
Submitted:
01 January 2025
Posted:
02 January 2025
You are already at the latest version
Abstract
Tuberculosis (TB) remains a major cause of ill health and one of the leading causes of death worldwide, with about 1.25 million deaths estimated in 2023. Control measures have focused principally on early diagnosis, treatment of active TB, and vaccination. However, the widespread emergence of anti-tuberculosis drug resistance remains the major public health threat to progress made in global TB care and control. Also, the Bacille Calmette-Guérin (BCG) vaccine, the only licensed vaccine against TB in children, has been in use for over a century, and there have been considerable debates concerning its effectiveness in TB control. A multi-epitope vaccine against TB would be an invaluable tool to attain the Global Plan to End TB, 2023-2030 target. A rational approach that combines several B-cell and T-cell epitopes from key lipoproteins was adopted to design a novel multi-epitope vaccine candidate. In addition, interactions with TLR4 were done to assess its ability to elicit an innate immune response. Conservation of the selected proteins suggests the possibility of cross-protection in line with the one-health approach to disease control. The vaccine candidate was predicted to be both antigenic and immunogenic, and immune simulation analyses demonstrated its ability to elicit both humoral and cellular immune responses. Protein-protein docking and normal mode analyses of the vaccine candidate with TLR4 predicted efficient binding and stable interaction. This study provides a promising one-health approach for the design of multi-epitope vaccines against human and livestock tuberculosis. Overall, the designed vaccine candidate demonstrated immunogenicity and safety features that warrant further experimental validation in vitro and in vivo.
Keywords:
Tuberculosis
; Immuno-informatics
; Lipoproteins
; TB-MEVA-1
; Multi-epitope vaccine candidate
1. Introduction
Tuberculosis (TB), caused by species of the Mycobacterium tuberculosis complex (MTBC), remains a major cause of ill health and one of the leading causes of death worldwide [1]. While TB in humans is caused principally by Mycobacterium tuberculosis sensu stricto, other highly related subspecies of the Mycobacterium tuberculosis complex, such as the bovine Mycobacterium bovis can also cause human TB [2]. It has been estimated that 1.4% of all human TB cases in the world and 2.8% of all cases in the African population are attributed to M. bovis [3]. Since bovine TB can spread from cattle to humans and from humans to cattle, there is an urgent need to adopt the One Health Approach to control the disease. M. bovis has one of the broadest host ranges of all known pathogens, affecting many groups of mammals [4]. Susceptible species include cattle, humans, non-human primates, goats, cats, dogs, pigs, buffalo, badgers, possums, deer, and bison [5]. TB is prevalent in all countries, affecting all age groups, though mostly adults in their productive years, leading to a significant social and economic impact [6]. According to the WHO, in 2023, an estimated 10.8 million people were affected by TB worldwide, a majority of whom were men (about 56%). In addition, a total of 1.25 million deaths were reported (including 161,000 people with HIV). Furthermore, 1.3 million cases were children [7], representing a significant number since TB in children and adolescents is often overlooked by health providers and is also difficult to diagnose and treat [8].
TB is curable and preventable, and control measures have focused principally on early diagnosis and treatment of active TB (to stop infectiousness), and the prevention of active disease in exposed or known latently infected individuals [9]. It is estimated that up to 66 million lives were saved through TB diagnosis and treatment between 2000 and 2020 [7]. Currently approved TB diagnostic tests encompassing smear microscopy, microbiological cultures, and molecular methods all rely on sputum but suffer poor diagnostic accuracy [10]. In addition, treatment is focused on using a combination of antibiotics for periods of between six and 12 months [11]. However, the long periods needed for treatment raise concerns about compliance and increase the emergence and persistence of antimicrobial resistance [12]. In fact, anti-TB drug resistance remains the major public health threat to the progress made in TB care and control worldwide [12,13].
To further compound the challenges faced by control measures, the Bacille Calmette-Guérin (BCG) vaccine, the only licensed vaccine against TB in children has been in use for over a century [14], and there have been considerable debates concerning its effectiveness in TB control [15]. It has been reported that while infant BCG is moderately effective in preventing severe, extrapulmonary forms of TB in young children [16], efficacy in preventing TB in adolescents and adults in multiple clinical trials has shown lots of variation, and the vaccine has been largely ineffective in controlling the global epidemic [17].
The achievement of WHO’s End TB ambitious strategic goal of ending the TB epidemic by 2035 urgently requires novel, safe, and effective vaccines [18]. Multi-epitope vaccines (MEVs) represent an innovative strategy based on immunoinformatics methods to design more robust vaccines for infectious diseases with several advantages [19]. Compared to classical vaccine approaches, which have been largely empirical, reverse vaccinology has several advantages, including the selection of epitopes that can induce strong cellular and humoral immune responses simultaneously, the elimination of unwanted antigen components that could trigger either pathological immune reactions or adverse effects and the possibility to polarize the immune response through the introduction of specific agonist for pathogen recognition receptors [20]. Several MEVs have been designed for various diseases, including TB [21,22,23]. The development of a robust multi-epitope vaccine depends primarily on the selection of appropriate candidate antigens and their immunodominant epitopes [24]. Bacterial lipoproteins play several functions, including nutrient uptake, signal transduction, virulence, adhesion, conjugation, and sporulation, and participate in antibiotic resistance and transport [25,26]. The development of immune responses against Mycobacterium tuberculosis lipoproteins could block vital physiological processes in the bacteria. This work focused on the deployment of immunoinformatics tools to design a novel MEV candidate for MTB based on selected lipoproteins reported to be implicated in pathogenesis and virulence.
2. Results
2.1. Protein Retrieval and Preliminary Characterization
The sequences of 14 lipoproteins (P9WIF5, O53692, Q79FB3, P9WIB5, P9WJE1, P9WK61, P9WK65, P9WNF3, P9WK45, P9WG29, O53859, I6Y3P1, A5TZX4 and P9WGT7) previously reported to be involved in pathogenesis or virulence constituted the initial dataset [27,28]. Amongst the obtained sequences were proteins that have already been investigated as vaccine candidates, including ESAT-6-like protein (O53692) [29] and PE_PGRS33 (P9WIF5) [30]. The sequences of all the selected lipoproteins were downloaded from the UniProtKB database and used for preliminary characterization to down-select the antigens to be employed for epitope prediction and chimeric antigen design. SignalP 6.0 predicted the presence of signal peptides in nine of the selected proteins, while the TOPCONS server predicted signal peptides in 12 of the selected proteins. For TM domain prediction, only one protein (O53859) was predicted by consensus using TOPCONS to have 1 TM region. The DeepTMHMM server 1.0, on the other hand, predicted no TM domains in all the selected proteins. The DeepLocPro-1.0 server predicted six proteins to be localized in the extracellular space. Seven proteins were predicted to be localized in the cytoplasmic membrane, while one (P9WK65) was predicted to be localized on both the cell wall and cytoplasmic membrane. Meanwhile, on the TBpred server, most of the proteins were predicted to be integral membrane proteins (6). Also, some proteins were predicted to be attached to the membrane by a lipid anchor (4). In addition, others were predicted to be localized in the cytoplasm (2) or secreted (2) (Supplementary Table S1). Following these analyses, seven antigens (P9WK61, P9WK65, P9WNF3, P9WK45, I6Y3P1, A5TZX4, and P9WGT7) were predicted to have a signal peptide and be localized in the extracellular space by the DeepLocPro-1.0 server. They were then selected for epitope prediction analyses (Table 1). The Mycobacterium tuberculosis 50S ribosomal L7/L12 (Locus RL7_MYCTU), P9WHE3, which is reported to be a TLR4 agonist [31], was downloaded from the UniProt database to be used as a built-in adjuvant.
2.2. Protein Conservation in Other Mycobacterium Species
A high degree of conservation for all the selected proteins in related species of the Myc bacterium genus (ranging from 30.9 – 100%) was observed based on the BLAST search done against the UniProtKB database and the BLASTp search on NCBI. For 3 of the selected proteins (I6Y3P1, A5TZX4, and P9WNF3), no homologs were found in at least one of the target Mycobacterium species. The conservation of the proteins against their homologs in M. decipiens, M. leprae, M. lacus, M. gordonae, M. asiaticum, M. bovis, M. riyadhense, and M. pseudokansasii) was performed. The highest level of conservation was observed for the M. bovis, with 6 of the seven proteins showing 100% conservation and the last protein showing 99.8% conservation (Table 2).
2.3. Cytotoxic T Lymphocyte Epitope Prediction
High-scoring CD8+ (9-mer) epitopes were predicted from all the selected antigens on the NetCTL 1.2 server using a threshold set at 1.0. From the predicted epitope sequences, sixteen CD8+ epitopes were selected to be incorporated into the chimera based on their antigenicity or their overlap with some of the HTL or LBL epitopes predicted (Table 3).
2.4. Helper T Lymphocyte Epitope Prediction
High-affinity MHC-II epitopes (based on IC50 scores) for the HLA-DR, HLA-DQ, and HLA-DP human alleles, predicted on the NetMHCII 2.3 web server, were selected as HTL epitopes. In total, eight high-affinity HTL epitopes were chosen to design the chimeric vaccine candidate. It was observed that some of the predicted HTL epitopes and linear B-cell epitopes had overlapping regions (Table 3).
2.5. Linear B-Lymphocyte Epitopes Prediction
Linear B-cell epitopes of varying residue lengths were predicted using two servers, and epitopes simultaneously predicted by both servers were selected for the final chimeric vaccine candidate. It was noticed that one of the predicted linear B-cell epitopes had a region of overlap with a predicted HTL epitope. A total of 9 linear B-epitopes were selected (Table 3).
2.6. Antibody Class Prediction
All the nine selected linear B-lymphocyte epitopes initially predicted to be antigenic and chosen for incorporation in the designed chimeric antigen were subjected to the antibody class prediction using the AbCPE server. All the epitopes were predicted to bind to IgG. None of the selected epitopes was predicted to bind to IgE or IgM, while two (from proteins A5TZX4 and P9WK65) were predicted to induce IgA. The prediction probabilities ranged from 66-100%, with 4 LBLs demonstrating a 100% probability of inducing antibody production (Table 4).
2.7. Design of the Chimeric Multi-Epitope Vaccine Candidate
Predicted epitopes containing overlapping amino acids were merged to form contiguous sequences (Table 1). As a consequence, the multi-epitope chimeric vaccine candidate was designed from 11 CTL epitopes, 7 HTL epitopes, and 9 linear B-lymphocyte epitopes in total. The chimera was designed by incorporating KK, AAY, and GPGPG linkers to merge the linear B-cell epitopes CD8+ T-epitopes and CD4+ T-epitopes, respectively. In addition, the TLR4 (PDB ID: 4G8A) agonist, RL7_MYCTU from Mycobacterium tuberculosis, (RPFE_MYCTU) (accession number P9WHE3) was chosen as a built-in adjuvant and incorporated at the N-terminal of the designed vaccine candidate using the EAAAK linker to boost immune elicited responses. In addition, the universal Pan DR Epitope (PADRE) sequence (AKFVAAWTLKAAA), a potent stimulator of CD4+ responses, was added after the M. tuberculosis RpfE sequence. Furthermore, the TAT sequence (GRKKRRQRRRPQ) was added after the PADRE sequence using the GGGS linker. The 10xHis tag was incorporated at the carboxyl terminal of the designed chimeric antigen to aid in purification and identification procedures. Summarily, the chimeric vaccine candidate (designated TB-MEVA-1) comprised 683 amino acid residues that included 27 M. tuberculosis peptides (predicted epitopes), the immune potentiators (TLR4 agonist and PADRE) added, and different linkers (Figure 1).
2.8. Physicochemical Properties and Solubility Analysis
The designed chimeric antigen, composed of 683 amino acids, was predicted to have a molecular weight (MW) of 69.7 kDa and a theoretical pI of 9.36, indicating that the protein is basic. In addition, physicochemical properties analyses revealed the protein to be rich in Ala (15.2%) and Gly (12.7%). The protein had a predicted half-life of 30 hours, >20 hours, and >10 hours in mammalian reticulocytes in vitro, yeast, and E. coli in vivo, respectively. The instability index (II) was predicted to be 27.03. This score classifies the protein as stable since it is below the threshold score of 40.00. The GRand AVerage of hydropathicitY (GRAVY) was predicted to be −0.295, indicating that the protein can interact with water molecules. An aliphatic index of 76.31 was predicted, suggesting that the protein exhibits thermostability. The predicted solubility score was 0.4811 (slightly below the 0.5 threshold) on the NetSolP 1.0 server and 0.4218 on the DeepSoluE server, predicting the protein to only be marginally soluble upon expression in bacteria (Table 5).
2.9. Secondary Structure and Intrinsic Disorder Prediction
The final chimeric protein was predicted to be composed of 27% alpha helix, 13% beta-strand, and 58% coil (Figure 2A). Regarding solvent accessibility, 59% of amino acid residues were predicted as being exposed, 16% as medium-exposed, and 23% were predicted as being buried. A total of 217 amino acid residues (31%) were predicted to be located in intrinsically disordered regions. (Figure 2B). Figure 2C shows the pictorial representation of the secondary structure by the color of the designed vaccine candidate.
2.10. Antigenicity, Allergenicity, and Toxigenicity Prediction
The predicted antigenicity score of the final sequence on the VaxiJen 2.0 server was 1.1314 (bacteria model with a threshold of 0.4). The antigenicity score on the ANTIGENpro server was 0.9218. In addition, the Vaxi-DL server predicted the designed chimera to be a vaccine candidate with a 96.66% probability. The results indicate that the generated protein is antigenic. The AllerTOP v.2 and AllergenFP servers predicted the protein to be non-allergenic. While the ToxinPred and ToxDL servers predicted TB-MEVA-1 did not contain any toxic peptides, with a score of 0.0001 predicted on the ToxDL server (Table 6).
2.11. IFN-γ-Inducing Epitope Prediction
Of the 122 potential IFN-γ inducing epitopes (15-mer) predicted for the built-in adjuvant, 62 scored above the epitope prediction threshold. Meanwhile, of the 486 potential epitopes predicted for the main vaccine sequence, a total of 86 IFN-γ inducing epitopes scored above the default threshold. The number of IFN-inducing epitopes predicted correlated with the simulated levels of IFN-γ induction following injection with the chimeric vaccine by immune simulation on the C-ImmSim server (Figure 3).
2.12. Prediction of Epitopes for Mouse MHC II Alleles
From the 669 9-mer epitopes that could be derived from the antigen for each allele, 168 peptides capable of binding to six mouse MHC II alleles were predicted for TB-MEV-1 with the H-2-IAb allele having the highest number of epitopes, and the H-2-IEk allele having no binding epitopes (Table 7).
2.13. Prediction, Refinement, and Validation of Modeled Tertiary Structure
AlphaFold2 was used to predict the 3D structure of the designed multi-epitope vaccine candidate on the ColabFold interface (Figure 4A). Refinement of the designed vaccine candidate 3D structure generated initially on the ModRefiner server, followed by the GalaxyRefine server yielded 5 models. From the quality scores model of the refined models, “model 4”, with parameters GDT-HA (0.8679), RMSD (0.682), and MolProbity (1.072) was selected as the final 3D model for further characterization (Figure 4B). In addition, the model had a clash score of 1.6, a poor rotamers score of 0.0, and a Ramachandran plot score of 97.1%. The Ramachandran plot analysis of the selected model protein predicted 97.06% of residues to be located in favored regions. This percentage is very similar to the score obtained from the GalaxyRefine analysis. In addition, 2.1% of residues were predicted to be located in allowed regions, while only 0.2% were predicted in disallowed regions (Figure 4C). Following refinement, the 3D model quality was tested on the ProSA-web and ERRAT servers. The selected model had a Z-score of −4.92 on the ProSA webserver (Figure 4D) and an overall quality factor of 94.444 on the ERRAT server (Figure not shown). The ProSA-web score was outside the typical score range for native proteins of similar size.
2.14. Discontinuous B-Cell Epitope Prediction
A total of 371 residues (53.1%) were predicted by the ElliPro server to be found in 25 conformational B-cell epitopes, with epitope prediction scores in the range of 0.517 to 0.983. The predicted discontinuous epitope residues ranged from 3 to 68 amino acid residues (Supplementary Table S2).
2.15. Protein-Protein Docking Between the Designed Vaccine Candidate and TLR4 Receptor Dimer
An appropriate immune response relies on well-coordinated recognition and interaction between antigenic determinants and specific immune receptors. Several studies have reported the involvement of toll-like receptor-4 (TLR-4) in protective immunity against M. tuberculosis [32,33]. Molecular docking investigated the binding interaction between the refined and validated 3D of the chimeric vaccine candidate and TLR4 after a protein-protein interaction pocket was predicted in the designed chimera (Figure 5A). Thirty models were generated, displayed, and ranked according to the cluster size generated by the ClusPro 2.0 server, the docking complex with the largest cluster size was selected. The TB-MEVA-1-TLR4 complex with the largest cluster size exhibited a center binding score of −1388.4, with the lowest energy identified at −1497.4 (Figure 5B). For further characterization of vaccine-receptor interactions, the selected model of the vaccine-TLR4 complex was analyzed for binding affinity using PrODIGY and the interactions at the interface using PDBsum. The relative binding free energy (ΔG) of the vaccine-TLR4 complex was predicted to be −15.3 Kcal/mol while the predicted Kd was 1.7e-11. Consistently, the number of contacts made at the interface (IC) per property (ICs charged-charged:16, ICs charged-polar:17, ICs charged-apolar:35, ICs polar-polar: 6, ICs apolar-apolar: 30. For the vaccine-TLR4 complex, the PDBsum server predicted that 22 hydrogen bonds, one salt bridge and 214 non-bonded contacts were formed between 38 residues from TLR4 and 32 residues from the chimeric vaccine candidate (Figure 5C). Above all, the vaccine candidate exhibited favorable interactions with TLR4.
2.16. Normal Mode Analyses
The normal mode analysis (NMA) was performed to investigate the molecular stability and functional motions of the vaccine candidate-TLR4 complex (Figure 6). The deformability graph showed peak points that represent the main chain residues deformed regions in the vaccine candidate-TLR4 complex. The locations with hinges are regions with high deformability (Figure 6A). The B-factor plot demonstrates the association between the NMA mobility and TB-MEVA-1-TLR4 complex, representing the average RMSD values of the docked complex (Figure 6B). B-factor values calculate the uncertainty of each atom. The calculated eigenvalue of the vaccine candidate-TLR4 complex was 2.000137-e07, reflecting the motion stiffness related to each normal mode (Figure 6C). The eigenvalue is an estimate of the energy required to deform the structure. Each normal mode of the complex is represented by an individual (purple) and cumulative (green) variance in the variance bar. The variance and eigenvalue showed a negative correlation (Figure 6D). In addition, the interacting motions between the vaccine candidate and the TLR4 in the complex are represented by a covariance matrix. The interrelated motions among different pairs of residues were specified by correlated (red), uncorrelated (white), and anti-correlated (blue) atomic motions in the vaccine candidate-TLR4 complex (Figure 6E). The elastic network map, which represents atom pairs connected by spring in the vaccine candidate-TLR4 complex, was also generated. Each dot in the graph denotes a spring connecting the corresponding pair of atoms. The stiffness of the dots is indicated by their color; darker greys denote stiffer parts, whereas the lighter dots denote more flexible sections (Figure 6F). All the data from the normal mode analyses further indicate the favourable interaction and stability in the vaccine candidate-TLR4 complex.
2.17. Immune Simulation for Vaccine Candidate Immunogenicity Analyses
Immune simulation predictions demonstrated an expansion in the elicited secondary responses. Theoretically, the observed trend portrays the operational development of immunological responses (Figure 7A). Elevated IgM levels characterized the immune response generated to the prime dose. In addition, significant increases in the B-cells population together with corresponding increases in IgM, total IgG + IgM, IgG1, and IgG2 antibody levels for the two vaccination booster doses (Figure 7A,B). The observed pattern suggests the development of immune memory, as depicted in the development of a sustained population of memory B-cells (Figure 7C). Similarly, the helper T-cell (TH) and cytotoxic T-cell (TC) populations were also predicted to elicit more potent responses to the antigen following inoculation of the booster doses. These responses led to sustained increases in the active T-cell populations and the development of immunological memory (Figure 7D–F). Furthermore, sustained increases in dendritic cell, macrophage, and NK cell populations were observed during the immunization period (Figure 7G–I). In summary, the designed vaccine candidate elicited both cellular and humoral immune responses, which have been reported to be critical in protection against tuberculosis.
2.18. Codon Optimization and In-Silico Cloning
The gene coding for the designed vaccine candidate was codon-optimized using the Java Codon Adaptation Tool (JCat) to ensure high protein expression levels in E. coli. The gene sequence contained 2,079 nucleotides in total, with a Codon Adaptation Index (CAI) of 1.0 and a mean GC content of 50.7%. These results suggest the possibility of suitable expression in the E. coli bacterial host. In general, a GC content in the range of 30% to 70% is preferred for optimal protein expression [34]. Lastly, the adapted codon sequence was inserted in the appropriate sites of the pET30a (+) vector using SnapGene software to generate a recombinant plasmid in silico (Figure 8).
3. Discussion
Vaccines remain one of the most valuable and reliable tools in the current End TB Strategy target of a 90% reduction in TB deaths and an 80% reduction in TB incidence by 2030 [35]. Considering the monetized value of health gains, modeling studies have estimated that the introduction of an adolescent/adult vaccine could generate $283 to 474 billion in economic benefits by 2050 [36]. However, at the moment, the only TB vaccine, BCG, has been in use for over 100 years, and several reports have indicated that BCG has only moderate effectiveness in preventing severe, extrapulmonary forms of TB in young children [16], shows lots of variation in efficacy to prevent TB in adolescents and adults [17]. This has resulted in BCG being largely ineffective in controlling the global TB epidemic [16]. In the search for potential alternatives to BCG, several vaccine candidates (both single antigens and antigen cocktails) have been evaluated in clinical trials [37]. These vaccines (which can be divided into four categories: viral vector, protein/adjuvant, inactivated, and attenuated live vaccines) include ID93/GLA-SE (consisting of Rv2608, Rv1813, ESXV, and ESXW), M72 (consisting Mtb39A and Mtb32A) GamTBvac (a fusion between Ag85a and ESAT6-CFP10), AEC/BC02, Ad5 Ag85A, and others [38]. However, none of these is yet to be licensed by the WHO. Multi-epitope vaccine candidates offer numerous advantages including the generation of a targeted immune response and the elimination or mitigation of potential unwanted (allergenic or tolerigenic) responses [39]. Several multi-epitope vaccine candidates have been designed for different infectious diseases including onchocerciasis [40], schistosomiasis [20], malaria [41], and others.
In addition, chimeric antigens have been designed for tuberculosis, with some of them evaluated in animal models [21,38]. The design of TB-MEVA-1 takes a unique approach to targeting protein antigens exposed on the surface of the bacteria and plays roles in pathogenicity and virulence. Extracellularly or cell surface localized proteins, due to their increased accessibility to the immune system, have been reported to be good vaccine candidates [42]. Since the selected lipoprotein antigens used for TB-MEVA-1 design are implicated in virulence and pathogenicity, blocking their activities could help in both reducing the infectivity of the pathogens and also reducing disease severity in persons who are already infected. Previously, the Shigella virulence antigen VirG (also known as IcsA) has been characterized as a vaccine candidate and provoked vigorous immune responses that included antibodies capable of blocking bacterial adhesion and invasion and provided high levels of protection against S. flexneri 2a or S. sonnei in mice models [43]. In addition, BauA and OmpA, which are proteins involved in the pathogenicity of A. baumannii, have been targeted as vaccine candidates and showed significant decreases in bacterial loads in organs of vaccinated mice [44].
Importantly, the selected antigens used in designing the chimeric vaccine candidate also exhibited the potential for cross-protection with high levels of conservation observed in homologous proteins in Mycobacterium species that affect humans and livestock. The current TB vaccine has been reported to promote immunity against other non-tuberculosis mycobacteria, such as Mycobacterium leprae, Mycobacterium ulcerans, Mycobacterium avium, Mycobacterium intracellulare, and Mycobacterium abscessus [45]. In addition, recent studies have reported that BCG can also be used to protect livestock against M. bovis, with a 74% reduction in bovine TB transmission and a substantial reduction in lesions of vaccinated animals [46]. A high level of conservation observed in M. bovis (Table 2), the causative agent for bovine TB which also affects humans [47], implies that the designed vaccine candidate can be a useful tool in the context of the current one-health approach to prevent human and livestock tuberculosis [48].
The roles of both B- and T-cells have been reported in protective immunity to tuberculosis with the involvement of different immune mechanisms [49,50,51]. The selected linear B-epitopes were predicted to potentially elicit both IgG and IgA responses, which have been reported to be important in immune protective mechanisms against TB [52,53]. Besides adaptive immune responses, innate immune responses are also vital in protective immune responses against TB, with the important role of TLRs also reported [54]. Studies in mice have suggested the vital protective role TLR4 plays a role in host defense against lung infection by M. tuberculosis, with splenocytes from infected TLR4 mutant mice demonstrating a reduced capacity to produce the protective type 1 cytokine IFN-γ upon antigen-specific stimulation [55]. Given the role of TLR4 in generating protective immune responses, a TLR4 agonist (M. tuberculosis 50S ribosomal protein L7/L12) was added at the N-terminal of the designed chimeric as a built-in adjuvant. The adjuvant has been reported to potentiate immune responses through DC maturation and pro-inflammatory cytokine production. Therefore, DCs activated by the vaccine candidate could activate naïve T cells, effectively polarize CD4+ and CD8+ T cells to secrete IFN-gamma and induce T cell-mediated-cytotoxicity [31]. These additional immune-stimulating effects of the adjuvant could be a valuable addition to the immune responses elicited by the selected epitopes.
Predicted B-cell and T-cell epitopes in TB-MEVA-1 were fused using specialized linkers to design a multi-epitope chimeric vaccine candidate. Linker sequences are a key component of epitope-based vaccine design and have been used to boost immune responses by minimizing the formation of neo-epitopes, thereby eliminating junctional immunogenicity and improving epitope processing and presentation [56,57]. In addition to linkers between the different epitopes (AAY, GPGPG, KK, and GGGS) the EAAAK linker was added between the built-in adjuvant sequence and the fused epitopes. The EAAAK linker has been reported to augment the expression levels of bioactivity bifunctional proteins [58]. Moreover, the TAT sequence which is a cell-penetrating peptide (CPP), and the Pan DR epitope, PADRE (a universal synthetic 13-mer peptide that activates CD4+ T cells [59]) were added to the design to further potentiate the immune response elicited by TB-MEVA-1. Cell-penetrating peptides have been successfully used in the delivery of a large variety of cargoes, from small particles to proteins, peptides, and nucleic acids [60]. They have been reported to be important in enhancing peptide vaccine accumulation and persistence in lymph nodes, enhancing immunogenicity [61]. [59], on the other hand, is a potent stimulator of CD4+ responses, which has been reported to be critical for the control of M. tuberculosis infection [62]. Immuno-informatics analyses of the generated TB-MEVA-1 revealed the presence of large numbers of IFN-γ epitopes which have been reported by multiple studies to play an essential role in host defense against infection with intracellular pathogens, including M. tuberculosis [63]. Further analyses of the designed chimeric antigen using different servers revealed that it was not only antigenic but also non-allergenic and non-toxigenic. These characterizations are important since efficacy and safety are key components of the vaccine research and development pipeline [64].
The designed chimeric antigen was predicted to have a molecular weight of 69.7 kDa. The predicted solubility score suggests that the protein may only be slightly soluble upon expression in E. coli. E. coli is a common host for recombinant protein expression, and the soluble expression and purification of protein is a vital step in many biochemical and functional investigations [65]. The predicted theoretical pI of 9.36 indicates that the designed protein is basic. The predicted instability index of 27.03 and the estimated half-life upon expression in different host organisms suggest that the vaccine candidate will be stable upon expression. Moreover, the high aliphatic index predicted for the designed antigen suggests the thermostability of the protein [66] , which indicates its suitability for use in low-resource settings.
Information on the secondary and tertiary structural features of the target protein is essential in vaccine design [67]. Secondary structure analyses predicted the vaccine candidate to consist predominantly of coils (58%). In addition, 31% of amino acid residues were predicted to be located in intrinsically disordered regions (IDRs). IDRs and alpha-helical coiled coils are reported to consist important forms of “structural antigens” with important biomedical applications, including vaccine delivery systems and drug development [68]. In addition, IDRs have been reported to be a major component of several vaccine candidates, including the circumsporozoite protein (CSP) used in RTS,S against malaria [69]. The Ramachandran plot following 3D structure and refinement revealed that most of the residues are found in the favoured and allowed regions (99.0%), with very few residues in the outlier region (Figure 4); this indicates that the quality of the overall model is satisfactory.
Since a TLR4 agonist was included in the designed chimera, protein-protein docking analysis and normal mode analyses investigated the potential interaction between TLR4 and TB-MEVA-1. Obtained findings reveal the presence of a large pocket in TB-MEVA-1 and favourable interactions between TLR4 with the presence of different bond types (Figure 5). Since the docking and NMA investigations predicted a stable interaction, the chimeric antigen is expected to interact with the TLR4 on professional antigen-presenting cells like dendritic cells to stimulate a protective immune response. This possibility was also observed in the immune simulation analyses which predicted an overall increase in the elicited immune responses following the injection with primer and booster doses. The immune simulation analyses also demonstrated the development of memory B-cells and T-cells which were sustained for several months. These memory cell pools are vital to the rapid clearance of the pathogen upon exposure of the host to M. tuberculosis [49,70].
All of the observations reported here are generated from computational studies. The validation of this data through both in-vitro and in-vivo investigations remains a dire necessity. The immediate next step of this project will involve the expression of TB-MEVA-1 in a suitable host for serological analyses using samples from infected persons or exposed to assess antibody recognition since the vital role of antibodies has been reported in the action of the current BCG vaccine [71]. Further investigations could include immunogenicity and protective studies in mice models, which will provide valuable information on the potential efficacy of the designed vaccine candidate in clinical trials. With the recent development of humanized mice models (some of which have already been tested in tuberculosis vaccine studies [72]), it is expected that results from studies generated in these mice would be more comparable to human studies. Regarding TB-MEVA-1, its potential to elicit an immune response in preclinical studies is demonstrated by its ability to bind to mouse alleles (Table 4). Results generated from both in-vitro and in-vivo studies of TB-MEVA-1 will provide the requisite pilot data to decide whether the vaccine candidate can be further evaluated in clinical trials.
4. Materials and Methods
The methodology used in this study comprises the following parts: (1) Lipoprotein selection and preliminary analyses; (2) T-cell and B-cell epitopes prediction from selected proteins; (3) Antigenicity prediction for selected epitopes; (4) design of a multi-epitope chimeric antigen using appropriate linkers; (5) preliminary characterization of the designed chimera; (6) 3D structure prediction, refinement and validation of the designed vaccine candidate; (7) molecular docking with TLR-4 and normal mode analysis; (8) immune simulation prediction and (9) codon optimization and in-silico cloning for bacterial expression. Figure 9 summarizes the methodology adopted in this study in a flowchart.
4.1. Protein Sequence Retrieval and Preliminary Analyses
The sequences of Mycobacterium 14 lipoproteins (accession numbers: P9WIF5, O53692, Q79FB3, P9WIB5, P9WJE1, P9WK61, P9WK65, P9WNF3, P9WK45, P9WG29, O53859, I6Y3P1, A5TZX4, and P9WGT7) previously reported to be involved in pathogenicity and virulence [27,28] were retrieved from the UniProtKB database (https://www.uniprot.org/) in the Fasta format unto a .txt file. Preliminary characterization of the sequences included analyses for: 1. Localization (using the DeepLocPro 1.0 server; https://services.healthtech.dtu.dk/services/DeepLocPro-1.0/ and the TBpred server; https://webs.iiitd.edu.in/raghava/tbpred); 2. Signal peptide presence (using the SignalP 6.0; https://services.healthtech.dtu.dk/services/SignalP-6.0/ and TOPCONS; https://topcons.cbr.su.se/servers); and 3. The presence of transmembrane domains (using the DeepTMHMM 1.0; https://services.healthtech.dtu.dk/services/DeepTMHMM-1.0/ and TOPCONS; https://topcons.cbr.su.se/servers). All proteins predicted to be localized in the extracellular space were selected for further analyses.
4.2. Protein Conservation Analyses
A BLAST search was performed on the UniProtKB database (https://www.uniprot.org/) to investigate the level of the conservation of each of the selected proteins amongst other species of the Mycobacterium genus. This is because a high degree of conservation across the genus suggests the possibility of cross-protection [73]. The UniProtKB is a central database for storing and interconnecting information from large and disparate sources. It is the most comprehensive catalog of protein sequence and functional annotation [74]. The degree of relatedness between the selected proteins and their homologs in M. decipiens (human), M. leprae (human), M. lacus (human), M. gordonae (human), M. asiaticum (primates), M. bovis (human, cattle and other livestock), M. riyadhense (humans), and. pseudokansasii (humans) was investigated.
4.3. Linear B-Cell Epitope Prediction
The sequences of the selected proteins were subjected to linear B-lymphocyte epitope (LBE) prediction using two servers: firstly, the BepiPred-3.0 webserver (https://services.healthtech.dtu.dk/service.php?BepiPred-3.0) and then the LBtope server (https://webs.iiitd.edu.in/raghava/lbtope/index.php). The BepiPred server is a sequence-based tool, that uses numerical representations from protein language model (LM) embeddings, to vastly improve prediction accuracy for linear and conformational B-cell epitope prediction on several independent test sets [75]. Employing the default server threshold of 0.1512, linear B-epitopes of various lengths for the selected lipoproteins were predicted. LBtope on the other hand, is an epitope prediction model that uses a larger dataset of validated B-cell epitopes and non-epitopes (12063 epitopes and 20589 non-epitopes obtained from the IEDB database) for epitope prediction. The LBtope server, which is based on SVM, is reported to have a predictive accuracy that can go up to ~ 81% [76]. The server assigns scores (ranging from 0 – 100%) to each of the predicted epitopes with a higher score indicating a higher probability of being an epitope.
4.4. T-Cell Epitope Prediction
The NetMHCII 2.3 server (http://www.cbs.dtu.dk/services/NetMHCII/) whose functioning is based on artificial neural networks (ANNs) was used to predict 15-mer helper T-lymphocyte (HTL) epitopes for human alleles (HLA-DR, HLA-DQ, and HLA-DP) using the default server parameter for strong binders (SB) and weak binders (WB) [77]. Epitopes for HLA-DR, HLA-DQ, and HLA-DP alleles were predicted for all selected proteins, and only the highly promiscuous epitopes (capable of binding to several alleles) classified as strong binders were selected for chimera for design.
The NetCTL 1.2 server (http://www.cbs.dtu.dk/services/NetCTL/) was used for cytotoxic T-lymphocyte (CTL) epitope prediction. The server works on an algorithm that implements epitope prediction by integrating class I MHC binding, proteasomal cleavage, and TAP (Transporter Associated with Antigen Processing) transport efficiency and predicts CTL epitopes for 12 MHC class I supertypes [78]. For this study, the epitopes restricted to the A2, A3, and B7 supertypes were predicted to achieve approximately 90% coverage of phenotypic frequency in the vaccinated population [57]. Though the default threshold for CTL epitope prediction is 0.75, a threshold of 1.0 was used (to improve the epitope specificity). Predicted epitopes were sorted by their combined scores.
4.5. Epitope Antigenicity Prediction
To design a multi-epitope chimeric vaccine candidate capable of stimulating a robust protective immune response, the VaxiJen v2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was used to assess the antigenicity of all the predicted epitopes. This server uses an alignment-independent approach based on the physicochemical characteristics of peptides and proteins to classify antigens. The server algorithm functions based on the autocross-covariance (ACC) transformation of protein sequences into uniform vectors of principal amino acid properties to predict the antigenicity of proteins from bacteria, fungi, parasites, viruses, and tumors [79]. The default threshold for the bacteria model (0.4) was used to evaluate epitope antigenicity, and only those with scores above 0.5 were selected to constitute the chimeric antigen. Figure 9 provides a summary flowchart showing the steps involved in this study.
4.6. Multi-Epitope Vaccine Candidate Design
The selected high-scoring CTL epitopes (with combined scores above the 1.0 threshold), promiscuous strong-binding HTL epitopes, and selected linear-B cell epitopes predicted to be antigenic were merged using AAY, GPGPG, and KK linkers, respectively. The EAAAK linker sequence was used to include the Mycobacterium tuberculosis 50S ribosomal protein L7/L12 (UniProt ID: P9WHE3), a TLR4 agonist at the N-terminal of the designed chimeric antigen to serve as a built-in adjuvant [31]. The EAAAK linker has been reported to improve protein expression, stability, and biological activity [80]. GPGPG linkers have been used to minimize the creation of junctional epitopes in epitope-based vaccines – facilitating antigen processing and presentation [56]. The pan HLA-DR epitope (PADRE) and cell-penetrating TAT peptide sequences were also embedded between the adjuvant and epitopes using the GGGS linker. The linker is used to ensure flexibility and ensure effective separation of individual epitopes [81]. Similarly, for the linear B-epitopes, KK linkers which have been reported to maintain the independent immune reactivity of each epitope were used [82]. In summary, all the incorporated linkers play pivotal roles in providing an extended conformation (flexibility), assisting folding, separating protein domains, and generally making the recombinant multi-epitope vaccine structure more stable [83]. Finally, a 10xHis-tag was added at the C-terminal of the multi-epitope vaccine candidate for application in identification and purification procedures after the HRV 3C Protease cleavage sequence.
4.7. Physicochemical Properties and Solubility Prediction
The physicochemical properties of the designed multi-epitope chimeric antigen were predicted using the ProtParam webserver (https://web.expasy.org/protparam/). The server generates information on the molecular weight, theoretical pI, aliphatic index, and predicted half-lives of the protein of interest upon expression in Escherichia coli, yeast, and mammal cells. In addition, information on the instability index, amino acid, and atomic composition is predicted [84]. Since E. coli is a common first choice for protein expression [85], chimera solubility upon expression in bacteria was also predicted using the NetSolP - 1.0 server (https://services.healthtech.dtu.dk/services/NetSolP-1.0/). The NetSolP - 1.0 server is used to predict the solubility and usability of purification of proteins expressed in Escherichia coli directly from the sequence. The working algorithm deployed in NetSolP is based on deep-learning protein language models and the server achieves state-of-the-art performance and improves extrapolation across datasets [86]. In addition, the DeepSoluE (http://lab.malab.cn/~wangchao/softs/DeepSoluE/), which predicts protein solubility using a long-short-term memory (LSTM) network with hybrid features composed of physicochemical patterns and distributed representation of amino acids was also used to predict solubility of the vaccine candidate upon expression in bacteria [87].
4.8. Antigenicity, Immunogenicity, Allergenicity, and Toxigenicity prediction
The VaxiJen v2.0 (previously described) and the ANTIGENpro server from Scratch Proteomic Predictor (https://scratch.proteomics.ics.uci.edu/) were used to predict the antigenicity of the designed multi-epitope chimeric antigen. The ANTIGENpro server, like VaxiJen v2.0, is a sequence-based, alignment-free prediction tool trained using a large, non-redundant dataset primarily obtained from protein microarray data analysis. The server achieved an estimated accuracy of 76% using the combined dataset in cross-validation experiments [88]. In addition, the Vaxi-DL server (https://vac.kamalrawal.in/vaxidl/) was used to further assess the potential of the designed candidate to stimulate protective immune responses. The Vaxi-DL server uses Deep Learning (DL) algorithms to classify given protein sequences into vaccine candidates and non-vaccine candidates [89].
The AllergenFP v1.0 (https://ddg-pharmfac.net/AllergenFP/) and AllerTop v.2.0 (https://www.ddg-pharmfac.net/AllerTOP/) servers were used to predict the potential of the designed multi-epitope antigen to stimulate allergenic responses. The AllergenFP server uses an alignment-independent descriptor-based fingerprint approach to classify proteins based on their allergenicity by implementing a four-step process. The AllergenFP server achieved an accuracy of 88% with a Matthews correlation coefficient of 0.759 when tested using a dataset of 2427 known allergens and 2427 non-allergens [90]. The AllerTop v2.0 server operates using amino acid E-descriptors, auto-, and cross-covariance transformation, and several machine learning methods with the k nearest neighbours (kNN) performing the best (85.3% accuracy at 5-fold cross-validation) [91].
Meanwhile, the ToxinPred3 (https://webs.iiitd.edu.in/raghava/toxinpred3/) and ToxDL (http://www.csbio.sjtu.edu.cn/bioinf/ToxDL/) servers were used to predict the presence of potential toxic peptides in the designed chimeric antigen. The ToxinPred3 server is based on machine learning (ML) and deep learning methods and has been trained using a large dataset of 5518 toxic and 5518 non-toxic experimentally validated peptides [92]. The ToxDL server uses a deep learning-based alignment-free approach for in silico prediction of protein toxicity, and independent test results showed that ToxDL outperformed servers based on traditional homology-based approaches and state-of-the-art machine-learning techniques [93].
4.9. Secondary Structure and Intrinsic Disorder Prediction
The PSIPRED v4.0 server (http://bioinf.cs.ucl.ac.uk/psipred/) was used to predict the secondary of the designed chimeric antigen. The PSIPRED algorithm incorporates two feed-forward neural networks that analyze the output of a PSI-BLAST (Position-Specific Iterated BLAST) to accurately predict protein secondary structure [94]. The PSIPRED v4.0 server has a Q3 prediction accuracy of 84.2.5% [95].
Disordered proteins constitute an important class of antigens in a wide range of human pathogens, and are frequently targets of protective immune responses [96]. Intrinsically disordered regions (IDRs) in the designed chimeric vaccine candidate were predicted using the IUPred3 (https://iupred.elte.hu/) and PrDOS (https://prdos.hgc.jp/cgi-bin/top.cgi) servers. The IUPred web server algorithm employs the amino acid sequence or UniProt ID to predict the tendency for each amino acid to be in a disordered region using a unique energy estimation approach [97]. The PrDOS server algorithm is composed of two predictors (one based on the local amino acid sequence and the other based on template proteins) to predict protein disorder using support vector machine (SVM) and PSI-BLAST [98].
4.10. Antibody Class Prediction
Specific immunoglobulins, predominantly IgG, have been reported to be implicated in immune protection against Mycobacterium tuberculosis [99]. The Antibody Class(es) Predictor for Epitopes (AbCPE) server (http://bioinfo.unipune.ac.in/AbCPE/Home.html) was used to investigate the specific antibody classes elicited by the predicted linear B-epitope sequences. The AbCPE server is a novel method based on a multi-label classification algorithm (including Binary Relevance, Label Powerset, Random Forest, and AdaBoost) for antibody class(es) prediction and the binding of peptide sequences to IgG, IgE, IgA, and IgM [100].
4.11. IFN-γ-Inducing Epitope Prediction
Interferon-gamma (IFN-γ), which is vital for the generation of protective responses against intracellular pathogens like M. tuberculosis is reported to be important in the development of protective anti-TB immune responses [101]. IFN-γ epitopes present in the designed chimeric antigen were predicted using the hybrid motif and SVM approach on the IFNepitope server (https://webs.iiitd.edu.in/raghava/ifnepitope/scan.php). The IFNepitope server algorithm functions by constructing overlapping 15-mer sequences, which are used to predict the IFN-γ epitopes. The server was developed using a training dataset of experimentally validated MHC class II binders obtained from the Immune Epitope Database (IEDB) comprising 3705 IFN-γ inducing and 6728 non-inducing MHC class-II binders [102].
4.12. Prediction of Epitopes for Mouse MHC II Alleles
To probe into the potential of the designed vaccine candidate to stimulate immune responses in preclinical studies, MHC II binding peptides (15-mer) for mice alleles in the designed vaccine candidate were predicted using the NetMHCII 2.3 server (http://www.cbs.dtu.dk/services/NetMHCII/). The server uses artificial neural networks to predict binding affinities to molecules covering human alleles (HLA-DR, HLA-DQ, and HLA-DP) as well as 7 mouse alleles. The functioning of the server has previously been described.
4.13. Immune Simulation Analyses
The C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php) which performs in-silico immune simulations was used to predict the immune response elicited by the chimeric vaccine candidate. The C-ImmSim server is an agent-based model which employs a position-specific scoring matrix (PSSM) for epitope prediction and machine-learning approaches to predict immunological interactions. The server “simultaneously simulates three compartments that represent three separate anatomical regions found in mammals: (i) the bone marrow, where hematopoietic stem cells are simulated and produce new lymphoid and myeloid cells; (ii) the thymus, where naive T cells are selected to avoid autoimmunity; and (iii) a tertiary lymphatic organ, such as a lymph node” [103]. A total of 500 simulation steps were set. The default settings of simulation parameters were used, and the time steps were set to 1, 84, and 168 for the three injections (each time step is 8 hours, and time step 1 is injection at time = 0). These settings were based on a slight modification of the protocol used in phase II clinical trials for the M72/AS01E vaccine candidate [104].
4.14. Tertiary Structure Prediction, Refinement, and Validation
The 3D structure of the designed chimeric antigen was predicted using ColabFold, an easy-to-use interface that uses the AlphaFold2 [105] technology within the Google Colab environment. ColabFold offers an accelerated prediction of protein structures and complexes by combining the fast homology search of MMseqs2 with AlphaFold2 or RoseTTAFold [106]. The 3D structure obtained was refined using the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE). The GalaxyRefine server functions based on a refining technique that was successfully evaluated in CASP10 community-wide tests and found to greatly improve local structure quality compared to other servers. To accomplish overall structure relaxation, this technique first rebuilds side chains, performs side chain repacking and then employs a molecular dynamics simulation [107]. The overall quality score of the refined chimera 3D structure was assessed using the ProSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) and ERRAT (http://services.mbi.ucla.edu/ERRAT/) servers. ProSA is widely used to examine 3D models of protein structures for potential errors and is used for error recognition in experimentally determined structures, theoretical models, and protein engineering [108]. The ERRAT server, on the other hand, analyzes non-bonded atom-atom interactions compared to reliable high-resolution crystallography structures [109].
4.15. Discontinuous B-Cell Epitope Prediction
Following 3D structure validation of the modeled chimeric vaccine candidate, the ElliPro (http://tools.iedb.org/ellipro/) server was used to predict discontinuous B-cell epitope using default parameters. The ElliPro server “implements a modified version of Thornton's method and, together with a residue clustering algorithm, the MODELLER program and the Jmol viewer, allows the prediction and visualization of antibody epitopes in protein sequences and structures”. The ElliPro server has been tested on a benchmark dataset of conformational epitopes deduced from 3D structures of antibody-protein complexes. ElliPro produced the best performance, when compared with other structure-based epitope prediction algorithms, giving an AUC value of 0.732 when considering the most significant prediction for each protein [110].
4.16. Binding Pocket Prediction, Molecular Docking, Binding Affinity, and Interaction Analyses
To assess the binding pockets in the designed chimera, the CASTp 3.0 (http://sts.bioe.uic.edu/castp/index.html?2was) server was used. CASTp is a web server that uses the alpha shape method developed in computational geometry to locate, delineate, and measure different geometric and topological properties of protein structures [111].
To assess the interaction between the designed chimera and TLR4, protein-protein docking analysis was executed using the ClusPro 2.0 server (https://cluspro.bu.edu/login.php). The ClusPro 2.0 server has consistently been one of the best docking servers, as demonstrated in Critical Assessment of Predicted Interactions (CAPRI), providing high predictive performance for the docking of protein-protein complexes [112]. The server performs docking through the following computational steps: (1) rigid body docking by sampling billions of conformations, (2) root-mean-square deviation (RMSD) based clustering of the 1000 lowest energy structures generated to find the largest clusters that will represent the most likely models of the complex, and lastly (3) refinement of selected structures through energy minimization. The rigid body docking step uses PIPER, a docking program based on the Fast Fourier Transform (FFT) correlation approach. The server performs similarity-based docking by retrieving templates from the database of experimentally determined structures and building models using energy-based optimization, which allows for structural flexibility [112].
The binding affinity of the predicted interaction between the vaccine candidate and the TLR4 immune receptor was assessed using the PROtein binDIng enerGY (PRODIGY) prediction server (https://wenmr.science.uu.nl/prodigy/). The server implements a simple but very efficient predictive model based on intermolecular contacts and properties derived from the non-interface surface [33]. The docking results were further analyzed by the PDBsum server ( http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html), which generates a 2D picture of the interactions at the ligand-protein interface from the 3D coordinates [113].
4.17. Normal Mode Analyses
Normal mode analysis (NMA) was performed for the docked vaccine candidate-TLR4 complex using the iMODS server (http://imods.Chaconlab.org/). This analysis was performed to demonstrate the internal dihedral coordinates and measure the cooperative functional motions of the docked complex. The essential dynamic simulation program of the iMODS server was deployed to ensure the energy minimization, molecular stability, and atomic mobility of the vaccine candidate in the docked complex. The server determines the possible motions of the vaccine candidate-TLR4 complex based on specialized parameters, including B-factors, RMSD, eigenvalues, deformability of the complex, covariance values, and elastic models [114].
4.18. Codon Optimization and In-Silico Cloning
The E. coli K12 strain was selected to express the multi-epitope chimeric antigen since bacteria is often the primary host for protein expression experiments. The Java Codon Adaptation Tool (https://www.jcat.de/) was used to reverse-translate the protein sequence into a DNA sequence and perform codon optimization. The JCat server offers the flexibility to adapt the codon usage for any gene the user provides to that of any host organism. The server also provides information on the codon adaptation index (CAI) and GC% content, which have both been reported to affect protein expression in bacteria [115,116]. For expression of the designed antigen in E. coli, the codon-optimized gene sequence was cloned between the Nde I and Xho I restriction sites of the pET30a+ vector using the Snapgene tool (https://www.snapgene.com/).
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Preliminary characterization of selected lipoproteins; Table S2: Characteristics of predicted conformational epitopes.
Author Contributions
Conceptualization, R.A.S., G.T.N., and T.Y.A.S.; Methodology, R.A.S., G.T.N., T.Y.A.S., N.E.Y., C.M.S., B.N.Y., D.N.N., J.A.C. and M.T.E.; software, R.A.S., G.T.N., A.B.A, G.K.Y., D.N.N., J.A.C. and A.A.L.; formal analysis, T.Y.A.S., N.E.Y., C.M.S., B.N.Y., M.T.E., A.B.A, G.K.Y. and A.A.L.; writing—original draft preparation, R.S, and G.T.N.; writing—review and editing, All authors.; visualization, R.A.S., G.T.N., and T.Y.A.S.; supervision, SM.G, L.V., JS.; project administration, S.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Data are contained within the article figures, tables and Supplementary Materials
Acknowledgments
The authors acknowledge all the open-source and globally available databases that were utilized for data mining. LV is the Director of Research at the Belgian National Fund for Scientific Research (FRS-FNRS).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ayalew, S.; Habtamu, G.; Melese, F.; Tessema, B.; Ashford, R.T.; Chothe, S.K.; Aseffa, A.; Wood, J.L.N.; Berg, S.; Mihret, A. Zoonotic tuberculosis in a high bovine tuberculosis burden area of Ethiopia. Front Public Health 2023, 11, 1204525. [Google Scholar] [CrossRef] [PubMed]
- Collins, Á.B.; Floyd, S.; Gordon, S.V.; More, S.J. Prevalence of Mycobacterium bovis in milk on dairy cattle farms: An international systematic literature review and meta-analysis. Tuberculosis 2022, 132, 102166. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Dürr, S.; Alonso, S.; Hattendorf, J.; Laisse, C.J.; Parsons, S.D.; van Helden, P.D.; Zinsstag, J. Zoonotic Mycobacterium bovis-induced tuberculosis in humans. Emerg Infect Dis 2013, 19, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.S.; Sweeney, S.J. Mycobacterium bovis (bovine tuberculosis) infection in North American wildlife: current status and opportunities for mitigation of risks of further infection in wildlife populations. Epidemiol Infect 2013, 141, 1357–1370. [Google Scholar] [CrossRef]
- O'Reilly, L.M.; Daborn, C.J. The epidemiology of Mycobacterium bovis infections in animals and man: a review. Tuber Lung Dis 1995, 76 Suppl 1, 1–46. [Google Scholar] [CrossRef]
- Millet, J.P.; Moreno, A.; Fina, L.; del Baño, L.; Orcau, A.; de Olalla, P.G.; Caylà, J.A. Factors that influence current tuberculosis epidemiology. Eur Spine J 2013, 22 Suppl 4, 539–548. [Google Scholar] [CrossRef]
- WHO. Tuberculosis. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis.
- An, Y.; Teo, A.K.J.; Huot, C.Y.; Tieng, S.; Khun, K.E.; Pheng, S.H.; Leng, C.; Deng, S.; Song, N.; Nonaka, D.; et al. Barriers to childhood tuberculosis case detection and management in Cambodia: the perspectives of healthcare providers and caregivers. BMC Infect Dis 2023, 23, 80. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.M.; Dolan, L.; Piggott, L.M.; McLaughlin, A.M. New developments in tuberculosis diagnosis and treatment. Breathe (Sheff) 2022, 18, 210149. [Google Scholar] [CrossRef]
- Hiza, H.; Hella, J.; Arbués, A.; Magani, B.; Sasamalo, M.; Gagneux, S.; Reither, K.; Portevin, D. Case–control diagnostic accuracy study of a non-sputum CD38-based TAM-TB test from a single milliliter of blood. Scientific Reports 2021, 11, 13190. [Google Scholar] [CrossRef]
- Dartois, V.A.; Rubin, E.J. Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nature Reviews Microbiology 2022, 20, 685–701. [Google Scholar] [CrossRef]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: causes, consequences, and management. Front Public Health 2014, 2, 145. [Google Scholar] [CrossRef] [PubMed]
- Migliori, G.B.; Dheda, K.; Centis, R.; Mwaba, P.; Bates, M.; O’Grady, J.; Hoelscher, M.; Zumla, A. Review of multidrug-resistant and extensively drug-resistant TB: global perspectives with a focus on sub-Saharan Africa. Tropical Medicine & International Health 2010, 15, 1052–1066. [Google Scholar] [CrossRef]
- dos Santos, P.C.P.; Messina, N.L.; de Oliveira, R.D.; da Silva, P.V.; Puga, M.A.M.; Dalcolmo, M.; dos Santos, G.; de Lacerda, M.V.G.; Jardim, B.A.; de Almeida e Val, F.F.; et al. Effect of BCG vaccination against <em>Mycobacterium tuberculosis</em> infection in adult Brazilian health-care workers: a nested clinical trial. The Lancet Infectious Diseases 2024, 24, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Bannon, M.J. BCG and tuberculosis. Archives of Disease in Childhood 1999, 80, 80. [Google Scholar] [CrossRef] [PubMed]
- Scriba, T.J.; Netea, M.G.; Ginsberg, A.M. Key recent advances in TB vaccine development and understanding of protective immune responses against Mycobacterium tuberculosis. Semin Immunol 2020, 50, 101431. [Google Scholar] [CrossRef]
- Brandt, L.; Feino Cunha, J.; Weinreich Olsen, A.; Chilima, B.; Hirsch, P.; Appelberg, R.; Andersen, P. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect Immun 2002, 70, 672–678. [Google Scholar] [CrossRef] [PubMed]
- García, J.I.; Allué-Guardia, A.; Tampi, R.P.; Restrepo, B.I.; Torrelles, J.B. New Developments and Insights in the Improvement of Mycobacterium tuberculosis Vaccines and Diagnostics Within the End TB Strategy. Curr Epidemiol Rep 2021, 8, 33–45. [Google Scholar] [CrossRef]
- Maleki, A.; Russo, G.; Parasiliti Palumbo, G.A.; Pappalardo, F. In silico design of recombinant multi-epitope vaccine against influenza A virus. BMC Bioinformatics 2022, 22, 617. [Google Scholar] [CrossRef] [PubMed]
- Sanches, R.C.O.; Tiwari, S.; Ferreira, L.C.G.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.F.; Maia, E.H.B.; Taranto, A.G.; Kato, R.; Azevedo, V.A.C.; et al. Immunoinformatics Design of Multi-Epitope Peptide-Based Vaccine Against Schistosoma mansoni Using Transmembrane Proteins as a Target. Frontiers in immunology 2021, 12, 621706–621706. [Google Scholar] [CrossRef]
- Andongma, B.T.; Huang, Y.; Chen, F.; Tang, Q.; Yang, M.; Chou, S.H.; Li, X.; He, J. In silico design of a promiscuous chimeric multi-epitope vaccine against Mycobacterium tuberculosis. Comput Struct Biotechnol J 2023, 21, 991–1004. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Shintouo, C.M.; Nkemngo, F.N.; Nebangwa, D.N.; Esoh, K.; Yaah, N.E.; Manka'aFri, M.; Nguve, J.E.; Ngwese, R.A.; et al. Computational Design and Preliminary Serological Analysis of a Novel Multi-Epitope Vaccine Candidate against Onchocerciasis and Related Filarial Diseases. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Nebangwa, D.N.; Shintouo, C.M.; Yaah, N.E.; Yengo, B.N.; Nkemngo, F.N.; Esoh, K.K.; Tchatchoua, N.M.T.; Mbachick, T.T.; et al. Rational design of a novel multi-epitope peptide-based vaccine against Onchocerca volvulus using transmembrane proteins. Frontiers in Tropical Diseases 2022, 3. [Google Scholar] [CrossRef]
- Zhang, L. Multi-epitope vaccines: a promising strategy against tumors and viral infections. Cellular & Molecular Immunology 2018, 15, 182–184. [Google Scholar] [CrossRef]
- Kovacs-Simon, A.; Titball, R.W.; Michell, S.L. Lipoproteins of bacterial pathogens. Infect Immun 2011, 79, 548–561. [Google Scholar] [CrossRef]
- Khandavilli, S.; Homer, K.A.; Yuste, J.; Basavanna, S.; Mitchell, T.; Brown, J.S. Maturation of Streptococcus pneumoniae lipoproteins by a type II signal peptidase is required for ABC transporter function and full virulence. Mol Microbiol 2008, 67, 541–557. [Google Scholar] [CrossRef]
- Gabriela, E.-V.; Susana, F.-V.; Clara, I.E. Virulence Factors and Pathogenicity of Mycobacterium. In Mycobacterium, Wellman, R., Ed. IntechOpen: Rijeka, 2017; 10.5772/intechopen.72027p. Ch. 12.
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.H.; Sun, R.F.; Lin, C.; Chen, F.Z.; Mai, J.T.; Liu, Y.X.; Xu, Z.Y.; Zhang, L.; Liu, J. Immunogenicity and Protective Efficacy of a Fusion Protein Tuberculosis Vaccine Combining Five Esx Family Proteins. Front Cell Infect Microbiol 2017, 7, 226. [Google Scholar] [CrossRef]
- Minerva, M.; De Maio, F.; Camassa, S.; Battah, B.; Ivana, P.; Manganelli, R.; Sanguinetti, M.; Sali, M.; Delogu, G. Evaluation of PE_PGRS33 as a potential surface target for humoral responses against Mycobacterium tuberculosis. Pathogens and Disease 2017, 75. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One 2014, 9, e104351. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.; Kwon, K.W.; Choi, H.H.; Kang, S.M.; Hong, J.J.; Shin, S.J. Toll-like receptor 4 signaling-mediated responses are critically engaged in optimal host protection against highly virulent Mycobacterium tuberculosis K infection. Virulence 2020, 11, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Abel, B.; Thieblemont, N.; Quesniaux, V.J.; Brown, N.; Mpagi, J.; Miyake, K.; Bihl, F.; Ryffel, B. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol 2002, 169, 3155–3162. [Google Scholar] [CrossRef]
- Akhtar, H.; Akhtar, S.; Jan, S.U.; Khan, A.; Zaidi, N.; Qadri, I. Over expression of a synthetic gene encoding interferon lambda using relative synonymous codon usage bias in Escherichia coli. Pak J Pharm Sci 2013, 26, 1181–1188. [Google Scholar] [PubMed]
- Arinaminpathy, N.; Mukadi, Y.D.; Bloom, A.; Vincent, C.; Ahmedov, S. Meeting the 2030 END TB goals in the wake of COVID-19: A modelling study of countries in the USAID TB portfolio. PLOS Glob Public Health 2023, 3, e0001271. [Google Scholar] [CrossRef]
- Portnoy, A.; Clark, R.A.; Quaife, M.; Weerasuriya, C.K.; Mukandavire, C.; Bakker, R.; Deol, A.K.; Malhotra, S.; Gebreselassie, N.; Zignol, M.; et al. The cost and cost-effectiveness of novel tuberculosis vaccines in low- and middle-income countries: A modeling study. PLoS Med 2023, 20, e1004155. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Samperio, P. Development of tuberculosis vaccines in clinical trials: Current status. Scandinavian Journal of Immunology 2018, 88, e12710. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Xue, Y.; Wang, J.; Jia, Z.; Wang, L.; Gong, W. Evaluation of the consistence between the results of immunoinformatics predictions and real-world animal experiments of a new tuberculosis vaccine MP3RT. Frontiers in Cellular and Infection Microbiology 2022, 12. [Google Scholar] [CrossRef]
- Terry, F.E.; Moise, L.; Martin, R.F.; Torres, M.; Pilotte, N.; Williams, S.A.; De Groot, A.S. Time for T? Immunoinformatics addresses vaccine design for neglected tropical and emerging infectious diseases. Expert Rev Vaccines 2015, 14, 21–35. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Scientific Reports 2019, 9, 4409. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, L.; Adeleke, V.T.; Fatoba, A.J.; Adeniyi, A.A.; Tshilwane, S.I.; Adeleke, M.A.; Maharaj, R.; Okpeku, M. Immunoinformatics approach for multi-epitope vaccine design against P. falciparum malaria. Infect Genet Evol 2021, 92, 104875. [Google Scholar] [CrossRef]
- Monterrubio-López, G.P.; González-Y-Merchand, J.A.; Ribas-Aparicio, R.M. Identification of Novel Potential Vaccine Candidates against Tuberculosis Based on Reverse Vaccinology. BioMed Research International 2015, 2015, 483150. [Google Scholar] [CrossRef]
- Desalegn, G.; Tamilselvi, C.S.; Lemme-Dumit, J.M.; Heine, S.J.; Dunn, D.; Ndungo, E.; Kapoor, N.; Oaks, E.V.; Fairman, J.; Pasetti, M.F. Shigella virulence protein VirG is a broadly protective antigen and vaccine candidate. npj Vaccines 2024, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Tamehri, M.; Rasooli, I.; Pishgahi, M.; Jahangiri, A.; Ramezanalizadeh, F.; Banisaeed Langroodi, S.R. Combination of BauA and OmpA elicit immunoprotection against Acinetobacter baumannii in a murine sepsis model. Microbial Pathogenesis 2022, 173, 105874. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.A.; Gálvez, N.M.S.; Andrade, C.A.; Ramírez, M.A.; Riedel, C.A.; Kalergis, A.M.; Bueno, S.M. BCG vaccination induces cross-protective immunity against pathogenic microorganisms. Trends in Immunology 2022, 43, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Fromsa, A.; Willgert, K.; Srinivasan, S.; Mekonnen, G.; Bedada, W.; Gumi, B.; Lakew, M.; Tadesse, B.; Bayissa, B.; Sirak, A.; et al. BCG vaccination reduces bovine tuberculosis transmission, improving prospects for elimination. Science 2024, 383, eadl3962. [Google Scholar] [CrossRef]
- van der Heijden, E.M.D.L.; Cooper, D.V.; Rutten, V.P.M.G.; Michel, A.L. Mycobacterium bovis prevalence affects the performance of a commercial serological assay for bovine tuberculosis in African buffaloes. Comparative Immunology, Microbiology and Infectious Diseases 2020, 70, 101369. [Google Scholar] [CrossRef] [PubMed]
- Villa, S.; Carugati, M.; Rubach, M.P.; Cleaveland, S.; Mpagama, S.G.; Khan, S.S.; Mfinanga, S.; Mmbaga, B.T.; Crump, J.A.; Raviglione, M.C. 'One Health´ approach to end zoonotic TB. Int J Tuberc Lung Dis 2023, 27, 101–105. [Google Scholar] [CrossRef]
- Stewart, P.; Patel, S.; Comer, A.; Muneer, S.; Nawaz, U.; Quann, V.; Bansal, M.; Venketaraman, V. Role of B Cells in Mycobacterium Tuberculosis Infection. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Hosseinian, K.; Gerami, A.; Bral, M.; Venketaraman, V. Mycobacterium tuberculosis–Human Immunodeficiency Virus Infection and the Role of T Cells in Protection. Vaccines 2024, 12, 730. [Google Scholar] [CrossRef]
- Behar, S.M. Antigen-specific CD8(+) T cells and protective immunity to tuberculosis. Adv Exp Med Biol 2013, 783, 141–163. [Google Scholar] [CrossRef]
- Rodríguez, A.; Tjärnlund, A.; Ivanji, J.; Singh, M.; García, I.; Williams, A.; Marsh, P.D.; Troye-Blomberg, M.; Fernández, C. Role of IgA in the defense against respiratory infections IgA deficient mice exhibited increased susceptibility to intranasal infection with Mycobacterium bovis BCG. Vaccine 2005, 23, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, H.A.; Snowden, M.A.; Landry, B.; Rida, W.; Satti, I.; Harris, S.A.; Matsumiya, M.; Tanner, R.; O'Shea, M.K.; Dheenadhayalan, V.; et al. T-cell activation is an immune correlate of risk in BCG vaccinated infants. Nat Commun 2016, 7, 11290. [Google Scholar] [CrossRef] [PubMed]
- Quesniaux, V.; Fremond, C.; Jacobs, M.; Parida, S.; Nicolle, D.; Yeremeev, V.; Bihl, F.; Erard, F.; Botha, T.; Drennan, M.; et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes and Infection 2004, 6, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Branger, J.; Leemans, J.C.; Florquin, S.; Weijer, S.; Speelman, P.; van der Poll, T. Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. International Immunology 2004, 16, 509–516. [Google Scholar] [CrossRef]
- Livingston, B.; Crimi, C.; Newman, M.; Higashimoto, Y.; Appella, E.; Sidney, J.; Sette, A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J Immunol 2002, 168, 5499–5506. [Google Scholar] [CrossRef]
- Sette, A.; Livingston, B.; McKinney, D.; Appella, E.; Fikes, J.; Sidney, J.; Newman, M.; Chesnut, R. The development of multi-epitope vaccines: epitope identification, vaccine design and clinical evaluation. Biologicals 2001, 29, 271–276. [Google Scholar] [CrossRef]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Alexander, J.; del Guercio, M.F.; Frame, B.; Maewal, A.; Sette, A.; Nahm, M.H.; Newman, M.J. Development of experimental carbohydrate-conjugate vaccines composed of Streptococcus pneumoniae capsular polysaccharides and the universal helper T-lymphocyte epitope (PADRE). Vaccine 2004, 22, 2362–2367. [Google Scholar] [CrossRef]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Scientific Reports 2019, 9, 6298. [Google Scholar] [CrossRef] [PubMed]
- Backlund, C.M.; Holden, R.L.; Moynihan, K.D.; Garafola, D.; Farquhar, C.; Mehta, N.K.; Maiorino, L.; Pham, S.; Iorgulescu, J.B.; Reardon, D.A.; et al. Cell-penetrating peptides enhance peptide vaccine accumulation and persistence in lymph nodes to drive immunogenicity. Proceedings of the National Academy of Sciences 2022, 119, e2204078119. [Google Scholar] [CrossRef] [PubMed]
- Du Bruyn, E.; Ruzive, S.; Lindestam Arlehamn, C.S.; Sette, A.; Sher, A.; Barber, D.L.; Wilkinson, R.J.; Riou, C. Mycobacterium tuberculosis-specific CD4 T cells expressing CD153 inversely associate with bacterial load and disease severity in human tuberculosis. Mucosal Immunology 2021, 14, 491–499. [Google Scholar] [CrossRef]
- Kawakami, K.; Kinjo, Y.; Uezu, K.; Miyagi, K.; Kinjo, T.; Yara, S.; Koguchi, Y.; Miyazato, A.; Shibuya, K.; Iwakura, Y.; et al. Interferon-γ production and host protective response against Mycobacterium tuberculosis in mice lacking both IL-12p40 and IL-18. Microbes and Infection 2004, 6, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Curlin, G.; Landry, S.; Bernstein, J.; Gorman, R.L.; Mulach, B.; Hackett, C.J.; Foster, S.; Miers, S.E.; Strickler-Dinglasan, P. Integrating safety and efficacy evaluation throughout vaccine research and development. Pediatrics 2011, 127 Suppl. 1, S9–15. [Google Scholar] [CrossRef]
- Pouresmaeil, M.; Azizi-Dargahlou, S. Factors involved in heterologous expression of proteins in E. coli host. Archives of Microbiology 2023, 205, 212. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Maheshwari, N.; Chauhan, R.; Sen, N.K.; Sharma, A. Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation 2011, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.O.; McLellan, J.S. Principles and practical applications of structure-based vaccine design. Curr Opin Immunol 2022, 77, 102209. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Collier, J.H. α-Helical coiled-coil peptide materials for biomedical applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Ameri, M.; Nezafat, N.; Eskandari, S. The potential of intrinsically disordered regions in vaccine development. Expert Review of Vaccines 2022, 21, 1–3. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.; Li, S.; Yuan, J.; Pang, Y. Maintenance and recall of memory T cell populations against tuberculosis: Implications for vaccine design. Front Immunol 2023, 14, 1100741. [Google Scholar] [CrossRef]
- Dieli, F.; Ivanyi, J. Role of antibodies in vaccine-mediated protection against tuberculosis. Cellular & Molecular Immunology 2022, 19, 758–760. [Google Scholar] [CrossRef]
- Gong, W.; Liang, Y.; Mi, J.; Jia, Z.; Xue, Y.; Wang, J.; Wang, L.; Zhou, Y.; Sun, S.; Wu, X. Peptides-Based Vaccine MP3RT Induced Protective Immunity Against Mycobacterium Tuberculosis Infection in a Humanized Mouse Model. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Tarke, A.; Zhang, Y.; Methot, N.; Narowski, T.M.; Phillips, E.; Mallal, S.; Frazier, A.; Filaci, G.; Weiskopf, D.; Dan, J.M.; et al. Targets and cross-reactivity of human T cell recognition of common cold coronaviruses. Cell Reports Medicine 2023, 4, 101088. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. The universal protein resource (UniProt). Nucleic Acids Res 2008, 36, D190–D195. [Google Scholar] [CrossRef]
- Clifford, J.N.; Høie, M.H.; Deleuran, S.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-3.0: Improved B-cell epitope prediction using protein language models. Protein Sci 2022, 31, e4497. [Google Scholar] [CrossRef]
- Singh, H.; Ansari, H.R.; Raghava, G.P. Improved method for linear B-cell epitope prediction using antigen's primary sequence. PLoS One 2013, 8, e62216. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 2007, 8, 424. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Engineering, Design and Selection 2001, 14, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Yano, A.; Onozuka, A.; Asahi-Ozaki, Y.; Imai, S.; Hanada, N.; Miwa, Y.; Nisizawa, T. An ingenious design for peptide vaccines. Vaccine 2005, 23, 2322–2326. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Advanced Drug Delivery Reviews 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 2014, 5, 172–172. [Google Scholar] [CrossRef] [PubMed]
- Thumuluri, V.; Martiny, H.-M.; Almagro Armenteros, J.J.; Salomon, J.; Nielsen, H.; Johansen, A.R. NetSolP: predicting protein solubility in Escherichia coli using language models. Bioinformatics 2021, 38, 941–946. [Google Scholar] [CrossRef]
- Wang, C.; Zou, Q. Prediction of protein solubility based on sequence physicochemical patterns and distributed representation information with DeepSoluE. BMC Biology 2023, 21, 12. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Rawal, K.; Sinha, R.; Abbasi, B.A.; Chaudhary, A.; Nath, S.K.; Kumari, P.; Preeti, P.; Saraf, D.; Singh, S.; Mishra, K.; et al. Identification of vaccine targets in pathogens and design of a vaccine using computational approaches. Scientific Reports 2021, 11, 17626. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2--a server for in silico prediction of allergens. J Mol Model 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Anand Singh Rathore, A.A., Shubham Choudhury, Purva Tijare, Gajendra P. S. Raghava. ToxinPred 3.0: An improved method for predicting the toxicity of peptides. bioRxiv 2023. [CrossRef]
- Pan, X.; Zuallaert, J.; Wang, X.; Shen, H.-B.; Campos, E.P.; Marushchak, D.O.; De Neve, W. ToxDL: deep learning using primary structure and domain embeddings for assessing protein toxicity. Bioinformatics 2020, 36, 5159–5168. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- MacRaild, C.A.; Seow, J.; Das, S.C.; Norton, R.S. Disordered epitopes as peptide vaccines. Pept Sci (Hoboken) 2018, 110, e24067. [Google Scholar] [CrossRef]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res 2021, 49, W297–W303. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res 2007, 35, W460–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Nag, D.; Baldwin, S.L.; Coler, R.N.; McNamara, R.P. Antibodies as key mediators of protection against Mycobacterium tuberculosis. Front Immunol 2024, 15, 1430955. [Google Scholar] [CrossRef] [PubMed]
- Kadam, K.; Peerzada, N.; Karbhal, R.; Sawant, S.; Valadi, J.; Kulkarni-Kale, U. Antibody Class(es) Predictor for Epitopes (AbCPE): A Multi-Label Classification Algorithm. Frontiers in Bioinformatics 2021, 1. [Google Scholar] [CrossRef] [PubMed]
- Lalvani, A.; Millington, K.A. T Cells and Tuberculosis: Beyond Interferon-γ. The Journal of Infectious Diseases 2008, 197, 941–943. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct 2013, 8, 30. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Meeren, O.V.D.; Hatherill, M.; Nduba, V.; Wilkinson, R.J.; Muyoyeta, M.; Brakel, E.V.; Ayles, H.M.; Henostroza, G.; Thienemann, F.; Scriba, T.J.; et al. Phase 2b Controlled Trial of M72/AS01<sub>E</sub> Vaccine to Prevent Tuberculosis. New England Journal of Medicine 2018, 379, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: making protein folding accessible to all. Nat Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 2008, 9, 514–514. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat Protoc 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem Sci 1997, 22, 488–490. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 2014, 42, W271–276. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Wernisch, L.; Savva, R. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res 2003, 31, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R. Eukaryotic dinucleotide preference rules and their implications for degenerate codon usage. Journal of Molecular Biology 1981, 149, 125–131. [Google Scholar] [CrossRef]
Figure 1.
Schematic presentation of the final multi-epitope vaccine antigen, TB-MEV-1. The 683-amino acid long polypeptide sequence containing a built-in adjuvant (green) at the N-terminal linked with the multi-epitope sequence through an EAAAK linker (cyan) to the PADRE and TAT peptides which are linked through GGGS linkers. The CTL, HTL epitopes, and linear-B cell (high-scoring), epitopes predicted to be antigenic are merged by AAY (yellow), GPGPG (orange), and KK linkers (blue), respectively). A 10x-His tag is added at the C-terminal for purification and identification purposes, separated from the peptide epitopes using the HRV 3C Protease cleavage sequence.
Figure 1.
Schematic presentation of the final multi-epitope vaccine antigen, TB-MEV-1. The 683-amino acid long polypeptide sequence containing a built-in adjuvant (green) at the N-terminal linked with the multi-epitope sequence through an EAAAK linker (cyan) to the PADRE and TAT peptides which are linked through GGGS linkers. The CTL, HTL epitopes, and linear-B cell (high-scoring), epitopes predicted to be antigenic are merged by AAY (yellow), GPGPG (orange), and KK linkers (blue), respectively). A 10x-His tag is added at the C-terminal for purification and identification purposes, separated from the peptide epitopes using the HRV 3C Protease cleavage sequence.
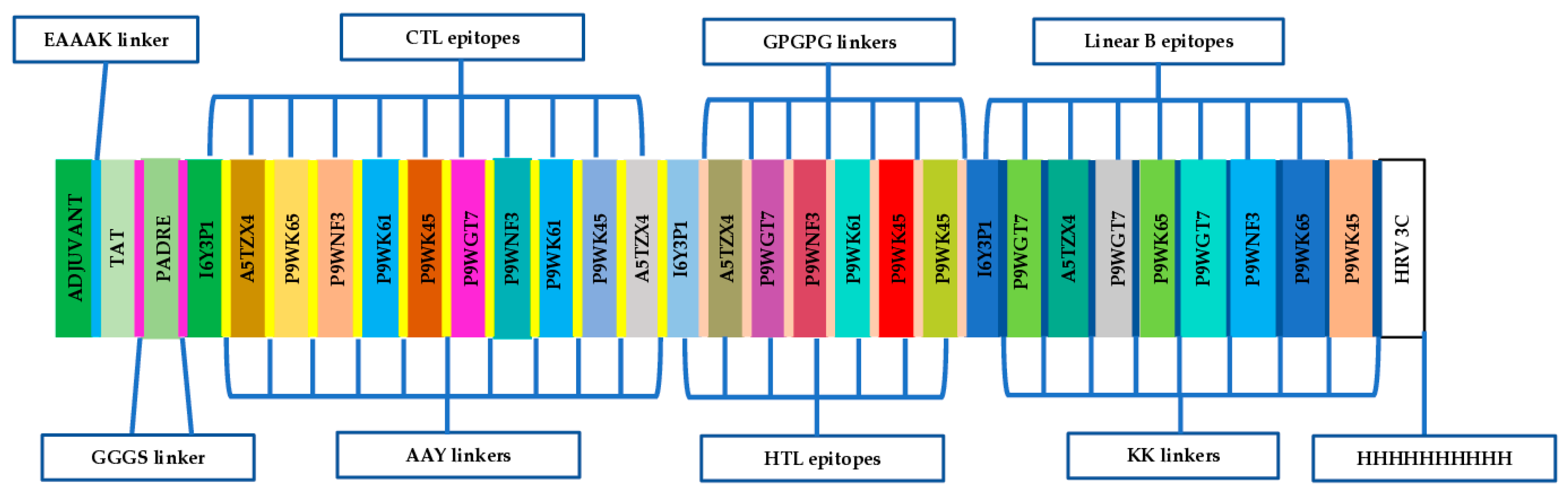
Figure 2.
Graphical representation of secondary structure features of the final subunit vaccine sequence. (A) The protein is predicted to comprise alpha-helices (28.0%), beta strands (7.0%), and coils (65.0%), and (B) 20% of positions are predicted to be in intrinsically disordered regions as predicted by DISOPRED3. (C). The vaccine candidate 3D structure shows the different secondary structure elements with the helices (cyan), coils (red), and strands (magenta).
Figure 2.
Graphical representation of secondary structure features of the final subunit vaccine sequence. (A) The protein is predicted to comprise alpha-helices (28.0%), beta strands (7.0%), and coils (65.0%), and (B) 20% of positions are predicted to be in intrinsically disordered regions as predicted by DISOPRED3. (C). The vaccine candidate 3D structure shows the different secondary structure elements with the helices (cyan), coils (red), and strands (magenta).
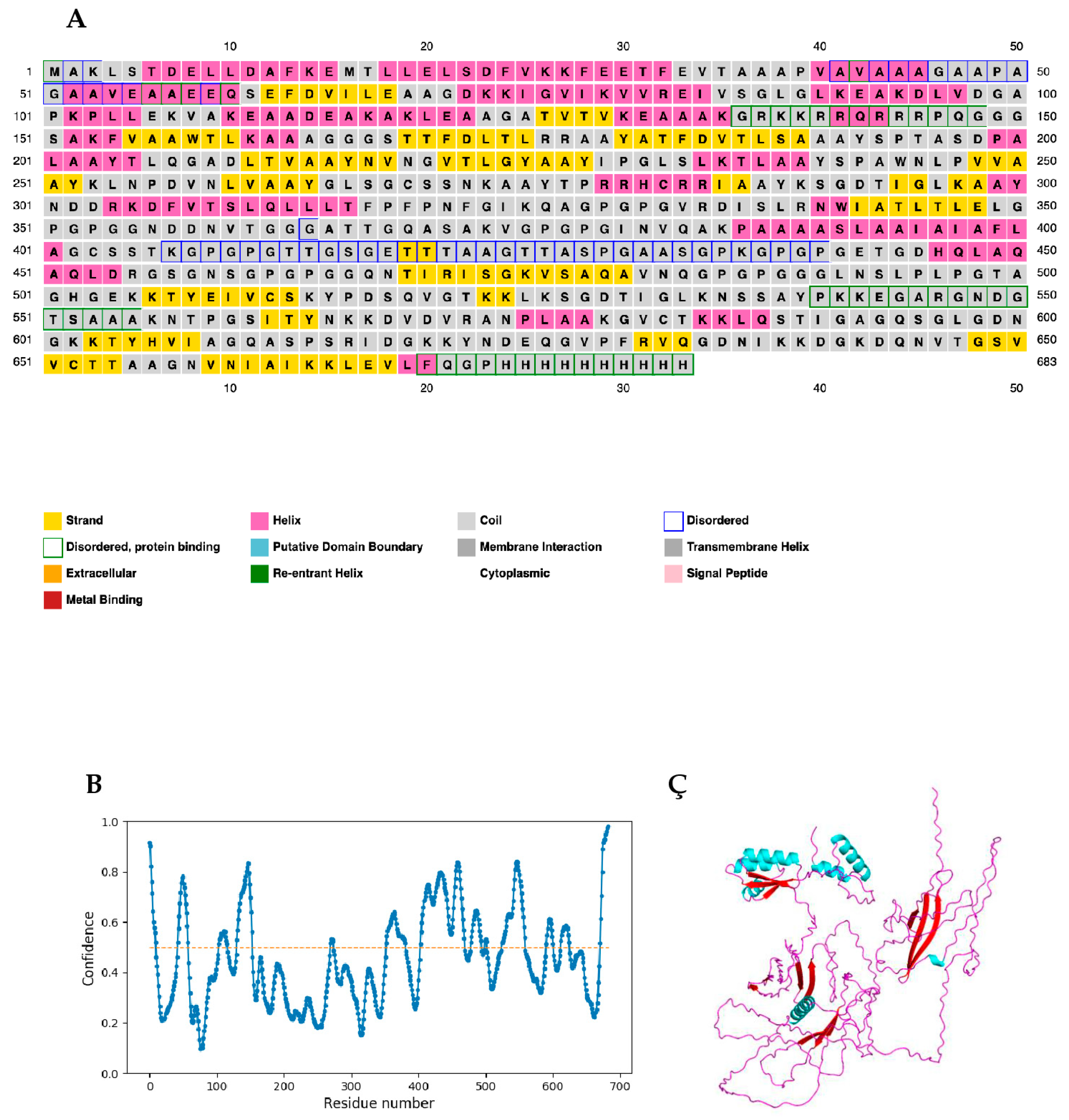
Figure 3.
Immune simulation of the cytokine profile induced by three injections of TB-MEVA-1 given 28 days apart. The main plot shows different cytokine levels after the three injections. The insert plot shows the IL-2 level and the Simpson index, D indicated by the blue line. D is a measure of diversity. An increase in D with time indicates the emergence of different epitope-specific dominant clones of T-cells. The smaller the D value, the lower the diversity.
Figure 3.
Immune simulation of the cytokine profile induced by three injections of TB-MEVA-1 given 28 days apart. The main plot shows different cytokine levels after the three injections. The insert plot shows the IL-2 level and the Simpson index, D indicated by the blue line. D is a measure of diversity. An increase in D with time indicates the emergence of different epitope-specific dominant clones of T-cells. The smaller the D value, the lower the diversity.
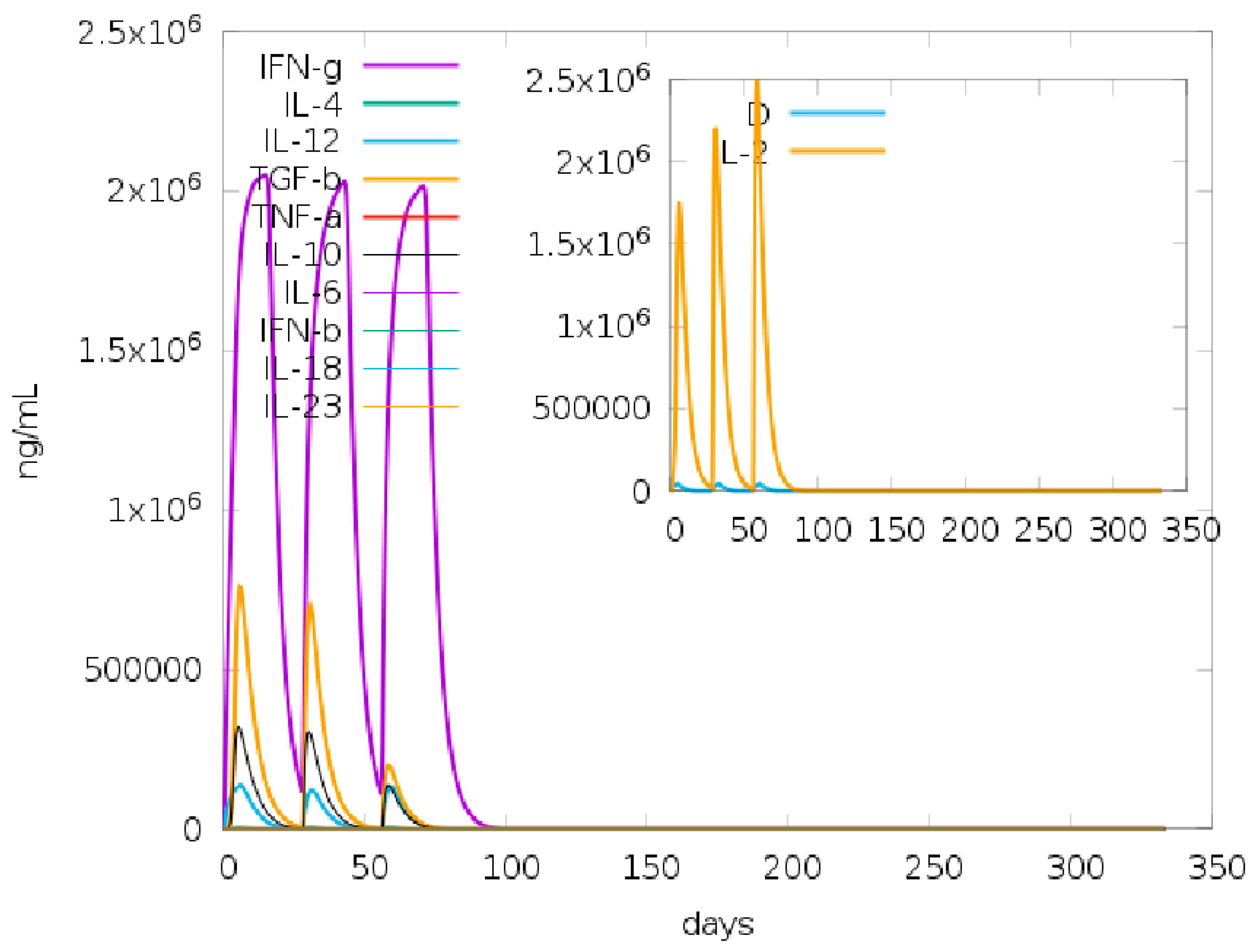
Figure 4.
Protein modeling, refinement, and validation. (A) The initial 3D model of the multi-epitope vaccine candidate predicted on the ColabFold server. (B) Superimposition of the refined 3D structure (colored) on the initial ‘crude model’ (gray) by the GalaxyRefine server. Validation of the refined model showing (C) Ramachandran plot analysis with 97.1% of residues in favoured regions (A, B, L), 2.1% in allowed regions (a, b, l, p and ~a, ~b, ~l, ~p), and 0.2% (red circles) of protein residues in favored, allowed, and disallowed (outlier) regions respectively, and (D) ProSA-web, giving a Z-score of -4.92.
Figure 4.
Protein modeling, refinement, and validation. (A) The initial 3D model of the multi-epitope vaccine candidate predicted on the ColabFold server. (B) Superimposition of the refined 3D structure (colored) on the initial ‘crude model’ (gray) by the GalaxyRefine server. Validation of the refined model showing (C) Ramachandran plot analysis with 97.1% of residues in favoured regions (A, B, L), 2.1% in allowed regions (a, b, l, p and ~a, ~b, ~l, ~p), and 0.2% (red circles) of protein residues in favored, allowed, and disallowed (outlier) regions respectively, and (D) ProSA-web, giving a Z-score of -4.92.
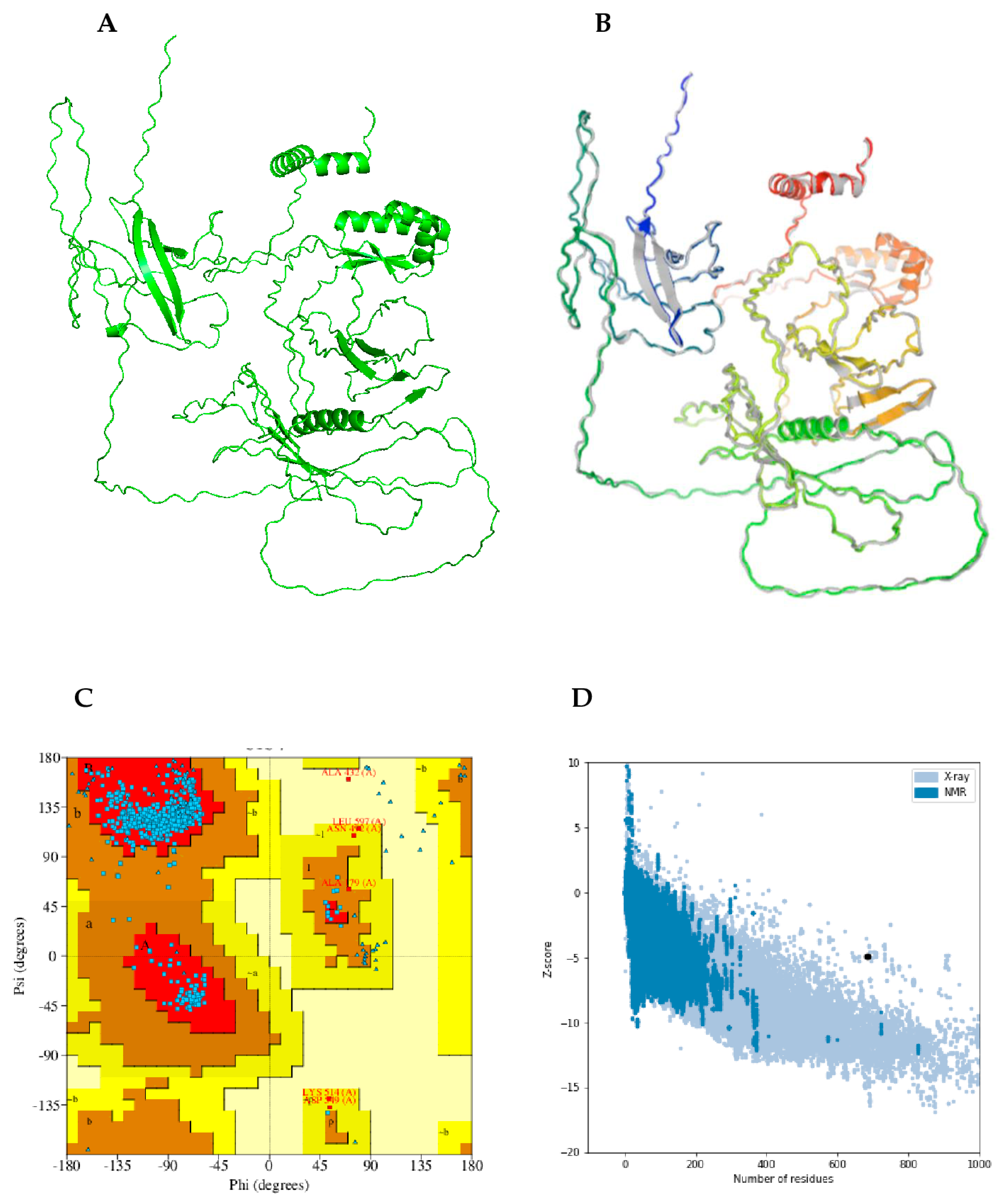
Figure 5.
Protein-protein interaction pocket prediction and molecular docking of vaccine candidate with the TLR4 receptor. (A) The largest predicted PPI pocket located at the interface of the designed chimeric vaccine candidate (red), with a surface area of 29113.275 Å2 and a surface volume of 129803.080 Å3. (B) The docked complex of the designed vaccine candidate (red) with the Toll-like receptor 4 chain (green). (C) Interaction network of the vaccine candidate and TLR4, respectively; hydrogen bonding interactions are shown in blue. Non-bonded interactions and salt bridges are shown in orange. The positive, negative, neutral, aliphatic, and aromatic charged residues are shown in blue, red, green, grey, and purple colors, respectively.
Figure 5.
Protein-protein interaction pocket prediction and molecular docking of vaccine candidate with the TLR4 receptor. (A) The largest predicted PPI pocket located at the interface of the designed chimeric vaccine candidate (red), with a surface area of 29113.275 Å2 and a surface volume of 129803.080 Å3. (B) The docked complex of the designed vaccine candidate (red) with the Toll-like receptor 4 chain (green). (C) Interaction network of the vaccine candidate and TLR4, respectively; hydrogen bonding interactions are shown in blue. Non-bonded interactions and salt bridges are shown in orange. The positive, negative, neutral, aliphatic, and aromatic charged residues are shown in blue, red, green, grey, and purple colors, respectively.
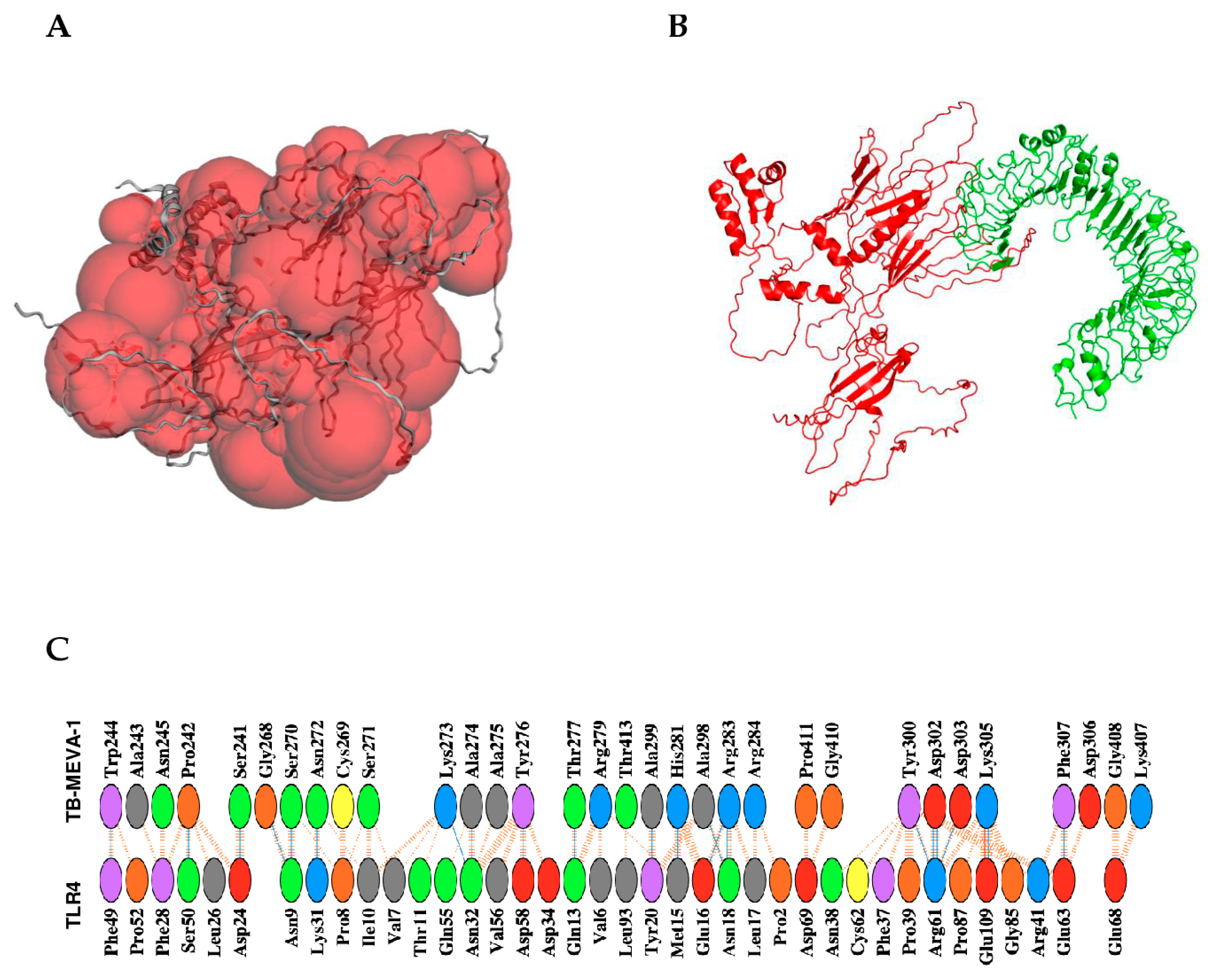
Figure 6.
The molecular dynamics simulation of the vaccine candidate-TLR4 docked complex. (A) The main-chain deformability of the TBMEVA-1-TLR4 complex. (B) The B-factor quantifies the uncertainty of each atom. (C) The eigenvalue specifies the motion stiffness linked to each normal mode. (D) The variance map is associated with individual (red) and cumulative (green) variances. (E) The covariance graph between pairs of residues shows correlated (red), uncorrelated (white), or anti-correlated (blue) mobility of the pairs. (F) The elastic network model describes the pairs of atoms connected by springs, and the darker greys indicate the stiffness of the springs.
Figure 6.
The molecular dynamics simulation of the vaccine candidate-TLR4 docked complex. (A) The main-chain deformability of the TBMEVA-1-TLR4 complex. (B) The B-factor quantifies the uncertainty of each atom. (C) The eigenvalue specifies the motion stiffness linked to each normal mode. (D) The variance map is associated with individual (red) and cumulative (green) variances. (E) The covariance graph between pairs of residues shows correlated (red), uncorrelated (white), or anti-correlated (blue) mobility of the pairs. (F) The elastic network model describes the pairs of atoms connected by springs, and the darker greys indicate the stiffness of the springs.
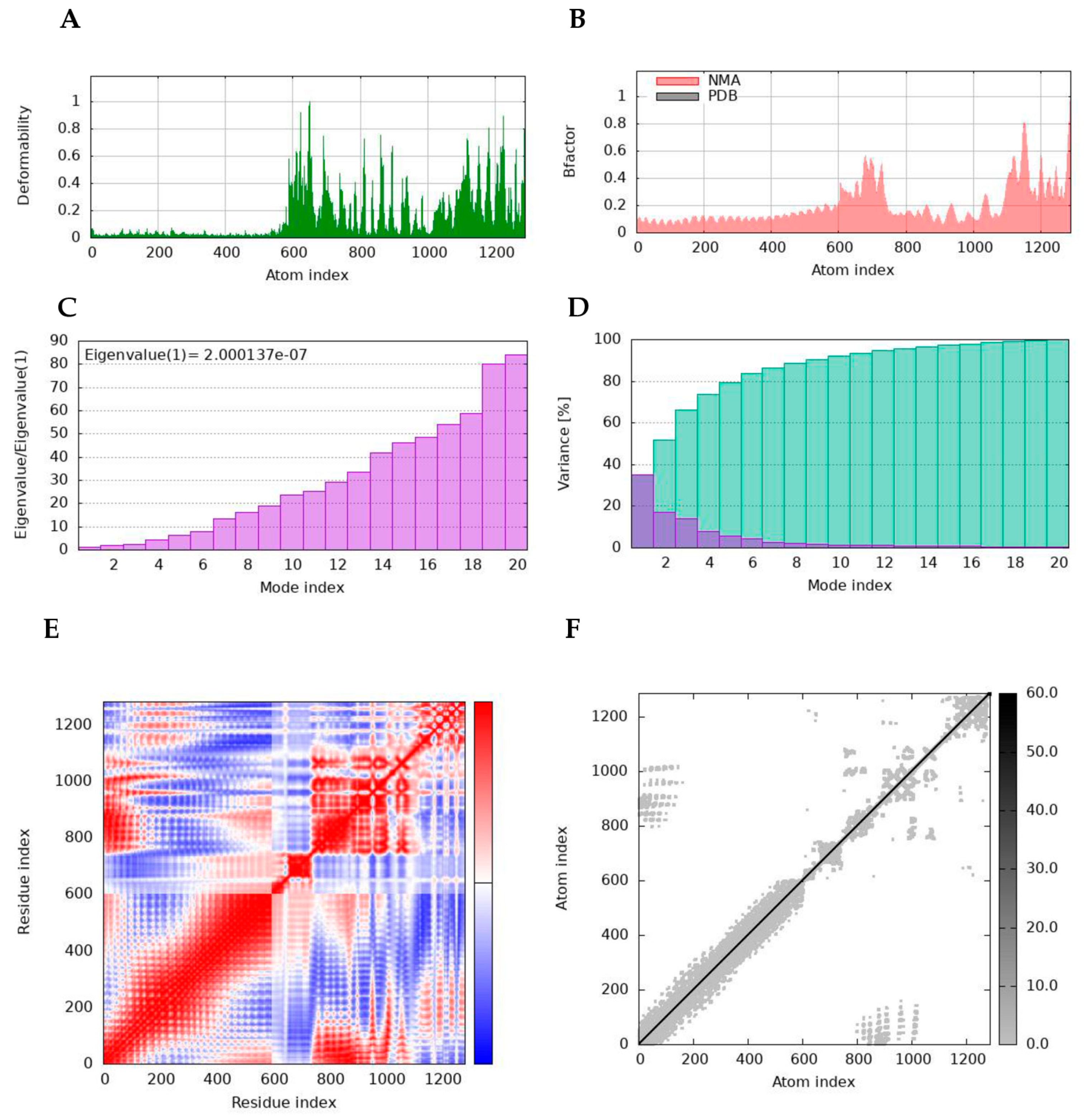
Figure 7.
In silico simulation of immune response induced by the designed vaccine candidate: (A) Antigen and immunoglobulins, (B) B cell population per state, (C) B cell population, (D) TH cell population, (E) TH cell population per state, (F) TC cell population per state, (G) MA population per state, (H) DC population per state, and (I) NK cell population.
Figure 7.
In silico simulation of immune response induced by the designed vaccine candidate: (A) Antigen and immunoglobulins, (B) B cell population per state, (C) B cell population, (D) TH cell population, (E) TH cell population per state, (F) TC cell population per state, (G) MA population per state, (H) DC population per state, and (I) NK cell population.
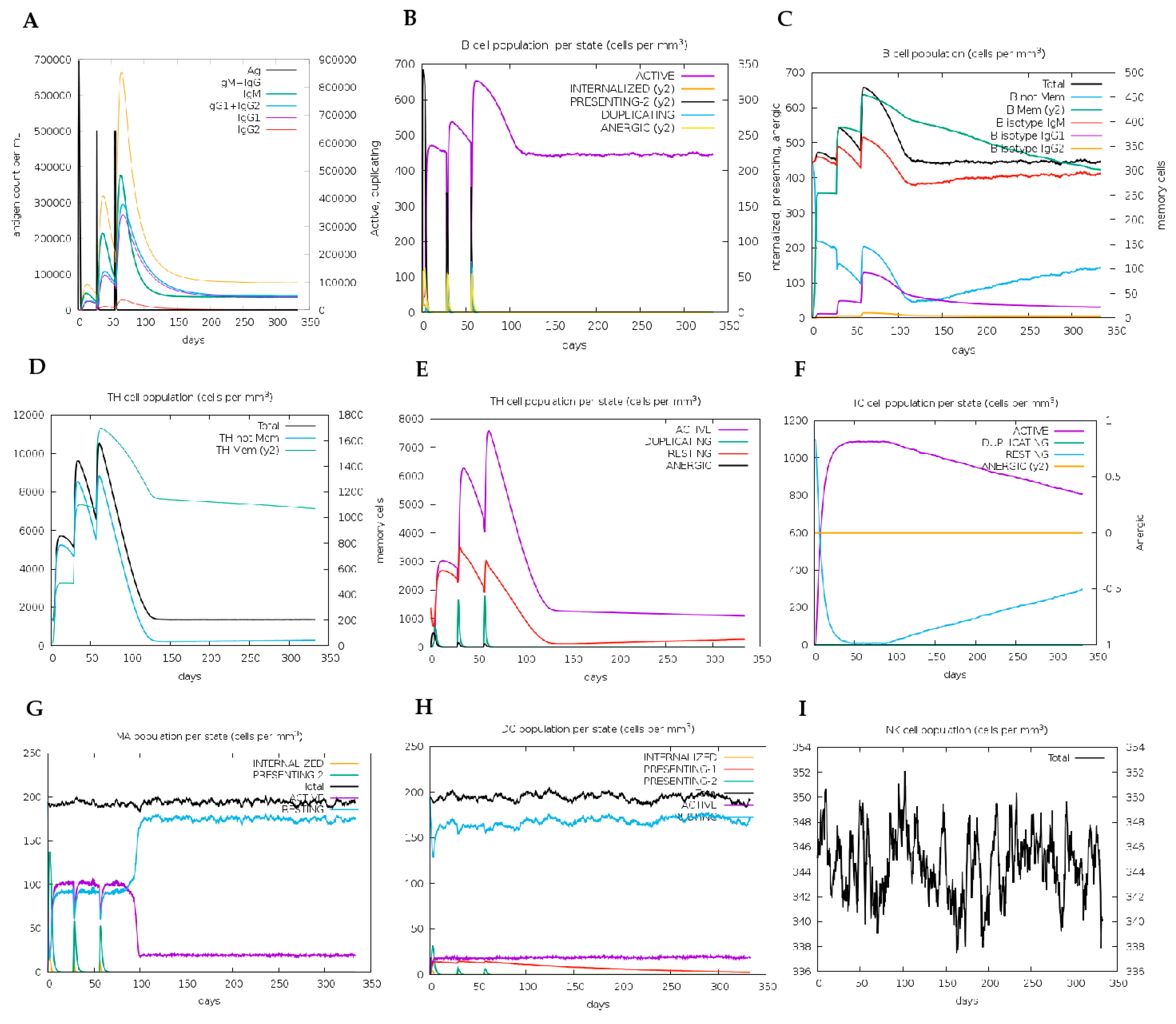
Figure 8.
In silico restriction cloning of the final vaccine sequence into the pET30a (+) expression vector where the red part represents the gene coding for the vaccine and the black circle represents the vector backbone. The His-tag is located at the Carboxy-terminal end.
Figure 8.
In silico restriction cloning of the final vaccine sequence into the pET30a (+) expression vector where the red part represents the gene coding for the vaccine and the black circle represents the vector backbone. The His-tag is located at the Carboxy-terminal end.
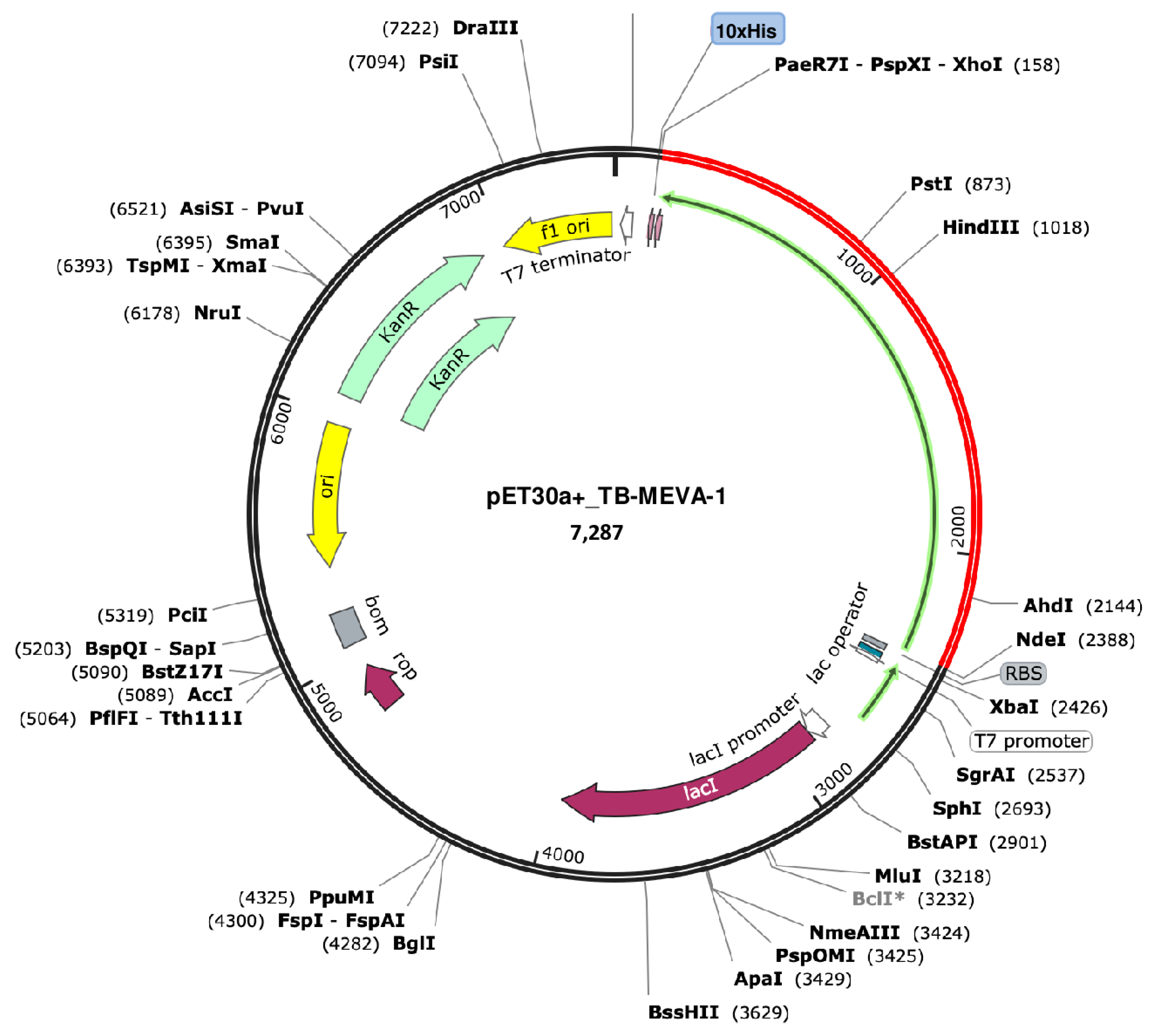
Figure 9.
Schematic presentation of the final multi-epitope vaccine antigen, TB-MEV-1. The 683-amino acid long polypeptide sequence containing a built-in adjuvant (green) at the N-terminal linked with the multi-epitope sequence through an EAAAK linker (cyan) to the PADRE and TAT peptides which are linked through GGGS linkers. The CTL, HTL epitopes, and linear-B cell (high-scoring) epitopes predicted to be antigenic are merged by AAY (yellow), GPGPG (orange), and KK linkers (blue), respectively). A 10x-His tag is added at the C-terminal for purification and identification purposes, separated from the peptide epitopes using the HRV 3C Protease cleavage sequence.
Figure 9.
Schematic presentation of the final multi-epitope vaccine antigen, TB-MEV-1. The 683-amino acid long polypeptide sequence containing a built-in adjuvant (green) at the N-terminal linked with the multi-epitope sequence through an EAAAK linker (cyan) to the PADRE and TAT peptides which are linked through GGGS linkers. The CTL, HTL epitopes, and linear-B cell (high-scoring) epitopes predicted to be antigenic are merged by AAY (yellow), GPGPG (orange), and KK linkers (blue), respectively). A 10x-His tag is added at the C-terminal for purification and identification purposes, separated from the peptide epitopes using the HRV 3C Protease cleavage sequence.
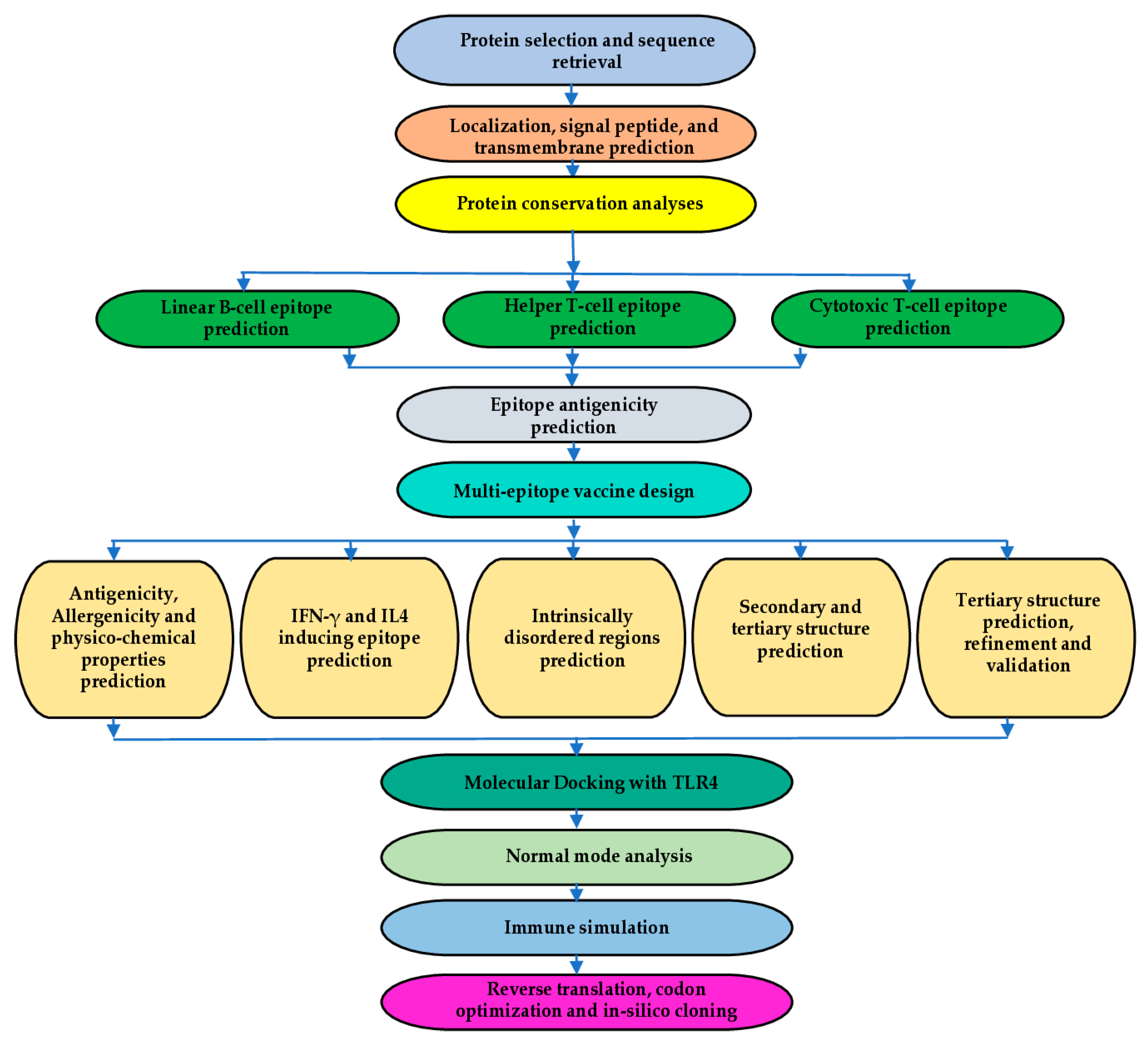

Table 1.
Protein localization prediction.
| Protein | Localization | |
| DeepLocPro-1.0 | TBpred | |
| P9WK45 | Cytoplasmic membrane | Protein attached to membrane by lipid anchor |
| P9WNF3 | Cytoplasmic membrane | Secreted protein |
| P9WK65 | Cytoplasmic membrane and cell surface | Protein attached to membrane by lipid anchor |
| A5TZX4 | Cytoplasmic membrane | Integral membrane protein |
| I6Y3P1 | Cytoplasmic membrane | Cytoplasmic protein |
| P9WGT7 | Cytoplasmic membrane | Protein attached to membrane by lipid anchor |
| P9WK61 | Cytoplasmic membrane | Protein attached to membrane by lipid anchor |
Table 2.
Conservation of target proteins in selected Mycobacterium species.
| Protein | Percentage Identity (%) | |||||||
| M. decipiens | M. leprae | M. lacus | M. gordonae | M. asiaticum | M. bovis | M. riyadhense | M. pseudokansasii | |
| P9WK45 | 91.5 | 68.1 | 81.0 | 70.6 | 72.5 | 100 | 77.5 | 81.8 |
| P9WNF3 | 86.0 | Not found | 83.3 | 77.2 | 75.0 | 100 | 65.8 | 75.0 |
| P9WK65 | 71.6 | 76.4 | 75.9 | 66.4 | 30.9 | 100 | 67.3 | 62.8 |
| A5TZX4 | 84.3 | 61.2 | 37.7 | 63.5 | 60.5 | 99.8 | 62.0 | 60.0 |
| I6Y3P1 | 83.1 | Not found | 85.6 | 79.9 | 79.7 | 100 | 85.3 | 79.1 |
| P9WGT7 | 91.1 | 77.6 | 85.1 | 81.5 | 80.5 | 100 | 84.9 | 82.5 |
| P9WK61 | 87.6 | 43.0 | 81.1 | 80.5 | 69.8 | 100 | 85.7 | 82.5 |
Table 3.
Predicted epitopes selected for the multi-epitope vaccine candidate design.
| Protein | LBL | CTL | HTL |
| I6Y3P1 | GGLNSLPLPGTAGHGE | TTFDLTLRR | NDDRKDFVTSLQLLLTFPFPNFGIKQA |
| A5TZX4 | LKSGDTIGLKNSSAYP | ATFDVTLSA WIATLTLEL KSGDTIGLK |
VRDISLRNWIATLTLEL |
| P9WGT7 | TYEIVCSKYPDSQVGT EGARGNDGTSAAAKNTPGSITYN LQSTIGAGQSGLGDNG |
ATTGQASAK NLDGPTLAK SPAWNLPVV |
GNDDNVTGGGATTGQASAKV |
| P9WK65 | DVDVRANPLAAKGVCT YNDEQGVPFRVQGDNI |
SPTASDPAL | |
| P9WNF3 | TYHVIAGQASPSRIDG | TLQGADLTV KLNPDVNLV FLAGCSSTK TLTSALSGK |
INVQAKPAAAASLAAIAIAFLAGCSSTK |
| P9WK61 | GSVVCTTAAGNVNIAI | NVNGVTLGY GLSGCSSNK |
DGKDQNVTGSVVCTTAAGNV TTGSGETTTAAGTTASPGAASGPK |
| P9WK45 | IPGLSLKTL TPRRHCRRI |
ETGDHQLAQAQLDRGSGNS GQNTIRISGKVSAQAVNQ |
Overlapping sequences are underlined.
Table 4.
Predicted immunoglobulin (Ig) class induction by linear B-epitopes.
| Predicted Linear B-Cell Epitope | Predicted epitope probability (%) | Predicted Antibody Class | |||
| IgG | IgE | IgA | IgM | ||
| GGLNSLPLPGTAGHGE | 66 | + | - | - | - |
| TYEIVCSKYPDSQVGT | 66 | + | - | - | - |
| LKSGDTIGLKNSSAYP | 100 | + | - | + | - |
| EGARGNDGTSAAAKNTPGSITYN | 100 | + | - | - | - |
| DVDVRANPLAAKGVCT | 66 | + | - | + | - |
| LQSTIGAGQSGLGDNG | 100 | + | - | - | - |
| TYHVIAGQASPSRIDG | 66 | + | - | - | - |
| YNDEQGVPFRVQGDNI | 100 | + | - | - | - |
| GSVVCTTAAGNVNIAI | 66 | + | - | - | - |
+: Epitope/peptide binds to the respective antibody class. -: Epitope/peptide does not bind to the respective antibody class.
Table 5.
The physicochemical properties of the designed chimeric vaccine.
| Property | Measurement |
| Number of Amino Acids | 683 |
| Molecular Weight | 69.7kDa |
| Formula | C3060H4917N885O959S8 |
| Theoretical pI | 9.36 |
| Instability Index | 27.03 |
| Aliphatic Index (AI) | -0.295 |
| Grand Average of Hydropathicity (GRAVY) | 76.31 |
| Solubility on expression (DeepSoluE) | 0.4218 |
| Solubility on expression (NetSolP 1.0) | 0.4811 |
Table 6.
The antigenicity, allergenicity, and toxigenicity of the designed vaccine candidate.
| Property | Measurement | Remark |
| Antigenicity (VaxiJen v2.0) | 1.1314 | Antigenic |
| Antigenicity (ANTIGENpro) | 0.9218 | Antigenic |
| Antigenicity (VaxiDL) | 96.66% | Antigenic |
| Allergenicity (AllerTOP v2.0) | Probable non-allergen | Probable non-allergen |
| Allergenicity (AllergenFP 2.0) | Probable non-allergen | Probable non-allergen |
| Toxicity (ToxinPred) | Non-toxin | Non-toxigenic |
| Toxicity (ToxDL) | 0.0001 | Non-toxigenic |
Table 7.
Number of predicted peptides binding to mouse H2 class II alleles.
| Allele | H-2-IAb | H-2-IAd | H-2-IAk | H-2-IAs | H-2-IAu | H-2-IEd | H-2-IEk |
| Number of epitopes | 78 | 32 | 9 | 22 | 24 | 3 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



