
Submitted:
19 October 2025
Posted:
20 October 2025
You are already at the latest version
Abstract
Receptor for Advanced Glycation Endproducts, RAGE, is a transmembrane multi-ligand receptor, which plays key role in amplifying immune response in numerous inflammatory processes and serious diseases. RAGE fulfills its function through binding of a variety of ligands. The structure of RAGE includes one transmembrane domain, one immunoglobulin V-like domain and two immunoglobulin C-like domains. Here, we have used combination of methods of domain structure analysis, fold classification, structure comparison and analysis of protein/ligand interactions with a chosen set of domains of RAGE and other immunoglobulin-like proteins from several different structural databases to describe structure-functional core and ligand recognition by the V-type domain of RAGE, and compare those to the other immunoglobulin-like proteins. We show that from one hand, similar to the other immunoglobulin-like proteins, RAGE contains a conserved substructure, the cross-β zone, with the cross-β motif at its core. The cross-β zone incorporates the hydrophobic core of RAGE and two multivalent surface ligand-binding sites for small molecules and proteins, one on the surface of the C-F-G β-sheet of the β-sheet sandwich, and the other formed by the CE-loop region. RAGE oligomerization allows RAGE to bind nucleic acids at a site different from the two mentioned above, leading to at least four distinct sites on the surface of RAGE: two for binding small molecules and proteins, one for binding nucleic acids, and one for RAGE dimerization. From the other hand, regardless of structural similarities with immunoglobulins, RAGE does not contain ligand-binding regions analogous to Complementary-Determining Regions in immunoglobulins, thus making RAGE and immunoglobulins functionally different.
Keywords:
RAGE
; Ig-like
; V-type
; cross-β zone
; hydrophobic core
; ligand-binding site
1. Introduction
The Receptor for Advanced Glycation Endproducts (RAGE) is a transmembrane multi-ligand receptor, which serves as a progression factor amplifying immune and inflammatory responses in (1) pathogenesis and therapy of cancer, (2) neurological disorders, inflammation and apoptosis, including kidney disease and Alzheimer’s disease (AD), (3) diabetes-related cardiovascular complications, such as thrombosis and myocardial infarctions, and (4) in impairing the physiological function of stem cells, all due to the buildup of numerous Advanced Glycation Endproducts (AGEs), RAGE agonists and other RAGE ligands, during these conditions [1,2,3,4].
RAGE is expressed in many cell types and its expression increases as a direct result of ligand accumulation. In therapy of cancer invasion and metastasis, the role of various AGEs together with another group of RAGE ligands, the S100 proteins, is particularly important [5,6,7,8,9]. There, it was demonstrated that RAGE interactions with AGEs and S100 proteins in tumor cells increased activity of NF-κB and expression of proinflammatory cytokines TNF-α and IL-6 [10,11,12]. In inflammation and neurological disorders, RAGE is involved on multiple levels [13,14,15,16,17]. For example, during development of AD there is buildup of amyloid beta (Aβ) peptides, which is a known group of RAGE agonists [13,16]. Here, apart from being a multi-ligand receptor, RAGE also functions in tandem with other receptors, such as Toll-Like Receptors (TLR), where RAGE enhances TLR response [18,19]. In diabetes, RAGE signaling was recently shown to regulate progression of diabetic complications [20,21]. Independently from diabetes-induced cardiovascular complications, the AGE-RAGE axis was found to be involved in initiation and progression of other types of cardiovascular diseases [22]. Thus, it is not surprising that both RAGE and its various ligands show significant potential as drug targets.
The sequence-based domain structure of RAGE, which includes one Transmembrane domain (TM), one possible Immunoglobulin V-like (IgVRAGE) domain and two possible Immunoglobulin C-like (IgCRAGE) domains, had been described in Neeper et al. [23] even before the structural immunoglobulin fold was officially reviewed and defined by Bork et al. [24], and Harpaz and Chothia [25]. The RAGE domain structure can be schematically described as Nterminus-IgVRAGE-IgC1RAGE-IgC2RAGE-TM-Cterminus. Topologies of IgVRAGE, IgC1RAGE and IgC2RAGE domains within the Immunoglobulin-like fold (Ig-like fold; full name Immunoglobulin-like β-sandwich fold; SCOP ID: 2000051; InterPro ID: IPR013783) are as defined in Bork et al. [24] for the respective classical V-type Ig-like and C-type Ig-like domains, IgV and IgC domains, respectively [26]. Similar to classical IgV domains, the IgVRAGE domain contains a disulfide bridge between Cys38 and Cys99 [27]. The link between the IgVRAGE and IgC1RAGE domains is rigid, creating a “V-C1RAGE” structural unit, which is predominantly non-polar/positively charged, and thus can recognize negatively-charged ligands or surfaces [26,28,29]. The IgC2RAGE-domain is different. It carries a negatively-charged surface [30], and it is bound to the V-C1RAGE unit by a flexible seven amino acid linker [29,31].
Along with RAGE, there is a large variety of unrelated proteins that include one or several domains with the Ig-like fold. Antigen receptors, which include various antibodies, i.e immunoglobulins, and T-cell receptors (TCRs) constitute the most populated group of proteins of the immunoglobulin superfamily with domains having the Ig-like fold. Similar to antibodies, all TCR chains contain one IgV domain at the N-terminus. For both TCRs and immunoglobulins, the main antigen recognition element is a set of canonical Complementary-Determining Region (CDR) loops, CDR1, CDR2 and CDR3, which were already compared based on their sequence and structure, and classified for all known antigen receptors [32]. Analysis of CDR-equivalent loops in RAGE (CDRRAGE loops) would bring insight into its multi-ligand recognition abilities.
RAGE undergoes oligomerization with its transmembrane and extracellular regions prior to ligand binding and signaling activity [33,34]. For example, it has been demonstrated that in the soluble version of RAGE (sRAGE) the V/V-domain homodimerization is required for AGE and S100B binding [35,36]. At the same time, recognition of S100A12 requires formation of the C1/C1-domain interface [37], and existence of the C2/C2-domain disulfide crosslinked dimer is needed for receptor stability [38]. For the binding of S100 proteins, it was concluded that they may require RAGE oligomerization for signal transduction [39], and as shown by the example of S100B, an S100 protein might need to be Ca2+-loaded [26]. The NMR structure of the RAGE-S100B complex showed a bound Zn2+ ion at the C1/C1-domain interface coordinating conserved residues from both sides, implying the possibility of a divalent cation requirement for RAGE multimerization [26]. Additionally, given multitude of bound ligands, existence of several RAGE oligomerization schemes should not be ruled out [34]. Taking into consideration all oligomerization data shown above, it is not difficult to see structural similarity between RAGE and heavy chain-only antibodies, with their standalone IgV domains mimicking nanobodies [40], which gives not only a better understanding of the multiligand functionality of RAGE, but also opens opportunities for search of novel therapeutic molecules [3,7].
Here, we aim to structurally align RAGE with the rest of the classical Ig-like domains, to determine and compare structural elements between RAGE and the other Ig-like domains, which are responsible for ligand recognition, and consequently protein function in these proteins.
2. Materials and Methods
The Protein Data Bank (PDB, http://www.rcsb.org/; 15 March 2025 [41]) and the PDBsum database (PDBsum, https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/; 15 March 2025 [42]) were used to retrieve Ig-like proteins, including RAGE, and also PDB entries of RAGE complexes with ligands. The SCOP classification database (SCOP2, https://www.ebi.ac.uk/pdbe/scop/; 15 March 2025 [43]) and InterPro classification of protein families (https://www.ebi.ac.uk/interpro/; 15 March 2025 [44]) were used to determine whether the 3D structures of the analyzed proteins belong to the Ig-like beta-sandwich fold. Detailed structural information from the above set of PDB files are given in Section 3.1 and Table S3.
Structure visualization and structural analysis of interactions between amino acids in proteins (hydrogen bonds, hydrophobic, other types of weak interactions) were conducted using PyMOL molecular graphics system (https://pymol.org/; 17 March 2025), and software [45] to determine interatomic contacts, i.e., of ligand–protein contacts (LPCs) and contacts of structural units (CSUs). Alignment of primary structures was carried out through the Dali software [46]. The localization of the complementary determining regions (CDRs) of the light chain fragment of the Bence-Jones protein (Rhe) was determined using the PyIgClassify2 server (https://dunbrack2.fccc.edu/PyIgClassify2/default.aspx; 17 March 2025) [47].
Weak hydrogen bonds were identified based on geometrical criteria [48]. Figures were drawn with PyMOL molecular graphics system (https://pymol.org/; 17 March 2025).
3. Results and Discussion
As it is described in Introduction, the immunoglobulin-like (Ig-like) domains show several key structural features, such as the overall β-strand topology, existence of S-S bonds and CDR regions that define ligand recognition by these domains. The aim of our analysis was to take different Ig-like domains as templates, divide into conserved and variable structural elements as bricks of a puzzle, derive their functional purpose, and then “wrap” respective RAGE domains around templates to describe functional attributes of different structural units of RAGE.
3.1. Creating a Dataset of the Analyzed Ig-Like Proteins
Extracellular part of the human RAGE structure (V and C1 domains, PDB ID: 6XQ1 [49]; C2 domain, PDB ID: 4P2Y [50]) has been compared with topologies of five different types of Ig-like domains, the V-type (IgV), C-type (IgC), S-type (IgS), H-type (IgH) and I-type (IgI), where the IgV domain topology is represented by the light chain fragment of the Bence-Jones protein (Rhe), PDB ID: 2RHE [51]; the IgC by the IgG2a-kappa 4-4-20 Fab (heavy chain), PDB ID: 4FAB [52]; the IgS by the tenansin (fibronectin type III domain), PDB ID: 1TEN [53]; the IgH by the galactose oxidase, PDB ID: 1GOF [54]; and the IgI by telokin, PDB ID: 1FHG [55]. In addition to these six known Ig-like types, five Ig-like structures of unknown type exist and have been investigated too: 1) chaperone protein PapD, PDB ID: 2J2L [56]; 2) cytochrome f, PDB ID: 1CTM [57]; 3) arthropodan haemocyanin, PDB ID: 1HC1 [58]; 4) superoxide dismutase [Cu-Zn], PDB ID: 1Q0E [59] and 5) synaptotagmin I, PDB ID: 1RSY [60].
3.2. Structural Homology between IgVRAGE and IgV, and the Overall Topology of Ig-Like Domains
As it is described in the Introduction section, the extracellular part of human RAGE protein consists of three Ig-like domains, IgVRAGE, IgC1RAGE and IgC2RAGE, with the total length of about 300 amino acids. Recently, nine related X-ray complexes of the same two-domain IgVRAGE-IgC1RAGE (or V-C1RAGE) configuration, but with nine different bound inhibitors, were reported [49]. We chose one of these nine structures, PDB ID: 6XQ1 (resolution 1.51 Å), as the RAGE domain representative of the V-C1RAGE complex. From this complex we extracted representative structure of the IgVRAGE domain.
Generally, the IgV domain is the main ligand binding domain in any antigen receptor and also in RAGE. As the first step, we checked if the IgVRAGE domain indeed, by homology, is similar to the other known IgV domains, and also, we created a structural superposition of different IgV domains that could be used as the background superposition for comparative purposes. The V-type domain topology of the Ig-like domains [24] was historically taken from the light chain fragment of the Bence-Jones protein (Rhe; PDB ID: 2RHE [51]). A structural alignment between IgVRAGE (6XQ1_A) and the IgV domain from the Bence-Jones protein (2RHE_A) has been done using the Dali software [46] (Figure 1). The Dali superposition resulted indeed in suggested structural homology between IgVRAGE and IgV with the Z-score of 9.6 and RMSD of 2.9 Å over 114 residues.
Sixteen alignment positions were found to be identical, of which three positions contained two key cysteines and a tryptophan. The two key cysteines form S-S bond in both IgVRAGE and IgV, linking the two β-sheets of the standard IgV topology together, and the key tryptophanes form similar stacking interactions with the S-S bonds in both types of structures. The Dali comparison also demonstrated similar arrangement of eight β-strands (A-G) in both domain types, underlying similar secondary structure topology (Figure 1). Earlier, it was suggested that in the immunoglobulin fold, a prototype Ig-like domain gene may had been derived from gene duplication, which in turn suggested possible existence of a prototype “half-domain” structure formed by either β-sheet ABCC’ or β-sheet GFED [61]. From the structural point of view, however, this hypothesis might work only for IgV domains, because of the resulting significant change in structure moving from two four-stranded β-sheets in IgV to “four-stranded plus three-stranded” β-sheets for IgC and IgS, where IgC lacks the C’ β-strand and IgS lacks the D β-strand (Figure 2) [24]. Thus, we did consider structures of the entire domains. We further “disassembled” different types of Ig-like domains into logical structural units in order to identify conserved structural blocks common to all types (IgV, IgC, IgS and IgH) of the Ig-like domains and variable structural blocks that are specific only to certain domains. Then, we determined roles of the conserved and variable structural blocks in ligand binding.
3.3. The Cross-β Zone of the IgVRAGE Domain
While different domains within the Ig-like fold contain 3x4, 4x4 or 5x4 β-sheet sandwiches, they all contain four central β-strands B, C, E and F (Figure 2) [24]. The topology of these four strands is known as the “cross-β” structural motif [62]. In IgVRAGE the respective cross-β motif is shown in Figure 3. Because IgVRAGE is structurally homologous to IgV, all the respective β-strands exist also in IgVRAGE, however their length is different, as it is seen from Figure 1. In Figure 3, we have accepted five amino acids as boundaries for these four β-strand segments in IgVRAGE, because (1) five is the average length of β-strands in IgVRAGE [49], and (2) five is the minimum length to keep the entire hydrophobic core of the cross-β Motif in IgV and IgVRAGE (Figure 3).
In IgVRAGE, fragments Leu34-Leu53 and Gly82-Met102 contain βB-loop-βC and βE-loop-βF super-secondary structures, where βB, βC, βE and βF designate β-strands B, C, E and F, respectively (Figure 3). β-Strands B and C lie in the opposing β-sheets of the β-sandwich and are antiparallel to each other (Figure 2). The same is true for β-strands E and F. In order to create the cross-β structural motif, the βB-loop-βC and βE-loop-βF super-secondary structures are put together antiparallel to each other, so that the loop regions reside on the opposite sides of the motif (Figure 4A).
Similar to IgV, in IgVRAGE the cross-β structural motif takes form of an antiparallel β-sheet sandwich, where β-strands B and E and β-strands C and F become connected by characteristic “antiparallel bridges” (Table S1). An “antiparallel bridge” is a known binding pattern, observed in the secondary structure of proteins, and specifically β-sheets [64], where pairs of hydrogen bonds occur between main chain atoms of β-strands within a β-sheet [65]. In IgVRAGE, β-Strands B and E are connected by two bridges, and β-strands C and F - by three (Table S1). Taken all the structural features together, we come to the description of the “cross-β structural zone” in IgV and IgVRAGE. The cross-β structural zone consists of approximately 40 amino acids, which incorporate four strands of two β-sheets totaling to 20 residues, the BC loop (in IgVRAGE) or the CDR1 loop (in IgV) of about 10 residues each, and the EF loop (in both IgV and IgVRAGE) of approximately 10 residues long (Table S2). Amino acids of the zone form a compact standalone sub-structure, and interact with each other by means of antiparallel bridges and a network of other weak interactions. Two interacting β-sheets form hydrophobic core and two different outer surfaces. The cross-β zone of the IgVRAGE domain plays central role for protein function.
3.4. Hydrophobic Core of Ig-Like Structures
Comparison between IgV and three other types of Ig-like domains, IgC, IgS and IgH, is given in Bork at al., 1994 [24], where the authors concluded that the cross-β motif was structurally conserved in all the four Ig-like domains. Here, we showed that the cross-β zone of the IgVRAGE domain does also belong to that group (Figure 3), and the same is also true for the two C-type domains of RAGE, IgC1RAGE and IgC2RAGE, where two β-loop-β motifs form a cross-β substructure with the hydrophobic core similar to that seen in the other Ig-like domains (Table 1). Even the length of the respective β-strand and loop regions of the two β-loop-β units in the cross-β motif are equivalent (Table S2), which supports the hypothesis of gene duplication in a prototype Ig-like domain [61]. Also, same arrangement can be seen in the I-type Ig-like (IgI) domain (PDB ID: 1FHG, telokin).
Unlike classical V-, C-, S-, H- and I-types of Ig-like domains, several unclassified Ig-like domains taken from the work of Halaby et al., 1999 [66]: 2J2L, chaperone protein PapD; 1CTM, cytochrome f; 1HC1, arthropodan haemocyanin; 1Q0E, superoxide dismutase [Cu-Zn]; and 1RSY, synaptotagmin I (Table S2), show variations in length of the β-loop-β fragments BC and EF, and the long loop CE. Nevertheless, according to the SCOP2 database [43], three out of the five structures, 2J7L, 1HC1, and 1Q0E, still have the Ig-like beta-sandwich fold (SCOP ID: 2000051), and 1RSY has fold, which is topologically similar to Ig-like beta-sandwich. Because all five of them have similar hydrophobic cores of the cross-β zone, their 3D structures can be potentially put together as new M(Mixed)-type of Ig-like domains.
3.5. Two Types of Ligand Binding in RAGE
We have analyzed sixteen available ligand-bound complexes of RAGE [27,49,67,68,69,70,71,72] for surfaces and amino acids involved in ligand binding using the PDBsum database [42]. All residues involved in ligand recognition in these structures of IgVRAGE are summarized in Table S3. Additionally, Table S3 contains highlighted information whether the ligand-interacting residues belong to the cross-β zone strands and loops (marked in red and yellow color, respectively) or to the rest of the IgVRAGE domain, such as the CE-loop, etc. The summary figure of all amino acids involved in ligand binding in RAGE is shown in Table S4. All ligand-interacting amino acids in RAGE (the IgVRAGE) domain can be roughly divided into two spatial surface-exposed clusters.
(1) β-Sheet based ligand-binding cluster: The core of the first ligand-binding cluster in RAGE consists of six large polar amino acids, Glu50, Lys52, Arg98, Gln100, Lys110 and Asn112, which reside on the outer surface of the larger β-sheet of the IgVRAGE domain β-sandwich (β-strands C, F and G; Figure 4A). β-Strands C and F belong to the cross-β zone. β-Strand G does not belong to the cross-β zone, but in all five standard Ig-like domain types the β-strand G forms a conserved β-hairpin with the β-strand F of the cross-β zone, creating an antiparallel β-sheet.
(2) CE-loop based ligand-binding cluster: The core of the second ligand-binding cluster in RAGE consists of seven amino acids: Asp73, Ser74, Arg77, Val78, Leu79, Phe85 and Pro87, which are predominantly located in the CE-loop of RAGE that lies outside the cross-β zone and on the side of the central β-sheet (Figure 4B).
Amino acids of the two cluster above are the most frequent amino acids repeatedly found in different RAGE complexes to bind different ligands (see numerical values in Table S4). At the same time the full set of ligand-interacting residues is listed in Table S4, where it is seen that almost all amino acids of the long CE-loop do interact with ligands in one complex or the other, which makes CE-loop a unique ligand-binding substructure.
The structure of the β-sheet based ligand-binding cluster is more rigid, because it is formed by conserved secondary structure elements of the cross-β motif, and its ligand-binding properties depend on the type of side-chain groups of interacting amino acids. The CE-loop based ligand-binding cluster is more flexible, and its ligand binding capabilities are determined by side-chain and main-chain groups of interacting residues, and by their secondary structure/conformation. The two ligand-binding clusters of RAGE shown above recognize both large molecules (proteins) and small molecule ligands (Table S3). At the same time, these ligand-binding clusters in RAGE do not coincide as a whole or in part with the three CDR peptides of the IgV domain (PDB ID: 2RHE), thus functionally distinguishing RAGE from immunoglobulins.
3.6. RAGE Dimerization and Binding of DNA
In the introduction section we showed that RAGE oligomerization is known to be required for binding a multitude of different ligands. If we look from the other side, it was also said that there may be several oligomerization techniques that RAGE employs to recognize those ligands [34]. Here, we demonstrate that IgVRAGE homodimer uses sites different from either the C-F-G β-sheet or the CE-loop described above (section 2.5) to bind nucleic acids, and thus can be roughly considered as the third type of ligand binding by IgVRAGE, complementary to the two types of binding shown above.
Recognition of DNA and RNA and the subsequent activation of signaling receptors by nucleic acids is a mechanism of detection of infection and tissue damage. DNA recognition by RAGE affects immune activation of Toll-like receptor 9 (TLR 9), the principal DNA-recognizing signaling receptor [63]. The binding of a nucleic acid requires homodimerization of the IgVRAGE domain by “stacking” two A-B-E β-sheets, which are opposite to the ligand-binding C-F-G β-sheets, from two respective IgVRAGE domains, as it is shown by the crystal structure PDB ID: 4OI7 [63] in Figure 4C. The stacking of β-sheets from two IgVRAGE domains happens through interactions of two hydrophobic clusters, Leu79, Pro80, Phe85 and Pro87, around the Phe85-Phe85 hydrophobic link from each IgVRAGE domain of the dimer. The DNA recognition comes with the help of electrostatic attraction by means of the triple-lysine Lys37-Lys39-Lys43 cluster on each of the IgVRAGE domain [63].
Such nucleic acid-binding mechanism leaves the CE-loop and the C-F-G β-sheet exposed for binding other macromolecules or small-molecule inhibitors.
3.7. Antiparallel Homodimerization of Two β-Loop-β Fragments to Create Functional Core of RAGE
Formation of a protein structural catalytic core (SCC) or a core zone of a protein through antiparallel dimerization of two similar fragments of protein structure has already been seen by us in structural analysis of the EF-hand calcium-binding proteins [73] and acid proteases [74]. In the EF-hand proteins [73], two “helix – binding loop – strand – helix” segments were locked together in an antiparallel manner by the two “locks”, called the “black” and “grey” clusters, which situated at the α-helical regions on both sides of the motif. The third “lock” was the resulting two-stranded antiparallel β-sheet in the middle of the motif. Here, in Ig-like domains, two “strand – loop – strand” segments are locked together in an antiparallel manner by the two “locks” of formed antiparallel β-sheets on the two ends, thus forming the cross-β functional motif. In acid proteases [74], the dimer is formed by two sets of equivalent structural segments, the DD-link, D-loop, and G-loop, around two catalytic aspartates in each individual chain. In immunoglobulins, such organization created an argument in favor of gene duplication [61]. However, as we have shown above, RAGE does have homologous IgVRAGE domain with the cross-β motif, but it is not immunoglobulin with respect to CDR loops or the other ligand recognition or functional elements, thus giving possibility that if gene duplication in a prototype Ig-like domain existed, it could not be an attribute of immunoglobulins, but something larger.
4. Conclusions
Receptor for Advanced Glycation Endproducts (RAGE) is a transmembrane multi-ligand receptor, which incorporates one Transmembrane domain (TM), one possible Immunoglobulin V-like (IgVRAGE) domain and two Immunoglobulin C-like (IgCRAGE) domains. Here, we showed that the ligand-binding IgVRAGE domain of RAGE is structurally homologous to the standard V-type Ig-like domain, and similarly contains respective disulfide bridge and the cross-β motif. Moreover, there is a wider conserved structure, which we call the cross-β zone. The cross-β zone in its inner side contains the hydrophobic core of the domain, and in its outer side contains main ligand-binding sites of the protein: one ligand-binding site on the surface of the larger β-sheet C-F-G of the β-sheet sandwich; and the other on the long loop fragment CE. Based on the data observed, we could speculate that the β-sheet based binding site could be more “universal” for Ig-like domains, while the CE-loop based was more “specific” for a particular domain. As opposed to the C-F-G β-sheet, the A-B-E β-sheet is used for RAGE dimerization through the hydrophobic cluster on its surface. Oligomerization allows RAGE to bind even larger variety of ligands, such as for example nucleic acids, and participate in even more immune signaling mechanisms. However, regardless of structural similarities and features mentioned above, RAGE does not show ligand-binding regions analogous to the Complementary-Determining Regions (CDR) in immunoglobulins, thus making RAGE and immunoglobulins functionally different.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1, Table S1. Antiparallel bridges connecting β-strand pairs in the cross-β zone of the Ig-like domains. Table S2. Approximate boundaries of amino acid fragments constituting the cross-β zone (β-loop-β fragments BC and EF) plus the intermediate segment CE (see Figure 4A) in thirteen representative structures of Ig-like domains. “N” designates fragment lengths. Table S3. Amino acids in contact with the ligand in different known ligand-bound complexes of RAGE (the IgVRAGE domain). Amino acids from the four β-strands, B, C, E, and F (belong to the cross-β zone) are marked in red, the β-strand G (do not belong to the cross-β zone) in dark red, (similar as in Figure 4A). The CE-loop is shown in green. Amino acids from the two loops, B-C and E-F are marked in gray. Table S4. Summary of amino acids, which are involved in ligand binding in 16 ligand-bound IgVRAGE complexes from Table S3. Color designations are as in Table S3.
Author Contributions
K.D.: Study design, Formal analysis, Methodology, Visualization, Writing – Original Draft, Writing – Review & Editing; V.N.U.: Formal analysis, Methodology, Writing – Original Draft, Writing – Review & Editing; E.A.P.: Formal analysis, Methodology, Visualization, Writing – Original Draft; S.E.P.: Study design, Formal analysis, Methodology, Visualization, Investigation, Writing – Original Draft, Writing – Review & Editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting reported results can be found in the Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dobrucki, I.T.,; Miskalis, A.,; Nelappana, M.,; Applegate, C.,; Wozniak, M.,; Czerwinski, A.,; Kalinowski, L.,; Dobrucki, L.W. Receptor for advanced glycation end-products: Biological significance and imaging applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2024, 16, e1935. [CrossRef]
- Khalid, M.,; Adem, A. The dynamic roles of advanced glycation end products. Vitam. Horm. 2024, 125, 1–29. [CrossRef]
- Sarkar, S. Pathological role of RAGE underlying progression of various diseases: its potential as biomarker and therapeutic target. Naunyn-Schmiedeberg's Arch. Pharmacol. 2025, 398, 3467–3487. [CrossRef]
- Zheng, Z.,; Zhou, H.,; Zhang, W.,; Wang, T.,; Swamiappan, S.,; Peng, X.,; Zhou, Y. Effects of advanced glycation end products on stem cell. Front. Cell Dev. Biol. 2024, 12, 1532614. [CrossRef]
- Roth, J.,; Vogl, T.,; Sorg, C.,; Sunderkötter, C. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003, 24, 155–158. [CrossRef]
- Delrue, C.,; Speeckaert, R.,; Delanghe, J.R.,; Speeckaert, M.M. Breath of fresh air: Investigating the link between AGEs, sRAGE, and lung diseases. Vitam. Horm. 2024, 125, 311–365. [CrossRef]
- Radziszewski, M.,; Galus, R.,; Łuszczyński, K.,; Winiarski, S.,; Wąsowski, D.,; Malejczyk, J.,; Włodarski, P.,; Ścieżyńska, A. The RAGE Pathway in Skin Pathology Development: A Comprehensive Review of Its Role and Therapeutic Potential. Int. J. Mol. Sci. 2024, 25, 13570. [CrossRef]
- Zglejc-Waszak, K.,; Jozwik, M.,; Thoene, M.,; Wojtkiewicz, J. Role of Receptor for Advanced Glycation End-Products in Endometrial Cancer: A Review. Cancers 2024, 16, 3192. [CrossRef]
- Coser, M.,; Neamtu, B. M.,; Pop, B.,; Cipaian, C. R.,; Crisan, M. RAGE and its ligands in breast cancer progression and metastasis. Oncol. Rev. 2025, 18, 1507942. [CrossRef]
- Sparvero, L.J.,; Asafu-Adjei, D.,; Kang, R.,; Tang, D.,; Amin, N.,; Im, J.,; Rutledge, R.,; Lin, B.,; Amoscato, A.A.,; Zeh, H.J.,; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [CrossRef]
- Palanissami, G.,; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer-a Review. Horm. Cancer. 2018, 9, 295–325. [CrossRef]
- Deepu, V.,; Rai, V.,; Agrawal, D.K. Quantitative Assessment of Intracellular Effectors and Cellular Response in RAGE Activation. Arch. Intern. Med. Res. 2024, 7, 80–103. [CrossRef]
- Paudel, Y.N.,; Angelopoulou, E.,; Piperi, C.,; Othman, I.,; Aamir, K.,; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer's Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [CrossRef]
- Cross, K.,; Vetter, S.W.,; Alam, Y.,; Hasan, M.Z.,; Nath, A.D.,; Leclerc, E. Role of the Receptor for Advanced Glycation End Products (RAGE) and Its Ligands in Inflammatory Responses. Biomolecules 2024, 14, 1550. [CrossRef]
- Raghavan, C.T. Advanced Glycation End Products in Neurodegenerative Diseases. J. Mol. Neurosci. 2024, 74, 114. [CrossRef]
- Li, W.,; Chen, Q.,; Peng, C.,; Yang, D.,; Liu, S.,; Lv, Y.,; Jiang, L.,; Xu, S.,; Huang, L. Roles of the Receptor for Advanced Glycation End Products and Its Ligands in the Pathogenesis of Alzheimer's Disease. Int. J. Mol. Sci. 2025, 26, 403. [CrossRef]
- Ma, Y.,; Wang, X.,; Lin, S.,; King, L.,; Liu, L. The Potential Role of Advanced Glycation End Products in the Development of Kidney Disease. Nutrients 2025, 17, 758. [CrossRef]
- Gąsiorowski, K.,; Brokos, B.,; Echeverria, V.,; Barreto, G.E.,; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [CrossRef]
- Jangde, N.,; Ray, R.,; Rai, V. RAGE and its ligands: from pathogenesis to therapeutics. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 555–575. [CrossRef]
- Taguchi, K.,; Fukami, K. RAGE signaling regulates the progression of diabetic complications. Front. Pharmacol. 2023, 14, 1128872. [CrossRef]
- Pedreañez, A.,; Mosquera-Sulbaran, J.A.,; Tene, D. Role of the receptor for advanced glycation end products in the severity of SARS-CoV-2 infection in diabetic patients. Diabetol. Int. 2024, 15, 732–744. [CrossRef]
- Wang, B.,; Jiang, T.,; Qi, Y.,; Luo, S.,; Xia, Y.,; Lang, B.,; Zhang, B.,; Zheng, S. AGE-RAGE Axis and Cardiovascular Diseases: Pathophysiologic Mechanisms and Prospects for Clinical Applications. Cardiovasc. Drugs Ther. 2024, 10.1007/s10557-024-07639-0. Advance online publication. [CrossRef]
- Neeper, M.,; Schmidt, A.M.,; Brett, J.,; Yan, S.D.,; Wang, F.,; Pan, Y.C.,; Elliston, K.,; Stern, D.,; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. https://pubmed.ncbi.nlm.nih.gov/1378843/.
- Bork, P.,; Holm, L.,; Sander, C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [CrossRef]
- Harpaz, Y.,; Chothia, C. Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J. Mol. Biol. 1994, 238, 528–539. [CrossRef]
- Koch, M.,; Chitayat, S.,; Dattilo, B.M.,; Schiefner, A.,; Diez, J.,; Chazin, W. J.,; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [CrossRef]
- Xue, J.,; Rai, V.,; Singer, D.,; Chabierski, S.,; Xie, J.,; Reverdatto, S.,; Burz, D.S.,; Schmidt, A.M.,; Hoffmann, R.,; Shekhtman, A. Advanced glycation end product recognition by the receptor for AGEs. Structure 2011, 19, 722–732. [CrossRef]
- Hudson, B.I.,; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [CrossRef]
- Kim, H.J.,; Jeong, M.S.,; Jang, S.B. Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors. Int. J. Mol. Sci. 2021, 22, 6904. [CrossRef]
- Hudson, B.I.,; Carter, A.M.,; Harja, E.,; Kalea, A.Z.,; Arriero, M.,; Yang, H.,; Grant, P.J.,; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22 1572–1580. [CrossRef]
- Bongarzone, S.,; Savickas, V.,; Luzi, F.,; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [CrossRef]
- Wong, W.K.,; Leem, J.,; Deane, C.M. Comparative Analysis of the CDR Loops of Antigen Receptors. Front Immunol. 2019, 10, 2454. [CrossRef]
- Xie, J.,; Reverdatto, S.,; Frolov, A.,; Hoffmann, R.,; Burz, D.S.,; Shekhtman, A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 2008, 283, 27255–27269. [CrossRef]
- Yatime, L.,; Andersen, G.R. Structural insights into the oligomerization mode of the human receptor for advanced glycation end-products. FEBS J. 2013, 280, 6556–6568. [CrossRef]
- Zong, H.,; Madden, A.,; Ward, M.,; Mooney, M.H.,; Elliott, C.T.,; Stitt, A.W. Homodimerization is essential for the receptor for advanced glycation end products (RAGE)-mediated signal transduction. J. Biol. Chem. 2010, 285, 23137–23146. [CrossRef]
- Sárkány, Z.,; Ikonen, T.P.,; Ferreira-da-Silva, F.,; Saraiva, M.J.,; Svergun, D.,; Damas, A.M. Solution structure of the soluble receptor for advanced glycation end products (sRAGE). J. Biol. Chem. 2011, 286, 37525–37534. [CrossRef]
- Xie, J.,; Burz, D.S.,; He, W.,; Bronstein, I.B.,; Lednev, I.,; Shekhtman, A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J. Biol. Chem. 2007, 282, 4218–4231. [CrossRef]
- Wei, W.,; Lampe, L.,; Park, S.,; Vangara, B.S.,; Waldo, G.S.,; Cabantous, S.,; Subaran, S.S.,; Yang, D.,; Lakatta, E.G.,; Lin, L. Disulfide bonds within the C2 domain of RAGE play key roles in its dimerization and biogenesis. PLoS One 2012, 7, e50736. [CrossRef]
- Fritz, G. RAGE: a single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [CrossRef]
- Truong, T.T.T.,; Huynh, V.Q.,; Vo, N.T.,; Nguyen, H.D. Studying the characteristics of nanobody CDR regions based on sequence analysis in combination with 3D structures. J. Genet. Eng. Biotechnol. 2022, 20, 157. [CrossRef]
- Berman, H.M.,; Westbrook, J.,; Feng, Z.,; Gilliland, G.,; Bhat, T.N.,; Weissig, H.,; Shindyalov, I.N.,; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [CrossRef]
- Laskowski, R.A.,; Hutchinson, E.G.,; Michie, A.D.,; Wallace, A.C.,; Jones, M.L.,; Thornton, J.M. PDBsum: a Web-based database of summaries and analyses of all PDB structures. Trends Biochem. Sci. 1997, 22, 488–490. [CrossRef]
- Andreeva, A.,; Kulesha, E.,; Gough, J.,; Murzin, A.G. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020, 48, D376–D382. [CrossRef]
- Blum, M.,; Andreeva, A.,; Florentino, L.C.,; Chuguransky, S.R.,; Grego, T.,; Hobbs, E.,; Pinto, B.L.,; Orr, A.,; Paysan-Lafosse, T.,; Ponamareva, I.,; et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [CrossRef]
- Sobolev, V.,; Sorokine, A.,; Prilusky, J.,; Abola, E.E.,; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [CrossRef]
- Holm, L.,; Sander, C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 1995, 20, 478–480. [CrossRef]
- Kelow, S.,; Faezov, B.,; Xu, Q.,; Parker, M.,; Adolf-Bryfogle, J.,; Dunbrack R.L. A penultimate classification of canonical antibody CDR conformations. bioRxiv 2022. [CrossRef]
- Derewenda, Z.S.,; Derewenda, U.,; Kobos, P.M. (His)C epsilon-H...O=C < hydrogen bond in the active sites of serine hydrolases. J. Mol. Biol. 1994, 241, 83–93. [CrossRef]
- Kozlyuk, N.,; Gilston, B.A.,; Salay, L.E.,; Gogliotti, R.D.,; Christov, P.P.,; Kim, K.,; Ovee, M.,; Waterson, A.G.,; Chazin, W.J. A fragment-based approach to discovery of Receptor for Advanced Glycation End products inhibitors. Proteins 2021, 89, 1399–1412. [CrossRef]
- Yatime, L.,; Betzer, C.,; Jensen, R.K.,; Mortensen, S.,; Jensen, P.H.,; Andersen, G.R. The Structure of the RAGE:S100A6 Complex Reveals a Unique Mode of Homodimerization for S100 Proteins. Structure 2016, 24, 2043–2052. [CrossRef]
- Furey, W.,; Jr, Wang, B.C.,; Yoo, C.S.,; Sax, M. Structure of a novel Bence-Jones protein (Rhe) fragment at 1.6 Å resolution. J. Mol. Biol. 1983, 167, 661–692. [CrossRef]
- Herron, J.N.,; He, X.M.,; Mason, M.L.,; Voss, E.W. Jr.,; Edmundson, A.B. Three-dimensional structure of a fluorescein-Fab complex crystallized in 2-methyl-2,4-pentanediol. Proteins 1989, 5, 271–280. [CrossRef]
- Leahy, D.J.,; Hendrickson, W.A.,; Aukhil, I.,; Erickson, H.P. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science 1992, 258, 987–991. [CrossRef]
- Ito, N.,; Phillips, S.E.,; Stevens, C.,; Ogel, Z.B.,; McPherson, M.J.,; Keen, J.N.,; Yadav, K.D.,; Knowles, P.F. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90. [CrossRef]
- Holden, H.M.,; Ito, M.,; Hartshorne, D.J.,; Rayment, I. X-ray structure determination of telokin, the C-terminal domain of myosin light chain kinase, at 2.8 Å resolution. J. Mol. Biol. 1992, 227, 840–851. [CrossRef]
- Pinkner, J.S.,; Remaut, H.,; Buelens, F.,; Miller, E.,; Aberg, V.,; Pemberton, N.,; Hedenström, M.,; Larsson, A.,; Seed, P.,; Waksman, G.,; et al. Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc. Natl. Acad. Sci. U S A 2006, 103, 17897–17902. [CrossRef]
- Martinez, S.E.,; Huang, D.,; Szczepaniak, A.,; Cramer, W.A.,; Smith, J.L. Crystal structure of chloroplast cytochrome f reveals a novel cytochrome fold and unexpected heme ligation. Structure 1994, 2, 95–105. [CrossRef]
- Volbeda, A.,; Hol, W.G. Crystal structure of hexameric haemocyanin from Panulirus interruptus refined at 3.2 Å resolution. J. Mol. Biol. 1989, 209, 249–279. [CrossRef]
- Hough, M.A.,; Hasnain, S.S. Structure of fully reduced bovine copper zinc superoxide dismutase at 1.15 Å. Structure 2003, 11, 937–946. [CrossRef]
- Sutton, R.B.,; Davletov, B.A.,; Berghuis, A.M.,; Südhof, T.C.,; Sprang, S.R. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 1995, 80, 929–938. [CrossRef]
- Tawfeeq, C.,; Song, J.,; Khaniya, U.,; Madej, T.,; Wang, J.,; Youkharibache, P.,; Abrol, R. Towards a structural and functional analysis of the immunoglobulin-fold proteome. Adv. Protein Chem. Struct. Biol. 2024, 138, 135–178. [CrossRef]
- Chidyausiku, T.M.,; Mendes, S.R.,; Klima, J.C.,; Nadal, M.,; Eckhard, U.,; Roel-Touris, J.,; Houliston, S.,; Guevara, T.,; Haddox, H.K.,; Moyer, A.,; et al. De novo design of immunoglobulin-like domains. Nat. Commun. 2022, 13, 5661. [CrossRef]
- Sirois, C.M.,; Jin, T.,; Miller, A.L.,; Bertheloot, D.,; Nakamura, H.,; Horvath, G.L.,; Mian, A.,; Jiang, J.,; Schrum, J.,; Bossaller, L.,; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [CrossRef]
- Kabsch, W.,; Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [CrossRef]
- Hutchinson, E.G.,; Sessions, R.B.,; Thornton, J.M.,; Woolfson, D.N. Determinants of strand register in antiparallel beta-sheets of proteins. Protein Sci. 1998, 7, 2287–2300. [CrossRef]
- Halaby, D.M.,; Poupon, A.,; Mornon, J. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng. 1999, 12, 563–571. [CrossRef]
- Xue, J.,; Ray, R.,; Singer, D.,; Böhme, D.,; Burz, D.S.,; Rai, V.,; Hoffmann, R.,; Shekhtman, A. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 2014, 53, 3327–3335. [CrossRef]
- Kim, H.J.,; Han, C.W.,; Jeong, M.S.,; Kwon, T.J.,; Choi, J.Y.,; Jang, S.B. Cryo-EM structure of HMGB1-RAGE complex and its inhibitory effect on lung cancer. Biomed. Pharmacother. 2025, 187, 118088. [CrossRef]
- Mohan, S.K.,; Gupta, A.A.,; Yu, C. Interaction of the S100A6 mutant (C3S) with the V domain of the receptor for advanced glycation end products (RAGE). Biochem. Biophys. Res. Commun. 2013, 434, 328–333. [CrossRef]
- Penumutchu, S.R.,; Chou, R.H.,; Yu, C. Structural insights into calcium-bound S100P and the V domain of the RAGE complex. PLoS One 2014, 9(8), e103947. [CrossRef]
- Jensen, J.L.,; Colbert, C.L. X-ray structure of Ca(2+)-S100B with human RAGE-derived W72 peptide. To be published. 2016. [CrossRef]
- Jensen, J.L.,; Indurthi, V.S.,; Neau, D.B.,; Vetter, S.W.,; Colbert, C.L. Structural insights into the binding of the human receptor for advanced glycation end products (RAGE) by S100B, as revealed by an S100B-RAGE-derived peptide complex. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1176–1183. [CrossRef]
- Denessiouk, K.,; Permyakov, S.,; Denesyuk, A.,; Permyakov, E.,; Johnson, M.S. Two structural motifs within canonical EF-hand calcium-binding domains identify five different classes of calcium buffers and sensors. PLoS One 2014, 9, e109287. [CrossRef]
- Denesyuk, A.I.,; Denessiouk, K.,; Johnson, M.S.,; Uversky, V.N. Structural Catalytic Core of the Members of the Superfamily of Acid Proteases. Molecules 2024, 29, 3451. [CrossRef]
Figure 1.
DALI comparison between IgVRAGE (RAGE; 6XQ1_A; V-domain) and IgV (Bence-Jones protein Rhe; 2RHE_A; V-domain), which supports structural homology between the two domains. Localization of β-strands (DSSP) and CDRs are given in accordance with Dali and PyIGClassify2 software, respectively.
Figure 1.
DALI comparison between IgVRAGE (RAGE; 6XQ1_A; V-domain) and IgV (Bence-Jones protein Rhe; 2RHE_A; V-domain), which supports structural homology between the two domains. Localization of β-strands (DSSP) and CDRs are given in accordance with Dali and PyIGClassify2 software, respectively.

Figure 2.
Four types of classical Ig-like domain topologies. The figure is adapted from Bork et al. [24].
Figure 2.
Four types of classical Ig-like domain topologies. The figure is adapted from Bork et al. [24].
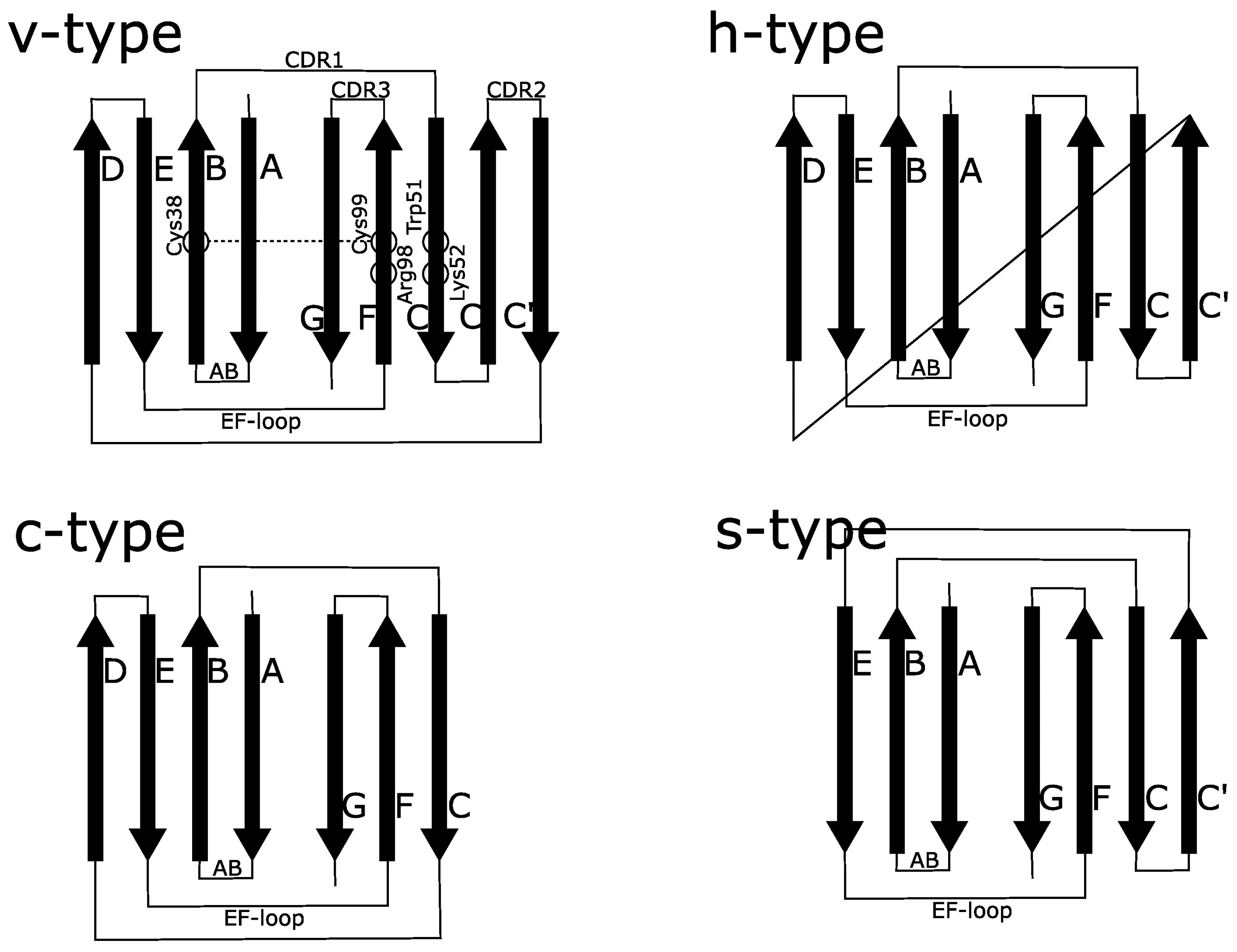
Figure 3.
The cross-β motif structural alignment and alignment of the respective hydrophobic core residues in IgV and IgVRAGE.
Figure 3.
The cross-β motif structural alignment and alignment of the respective hydrophobic core residues in IgV and IgVRAGE.
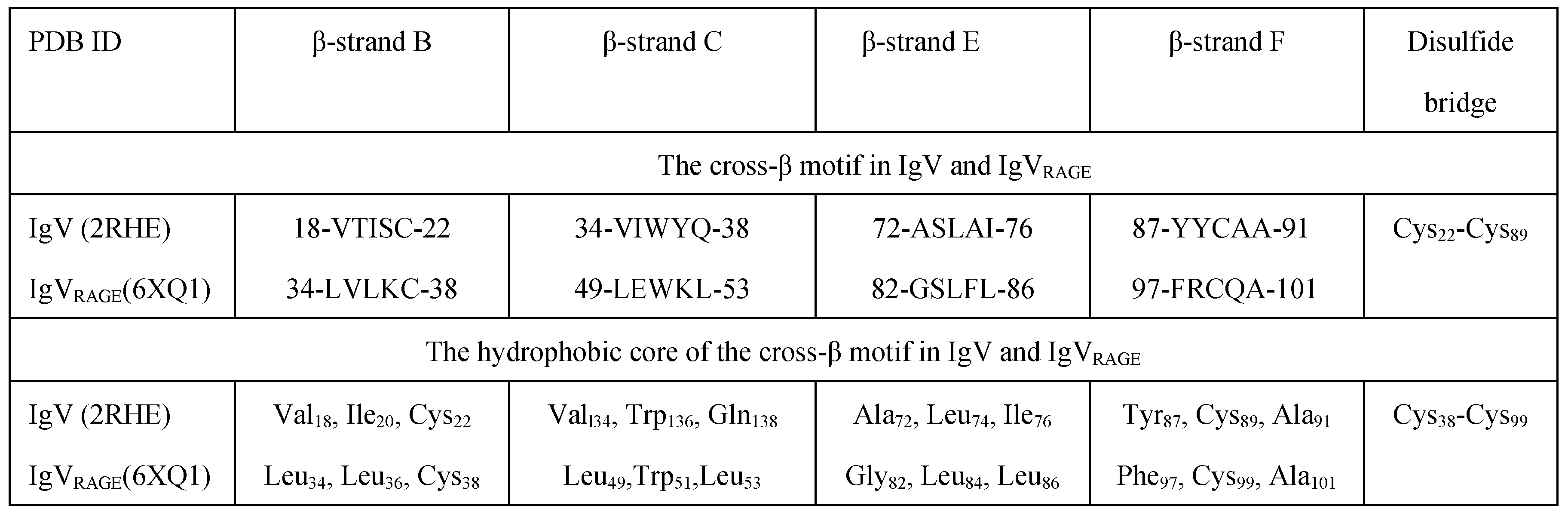
Figure 4.
Three ligand-binding modes observed on the surface of IgVRAGE (RAGE; 6XQ3 and 4OI7; V-domain): (A) The C-F-G β-sheet surface based mode; (B) the CE-loop based mode; and (C) the nucleic acid binding mode. The CE-loop is shown in green, the β-strands B, C, E and F (belong to the cross-β zone) in red, the β-strands A and G (do not belong to the cross-β zone) in dark red, and all the other loops in gray. In (A) and (B), a molecule of the same inhibitor V6Y bound to the two sites is shown in blue, and positions and names of the key ligand-binding amino acids are shown. In (C), positions of the key DNA-binding lysine triad, Lys37/Lys39/Lys43 [63], are shown in yellow in both domains of the dimer. Phe85-Phe85 designates central interaction of the non-polar dimerization interface between two IgVRAGE monomers. Approximate positions of the β-sheet ligand-binding mode and the CE-loop ligand-binding mode are also shown with respect to the nucleic acid binding location.
Figure 4.
Three ligand-binding modes observed on the surface of IgVRAGE (RAGE; 6XQ3 and 4OI7; V-domain): (A) The C-F-G β-sheet surface based mode; (B) the CE-loop based mode; and (C) the nucleic acid binding mode. The CE-loop is shown in green, the β-strands B, C, E and F (belong to the cross-β zone) in red, the β-strands A and G (do not belong to the cross-β zone) in dark red, and all the other loops in gray. In (A) and (B), a molecule of the same inhibitor V6Y bound to the two sites is shown in blue, and positions and names of the key ligand-binding amino acids are shown. In (C), positions of the key DNA-binding lysine triad, Lys37/Lys39/Lys43 [63], are shown in yellow in both domains of the dimer. Phe85-Phe85 designates central interaction of the non-polar dimerization interface between two IgVRAGE monomers. Approximate positions of the β-sheet ligand-binding mode and the CE-loop ligand-binding mode are also shown with respect to the nucleic acid binding location.
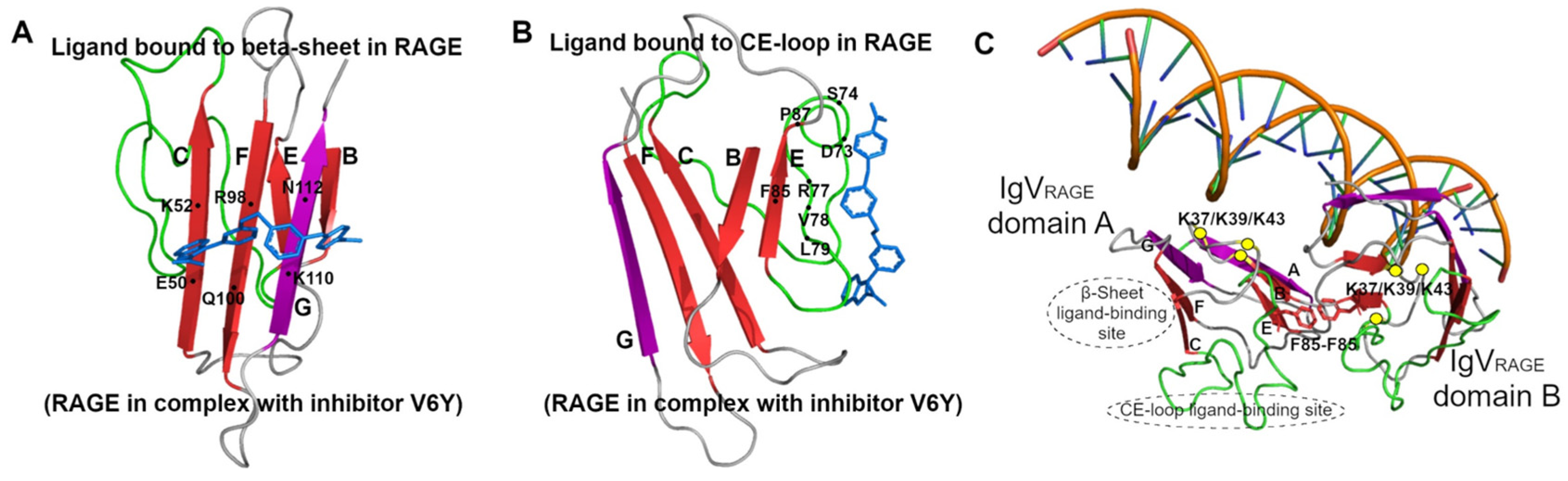

Table 1.
Hydrophobic core of the cross-β zone in different Ig-like domains.
| PDB ID | β-strand B | β-strand C | β-strand E | β-strand F |
Disulfide bridge / polar interactions |
Ref. # |
| IgVRAGE (6XQ1_A) | Leu34, Leu36, Cys38 | Leu49, Trp51, Leu53 | Gly82, Leu84, Leu86 | Phe97, Cys99, Ala101 |
Cys38-Cys99 NE1/Trp51-O/Gly82 |
49 |
| IgC1RAGE (6XQ1_A) | Asn139, Val141, Cys144 | Leu155, Trp157, Leu159 | Ser190, Leu192, Val194 | Phe206, Cys208, Phe210 |
Cys144-Cys208 OD1/Asn139-N/Thr134 ND2/Asn139-O/Thr134 OG/Ser190-NE1/Trp157 |
49 |
| IgC2RAGE (4P2Y_A) | Val255, Leu257, Cys259 | Ile269, Trp271, Lys273 | Pro284, Leu286, Leu288 | Tyr299, Cys301, Ala303 |
Cys259-Cys301 NE1/Trp271-O/Ser283 NZ/Lys273-O/Gln294 OH/Tyr299-O/Asp295 |
50 |
| IgV (2RHE_A) | Val18, Ile20, Cys22 | Val34, Trp36, Gln38 | Ala72, Leu74, Ile76 | Tyr87, Cys89, Ala91 |
Cys22-Cys89 NE2/Gln38-OH/Tyr87 |
51 |
| IgC (4FAB_H) | Leu143, Cys145, Val147 | Val155, Leu157, Trp159 | Leu182, Ser184, Val186 | Cys200,Val202, His204 |
Cys145-Cys200 NE1/Trp159-OG/Ser184 ND1/His204-OG/Ser208 NE2/His204-O/Phe151 |
52 |
| IgS (1TEN_A) | Ala819, Ile821, Trp823 | Ile833, Leu835, Tyr837 | Asn856, Tyr858, Ile860 | Val871, Leu873, Ser875 | O/Trp823-ND2/Asn856 OG/Ser875-O/Asp804 |
53 |
| IgH (1GOF_A) | Gly558, Ile560, Ile562 | Ala571, Leu573, Arg575 | Tyr602, Phe604, Val606 | Trp618, Leu620, Val622 | NH1/Arg575-OD1/Asp586 NH2/Arg575-O/Met233 NH2/Arg575-O/Pro615 OH/Tyr602-N/Ile568 NE1/Trp618-O/Leu614 |
54 |
| IgI (1FHG_A) | Ala59, Phe61, Cys63 | Val73, Trp75, Lys77 | Cys98, Leu100, Ile102 | Tyr113, Cys115, Ala117 |
Cys63-Cys115 NZ/Lys77-O/Asp108 OH/Tyr113-O/Asp109 |
55 |
| 2J7L_A | Met18, Leu20, Ile22 | Ala33, Ala35, Ile37 | Ser65, Val67, Leu69 | Phe88, Leu90, Glu92 | OG/Ser65-OE1/Gln57 OE1/Glu92-N/Ala1 OE2/Glu92-N/Ala1 |
56 |
| 1CTM_A | Phe45, Ala47, Val49 | Ala73, Leu75, Leu77 | Ile126, Phe128, Ile130 | Ile148, Val150, Gly152 | N/A | 57 |
| 1HC1_A | Phe460, Tyr462, Ile464 | Phe478, Ile480, Leu482 | Ile519, Arg521, Ser523 | Leu582, Val584, Val586 | NH2/Arg521-O/Asp525 NH2/Arg521-OD2/Asp506 |
58 |
| 1Q0E_A | Val29, Gly31, Ile33 | His41, Phe43, Val45 | Ala93, Val95, Ile97 | Met115, Val117, Glu119 | ND1/His41-O/His118 NE2/His41-O/Thr37 OE1/Glu119-N/Ser140 |
59 |
| 1RSY_A | Leu158, Val160, Ile162 | Val181, Val183, Leu185 | Glu208, Phe210, Phe212 | Leu224, Met226, Val228 | OE2/Glu208-N/Lys196 | 60 |
If a hydrophilic amino acid is found in any invariant hydrophobic position, the “polar interactions” comment shows how the charge of the side chain group of that amino acid is neutralized. N/A – Not Available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



