
Submitted:
23 October 2025
Posted:
24 October 2025
You are already at the latest version
Abstract
Chronic kidney disease is a significant global health burden and a leading cause of cardiovascular morbidity and mortality. Diabetes mellitus is the primary cause of kidney disease, driving the progression of both micro- and macrovascular complications. Sustained hyperglycemia initiates a cascade of deleterious molecular and cellular events, including mitochondrial dysfunction, inflammation, oxidative stress, and dysregulated apoptosis and autophagy, that collectively contribute to the progression of renal injury. Beyond these well-established mechanisms, a compelling body of evidence highlights the pivotal role of epigenetic alterations (such as DNA methylation, histone post-translational modifications, and non-coding RNAs) in mediated long-term kidney damages. The interplay between transcriptional and epigenetic regulation underlies the phenomenon of the “metabolic memory”, wherein cellular dysfunction persists even after glycemic control is achieved. This review synthesizes the current knowledge on mechanisms sustaining metabolic and epigenetic memory, with a particular focus on the epigenetic machinery that establishes and maintains these signals, a concept increasingly termed "epigenetic memory." Given their reversible nature, epigenetic determinants are emerging as promising biomarkers and a compelling therapeutic avenue. Targeting these "epifactors" offers a novel strategy to halt progression to end-stage renal disease, thereby paving the way for precision medicine approaches in diabetes-related renal disease.
Keywords:
chronic kidney disease
; cardiovascular complications
; oxidative stress
; mitochondrial alterations
; metabolic memory
; epigenetics
; epigenetic modifications
; epigenetic memory
1. Introduction
Kidney diseases, encompassing both acute kidney injury (AKI) and chronic kidney disease (CKD), represent a major global health burden and and recognized as leading contributors to cardiovascular morbidity and mortality [1,2]. CKD is frequently associated with a higher incidence of comorbid conditions, such as hypertension, diabetes mellitus, glomerulonephritis, chronic pyelonephritis, autoimmune diseases, and recurrent urinary tract infections, whose progression is associated with the development of renal impairment and eventual kidney failure [1,3]. Conversely, diabetes mellitus and hypertension themselves are the primary causes of CKD, accounting for approximately 60% of all cases [4]. Both AKI and CKD arise from complex systemic interactions and cellular disturbances, involving hemodynamic alterations, inflammatory responses, and maladaptive repair mechanisms that perpetuate the renal injury. CKD progression, in particular, is driven by endothelial dysfunction, vascular rarefaction, inflammation, and progressive nephron loss [5]. Recent guideline frameworks have reframed CKD as a highly prevalent condition with a continuum of severity, no longer viewed solely as a specialist disease but as a public health priority requiring early detection, preventive strategies, and multidisciplinary management [6]. According to the Kidney Disease Outcomes Quality Initiative (KDOQI) and Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, CKD is defined by the presence of kidney damage or a reduction in glomerular filtration rate (GFR) for at least three months, regardless of etiology. Decreased kidney function is typically assessed through estimated GFR (eGFR) derived from serum creatinine–based equations. CKD often progresses silently, with patients remaining asymptomatic until substantial functional decline occurs. In a subset of individuals, disease progression culminates in end-stage renal disease (ESRD), characterized by severely reduced kidney function or dependence on dialysis. Despite advances in therapeutic strategies, the coexistence of diabetes remains a critical determinant of adverse cardiovascular and renal outcomes. Chronic hyperglycemia, the hallmark of diabetes mellitus, orchestrates a complex and multifactorial cascade involving mitochondrial dysfunction, oxidative stress, inflammation, and dysregulated apoptosis and autophagy - processes that collectively promote cellular injury and fibrosis [7,8,9].
Although traditional biochemical pathways underlying diabetic kidney disease (DKD) are extensively documented, emerging evidence highlights the pivotal contribution of epigenetic mechanisms (such as DNA methylation, chromatin histone post-translational modifications, non-coding RNAs, and so on) in initiating and sustaining renal dysfunctions, and the onset of related complications [10]. The interaction between transcriptional and epigenetic factors lead to aberrant signalling, which underpins the phenomenon of the “metabolic memory”, term coined by Michael Brownlee to describe the long-term and detrimental effects of a poor metabolic control even after the normoglycemia has been achieved [11].
This review aims to summarize the current understanding of the molecular bases underlining the metabolic memory, emphasizing on epigenetic machinery mechanisms responsible for its establishment and maintenance – collectively referred to as “epigenetic memory”. Given the reversible nature of epigenetic modifications, “epifactors” hold promise as both biomarkers and therapeutic targets for hyperglycemia-related CKD. Modulating epigenetic regulators may open new avenues for halting disease progression, preventing ESRD, and advancing precision medicine approaches in DKD.
2. The Metabolic Memory
Exposure to sustained glucose levels induces a cascade of intricate and deleterious molecular and cellular events, culminating in widespread tissue damage. The disruptive effects of glucotoxicity involve widespread and interconnected mechanisms, generating advanced glycation end products (AGEs), overproduction of reactive oxygen species (ROS), and profound bioenergetic dysfunctions, culminating in epigenetic changes, and inflammatory memory [12] (Figure 1). The accumulation of AGEs is accelerated by chronic hyperglycemia and it is directly associated with cardiovascular complications of hyperglycemia [11,13]; when AGE bind the advanced glycation end-products receptor (RAGE), it triggers different pathways involved in response to inflammation, proliferation, apoptosis, and angiogenesis, generating ROS and exacerbating the oxidative stress through the activation of MAPK, ERKs, JK, NF-κB, TGF-α, and NOX-1 signaling [13,14,15]. Critically, transient or prolonged hyperglycemic insults can engender a persistent fingerprint that can establish long-lasting and perpetuating stimuli, sustaining the progression of diabetic implications even after the re-establishment of normoglycemia [16,17,18,19,20]. One of the first experimental studies trying to elucidate more on this metabolic memory phenomenon can be traced back to several years ago, when studies conducted in vitro demonstrated the existence of self-perpetuating changes in the gene expression induced and maintained by hyperglycemia. Cultured human endothelial cells, treated for 14 days with high glucose levels and followed by 7 days of normal growing conditions, have proven the overexpression of fibronectin mRNA (important in the pathogenesis of diabetic retinopathy) that persisted for weeks after restoration of near normoglycemia [21]. Over the years, a lot of studies have been conducted, either in experimental or in the clinical setting, to sustain this hypothesis, also known as “legacy effect” or “glycemic effect”.
The concept of metabolic memory is not theoretical but is strongly supported by longitudinal follow-up studies of two landmark randomized controlled trials in diabetes: the Epidemiology of Diabetes Interventions and Complications (EDIC) trial and the UK Prospective Diabetes Study (UKPDS). The Diabetes Control and Complications Trial (DCCT) compared intensive versus conventional glucose control in patients with type 1 diabetes. Following the completion of the active trial phase, participants were enrolled in the observational EDIC study. During EDIC, the glycemic control, in terms of glycated haemoglobin (HbA1c) levels, in the former intensive and conventional groups rapidly converged. Crucially, however, the initial benefit of intensive therapy on reducing the risk of microvascular complications (retinopathy, nephropathy, and neuropathy), not only persisted, but often widened over the subsequent two decades [22]. This sustained benefit, rooted in the initial period of superior glycemic control, is the core definition of metabolic memory. A similar effect, often referred to as the "legacy effect," was observed in the follow-up of the UKPDS cohort. In type 2 diabetes patients, the initial period of intensive glucose control reduced the risk of myocardial infarction and death from any cause over the long term, even after the glucose control difference between the intensive and conventional groups vanished. These trials collectively established that early, tight glycemic management provides a persistent, protective advantage against both micro- and macrovascular disease, underscoring the urgency of early intervention to prevent this pathogenic "memory" from being established [22].
From the experimental point of view, overall, we know that memory process identifies a specific mark of vascular stress, characterized by crucial event, principally increased ROS, inactivation of anti-atherosclerosis enzymes, induction of endoplasmic reticulum stress, which persist in a prolonged manner over time even after glucose normalization [23]. Well-established evidence confirmed the association between oxidative stress and diabetic complications, deriving from multiple studies conducted over the last twenty years and more [11,24,25]. The role of the excessive production of ROS is a key initiator in the pathology of diabetic vascular complications, covering a critical significance especially at the mitochondrial level [18,26,27,28]. Mitochondrial-derived ROS (mtROS) highly influence endothelial fitness and vascular homeostasis, participating in nitric oxide levels regulation and ROS-dependent signaling cascades [28,29]. Also, Ceriello et al. pointed out the role of mitochondria, highlighting that the establishment of the memory phenomenon originated at the level of glycated mitochondrial proteins, but involved also the non-enzymatic glycation of cellular proteins, lipids, and nucleic acids [16]. In support of these observations, experimental studies have shown that the progression of diabetic complications can be mitigated through the potent inhibition of oxidative stress with selected antioxidant agents [11,24,26]. Normalizing this mtROS has been shown to alleviate major pathways of hyperglycemic damage, including the activation of the polyol pathway, protein kinase C (PKC) activation, and the accumulation of AGEs [26]. In this context, the reduction of intracellular production of mitochondria-derived free radicals can switch off the metabolic memory induced by the high glucose stress signaling, consolidating the role of mitochondrial alterations in the glycemic memory phenomenon [17]. Glycated mitochondria can initiate a deleterious cascade, characterized by damage to mitochondrial DNA (mtDNA) and a subsequent decline in mitochondrial function, which in turn perpetuates the generation of reactive oxygen species and cellular injury [30]. In a recent investigation, Kowluru and colleagues delineates that a hyperglycemic environment induces an imbalance in mitochondrial dynamics, characterized by a reduction in the fusion protein Mitofusin 2 (Mfn2) and an elevation in the fission protein Dynamin-Related Protein 1 (Drp1), resulting in mitochondrial fragmentation; moreover, the re-institution of normal glycemic conditions is insufficient to reverse this pathological fragmentation and the accumulation of dysfunctional mitochondria. The research examined the potential therapeutic efficacy of modulating the mitochondrial fusion process during a normoglycemic period following a high-glucose insult to ameliorate deficits in mitochondrial quality control [31]. The disruption of mitochondria quality control induced by hyperglycemia can induce a state of cellular aging, or senescence, which is associated with a reduced capacity for mitophagy, as observed in retinal endothelial cells in diabetic retinopathy models [31,32,33]. The collapse of mitochondrial quality control has, as might have guessed, a critical role also in the pathogenesis and progression of CKD, being an organ with high-energy demand, of profound metabolic activity and rich in mitochondria. Indeed, chronic nephrotoxic insults are notably linked to oxidative stress, damages of energy-demanding renal cells, and different mitochondrial alterations, such as respiratory chain-derived oxidative stress, defects of biogenesis, fission and fusion imbalance, defects of clearance, and oxidative phosphorylation disturbance [34]. Thus, mtROS not only influence kidney activity, but also cells of other compartments bear their consequences, inducing a more complex metabolic framework in CKD patients, sustaining a pro-inflammatory milieu, activating pro-atherogenic pathways, and inducing endothelial dysfunctions and insulin resistance [35,36,37]. Targeting these mitochondrial quality control pathways and metabolic memory regulatory mechanisms, therefore, represents a highly promising avenue for novel therapeutic interventions to mitigate or prevent the relentless progression of the disease and their complications [38,39,40].
Figure 1.
Molecular pathophysiology of hyperglycemic memory in the development of vascular complications. The process is principally driven by four arms. (A) Initiation by formation of Advanced Glycation End Products: hyperglycemia promotes the non-enzymatic formation of advanced end products, followed by the binding to their specific receptor, subsequently activating a cascade of pro-inflammatory and pro-oxidant signals; (B) ROS overproduction and mitochondrial damage: mitochondrial dysfunctions, characterized by increased fission, decreased fusion, impaired quality control via mitophagy, and compromised respiratory efficiency are induced and result in the overproduction of reactive oxygen species; (C) Epigenetic pathological modification: the cumulative cellular stress from AGE overproduction and excessive reactive oxygen species induce an epigenetic reprogramming, including alteration of DNA methylation, aberrant post-translational histone modifications, and dysregulation of non-coding RNAs; (D) Persistent altered gene expression: transient or sustained hyperglycemia induces durable cellular and epigenetic reprogramming characterized by endoplasmic reticulum stress and the chronic activation of transcription factors, which drives the expression of pro-inflammatory mediators, promotes extracellular matrix remodeling, and increases endothelium stiffness. AGE: Advanced Glycation End Products; RAGE: Receptor for Advanced Glycation End Products; mtDNA: mitochondrial DNA; ROS: reactive oxygen species; DNMT: DNA methyltransferases; ncRNA: non-coding RNA; ER: endoplasmic reticulum; ECM: extracellular matrix.
Figure 1.
Molecular pathophysiology of hyperglycemic memory in the development of vascular complications. The process is principally driven by four arms. (A) Initiation by formation of Advanced Glycation End Products: hyperglycemia promotes the non-enzymatic formation of advanced end products, followed by the binding to their specific receptor, subsequently activating a cascade of pro-inflammatory and pro-oxidant signals; (B) ROS overproduction and mitochondrial damage: mitochondrial dysfunctions, characterized by increased fission, decreased fusion, impaired quality control via mitophagy, and compromised respiratory efficiency are induced and result in the overproduction of reactive oxygen species; (C) Epigenetic pathological modification: the cumulative cellular stress from AGE overproduction and excessive reactive oxygen species induce an epigenetic reprogramming, including alteration of DNA methylation, aberrant post-translational histone modifications, and dysregulation of non-coding RNAs; (D) Persistent altered gene expression: transient or sustained hyperglycemia induces durable cellular and epigenetic reprogramming characterized by endoplasmic reticulum stress and the chronic activation of transcription factors, which drives the expression of pro-inflammatory mediators, promotes extracellular matrix remodeling, and increases endothelium stiffness. AGE: Advanced Glycation End Products; RAGE: Receptor for Advanced Glycation End Products; mtDNA: mitochondrial DNA; ROS: reactive oxygen species; DNMT: DNA methyltransferases; ncRNA: non-coding RNA; ER: endoplasmic reticulum; ECM: extracellular matrix.
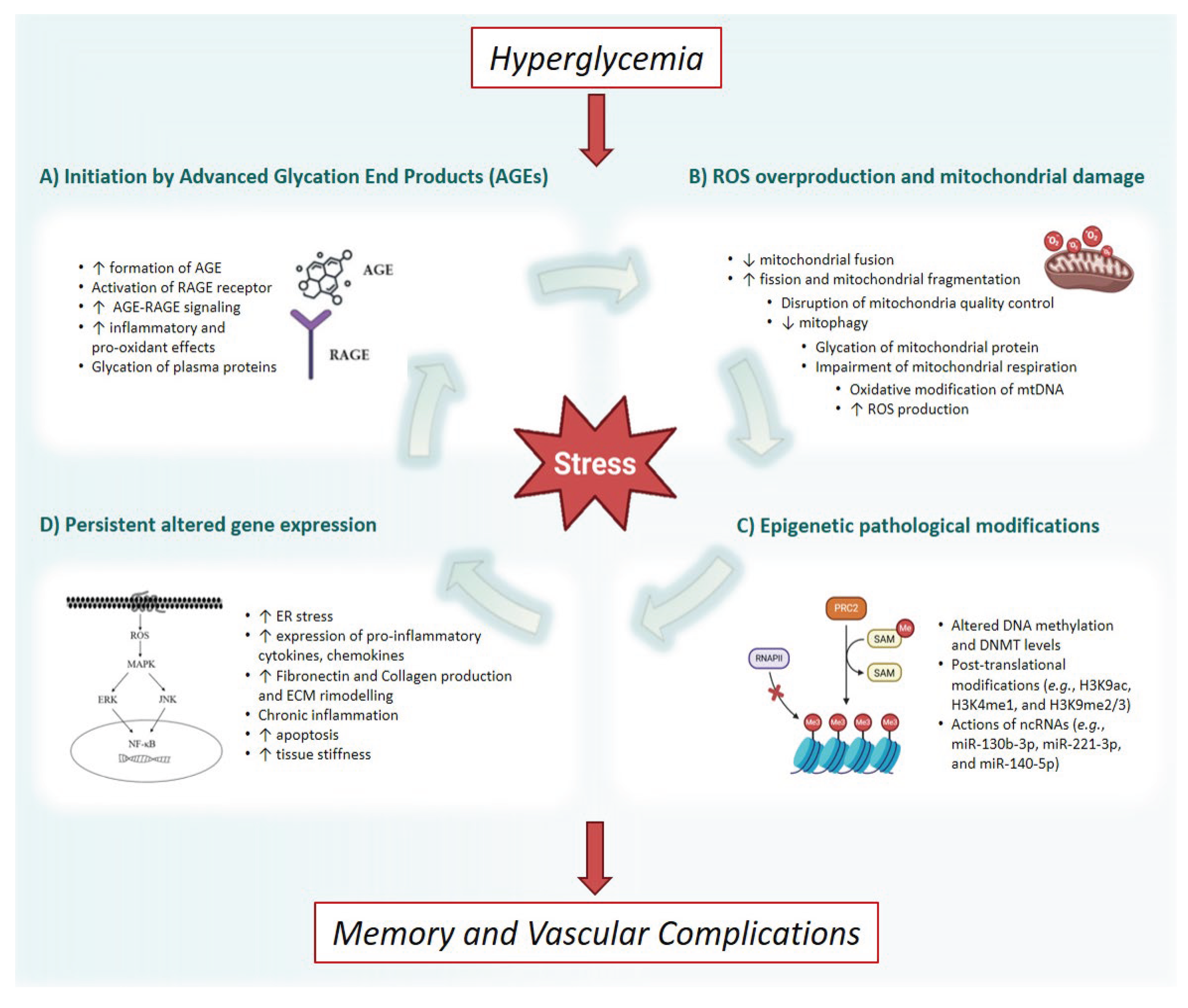
An interesting point has been singled out about oscillations of glucose concentrations, in terms of damages induced by exposure to transient hyperglycemia. In fact, intermittent episodes can be significantly more pernicious than sustained and chronic high glucose levels. Through mechanisms including exacerbated oxidative stress and persistent epigenetic programming, these glucose spikes promote a sustained pro-inflammatory and dysfunctional endothelial phenotype, making the picture complex and intricate [17,41]. This translates clinically to the concept that high glycemic variability might represent a powerful, HbA1c-independent predictor of hyperglycemia-related complications and mortality, representing a uniquely insidious aspect of dysglycemia. Moreover, many experimental results confirmed that transient episodes of hyperglycemia can induce changes in gene expression that are dependent on epigenetic modifications of histone tails (e.g., methylation and acetylation), and that these changes persist after return to normoglycemia [42,43]. The involvement of persistent epigenetic changes in transient high glucose have gained much attention in explaining the long-lasting detrimental consequences of hyperglycemia causing the development of chronic complications in the pathophysiology of CKD and of AKI-to-CKD transition [44,45,46]. From a clinical perspective, enhanced elucidations of the underlying mechanisms governing these processes may provide a compelling rationale for an early intensive therapeutic intervention for glycemic control, with the primary objective of rapidly restoring the metabolic homeostasis; moreover, the to reduce long-term complications of diabetes, it could be of great advantage, in concert with the stringent management of blood glucose levels, the administration of therapeutic agents capable of attenuating oxidative stress and inhibiting non-enzymatic glycation processes, in order to set up a more effective strategy for mitigating the long-term sequelae of hyperglycemia.
3. Epigenetic Regulation of the Gene Expression
Understanding the hierarchical packaging of DNA into chromatin provides the fundamental framework for exploring the mechanisms and implications of epigenetic regulation. First, epigenetic mechanisms refer to a series of potentially reversible and heritable changes in chromatin structure and in gene expression, which address the tissue-specific transcription and conserve cell identity, inducing changes in the phenotype, without altering the underlying DNA backbone [47]. Through interconnected biochemical modifications of DNA and histone/non-histone proteins, as well as by the activity of highly controlled remodelers/regulators, specific genes can be switched “on” or “off”, determining which proteins are transcribed at a specific time and in a particular cell type, in response to extracellular signals. Histone proteins are key components of chromatin, whose fundamental unit is the nucleosome. In eukaryotes, it consists of 147 base pairs of DNA wrapped around a histone octamer, the histone core, comprising two copies for each histone H2A, H2B, H3, and H4, while H1 acts as linker protein (Figure 2A).
In higher-order eukaryotes, epigenetic marks include principally DNA methylation, post-translational modifications (PTMs) of histone proteins, chromatin conformational changes, histone variant exchange, and non-coding RNAs. The most studied mechanism of epigenetic regulation is the DNA methylation, elicited by the addition of a methyl (-CH3) group to the 5′ carbon of cytosine to become 5-methylcytosines (Figure 2B). It affects especially small regions of DNA (<500 bp) enriched in cytosine-guanosine dinucleotides (CpG islands), owning a CG content greater than 55%. CpG-rich DNA is usually clustered around the promoter region of the gene and, thus, the methylation process can affect the transcriptional regulation. CpG sites are methylated by DNA methyltransferases (DNMT) enzymes that establish the marks, crucial for the normal development of tissues, as evidenced by research in the field of embryology [48,49] (Fig. 2B). DNMTs are mainly grouped in enzymes performing de novo methylation and those in charge to stably maintain the methylation patterns, recognizing hemi-methylated sites; all of them catalyze the reaction through the s-adenosyl-l-methionine (SAM) as methyl groups donor. There are five known DNMTs and DNMT-like proteins: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L [50,51]. DNMT3A and DNMT3B are responsible for the establishment of novel methylation patterns (both in vitro and in vivo), DNMT1 maintains the methylation mark during DNA replication, while DNMT3L is enzymatically inactive and acts as regulatory factor [51,52]. Moreover, it seems that the five known DNMTs and their splice variants/isoforms are differentially expressed across tissues and organs. Indeed, Tóth et al. have found that DNMT3A primarily regulated hematopoietic differentiation, while DNMT3B was crucial for cartilage homeostasis and ossification [53]. The deep understanding of the context-dependent role of DNMTs is of crucial importance, both to refine our knowledge of their involvement in pathophysiologic clinical conditions and to provide insights into potential therapeutic applications. The reversible arrangement of methylation occurred either with passive mechanisms or with active processes. Ten-Eleven Translocation (TET) enzyme family proteins (TET1, TET2, and TET3) are responsible for the active demethylation, catalyzing the iterative oxidation of 5-methylcytosine (5mC) to form the intermediate products, which are subsequently removed (Figure 2B); finally, the locus is restored to an unmethylated state through the base excision repair pathway [54,55,56].
Another important mechanism of epigenetic regulation involves PTMs of histones, which have got a specific structural and functional organization due to their role in the dynamic regulation of gene transcription, by controlling accessibility of nuclear transcription factors and RNA polymerase to regulatory DNA elements. Different amino acids of histone amino-terminal tails are subject to PTMs, including (more commonly) methylation, acetylation, and phosphorylation, but also ubiquitylation, SUMOylation, lactylation, crotonylation, and others [57,58,59,60]. These covalent modifications can shape chromatin conformation in different ways: for example, the acetylation of lysine residues is responsible for breaking down the positive charge of lysines, so that the chromatin structure is released, facilitating the transcription; conversely, if the affinity between histones and DNA is enhanced, chromatin is condensed in form of heterochromatin, resulting in the inactivation of transcription [61] (Figure 2C).
Figure 2.
Schematic representation of the organization and packaging of elements of the nucleosome. (A) Nucleosome as the fundamental unit of the epigenome: the structure of the nucleosome consists of an octamer of core histone proteins (two copies each of H2A, H2B, H3, and H4) and the H1 linker histone, around which DNA is wrapped. Histone tails undergo post-translational modifications at different amino acid residues and biochemical groups added at the N-terminal region are represented (e.g., acetyl, methyl, and phosphoryl group); (B) Dinamics of DNA methylation: enzymatic processes governing DNA methylation and demethylation. DNA methyltransferases (DNMT1, DNMT3A, DNMT3B) catalyze the addition of a methyl group to the 5' position of cytosine, forming 5-methylcytosine (5-mC); conversely, the ten-eleven translocation family of dioxygenases initiates active demethylation by oxidizing 5-mC to 5-hydroxymethylcytosine; (C) Chromatin architecture and function as a consequence of epigenetic marks deposition: covalent modifications of histone tails can shape chromatin conformation, impacting on gene expression The acetylation of lysine residues break down the affinity between positive charge of lysines and negative charge of DNA, inducing relaxation of the chromatin structure and facilitating gene transcription; conversely, DNA methylation enhances affinity between DNA and histones, resulting in condensed chromatin (in form of heterochromatin) and inactivation of the transcription. SAM: S-adenosyl-methyionin; TET: ten-eleven translocation.
Figure 2.
Schematic representation of the organization and packaging of elements of the nucleosome. (A) Nucleosome as the fundamental unit of the epigenome: the structure of the nucleosome consists of an octamer of core histone proteins (two copies each of H2A, H2B, H3, and H4) and the H1 linker histone, around which DNA is wrapped. Histone tails undergo post-translational modifications at different amino acid residues and biochemical groups added at the N-terminal region are represented (e.g., acetyl, methyl, and phosphoryl group); (B) Dinamics of DNA methylation: enzymatic processes governing DNA methylation and demethylation. DNA methyltransferases (DNMT1, DNMT3A, DNMT3B) catalyze the addition of a methyl group to the 5' position of cytosine, forming 5-methylcytosine (5-mC); conversely, the ten-eleven translocation family of dioxygenases initiates active demethylation by oxidizing 5-mC to 5-hydroxymethylcytosine; (C) Chromatin architecture and function as a consequence of epigenetic marks deposition: covalent modifications of histone tails can shape chromatin conformation, impacting on gene expression The acetylation of lysine residues break down the affinity between positive charge of lysines and negative charge of DNA, inducing relaxation of the chromatin structure and facilitating gene transcription; conversely, DNA methylation enhances affinity between DNA and histones, resulting in condensed chromatin (in form of heterochromatin) and inactivation of the transcription. SAM: S-adenosyl-methyionin; TET: ten-eleven translocation.
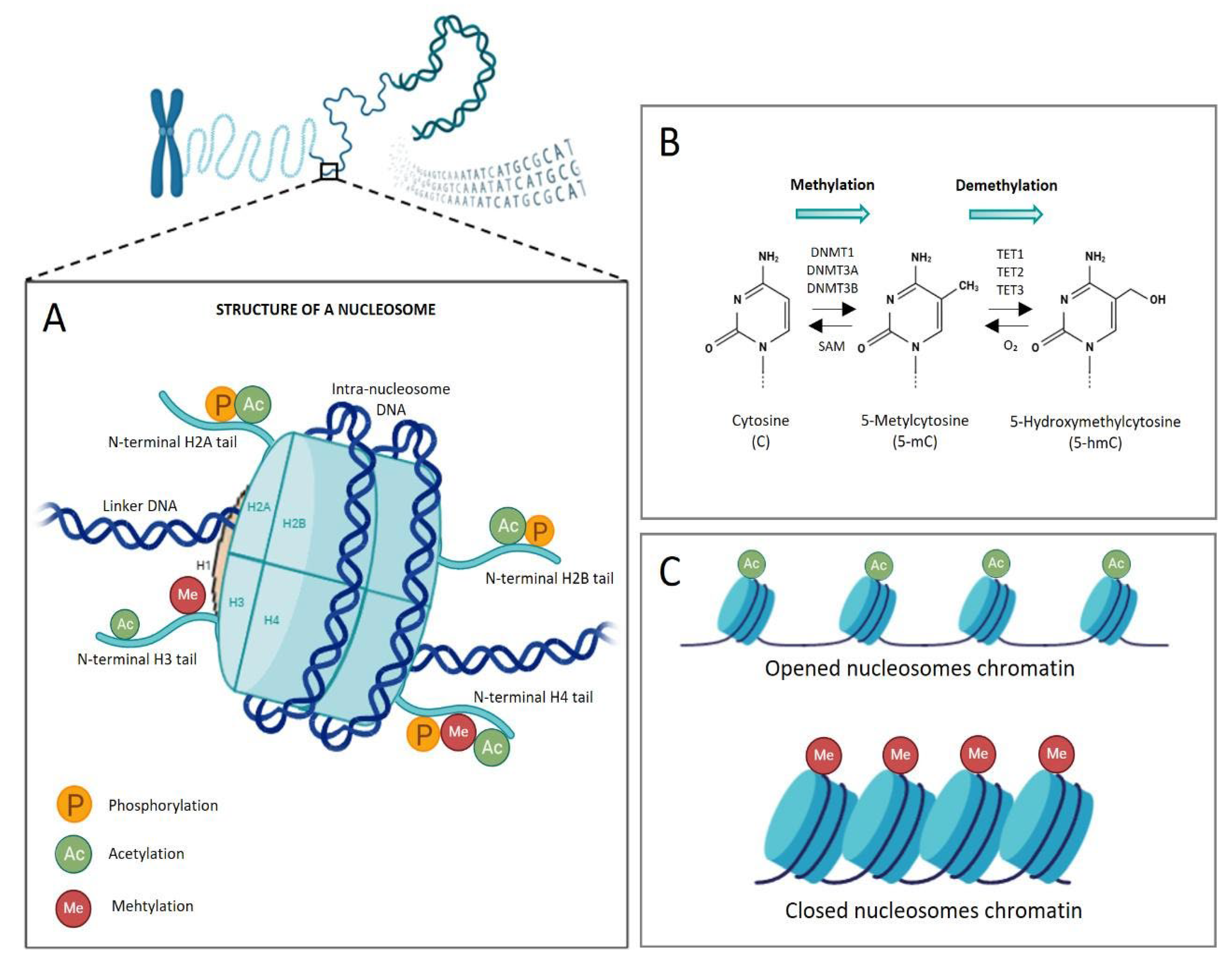
Besides acetylation, methylation of histone tails is one of the major epigenetic PTMs, interesting arginine or, more commonly, lysine (K) residues. The effect of histone methylation depends on histone isoforms and lysine position at the level of N-terminal residue, as well as on the extent of methylation. Methylation of histone lysines is catalyzed by lysine methyltransferase (KMT) and removed by lysine demethylase (KDM) enzymes. KDMs play a role both in repression and activation of transcription, and it has been reported their involvement in mental disorder, epilepsy, inflammation, as well as cancer and other pathologies [62,63,64]. Canonical lysine methylation sites are found commonly on histone 3 at lysine 4 (H3K4), 9 (H3K9), 27 (H3K27), 36 (H3K36), or 79 (H3K79), and on histone H4 at lysine 20 (H4K20), regulating chromatin structure [65]; also, other non-canonical sites have been described, but are much less characterized. The stable propagation of silenced gene expression states, a process initiated during early embryogenesis, is critically dependent on the recruitment of repressive proteins/complexes, such as methyl-CpG-binding proteins (e.g., MeCP2) or the Polycomb Repressive Complex 2 (PRC2) [66,67]. The latter is a multi-component histone methyltransferase complex able to catalyze the deposition of mono-, di-, and trimethyl marks on the lysine 27 residue within the N-terminal tail of histone H3 (H3K27me1/2/3); a central, yet incompletely understood, aspect of PRC2 function is how the recruitment to specific regions of the genome take place, conferring target-site specificity to PRC2 [67,68]. Methylated sites on histones are recognized by chromodomains of proteins associated with chromatin remodeling. Their binding to these epigenetic marks can trigger a cascade of events leading to either the silencing or activation of genes, depending on the specific chromodomain-containing protein and the cellular context [69]. In the case of PRC2, the Polycomb complex is recognized by CBX protein chromodomain and this leads to the monoubiquitination of histone H2A at lysine 119 (H2AK119ub) [70]. These histone modifications result in the compaction of chromatin, making it inaccessible to the transcription machinery, enforcing gene silencing, and emphasizing the complex interplay among these biochemical modifications.
In addition to methylation and demethylation, histone acetylation and deacetylation are highly prevalent and critical PTMs. These modifications are dynamically regulated by dedicated epigenetic enzymes: histone acetyl-transferases (HATs) and histone deacetylases (HDACs). HATs catalyze the transfer of an acetyl (CH3CO–) moiety from acetyl-CoA to the ε-amino group of a specific histone lysine residue. These enzymes, which possess characteristic HAT domains and can target both histone and non-histone proteins, install the acetyl mark, which is subsequently recognized by bromodomains to facilitate signal activation. Conversely, HDACs remove the acetyl group from lysine residues, restoring the positive charge. This process can lead to the reorganization of adjacent lysine PTMs and is generally associated with the suppression of gene transcription [71]. The 18 known HDACs are broadly categorized into four classes: Class I (HDAC1, 2, 3, 8), Class II (HDAC4, 5, 6, 7, 9, 10), Class III (SIRT1, 2, 3, 4, 5, 6, 7, known as Sirtuins), and Class IV (HDAC11). Classes I, II, and IV are collectively referred to as "classical" HDACs due to their shared structural and functional properties. In contrast, Class III sirtuins utilize a unique NAD+-dependent deacetylation mechanism via a NAD/FAD-binding domain, generating nicotinamide and 2′-O-acetyl-ADP-ribose as metabolic products [71] (Table 1).
Overall, despite DNA methylation and histone PTMs are executed by different cellular machinery, both are dynamically linked and act synergistically to regulate transcription. Therefore, it is now widely recognized that a significant interplay exists among these epigenetic modulation events. Histone modifications have been shown to induce DNA methylation, a process especially observed during the early development, regulating also the stability of DNMT enzymes [72,73]. Once established, methylated CpGs are recognized by methyl-CpG-binding proteins, such as the aforementioned MeCP2, which form a complex with histone deacetylase and histone methyltransferases, acting as transcription co-repressive complexes [72,74]. Additionally, to explicate the local histone code, epigenetic rearrangements might be required: for example, if PRC2 is commissioned to methylate H3K27 but this lysine residue is acetylated, HDACs are recruited, making the amino-group of lysine side chain available for the PRC2-mediated methylation [75].
In the last decade, many studies have focused on microRNAs (miRNA), known to be implicated in the repression of gene expression, as novel clinical targets or diagnostic tools. Currently, a lot of miRNAs are in the spotlight for their role as biomarkers for diagnosis and prognosis, perceived also as a new generation of drugs in different human disorders [34,76]. miRNAs are small endogenous, non-coding, and single-stranded RNA molecules (with a length of approximately 18 to 25 nucleotides), master regulator of the gene expression at post-transcriptional level. These fragments originated from a hairpin loop cleaved into small dimers, following a long series of modifications, until the expulsion from the nucleus. Then, they are mainly transcribed by RNA polymerase II, forming the hairpin structures, to create the primary miRNA transcript, called pri-miRNA); it can be hundreds to thousands of nucleotides long, containing the sequence of the mature future miRNA, subsequently cleaved into small dimers [77]. Still in the nucleus, the Drosha and Pasha enzymes cleave the pri-miRNA, generating the pre-miRNA, transported lately from the nucleus to the cytoplasm. Here, they are further processed by the RNAase III Dicer which cleaves the hairpin loop, creating the miRNA duplex; the latter is loaded in the Argonaute protein to produce the RNA-induced silencing complex (RISC) and tolled out. The silencing functions are exerted by two main mechanisms, cleaving target transcripts, or arresting translation [76].
Given that histone PTMs modulates chromatin structure and gene expression, it is not surprising that abnormal epigenetic events are associated with multiple diseases, including cancer, interstitial fibrosis, autoimmune and inflammatory diseases, and metabolic disorders [78,79]. Given that epigenetic marks can persist even after the initial trigger, it can lead to sustained abnormal gene expression and contribute also to the progressive nature of CKD, accumulating evidences on the keystone role of epigenetics on the establishment of the memory phenomenon; at the same time, it is important to underline that epigenetic modifications are notoriously and potentially reversible, representing a potential therapeutic and prognostic avenue to delay disease progression and catalyzing the advent of next-generation diagnostics in nephrology.
4. The Role of the Epigenetic Memory in Hyperglycemia-Related Ckd Progression
Epigenetics serves as a crucial mediator between genetic predisposition and environmental or lifestyle factors - such as diet, exercise, oxidative stress, toxins, metabolic alterations, and inflammation - thereby influencing disease onset and progression. In the context of renal disease, oxidative stress, inflammation, and metabolic dysfunction are tightly linked to extensive epigenetic remodeling, which heightens the risk of developing ESRD and related complications. This association is particularly pronounced in the presence of hyperglycemia or diabetes. The investigation of epigenetic processes contributing to CKD progression and its cardiovascular complications is an emerging and rapidly expanding field. Multiple biochemical and signaling pathways are disrupted in CKD, including those governing inflammation, immune modulation, epithelial-to-mesenchymal transition (EMT), and metabolic homeostasis, key drivers of renal fibrosis and vasculopathy, including accelerated atherosclerosis and vascular calcification [80,81].
4.1. Histone Modifications and the Activation of Pro-Inflammatory Signaling
Epigenetic modulation involves alterations to DNA accessibility, determined in part by post-translational modifications of core histones. These histone modifications are intrinsically linked to the development and progression of DKD, contributing to localized oxidative stress, fibrosis, and inflammation within the renal environment [43]. Specifically, certain histone modifications can activate pro-inflammatory signaling pathways, thereby initiating sustained inflammation and structural damage within the kidney parenchyma [82]. In diabetic mouse models, reduced Suv39h1 levels led to decreased H3K9me3 deposition at inflammatory gene promoters, promoting gene derepression and persistent vascular inflammation [83]. Hyperglycemia enhances activating histone marks (H3K9ac, H3K4me3, H3K4me1) while diminishing repressive marks (H3K27me3) at promoters such as TXNIP, a key regulator of oxidative stress and inflammation [84]. Hyperglycemia also induces sustained DNMT1 overexpression and histone methylation changes, even after glucose normalization, establishing an epigenetic “memory” [17,85]. Short-term hyperglycemic exposure can trigger long-lasting H3K4 monomethylation at the NF-κB p65 promoter, maintaining pro-inflammatory gene expression and vascular dysfunction through persistent VCAM-1 and MCP-1 expression [17]. These effects are prevented by suppressing mitochondrial superoxide production. Similarly, decreased H3K9me2/3 at the NF-κB promoter sustains its expression, promoting inflammation [43]. Hyperglycemia-induced PTMs at NOX4 and eNOS promoters perpetuate ROS production and vascular damage [86]. Epigenetic enzymes such as HMTs, HDMs, and DNMTs interact antagonistically to maintain inflammatory gene expression [17,87,88]. The HMT EZH2, a PRC2 complex component, contributes to H3K27me3 deposition on the HIC1 gene in glucose-stimulated tubular endothelial cells, repressing SIRT1, a key antioxidant factor, and promoting ROS accumulation [88].
4.2. Dna Methylation Status Connected to Renal Disease Progression
Likewise histone methylation, altered DNA methylation patterns influence the expression of cytokines and chemokines. Hyperglycemia induces dramatic changes in DNA methylation status that coincide with the activation of pathways associated with renal disease progression [10]. Mechanistically, high glucose exposure leads to the accumulation of ROS, which not only causes direct damage to glomerular cells but also alters gene expression via epigenetic changes [10]. Emerging evidence indicates that oxidative stress, a known driver of tubular damage, directly modulates DNA methylation patterns by altering the function of key methylation enzymes, specifically DNMTs and TET proteins [89]. This interplay provides a direct causal mechanism linking environmental stress (oxidative damage) to sustained epigenetic outcomes that drive disease. Changes in DNA methylation were specifically observed in the promoter regions of genes related to oxidative stress, inflammation, and renal fibrosis in both glomerular and tubular cells under diabetic conditions [10].
Studies focusing on advanced CKD patients, particularly those undergoing dialysis, have established a strong relationship between global DNA methylation status in circulating immune cells and poor clinical outcomes [90,91]. Global DNA hypermethylation correlates with systemic inflammation and poor clinical outcomes [92,93]. In contrast to systemic methylation markers, highly localized, tissue-specific methylation changes offer critical information for predicting organ-specific progression. Gluck et al. identified 471 methylation probes associated with renal failure, enriched within kidney regulatory regions, including the epidermal growth factor (EGF) gene locus, capable of predicting renal function decline in diabetic kidney disease [94,95]. Additional evidence indicates that diabetes-related DNA methylation changes in genes regulating oxidative stress, inflammation, and fibrosis occur in glomerular and tubular cells [96]. Similarly, hyperglycemia-induced DNA methylation alterations parallel the activation of pathways driving renal disease progression. A study published in Diabetic Medicine revealed specific DNA methylation patterns in mitochondrial protein-coding genes associated with kidney disease in type 1 diabetes, highlighting 51 genes, such as TAMM41 and COX6A1, with both genetic and epigenetic associations to diabetic kidney disease [97]. Differential methylation of genes crucial for mitochondrial metabolism (e.g., PMPCB, TSFM, and AUH) further links epigenetic dysregulation to diabetic kidney disease and end-stage renal failure [97].
4.3. The Role of Sirtuins in the Hyperglycemia-Induced Epigenetic Memory
Among epigenetic regulators, the NAD⁺-dependent histone deacetylases known as Sirtuins have drawn substantial attention. In particular, SIRT1 regulates mitochondrial energy metabolism and redox balance [98]. Experimental studies demonstrate that SIRT1 activation stimulates the LKB1/AMPK/ROS pathway, suppressing ROS production and inflammation [99]. Conversely, hyperglycemia-induced PARP activation inhibits SIRT1, forming a self-perpetuating cycle of oxidative stress. SIRT1 also deacetylates endothelial nitric oxide synthase (eNOS), maintaining endothelial integrity and preventing vascular senescence and macrophage foam cell formation [100]. Downregulation of SIRT1 elevates miR-34a levels, which enhances p66shc-mediated ROS production and endothelial dysfunction [101]. Overexpression of SIRT1 activates the NRF2/ARE antioxidant pathway, increasing expression of HO-1 and SOD, and reducing AGE-induced ROS in mesangial cells [102].
Other Sirtuins, including SIRT3 and SIRT7, are critical for mitochondrial dynamics and renal protection. SIRT3 mitigates oxidative stress and metabolic dysfunction in proximal tubular cells [103,104]. Reduced SIRT3 expression correlates with increased mitochondrial ROS (mtROS) and cardiovascular risk [105,106]. The mtROS accumulation activates PARP, leading to endothelial dysfunction—a key event in diabetic complications [107]. SIRT7, responsible for H3K18 deacetylation, plays a protective role against hyperglycemia-induced endothelial-to-mesenchymal transition (EndMT) and podocyte apoptosis [108,109,110]. Its downregulation after transient hyperglycemia perpetuates inflammation via the ELK1/DAPK3 axis and enhances vascular injury [111,112]. Notably, miR-20b directly targets SIRT7, and its overexpression induces podocyte apoptosis through caspase-3 activation, whereas SIRT7 restoration mitigates this damage [109].
4.5. Micrornas and the Epigenetic Memory of Hyperglycemia
MicroRNAs have emerged as pivotal epigenetic regulators in hyperglycemia-induced kidney disease. They fine-tune gene expression controlling fibrosis, inflammation, and apoptosis [113,114]. Dysregulated miRNAs contribute to atherosclerosis, retinopathy, and microvascular dysfunction [115,116]. For instance, miR-130b-3p, miR-221-3p, and miR-140-5p are glucose-responsive and target genes linked to apoptosis and angiogenesis [117]. Persistent upregulation of miR-125b activates NF-κB signaling, while reduced miR-146a-5p amplifies inflammatory gene expression [118]. miR-214 downregulates PTEN, promoting AKT/mTORC activation and renal damage [119], while miR-22 suppresses PTEN and autophagy, driving fibrosis [120]. Circulating miR-15/-16-loaded extracellular vesicles from diabetic patients preserve a “metabolic memory” signal, perpetuating endothelial CaMK2a activation and cardiac dysfunction [121]. Yao et al. demonstrated that transient hyperglycemia establishes long-lasting inflammation via the NF-κB/miR-27a-3p/NRF2/ROS/TGF-β/EndMT loop, and that NRF2 activation reverses these effects. NRF2, a master regulator of antioxidant defense, controls genes such as SOD2 and GPX, maintaining redox balance [122]. Its dysregulation across CKD stages reflects both compensatory activation and subsequent exhaustion, potentially mediated by epigenetic memory mechanisms [123,124].
The progressive elucidation of epigenetically regulated networks in CKD provides profound insights into disease mechanisms and potential therapeutic interventions. Sustained endothelial dysfunction - driven by oxidative stress, inflammation, EndMT, and disrupted metabolic homeostasis - is perpetuated by epigenetic “memory” mechanisms that stabilize pathological phenotypes [125]. Consequently, therapeutic strategies for CKD should extend beyond glycemic control to specifically target molecular and epigenetic memory pathways, offering new avenues to halt or reverse renal decline.
Figure 4.
The central role of hyperglycemia-induced changes in driving the progression of chronic kidney disease. Transient or persistent hyperglycemia acts as the primum movens to establish the memory phenomenon and to initiate the cascade of downstream pathological events, centered around mitochondrial, endothelial, and vascular dysfunctions. At the cellular level, high glucose impacts on mitochondrial functions, inducing overproduction of reactive oxygen species through oxidative phosphorylation, resulting in fission/fusion imbalance and damage to the mitochondrial DNA. These processes are regulated by an epigenetic memory, able to create changes in DNA methylation, post-translational modification (e.g., Sirtuin activation/repression), and miRNA deregulations. Hyperglycemia also promotes endothelial dysfunctions, with activation of inflammatory pathways with involvement of molecular and epigenetic targets, resulting in imbalance in vascular homeostasis. The initial stimulus, coupled with endothelial and mitochondrial dysfunction, triggers a vicious cycle within the kidney, producing oxidative stress, accumulation of nephrotoxins, and chronic inflammation. These factors could contribute to the progression of chronic kidney disease, with fibrosis, senescence, apoptosis, ultimately predisposing to the onset of end-stage renal disease. Moreover, the systemic nature of endothelial dysfunction gives rise to widespread vascular complications, classified in microvascular and macrovascular complications, such as retinopathy and artery disease. The dysfunction of cardiomyocytes and of vascular smooth muscle cells can be involved, finally, in structural damages and adverse cardiac outcomes, such as atherosclerotic cardiovascular disease, left ventricular hypertrophy, and heart failure. CKD, chronic kidney disease; ESRD, end-stage renal disease; miRNA, microRNA; DNMT, DNA methyltransferase; SOD2, superoxide dismutase-2; Mfn2, mitofusin-2; EZH2, Enhancer of Zeste Homolog-2; SET7, SET Domain Containing 7; mtDNA, mitochondrial DNA; ROS, reactive oxygen species; TCA, tricarboxylic acid; ASCVD, atherosclerotic cardiovascular disease; LVH, left ventricular hypertrophy; HF, heart failure.
Figure 4.
The central role of hyperglycemia-induced changes in driving the progression of chronic kidney disease. Transient or persistent hyperglycemia acts as the primum movens to establish the memory phenomenon and to initiate the cascade of downstream pathological events, centered around mitochondrial, endothelial, and vascular dysfunctions. At the cellular level, high glucose impacts on mitochondrial functions, inducing overproduction of reactive oxygen species through oxidative phosphorylation, resulting in fission/fusion imbalance and damage to the mitochondrial DNA. These processes are regulated by an epigenetic memory, able to create changes in DNA methylation, post-translational modification (e.g., Sirtuin activation/repression), and miRNA deregulations. Hyperglycemia also promotes endothelial dysfunctions, with activation of inflammatory pathways with involvement of molecular and epigenetic targets, resulting in imbalance in vascular homeostasis. The initial stimulus, coupled with endothelial and mitochondrial dysfunction, triggers a vicious cycle within the kidney, producing oxidative stress, accumulation of nephrotoxins, and chronic inflammation. These factors could contribute to the progression of chronic kidney disease, with fibrosis, senescence, apoptosis, ultimately predisposing to the onset of end-stage renal disease. Moreover, the systemic nature of endothelial dysfunction gives rise to widespread vascular complications, classified in microvascular and macrovascular complications, such as retinopathy and artery disease. The dysfunction of cardiomyocytes and of vascular smooth muscle cells can be involved, finally, in structural damages and adverse cardiac outcomes, such as atherosclerotic cardiovascular disease, left ventricular hypertrophy, and heart failure. CKD, chronic kidney disease; ESRD, end-stage renal disease; miRNA, microRNA; DNMT, DNA methyltransferase; SOD2, superoxide dismutase-2; Mfn2, mitofusin-2; EZH2, Enhancer of Zeste Homolog-2; SET7, SET Domain Containing 7; mtDNA, mitochondrial DNA; ROS, reactive oxygen species; TCA, tricarboxylic acid; ASCVD, atherosclerotic cardiovascular disease; LVH, left ventricular hypertrophy; HF, heart failure.
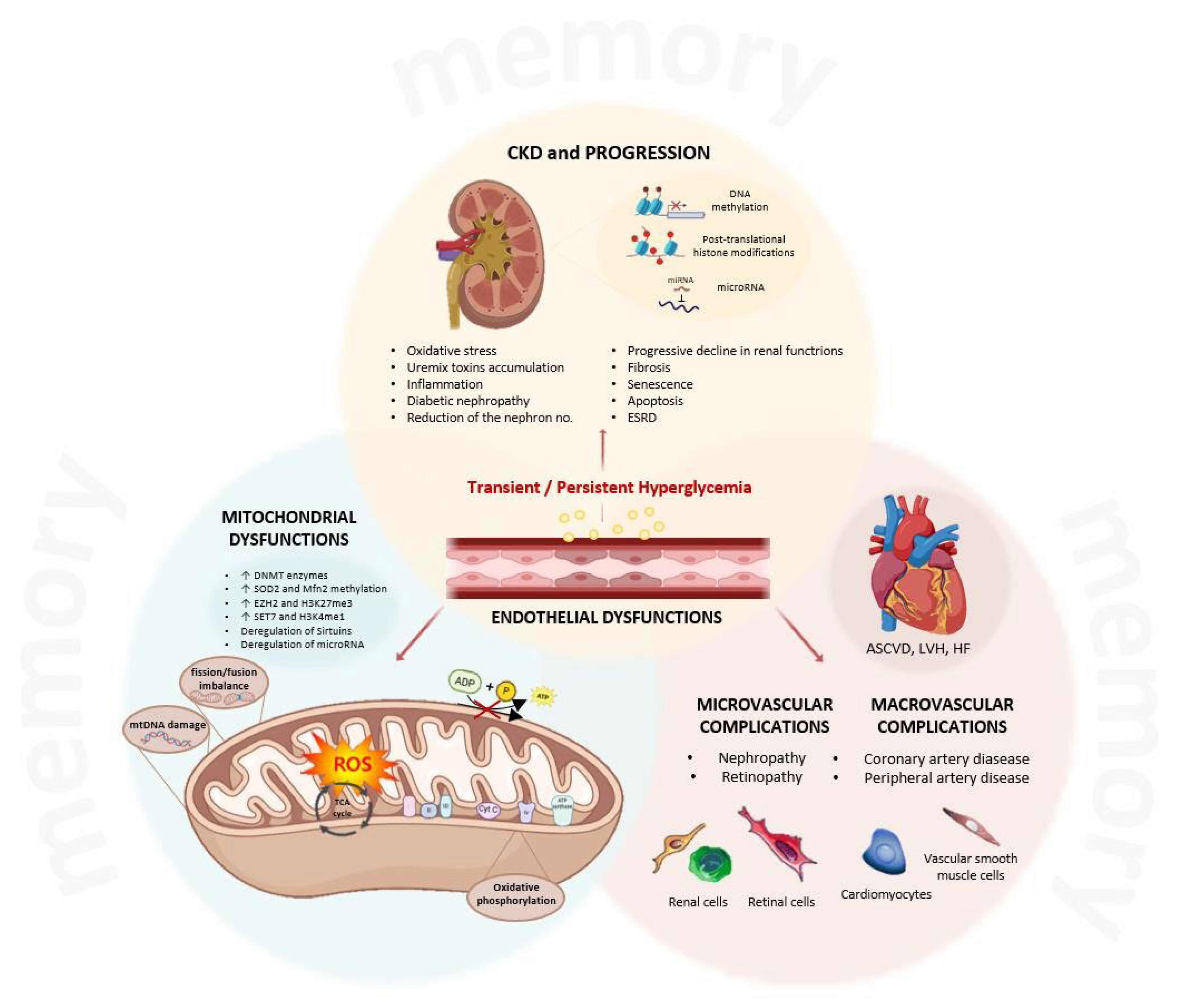
5. Epigenetic Players as Therapeutic Targets and Biomarkers for Ckd Patients’ Stratification
Epigenetic molecules act cooperatively to preserve the integrity of the epigenetic machinery and maintain cell identity. They can be categorized functionally as: i) Writers, which add chemical marks (e.g., methyl or acetyl groups) to DNA or histones, including DNA methyltransferases (DNMTs), histone acetyltransferases (HATs), and histone methyltransferases (HMTs); ii) Readers, which recognize and bind specific epigenetic marks, such as bromodomains (acetyl-lysine readers) and chromodomains (methyl-lysine readers); and iii) Erasers, which remove post-translational modifications, including histone deacetylases (HDACs) and histone demethylases (HDMs) [126,127].
The therapeutic targeting of these regulators has emerged as a promising pharmacological avenue. Several epigenetic modulators—collectively known as epidrugs—have already achieved clinical success in oncology. DNMT inhibitors (DNMTi; e.g., azacytidine, decitabine) and HDAC inhibitors (HDACi; e.g., vorinostat, panobinostat) have been approved for the treatment of myelodysplastic syndromes, leukemias, and other hematologic malignancies [127,128,129,130]. The current landscape of FDA-approved epidrugs includes a wide range of compounds (Table 2), and numerous inhibitors (e.g., ivaltinostat, AR-42, abexinostat, bisthianostat, valproic acid) are undergoing clinical evaluations.
Combination strategies integrating epidrugs with traditional chemotherapeutics are being explored to curb both solid and hematologic tumor progression [131,132,133,134]. Beyond oncology, epigenetic modulators are being investigated for a variety of conditions, including cardiovascular, neurodevelopmental, and neurodegenerative disorders [135,136]. Originally designed for cancer, these compounds also exhibit potential for mitigating chronic inflammation, oxidative stress, and fibrotic remodeling - key mechanisms underpinning metabolic memory and CKD progression [137,138,139,140]. Because epigenetic marks are reversible, targeting the enzymes that write, read, and erase them, including through miRNA mimics or inhibitors, offers a promising therapeutic strategy for managing CKD and its complications, and supports the development of precision medicine approaches for high-risk patients.
5.1. Epigenetic Biomarkers in Ckd and Diabetes
Early biomarker identification is critical for timely diagnosis, intervention, and prevention of progression to ESRD. Epigenome-wide association studies (EWAS) in individuals newly diagnosed with diabetes have uncovered blood-based epigenetic biomarkers capable of: i) early risk stratification, ii) predicting CKD development, and iii) monitoring disease progression. To elucidate the relationship between metabolic memory and epigenetic modifications, several genome-wide studies based on samples from the DCCT/EDIC cohort have been conducted [141]:
1) In a first study, patients exhibited elevated levels of the active chromatin mark H3K9ac in monocytes, correlated with baseline hyperglycemia and the upregulation of genes linked to inflammation and vascular complications. H3K9ac thus emerged as a potential biomarker for metabolic memory [142];
2) In a second study, DNA methylation profiling at two time points revealed 12 persistently differentially methylated loci, notably TXNIP, which was hypomethylated in patients with more complications [143]. Other validations confirmed a strong inverse correlation between TXNIP methylation and glycemic control, suggesting that TXNIP hypomethylation reflects chronic hyperglycemia and may predict renal and vascular damage [144];
3) In a third study, longitudinal analysis (18-year follow-up) showed that specific HbA1c-associated CpGs predicted diabetic kidney disease (DKD) risk, again highlighting TXNIP as a key biomarker [145,146].
These findings, corroborated by studies across tissues such as kidney, nerves, and retina, suggest TXNIP as a pivotal player in metabolic memory and a target for intervention [84,147,148].
A longitudinal study from Lund University followed 752 newly diagnosed type 2 diabetes patients, identifying over 400 differentially methylated sites associated with cardiovascular events, 87 of which were used to build a methylation risk score (MRS) for cardiovascular risk prediction [149]. Similar EWAS have identified differential methylation in genes regulating mitochondrial function, fibrosis, and inflammation, molecular hallmarks of persistent renal injury [97,150,151,152,153].
A methylation score proposed by Marchiori et al., derived from 37 methylation sites, predicted CKD incidence during an 11.5-years follow-up [154]. Moreover, aberrant DNA methylation patterns in key genes (mTOR, RPTOR, IRS2, GRK5, LCAT, SLC27A3, SLC1A5) have been linked to DKD progression [155]. Elevated DNMT1 expression in DKD mononuclear cells induces mTOR-related methylation changes and inflammation, reversible through DNMT1 inhibition with 5-aza-2’-deoxycytidine, offering a new therapeutic perspective [156].
Different compounds have been proven to act on the key mechanisms of hyperglycemic memory, such as epigenetic modifications, inflammation, and senescence, but none have been approved for CKD and complications. The DNMT1 inhibitor SGI-1027 disrupts p21 methylation, attenuating senescence and fibrosis in DKD models [157,158]. Histone methylation also contributes to metabolic memory. Downregulation of the demethylase JMJD3 (also called KDM6B) correlates with neointimal hyperplasia in CKD-related vascular lesions [159]. JMJD3 loss promotes EndMT, vascular remodeling, and nitric oxide deficiency. Additionally, EZH2 upregulation, with concurrent loss of UTX/JMJD3, represses antioxidant genes (SOD1/2, JunD), exacerbating oxidative stress [160]. In fact, its upregulation is linked to tubular cell injury, podocyte dedifferentiation, fibroblast proliferation, inflammatory cytokines/chemokines production and infiltration of inflammatory cells [161]; moreover, depleting EZH2 from podocyte culture in high-glucose conditions derepressed the endogenous antioxidant TXNIP, abrogating ROS accumulation [162]. These findings make EZH2 as a legitimate therapeutic target; however, no current clinical trials of EZH2 inhibitors are available for treating hyperglycemia-induced CKD.
Aberrant activity of BET (bromodomain and extra-terminal domain) proteins—particularly BRD2, BRD3, and BRD4—also sustains vascular and renal injury. BET inhibitors (BETi) such as JQ1 and Apabetalone (RVX-208) have shown preclinical efficacy in DKD and diabetic cardiomyopathy. The selective pharmacological inhibition of BRD4 with JQ1 restored mitochondrial functions via the PTEN-induced PINK1/Parkin mitophagy pathway [163,164]. Besides, the phase 3 BETonMACE trial (ClinicalTrials.gov ID: NCT02586155) tested Apabetalone in type 2 diabetes, with or without CKD, focusing on cardiovascular complications. While it did not significantly reduce major adverse cardiac events overall, it markedly reduced heart failure hospitalizations, particularly among CKD patients’ subgroup [165]. Further clinical studies are needed to confirm these brilliant results in a larger patients’ sample; nevertheless, future clinical settings must be adjusted to reduce side effects [166].
5.2. Histone Deacetylase Inhibitors and Mirna-Based Therapies
HDAC inhibitors are gaining interest in diabetic nephropathy [43,167]. Non-selective inhibitors such as vorinostat (SAHA) improved renal function and reduced fibrosis in animal models [168]. Another HDACi Valproic acid (VPA), used for epilepsy and neurologic disorders, was demonstrated to repress the NF-κB pathway, inactivating inflammation and alleviating podocytes and renal cell injury [169]. The same effects on inflammation and fibrosis have been observed with the HDACi trichostatin A (TSA); this compound was able to influence the metabolic memory through the inhibition of TGF-β1, the decrease of extracellular matrix deposition and the escape from the phenomenon of EMT [170]. SIRT1, a class III HDAC, regulates metabolic memory by deacetylating p53 and modulating apoptosis, inflammation, and oxidative stress [99,171]; its activity is negatively regulated by miR-200a-3p, whose inhibition mitigates tubular injury, suggesting its biomarker potential for diabetes- and hypertension-related nephropathy [172].
MicroRNAs (miRNAs) are both biomarkers and therapeutic targets in DKD. Two strategies have emerged:
· Inhibition of overexpressed miRNAs (via antisense oligonucleotides, gene knockouts, or “sponges”);
· Restoration of protective miRNAs (via doublestranded mimics or expression vectors).
Examples include i) miR-29b mimics, that interfere with Sp1-dependent, TGF-β/Smad3-mediated renal fibrosis, NF-κB–driven renal inflammation, and Th1-associated immune injury [173,174]; and ii) inhibitors of miR-21 and miR-192, which reduce albuminuria and renal fibrosis, maintaining the structural and functional integrity of the kidney [175,176]. Circulating miRNAs such as miR-1, miR-133a, and miR-126 serve as non-invasive biomarkers for diabetic cardiomyopathy and cardiovascular risk [177,178].
In a study by Al-Kafaji et al., diabetic nephropathy was associated with upregulation of miR-377 and downregulation of miR-192, correlating with albuminuria severity and identifying miR-377 as a positive, and miR-192 as a negative, biomarker of DKD [179]. Recent approaches advocate miRNA panels (e.g., miR-21, miR-34a, miR-133a) to enhance diagnostic and prognostic power, bridging therapy and diagnostics - theranostics - for patient stratification [151,180].
5.3. Integrating Machine Learning and Multi-Omics
Advances in artificial intelligence now enable integration of epigenetic, genomic, transcriptomic, and metabolomic data to predict disease risk. For instance, in the MESA study (ClinicalTrials.gov ID: NCT00005487), machine learning models combining epigenetic and imaging data successfully predicted cardiovascular outcomes [181].
6. Conclusions
Further studies are urgently needed to validate candidate biomarkers and identify safer, more effective epigenetic drugs. Integrating multi-omics data will allow stratification of patients predisposed to vascular complications and enhance precision medicine in CKD. Understanding the dynamic role of epigenetic regulation is pivotal for predicting disease trajectory, preventing toxin-induced CKD progression, and overcoming micro- and macrovascular complications.
Author Contributions
Conceptualization, S.C. and I. G.; writing - original draft preparation, S.C., I.G., and A.P.; writing - review and editing, S.C. and I.G.; visualization, M.D., F.S., and A.R.; supervision, A.P., I.G. and M.M-P.; All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to this work has received funding from the European Union—Next Generation EU through the Italian Ministry of University and Research under PNRR—M4C2- I1.3 Project PE_00000019 “HEAL ITALIA” to Lorenzo Lo Muzio CUP UNIFG D73C22001230006. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
The figures were created with BioRender.com
Abbreviations
The following abbreviations are used in this manuscript:
| AGE | Advanced glycation end products |
| AMPK | AMP-activated protein kinase |
| AKI | Acute kidney injury |
| AKT | Protein kinase B |
| AR-42 | Histone Deacetylase Inhibitor AR-42 (or HDAC-42) |
| ARE | Antioxidant Response Element |
| AUH | AU RNA binding methylglutaconyl-CoA hydratase |
| BD1 | Bromodomain1 |
| BD2 | Bromodomain2 |
| BET | Bromodomain and extra-terminal domain |
| BRD | Bromodomain containing protein |
| CBX | Polycomb group protein chromobox |
| CaMK2a | Calcium/Calmodulin-Dependent Protein Kinase II alpha |
| CKD | Chronic Kidney Disease |
| COX6A1 | Cytochrome C Oxidase Subunit 6A1 |
| CpG | Cytosine-phosphate-guanine |
| DAPK3 | Death-Associated Protein Kinase 3 |
| DCCT | Diabetes Control and Complications Trial |
| DMNP | DNA Methyltransferases |
| EDIC | Epidemiology of Diabetes Interventions and Complications Study |
| ELK1 | ETS Transcription Factor ELK1 |
| eGFR | Estimated Glomerular Filtration Rate |
| EndMT | Endothelial-to-mesenchymal transition |
| eNOS | Endothelial nitric oxide synthase |
| ERKs | Extracellular signal-regulated kinases |
| ESRD | End-stage renal disease |
| EWAS | Epigenetic-wide association study |
| EZH2 | Enhancer of Zeste Homolog-2 |
| GFR | Glomerular filtration rate |
| GRK5 | G protein-coupled receptor kinase 5 |
| HATs | Histone acetyltransferases |
| H2A | Histone 2A |
| H2B | Histone 2B |
| H3 | Histone 3 |
| H4 | Histone 4 |
| HbA1c | Glycated haemoglobin |
| HDAC | Histone deacetylase |
| H3K9ac | Histone H3 Lysine 9 acetylation |
| H3K4me3 | Histone H3 lysine 4 trimethylation |
| H3K4me1 | Histone H3 Lysine 4 monomethylation |
| H3K27me3 | Histone H3 Lysine 27 trimethylation |
| H3K9me3 | Histone H3 Lysine 9 trimethylation |
| HO-1 | Heme oxygenase-1 |
| IRS2 | Insulin Receptor Substrate 2 |
| JK | Janus Kinase |
| JMJD3 | Lysine demethylase 6B |
| KDIGO | Kidney Disease Improving Global Outcomes Guidelines |
| KDM | Lysine demethylase |
| KDOQI | Kidney Disease Outcomes Quality Initiative Guidelines |
| KMT | Lysine methyltransferase |
| LCAT | Lecithin Cholesterol Acyltransferase |
| LKB1 | Liver kinase B1 |
| MeCP2 | Methyl-CpG-binding protein |
| MAPK | Mitogen-Activated Protein Kinase |
| MESA | Multi-Ethnic Study of Atherosclerosis |
| miRna | microRNA |
| mtDNA | Mitochondrial DNA |
| mtROS | Mitochondrial Reactive Oxygen Species |
| mTor | Mechanistic Target of Rapamycin |
| mTORC | Target of rapamycin complex |
| NAD+ | Nicotinamide Adenine Dinucleotide |
| Nox1 | NADPH oxidase 1 |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cell |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| PARP | Poly (ADP-ribose) Polymerase |
| P21 | cyclin-dependent kinase inhibitor 1 |
| PINK | PTEN-induced kinase 1 |
| PMPCB | Mitochondrial-processing peptidase subunit beta |
| PRC2 | Polycomb Repressive Complex 2 |
| PTEN | Phosphatase and TENsin homolog |
| PTMs | Post-Translational Modifications |
| RAGE | glycation end-products receptor |
| ROS | Reactive Oxygen Species |
| RPTOR | Regulatory Associated Protein of MTOR complex 1 |
| SAM | S-adenosyl-l-methionine |
| SGI-1027 | DNA Methyltransferase Inhibitor II |
| SIRT | Sirtuin, Silent Mating Type Information Regulation 2 Homolog |
| SLC1A5 | Solute Carrier Family 1 (neutral amino acid transporter), Member 5 |
| SLC27A3 | Solute Carrier Family 27 Member 3 |
| SOD | Superoxide Dismutase |
| TAMM41 | TAM41 Mitochondrial Translocator Assembly and Maintenance Homolog |
| TET | Ten-eleven translocation |
| TSA | Trichostatin A |
| TSFM | Ts translation elongation factor, mitochondrial |
| TXNIP | Thioredoxin-interacting protein |
| UKPDs | United Kingdom Prospective Diabetes Study |
| VPA | Valproic acid |
References
- Inker, L. A.; Astor, B. C.; Fox, C. H.; Isakova, T.; Lash, J. P.; Peralta, C. A.; Kurella Tamura, M.; Feldman, H. I., KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis 2014, 63, (5), 713-35. [CrossRef]
- Rysz, J.; Franczyk, B.; Rysz-Gorzynska, M.; Gluba-Brzozka, A., Are Alterations in DNA Methylation Related to CKD Development? Int J Mol Sci 2022, 23, (13). [CrossRef]
- Gilg, J.; Rao, A.; Fogarty, D., UK Renal Registry 16th annual report: chapter 1 UK renal replacement therapy incidence in 2012: national and centre-specific analyses. Nephron Clin Pract 2014, 125, (1-4), 1-27. [CrossRef]
- Coffman, T. M., Under pressure: the search for the essential mechanisms of hypertension. Nat Med 2011, 17, (11), 1402-9. [CrossRef]
- Ho, H. J.; Shirakawa, H., Oxidative Stress and Mitochondrial Dysfunction in Chronic Kidney Disease. Cells 2022, 12, (1), 88. [CrossRef]
- Levey, A. S.; Stevens, L. A.; Coresh, J., Conceptual model of CKD: applications and implications. Am J Kidney Dis 2009, 53, S4-16. [CrossRef]
- Amorim, R. G.; Guedes, G. D. S.; Vasconcelos, S. M. L.; Santos, J. C. F., Kidney Disease in Diabetes Mellitus: Cross-Linking between Hyperglycemia, Redox Imbalance and Inflammation. Arq Bras Cardiol 2019, 112, (5), 577-587. [CrossRef]
- Reidy, K.; Kang, H. M.; Hostetter, T.; Susztak, K., Molecular mechanisms of diabetic kidney disease. J Clin Invest 2014, 124, (6), 2333-40. [CrossRef]
- Turkmen, K., Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: the Four Horsemen of the Apocalypse. Int Urol Nephrol 2017, 49, (5), 837-844. [CrossRef]
- Kato, M.; Natarajan, R., Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol 2019, 15, (6), 327-345. [CrossRef]
- Giacco, F.; Brownlee, M., Oxidative stress and diabetic complications. Circ Res 2010, 107, (9), 1058-70.
- Yapislar, H.; Gurler, E. B., Management of Microcomplications of Diabetes Mellitus: Challenges, Current Trends, and Future Perspectives in Treatment. Biomedicines 2024, 12, (9), 1958. [CrossRef]
- Pal, R.; Bhadada, S. K., AGEs accumulation with vascular complications, glycemic control and metabolic syndrome: A narrative review. Bone 2023, 176, 116884. [CrossRef]
- Ng, Z. X.; Kuppusamy, U. R.; Iqbal, T.; Chua, K. H., Receptor for advanced glycation end-product (RAGE) gene polymorphism 2245G/A is associated with pro-inflammatory, oxidative-glycation markers and sRAGE in diabetic retinopathy. Gene 2013, 521, (2), 227-33. [CrossRef]
- de Souza Ferreira, C.; Pennacchi, P. C.; Araujo, T. H.; Taniwaki, N. N.; de Araujo Paula, F. B.; da Silveira Duarte, S. M.; Rodrigues, M. R., Aminoguanidine treatment increased NOX2 response in diabetic rats: Improved phagocytosis and killing of Candida albicans by neutrophils. Eur J Pharmacol 2016, 772, 83-91. [CrossRef]
- Ceriello, A., Hypothesis: the "metabolic memory", the new challenge of diabetes. Diabetes Res Clin Pract 2009, 86, (1), S2-6.
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P. L.; Roeder, R. G.; Cooper, M. E.; Brownlee, M., Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 2008, 205, (10), 2409-17.
- Luna, P.; Guarner, V.; Farias, J. M.; Hernandez-Pacheco, G.; Martinez, M., Importance of Metabolic Memory in the Development of Vascular Complications in Diabetic Patients. J Cardiothorac Vasc Anesth 2016, 30, (5), 1369-78. [CrossRef]
- Testa, R.; Bonfigli, A. R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A., The "Metabolic Memory" Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, (5), 437. [CrossRef]
- Dong, H.; Sun, Y.; Nie, L.; Cui, A.; Zhao, P.; Leung, W. K.; Wang, Q., Metabolic memory: mechanisms and diseases. Signal Transduct Target Ther 2024, 9, (1), 38. [CrossRef]
- Roy, S.; Sala, R.; Cagliero, E.; Lorenzi, M., Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci U S A 1990, 87, (1), 404-8. [CrossRef]
- Lachin, J.M.; Bebu, I.; Nathan, D.M.; DCCT/EDIC Research Group, The Beneficial Effects of Earlier Versus Later Implementation of Intensive Therapy in Type 1 Diabetes. Diabetes Care 2021, 44, (10), 2225–30. [CrossRef]
- Wilson-Verdugo, M.; Bustos-Garcia, B.; Adame-Guerrero, O.; Hersch-Gonzalez, J.; Cano-Dominguez, N.; Soto-Nava, M.; Acosta, C. A.; Tusie-Luna, T.; Avila-Rios, S.; Noriega, L. G.; Valdes, V. J., Reversal of high-glucose-induced transcriptional and epigenetic memories through NRF2 pathway activation. Life Sci Alliance 2024, 7, (8), e202302382. [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X. L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M. A.; Beebe, D.; Oates, P. J.; Hammes, H. P.; Giardino, I.; Brownlee, M., Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, (6779), 787-90. [CrossRef]
- Ihnat, M.A.; Thorpe, J.E.; Kamat, C.D.; Szabó, C.; Green, D.E.; Warnke, L.A.; Lacza, Z.; Cselenyák, A.; Ross, K.; Shakir, S.; Piconi, L.; Kaltreider, R.C.; Ceriello, A., Reactive oxygen species mediate a cellular 'memory' of high glucose stress signalling. Diabetologia 2007, 50, (7), 1523-31. [CrossRef]
- Brownlee, M., The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005, 54, (6), 1615-25.
- Araki, E.; Nishikawa, T., Oxidative stress: A cause and therapeutic target of diabetic complications. J Diabetes Investig 2010, 1, (3), 90-6. [CrossRef]
- Caja, S.; Enríquez, J.A., Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol 2017, 12, 821-827. [CrossRef]
- Cannito, S.; Giardino, I.; d'Apolito, M.; Pettoello-Mantovani, M.; Scaltrito, F.; Mangieri, D.; Piscazzi, A., The Multifaceted Role of Mitochondria in Angiogenesis. Int J Mol Sci 2025, 26, (16), 7960. [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J., Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006, 1757, (5-6), 509-17. [CrossRef]
- Kowluru, R.A.; Alka, K., Mitochondrial Quality Control and Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Int J Mol Sci 2023, 24, (9), 8076. [CrossRef]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H., Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight 2019, 5, 4, (23), e129760. [CrossRef]
- Kowluru, R.A.; Mohammad, G.; Kumar, J., Impaired Removal of the Damaged Mitochondria in the Metabolic Memory Phenomenon Associated with Continued Progression of Diabetic Retinopathy. Mol Neurobiol 2024, 61, (1), 188-199. [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T., MicroRNA-Based Diagnosis and Therapy. Int J Mol Sci 2022, 23, (13), 7167.
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Spitler, K.; Goulopoulou, S.; Webb, R.C., Working Group on DAMPs in Cardiovascular Disease. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J 2014, 35, 1172–1177. [CrossRef]
- Giardino, I.; d'Apolito, M.; Brownlee, M.; Maffione, A.B.; Colia, A.L.; Sacco, M.; Ferrara, P.; Pettoello-Mantovani, M., Vascular toxicity of urea, a new "old player" in the pathogenesis of chronic renal failure induced cardiovascular diseases. Turk Pediatri Ars 2017, 52, (4), 187-193. [CrossRef]
- Vercellino, I.; Sazanov, L.A., The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol 2022, 23, (2), 141-161. [CrossRef]
- Dai, W.; Lu, H.; Chen, Y.; Yang, D.; Sun, L.; He, L., The Loss of Mitochondrial Quality Control in Diabetic Kidney Disease. Front Cell Dev Biol 2021, 9, 706832. [CrossRef]
- Dong, H.; Sun, Y.; Nie, L.; Cui, A.; Zhao, P.; Leung, W.K.; Wang, Q., Metabolic memory: mechanisms and diseases. Signal Transduct Target Ther 2024, 9, (1), 38. [CrossRef]
- Yang, T.; Qi, F.; Guo, F.; Shao, M.; Song, Y.; Ren, G.; Linlin, Z.; Qin, G.; Zhao, Y., An update on chronic complications of diabetes mellitus: from molecular mechanisms to therapeutic strategies with a focus on metabolic memory. Mol Med 2024, 30, (1), 71. [CrossRef]
- Schisano, B.; Tripathi, G.; McGee, K.; McTernan, P.G.; Ceriello, A., Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, (5), 1219-26. [CrossRef]
- Tonna, S.; El-Osta, A.; Cooper, M.E.; Tikellis, C., Metabolic memory and diabetic nephropathy: potential role for epigenetic mechanisms. Nat Rev Nephrol 2010, 6, (6), 332-41. [CrossRef]
- Kushwaha, K.; Garg, S. S.; Gupta, J., Targeting epigenetic regulators for treating diabetic nephropathy. Biochimie 2022, 202, 146-158. [CrossRef]
- Siebel, A. L.; Fernandez, A. Z.; El-Osta, A., Glycemic memory associated epigenetic changes. Biochem Pharmacol 2010, 80, (12), 1853-9. [CrossRef]
- Reddy, M. A.; Zhang, E.; Natarajan, R., Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, (3), 443-55. [CrossRef]
- Tanemoto, F.; Nangaku, M.; Mimura, I., Epigenetic memory contributing to the pathogenesis of AKI-to-CKD transition. Front Mol Biosci 2022, 9, 1003227. [CrossRef]
- Esteller, M., Epigenetics in cancer. N Engl J Med 2008, 358, (11), 1148-59. [CrossRef]
- Li, E.; Bestor, T. H.; Jaenisch, R., Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, (6), 915-26. [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P. A., Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, (6990), 457-63. [CrossRef]
- Robertson, K. D., DNA methylation and chromatin - unraveling the tangled web. Oncogene 2002, 21, (35), 5361-79. [CrossRef]
- Okano, M.; Bell, D. W.; Haber, D. A.; Li, E., DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, (3), 247-57. [CrossRef]
- Dabrowski, M. J.; Wojtas, B., Global DNA Methylation Patterns in Human Gliomas and Their Interplay with Other Epigenetic Modifications. Int J Mol Sci 2019, 20, (14), 3478. [CrossRef]
- Toth, D. M.; Szeri, F.; Ashaber, M.; Muazu, M.; Szekvolgyi, L.; Aranyi, T., Tissue-specific roles of de novo DNA methyltransferases. Epigenetics Chromatin 2025, 18, (1), 5. [CrossRef]
- Yang, L.; Yu, S. J.; Hong, Q.; Yang, Y.; Shao, Z. M., Reduced Expression of TET1, TET2, TET3 and TDG mRNAs Are Associated with Poor Prognosis of Patients with Early Breast Cancer. PLoS One 2015, 10, (7), e0133896. [CrossRef]
- Zhang, X.; Zhang, Y.; Wang, C.; Wang, X., TET (Ten-eleven translocation) family proteins: structure, biological functions and applications. Signal Transduct Target Ther 2023, 8, (1), 297. [CrossRef]
- Sufiyan, S.; Salam, H.; Ilyas, S.; Amin, W.; Arshad, F.; Fatima, K.; Naeem, S.; Laghari, A. A.; Enam, S. A.; Mughal, N., Prognostic implications of DNA methylation machinery (DNMTs and TETs) expression in gliomas: correlations with tumor grading and patient survival. J Neurooncol 2025, 173, (3), 667-682. [CrossRef]
- Kouzarides, T., Chromatin modifications and their function. Cell 2007, 128, (4), 693-705.
- Hermann, J.; Schurgers, L.; Jankowski, V., Identification and characterization of post-translational modifications: Clinical implications. Mol Aspects Med 2022, 86, 101066. [CrossRef]
- Ji, Y.; Liu, S.; Zhang, Y.; Min, Y.; Wei, L.; Guan, C.; Yu, H.; Zhang, Z., Lysine crotonylation in disease: mechanisms, biological functions and therapeutic targets. Epigenetics Chromatin 2025, 18, (1), 13. [CrossRef]
- Raju, C.; Sankaranarayanan, K., Insights on post-translational modifications in fatty liver and fibrosis progression. Biochim Biophys Acta Mol Basis Dis 2025, 1871, (3), 167659. [CrossRef]
- Shiio, Y.; Eisenman, R. N., Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A 2003, 100, (23), 13225-30. [CrossRef]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y., The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, (7144), 601-5. [CrossRef]
- Kaniskan, H. U.; Martini, M. L.; Jin, J., Inhibitors of Protein Methyltransferases and Demethylases. Chem Rev 2018, 118, (3), 989-1068. [CrossRef]
- Bu, C.; Xie, Y.; Weng, J.; Sun, Y.; Wu, H.; Chen, Y.; Ye, Y.; Zhou, E.; Yang, Z.; Wang, J., Inhibition of JMJD3 attenuates acute liver injury by suppressing inflammation and oxidative stress in LPS/D-Gal-induced mice. Chem Biol Interact 2025, 418, 111576. [CrossRef]
- Cao, Y.; Li, Y.; Zhang, N.; Hu, J.; Yin, L.; Pan, Z.; Li, Y.; Du, X.; Zhang, W.; Li, F., Quantitative DNA hypomethylation of ligand Jagged1 and receptor Notch1 signifies occurrence and progression of breast carcinoma. Am J Cancer Res 2015, 5, (5), 1621-34.
- Tycko, B., Epigenetic gene silencing in cancer. J Clin Invest 2000, 105, (4), 401-7.
- Urli, T.; Greenberg, M. V. C., Epigenetic relay: Polycomb-directed DNA methylation in mammalian development. PLoS Genet 2025, 21, (9), e1011854. [CrossRef]
- Wiles, E. T.; Selker, E. U., H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev 2017, 43, 31-37. [CrossRef]
- Sharma, S.; Hampton, J. T.; Kutateladze, T. G.; Liu, W. R., Epigenetic reader chromodomain as a potential therapeutic target. RSC Chem Biol 2025, 6, (6), 833-844.
- Angrand, P. O., Structure and Function of the Polycomb Repressive Complexes PRC1 and PRC2. Int J Mol Sci 2022, 23, (11). [CrossRef]
- Seto, E.; Yoshida, M., Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014, 6, (4), a018713. [CrossRef]
- Cedar, H.; Bergman, Y., Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009, 10, (5), 295-304. [CrossRef]
- Esteve, P. O.; Chin, H. G.; Benner, J.; Feehery, G. R.; Samaranayake, M.; Horwitz, G. A.; Jacobsen, S. E.; Pradhan, S., Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc Natl Acad Sci U S A 2009, 106, (13), 5076-81. [CrossRef]
- Feng, Q.; Zhang, Y., The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev 2001, 15, (7), 827-32.
- Tan, J. Z.; Yan, Y.; Wang, X. X.; Jiang, Y.; Xu, H. E., EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin 2014, 35, (2), 161-74. [CrossRef]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y. Y., MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta 2010, 1803, (11), 1231-43. [CrossRef]
- Macgregor-Das, A. M.; Das, S., A microRNA's journey to the center of the mitochondria. Am J Physiol Heart Circ Physiol 2018, 315, (2), H206-H215. [CrossRef]
- Tang, J.; Yan, H.; Zhuang, S., Histone deacetylases as targets for treatment of multiple diseases. Clin Sci (Lond) 2013, 124, (11), 651-62. [CrossRef]
- Ibanez-Cabellos, J. S.; Pallardo, F. V.; Garcia-Gimenez, J. L.; Seco-Cervera, M., Oxidative Stress and Epigenetics: miRNA Involvement in Rare Autoimmune Diseases. Antioxidants (Basel) 2023, 12, (4), 800. [CrossRef]
- Dwivedi, R. S.; Herman, J. G.; McCaffrey, T. A.; Raj, D. S., Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int 2013, 79, (1), 23-32. [CrossRef]
- Wing, M. R.; Ramezani, A.; Gill, H. S.; Devaney, J. M.; Raj, D. S., Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol 2014, 33, (4), 363-74. [CrossRef]
- Wang, J.; Shen, F.; Liu, F.; Zhuang, S., Histone Modifications in Acute Kidney Injury. Kidney Dis (Basel) 2022, 8, (6), 466-477. [CrossRef]
- Villeneuve, L. M.; Reddy, M. A.; Lanting, L. L.; Wang, M.; Meng, L.; Natarajan, R., Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A 2008, 105, (26), 9047-52. [CrossRef]
- De Marinis, Y.; Cai, M.; Bompada, P.; Atac, D.; Kotova, O.; Johansson, M. E.; Garcia-Vaz, E.; Gomez, M. F.; Laakso, M.; Groop, L., Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 2016, 89, (2), 342-53. [CrossRef]
- Mishra, M.; Kowluru, R. A., The Role of DNA Methylation in the Metabolic Memory Phenomenon Associated With the Continued Progression of Diabetic Retinopathy. Invest Ophthalmol Vis Sci 2016, 57, (13), 5748-5757. [CrossRef]
- Liao, Y.; Gou, L.; Chen, L.; Zhong, X.; Zhang, D.; Zhu, H.; Lu, X.; Zeng, T.; Deng, X.; Li, Y., NADPH oxidase 4 and endothelial nitric oxide synthase contribute to endothelial dysfunction mediated by histone methylations in metabolic memory. Free Radic Biol Med 2018, 115, 383-394. [CrossRef]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E. K.; Calkin, A. C.; Brownlee, M.; Cooper, M. E.; El-Osta, A., Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, (5), 1229-36. [CrossRef]
- Zeng, S.; Wu, X.; Chen, X.; Xu, H.; Zhang, T.; Xu, Y., Hypermethylated in cancer 1 (HIC1) mediates high glucose induced ROS accumulation in renal tubular epithelial cells by epigenetically repressing SIRT1 transcription. Biochim Biophys Acta Gene Regul Mech 2018, 1861, (10), 917-927. [CrossRef]
- Rossi, M. N.; Ciolfi, A.; Matteo, V.; Pedace, L.; Nardini, C.; Loricchio, E.; Caiello, I.; Bellomo, F.; Taranta, A.; De Leo, E.; Tartaglia, M.; Emma, F.; De Benedetti, F.; Miele, E.; Prencipe, G., Genome-wide DNA methylation analysis identifies kidney epigenetic dysregulation in a cystinosis mouse model. Front Cell Dev Biol 2025, 13, 1638123. [CrossRef]
- Stenvinkel, P.; Ekstrom, T. J., Does the uremic milieu affect the epigenotype? J Ren Nutr 2009, 19, (1), 82-5. [CrossRef]
- Kato, S.; Lindholm, B.; Stenvinkel, P.; Ekstrom, T. J.; Luttropp, K.; Yuzawa, Y.; Yasuda, Y.; Tsuruta, Y.; Maruyama, S., DNA hypermethylation and inflammatory markers in incident Japanese dialysis patients. Nephron Extra 2012, 2, (1), 159-68. [CrossRef]
- Wielscher, M.; Mandaviya, P. R.; Kuehnel, B.; Joehanes, R.; Mustafa, R.; Robinson, O.; Zhang, Y.; Bodinier, B.; Walton, E.; Mishra, P. P.; Schlosser, P.; Wilson, R.; Tsai, P. C.; Palaniswamy, S.; Marioni, R. E.; Fiorito, G.; Cugliari, G.; Karhunen, V.; Ghanbari, M.; Psaty, B. M.; Loh, M.; Bis, J. C.; Lehne, B.; Sotoodehnia, N.; Deary, I. J.; Chadeau-Hyam, M.; Brody, J. A.; Cardona, A.; Selvin, E.; Smith, A. K.; Miller, A. H.; Torres, M. A.; Marouli, E.; Gao, X.; van Meurs, J. B. J.; Graf-Schindler, J.; Rathmann, W.; Koenig, W.; Peters, A.; Weninger, W.; Farlik, M.; Zhang, T.; Chen, W.; Xia, Y.; Teumer, A.; Nauck, M.; Grabe, H. J.; Doerr, M.; Lehtimaki, T.; Guan, W.; Milani, L.; Tanaka, T.; Fisher, K.; Waite, L. L.; Kasela, S.; Vineis, P.; Verweij, N.; van der Harst, P.; Iacoviello, L.; Sacerdote, C.; Panico, S.; Krogh, V.; Tumino, R.; Tzala, E.; Matullo, G.; Hurme, M. A.; Raitakari, O. T.; Colicino, E.; Baccarelli, A. A.; Kahonen, M.; Herzig, K. H.; Li, S.; Conneely, K. N.; Kooner, J. S.; Kottgen, A.; Heijmans, B. T.; Deloukas, P.; Relton, C.; Ong, K. K.; Bell, J. T.; Boerwinkle, E.; Elliott, P.; Brenner, H.; Beekman, M.; Levy, D.; Waldenberger, M.; Chambers, J. C.; Dehghan, A.; Jarvelin, M. R., DNA methylation signature of chronic low-grade inflammation and its role in cardio-respiratory diseases. Nat Commun 2022, 13, (1), 2408. [CrossRef]
- Vujcic, S.; Kotur-Stevuljevic, J.; Vujcic, Z.; Stojanovic, S.; Beljic Zivkovic, T.; Vuksanovic, M.; Marjanovic Petkovic, M.; Perovic Blagojevic, I.; Koprivica-Uzelac, B.; Ilic-Mijailovic, S.; Rizzo, M.; Zeljkovic, A.; Stefanovic, T.; Bosic, S.; Vekic, J., Global DNA Methylation in Poorly Controlled Type 2 Diabetes Mellitus: Association with Redox and Inflammatory Biomarkers. Int J Mol Sci 2025, 26, (14) 6716. [CrossRef]
- Gluck, C.; Qiu, C.; Han, S. Y.; Palmer, M.; Park, J.; Ko, Y. A.; Guan, Y.; Sheng, X.; Hanson, R. L.; Huang, J.; Chen, Y.; Park, A. S. D.; Izquierdo, M. C.; Mantzaris, I.; Verma, A.; Pullman, J.; Li, H.; Susztak, K., Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat Commun 2019, 10, (1), 2461. [CrossRef]
- Ingrosso, D.; Perna, A. F., DNA Methylation Dysfunction in Chronic Kidney Disease. Genes (Basel) 2020, 11, (7), 811. [CrossRef]
- Mimura, I.; Chen, Z.; Natarajan, R., Epigenetic alterations and memory: key players in the development/progression of chronic kidney disease promoted by acute kidney injury and diabetes. Kidney Int 2025, 107, (3), 434-456. [CrossRef]
- Swan, E. J.; Maxwell, A. P.; McKnight, A. J., Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet Med 2015, 32, (8), 1110-5. [CrossRef]
- Imai, S.; Guarente, L., NAD+ and sirtuins in aging and disease. Trends Cell Biol 2014, 24, (8), 464-71. [CrossRef]
- Zheng, Z.; Chen, H.; Li, J.; Li, T.; Zheng, B.; Zheng, Y.; Jin, H.; He, Y.; Gu, Q.; Xu, X., Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes 2012, 61, (1), 217-28. [CrossRef]
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I., Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol 2009, 29, (6), 889-94. [CrossRef]
- Li, Q.; Kim, Y. R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S. K.; Jacobs, J. S.; Irani, K., P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler Thromb Vasc Biol 2017, 36, (12), 2394-2403.
- Huang, K.; Huang, J.; Xie, X.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H., Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-beta1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med 2013, 65, 528-540. [CrossRef]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; Maegawa, H.; Uzu, T., SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med 2011, 51, (6), 1258-67. [CrossRef]
- Juszczak, F.; Arnould, T.; Decleves, A. E., The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases. Int J Mol Sci 2024, 25, (13) 6936. [CrossRef]
- Dikalova, A. E.; Itani, H. A.; Nazarewicz, R. R.; McMaster, W. G.; Flynn, C. R.; Uzhachenko, R.; Fessel, J. P.; Gamboa, J. L.; Harrison, D. G.; Dikalov, S. I., Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ Res 2017, 121, (5), 564-574.
- Dikalova, S.; Dikalova, A., Mitochondrial deacetylase Sirt3 in vascular dysfunction and hypertension. Curr Opin Nephrol Hypertens 2024, 31, (2), 151-156.
- Pacher, P.; Szabo, C., Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal 2008, 7, (11-12), 1568-80. [CrossRef]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S. P.; Koya, D., Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, (6), 2120-31. [CrossRef]
- Wang, X.; Lin, B.; Nie, L.; Li, P., microRNA-20b contributes to high glucose-induced podocyte apoptosis by targeting SIRT7. Mol Med Rep 2017, 16, (4), 5667-5674. [CrossRef]
- Jin, Q.; Ma, F.; Liu, T.; Yang, L.; Mao, H.; Wang, Y.; Peng, L.; Li, P.; Zhan, Y., Sirtuins in kidney diseases: potential mechanism and therapeutic targets. Cell Commun Signal 2024, 22, (1), 114. [CrossRef]
- Zhang, Y.; Zhang, C.; Zhang, H.; Zeng, W.; Li, S.; Chen, C.; Song, X.; Sun, J.; Sun, Z.; Cui, C.; Cao, X.; Zheng, L.; Wang, P.; Zhao, W.; Zhang, Z.; Xu, Y.; Zhu, M.; Chen, H., ZIPK mediates endothelial cell contraction through myosin light chain phosphorylation and is required for ischemic-reperfusion injury. Faseb J 2019, 33, (8), 9062-9074. [CrossRef]
- Li, X.; Liu, J.; Lu, L.; Huang, T.; Hou, W.; Wang, F.; Yu, L.; Wu, F.; Qi, J.; Chen, X.; Meng, Z.; Zhu, M., Sirt7 associates with ELK1 to participate in hyperglycemia memory and diabetic nephropathy via modulation of DAPK3 expression and endothelial inflammation. Transl Res 2022, 247, 99-116. [CrossRef]
- Wegner, M.; Neddermann, D.; Piorunska-Stolzmann, M.; Jagodzinski, P. P., Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res Clin Pract 2014, 105, (2), 164-75. [CrossRef]
- Yang, Y.; Wang, Y.; Fan, X.; Xu, X.; Wang, H.; Wang, X.; Shi, T.; Tang, J.; Guan, Y.; Li, S.; Wang, A., Role of DNA methylation transferase in urinary system diseases: From basic to clinical perspectives (Review). Int J Mol Med 2025, 55, (2) 19. [CrossRef]
- McClelland, A. D.; Kantharidis, P., microRNA in the development of diabetic complications. Clin Sci (Lond) 2014, 126, (2), 95-110. [CrossRef]
- Prattichizzo, F.; Giuliani, A.; De Nigris, V.; Pujadas, G.; Ceka, A.; La Sala, L.; Genovese, S.; Testa, R.; Procopio, A. D.; Olivieri, F.; Ceriello, A., Extracellular microRNAs and endothelial hyperglycaemic memory: a therapeutic opportunity? Diabetes Obes Metab 2016, 18, (9), 855-67. [CrossRef]
- Jin, J.; Wang, X.; Zhi, X.; Meng, D., Epigenetic regulation in diabetic vascular complications. J Mol Endocrinol 2019, 63, (4), R103-R115. [CrossRef]
- Zhong, X.; Liao, Y.; Chen, L.; Liu, G.; Feng, Y.; Zeng, T.; Zhang, J., The MicroRNAs in the Pathogenesis of Metabolic Memory. Endocrinology. 2015, 156, (9), 3157-68. [CrossRef]
- Bera, A.; Das, F.; Ghosh-Choudhury, N.; Mariappan, M. M.; Kasinath, B. S.; Ghosh Choudhury, G., Reciprocal regulation of miR-214 and PTEN by high glucose regulates renal glomerular mesangial and proximal tubular epithelial cell hypertrophy and matrix expansion. Am J Physiol Cell Physiol 2017, 313, (4), C430-C447. [CrossRef]
- Zhang, Y.; Zhao, S.; Wu, D.; Liu, X.; Shi, M.; Wang, Y.; Zhang, F.; Ding, J.; Xiao, Y.; Guo, B., MicroRNA-22 Promotes Renal Tubulointerstitial Fibrosis by Targeting PTEN and Suppressing Autophagy in Diabetic Nephropathy. J Diabetes Res 2018, 2018, 4728645. [CrossRef]
- Ding, M.; Shi, R.; Du, Y.; Chang, P.; Gao, T.; De, D.; Chen, Y.; Li, M.; Li, J.; Li, K.; Cheng, S.; Gu, X.; Li, J.; Zhang, S.; Feng, N.; Liu, J.; Jia, M.; Fan, R.; Pei, J.; Gao, C.; Fu, F., O-GlcNAcylation-mediated endothelial metabolic memory contributes to cardiac damage via small extracellular vesicles. Cell Metab 2025, 37, (6), 1344-1363 e6. [CrossRef]
- Yao, Y.; Song, Q.; Hu, C.; Da, X.; Yu, Y.; He, Z.; Xu, C.; Chen, Q.; Wang, Q. K., Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc Res 2022, 118, (1), 196-211. [CrossRef]
- Pedruzzi, L. M.; Cardozo, L. F.; Daleprane, J. B.; Stockler-Pinto, M. B.; Monteiro, E. B.; Leite, M., Jr.; Vaziri, N. D.; Mafra, D., Systemic inflammation and oxidative stress in hemodialysis patients are associated with down-regulation of Nrf2. J Nephrol 2015, 28, (4), 495-501. [CrossRef]
- Bondi, C. D.; Hartman, H. L.; Tan, R. J., NRF2 in kidney physiology and disease. Physiol Rep 2024, 12, (5), e15961. [CrossRef]
- Intine, R. V.; Sarras, M. P., Jr., Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Curr Diab Rep 2013, 12, (5), 551-9. [CrossRef]
- Yang, A. Y.; Kim, H.; Li, W.; Kong, A. N., Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr Top Med Chem 2017, 16, (7), 697-713. [CrossRef]
- Biswas, S.; Rao, C. M., Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol 2018, 837, 8-24. [CrossRef]
- Contieri, B.; Duarte, B. K. L.; Lazarini, M., Updates on DNA methylation modifiers in acute myeloid leukemia. Ann Hematol 2020, 99, (4), 693-701. [CrossRef]
- Pinto, V.; Bergantim, R.; Caires, H. R.; Seca, H.; Guimaraes, J. E.; Vasconcelos, M. H., Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers (Basel) 2020, 12, (2), 407. [CrossRef]
- Damiani, E.; Duran, M. N.; Mohan, N.; Rajendran, P.; Dashwood, R. H., Targeting Epigenetic 'Readers' with Natural Compounds for Cancer Interception. J Cancer Prev 2020, 25, (4), 189-203. [CrossRef]
- Arce, C.; Perez-Plasencia, C.; Gonzalez-Fierro, A.; de la Cruz-Hernandez, E.; Revilla-Vazquez, A.; Chavez-Blanco, A.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Bargallo, E.; Villarreal, P.; Ramirez, T.; Vela, T.; Candelaria, M.; Camargo, M. F.; Robles, E.; Duenas-Gonzalez, A., A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One 2006, 1, (1), e98. [CrossRef]
- Candelaria, M.; Gallardo-Rincon, D.; Arce, C.; Cetina, L.; Aguilar-Ponce, J. L.; Arrieta, O.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; de la Cruz-Hernandez, E.; Camargo, M. F.; Trejo-Becerril, C.; Perez-Cardenas, E.; Perez-Plasencia, C.; Taja-Chayeb, L.; Wegman-Ostrosky, T.; Revilla-Vazquez, A.; Duenas-Gonzalez, A., A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol 2007, 18, (9), 1529-38.
- Coronel, J.; Cetina, L.; Pacheco, I.; Trejo-Becerril, C.; Gonzalez-Fierro, A.; de la Cruz-Hernandez, E.; Perez-Cardenas, E.; Taja-Chayeb, L.; Arias-Bofill, D.; Candelaria, M.; Vidal, S.; Duenas-Gonzalez, A., A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med Oncol 2011, 28 Suppl 1, S540-6. [CrossRef]
- Candelaria, M.; Herrera, A.; Labardini, J.; Gonzalez-Fierro, A.; Trejo-Becerril, C.; Taja-Chayeb, L.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Arias-Bofill, D.; Vidal, S.; Cervera, E.; Duenas-Gonzalez, A., Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann Hematol 2011, 90, (4), 379-87. [CrossRef]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S., Use of epigenetic drugs in disease: an overview. Genet Epigenet 2014, 6, 9-19. [CrossRef]
- Farsetti, A.; Illi, B.; Gaetano, C., How epigenetics impacts on human diseases. Eur J Intern Med 2023, 114, 15-22. [CrossRef]
- Zhao, H.; Han, Z.; Ji, X.; Luo, Y., Epigenetic Regulation of Oxidative Stress in Ischemic Stroke. Aging Dis 2016, 7, (3), 295-306. [CrossRef]
- Guzik, T. J.; Touyz, R. M., Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, (4), 660-667. [CrossRef]
- Marchant, V.; Trionfetti, F.; Tejedor-Santamaria, L.; Rayego-Mateos, S.; Rotili, D.; Bontempi, G.; Domenici, A.; Mene, P.; Mai, A.; Martin-Cleary, C.; Ortiz, A.; Ramos, A. M.; Strippoli, R.; Ruiz-Ortega, M., BET Protein Inhibitor JQ1 Ameliorates Experimental Peritoneal Damage by Inhibition of Inflammation and Oxidative Stress. Antioxidants (Basel) 2023, 12, (12), 2055. [CrossRef]
- Fraile-Martinez, O.; De Leon-Oliva, D.; Boaru, D. L.; De Castro-Martinez, P.; Garcia-Montero, C.; Barrena-Blazquez, S.; Garcia-Garcia, J.; Garcia-Honduvilla, N.; Alvarez-Mon, M.; Lopez-Gonzalez, L.; Diaz-Pedrero, R.; Guijarro, L. G.; Ortega, M. A., Connecting epigenetics and inflammation in vascular senescence: state of the art, biomarkers and senotherapeutics. Front Genet 2024, 15, 1345459. [CrossRef]
- Chen, Z.; Malek, V.; Natarajan, R., Update: the role of epigenetics in the metabolic memory of diabetic complications. Am J Physiol Renal Physiol 2024, 327, (3), F327-F339. [CrossRef]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S. M.; Cleary, P.; Riggs, A.; Harlan, D. M.; Lorenzi, G.; Kolterman, O.; Sun, W.; Lachin, J. M.; Natarajan, R., Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, (5), 1748-62. [CrossRef]
- Chen, Z.; Miao, F.; Paterson, A. D.; Lachin, J. M.; Zhang, L.; Schones, D. E.; Wu, X.; Wang, J.; Tompkins, J. D.; Genuth, S.; Braffett, B. H.; Riggs, A. D.; Natarajan, R., Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc Natl Acad Sci U S A 2016, 113, (21), E3002-11. [CrossRef]
- Soriano-Tarraga, C.; Jimenez-Conde, J.; Giralt-Steinhauer, E.; Mola-Caminal, M.; Vivanco-Hidalgo, R. M.; Ois, A.; Rodriguez-Campello, A.; Cuadrado-Godia, E.; Sayols-Baixeras, S.; Elosua, R.; Roquer, J., Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum Mol Genet 2016, 25, (3), 609-19. [CrossRef]
- Cooper, M. E.; El-Osta, A.; Allen, T. J.; Watson, A. M. D.; Thomas, M. C.; Jandeleit-Dahm, K. A. M., Metabolic Karma-The Atherogenic Legacy of Diabetes: The 2017 Edwin Bierman Award Lecture. Diabetes 2018, 67, (5), 785-790. [CrossRef]
- Chen, Z.; Natarajan, R., Epigenetic modifications in metabolic memory: What are the memories, and can we erase them? Am J Physiol Cell Physiol 2022, 323, (2), C570-C582. [CrossRef]
- Zhang, X.; Zhao, S.; Yuan, Q.; Zhu, L.; Li, F.; Wang, H.; Kong, D.; Hao, J., TXNIP, a novel key factor to cause Schwann cell dysfunction in diabetic peripheral neuropathy, under the regulation of PI3K/Akt pathway inhibition-induced DNMT1 and DNMT3a overexpression. Cell Death Dis 2021, 12, (7), 642. [CrossRef]
- Singh LP. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J Clin Exp Ophthalmol 2013, 4, (10), 4172.
- Garcia-Calzon, S.; Maguolo, A.; Eichelmann, F.; Edsfeldt, A.; Perfilyev, A.; Maziarz, M.; Lindstrom, A.; Sun, J.; Briviba, M.; Schulze, M. B.; Klovins, J.; Ahlqvist, E.; Goncalves, I.; Ling, C., Epigenetic biomarkers predict macrovascular events in individuals with type 2 diabetes. Cell Rep Med 2025, 6, (8), 102290. [CrossRef]
- Park, J.; Guan, Y.; Sheng, X.; Gluck, C.; Seasock, M. J.; Hakimi, A. A.; Qiu, C.; Pullman, J.; Verma, A.; Li, H.; Palmer, M.; Susztak, K., Functional methylome analysis of human diabetic kidney disease. JCI Insight 2019, 4, (11), e128886. [CrossRef]
- Kuo, F. C.; Chao, C. T.; Lin, S. H., The Dynamics and Plasticity of Epigenetics in Diabetic Kidney Disease: Therapeutic Applications Vis-a-Vis. Int J Mol Sci 2022, 23, (2), 843. [CrossRef]
- Smyth, L. J.; Patterson, C. C.; Swan, E. J.; Maxwell, A. P.; McKnight, A. J., DNA Methylation Associated With Diabetic Kidney Disease in Blood-Derived DNA. Front Cell Dev Biol 2020, 8, 561907. [CrossRef]
- Smyth, L. J.; Dahlstrom, E. H.; Syreeni, A.; Kerr, K.; Kilner, J.; Doyle, R.; Brennan, E.; Nair, V.; Fermin, D.; Nelson, R. G.; Looker, H. C.; Wooster, C.; Andrews, D.; Anderson, K.; McKay, G. J.; Cole, J. B.; Salem, R. M.; Conlon, P. J.; Kretzler, M.; Hirschhorn, J. N.; Sadlier, D.; Godson, C.; Florez, J. C.; Forsblom, C.; Maxwell, A. P.; Groop, P. H.; Sandholm, N.; McKnight, A. J., Epigenome-wide meta-analysis identifies DNA methylation biomarkers associated with diabetic kidney disease. Nat Commun 2022, 13, (1), 7891. [CrossRef]
- Marchiori, M.; Maguolo, A.; Perfilyev, A.; Maziarz, M.; Martinell, M.; Gomez, M. F.; Ahlqvist, E.; Garcia-Calzon, S.; Ling, C., Blood-Based Epigenetic Biomarkers Associated With Incident Chronic Kidney Disease in Individuals With Type 2 Diabetes. Diabetes 2025, 74, (3), 439-450. [CrossRef]
- Akhouri, V.; Majumder, S.; Gaikwad, A. B., Targeting DNA methylation in diabetic kidney disease: A new perspective. Life Sci 2023, 335, 122256. [CrossRef]
- Chen, G.; Chen, H.; Ren, S.; Xia, M.; Zhu, J.; Liu, Y.; Zhang, L.; Tang, L.; Sun, L.; Liu, H.; Dong, Z., Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int 2019, 96, (2), 409-420. [CrossRef]
- Al-Dabet, M. M.; Shahzad, K.; Elwakiel, A.; Sulaj, A.; Kopf, S.; Bock, F.; Gadi, I.; Zimmermann, S.; Rana, R.; Krishnan, S.; Gupta, D.; Manoharan, J.; Fatima, S.; Nazir, S.; Schwab, C.; Baber, R.; Scholz, M.; Geffers, R.; Mertens, P. R.; Nawroth, P. P.; Griffin, J. H.; Keller, M.; Dockendorff, C.; Kohli, S.; Isermann, B., Reversal of the renal hyperglycemic memory in diabetic kidney disease by targeting sustained tubular p21 expression. Nat Commun 2020, 13, (1), 5062. [CrossRef]
- Yang, T.; Qi, F.; Guo, F.; Shao, M.; Song, Y.; Ren, G.; Linlin, Z.; Qin, G.; Zhao, Y., An update on chronic complications of diabetes mellitus: from molecular mechanisms to therapeutic strategies with a focus on metabolic memory. Mol Med 2024, 30, (1), 71. [CrossRef]
- Feng, S.; Peden, E. K.; Guo, Q.; Lee, T. H.; Li, Q.; Yuan, Y.; Chen, C.; Huang, F.; Cheng, J., Downregulation of the endothelial histone demethylase JMJD3 is associated with neointimal hyperplasia of arteriovenous fistulas in kidney failure. J Biol Chem 2022, 298, (5), 101816. [CrossRef]
- Sanchez-Ceinos, J.; Hussain, S.; Khan, A. W.; Zhang, L.; Almahmeed, W.; Pernow, J.; Cosentino, F., Repressive H3K27me3 drives hyperglycemia-induced oxidative and inflammatory transcriptional programs in human endothelium. Cardiovasc Diabetol 2024, 23, (1), 122. [CrossRef]
- Li, T.; Yu, C.; Zhuang, S., Histone Methyltransferase EZH2: A Potential Therapeutic Target for Kidney Diseases. Front Physiol 2021, 12, 640700. [CrossRef]
- Siddiqi, F. S.; Majumder, S.; Thai, K.; Abdalla, M.; Hu, P.; Advani, S. L.; White, K. E.; Bowskill, B. B.; Guarna, G.; Dos Santos, C. C.; Connelly, K. A.; Advani, A., The Histone Methyltransferase Enzyme Enhancer of Zeste Homolog 2 Protects against Podocyte Oxidative Stress and Renal Injury in Diabetes. J Am Soc Nephrol 2015, 27, (7), 2021-34. [CrossRef]
- Sekine, S.; Youle, R. J., PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 2018, 16, (1), 2. [CrossRef]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M. H., BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J Mol Cell Cardiol 2021, 149, 1-14. [CrossRef]
- Ray, K. K.; Nicholls, S. J.; Buhr, K. A.; Ginsberg, H. N.; Johansson, J. O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P. P.; Wong, N.; Sweeney, M.; Schwartz, G. G., Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. Jama 2020, 323, (16), 1565-1573.
- Brandts, J.; Ray, K. K., Apabetalone - BET protein inhibition in cardiovascular disease and Type 2 diabetes. Future Cardiol 2020, 16, (5), 385-395. [CrossRef]
- Sasaki, K.; Masaki, T., Epigenetic histone modifications in kidney disease and epigenetic memory. Clin Exp Nephrol 2025, 29, (9), 1129-1138. [CrossRef]
- Liu, M.; Qiao, Z.; Zhang, Y.; Zhan, P.; Yi, F., Histone Deacetylases Take Center Stage on Regulation of Podocyte Function. Kidney Dis (Basel) 2020, 6, (4), 236-246. [CrossRef]
- Khan, S.; Jena, G.; Tikoo, K.; Kumar, V., Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-kappaB/iNOS signaling in diabetic rat. Biochimie 2015, 110, 1-16. [CrossRef]
- Yoshikawa, M.; Hishikawa, K.; Marumo, T.; Fujita, T., Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells. J Am Soc Nephrol 2007, 18, (1), 58-65. [CrossRef]
- Huang, K.; Gao, X.; Wei, W., The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-beta1 expressions in rat glomerular mesangial cells. Exp Cell Res 2017, 361, (1), 63-72. [CrossRef]
- Martinez-Arroyo, O.; Flores-Chova, A.; Mendez-Debaets, M.; Garcia-Ferran, L.; Escriva, L.; Forner, M. J.; Redon, J.; Cortes, R.; Ortega, A., Inhibiting miR-200a-3p Increases Sirtuin 1 and Mitigates Kidney Injury in a Tubular Cell Model of Diabetes and Hypertension-Related Renal Damage. Biomolecules 2025, 15, (7), 995. [CrossRef]
- Chen, H. Y.; Zhong, X.; Huang, X. R.; Meng, X. M.; You, Y.; Chung, A. C.; Lan, H. Y., MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther 2014, 22, (4), 842-53. [CrossRef]
- Gondaliya, P.; Dasare, A.; Srivastava, A.; Kalia, K., Correction: miR29b regulates aberrant methylation in In-Vitro diabetic nephropathy model of renal proximal tubular cells. PLoS One 2019, 14, (1), e0211591.
- Putta, S.; Lanting, L.; Sun, G.; Lawson, G.; Kato, M.; Natarajan, R., Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 2012, 23, (3), 458-69. [CrossRef]
- Dhas, Y.; Arshad, N.; Biswas, N.; Jones, L. D.; Ashili, S., MicroRNA-21 Silencing in Diabetic Nephropathy: Insights on Therapeutic Strategies. Biomedicines 2023, 11, (9), 2583. [CrossRef]
- de Gonzalo-Calvo, D.; van der Meer, R. W.; Rijzewijk, L. J.; Smit, J. W.; Revuelta-Lopez, E.; Nasarre, L.; Escola-Gil, J. C.; Lamb, H. J.; Llorente-Cortes, V., Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep 2017, 7, (1), 47. [CrossRef]
- Napoli, C.; Benincasa, G.; Schiano, C.; Salvatore, M., Differential epigenetic factors in the prediction of cardiovascular risk in diabetic patients. Eur Heart J Cardiovasc Pharmacother 2020, 6, (4), 239-247. [CrossRef]
- Al-Kafaji, G.; Al-Muhtaresh, H. A., Expression of microRNA377 and microRNA192 and their potential as blood-based biomarkers for early detection of type 2 diabetic nephropathy. Mol Med Rep 2018, 18, (1), 1171-1180. [CrossRef]
- De, A.; Sarkar, A.; Banerjee, T.; Bhowmik, R.; Sar, S.; Shaharyar, M. A.; Karmakar, S.; Ghosh, N., MicroRNA: unveiling novel mechanistic and theranostic pathways in diabetic cardiomyopathy. Front Pharmacol 2025, 16, 1613844. [CrossRef]
- Zhang, M.; Su, Q.; Lu, Y.; Zhao, M.; Niu, B., Application of machine learning approaches for protein-protein interactions prediction. Med Chem 2017, 13, (6), 506–514. [CrossRef]

Table 1.
HDAC enzymes classes.
| Class | Enzymes | Common Name / Characteristics | Mechanism |
| I | HDAC1, HDAC2, HDAC3, HDAC8 | "Classical" HDACs | Traditional deacetylation mechanism |
| II | HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDAC10 | “Classical" HDACs | Traditional deacetylation mechanism |
| III | SIRT 1-7 | Sirtuins | NAD+-dependent mechanism |
| IV | HDAC11 | "Classical" HDACs | Shares only weak homology with Class I and II |
HDAC, histone deacetylases; SIRT, silent information regulator Sirtuin; NAD+, nicotinamide adenine dinucleotide.
Table 2.
List of FDA-approved epigenetic drugs.
| Drug name | Target | Indication | Year |
| Azacitidine (Vidaza®) | DNMTi | Myelodysplastic syndromes, Chronic myelomonocytic leukemia, Acute myeloid leukemia |
2004 2004 2007 |
| Decitabine (Dacogen®) Decitabine-Cedazuridine (Inqovi®) |
DNMTi | Myelodysplastic syndromes, Intermediate/high-risk myelodysplastic syndromes |
2006 2020 |
| Valproic Acid (Depakin®) | Class I/II HDAC | Epilepsy, bipolar disorder, migraine | 2010 |
| Vorinostat (Zolinza®) | Class I/II HDAC | Cutaneous T-cell lymphoma | 2006 |
| Romidepsin (Istodax®) | HDAC6 | Cutaneous T-cell lymphoma, Peripheral T-cell lymphoma (withdrawn 2021) |
2009 2011 |
| Belinostat (Beleodaq®) | Non-selective HDACi | Relapsed or refractory peripheral T-cell lymphoma | 2014 |
| Panobinostat (Farydak®) | Non-selective HDACi | Relapsed multiple myeloma (discontinued 2021) | 2015 |
| Tazemetostat (Tazverik®) | EZH2i | Metastatic or locally advanced epithelioid sarcoma, Relapsed or refractory follicular lymphoma |
2020 2020 |
DNMT, DNA methyltransferase; HDAC, histone deacetylases; EZH2, enhancer of zeste homolog 2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



