Submitted:
06 November 2025
Posted:
07 November 2025
You are already at the latest version
Abstract
In all arrestins the gate loop is the central part of the lariat loop, which has an unusual shape and participates in maintaining the basal conformation. Gate loop supplies two out of five charges that constitute a stabilizing intramolecular interaction, the polar core between the two domains. To elucidate the functional role of individual gate loop residues we performed comprehensive site-directed mutagenesis and tested the effects of mutations on arrestin-1 binding to its preferred target, phosphorylated light-activated rhodopsin, and unphosphorylated activated form. Out of 34 mutations tested, 24 and 25 affected the binding to phosphorylated and unphosphorylated rhodopsin, respectively. Manipulation of residues following polar core aspartates reduced preference for phosphorylated over unphosphorylated light-activated rhodopsin as dramatically as replacing these negatively charged aspartates with positively charged arginine. The data show that numerous lariat loop residues play distinct roles in arrestin-1 binding and its exquisite preference for phosphorylated light-activated rhodopsin.
Keywords:
GPCR
; desensitization
; arrestin
; receptor binding
; conformational change
; structure-function
1. Introduction
Mammals express hundreds of different G protein-coupled receptors (GPCRs), with ~800 distinct subtypes in humans [1], but only four arrestin proteins [2]. Two of these (arrestin-1a and -4) are expressed in the photoreceptors in the retina, while the two non-visual subtypes (arrestin-2 and -3; a.k.a. b-arrestin1 and 2, respectively) are ubiquitous. Activated GPCRs signal by catalyzing GDP/GTP exchange on cognate heterotrimeric G proteins [3]. The signaling of most GPCRs is stopped by a conserved two-step mechanism of homologous desensitization: active receptors are phosphorylated by specific kinases, whereupon arrestins bind active phosphoreceptors and block (arrest) their coupling to G proteins [4]. Arrestin-1 was the first member of the family discovered [5] and cloned [6]. It is expressed at very high levels in both rod [7,8] and cone [9] photoreceptors. Arrestin-1 demonstrates much greater preference for the activated phosphorylated rhodopsin (R-Rh*) over activated unphosphorylated (Rh*) than other arrestin family members have for the activated phosphorylated forms of their cognate receptors [10]. Vertebrate arrestins are held in their basal conformation by two intramolecular interactions, the polar core between the two domains, consisting of five interacting charged residues, and three-element interaction of the C-terminus with b-strand I and a-helix I in the N-domain that involves bulky hydrophobic side chains in all three elements [11,12,13,14,15] (Figure 1). Arrestin transition into receptor-bound conformation, which was revealed by the structure of rhodopsin-bound arrestin-1 [16,17] and several structures of arrestin-2 bound to non-visual GPCRs [18,19,20,21,22,23,24], and of both arrestin-2 and -3 bound to atypical chemikine receptor ACKR3 [25], requires the disruption of both autoinhibitory intramolecular interactions [26].
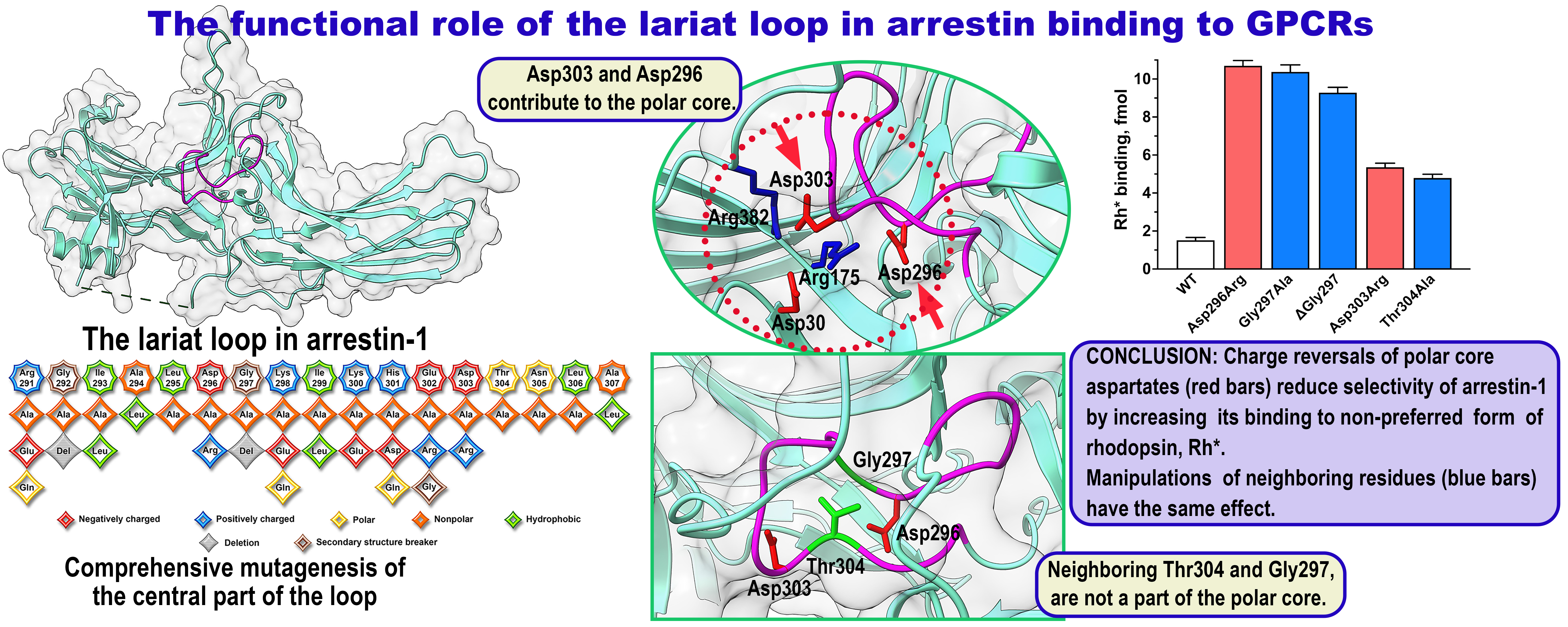
The lariat loop (the term is from [11]; it reflects its peculiar shape, which is conserved in all arrestins crystallized thus far [11,12,13,14,15,27,28,29]) is located between b-strands XVII and XVIII of the C-domain, encompassing residues 281-322 in bovine arrestin-1. This loop is a long stretch of residues without clear secondary structure flanked by two very short a-helices [11] (Figure 1). Its central part (usually called the gate loop), encompassing residues 291-307 in bovine arrestin-1 (Figure 1 and Figure 2), supplies two negative charges to the polar core, including Asp296 that was shown to be critical for the selectivity of arrestin-1 for its preferred binding partner, P-Rh* [30]. Despite undisputed functional importance of the gate loop, only the role of the two polar core aspartates in it, Asp296 and Asp303, was tested experimentally [30]. Here we used site-directed mutagenesis to determine the functional role of all residues in the gate loop. The data showed that changing fourteen out of seventeen residues has profound effects on arrestin-1 binding to rhodopsin. In particular, mutations of the residues following the polar core aspartates in the linear sequence yielded as dramatic increase in Rh* binding as mutations of these aspartates.
2. Results
To obtain deeper insight into the functional role of individual gate loop residues, in addition to conventional alanine scanning, we introduced charge reversals (Arg, Lys->Glu; Asp, Glu->Arg), replaced Ala with Leu that has bulky hydrophobic side chain, and deleted glycine residues that usually break secondary structures. In addition, we replaced some residues in bovine arrestin-1 gate loop with those found in homologous positions in other arrestin family members. Thus, the total number of tested mutations was thirty-four (Figure 3).
In the direct binding assay we used 1 nM bovine arrestin-1 produced in cell-free translation and labeled by incorporation of [14C]leucine. Human, mouse and bovine arrestin-1 oligomerizes, forming dimers and tetramers [38]. As experimentally determined dimerization and tetramerization constants of bovine arrestin-1 are 37 mM and 7.5 mM, respectively [39], at 1 nM concentration used in the assay >99.9% of it would be monomeric. Monomer is the form competent to bind rhodopsin [39]. Thus, possible effects of introduced mutations on arrestin-1 self-association could not have affected the binding results.
We use bovine arrestin-1 residue numbers throughout for consistency: we mutated the bovine protein and there are fairly high resolution crystal structures of wild type (WT) bovine arrestin-1 in the basal conformation (PDB 1CF1 [11] and 7JSM [27]. Note that the structure of rhodopsin-bound arrestin-1 contains mouse protein [16,17] (Figure 2), in which residue numbers are N+1 relative to the bovine homologue.
Although Asp296Ala, Asp296Arg, Asp303Ala, and Asp303Arg were characterized previously [30], these mutants were included along with others for comparison (Figure 4). We confirmed previous reports that all four of these mutations dramatically increase arrestin-1 binding to Rh*, thereby reducing arrestin-1 preference for P-Rh* [11,30]. The effects of substitutions of Asp296 were more prominent than those of Asp303 (Figure 4). Unexpectedly, alanine substitution or deletion of Gly297 yielded effects as strong as those of mutations of neighboring Asp296; Thr304Ala mutation yielded a phenotype similar to that of proteins with Asp303 substitutions (Figure 4). The effects of manipulation of Gly297 suggest that changes in the gate loop conformation destabilize the polar core as effectively as the replacement of the adjacent negatively charged Asp296.
Both substitutions of Arg291 (Arg291Ala and Arg291Glu) increased the binding to P-Rh* by 20-30% and more than doubled the binding to Rh* (Figure 4). The deletion of Gly292 increased P-Rh* and Rh* binding by 38% and 156%, respectively, while Gly292Ala mutation did not affect P-Rh* binding but enhanced Rh* binding by ~60% (Figure 4). These differential effects suggest that the change of the gate loop conformation, which deletion affects more significantly than alanine substitution, is the key. Interestingly, dramatic changes of the size of the side chain of the next two residues (Ile293Ala and Ala294Leu) did not significantly affect arrestin-1 function (Figure 4). Lys298Ala substitution slightly increased P-Rh* binding, while Lys298Glu reduced it.
The Ile299Ala mutation was detrimental for the binding to both forms of rhodopsin. The structure of the complex [17] suggests that Lys300 engages phosphorylated Ser338 in the C-terminus of rhodopsin. Charge reversal would preclude this interaction. However, similarly strong negative impact on P-Rh* and Rh* binding suggests that possible interaction with rhodopsin-attached phosphate (which is absent in Rh*) does not underlie this effect. Alanine substitution of Lys300 did not reduce the binding to WT P-Rh* (Figure 4), also suggesting limited role of this interaction. Both substitutions of His301 facilitated Rh* binding, but only His302Glu enhanced the binding to P-Rh* (Figure 4).
Mutations can change thermal stability of proteins, which would affect the results of the binding tests. Although the yields of soluble translated mutants did not reveal any significant changes (Supplemental Table S1), we additionally tested thermal stability by incubating the proteins at assay temperature (37oC) for up to 30 min, which is six times longer than the binding assay (Figure 5). Previously we found that charge reversals of the residues forming the polar core (Figure 1) affect thermal stability of arrestin-1 most dramatically [40]. Therefore, we tested D296R and D303R mutants as well as their functional mimics G297A, DG297, and T304A (Figure 4). We found that at 37oC arrestin-1-D296R was somewhat less stable than WT (p<0.01, as determined by time-protein interaction by two-way ANOVA analysis), whereas the stability of other mutants was not significantly different from WT (Figure 5). Thus, the difference in thermal stability was unlikely to significantly affect the results of the binding assay (Figure 4).
Based on sequence alignment of arrestin proteins from various species (Figure 6), we introduced several additional mutations in the gate loop of arrestin-1 to assess possible functional effects of non-conserved residues in these positions in some members of the arrestin family. These included Arg291Gln (as in bovine and mouse arrestin-4), Ile293Leu, Ile299Leu, and Ile293Leu+Ile299Leu (as in mammalian arrestin-2, -3, and -4, as well as in arrestins from C. intestinalis, C. elegance, and Drosophila kurtz), His301Gln, Glu302Gly, and His301Gln+Glu302Gly (as in bovine arrestin-4), and Lys298Gln (as in mouse arrestin-3, and arrestins from C. elegans, C. intestinalis, and Drosophila kurtz). For the sake of comparing these substitutions with previously tested ones in the same experiment, we repeated the binding of relevant mutants shown in Figure 4 along with new ones changing the residues in bovine arrestin-1 to those found in homologous positions in other family members (Figure 6).
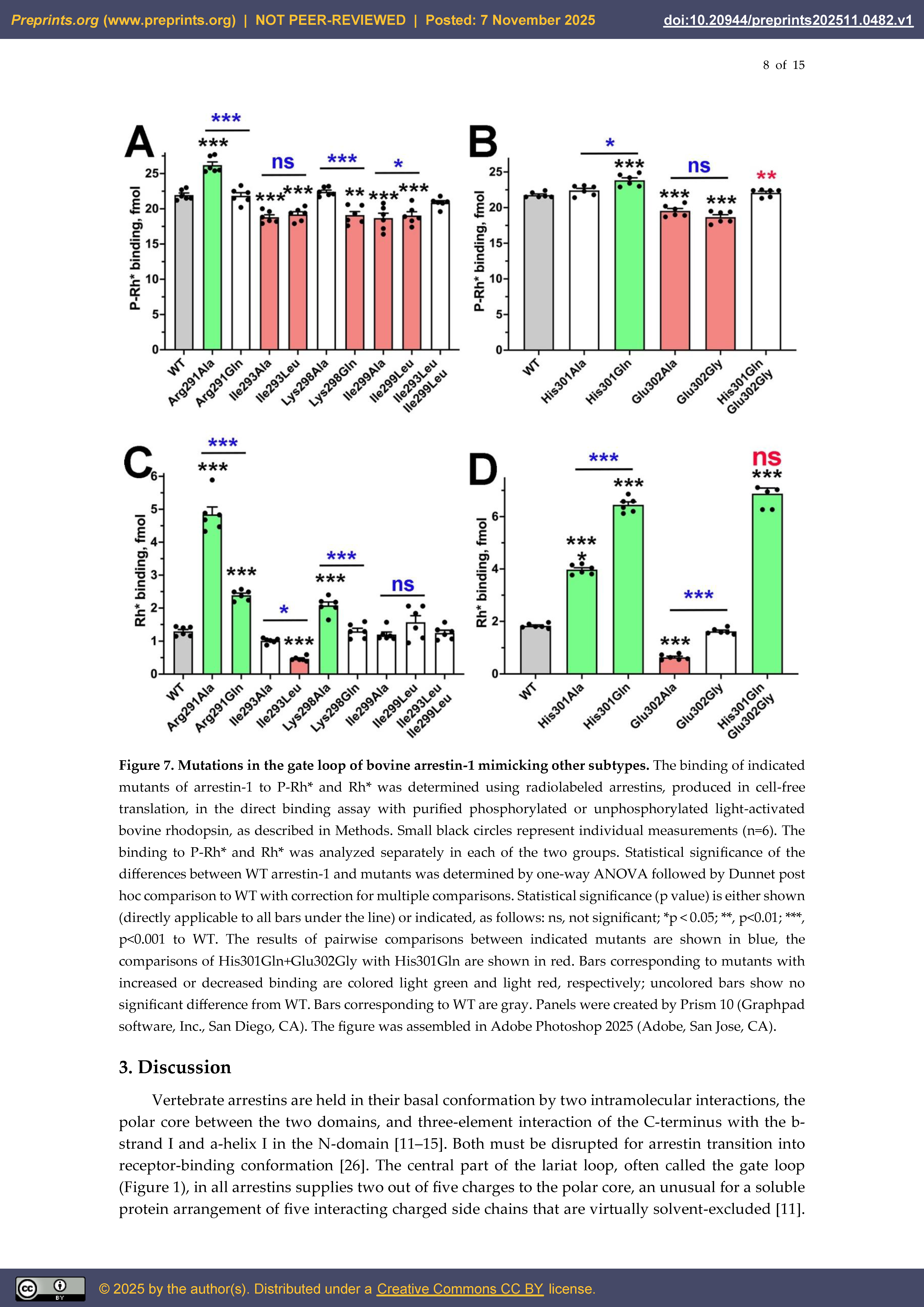
Arrestin-4 demonstrates significantly lower preference for the phosphorylated form of its cognate receptor over unphosphorylated than arrestin-1 [14]. As could be expected, replacement of Arg291 native for arrestin-1 with Gln characteristic for arrestin-4 reduced arrestin-1 preference for P-Rh* over Rh* (Figure 7). However, the substitution of Arg for Gln did not produce an effect like alanine substitution, which enhanced the binding to P-Rh*, and even more so to Rh* (Figure 4 and Figure 7). Thus, the presence of Gln in some arrestin-4 proteins reduces selectivity as compared to Arg in arrestin-1 but does not significantly affect the ability to bind the receptor. The effects of replacing Ile293 and Ile299 found in human, bovine and mouse arrestin-1 with Leu turned out to be similar to alanine substitutions: both Leu mutants yielded somewhat lower binding to P-Rh*, with Ile293Ala also reducing the binding to Rh* (Figure 7). Ile299Leu mutant demonstrated the same selectivity as WT; unexpectedly the selectivity of Ile293Leu was even increased (Figure 6). However, when both Ile residues were replaced with Leu, as in vertebrate arrestin-2, -3, -4, and invertebrate arrestins (Figure 6), rhodopsin binding of the double mutant was not significantly different from WT arrestin-1 (Figure 7), suggesting that these two Leu residues in other subtypes might play a role in the function(s) arrestin-1 does not have. The mutant with the substitution of positively charged Lys298 for uncharged Gln (as in some mammalian and all invertebrate arrestins; Figure 6) demonstrated WT level binding to Rh* and slightly lower binding to P-Rh* (Figure 5). Notably, the Lys298Gln mutant displayed lower binding to both forms of rhodopsin than Lys298Ala, (Figure 7). Thus, it appears that the presence of uncharged Gln in position 298 instead of Lys in arrestin-1 does not dramatically affect receptor interactions. Interestingly, the effects of neutral Gln in position 298 were similar to those of negatively charged Glu (Figure 4 and Figure 7), suggesting that H-bonding, rather than charge-charge interaction involving this residue, is functionally important. This hypothesis is supported by the finding that the substitution of Lys298 for an alanine (small side chain lacking H-bonding capability) increased the binding to P-Rh* (Figure 4 and Figure 7). Replacing His301 with Gln in this position (as in invertebrate arrestins; Figure 6) enhanced the binding to both forms of rhodopsin, but more strongly to Rh*, thereby reducing arrestin-1 preference for P-Rh*. The effect was even stronger than in case of His301Ala substitution (Figure 4 and Figure 7). Arrestin-1 with glycine in position 302 (as in bovine arrestin-4, instead of Glu conserved in almost all other family members; Figure 6) showed reduced P-Rh* binding but bound Rh* like WT (Figure 7). The negative impact of Glu302Ala substitution is stronger than that of Glu302Gly, reducing the binding to both P-Rh* and Rh* (Figure 4 and Figure 7). Arrestin-1 with double mutation His301Gln+Glu302Gly (as in bovine arrestin-4; Figure 6) bound P-Rh* like WT, but demonstrated greatly enhanced Rh* binding, similar to the single His301Gln mutant (Figure 7). Apparently, His301Gln substitution largely determined the effect observed in the double mutant. Notably, the performance of His301Gln+Glu302Gly resembled that of arrestin-4 [14] that these mutations were designed to mimic (Figure 7).
3. Discussion
Vertebrate arrestins are held in their basal conformation by two intramolecular interactions, the polar core between the two domains, and three-element interaction of the C-terminus with the b-strand I and a-helix I in the N-domain [11,12,13,14,15]. Both must be disrupted for arrestin transition into receptor-binding conformation [26]. The central part of the lariat loop, often called the gate loop (Figure 1), in all arrestins supplies two out of five charges to the polar core, an unusual for a soluble protein arrangement of five interacting charged side chains that are virtually solvent-excluded [11]. The peculiar shape of the lariat loop, including its central part, the gate loop, is conserved in the basal conformation of all four subtypes of vertebrate arrestins [11,12,13,14,15,27,29]. Its conformation in receptor-bound arrestin-1 [16,17], -2 [18,19,20,21,22,23,24,25,55,56], and -3 [25] as well as in the receptor bound-like conformation of arrestin-3 in the trimer [57], is significantly different. Thus, the gate loop likely plays an important role in the conformational rearrangement accompanying receptor binding. Indeed, the reversal of the polar core negative charges supplied by the lariat loop “pre-activated” arrestins, yielding mutants that readily bind active unphosphorylated forms of their cognate receptors [30,35,58,59,60]. However, this critically important element of arrestins remains understudied: the functional role of the other residues in the gate loop was never tested experimentally. Unfortunately, the only comprehensive studies of the effects of alanine substitutions of all arrestin-1 residues on P-Rh* [61] and phospho-opsin [62] binding used relatively low sensitivity assay and yielded inconclusive results.
Arrestin-1 demonstrates much greater preference for P-Rh* than other family members for the active phosphorylated form of their cognate receptors (reviewed in [10]). Among 34 mutations tested here eight did not significantly affect this selectivity, eight even increased it, whereas the majority (18 mutations) decreased it, in many cases dramatically (Figure 4 and Figure 7). Our recent studies suggest that the function of many native residues in arrestin-1 is to maintain high selectivity for P-Rh* by suppressing Rh* binding [63,64]. Thus, the data raise the question why evolution did not favor selectivity-enhancing residues in arrestin proteins (see alignments in [2,65]). Among the eight substitutions that increased the preference for P-Rh* over Rh*, five reduced absolute P-Rh* binding, so in these cases the answer appears to be clear: these are detrimental for the binding to the preferred arrestin-1 partner, P-Rh* (Figure 4). However other selectivity enhancers, Ala294Leu, Glu302Arg, and Asn305Ala, did not reduce P-Rh* binding; in fact, Glu302Arg even increased it (Figure 4). However, the analysis of multiple arrestins from a variety of vertebrate and invertebrate species [65] showed that the residues we introduced are never found in these positions (the only exception is Drosophila visual arrestin1, which carries alanine in position homologous to Asn305). All analyzed homologs harbor an alanine in position equivalent to Ala294. The majority carries Glu and Asn in positions homologous to Glu302 and Asn305, respectively, with some variation in two mammalian (bovine and pig) and one frog (R. pipiens) arrestin-4 and several invertebrate arrestins [65]. It is tempting to speculate that Ala, Glu, and Asn in respective positions are advantageous for the thermal stability (which we did not test extensively): WT arrestin-1 is remarkably stable, it did not lose activity even after prolonged incubation at 39oC [40,66]. We found that the mutants used are also stable in the assay conditions (37oC)(Figure 5).
The effects of substitutions mimicking other arrestin subtypes yielded answers only in some cases. Among arrestin-4-inspired mutations (Figure 6), His301Gln dramatically changed the selectivity of arrestin-1 towards that of arrestin-4, Arg291Gln acted in the same direction to a lesser extent, whereas individual Glu302Gly substitution even increased selectivity (while reducing the binding to both forms of rhodopsin). Importantly, double mutant His301Gln+Glu302Gly, mimicking bovine arrestin-4 (these residues in mouse and human arrestin-4 are the same as in arrestin-1 (Figure 6) was less selective than parental arrestin-1, as could be expected (Figure 7). Lys298Gln, as well as double substitution Ile293Leu+Ile299Leu, only marginally affected receptor binding, suggesting that these residues might play a role in other functions of vertebrate non-visual and invertebrate arrestins (Figure 7). It should be noted, however, that relatively long side chain with H-bonding capability was preserved by Lys298Gln substitution, while in the latter case residues with bulky hydrophobic side chains were substituted with alternatives of similar size and chemical nature. Thus, receptor binding might require these chemical characteristics, rather than residue identity.
Overall, the conservation of the sequence of the gate loop in arrestins from round worm C. elegans, fly Drosophila melanogaster, tunicate Ciona officinalis, squid Loligo pealei, and mammals, i.e., animals that had common ancestors more than 600,000,000 years ago, is striking: among seventeen residues in the gate loop, eleven are strictly conserved, and the other six are either conserved in most, or underwent conservative substitutions (Figure 6). This sequence contains seven residues carrying charges, three with bulky hydrophobic side chains, two glycines, two alanines with very small side chains, and only three residues that do not belong to either of these categories. Available structures suggest that charged and bulky hydrophobic side chains in the core of proteins usually mediate intramolecular interactions, glycines disallow the formation of b-strands and a-helices, whereas due to small size of the side chain alanines preclude steric clashes with nearby elements.
Both polar core aspartates and several other lariat loop residues contribute to exceptional selectivity of arrestin-1 for P-Rh* over Rh*. However, the positioning of the side chains with different chemical nature in the finger loop is virtually identical in arrestins with high selectivity (arrestin-1 from different species) and in less selective isoforms, such as non-visual or cone arrestins, and even in invertebrate visual arrestins that bind unphosphorylated rhodopsins [67,68,69]. Thus, it appears likely that the function of the gate loop goes beyond proper positioning of the polar core aspartates. Its conserved sequence and unusual shape appear to be important for the function shared by all arrestin proteins, the binding to GPCRs.
4. Materials and Methods
Materials. [g-32P]ATP and [14C]leucine were from Perkin-Elmer (Waltham, MA). Restriction endonucleases, Vent DNA polymerase, and Quick T4 DNA ligase were from New England Biolabs (Ipswich, MA). Rabbit reticulocyte lysate was made in bulk by Ambion (Austin, TX). SP6 RNA polymerase was overexpressed in E. coli and purified, as described [70]. DNA purification kits (mini-, midi-, and maxi; 3 ml, 50 ml, and 100 ml of bacterial culture, respectively) were from Zymo Research (Irvine, CA). All other reagents were from Sigma-Aldrich (St. Louis, MO).
Mutagenesis and plasmid construction. For in vitro transcription by SP6 RNA polymerase bovine arrestin-1 cDNA was subcloned into pGEM2 vector (Promega; Madison, WI) with “idealized” 5-UTR that ensures efficient translation of uncapped mRNAs [70] between Eco RI and Hind III sites, as described [71]. Mutations were introduced by PCR. Unique restriction sites Msc I (codons 284-286) and Spe I (codons 323-325) in the reengineered by the introduction of silent mutations coding sequence of bovine arrestins-1 (described in [72]) were used to subclone generated mutant fragments. All mutations were confirmed by dideoxy sequencing (GenHunter Corporation, Nashville, TN).
In vitro transcription [70], translation and calculation of specific activity of synthesized radiolabeled arrestin proteins [73], preparation of unphosphorylated and phosphorylated bovine rhodopsin and quantification of the level of its phosphorylation [74,75] were performed as described. Translation yields and the fraction of translated protein that remains in the supernatant after 1 h centrifugation at 357,000xg (to remove ribosomes and aggregated proteins) (100,000 rpm in TLA120.1 rotor) suggests that none of the mutants used had folding problems (Supplemental Table S1). Experiments in vitro [76] and in vivo [77] demonstrate that three rhodopsin-attached phosphates are necessary for tight arrestin-1 binding. P-Rh* used in binding experiments had on average 2.6 phosphates per rhodopsin, which suggests that a large fraction of it carries three or more phosphates. To ensure that rhodopsin is not rate-limiting, very high molar excess was used in the assay. Rhodopsin was phosphorylated in bright light by endogenous rhodopsin kinase (systematic name GRK1), as described [78]. Thus, it is likely that biologically relevant sites (the C-terminus of bovine rhodopsin has seven phosphorylatable serines and threonines) were phosphorylated. Phosphorylated rhodopsin has been regenerated by two additions of 3-fold molar excess of 11-cis-retinal, as described [79].
Direct binding assay was performed, as described [71,80]. Briefly, 1 nM arrestin-1 (50 fmol, specific activity 12.1 – 12.6 dpm/fmol) was incubated with 0.3 mg of P-Rh* or Rh* (7.8 pmol; concentration in the assay 156 pM) in 50 ml of 50 mM Tris-HCL, pH 7.4, 100 mM potassium acetate, 1 mM EDTA, 1 mM DTT for 5 min at 37oC under room light. Samples were then cooled on ice for 2-3 min, whereupon bound arrestin-1 was separated from free arrestin-1 and unincorporated [14C]leucine at 4oC by gel-filtration chromatography on 2-ml column of Sepharose CL-2B. Bound arrestin-1 eluted with rhodopsin-containing membranes was quantified by liquid scintillation counting on Tri-Carb (PerkinElmer, Waltham, MA). Non-specific “binding” (likely reflecting the aggregation of arrestin-1 proteins during the assay) was determined in samples without rhodopsin and subtracted.
Thermal stability test. Translation mixes obtained after centrifugation were kept on ice (0, control) or incubated at assay temperature (37oC) for 5, 15, and 30 min and then cooled on ice. The binding to P-Rh* of all samples was performed, as described above.
Data Analysis and Statistics. Statistical significance of the differences between mutants and WT arrestin-1 in each group was determined by one-way ANOVA (analysis of variance) with Dunnett’s multiple comparison test using Prism 10 software (GraphPad, Boston, MA). Stability data were analyzed by two-way ANOVA, where protein-time interaction reflected the difference in the survival rate. P values < 0.05 were considered statistically significant and indicated, as follows: *p < 0.05; **, p<0.01; ***p < 0.001.
Footnote
aWe use systematic names of arrestin proteins. The number after the dash indicates the order of cloning: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin; SAG in HUGO database), arrestin-2 (β-arrestin or β-arrestin1; ARRB1 in HUGO database), arrestin-3 (β-arrestin2 or hTHY-ARRX; ARRB2 in HUGO database), and arrestin-4 (cone or X-arrestin; ARR3 in HUGO database).
Supplementary Materials
The following are available online at Preprints.org, Table S1: Cell-free translation results for gate loop mutants.
Author Contributions
Conceptualization, S.A.V and V.V.G.; formal analysis, S.A.V. and E.V.G.; investigation, S.A.V. and D.G.; writing—original draft preparation, S.A.V. and V.V.G.; writing—review and editing, S.A.V., V.V.G., and E.V.G.; visualization, S.A.V and V.V.G.; project administration, V.V.G.; funding acquisition, V.V.G.
Funding
This research was funded by NIH grant RO1 EY011500 and Cornelius Vanderbilt Chair, Vanderbilt University (V.V.G.). Funding sources had no influence over the study design, collection, analysis and interpretation of data, writing of the report and decision to submit the article for publication.
Data Availability Statement
The data are presented in the manuscript. Raw binding data obtained in each experiment are available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Indrischek, H.; Prohaska, S.J.; Gurevich, V.V.; Gurevich, E.V.; Stadler, P.F. Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol 2017, 17, 163. [Google Scholar] [CrossRef]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.W.; Park, J.H.; Heck, M.; Ernst, O.P. A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem Sci 2009, 34, 540–552. [Google Scholar] [CrossRef]
- Carman, C.V.; Benovic, J.L. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol 1998, 8, 335–344. [Google Scholar] [CrossRef]
- Kuhn, H. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry 1978, 17, 4389–4395. [Google Scholar] [CrossRef]
- Shinohara, T.; Dietzschold, B.; Craft, C.M.; Wistow, G.; Early, J.J.; Donoso, L.A.; Horwitz, J.; Tao, R. Primary and secondary structure of bovine retinal S antigen (48 kDa protein). Proceedings of the National Academy of Sciences 1987, 84, 6975–6979. [Google Scholar] [CrossRef]
- Song, X.; Vishnivetskiy, S.A.; Seo, J.; Chen, J.; Gurevich, E.V.; Gurevich, V.V. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience 2011, 174, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.M.; Gurevich, E.V.; Vishnivetskiy, S.A.; Ahmed, M.R.; Song, X.; Gurevich, V.V. Each rhodopsin molecule binds its own arrestin. Proc Nat Acad Sci USA 2007, 104, 3125–3128. [Google Scholar] [CrossRef] [PubMed]
- Nikonov, S.S.; Brown, B.M.; Davis, J.A.; Zuniga, F.I.; Bragin, A.; Pugh, E.N., Jr.; Craft, C.M. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron 2008, 59, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V. Arrestins: A Small Family of Multi-Functional Proteins. Int J Mol Sci 2024, 25, 6284. [Google Scholar] [CrossRef]
- Hirsch, J.A.; Schubert, C.; Gurevich, V.V.; Sigler, P.B. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell 1999, 97, 257–269. [Google Scholar] [CrossRef]
- Han, M.; Gurevich, V.V.; Vishnivetskiy, S.A.; Sigler, P.B.; Schubert, C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure 2001, 9, 869–880. [Google Scholar] [CrossRef]
- Milano, S.K.; Pace, H.C.; Kim, Y.M.; Brenner, C.; Benovic, J.L. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 2002, 41, 3321–3328. [Google Scholar] [CrossRef]
- Sutton, R.B.; Vishnivetskiy, S.A.; Robert, J.; Hanson, S.M.; Raman, D.; Knox, B.E.; Kono, M.; Navarro, J.; Gurevich, V.V. Crystal Structure of Cone Arrestin at 2.3Å: Evolution of Receptor Specificity. J Mol Biol 2005, 354, 1069–1080. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol 2011, 406, 467–478. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin determined by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; Barty, A.; Latorraca, N.R.; Chapman, H.N.; Hubbell, W.L.; Dror, R.O.; Stevens, R.C.; Cherezov, V.; Gurevich, V.V.; Griffin, P.R.; Ernst, O.P.; Melcher, K.; Xu, H.E. Identification of Phosphorylation Codes for Arrestin Recruitment by G protein-Coupled Receptors. Cell 2017, 170, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Li, Z.; Jin, M.; Yin, Y.L.; de Waal, P.W.; Pal, K.; Yin, Y.; Gao, X.; He, Y.; Gao, J.; Wang, X.; Zhang, Y.; Zhou, H.; Melcher, K.; Jiang, Y.; Cong, Y.; Zhou, X.E.; Yu, X.; Xu, H.E. A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res 2019, 29, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; Tate, C.G. Molecular basis of β-arrestin coupling to formoterol-bound β(1)-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef]
- Liao, Y.Y.; Zhang, H.; Shen, Q.; Cai, C.; Ding, Y.; Shen, D.D.; Guo, J.; Qin, J.; Dong, Y.; Zhang, Y.; Li, X.M. Snapshot of the cannabinoid receptor 1-arrestin complex unravels the biased signaling mechanism. Cell 2023, 186, 5784–5797. [Google Scholar] [CrossRef]
- Huang, W.; Masureel, M.; Qianhui, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; Kobilka, B.K. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Bous, J.; Fouillen, A.; Orcel, H.; Trapani, S.; Cong, X.; Fontanel, S.; Saint-Paul, J.; Lai-Kee-Him, J.; Urbach, S.; Sibille, N.; Sounier, R.; Granier, S.; Mouillac, B.; Bron, P. Structure of the vasopressin hormone-V2 receptor-β-arrestin1 ternary complex. Sci Adv 2022, 8, eabo7761. [Google Scholar] [CrossRef]
- Cao, C.; Barros-Álvarez, X.; Zhang, S.; Kim, K.; Dämgen, M.A.; Panova, O.; Suomivuori, C.M.; Fay, J.F.; Zhong, X.; Krumm, B.E.; Gumpper, R.H.; Seven, A.B.; Robertson, M.J.; Krogan, N.J.; Hüttenhain, R.; Nichols, D.E.; Dror, R.O.; Skiniotis, G.; Roth, B.L. Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron 2022, 110, 3154–3167. [Google Scholar] [CrossRef]
- Chen, Q.; Schafer, C.T.; Mukherjee, S.; Wang, K.; Gustavsson, M.; Fuller, J.R.; Tepper, K.; Lamme, T.D.; Aydin, Y.; Agrawal, P.; Terashi, G.; Yao, X.Q.; Kihara, D.; Kossiakoff, A.A.; Handel, T.M.; Tesmer, J.J.G. Effect of phosphorylation barcodes on arrestin binding to a chemokine receptor. Nature 2025, 643, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Sente, A.; Peer, R.; Srivastava, A.; Baidya, M.; Lesk, A.M.; Balaji, S.; Shukla, A.K.; Babu, M.M.; Flock, T. Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat Struct Mol Biol 2018, 25, 538–545. [Google Scholar] [CrossRef]
- Sander, C.L.; Luu, J.; Kim, K.; Furkert, D.; Jang, K.; Reichenwallner, J.; Kang, M.; Lee, H.J.; Eger, B.T.; Choe, H.W.; Fiedler, D.; Ernst, O.P.; Kim, Y.J.; Palczewski, K.; Kiser, P.D. Structural evidence for visual arrestin priming via complexation of phosphoinositols. Structure 2022, 30, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Van Eps, N.; Eger, B.T.; Rauscher, S.; Yedidi, R.S.; Moroni, T.; West, G.M.; Robinson, K.A.; Griffin, P.R.; Mitchell, J.; Ernst, O.P. A Novel Polar Core and Weakly Fixed C-Tail in Squid Arrestin Provide New Insight into Interaction with Rhodopsin. J Mol Biol 2018, 430, 4102–4118. [Google Scholar] [CrossRef]
- Granzin, J.; Wilden, U.; Choe, H.W.; Labahn, J.; Krafft, B.; Buldt, G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature 1998, 391, 918–921. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Paz, C.L.; Schubert, C.; Hirsch, J.A.; Sigler, P.B.; Gurevich, V.V. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem 1999, 274, 11451–11454. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Huh, E.K.; Gurevich, E.V.; Gurevich, V.V. The finger loop as an activation sensor in arrestin. J Neurochem 2021, 157, 1138–1152. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Zheng, C.; May, M.B.; Karnam, P.C.; Gurevich, E.V.; Gurevich, V.V. Lysine in the lariat loop of arrestins does not serve as phosphate sensor. J Neurochem 2021, 156, 435–444. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Schubert, C.; Climaco, G.C.; Gurevich, Y.V.; Velez, M.-G.; Gurevich, V.V. An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J. Biol. Chem. 2000, 275, 41049–41057. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem 1998, 273, 15501–15506. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Francis, D.J.; Van Eps, N.; Kim, M.; Hanson, S.M.; Klug, C.S.; Hubbell, W.L.; Gurevich, V.V. The role of arrestin alpha-helix I in receptor binding. J. Mol. Biol. 2010, 395, 42–54. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Kim, M.; Hanson, S.M.; Vishnivetskiy, S.A.; Song, X.; Cleghorn, W.M.; Hubbell, W.L.; Gurevich, V.V. Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry 2011, 50, 2235–2242. [Google Scholar] [CrossRef]
- Hanson, S.M.; Van Eps, N.; Francis, D.J.; Altenbach, C.; Vishnivetskiy, S.A.; Arshavsky, V.Y.; Klug, C.S.; Hubbell, W.L.; Gurevich, V.V. Structure and function of the visual arrestin oligomer. EMBO J 2007, 26, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Vishnivetskiy, S.A.; Gross, O.P.; Emelianoff, K.; Mendez, A.; Chen, J.; Gurevich, E.V.; Burns, M.E.; Gurevich, V.V. Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Current biology: CB 2009, 19, 700–705. [Google Scholar] [CrossRef]
- Smith, W.C. A splice variant of arrestin from human retina. Exp Eye Res 1996, 62, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Syed, M.; Bugra, K.; Whelan, J.P.; McGinnis, J.F.; Shinohara, T. Structural analysis of mouse S-antigen. Gene 1988, 73, 11–20. [Google Scholar] [CrossRef]
- Sterne-Marr, R.; Gurevich, V.V.; Goldsmith, P.; Bodine, R.C.; Sanders, C.; Donoso, L.A.; Benovic, J.L. Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem 1993, 268, 15640–15648. [Google Scholar] [CrossRef] [PubMed]
- Parruti, G.; Peracchia, F.; Sallese, M.; Ambrosini, G.; Masini, M.; Rotilio, D.; De Blasi, A. Molecular analysis of human beta-arrestin-1: cloning, tissue distribution, and regulation of expression. Identification of two isoforms generated by alternative splicing. J Biol Chem 1993, 268, 9753–9761. [Google Scholar] [CrossRef]
- Kingsmore, S.F.; Peppel, K.; Suh, D.; Caron, M.G.; Lefkowitz, R.J.; Seldin, M.F. Genetic mapping of the beta-arrestin 1 and 2 genes on mouse chromosomes 7 and 11 respectively. Mamm Genome 1995, 6, 306–307. [Google Scholar] [CrossRef]
- Rapoport, B.; Kaufman, K.D.; Chazenbalk, G.D. Cloning of a member of the arrestin family from a human thyroid cDNA library. Mol Cell Endocrinol 1992, 84, R39–R43. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Ohguro, H.; Sohma, H.; Kuroki, Y.; Wada, H.; Okisaka, S.; Murakami, A. Purification and characterization of bovine cone arrestin (cArr). FEBS Lett. 2000, 470, 336–340. [Google Scholar] [CrossRef]
- Murakami, A.; Yajima, T.; Sakuma, H.; McLaren, M.J.; Inana, G. X-arrestin: a new retinal arrestin mapping to the X chromosome. FEBS Lett. 1993, 334, 203–209. [Google Scholar] [CrossRef]
- Hyde, D.R.; Mecklenburg, K.L.; Pollock, J.A.; Vihtelic, T.S.; Benzer, S. Twenty Drosophila visual system cDNA clones: one is a homolog of human arrestin. Proc Natl Acad Sci U S A 1990, 87, 1008–1012. [Google Scholar] [CrossRef]
- Yamada, T.; Takeuchi, Y.; Komori, N.; Kobayashi, H.; Sakai, Y.; Hotta, Y.; Matsumoto, H. A 49-kilodalton phosphoprotein in the Drosophila photoreceptor is an arrestin homolog. Science 1990, 246, 483–486. [Google Scholar] [CrossRef]
- Mayeenuddin, L.H.; Mitchell, J. Squid visual arrestin: cDNA cloning and calcium-dependent phosphorylation by rhodopsin kinase (SQRK). J Neurochem 2003, 85, 592–600. [Google Scholar] [CrossRef]
- Johnson, E.C.; Tift, F.W.; McCauley, A.; Liu, L.; Roman, G. Functional characterization of kurtz, a Drosophila non-visual arrestin, reveals conservation of GPCR desensitization mechanisms. Insect Biochem Mol Biol 2008, 38, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Palmitessa, A.; Hess, H.A.; Bany, I.A.; Kim, Y.M.; Koelle, M.R.; Benovic, J.L. Caenorhabditus elegans arrestin regulates neural G protein signaling and olfactory adaptation and recovery. J Biol Chem 2005, 280, 24649–24662. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Orii, H.; Yoshida, N.; Jojima, E.; Horie, T.; Yoshida, R.; Haga, T.; Tsuda, M. Ascidian arrestin (Ci-arr), the origin of the visual and nonvisual arrestins of vertebrate. Eur J Biochem 2002, 269, 5112–5118. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Lin, S.; Yan, X.; Cai, H.; Yi, C.; Ma, L.; Chu, X.; Liu, Y.; Zhu, Y.; Han, S.; Zhao, Q.; Wu, B. Tail engagement of arrestin at the glucagon receptor. Nature 2023, 620, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, X.; Yuan, Q.; Wang, Y.; Shi, P.; Zhang, H.; Wang, T.; Sun, W.; Ling, S.; Liu, Y.; Lai, J.; Xie, W.; Yin, W.; Liu, L.; Xu, H.E.; Tian, C. Molecular mechanism of the arrestin-biased agonism of neurotensin receptor 1 by an intracellular allosteric modulator. Cell Res 2025, 35, 284–295. [Google Scholar] [CrossRef]
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; Brautigam, C.A.; Klug, K.S.; Gurevich, V.V.; Iverson, T.M. Structural basis of arrestin-3 activation and signaling. Nat Commun 2017, 8, 1427. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Pals-Rylaarsdam, R.; Benovic, J.L.; Hosey, M.M.; Onorato, J.J. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem 1997, 272, 28849–28852. [Google Scholar] [CrossRef]
- Celver, J.; Vishnivetskiy, S.A.; Chavkin, C.; Gurevich, V.V. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 2002, 277, 9043–9048. [Google Scholar] [CrossRef]
- Kovoor, A.; Celver, J.; Abdryashitov, R.I.; Chavkin, C.; Gurevich, V.V. Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 1999, 274, 6831–6834. [Google Scholar] [CrossRef]
- Ostermaier, M.K.; Peterhans, C.; Jaussi, R.; Deupi, X.; Standfuss, J. Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A 2014, 111, 1825–1830. [Google Scholar] [CrossRef]
- Peterhans, C.; Lally, C.C.; Ostermaier, M.K.; Sommer, M.E.; Standfuss, J. Functional map of arrestin binding to phosphorylated opsin, with and without agonist. Sci Rep 2016, 6, 28686. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Huh, E.K.; Karnam, P.C.; Oviedo, S.; Gurevich, E.V.; Gurevich, V.V. The Role of Arrestin-1 Middle Loop in Rhodopsin Binding. Int J Mol Sci 2022, 23, 13887. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Weinstein, L.D.; Zheng, C.; Gurevich, E.V.; Gurevich, V.V. Functional Role of Arrestin-1 Residues Interacting with Unphosphorylated Rhodopsin Elements. Int J Mol Sci 2023, 24, 8903. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol 2006, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Vishnivetskiy, S.A.; Chen, Q.; Palazzo, M.C.; Brooks, E.K.; Altenbach, C.; Iverson, T.M.; Hubbell, W.L.; Gurevich, V.V. Engineering visual arrestin-1 with special functional characteristics. J Biol Chem 2013, 288, 11741–11750. [Google Scholar] [CrossRef]
- Alloway, P.G.; Dolph, P.J. A role for the light-dependent phosphorylation of visual arrestin. Proc Natl Acad Sci U S A 1999, 96, 6072–6077. [Google Scholar] [CrossRef]
- Bentrop, J.; Paulsen, R. Light-modulated ADP-ribosylation, protein phosphorylation and protein binding in isolated fly photoreceptor membranes. Eur J Biochem 1986, 161, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.K.; Xia, H.; Yan, L.; Liu, C.H.; Hardie, R.C.; Ready, D.F. Arrestin translocation is stoichiometric to rhodopsin isomerization and accelerated by phototransduction in Drosophila photoreceptors. Neuron 2010, 67, 997–1008. [Google Scholar] [CrossRef]
- Gurevich, V.V. Use of bacteriophage RNA polymerase in RNA synthesis. Methods Enzymol 1996, 275, 382–397. [Google Scholar]
- Vishnivetskiy, S.A.; Lee, R.J.; Zhou, X.E.; Franz, A.; Xu, Q.; Xu, H.E.; Gurevich, V.V. Functional role of the three conserved cysteines in the N domain of visual arrestin-1. J Biol Chem 2017, 292, 12496–12502. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Hosey, M.M.; Benovic, J.L.; Gurevich, V.V. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem 2004, 279, 1262–1268. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Benovic, J.L. Visual arrestin interaction with rhodopsin: Sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J. Biol. Chem. 1993, 268, 11628–11638. [Google Scholar] [CrossRef]
- McDowell, J.H. Preparing Rod Outer Segment Membranes, Regenerating Rhodopsin, and Determining Rhodopsin Concentration. Methods in Neurosciences 1993, 15, 123–130. [Google Scholar]
- McDowell, J.H.; Nawrocki, J.P.; Hargrave, P.A. Phosphorylation sites in bovine rhodopsin. Biochemistry 1993, 32, 4968–4974. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Raman, D.; Wei, J.; Kennedy, M.J.; Hurley, J.B.; Gurevich, V.V. Regulation of arrestin binding by rhodopsin phosphorylation level. J Biol Chem 2007, 282, 32075–32083. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.; Burns, M.E.; Roca, A.; Lem, J.; Wu, L.W.; Simon, M.I.; Baylor, D.A.; Chen, J. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 2000, 28, 153–164. [Google Scholar] [CrossRef]
- Wilden, U.; Kühn, H. Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry 1982, 21, 3014–3022. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem 1992, 267, 21919–21923. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Sullivan, L.S.; Bowne, S.J.; Daiger, S.P.; Gurevich, E.V.; Gurevich, V.V. Molecular Defects of the Disease-Causing Human Arrestin-1 C147F Mutant. Invest Ophthalmol Vis Sci 2018, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Key elements in arrestin-1. The structure of arrestin-1 (PDB ID 1CF1 [11]) with functionally important elements is shown, as follows: the gate loop (central part of the lariat loop, residues 291-307), magenta; the remaining part of the lariat loop (residues 281-290 and 308-322), light brown; the finger loop (residues 66-81; putative activation sensor [31]), green; the lysines in b-strand I (Lys14 + Lys15, putative phosphate sensor [32,33]), dark blue stick models; polar core residues (Asp30, Arg175, Asp296, Asp303, Arg382 [11,30]), red stick models; hydrophobic residues anchoring the C-terminus to the N-domain (Val11, Ile12, Phe13 in the b-strand I; Leu103, Leu107, Leu111 in the a-helix; Phe375, Val376, Phe377, Phe380 in the C-terminus [11,34,35]), yellow stick models. Dashed line indicates long loop between b-strands XIX and XX not resolved in crystal. The image was created in UCSF ChimeraX [36,37] and labeled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 1.
Key elements in arrestin-1. The structure of arrestin-1 (PDB ID 1CF1 [11]) with functionally important elements is shown, as follows: the gate loop (central part of the lariat loop, residues 291-307), magenta; the remaining part of the lariat loop (residues 281-290 and 308-322), light brown; the finger loop (residues 66-81; putative activation sensor [31]), green; the lysines in b-strand I (Lys14 + Lys15, putative phosphate sensor [32,33]), dark blue stick models; polar core residues (Asp30, Arg175, Asp296, Asp303, Arg382 [11,30]), red stick models; hydrophobic residues anchoring the C-terminus to the N-domain (Val11, Ile12, Phe13 in the b-strand I; Leu103, Leu107, Leu111 in the a-helix; Phe375, Val376, Phe377, Phe380 in the C-terminus [11,34,35]), yellow stick models. Dashed line indicates long loop between b-strands XIX and XX not resolved in crystal. The image was created in UCSF ChimeraX [36,37] and labeled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
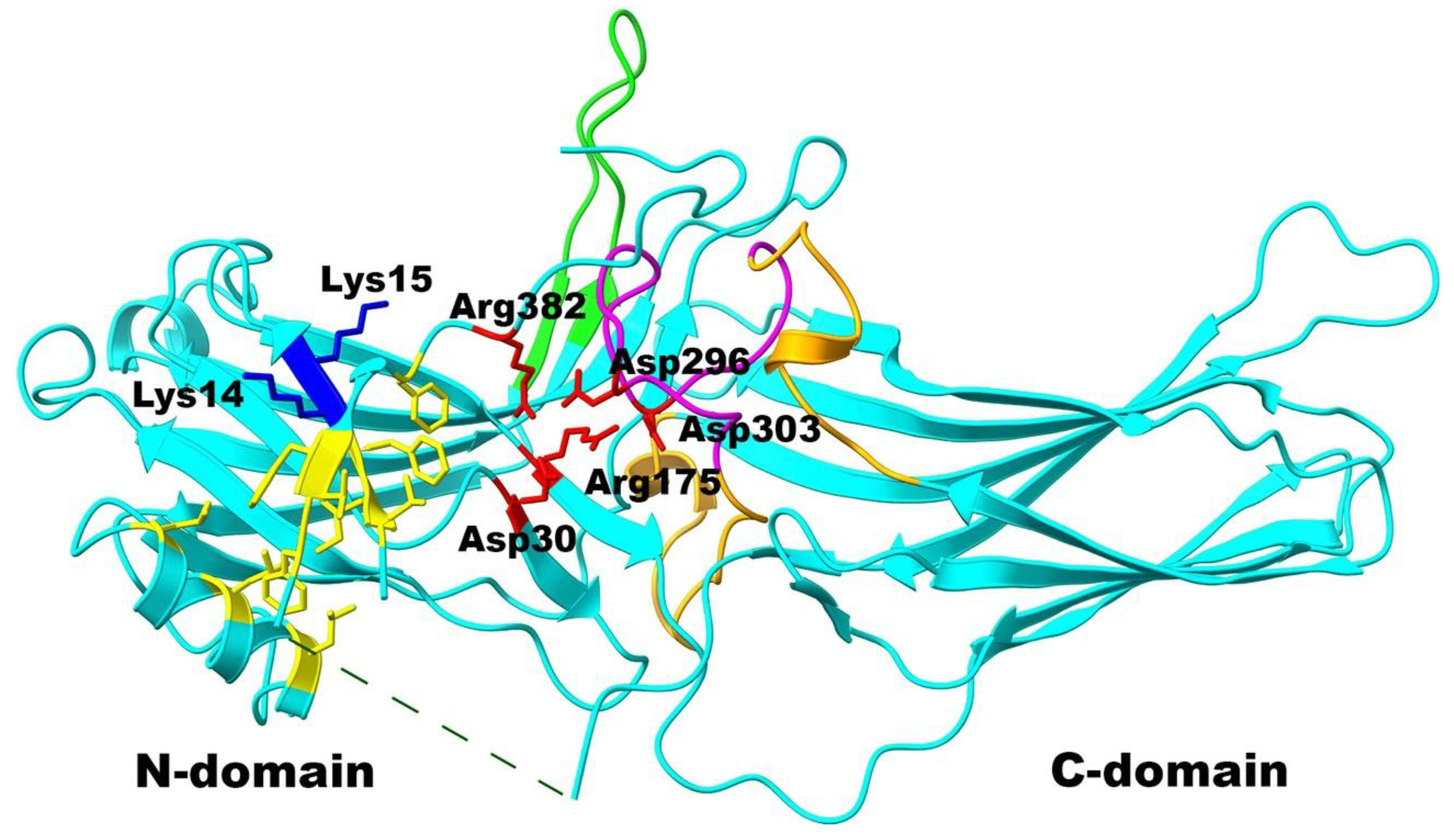
Figure 2.
Gate loop of arrestin-1. A. Crystal structure of basal bovine arrestin-1 (PDB ID 1CF1 [11]). B. Enlarged area shows the residues targeted in this study. C. Crystal structure of mouse arrestin-1-3A bound to rhodopsin (PDB ID 5W0P [17]). D. Enlarged area of the complex shows the residues targeted in this study. In panels A, B, C, and D gate loop is shown in magenta, with side chains of residues shown as stick models. The rest of the protein is shown in green. Surface of arrestin-1 is shown in pale gray. Rhodopsin in panels C and D is shown in yellow. Note that the numbers in mouse arrestin-1 (panel D) are N+1 relative to the bovine protein (panel B). Images were created in UCSF ChimeraX [36,37]. The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 2.
Gate loop of arrestin-1. A. Crystal structure of basal bovine arrestin-1 (PDB ID 1CF1 [11]). B. Enlarged area shows the residues targeted in this study. C. Crystal structure of mouse arrestin-1-3A bound to rhodopsin (PDB ID 5W0P [17]). D. Enlarged area of the complex shows the residues targeted in this study. In panels A, B, C, and D gate loop is shown in magenta, with side chains of residues shown as stick models. The rest of the protein is shown in green. Surface of arrestin-1 is shown in pale gray. Rhodopsin in panels C and D is shown in yellow. Note that the numbers in mouse arrestin-1 (panel D) are N+1 relative to the bovine protein (panel B). Images were created in UCSF ChimeraX [36,37]. The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
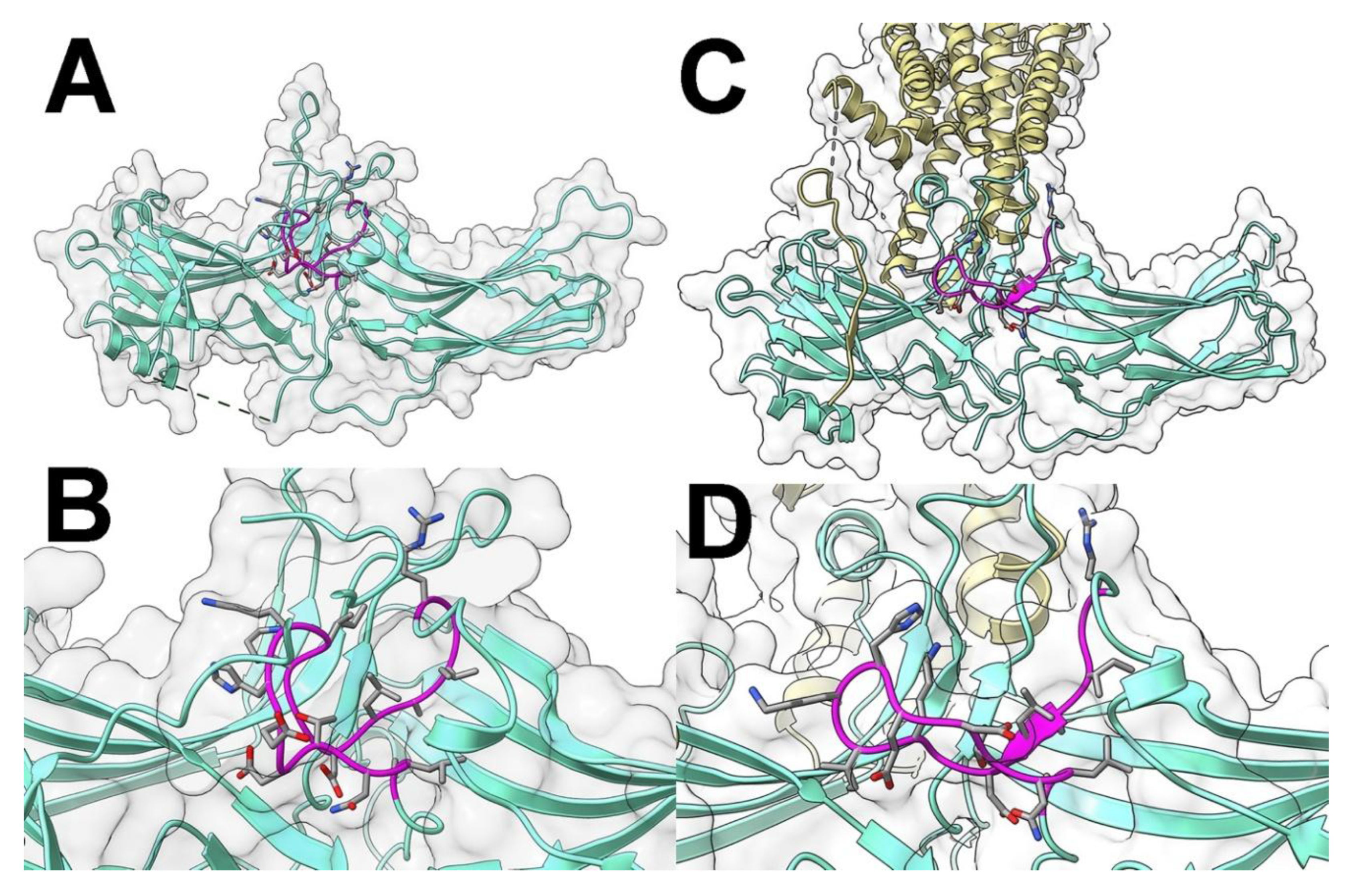
Figure 3.
The native sequence of the gate loop of bovine arrestin-1 and mutations introduced. All residues are color coded, as follows: small non-polar, orange; uncharged polar, yellow; positively charged, blue; negatively charged, red. Glycine residues breaking secondary structures are shown in brown. Deletions are shown in gray.
Figure 3.
The native sequence of the gate loop of bovine arrestin-1 and mutations introduced. All residues are color coded, as follows: small non-polar, orange; uncharged polar, yellow; positively charged, blue; negatively charged, red. Glycine residues breaking secondary structures are shown in brown. Deletions are shown in gray.
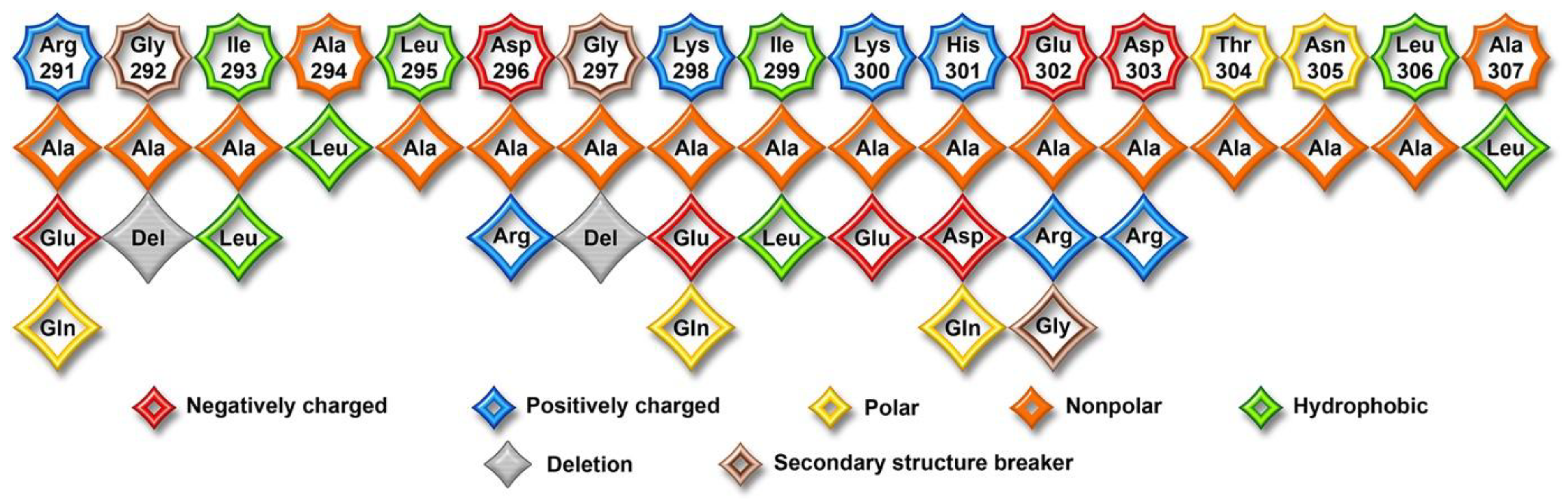
Figure 4.
The effect of gate loop mutations on arrestin-1 binding to rhodopsin. The binding of indicated mutants of arrestin-1 to P-Rh* and Rh* was determined using radiolabeled arrestins, produced in cell-free translation, in the direct binding assay with purified phosphorylated or unphosphorylated light-activated bovine rhodopsin, as described in Methods. Small black circles represent individual measurements (n=6). The binding to P-Rh* and Rh* was analyzed separately in each of the two groups. Statistical significance of the differences between WT arrestin-1 and mutants was determined by one-way ANOVA followed by Dunnet post hoc comparison to WT with correction for multiple comparisons. Statistical significance (p value) is either shown (directly applicable to all bars under the line) or indicated, as follows: *p < 0.05; **, p<0.01; ***, p<0.001 to WT. Bars corresponding to mutants with increased or decreased binding are colored light green and light red, respectively; uncolored bars show no significant difference from WT. Bars corresponding to WT are gray. Panels were created by Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 4.
The effect of gate loop mutations on arrestin-1 binding to rhodopsin. The binding of indicated mutants of arrestin-1 to P-Rh* and Rh* was determined using radiolabeled arrestins, produced in cell-free translation, in the direct binding assay with purified phosphorylated or unphosphorylated light-activated bovine rhodopsin, as described in Methods. Small black circles represent individual measurements (n=6). The binding to P-Rh* and Rh* was analyzed separately in each of the two groups. Statistical significance of the differences between WT arrestin-1 and mutants was determined by one-way ANOVA followed by Dunnet post hoc comparison to WT with correction for multiple comparisons. Statistical significance (p value) is either shown (directly applicable to all bars under the line) or indicated, as follows: *p < 0.05; **, p<0.01; ***, p<0.001 to WT. Bars corresponding to mutants with increased or decreased binding are colored light green and light red, respectively; uncolored bars show no significant difference from WT. Bars corresponding to WT are gray. Panels were created by Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
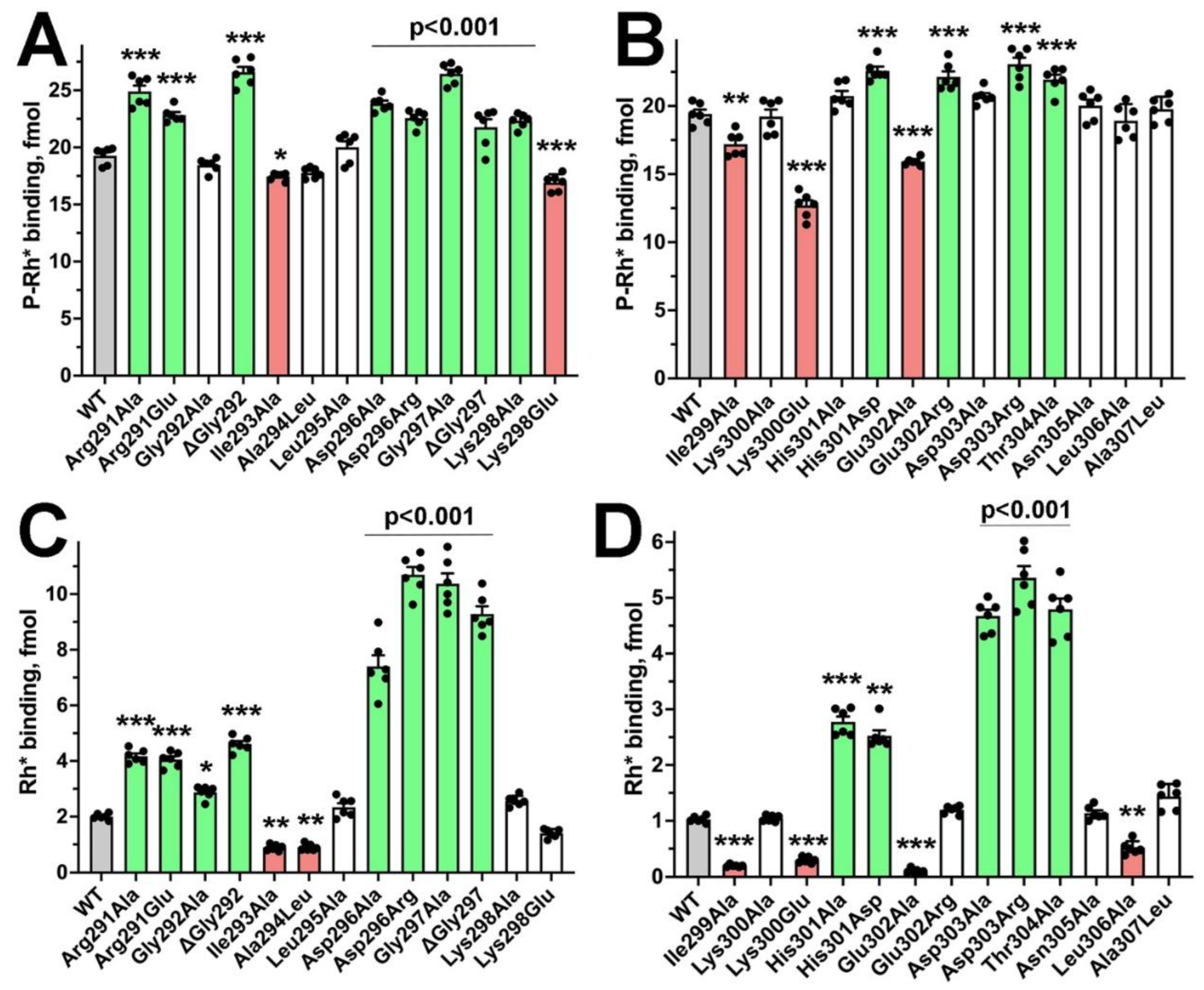
Figure 5.
Thermal stability of mutants. Translation mix containing WT arrestin-1 and indicated mutants was kept on ice (0, control) or incubated at 37oC for indicated times and then cooled on ice, whereupon the binding of all samples to P-Rh* was measured. Plotted survival curves (as % of control) show means +/- SEM of three independent experiments. Statistical analysis of the binding data (in fmol bound) showed that the stability (protein-time interaction in two-way ANOVA) of only D296R mutant was different from WT (p<0.01). Statistical analysis was performed and panels were created in Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 5.
Thermal stability of mutants. Translation mix containing WT arrestin-1 and indicated mutants was kept on ice (0, control) or incubated at 37oC for indicated times and then cooled on ice, whereupon the binding of all samples to P-Rh* was measured. Plotted survival curves (as % of control) show means +/- SEM of three independent experiments. Statistical analysis of the binding data (in fmol bound) showed that the stability (protein-time interaction in two-way ANOVA) of only D296R mutant was different from WT (p<0.01). Statistical analysis was performed and panels were created in Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).

Figure 6.
Conservation of the gate loop sequence. The numbers of the first and last residue in each arrestin are shown in parentheses before and after the sequence in single-letter code, respectively. Conserved residues are shown in red, conservative substitutions in light blue, residues conserved in most, but not all arrestins are shown in dark blue, residues conserved only in arrestin-1 from different mammalian species are shown in green. Three visual arrestins from two invertebrate species, fruit fly Drosophila melanogaster and squid Loligo pealei, as well as arrestins from the round worm C. elegans and tunicate C. intestinalis, are shown for comparison. Black arrows point to the two aspartates of the polar core. The sequences are from: arrestin-1 bovine [6], human [41], mouse [42]; arrestin-2 bovine [43], human [44], mouse [45], arrestin-3 bovine [43], human [46], mouse [45], arrestin-4 bovine [47], human [48], mouse (GeneBank AF156979); Drosophila arrestin1 [49], arrestin2 [50], squid visual arrestin [51]; Drosophila non-visual arrestin kurtz [52]; Caenorhabditis elegans arrestin [53]; Ciona intestinalis arrestin [54]. The figure was created in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 6.
Conservation of the gate loop sequence. The numbers of the first and last residue in each arrestin are shown in parentheses before and after the sequence in single-letter code, respectively. Conserved residues are shown in red, conservative substitutions in light blue, residues conserved in most, but not all arrestins are shown in dark blue, residues conserved only in arrestin-1 from different mammalian species are shown in green. Three visual arrestins from two invertebrate species, fruit fly Drosophila melanogaster and squid Loligo pealei, as well as arrestins from the round worm C. elegans and tunicate C. intestinalis, are shown for comparison. Black arrows point to the two aspartates of the polar core. The sequences are from: arrestin-1 bovine [6], human [41], mouse [42]; arrestin-2 bovine [43], human [44], mouse [45], arrestin-3 bovine [43], human [46], mouse [45], arrestin-4 bovine [47], human [48], mouse (GeneBank AF156979); Drosophila arrestin1 [49], arrestin2 [50], squid visual arrestin [51]; Drosophila non-visual arrestin kurtz [52]; Caenorhabditis elegans arrestin [53]; Ciona intestinalis arrestin [54]. The figure was created in Adobe Photoshop 2025 (Adobe, San Jose, CA).

Figure 7.
Mutations in the gate loop of bovine arrestin-1 mimicking other subtypes. The binding of indicated mutants of arrestin-1 to P-Rh* and Rh* was determined using radiolabeled arrestins, produced in cell-free translation, in the direct binding assay with purified phosphorylated or unphosphorylated light-activated bovine rhodopsin, as described in Methods. Small black circles represent individual measurements (n=6). The binding to P-Rh* and Rh* was analyzed separately in each of the two groups. Statistical significance of the differences between WT arrestin-1 and mutants was determined by one-way ANOVA followed by Dunnet post hoc comparison to WT with correction for multiple comparisons. Statistical significance (p value) is either shown (directly applicable to all bars under the line) or indicated, as follows: ns, not significant; *p < 0.05; **, p<0.01; ***, p<0.001 to WT. The results of pairwise comparisons between indicated mutants are shown in blue, the comparisons of His301Gln+Glu302Gly with His301Gln are shown in red. Bars corresponding to mutants with increased or decreased binding are colored light green and light red, respectively; uncolored bars show no significant difference from WT. Bars corresponding to WT are gray. Panels were created by Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Figure 7.
Mutations in the gate loop of bovine arrestin-1 mimicking other subtypes. The binding of indicated mutants of arrestin-1 to P-Rh* and Rh* was determined using radiolabeled arrestins, produced in cell-free translation, in the direct binding assay with purified phosphorylated or unphosphorylated light-activated bovine rhodopsin, as described in Methods. Small black circles represent individual measurements (n=6). The binding to P-Rh* and Rh* was analyzed separately in each of the two groups. Statistical significance of the differences between WT arrestin-1 and mutants was determined by one-way ANOVA followed by Dunnet post hoc comparison to WT with correction for multiple comparisons. Statistical significance (p value) is either shown (directly applicable to all bars under the line) or indicated, as follows: ns, not significant; *p < 0.05; **, p<0.01; ***, p<0.001 to WT. The results of pairwise comparisons between indicated mutants are shown in blue, the comparisons of His301Gln+Glu302Gly with His301Gln are shown in red. Bars corresponding to mutants with increased or decreased binding are colored light green and light red, respectively; uncolored bars show no significant difference from WT. Bars corresponding to WT are gray. Panels were created by Prism 10 (Graphpad software, Inc., San Diego, CA). The figure was assembled in Adobe Photoshop 2025 (Adobe, San Jose, CA).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



