.1003 [M–H]–. C8H15N4O3 requires m/z = 213.0993. νmax (ATR) 2937, 2109 (N3), 1708 (C=O), 1631 (C=O), 1545, 1411, 1194, 1102, 927, 789, 639 cm–1.
4.8. Synthesis of Tert-butyl (2-azidoethyl)carbamate (14) [42,43]
Compound 14 was prepared following slightly modified literature procedures [42,43]. A mixture of tert-butyl (2-bromoethyl)carbamate (13) (1.12 g, 5 mmol), sodium azide (357 mg, 5.5 mmol), and anh. DMF (10 mL) was stirred under argon at 80 °C for12 h. The mixture was cooled to room temperature, Et2O (50 mL) was added, and the solution was washed with brine (5×10 mL). The organic phase was evaporated in vacuo to give 14. Yield: 723 mg (78%) of colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.89 (br s, 1H), 3.39 (t, J = 5.6 Hz, 2H), 3.28 (q, J = 5.9 Hz, 2H), 1.43 (s, 9H). νmax (ATR) 3349, 2978, 2933, 2095 (N3), 1687 (C=O), 1511, 1451, 1391, 1366, 1247, 1162, 1099, 1039, 992, 861, 781, 758, 637. cm–1. Spectral data are in agreement with the literature data [42,43].
4.9. 2-Azidoethan-1-aminium chloride (15) [44]
Compound 15 was prepared following modified literature procedure [44]. Compound 14 (723 mg, 3.88 mmol) was dissolved in EtOAc (6 mL) and cooled to 0 °C (ice-bath). While stirring at 0 °C, 2 M HCl (6 mL, 12 mmol) was added and stirring at 0 °C was continued for 2.5 h. The precipitate was collected by filtration, washed with EtOAc (2× 5 mL), and dried over NaOH pellets in vacuo at room temperature for 24 h to give 14. Yield: 250 mg (68%) of white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.26 (br s, 3H), 3.75 – 3.55 (m, 2H), 2.95 (dd, J = 6.6, 5.0 Hz, 2H). Spectral data are in agreement with the literature data [44,45].
4.10. Synthesis of 4-[(2-Azidoethyl)amino]-4-oxobutanoic Acid (8d) [46]
This compound was prepared following modified literature procedure [46]. Succinic anhydride (16) (50 mg, 0.5 mmol) and N-methylmorpholine (NMM) (110 μL, 1 mmol) were added to a stirred suspension of compound 15 (60 mg, 0.5 mmol) in a mixture of anh. CH2Cl2 (1 mL) and anh. THF (1 mL) for 1 h at room temperature. KOH (28 mg, 0.5 mmol) was added and stirring at room teperature was continued for 24 h. Then, CH2Cl2 (10 mL) was added and the mixture was washed with 1 M aq. HCl (5 mL) and brine (2×5 mL). The organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 8d. Yield: 15 mg (16%) of yellow resin. 1H NMR (500 MHz, CDCl3) δ 6.01 (br s, 1H), 3.49 – 3.41 (m, 4H), 2.73 (dd, J = 7.3, 5.7 Hz, 2H), 2.53 (dd, J = 7.3, 5.7 Hz, 2H). Spectral data are in agreement with the literature data [46].
4.11. Synthesis of Ethyl (1R*,8S*,4Z)-Bicyclo[6.1.0]non-4-ene-9-carboxylate (20) [47]
This compound was prepared following literature procedure [47]. A solution of ethyl diazoacetate (2.1 mL, 20 mmol) in CH2Cl2 (10 mL) was added slowly (dropwise over ~1 h) to a stirred mixture of Z,Z-1,5-cyclooctadiene (19.6 mL, 160 mmol), Rh(OAc)4 (380 mg, 0.86 mmol), and CH2Cl2 (10 mL) and the mixture was stirred at r.t. for 40 h. Insoluble material was removed by filtration through a glass frit and the filtrate was evaporated in vacuo. The residue was purified by CC (silica gel, petroleum ether). Fractions containing the product were combined and evaporated in vacuo to afford 20 as a C(9)-endo/exo-mixture of epimers. Yield: 3.22 g (83%) of colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.63 and 5.60 (dq and td, 3:2, J = 4.1, 2.1 and 4.0, 2.3 Hz, 2H), 4.11 and 4.09 (2q, 2:3, J = 7.1 Hz, 2H), 2.50 and 2.30 (2 ddt, 2:3, J = 15.8, 8.0, 3.7 and 15.1, 7.6, 3.5 Hz, 1H), 2.24 – 2.15 (m, 2H), 2.12 – 2.01 (m, 2H), 1.82 (dddd, J = 14.4, 9.8, 7.1, 5.1 Hz, 1H), 1.71 and 1.18 (2t, 3:3, J = 8.8 and 4.6 Hz, 1H), 1.59 – 1.53 (m, 1H), 1.52 – 1.43 (m, 1H), 1.42 – 1.36 (m, 1H), 1.28 – 1.23 (m, 4H). Spectral data are in agreement with the literature data [47,52].
4.12. Synthesis of Ethyl (1R*,4R*,5R*,8S*)-4,5-Dibromobicyclo[6.1.0]non-4-ene-9-carboxylate (21) [48]
A solution of Br2 (320 mg, 2 mmol) in CH2Cl2 (2 mL) was added to a stirred solution of 20 (388 mg, 2 mmol) in CH2Cl2 (20 mL) and the solution was stirred at room temperature for 15 min. Then, the reaction was quenched by the addition of 10% aq. Na2S2O3 (5 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2×10 mL). The combined organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 21 as a mixture of isomers. Yield: 642 mg (90%) of yellow resin. 1H NMR (500 MHz, CDCl3) δ 4.87 – 4.81 (m, 1H), 4.80 – 4.75 (m, 1H), 4.12 and 4.11 (2 q, 1:1, J = 7.1Hz, 2H), 2.77 – 2.68 and 2.68 – 2.60 (2 m, 3:2, 2H), 2.37 – 2.24 (m, 2H), 2.23 – 2.06 (m, 2H), 1.88 – 1.60 (m, 3H), 1.53 – 1.37 (m, 1H), 1.29 – 1.18 (m, 4H). Spectral data are in agreement with the literature data [48].
4.13. Synthesis of (1R*,8S*,4Z)-Bicyclo[6.1.0]non-4-ene-9-carboxylic Acid (23) [49,50]
2 M aq. NaOH (2 mL, 4 mmol) was added to a solution of 20 (136 mg, 0.7 mmol) in EtOH (3 mL), the mixture was stirred at room temperature for 3 h, and acidified with 1 M aq. HCl to pH 2. The product was extracted with EtOAc (3×20 mL), the organic phases were combined, dried over anh. Na2SO4, filtered, and the filtrate was evaporated in vacuo to give 23 as a C(9)-endo/exo-mixture of epimers. Yield: 108 mg (93%) of colorless oil. 1H NMR (500 MHz, CDCl3) δ 11.77 (s, 1H), 5.60 – 5.54 (m, 2H), 2.42 – 2.34 (m, 1H), 2.29 – 2.21 (m, 1H), 2.14 – 2.06 (m, 2H), 2.06 – 1.95 (m, 2H), 1.79 – 1.70 (m, 1H), 1.59 and 1.14 (2 t, 1:2, J = 8.7 and 4.6 Hz, 1H), 1.52 – 1.42 (m, 1H), 1.41 – 1.29 (m, 2H). Spectral data are in agreement with the literature data [49,50].
4.14. Synthesis of (1R*,4R*,5R*,8S*)-4,5-Dibromobicyclo[6.1.0]non-4-ene-9-carboxylic Acid (24) [51]
A solution of Br2 (431 mg, 2.7 mmol) in CH2Cl2 (2 mL) was added to a stirred solution of 23 (450 mg, 2.7 mmol) in CH2Cl2 (20 mL) and the solution was stirred at room temperature for 15 min. Then, the reaction was quenched by the addition of 10% aq. Na2S2O3 (5 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2×10 mL). The combined organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 24 as a mixture of isomers. Yield: 720 mg (82%) of colorless resin. 1H NMR (500 MHz, CDCl3) δ 11.92 (br s, 1H), 5.04 – 4.92 (m, 2H), 2.64 – 2.51 (m, 2H), 2.24 – 2.09 (m, 2H), 2.08 – 1.99 (m, 2H), 1.78 – 1.61 (m, 1H), 1.69 and 1.17 (2 t, 2:3, J = 8.7 and 4.1 Hz, 1H), 1.52 – 1.30 (m, 3H). Spectral data are in agreement with the literature data [51].
4.15. Synthesis of (1R*,8S*,9RS,4Z)-Bicyclo[6.1.0]non-4-en-9-ylmethanol (25) [47]
This compound was prepared following literature procedure [47]. Reaction was carried out under argon in a flame-dried flask. A solution of LiAlH4 in anh. THF (2.4 M, 500 μL, 1.2 mmol) was added via syringe to a stirred cold (0 °C, ice-bath) solution of 20 (194 mg, 1 mmol) in anh. Et2O (2 mL) and the mixture was stirred at 0 °C for 15 min. and then at 45 °C for 1 h. The mixture was cooled to 0 °C and the reaction was quenched by slow (dropwise) addition of water (3 mL) to the stirred mixture. The obtained solution was diluted with THF (20 mL), dried over anh. Na2SO4, filtered, and the filtrate was evaporated in vacuo to give 25. Yield: 111 mg (73%) of pale yellow resin. 1H NMR (500 MHz, CDCl3) δ 5.63 (td, J = 4.2, 2.1 Hz, 2H), 3.71 (d, J = 7.6 Hz, 1H), 3.47 (d, J = 6.9 Hz, 1H), 2.45 – 2.24 (m, 2H), 2.22 – 1.93 (m, 4H), 1.91 – 1.70 (m, 1H), 1.57 (m, 1H), 1.47 – 1.32 (m, 1H), 1.29 – 1.06 (m, 1H), 1.05 – 0.97 (m, 1H), 0.80 – 0.63 (m, 1H). Spectral data are in agreement with the literature data [47].
4.16. Synthesis of (1R*,4R*,5R*,8S*,9RS)-4,5-Dibromo bicyclo[6.1.0]non-4-en-9-ylmethanol (26) [47,52]
This compound was prepared following literature procedure [47]. A solution of Br2 (37 μL, 115 mg, 0.73 mmol) in CH2Cl2 (2 mL) was added to a stirred solution of 25 (111 mg, 0.7 mmol) in CH2Cl2 (5 mL) and the solution was stirred at room temperature for 15 min. Then, the reaction was quenched by the addition of 10% aq. Na2S2O3 (5 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 (2×10 mL). The combined organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo to give 25. Yield: 167 mg (77%) of yellowish resin. 1H NMR (500 MHz, CDCl3) δ 4.89 – 4.69 (m, 2H), 3.76 (dd, J = 7.5, 1.3 Hz, 1H), 3.52 (dd, J = 7.1, 1.9 Hz, 1H), 2.79 – 2.56 (m, 2H), 2.36 – 1.84 (m, 4H), 1.70 – 1.52 (m, 2H), 1.48 – 1.31 (m, 1H), 1.30 – 1.03 (m, 1H), 1.00 – 0.81 (m, 1H), 0.68 (m, 1H). Spectral data are in agreement with the literature data [47,52].
4.17. 2-Azido-N-(3',6'-Dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9'-xanthen]-6-yl)acetamide (5) [33,34]
NHS azidoacetate 1a (99 mg, 0.5 mmol) was added to a stirred solution of 28 (174 mg, 0.5 mmol) in MeCN (3 mL) and the mixture was stirred at room temperature for 24 h. EtOAc (15 mL) was added, and the solution was washed with 1 M aq. NaHSO4 (5 mL) and brine (5 mL). The organic phase was dried over anh. MgSO4, filtered, and evaporated in vacuo. The residue (crude compound 5, 165 mg) was suspended in CH2Cl2 (100 mL) and stirred at room temperature for 2 h. Then, stirring was stopped and the suspension was left to settle down. The supernatant was decanted and the solid residue was dried in vacuo at room temperature to give 5. Yield: 52 mg (24%)(G.P.B) of brown solid. 1H NMR (500 MHz, DMSO-d6) δ 10.64 (s, 1H), 10.17 (br s, 2H), 8.30 (d, J = 2.0 Hz, 1H), 7.84 (dd, J = 8.3, 2.0 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H), 6.68 (d, J = 2.4 Hz, 2H), 6.60 (d, J = 8.6 Hz, 2H), 6.55 (dd, J = 8.6, 2.4 Hz, 2H), 4.14 (s, 2H). 13C NMR (126 MHz, CD3OD): δ 13C NMR (126 MHz, CD3OD) δ 165.4, 161.7, 159.5, 151.9, 144.7, 131.7, 120.7, 119.6, 118.8, 116.4, 106.9, 104.1, 101.9, 94.0, 43.8, 16.8. m/z (HRMS) Found: 431.0981 (MH+). C22H15N4O6 requires m/z = 431.0986. νmax (ATR) 2208, 2104, 1695, 1584, 1531, 1453, 1384, 1237, 1204, 1169, 1109, 992, 909, 843, 759, 662 cm–1. Physical and spectral data are in agreement with the literature data [33,34].
4.18. N-(3',6'-Dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9'-xanthen]-6-yl)pent-4-ynamide (6a) [35]
This compound was prepared following modified literature procedure [35]. The reaction was carried out under argon in a flame-dried flask. A mixture of 28 (174 mg, 0.5 mmol), 4-pentynoic acid (29) (60 mg, 0.5 mmol), and anh. DMF (4 mL) was stirred at 0 °C (ice-bath) for 5 min. Then, EDCI (115 mg, 0.6 mmol) was added and stirring was continued for 10 min. at 0 °C and then at room temperature for 24 h. EtOAc (20 ml) was added, and the solution was washed with 1 M aq. NaHSO4 (10 mL) and brine (10 mL). The organic phase was dried over anh. MgSO4, filtered, and the filtrate was evaporated in vacuo. The residue was purified by CC (hexanes–EtOAc, first 2:1 toe elute non-polar impurities, then 1:3 to elute the product). Fractions containing the product were combined and evaporated in vacuo to give 6a. Yield: 52 mg (24%) of orange-brown solid. 1H NMR 1H NMR (500 MHz, MeOD) δ 7.15 (d, J = 2.0 Hz, 1H), 7.06 (dd, J = 8.3, 2.2 Hz, 1H), 6.88 (d, J = 8.2 Hz, 1H), 6.69 – 6.59 (m, 4H), 6.53 (dd, J = 8.7, 2.4 Hz, 2H), 2.82 and 2.66 (2 br t, 2:3, 1H), 2.62 – 2.57 (m, 1H), 2.51 – 2.47 (m, 1H), 2.46 – 2.41 (m, 1H), 2.36, 2.32, and 2.24 (3 t, 2:3:5, J = 2.6 Hz, 1H). Physical and spectral data are in agreement with the literature data [35].
4.19. 4-(11,12-Didehydro-5,6-dihydrodibenzo[b,f]azocin-5-yl)-N-(3',6'-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9'-xanthen]-6-yl)-4-oxobutanamide (6b)
The reaction was carried out under argon in a flame-dried flask. A mixture of DBCO-acid 17 (153 mg, 0.5 mmol) and anh. DMF was stirred at 0 °C (ice-bath) for 5 min. Then, EDCI (115 mg, 0.6 mmol) was added and stirring at 0 °C was continued for 10 min. Next, 6-aminofluoresceine (28) (174 mg, 0.5 mmol) was added and the mixture was stirred at room temperature for 24 h. Most of DMF was evaporated in vacuo until approximately 1 mL of DMF left. EtOAc (20 ml) was added, and the solution was washed with 1 M aq. NaHSO4 (2×10 mL), brine (10 mL), and sat. aq. NaHCO3 (20 mL). The aqueous NaHCO3 phase was acidified with 2 M aq. HCl until pH 1 and the precipitate was collected by filtration to give 6b. Yield: 25 mg (8%) of brown solid. 1H NMR (500 MHz, DMSO-d6) δ 10.35, 10.32, 8.28 and 8.23 (4 s, 1:3:1:3, 1H), 10.13 (s, 1H), 7.77 – 7.61 (m, 3H), 7.58 – 7.43 (m, 3H), 7.42 – 7.25 (m, 3H), 7.25 – 7.15 (m, 2H), 7.08 – 6.85 (m, 2H), 6.83 – 6.47 (m, 5H), 5.06 (dd, J = 14.1, 5.3 Hz, 1H), 3.65 (d, J = 14.1 Hz, 1H), 2.81 – 2.56 (m, 3H), 2.41 – 2.24 (m, 1H). 13C NMR (126 MHZ, CD3OD) δ 164.6, 163.8, 151.8, 151.4, 147.9, 147.7, 143.2, 143.1, 143.0, 139.9, 137.1, 125.1, 124.0, 123.9, 121.2, 121.1, 120.5, 120.5, 120.2, 120.2, 119.6, 119.4, 118.7, 117.0, 117.0, 114.4, 106.1, 104.3, 97.0, 94.0, 47.2, 23.3, 21.3. m/z (HRMS) Found: 635.1808 (MH+). C39H27N2O7 requires m/z = 635.1813. νmax (ATR) 2151, 2043, 1754 (C=O), 1603, 1481, 1425, 1253, 1204, 1160, 1110, 1072, 993, 848, 752 cm–1.
4.20. 3',6'-Dihydroxy-3-oxo-N-(prop-2-yn-1-yl)-3H-spiro[isobenzofuran-1,9'-xanthene]-6-carboxamide (6c) [36,37]
This compound was prepared according to slightly modified literature procedure for the preparation of closely related fluorescein-6-carboxamides [53]. Et3N (418 μL, 303 mg, 3 mmol) and DMAP (6 mg, 50 μmol) were added to a stirred solution of carboxyfluorescein 30 (188 mg, 0.53 mmol) and DSC (282 mg, 1.1 mmol) in anh. DMF (8 mL) and the mixture was stirred at room temperature in dark for 1 h. Then, propargylamine (31) (86 μL, 69 mg, 1.25 mmol) was added and stirring in dark at room temperature was continued for 2 h. Volatile components were evaporated in vacuo, the residue was dissolved in EtOAc (20 mL), and the solution was washed with 1 M aq. NaHSO4 (2×10 mL) and brine (1×10 mL). The organic phase was dried over anh. Na2SO4, filtered, the filtrate was evaporated in vacuo, and the residue was purified by CC (silica gel, CH2Cl2–MeOH, 8:1). Fractions containing the product were combined and evaporated in vacuo. The crude product 6c (25 mg, 12%) was dissolved in EtOAc (30 mL), the solution was transferred to a separatory funnel and shaken with sat. aq. NaHCO3 (3×15 mL). The combined aqueous phase was transferred to a separatory funnel, acidified with 2 M aq. HCl to pH 1, EtOAc (50 mL) was added, and the biphasic system was shaken and left to settle. The precipitate, which was formed between the organic and the aqueous phase, was collected by filtration to give 6c. Yield: 12 mg (6%) of red solid. 1H NMR (500 MHz, DMSO-d6) δ 10.27 (br s, 2H), 9.35 (br t, J = 5.5 Hz, 1H), 8.47 (s, 1H), 8.26 (br d, J = 8.1 Hz, 1H), 7.38 (br d, J = 8.1 Hz, 1H), 6.72 (br s, 2H), 6.56 (br q, J = 8.7 Hz, 4H), 4.11 (br t, J = 3.8 Hz, 2H), 3.16 and 2.89 (2 br s, 1:1, 1H). 13C NMR (126 MHz, DMSO-d6) δ 169.0, 165.8, 159.9, 155.2, 152.3, 135.9, 135.2, 129.6, 126.9, 124.8, 124.1, 113.3, 109.4, 102.8, 90.4, 81.1, 73.3, 29.3. m/z (HRMS) Found: 414.0969 (MH+). C24H16NO6 requires m/z = 414.0972. Spectral data are in agreement with the literature data [36,37].
4.21. N,N'-[(Ethane-1,2-diylbis(oxy)bis(ethane-2,1-diyl)]bis(4-azidobutanamide) (3)
Et3N (333 μL, 242 mg, 2.4 mmol) was added to a stirred solution of 2,2'-[ethane-1,2-diylbis(oxy)]bis(ethan-1-amine) (32) (147 μL, 148 mg, 1 mmol) and N-(4-azidobutanoyl)benzotriazole (1d) (470 mg, 2 mmol) in MeCN (10 mL) and the mixture was stirred at room temperature for 24 h. Volatile components were evaporated in vacuo and the residue was purified by CC. First benzotriazole was eluted with EtOAc–hexanes 1:1, followed by elution of the product with MeOH. Fractions containing the product were combined and evaporated in vacuo to give 3. Yield: 316 mg (85%) of a yellowish solid, m.p. 40–55 °C. 1H NMR (500 MHz, CDCl3) δ 6.12 (br s, 1H), 3.60 (br s, 2H), 3.55 (br t, J = 5.2 Hz, 2H), 3.45 (br q, J = 5.4 Hz, 2H), 3.35 (br t, J = 6.6 Hz, 2H), 2.28 (t, J = 7.2 Hz, 2H), 1.92 (p, J = 7.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 171.9, 70.3, 69.9, 50.9, 39.3, 33.2, 24.9. m/z (HRMS) Found: 371.2153 (MH+). C14H27N8O4 requires m/z = 371.2150. νmax (ATR) 3252, 2866, 2094, 1625 (C=O), 1564, 1457, 1422, 1341, 1253, 1122, 1031, 893, 853, 743 cm–1.
4.22. N,N'-[(Ethane-1,2-diylbis(oxy))bis(ethane-2,1-diyl)]bis[4-oxo-4-(11,12-didehydro-5,6-dihydrodibenzo[b,f]azocin-5-yl]butanamide) (4)
Et3N (384 μL, 278 mg, 2.76 mmol) was added to a stirred solution of 2,2'-[ethane-1,2-diylbis(oxy)]bis(ethan-1-amine) (32) (169 μL, 170 mg, 1.15 mmol) and N-[4-oxo-4-(5-aza-3,4:7,8-dibenzocyclooctyn-5-yl)butananoyl]benzotriazole (2b) (934 mg, 2.3 mmol) in DMF (10 mL) and the mixture was stirred at room temperature for 24 h. Volatile components were evaporated in vacuo and the residue was purified by CC. First benzotriazole was eluted with EtOAc–EtOH 10:1, followed by elution of the product with MeOH. Fractions containing the product were combined and evaporated in vacuo to give 4. Yield: 805 mg (97%) of yellow-brown resin. 1H NMR (500 MHz, CDCl3) δ 7.70 – 7.56 (m, 2H), 7.51 – 7.46 (m, 2H), 7.39 – 7.32 (m, 6H), 7.30 – 7.15 (m, 6H), 6.36 and 6.31 (2t, 1:1, J = 5.5 Hz, 2H), 5.11 and 5.08 (2s, 1:1, 2H), 3.61 (dd, J = 13.8, 2.6 Hz, 2H), 3.57 – 3.48 (m, 4H), 3.47 – 3.36 (m, 4H), 3.35 – 3.23 (m, 4H), 2.81 – 2.70 (m, 2H), 2.41 – 2.31 (m, 2H), 2.12 and 2.03 (2dt, 1:1, J = 15.1, 6.1 Hz, 2H), 1.91 and 1.84 (2dt, 1:1, J = 16.8, 6.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 172.4, 172.4, 172.3, 172.3, 162.7, 151.5, 148.2, 148.2, 132.3, 132.3, 129.5, 129.5, 128.8, 128.7, 128.2, 128.2, 128.2, 127.8, 127.8, 127.1, 127.1, 125.6, 125.5, 123.3, 123.2, 122.6, 114.8, 114.8, 108.0, 70.3, 69.9, 69.8, 55.6, 50.8, 39.3, 39.3, 36.6, 31.5, 31.3, 31.1, 30.3, 30.2. Multiple signals for carbon nuclei are due to the presence of isomers (rotamers). m/z (HRMS) Found: 723.3162 (MH+). C44H43N4O6 requires m/z = 723.3177.
4.23. Synthesis of 1,4-Dioxa-7,14-diazacyclohexadecane-8,13-dione (34) [55,56]
The reaction was carried out under argon using a flame-dried flask and a rubber septum. Adipoyl chloride (33) (145 μL, 181 mg, 1 mmol) was dissolved in anh. CH2Cl2 (10 mL) and the stirred solution was cooled to 0 °C (ice-bath). Diamine 32 (292 μL, 296 mg, 2 mmol) was then added slowly via syringe at 0 °C. Ice-barh was removed and the obtained suspension was stirred at room temperature for 16 h. The precipitate was collected by filtration and dried in vacuo over NaOH pellets at room temperature for 24 h to give 34. Yield: 245 mg (95%) of a yellowish solid, m.p. 111–131 °C, lit. [55] m.p. 153–155 °C. 1H NMR (500 MHz, CDCl3) δ 6.07 (br s, 2H), 3.62 (br s, 4H), 3.59 – 3.54 (m, 4H), 3.49 – 3.43 (m, 4H), 2.27 – 2.22 (m, 4H), 1.72 – 1.65 (m, 4H). m/z (HRMS) Found: 259.1652 (MH+). C12H23N2O4 requires m/z = 259.1650. νmax (ATR) 3291, 2868, 1635, 1552, 1460, 1350, 1296, 1177, 1139, 1097, 1033, 982, 937, 864, 823, 696 cm–1. Spectral data are in agreement with the literature data [55,56].
4.24. Synthesis of 1,1'-(Butane-1,4-diyl)bis(3-{2-[2-(2-aminoethoxy)ethoxy]ethyl}thiourea) (36)
Diamine 32 (292 μL, 296 mg, 2 mmol) was added to a solution of 1,4-diisothiocyanatobutane (35) (172 mg, 1 mmol) in Et2O (2 mL), the mixture was stirred at room temperature for 16 h, and volatile components were evaporated in vacuo to give 36. Yield: 456 mg (97%) of a yellow resin. 1H NMR (500 MHz, CDCl3) δ 7.68 and 7.09 (2s, 1:2, 2H), 3.89 – 3.36 (m, 24H), 3.01 – 2.90 (m, 2H), 2.87 (t, J = 5.2 Hz, 2H), 2.47 br (s, 6H), 1.72 – 1.58 (br m, 4H). 13C NMR (126 MHz, CDCl3) δ 13C NMR (126 MHz, CDCl3) δ 183.6, 72.6, 70.3, 70.2, 70.1, 70.0, 68.1, 44.8, 44.2, 41.5, 40.6, 31.0, 26.4, 26.2, 25.7. Multiple signals for carbon nuclei are due to the presence of isomers (rotamers). m/z (HRMS) Found: 468.2555 (M+). C18H40N6O4S2 requires m/z = 468.2552. νmax (ATR) 3260, 3067, 2863, 1544, 1344, 1283, 1096, 698 cm–1.
4.25. Preparation of Stock Solutions of Azide- and Cyclooctyne-Functionalized Proteins BSA-1 and BSA-2 and Determination of Molar Loading (FG/BSA) from Molar Absorbances (Method A)
First, stock solutions of BSA-1a, BSA-1b, BSA-1e, BSA-2a, and BSA-2c were prepared. Five 1.5 mL PP vials were charged with reagents 1a, 1b, 1e, 2a, and 2c (1 mg, 5.0, 4.4, 4.0, 3.4, and 2.5 μmol, 10 equiv.) and a solution of BSA (33.2, 29.2, 26.4, 22.6, and 16.6 mg, 0.50, 0.44, 0.40, 0.34, and 0.25 μmol, 1 equiv.) in PBS buffer (1 mL) was added to each PP vial. The mixtures were stirred (400 min.–1) at 20 °C for 24 h. Reagent 1e dissolved completely, while reagents 1a, 1b, 2a, and 2c remained partially undissolved. The obtained stock solutions of BSA-1a, BSA-1b, BSA-1e, BSA-2a, and BSA-2c were stored at 4 °C. Next, aliquots (130 μL) of the crude BSA-1 and BSA-2 solutions were taken, and excess small molecular reagents and byproducts were removed by desalting. To purified cyclooctyne-conjugates BSA-2a (44.2 nmol) and BSA-2c (32.5 nmol) excess fluorescent probe 5 was added (1.5 mM, 295 and 220 μL, 443 and 330 nmol respectively, 10 equiv.). To purified azide-conjugates BSA-1a (65 nmol), BSA-1b (57.2 nmol), and BSA-1e (52 nmol) excess alkyne-fluorescent probe 6a (1.5 mM, 433, 370, and 345 μL, 650, 572, and 520 nm, respectively, 10 equiv.) and 1.25 μL of an aqueous solution of CuSO4 (10 mM, 12.5 nmol) and ascorbic acid (50 mM, 62.5 nmol) were added. The reaction mixtures were stirred (400 min.–1) at 20 °C for 3 days, aliquots (350 μL) were taken, and purified by SEC-FPLC. A Superdex 200 10/300 GL size exclusion column (formerly GE Healthcare Life Sciences, now Cytiva), equilibrated in PBS buffer (pH 7.4) and a flow rate of 0.5 mL·min−1 was used to purify the samples. Fractions containing the highest concentration of protein were collected to afford the fluorescently labeled proteins BSA-1a-6a, BSA-1b-6a, BSA-1e-6a, BSA-2a-5, and BSA-2c-5. Next, molar loading FG/BSA was determined for each labeled protein on the basis of absorbances measured at 280 nm and 498 nm (for FG/BSA values see Scheme 5, Method A). In a separate run, a solution of BSA-2a-6a with FG/BSA = 2.2 was prepared again for the use as a standard reference solution for rapid determination of FG/BSA from fluorescence intensities (see Method B, see Section 4.26).
4.26. Procedure for Rapid Determination of Molar Loading (FG/BSA) of Functionalized Proteins BSA-1 and BSA-2 from Fluorescence Intensities (Method B)
Stock solutions of the crude functionalized proteins BSA-1a,b,d,e and BSA-2b,c were prepared as described previously (see Section 4.25) by treatment of BSA with 10-fold excess reagents 1 and 2. Also this time, only reagent 1e dissolved completely, while reagents 1a, 1b, 2a, 2b, and 2c remained partially undissolved. Aliquots (100 μL) of the crude BSA-1 and BSA-2 solutions were taken, excess small molecular reagents and byproducts were removed by desalting, the purified BSA-1 and BSA-2 solutions were diluted with water to c ~ 2 mg mL–1 (~30 μM), and aliquoted (6 × 5 μL, 6 × ~0.15 nmol). To each aliquot PBS buffer containing 1% SDS (39 μL) was added, the mixture was heated at 90 °C for 10 min., and cooled to room temperature. To each aliquot of cyclooctyne-conjugates BSA-2b,c large excess of azide-fluorescent probe 5 (2.5 μL, 50 mM, 125 nmol) was added. To each aliquot of azide-conjugates BSA-1a,b,d,e large excess of alkyne-probe 6a (2.5 μL, 50 mM, 125 nmol) and 3.5 μL of an aqueous solution of CuSO4 (50 mM, 165 nmol), TCEP (100 mM, 330 nmol), and TBTA (3.4 mM, 11.9 nmol) was added. The reaction mixtures were stirred (400 min.–1) in dark for 1 h, followed by addition of 4V (200 μL) of cold (0 °C) acetone, and centrifugation (13000 G, 4 °C, 2 min.). The supernatants were decanted, and the proteins were analyzed by SDS-PAGE. To each fluorescently labeled protein 5 μL 4× SDS sample buffer, supplemented with 10 % v/v β-mercaptoethanol (4× SDS+R) was added, the mixtures were stirred (400 min.–1) at 37 °C for 30 min. and labeled proteins BSA-1-5 and BSA-2-6 and the reference standard solution BSA-2a-6a with known molar loading (FG/BSA = 2.2, see Section 4.25) were analyzed by SDS-PAGE. Afterwards, gels were briefly washed with deionized H2O and scanned with a ChemiDoc MP Imaging System (Bio-Rad) using settings for ProQ Emerald 300 detection. The same gels were subsequently stained with Coomassie Brilliant Blue, destained and photographed using the same imager. The molar loading FG/BSA was then determined by analyzing the two digital image data obtained (Emerald 300 and Coomasie Brilliant Blue) using Image Lab Software (Bio-Rad).
4.27. Procedure for Direct Covalent Cross-Linking of Functionalized Proteins BSA-1 and BSA-2 (Method C)
Solutions of the purified functionalized proteins BSA-1a, BSA-1b, BSA-1e, BSA-2a, and BSA-2c were prepared as described above (see Section 4.25) and their FG/BSA (0.2, 0.1, 0.1, 5.0, and 1.4, respectively) were determined by Method A (see Section 4.25 and Scheme 5). Twelve 1.5 mL PP vials were charged with purified cyclooctyne-conjugates BSA-2a and BSA-2c (6 × 100 μL each) and mixed with azide-conjugates BSA-1a, BSA-1b, and BSA-1e (2 × 100 μL each and 2 × 500 μL each). The mixtures were stirred at 20 °C for 4 hours and then analyzed by SDS-PAGE. Afterwards, gels were briefly washed with deionized H2O, stained with Coomassie Brilliant Blue, destained and photographed with a ChemiDoc MP Imaging System (Bio-Rad).
4.28. Procedure for Covalent Cross-Linking of Functionalized Proteins BSA-1b and BSA-2a Using Bifunctional Linkers 3 and 4 (Method D)
Solutions of the purified functionalized proteins BSA-1b and BSA-2a were prepared (see Section 4.25) and their FG/BSA (4.6 and 2.2, respectively) were determined by Method B (see Section 4.26 and Scheme 5). An 1.5 mL PP vial was charged with the purified azide-conjugate BSA-1b (200 μL, 439 μM, 87.8 nmol; FG/BSA = 4.6, n(azide) = 404 nmol, 1 equiv.) and a solution of bis-cyclooctyne 4 in DMSO (72.9 μL, 2.77 mM in DMSO, 202 nmol, 0.5 equiv.). In the same manner, cyclooctyne conjugate BSA-2a (200 μL, 341 μM, 68.3 nmol; FG/BSA = 2.2, n(cyclooctyne) = 150.3 nmol, 1 equiv.) was mixed with bis-azide 3 in PBS buffer (13.9 μL, 5.4 mM, 75.1 nmol, 0.5 equiv.). Both reaction mixtures were shaken at room temperature for 24 h and SDS-PAGE analysis was performed. Afterwards, gels were briefly washed with deionized H2O, stained with Coomassie Brilliant Blue, destained and photographed with a ChemiDoc MP Imaging System (Bio-Rad).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Copies of 1H and 13C NMR spectra.
Author Contributions
Conceptualization, M.N. and J.S.; methodology, N.S., M.N. and J.S.; software, N.S., M.N. and J.S.; validation, N.S., M.N. and J.S.; formal analysis, N.S., L.C.; N.P., M.N. and J.S.; investigation, N.S. and J.S.; resources, J.S.; data curation, N.S. and J.S.; writing—original draft preparation, L.C., U.G., N.P., M.N., B.Š. and J.S.; writing—review and editing, N.S., L.C., U.G., N.P., M.N., B.Š., and J.S.; visualization, J.S. L.C., U.G., N.P., M.N. and B.Š.; supervision, J.S. and M.N.; project administration, J.S.; funding acquisition, M.N. and J.S. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This research was funded by Slovenian Research and Innovation Agency (ARIS), research core funding No. P1-0179, research project No. N1-0211, and infrastructure programme No. I0-0022.
Data Availability Statement
The data presented in this study are available in the main manuscript and in the Supplementary Material of this manuscript.
Acknowledgments
NMR and LC-HRMS characterization and elemental analyses of compounds were performed at the Centre for Research Infrastructure at the Faculty of Chemistry and Chemical Technology, University of Ljubljana (IC UL FCCT).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hermanson, G. T. Bioconjugate Techniques, 3rd ed.; Academic Press: Amsterdam, 2013. [Google Scholar]
- Chauhan, P.V.R.; Kumar, M.; Molla, R.; Mishra, S. D.; Basa, S.; Rai, V. Chemical Technology Principles for Selective Bioconjugation of Proteins and Antibodies. Chem. Soc. Rev. 2024, 53, 380–449. [Google Scholar] [CrossRef]
- Cao, L.; Wang, L. Biospecific Chemistry for Covalent Linking of Biomacromolecules. Chem. Rev. 2024, 124, 8516–8549. [Google Scholar] [CrossRef]
- Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angewandte Chemie International Edition 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornøe, C. W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angewandte Chemie International Edition 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C. W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Fokin, V. V.; Matyjaszewski, K. CuAAC: The Quintessential Click Reaction. In Organic Chemistry – Breakthroughs and Perspectives; John Wiley & Sons, Ltd, 2012; pp. 247–277. [Google Scholar] [CrossRef]
- Berg, R.; Straub, B. F. Advancements in the Mechanistic Understanding of the Copper-Catalyzed Azide–Alkyne Cycloaddition. Beilstein J. Org. Chem. 2013, 9, 2715–2750. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M. C.; Pérez, P. J. Copper-Catalysed Azide–Alkyne Cycloadditions (CuAAC): An Update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Agard, N. J.; Prescher, J. A.; Bertozzi, C. R. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Alabugin, I. V. 4.1 Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC): Background, Substrate Preparation, and Reactivity, 1st edition.; Thieme Verlag, 2022. [CrossRef]
- Upadhyay, R.; Rastogi, S.; Mishra, A. K.; Yadav, S.; Yadav, A. K.; Maurya, S. K. Progress in Strain Promoted Azide-Alkyne Cycloaddition (SPAAC) Reaction and Their Applications. Asian Journal of Organic Chemistry 2025, e00505. [Google Scholar] [CrossRef]
- Yoshikawa, R.; Hamada, S.; Matsuo, J. Strain-Promoted Azide–Alkyne Cycloaddition Enhanced by Secondary Interactions. Org. Biomol. Chem. 2025, 23, 1837–1840. [Google Scholar] [CrossRef]
- Feast, G. C.; Hutt, O. E.; Mulet, X.; Conn, C. E.; Drummond, C. J.; Savage, G. P. The High-Throughput Synthesis and Phase Characterisation of Amphiphiles: A Sweet Case Study. Chemistry – A European Journal 2014, 20, 2783–2792. [Google Scholar] [CrossRef]
- Hutt, O. E.; Mulet, X.; Savage, G. P. Click-Chemistry as a Mix-and-Match Kit for Amphiphile Synthesis. ACS Comb. Sci. 2012, 14, 565–569. [Google Scholar] [CrossRef]
- Wang, X.; Huang, B.; Liu, X.; Zhan, P. Discovery of Bioactive Molecules from CuAAC Click-Chemistry-Based Combinatorial Libraries. Drug Discovery Today 2016, 21, 118–132. [Google Scholar] [CrossRef]
- Hong, V.; Presolski, S. I.; Ma, C.; Finn, M. G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angewandte Chemie International Edition 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Lallana, E.; Riguera, R.; Fernandez-Megia, E. Reliable and Efficient Procedures for the Conjugation of Biomolecules through Huisgen Azide–Alkyne Cycloadditions. Angewandte Chemie International Edition 2011, 50, 8794–8804. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Chung, H. W.; Kluger, R. Conjugating Hemoglobin and Albumin by Strain-Promoted Azide- Alkyne Cycloaddition. ChemBioChem 2024, 25, e202400206. [Google Scholar] [CrossRef]
- Kechkeche, D.; El Mousli, S.; Poujouly, C.; Secret, E.; Dupuis, V.; Le Potier, I.; Goriot, M.-E.; Siracusa, J.; Banzet, S.; Gamby, J.; Siaugue, J.-M. Strain Promoted Azide Alkyne Cycloaddition, an Efficient Surface Functionalization Strategy for microRNA Magnetic Separation. Next Materials 2025, 6, 100409. [Google Scholar] [CrossRef]
- Li, K.; Fong, D.; Meichsner, E.; Adronov, A. A Survey of Strain-Promoted Azide–Alkyne Cycloaddition in Polymer Chemistry. Chemistry – A European Journal 2021, 27, 5057–5073. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F. 1,3-Dipolar Cycloadditions of Azides and Alkynes: A Universal Ligation Tool in Polymer and Materials Science. Angewandte Chemie International Edition 2007, 46, 1018–1025. [Google Scholar] [CrossRef]
- Jurajevčič, M.; Grošelj, L.; Ciber, L.; Grošelj, U.; van Midden, K. P.; Petek, N.; Štefane, B.; Novinec, M.; Svete, J. Application of (Aza)Quinolizinyl-3-Diazonium Tetrafluoroborates in Photocatalytic Sulfanylation and in Fluorescent Labeling of Proteins. Bioorganic Chemistry 2025, 163, 108686. [Google Scholar] [CrossRef]
- Petek, N.; Erjavec, B.; Slapšak, D.; Gaber, A.; Grošelj, U.; Požgan, F.; Ričko, S.; Štefane, B.; Klemenčič, M.; Svete, J. 2-Acyl-1-Aryl-6,7-Dihydro-1H,5H-Pyrazolo[1,2-a]Pyrazole Derivatives: Versatile Fluorescent Probes with Remarkably Large Stokes Shift. Dyes and Pigments 2022, 201, 110224. [Google Scholar] [CrossRef]
- Vogel, K.; Glettenberg, M.; Schroeder, H.; Niemeyer, C. M. DNA-Modification of Eukaryotic Cells. Small 2013, 9, 255–262. [Google Scholar] [CrossRef]
- van der Peet, P.; Gannon, C. T.; Walker, I.; Dinev, Z.; Angelin, M.; Tam, S.; Ralton, J. E.; McConville, M. J.; Williams, S. J. Use of Click Chemistry to Define the Substrate Specificity of Leishmania β-1,2-Mannosyltransferases. ChemBioChem 2006, 7, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Beagle, L.; Hansen, F.; Monbaliu, J.-C.; DesRosiers, M.; Phillips, A.; Stevens, C.; Katritzky, A. Efficient Synthesis of 2,5-Diketopiperazines by Staudinger-Mediated Cyclization. Synlett 2012, 23, 2337–2340. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, X.; Ong, S. Y.; Yao, S. Q. Live-Cell Imaging of Survivin mRNA by Using a Dual-Color Surface-Cross-Linked Nanoquencher. Anal. Chem. 2021, 93, 12081–12089. [Google Scholar] [CrossRef]
- DeForest, C. A.; Tirrell, D. A. A Photoreversible Protein-Patterning Approach for Guiding Stem Cell Fate in Three-Dimensional Gels. Nature Mater 2015, 14, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.-F.; Qin, H.-J.; Yu, Y.; Yang, M.-B.; Chang, H.; Guo, N.; He, Y.; Yang, Y.; Yu, P. Multivalent Zanamivir-Bovine Serum Albumin Conjugate as a Potent Influenza Neuraminidase Inhibitor. Journal of Carbohydrate Chemistry 2017, 36, 235–246. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L. S.; Mirfeizi, L.; Schoonen, A. K.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Strain-Promoted Copper-Free “Click” Chemistry for 18F Radiolabeling of Bombesin. Angewandte Chemie International Edition 2011, 50, 11117–11120. [Google Scholar] [CrossRef]
- Ruivo, E. F. P.; Gonçalves, L. M.; Carvalho, L. A. R.; Guedes, R. C.; Hofbauer, S.; Brito, J. A.; Archer, M.; Moreira, R.; Lucas, S. D. Clickable 4-Oxo-β-Lactam-Based Selective Probing for Human Neutrophil Elastase Related Proteomes. ChemMedChem 2016, 11, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sanfrutos, J.; Lopez-Jaramillo, F. J.; Hernandez-Mateo, F.; Santoyo-Gonzalez, F. Vinyl Sulfone Bifunctional Tag Reagents for Single-Point Modification of Proteins. J. Org. Chem. 2010, 75, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R. K.; Joralemon, M. J.; Wooley, K. L.; Hawker, C. J. Functionalization of Micelles and Shell Cross-Linked Nanoparticles Using Click Chemistry. Chem. Mater. 2005, 17, 5976–5988. [Google Scholar] [CrossRef]
- Brun, M. A.; Tan, K.-T.; Nakata, E.; Hinner, M. J.; Johnsson, K. Semisynthetic Fluorescent Sensor Proteins Based on Self-Labeling Protein Tags. J. Am. Chem. Soc. 2009, 131, 5873–5884. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, Y. L.; Zhang, X.; Bhabha, G.; Ekiert, D. C.; Genereux, J. C.; Cho, Y.; Kipnis, Y.; Bjelic, S.; Baker, D.; Kelly, J. W. Small Molecule Probes to Quantify the Functional Fraction of a Specific Protein in a Cell with Minimal Folding Equilibrium Shifts. Proceedings of the National Academy of Sciences 2014, 111, 4449–4454. [Google Scholar] [CrossRef]
- Maisonial, A.; Serafin, P.; Traïkia, M.; Debiton, E.; Théry, V.; Aitken, D. J.; Lemoine, P.; Viossat, B.; Gautier, A. Click Chelators for Platinum-Based Anticancer Drugs. Eur J Inorg Chem 2008, 2008, 298–305. [Google Scholar] [CrossRef]
- Öberg, K.; Ropponen, J.; Kelly, J.; Löwenhielm, P.; Berglin, M.; Malkoch, M. Templating Gold Surfaces with Function: A Self-Assembled Dendritic Monolayer Methodology Based on Monodisperse Polyester Scaffolds. Langmuir 2013, 29, 456–465. [Google Scholar] [CrossRef]
- Lomant, A. J.; Fairbanks, G. Chemical Probes of Extended Biological Structures: Synthesis and Properties of the Cleavable Protein Cross-Linking Reagent [35S]Dithiobis(Succinimidyl Propionate). Journal of Molecular Biology 1976, 104, 243–261. [Google Scholar] [CrossRef]
- Katritzky, A. R.; Zhang, Y.; Singh, S. K. Efficient Conversion of Carboxylic Acids into N -Acylbenzotriazoles <b/>. Synthesis 2003, No. 18, 2795–2798. [Google Scholar] [CrossRef]
- Lynch, S. M.; Neidhart, W.; Plancher, J.-M.; Schulz-Gasch, T. Substituted Triazole Boronic Acid Compounds. WO2014086664A1 2003. [Google Scholar]
- Bosch-Sanz, O.; Rabadà, Y.; Biarnés, X.; Pedreño, J.; Caveda, L.; Balcells, M.; Martorell, J.; Sánchez-García, D. 1,2,3-Triazole Derivatives as Novel Antifibrinolytic Drugs. International Journal of Molecular Sciences 2022, 23, 14942. [Google Scholar] [CrossRef] [PubMed]
- Klaić, L.; Morimoto, R. I.; Silverman, R. B. Celastrol Analogues as Inducers of the Heat Shock Response. Design and Synthesis of Affinity Probes for the Identification of Protein Targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef]
- Pyta, K.; Klich, K.; Domagalska, J.; Przybylski, P. Structure and Evaluation of Antibacterial and Antitubercular Properties of New Basic and Heterocyclic 3-Formylrifamycin SV Derivatives Obtained via ‘Click Chemistry’ Approach. European Journal of Medicinal Chemistry 2014, 84, 651–676. [Google Scholar] [CrossRef]
- Duan, L.; Zangiabadi, M.; Zhao, Y. Synthetic Lectins for Selective Binding of Glycoproteins in Water. Chem. Commun. 2020, 56, 10199–10202. [Google Scholar] [CrossRef]
- Oehrl, A.; Schötz, S.; Haag, R. Systematic Screening of Different Polyglycerin-Based Dienophile Macromonomers for Efficient Nanogel Formation through IEDDA Inverse Nanoprecipitation. Macromol. Rapid Commun. 2020, 41, 1900510. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Mashayekhi, G. A Simple and Efficient Approach to the Synthesis of Endo and Exo Bicyclo[6.1.0]Nona-3,5-Diene-9-Carboxaldehyde. Molecules 2007, 12, 2427–2433. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J. G. K.; Chintala, S. R.; Fox, J. M. Stereoselective Synthesis of Bicyclo[6.1.0]Nonene Precursors of the Bioorthogonal Reagents s-TCO and BCN. J. Org. Chem. 2018, 83, 7500–7503. [Google Scholar] [CrossRef]
- Dehmlow, E. V.; Plückebaum, O. Preparation of Diastereomerically Pure 9-Carboxybicyclo[6.1.0]Nonane Derivatives. Journal für Praktische Chemie/Chemiker-Zeitung 1996, 338, 303–306. [Google Scholar] [CrossRef]
- Rady, T.; Mosser, M.; Nothisen, M.; Erb, S.; Dovgan, I.; Cianférani, S.; Wagner, A.; Chaubet, G. Bicyclo[6.1.0]Nonyne Carboxylic Acid for the Production of Stable Molecular Probes. RSC Adv. 2021, 11, 36777–36780. [Google Scholar] [CrossRef] [PubMed]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L. J. A.; Rutjes, F. P. J. T.; van Hest, J. C. M.; Lefeber, D. J.; Friedl, P.; van Delft, F. L. Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three-Dimensional Imaging of Living Cells. Angewandte Chemie International Edition 2010, 49, 9422–9425. [Google Scholar] [CrossRef]
- Montiel, L.; Spada, F.; Crisp, A.; Serdjukow, S.; Carell, T.; Frischmuth, T. Divergent Synthesis of Ultrabright and Dendritic Xanthenes for Enhanced Click-Chemistry-Based Bioimaging. Chemistry – A European Journal 2023, 29, e202202633. [Google Scholar] [CrossRef]
- Nishat, N.; Asma; Dhyani, S. Synthesis, Spectral and Antimicrobial Studies of Transition Metal Complexes with Novel Macrocyclic Ligand Containing C=N and CO–NH Group. Journal of Coordination Chemistry 2009, 62, 3003–3011. [Google Scholar] [CrossRef]
- Gluziński, P.; Kasprzyk, S.; Krajewski, J. W.; Sałański, P.; Stankiewicz, T.; Jurczak, J. Synthesis, Crystal and Molecular Structures of 1,8-Diaza-11,14-Dioxacyclohexadeca-2,7-Dione and 1,10-Diaza-4,7-Dioxacyclohexadeca-2,9-Dione. Journal of Crystallographic and Spectroscopic Research 1991, 21, 357–363. [Google Scholar] [CrossRef]
- Jurczak, J.; Kasprzyk, S.; Sałański, P.; Stankiewicz, T. A General Method for the Synthesis of Diazacoronands. J. Chem. Soc., Chem. Commun. 1991, 14, 956–957. [Google Scholar] [CrossRef]
- For details dee the Supporting Information.
Figure 1.
The planned method for studying covalent binding of native proteins.
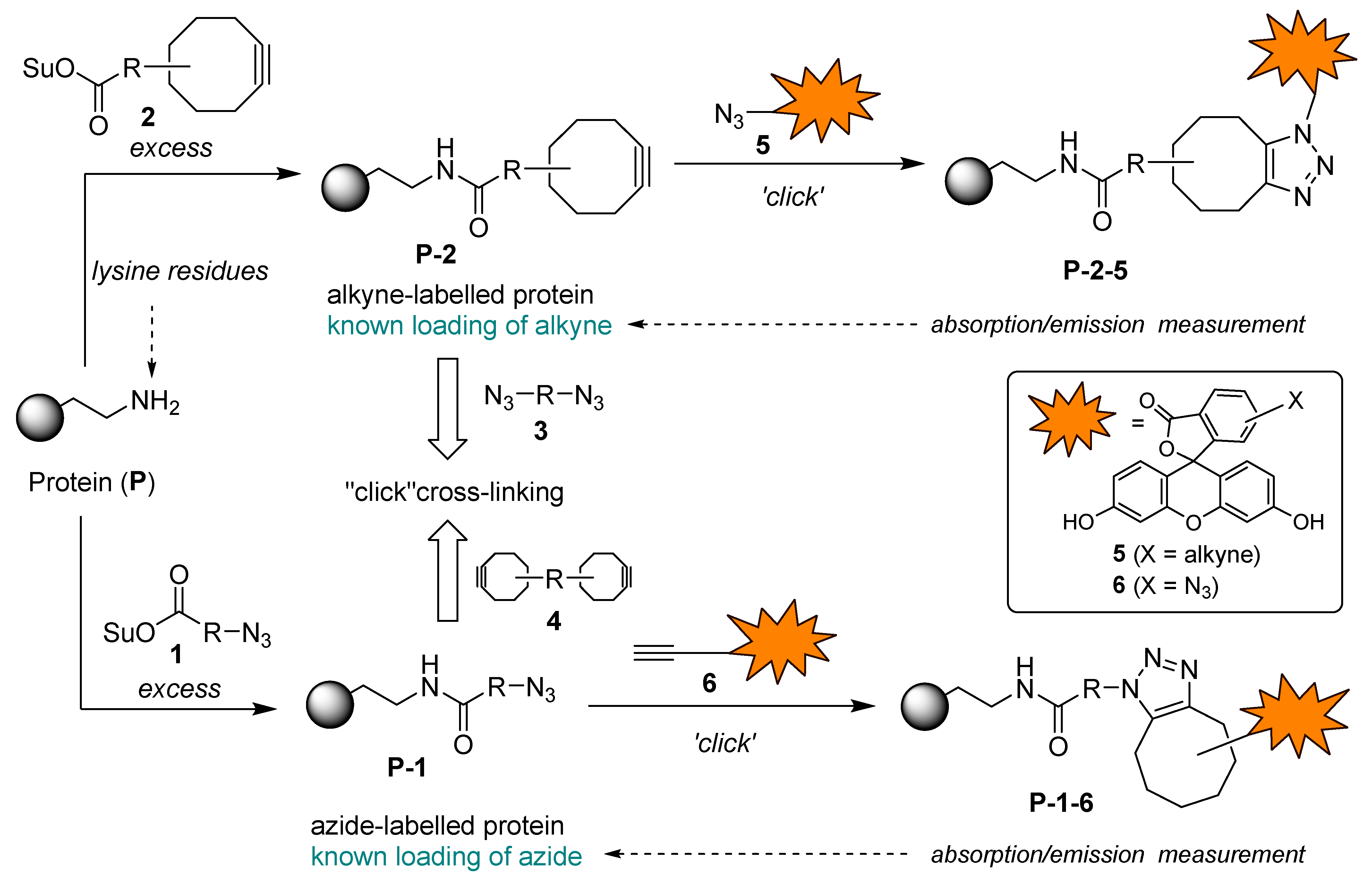
Figure 2.
Azide- and alkyne-functionalized bioconjugation reagents 1a–e and 2a–c, homobifunctional linkers 3 and 4, and fluorescein derivatives 5 and 6a–c selected for the use in this study.
Figure 2.
Azide- and alkyne-functionalized bioconjugation reagents 1a–e and 2a–c, homobifunctional linkers 3 and 4, and fluorescein derivatives 5 and 6a–c selected for the use in this study.
Scheme 1.
Synthesis of azido acids 8a–d and azide-functionalized acylation reagents 1a–c.
Scheme 2.
Synthesis of cyclooctyne-functionalized acylation reagent 2b and synthesis of bicyclo[6.1.0]non-4-yne derivatives 20, 21, and 23–26.
Scheme 2.
Synthesis of cyclooctyne-functionalized acylation reagent 2b and synthesis of bicyclo[6.1.0]non-4-yne derivatives 20, 21, and 23–26.
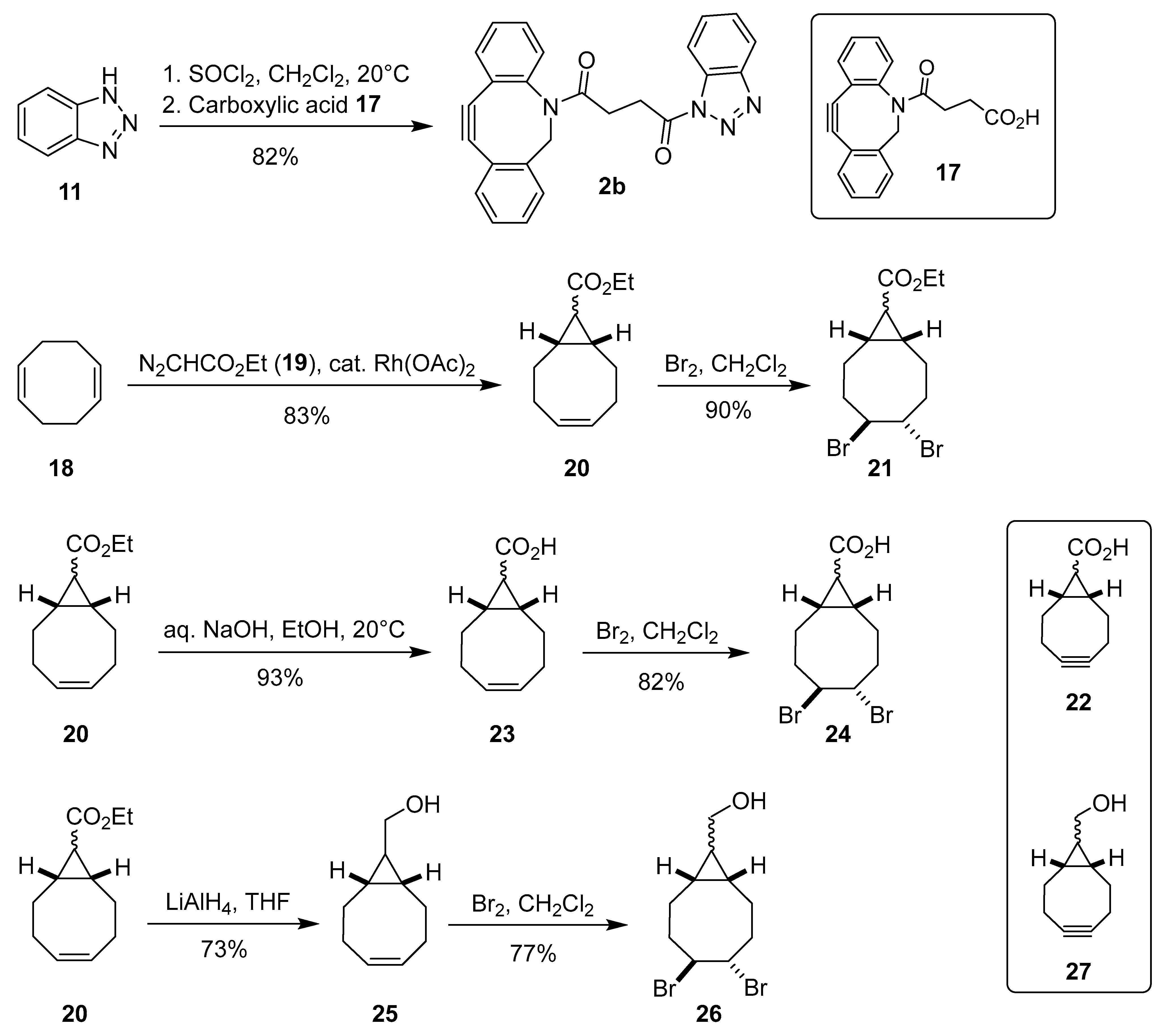
Scheme 3.
Synthesis of azide- and alkyne-functionalized fluorescein derivatives 5 and 6a–c.
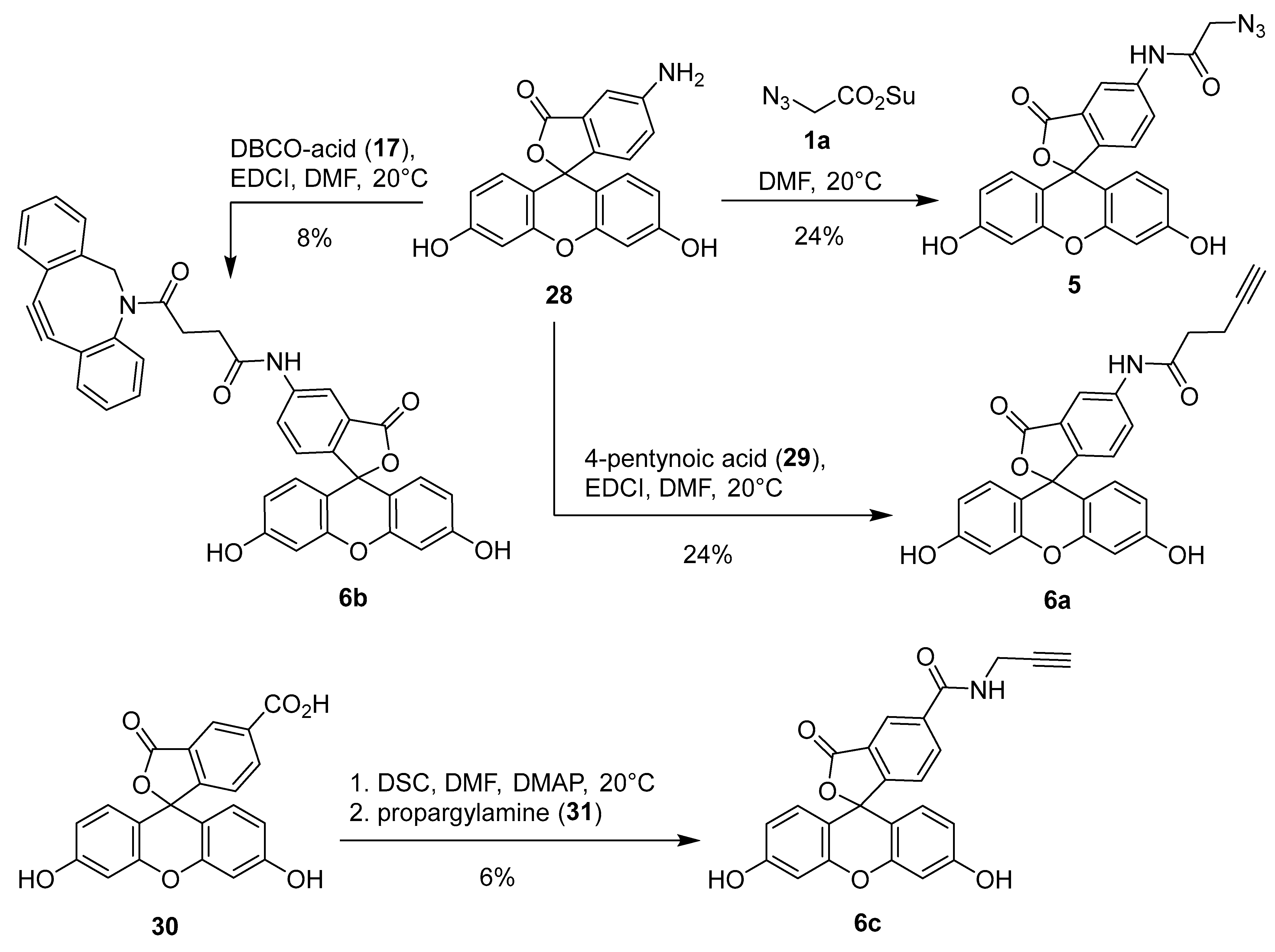
Scheme 4.
Synthesis of homobifunctional linkers 3, 4, 36, and a macrocycle 34.
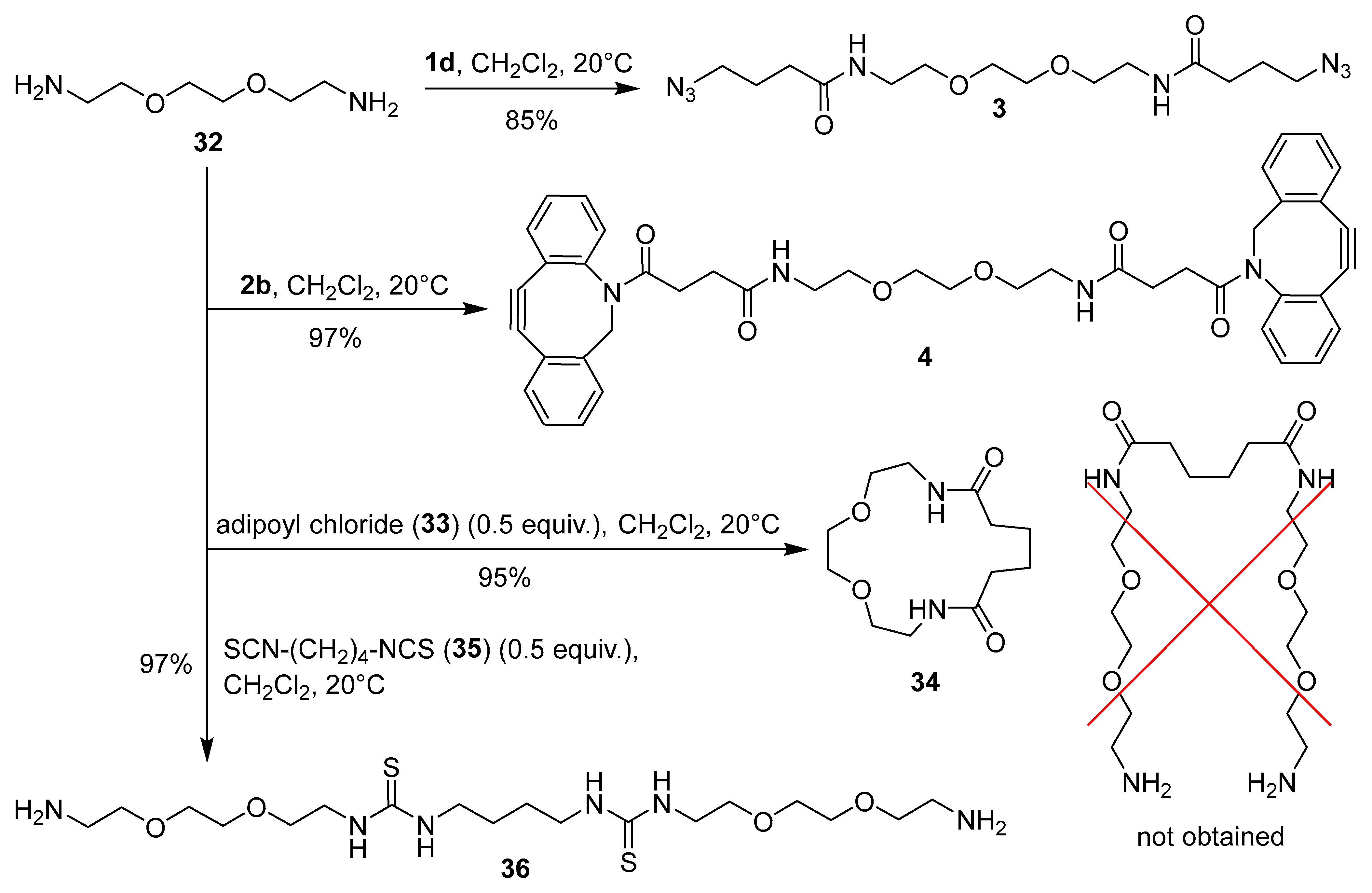
Scheme 5.
Functionalizing of BSA with azide and cyclooctyne functional groups and determination of molar loading of functional grops (FG/BSA) by Methods A and B.
Scheme 5.
Functionalizing of BSA with azide and cyclooctyne functional groups and determination of molar loading of functional grops (FG/BSA) by Methods A and B.
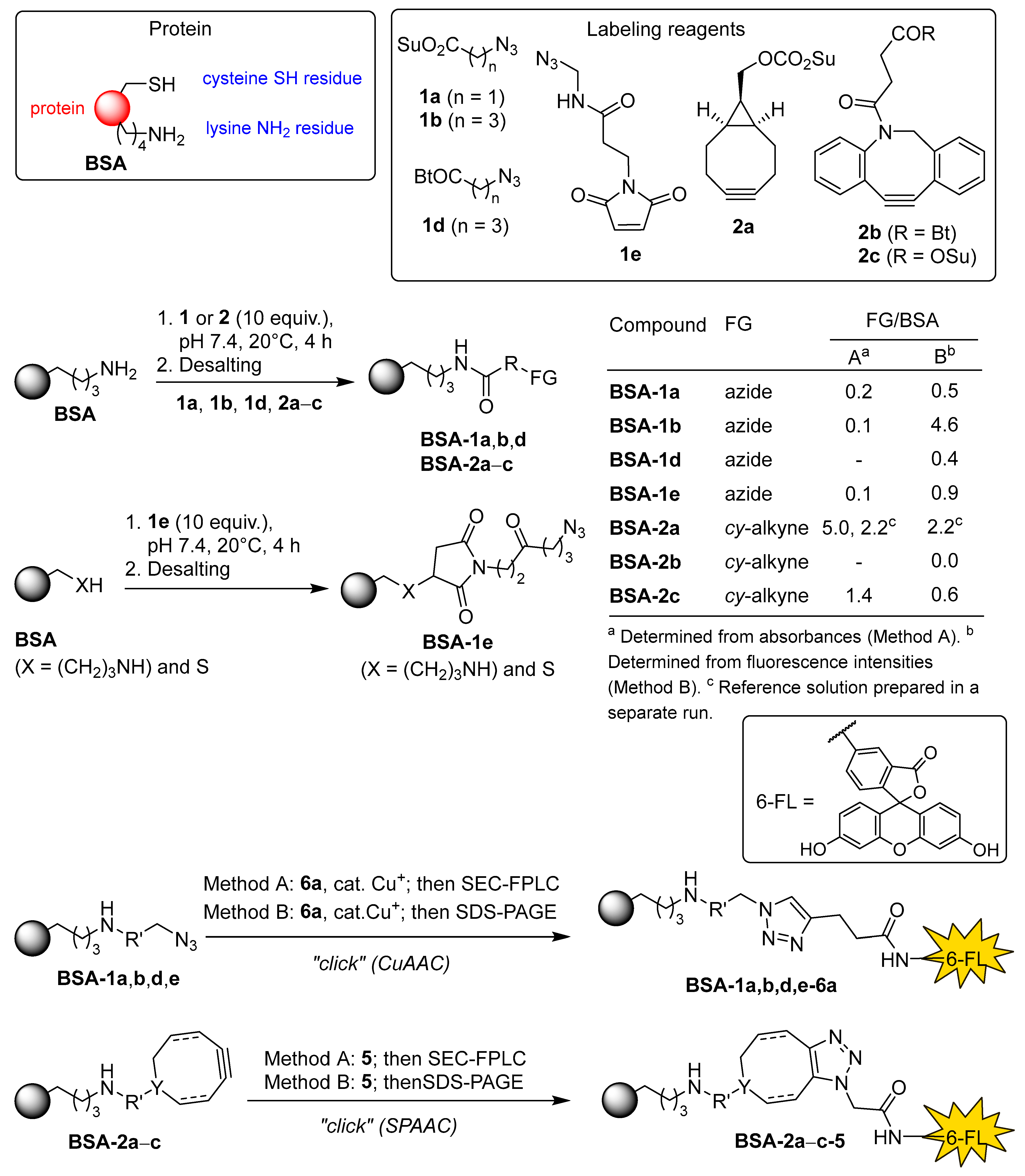
Figure 3.
A: SDS-PAGE of (a) BSA control, (b) reference standard BSA-2a-6a (FG/BSA = 2.2), (c) BSA-1b-5, (d) BSA-1e-5, (e) BSA-1a-5, (f) BSA-1d-5, (g) BSA-2c-6a, and (h) BSA-2b-6a. B, C: SDS-PAGE (12% cross-linked gel) of (a) BSA control, (b) BSA-1b-5, (c) BSA-1e-5, and (d) BSA-1a-5. D, E: Zoomed SDS-PAGE (7% cross-linked gel) of (a) unlabeled BSA control, (b) BSA-1b-5, (c) BSA-1e-5, and (d) BSA-1a-5. B, D: Detection using setting for ProQ Emerald 300 detection. C, E: Detection after staining with Coomassie Brilliant Blue using the corresponding detection settings. Red dotted line marks the lower edge of Mw of BSA control.
Figure 3.
A: SDS-PAGE of (a) BSA control, (b) reference standard BSA-2a-6a (FG/BSA = 2.2), (c) BSA-1b-5, (d) BSA-1e-5, (e) BSA-1a-5, (f) BSA-1d-5, (g) BSA-2c-6a, and (h) BSA-2b-6a. B, C: SDS-PAGE (12% cross-linked gel) of (a) BSA control, (b) BSA-1b-5, (c) BSA-1e-5, and (d) BSA-1a-5. D, E: Zoomed SDS-PAGE (7% cross-linked gel) of (a) unlabeled BSA control, (b) BSA-1b-5, (c) BSA-1e-5, and (d) BSA-1a-5. B, D: Detection using setting for ProQ Emerald 300 detection. C, E: Detection after staining with Coomassie Brilliant Blue using the corresponding detection settings. Red dotted line marks the lower edge of Mw of BSA control.
Scheme 6.
Methods C and D used in attepts to achieve covalent cross-linking of functionalized proteins BSA-1 and BSA-2.
Scheme 6.
Methods C and D used in attepts to achieve covalent cross-linking of functionalized proteins BSA-1 and BSA-2.
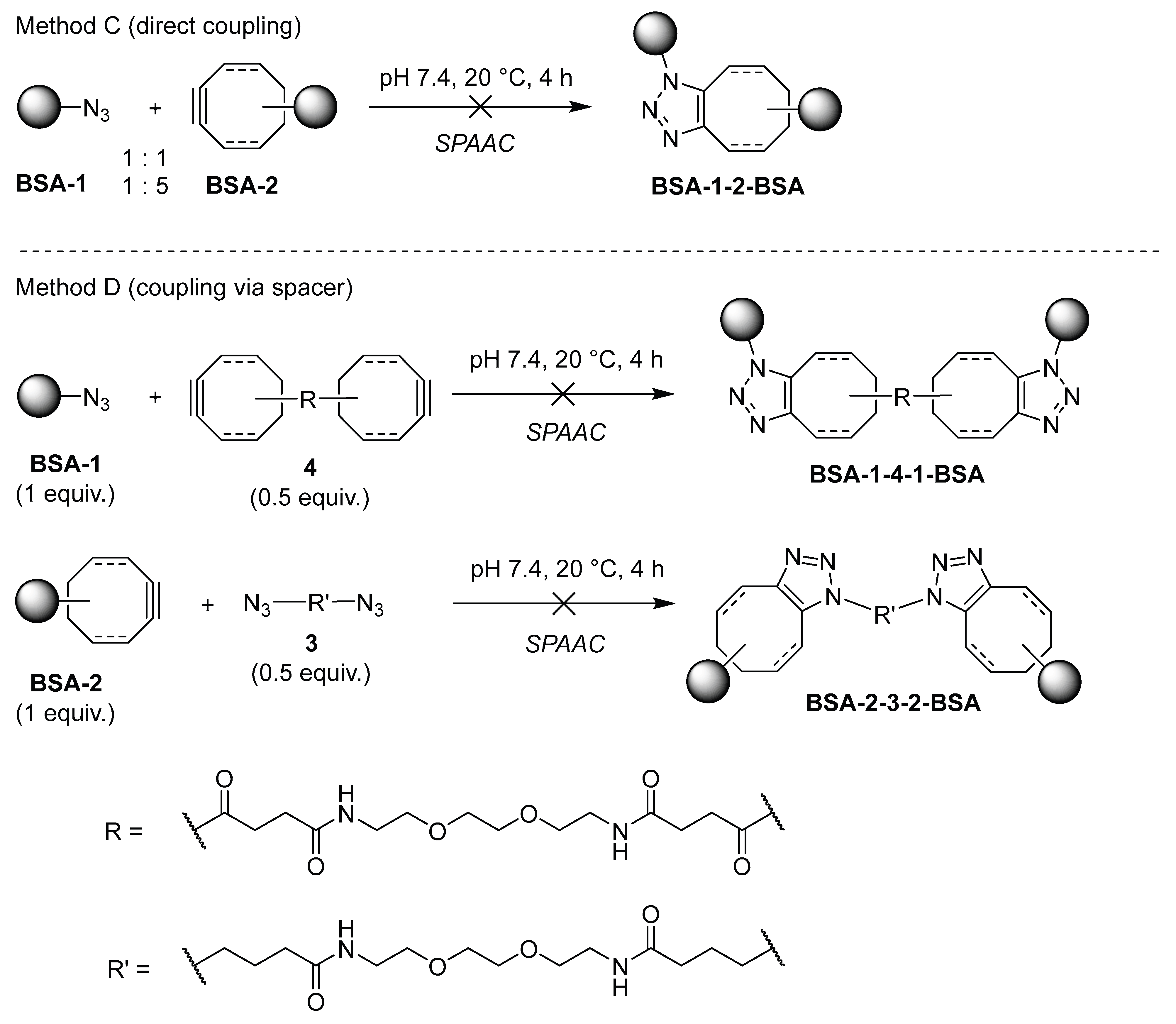
Figure 4.
A: SDS-PAGE of (a) BSA control, (b) BSA-2a + BSA-1b (1:1), (c) BSA-2a + BSA-1e (1:1), (d) BSA-2a + BSA-1a (1:1), (e) BSA-2a + BSA-1b (1:5), (f) BSA-2a + BSA-1e (1:5), and (g) BSA-2a + BSA-1a (1:5). B: SDS-PAGE of (a) BSA control, (b) BSA-2c + BSA-1b (1:1), (c) BSA-2c + BSA-1e (1:1), (d) BSA-2c + BSA-1a (1:1), (e) BSA-2c + BSA-1b (1:5), (f) BSA-2c + BSA-1e (1:5), and (g) BSA-2c + BSA-1a (1:5). C: SDS-PAGE of (a) BSA control, (b) BSA-2c + 3 (2:1), (c) BSA-1b + 4 (2:1). A–C: Detection after staining with Coomassie Brilliant Blue using the corresponding detection settings. Red arrows point at smeared spots at ~120 kDa corresponding to dimers of BSA.
Figure 4.
A: SDS-PAGE of (a) BSA control, (b) BSA-2a + BSA-1b (1:1), (c) BSA-2a + BSA-1e (1:1), (d) BSA-2a + BSA-1a (1:1), (e) BSA-2a + BSA-1b (1:5), (f) BSA-2a + BSA-1e (1:5), and (g) BSA-2a + BSA-1a (1:5). B: SDS-PAGE of (a) BSA control, (b) BSA-2c + BSA-1b (1:1), (c) BSA-2c + BSA-1e (1:1), (d) BSA-2c + BSA-1a (1:1), (e) BSA-2c + BSA-1b (1:5), (f) BSA-2c + BSA-1e (1:5), and (g) BSA-2c + BSA-1a (1:5). C: SDS-PAGE of (a) BSA control, (b) BSA-2c + 3 (2:1), (c) BSA-1b + 4 (2:1). A–C: Detection after staining with Coomassie Brilliant Blue using the corresponding detection settings. Red arrows point at smeared spots at ~120 kDa corresponding to dimers of BSA.
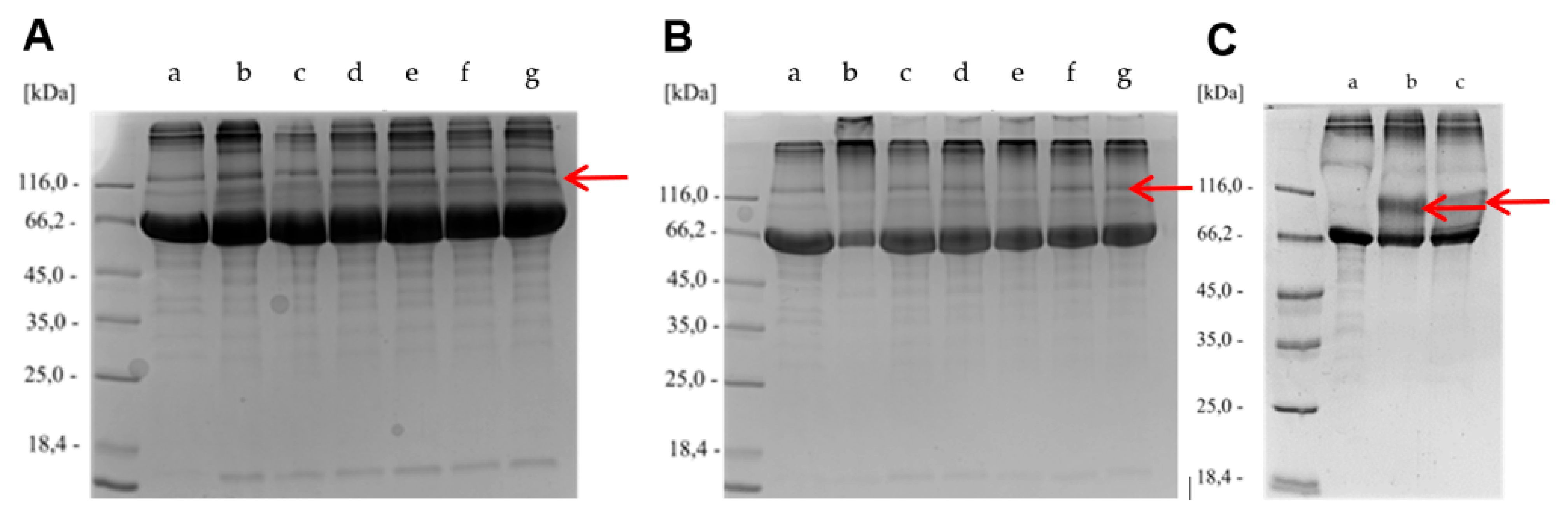
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



