Submitted:
24 October 2025
Posted:
29 October 2025
You are already at the latest version
Abstract
The current study explored the martensite structures of Fe3Pt alloys, specifically Cmmm-Fe3Pt, P63/mmc-Fe3Pt, P4/mmm-Fe3Pt, and R\( \overline{3} \)m-Fe3Pt, aiming to provide a comprehensive understanding of the mechanisms that govern their physical and chemical properties. We have focused on their structural, thermodynamical, magnetic, electronic, and mechanical characteristics, utilizing the Density Functional Theory (DFT) technique. Our study revealed that in addition to the previously reported austenitic cubic Pm\( \overline{3} \)m-Fe3Pt and martensite tetragonal I4/mmm-Fe3Pt with L12 structure, there exist additional Fe3Pt phases that exhibit excellent structural, thermodynamic, magnetic and mechanical properties. The calculated enthalpies of formation were found to be negative and less than -0.39 eV in all the structures considered, indicating thermodynamic stability and formation under experimental synthetic conditions. Moreover, the computed magnetic moments are in the range 2.94 to 3.04 μB, which is relatively comparable to 3.24 μB of the widely reported Pm\( \overline{3} \)m-Fe3Pt alloy. The analysis of the electronic structure also revealed strong magnetism due to the presence of asymmetry in the spin up and down states of the density of states (DOS) plots. To determine the mechanical response of Fe3Pt structures under loading conditions, we computed the independent elastic constants, macroscopic properties and stress-strain relationship under hydrostatic stress. All four phases, but the hypothetical P63/mmc-Fe3Pt showed excellent mechanical stability at ambient conditions and exceptional hardness and resistance to compression in the elastic region 0% ≤ strain ≤ 10%. This evidence is provided by satisfying the Born necessary stability conditions, large bulk modulus and a strong linear relationship fit (R2) of greater than 0.94.
Keywords:
martensite Fe3Pt
; thermodynamic stability
; magnetic moments
; mechanical stability
; spin asymmetry
1. Introduction
The exploration of bimetallic Fe-Pt alloys has long garnered significant attention due to their intriguing properties and promising applications in various fields such as ultra-high density data storage, spintronics, catalysis, and nanotechnology [1, 2, 3, 4]. As indicated by their binary stability phase diagram [5], there have been three well-known and widely reported structures of Fe-Pt alloys which are chemically ordered. These include the tetragonal L10 -FePt phase which exhibits ferromagnetic ordering, and the face-centered cubic (FCC) phases FePt3 and Fe3Pt, both crystallizing in the space group with L12-type crystal symmetry. The latter two phases support both antiferromagnetic and ferromagnetic magnetic ordering [6]. These alloys have been previously investigated both experimentally and computationally and have been found to exhibit excellent thermodynamic, vibrational and mechanical stability. Moreover, they possess desirable magnetic properties due to large magnetic moments, large uniaxial magnetocrystalline anisotropy (MCA) energies originating from spin-orbit interactions and asymmetry of spin up and down in the electronic density of states [7, 8, 9]. Variations in crystallization and chemical composition significantly influence their magnetic behavior, electronic structure, and lattice dynamics. For instance, although both Fe and Pt contribute to the magnetic moments, Fe contributes approximately eight times more than Pt, thereby making the magnetic behavior predominantly dependent on the Fe content [10]. The -FePt phase crystallizes at temperatures below 1570 K in equiatomic Fe:Pt concentration, while the FCC structures crystallize around 25 % and 75 % Fe contents at temperatures below 1120 K and 1620 K, respectively [11]. The tetragonal L10 FePt shows one of the highest MCA energy ranging from to along the magnetically easy axis [001], making it a promising candidate to overcome superparamagnetic limit, which is the loss data due to thermally activated fluctuations of magnetization [12, 13]. It consists of alternating stacked atomic layers of Fe and Pt atoms along [100] direction similar to the AuCu structure, leading to a tetragonal distortion along the -axis. As per the previous communication, the cubic Fe3Pt and FePt3 alloys exhibit the highest MCA energies of (0.375 meV) and (0.055 meV) along the [100] easy axis [12]. In the Fe-rich Fe3Pt alloy, Fe atoms occupy the corner lattice positions, while Pt atoms are located at face-centered positions. Conversely, in the Pt-rich FePt3 alloy, Pt atoms occupy the corner positions, and Fe atoms are situated at the face-centered positions.
In addition, the L12 face-centered cubic (austenitic) Fe3Pt alloy is reported to exhibit strong magnetoelastic coupling and undergoes a thermoelastic martensitic transformation from the ordered L1₂ phase into low-temperature martensitic structures, which inherently depend on the degree of order of the L12 structure [14, 15]. One such martensite structure is the body-centered tetragonal (BCT) Fe3Pt phase, which crystallizes in the I4/mmm space group [16, 17]. The equilibrium cell parameters of the I4/mmm bulk structure as produced by X-ray diffraction are and [18]. It was found that the spin up Fe states in the density of states (DOS) in this martensite structure are situated below the Fermi level, while the spin down states overlaps above the Fermi, indicating a pronounced spin asymmetry. Consequently, this spin polarization results in relatively large magnetic moments of 2.61 and 2.82 for Fe1 and Fe2 atoms, respectively. Moreover, Pt atoms were also found to exhibit a finite magnetic moment of 0.45 , suggesting a degree of spin polarization induced by hybridization with neighboring Fe atoms [16]. Our previous communication further reported magnetic moments of 2.744 for Fe and 0.45 Pt along with the MCA energy of 1.364 meV along the [001] easy axis [12], which are higher than those of the austenitic Fe3Pt.
The structural models of other Fe3Pt martensite phases have long been proposed, however, their properties remain unstudied [19]. According to the Materials Explorer within the Materials Project database [20, 21], three additional martensitic phases of Fe3Pt are predicted to exist whose structural, thermodynamic, magnetic and mechanical properties are not yet fully explored. These correspond to the P4/mmm, , and R3m space groups as shown in Figure 1. The –Fe3Pt phase crystallizes at approximately 55 K in an orthorhombic lattice characterized by primitive lattice parameters as determined by XRD. The structure comprises two crystallographically distinct Fe sites. This orthorhombic structure is an intermediate phase, appearing between face-centered tetragonal (FCT1) () and FCT2 () [15]. The system considered in this current work is a conventional -centered (centering doubles the number of lattice points along one direction) orthorhombic Fe3Pt which is equivalent, but twice the volume and number of atoms (eight) as the primitive (four atoms) reported in literature. In the first site, Fe atoms adopt a two-coordinate geometry, bonded to two Fe and two Pt atoms, whereas the second Fe site exhibits eightfold coordination with neighboring Fe atoms. The Pt atoms display a four-coordinate geometry, each bonded to four Fe atoms, completing the orthorhombic network. The –Fe3Pt phase exhibits a USi2-type structure and crystallizes in a body centered tetragonal (BCT) lattice. This phase forms through a thermoelastic transformation. During the formation, the tetragonality ratio shifts from 1 to 0.79 at the transformation temperature, signifying a discontinuous lattice distortion characteristic of a first-order martensitic transformation [15, 17]. The structure contains two inequivalent Fe sites. Each Fe atom is coordinated by eight Fe and four Pt atoms, forming distorted FeFe8Pt4 cuboctahedra. These polyhedra interconnect through corner-, edge-, and face-sharing with neighboring FeFe8Pt4 and PtFe12 units, forming a highly integrated three-dimensional framework. The Pt atoms are twelvefold coordinated by Fe atoms, generating PtFe12 cuboctahedra that are similarly interconnected through shared corners, edges, and faces with surrounding FeFe8Pt4 polyhedra. The –Fe3Pt phase crystallizes in a rhombohedral centered hexagonal or trigonal lattice. Each Fe atom is surrounded by nine Fe and three Pt atoms, forming distorted FeFe9Pt3 cuboctahedra that share corners, edges, and faces with adjacent FeFe9Pt3 and PtFe6Pt6 polyhedra. The Pt atoms are coordinated by six Fe and six Pt atoms, creating PtFe6Pt6 cuboctahedra that link through corner-, edge-, and face-sharing, producing a robust trigonal framework. Lastly, the -Mn3Ir structure served as a prototype for Fe3Pt, since Fe and Mn as well as Pt and Ir are chemically analogous neighboring transition metals with comparable atomic radii and electronic configurations. Such substitution preserves the symmetry and coordination environment, enabling reliable DFT modeling of synthetical Fe–Pt system within the same crystallographic framework. This is beta Cu3Ti and Mn3Ga type structure which crystallizes in the hexagonal lattice with eight atoms in a primitive cell [22, 23]. Fe is bonded to four equivalent Pt atoms to form a mixture of distorted corner, edge, and face-sharing FePt4 cuboctahedra. Pt is bonded to twelve equivalent Fe atoms to form a mixture of corner and face-sharing PtFe12 cuboctahedra.
The magnetic, thermodynamic and mechanical properties of these four martensite phases are yet to be systematically explored either experimentally or theoretically, which limits a comprehensive understanding of the structure–property relationships and the potential functional applicability in advanced magnetic and electronic technologies. Experimental exploration of these alloys may be challenging due to the complexities associated with detecting such phases arising from dynamical effects on diffraction reflections [16]. However, the development of density functional theory (DFT)-based first-principles methods enables theoretical exploration and optimization of L12 bimetallic alloys by accurately calculating the interatomic forces acting on the nuclei [24, 25, 26]. The current study focuses on the structural, thermodynamic, magnetic, electronic and mechanical properties of the four martensite Fe3Pt bimetallic alloys at 0K to assess their stability and deduce their suitability as magnetic materials. We have employed the density functional theory (DFT) quantum mechanical method embedded in the CASTEP code to perform first-principles quantum mechanical calculations on Fe3Pt alloys. Our computational findings revealed that three martensite Fe3Pt alloys exhibit excellent thermodynamic and mechanical stability and magnetic moments comparable to those of the extensively studied Fe-Pt alloys.
2. Computational Approach
The present study utilized the Cambridge Serial Total Energy Package (CASTEP) [27] which employs the plane-wave density functional theory (DFT) plane-wave pseudopotential method to perform first-principles calculations on Fe3Pt alloys. The Generalized Gradient Approximation (GGA) and Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional were preferred since they account for inhomogeneities in the electron distribution [28, 29]. To determine the minimum optimal plane-wave cut-off energies that converge the total energy, single-point energy calculations were performed for all the systems at several increasing cut-off values without relaxing the atomic positions and cell parameters. This was done until the total energy between successive steps was below 1 meV. The cut-off energies 400 eV, 300 eV, 400 eV and 300 eV were respectively found sufficiently to converge the total energies of , , and systems. Moreover, the customized Monkhorst-Pack [30] k-points grid size of 9x7x4, 4x4x6, 5x5x6 and 8x8x1 for self-consistent field (SCF) calculations were applied for Brillouin zone sampling. Before subsequent calculations of electronic structure and elastic properties, full geometry optimization was performed to obtain the arrangement of atoms and cell parameters corresponding to the minimum total energy and zero net forces on atoms, i.e. the ground-state structures. Both cell volume and shape were allowed to change until the final energy between two iterations was less than 5.7 meV. To allow separation of majority and minority spin projections, the spin density was treated by the collinear spin polarization using formal spin as initial. The low-memory Brayden-Fletcher-Goldfarb-Shanno (LBFGS) algorithm [31] that scales linearly in memory and performs better for large systems was preferred for cell optimization over the normal BFGS [32] that scales as a square of the system size for cell. Furthermore, the LBFGS algorithm employs a general sparse preconditioner that enhances the efficiency of geometry optimization, achieving up to a twofold speed increase for small systems and up to tenfold for large systems [33]. Subsequent computation of density of states and elastic constants were done using the equilibrium structures. Calculation of the elastic properties was performed using the Elastic Constants task with a maximum strain amplitude of 0.003, which is adequately low for the material to stay in the linear elasticity. The calculation of electronic band structure and density of states was done with and band energy tolerance of eV.
3. Results and Discussion
3.1. Structural Properties
The equilibrium cell parameters, densities and enthalpies of formation of the four martensite Fe3Pt structure as shown in Table 1 were obtained by performing full geometry optimization. The experimental volume for the orthorhombic -Fe3Pt and lattice parameters of the previously reported austenitic -Fe3Pt are given in parenthesis to validate DFT calculations against existing experimental data. The calculated lattice parameters of -Fe3Pt are more than 99 % in agreement with experimental data [34] and previous calculations [8], while the optimized volume of -Fe3Pt in 96 % in agreement with the XRD results [15]. These agreements indicate that the employed approach correctly predicts the ground state properties of Fe3Pt alloys. The conventional hexagonal -Fe3Pt structure shows the largest unit cell volume and smallest density corresponding to weaker interatomic interactions. The enthalpies of formation () were computed according to equation 1,
where is the total ground state energy of Fe3Pt formula unit and the elemental energies of Fe and Pt atoms. Negative enthalpies of formation indicate the release of energy when forming a compound (exothermic reaction) and thus thermodynamic stability of the compounds in relation to its constituent elements, i.e. . Our DFT calculations predict that all the four martensite Fe3Pt alloys are thermodynamically stable due to negative enthalpies of formation. Expectedly, the enthalpy of formation values of austenitic and martensite , , and are similar since they have the same stoichiometry and elemental reference state.
3.2. Magnetic Properties
Table 2 presents the calculated spin polarized magnetic moments and charge distribution characteristics of the four considered Fe3Pt crystal structures derived from Mulliken atomic population analysis. The magnetic moments are calculated as the difference between spin up and spin down () charge densities. Moreover, the average of Fe1 and Fe2 were taken when quantifying the total spin moment per formula unit. The calculated total moments for , , and are 3.01 , 3.04 , 2.94 and 2.97 , respectively. These values are comparable with those reported for other Fe-Pt alloys and bimetallic systems of similar composition [10, 35, 36]. For all space groups, the Fe atoms exhibit substantial local magnetic moments compared to Pt, indicative of strong ferromagnetic ordering within the Fe sublattice and large spin–orbit effects from Pt. Moreover, in all the structures, the spin up () for both Fe1 and Fe2 are more densely than the spin down () revealing pronounced spin asymmetry in the electronic density of states and confirming strong magnetic polarization within the system.
Our DFT calculations reveal that the orthorhombic space group possesses the largest Fe1 magnetic moment (2.98 ), followed by the hexagonal (), while the trigonal and tetragonal display relatively decreased moment values of 2.87 and 2.83 , respectively. This may be attributed to the increase of restrictions on independent lattice parameters and angles, i.e. symmetry from orthorhombic to tetragonal, which slightly decreases the exchange splitting, consistent with increased Fe–Pt orbital hybridization. The smaller variation in Fe2 moments indicates comparable local environments across the structures, except for the P6₃/mmc phase, where Fe occupies only one unique crystallographic site due to higher symmetry. The Pt atoms exhibit relatively small induced magnetic moments, ranging from 0.12 to 0.30 , arising from Fe–Pt hybridisation and spin polarization of Pt 5d orbitals. -Fe3Pt shows the largest Pt moment (0.30 ), whereas the phase displays the smallest (0.12 ).
The Hirshfeld partitioning of electron density into atomic contributions further confirms the dominance of Fe atoms in the overall magnetic behavior. The Fe1 moments range from 2.72 to 2.83 , while Fe2 moments vary between 2.45 to 2.68 In contrast, Pt contributes only 0.35-0.57 , consistent with the induced magnetization mechanism. The structure again exhibits the highest overall Fe magnetization, while the phase shows a marginally reduced value, reflecting differences in local coordination and exchange interactions.
Hirshfeld charge analysis indicates notable charge redistribution between Fe and Pt atoms. Fe atoms possess positive charges (+0.07 to +0.27 e), signifying electron donation, while Pt atoms carry negative charges (−0.46 to −0.79 e), confirming their role as electron acceptors. The phase shows the highest charge transfer (-0.79 e), suggesting stronger Fe–Pt bonding and more effective hybridization.
3.3. Electronic Structure
Figure 2 presents the electronic total and orbital-projected spin polarized density of states (DOS) for the martensitic Fe3Pt, with the Fermi level set as the origin of the energy scale. As anticipated, all the investigated Fe3Pt alloys display metallic behavior, as indicated by the finite density of states at the Fermi level arising from the overlap of valence and conduction bands, most prominently in the spin-down states. In all total DOS plots, the spin-up states are predominantly located below the Fermi level, whereas the spin-down states extend into the conduction band above the Fermi level, reflecting a pronounced spin asymmetry which indicates spin polarization and gives rise to substantial magnetic moments and strong ferromagnetic character consistent with previous experimental findings on Fe3Pt thin films [37]. Notably, the phase exhibits a further strong asymmetry (enhanced spin polarization) in the valence states between −4 eV and −2 eV, consistent with its highest calculated magnetic moment. There exist shallow pseudo-gaps near the Fermi level, indicating enhanced Fe-Pt hybridization and electronic stability for all phases.
The orbital-projected DOS shows a dominant contribution from Fe 3d states in the total plots, with minor contribution from the Pt 5d states hybridizing with Fe d-bands. Moreover, there exist strong spin asymmetry in Fe states and weaker in Pt. This strong exchange splitting reflects localized Fe moments stabilized by the low structural symmetry. Furthermore, the strong spin polarization arises from the Fe 3d orbitals, while Pt-5d orbitals show small but non-zero induced moments, contributing slightly to the total magnetization.
The electronic properties of these systems were further analyzed using the band structure along high symmetry direction in the Brillouin zone as presented in Figure 3. All four structures exhibit metallic characteristics, as evidenced by the continuous overlap of energy bands at the Fermi level and the finite DOS at and transitioning of electron wave highest to lowest energy levels through the Brillouin zone [38]. Moreover, there exists a clear spin splitting: red and blue bands diverge near the Fermi level. Interestingly, the phase shows almost similar spin up (red) and spin down (blue) bands, indicating nearly degenerate spins.
3.4. Mechanical Properties
Table 3 shows the calculated independent elastic constants () and derived macroscopic properties of the orthorhombic, hexagonal, tetragonal and trigonal Fe3Pt alloys at 0 K. CASTEP uses the generalized Hooke’s law shown in equation 2) to fit the linear relationship between stress and strain components in the elastic regime to compute the independent elastic constants.
where and are the Voigt notation components of the stress and strain tensor, respectively. Calculation and elastic constants and macroscopic mechanical properties are crucial in understanding the intrinsic mechanical stability, bonding nature and deformation behavior of solid-state materials. For any solid-state crystal lattice to be considered mechanically stable, certain stability conditions as postulated by Born must be satisfied [39].
Hexagonal crystals possess five independent elastic constants, while the tetragonal contain six due to the added . Since the Laue classes of the hexagonal and tetragonal crystal lattice have similar elastic matric, their Born necessary stability conditions are also similar and are given as [40]:
The condition is redundant for hexagonal crystal lattices. The trigonal or rhombohedral lattice has 6 independent elastic constants and like the hexagonal one dependent. The following four conditions are sufficient for stability:
The orthorhombic crystal lattice with lower symmetry contains larger number (nine) independent elastic constants and the born stability conditions are:
The independent elastic constants of the orthorhombic , tetragonal and trigonal structures are positive and obey all the necessary Born stability conditions, which indicates mechanical stability. In contrast, the phase exhibits anomalous elastic constants, due to a negative value which violates the Born stability criteria. Such behavior signifies mechanical instability, suggesting that this hypothetical hexagonal structure cannot exist under ambient conditions and would spontaneously transform into a lower-symmetry, more stable phase with space group [11, 41]. The elastic stiffness coefficients , , and for the -Fe3Pt are relatively high, above 224.53 GPa, which signifies a strong resistance to axial deformation along all crystallographic directions. Furthermore, the off-diagonal and are also large, indicating considerable interplanar coupling, while the comparatively smaller shear constant (36.270 GPa) reveals moderate shear anisotropy. The -Fe3Pt structure shows moderately high values of and , indicating fair axial stiffness. However, its small value (12.11 GPa) reflects weak shear resistance, implying significant anisotropy in its mechanical response. The -Fe3Pt phase displays intermediate stiffness with and , while the relatively small off-diagonal value (5.36 GPa) indicates limited shear coupling.
The corresponding macroscopic mechanical properties representing a polycrystalline aggregate using Voigt–Reuss–Hill (VRH) averaging schemes were computed from the independent elastic constants to determined deformation characteristics [42]. Table 4 summarizes the expressions and descriptions of the calculated macroscopic properties.
The Pphase shows large bulk modulus and shear modulus further confirming its robustness and greater resistance to compression, while the relatively high Young’s modulus indicates good stiffness and metallic bonding characteristics. Moreover, the Pugh () ratio greater than the critical value 1.5 of ductility and brittleness and Poisson’s ratio () classifies this phase as ductile, while the positive anisotropy factor (A = 1.97) that diverge from unity suggests moderate elastic anisotropy.
The calculated Poisson’s ratio ranges from 0.33 to 0.40, except for are within the error limit (0.320.09) and similar to recorded in most alloys [11, 43]. Consistent with the negative , the hexagonal , shows a negative shear modulus and Pugh ration. Its Poisson ration is 0.76 which is beyond the metallic limit (~0.5), indicating that this phase exhibits extreme lateral expansion when compressed (or contraction when stretched), very high ductility, low shear resistance, and strong anisotropic bonding or magnetostrictive behavior. This phase likely reflects magnetoelastic softening, not purely mechanical elasticity. The bulk modulus (179.09 GPa) of the phase is relatively larger than the shear (38.75 GPa), leading and larger Pugh ratio, confirming a ductile but less rigid nature relative to the orthorhombic phase. The moderate Poisson’s ratio (0.40) also supports a metallic and ductile bonding character. Similar behavior is observed for the .
To further analyze the mechanical properties of the martensite Fe3Pt alloys, we plotted stress vs strain profiles which represent the elastic response of crystal systems under applied strain, from which elastic constants and mechanical stability are evaluated. Figure 4 illustrates the calculated stress vs strain relationships for the , , , and phases of Fe3Pt. The , and structures exhibit smooth and well-defined linear elastic regions, 0% ≤ Strain ≤ 10% (red line) with strong correlations between the fitted and computed data ( and 0.970, respectively). This excellent agreement confirms excellent elastic behavior, numerical accuracy, mechanical stability with the elastic regime and validates the reliability of the derived elastic constants. Conversely, the hypothetical -Fe₃Pt phase displays a noticeable deviation from linearity even at small strains, reflected by a relatively low . The irregular stress response suggests internal atomic relaxations or numerical instabilities during deformation, which consistent with the observed negative elastic constant and shear modulus values. This behavior indicates that although predicted to be thermodynamically stable, the hypothetical –Fe3Pt phase is mechanically unstable and prone to soft-mode distortions under elastic perturbation. Therefore, the combination of stress vs strain analysis and correlation fitting provides a robust means of assessing the elastic consistency and mechanical resilience of crystal systems.
4. Conclusions
Density functional theory calculation on the martensite , , and Fe3Pt alloys were successfully carried out to determine the structural, thermodynamic, magnetic, electronic and mechanical properties. The negative enthalpies of formation showed that all the structures are thermodynamically stable. The substantial magnetic moments ranging from 2.94 to 3.04 and strong asymmetry of the spin up and down in the density of states plots, suggest strong magnetism and ferromagnetism behavior of the Fe3Pt alloys. Calculations of independent and macroscopic elastic constant predicted mechanical stability on the , and phases. However, the hypothetical phase possesses negative and shear modulus values, suggesting that it cannot exist under ambient conditions and would transform into a lower-symmetry and more stable phase. The observations collectively demonstrate that in addition to the -Fe₃Pt alloy, there exist other martensite , and phases which are thermodynamically and mechanically stable, highly magnetic, strongly ferromagnetic, ductile, and stiff, consistent with their martensitic character.
References
- R.L. Comstock, "Modern magnetic materials in data storage," J. Mater. Sci. Mater. Electron., vol. 13, p. 509, 2002.
- M.R. Visokay and R. Sinclair, "Direct formation of ordered CoPt and FePt compound thin films by sputtering," Appl. Phys. Lett., vol. 66, p. 1692, 1995.
- M. Ohtake, S. Ouchi, F. Kirino and M. Futamoto, "L10 ordered phase formation in FePt, FePd, CoPt, and CoPd alloy thin films epitaxially grown on MgO(001) single-crystal substrates," J. Appl. Phys., vol. 111, p. 07A708, 2012.
- R.A. Ristau, K. Barmak, L.H. Lewis, K.R. Coffey and J.K. Howard, "On the relationship of high coercivity and L10 ordered phase in CoPt and FePt thin films Available," J. Appl. Phys., vol. 86, p. 4527, 1999.
- T.B. Massalski, Binary Alloy Phase Diagrams, Ohio: ASM International, 1990, p. 1752.
- B. Wang; D. C. Berry; Y. Chiari; K. Barmak, "Experimental measurements of the heats of formation of Fe3Pt, FePt, and FePt3 using differential scanning calorimetry," J. Appl. Phys., vol. 110, p. 013903, 2011.
- N.L. Lethole and P. Mukumba, "Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys," Materials, vol. 17, p. 3879, 2024.
- M. Sternik, S. Couetm, J. Ła_zewski, P.T. Jochym, K. Parlinski and A. Vantomme, "Dynamical properties of ordered Fe-Pt alloys," J. Alloys Compd., vol. 651, p. 528, 2015.
- Y. Kota and A. Sakuma, "Relationship between magnetocrystalline anisotropy and orbital magnetic moment in L10-type ordered and disordered alloys," J. Phys. Soc. Jpn., vol. 81, p. 84705, 2012.
- J. Lyubina, I. Opahle, M. Richter, O. Gutfleisch, K.H. Müller, L. Schultz and O. Isnard, "Influence of composition and order on the magnetism of Fe–Pt alloys: Neutron powder diffraction and theory," Appl. Phys. Lett., vol. 89, p. 032506, 2006.
- N. Zotov and A. Ludwig, "First-principles calculations of the elastic constants of FeePt alloys," Intermetallics, vol. 16, p. 113, 2008.
- N. Lethole, P. N. Lethole, P. Ngoepe and H. Chauke, "Compositional Dependence of Magnetocrystalline Anisotropy, Magnetic Moments, and Energetic and Electronic Properties on Fe-Pt Alloys," Materials, vol. 15, p. 5679, 2022.
- T. Shima, K. Takanashi, Y.K. Takahashi and K. Hono, "Coercivity exceeding 100 kOe in epitaxially grown FePt sputtered films," Appl. Phys. Lett., vol. 85, p. 2571, 2004.
- S. Muto, R. Oshima and F.-E. Fujita, "Nucleation and growth in martensitic transformations of ordered Fe3Pt alloys," Metall.Trans., vol. 19, p. 2931, 1988.
- M. Yamamoto, T. Fukuda, T. Kakeshita and K. Takahashi, "Orthorhombic martensiteformedinL12-type Fe3Pt Invaralloy," J. Alloys Compd., vol. 577S, p. S503, 2013.
- Y. Nakata, "The Crystal Structure and Magnetic Properties of Fe3Pt Martensite Determined by First Principle Calculations," Mater. Trans., vol. 44, p. 1706, 2003.
- T. Fukuda and T. Kakeshita, "Lattice Softening in Fe3Pt Exhibiting Three Types of Martensitic Transformations," Metals, vol. 7, p. 156, 2017.
- T. Tadaki, Y. Nakata and K. Shimizu, "Anomalous reflections in low temperature electron diffraction from the thermoelastic martensite in a fully ordered Fe-Pt Alloy," Mater. Sci. Forum, vol. 169, p. 56, 1990.
- T. Tadaki, Y. Nakata and K. Shirnizu, "Occupancy Sites of Constituent Atoms and their Effects on the Martensitic Transformations in some Cu-Based and Ti-Ni-Based Ternary Alloys," J. Phys. IV, vol. 5, pp. C8-81, 1995.
- M.K. Horton et al., "Accelerated data-driven materials science with the Materials Project," Nat. Mater, vol. 24, p. 1522, 2025.
- Jain, S.P. Ong, G. Hautier, W. Chen, W.D. Richards, S. Dacek, S. Cholia; D. Gunter, D. Skinner, G. Ceder and K.A. Persson, "Commentary: The Materials Project: A materials genome approach to accelerating materials innovation," APL Mater., vol. 1, p. 011002, 2013. [Google Scholar]
- Z.H. Liu, Y.J. Zhang, G.D. Liu, B. Ding, E.K. Liu, H.M. Jafri, Z.P. Hou, W.H. Wang, X.Q. Ma and G.H. Wu, "Transition from Anomalous Hall Effect to Topological Hall Effect in Hexagonal Non-Collinear Magnet Mn3Ga," Sci. Rep., vol. 7, p. 515, 2017.
- K. Zellagui, M. Khedidji and H. Yousfi, "First-principles study of the structural, electronic, dielectric, and dynamical properties of gallium nitride in the graphite-like hexagonal P63/mmc phase," Theor. Chem. Acc., vol. 143, p. 79, 2024.
- B.O. Mnisi, E.M. Benecha and M.M. Tibane, "Density functional theory study of phase stability and electronic properties for L12 X3Ru and XRu3 alloys," Eur. Phys. J. B, vol. 98, p. 106, 2025.
- B. Thacker, T. Akhani, M.B. Solanki and R.N. Kharatmol, "Theoretical investigation in structural, electronic, vibrational, thermophysical, thermoelectric, and elastic properties of L12 and L12 FePd3 alloys by a DFT approach," Theor. Chem. Acc., vol. 144, p. 78, 2025.
- B. Thacker, M.B. Solanki, R. Kharatmol, Y.D. Kale and T. Akhani, "Exploring Structural, Electronic, Vibrational, and Thermophysical Properties of Fe–Pt, Fe3–Pt, and Fe–Pt3 Alloys: A Density Functional Theory Study," Phys. Status Solidi B, vol. 262, p. 2400160, 2024.
- S.J. Clark, M.D. Segall, C.J. Pickard, P.J. Hasnip, M.I.J. Probert, K. Refson and M.C. Payne, "First principles methods using CASTEP," Z. Kristallogr., vol. 220, p. 567, 2005.
- J.P. Perdew, K. Burke and M. Ernzerhof, "Generalized Gradient Approximation Made Simple," Phys. Rev. Lett., vol. 77, p. 3865, 1996.
- P. Hohenberg and W. Kohn, "Inhomogeneous electron gas," Phys. Rev., vol. 136, p. B964, 1964.
- H.J. Monkhorst and J.D. Pack, "Special points for Brillouin-zone integrations," Phys. Rev. B, vol. 16, p. 1748, 1977.
- J. Aarons, "A New CASTEP and ONETEP Geometry Optimiser," http://www.hector.ac.uk/cse/distributedcse/reports/castep-geom/castep-geom/HTML/dCSE_project.html.
- B.G. Pfrommer, M. Cote, S.G. Louie and M.L. Cohen, "Relaxation of Crystals with the Quasi-Newton Method," J. Comput. Phys., vol. 131, p. 233, 1997.
- D. Packwood, J. Kermode, L. Mones, N. Bernstein, J. Woolley, N. Gould, C. Ortner and G. Csányi, "A universal preconditioner for simulating condensed phase materials," J. Chem. Phys., vol. 144, p. 164109, 2016.
- K.H.J. Buschow, P.G. van Engen and R. Jongebreur, "Magneto-optical properties of metallic ferromagnetic materials," J. Magn. Magn. Mater., vol. 38, p. 1, 1983.
- J.M. MacLaren, R.R. Duplessis, R.A. Stern and S. Willoughby, "First Principles Calculations of FePt, CoPt, Co3Pt, and Fe3Pt Alloys," IEEE Trans. Magn., vol. 41, p. 4374, 2005.
- T. Cheng, G. Yu, Y. Su, C. Ge, X. Zhang, L. Zhu and L. Li, "Lattice dynamics, elasticity and magnetic abnormality in ordered crystalline alloys Fe3Pt at high pressures," J. Magn. Magn. Mater., vol. 453, p. 67, 2018.
- M. Li, H. Pi, Y. Zhao, T. Lin, Q. Zhang, X. Hu, C. Xiong, Z. Qiu, L. Wang, Y. Zhang, J. Cai, W. Liu, J. Sun, F. Hu, L. Gu, H. Weng, Q. Wu, S. Wang, Y. Chen and B. Shen, "Large Anomalous Nernst Effects at Room Temperature in Fe3Pt Thin Films," Adv. Mater., vol. 35, p. 2301339, 2023.
- K. Aledealat, B. Aladerah, A. Obeidat and M. Gharaibeh, "First-principles study of electronic structure and magnetic properties of L10-ordered FeNi, FePd, and FePt alloys," Heliyon, vol. 7, p. e08639, 2021.
- M. Born and K. Huang, "Dynamics Theory of Crystal Lattices," Oxford University Press, 1954.
- F. Mouhat and F.X. Coudert, "Necessary and Sufficient Elastic Stability Conditions in Various Crystal Systems," Phys. Rev. B, vol. 90, p. 224104, 2014.
- T. Paszkiewicz, M. Pruchnik and P. Zieliński, "Unified description of elastic and acoustic properties of cubic media: elastic instabilities, phase transitions and soft modes," Eur. Phys. J. B, vol. 24, p. 327, 2001.
- R. Hill, "The Elastic Behaviour of a Crystalline Aggregate," Proc. Phys. Soc., vol. A65, p. 349, 1952.
- Al-Ghaferi, P. Mullner, H. Heinrich, G. Kostorz and J.M.K. Wiezorek, "Elastic Constants of Equiatomic L10Ordered FePd Single Crystals," Acta Materialia, vol. 54, p. 881, 2006.
Figure 1.
Schematic representation of atomic arrangements in (a) (b) (c) P4/mmm and (d) R3m Fe3Pt alloys.
Figure 1.
Schematic representation of atomic arrangements in (a) (b) (c) P4/mmm and (d) R3m Fe3Pt alloys.
Figure 2.
Total and partial density of states for , , and Fe3Pt alloys. The Fermi level is taken as the origin of the energy axis ().
Figure 2.
Total and partial density of states for , , and Fe3Pt alloys. The Fermi level is taken as the origin of the energy axis ().
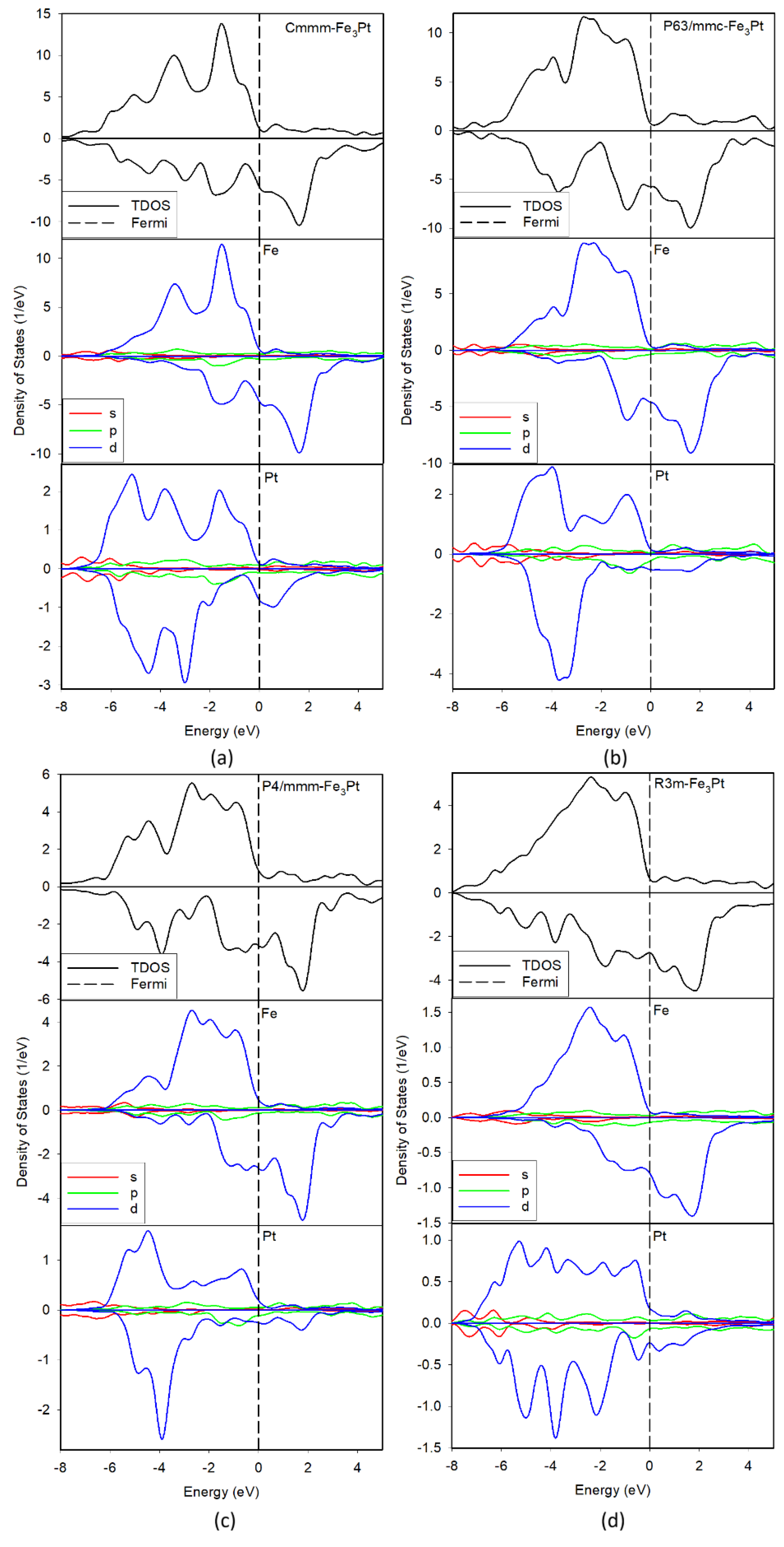
Figure 3.
Electronic band structure plots of or , , and of Fe3Pt along high symmetry line in the Brillouin zone. The Fermi level is taken as the zero line ().
Figure 3.
Electronic band structure plots of or , , and of Fe3Pt along high symmetry line in the Brillouin zone. The Fermi level is taken as the zero line ().
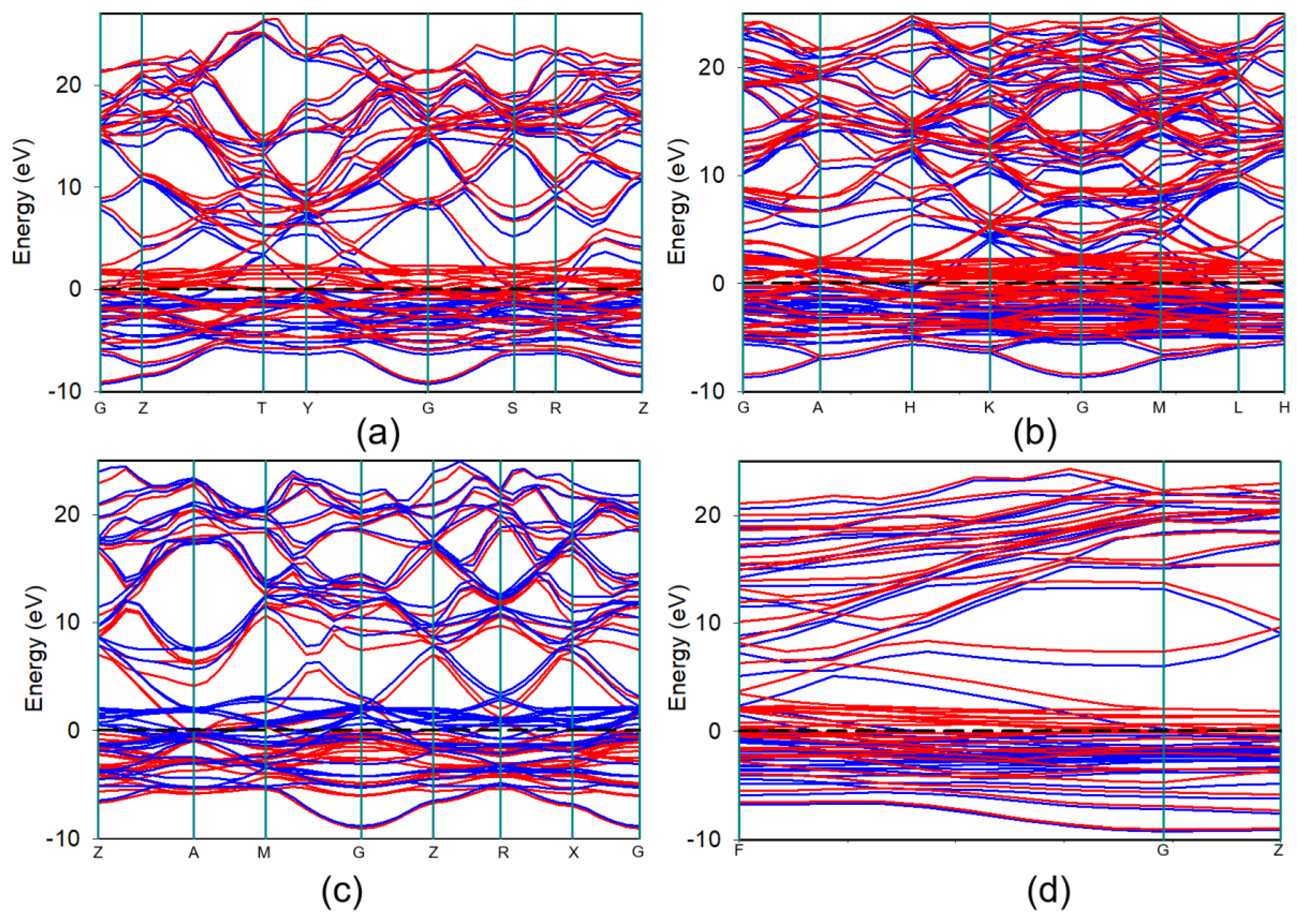
Figure 4.
Stress vs strain curve with linear fit in the elastic region for Fe3Pt alloys.
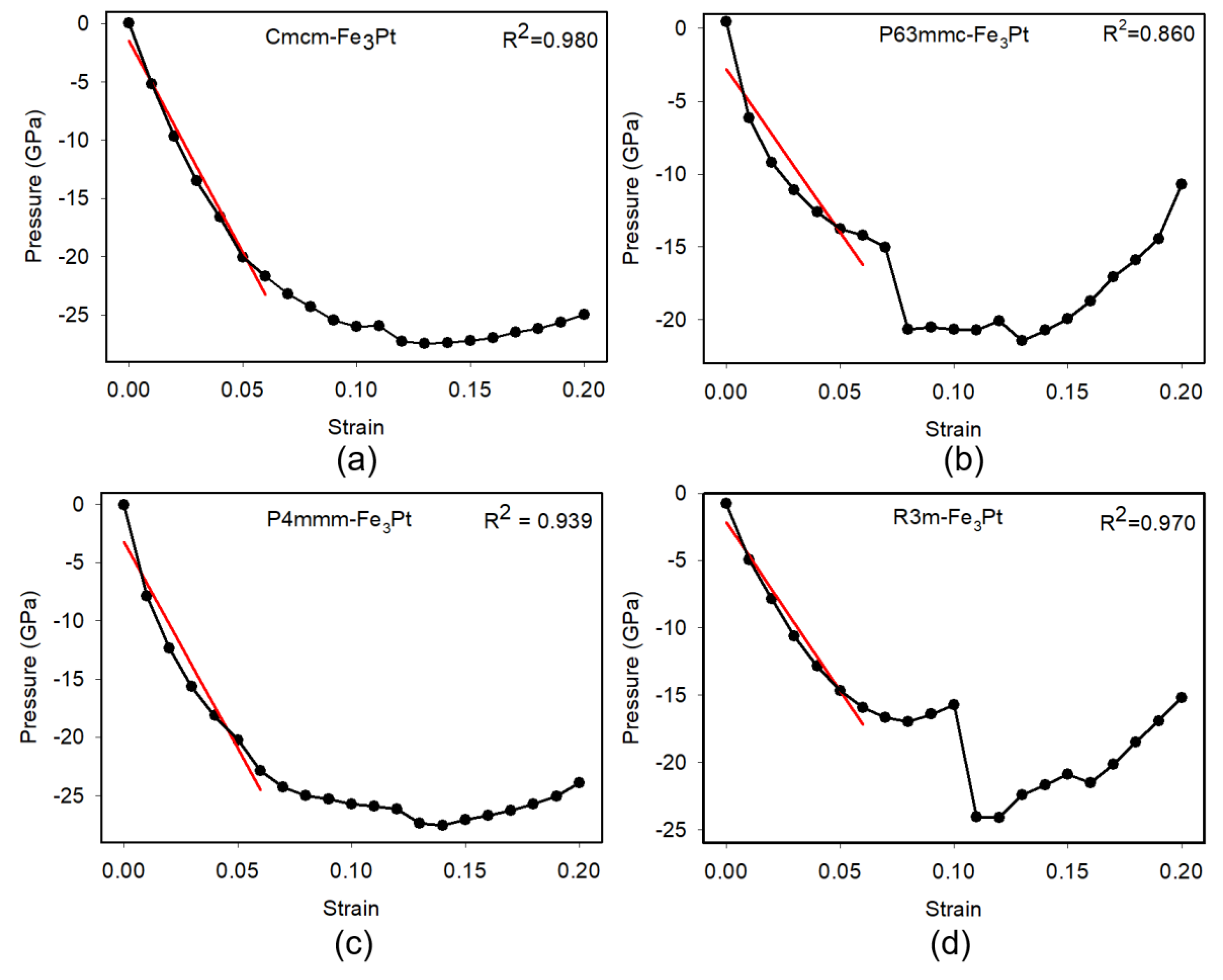

Table 1.
Calculated equilibrium cell parameters, densities and enthalpies of formation and density of Fe3Pt alloys.
Table 1.
Calculated equilibrium cell parameters, densities and enthalpies of formation and density of Fe3Pt alloys.
| Parameter | |||||
|---|---|---|---|---|---|
| a (Å) | 3.756 (3.73) [34] | 4.089 | 5.262 | 3.827 | 8.550 |
| b (Å) | - | 4.092 | |||
| c (Å) | 6.197 | 4.685 | 3.593 | ||
| V (Å3) | 53.000 | 103.694 | 112.34 | 52.620 | 54.330 |
| -0.391 | -0.391 | -0.391 | -0.391 | -0.391 | |
| 11.361 | 11.614 | 10.720 | 11.440 | 11.082 |
Table 2.
Calculated magnetic moments, Hirshfield spins and charges Fe3Pt alloys.
| 5.35 | 5.33 | 5.29 | 5.34 | |
| 2.38 | 2.41 | 2.47 | 2.46 | |
| () | 2.98 | 2.92 | 2.83 | 2.87 |
| 5.18 | - | 5.26 | 5.29 | |
| 2.74 | - | 2.50 | 2.64 | |
| 2.43 | - | 2.75 | 2.65 | |
| 16.46 | 16.45 | 16.43 | 16.34 | |
| 16.16 | 16.34 | 16.29 | 16.13 | |
| () | 0.30 | 0.12 | 0.15 | 0.21 |
| Total ( | 3.01 | 3.04 | 2.94 | 2.97 |
| Hirshfeld Analysis( | ||||
| 2.83 | 2.81 | 2.72 | 2.82 | |
| 2.45 | - | 2.68 | 2.61 | |
| 0.57 | 0.45 | 0.44 | 0.35 | |
| Charge (e) | ||||
| 0.27 | 0.26 | 0.24 | 0.20 | |
| 0.08 | - | 0.24 | 0.07 | |
| -0.62 | -0.79 | -0.72 | -0.46 | |
Table 3.
Calculated independent elastic constants and macroscopic properties of Fe3Pt alloys.
| Structure | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 300.43 | 298.95 | 224.53 | 120.12 | 119.83 | 36.27 | 92.71 | 161.85 | ||||||||
| 249.31 | 22.23 | -302.21 | 92.47 | 41.74 | |||||||||||
| 198.88 | 166.50 | 46.35 | 12.11 | 141.24 | 190.46 | ||||||||||
| 239.92 | 152.93 | 48.48 | 125.14 | 88.28 | 5.36 | ||||||||||
| Macroscopic Properties (GPa) | |||||||||||||||
| B | G | E | |||||||||||||
| 183.77 | 70.85 | 188.34 | 0.33 | 2.59 | 1.97 | ||||||||||
| 57.87 | -25.51 | 74.88 | 0.76 | -2.27 | -14.01 | ||||||||||
| 179.09 | 38.75 | 108.44 | 0.40 | 4.62 | -2.79 | ||||||||||
| 131.99 | 52.15 | 138.23 | 0.33 | 2.53 | 0.24 | ||||||||||
Table 4.
Mathematical expressions and descriptions of the macroscopic mechanical properties of the polycrystalline material. .
Table 4.
Mathematical expressions and descriptions of the macroscopic mechanical properties of the polycrystalline material. .
| Property | Expression | Description |
|---|---|---|
| Bulk modulus (B) | Resistance to uniform volume compression | |
| Shear modulus (G) | Resistance to shape change | |
| Young’s modulus (E) | Stiffness | |
| Poisson’s ratio (σ) | Ductility and brittleness | |
| Pugh Ratio | Ductility | |
| Anisotropy factor (A) | Elastic anisotropy |
represents the compliance tensor, the inverse of .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



