
Submitted:
19 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
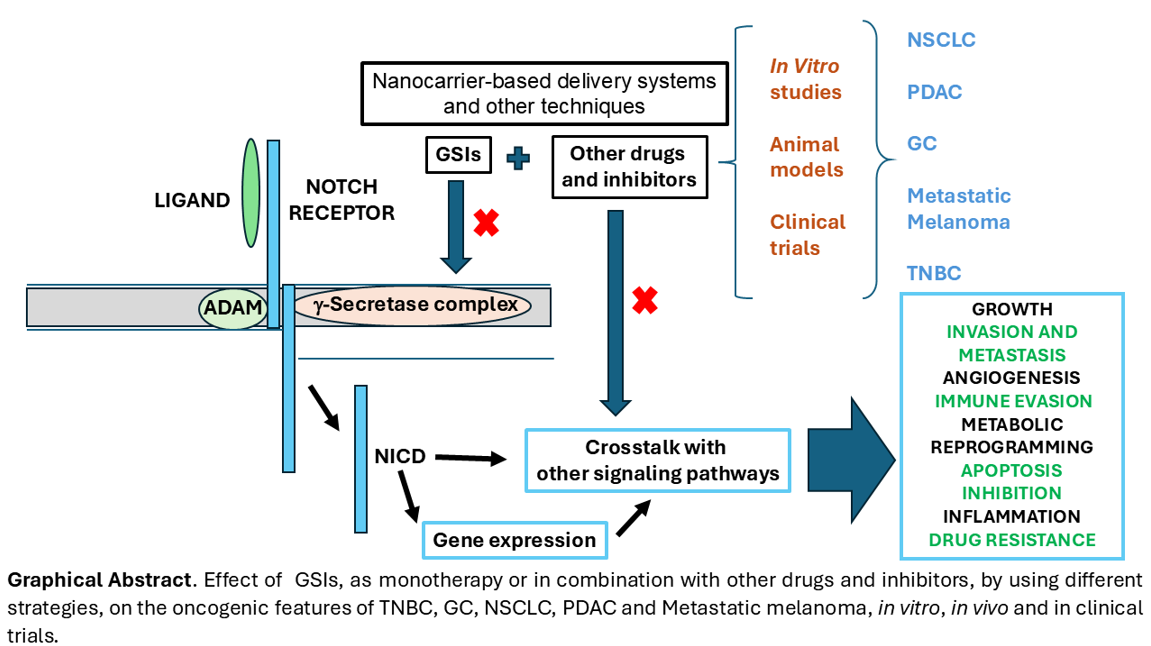
Background. NOTCH receptors play a pivotal role in carcinogenesis. Upon ligand binding, a cascade of proteolytic cleavages mediated by ADAM proteases and the γ-secretase complex activates the receptor, culminating in the release of the NOTCH intracellular domain (NICD). NICD translocates to the nucleus, where it modulates gene expression. The review aims to assess γ-secretase inhibitors (GSIs) as anticancer agents in preclinical and clinical settings, focusing on their ability to block tumor progression, target cancer stem cells, and overcome resistance to standard therapies. Methods. A systematic search was conducted in the ISI Web of Science, PubMed and Scopus databases, following the Preferred Reporting Items for Systematic Reviews (PRISMA) guidelines. The review included preclinical in vitro and in vivo studies and clinical trials on GSIs as monotherapy or in combination with other therapies for TNBC, metastatic melanoma, PDAC, GC, and NSCLC. Excluded were duplicates, non-English articles, articles with unknown authors in Scopus, studies before 2010, studies on non-cancer conditions, studies unrelated to NOTCH signaling, and studies outside the cancer types under study. Risk of bias was assessed through study design factors like randomization, blinding, and data handling. Findings were synthesized qualitatively across the selected cancer types. Results. This review analyzes therapeutic advances using GSIs in cancers driven by oncogenic NOTCH signaling, based on 61 sources covering NSCLC, TNBC, metastatic melanoma, gastric cancer, PDAC, and global epidemiology data. Preclinical studies show GSIs synergize with chemotherapy, radiotherapy, and novel agents, especially in NSCLC, melanoma, and TNBC, blocking EMT, overcoming resistance, and improving prognosis. Common GSIs include DAPT and RO4929097, which enhance drugs like gemcitabine (PDAC), paclitaxel, osimertinib, erlotinib (NSCLC), and 5-FU (GC, TNBC). Promising combinations include GSIs with SAHA, ATRA, and CB103 in TNBC. Clinical trials remain limited: PDAC trials with gemcitabine showed no benefit; melanoma trials had modest results; TNBC trials showed partial responses but overall low efficacy and severe adverse events. RO4929097 is the most clinically tested GSI. Discussion. Clinical trials with γ-secretase inhibitors (GSIs) have underperformed despite promising preclinical results, largely due to tumor heterogeneity, dosing challenges, and non-selective inhibition. Future strategies should prioritize receptor-specific GSIs, patient stratification based on NOTCH pathway activation, and combination therapies administered at optimized doses. Emerging approaches include integrating immunotherapy with advanced techniques such as CRISPR, CAR-T cells, and bispecific antibodies, alongside targeted delivery systems to enhance efficacy and minimize toxicity. Addressing the tumor microenvironment, which contributes to therapy resistance, and inhibiting tumor angiogenesis are also active areas of investigation. Finally, leveraging artificial intelligence and big data for personalized medicine, including male- and female-specific considerations, will be critical for improving patient outcomes.
Keywords:
NOTCH
; GSIs
; PDAC
; GC
; NSCLC
; TNBC
; metastatic melanoma
1. Introduction
1.1. Global Cancer Epidemiology Overview
Cancer remains one of the leading causes of death worldwide. According to estimates from the International Agency for Research on Cancer [1], based on the most reliable data available across countries in 2022, the global burden of cancer continues to grow. That year, there were approximately 20 million new cancer cases and 9.7 million cancer-related deaths [2]. Looking ahead, the projected number of new cancer cases worldwide between 2022 and 2040, across males and females, is expected to reach 29.9 million. During the same period, the estimated number of cancer-related deaths is anticipated to rise to 15.3 million.
According to the World Health Organization, and excluding non-melanoma skin cancers, the most frequently diagnosed tumors worldwide are breast cancer, followed by lung, colorectal, prostate, and stomach cancers [Available in [2]]. In terms of mortality, the most lethal cancers, ranked in descending order, are lung, colorectal, liver, stomach, breast and pancreas cancer [Available in [1,2]] (Figure 1).
The molecular and cellular mechanisms driving cancer development across different tissues and organs are highly complex. Intensive global research efforts, encompassing both basic and clinical studies, are focused on elucidating these mechanisms. Such investigations are essential for the development of novel therapeutic strategies that are not only more effective but also tailored to individual patients, with reduced adverse effects.
Among the many intracellular signaling pathways implicated in cancer, the NOTCH receptor signaling pathway stands out due to its pivotal role in the initiation and progression of various neoplasms [3,4,5]. Aberrant NOTCH signaling contributes to several hallmark features of cancer and is particularly associated with the emergence of highly aggressive tumors with poor prognosis [6,7]. Moreover, the NOTCH pathway forms an intricate network of interactions with other key signaling pathways involved in oncogenesis, further driving tumor development and progression in multiple malignancies [3,8].
1.2. Structure of NOTCH Receptors and Their Ligands
NOTCH receptors and their ligands constitute a highly conserved protein family across evolution [9,10,11]. From a biological standpoint, the NOTCH receptor signaling pathway acts as a key regulator in cell fate decisions, controlling cellular proliferation and differentiation, senescence, or apoptosis of progenitor and stem cells, among other biological processes [7,12]. Its function is critical in numerous cellular contexts such as neurogenesis, angiogenesis, hematopoiesis, epithelial morphogenesis, and the specification of cell lineages in organs like the digestive system, lungs, skin, immune system, and in cancer [3,7]. The NOTCH signaling pathway also plays a central role in maintaining the identity and plasticity of adult stem cells and in establishing spatial and temporal patterns of cell differentiation [7,12]. Furthermore, its involvement in phenomena such as epithelial-mesenchymal transition (EMT) and its interaction with other intracellular signaling pathways reinforces its importance in many complex biological processes in both healthy and pathological states, such as cancer [3,8]. In mammals, the main members of this protein family, both receptors and ligands, are differentially expressed depending on the cell type, physiological context, or tumor microenvironment, participating in essential cell-to-cell interactions that modulate cell fate [13,14,15].
In mammals, unlike in Drosophila melanogaster, where the first Notch gene was discovered [16,17,18], there are four NOTCH receptors: NOTCH1, NOTCH2, NOTCH3, and NOTCH4 (Figure 2) [7,16,19,20,21].
NOTCH1 and NOTCH2 are broadly expressed across adult tissues, while NOTCH3 and NOTCH4 expression is found in vascular smooth muscle, endothelial cells and different types of immune cells [7,13,14,15,16,21,22,23]. NOTCH receptors are heterodimeric transmembrane proteins composed of three main regions: an extracellular domain (NECD), a transmembrane domain (TMD), and an intracellular domain (NICD) [8,24,25,26]. The NECD contains multiple EGF-like repeats (epidermal growth factor-like) , the number of which varies depending on the receptor subtype and some of them specifically interact with the canonical ligands. Adjacent to the transmembrane domain, the NECD also includes a segment known as the negative regulatory region (NRR), composed of Lin12/NOTCH repeats. The active NICD region includes several functional domains: a RAM domain for protein binding, seven ankyrin repeat domains, nuclear localization signals, a transcriptional activation domain (TAD), and a PEST domain involved in protein degradation. There are five canonical activating ligands of NOTCH receptors: Delta-Like 1 (DLL1), Delta-Like 3 (DLL3), and Delta-Like 4 (DLL4), three homologs of the Drosophila Delta ligand, and Jagged1 (JAG1) and Jagged2 (JAG2), which are homologs of the Drosophila Serrate ligand [21,27,28,29,30,31] (Supplementary Figure S1). These ligands are transmembrane proteins, and their interaction with NOTCH receptors requires direct intercellular contact. Specifically, the binding occurs between specific EGF-like repeats on the NOTCH receptors and the DSL (Delta/Serrate/LAG2) domain present in the canonical ligands. In addition to canonical activating ligands, several non-canonical ligands with inhibitory functions have been identified, including DLK1 and DLK2 proteins (Supplementary Figure S2) [32,33,34,35]. DLK1 and DLK2 proteins possess six EGF-like repeat sequences in their extracellular region, a transmembrane region, and a short intracellular region [32,34]. The extracellular region of DLK1 is recognized by the TACE protease, which processes DLK1 and releases a soluble form containing the extracellular region [36,37,38,39]. DLK2 might be processed similarly to DLK1, although there is no evidence to confirm or refute this possibility. Despite lacking the DSL domain that canonical ligands possess at the N-terminal end, DLK1 and DLK2 can interact with NOTCH receptors through their N-terminal DOS domains, and function as non-canonical inhibitory ligands, competing with canonical ligands [33,35,40,41]. Regarding their tissue distribution, DLK proteins also show differences. DLK1 expression in adults is limited to the adrenal glands, while DLK2 has a broader expression in adults, being mainly expressed in the skin, prostate, esophagus, brain, and salivary glands, although its expression is not remarkably high [42,43].
1.3. Mechanism of NOTCH Receptor Activation and Downstream Signaling
To understand the functional dynamics of NOTCH receptors, it is essential to recognize that these receptors autonomously initiate a signaling cascade that culminates in the regulation of gene expression within the nucleus [6,12,44]. Activation of NOTCH receptors involves three sequential proteolytic cleavages at conserved sites known as S1, S2, and S3–S4 [8,11,15,45,46]. The first cleavage occurs during receptor maturation in the Golgi apparatus, where the enzyme Furin processes the receptor at the S1 site. This event generates a heterodimer composed of an extracellular domain, a transmembrane, and an intracellular domain, held together by non-covalent interactions. Once the receptor is transported to the plasma membrane, it remains inactive due to the presence of the Negative Regulatory Region (NRR) [47,48].
Activation is triggered by the interaction of a canonical ligand, expressed on the membrane of a neighboring cell, with the NOTCH receptor. This ligand–receptor binding exposes the S2 cleavage site, allowing ADAM10 or ADAM17 secretases to cleave the receptor. As a result, most of the extracellular domain is endocytosed by the ligand-expressing cell [47,48] (Figure 3). This S2 cleavage is the only ligand-dependent step and represents a critical event in the canonical NOTCH signaling pathway. Following S2 cleavage, the remaining membrane-bound fragment, known as NEXT (NOTCH extracellular truncation), undergoes further processing at the S3–S4 sites by the γ-secretase complex. This final cleavage releases the active NOTCH intracellular domain (NICD), which translocates to the nucleus [15,49,50,51,52].
Within the nucleus, NICD binds to the transcription factor CSL/RBPJκ (C-promoter-binding factor, CBF1 (in mammals; also known as Recombination Signal Binding Protein Jκ, RBPJκ)/Suppressor of hairless (in Drosophila melanogaster) /Lag1 (in Caenorhabditis elegans)) along with Mastermind-like (MAML) proteins and other cofactors. This complex displaces transcriptional repressors and forms the NOTCH Transcriptional Complex (NTC), which activates the expression of target genes, including members of the Hes and Hey families (Hairy and Enhancer-of-Split) [8,44,45,53,54]. These transcriptional responses define the canonical NOTCH signaling pathway.
1.4. Role of NOTCH Receptors and NOTCH Ligands in Carcinogenesis
Abnormalities in the NOTCH receptor signaling pathway are well-established contributors to carcinogenesis [3,4,5,6,7]. Depending on the context, aberrant NOTCH activation can promote tumor initiation and progression, functioning as an oncogene, or conversely, its inactivation can lead to tumor development, acting as a tumor suppressor. The dual role of NOTCH signaling is highly dependent on the specific receptor involved and the tumor type. For instance, NOTCH1 may act as an oncogene in certain hematological malignancies, while serving a tumor-suppressive role in skin cancers [3,4,6,55]. NOTCH3 also have a critical role in the developmental and functional pathways of immune cells and in sustaining oncogenic programs [56,57]. Despite extensive research, the molecular mechanisms underlying these opposing effects remain poorly understood. Elucidating these mechanisms is critical for the development of targeted anticancer therapies. Such therapies would aim to selectively inhibit the NOTCH receptor exhibiting oncogenic activity in a specific tumor type or counteract its role in mediating resistance to conventional chemotherapeutic agents.
NOTCH receptors can contribute to neoplastic transformation through three distinct mutational patterns [3,6,58]. The first mechanism involves chromosomal translocations that eliminate the Negative Regulatory Region (NRR), leading to constitutive activation of the receptor. This mechanism has been identified in triple-negative breast cancer. The second pattern consists of mutations in the PEST domain, which impair NICD degradation and prolong its activity. The third pattern includes mutations in the N-terminal region of the receptor, commonly observed in squamous cell carcinomas such as esophageal and lung cancers.
Beyond point mutations, two additional mechanisms can contribute to NOTCH receptor dysregulation: gene amplification and chromosomal rearrangements [6]. Gain-of-function alterations in NOTCH signaling have been associated with several cancers, including breast cancer and non-small cell lung cancer (NSCLC). Conversely, loss-of-function mutations, where NOTCH acts as a tumor suppressor, are primarily linked to squamous cell carcinomas of the esophagus, and lung [55] (Figure 4). In triple-negative breast cancer, activation of NOTCH1 has been associated with increased tumor aggressiveness, in contrast to NOTCH2–4, which may play more protective roles [59]. In highly aggressive cancers, the Int3 oncogene, a truncated form of the NOTCH4 receptor, has also been implicated in tumorigenesis [3,60].
Beyond its role as an oncogene or tumor suppressor, NOTCH signaling plays a critical role in different processes that lead to the generation and development of the tumors (Supplementary Figure S3). NOTCH signaling plays a role in the tumor microenvironment, particularly in the maintenance of cancer stem cells [6]. For example, in gastric cancer, overactivation of NOTCH signaling has been observed in tumor stem cell populations [61]. Increasing evidence has shown that the activation of the γ-secretase/NOTCH pathway is a key driver of drug resistance development. In breast cancer, NOTCH signaling induces cell cycle arrest in stem cells, promoting the emergence of chemoresistance [6]. NOTCH activity has also been linked to protection against drugs targeting the estrogen receptor or the MAPK pathway in certain melanomas and breast cancers [3].
NOTCH signaling is also involved in tumor infiltration and metastasis, particularly through its role in the epithelial–mesenchymal transition (EMT). During EMT, NOTCH activation leads to the downregulation of cell adhesion proteins such as E-cadherin, facilitating tumor cell migration and invasion [6]. Additionally, NOTCH receptors contribute to immune evasion, metabolic reprogramming, apoptosis inhibition, and tumor-associated inflammation [62].
In the context of angiogenesis, NOTCH ligands exhibit opposing roles. The canonical ligand DLL4 suppresses angiogenic sprouting, whereas JAG1 promotes angiogenesis, tumor growth, and the maintenance of cancer stem cells. In pancreatic ductal adenocarcinoma (PDAC), NOTCH overactivation has been linked to enhanced angiogenic sprouting [63]. Elevated JAG1 expression has also been reported in breast, lung, and pancreatic cancers [64].
Among non-canonical NOTCH ligands, DLK1 (Delta-like homolog 1) is minimally expressed in adult tissues but exhibits elevated levels in various neoplasms, where it appears to contribute to the maintenance of cancer stem cells [65]. In metastatic melanoma, NOTCH signaling plays an oncogenic role, and both DLK1 and DLK2 have been shown to promote tumor formation in nude mice in a dose-dependent manner by inhibiting NOTCH signaling [66]. In triple-negative breast cancer, high DLK1 expression has been associated with reduced tumor progression, whereas low DLK1 levels correlate with increased tumor size in vivo. Conversely, low DLK2 expression enhances tumor aggressiveness, while high DLK2 levels have been shown to prevent tumor formation in nude mice [67,68]. These findings suggest that DLK1 and DLK2 exert context-dependent regulatory effects on NOTCH signaling and tumor behavior, highlighting their potential as therapeutic targets in specific cancer subtypes.
1.5. Strategies for the Inhibition of NOTCH Receptor Signaling
Extensive evidence supports the hypothesis that NOTCH signaling is one of the most promising therapeutic targets in cancer treatment. In recent years, various pharmacological strategies have been developed to inhibit this pathway [69,70,71]. Key approaches to disrupting NOTCH receptor signaling include monoclonal antibodies (mAb) that block the receptors or their canonical and non-canonical ligands; peptides that interfere with the transcriptional activation complex; inhibitors of the α-secretases ADAM10 and ADAM17; and γ-secretase inhibitors (GSIs), which prevent the final proteolytic processing of NOTCH receptors. GSIs specifically target the γ-secretase complex, thereby blocking the release of the active NOTCH intracellular domain (NICD) (Figure 5).
1.6. The γ-Secretase Complex and γ-Secretase Complex Inhibitors (GSIs)
The γ-secretase complex (GSC) is a member of the intramembrane-cleaving proteases (I-CLiPs) family [50]. It plays a central role in the proteolytic processing of several substrates, including NOTCH receptors, the β-amyloid precursor protein, whose cleavage generates the β-amyloid peptide implicated in the formation of senile plaques in Alzheimer’s disease [69], and E-cadherin, a key molecule in cell adhesion [72]. Beyond its proteolytic functions, the GSC also participates in non-proteolytic processes such as calcium homeostasis, autophagy, and apoptosis [73], earning it the nickname “Transmembrane Cellular Proteasome”.
The GSC is composed of several transmembrane subunits (Figure 6): Presenilin (PS), with two isoforms (PS1 and PS2), is responsible for the catalytic activity of the complex [73,74]; Nicastrin (NCT) acts as a scaffolding protein that recognizes and binds substrates targeted for cleavage; Anterior pharynx defective 1 (APH-1), which exists in multiple isoforms, contributes to complex stabilization [75]; and Presenilin enhancer 2 (PEN-2) is involved in the heterodimerization of presenilin and the maturation of nicastrin.
γ-secretase inhibitors (GSIs) were initially developed as therapeutic agents for Alzheimer’s disease [69,76,77]. However, their clinical use was discontinued due to limited efficacy and significant adverse effects, including the emergence of non-melanoma skin tumors. Interest in GSIs was later rekindled in the field of oncology, driven by the role of NOTCH receptors in tumorigenesis. Currently, extensive research is underway to evaluate GSIs as potential anti-cancer agents [78,79,80]. Both preclinical and clinical studies have explored their ability to suppress tumor progression by targeting NOTCH signaling pathways in vitro and in vivo. Moreover, GSIs may hold promise in overcoming chemotherapy resistance by modulating the γ-secretase/NOTCH axis. In addition, GSIs are being investigated as candidates for therapies targeting cancer stem cells (CSCs), which are characterized by slow proliferation and resistance to conventional chemotherapy and radiotherapy, factors that contribute to treatment failure and disease recurrence. Eradicating CSCs is considered a key strategy for achieving long-term cancer remission. Promising studies have shown that GSIs, when combined with other anticancer agents, exert a stronger inhibitory effect on CSCs [81].
It is important to note that GSIs exhibit differential inhibition profiles across various NOTCH substrates. In some cases, GSIs enhance the cleavage of certain NOTCH substrates at concentrations that inhibit NOTCH1 cleavage [82]. This paradoxical effect may result from the direct action of low GSI concentrations on γ-secretase itself.
Nonetheless, numerous side effects have been reported in clinical trials, largely due to GSIs’ broad activity across multiple substrates and biological processes. For this reason, gamma-secretase inhibitors (GSIs) are associated with several side effects that remain a subject of debate among researchers. The most common adverse effects include gastrointestinal toxicity (diarrhea, nausea, vomiting) due to disruption of NOTCH signaling, and skin toxicity linked to impaired protein production for skin health. These symptoms are generally mild to moderate and manageable. Concerns about neurological effects exist, but clinical trials have not reported significant findings to date. Other reported side effects include fatigue, headache, and anemia. Overall, while GSIs show therapeutic potential, their risks must be carefully balanced against benefits, and further research is required to establish their safety and efficacy [78].
This review explores therapeutic strategies involving gamma-secretase inhibitors (GSIs), administered either as monotherapy or in combination with other agents, in highly aggressive malignancies such as non-small cell lung cancer (NSCLC), triple-negative breast cancer (TNBC), metastatic melanoma, gastric cancer, and pancreatic ductal adenocarcinoma (PDAC), all characterized by oncogenic NOTCH signaling.
2. Methods
A systematic search was conducted in the ISI Web of Science (http://www.webofknowledge.com), PubMed (https://pubmed.ncbi.nlm.nih.gov/), and Scopus (https://www.scopus.com/) databases to identify relevant studies published between 1 January 2010 and 15 December 2025 on the use of gamma-secretase inhibitors (GSIs) in triple-negative breast cancer (TNBC), metastatic melanoma, pancreatic ductal adenocarcinoma (PDAC), gastric cancer (GC), and non-small cell lung cancer (NSCLC) (Figure 7).
The search strategy employed the following keywords: “GSI”, “gamma-secretase complex”, “cancer”, “NOTCH”, “pancreatic adenocarcinoma (PDAC)”, “triple-negative breast cancer (TNBC)”, “metastatic melanoma”, “non-small cell lung cancer (NSCLC)”, and “gastric cancer (GC)”.
Articles were screened and filtered according to the inclusion and exclusion criteria outlined by the PRISMA methodology [83]. Eligible studies included preclinical research conducted in vitro or in vivo using animal models, as well as clinical trials in humans evaluating GSIs, either as monotherapy or in combination with other anticancer agents, targeting TNBC, metastatic melanoma, PDAC, GC, and NSCLC. Exclusion criteria comprised articles published before 2010, duplicates, non-English publications, studies with unknown authors in Scopus, and studies investigating GSIs in non-cancer pathologies or cancer types outside the scope of this review.
The initial search employed the keywords “GSIs (gamma-secretase inhibitors)” and “gamma-secretase complex” across the three databases. After applying preliminary exclusion parameters, the keywords “GSIs” and “NOTCH” and “cancer” were used. Once eligible studies from the Web of Science database addressing GSIs in cancer and their relationship to the NOTCH signaling pathway were identified, articles were further filtered using the key words GSIs and each of the five cancer types under study. Subsequently, titles and abstracts were screened to retain studies directly related to GSIs in the selected cancer types. Finally, a thorough reading of the selected studies was performed, and the most relevant information was extracted into a notebook, later incorporated into the Results section of the manuscript.
In total, 61 sources were included (Figure 7). Additional references were consulted for background information, including reviews on NOTCH signaling, GSIs in cancer, and emerging therapeutic approaches, which informed the Introduction and Discussion sections.
3. Results
Next subsections in this section analyze the recent findings that underscore advances in GSI-based therapies for the five aggressive malignancies under study with poor prognosis. In total, 72 sources were included: 16 on NSCLC, 22 on TNBC, 11 on metastatic melanoma, 10 on GC, 11 on PDAC, and 2 web-based sources on global cancer epidemiology. Evidence from both preclinical studies and clinical trials highlights the potential of GSIs, particularly when combined with established anticancer agents, to improve therapeutic efficacy and reduce adverse effects associated with their non-specific mechanisms of action.
3.1. The Combination of GSIs with Other Therapeutic Agents has Demonstrated Efficacy in Reducing Pancreatic Ductal Adenocarcinoma (PDAC) Progression
The development of pancreatic ductal adenocarcinoma (PDAC) is known to involve the progression of a precursor lesion termed pancreatic intraepithelial neoplasia (PanIN). A critical genetic alteration frequently identified in PanIN is the mutation of the KRAS gene, which appears to be indispensable for both the malignant transformation of pancreatic epithelial cells within PanIN and their subsequent progression to PDAC [84].
Regarding NOTCH signaling, current evidence indicates that it may exert dual functions, either tumor-suppressive or oncogenic, depending on the stage of tumor development [85]. Studies using mouse models have shown that the coactivation of KRAS and NOTCH1 markedly increases the number of PanIN lesions [85,86]. These findings support the hypothesis that NOTCH signaling may inhibit PanIN formation during the early phases of tumorigenesis, while facilitating its progression in later stages.
Cook and colleagues evaluated the γ-secretase inhibitor MRK003 in pancreatic ductal adenocarcinoma (PDAC) xenograft models, both as a monotherapy and in combination with gemcitabine. Their results indicated that MRK003 as monotherapy did not elicit significant therapeutic effects; however, its combination with gemcitabine led to a substantial improvement in mouse survival [63]. Treatment with MRK003 was associated with increased necrosis of neoplastic tissue, enhanced apoptosis, and reduced cellular proliferation. One of the principal mechanisms attributed to MRK003 was its capacity to inhibit intratumoral vascular proliferation, observed both as monothearapy treatment and in combination therapy. This vascular suppression induced hypoxic conditions within the tumor microenvironment, thereby potentiating the efficacy of the therapeutic agents. In a separate study, Mizuma and colleagues similarly demonstrated that the combination of MRK003 and gemcitabine effectively impeded tumor progression in PDAC mouse models [87].
GSI IX treatment induced apoptosis and selectively targeted epithelial–mesenchymal transition (EMT) markers. Notably, GSI IX suppressed the growth of pancreatic tumor-initiating CD44⁺/EpCAM⁺ cells both in vitro and in xenograft mouse models, underscoring its potential as a therapeutic strategy against pancreatic cancer by impairing tumor initiation and limiting epithelial plasticity [80]. In other work, Palagani and colleagues explored the therapeutic potential of combining GSI-IX with AG-490, a Janus Kinase 2 (JAK2) inhibitor that blocks activation of STAT3, signaling molecules implicated in PDAC pathogenesis. Using mouse models exhibiting pancreatic intraepithelial neoplasia (PanIN) and acinar-to-ductal metaplasia (ADM), they tested each compound as monotherapy. After six weeks, small tumors and microscopic foci of PDAC were observed. Remarkably, in the group treated with the combination of GSI-IX and AG-490, none of the five mice developed visible tumors, indicating a synergistic therapeutic effect [88].
The stroma component of pancreatic cancer makes up to 90% of the tumor mass and is thought to be one of the main reasons for the tumor’s high chemoresistance. Cancer associated fibroblasts (CAFs) have previously been identified to be the key stromal players. Neumann and coworkers study the effect of DAPT in PDAC cell lines and associated CAFs. Unlike PDAC cells, CAF monocultures hardly responded to any treatment which suggested that stroma (CAFs) itself is more resistant to standard chemo-treatments than the epithelial cancer cells. High levels of IL-6 were also associated with a reduced response to therapy [89].
The γ-secretase inhibitor PF-03084014 has shown promising results in a phase III clinical trial for desmoid tumors (aggressive fibromatosis), rare, non-malignant connective tissue growths that are locally invasive and prone to recurrence [90]. In the context of PDAC, Yabuuchi and colleagues assessed PF-03084014 in various xenograft mouse models, both as monotherapy and in combination with gemcitabine [86]. While PF-03084014 as monotherapy did not significantly inhibit tumor proliferation, its combination with gemcitabine resulted in notable antiproliferative effects and tumor regression. Flow cytometry analyses revealed that gemcitabine as monotherapy failed to eliminate tumor stem cells and even appeared to increase their prevalence. In contrast, PF-03084014, both as monotherapy and in combination, reduced the population of tumor stem cells. Moreover, the combination therapy significantly decreased distant metastasis, suggesting a broader impact on tumor aggressiveness and dissemination [86].
It is worth highlighting the development of the γ-secretase inhibitor (GSI) MRK-0752, which was evaluated by Cook and colleagues in a phase I clinical trial in combination with gemcitabine in patients with stage IV pancreatic ductal adenocarcinoma (PDAC). In this study, 14 out of 44 patients achieved disease stabilization. However, the outcomes were comparable to those observed with gemcitabine monotherapy [91]. Notably, several adverse effects associated with GSI treatment were reported, including gastrointestinal disturbances, thrombocytopenia, and anemia.
Finally, the γ-secretase inhibitor RO4929097 was evaluated in a phase II clinical trial involving patients with previously treated metastatic pancreatic adenocarcinoma; however, the trial was terminated due to the discontinuation of GSI synthesis. [92].
3.2. Treatment Resistance in Non-Small Cell Lung Cancer (NSCLC) Can Be Mitigated Through the Application of γ-Secretase Inhibitors as Monotherapy and Combined with Other Drugs
NOTCH signaling plays a critical role in the pathogenesis of non-small cell lung cancer (NSCLC). NOTCH3 overexpression has been reported in approximately 40–50% of cases (23), while elevated NOTCH1 levels are associated with poor prognosis and tumor initiation, particularly in KRAS-driven models [93]. Interestingly, in the squamous subtype of NSCLC, NOTCH signaling appears to be suppressed, suggesting a potential tumor-suppressive function in this context [94,95].
NOTCH pathway is essential for the development and maintenance of KrasG12V-driven NSCLC. Genetic evidence indicates that γ-secretase and RBPJκ are critical for tumor formation, and pharmacologic inhibition of γ-secretase effectively suppresses cancer growth in mice. HES1 was shown to repress the promoter of DUSP1, a dual phosphatase that targets phospho-ERK. Consequently, DAPT treatment upregulated DUSP1, leading to reduced phospho-ERK levels and revealing a pathway through which NOTCH inhibition suppresses tumor progression [96].
Liu and colleagues demonstrated in vitro that cisplatin treatment of NSCLC cell lines led to the enrichment of CD133⁺ and ALDH⁺ cells, markers of lung cancer stem cells (LCSCs) linked to chemoresistance. Pre-treatment with the γ-secretase inhibitor DAPT significantly inhibited the selection of these stem-like cells and reduced cisplatin resistance [97]. Treatment with GSI-34 of a CD166⁺Lin⁻ subpopulation with LCSC characteristics and intrinsic resistance to cisplatin in xenograft mouse models sensitized these cells to cisplatin, resulting in reduced tumor size, with the most pronounced effect observed when both agents were combined [93].
Xie and colleagues investigated strategies to overcome resistance to gefitinib, an EGFR inhibitor, in NSCLC. Using gefitinib-resistant cell lines, they applied GSI BMS-708163 and found that high doses reversed resistance. In 3D cultures, treated cells formed significantly smaller colonies, with enhanced effects when combined with gefitinib. In vivo experiments in xenograft models confirmed that the combination significantly inhibited tumor growth [98].
Osimertinib, a third-generation EGFR-TKI used in EGFR-mutated NSCLC. Despite its strong antitumor activity, many patients develop resistance through drug-tolerant persisted (DTP) cells. Researchers observed that NOTCH1 and its downstream targets are upregulated in DTP cells, implicating NOTCH signaling in resistance. In vitro and in vivo experiments demonstrated that combining osimertinib with GSI XX impaired drug-tolerant persistence, suppressed phospho-ERK, and enhanced DUSP1 expression. These findings suggest that co-administration of GSIs with osimertinib may represent a promising therapeutic strategy to overcome resistance in EGFR-mutated NSCLC [99].
BMS-906024 GSI was analyzed both as monotherapy and in combination with radiotherapy (RT). While no significant reduction in cell proliferation was observed in 2D cultures, combining BMS-906024 with paclitaxel or crizotinib, either with or without RT, led to a marked decrease in proliferation. In 3D cultures, BMS-906024 as monotherapy reduced spheroidal growth, with even greater effects when combined with crizotinib or RT. The most pronounced reduction was achieved when all three treatments were used together [95]. The use of BMS-906024 GSI in xenograft models demonstrated also enhanced cytotoxicity when combined with paclitaxel, particularly in tumors harboring KRAS or BRAF mutations. This effect was linked to mutant or null p53 status, contrasting with other studies that associated NOTCH1 activity with p53 expression [94,100].
Mizugaki and colleagues found that NSCLC cell lines exposed to RT exhibited increased expression of NOTCH1 and NOTCH3 at 48 hours. Treatment with GSI-I or GSI-XX in combination with RT led to higher levels of apoptosis compared to RT as monotherapy. In xenograft models, the combination of RT and GSI-XX significantly delayed tumor growth relative to either treatment as monotherapy [101].
GSI XX and ABT-737 independently inhibit cell proliferation in a dose-dependent manner, while their combination produces a synergistic antitumor effect in vitro and markedly suppresses tumor growth in vivo. Treatment with either agent regulated the expression of apoptosis proteins. The enhanced antitumor activity of GSI XX and ABT-737 in NOTCH-expressing non-small cell lung cancer leads to apoptosis [102].
NSCLC frequently develops resistance to conventional chemotherapy such as paclitaxel through NOTCH signaling–mediated mechanisms. Targeting NOTCH3 with specific inhibition via GSI-IX was shown to overcome this resistance. In combination with paclitaxel, NOTCH3 inhibition produced a synergistic antitumor effect by modulating the intrinsic apoptosis pathway and enhancing cell death. Moreover, GSI-IX reduced NOTCH3–induced chemoresistance in a concentration-dependent manner [103].
Survival of NSCLC cells in hypoxic tumor environments relies on NOTCH1 signaling, which contributes to chemotherapy resistance, recurrence, and metastasis. Using an orthotopic NSCLC model, researchers inhibited the NOTCH1/IGF-1R/AKT-1 axis with three agents: the γ-secretase inhibitor MRK-003, the IGF-1R antibody MK-0646, and the pan-AKT inhibitor MK-2206. All treatments, except AKT inhibition, significantly prolonged median survival in mice. MRK-003 specifically induced cell death in hypoxic tumors, reduced hypoxia markers, and decreased metastasis to the liver and brain. Sequential administration of MK-0646 followed by erlotinib improved survival, whereas simultaneous treatment was less effective [104].
Stromal-mediated resistance significantly limits the effectiveness of immunotherapy in lung adenocarcinoma. Single-cell RNA sequencing of over 250,000 cells from treatment-naïve patients identified tumor-enriched mesenchymal subsets of cancer-associated fibroblasts (CAFs) and ACTA2⁺MCAM⁺ pericytes, which interact closely with endothelial cells in the perivascular niche. Computational modeling revealed that these interactions are driven by NOTCH signaling, with CAFs and pericytes functioning as signal receivers and immature neovascular endothelial cells as signal senders. Inhibition of NOTCH signaling with GSI MRK-003 or depletion of NOTCH3 reduced collagen production and suppressed invasive behavior. A T cell–inflamed gene signature predicted survival only in patients with low NOTCH3 expression [105].
Gamma-secretase inhibitors, such as DAPT, play a significant role in angiogenesis because the NOTCH signaling pathway regulates vascular development. In experimental models using human lung adenocarcinoma xenografts in nude mice, DAPT reduced endothelial cell proliferation without inducing cellular toxicity, suppressed capillary structure formation in vitro, and inhibited microvessel sprouting in the rat aortic ring assay. Collectively, these findings demonstrate that DAPT potently blocks angiogenesis, leading to impaired tumor vascularization and reduced tumor growth. [106].
Recently, evodiamine (EVO), a low toxicity natural alkaloid, has emerged as a potential antitumor agent for NSCLC [107]. EVO reduced cell proliferation and metastasis and lowered expression of NOTCH3 and GSC. Although EVO did not bind directly to NOTCH3, it showed strong affinity for GSC, comparable to that of GSIs.
Finally challenges of applying particle therapy to NSCLC, particularly the impact of lung tissue heterogeneity on dose modulation have been performed. Lung modulation models into clinical workflows is critical for optimizing particle therapy outcomes in NSCLC. [108].
3.3. The Use of GSIs, ADAM Inhibitors and Other Combined Therapies Have Contributed to Elucidating the Role of NOTCH Signaling in Gastric Cancer (GC)
NOTCH signaling has been firmly established as a key regulator of gastric stem cell proliferation, differentiation, and maintenance. Moreover, it plays a role in glandular fission, a process in which a glandular unit is divided into two due to elevated cell proliferation, thereby contributing to tumor development. NOTCH signaling has also been implicated in the initiation of oncogenesis through crosstalk with other pathways and is associated with increased levels of CD44⁺ and CD133⁺ cells, which are recognized markers of gastric cancer stem cells (GCSCs) [61,109].
CD133⁺ gastric cancer stem cells (GCSCs) exhibited low RECK protein expression, a cysteine-rich protein with Kazal motifs known for its protease activity that inhibits metastasis and angiogenesis, and high NOTCH1 levels. RECK suppress ADAM-mediated NOTCH1 activation. Moreover, by using the γ-secretase inhibitor DAPT, they demonstrated that NOTCH signaling was oncogenic in these tumors, with DAPT reducing the formation of GCSC-rich spheres by 25% compared to the control group [110].
CD44⁺ GCSCs, which showed elevated NOTCH1 expression and increased resistance to 5-fluorouracil (5-FU) were compared to CD44⁻ cells. Treatment with DAPT selectively affected CD44⁺ cells, leading to reduced self-renewal, diminished tumor initiation and migration, and enhanced sensitivity to 5-FU. In xenograft models, intraperitoneal administration of DAPT significantly inhibited tumor growth and epithelial–mesenchymal transition (EMT) [111].
Barat and colleagues also studied CD44⁺ cells, hypothesizing a functional link between NOTCH1 and the WNT/β-catenin pathway, another key signaling axis in gastric carcinoma. Treatment with GSI-IX produced dose- and time-dependent reductions in cell proliferation, migration, invasion, and tumor sphere size, along with increased apoptosis. Treated cells showed decreased levels of active NOTCH intracellular domain 1 (NICD1) and WNT signaling components. Similar results were observed in xenograft mice, with reduced tumor growth and increased necrosis in the GSI-IX-treated group [112].
NOTCH signaling and PTEN, a tumor suppressor gene frequently inactivated in gastric cancer (GC), seem to be related. Treatment of GC cells with GSI-I led to reduced tumor activity and increased PTEN expression compared to controls [113]. In xenograft models, the combination of GSI-I and paclitaxel resulted in significantly greater tumor growth inhibition than either agent as monotherapy. Lee and colleagues further evaluated GSI-I in GC cell lines and xenograft mice, confirming its efficacy and lack of significant side effects. When combined with 5-FU, both in vitro and in vivo, the dual treatment produced markedly greater tumor suppression than monotherapy [114].
Yao and colleagues observed that DAPT treatment increased ERK1/2 phosphorylation in GC cells, suggesting that NOTCH signaling may suppress this oncogenic kinase. Combining DAPT with PD98059, an ERK1/2 MAPK inhibitor, led to reduced tumor growth and increased apoptosis in vitro, with similar synergistic effects observed in xenograft models [115]. Finally, a combination of DAPT and an anti-DLL4 antibody was tested in GC cell lines and xenograft mice. This dual approach significantly enhanced apoptosis, reduced cell invasion, and decreased tumor size compared to control treatments [116].
3.4. GSIs Enhance the Efficacy of Targeted Therapies for Metastatic Melanoma
NOTCH signaling has also been implicated in the oncogenic potential of melanoma stem cells (MSCs). Kumar and colleagues associated CD133⁺ melanoma cells with MSCs and demonstrated, both in vitro and in vivo, enhanced proliferation, angiogenesis, epithelial–mesenchymal transition (EMT), metastasis, and chemoresistance. These cells exhibited overactivation of NOTCH1, and treatment with GSIs (GSI-IX and GSI-X) significantly reduced the number of CD133⁺ MSCs, leading to decreased migration and interaction with vascular endothelium [117].
Approximately half of metastatic melanomas harbor mutations in the BRAF gene, for which targeted therapies using BRAF inhibitors (BRAFi) and MEK inhibitors (MEKi) are available. Zhu and colleagues explored the role of NOTCH signaling in acquired resistance to BRAFi. They found that combining DAPT with BRAFi reversed resistance in melanoma cell lines, suggesting that NOTCH signaling enables BRAFi-treated cells to escape senescence, a state that was re-induced by DAPT treatment [118]. Porcelli and colleagues tested the combination of MEKi and the GSI nirogacestat (PF-03084014), observing enhanced inhibition of cell proliferation and migration in vitro compared to monotherapy [119].
NOTCH signaling also appears to contribute to resistance against other targeted therapies. Krepler and colleagues combined the GSI RO4929097 with an ERK inhibitor (ERKi), finding that in ERKi-resistant cell lines, the combination significantly reduced cell viability and increased apoptosis compared to either agent as monotherapy. In xenograft models, the combination also produced superior tumor growth inhibition [120].
The combination of GSI-I with BCL-2 inhibitors (BCL2i) demonstrated greater efficacy than monotherapy in both melanoma cell lines and xenograft mice. The treatment increased apoptosis and reduced the population of ALDH⁺ MSCs [121].
Nueda and colleagues studied the dose-dependent effects of DAPT on metastatic melanoma cell lines. High doses reduced proliferation, while low doses paradoxically increased it. Nueda and colleagues investigated the combination of DAPT with high expression levels of DLK1 or DLK2, finding synergistic inhibition of NOTCH signaling and reduced proliferation of metastatic melanoma cells [66]. Keyghobadi and colleagues further observed that prolonged DAPT treatment led to increased tumor growth in vitro and in vivo [122].
This study demonstrates that FOXP3, a transcription factor associated with immune regulation and cancer, plays a critical role in melanoma progression. Its expression increases in metastatic melanoma cells under TGFβ stimulation, marking tumor aggressiveness and metastatic potential. Mechanistically, NOTCH1 was identified as the key driver of TGFβ-induced FOXP3 expression and blocking NOTCH1 with a GSI significantly reduced melanoma cell growth and survival. These findings highlight the importance of the TGFβ1/NOTCH1 signaling axis in regulating FOXP3 and advancing melanoma progression [123].
Among GSIs, RO4929097 is the only one to have entered clinical trials for metastatic melanoma. Tolcher and colleagues conducted a phase I trial in patients with metastatic or locally advanced tumors to determine the maximum tolerated dose (MTD), adverse effects, and preliminary efficacy. A total of 110 patients were enrolled and divided into three dosage groups (A, B, and C). Drug levels in all patients exceeded the threshold for antitumor activity. The most common adverse effects were gastrointestinal, with some cases of hypophosphatemia (Figure 9). Notably, one patient with melanoma showed a minor response, and 33% and 41% of patients in groups A and B, respectively, achieved disease stabilization [124].
Building on earlier findings, Lee and colleagues conducted a phase II clinical trial to assess the efficacy and tolerability of RO4929097 GSI in patients with metastatic melanoma. Among the 32 patients enrolled, most adverse effects were grade 1 or 2, with only six patients experiencing grade 3 toxicities, including hypophosphatemia. In terms of clinical response, one patient achieved a partial response lasting seven months and survived for over 28 months. Additionally, eight patients reached a stable disease state. However, overall efficacy was limited: the disease control rate at 12 weeks was 31%, the median progression-free survival (PFS) was 1.5 months, and the 6-month PFS rate was 9%. The 1-year survival rate was 50%, with a confidence interval of 23%-66%. The authors attributed the modest outcomes to subtherapeutic drug levels [125].
More recently, Jayaprakash and colleagues revisited the use of RO4929097 in melanoma, identifying a potential synergistic effect at low doses when combined with radiotherapy (RT). In vitro studies also showed reduced cell migration with this combination [126].
3.5. Various Works Explore the Use of GSIs as Monotherapy and in Combination Therapies for Triple-Negative Breast Cancer (TNBC)
As in other cancer types, tumor stem cells play a critical role in the tumorigenesis of triple-negative breast cancer (TNBC). Azzam and colleagues identified two subpopulations in TNBC cell lines, CD44⁺CD24low (hereafter CD24low) and CD44⁺CD24⁻ (hereafter CD24⁻), both exhibiting characteristics of breast cancer stem cells (BCSCs). Treatment with the GSI RO4929097 inhibited sphere formation in CD24low cells and significantly slowed tumor growth and metastasis in xenograft models, effects not observed in CD24⁻ cells [127]. Treatment of BCSC cell lines with DAPT decreased cell proliferation, increased apoptosis, reduced invasion, and diminished sphere formation. In xenograft mice, DAPT delayed tumor onset and slowed subsequent tumor growth [128].
An innovative nanoparticle-based approach to counter resistance in cancers driven by NOTCH–EGFR interactions has been developed. Researchers developed nanoparticles carrying the γ-secretase inhibitor DAPT (NP-EB/DART) to block NOTCH signaling, and enhanced tumor targeting with a specially engineered peptide (CF). This peptide links CREKA, which homes to tumors, with F3, a cell-penetrating peptide, via a pH-sensitive bond that prevents off-target activity. In the acidic tumor environment, the bond is cleaved, activating F3 to promote nanoparticle penetration into cancer cells [129].
Stoeck and colleagues explored the relationship between NOTCH gene mutations and sensitivity to the GSI MRK003 in TNBC models. They found that therapeutic response was more closely linked to levels of active NOTCH intracellular domain (NICD) than to the presence of NOTCH mutations. In xenograft mice with elevated NICD levels, MRK003 was more effective, and in vitro, its combination with paclitaxel showed significant antitumor activity [130].
The combined use of the GSI MK-0752 and the MET inhibitor (METi) SU11274 in two TNBC cell lines with differing NOTCH1 expression was also analyzed in other work. MK-0752 as monotherapy did not inhibit cell growth, whereas SU11274 was effective, and its efficacy was enhanced when combined with MK-0752. Interestingly, MK-0752 was more effective than SU11274 in blocking colony formation, and the combination yielded the strongest inhibitory effect [131].
MDA-MB-231 triple-negative breast cancer cells respond effectively to treatment with a γ-secretase inhibitor (GSI), both as monotherapy and when paired with doxorubicin in vitro and in vivo. Combination of GSI with doxorubicin was not only practical but also improved therapeutic outcomes, highlighting its potential as a promising approach for treating triple-negative breast cancer [132].
An innovative strategy was proposed by Paroni and colleagues [133]. All-trans retinoic acid (ATRA), an unconventional therapy for breast cancer, is often ineffective in TNBC. However, their study revealed a correlation between ATRA sensitivity and elevated levels of active NICD1 in certain TNBC subtypes. In various TNBC cell lines, the combination of DAPT and ATRA proved more effective in suppressing tumor growth than either agent as monotherapy. Similar results were obtained in TNBC xenograft mice treated with PF-03084014 and ATRA.
Recent literature highlights suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, as a promising therapeutic candidate for TNBC [68]. However, SAHA may promote epithelial–mesenchymal transition (EMT), potentially due to NOTCH pathway overactivation [134]. They combined SAHA with the GSI LY411575 in TNBC cell lines. This combination enhanced apoptosis, increased reactive oxygen species, induced mitochondrial depolarization, reduced EMT marker expression, and diminished breast cancer stem cell (BCSC) characteristics. Sen and colleagues also used a multi-targeting TACE/ADAM17 and gamma-secretase of NOTCH signaling pathway in TNBC via drug repurpose approach using lomitapide [135].
The limited success of GSIs in clinical trials is largely attributed to their intestinal toxicity and potential immunological side effects, given the critical role of NOTCH signaling in T-cell activation, including CD8+ T cells within tumors. To overcome these limitations, Hossain and colleagues explored alternative agents that lack systemic toxicity and preserve tumor immunity [136]. They identified sulindac sulfide (SS), the active metabolite of the FDA-approved NSAID sulindac, as a promising GSI substitute. SS significantly inhibited nanosphere formation across human and murine TNBC models in vivo, in vitro, and ex vivo. In a transplantable TNBC mouse model (C0321), SS demonstrated potent single-agent antitumor activity and effectively suppressed NOTCH1 protein expression in tumors [136].
CB-103, an orally available pan-NOTCH inhibitor, as a promising and safer alternative to traditional γ-secretase inhibitors (GSIs) in breast cancer therapy. Unlike GSIs, CB-103 avoids gastrointestinal side effects due to its distinct mechanism of action. Preclinical evidence shows strong potential in luminal endocrine-resistant and triple-negative breast cancers (TNBCs) with confirmed NOTCH activity. When combined with SERDs or CDK inhibitors in endocrine-resistant recurrent breast cancers, and with taxane-based chemotherapy in TNBC, CB-103 produced synergistic effects boosting paclitaxel’s impact in TNBC resistant xenografts models [137].
Through a genome-wide CRISPR-Cas9 screen, researchers found that SOX2 and NOTCH signaling form a reciprocal feedback loop: NOTCH signaling suppresses SOX2 via HEY target genes, while SOX2 inhibits NOTCH signaling by interacting with RBPJκ. This interplay creates distinct TNBC cell states: NOTCH-active cells with epithelial-like traits versus SOX2-driven cells with EMT features, stem cell properties, and drug resistance. Moreover, Paclitaxel to synergize with γ-secretase inhibitor Crenigacestat (LY3039478), leading to tumor growth and metastasis reduction in NOTCH1High/SOX2Low TNBC xenografts, while the synergistic combination Paclitaxel and Dasatinib is efficient in NOTCH1Low/SOX2High TNBC xenografts [138].
Schott and colleagues were the first to evaluate a GSI as a therapeutic agent for TNBC in humans. They studied chemotherapy-resistant BCSCs models treated with MK-0752 and found that, while tumors formed in 50% of control mice, none developed in the treated group. These findings led to a phase I clinical trial combining MK-0752 with docetaxel. Among 24 patients, one experienced grade 5 pneumonitis (likely due to docetaxel), while 11 had partial responses, 9 achieved stable disease, and 3 showed progression. Serial biopsies from six patients revealed a reduction in BCSCs populations [139].
A phase I trial combining the GSI PF-03084014 with docetaxel in 29 women with TNBC was also conducted. The study aimed to determine the maximum tolerated dose (MTD). Severe adverse events were reported, including one death from septic shock following febrile neutropenia. Grade 4 neutropenia occurred in 24 of the 29 patients, and hypophosphatemia was also observed (Figure 10). Treatment efficacy was limited [140].
In another phase I study, the MTD of RO4929097 in combination with paclitaxel and carboplatin in 14 patients with triple-negative breast cancer (TNBC) was analyzed. Similar to the previous study, several grade 4 adverse events were reported, the most significant being neutropenia and thrombocytopenia. In terms of clinical response, 5 patients showed a partial response, 4 achieved disease stabilization, and 5 exhibited residual disease [141]. RO4929097 has also been investigated in a phase Ib clinical trial for metastatic estrogen receptor-positive breast cancer (EPBCm). Means-Powell and colleagues administered RO4929097 alongside exemestane, an aromatase inhibitor, to 15 patients with EPBCm [142]. One dose-limiting grade 4 adverse event was observed, and grade 3 hypophosphatemia occurred in 13% of patients. Regarding efficacy, 7 patients demonstrated a partial response, and 7 maintained stable disease among the 14 evaluated.
4. Discussion
The use of γ-secretase inhibitors in vitro and in mouse xenograft models has emerged as a promising therapeutic strategy. These preclinical studies have shown encouraging results, particularly when GSIs are combined with other treatments. GSIs have demonstrated efficacy in enhancing the effects of various chemotherapeutic agents, for example, gemcitabine in pancreatic ductal adenocarcinoma (PDAC) [63,86], paclitaxel, osimertinib, erlotinib and ABT-737in non-small cell lung cancer (NSCLC) [99,100,102,103,104], 5-FU in gastric cancer (GC) [113] or TNBC [129,138]. Additionally, combinations with radiotherapy (RT) have shown effectiveness in NSCLC and metastatic melanoma [95,101,126]. The synergistic potential of GSIs with other drugs and their ability to sensitize tumors to treatment has been observed across all five cancer types reviewed, with particular emphasis on NSCLC and melanoma [66,67,68,95,98,115,116,119,120,131,142,143].
Research has also focused on the role of NOTCH signaling in cancer stem cells and its inhibition to overcome resistance to therapy and improve prognosis. A significant relationship has been identified between NOTCH signaling and gastric cancer stem cells (GCSCs), NSCLC, metastatic melanoma, and TNBC, suggesting promising therapeutic applications [93,97,109,110,111,112,117,121,127,128]. Another key area of investigation is the potential of GSIs to block epithelial-mesenchymal transition (EMT), a process implicated in GC, metastatic melanoma, and TNBC [111,117,134,144].
Recent studies have increasingly focused on combining GSIs with innovative therapeutic agents, yielding promising results. For instance, in TNBC, GSIs have been successfully combined with SAHA and ATRA [133,135,137,144]. Table 1 classifies the data analyzed by GSI used, cancer type, study model (in vitro or xenograft), and outcomes. DAPT was the most frequently used GSI, followed by RO4929097.
GSIs have also entered clinical trials, although results remain limited (Table 2). Despite the promising preclinical data, clinical trials have yet to meet expectations. In a phase I trial involving stage IV PDAC patients, combining GSI with gemcitabine did not yield superior outcomes compared to gemcitabine as monotherapy [91]. Another phase II trial in metastatic PDAC was discontinued due to the discontinuation of the GSI under investigation [92]. In metastatic melanoma, a phase I trial of GSI monotherapy showed promising results, prompting a phase II trial. However, the latter produced limited outcomes, likely due to subtherapeutic dosing. Overall tolerability was acceptable, although severe hypophosphatemia was reported as a significant adverse event [124,125].
TNBC has been the cancer type with the highest number of clinical trials involving GSIs. Three phase I trials tested different GSIs in combination with various chemotherapeutic agents, revealing several severe hematological and infectious adverse reactions. Although partial responses were observed in some patients, overall clinical efficacy was limited. A GSI was also tested in EPBCm with similar results [120,127,139,140,141]. Table 1 presents a classification of clinical trial data by cancer type and outcomes, showing RO4929097 as the most frequently tested GSI.
Identification of limitations of the studies
Despite promising preclinical findings, clinical trials have not yet delivered the expected results. One possible explanation is the reliance on cell lines with overactivated NOTCH signaling, which may not accurately represent the heterogeneity of human tumors. These cell lines were treated with GSIs and used to generate xenograft models in several cases [98,115,117,137,138]. However, human tumors consist of diverse cell populations with varying levels of NOTCH receptor expression and activation. A specific GSI may only affect certain cell types, and its efficacy may depend more on activation levels than expression levels.
Moreover, many studies have not accounted for the specificity of GSIs toward different NOTCH receptors. This non-selective inhibition may lead to adverse effects, such as increased incidence of non-melanoma skin neoplasms. For example, indiscriminate inhibition of all NOTCH receptors may suppress tumor-suppressive pathways, as seen with NOTCH2 in breast cancer [59]. Additionally, low levels of NOTCH inhibition by DLK1 and DLK2 proteins may paradoxically increase cell proliferation, as observed by Nueda and colleagues, Naranjo and Colleagues and others [66,67,68].
To enhance GSI efficacy and minimize adverse effects in human clinical trials, combined treatments of GSIs and other current drugs should be studied and drug delivery methods such as nanoparticles should be potentiated, although many delivery systems have already been performed in different types of cancer [145]. For example, Wan and co-workers used pH-sensitive peptide-functionalized nanoparticles to co-deliver erlotinib and DAPT, effectively restricting TNBC progression [129]. Zhou and coworkers employed DT7-modified lecithin nanoparticles loaded with a GSI and dexamethasone, achieving effective inhibition of T-cell acute lymphoblastic leukemia while reducing gastrointestinal toxicity [146].
Future prospects
CRISPR is reshaping oncology by enabling precise gene editing for modeling tumors, immune responses, and drug resistance. It identifies oncogenesis and chemoresistance mechanisms, offering new therapeutic opportunities beyond conventional NOTCH inhibition. CRISPR advances immune-based treatments like CAR T cells and direct antitumor agents with bispecific antibodies [147,148]. BCMA-targeted therapies (CAR T cells, bispecific antibodies) revolutionize multiple myeloma treatment but face relapse due to resistance mechanisms [149] [150].
Combination therapies show promise but can promote stem-like cells and EMT. GSIs counteract chemotherapy resistance by suppressing EMT, stem-like transformation, and survival signaling [151] [152]. γ-Secretase Inhibitors (GSIs) also act as anti-angiogenic agents, blocking endothelial growth and tumor vascularization cancer models [106] [153]. Tumor Microenvironment may generate resistance to drugs, such as cancer-associated fibroblasts (CAFs) in pancreatic cancer are highly chemoresistant, forming protective structures and secreting IL-6. Biomarkers like IL6/IL8 predict resistance to GSIs (e.g., RO4929097), guiding patient selection for better outcomes [89].
There are some clinical advances such as those performed with Nirogacestat (PF-03084014), which shows striking benefit in desmoid tumors (71.4% response rate, durable >6 years) . Other GSIs (e.g., AL-102) are in late-stage trials, expanding potential beyond desmoid tumors to multiple myeloma [154,155,156]. Preclinical work with GSI RO4929097 showed that tumor cell lines with high IL6 and IL8 expression were resistant, as the drug no longer impacted angiogenesis or fibroblast infiltration. Xenograft models confirmed that IL6/IL8 overexpression predicts in vivo resistance. Clinically, Phase I data revealed that patients with low baseline IL6 and IL8 levels were more likely to benefit from treatment. [157].
Innovative trials combine immunotherapy (Pembrolizumab) with carbon ion radiotherapy, aiming to enhance immune responses while sparing healthy cells. Combination therapies and precision strategies are key to long-term cancer control [158].
5. Conclusions
NOTCH signaling plays a key role in the five types of tumors under study and in cancer stem cells. 2. γ-secretase inhibitors (GSIs) show promise in vitro and mouse xenograft models and enhance the effects of chemotherapy and radiotherapy. GSIs can block epithelial-mesenchymal transition (EMT) and may help overcome therapy resistance and improve prognosis. However, clinical trials results remain limited despite preclinical promise. Future research should identify oncogenic NOTCH receptors/ligands to enable receptor-specific GSIs or alternative inhibitors (e.g., CB103, ADAM inhibitors, DLK proteins), combined with patient stratification based on NOTCH activation levels. Lower-dose combinations show promise but may induce stem-like traits and EMT, requiring careful monitoring. Trials are integrating new strategies such as immunotherapy with CRISPR, CAR T cells, and bispecific antibodies to model tumors and drug resistance more precisely. Studying microenvironment-driven resistance and targeting angiogenesis remains critical strategies. Finally, leveraging big data and AI can enable individualized treatments, including male- and female-specific considerations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Figures S1, S2 and S3.pptx. TENGO QUE CAMBIARLO POR LA FIGURA DESCENTRADA.
Author Contributions
Conceptualization, V.B and P.M-G; methodology, P.M-G.; software, V.B, M-L. N and P.M-G; validation, V.B, M-L.N and P.M-G.; formal analysis, V.B, M-L.N and P.M-G; investigation, P.M-G.; resources, V.B. and M-L.N; data curation, V.B, M-L.N and P.M-G.; writing original draft preparation, V.B and M-L.N.; writing review and editing, V.B, M-L.N and P.M-G.; visualization, V.B, M-L.N and P.M-G.; supervision, V.B and M-L.N.; project administration, V.B, M-L.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created. All data analyzed in this study are included in this published systematic review [and its Supplementary Information Files]. This work will be deposited in the RUIdeRA institutional repository at University of Castilla-La Mancha, Spain.
Acknowledgments
We thank Universidad de Castilla-La Mancha (Spain) for its institutional support.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this manuscript.
Use of Artificial Intelligence
During the preparation of this work the author(s) used ChatGPT 4 in order to edit English writing. After using this tool/service, the author(s) reviewed the English writing as needed and take(s) full responsibility for the content of the publication.
Abbreviations
The following abbreviations are used in this manuscript:
| ABT-737 | Bcl-2 inhibitor |
| ADAM | A Disintegrin And Metalloproteinase |
| AKT | Protein kinase B (PKB) |
| ALDH | aldehyde dehydrogenase |
| ATRA | All-trans retinoic acid. |
| BCL2i | BLC-2 inhibitors. |
| BCSCs | Breast cancer stem cells. |
| BCMA | B-cell maturation antigen |
| BRAFi | BRAF inhibitor. |
| BRCA1/2 | Breast cancer gene 1/2 |
| CAFs | Cancer-associated fibroblasts |
| CAR-T | Chimeric artificial T cell receptors |
| CB-103 | Non-gamma-secretase inhibitor |
| CD44 | Cell Surface Glycoprotein CD44 |
| CD133 | Transmembrane glycoprotein CD133 |
| CREKA | Pentapeptide lineal biologically active compound |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CSCs | cancer stem cells |
| CSL | CBF1/Suppressor of Hairless/LAG-1, also known as RBP-Jκ |
| DAPT | GSI-IX |
| DLK1 | Delta like homolog 1. |
| DLK2 | Delta like homolog 2. |
| DLL1 | Canonical Delta-Like1 ligand. |
| DLL3 | Canonical Delta-Like3 ligand. |
| DLL4 | Canonical Delta-Like4 ligand. |
| DOS | Delta and OSM-11 Motif. |
| DTP | Drug-tolerant persisted cells |
| DSL | Delta/Serrate/LAG-2 domain |
| DT | Desmoid tumors |
| DUSP1 | Dual specificity phosphatase 1 |
| EGF | Epidermal growth factor. |
| EGFR | |
| EMT | Epithelial-Mesenchymal Transition. |
| EPBCm | Estrogen receptor-positive metastatic breast cancer. |
| EpCAM | Epithelial cell adhesion molecule |
| ErbB-4 | EGFR subfamily of receptor tyrosine kinases |
| ERK | Extracellular Signal-Regulated Kinase |
| ERKi | ERK MAPK inhibitor. |
| EVO | Evodiamine. |
| 5-FU | 5-fluorouracil |
| FOXP3 | Forkhead box P3 |
| GC | Gastric cancer. |
| GCSCs | Gastric cancer stem cells. |
| GSC | γ-Secretase complex |
| GSI | γ-Secretase inhibitor. |
| Hes1 | Hairy and enhancer of split-1 |
| HEY | Hes related family BHLH transcription factor with YRPW motif |
| IL | Interleukin |
| IGF-1R | Receptor of growth factor similar to insulin 1. |
| JAG1 | Canonical Jagged 1 ligand. |
| JAG2 | Canonical Jagged 2 ligand. |
| KRAS | Kirsten rat Sarcoma |
| LCSCs | Lung cancer stem cells. |
| mAb | monoclonal antibodies |
| MAML | Mastermind-like protein. |
| MAPK | Mitogen-Activated Protein Kinase |
| MEK | Mitogen-Activated Protein Kinase 1 (MAP2K1) |
| MEKi | MEK inhibitor. |
| MET | Mesenchymal Epithelial Transition receptor tyrosine kinase |
| METi | MET inhibitor. |
| MM | Multiple myeloma |
| MTD | maximum tolerated dose |
| MSC | Melanoma stem cells. |
| NECD | NOTCH extracellular domain |
| NF-κB | Nuclear factor enhancing kappa light chains of activated B cells |
| NICD | NOTCH intracellular domain. |
| NRR | Negative regulatory region. |
| NSCLC | Non-small cell lung cancer. |
| PARP | Poly(ADP-ribose) polymerase |
| PDAC | Pancreatic ductal adenocarcinoma. |
| PEST | proline, glutamic acid, serine, and threonine domain |
| PD-1 | Programmed cell death protein 1 |
| PFS | Progression-free survival. |
| PI3K | Phosphoinositide 3-kinase |
| RBP-Jκ | Recombination signal binding protein for immunoglobulin kappa J region |
| RECK | Reversion-inducing cysteine-rich protein with Kazal motifs. |
| RT | Radiotherapy. |
| SAHA | Suberoylanilide hydroxamic acid. |
| SOX2 | SRY-related HMG-box 2 |
| SS | Sulindac sulfide. |
| TACE | Tumor necrosis factor (TNF)-converting enzyme |
| TGF-β | Transforming growth factor beta |
| TMD | transmembrane domain. |
| TNBC | Triple-negative breast cancer. |
| VEGF | Vascular endothelial growth factor |
| VEGFR1 | Receptor of vascular endothelial growth factor 1 |
| WNT | Wingless and Int-1. |
| 2D | Two dimensions. |
| 3D | Three dimensions. |
| WHO | World Health Organization. |
References
- International Agency for Research on Cancer 2025; Available from: https://www.iarc.who.int/cancer-topics/.
- World Health Organization. 2025; Available from: https://www.who.int/health-topics/cancer#tab=tab_1.
- Aster, J.C., W.S. Pear, and S.C. Blacklow, The Varied Roles of Notch in Cancer. Annu Rev Pathol, 2017. 12: p. 245-275. [CrossRef]
- Ferreira, A. and J.C. Aster, Notch signaling in cancer: Complexity and challenges on the path to clinical translation. Semin Cancer Biol, 2022. 85: p. 95-106. [CrossRef]
- Guo, M., et al., Notch signaling, hypoxia, and cancer. Front Oncol, 2023. 13: p. 1078768. [CrossRef]
- Zhou, B., et al., Notch signaling pathway: architecture, disease, and therapeutics. Signal Transduct Target Ther, 2022. 7(1): p. 95. [CrossRef]
- Siebel, C. and U. Lendahl, Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev, 2017. 97(4): p. 1235-1294. [CrossRef]
- Kopan, R. and M.X. Ilagan, The canonical Notch signaling pathway: unfolding the activation mechanism. Cell, 2009. 137(2): p. 216-33. [CrossRef]
- Bray, S., Notch. Curr Biol, 2000. 10(12): p. R433-5.
- Bray, S. and M. Furriols, Notch pathway: making sense of suppressor of hairless. Curr Biol, 2001. 11(6): p. R217-21. [CrossRef]
- Bray, S.J., Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol, 2006. 7(9): p. 678-89. [CrossRef]
- Zamfirescu, A.M., A.S. Yatsenko, and H.R. Shcherbata, Notch signaling sculpts the stem cell niche. Front Cell Dev Biol, 2022. 10: p. 1027222. [CrossRef]
- Artavanis-Tsakonas, S., M.D. Rand, and R.J. Lake, Notch signaling: cell fate control and signal integration in development. Science, 1999. 284(5415): p. 770-6.
- Weinmaster, G., Notch signal transduction: a real rip and more. Curr Opin Genet Dev, 2000. 10(4): p. 363-9. [CrossRef]
- Mumm, J.S. and R. Kopan, Notch signaling: from the outside in. Dev Biol, 2000. 228(2): p. 151-65.
- Lai, E.C., Notch signaling: control of cell communication and cell fate. Development, 2004. 131(5): p. 965-73. [CrossRef]
- Artavanis-Tsakonas, S. and M.A. Muskavitch, Notch: the past, the present, and the future. Curr Top Dev Biol, 2010. 92: p. 1-29.
- DEXTER, J.S., The Analysis of a Case of Continuous Variation in Drosophila by a Study of Its Linkage Relations. The American Naturalist, 1914. 48: p. 712-758. [CrossRef]
- Blaumueller, C.M., et al., Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell, 1997. 90(2): p. 281-91. [CrossRef]
- Logeat, F., et al., The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A, 1998. 95(14): p. 8108-12. [CrossRef]
- Czerwonka, A., J. Kalafut, and M. Nees, Modulation of Notch Signaling by Small-Molecular Compounds and Its Potential in Anticancer Studies. Cancers (Basel), 2023. 15(18). [CrossRef]
- Tsaouli, G., et al., Molecular Mechanisms of Notch Signaling in Lymphoid Cell Lineages Development: NF-kappaB and Beyond. Adv Exp Med Biol, 2020. 1227: p. 145-164.
- Lopez-Lopez, S., et al., NOTCH3 signaling is essential for NF-kappaB activation in TLR-activated macrophages. Sci Rep, 2020. 10(1): p. 14839. [CrossRef]
- Lubman, O.Y., et al., Quantitative dissection of the Notch:CSL interaction: insights into the Notch-mediated transcriptional switch. J Mol Biol, 2007. 365(3): p. 577-89. [CrossRef]
- Kopan, R., et al., Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A, 1996. 93(4): p. 1683-8. [CrossRef]
- Kopan, R. and R. Cagan, Notch on the cutting edge. Trends Genet, 1997. 13(12): p. 465-7. [CrossRef]
- Krebs, L.T., et al., Notch signaling is essential for vascular morphogenesis in mice. Genes Dev, 2000. 14(11): p. 1343-52.
- D’Souza, B., L. Meloty-Kapella, and G. Weinmaster, Canonical and non-canonical Notch ligands. Curr Top Dev Biol, 2010. 92: p. 73-129.
- D’Souza, B., A. Miyamoto, and G. Weinmaster, The many facets of Notch ligands. Oncogene, 2008. 27(38): p. 5148-67. [CrossRef]
- Hozumi, K., Distinctive properties of the interactions between Notch and Notch ligands. Dev Growth Differ, 2020. 62(1): p. 49-58. [CrossRef]
- Kuintzle, R., L.A. Santat, and M.B. Elowitz, Diversity in Notch ligand-receptor signaling interactions. bioRxiv, 2024.
- Laborda, J., et al., dlk, a putative mammalian homeotic gene differentially expressed in small cell lung carcinoma and neuroendocrine tumor cell line. J Biol Chem, 1993. 268(6): p. 3817-20. [CrossRef]
- Baladron, V., et al., dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Experimental Cell Research, 2005. 303(2): p. 343-359. [CrossRef]
- Nueda, M.L., et al., The novel gene EGFL9/Dlk2, highly homologous to Dlk1, functions as a modulator of adipogenesis. J Mol Biol, 2007. 367(5): p. 1270-80. [CrossRef]
- Sanchez-Solana, B., et al., The EGF-like proteins DLK1 and DLK2 function as inhibitory non-canonical ligands of NOTCH1 receptor that modulate each other’s activities. Biochim Biophys Acta, 2011. 1813(6): p. 1153-64. [CrossRef]
- Lee, Y.L., et al., dlk, pG2 and Pref-1 mRNAs encode similar proteins belonging to the EGF-like superfamily. Identification of polymorphic variants of this RNA. Biochim Biophys Acta, 1995. 1261(2): p. 223-32. [CrossRef]
- Wang, Y., et al., Pref-1 , a preadipocyte secreted factor that inhibits adipogenesis J Nutr, 2006. 136: p. 2953-2956.
- Smas, C., L. Chen, and H. Sul, Cleavage of membrane-associated pref-1 generates a soluble inhibitor of adipocyte differentiation. Mol Cell Biol, 1997. 17: p. 977-988. [CrossRef]
- Smas, C.M., D. Green, and H.S. Sul, Structural characterization and alternate splicing of the gene encoding the preadipocyte EGF-like protein pref-1. Biochemistry, 1994. 33(31): p. 9257-65. [CrossRef]
- Pittaway, J.F.H., et al., The role of delta-like non-canonical Notch ligand 1 (DLK1) in cancer. Endocr Relat Cancer, 2021. 28(12): p. R271-R287. [CrossRef]
- Nueda, M.L., et al., DLK proteins modulate NOTCH signaling to influence a brown or white 3T3-L1 adipocyte fate. Sci Rep, 2018. 8(1): p. 16923. [CrossRef]
- Yevtodiyenko, A. and J.V. Schmidt, Dlk1 expression marks developing endothelium and sites of branching morphogenesis in the mouse embryo and placenta. Dev Dyn, 2006. 235(4): p. 1115-23. [CrossRef]
- Garcia-Gallastegi, P., et al., Similarities and differences in tissue distribution of DLK1 and DLK2 during E16.5 mouse embryogenesis. Histochem Cell Biol, 2019. 152(1): p. 47-60. [CrossRef]
- Barrick, D. and R. Kopan, The Notch transcription activation complex makes its move. Cell, 2006. 124(5): p. 883-5. [CrossRef]
- Christopoulos, P.F., et al., Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front Immunol, 2021. 12: p. 668207. [CrossRef]
- Miele, L., Notch signaling. Clin Cancer Res, 2006. 12(4): p. 1074-9.
- Mumm, J.S., et al., A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell, 2000. 5(2): p. 197-206. [CrossRef]
- Brou, C., et al., A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell, 2000. 5(2): p. 207-16.
- Lai, E.C., Notch cleavage: Nicastrin helps Presenilin make the final cut. Curr Biol, 2002. 12(6): p. R200-2. [CrossRef]
- Wolfe, M.S., Substrate recognition and processing by gamma-secretase. Biochim Biophys Acta Biomembr, 2020. 1862(1): p. 183016. [CrossRef]
- Kimberly, W.T., et al., Notch and the amyloid precursor protein are cleaved by similar gamma-secretase(s). Biochemistry, 2003. 42(1): p. 137-44.
- Wong, E., G.R. Frost, and Y.M. Li, gamma-Secretase Modulatory Proteins: The Guiding Hand Behind the Running Scissors. Front Aging Neurosci, 2020. 12: p. 614690. [CrossRef]
- Schroeter, E.H., J.A. Kisslinger, and R. Kopan, Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature, 1998. 393(6683): p. 382-6. [CrossRef]
- Previs, R.A., et al., Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res, 2015. 21(5): p. 955-61. [CrossRef]
- Katoh, M. and M. Katoh, Precision medicine for human cancers with Notch signaling dysregulation (Review). Int J Mol Med, 2020. 45(2): p. 279-297.
- Bernasconi-Elias, P., et al., Characterization of activating mutations of NOTCH3 in T-cell acute lymphoblastic leukemia and anti-leukemic activity of NOTCH3 inhibitory antibodies. Oncogene, 2016. 35(47): p. 6077-6086. [CrossRef]
- Huang, K., et al., Notch3 signaling promotes colorectal tumor growth by enhancing immunosuppressive cells infiltration in the microenvironment. BMC Cancer, 2023. 23(1): p. 55. [CrossRef]
- Aster, J.C. and S.C. Blacklow, Targeting the Notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol, 2012. 30(19): p. 2418-20. [CrossRef]
- Chimento, A., et al., Notch Signaling in Breast Tumor Microenvironment as Mediator of Drug Resistance. Int J Mol Sci, 2022. 23(11). [CrossRef]
- Gallahan, D. and R. Callahan, The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene, 1997. 14(16): p. 1883-90. [CrossRef]
- Demitrack, E.S. and L.C. Samuelson, Notch as a Driver of Gastric Epithelial Cell Proliferation. Cell Mol Gastroenterol Hepatol, 2017. 3(3): p. 323-330.
- Gupta, S., P. Kumar, and B.C. Das, HPV: Molecular pathways and targets. Curr Probl Cancer, 2018. 42(2): p. 161-174. [CrossRef]
- Cook, N., et al., Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med, 2012. 209(3): p. 437-44. [CrossRef]
- Grochowski, C.M., K.M. Loomes, and N.B. Spinner, Jagged1 (JAG1): Structure, expression, and disease associations. Gene, 2015. 576(1 Pt 3): p. 381-4. [CrossRef]
- Grassi, E.S. and A. Pietras, Emerging Roles of DLK1 in the Stem Cell Niche and Cancer Stemness. J Histochem Cytochem, 2022. 70(1): p. 17-28. [CrossRef]
- Nueda, M.-L., et al., The proteins DLK1 and DLK2 modulate NOTCH1-dependent proliferation and oncogenic potential of human SK-MEL-2 melanoma cells. Biochimica Et Biophysica Acta-Molecular Cell Research, 2014. 1843(11): p. 2674-2684. [CrossRef]
- Nueda, M.L., et al., Different expression levels of DLK1 inversely modulate the oncogenic potential of human MDA-MB-231 breast cancer cells through inhibition of NOTCH1 signaling. FASEB J, 2017. [CrossRef]
- Naranjo, A.I., et al., Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo. Int J Mol Sci, 2022. 23(3). [CrossRef]
- Lundkvist, J. and J. Naslund, Gamma-secretase: a complex target for Alzheimer’s disease. Curr Opin Pharmacol, 2007. 7(1): p. 112-8.
- Moore, G., et al., Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells, 2020. 9(6). [CrossRef]
- Groth, C. and M.E. Fortini, Therapeutic approaches to modulating Notch signaling: current challenges and future prospects. Semin Cell Dev Biol, 2012. 23(4): p. 465-72. [CrossRef]
- Panelos, J., et al., Expression of Notch-1 and alteration of the E-cadherin/beta-catenin cell adhesion complex are observed in primary cutaneous neuroendocrine carcinoma (Merkel cell carcinoma). Mod Pathol, 2009. 22(7): p. 959-68. [CrossRef]
- Zhang, X., et al., The gamma-secretase complex: from structure to function. Front Cell Neurosci, 2014. 8: p. 427.
- Wolfe, M.S., Structure and Function of the gamma-Secretase Complex. Biochemistry, 2019. 58(27): p. 2953-2966.
- Kimberly, W.T. and M.S. Wolfe, Identity and function of gamma-secretase. J Neurosci Res, 2003. 74(3): p. 353-60.
- Hur, J.Y., gamma-Secretase in Alzheimer’s disease. Exp Mol Med, 2022. 54(4): p. 433-446.
- Mekala, S., G. Nelson, and Y.M. Li, Recent developments of small molecule gamma-secretase modulators for Alzheimer’s disease. RSC Med Chem, 2020. 11(9): p. 1003-1022.
- Song, C., et al., The critical role of gamma-secretase and its inhibitors in cancer and cancer therapeutics. Int J Biol Sci, 2023. 19(16): p. 5089-5103. [CrossRef]
- Lopez-Nieva, P., et al., More Insights on the Use of gamma-Secretase Inhibitors in Cancer Treatment. Oncologist, 2021. 26(2): p. e298-e305. [CrossRef]
- McCaw, T.R., et al., Gamma Secretase Inhibitors in Cancer: A Current Perspective on Clinical Performance. Oncologist, 2021. 26(4): p. e608-e621. [CrossRef]
- Ghanbari-Movahed, M., et al., Unlocking the Secrets of Cancer Stem Cells with gamma-Secretase Inhibitors: A Novel Anticancer Strategy. Molecules, 2021. 26(4). [CrossRef]
- Ran, Y., et al., gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med, 2017. 9(7): p. 950-966. [CrossRef]
- Page, M.J., et al., The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ, 2021. 372: p. n71.
- Avila, J.L. and J.L. Kissil, Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med, 2013. 19(5): p. 320-7. [CrossRef]
- Samore, W.R. and C.S. Gondi, Brief overview of selected approaches in targeting pancreatic adenocarcinoma. Expert Opin Investig Drugs, 2014. 23(6): p. 793-807. [CrossRef]
- Yabuuchi, S., et al., Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett, 2013. 335(1): p. 41-51.
- Mizuma, M., et al., The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther, 2012. 11(9): p. 1999-2009. [CrossRef]
- Palagani, V., et al., Combined inhibition of Notch and JAK/STAT is superior to monotherapies and impairs pancreatic cancer progression. Carcinogenesis, 2014. 35(4): p. 859-66. [CrossRef]
- Neumann, C.C.M., et al., Tumor-stromal cross-talk modulating the therapeutic response in pancreatic cancer. Hepatobiliary Pancreat Dis Int, 2018. 17(5): p. 461-472. [CrossRef]
- Gounder, M., et al., Nirogacestat, a gamma-Secretase Inhibitor for Desmoid Tumors. N Engl J Med, 2023. 388(10): p. 898-912. [CrossRef]
- Cook, N., et al., A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer, 2018. 118(6): p. 793-801. [CrossRef]
- De Jesus-Acosta, A., et al., A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs, 2014. 32(4): p. 739-45.
- Zhang, Y., et al., NOTCH1 Signaling Regulates Self-Renewal and Platinum Chemoresistance of Cancer Stem-like Cells in Human Non-Small Cell Lung Cancer. Cancer Res, 2017. 77(11): p. 3082-3091.
- Morgan, K.M., et al., Gamma Secretase Inhibition by BMS-906024 Enhances Efficacy of Paclitaxel in Lung Adenocarcinoma. Mol Cancer Ther, 2017. 16(12): p. 2759-2769. [CrossRef]
- Sosa Iglesias, V., et al., Synergistic Effects of NOTCH/gamma-Secretase Inhibition and Standard of Care Treatment Modalities in Non-small Cell Lung Cancer Cells. Front Oncol, 2018. 8: p. 460. [CrossRef]
- Maraver, A., et al., Therapeutic effect of gamma-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell, 2012. 22(2): p. 222-34. [CrossRef]
- Liu, Y.P., et al., Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res, 2013. 73(1): p. 406-16.
- Xie, M., et al., gamma Secretase inhibitor BMS-708163 reverses resistance to EGFR inhibitor via the PI3K/Akt pathway in lung cancer. J Cell Biochem, 2015. 116(6): p. 1019-27. [CrossRef]
- Takahashi, H., et al., Notch pathway regulates osimertinib drug-tolerant persistence in EGFR-mutated non-small-cell lung cancer. Cancer Sci, 2023. 114(4): p. 1635-1650.
- Pine, S.R., Rethinking Gamma-secretase Inhibitors for Treatment of Non-small-Cell Lung Cancer: Is Notch the Target? Clin Cancer Res, 2018. 24(24): p. 6136-6141.
- Mizugaki, H., et al., gamma-Secretase inhibitor enhances antitumour effect of radiation in Notch-expressing lung cancer. Br J Cancer, 2012. 106(12): p. 1953-9. [CrossRef]
- Sakakibara-Konishi, J., et al., Combined antitumor effect of gamma-secretase inhibitor and ABT-737 in Notch-expressing non-small cell lung cancer. Int J Clin Oncol, 2017. 22(2): p. 257-268. [CrossRef]
- He, F., et al., Synergistic Effect of Notch-3-Specific Inhibition and Paclitaxel in Non-Small Cell Lung Cancer (NSCLC) Cells Via Activation of The Intrinsic Apoptosis Pathway. Med Sci Monit, 2017. 23: p. 3760-3769. [CrossRef]
- Liang, S., et al., Multimodality Approaches to Treat Hypoxic Non-Small Cell Lung Cancer (NSCLC) Microenvironment. Genes Cancer, 2012. 3(2): p. 141-51. [CrossRef]
- Xiang, H., et al., Single-Cell Analysis Identifies NOTCH3-Mediated Interactions between Stromal Cells That Promote Microenvironment Remodeling and Invasion in Lung Adenocarcinoma. Cancer Res, 2024. 84(9): p. 1410-1425. [CrossRef]
- Paris, D., et al., Inhibition of angiogenesis and tumor growth by beta and gamma-secretase inhibitors. Eur J Pharmacol, 2005. 514(1): p. 1-15.
- Yang, X., et al., Evodiamine suppresses Notch3 signaling in lung tumorigenesis via direct binding to gamma-secretases. Phytomedicine, 2020. 68: p. 153176. [CrossRef]
- Witt, M., et al., On the Way to Accounting for Lung Modulation Effects in Particle Therapy of Lung Cancer Patients-A Review. Cancers (Basel), 2024. 16(21). [CrossRef]
- Demitrack, E.S., et al., NOTCH1 and NOTCH2 regulate epithelial cell proliferation in mouse and human gastric corpus. Am J Physiol Gastrointest Liver Physiol, 2016. 312(2): p. G133-G144. [CrossRef]
- Hong, K.J., et al., RECK inhibits stemness gene expression and tumorigenicity of gastric cancer cells by suppressing ADAM-mediated Notch1 activation. J Cell Physiol, 2014. 229(2): p. 191-201. [CrossRef]
- Li, L.C., et al., Gastric tumor-initiating CD44(+) cells and epithelial-mesenchymal transition are inhibited by gamma-secretase inhibitor DAPT. Oncol Lett, 2015. 10(5): p. 3293-3299. [CrossRef]
- Barat, S., et al., Gamma-Secretase Inhibitor IX (GSI) Impairs Concomitant Activation of Notch and wnt-beta-catenin Pathways in CD44+ Gastric Cancer Stem Cells. Stem Cells Transl Med, 2017. [CrossRef]
- Kim, S.J., et al., Activation of nuclear PTEN by inhibition of Notch signaling induces G2/M cell cycle arrest in gastric cancer. Oncogene, 2015. 35(2): p. 251-60. [CrossRef]
- Lee, H.W., et al., Targeting Notch signaling by gamma-secretase inhibitor I enhances the cytotoxic effect of 5-FU in gastric cancer. Clin Exp Metastasis, 2015. 32(6): p. 593-603. [CrossRef]
- Yao, J., et al., Combination treatment of PD98059 and DAPT in gastric cancer through induction of apoptosis and downregulation of WNT/beta-catenin. Cancer Biol Ther, 2013. 14(9): p. 833-9. [CrossRef]
- Kang, M., et al., Concurrent Treatment with Anti-DLL4 Enhances Antitumor and Proapoptotic Efficacy of a gamma-Secretase Inhibitor in Gastric Cancer. Transl Oncol, 2018. 11(3): p. 599-608. [CrossRef]
- Kumar, D., et al., Notch1-MAPK Signaling Axis Regulates CD133(+) Cancer Stem Cell-Mediated Melanoma Growth and Angiogenesis. J Invest Dermatol, 2016. 136(12): p. 2462-2474. [CrossRef]
- Zhu, G., et al., Combination with gamma-secretase inhibitor prolongs treatment efficacy of BRAF inhibitor in BRAF-mutated melanoma cells. Cancer Lett. 376(1): p. 43-52. [CrossRef]
- Porcelli, L., et al., BRAF(V600E;K601Q) metastatic melanoma patient-derived organoids and docking analysis to predict the response to targeted therapy. Pharmacol Res, 2022. 182: p. 106323. [CrossRef]
- Krepler, C., et al., Targeting Notch enhances the efficacy of ERK inhibitors in BRAF-V600E melanoma. Oncotarget, 2016. 7(44): p. 71211-71222. [CrossRef]
- Mukherjee, N., et al., Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget, 2016. 7(51): p. 84594-84607. [CrossRef]
- Keyghobadi, F., et al., Long-Term Inhibition of Notch in A-375 Melanoma Cells Enhances Tumor Growth Through the Enhancement of AXIN1, CSNK2A3, and CEBPA2 as Intermediate Genes in Wnt and Notch Pathways. Front Oncol, 2020. 10: p. 531. [CrossRef]
- Skarmoutsou, E., et al., FOXP3 expression is modulated by TGF-beta1/NOTCH1 pathway in human melanoma. Int J Mol Med, 2018. 42(1): p. 392-404.
- Tolcher, A.W., et al., Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol, 2012. 30(19): p. 2348-53. [CrossRef]
- Lee, S.M., et al., Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer, 2015. 121(3): p. 432-440. [CrossRef]
- Thippu Jayaprakash, K., et al., In Vitro Evaluation of Notch Inhibition to Enhance Efficacy of Radiation Therapy in Melanoma. Adv Radiat Oncol, 2021. 6(2): p. 100622. [CrossRef]
- Azzam, D.J., et al., Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in gamma-secretase inhibitor drug responses. EMBO Mol Med, 2013. 5(10): p. 1502-22. [CrossRef]
- Li, W., et al., Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol Rep, 2019. 41(1): p. 437-446. [CrossRef]
- Wan, X., et al., pH sensitive peptide functionalized nanoparticles for co-delivery of erlotinib and DAPT to restrict the progress of triple negative breast cancer. Drug Deliv, 2019. 26(1): p. 470-480. [CrossRef]
- Stoeck, A., et al., Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov, 2014. 4(10): p. 1154-67.
- Zhang, S., et al., Targeting Met and Notch in the Lfng-deficient, Met-amplified triple-negative breast cancer. Cancer Biol Ther, 2014. 15(5): p. 633-42. [CrossRef]
- Li, Z.L., et al., Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int J Clin Exp Pathol, 2015. 8(5): p. 4378-87.
- Paroni, G., et al., Retinoic Acid Sensitivity of Triple-Negative Breast Cancer Cells Characterized by Constitutive Activation of the notch1 Pathway: The Role of Rarbeta. Cancers (Basel), 2020. 12(10). [CrossRef]
- Sen, P. and S.S. Ghosh, gamma-Secretase Inhibitor Potentiates the Activity of Suberoylanilide Hydroxamic Acid by Inhibiting Its Ability to Induce Epithelial to Mesenchymal Transition and Stemness via Notch Pathway Activation in Triple-Negative Breast Cancer Cells. ACS Pharmacol Transl Sci, 2023. 6(10): p. 1396-1415. [CrossRef]
- Sen, P., T. Kandasamy, and S.S. Ghosh, Multi-targeting TACE/ADAM17 and gamma-secretase of notch signalling pathway in TNBC via drug repurposing approach using Lomitapide. Cell Signal, 2023. 102: p. 110529. [CrossRef]
- Hossain, F., et al., Sulindac sulfide as a non-immune suppressive gamma-secretase modulator to target triple-negative breast cancer. Front Immunol, 2023. 14: p. 1244159. [CrossRef]
- Vigolo, M., et al., The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers (Basel), 2023. 15(15). [CrossRef]
- Fournier, M., et al., Reciprocal inhibition of NOTCH and SOX2 shapes tumor cell plasticity and therapeutic escape in triple-negative breast cancer. EMBO Mol Med, 2024. 16(12): p. 3184-3217. [CrossRef]
- Schott, A.F., et al., Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res, 2013. 19(6): p. 1512-24.
- Locatelli, M.A., et al., Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget, 2017. 8(2): p. 2320-2328. [CrossRef]
- Sardesai, S., et al., A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Invest New Drugs, 2020. 38(5): p. 1400-1410. [CrossRef]
- Means-Powell, J.A., et al., A Phase Ib Dose Escalation Trial of RO4929097 (a gamma-secretase inhibitor) in Combination with Exemestane in Patients with ER + Metastatic Breast Cancer (MBC). Clin Breast Cancer, 2022. 22(2): p. 103-114. [CrossRef]
- Mukherjee, A., et al., Development and Application of a Novel Model System to Study “Active” and “Passive” Tumor Targeting. Mol Cancer Ther, 2016. 15(10): p. 2541-2550.
- Sen, P. and S.S. Ghosh, The Intricate Notch Signaling Dynamics in Therapeutic Realms of Cancer. ACS Pharmacol Transl Sci, 2023. 6(5): p. 651-670. [CrossRef]
- Li, S.Y., et al., Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J Control Release, 2015. 205: p. 7-14. [CrossRef]
- Zhou, Y., et al., DT7 peptide-modified lecithin nanoparticles co-loaded with gamma-secretase inhibitor and dexamethasone efficiently inhibit T-cell acute lymphoblastic leukemia and reduce gastrointestinal toxicity. Cancer Lett, 2022. 533: p. 215608. [CrossRef]
- Wang, K.C., T. Zheng, and B.P. Hubbard, CRISPR/Cas technologies for cancer drug discovery and treatment. Trends Pharmacol Sci, 2025. 46(5): p. 437-452. [CrossRef]
- Loganathan, S.K. and D. Schramek, In vivo CRISPR screens reveal potent driver mutations in head and neck cancers. Mol Cell Oncol, 2020. 7(4): p. 1758541. [CrossRef]
- Tedder, B. and M. Bhutani, Resistance Mechanisms to BCMA Targeting Bispecific Antibodies and CAR T-Cell Therapies in Multiple Myeloma. Cells, 2025. 14(14). [CrossRef]
- Cowan, A.J., et al., gamma-Secretase inhibitor in combination with BCMA chimeric antigen receptor T-cell immunotherapy for individuals with relapsed or refractory multiple myeloma: a phase 1, first-in-human trial. Lancet Oncol, 2023. 24(7): p. 811-822. [CrossRef]
- Fu, W., et al., Bispecific antibodies targeting EGFR/Notch enhance the response to talazoparib by decreasing tumour-initiating cell frequency. Theranostics, 2023. 13(11): p. 3641-3654. [CrossRef]
- Feng, M., et al., Inhibition of gamma-secretase/Notch pathway as a potential therapy for reversing cancer drug resistance. Biochem Pharmacol, 2024. 220: p. 115991. [CrossRef]
- Kalantari, E., et al., Effect of DAPT, a gamma secretase inhibitor, on tumor angiogenesis in control mice. Adv Biomed Res, 2014. 2: p. 83. [CrossRef]
- Tansir, G., S. Rastogi, and M.M. Gounder, Repurposing nirogacestat, a gamma secretase enzyme inhibitor in desmoid tumors. Future Oncol, 2025. 21(23): p. 2985-2993. [CrossRef]
- Federman, N., Molecular pathogenesis of desmoid tumor and the role of gamma-secretase inhibition. NPJ Precis Oncol, 2022. 6(1): p. 62. [CrossRef]
- Zheng, C., et al., The Notch signaling pathway in desmoid tumor: Recent advances and the therapeutic prospects. Biochim Biophys Acta Mol Basis Dis, 2024. 1870(1): p. 166907. [CrossRef]
- He, W., et al., High tumor levels of IL6 and IL8 abrogate preclinical efficacy of the gamma-secretase inhibitor, RO4929097. Mol Oncol, 2011. 5(3): p. 292-301. [CrossRef]
- Cavalieri, S., et al., Immune checkpoint inhibitors and Carbon iON radiotherapy In solid Cancers with stable disease (ICONIC). Future Oncol, 2023. 19(3): p. 193-203. [CrossRef]
Figure 1.
Absolute numbers of cancer-related deaths worldwide in 2022, for males and females and all age groups [1]. Data analyzed by authors.
Figure 1.
Absolute numbers of cancer-related deaths worldwide in 2022, for males and females and all age groups [1]. Data analyzed by authors.
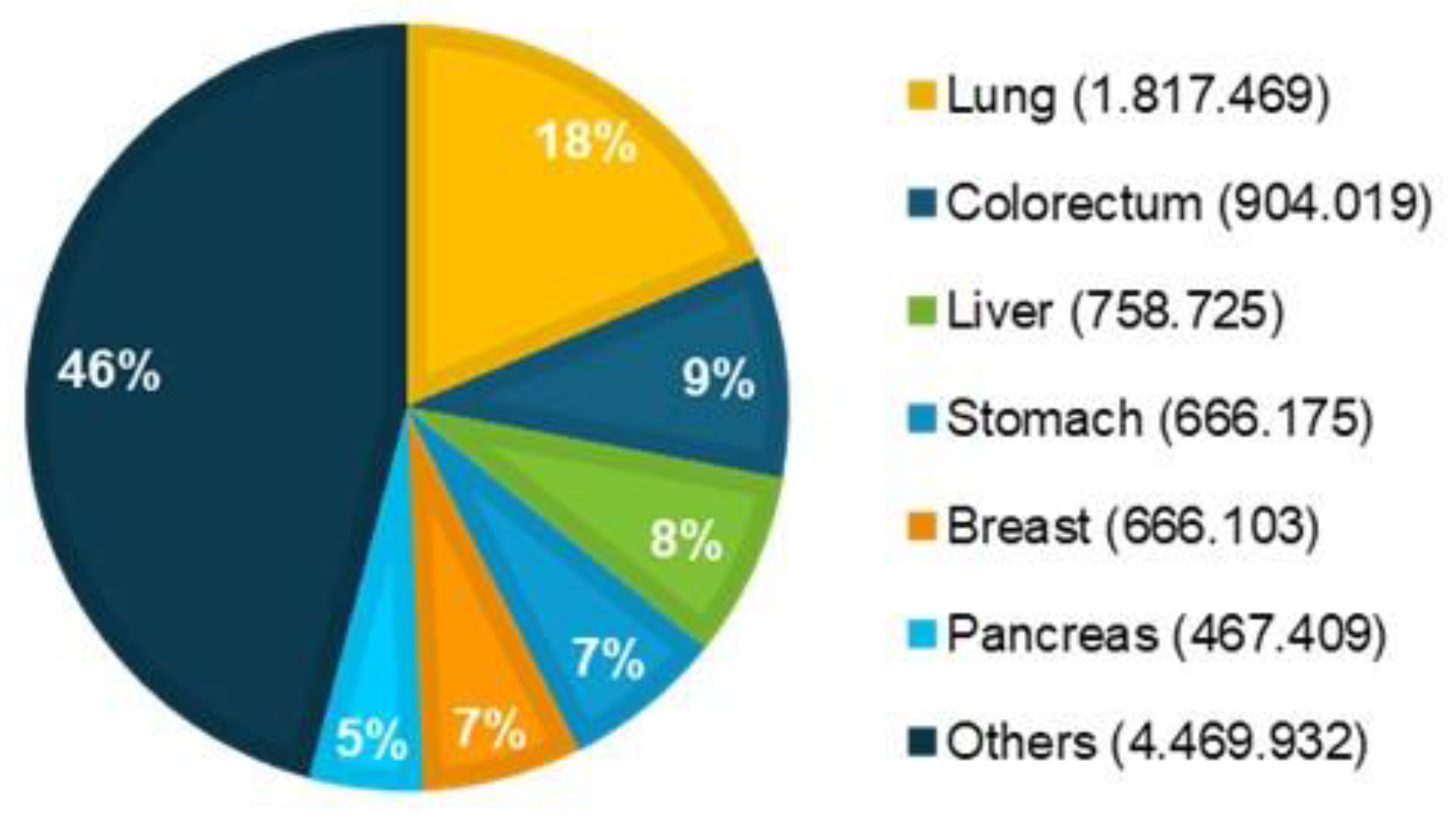
Figure 2.
Structure of NOTCH receptors in mammals. The four NOTCH receptors differ primarily in the number of EGF-like repeats, with NOTCH1 and NOTCH2 containing the highest number (36 and 35 repeats, respectively). The EGF-like repeats 11–12 of NOTCH receptors interact with the DSL (Delta/Serrate/LAG2) domain of canonical activating ligands. The Negative Regulatory Region (NRR) consists of three LNR repeats and a heterodimerization (HD) domain. The NOTCH intracellular domain (NICD) includes the RAM domain, ankyrin repeats (ANK), nuclear localization signals (NLS), and the PEST domain. The S2 (Ala1710- Val1711) and S3 (Gly1753-Val1754) sites represent the proteolytic cleavage points involved in receptor processing and activation at the plasma membrane upon ligand interaction.
Figure 2.
Structure of NOTCH receptors in mammals. The four NOTCH receptors differ primarily in the number of EGF-like repeats, with NOTCH1 and NOTCH2 containing the highest number (36 and 35 repeats, respectively). The EGF-like repeats 11–12 of NOTCH receptors interact with the DSL (Delta/Serrate/LAG2) domain of canonical activating ligands. The Negative Regulatory Region (NRR) consists of three LNR repeats and a heterodimerization (HD) domain. The NOTCH intracellular domain (NICD) includes the RAM domain, ankyrin repeats (ANK), nuclear localization signals (NLS), and the PEST domain. The S2 (Ala1710- Val1711) and S3 (Gly1753-Val1754) sites represent the proteolytic cleavage points involved in receptor processing and activation at the plasma membrane upon ligand interaction.
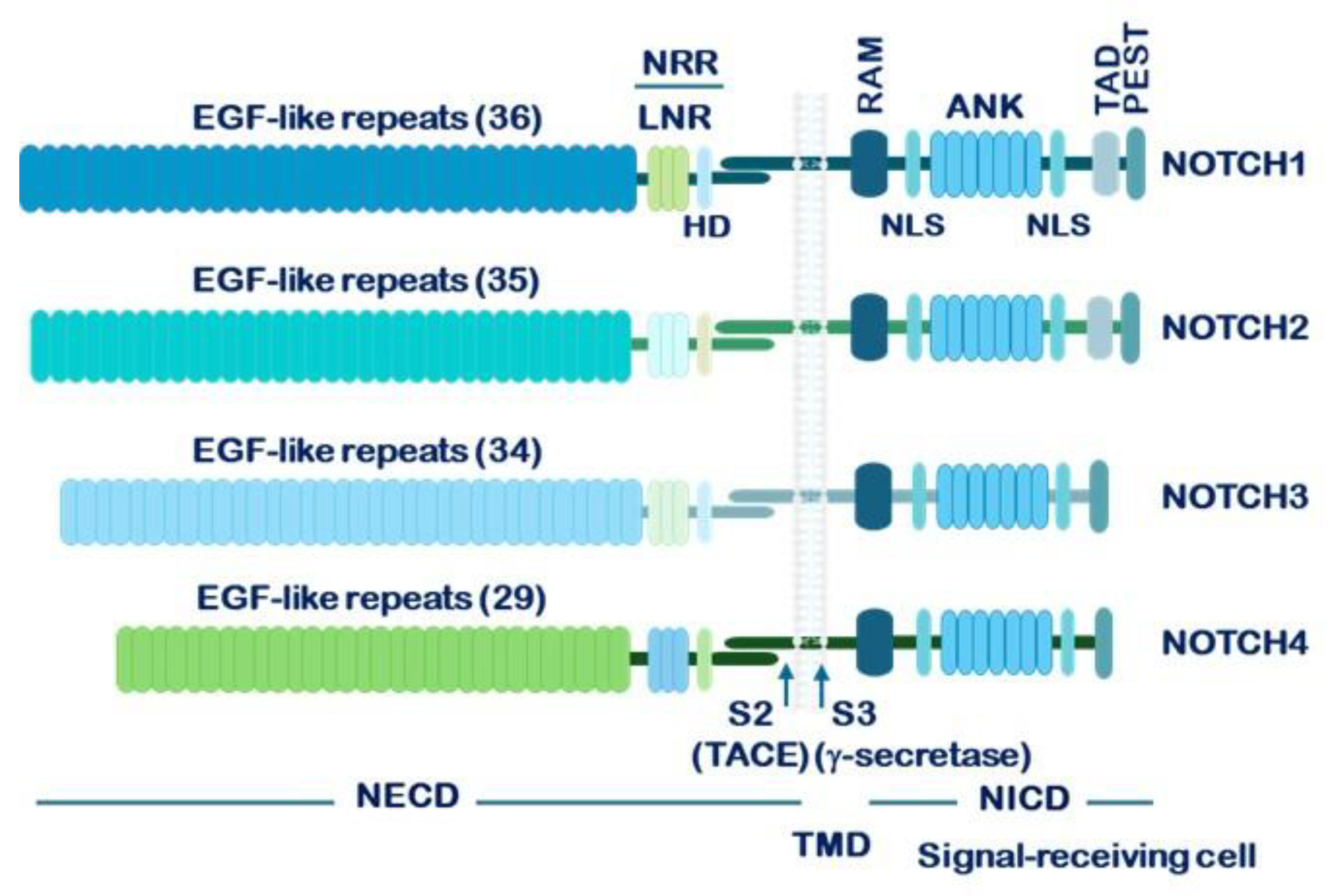
Figure 3.
Canonical NOTCH receptors signaling pathway. This figure depicts the sequential proteolytic processing of a NOTCH receptor at cleavage sites S2 and S3–S4, culminating in the release of the active form, NICD (NOTCH intracellular domain). Once released, NICD translocates to the nucleus, where it interacts with transcriptional regulators to activate gene expression. Abbreviations: Co-R: co-repressor; Co-A: co-activator; CSL: CSL/RBPJκ transcription factor (CBF1/Suppressor of hairless/Lag1/Recombination signal binding protein-Jκ); MAML: Mastermind-like proteins.
Figure 3.
Canonical NOTCH receptors signaling pathway. This figure depicts the sequential proteolytic processing of a NOTCH receptor at cleavage sites S2 and S3–S4, culminating in the release of the active form, NICD (NOTCH intracellular domain). Once released, NICD translocates to the nucleus, where it interacts with transcriptional regulators to activate gene expression. Abbreviations: Co-R: co-repressor; Co-A: co-activator; CSL: CSL/RBPJκ transcription factor (CBF1/Suppressor of hairless/Lag1/Recombination signal binding protein-Jκ); MAML: Mastermind-like proteins.
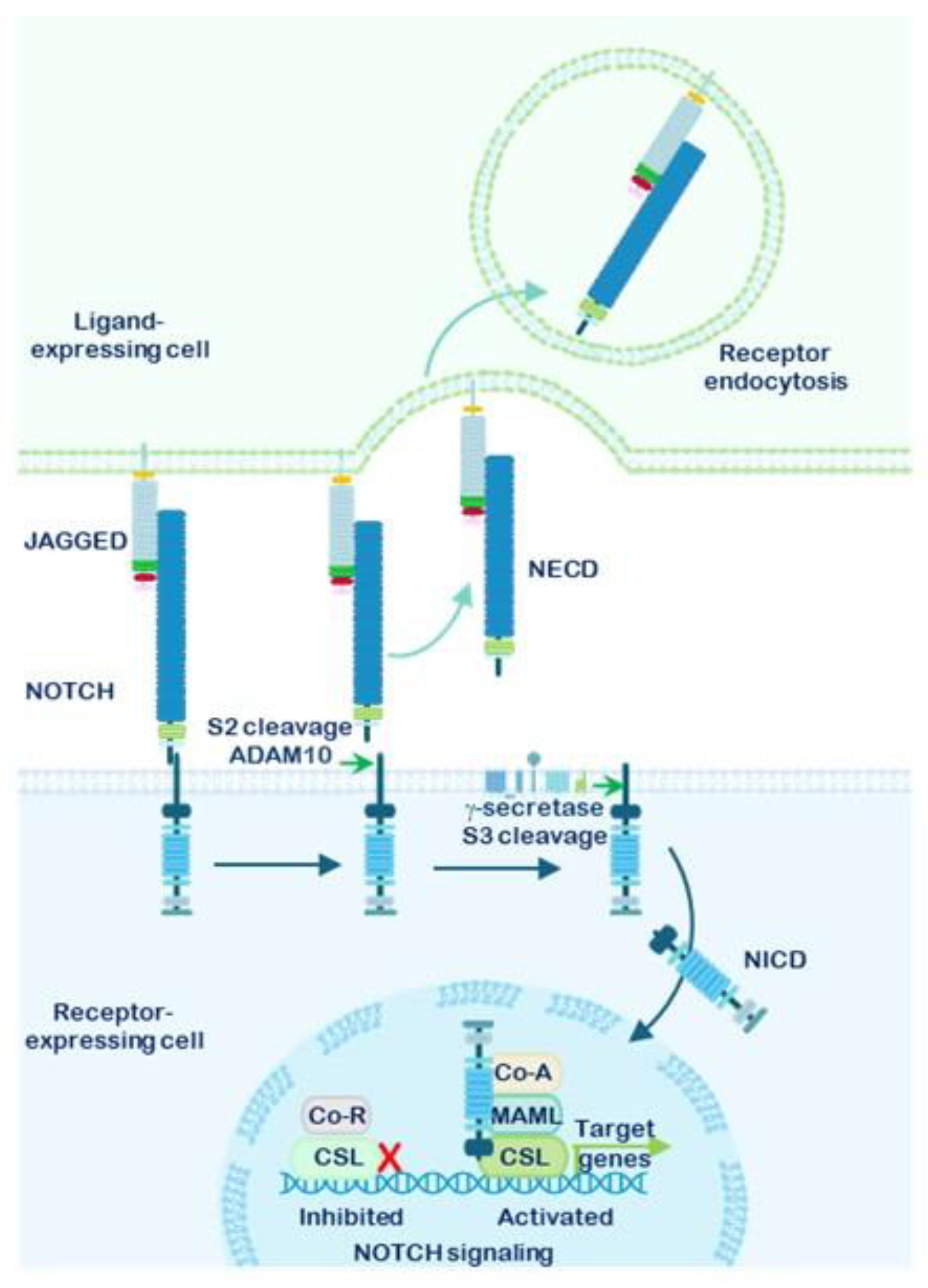
Figure 4.
NOTCH receptors in human cancer. Various cancer types are classified based on whether NOTCH functions predominantly as an oncogene or as a tumor suppressor. Analysis made by authors.
Figure 4.
NOTCH receptors in human cancer. Various cancer types are classified based on whether NOTCH functions predominantly as an oncogene or as a tumor suppressor. Analysis made by authors.
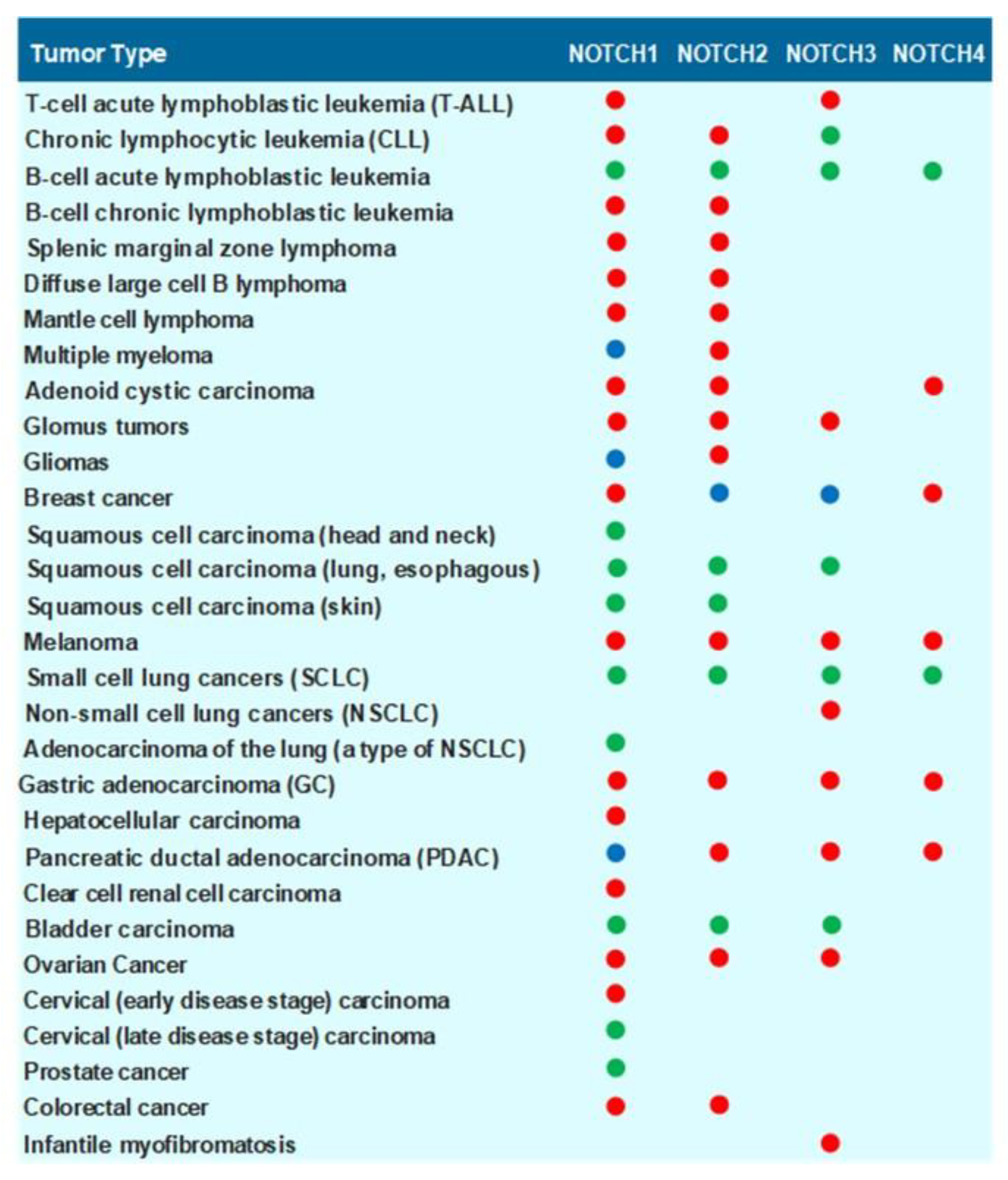
Figure 5.
Strategies for Inhibiting NOTCH Receptor Signaling. Figure 5 illustrates several therapeutic strategies designed to modulate NOTCH signaling. These include immunological approaches using antibodies that target NOTCH receptors and their ligands, chemical inhibition of ADAM proteases and the γ-secretase complex, and disruption of transcriptional regulators such as CSL (CBF1/Suppressor of Hairless/LAG-1, also known as RBP-Jκ) and Mastermind-like proteins. Collectively, these interventions aim to suppress the transcription of NOTCH target genes.
Figure 5.
Strategies for Inhibiting NOTCH Receptor Signaling. Figure 5 illustrates several therapeutic strategies designed to modulate NOTCH signaling. These include immunological approaches using antibodies that target NOTCH receptors and their ligands, chemical inhibition of ADAM proteases and the γ-secretase complex, and disruption of transcriptional regulators such as CSL (CBF1/Suppressor of Hairless/LAG-1, also known as RBP-Jκ) and Mastermind-like proteins. Collectively, these interventions aim to suppress the transcription of NOTCH target genes.
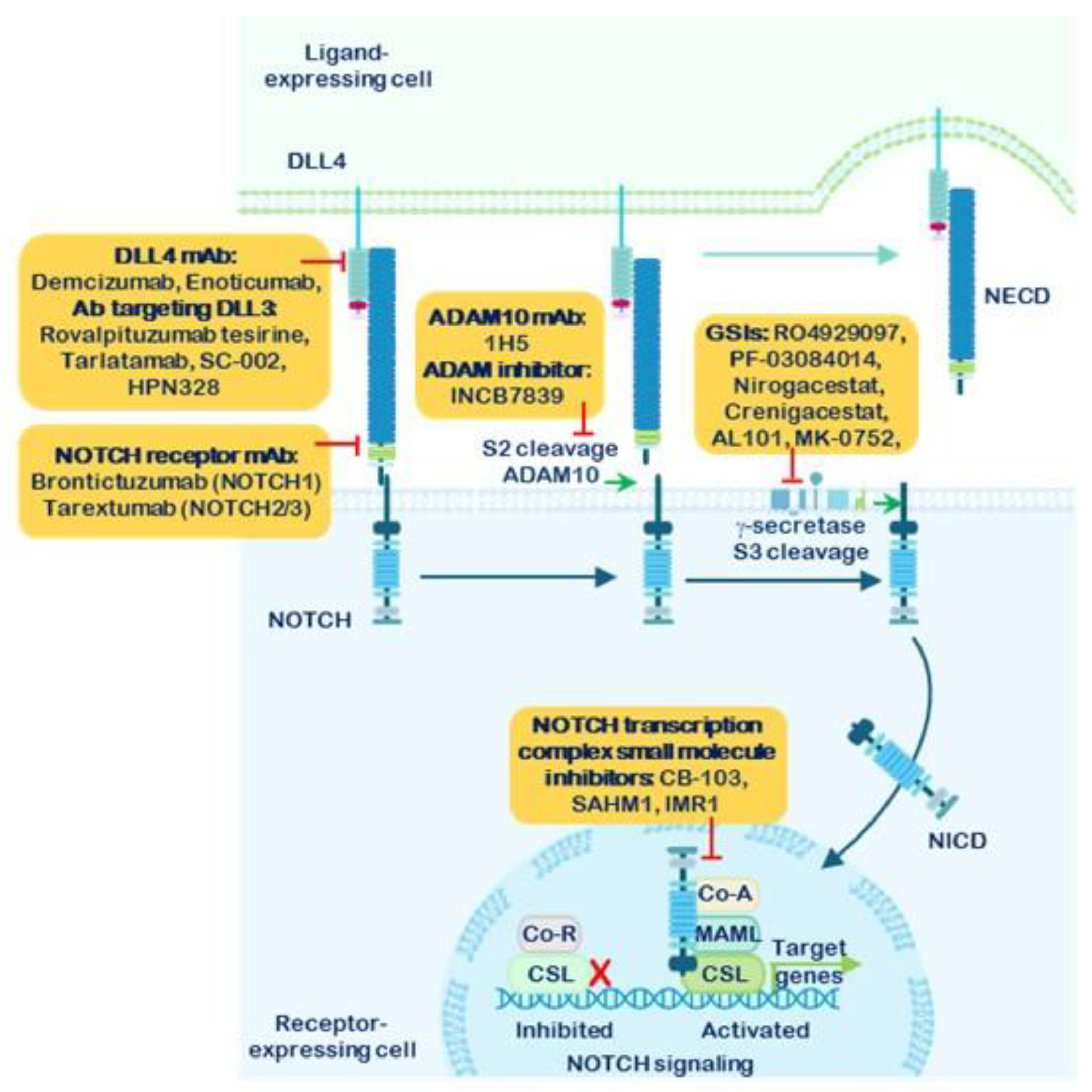
Figure 6.
Schematic representation of the γ-secretase complex (GSC). This diagram illustrates the structural organization of the γ-secretase complex, with each component traversing the membrane multiple times. Presenilin, the catalytic core of the complex, is depicted as comprising two subunits. Each subunit contains a critical aspartate residue (indicated by stars), which is essential for the proteolytic activity of the enzyme.
Figure 6.
Schematic representation of the γ-secretase complex (GSC). This diagram illustrates the structural organization of the γ-secretase complex, with each component traversing the membrane multiple times. Presenilin, the catalytic core of the complex, is depicted as comprising two subunits. Each subunit contains a critical aspartate residue (indicated by stars), which is essential for the proteolytic activity of the enzyme.
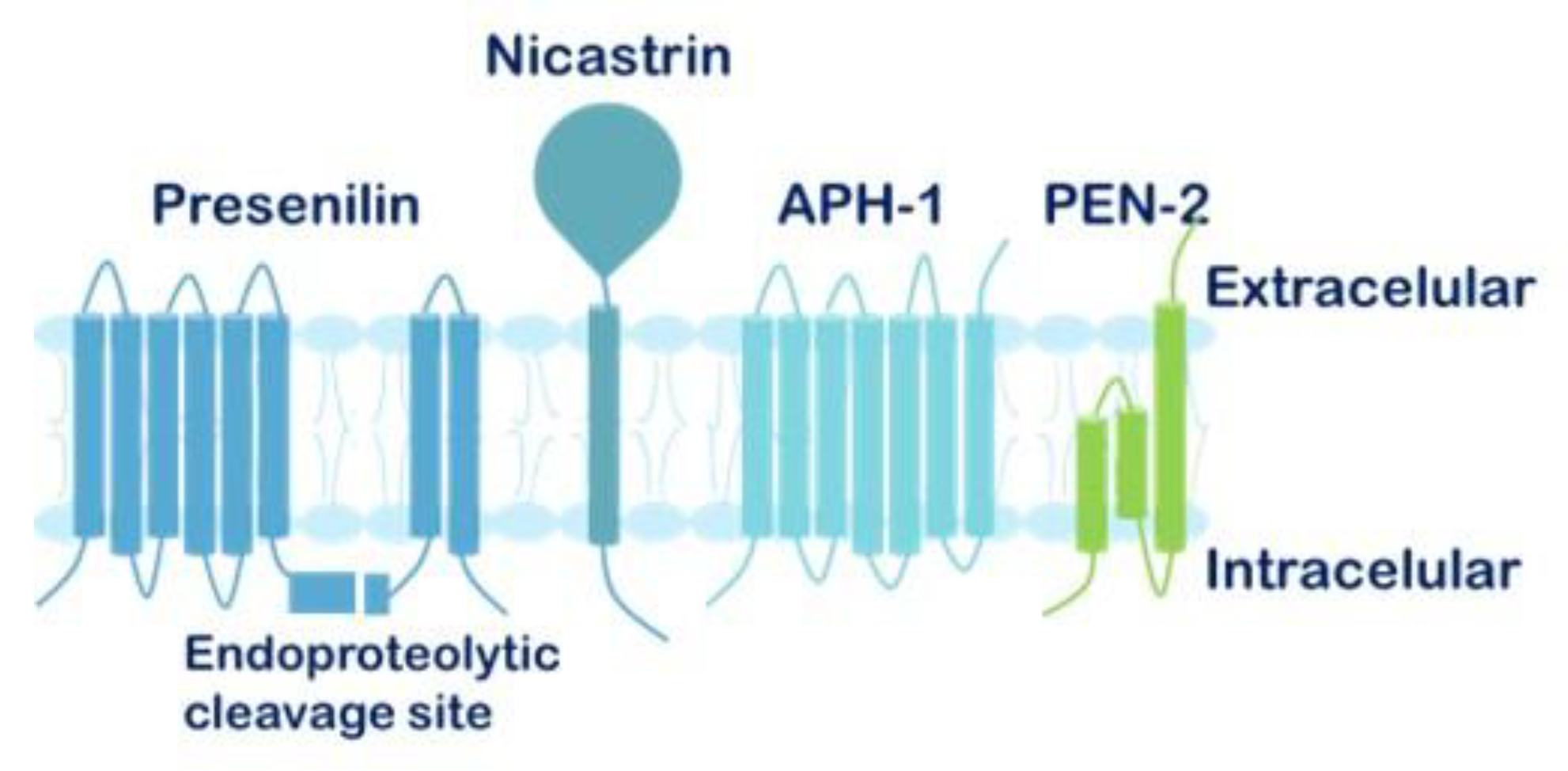
Figure 7.
Flow diagram of the systematic review. This diagram illustrates the database search process, the number of publications identified, screened, and the final full-text articles included in the systematic review. The review evaluated the main therapeutic advances achieved with GSIs in vitro, in vivo, and in clinical trials across five highly aggressive cancers driven by oncogenic NOTCH signaling: Pancreatic Adenocarcinoma (PDAC), Triple-Negative Breast Cancer (TNBC), Non-Small Cell Lung Cancer (NSCLC), and Gastric Cancer (GC).
Figure 7.
Flow diagram of the systematic review. This diagram illustrates the database search process, the number of publications identified, screened, and the final full-text articles included in the systematic review. The review evaluated the main therapeutic advances achieved with GSIs in vitro, in vivo, and in clinical trials across five highly aggressive cancers driven by oncogenic NOTCH signaling: Pancreatic Adenocarcinoma (PDAC), Triple-Negative Breast Cancer (TNBC), Non-Small Cell Lung Cancer (NSCLC), and Gastric Cancer (GC).
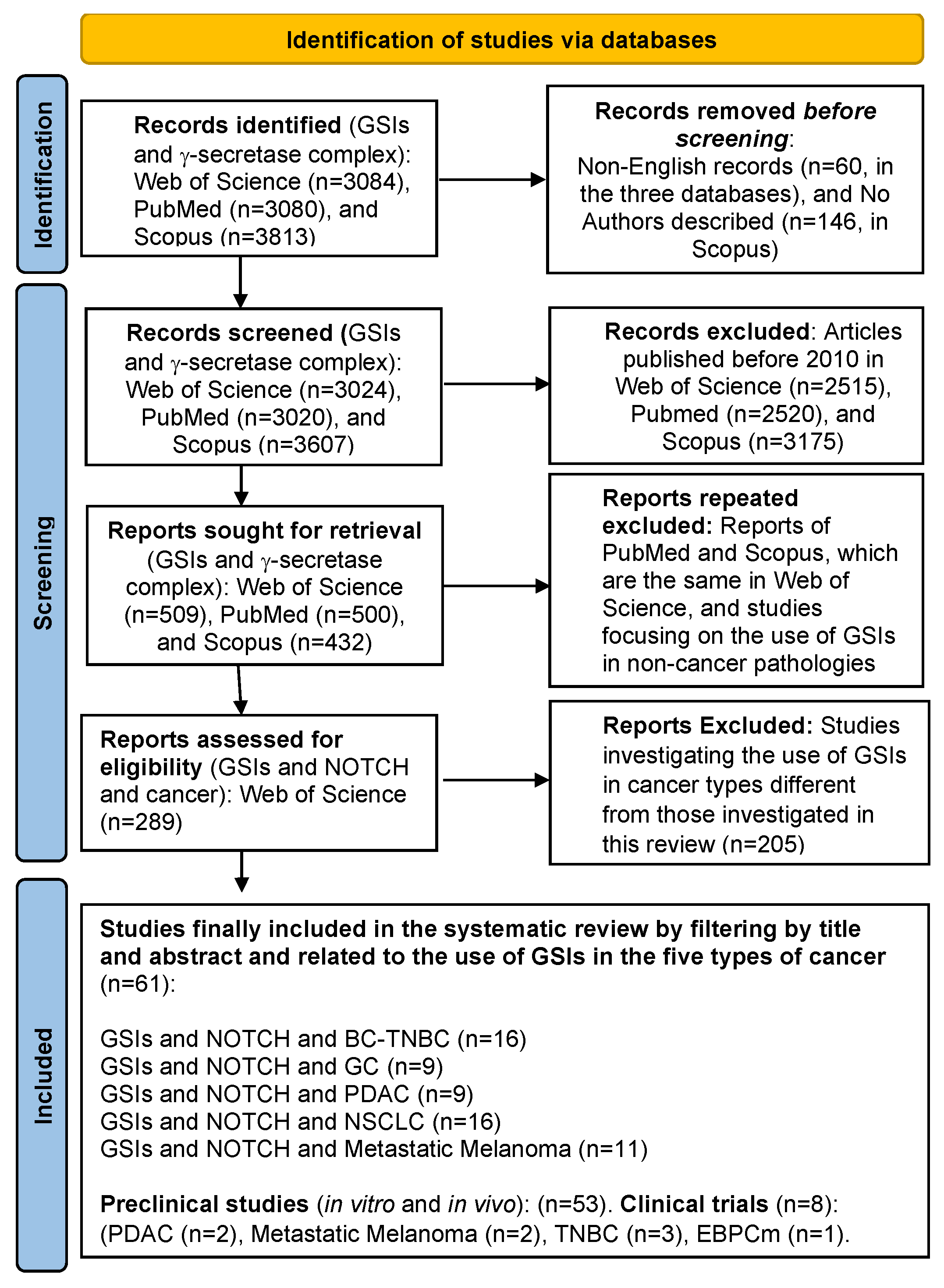

Table 1.
Summary of the effect of different GSIs in vitro studies and xenograft mice in the cancer types under study in this review. PDAC: pancreatic ductal adenocarcinoma. TNBC: triple-negative breast cancer. GC: gastric carcinoma. NSCLC: non-small cell lung cancer.
Table 1.
Summary of the effect of different GSIs in vitro studies and xenograft mice in the cancer types under study in this review. PDAC: pancreatic ductal adenocarcinoma. TNBC: triple-negative breast cancer. GC: gastric carcinoma. NSCLC: non-small cell lung cancer.
| GSI | Type of cancer | Type of study | Main results | Reference | |
|---|---|---|---|---|---|
| MRK003 | PDAC | In vivo (xenograft) +/- gemcitabine | The combination blocked tumor progression | [63], [87] | |
| TNBC | In vitro e in vivo + placitaxel |
Greater antitumor activity of the combination in cells with higher NICD levels | [130] | ||
| NSCLC | In vivo + erlotinib | Induced cell death in hypoxic tumors and decreased metastasis to the liver and brain. Prolonged median survival in mice. | [104] | ||
| Mesenchymal cells and 9 treatment-naïve patients | Reduced collagen production and suppressed invasive behavior. | [105] | |||
| GSI-IX | PDAC | In vivo (xenograft) + AG-490 |
Mice treated with the combination showed no visible tumors | [88] | |
| In vitro and in a xenograft mouse model | Reduced the growth of pancreatic tumor-initiating CD44+/EpCAM+ cells | [80] | |||
| GC | In vitro, in CD44+ cells | Smaller tumor spheres and increased apoptosis | [112] | ||
| In vivo (xenograft) | Reduced tumor growth and increased necrosis | ||||
| NSCLC | In vitro + paclitaxel | Synergistic antitumor effect by modulating the intrinsic apoptosis pathway and enhancing cell death. Reduced NOTCH3–induced chemoresistance in a concentration-dependent manner | [103] | ||
| Metastatic melanoma | In vitro | GSI decreased CD133+ cells (MSCs) | [117] | ||
| GSI-X | |||||
| PF-03084014 (Nirogacestat) | PDAC | In vivo (xenograft) +/- gemcitabine |
Only in combination did it show antiproliferative activity and reduce cancer stem cells | [86] | |
| Metastatic melanoma | In vitro + MEKi | The combination was more effective in stopping proliferation and migration | [119] | ||
| DAPT | PDAC | In vitro | CAF monocultures hardly responded to DAPT which suggested that CAFs are more resistant to standard chemo-treatments than the epithelial cancer cells. High levels of IL-6 were also associated with a reduced response to therapy | [89] | |
| NSCLC | In vitro + cisplatin | Decrease in the appearance of CD133+, ALDH+ LCSC cells, with lower resistance to cisplatin | [97] | ||
| KrasG12V-driven NSCLC | GSI treatment upregulated DUSP1, leading to reduced phospho-ERK levels | [96] | |||
| In vitro and lung adenocarcinoma tumors xenotransplanted into nude mice. | Reduced endothelial cell proliferation, suppressed the formation of capillary structures, opposed the sprouting of microvessel outgrowths and potently inhibit the growth and vascularization | [106] | |||
| GC | In vitro | Inhibited the formation of GCSC-rich spheres by 25% | [110] | ||
| In vitro, in CD44+ and CD44- cells | CD44+ cells, behaving as GCSCs, showed greater antitumor response to GSI. Enhanced sensitivity to 5-FU | [111] | |||
| In vivo (xenograft) | Significant inhibition of tumor growth and EMT | ||||
| In vitro +/- PD98059 In vivo (xenograft) +/- PD98059 |
Reduced tumor growth and increased apoptosis in combination | [115] | |||
| In vitro +/- anti-DLL4 | Significant increase in apoptosis and reduced invasion and tumor size | [116] | |||
| Metastatic melanoma | In vivo (xenograft) +/- BRAFi |
Reversal of melanoma cell resistance to BRAFi | [118] | ||
| DAPT | Metastatic melanoma | In vitro +/- DLK1 and/or DLK2 levels |
Dose-dependent effect of DAPT: decreased proliferation at high doses, increased at low doses. The combination reduced cell proliferation | [66] | |
| In vitro e In vivo (Xenograft) | Long-term use of DAPT increased tumor growth | [122] | |||
| TNBC | In vitro, in BCSCs cells | Reduced proliferation and increased apoptosis | [128] | ||
| In vivo (xenograft) | Delay in tumor formation and reduced subsequent growth | ||||
| Nanoparticles carrying the γ-secretase inhibitor DAPT In vivo (xenograft) + Erlotinib + Director peptide |
The nanoparticle reduced tumor growth and cell migration | [129] | |||
| In vitro + ATRA | The combination was more effective in inhibiting tumor growth | [133] | |||
| GSI-34 | NSCLC | In vivo (xenograft) with CD166+Lin- LCSC cells +/- cisplatin |
CD166+Lin- showed intrinsic resistance to cisplatin, which was reversed with GSI. The combination effectively reduced tumor size | [93] | |
| BMS-708163 | In vitro, in NSCLC-gefitinb resistant cells + gefitinib |
High doses of GSI reversed resistance to gefitinib and formed smaller colonies | [98] | ||
| In vivo (xenograft) + gefitinib |
The combination produced considerable inhibition of tumor growth | ||||
| BMS-906024 | In vitro, in NSCLC cells + RT +/- placitaxel y crizotinib |
Monotherapy + RT did not show significant reduction. It was observed with the combination | [95] | ||
| In vivo (xenograft) + placitaxel |
The combination enhanced the cytotoxic effect of paclitaxel | [94,100] | |||
| GSI-XX | In vivo (xenograft) + RT |
The combination caused a significant delay in tumor growth | [101] | ||
| In vitro and in vivo experiments + osimertinib | Impaired drug-tolerant persistence, suppressed phospho-ERK, and enhanced DUSP1 expression. | [99] | |||
| In vitro and in vivo +ABT-737 |
Treatment with either agent and in combination inhibit cell proliferation in a dose-dependent manner and regulated the expression of apoptosis proteins. | [102] | |||
| GSI-I | In vitro + RT | Higher level of apoptosis than isolated RT | [101] | ||
| GC | In vitro and in vivo (xenograft) + placitaxel or 5-FU |
Increased activity of PTEN, a tumor suppressor gene. Both combinations were more effective than monotherapy |
[113], [114] | ||
| Metastatic melanoma | In vivo (xenofraft) and in vitro + BCL2i |
The combination was more effective than monotherapy | [121] | ||
| RO4929097 | In vitro + ERKi | Sensitization to ERKi in cell lines that did not respond to it in monotherapy | [120] | ||
| In vivo (xenograft) +ERKi | The combination was more effective than monotherapy | ||||
| In vitro + RT | Synergism at low doses in combination | [126] | |||
| TNBC | In vitro, in CD24 low and CD24- (BCSCs) cells | Inhibition of CD24low sphere growth | [127] | ||
| In vivo (xenograft) with CD24 low and CD24- (BCSCs) cells | Halted tumor growth and metastasis in CD24 low models | ||||
| MK-0752 | In vitro. Different levels of NOTCH expression + METi |
The combination showed synergism in halting cell growth | [131] | ||
| In vivo (xenograft) | Injecting BCSC cells treated with GSI did not reproduce the tumor | [139] | |||
| LY411575 | In vitro + SAHA | SAHA in monotherapy was seen to promote EMT. The combination reduced EMT and increased apoptosis | [134] | ||
|
LY3039478 (Crenigacestat) |
TNBC | TNBC xenografts + Paclitaxel + Dasatinib | Tumor growth and metastasis reduction | [138] | |
|
OTHERS (Evodiamine) |
NSCLC | In vitro | Not a GSI but behaves like one. It reduced cell proliferation and metastasis | [107] | |
|
OTHERS (NSAID sulindac (SS)) |
TNBC | In vitro, in vivo and ex vivo | Significantly inhibited nanosphere growth in all human and murine TNBC models. Eliminated NOTCH1 protein expression in tumors |
[136] | |
|
OTHERS (CB103, a pan-Notch inhibitor) |
Endocrine-resistant BC xenografts | When combined with SERDs or CDK inhibitors in endocrine-resistant recurrent breast cancers, and with taxane-based chemotherapy in TNBC, CB-103 produced synergistic effects boosting paclitaxel’s impact | [137] | ||
|
OTHERS (Lomitapide) |
In vitro | Multi-targeting TACE/ADAM17 and gamma-secretase complex of NOTCH signaling pathway | [135] |
Table 2.
Summary of the different clinical trials conducted with GSIs in the cancer types under study in this review. PDAC: pancreatic ductal adenocarcinoma. TNBC: triple-negative breast cancer. Table created by the author. EPBCm: metastatic estrogen receptor-positive breast cancer. GC: gastric cancer.
Table 2.
Summary of the different clinical trials conducted with GSIs in the cancer types under study in this review. PDAC: pancreatic ductal adenocarcinoma. TNBC: triple-negative breast cancer. Table created by the author. EPBCm: metastatic estrogen receptor-positive breast cancer. GC: gastric cancer.
| Type of cancer | GSI | Phase | Results | Reference |
|---|---|---|---|---|
| PDAC | MK-0752 + gemcitabine |
I | 14 out of 44 patients reached a stable condition, in both monotherapy and combination therapy. Gastrointestinal disorders and anemia were observed. ClinicalTrials.gov identifier: NCT01098344. | [91] |
| RO4929097 | II | The trial could not be completed because GSI synthesis was discontinued. Clinicaltrials.gov identifier NCT01232829 | [92] | |
| Metastatic melanoma | RO4929097 |
I | In two groups of 110 patients, 33% and 41% reached a stable condition. Hypophosphatemia was noted. Cancer Therapy Evaluation Program (CTEP) | [124] |
| II | Of 32 evaluated patients, 1 had a partial response and 8 reached a stable condition. Hypophosphatemia was also observed, ClinicalTrials.gov identifier NCT01120275 | [125] | ||
| TNBC | MK-0752 + docetaxel |
I | Among 24 patients, 11 had a partial response, 9 reached a stable condition, and 3 showed tumor progression. There was one case of severe pneumonitis. ClinicalTrials.gov identifier: NCT00645333 | [139] |
| PF-03084014 + docetaxel |
I | 29 women showed limited treatment efficacy, with severe hematologic and infectious reactions. ClinicalTrials.gov identifier NCT01876251 | [140] | |
| RO4929097 + placitaxel + carboplatin |
I | Of 14 evaluated patients, 5 had a partial response, 4 reached a stable condition, and 5 had residual disease. Neutropenia was reported (http://ctep.cancer.gov/protocol). | [141] | |
| EPBCm | RO4929097 + exemestane |
Ib | Among 14 evaluated patients, 7 had a partial response and 7 reached a stable condition, ClinicalTrials.gov identifier NCT01149356 | [142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



