
Submitted:
29 October 2025
Posted:
31 October 2025
You are already at the latest version
Abstract
One open question regarding plant-microbe interactions is how a plant interacts molecularly with both a beneficial microbe and a pathogenic fungus. This study used RNA-seq to investigate molecular responses in maize roots during a tripartite interaction with the fungal pathogen Fusarium verticillioides (Fv), which causes stalk, ear, and root rot, and the endophytic biocontrol agent Bacillus cereus sensu lato B25, known to suppress Fv and promote plant growth. Roots of seven-day-old maize inoculated with Fv (Zm-Fv), B25 (Zm-B25), and co-inoculated (Zm-Fv-B25) were compared to uninoculated control (Zm). Differential Gene Expression (DEG), Gene Ontology (GO) and KEGG analysis revealed distinct molecular responses. Fv suppressed plant pathways related to DNA and protein synthesis and impaired root development. In contrast, B25 triggered defense preparedness and growth-related responses. In the co-inoculation (Zm-Fv-B25), upregulated DEGs were associated with defense-related metabolic pathways, including jasmonic acid signaling and secondary metabolite biosynthesis, while also promoting plant growth. Co-expression networks using Arabidopsis orthologs supported these findings. This study is the first RNA-seq analysis of maize root molecular responses during the tripartite interaction with a fungal pathogen and its bacterial biocontrol agent, providing new directions for further research to understand the detailed molecular mechanisms underlying this interaction fully.
Keywords:
RNA-seq
; maize transcriptome
; endophyte
; Bacillus cereus
; Fusarium verticillioides
; differentially expressed genes
; biological control
; plant-microbe interactions
1. Introduction
Currently, over a billion tons of maize are produced every year, making it the most abundant cereal crop globally [1]. Maize is susceptible to different fungal pathogens, with Fusarium verticillioides (Fv) being the most common cause of stalk, ear and root rot (SERR) [2,3]. Fv is a versatile fungus that can infect maize either through seeds (vertical transmission), or by environmental causes such as dissemination through the soil, air or natural wounds inflicted by herbivores (horizontal transmission). As a hemibiotroph, Fv can either live harmlessly inside maize as a symptomless endophyte or cause disease as a necrotrophic pathogen [4,5]. Fv not only affects maize yield but also grain quality, due to contamination by mycotoxins [6].
Due to increasing global concerns regarding the environment and the human health effects of synthetic agricultural chemicals (such as pesticides, fungicides, insecticides, and herbicides), there is a strong drive to identify sustainable ways to manage crop diseases [7]. Among such alternatives, the use of biological control agents (BCAs) is not only efficient in controlling target pests and diseases, but they also improve plant growth, development, and ultimately crop yields [8,9]. Bacterial endophytes from the genera Bacillus, Paenibacillus, Burkholderia, Pseudomonas and Staphylococcus have been successfully employed as BCAs to effectively control SERR caused by Fv on maize [10,11,12,13]. Endophytes are non-pathogenic microorganisms that inhabit the plant internal parts for at least part of their life cycle and are generally considered beneficial microorganisms due to their plant growth promotion and plant protection properties [14,15]. These beneficial microorganisms use several mechanisms to promote plant growth and act as biocontrol agents. To enhance plant growth, endophytes can fix nitrogen, solubilize phosphate and potassium, produce siderophores, and synthesize various phytohormones like auxins, cytokinins and gibberellins [14]. On the other hand, these endophytes can employ direct and indirect antagonism mechanisms for biocontrol. Indirect antagonism in biocontrol works by priming the host plant defenses, making them react faster and more powerfully when pathogens attack. In contrast, direct antagonism is achieved through mechanisms like hyperparasitism, predation, and the production of lytic enzymes and antibiotics [14,16].
The advantage that endophytes have over other biocontrol agents is their ability to colonize the internal tissues of plants, which allows them to share the same ecological niche as some phytopathogens inside the plant host, increasing their value as biocontrol candidates [14,15]. As an example of this, the maize endophytic biocontrol agent Bacillus cereus strain B25 colonizes the maize root vasculature [17], just like the phytopathogen Fv [4].
B25 has demonstrated effective control of Fv through in vitro, greenhouse, and field trials [9,13,18]. The genome of B25 possesses genes related to several antagonistic mechanisms against fungal pathogens, including the production of lytic enzymes (chitinases A and B, chitosanase and glycoside hydrolase), siderophores (petrobactin and bacillibactin), antibiotics (surfactin), and biofilm production [19]. These mechanisms have been biochemically and molecularly characterized and appear to be involved in the direct biocontrol of B25 over Fv, as all of them are induced during their direct interaction in vitro [13,20,21,22]. In addition, B25 contributes to the reduction of fumonisin levels and increases maize yield [9].
Although we understand the antagonism mechanisms used by B25 to control Fv (bacteria-fungus interaction) and the maize physiological responses when inoculated with B25 and challenged with Fv, the molecular mechanisms involved in the maize-B25-Fv tripartite interaction have not been investigated at the transcriptional level. To understand this tripartite interaction, and due to the complexity of capturing the molecular communication (mRNA) of microorganisms due to their proportion with respect to maize, this work focuses on the description of the molecular responses of maize regarding its interaction with the bacterial biocontrol agent and the fungal pathogen.
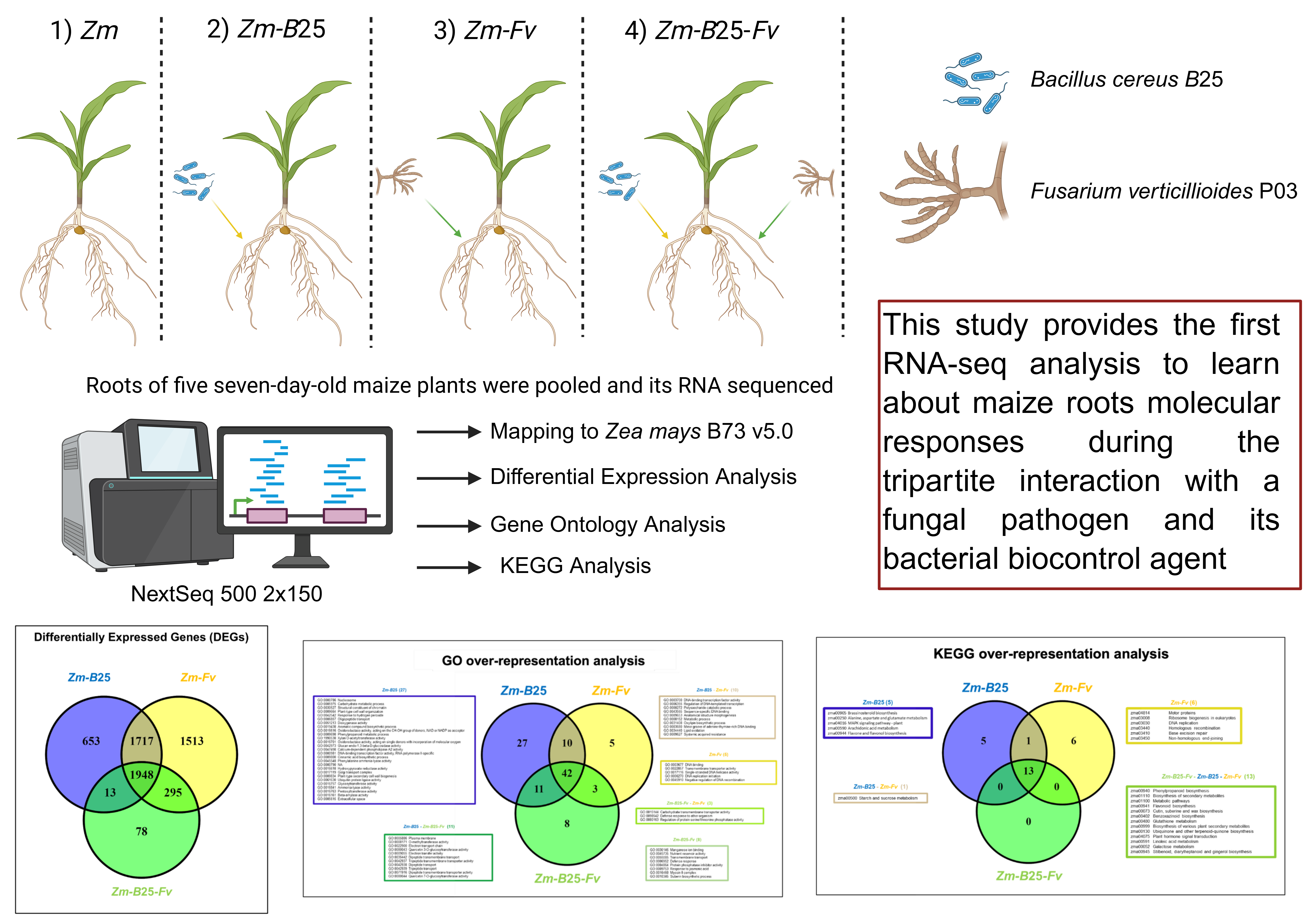
Here, we address the question of how a plant could simultaneously interact with a phytopathogenic fungus and a beneficial organism. Because studying interactions between three organisms is particularly challenging, we developed a model of the tripartite interaction between maize, the phytopathogenic fungus Fv (which causes SERR) and its biological control agent, the endophytic bacterium B25. RNA-seq is a method for understanding the molecular mechanisms underlying certain host-microorganism interactions [23]. By analyzing the maize transcriptome in response to inoculation with 1) the biocontrol endophyte B25 (Zm-B25), 2) the fungal pathogen Fv (Zm-Fv), and 3) both B25 and Fv (Zm-B25-Fv), we have identified key genes and molecular mechanisms involved in the maize-B25-Fv (Zm-B25-Fv) tripartite interaction.
2. Results
2.1. B25 Mediated the Biocontrol of Fv and Enhanced Maize Plant Growth in the Tripartite Interaction
To confirm that the rolled paper seedling bioassay enabled initial host plant colonization, and to assess the physiological responses of the plant to each organism in bi- and tri-partite interactions, we microbiologically verified colonization and measured plantlet biomass and length. Microbial colonization was visually confirmed on disinfected maize roots (Supplementary Figure 1), with no growth on controls (Zm). Bipartite associations (Zm-B25 and Zm-Fv) showed greater microbial growth than the tripartite interaction (Zm-B25-Fv). Fv inoculation (Zm-Fv) significantly reduced root length and fresh weight compared to other treatments (Zm, Zm-B25, Zm-B25-Fv) (Table 1). However, co-inoculation with Fv and B25 (Zm-B25-Fv) promoted a significant increase in leaf length and fresh weight compared to all other conditions (Zm, Zm-B25, Zm-Fv).
2.2. RNA Sequencing of the Tripartite Assay
More than 162 million raw paired-end reads were obtained from the sequencing of the whole tripartite experiment, with an average of 20 million reads per sample (Table 2). After quality trimming (>Q20), an average of 98.49% of the reads was retained per library. Additionally, 95.93% of these filtered reads also achieved a quality score of >Q30. The mean values of unique and multiple mapped reads were 17,703,196 (87.28%) and 758,808 (3.71%), respectively.
A principal component analysis (PCA) was carried out to confirm consistency between biological replicates. One biological replicate from each of the four conditions (Zm, Zm-B25, Zm-Fv and Zm-B25-Fv) showed an atypical behavior and were thus removed, keeping two biological replicates from each condition as described in Materials and Methods section 2.5 (Supplementary Figure 2).
2.3. Global Transcriptome Profiles of Maize Roots from the Tripartite Assay
Expressed genes (EGs) were defined as those averaging at least 10 read counts across two biological replicates. Maize roots in the tripartite assay expressed 24,628 out of 44,303 predicted genes from the Zea mays B73 v5 genome, representing 55.59% of the genome (Table 3). Inoculation of maize roots with B25 and/or Fv induced the expression of a higher percentage of the genome compared to the control (Zm). Interestingly, maize root colonization by B25 (Zm-B25) caused the highest percentage of gene expression, with 57.36% (25,412 genes) of the genome. A comparative analysis across all conditions revealed a core of 22,576 (86.6%) common EGs. However, individual conditions showed distinct profiles: Zm-B25 exhibited the largest number of unique EGs (454, 1.7%), followed by Zm with 254 (1%), Zm-Fv with 215 (0.8%), and Zm-B25-Fv with 189 (0.7%) (Figure 1).

Figure 1.
Venn diagram of common and uniquely expressed genes in the transcriptome of maize roots of the tripartite assay. Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03. .
Figure 1.
Venn diagram of common and uniquely expressed genes in the transcriptome of maize roots of the tripartite assay. Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03. .
Unique EGs from the interaction conditions were subjected to GO analysis. The results showed 11, 8 and 4 overrepresented terms for Zm-B25, Zm-Fv, and Zm-B25-Fv, respectively (Supplementary Table 3). Overrepresented terms within the unique EGs of Zm-B25 were associated with the regulation of transcription and negative regulation of catalytic activity. Additionally, response to auxin also appeared as an overrepresented term in this interaction. On the other hand, overrepresented terms within the set of unique EGs in Zm-Fv were related to photosynthesis regulation, primary metabolic process, cell fate determination, and regulation of transcription. Finally, the terms overrepresented in Zm-B25-Fv unique EGs were associated with restructuring and growth of the actin cytoskeleton (Supplementary Table 3).
2.4. Differentially Expressed Genes Within and Among Maize Interactions
The number of DEGs for the Zm-B25, Zm-Fv, and Zm-B25-Fv conditions was 4,331, 5,473 and 2,334, respectively (Table 4, Supplementary Table 2). The three interactions shared a common core of 1,948 DEGs (Figure 2). The tripartite Zm-B25-Fv interaction showed the fewest unique DEGs (78), in contrast to the bipartite Zm-B25 (653) and Zm-Fv (1,513) interactions, which exhibited more unique DEGs. Furthermore, Zm-B25-Fv shared 295 DEGs exclusively with the Zm-Fv interaction and only 13 with the Zm-B25 interaction, while Zm-B25 and Zm-Fv shared a substantial 1,717 DEGs.
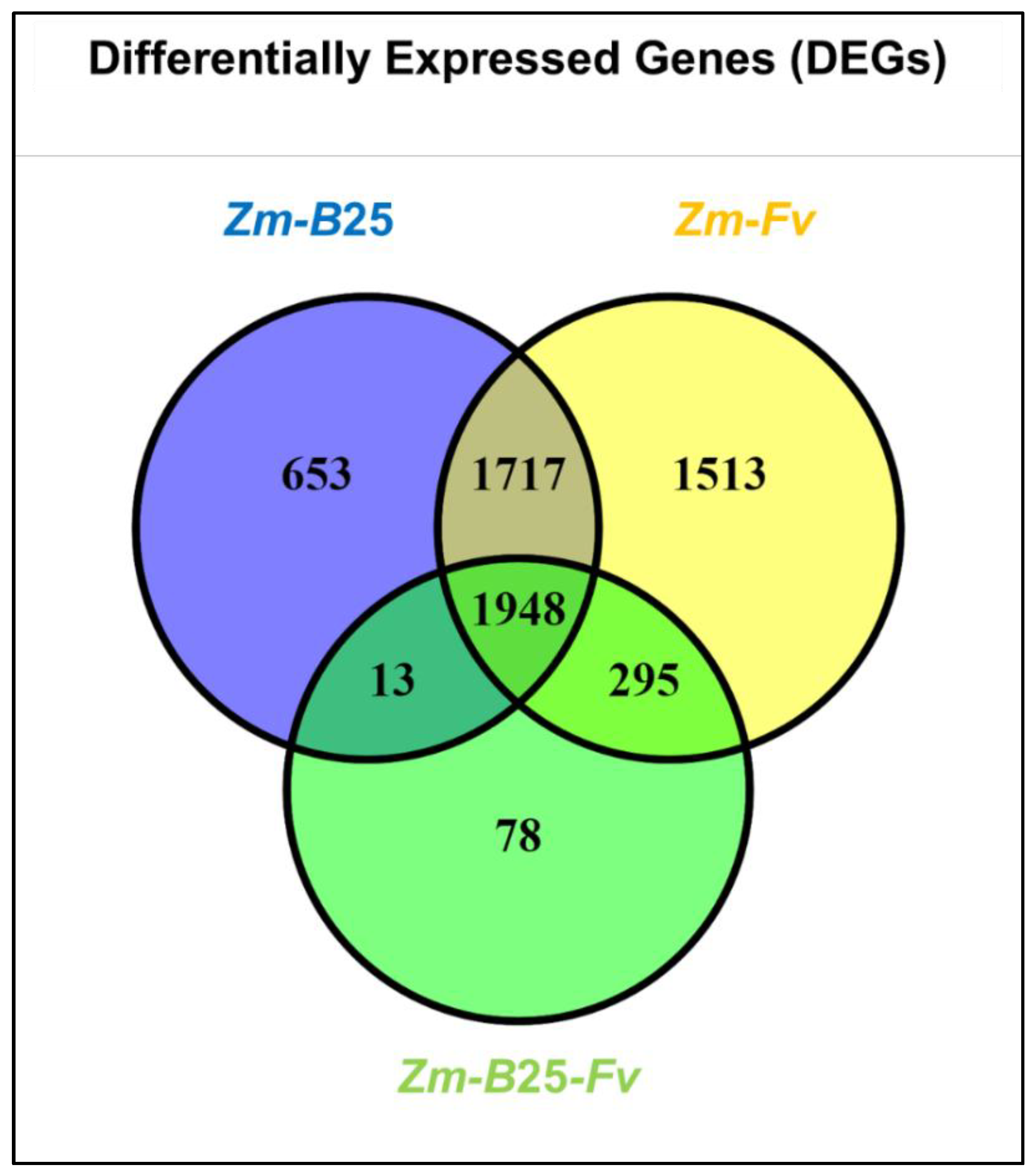
Figure 2.
Differentially expressed genes (DEGs) in the tripartite interaction. Venn diagrams of common and unique DEGs between bi- and tri-partite interactions. Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03.
Figure 2.
Differentially expressed genes (DEGs) in the tripartite interaction. Venn diagrams of common and unique DEGs between bi- and tri-partite interactions. Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03.
In general, the proportion of upregulated genes (>74%) was higher than the downregulated genes for all conditions (Table 4). Bacterial (Zm-B25) and bacterial-pathogen (Zm-B25-Fv) inoculation on maize resulted in gene expression patterns with approximately 89% of upregulated and only 10-11% of downregulated genes. In contrast, 25.64% of genes were repressed in Zm-Fv (Table 4).
By analyzing the number of up- and downregulated genes (both unique and shared) within each interaction, we gained a comprehensive understanding of the transcriptome’s overall behavior in this study. Whereas 6 out of 7 Venn diagram sections displayed a common trend of more upregulated than downregulated genes (data not shown), only unique DEGs in Zm-Fv showed the opposite trend. In Zm-Fv, downregulated genes were more numerous (972; 64.24%) than upregulated genes (541; 35.75%). In summary, Zm-Fv had the highest number of DEGs, accumulating most of the unique DEGs as well as those shared with Zm-B25 and Zm-B25-Fv.
2.5. Overrepresentation Analysis: A General Approach
GO analysis revealed a significant overrepresentation (adjusted p-value <0.05) of 64 categories in the Zm-B25-Fv interaction, 90 categories in the Zm-B25 interaction, and 60 categories in the Zm-Fv interaction (Figure 3; Supplementary Table 4). The Zm-Fv interaction contained the highest number of DEGs (5,473), but it showed the lowest number of overrepresented categories. Overall, the overrepresented categories belonged mainly to Molecular Function (MF) with 54, followed by Biological Process (BP) with 38, and Cellular Compartment (CC) with 10. The three conditions shared 42 overrepresented categories. Notably, Zm-B25 exhibited the highest number of unique overrepresented categories with 27, followed by Zm-B25-Fv with 8, and Zm-Fv with 5 (Figure 3).
Among the eight unique and overrepresented GO terms for Zm-B25-Fv, four corresponded to the BP category defense response (GO:0006952), and response to jasmonic acid (GO:0009753) appeared together with suberin biosynthetic process (GO:0010345) and transmembrane transport (GO:0055085) (Supplementary Table 4). The myosin II complex (GO:0016460) was the sole overrepresented term identified in the CC category, whereas nutrient reservoir activity (GO:0045735), protein phosphatase inhibitor activity (GO:0004864) and manganese ion binding (GO:0030145) were found in the MF category (Supplementary Table 4).
For the Zm-B25 interaction, the unique overrepresented GO terms in the BP category included those related to plant growth and development, such as aromatic compound biosynthetic process (GO:0019438), phenylpropanoid and carbohydrate metabolic process (GO:0009698 and GO:0005975) and plant-type secondary cell wall biogenesis (GO:0009834). In the MF category, a diverse set of overrepresented terms were identified, primarily related to transcription regulation (GO:0000981), catalysis of O-glycosil compounds (GO:001616 and GO:0042973), and transferase activity (ubiquitin (GO:0061630), as well as the pentosyl (GO:0016763), glycosil (GO:0016757) and acyl groups (GO:1990538), oxido-reductase activity (GO:0016616, GO:0016618, GO:0016701 and GO:0051213), calcium-dependent phospholipase A2 activity (GO:0047498), structural constituent of chromatin (GO:0030527), and ammonia-lyase activity (GO:0016841). The overrepresented terms for the CC category included extracellular space (GO:0005615), nucleosome (GO:0000786), chromatin (GO:0000785), and Golgi transport complex (GO:0017119).
In the Zm-Fv interaction, four out of the five unique overrepresented GO terms were associated with DNA replication (GO:0003677, GO:0017116, GO:0006270 and GO:0045910).
Interestingly, Zm-B25 and Zm-Fv shared 10 overrepresented GO terms (Figure 3, brown box). Several terms stood out within these overrepresented GO terms, such as those associated with transcription (GO:0003700, GO:0006355, GO:0043565), oxylipin biosynthesis (GO:0031408), and the systemic acquired resistance response (GO:0009627).
2.6. KEGG Analysis
KEGG analysis revealed 13, 19, and 20 overrepresented pathways in the Zm-B25-Fv, Zm-B25, and Zm-Fv interactions, respectively (Figure 4, Supplementary Table 5). Only Zm-B25 (5) and Zm-Fv (6) exhibited uniquely overrepresented KEGG pathways. The five unique pathways identified in Zm-B25 were associated with plant growth and development (zma00905: brassinosteroid biosynthesis), signaling (zma00590: arachidonic acid metabolism, zma04016: MAPK signaling pathway - plant), amino acid metabolism (zma00250: alanine, aspartate and glutamate metabolism), and production of secondary metabolites (zma00944: flavone and flavonol biosynthesis). On the other hand, five out of the six unique overrepresented KEGG pathways in the Zm-Fv interaction were related to DNA replication and maintenance, including DNA replication (zma03030), homologous recombination (zma03440), base excision repair (zma03410), and non-homologous end-joining (zma03450). Additionally, ribosome biogenesis in eukaryotes (zma03008) was a unique overrepresented pathway. The remaining unique KEGG pathway was motor proteins (zma04814) (Figure 4, Supplementary Table 5).
The 13 KEGG overrepresented pathways common to all conditions were primarily linked to the biosynthesis of different secondary metabolites and the metabolism of glutathione, linoleic acid, and galactose. The starch and sucrose metabolism (zma00500) pathway was only altered when maize interacted with either B25 or Fv individually, but not when both microorganisms were present within the plant.
2.7. Functional Gene Association Networks: Enriched Biological PROCESSES using A. thaliana Orthologous Genes
Although the GO overrepresentation analysis based on maize gene annotations from BioMart provided insight into each interaction as described above, an additional GO enrichment analysis was conducted using A. thaliana orthologous genes (Supplementary methods, Supplementary Table 6) to examine the governing mechanisms behind each interaction. Using up- and downregulated genes from each interaction and intersection between them (Figure 2), we built gene network associations to understand the functional relationship among DEGs. Supplementary Table 7 shows the main results obtained from the analysis of NRGIs using String.
Analysis of NRGIs revealed the formation of diverse networks for each maize interaction (Supplementary Table 7). Even though the Zm-B25-Fv/Zm-Fv/Zm-B25 (UP) gene set harbored the highest number of nodes (874), the Zm-Fv (DOWN) gene set (678) exhibited a higher number of edges (5866) than the former one (2560). Gene sets that produced a main network with fewer than 200 nodes were either expanded via String or submitted to Genemania as a complementary approach to identify enriched pathways. Significant associations for Zm-B25-Fv (UP), Zm-B25-Fv/Zm-B25 (UP) and Zm-B25-Fv/Zm-B25 (DOWN) were only detected after the String network expansion and Genemania analysis (Supplementary Table 7).
Overall, instead of evaluating individual genes, obtaining BPs from gene association networks allowed us to clearly and comprehensively identify the most prominent genes associated with each maize interaction (Figure 5). For all cases in which Zm-Fv appeared (4 out of 7; see sections Zm-B25-Fv/Zm-Fv/Zm-B25, Zm-Fv/Zm-B25, Zm-Fv and Zm-B25-Fv/Zm-Fv), the response to the stress-related stimulus (chemical) was the prevalent process among upregulated genes, whereas the process of DNA-related metabolism stood out among downregulated genes (Supplementary Table 7). Both processes covered 5473 DEGs (88.03%) out of the total 6217 DEGs. Even though the Zm-B25-Fv/Zm-Fv/Zm-B25, Zm-Fv/Zm-B25, Zm-Fv and Zm-B25-Fv/Zm-Fv interactions shared similar BPs within upregulated genes, Zm-B25-Fv/Zm-Fv exhibited exclusive BPs such as photosynthesis, generation of precursor metabolites and energy, and small molecule metabolic process, reinforcing the previous observation that Zm-B25-Fv/Zm-Fv differed slightly from the other interaction sections in which Zm-Fv was involved.
The remaining 744 DEGs (11.97%) clustered into the last three sections (Zm-B25, Zm-B25-Fv and Zm-B25-Fv/Zm-B25). BPs associated with upregulated genes were the most contrasting compared to 88.03% of DEGs. For example, Zm-B25 harbored exclusive BPs such as carbohydrate metabolic process, cell wall organization or biogenesis, and regulation of macromolecule biosynthetic process, whereas Zm-B25-Fv/Zm-B25 showed overrepresented processes such as phospholipid biosynthetic process and inositol metabolic process (Supplementary Table 7). On the other hand, Zm-B25-Fv featured BPs such as ribosome biogenesis, gene expression, rRNA processing, cellular nitrogen compound metabolic process, ribosome assembly, macromolecule metabolic process and translation. Finally, it is important to note that Zm-B25 and Zm-B25-Fv also showed DNA-related metabolism processes among their downregulated genes, which differed sharply from Zm-B25-Fv/Zm-B25 that featured BPs such as phosphatidylinositol-3-phosphate biosynthetic process, protein targeting mitochondrion, protein transmembrane transport, vacuole organization, photomorphogenesis and autophagy-related processes (Supplementary Table 7). Functional gene association networks therefore provide an overall insight into the transcriptomic behavior covering each section and intersection of maize interactions.
Figure 5.
Enriched biological processes emerging after applying the Markov clustering (MCL) based on co-expression.
Figure 5.
Enriched biological processes emerging after applying the Markov clustering (MCL) based on co-expression.
2.8. Gene Interaction Networks Based on Co-Expression
Gene associations often result in highly connected networks, with little informative power. Therefore, clustering them through algorithms allows for a better interpretation (Supplementary methods). The largest identified clusters turn out to be highly relevant to the main findings of the original network. Therefore, to gain more insight into previously described functional networks (Supplementary Table 7), Markov clustering (MCL) was performed based on co-expression using A. thaliana orthologs. Interactions involving Zm-B25-Fv, Zm-B25 and Zm-Fv, as well as their corresponding intersections (Zm-B25-Fv/Zm-B25, Zm-B25-Fv/Zm-Fv and Zm-B25/Zm-Fv) were considered for this analysis.
The Zm-B25/Zm-Fv intersection harbored a main cluster among its upregulated genes related to stress response (response to chemicals and chitin) (Supplementary Table 7), cell wall remodeling (biosynthesis of secondary cell wall), phenylpropanoid metabolic process, oxoacid metabolic process, hormone-mediated signaling pathway (auxin) and photosynthesis. In Zm-B25, chromatin remodeling, gene expression, protein metabolic process (translation), nucleocytoplasmic trafficking of RNAs and proteins, phenylpropanoid-related metabolic process, hormone-mediated signaling pathway (auxin), protein phosphorylation (involved in growth and development), and thermotolerance were all exclusive to this interaction. BPs exclusive to the Zm-Fv interaction were found, such as response to external stimuli (bacteria and fungi), glycolysis, photosynthesis, carboxylic acid metabolic process (oxidative phosphorylation), very-long-chain fatty acid (VLCFA) biosynthesis, transition metal ion transport, pigment metabolic process, nitrate assimilation, cellular detoxification, proteasomal protein catabolic process, and circadian rhythm (Supplementary Table 7). Noteworthy, the cell wall biogenesis process, specifically the biosynthesis of secondary cell wall, was shared between Zm-B25 and Zm-Fv.
Zm-B25-Fv exclusive BPs as well as BPs shared with Zm-B25 and Zm-Fv were analyzed. First, upregulated Zm-B25-Fv genes exhibited overlapping BPs with Zm-B25, such as lipid metabolic processes (phospholipid biosynthetic process and inositol metabolic process), L-ascorbic acid biosynthetic process, transcription regulation of SA- (positive) and JA- (negative) related genes, and melatonin biosynthetic process. Secondly, the BPs shared with Zm-Fv included cellular response to oxidative stress (chemical), generation of precursor metabolites and energy (photosynthesis and glycolysis), cellular aldehyde metabolic process (related to oxidative stress tolerance), cellular detoxification, mRNA polyadenylation, VLCFA biosynthesis, and plant hormone signal transduction (ABA). Altogether, the above-described BPs allowed us to identify those exclusive BPs corresponding to Zm-B25-Fv, such as ribosome biogenesis (ribosome assembly), actin filament depolymerization and polysaccharide catabolic process (stress-induced starch degradation) (Supplementary Table 7). On the other hand, protein metabolic (translation) and lipid metabolic (phospholipid biosynthetic process) processes found in the Zm-B25-Fv interaction were also present in Zm-B25 and Zm-B25-Fv/Zm-B25, respectively. However, it is important to note that the NRGIs involved in both BPs were not the same as between Zm-B25-Fv and Zm-B25, or between Zm-B25-Fv and Zm-B25-Fv/Zm-B25.
Although clusters formed by downregulated genes involved DNA metabolic processes in general, peculiarities of their interactions and intersections could be observed when compared to cluster-associated BPs of upregulated genes. For example, whereas Zm-Fv exhibited BPs associated with response to heat and protein folding in their downregulated genes, such BPs were among the upregulated Zm-B25 genes. Furthermore, BPs associated with the process of translation (ribosome biogenesis, rRNA processing, and ncRNA metabolic process) identified among the downregulated genes of Zm-Fv and Zm-B25 (as well as the Zm-B25/Zm-Fv intersection) were found among the upregulated genes of Zm-B25-Fv.
In addition to the identified exclusive BPs, other processes were also associated with the Zm-B25-Fv tripartite interaction, such as cellular response to oxidative stress and its corresponding counteracting mechanisms (transcription regulation of hormone-related genes [SA and JA], plant hormone signal transduction [ABA], cellular aldehyde metabolic process, and cellular detoxification involving the glutathione-ascorbic acid redox cycle). Changes occurring in the plasma membrane and plant cuticle are suggested by the identification of lipid metabolic processes (phospholipid biosynthetic process, inositol metabolic process and VLCFAs). Finally, the generation of precursor metabolites and energy (photosynthesis, glycolysis, and stress-induced starch degradation) also emerged as important processes occurring in this interaction. In summary, the Zm-B25-Fv interaction seems to harbor BPs related to plant growth and development (gene expression, RNA metabolic process, ribosome biogenesis, translation, actin filament depolymerization, photosynthesis and glycolysis), as well as BPs associated with plant protection triggered by pathogens (changes in plasma membrane and cuticle, as well as cellular response to oxidative stress), which may help explain the B25-mediated maize protection against Fv.
2.9. Validation of RNA Sequencing by qRT-PCR
To validate the RNA-seq data, eleven unique DEGs were selected based on their FC (high and/or low) from the interactions, and their expression profiles were evaluated by qRT-PCR. As indicated in Table 5, FC values obtained by qRT-PCR analysis displayed similar expression trends as in the RNA-seq data.
3. Discussion
Bacterial endophytes, which are abundant in plants, are currently being extensively studied due to their plant growth-promoting effect and their ability to protect plants against diseases. A deeper understanding of not only the bacterial endophyte-host plant interaction, but also the fungal pathogen-host plant interaction and the tripartite interaction between the bacterial endophyte, the fungal pathogen and the host plant can enhance the use of bacterial endophytes to overcome plant disease and improve agricultural production [15,16,24].
Previous studies have demonstrated the potential of B25 to protect maize plants against Fv, and to promote their growth at early [13,18] and late growth stages [9]. To better understand the molecular basis of the biocontrol and plant growth promotion induced by B25 on maize against the fungal pathogen Fv, we analyzed the transcriptomic changes of maize roots obtained at seven days after seed inoculation with B25 and Fv, either individually or combined. The expression patterns of a number of DEGs were validated (Table 5) in this work, complementary to a previous report in which ten induced maize chitinase DEGs were validated from the Zm-Fv condition [25]. To the best of our knowledge, this study represents the first report of a maize root transcriptome in response to the dual inoculation of two endophytes, i.e. a bacterial biocontrol agent and a fungal pathogen.
Plant-bacteria interaction (Zm-B25): B25 is recognized by maize as a beneficial microorganism
A core of 1,961 DEGs was shared among all conditions. Of these, only the gene Zm00001eb141540 (geb1-glucan endo-1,3-beta-glucosidase homolog1), a β-1,3-glucanase, presented a contrasting pattern of expression in maize interacting with the different microorganisms (Supplementary Table 2). This gene was upregulated in the Zm-B25-Fv and Zm-Fv interactions, where Fv was present (Log2FC of 2.46 and 2.76, respectively), whereas it was downregulated in the Zm-B25 interaction (Log2FC of -2.93). Plant β-1,3-glucanases have a key role in plant–microbe interactions, and several β-1,3-glucanases are included in the pathogenesis related (PR) group 2 of proteins. This type of β-1,3-glucanase accumulates during a pathogen attack, and some of them have been directly involved in the hydrolysis of pathogen cell walls, since β-1,3-glucans are found in bacteria, metazoa, viruses, and particularly fungi [26]. Thus, the upregulation of Zm00001eb141540 in maize roots in the presence of the fungus, and its downregulation when bacteria are present, could suggest that maize perceives the endophytic bacteria B25 as a non-threatening microorganism. Further analysis is necessary to demonstrate the role of Zm00001eb141540 in the Zm-B25 association.
Moreover, overrepresented unique GO terms in Zm-B25 were related to the establishment of a beneficial plant-bacteria interaction (Figure 3), including carbohydrate metabolic processes (GO:0005975), oligopeptide transport (GO:0006857), and phenylpropanoid metabolic processes (GO:0009698); these compounds have been reported as chemo attractants in plant roots (Villarino et al., 2021). Plant roots can secrete organic exudates into their surroundings, which attract beneficial microorganisms, establishing a favorable plant-bacteria interaction (Sasse et al., 2018). Furthermore, the GO term calcium-dependent phospholipase A2 activity (GO:0047498) was exclusively overrepresented in Zm-B25. A key process in the beneficial plant-bacteria interaction is the increase in calcium flow and reactive oxygen species, leading to lipid oxidation in the cell membrane, indicating a favorable relationship (Gowtham et al., 2022). The GO term lipid oxidation (GO:0034440) appeared between the overrepresented GO terms in Zm-B25, but also in Zm-Fv. Quercetin 3-O-glucosyltransferase (GO:0080043) and quercetin 7-O-glucosyltransferase (GO:0080044) activities appeared as overrepresented GO terms only in those interactions where B25 was present (Zm-B25 and Zm-B25-Fv) (Figure 3). Quercetin is a flavonol from the flavonoid subclass that is linked to the establishment of plant interactions with other beneficial organisms, including the arbuscular mycorrhizal association and the symbiosis between legumes and nitrogen-fixing rhizobia (Singh et al., 2021; Zhang et al., 2013). Flavonoids also work as communication mediators between PGPRs and plants, suggesting that they might play a key role in these interactions (Wang et al., 2022). Taking these biological processes together may help to explain how maize plants perceive the B25 bacterium as a beneficial microorganism.
Plant-bacteria interaction (Zm-B25): B25 induces the activation of pathways related to plant growth, development, and the defense response
Our study revealed the induction of several unique KEGG pathways during the interaction between maize and B25 (Zm-B25) at 7 dpi. These pathways, mainly related to plant growth and development and defense responses, include brassinosteroid biosynthesis; alanine, aspartate and glutamate metabolism; flavone/flavonol biosynthesis; plant MAPK signaling pathway; and arachidonic acid metabolism.
Zhang et al., (2022) showed that the exogenous addition of brassinosteroids (BRs) to maize promotes root growth, enhances stress tolerance, and increases grain yield. Specifically, root development was attributed to the regulation of BRs via auxin, implicating the upregulation of the ZmWOX5, ZmBBM1 and ZmBBM2 homologs of PLT1 and PLT2, as well as the auxin transporter genes ZmPIN2, ZmPIN3a and ZmPIN3b and the positive auxin response factors (ARF) that promote the initiation of lateral root primordia in ZmARF7 and ZmARF19. None of these genes were induced within the unique DEGs in Zm-B25; instead, Zm00001eb433020 (arftf29-ARF-transcription factor 29) was downregulated (Log2FC= -1.55), and Zm00001eb026490 (aic1-auxin import carrier1) was upregulated (Log2FC= 1.23). Nonetheless, four genes related to auxin response were identified within the unique EGs in Zm-B25: Zm00001eb321090 (saur66 - small auxin up RNA66), Zm00001eb070660 (saur17 - small auxin up RNA17), Zm00001eb321100 (saur67 - small auxin up RNA67), and Zm00001eb218080 (umc2293 - auxin responsive protein). These four genes belong to the small auxin-up RNA (SAUR) gene family, which itself forms part of three different early auxin-responsive gene families [31]. SAUR genes are involved in the auxin signaling pathway, and their expression can be induced within 2–5 min by active auxin [31,32]. These observations suggest that the expression of SAUR genes mediated by B25 could play a key role in the regulation of BRs, to promote maize growth.
Amino acids play vital roles in the central metabolism of plants, as they are involved in physiological processes that are directly or indirectly linked to the synthesis of diverse metabolites [33]. Amino acids and their derivatives have prominent functions in plants, such as protein synthesis, growth and development, nutrition, and stress responses (Hildebrandt et al., 2015). Among other amino acids, glutamate, aspartate, and alanine are used in the formulation of biostimulants [34]. Alfosea-Simón et al., 2021 studied the effects of exogenous applications of these three amino acids on tomato growth, and found a positive effect of aspartic and glutamic acid on plant growth. Foliar application of glutamine (Gln) at the V6, V8, V12, R1 (silk), and R3 (dough) stages in maize hybrid ZD958 impacts grain yield and nutritional quality, enhancing grain yield by 20% under sufficient N, and by 38% under low N (Slam et al., 2024). In this work, non-significant differences at an early stage of maize development (7 dpi) were observed between Zm-B25 and the control condition (Zm), related to the evaluated growth parameters (Table 1). This is possibly due to the developmental stage of the evaluated plantlets. Nonetheless, previous field work demonstrated that maize inoculation with the biocontrol agent B25 significantly increased plant growth and grain yield in several field sites and crop seasons, compared to untreated (Zm) and Fv-inoculated controls (Zm-Fv) [9]. B25 may induce the activation of glutamate, aspartate and alanine metabolic pathways as a key mechanism in maize to promote growth and increase grain yield. It is possible that B25-mediated induction of amino acid metabolism at an early growth stage (7 dpi) remains induced at later maize developmental stages, but further maize transcriptome analysis at later developmental stages will be necessary to confirm this hypothesis.
Mitogen-activated protein kinases (MAPKs) play essential roles in plant growth and development by regulating both cell differentiation and proliferation [37]. MAPKs are also important signaling modules in eukaryotes, with key roles in regulating responses to four major environmental stresses (high salinity, drought, extreme temperature, and insect/pathogen infections) via gene expression regulation, plant hormone production, and crosstalk between environmental stresses (Lin et al., 2021). In the Zm-B25 interaction, 29 DEGs were found in the overrepresented MAPK plant signaling pathway, with most of them related to stress adaptation, defense response, pathogen defense and late pathogen defense response (data not shown). All of these DEGs were upregulated, except for Zm00001eb133330 (Mitogen-activated protein kinase kinase 3).
In addition to the MAPK plant signaling pathway, arachidonic acid metabolism also appeared as an overrepresented KEGG pathway in the Zm-B25 interaction, with five upregulated DEGs. Arachidonic acid (AA) is an evolutionarily conserved signaling molecule that modulates plant stress signaling networks, and which acts as an elicitor of pathogen defense response and plant programmed cell death [39,40]. Dye & Bostock, (2021) demonstrated that the exogenous addition of AA to the roots of tomato and pepper seedlings protects them against root and crown rot caused by Phytophtora capsici. Lewis et al., (2023) showed that tomato roots treated with AA altered the tomato metabolome with an enrichment of chemical classes and the accumulation of metabolites associated with defense-related secondary metabolism. Among the five upregulated DEGs found in this overrepresented KEGG pathway, Zm00001eb184300 ((+)-neomenthol dehydrogenase) was the second-most upregulated gene (log2FC= 3.2). The menthone reductase gene CaMNR1 in Capsicum annuum and its ortholog AtSDR1 in Arabidopsis thaliana were observed to positively regulate plant defenses against a broad spectrum of pathogens. Specifically, CaMNR1-overexpressing Arabidopsis plants exhibited enhanced resistance to the hemibiotrophic pathogen Pseudomonas syringae pv. tomato DC3000 and the biotrophic pathogen Hyaloperonospora parasitica isolate Noco2, whereas a mutation in AtSDR1 significantly enhanced susceptibility to both pathogens [43]. This suggests that the maize gene Zm00001eb184300 in the Zm-B25 interaction could be implicated in preparing plant defense responses against pathogen attack.
The induction of flavone/flavonol biosynthesis detected by KEGG analysis in the Zm-B25 interaction could contribute to plant growth and development as well as the defense response, as this plant-specialized secondary metabolite performs many functions in plants, including plant color development, cell growth regulation, attraction to pollinators, and protection against biotic and abiotic stresses (Alseekh et al., 2020; Dias et al., 2021; Wang et al., 2021).
Some other unique GO terms significantly overrepresented in Zm-B25 were related to plant defense and growth promotion. These include the cinnamic acid biosynthetic process (GO:0009800), which is involved in the biosynthesis of secondary metabolites related to plant defense and acts as an antioxidant, promoting plant growth (Jiménez-Gómez et al., 2020); the plant-type secondary cell wall biogenesis (GO:0009834); and plant-type cell wall organization (GO:0009664). The last two GO terms may contribute to optimal cell wall development and the providing of mechanical resistance (Dauwe et al., 2007). Zm00001eb399600 (allene oxide synthase, aos1) was a unique upregulated DEG (Log2FC of 2.09) in Zm-B25. This defense-related gene is overexpressed in maize plants when they come into contact with the PGPR Klebsiella jilinsis (Zhang et al., 2021).
Plant-pathogenic fungus interaction (Zm-Fv): Fv turns off DNA replication and repair, and negatively affects plant growth and development
The genomic integrity of every organism is constantly challenged by endogenous and exogenous DNA-damaging factors, and maintaining this integrity is crucial to the proper development of all living organisms and the faithful transmission of genetic information from one generation to the next [47,48]. The most important sources of DNA damage are endogenous, including oxidative metabolism, spontaneous hydrolytic reactions, and replication machinery errors, although environmental factors such as ionizing and UV radiation, and genotoxic biogenic or industrial chemicals, also contribute significantly [49]. Because plants are attached to their substrate, they cannot avoid these stresses. Therefore, proper sensing of DNA damage and the precise activation and functioning of the DNA repair machinery is of foremost importance in preserving genome integrity [50]. In the present work, our results highlight that the main effect of Fv in maize roots is the downregulation of genes related to DNA replication and maintenance. Both KEGG and GO enrichment analyses (Figure 3 and Figure 4) confirmed the enrichment of unique pathways (motor proteins, ribosome biogenesis in eukaryotes, DNA replication, homologous recombination, base excision repair, and non-homologous end-joining) and GO terms (DNA binding, transmembrane transporter activity, single-stranded DNA helicase activity, DNA replication initiation, and negative regulation of DNA recombination) associated with DNA replication and repair processes. Co-expression network analysis further supports this observation, as it revealed DNA replication and maintenance within the modules with downregulated, significantly enriched terms only in the Zm-Fv condition (Supplementary Tables 6 and 7). Moreover, an independent GO analysis assessing the unique downregulated DEGs (809 out of 1,513 DEGs) in this condition confirmed the overrepresentation of the following GO terms related to DNA metabolism: organelle organization, chromosome organization, DNA metabolic, nucleic acid metabolic process, and cellular component organization or biogenesis (Supplementary Table 7).
One key mechanism of DNA repair processes is the DNA Damage Response (DDR). This mechanism activates the DNA repair pathways BER (base excision repair), NER (nucleotide excision repair), and MMR (mismatch repair mechanism) for single-strand breaks, as well as HR (Repair–Homologous Recombination) and NHEJ (Non-Homologous End Joining) for double-strand breaks (DSB) [48,49,50]. We observed that both single-strand and double-strand DNA repair processes were affected by Fv in maize roots in the Zm-Fv condition. Five DEGs associated with the Non-Homologous End Joining Pathway were downregulated: Zm00001eb116810 (ATP-dependent DNA helicase 2 subunit KU70), Zm00001eb371940 (DNA polymerase lambda (POLL)), Zm00001eb181120 (DNA repair protein RAD50), Zm00001eb069270 (double-strand break repair protein MRE11), and Zm00001eb071490 (putative DNA ligase 4). Similarly, a total of 18 DEGs in the Homologous Recombination Pathway were also downregulated, including genes coding for DNA polymerase I A (Zm00001eb431790) and delta (Zm00001eb201340 and Zm00001eb002170), DNA repair proteins (Zm00001eb255780 and Zm00001eb181120), and DNA topoisomerases (Zm00001eb005010 and Zm00001eb317420), as well as 17 DEGs in the DNA Replication Pathway.
Fv affected maize root growth and development by turning off the pathways of ribosome biogenesis and motor proteins (Figure 4). From a total of 31 DEGs related to the ribosome biogenesis pathway in the Zm-Fv condition, only Zm00001eb332570 (eukaryotic translation initiation factor 6) appeared as upregulated, while the rest were downregulated. Given the central function of ribosomes as the molecular machines responsible for protein synthesis, ribosome biogenesis is tightly linked with plant growth and development [51] and thus also the correct development of plant roots. Moreover, DEGs coding for kinesin (1 DEG), kinesin like-protein (11) and kinesin-related protein (4) were downregulated, along with two myosin-17 genes. Kinesins and myosins are motor proteins that can move actively along microtubules and actin filaments, respectively, coordinating diverse cellular functions in plants [52]. Kinesins are responsible for unidirectionally transporting various cargos, including membranous organelles, protein complexes, and mRNAs; they also play critical roles in mitosis, morphogenesis, and signal transduction, and contribute directly or indirectly to cell division and cell growth in various tissues (Li et al., 2012). In contrast, myosin drives cytoplasmic streaming, actin organization, and cell expansion, thus performing supporting roles in plant growth, environmental responses, and the defense against pathogens[54].
The tripartite interaction Zm-B25-Fv: induction of defense response genes and plant growth development
A common molecular response was found to be derived from the interactions of maize roots with 1) B25 (Zm-B25); 2) Fv (Zm-Fv); and 3) both microorganisms (Zm-B25-Fv). This response included 22,576 core EGs (Figure 1), 1,948 core DEGs (Figure 2), 42 core overrepresented GO terms (Figure 3, Supplementary Table 4), and 13 core overrepresented KEGG pathways (Figure 4). In contrast, a differential and unique response was observed for each interaction, which was stronger when microorganisms interacted separately with maize than at the same time (i.e. the tripartite interaction).
The Zm-B25-Fv tripartite interaction did not present any unique overrepresented KEGG pathways (Figure 4) that could explain the molecular mechanisms involved in the plant response during this interaction, aside from a few unique overrepresented GO terms and unique DEGs. From a total of 78 unique DEGs in Zm-B25-Fv, 34 of them were upregulated and 44 were downregulated. The top ten upregulated DEGs include four genes that are directly and/or indirectly (orthologous genes) involved in plant resistance against different pathogens. These four genes are Zm00001eb282040 (glp1 - germin-like protein1; log2FC= 4.37); Zm00001eb339590 (ABC transporter G family member 34; log2FC= 3.87), the most induced DEG; Zm00001eb393680 (peroxidase 5; log2FC= 3.18); and Zm00001eb009380 (LysM-containing Receptor-Like Kinases LYK2; logFC=2.22).
When overexpressed in Arabidopsis, Zm00001eb282040 (glp1) improves the resistance to the biotrophic P. syringae pv. tomato DC3000 (PstDC3000) and the necrotrophic Sclerotinia sclerotiorum pathogens by inducing the expression of JA signaling-related genes [55]. In support of this, the GO term response to jasmonic acid (GO:0009753) was only found as an overrepresented GO term in the Zm-B25-Fv interaction (Figure 3). AtABCG34, the Arabidopsis ortholog of Zm00001eb339590 (ABC transporter G family member 34 [LOC103635032]), mediates camalexin secretion to defend against the necrotrophic pathogens Alternaria brassicicola and Botrytis cinerea [56]. Compared to the susceptible maize inbred line PH4CV, the resistant inbred maize line 9D207 exhibits significantly induced expression of the phenylpropanoid biosynthesis-related genes Zm00001eb393680 (peroxidase 5 [LOC103639086]), Zm00001eb381290 (peroxidase), Zm00001eb225390 (EC 3.2.1.21), Zm00001eb318150, Zm00001eb329520 (bglB), Zm00001eb160830 (REF1) and Zm00001eb158880 (TOGT1) when infected by spray-inoculation of an Exserohilum turcicum spore suspension (Li et al., 2024). This suggests a possible plant defense role for these phenylpropanoid biosynthesis-related genes. Finally, gene Zm00001eb009380 ([LOC103633076] Protein LYK2) is a member of the LysM-containing Receptor-Like Kinases (LYKs), which play a key role in the plant defense response. LysM and LYK proteins are the second major class of plant recognition receptors (PRPs) after LRR-RPs/LRR-RLKs, playing a significant role in the plant immune response [58]. In A. thaliana, LYK2 proved to be dispensable for chitin perception and early signaling events, but necessary for enhanced resistance to B. cinerea and P. syringae induced by flagellin, and for elicitor-induced priming of defense gene expression during fungal infection. LYK2 overexpression in Arabidopsis enhances resistance to B. cinerea and P. syringae and results in increased expression of defense-related genes during fungal infection (Giovannoni et al., 2021). Thus, due to the crucial role of LYK2 in Arabidopsis for chitin perception, it is possible that Zm00001eb009380 is essential for Fv detection, since chitin is a major component of the fungal cell wall (Elieh-Ali-Komi et al., 2016).
Other defense response-related genes were found among the Zm-B25-Fv unique upregulated DEGs, including Zm00001eb371160 (pme39 - pectin methylesterase39), Zm00001eb415440 (indole-2-monooxygenase-like [LOC103641249]), Zm00001eb001120 (ATPP2-A13 [LOC100286177]), and Zm00001eb259950 (NDR1/HIN1-like protein 1 [LOC100284275]). Gene Zm00001eb371160 (pme39) is a member of the pectin methylesterases (PMEs), which modify pectin, a complex polysaccharide constituent of plant cell walls. PMEs are implicated in various biological processes, including fruit ripening, pathogen defense, and cell wall remodeling [60]. In A. thaliana, AtPME17 activity triggers the synthesis of plant defensin 1.2 (PDF1.2) through the jasmonic acid-ethylene signaling pathway, conferring resistance to the fungal pathogen B. cinerea, while in wheat, the expression of several PME genes was associated with resistance to F. graminearum, the causal agent of head blight [60].
Zm00001eb415440, an indole-2-monooxygenase-like gene, is involved in the biosynthesis of benzoxazinoids, which are protective and allelopathic compounds found in some plants such as maize, wheat, rye and other wild and cultivated Poaceae. Benzoxazinoids have been associated with biochemical defense against a variety of biotic stresses, including insect herbivory, microbial pathogens, and competition with other plant species (Zhou et al., 2018).
The A. thaliana AtPP2-A1 gene ortholog of the maize Zm00001eb001120 (ATPP2-A13) gene exhibits molecular chaperone and antifungal activities against F. moniliforme, F. solani, Rhizoctonia solani and Trichoderma harzianum [62]. Overexpressing AtPP2-A1 in A. thaliana inhibited the phloem-feeding behavior of the aphid Myzus persicae, indicating a potential role for PP2 genes in mediating plant defense against insect herbivory [63]. Finally, the gene Zm00001eb259950 (NDR1/HIN1-like protein 1) is a member of the NHL protein family, involved in plant resistance to biotic and abiotic stresses. In the Arabidopsis genome, 45 NHL (NDR1/HIN1-like) genes that are homologous to NDR1 (non-race-specific disease resistance) or HIN1 (harpin-induced) genes have been identified, and their functions in pathogen perception have been extensively studied. Overexpression of NHL2 in Arabidopsis induced the expression of PR1 (pathogenesis-related gene 1) and light-dependent ‘speck disease-like’ symptoms in the leaves of transgenic plants. Likewise, overexpression of NHL3 increased resistance to P. syringae pv. tomato DC3000 [64]. Moreover, the overexpression of the A. thaliana genes NHL1 and NHL8 in soybeans enhanced resistance to the pathogen Heterodera glycines by activating the jasmonic acid (JA) and ethylene (ET) pathways [65].
Other upregulated unique DEGs found in the Zm-B25-Fv tripartite interaction relevant to plant defense are: Zm00001eb153350, a receptor-like protein 51 (LRR6) that can recognize microbial proteins or peptides [66]; Zm00001eb426640 (nactf65 - NAC-transcription factor 65), which is part of the NAC transcription factor family and plays crucial regulatory roles in plant immunity [67]; Zm00001eb249860, an actin depolymerizing factor (adf13) involved in actin dynamics [68]; Zm00001eb333050 (α-xylosidase 1 [LOC107548101]), an α-xylosidase that participates in remodeling xyloglucan, involved in the mechanical integrity of the primary cell wall of growing tissues, cell expansion, and seed germination (Shigeyama et al., 2016); and Zm00001eb072230, an S-norcoclaurine synthase 2. Norcoclaurine synthase (NCS) from Thalictrum flavum (Tf NCS), Papaver somniferum (Ps NCS1 and Ps NCS2), and Coptis japonica (Cj PR10A) share substantial identity with pathogen-related 10 (PR10) and Bet v1 proteins [62]. In support of the possible function of these unique upregulated DEGs, the term defense response (GO:0006952) was only found in the Zm-B25-Fv interaction as an overrepresented GO-term.
On the other hand, genes Zm00001eb102960 (Log2FC= -3.37) and Zm00001eb311640 (Log2FC= -3.64) were found as annotated genes within the top ten most downregulated DEGs. Zm00001eb311640 is an elongation factor 2 protein that helps ribosomes move along messenger RNA during protein synthesis (White-Gilbertson et al., 2008);, while Zm00001eb102960 is an SH3 domain-containing protein 3. Overexpression of SH3P2 in rice variety japonica YY compromised Pib-mediated resistance to M. oryzae isolates carrying AvrPib and Pib-AvrPib recognition-induced cell death [71]. Regarding the remaining downregulated DEGs, the genes Zm00001eb296580 (GTP-binding nuclear protein Ran-2), Zm00001eb156510 (9DNA ligase 6), Zm00001eb341090 (parp1 - poly(ADP-ribose) polymerase1), Zm00001eb029490 (TFIID basal transcription factor complex) and Zm00001eb226110 (DNA repair protein REV1) were related to synthesis and/or DNA repair and transcription. The genes Zm00001eb101530 (probable galacturonosyl-transferase 4), Zm00001eb325370 (pdcb44 - plasmodesmata callose binding protein44) and Zm00001eb336480 (rhamnogalacturonan I rhamnosyl-transferase 1) were related to the plant cell wall. The genes Zm00001eb230490 (mate6 - multidrug and toxic compound extrusion6), Zm00001eb293110 (trps12 - trehalose-6-phosphate synthase12), Zm00001eb251380 (jmj15 - JUMONJI-transcription factor 15), Zm00001eb332340 (mybr2 - MYB-related-transcription factor 2), Zm00001eb182690 (prh37 - protein phosphatase homolog37) and Zm00001eb154830 (type I inositol 1,4,5-trisphosphate 5-phosphatase 1-like) were reported to be associated with salinity, drought, chilling and cold stress. Finally, Zm00001eb395630 (gpdh3 - glucose-6-phosphate dehydrogenase3) was associated with ROS homeostasis; Zm00001eb203230 (naat1 - nicotianamine aminotransferase1) with iron (Fe) acquisition; and Zm00001eb151610 (inositol-hexakisphosphate kinase/IP6K) with plant defense mechanisms against bacterial, fungal and viral infections. Overall, despite a considerable number of unique downregulated DEGs related to different mechanisms relevant to maize growth and development, no DEGs stood out in the tripartite interaction. Nevertheless, extended gene network analysis (Supplementary Table 7) shows that biological processes involved in plant growth and development such as gene expression, RNA metabolic process, ribosome biogenesis, translation, actin filament depolymerization, photosynthesis and glycolysis may be relevant to the tripartite interaction. This is in agreement with observations in this work and in the field where maize seed inoculation with B25 results in a growth response and even an increased grain production (Lizárraga-Sánchez et al., 2015). Together, the gene interaction network results based on co-expression, using A. thaliana orthologous genes and focusing on BPs, support the GO and KEGG findings, as addressed in this Discussion section.
The present work reveals molecular mechanisms used by maize when interacting simultaneously with two quite different microbes: a beneficial bacterium and a fungal pathogen. However, a complete understanding of the molecular mechanisms triggered by microbial inoculation in plants will require more research devoted to understanding the tripartite interaction, as well as the more complex plant-microbe interactions in the rhizosphere.
4. Materials and Methods
4.1. Microorganisms and Inoculum Preparation
The biocontrol maize endophyte Bacillus cereus sensu lato strain B25, isolated from the rhizosphere [13] and the maize pathogenic fungus Fusarium verticillioides strain P03, isolated from maize roots [4] were used in this study. Both microorganisms are kept as cryopreserved frozen stocks (-70 °C) at the CIIDIR-IPN Sinaloa Unit. For bacterial inoculum, a powder formulation based on B25 spores obtained according to Martínez-Álvarez et al., (2016) was used. The fungal inoculum Fv P03 was reactivated from a frozen stock by growing it on PDA plates at 25 °C for 10 days. Fv conidia were harvested using sterile distilled water and adjusted to a concentration of 1 x 106 conidia mL-1. Harvested conidia were used as fungal inoculum.
4.2. Tripartite Assay: Maize-Bacteria-Fungus Interaction
Four conditions were established in this experiment: 1) maize without microorganisms (Zm), as a control; 2) maize inoculated with B25 (Zm-B25); 3) maize inoculated with Fv (Zm-Fv); and 4) maize inoculated with both B25 and Fv (Zm-B25-Fv). Commercial maize seeds (Garañón hybrid, Asgrow) were surface-disinfected using a hydrothermal treatment [4]. Subsequently, seeds were immersed in a Tween-20 solution (5 drops of Tween-20 per 100 mL of sterile distilled water) and sonicated for 5 min. The Tween solution was then decanted, and seeds were immersed in a 0.75% sodium hypochlorite solution and placed in a thermal bath at 52 °C for 20 min. Finally, the seeds were washed three times with sterile distilled water and allowed to air dry in a laminar flow hood.
For Zm-B25 and Zm-B25-Fv conditions, disinfected maize seeds were inoculated with B25 powder formulation as in Martínez-Álvarez et al., (2016). Seeds were moistened with 0.4 mL of 1% CMC (w/v) and then coated with 2 g of powder formulation (1 x 109 spores mL-1). The mixture was then stirred until the formulation completely covered the seeds, and the excess formulation was removed. The final inoculum concentration was 1 x 106 CFU of formulated B25 spores per seed. For Zm and Zm-Fv conditions, the seeds were coated with the same powder formulation but without B25 spores.
The rolled paper towel technique (Warham et al., 1996) was used with modifications to assay the interaction of maize with the microorganisms. Four pieces (1 cm2) of sterile filter paper were placed on a 36 x 19.5 cm sterile paper towel previously moistened with sterile distilled water. For Zm-Fv and Zm-B25-Fv conditions, the sterile filter papers were inoculated with 1 x 104 Fv conidia, whereas for the Zm and Zm-B25 conditions, the sterile filter papers were inoculated with sterile distilled water. A single maize seed was placed on each of the four sterile filter papers (four seeds per paper towel), and the paper towel was rolled and maintained in sterile plastic bags. Each plastic bag contained three rolls (12 seeds in total) and represented one biological replicate for the fungal biocontrol and plant growth assays. Three plastic bags with three rolls each were placed in an acrylic sterile box and sealed with Micropore tape (3MTM MicroporeTM surgical tape 1530-1) to avoid possible contamination. Acrylic boxes were then placed in a growth chamber at 28 °C, 60% relative humidity (RH), and a 16:8 h (light:darkness) photoperiod for seven days. Acrylic boxes were opened once per day in a laminar flow hood to remove excess humidity inside the boxes, and to moisten the rolled paper towels with sterile distilled water as needed. Subsequently, the boxes were resealed with micropore tape.
4.3. Maize Root Colonization and Plant Protection Mediated by B25 Against Fv
To confirm the protective effect of B25 on maize plants against Fv and the colonization of maize roots by both microorganisms during the tripartite assay, growth parameters were evaluated in five plants. Superficially disinfected root pieces were then used to re-isolate the microorganisms on LB and PD agar plates. Immediately after harvesting the five plants, fresh weight and shoot and root length were measured using an analytical balance (HR-150AZ, A&D Company, Tokyo, Japan) and a graduated ruler, respectively. Later, roots were superficially disinfected as in Jasim et al., (2014) with modifications. Roots were washed with tap water and cut into 1-2-cm long pieces. Root pieces were submerged in 70% ethanol for 1 min and in 1% sodium hypochlorite for 10 min. Next, the samples were dipped into a 10% (v/v) Tween-20 solution (Sigma, Cat. P7949) for 1 min and then washed three times with sterile distilled water. Disinfected root pieces were air-dried in a laminar flow hood, longitudinally cut, and placed on LB and PD agar plates, and finally incubated at 30 °C for 24-48 h.
4.4. RNA Extraction and Sequencing
Five out of twelve maize plants from a single bag were randomly selected. Entire root systems were pooled, immediately frozen in liquid nitrogen, and subsequently ground using a sterile pestle and mortar. The root pool from the five plants was used as a biological replicate for RNA extractions. The total RNA of three biological replicates was extracted using TRIzol® reagent (Thermo Fisher Scientific, Cat. No. 15596-026, Waltham, MA). The concentration and quality (260/280 nm ratio) of the total RNA were estimated by spectrophotometry using a Nanodrop 2000c (Thermo Fisher Scientific, Wilmington, DE), and RNA integrity was determined by agarose gel electrophoresis [73]. RNA samples were sent to the Genomic Services Laboratory (LabServGen CINVESTAV-IPN Irapuato, México) for sequencing. Before processing, the RNA integrity number (RIN) was verified using the Agilent RNA 6000 Nano Kit. All samples presented an RIN value >7. Twelve independent libraries, corresponding to three biological replicates from each condition (Zm, Zm-B25, Zm-Fv, and Zm-B25-Fv) were constructed using the Illumina® TruSeq RNA sample prep v2 kit. Libraries were sequenced in paired-end format (2x150 bp) in the Illumina NextSeq 500 platform.
4.5. Bioinformatic Analysis
Bioinformatic analyses were performed using the OOREAM computing server at the CIIDIR-IPN Sinaloa Unit. The quality of reads was examined before and after the trimming process with FastQC v0.11.4 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc). Raw reads were filtered using Trimmomatic v0.39 [74] with a minimum quality score of 20 and a minimum length of 50 bp. Illumina adapters were also removed during this step. Trimmed reads were mapped to the reference genome of Zea mays B73 v5.0 [75] using the STAR v2.7.0 software [76] with default parameters. Gene expression levels of SAM files were estimated based on the number of uniquely mapped reads using the HTSeq-count v0.11.1 software [77]. Raw counts were imported into the R/DESeq2 v1.32 package [78] for differential expression analysis. For principal component analysis (PCA), raw counts from all 12 libraries (4 conditions with 3 replicates) were normalized using the variance-stabilizing transformation (vst) method. One atypical replicate per condition was identified and removed for further analysis. The remaining 8 libraries (4 conditions with 2 replicates) were then normalized using the median of ratios method to identify differentially expressed genes (DEGs) between two selected conditions. DEGs were defined as genes with an adjusted P-value <0.01 and |Log2 fold change| >1. The interaction conditions Zm-B25, Zm-Fv and Zm-B25-Fv were individually compared against the control Zm to obtain a total of three sets of DEGs, corresponding to each comparison. Next, DEGs from the three interaction conditions (Zm-B25, Zm-Fv and Zm-B25-Fv) were compared in a Venn diagram to identify unique and common DEGs among them.
4.6. Gene Function Annotation and Overrepresentation Analysis
Gene ontology (GO) terms were obtained from the Ensembl database [79] using the BioMart tool (https://plants.ensembl.org/biomart/) and assigned to the DEGs of the three interaction conditions. Next, DEGs of each interaction condition were subjected to GO and KEGG overrepresentation analyses independently.
GO overrepresentation analysis was performed using the R/goseq v1.44 package [80] with the Wallenius method. GO terms with an adjusted p-value <0.05 were considered as significantly overrepresented.
KEGG overrepresentation analysis was performed in the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/). The DEG lists were loaded into the database and gene identifiers were linked to ENSEMBL_GEN_ID. KEGG pathways with an adjusted p-value <0.05 were considered as significantly overrepresented.
4.7. Gene Set Enrichment Analyses: Networks and Clustering
To investigate whether the set of genes from each section and intersection of maize interactions were associated with certain biological pathways, non-redundant gene identifiers (NRGIs) from A. thaliana orthologous genes corresponding to up- and downregulated maize genes were submitted to AgriGO [81], as well as to String [82] and Genemania [83]. The functional enrichment analyses generated GO terms belonging to biological processes (BPs), molecular functions (MFs), and cellular components (CCs). The most significant GO terms and pathways were those with an FDR threshold of 5% and a redundancy score set at 0.5. In the case of network analysis, all gene sets that generated a main network containing less than 200 nodes were expanded via String. Moreover, the same former gene sets were also submitted to Genemania as a complementary approach to identify enriched pathways. For network clustering based on co-expression, the Markov clustering (MCL) method of StringApp was employed with an inflation value set to 2.5 and STRING interaction scores of 0.4. Only the largest clusters were further used for functional enrichment analysis. Network associations were visualized and analyzed in Cytoscape v3.9.1 [84].
4.8. RNA-Seq Validation by qRT-PCR
Quantitative RT-PCR experiments were carried out on the same RNA samples used for RNA-seq. Total RNA was DNase treated (RQ1 DNase). First-strand cDNA was synthesized from 1 µg of total RNA using SuperScript III reverse transcriptase (Thermo Fisher Scientific, Cat. No.18080-044, Waltham, MA) following the manufacturer’s instructions. qRT-PCR reactions were conducted in a Rotor Gene-Q Real time PCR system (Qiagen, Cat. No. 9001550, Hilden, Germany); four technical replicates were performed for each of the two biological replicates. Reactions included 5 μL of SYBR Green master mix (Qiagen, Cat. no. 204074, Hilden, Germany), 1 µM of each primer, 10 ng of cDNA, and RNase-free water for a final volume of 10 μL. For PCR amplification, the thermocycler program included a preheating step at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s, 30 s annealing at 59 °C, and 30 s extension at 72 °C. Dissociation curves were performed at the end of each run to confirm single-product amplifications. Gene-specific primers were designed with Primer3 software (Supplementary Table 1); only primers for the housekeeping gene CDK [85] were downloaded from the literature. PCR amplification efficiency of the housekeeping and target genes was determined from standard curves constructed from serial dilutions of cDNA (from 1 to 100 ng). Relative quantification of maize genes was normalized to the housekeeping gene CDK, and fold change (FC) values in gene expression were calculated using the comparative threshold cycle method 2−ΔΔCt [86].
4.9. Statistical Analysis
Data on plant growth parameters from the tripartite assay samples were assessed for normality by the Kolmogorov–Smirnov test and then analyzed by one-way ANOVA using the IBM SPSS Statistics v25 software. Differences among treatments were determined by Tukey’s test (P = 0.05).
5. Conclusion
The transcriptomic analysis of a tripartite interaction model system allowed us to dissect the molecular mechanisms involved in maize roots when interacting with the biocontrol endophytic bacteria B25, the fungal phytopathogen Fv, and both microorganisms simultaneously (Figure 6). In the plant-bacterium interaction (Figure 6a), maize plants appear to recognize B25 as a non-threatening organism, after which the bacterium induces key mechanisms that promote plant growth, preparing the plant for pathogen attack. In the plant-fungus interaction (Figure 6b), the fungal pathogen strongly affected the proper function of plant DNA and protein synthesis machinery, turning off pathways related to these mechanisms. Finally, during the tripartite interaction (Figure 6c), a core of unique genes related to plant defense responses were involved in the successful control of Fv infection, as well as the involvement of genes positively affecting plant growth and development.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Methods. Figure S1: Superficially disinfected maize root pieces showing the growth of B25 (red arrows) or Fv (green arrows) at 7 dpi, 48 h after incubation on media. Figure S2: Principal component analysis of maize root transcriptional profiles using normalized expression values.; Table S1: Principal component analysis of maize root transcriptional profiles using normalized expression values. Table S2: List of total, common and unique DEGs with log2FoldChange value in and between the interaction conditions from the tripartite experiment. Table S3: Overrepresented GO terms for unique expressed genes from interaction conditions. Table S4: GO Enriched Terms from DEGs of interaction conditions. Table S5: KEGG over-represented Terms of interaction conditions. Table S6: Integration of B73 RefGen_v4 and RefGen_v5 annotations. Table S7: Analysis of non-redundant gene identifiers (NRGIs) using String and Genemania.
Author Contributions
Conceptualization, PABA, ACM and IEMM; methodology, PABA, JECA and ACM; software, ACM and JGJ; validation, PABA, JLFC, IGLS and ACM; formal analysis, PABA, ACM, JLFC, IGLS and JGJ; investigation, PABA, JLFC, IGLS, JECA and ACM; resources, IEMM, ACM and CLCV; data curation, PABA, ACM and IEMM; writing—original draft preparation, PABA, ACM and IEMM; writing—review and editing, JGJ, JLFC, IGLS, CLCV and JECA; visualization, PABA, ACM, IEMM, JLFC and IGLS; supervision, IEMM and ACM.; project administration, IEMM; funding acquisition, IEMM. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by SECIHTI FOINS Fronteras de la Ciencia Grant No. 2016-01-2510. PABA was granted a PhD fellowship (No. 70883) from SECIHTI, and JECA received Master’s and PhD fellowships (No. 934735) from SECIHTI. PABA and JECA were both granted complementary fellowship support from IPN (BEIFI fellowship program). The OOREAM server at the CIIDIR-IPN Sinaloa was funded by SECIHTI (project 302541).
Data Availability Statement
Raw sequence reads of this study can be downloaded download from https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1345406. The rest of data related to this work are contained within the article.
Acknowledgments
We thank Dr. Brandon Loveall from Improvence editing services for English proofreading of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Erenstein, O.; Jaleta, M.; Sonder, K.; Mottaleb, K.; Prasanna, B.M. Global Maize Production, Consumption and Trade: Trends and R&D Implications. Food Secur 2022. [Google Scholar] [CrossRef]
- Butrón, A.; Reid, L.M.; Santiago, R.; Cao, A.; Malvar, R.A. Inheritance of Maize Resistance to Gibberella and Fusarium Ear Rots and Kernel Contamination with Deoxynivalenol and Fumonisins. Plant Pathol 2015, 64, 1053–1060. [Google Scholar] [CrossRef]
- Subedi, S. A Review on Important Maize Diseases and Their Management in Nepal. Journal of Maize Research and Development 2015, 1, 28–52. [Google Scholar] [CrossRef]
- Leyva-Madrigal, K.Y.; Larralde-Corona, C.P.; Apodaca-Sánchez, M.A.; Quiroz-Figueroa, F.R.; Mexia-Bolaños, P.A.; Portillo-Valenzuela, S.; Ordaz-Ochoa, J.; Maldonado-Mendoza, I.E. Fusarium Species from the Fusarium Fujikuroi Species Complex Involved in Mixed Infections of Maize in Northern Sinaloa, Mexico. Journal of Phytopathology 2015, 163, 486–497. [Google Scholar] [CrossRef]
- Bacon, C.W.; Glenn, A.E.; Yates, I.E. Fusarium Verticillioides: Managing the Endophytic Association with Maize for Reduced Fumonisins Accumulation. Toxin Rev 2008, 27, 411–446. [Google Scholar] [CrossRef]
- Deepa, N.; Nagaraja, H.; Sreenivasa, M.Y. Prevalence of Fumonisin Producing Fusarium Verticillioides Associated with Cereals Grown in Karnataka (India). Food Science and Human Wellness 2016, 5, 156–162. [Google Scholar] [CrossRef]
- Koul, B.; Chopra, M.; Lamba, S. Microorganisms as Biocontrol Agents for Sustainable Agriculture. Relationship Between Microbes and the Environment for Sustainable Ecosystem Services, Volume 1 2022, 45–68. [CrossRef]
- De los Santos Villalobos, S.; Parra Cota, F.I.; Herrera Sepúlveda, A.; Valenzuela Aragón, B.; Estrada Mora, J.C. Colmena: Colección de Microorganismos Edáficos y Endófitos Nativos, Para Contribuir a La Seguridad Alimentaria Nacional. Rev Mex De Cienc Agric 2018, 9, 191–202. [Google Scholar] [CrossRef]
- Lizárraga-Sánchez, G.J.; Leyva-Madrigal, K.Y.; Sánchez-Peña, P.; Quiroz-Figueroa, F.R.; Maldonado-Mendoza, I.E. Bacillus Cereus Sensu Lato Strain B25 Controls Maize Stalk and Ear Rot in Sinaloa, Mexico. Field Crops Res 2015, 176, 11–21. [Google Scholar] [CrossRef]
- Bacon, C.W.; Hinton, D.M. In Planta Reduction of Maize Seedling Stalk Lesions by the Bacterial Endophyte Bacillus Mojavensis. Can J Microbiol 2011, 57, 485–492. [Google Scholar] [CrossRef]
- Pal, G.; Kumar, K.; Verma, A.; Verma, S.K. Seed Inhabiting Bacterial Endophytes of Maize Promote Seedling Establishment and Provide Protection against Fungal Disease. Microbiol Res 2022, 255, 126926. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, R.; Wu, X.; Xu, T.; Ahmad, S.; Zhang, X.; Zhao, J.; Liu, Y. An Endophytic Strain of the Genus Bacillus Isolated from the Seeds of Maize (Zea Mays L.) Has Antagonistic Activity against Maize Pathogenic Strains. Microb Pathog 2020, 142, 104074. [Google Scholar] [CrossRef]
- Figueroa-López, A.M.; Cordero-Ramírez, J.D.; Martínez-Álvarez, J.C.; López-Meyer, M.; Lizárraga-Sánchez, G.J.; Félix-Gastélum, R.; Castro-Martínez, C.; Maldonado-Mendoza, I.E. Rhizospheric Bacteria of Maize with Potential for Biocontrol of Fusarium Verticillioides. Springerplus 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Fadiji, A.E.; Babalola, O.O. Elucidating Mechanisms of Endophytes Used in Plant Protection and Other Bioactivities With Multifunctional Prospects. Front Bioeng Biotechnol 2020, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bolivar-Anillo, H.J.; González-Rodríguez, V.E.; Cantoral, J.M.; García-Sánchez, D.; Collado, I.G.; Garrido, C. Endophytic Bacteria Bacillus Subtilis, Isolated from Zea Mays, as Potential Biocontrol Agent against Botrytis Cinerea. Biology (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Liu, H.; Carvalhais, L.C.; Crawford, M.; Singh, E.; Dennis, P.G.; Pieterse, C.M.J.; Schenk, P.M. Inner Plant Values: Diversity, Colonization and Benefits from Endophytic Bacteria. Front Microbiol 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Figueroa-López, A.M. Caracterización de Los Mecanismos de Antagonismo Que Emplea Bacillus Cereus Seleccionado Para El Control de Fusarium Verticillioides. Doctoral Thesis, Instituto Politécnico Nacional: Guasave, Sinaloa, 2016. [Google Scholar]
- Martínez-Álvarez, J.C.; Castro-Martínez, C.; Sánchez-Peña, P.; Gutiérrez-Dorado, R.; Maldonado-Mendoza, I.E. Development of a Powder Formulation Based on Bacillus Cereus Sensu Lato Strain B25 Spores for Biological Control of Fusarium Verticillioides in Maize Plants. World J Microbiol Biotechnol 2016, 32. [Google Scholar] [CrossRef]
- Douriet-Gámez, N.R.; Maldonado-Mendoza, I.E.; Ibarra-Laclette, E.; Blom, J.; Calderón-Vázquez, C.L. Genomic Analysis of Bacillus Sp. Strain B25, a Biocontrol Agent of Maize Pathogen Fusarium Verticillioides. Curr Microbiol 2018, 75, 247–255. [Google Scholar] [CrossRef]
- Morales-Ruiz, E.; Priego-Rivera, R.; Figueroa-López, A.M.; Cazares-Álvarez, J.E.; Maldonado-Mendoza, I.E. Biochemical Characterization of Two Chitinases from Bacillus Cereus Sensu Lato B25 with Antifungal Activity against Fusarium Verticillioides P03. FEMS Microbiol Lett 2021, 368, 1–8. [Google Scholar] [CrossRef]
- Báez-Astorga, P.A.; Cázares-Álvarez, J.E.; Cruz-Mendívil, A.; Quiroz-Figueroa, F.R.; Sánchez-Valle, V.I.; Maldonado-Mendoza, I.E. Molecular and Biochemical Characterisation of Antagonistic Mechanisms of the Biocontrol Agent Bacillus Cereus B25 Inhibiting the Growth of the Phytopathogen Fusarium Verticillioides P03 during Their Direct Interaction in Vitro. Biocontrol Sci Technol 2022, 1–21. [Google Scholar] [CrossRef]
- Figueroa-López, A.M.; Leyva-Madrigal, K.Y.; Cervantes-Gámez, R.G.; Beltran-Arredondo, L.I.; Douriet-Gámez, N.R.; Castro-Martínez, C.; Maldonado-Mendoza, I.E. Induction of Bacillus Cereus Chitinases as a Response to Lysates of Fusarium Verticillioides. 2017, 22, 12722–12731. [Google Scholar]
- Park, Y.S.; Park, J.S.; Lee, S.; Jung, S.H.; Kim, S.K.; Ryu, C.M. Simultaneous Profiling of Arabidopsis Thaliana and Vibrio Vulnificus MO6-24/O Transcriptomes by Dual RNA-Seq Analysis. Comput Struct Biotechnol J 2021, 19, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Gheler, C.F.; Domingues Zucchi, T.; Soares de Melo, I. Biological Control of Phytopathogenic Fungi by Endophytic Actinomycetes Isolated from Maize (Zea Mays L.). Brazilian archives of Biology and Technology 2013, 56, 948–955. [Google Scholar] [CrossRef]
- Cazares-Álvarez, J.E.; Báez-Astorga, P.A.; Arroyo-Becerra, A.; Maldonado-Mendoza, I.E. Genome-Wide Identification of a Maize Chitinase Gene Family and the Induction of Its Expression by Fusarium Verticillioides (Sacc.) Nirenberg (1976) Infection. Genes (Basel) 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Perrot, T.; Pauly, M.; Ramírez, V. Emerging Roles of β-Glucanases in Plant Development and Adaptative Responses. Plants 2022, 11. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, X.; Qiu, H. Flavones and Flavonols: Phytochemistry and Biochemistry. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; pp. 1821–1847. ISBN 978-3-642-22144-6. [Google Scholar]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The Role of Quercetin in Plants. Plant Physiology and Biochemistry 2021, 166, 10–19. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Lam, P.Y.; Dini-Andreote, F.; Dai, L.; Wei, Z. Multifaceted Roles of Flavonoids Mediating Plant-Microbe Interactions. Microbiome 2022, 10, 1–13. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, D.; Tang, Z.; Zhang, Y.; Zhang, K.; Dong, J.; Wang, F. Exogenous Brassinosteroids Promotes Root Growth, Enhances Stress Tolerance, and Increases Yield in Maize. Plant Signal Behav 2022, 17, 1–14. [Google Scholar] [CrossRef]
- Hagen, G.; Guilfoyle, T. Auxin-Responsive Gene Expression: Genes, Promoters and Regulatory Factors. Plant Mol Biol 2002, 49, 373–385. [Google Scholar] [CrossRef]
- Franco, A.R.; Gee, M.A.; Guilfoyle, T.J. Induction and Superinduction of Auxin-Responsive MRNAs with Auxin and Protein Synthesis Inhibitors. J Biol Chem 1990, 265, 15845–15849. [Google Scholar] [CrossRef]
- Sowmya, R.S.; Warke, V.G.; Mahajan, G.B.; Annapure, U.S. Effect of Amino Acids on Growth, Elemental Content, Functional Groups, and Essential Oils Composition on Hydroponically Cultivated Coriander under Different Conditions. Ind Crops Prod 2023, 197, 116577. [Google Scholar] [CrossRef]
- Colla, G.; Nardi, S.; Cardarelli, M.; Ertani, A.; Lucini, L.; Canaguier, R.; Rouphael, Y. Protein Hydrolysates as Biostimulants in Horticulture. Sci Hortic 2015, 196, 28–38. [Google Scholar] [CrossRef]
- Alfosea-Simón, M.; Simón-Grao, S.; Zavala-Gonzalez, E.A.; Cámara-Zapata, J.M.; Simón, I.; Martínez-Nicolás, J.J.; Lidón, V.; García-Sánchez, F. Physiological, Nutritional and Metabolomic Responses of Tomato Plants After the Foliar Application of Amino Acids Aspartic Acid, Glutamic Acid and Alanine. Front Plant Sci 2021, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Hassan, M.U.; Ishfaq, M.; Ripa, F.A.; Nadeem, F.; Ahmad, Z.; Xu, J.; Ning, P.; Li, X. Foliar Glutamine Application Improves Grain Yield and Nutritional Quality of Field-Grown Maize (Zea Mays L.) Hybrid ZD958. J Plant Growth Regul 2024, 43, 624–637. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S. Mitogen-Activated Protein Kinase Cascades in Signaling Plant Growth and Development. Trends Plant Sci 2015, 20, 56–64. [Google Scholar] [CrossRef]
- Lin, L.; Wu, J.; Jiang, M.; Wang, Y. Plant Mitogen-activated Protein Kinase Cascades in Environmental Stresses. Int J Mol Sci 2021, 22, 1–24. [Google Scholar] [CrossRef]
- Dedyukhina, E.G.; Kamzolova, S. V.; Vainshtein, M.B. Arachidonic Acid as an Elicitor of the Plant Defense Response to Phytopathogens. Chemical and Biological Technologies in Agriculture 2014, 1, 2–7. [Google Scholar] [CrossRef]
- Savchenko, T.; Walley, J.W.; Chehab, E.W.; Xiao, Y.; Kaspi, R.; Pye, M.F.; Mohamed, M.E.; Lazarus, C.M.; Bostock, R.M.; Dehesh, K. Arachidonic Acid: An Evolutionarily Conserved Signaling Molecule Modulates Plant Stress Signaling Networks. Plant Cell 2010, 22, 3193–3205. [Google Scholar] [CrossRef]
- Dye, S.M.; Bostock, R.M. Eicosapolyenoic Fatty Acids Induce Defense Responses and Resistance to Phytophthora Capsici in Tomato and Pepper. Physiol Mol Plant Pathol 2021, 114, 101642. [Google Scholar] [CrossRef]
- Lewis, D.C.; van der Zwan, T.; Richards, A.; Little, H.; Coaker, G.L.; Bostock, R.M. The Oomycete Microbe-Associated Molecular Pattern, Arachidonic Acid, and an Ascophyllum Nodosum-Derived Plant Biostimulant Induce Defense Metabolome Remodeling in Tomato. Phytopathology 2023, 113, 1084–1092. [Google Scholar] [CrossRef]
- Hyong, W.C.; Byung, G.L.; Nak, H.K.; Park, Y.; Chae, W.L.; Hyun, K.S.; Byung, K.H. A Role for a Menthone Reductase in Resistance against Microbial Pathogens in Plants. Plant Physiol 2008, 148, 383–401. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, L.J.; Wang, Y.; Liu, S.; Geng, Z.; Song, A.; Jiang, J.; Chen, S.; Chen, F. Functional Identification of a Flavone Synthase and a Flavonol Synthase Genes Affecting Flower Color Formation in Chrysanthemum Morifolium. Plant Physiology and Biochemistry 2021, 166, 1109–1120. [Google Scholar] [CrossRef]
- Alseekh, S.; Perez de Souza, L.; Benina, M.; Fernie, A.R. The Style and Substance of Plant Flavonoid Decoration; towards Defining Both Structure and Function. Phytochemistry 2020, 174. [Google Scholar] [CrossRef] [PubMed]
- Manova, V.; Gruszka, D. DNA Damage and Repair in Plants – From Models to Crops. Front Plant Sci 2015, 6, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nisa, M.U.; Huang, Y.; Benhamed, M.; Raynaud, C. The Plant DNA Damage Response: Signaling Pathways Leading to Growth Inhibition and Putative Role in Response to Stress Conditions. Front Plant Sci 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Grin, I.R.; Petrova, D. V.; Endutkin, A. V.; Ma, C.; Yu, B.; Li, H.; Zharkov, D.O. Base Excision DNA Repair in Plants: Arabidopsis and Beyond. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Szurman-Zubrzycka, M.; Jędrzejek, P.; Szarejko, I. How Do Plants Cope with DNA Damage? A Concise Review on the DDR Pathway in Plants. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, X.; Li, Y.; Xing, A.; Wu, C.; Li, D.; Wang, C.; de Bures, A.; Zhang, Y.; Guo, S.; et al. Arabidopsis SMO2 Modulates Ribosome Biogenesis by Maintaining the RID2 Abundance during Organ Growth. Plant Journal 2023, 114, 96–109. [Google Scholar] [CrossRef]
- Nebenführ, A.; Dixit, R. Kinesins and Myosins: Molecular Motors That Coordinate Cellular Functions in Plants. Annu Rev Plant Biol 2018, 69, 329–361. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Chong, K. The Novel Functions of Kinesin Motor Proteins in Plants. Protoplasma 2012, 249, 95–100. [Google Scholar] [CrossRef]
- Ryan, J.M.; Nebenführ, A. Update on Myosin Motors: Molecular Mechanisms and Physiological Functions. Plant Physiol 2018, 176, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Ge, L.; Ye, X.; Xu, L.; Si, W.; Ding, T. ZmGLP1, a Germin-like Protein from Maize, Plays an Important Role in the Regulation of Pathogen Resistance. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Khare, D.; Choi, H.; Huh, S.U.; Bassin, B.; Kim, J.; Martinoia, E.; Sohn, K.H.; Paek, K.H.; Lee, Y.; Chrispeels, M.J. Arabidopsis ABCG34 Contributes to Defense against Necrotrophic Pathogens by Mediating the Secretion of Camalexin. Proc Natl Acad Sci U S A 2017, 114, E5712–E5720. [Google Scholar] [CrossRef]
- Li, M.R.; Qi, X.; Li, D.; Wu, Z.; Liu, M.; Yang, W.; Zang, Z.; Jiang, L. Comparative Transcriptome Analysis Highlights Resistance Regulatory Networks of Maize in Response to Exserohilum Turcicum Infection at the Early Stage. Physiol Plant 2024, 176. [Google Scholar] [CrossRef]
- Shumayla; Madhu; Singh, K.; Upadhyay, S.K. LysM Domain-Containing Proteins Modulate Stress Response and Signalling in Triticum Aestivum L. Environ Exp Bot. 2021, 189. [Google Scholar] [CrossRef]
- Giovannoni, M.; Lironi, D.; Marti, L.; Paparella, C.; Vecchi, V.; Gust, A.A.; De Lorenzo, G.; Nürnberger, T.; Ferrari, S. The Arabidopsis Thaliana LysM-Containing Receptor-Like Kinase 2 Is Required for Elicitor-Induced Resistance to Pathogens. Plant Cell Environ 2021, 44, 3545–3562. [Google Scholar] [CrossRef]
- Kumar, R.; Meghwanshi, G.K.; Marcianò, D.; Ullah, S.F.; Bulone, V.; Toffolatti, S.L.; Srivastava, V. Sequence, Structure and Functionality of Pectin Methylesterases and Their Use in Sustainable Carbohydrate Bioproducts: A Review. Int J Biol Macromol 2023, 244. [Google Scholar] [CrossRef]
- Zhou, S.; Richter, A.; Jander, G. Beyond Defense: Multiple Functions of Benzoxazinoids in Maize Metabolism. Plant Cell Physiol 2018, 59, 1528–1533. [Google Scholar] [CrossRef]
- Lee, J.R.; Boltz, K.A.; Lee, S.Y. Molecular Chaperone Function of Arabidopsis Thaliana Phloem Protein 2-A1, Encodes a Protein Similar to Phloem Lectin. Biochem Biophys Res Commun 2014, 443, 18–21. [Google Scholar] [CrossRef]
- Zheng, L.; Zheng, H.; Zheng, X.; Duan, Y.; Yu, X. PP2 Gene Family in Phyllostachys Edulis: Identification, Characterization, and Expression Profiles. BMC Genomics 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Song, W.M.; Pan, J.; Jiang, C.M.; Srivastava, R.; Li, B.; Zhu, L.Y.; Su, H.Y.; Gao, X.S.; Liu, H.; et al. Overexpression of the NDR1/HIN1-Like Gene NHL6 Modifies Seed Germination in Response to Abscisic Acid and Abiotic Stresses in Arabidopsis. PLoS One 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.; Youssef, R.; McDonald, M.; Brewer, E.; Beard, H.; Matthews, B. Overexpression of Four Arabidopsis Thaliana NHL Genes in Soybean (Glycine Max) Roots and Their Effect on Resistance to the Soybean Cyst Nematode (Heterodera Glycines). Physiol Mol Plant Pathol 2014, 86, 1–10. [Google Scholar] [CrossRef]
- Block, A.K.; Tang, H. V.; Hopkins, D.; Mendoza, J.; Solemslie, R.K.; du Toit, L.J.; Christensen, S.A. A Maize Leucine-Rich Repeat Receptor-like Protein Kinase Mediates Responses to Fungal Attack. Planta 2021, 254. [Google Scholar] [CrossRef]
- Ding, N.; Zhao, Y.; Wang, W.; Liu, X.; Shi, W.; Zhang, D.; Chen, J.; Ma, S.; Sun, Q.; Wang, T.; et al. Transcriptome Analysis in Contrasting Maize Inbred Lines and Functional Analysis of Five Maize NAC Genes under Drought Stress Treatment. Front Plant Sci 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shi, M.; Wang, D.; Gong, Y.; Sha, Q.; Lv, P.; Yang, J.; Chu, P.; Guo, S. Research Progress on the Roles of Actin-Depolymerizing Factor in Plant Stress Responses. Front Plant Sci 2023, 14. [Google Scholar] [CrossRef]
- Shigeyama, T.; Watanabe, A.; Tokuchi, K.; Toh, S.; Sakurai, N.; Shibuya, N.; Kawakami, N. α-Xylosidase Plays Essential Roles in Xyloglucan Remodelling, Maintenance of Cell Wall Integrity, and Seed Germination in Arabidopsis Thaliana. J Exp Bot 2016, 67, 5615–5629. [Google Scholar] [CrossRef]
- White-Gilbertson, S.; Rubinchik, S.; Voelkel-Johnson, C. Transformation, Translation and TRAIL: An Unexpected Intersection. Cytokine Growth Factor Rev 2008, 19, 167–172. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, Y.; Yu, X.; Lin, Y.; Zhu, Y.; Chen, J.; Xie, H.; Zhang, Q.; Wang, L.; Wei, Y.; et al. SH3P2, an SH3 Domain-Containing Protein That Interacts with Both Pib and AvrPib, Suppresses Effector-Triggered, Pib-Mediated Immunity in Rice. Mol Plant 2022, 15, 1931–1946. [Google Scholar] [CrossRef]
- Jasim, B.; Joseph, A.A.; John, C.J.; Mathew, J.; Radhakrishnan, E.K. Isolation and Characterization of Plant Growth Promoting Endophytic Bacteria from the Rhizome of Zingiber Officinale. 3 Biotech 2014, 4, 197–204. [Google Scholar] [CrossRef]
- Aranda, P.S.; Lajoie, D.M.; Jorcyk, C.L. Bleach Gel: A Simple Agarose Gel for Analyzing RNA Quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Woodhouse, M.R.; Cannon, E.K.; Portwood, J.L.; Harper, L.C.; Gardiner, J.M.; Schaeffer, M.L.; Andorf, C.M. A Pan-Genomic Approach to Genome Databases Using Maize as a Model System. BMC Plant Biol 2021, 21. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res 2023, 51, D933–D941. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol 2010, 11. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO Analysis Toolkit for the Agricultural Community, 2017 Update. Nucleic Acids Res 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lin, F.; Jiang, L.; Liu, Y.; Lv, Y.; Dai, H.; Zhao, H. Genome-Wide Identification of Housekeeping Genes in Maize. Plant Mol Biol 2014, 86, 543–554. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
Figure 3.
Venn diagram of unique and shared overrepresented Gene Ontology (GO) terms between the interactions. Boxes display the list of the overrepresented GO terms exclusive to one or shared by two interactions. The list of common GO terms (42) shared between the three interactions is shown in Supplementary Table 4.
Figure 3.
Venn diagram of unique and shared overrepresented Gene Ontology (GO) terms between the interactions. Boxes display the list of the overrepresented GO terms exclusive to one or shared by two interactions. The list of common GO terms (42) shared between the three interactions is shown in Supplementary Table 4.
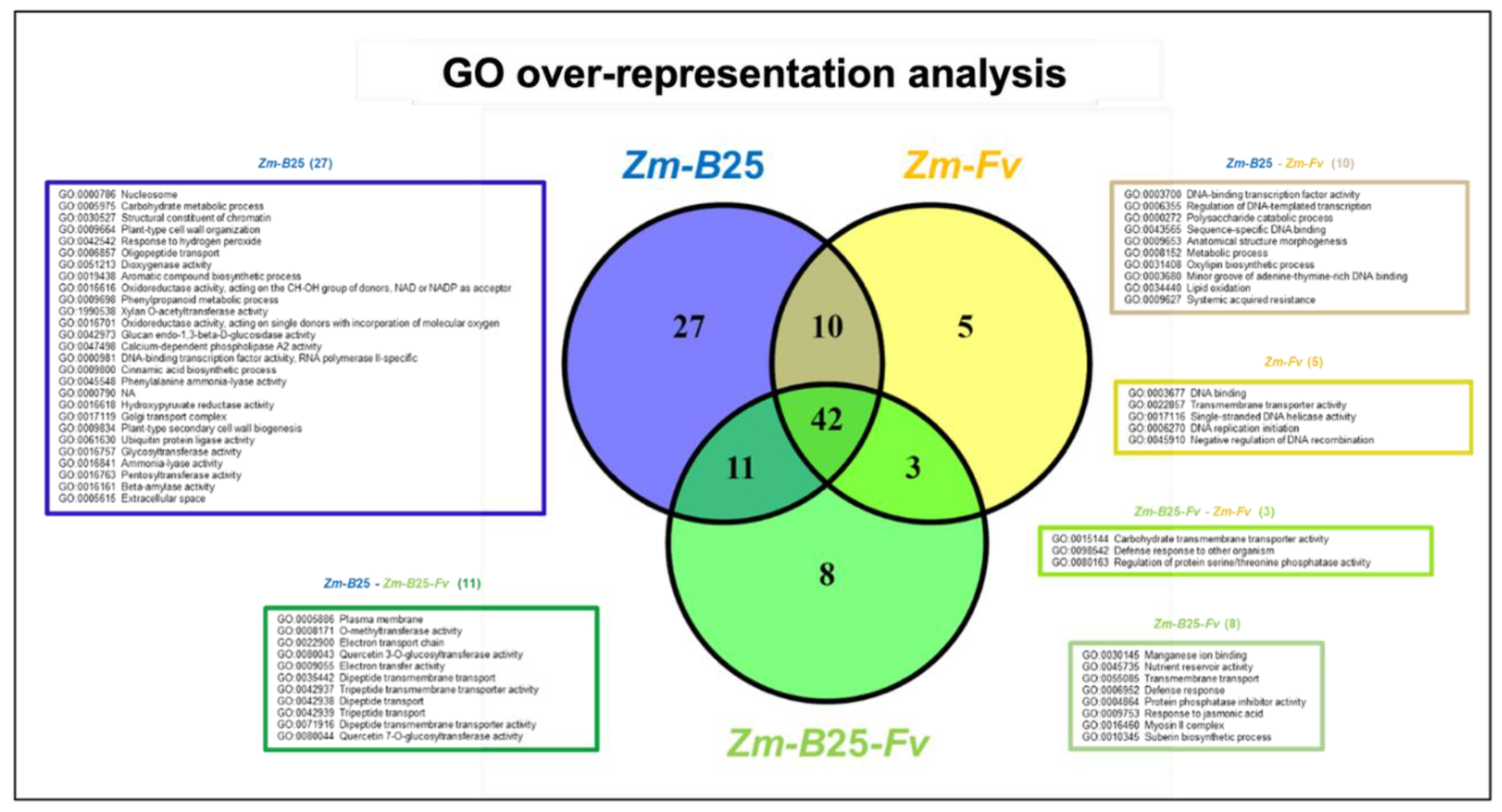
Figure 4.
Venn diagram of unique and shared overrepresented KEGG pathways between the interaction conditions. Boxes display the list of overrepresented KEGG terms exclusive to one or shared by two or three interactions.
Figure 4.
Venn diagram of unique and shared overrepresented KEGG pathways between the interaction conditions. Boxes display the list of overrepresented KEGG terms exclusive to one or shared by two or three interactions.
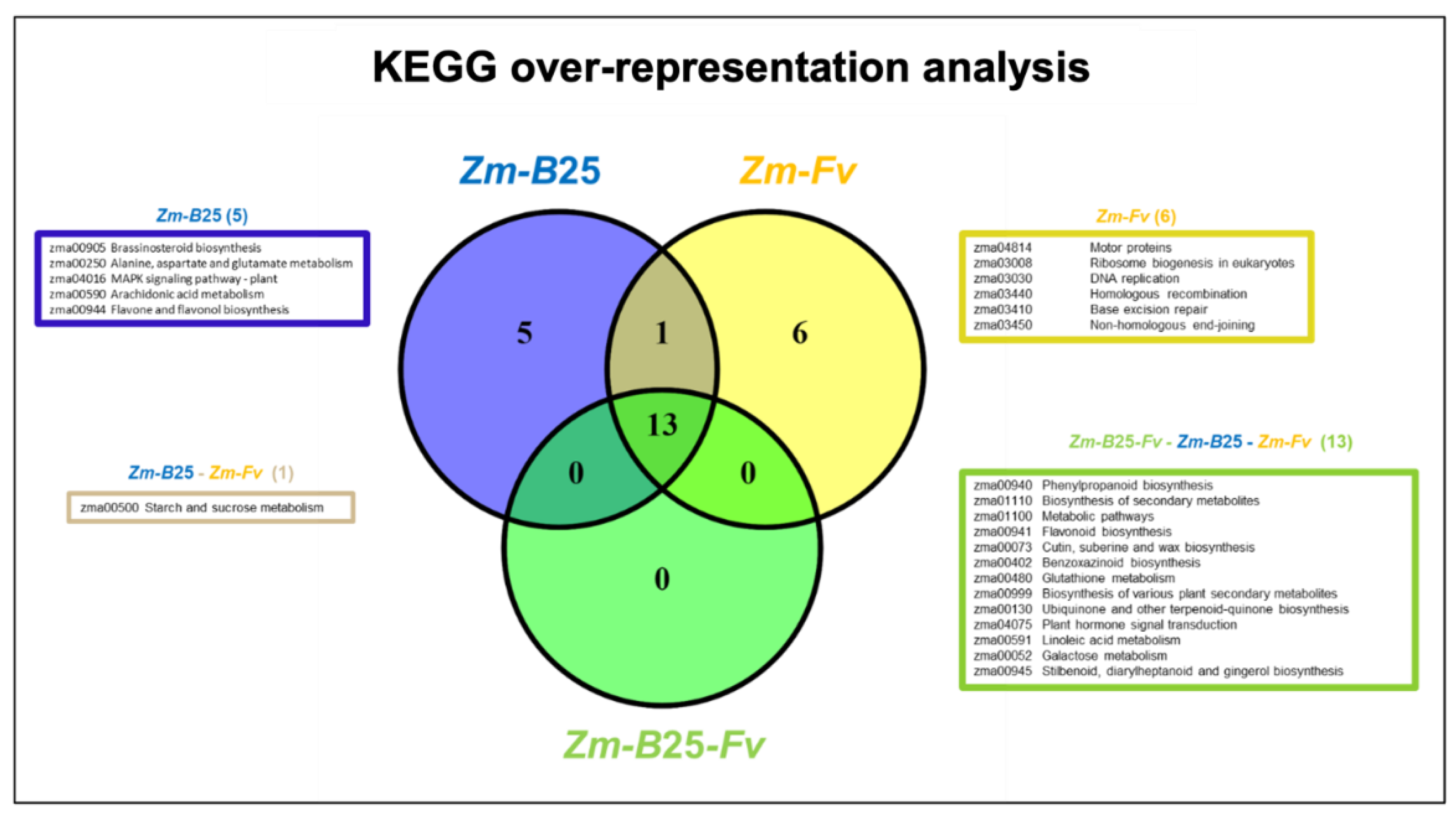
Figure 6.
Graphic summary of the main molecular mechanisms involved in maize roots when interacting with the biocontrol endophytic bacterium B25, the fungal phytopathogen Fv, or both microorganisms (tripartite interaction). a) During the plant-bacterium interaction, B25 is recognized by maize as a beneficial microorganism and induces the activation of pathways related to plant growth and development as well as defense responses, preparing the plant for pathogen attack. b) During the plant-fungus interaction, Fv turns off key mechanisms of plant DNA replication and repair, as well as pathways of ribosome biogenesis and motor proteins; additionally, it negatively affects plant growth and development, decreasing root length. c) During the maize-bacterium-fungus tripartite interaction, several unique DEGs related to defense response and plant growth development are induced. The most induced maize DEGs in the tripartite interaction are shown in the red box.
Figure 6.
Graphic summary of the main molecular mechanisms involved in maize roots when interacting with the biocontrol endophytic bacterium B25, the fungal phytopathogen Fv, or both microorganisms (tripartite interaction). a) During the plant-bacterium interaction, B25 is recognized by maize as a beneficial microorganism and induces the activation of pathways related to plant growth and development as well as defense responses, preparing the plant for pathogen attack. b) During the plant-fungus interaction, Fv turns off key mechanisms of plant DNA replication and repair, as well as pathways of ribosome biogenesis and motor proteins; additionally, it negatively affects plant growth and development, decreasing root length. c) During the maize-bacterium-fungus tripartite interaction, several unique DEGs related to defense response and plant growth development are induced. The most induced maize DEGs in the tripartite interaction are shown in the red box.
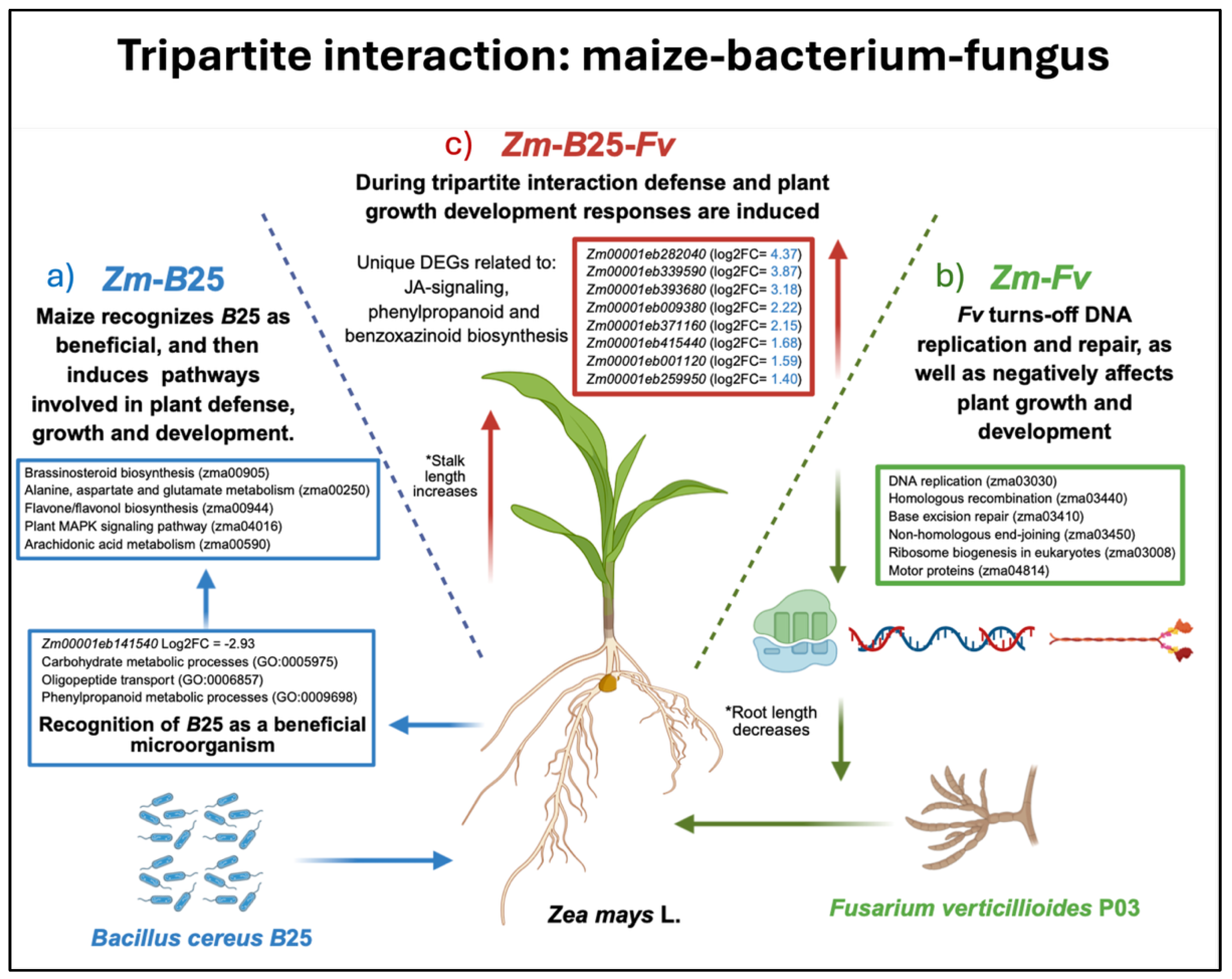

Table 1.
Growth parameters of maize plants from the tripartite assay seven days post infection (dpi).
Table 1.
Growth parameters of maize plants from the tripartite assay seven days post infection (dpi).
| Condition | Length (cm) | Fresh weight (g) | |||
| Leaf | Root | Leaf | Root | ||
| Zm | 11.92b | 13.07a | 0.38b | 0.78a | |
| Zm-B25 | 11.27b | 12.52a | 0.34b | 0.78a | |
| Zm-Fv | 11.52b | 9.86b | 0.34b | 0.64b | |
| Zm-B25-Fv | 14.28a | 13.80a | 0.47a | 0.77a | |
Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03. Values indicate the average of five plants. Different letters indicate significant differences (Tukey’s test P ≤ 0.05). Bold values highlight statistically significant differences as described in the text.
Table 2.
Summary of filtered and mapped reads obtained from the RNA-seq of maize root samples.
| Sample | Raw Reads | Filtered Reads (Q>20) | Unique Mapped Reads | ||
| Zm-2 | 13,742,216 | 13,534,709 | (98.49%) | 12,542,341 | (91.27%) |
| Zm-3 | 21,273,970 | 20,839,981 | (97.96%) | 17,042,555 | (80.11%) |
| Zm-B25-1 | 22,841,141 | 22,498,524 | (98.50%) | 19,719,075 | (86.33%) |
| Zm-B25-2 | 27,395,643 | 27,003,885 | (98.57%) | 24,442,305 | (89.22%) |
| Zm-Fv-1 | 19,155,891 | 18,880,046 | (98.56%) | 17,497,335 | (91.34%) |
| Zm-Fv-3 | 18,080,786 | 17,815,902 | (98.54%) | 14,513,113 | (80.27%) |
| Zm-B25-Fv-1 | 18,442,655 | 18.199,212 | (98.68%) | 16,495,370 | (89.44%) |
| Zm-B25-Fv-3 | 21,465,145 | 21,166,779 | (98.61%) | 19,373,477 | (90.26%) |
| Average | 20,299,681 | 19,992,380 | (98.49%) | 17,703,196 | (87.28%) |
Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03; Q: Phred quality score. Numbers after the condition name indicate the biological replicate. Filtered reads were mapped to the reference genome of maize B73 v5.
Table 3.
Expressed genes (EGs) in maize roots of the tripartite assay.
| Condition | Total EGs | Percentage of the genomea |
| Zm | 23,625 | 53.33% |
| Zm-B25 | 25,412 | 57.36% |
| Zm-Fv | 24,668 | 55.68% |
| Zm-B25-Fv | 24,808 | 56.00% |
| Average | 24,628 | 55.59% |
Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03. a Refers to the Zea mays B73 v5.0 genome (44,303 genes).
Table 4.
Differentially expressed genes (DEGs) in the interaction conditions (vs. control).
| Condition | Total DEGs | Up-regulated | Down-regulated |
| Zm-B25 | 4331 | 3884 (89.68%) | 447 (10.32%) |
| Zm-Fv | 5473 | 4076 (74.47%) | 1397 (25.53%) |
| Zm-B25-Fv | 2334 | 2077 (88.98%) | 257 (11.02%) |
Zm: Zea mays; B25: Bacillus cereus B25; Fv: Fusarium verticillioides P03.
Table 5.
Comparison of relative expression levels obtained by RNA-seq and qRT-PCR for selected genes from the unique DEGs of the interaction conditions.
Table 5.
Comparison of relative expression levels obtained by RNA-seq and qRT-PCR for selected genes from the unique DEGs of the interaction conditions.
| Condition | IDa | Gene | Fold Change vs control | |
| FC RNA-seq | FC qRT-PCR | |||
| Zm-B25-Fv | Zm00001eb375460 | Glutaredoxin | 11.49 | 4.11 |
| Zm00001eb229260 | Protein kinase domain | 2.18 | 2.24 | |
| Zm00001eb251380 | Cyclic nucleotide-binding domain | -2.23 | -7.56 | |
| Zm00001eb029490 | Gdt1 family | -2.41 | -4.37 | |
| Zm00001eb397080 | CRAL/TRIO, N-terminal domain | -2.61 | -2.74 | |
| Zm-B25 | Zm00001eb124940 | Multi antimicrobial extrusion protein | 2.87 | 2.92 |
| Zm-Fv | Zm00001eb041100 | Hydroxycinnamoyltransferase 9 | 13.1 | 7.3 |
| Zm00001eb419890 | Deoxymugineic acid synthase 6 | 5.09 | 5.57 | |
| Zm00001eb285030 | Putative cytochrome P450 superfamily protein | 7.07 | 3.72 | |
| Zm00001eb241870 | Small auxin up RNA54 | 9.69 | 24.98 | |
a Maize reference identifier (Zea mays B73 v5.0).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



