
Submitted:
29 October 2025
Posted:
29 October 2025
You are already at the latest version
Abstract
Mycobacterium tuberculosis (M.tb), the causative agent of tuberculosis (TB) is responsible for millions of deaths annually. BCG, the only vaccine against TB, fails to evoke memory response and is ineffective to provide protection among adults. The emergence of multi drug resistant strains of M.tb has necessitated development of new vaccines that can confer wider degree and long-term protection against TB. In this study, we have designed a novel multi-epitope vaccine (MEV) using the M.tb proteins, Rv1507A, Rv1509, Rv1954A and Rv2231A that are not only unique to this pathogen but are hallmark as these modulate different aspects of M.tb physiology. Rv1507A and Rv1954A elicit memory response in host while Rv1509 and Rv2231A have roles in virulence. These four proteins were chosen so as to develop a MEV that fills the gaps in existing vaccine against TB. We carried out in-silico analyses of various combinations of epitopes from these proteins, after codon optimization, to ensure that there is no toxicity and allergenicity while maintaining the highest level of immunogenicity of the MEV construct. Two MEV constructs were designed which can pair with different TLR-binding adjuvants: Laterosporulin (TLR-4) and PorB (TLR-2). In silico analysis of the MEV constructs revealed favorable biophysical and physicochemical properties, stability in structure and strong interactions with TLR receptors, suggesting their potential to trigger a robust immune response. Immunological simulations showed that MEV elicits robust immune response and exhibits nearly 90% coverage for global population due to its conserved nature. This study provides an important lead that can be validated through in vitro and in vivo assays, which may translate into tangible vaccine against TB.
Keywords:
adjuvant
; immunoinformatic
; in silico cloning
; MDR-TB
; MD simulation
; memory response
; signature protein
; TLR-2
; TLR-4
Introduction
Mycobacterium tuberculosis (M.tb) is a deadly pathogen responsible for causing tuberculosis (TB). TB continues to pose a significant health threat, contributing to 1.25 million deaths and nearly 10.8 million individuals being infected by M.tb annually (WHO TB report 2024). The COVID-19 pandemic has worsened the management of TB, augmenting the rise in TB-related deaths by 4.5% (Shariq et al., 2022). Rapid emergence of drug-resistant M.tb strains (Siddiqi et al., 2002) and limitations in accurate diagnosis of latent tuberculosis infection (LTBI) (Ahmed and Hasnain, 2011) have outpaced the existing pursuit for TB eradication programmes globally. Vaccination is the viable option as it can provide protection to M.tb infections across a wider group of population (Ahmed and Hasnain, 2011). BCG, the only approved vaccine against TB, provides protection against pulmonary TB among children. However, BCG does not elicit memory response and hence fails to remain effective among previously immunized adults (Bali et al., 2015; Romano and Huygen, 2012; Naqvi et al., 2025). TB vaccination strategies including the use of M72/AS01E are undergoing clinical trials, however they exhibit nearly 49.7% protective efficacy due to their failure to stimulate specific immune cells (Gillard et al., 2016). The potency of subunit vaccine hinges on activating B lymphocytes, CD8+ and CD4+ T lymphocyte cells, which can trigger Th1 response required for M.tb clearance. Conventional methods for developing subunit vaccines are based on one or more immunodominant antigens. Due to lack of predictability of the dominant epitopes on the subunit vaccine, functional assessment of B cells, helper T cells, and cytotoxic T cells response remained a challenging task (Ong, He and Yang, 2020; Selvapandiyan et al., 2023). In silico tools for designing multi-epitope vaccine (MEV) can reliably predict antigenic epitopes, screen suitable adjuvants and linkers that can provide preliminary leads for in vitro assays (Lindestam Arlehamn et al., 2016). Molecular simulation to assess immunogenic potential, including the capacity to activate effector cells and induction of cytokine provide critical inputs that aid in channelizing assays, thereby reducing inadvertent side effects during in vivo testing (Oli et al., 2020). Research on development of MEV’s has notably surged since COVID-19, and it is evident that immunoinformatics based MEV designing can be a robust and reliable tool for providing impetus to development of vaccine (Bibi et al., 2021) (Jiang et al., 2023, Ruaro-Moreno et al., 2023) against infectious diseases and more complex co-morbid conditions.
Out of the 4,173 genes in the M.tb genome, the functional role of around 25% are yet unknown. In the present study we have used four different M.tb proteins which are hallmark due to their exclusive presence in mycobacteria and their immunological properties; and are suitable candidates for MEV (Rahman et al., 2014; Grover et al., 2016; Ahmad et al., 2022; Arora, Alam, et al., 2020a; Arora, Naqvi, et al., 2020a; Chen et al., 2021; Manjunath et al., 2021, 2024a; Rastogi et al., 2023a)(Rani et al., 2022; Manjunath et al., 2024b)(Quadir et al., 2021). Rv1509 is unique to M.tb and is absent in other mycobacterial species, including BCG. Rv1509 induces proliferation of multifunctional CD8+ T and CD4+ cells, stimulates effector memory response and upregulates Th1 response in vivo (Manjunath P et al., 2021). A previous study based on the comparative genomic and proteomic analysis showed that Rv1507a is also unique to M.tb. Rv1507A elicits increased IL-6, IL-12, and TNF-α production, pro-inflammatory Th1 response, and improved survival under stress in mice. Rv1507A also triggers B cell response and generates robust memory responsive immune cells (Arora et al. 2020b). Rv1954A is a secretory protein of M.tb and possess intrinsically disordered regions that bear epitopes for B and T cells. Rv1954A triggers immunogenic response through the TLR-4 receptor and generates robust production of pro-inflammatory cytokines. Functional characterization of Rv1954A, carried out by generation of recombinants expressing M.tb-Rv1954a, showed that this protein enables the recombinant bacilli to upregulate the production of CD86, CD80, MHC I/II and CD40 markers in macrophage, thereby pointing to its role in antigen presentation pathways. In vivo assessment of antibody titers in mice showed that Rv1954A elicits IgG in the sera (Arora, Naqvi, et al., 2020b). TB patients’ sera exhibit enhanced B-cell response including higher level of IgG titers against Rv1509 and Rv1507A (Arora et al 2020a, Arora et al 2020b, Manjunath P et al 2021). Rv2231A, aids the mycobacteria in forming biofilms, which imparts drug tolerance and allows the bacilli to persist intracellularly (Rastogi et al., 2023b). Rv2231a protein interacts with the TLR-4 receptor and elicits pro-inflammatory cytokines. Rv1507A, Rv1509, Rv1954A, and Rv2231A proteins exhibit common mechanism to elicit immune response and also exhibit functional uniqueness which are utilized in our study to design MEV (Arora, et al., 2020a; Arora, Naqvi, et al., 2020a; Chen et al., 2021; Manjunath et al., 2024, 2024a; Rastogi et al., 2023). This new MEV designed against M.tb exhibits several noteworthy improvements over existing documented TB vaccines, such as a greater diversity of epitopes, increased immunogenicity, higher population coverage, and enhanced immune simulation.
Materials and Methods
Prediction of allergenicity, antigenicity and toxicity of epitopes for B cells, CTL and HTL
The NCBI database and Mycobrowser were utilized to retrieve the FASTA sequences of M.tb proteins, Rv1507A, Rv1509, Rv1954A and Rv2231A (Kapopoulou et al. 2011). The NetMHC 4 server having the artificial neural network (ANN) algorithms for sequence alignment was used (Martinelli, 2022) (Andreatta et al. 2016). B cell Epitopes were predicted using the IEDB online webserver and the retrieved sequences were then searched for linear B-cell epitopes (Prawiningrum et al. 2022) keeping the threshold value of 0.51. CTL (MHC-I) and HTL (MHC-II) epitopes were predicted using the IEDB server and reference set of HLA alleles were examined (Kumar and Hasija, 2022; Martinelli, 2022; Yan et al., 2024). CTL epitopes with IC50 value below 50 were selected with the epitope length of 9 mer. The ‘HLA-DR’ allele and a 7-allele (HLA) reference set were included in the search (Martinelli, 2022; Yan et al., 2024). The top 10 epitopes that showed the highest binding potential for MHC-II cells were chosen based on their non-overlapping score. A low percentile score indicating a stronger binding for the HTL/MHC-II cell epitope (Wang et al., 2010) was considered. VaxiJen was used to assess antigenicity (threshold value of 0.5), AllerTOP was used to study allergenicity and ToxinPred was used to examine toxicity (Gupta et al., 2013; Sunita et al., 2020; Pillay et al., 2024). The link to the various webservers or online programs used in the study is provided in the Supplementary Table S1. Only those epitopes that passed the filters for non-toxicity, non-allergenicity, and antigenic profile were used for constructing the MEV. HTL epitopes from each of the M.tb target proteins were docked with the TLR4 and TLR2 receptors using webservers MDOCKPEP (Xu, Yan and Zou, 2018) and HPEPDOCK (Zhou et al., 2018).
Construction of the Multi epitope vaccine
The components of the MEV includes adjuvants, epitopes, and linkers. Adjuvants were attached with CTL, HTL and B cell epitopes using the linkers. EAAAK linker and AAY linkers were attached with CTL epitopes, GPGPG linkers were attached with HTL epitopes and flexible KK linkers were attached with B cell epitopes. Two types of TLR agonists were used for vaccine constructs (Baindara et al., 2016, Dinata and Baindara, 2023). For TLR-4 and TLR-2 receptors, Laterosporulin and Porin B (PorB) were used respectively, as an adjuvant and agonist (Dinata and Baindara, 2023, Yuen et al., 2019).
The physicochemical properties of the MEV vaccine including its molecular weight, aliphatic index, isoelectric point, GRAVY and instability index were evaluated using the Expasy-ProtPram webserver (Sharma et al., 2021; Kumar and Hasija, 2022; Martinelli, 2022). The solubility was assessed using the Protein sol webserver (Almofti, Abdelrahman and Eltilib, 2021).
Structure Modelling, Molecular docking and Molecular dynamics simulations
The 3D and the 2D structures of the MEV were predicted using the PDBsum and PSIPRED web server, which provide information about the type of vaccine being built and display charts showing the proportions of amino acids with aromatic, non-polar, polar chains, alpha-helix, coil, and beta-strand structures (Sharma et al., 2021). To design effective vaccine constructs, we used the Robetta webserver. We further refined these models using Galaxy Refine webserver to enhance their structural quality. SAVES v6.0 webserver was used to validate the tertiary structure of the designed vaccine (Sunita et al., 2020). Vadar and PDBsum were used to generate Ramachandran plots, which assess the stereochemical properties and structural geometry of protein residues. The MEV tertiary structure and quality were evaluated using ERRAT, which examines the interactions between atoms that are not directly bonded to each other (Colovos and Yeates, 1993).
The ClusPro 2.0 webserver was used to conduct a comprehensive analysis of the interactions between the designed MEV and the TLR receptors (Sharma et al., 2021). This analysis involved the identification and characterization of potential binding sites, the assessment of binding affinity, and the exploration of the structural and functional implications of these interactions. MD simulation was carried out using the GROMACS software suite to determine the structural and dynamical features of MEV. The protein structures were calibrated using the GROMOS43a1 force field (Van Der Spoel et al., 2005). To minimize artifacts at the solvent-vacuum interface, the MEV complex was positioned in the center of a cubic simulation box with periodic boundary conditions set at 1.5 nm. To simulate biological conditions, the systems were neutralized with ions, solvated with Simple Point Charge, and water molecules. The steepest-descent approach was used to minimize energy until the maximal force on any given atom was less than 1000 kJ/mol/nm. After minimization, the systems were equilibrated twice: once at 300 K under the NVT ensemble and once at 1 bar under the NPT ensemble. Each equilibration phase ran for 1 ns followed by 200 ns MD simulation with a 2-fs time step. Post-simulation processing for aligning trajectories and fixation of periodicity errors were done using the gmx_trjconv module. The stability and conformational alterations of the TLR-2 and TLR-4 with the MEV complex with respect to time was evaluated. The gmx_rmsd tool was utilized to measure the RMSD to ascertain the average deviation of the complex atoms from their initial positions. The Radius of Gyration (Rg) was computed using the gmx gyrate tool to determine the compactness of the protein. Solvent-Accessible Surface Area (SASA), to determine the degree of solvent exposure of vaccine, was done using the gmx sasa tool (Durham et al., 2009) (Li et al., 2023).
Host immune response, safety analysis and population coverage
The C-ImmSim web tool was utilized to assess potential immune responses triggered by the MEV (Sharma et al 2021). NCBI BLAST was used to compare the MEV sequence with the human proteome, obtained from the NCBI UniProt database (Rasheed et al., 2021). The suitability of the MEV for global population (Kumar and Hasija, 2022) was assessed for compatibility using diverse genetic markers, specifically HLA alleles, present in various populations worldwide. The IEDB database was used to examine the efficacy of the MEV by analyzing their sequences and their associated HLA class I and II alleles, thereby estimating the population coverage that may be protected (Bui et al., 2006; Yan et al., 2024).
Codon optimization and In-silico cloning
The protein sequence of the proposed vaccine was reverse translated into a nucleotide sequence. The nucleotide sequence was optimized using the JCat Codon Optimization web server, to ensure that the MEV exhibits highest gene expression in the host. Restriction enzyme cleavage sites, rho-independent transcription terminators and prokaryotic ribosome binding sites were removed during codon optimization. The JCat webserver was used to improve the GC content of the nucleotide sequence, elevate the CAI score, and eliminate any errors. The SnapGene in-silico cloning tool was used to insert the optimized nucleotide sequence into the pET28a (+) vector at the NdeI and XhoI restriction sites (Sharma et al., 2021)(Grote et al., 2005, Sunita et al., 2020). The optimized nucleotide sequence was then used for in silico cloning in the E. coli K12 strain.
Results
Selection of B cell and T cell epitopes exhibiting no toxicity and allergenicity
Four M.tb proteins (Rv1507A, Rv1509, Rv1954A, and Rv2231A), which have hallmark sequences that are absent in any other mycobacterial species, were selected based on the functional characterization of these proteins done in previous studies (Arora et al. 2020a, Arora et al. 2020b, Manjunath et al. 2021, Rastogi et al. 2023). The NetMHC 4 server having the artificial neural network (ANN) algorithms for sequence alignment was used as this method is resilient to insertions and deletions, leading to better performance compared to other approaches (Martinelli, 2022) (Andreatta and Nielsen, 2016). The FASTA format of the M.tb proteins Rv1507A, Rv1509, Rv1954A, and Rv2231A (Supplementary Table S2) were evaluated using online tools ToxinPred, and VaxiJen, for toxicity, and antigenicity, respectively (Supplementary Table S3). The physicochemical properties, including molecular weight, isoelectric point, instability index, aliphatic index and GRAVY scores, were obtained to assess their suitability for vaccine development (Supplementary Table S1).
Several epitopes like IEQSRLSSTRC (0.91), RALTDKEFAGRRAQW (0.62), RSVSGFVLM (0.52), VPATLQPIM (0.6), SAWEIAINY (1.64), SVSGFVLMIKSASVH (0.62), LARIRSHVDAHAPEL (0.6), PGQKWVNVDPTKNKS (0.58), TSPLSVDHAHTAHRR (0.85) within these M.tb proteins were found to be antigenic and showed non allergenicity. These epitopes exhibited a negative GRAVY value, indicating their hydrophilicity and potential for effective interaction with the immune system. The BepiPred algorithm from the IEDB webserver identified four B-cell epitopes within our target proteins. The four epitopes were subjected to individual docking with TLR receptors, and two epitopes IEQSRLSSTRC and RALTDKEFAGRRAQW were shortlisted based on high docking scores, antigenicity, and non-allergenicity (Supplementary Table S5) (Gupta et al., 2013; Sunita et al., 2020; Pillay et al., 2024).
For each protein, the top 10 non-overlapping HTL epitopes with the lowest ranking scores, and CTL epitopes with an IC50 value below 50 were evaluated for potential allergenicity, antigenicity, and toxicity (Almofti, Abd-elrahman and Eltilib, 2021; Sharma et al., 2021; Prawiningrum, Paramita and Panigoro, 2022; Pillay et al., 2024). The 4 CTL epitopes and 11 HTL epitopes from each of the proteins were further shortlisted based on their high antigenic score, lack of allergenicity and no toxicity. The docking score of each epitope with the TLR4 / TLR2 receptors, 3 CTL epitopes- RSVSGFVLM (-171.1 kcal/mol), SAWEIAINY (-178.3 kcal/mol) and VPATLQPIM (-151.2 kcal/mol) epitopes; and 4 HTL epitopes- SVSGFVLMIKSASVH (-202.7 kcal/mol), LARIRSHVDAHAPEL (-215.9 kcal/mol), PGQKWVNVDPTKNKS (-208.1 kcal/mol) and TSPLSVDHAHTAHRR (-217.6 kcal/mol) were shortlisted for the final construct of MEV (Supplementary Table S5). The two MEV constructs were evaluated using the AllerTop, which is an innovative alignment-free method that determines allergenicity based on the chemical composition of amino acid sequences within the vaccine (Kumar and Hasija, 2022; Martinelli, 2022). This method operates by transforming amino acid sequences into a fixed-length vector through auto-cross-covariance. By analyzing the resulting vector, AllerTop can predict the likelihood of an allergic response to the vaccine and aid in early identification and mitigation of potential risks. (Dimitrov et al., 2014; Sunita et al., 2020; Pillay et al., 2024). The MEV constructs were found to be non-allergenic. We selected two specific vaccine constructs, one for TLR-4 (MEV-1) and one for TLR-2 (MEV-2) receptors, for further study.
Construction of MEV using the Epitopes, Adjuvants and Linkers
B cell and T cell epitopes were randomly shuffled and TLR-4 / TLR-2 adjuvant were added to the C-terminals of the construct (Supplementary Table S6). The overview of constructing the MEV is depicted in (Figure 1) (Sharma et al., 2021). Adjuvants and linkers are indispensable components in the development of MEV (Martinelli, 2022). Adjuvants act as immune system potentiators, amplifying the body’s immunological response to vaccine antigens. Laterosporulin is an anti-TB peptide and used as TLR-4 adjuvant (Figure 2a). Porin is an outer membrane protein from Neisseria meningitidis and used as a TLR-2 adjuvant (Figure 2b). Linkers are short amino acid segments that act as molecular bridges between adjuvant and epitopes, enhance immune responses, and prevent epitope misfolding. Adjuvants were linked to T cell and B cell epitopes via a variety of immunogenic linkers, including KK, GPGPG, AAY, and EAAAK. By strategically positioning antigens and adjuvants, linkers can optimize the presentation of antigens to the immune system, thereby promoting a more robust and targeted immune response (Sharma et al., 2021). The adjuvants were linked with epitopes using the α-helical EAAAK linker to stabilize their interaction. AAY linker, which joins two proteins, was used to enhance the integrity of the MEV construct and allows immune system to recognize the antigenic epitopes on the MEV. GPGPG linker, which is high in glycine-proline residues, was used to aid in correct folding of the MEV antigens, provide stability to MEV, minimize aggregation, and maintain the immunogenicity of the MEV construct. The KK linkers bridge the B cell epitopes.
MEV constructs exhibits favourable Physiochemical properties
The physiochemical properties of the MEV-1 and MEV-2 construct were evaluated. The aliphatic index, molecular weight, theoretical pI, instability index and GRAVY score of the designed MEV constructs were evaluated using the Expasy ProtPram web server (Almosfti, Abd-elrahman and Eltilib, 2021; Sharma et al., 2021). The functioning of the Expasy is based on the amino acid sequence and pKa values of the MEV construct (Yang et al., 2024). Molecular weight influences the vaccine antigenicity, the pI value indicates the alkaline nature of the designed constructs, the aliphatic index validates thermostability, the GRAVY index determines polar or non-polar nature, and solubility guarantees homogenous vaccine dissolution. The constructs have basic nature, with pI value of 9.49 (MEV-1) and 9.69 (MEV-2). Both constructs MEV-1 and MEV-2 were also stable, with instability scores of 37.61 and 30.85, respectively, which are well below the threshold of 40. Additionally, the vaccine’s aliphatic index, which measures its thermostability, was found to be 66.5 for the MEV-1 and 80.4 for the MEV-2 constructs, indicating they were thermostable. The constructs were hydrophilic, and their solubility was evaluated using the protein sol webserver, with results ranging from 0.63 (MEV-1) to 0.66 (MEV-2), demonstrating these were soluble (Supplementary Figure S1). The antigenicity of the MEV constructs were assessed using the VaxJen 2.0 webserver. The physicochemical features of the MEV constructs (Table 1) showed that these are stable, hydrophilic and soluble, making them promising candidates for further vaccine development (Martinelli, 2022).
MEV exhibits structural stability
Analyzing the functional characteristics of a vaccine involves predicting its tertiary structure, which is a crucial step in the development of a MEV. The PDBSum and PSIPRED web servers were utilized to estimate the secondary structures of the MEV constructs (Rasheed et al., 2021; Kumar and Hasija, 2022; Yang et al., 2024) (Sharma et al., 2021). The two designed MEV constructs have distinct secondary structures with different proportions of alpha-helices, random coils, and beta-strands. The Robetta Baker lab webserver was used to create 3D models of both the MEVs, which were then refined using the GalaxyRefine webserver. The ERRAT and Ramachandran plot analyses were utilized to assess the overall quality of the vaccine constructs (Table 2). For the MEV-1 model, an estimated 93.4% of residues occupy the most stable conformations, with approximately (~88.1%) in the favored region and (9.4~%) in the allowed region. This model contained 6 residues (~1.2%) in disallowed regions. The MEV-2 model demonstrated an estimated 96.0% of its residues in stable conformations, breaking down to approximately (~88.2%) in the favored region and (~9.6%) in the allowed region. MEV-2 model had 4 residues (~1.1%) in disallowed regions. Our results showed that both MEVs have amino acid residues in favoured regions, indicating minimal errors in protein folding and structure. We observed that the MEV-1 is composed of 10 α-helices and 3 β-strands, possesses a heterogeneous and flexible architecture; this structural variation is hypothesized to facilitate the presentation of a diverse repertoire of both linear and conformational epitopes (Figure 3a). The MEV-2 construct, comprising 11 α-helices and a predominance of 7 β-strands, adopts a more complex and ordered conformation dominated by β-sheets. Our results suggest that MEV-1 may have a more stable structure and resistant to degradation. On the other hand, the MEV-2 (Figure 3b) has a higher proportion of beta-strands, which may favour protein-protein interactions.
Molecular docking of MEV construct shown stable interaction with TLR
It is important for vaccines to interact with human receptors, such as TLRs, as a consequence of which host cells can produce antibodies that stimulate and attract more immune cells at the site of infection. We evaluated the interaction of both MEV constructs with TLR-4 and TLR-2 (Martinelli, 2022). The crystal structure of human TLR-4 and TLR-2 were retrieved from the RCSB Protein Data Bank (PDB). The monomeric form of the receptors was prepared using PyMOL and structures were refined with Galaxy Refine webserver (Sunita et al., 2020; Almofti, Abdelrahman and Eltilib, 2021; Sharma et al., 2021). Protein-protein docking was carried out using ClusPro, and the PDB files of both the receptors and ligands were considered (Figure 4a and Figure 4b). The docking model with the lowest energy and the highest number of clusters was selected for further analysis. The binding energy (ΔG) was calculated using the PRODIGY webserver. Protein-protein docking simulations utilizing the ClusPro webserver revealed a favorable binding affinity between the MEV constructs and their corresponding TLR-2/TLR-4 receptors. Molecular docking simulations predicted stable and high-affinity interactions for both vaccine constructs with their respective receptors. The MEV-1-TLR4 complex yielded a binding score of (-17.9 kcal/mol), indicating a more energetically favorable interaction than the MEV-2-TLR2 complex, which scored (-15.5 kcal/mol). This higher affinity is structurally supported by an extensive interface, featuring key interactions such as those between Gln39 of MEV-1 and residues His159, Asp209, and Asp181 of TLR4 receptor, alongside a salt bridge between Arg139 (MEV-1) and Glu605 (TLR4). The MEV-2-TLR2 complex is also stabilized by significant contacts, including a salt bridge between Lys130 (MEV-2) and Glu369 (TLR2). These interactions suggest that the MEV construct can effectively engage thermodynamically with the TLR-2 and TLR-4 receptors. The protein-protein interaction of both vaccine constructs with the TLR receptors demonstrated a strong interaction of -17.9 ΔG for the TLR-4 vaccine construct and -15.5 ΔG for the TLR-2 vaccine construct. Our results suggest that the MEV can favorably interact with TLR and may potentially activate downstream TLR signaling pathways that may modulate the outcome of the immune response.
Molecular Dynamic simulation and binding affinity shows conformational stability of MEV with TLR
Molecular dynamic simulation was performed to assess the conformational stability of the MEV during their interaction with TLR (Van Der Spoel et al., 2005; Durham et al., 2009). The RMSD analysis, which measures the average distance between the atoms of protein structures, showed that the conformation of the MEV construct was steady throughout the whole simulation, with slight RMSD value fluctuation. The MEV-1-TLR4 complex demonstrated exceptional stability with a lower average RMSD of 0.53 nm compared to the MEV-2-TLR2 complex’s 0.83 nm. The gmx_rmsf tool was utilized to conduct the RMSF analysis, which reveals the area of structural variability and computes the average displacement of atoms from their starting positions. This enabled an evaluation of the flexibility of various regions within the complex. Using the data of RMSF, which describes the flexibility of each amino acid residue in the protein, it was evident that the MEV construct was structurally stable during 200 ns of the dynamic simulation. The MD simulation results (Figure 5a and Figure 5b) point that the MEV construct remains stable, with some regions showing some flexibility while retaining the compactness and solvent accessibility of the structures. The average RMSD values for both the MEV (TLR-4 construct and TLR-2 construct) showed minor deviations in the initial structures in the initial duration of MD simulation, further suggesting conformational equilibration with only minor deviations during the starting period of the simulation. The RMSF analysis highlighted regions in the MEV constructs that have flexible loops and terminal regions portrayed a more significant atomic displacement than relatively stable regions. The difference in flexibility points that specific regions within the TLR-4 and TLR-2 vaccine complexes are more dynamic which may contribute to their functional roles. This was reflected in the average RMSF values of 0.23 nm for MEV-1-TLR4 and 0.29 nm for MEV-2-TLR2. The compactness of the MEV structure was computed using the gmx gyrate tool that analyses the distribution of atoms around the complex’s center of mass, the Radius of Gyration (Rg), which provides insights into folding and structural integrity. The steady Rg values of 2.92 nm (MEV-1-TLR4) and 3.42 nm (MEV-2-TLR2) was observed. The results show that both constructs are compact and relatively stable during the simulation time, their respective Rg values remain steady, with no indication of unfolding events. Thus, the overall structural integrity of the TLR4-vaccine (Figure 6a) and the TLR2-vaccine (Figure 6b) constructs was stable. The SASA measurements which assess solvent exposure into both protein complexes, recorded values of 302.75 nm² (MEV-1-TLR4) and 314.63 nm² (MEV-2-TLR2) which confirmed the conformational stability and compactness observed. Overall, the MD results confirm that the TLR4-MEV-1 and TLR2-MEV-2 complexes are conformationally stable (Table 3).
Pro-inflammatory response predicted for the MEV-TLR2/4 complex
Effective vaccine development necessitates a comprehensive understanding of immune responses. In order to evaluate the potential immunogenicity of the novel MEV constructs, we carried out in silico immune simulation using the C-ImmSim webserver. This web server predicts the production of interferons, cytokines, and antibodies in response to the synthetic MEV (M.tb antigen), as well as the levels of helper (Th1 and Th2) cells (Sharma et al., 2021). Simulation studies predicted that MEV-1 and MEV-2 can mount a robust immune response which may influence the production of pro-inflammatory and regulatory cytokines, including IFN-γ, TGF-β, TNF-α, IL-12, IL-6, IL-4, IL-18, IL-10, IL-28, and IFN-β (Sharma et al., 2021; Kumar and Hasija, 2022; Pillay et al., 2024). Production of diverse antibody classes (IgM, IgG, IgG1, and IgG2) by activated B cells was also predicted. A significant expansion and activation of both B and T cells is indicative of a robust immune response. Our results (Figure 7) imply that both MEV-1 and MEV-2 constructs may induce robust immune response which may be effective in conferring protection against M.tb.
Wider population coverage and safety predicted for the MEV-TLR2/4 complex
In order to assess the potential safety of the novel MEV constructs we compared the protein sequences of MEV construct with the extensive database of human proteins and found no significant sequence homology between the two. Worldwide, there could be small variations in the expression of HLA alleles due to geographical and ethnic variances. Our MEV-1 and MEV-2 constructs were meticulously engineered using specific CTL and HTL epitopes, paired with their corresponding HLA alleles. These components were carefully selected to ensure broad global compatibility. By utilizing the IEDB webserver for population coverage analysis (Supplementary Figure S2), we determined that our vaccine constructs are highly effective in a significant portion of the worldwide population, achieving an impressive coverage of 89.3% (Table 4) This observation underscores the MEV potential to provide immunity to a diverse range of individuals across the globe (Martinelli, 2022).
Codon optimization and in-silico cloning of the MEV construct
In silico codon optimization of the heterologous expression of MEV-1 and MEV-2 constructs for the E. coli K12 strain was carried out (Almofti, Abd-elrahman and Eltilib, 2021; Rasheed et al., 2021; Sharma et al., 2021; Kumar and Hasija, 2022; Pillay et al., 2024; Yang et al., 2024). This method involves modifying the codon usage of the gene sequence to align with the host organism’s codon bias, thereby increasing protein production efficiency. The optimal GC content and CAI values for the constructs were determined using the JCat webserver. Following optimization, the MEV-1 construct exhibited a GC content of nearly 58% and a CAI of 0.75, while the MEV-2 construct had a GC content of 57.5% and a CAI of 0.76. A high CAI value, typically considered to be 0.8 or above, indicates favorable codon usage for efficient protein synthesis in E. coli. In silico cloning of the optimized constructs was performed using SnapGene software. The gene sequences were inserted into the pET28a(+) vector at the XhoI and NdeI restriction sites, resulting in final genome sizes of 5908bp for MEV-1 vaccine construct (Figure 8a) and 5974bp for MEV-2 vaccine construct (Figure 8b).
Discussion
The only approved vaccine against TB is the BCG, which is effective against extrapulmonary TB in children but largely ineffective against pulmonary TB among the adults. BCG fails to elicit memory response and is also deficient in many essential antigens in the region of deletion (RD) when compared to M.tb. There is a need for development of new and affective forms of vaccine against TB given the growing number of MDR-TB cases. Conventional method for developing vaccine is cumbersome and time consuming as it involves various steps including in vitro and in vivo validation that sometimes fail to get desired outcome (Shi et al., 1999). Recent advancements in the field of immunoinformatics and data driven online resources have catapulted the speed of vaccine development. In silico construction of vaccine relies on database and online servers that can simulate the biophysical aspects of structure based testing of proteins. We ventured to develop a multi-epitope vaccine against TB, using unique and hallmark M.tb proteins, Rv1507A, Rv1509, Rv1954 and Rv2231A (Arora et al 2020a, Arora et al 2020b, , Rastogi et al 2023, Manjunath P et al. 2024) which have been functionally characterized to be immunogenic. These proteins were meticulously selected as these elicit humoral and cellular immune responses thereby modulating different aspects of M.tb physiology associated with pathogenicity (Arora, Alam, et al., 2020a; Arora, Naqvi, et al., 2020a; Chen et al., 2021; Manjunath et al., 2021, 2024a; Rastogi et al., 2023a). The presence of antibody titers against Rv1507A and Rv1954 in TB patients further underlines their importance in pathophysiology. The MEV vaccine has been designed in such a way that it can target both innate and adaptive immunity by using T-cell and B-cell epitopes. Epitopes are specific sites on antigenic protein molecules that can interact with human immune cell receptors and trigger a heightened immune response. HTL and CTL helps the host to establish a robust immunological defense against infections. To predict highly antigenic CTL epitopes, we used the IEDB webserver. IEDB webserver provides a single repository for a variety of epitope-related data connected to various immunological processes, including production of vaccines, infectious diseases, autoimmune disorders, and cancer immunotherapy (Larsen, Lund and Nielsen, 2006).
When developing a MEV, choosing the right B-cell epitopes is essential since they can trigger several immunogenic processes, such as complement system activation, T-cell cytotoxicity and antigen neutralization. B cell and T cell epitopes are non-self, non-allergic and antigenic, implying they induce immune response in the host (Kumar and Hasija, 2022). Through the incorporation of two B-cell epitopes, three helper T lymphocyte epitopes and four cytotoxic T lymphocytes generated from four M.tb proteins, this in-silico work describes a new MEV vaccine that may be effective against M.tb pathogen. It is essential that the various epitopes used for MEV construct are non-toxic and non-allergenic (Martinelli, 2022). Epitopes were thoroughly assessed for allergenicity, toxicity, and antigenicity in each step of funnel-down approach so as to ensure safety and efficacy of the screened MEV constructs. Linkers were used to join the epitopes such that they facilitate proper protein folding of MEV. The EAAAK linker’s rigid α-helical conformation provides benefits in molecular design by efficiently dividing functional domains. GPGPG linker can trigger helper T-cell responses and reduce junctional immunogenicity, while KK linker targets cathepsin B, a lysosomal protease essential for antigen processing and presentation. This approach prevents antibody-mediated degradation of the linearly connected peptide sequence, leading to increased overall immunogenicity. To further enhance immunogenicity, we incorporated adjuvants and TLR agonists like Laterosporulin and PorB. These molecules stimulate the immune system, amplifying the vaccine’s effectiveness. A flexible 3D structure was designed, incorporating linkers to facilitate protein folding and enhance immunogenicity. Molecular simulation studies validated the conformational stability of the MEV structure. Simulation of the immunogenic potential showed that the MEV can, in theory, activate both TLR2 and TLR4 mediated pathways. TLR 2 and TLR 4 recognize specific components of M.tb and initiate the cascade of signaling events that coordinate the pro-inflammatory response.
Development of vaccines depends on a thorough understanding of how HLA alleles are distributed throughout the world’s population (Almofti, Abdelrahman and Eltilib, 2021). It was observed that the MEV is compatible for 89.3% of the world’s population. Our MEV construct shows no homology to human proteins and hence is unlikely to interfere with the normal functioning of the human immune system. This is crucial for ensuring the effectiveness and safety of the vaccine, as it minimizes the risk of adverse reactions and ensures that the immune system can effectively mount a protective response against the target pathogen.
The escalating global burden of TB necessitates innovative strategies to enhance disease management and combat treatment resistance. In-silico cloning suggested that the designed MEV can be produced efficiently in microbial expression system. In silico results point that MEV is safe and can target both TLR-2 and TLR-4 pathways, and hence may be better as compared to existing TB vaccines in terms of its potential for wider population coverage. Recent reports show that M.tb can act as a carcinogen which can trigger pathways leading to cancer (Malik et al. 2022, 2023). This has widened the realm of designing the MEV that can confer protection against multiple diseases in co-morbid conditions such as TB and cancer, which surprisingly have various convergent mechanisms and pathways (Roy et al. 2021), utilization of immunoinformatics approach for designing MEV can provide a robust solution in development of vaccine effective against co-morbid conditions (Nalbandian et al., 2009; Roy, Ehtesham and Hasnain, 2021; Vashishth et al., 2023)(Malik et al., 2022). The M.tb protein Rv1509, has been implicated to cause multi-nucleated giant cells (Manjunath et al 2024) which possess features resembling the granuloma. We have incorporated Rv1509 in our MEV and are optimistic that this addition will improve the novelty of the MEV as it may widen the spectrum for targeting mechanisms where TB and Cancer converge. It is envisaged that MEV may possibly play role in host directed therapy for disrupting the shared mechanistic pathways of TB and Cancer, potentially offering hope for co-morbid conditions (Nalbandian et al., 2009; Gupta et al., 2016; Roy, Ehtesham and Hasnain, 2021; Sheikhpour et al., 2021; Malik et al., 2022; Qin et al., 2022; Vashishth et al., 2023; Simatupang et al., 2024; Wang, Zou and He, 2024). The leads from our study could pave the way for preclinical and clinical trials to evaluate the vaccine’s efficacy and safety in human populations. This MEV represents a promising advancement in the fight against TB. Its innovative design, incorporation of immunogenic epitopes, and rigorous validation demonstrate its potential to contribute significantly to global TB management. While our results underline the proof of concept, in vitro and in vivo studies are necessary to validate the vaccine’s efficacy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Conflict of Interest
Authors declare NO conflict of Interest.
Acknowledgement
SEH is the National Science Chair, ANRF, Department of Science & Technology, GoI and Fellow of Robert Koch Institute, Germany. AA is the member of the Programme Implementation Group of the DST-FIST at Sharda University, Greater Noida. The support of Indian Council of Medical Research-GoI for supporting the research work is acknowledged. The Support of Department of Biotechnology-GoI for supporting the research work is acknowledged.
References
- Ahmad, F. et al. (2022) ‘Macrophage: A Cell With Many Faces and Functions in Tuberculosis’, Frontiers in Immunology, 13, p. 747799. [CrossRef]
- Ahmed, N. and Hasnain, S.E. (2011) ‘Molecular epidemiology of tuberculosis in India: Moving forward with a systems biology approach’, Tuberculosis, 91(5), pp. 407–413. [CrossRef]
- Almofti, Y.A., Abd-elrahman, K.A. and Eltilib, E.E.M. (2021) ‘Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2)’, BMC Immunology, 22(1). [CrossRef]
- Andreatta, M. and Nielsen, M. (2016) ‘Gapped sequence alignment using artificial neural networks: application to the MHC class I system’, Bioinformatics, 32(4), pp. 511–517. [CrossRef]
- Arora, S.K., Alam, A., et al. (2020a) ‘Immunodominant Mycobacterium tuberculosis Protein Rv1507A Elicits Th1 Response and Modulates Host Macrophage Effector Functions’, Frontiers in Immunology, 11, p. 505895. [CrossRef]
- Arora, S.K., Alam, A., et al. (2020b) ‘Immunodominant Mycobacterium tuberculosis Protein Rv1507A Elicits Th1 Response and Modulates Host Macrophage Effector Functions’, Frontiers in Immunology, 11. [CrossRef]
- Arora, S.K., Naqvi, N., et al. (2020a) ‘Mycobacterium smegmatis Bacteria Expressing Mycobacterium tuberculosis-Specific Rv1954A Induce Macrophage Activation and Modulate the Immune Response’, Frontiers in Cellular and Infection Microbiology, 10, p. 564565. [CrossRef]
- Arora, S.K., Naqvi, N., et al. (2020b) ‘Mycobacterium smegmatis Bacteria Expressing Mycobacterium tuberculosis-Specific Rv1954A Induce Macrophage Activation and Modulate the Immune Response’, Frontiers in Cellular and Infection Microbiology, 10. [CrossRef]
- Baindara, P. et al. (2016) ‘Laterosporulin10: a novel defensin like Class IId bacteriocin from Brevibacillus sp. strain SKDU10 with inhibitory activity against microbial pathogens’, Microbiology, 162(8), pp. 1286–1299. [CrossRef]
- Bali, P. et al. (2015) ‘Strategies to Improve BCG Vaccine Efficacy’, Immunotherapy, 7(9), pp. 945–948. [CrossRef]
- Bibi, S. et al. (2021) ‘In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology’, Scientific Reports, 11(1), p. 1249. [CrossRef]
- Bui, H.-H. et al. (2006) ‘Predicting population coverage of T-cell epitope-based diagnostics and vaccines.’, BMC bioinformatics, 7, p. 153. [CrossRef]
- Chen, Y. et al. (2021) ‘Use of Rv0222-Rv2657c-Rv1509 Fusion Protein to Improve the Accuracy of an Antibody ELISA for Extra-Pulmonary Tuberculosis in Humans’, Pathogens 2021, Vol. 10, Page 828, 10(7), p. 828. [CrossRef]
- Colovos, C. and Yeates, T.O. (1993) ‘Verification of protein structures: patterns of nonbonded atomic interactions.’, Protein science : a publication of the Protein Society, 2(9), pp. 1511–9. [CrossRef]
- Dimitrov, I. et al. (2014) ‘AllerTOP v.2--a server for in silico prediction of allergens.’, Journal of molecular modeling, 20(6), p. 2278. [CrossRef]
- Dinata, R. and Baindara, P. (2023) ‘Laterosporulin25: A probiotically produced, novel defensin-like bacteriocin and its immunogenic properties’, International Immunopharmacology, 121, p. 110500. [CrossRef]
- Durham, E. et al. (2009) ‘Solvent accessible surface area approximations for rapid and accurate protein structure prediction’, Journal of Molecular Modeling, 15(9), pp. 1093–1108. [CrossRef]
- Gillard, P. et al. (2016) ‘Safety and immunogenicity of the M72/AS01E candidate tuberculosis vaccine in adults with tuberculosis: A phase II randomised study.’, Tuberculosis (Edinburgh, Scotland), 100, pp. 118–127. [CrossRef]
- Global Tuberculosis Report 2024 (no date). Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024 (Accessed: 24 November 2024).
- Grote, A. et al. (2005) ‘JCat: a novel tool to adapt codon usage of a target gene to its potential expression host.’, Nucleic acids research, 33(Web Server issue), pp. W526-31. [CrossRef]
- Grover, S. et al. (2016) ‘Analyses of methyltransferases across the pathogenicity spectrum of different mycobacterial species point to an extremophile connection’, Molecular BioSystems, 12(5), pp. 1615–1625. [CrossRef]
- Gupta, P.K. et al. (2016) ‘Mycobacterium tuberculosis H37Rv infected THP-1 cells induce epithelial mesenchymal transition (EMT) in lung adenocarcinoma epithelial cell line (A549)’, Cellular Immunology, 300, pp. 33–40. [CrossRef]
- Gupta, S. et al. (2013) ‘In Silico Approach for Predicting Toxicity of Peptides and Proteins’, PLoS ONE, 8(9). [CrossRef]
- Jiang, F. et al. (2023) ‘A comprehensive approach to developing a multi-epitope vaccine against Mycobacterium tuberculosis: from in silico design to in vitro immunization evaluation’, Frontiers in Immunology, 14. [CrossRef]
- Kapopoulou, A., Lew, J.M. and Cole, S.T. (2011) ‘The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes.’, Tuberculosis (Edinburgh, Scotland), 91(1), pp. 8–13. [CrossRef]
- Kumar, S. and Hasija, Y. (2022) ‘Immunoinformatics Tools: A boon in vaccine Development Against Covid-19’, 2022 IEEE Delhi Section Conference, DELCON 2022 [Preprint].
- Larsen, J.E.P., Lund, O. and Nielsen, M. (2006) ‘Improved method for predicting linear B-cell epitopes.’, Immunome research, 2, p. 2. [CrossRef]
- Li, Z. et al. (2023) ‘Assessment of the Binding of Pseudallecin A to Human Serum Albumin with Multi-Spectroscopic Analysis, Molecular Docking and Molecular Dynamic Simulation’, Chemistry & Biodiversity, 20(12). [CrossRef]
- Lindestam Arlehamn, C.S. et al. (2016) ‘A Quantitative Analysis of Complexity of Human Pathogen-Specific CD4 T Cell Responses in Healthy M. tuberculosis Infected South Africans.’, PLoS pathogens, 12(7), p. e1005760. [CrossRef]
- Malik, A.A. et al. (2022) ‘Can Mycobacterium tuberculosis infection lead to cancer? Call for a paradigm shift in understanding TB and cancer’, International Journal of Medical Microbiology, 312(5). [CrossRef]
- Manjunath, P. et al. (2021) ‘Mycobacterium tuberculosis Specific Protein Rv1509 Evokes Efficient Innate and Adaptive Immune Response Indicative of Protective Th1 Immune Signature’, Frontiers in Immunology, 12, p. 706081. [CrossRef]
- Manjunath, P. et al. (2024a) ‘Expression of a unique M. tuberculosis DNA MTase Rv1509 in M. smegmatis alters the gene expression pattern and enhances virulence’, Frontiers in Microbiology, 15, p. 1344857. [CrossRef]
- Manjunath, P. et al. (2024b) ‘Expression of a unique M. tuberculosis DNA MTase Rv1509 in M. smegmatis alters the gene expression pattern and enhances virulence’, Frontiers in Microbiology, 15, p. 1344857. [CrossRef]
- Martinelli, D.D. (2022) ‘In silico vaccine design: A tutorial in immunoinformatics’, Healthcare Analytics, 2, p. 100044. [CrossRef]
- Nalbandian, A. et al. (2009) ‘Lung carcinogenesis induced by chronic tuberculosis infection: The experimental model and genetic control’, Oncogene, 28(17), pp. 1928–1938. [CrossRef]
- Naqvi, N. et al. (2025) ‘BCG’s role in strengthening immune responses: Implications for tuberculosis and comorbid diseases’, Infection, Genetics and Evolution, 127, p. 105703. [CrossRef]
- Oli, A.N. et al. (2020) ‘Immunoinformatics and Vaccine Development: An Overview’, ImmunoTargets and Therapy, Volume 9, pp. 13–30. [CrossRef]
- Ong, E., He, Y. and Yang, Z. (2020) ‘Epitope promiscuity and population coverage of Mycobacterium tuberculosis protein antigens in current subunit vaccines under development.’, Infection, genetics and evolution : journal of molecular epidemiology and evolutionary genetics in infectious diseases, 80, p. 104186. [CrossRef]
- Pillay, K. et al. (2024) ‘In silico design of Mycobacterium tuberculosis multi-epitope adhesin protein vaccines’, Heliyon, p. e37536. [CrossRef]
- Prawiningrum, A.F., Paramita, R.I. and Panigoro, S.S. (2022) ‘Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine’, Diagnostics. Multidisciplinary Digital Publishing Institute (MDPI). [CrossRef]
- Qin, Y. et al. (2022) ‘The relationship between previous pulmonary tuberculosis and risk of lung cancer in the future’, Infectious Agents and Cancer. BioMed Central Ltd. [CrossRef]
- Quadir, N. et al. (2021) ‘Development and Validation of Signature Sequence–Based PCR for Improved Molecular Diagnosis of Tuberculosis’, The Journal of Molecular Diagnostics, 23(9), pp. 1138–1144. [CrossRef]
- Rahman, S.A. et al. (2014) ‘Comparative analyses of nonpathogenic, opportunistic, and totally pathogenic mycobacteria reveal genomic and biochemical variabilities and highlight the survival attributes of Mycobacterium tuberculosis’, mBio, 5(6). [CrossRef]
- Rani, A. et al. (2022) ‘Mycobacterium tuberculosis Methyltransferase Rv1515c Can Suppress Host Defense Mechanisms by Modulating Immune Functions Utilizing a Multipronged Mechanism’, Frontiers in Molecular Biosciences, 9, p. 906387. [CrossRef]
- Rasheed, M.A. et al. (2021) ‘Immunoinformatics based prediction of recombinant multi-epitope vaccine for the control and prevention of SARS-CoV-2’, Alexandria Engineering Journal, 60(3), pp. 3087–3097. [CrossRef]
- Rastogi, N. et al. (2023a) ‘Structural and Biophysical properties of therapeutically important proteins Rv1509 and Rv2231A of Mycobacterium tuberculosis’, International Journal of Biological Macromolecules, 245, p. 125455. [CrossRef]
- Rastogi, N. et al. (2023b) ‘Structural and Biophysical properties of therapeutically important proteins Rv1509 and Rv2231A of Mycobacterium tuberculosis’, International Journal of Biological Macromolecules, 245, p. 125455. [CrossRef]
- Romano, M. and Huygen, K. (2012) ‘An update on vaccines for tuberculosis - there is more to it than just waning of BCG efficacy with time.’, Expert opinion on biological therapy, 12(12), pp. 1601–10. [CrossRef]
- Roy, D., Ehtesham, N.Z. and Hasnain, S.E. (2021) ‘Is Mycobacterium tuberculosis carcinogenic to humans?’, FASEB Journal, 35(9). [CrossRef]
- Ruaro-Moreno, M. et al. (2023) ‘Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology.’, Microorganisms, 11(7). [CrossRef]
- Selvapandiyan, A. et al. (2023) ‘Zooming in on common immune evasion mechanisms of pathogens in phagolysosomes: potential broad-spectrum therapeutic targets against infectious diseases’, FEMS Microbiology Reviews, 47(1). [CrossRef]
- Shariq, M. et al. (2022) ‘COVID-19 and tuberculosis: the double whammy of respiratory pathogens’, European Respiratory Review, 31(164). [CrossRef]
- Sharma, R. et al. (2021) ‘An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins’, Scientific Reports, 11(1). [CrossRef]
- Sheikhpour, M. et al. (2021) ‘The Common miRNAs between Tuberculosis and Non-Small Cell Lung Cancer: A Critical Review’, Tanaffos, 20(3), pp. 197–208.
- Shi, Y.P. et al. (1999) ‘Immunogenicity and in vitro protective efficacy of a recombinant multistage Plasmodium falciparum candidate vaccine’, Proceedings of the National Academy of Sciences of the United States of America, 96(4), pp. 1615–1620. [CrossRef]
- Siddiqi, N. et al. (2002) ‘Molecular characterization of multidrug-resistant isolates of Mycobacterium tuberculosis from patients in North India’, Antimicrobial Agents and Chemotherapy, 46(2), pp. 443–450. [CrossRef]
- Simatupang, E.T.M. et al. (2024) ‘How Tuberculosis Scar Could Induce Lung Cancer?’, Journal of The Indonesian Medical Association, 73(6), pp. 261–264. [CrossRef]
- Van Der Spoel, D. et al. (2005) ‘GROMACS: Fast, flexible, and free’, Journal of Computational Chemistry, 26(16), pp. 1701–1718. [CrossRef]
- Sunita et al. (2020) ‘Computational approaches in epitope design using DNA binding proteins as vaccine candidate in Mycobacterium tuberculosis’, Infection, Genetics and Evolution, 83. [CrossRef]
- Vashishth, A. et al. (2023) ‘Mycobacterium Tubercular Mediated Inflammation and Lung Carcinogenesis: Connecting Links’, OBM Genetics. LIDSEN Publishing Inc. Available at: . [CrossRef]
- Wang, C., Zou, R.Q. and He, G.Z. (2024) ‘Progress in mechanism-based diagnosis and treatment of tuberculosis comorbid with tumor’, Frontiers in Immunology. Frontiers Media SA. [CrossRef]
- Wang, P. et al. (2010) ‘Peptide binding predictions for HLA DR, DP and DQ molecules.’, BMC bioinformatics, 11, p. 568. [CrossRef]
- Xu, X., Yan, C. and Zou, X. (2018) ‘MDockPeP: An ab-initio protein-peptide docking server.’, Journal of computational chemistry, 39(28), pp. 2409–2413. [CrossRef]
- Yan, Z. et al. (2024) ‘Next-generation IEDB tools: a platform for epitope prediction and analysis’, Nucleic Acids Research, 52(W1), pp. W526–W532. [CrossRef]
- Yang, Y. et al. (2024) ‘Bioinformatics analysis and immunogenicity assessment of the novel multi-stage DNA vaccine W541 against Mycobacterium tuberculosis’, Authorea Preprints [Preprint].
- Yuen, R. et al. (2019) ‘Neisserial PorB immune enhancing activity and use as a vaccine adjuvant’, Human Vaccines & Immunotherapeutics, 15(11), pp. 2778–2781. [CrossRef]
- Zhou, P. et al. (2018) ‘HPEPDOCK: a web server for blind peptide-protein docking based on a hierarchical algorithm.’, Nucleic acids research, 46(W1), pp. W443–W450. [CrossRef]
Figure 1.
An overview of the immunoinformatics based workflow used to develop MEV construct using four proteins of Mycobacterium tuberculosis.
Figure 1.
An overview of the immunoinformatics based workflow used to develop MEV construct using four proteins of Mycobacterium tuberculosis.
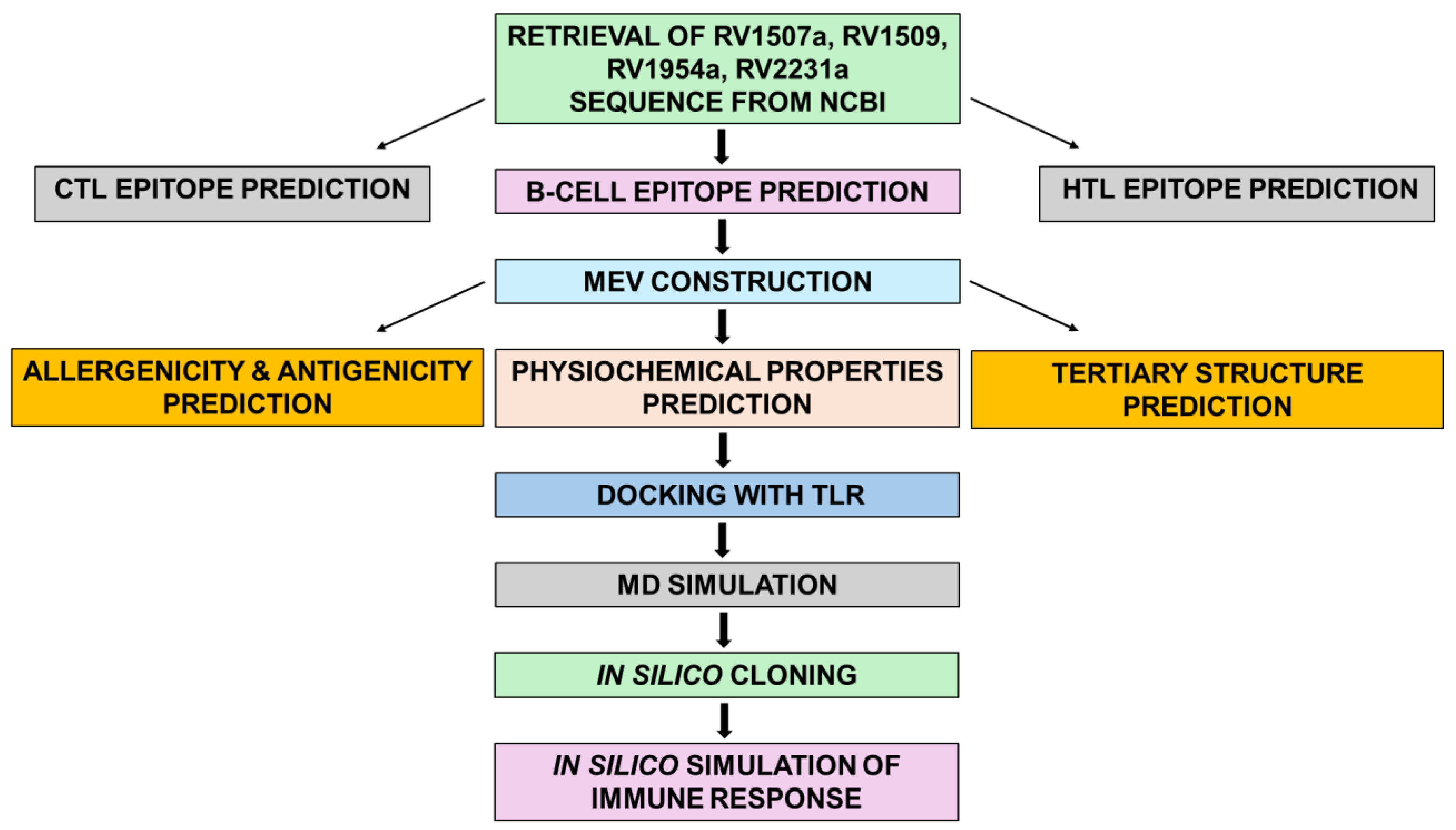
Figure 2.
Diagrammatic representation of the final components of the Multiepitope vaccine (MEV). (a) MEV-1 with Laterosporulin, a TLR4 adjuvant. (b) MEV-2 with PorB, a TLR2 adjuvant. B-cell epitopes (green color), CTL epitopes (pink color), and HTL epitopes (blue color) are connected using linkers. Two HTL-CTL epitopes are connected by GPGPG and AAY linkers, whereas two B cells are joined by KK linkers. TLR adjuvants and epitopes are connected by EAAAK linkers (yellow color).
Figure 2.
Diagrammatic representation of the final components of the Multiepitope vaccine (MEV). (a) MEV-1 with Laterosporulin, a TLR4 adjuvant. (b) MEV-2 with PorB, a TLR2 adjuvant. B-cell epitopes (green color), CTL epitopes (pink color), and HTL epitopes (blue color) are connected using linkers. Two HTL-CTL epitopes are connected by GPGPG and AAY linkers, whereas two B cells are joined by KK linkers. TLR adjuvants and epitopes are connected by EAAAK linkers (yellow color).
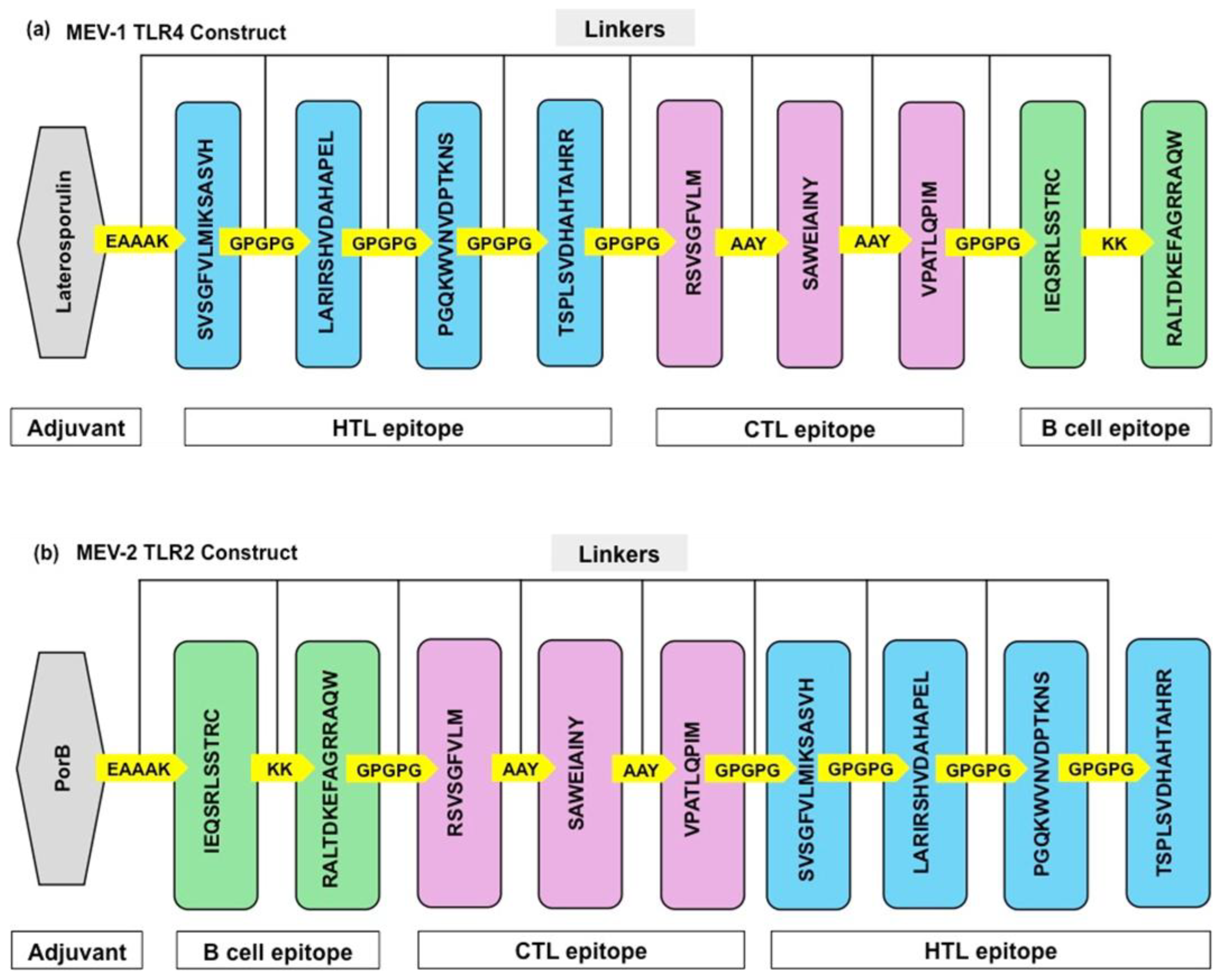
Figure 3.
Structural prediction of the MEV constructs. (A) Secondary structure of TLR4 vaccine construct predicted using PSIPRED webserver. Best model of TLR4 vaccine construct based on tertiary structure analysis using ROBETTA webserver. Quality index of tertiary structure of TLR4 vaccine construct predicted using VADAR webserver. Ramachandran plot analysis of TLR4 vaccine construct using PDBsum webserver. (B) Secondary structure of TLR-2 vaccine construct predicted using PSIPRED webserver. Best model of TLR2 vaccine construct based on tertiary structure analysis using ROBETTA webserver. Quality index of tertiary structure of TLR2 vaccine construct predicted using VADAR webserver. Ramachandran plot analysis of TLR2 vaccine construct using PDBsum webserver.
Figure 3.
Structural prediction of the MEV constructs. (A) Secondary structure of TLR4 vaccine construct predicted using PSIPRED webserver. Best model of TLR4 vaccine construct based on tertiary structure analysis using ROBETTA webserver. Quality index of tertiary structure of TLR4 vaccine construct predicted using VADAR webserver. Ramachandran plot analysis of TLR4 vaccine construct using PDBsum webserver. (B) Secondary structure of TLR-2 vaccine construct predicted using PSIPRED webserver. Best model of TLR2 vaccine construct based on tertiary structure analysis using ROBETTA webserver. Quality index of tertiary structure of TLR2 vaccine construct predicted using VADAR webserver. Ramachandran plot analysis of TLR2 vaccine construct using PDBsum webserver.
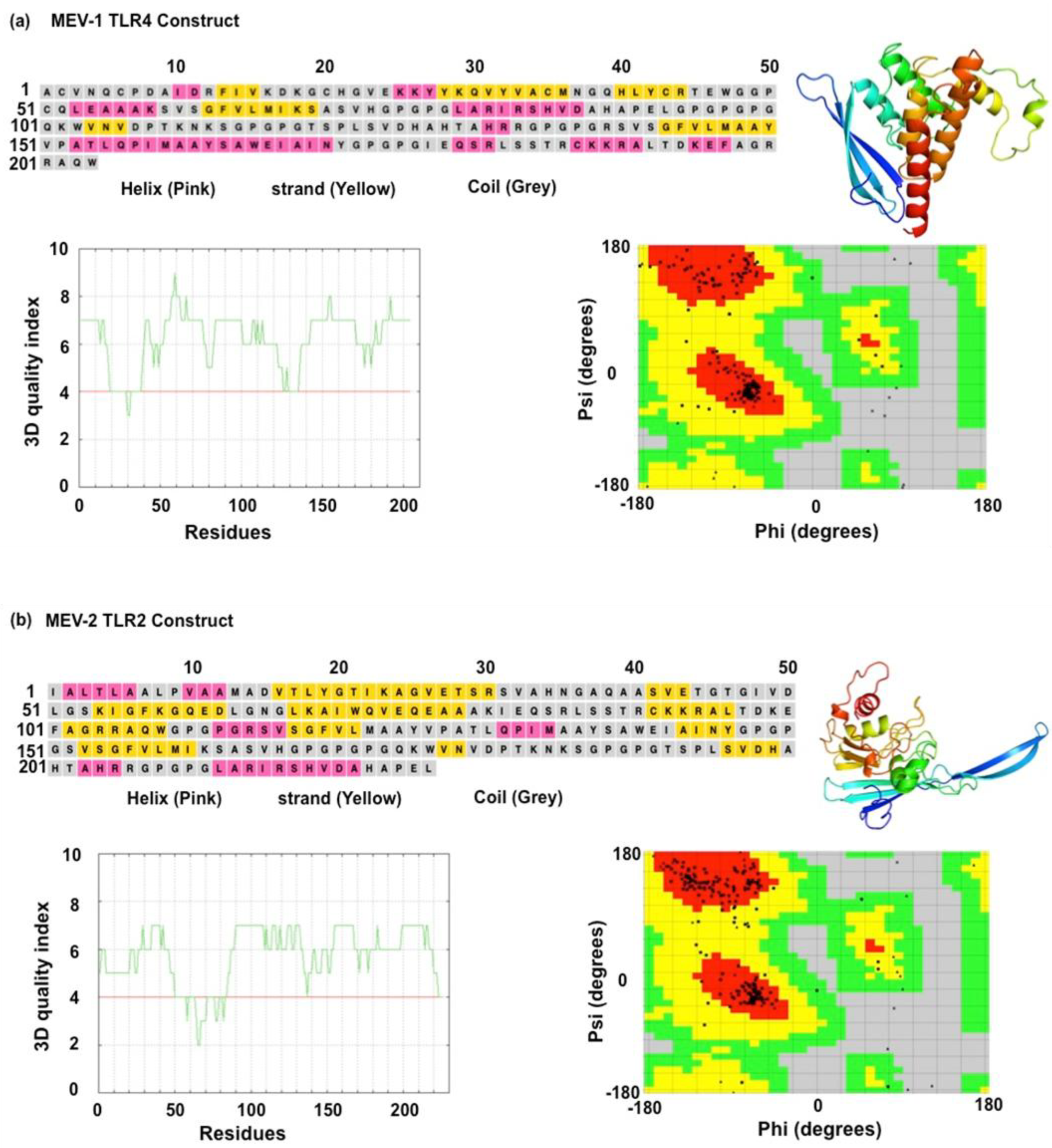
Figure 4.
(a) Protein-protein interaction between MEV-1 vaccine construct and its corresponding receptor (TLR4) predicted using prodigy webserver. Docked structure of the vaccine (Red color) with TLR4 receptor (multi-color) using Cluspro webserver. (b) Protein-protein interaction between MEV-2 vaccine construct and its corresponding receptor (TLR2) predicted using prodigy webserver. Docked structure of the vaccine (Yellow color) with TLR2 receptor (Pink color) were constructed using Cluspro webserver.
Figure 4.
(a) Protein-protein interaction between MEV-1 vaccine construct and its corresponding receptor (TLR4) predicted using prodigy webserver. Docked structure of the vaccine (Red color) with TLR4 receptor (multi-color) using Cluspro webserver. (b) Protein-protein interaction between MEV-2 vaccine construct and its corresponding receptor (TLR2) predicted using prodigy webserver. Docked structure of the vaccine (Yellow color) with TLR2 receptor (Pink color) were constructed using Cluspro webserver.
Figure 5.
(A) MD simulation analysis of MEV-1 TLR4 construct. (B) MD simulation analysis of MEV-2 TLR2construct analyzed after 200 ns of MD simulation.
Figure 5.
(A) MD simulation analysis of MEV-1 TLR4 construct. (B) MD simulation analysis of MEV-2 TLR2construct analyzed after 200 ns of MD simulation.
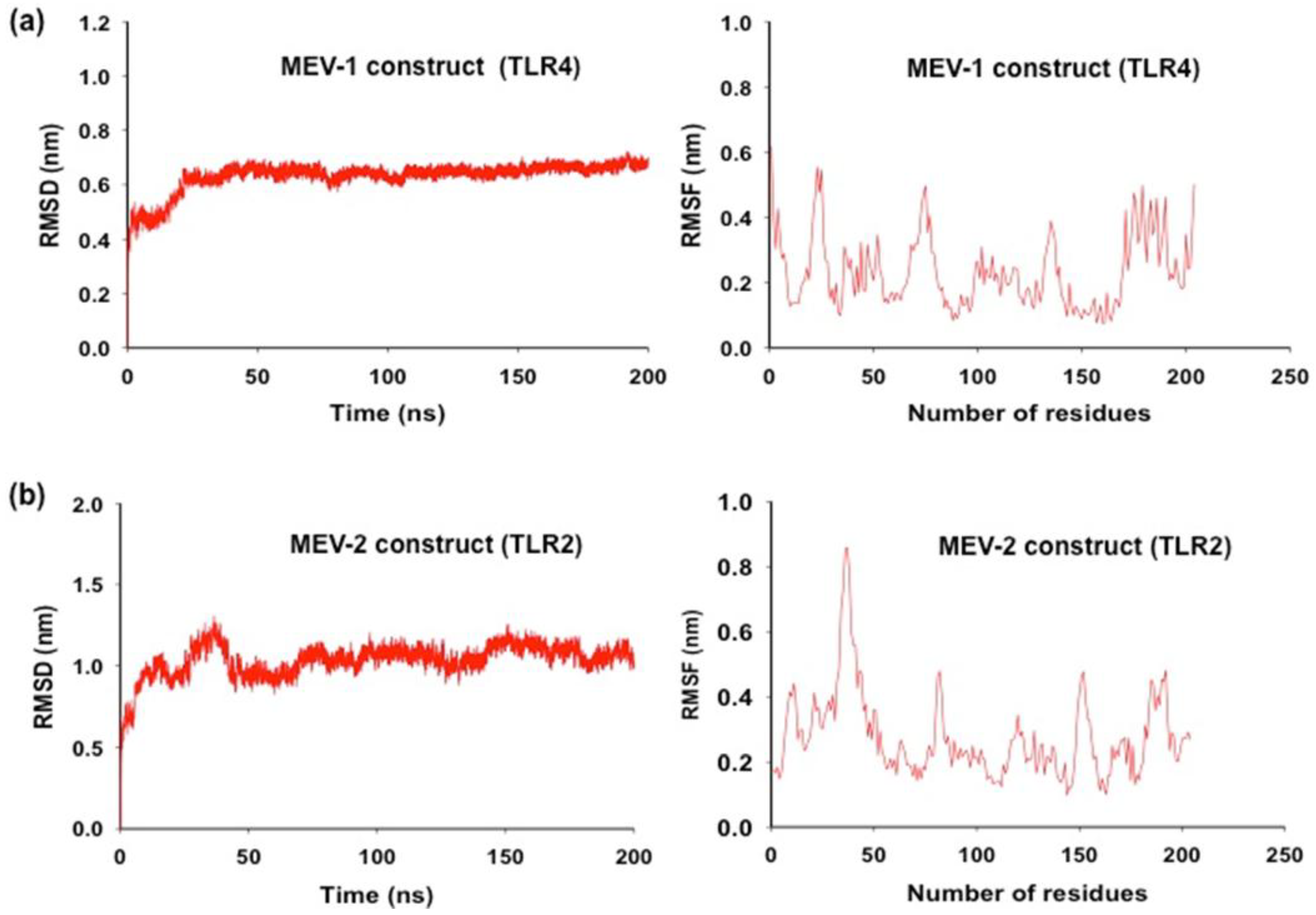
Figure 6.
(a) MD simulation of MEV-1 construct with TLR4 human receptor. RMSD, RMSF, SASA, and Rg analysis of MEV-1 with TLR4 human receptor was analyzed after 200 ns of MD simulation using Gromacs software. (b) MD simulation of MEV-2 construct with TLR2 human receptor. RMSD, RMSF, SASA, and Rg analysis of MEV-2 with TLR2 human receptor was analyzed after 200 ns of MD simulation using Gromacs software.
Figure 6.
(a) MD simulation of MEV-1 construct with TLR4 human receptor. RMSD, RMSF, SASA, and Rg analysis of MEV-1 with TLR4 human receptor was analyzed after 200 ns of MD simulation using Gromacs software. (b) MD simulation of MEV-2 construct with TLR2 human receptor. RMSD, RMSF, SASA, and Rg analysis of MEV-2 with TLR2 human receptor was analyzed after 200 ns of MD simulation using Gromacs software.
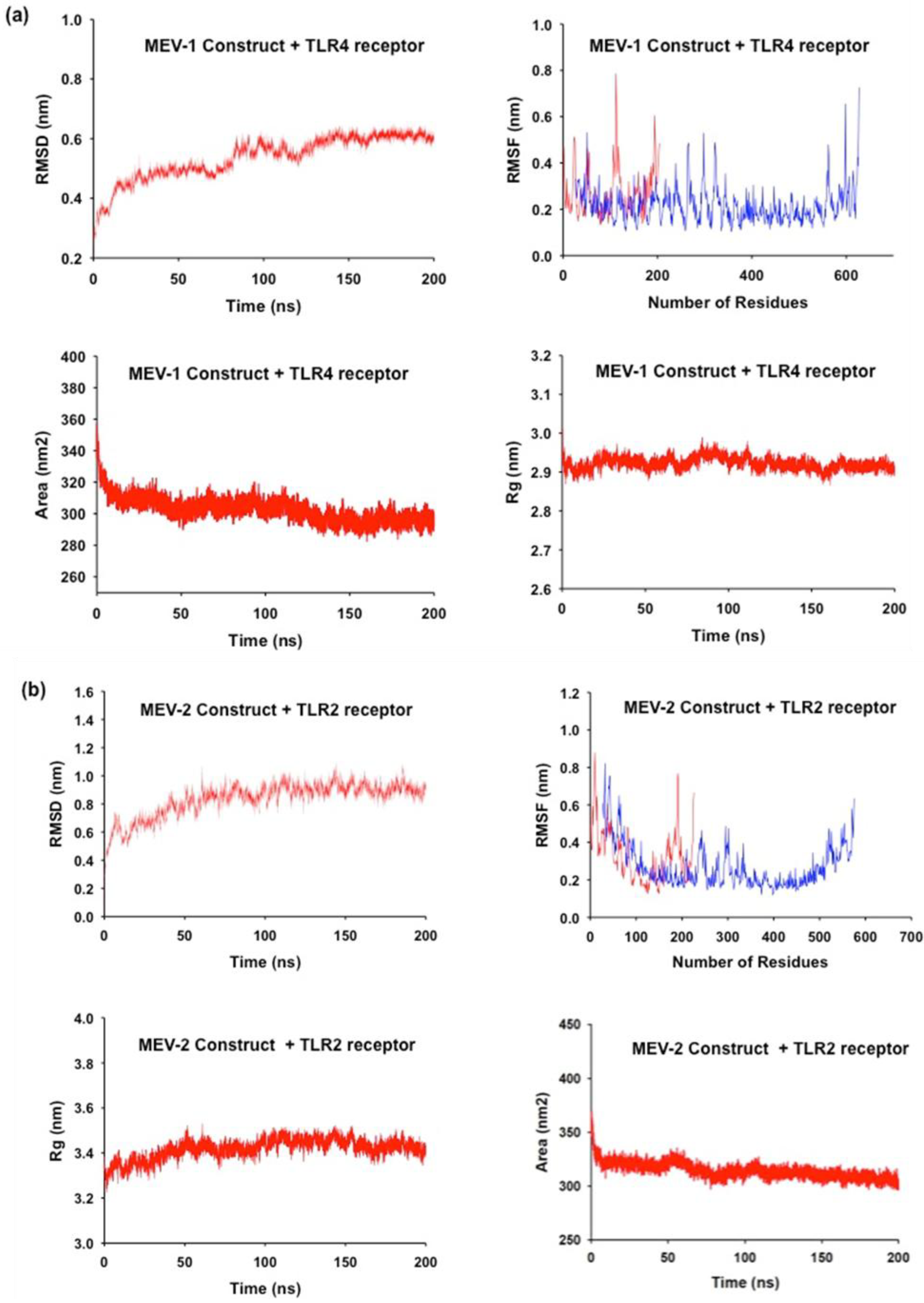
Figure 7.
Immune simulation of the MEV vaccine constructs using C-ImmSim webserver. (a) Active B-cell population; (b) Helper-T cells; (c) Cytotoxic-T cells; (d) Cytokine levels induced by the vaccine; (e) Antibodies produced; (f) Dendritic cell population per state; (g) NK-cell population.
Figure 7.
Immune simulation of the MEV vaccine constructs using C-ImmSim webserver. (a) Active B-cell population; (b) Helper-T cells; (c) Cytotoxic-T cells; (d) Cytokine levels induced by the vaccine; (e) Antibodies produced; (f) Dendritic cell population per state; (g) NK-cell population.
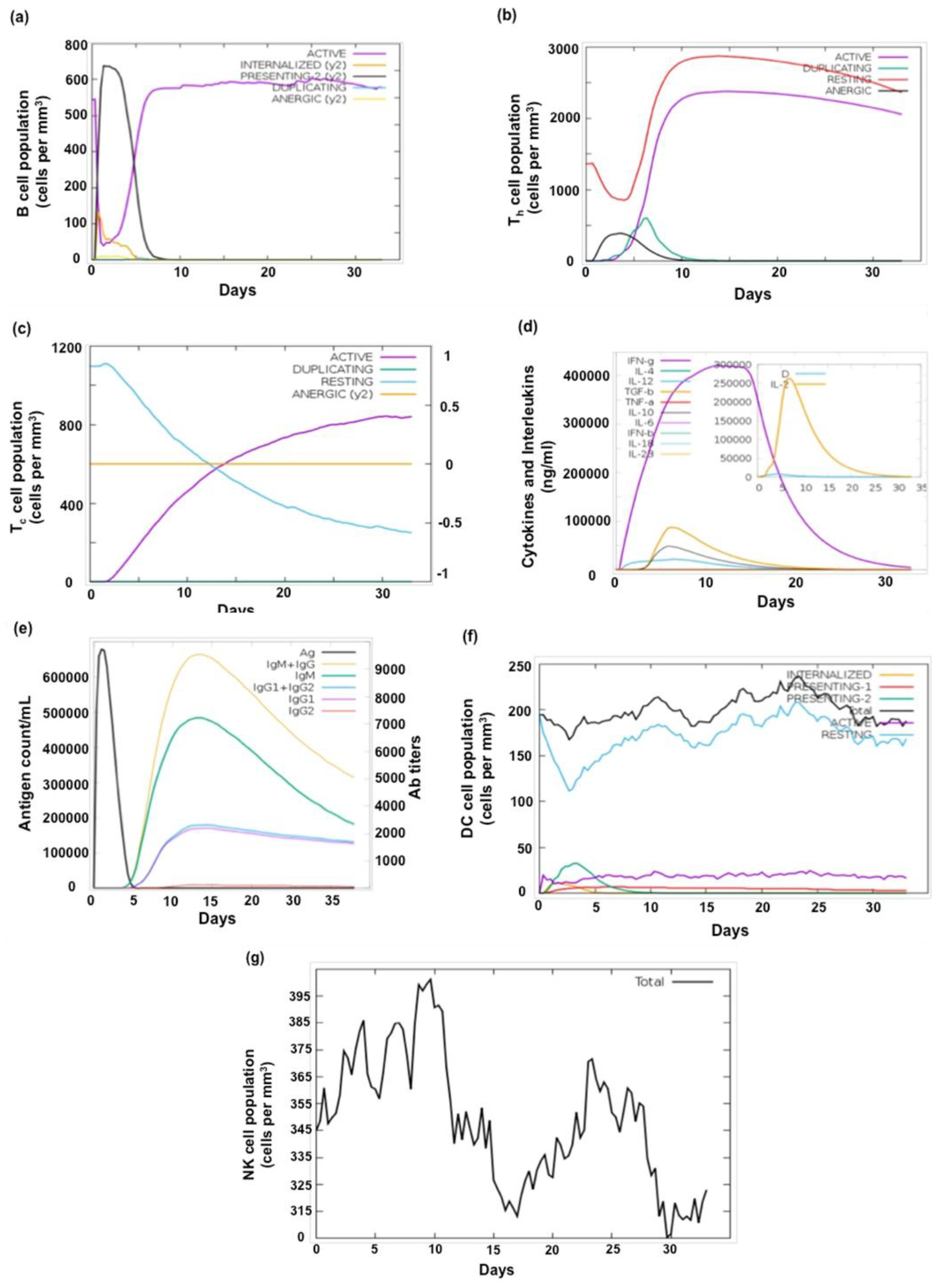
Figure 8.
(a) MEV-1 TLR-4(b) MEV-2 vaccine constructs were inserted into E.coli vector pET28a using restriction sites. The red color in the vector map represents the inserted DNA sequence of the vaccine construct.
Figure 8.
(a) MEV-1 TLR-4(b) MEV-2 vaccine constructs were inserted into E.coli vector pET28a using restriction sites. The red color in the vector map represents the inserted DNA sequence of the vaccine construct.
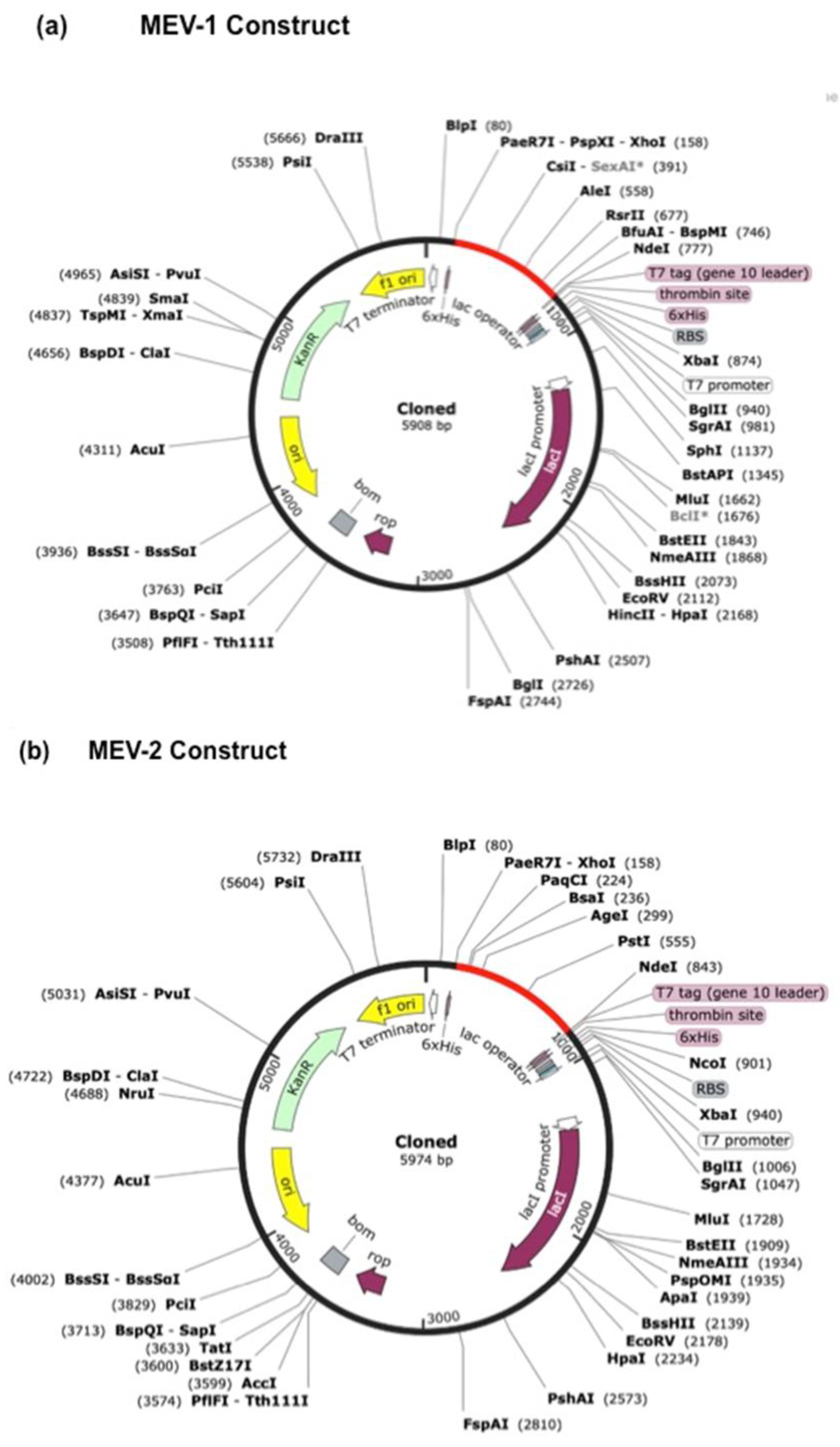

Table 1.
Physiochemical properties of the TLR-4 and TLR-2 multiepitope vaccine constructs predicted using ProtParam Expasy webserver.
Table 1.
Physiochemical properties of the TLR-4 and TLR-2 multiepitope vaccine constructs predicted using ProtParam Expasy webserver.
| Construct | pI | Aliphatic index | Molecular weight | Gravy score | Instability index | Solubility |
|---|---|---|---|---|---|---|
|
TLR-4 Vaccine construct |
9.49 | 66.52 | 21kDa | -0.388 | 37.61 | 0.636 |
|
TLR-2 Vaccine construct |
9.67 | 80.40 | 23kDa | -0.158 | 30.85 | 0.668 |
Table 2.
Ramachandran plot values of TLR-2 and TLR-4 vaccine construct generated using PDBsum webserver.
Table 2.
Ramachandran plot values of TLR-2 and TLR-4 vaccine construct generated using PDBsum webserver.
| Ramachandran statistics | TLR-4 construct | TLR-2 construct |
|---|---|---|
| Disallowed regions | 1.2% | 1.1% |
| Generously allowed regions | 1.2% | 1.1% |
| Most favored regions | 88.1% | 88.2% |
| Additional allowed regions | 9.4% | 9.6% |
Table 3.
MD simulation parameters RMSD, RMSF, SASA and Rg of multiepitope vaccine constructs and TLR receptors analyzed after 200 ns of MD simulation.
Table 3.
MD simulation parameters RMSD, RMSF, SASA and Rg of multiepitope vaccine constructs and TLR receptors analyzed after 200 ns of MD simulation.
| Parameters | TLR-2 and vaccine complex | TLR-4 and vaccine complex |
|---|---|---|
| RMSD (nm) | 0.83 | 0.53 |
| RMSF (nm) | 0.29 | 0.23 |
| SASA (nm2) | 314.63 | 302.75 |
| Rg (nm) | 3.42 | 2.92 |
Table 4.
The HTL and CTL epitopes of the vaccine construct were analyzed for population coverage using IEDB webserver.
Table 4.
The HTL and CTL epitopes of the vaccine construct were analyzed for population coverage using IEDB webserver.
| Population | MHC-I AND MHC-II class combined | ||
| Coverage (%) | Average hit | Pc90 | |
| World | 89.32 % | 11.85 | 6.55 |
| Standard deviation | 0.0 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



