
Submitted:
15 October 2025
Posted:
21 October 2025
You are already at the latest version
Abstract
Hypercoagulability, immunothrombosis, and protein misfolding are deeply interconnected processes that converge on cell membranes as central orchestrators of thrombo-inflammation. In health, membrane lipid asymmetry, intact glycocalyx, and regulated receptor activity maintain vascular homeostasis. During inflammation or cell death, however, phosphatidylserine (PS) externalization, protein unfolding, and damage to glycosaminoglycans expose negatively charged, amyloidogenic surfaces that attract coagulation factors, inflammatory mediators, and adhesion proteins. These events generate catalytic sites for prothrombinase assembly. We review how cellular debris, microparticles, immune complexes such as neutrophil extracellular traps, and amyloidogenic plasma proteins, including serum amyloid A, interact with fibrinogen to form circulating (heterogeneous) procoagulant complexes, we term fibrinaloid microclot complexes (FMCs). Distinct from canonical fibrin clots, these FMCs display β-sheet–rich features, ThT-binding, and resistance to fibrinolysis, implicating them as key drivers of vascular pathology in inflammatory (and post-viral) syndromes. Recognizing different FMC phenotypes, mechanisms, and biochemical composition of these circulating complexes provides new insights into the pathogenesis of systemic inflammatory diseases, and highlights their potential as both diagnostic markers and therapeutic targets.
Keywords:
inflammation
; cell death
; membrane damage
; procoagulant
; amyloidogenic
; microclotting
Introduction
It has long been recognized that hypercoagulability, immunothrombosis, and protein damage represent interconnected and complex pathophysiological processes [1,2,3,4]. The classical triggers of cellular injury and prothrombotic surface formation, together with the well-characterized coagulation cascades, culminating in the conversion of prothrombin to thrombin and fibrinogen to fibrin, are well understood [5]. Widespread vascular injury, characterized by endothelial disruption, platelet dysfunction, abnormalities of erythrocytes and white blood cells, and immune dysregulation at the cellular level, collectively drive hypercoagulability and immunothrombosis in conditions such as type 2 diabetes mellitus, stroke, cardiovascular disease, and post-viral syndromes. In such conditions, endothelial cells and blood cells may undergo apoptosis or necrosis as a consequence of inflammatory cascades. At the centre of these cell death processes is the cell membrane, that plays a fundamental role in orchestrating mechanisms of cell death [6,7].
The cell membranes of healthy endothelial cells, platelets, and blood cells consist of a phospholipid bilayer embedded with proteins that together ensure vascular integrity, immune surveillance, and hemostatic balance [8,9,10]. By weight, these membranes are close to a 50:50 ratio of proteins: lipids or more (and in many cases 3:1) [11,12] although when counted by molecules, for every single protein molecule there may be as many as 50 to 100 molecules [13]. Despite being fewer in number, proteins are bulky and can occupy nearly half of the membrane surface area [12,14], providing dynamic functions such as adhesion, signaling, and enzymatic activity[14,15] (See Figure 1).
Under normal conditions, phosphatidylserine (PS) is restricted to the inner leaflet of the bilayer by energy-dependent flippases, maintaining lipid asymmetry [16]. The ATP-dependent enzyme, flippase normally keeps PS inside the cell, but when a PS flop occurs in the opposite direction (Figure 1C), PS acts as an ‘eat me’ signal on dead cells, and creates a scaffold for blood-clotting factors on activated platelets [17].
When endothelial cells are injured, when platelets become hyperactivated, or when red and white blood cells undergo stress, apoptosis, or eryptosis, scramblases are activated and PS also flips to the outer leaflet [17,18,19,20,21]. This externalization exposes a strongly negatively charged surface [22]. Although the total protein-to-lipid mass ratio remains unchanged, the exposure of negatively charged PS on the cell surface alters the biochemical landscape of the membrane [23,24,25]. Simultaneously with this PS flip, proteins are damaged. Protein damage and loss of proteostasis during cell stress exposes β-sheet motifs that can aggregate into amyloid-like assemblies [26]. This does not mean every dying cell becomes “amyloid-rich,” but it does mean that the risk of β-sheet aggregation increases sharply as proteins denature and unfold.
The flip of PS to the outer membrane leaflet has profound consequences for the protein–lipid balance of the cell surface. Because PS carries a negative charge, its externalization exposes new electrostatic sites that strongly influence how proteins bind, attracting integrins, annexins, and coagulation factors such as prothrombin and factor Xa, which effectively increases the functional protein content at the surface [27,28]. In addition, PS redistribution alters lipid microdomains, promoting clustering of membrane proteins [29], including receptors and adhesion molecules [30], so that proteins occupy a larger proportion of the surface area even though their overall mass fraction within the membrane remains unchanged. This could create a shift in protein coverage, and in biophysical measurements it may appear that membranes are more protein-dense after PS exposure because cytoplasmic or plasma proteins are recruited to the newly exposed PS [31,32]. In some instances, however, PS redistribution (externalization) on apoptotic or activated cells serves as an “eat-me” or tolerogenic signal [33] and also facilitates binding of serum proteins, opsonins, and complement. Lipid microdomains may shift with PS exposure, favoring receptor clustering and increasing functional protein coverage at the membrane, although direct evidence for that remains less well established.Specific examples illustrating this transformation, are known in platelets [34], where PS externalization drives assembly of the tenase and prothrombinase complexes [7,35,36], producing a strongly procoagulant phenotype [37]. A procoagulant environment is also seen in erythrocytes undergoing eryptosis [21]. PS exposure also serves as a clearance signal for macrophages [38]; while simultaneously attracting annexins, immunoglobulins, and complement proteins and in endothelial cells [39]. PS externalization during apoptosis or injury thus facilitates binding of clotting proteins, including prothrombin and factor Xa, thereby further shifting the membrane toward a protein-dominated interface [7,28,40]. Similarly, tissue factor (TF) is important for a procoagulant surface, as it is the principal initiator of one arm of the coagulation cascade. Under normal conditions, TF is largely confined to subendothelial tissues, where its separation from circulating blood maintains tight control of clotting activation. When vascular damage occurs, TF becomes exposed and rapidly engages circulating factor VII, triggering downstream steps of the coagulation cascade [41]. In pathological contexts, TF expression can also be upregulated in monocytes and macrophages in response to inflammatory mediators, including lipopolysaccharide, tumor necrosis factor-α, and interleukin-1. As a glycosylated membrane protein, TF requires interaction with PS for its full procoagulant activity [42,43].
Glycosaminoglyans
Glycosaminoglycans (GAGs), and in particular heparan sulfate proteoglycans (HSPGs) of the endothelial glycocalyx, are central regulators of both inflammation and thrombosis [44]. In the context of inflammation, GAGs modulate leukocyte adhesion, chemokine binding, and vascular barrier function [44,45,46]. They act as reservoirs for cytokines and chemokines, regulating their bioavailability and signaling. In thrombo-inflammation, intact GAGs help preserve anticoagulant activity by binding and activating proteins such as antithrombin and heparin cofactor II, while their degradation or shedding exposes prothrombotic endothelial surfaces, promotes leukocyte adhesion, and facilitates complement and coagulation activation [47,48]. Loss of endothelial GAGs has been shown to directly trigger a procoagulant phenotype, whereas their preservation maintains vascular homeostasis [49]
Creating a Prothrombotic Surface
When PS externalizes, the lipid microenvironment of the membrane changes dramatically, and this directly affects embedded and peripheral proteins. The redistribution of PS alters local lipid packing, which drives clustering of transmembrane proteins and receptors (such as integrins, adhesion molecules, and glycoproteins) [50]. These clusters create microdomains where binding sites for circulating proteins are concentrated [51]. These newly exposed negative charges on PS attract plasma proteins that contain calcium-dependent binding motifs (e.g., the Gla domains of clotting factors) [40,52]. Functionally, this means that even though the intrinsic protein-to-lipid ratio in the membrane does not change, the effective protein density at the surface increases as both membrane proteins and newly bound plasma proteins accumulate in PS-enriched regions. In essence, therefore it can be assumed that the PS flip reorganizes membrane proteins into prothrombotic clusters while simultaneously transforming the surface into a catalytic platform for the coagulation cascade.
Prothrombin, factor Xa, and factor Va assemble at PS-rich membrane domains, where negatively charged phospholipids provide the Ca[2]+-dependent binding sites required for formation and stabilization of the prothrombinase complex, thereby markedly enhancing thrombin generation [28,53,54]. Annexins and complement components are also recruited, which crosslink PS with membrane proteins and further consolidate a surface that is highly adhesive for clotting factors [7]. Factor Xa and Factor Va assemble on PS-rich microdomains to form the prothrombinase complex, which catalyzes the rapid conversion of prothrombin to thrombin [28]. Thrombin then cleaves fibrinopeptides from circulating fibrinogen to form fibrin monomers. Because the prothrombinase complex is assembled on PS-rich membrane surfaces (e.g., activated platelets or PS-exposed cells), subsequent fibrin formation (polymerization) begins in close proximity to those membrane surfaces [53]. The PS flip area is essential for the formation of the ternary prothrombinase-prothrombin complex (discovered by [55], and this anionic phospholipid layer provides a binding surface for the ternary complex assembly and is mainly provided by the membranes of activated platelets and endothelial cells [53].
Fibrin monomers polymerize into protofibrils and fibers after thrombin cleavage of fibrinogen; fibrinogen also binds adhesion receptors such as integrin αIIbβ3 when they are activated, which can localize and concentrate fibrin(ogen) at membrane surfaces [56,57]. Although direct anchoring to PS-rich membrane fragments is less well quantified, these combined interactions bias fibrin deposition toward damaged (procoagulant) cell surfaces. This local enrichment of fibrinogen ensures that, once thrombin is generated, fibrin rapidly deposits at these procoagulant layers.
Another important molecule is fibronectin. It is a multifunctional adhesive glycoprotein that regulates clot formation, and orchestrates tissue repair and immune clearance [58]. Fibronectin also enhances thrombogenesis by cross-linking to fibrin [59] and promotes thrombus growth [60]. Fibronectin mostly binds to integrins, with α and β subunits located on the cell surface [61].
Although not a focus of this paper, Table 1 shows the various fibrinogen receptors on cells in the vasculature that interact with fibrinogen. In fact, cells of the vasculature are abundant in receptors for various inflammatory molecules. These molecules may interact with both healthy cells and the cellular debris resulting from cell death and inflammation, with both the functioning cells and debris creating binding surfaces for circulating inflammatory cells. One of the inflammatory molecules (serum amyloid A (SAA)) is known to be highly upregulated in various inflammatory diseases, and that is also well-recognized as an amyloid and amyloidogenic molecule [62,63]. SAA proteins, particularly SAA1 and SAA2, mediate functions such as chemoattraction, cytokine induction, and modulation of lipid metabolism through receptors like Formyl Peptide Receptor 2 (FPR2), Toll-like receptor 4 (TLR4), and the receptor for advanced glycation end products (RAGE) [63,64]. Binding of SAA to platelets is mediated through the integrin receptor αIIbβ3, whose ligands encompass a range of ECM proteins amongst which is fibronectin [64,65,66]. In particular, αIIbβ3 binds to talin, a cytoplasmic protein that is essential both for inside-out integrin activation and for linking integrin to the actin cytoskeleton [67]. Specifically, talin binds to the cytoplasmic tail of the integrin’s β3 subunit, with one binding site being a membrane-proximal region and another being the membrane-distal NPXY motif. The interaction of the amyloidogenic talin with αIIbβ3 is essential for proper activation of the integrin in platelets, thus playing a direct role in thrombosis and other amyloid-related processes [68,69].
SAA also binds to FPR2 on neutrophils [70,71]. FPR2 is present on human platelets, where it mediates responses to SAA and other ligands, contributing to platelet activation, aggregation, and thrombo-inflammatory signaling [64]. FPR2 is also abundantly expressed on neutrophils, monocytes, macrophages, and some lymphocyte subsets, where it regulates chemotaxis, phagocytosis, inflammatory processes. [72]. Interestingly, SAA also binds to membrane surfaces that has undergone the PS flip [73]
In healthy individuals, decaying cells undergo rapid clearance by phagocytes [81]. Interestingly, although the number of apoptotic cells produced and lost daily is known to be some 50-70 billion in a healthy adult human, apoptotic cells are rarely observed [82]. This absence of numerous decaying cells, is due to the existence of a cellular process called efferocytosis that efficiently clears apoptotic cells [81]. Apoptotic cells that expose PS are typically and rapidly recognized by phagocytic cells (macrophages,, etc.), via ‘eat-me’ signals [83]. This clearance helps to remove PS-exposed cells before they can nucleate coagulation.
Damaged Cell Membranes Can Interact with Various Circulating Inflammatory Molecules
When PS-exposed procoagulant membranes encounter circulating inflammatory molecules (such as SAA, CRP, VWF, complement, or histones), these proteins bind to the negative surface, amplifying coagulation factor assembly and stabilizing the prothrombotic phenotype [7]. This interaction represents a major link between inflammation and pathological clotting [2]. Inflammatory mediators such as SAA, VWF, histones, complement proteins, and C-reactive protein can directly interact with PS-rich membranes, reinforcing their prothrombotic activity [34,84,85]. Negatively charged PS patches act as high-affinity binding sites for these proteins. SAA is for example an acute-phase protein that is inherently amyloid in nature. It can bind to membrane receptors, including αIIbβ3 and is capable of forming oligomers (e.g., in amyloidosis [86]), It is also strongly amyloidogenic under pathological conditions [87,88]. As inflammatory molecules accumulate on PS-exposed membranes, they disrupt lipid microdomains, drive clustering of membrane proteins, and recruit coagulation factors such as prothrombin and factor Xa; together, these changes amplify and stabilize the procoagulant phenotype [89,90].
In healthy physiology, PS exposure alone is insufficient to sustain thrombin generation because apoptotic cells are rapidly cleared. In inflammatory states, however, mediators such as CRP, histones, and serum amyloid A bind to PS-rich surfaces, protecting them from clearance and converting them into scaffolds for fibrin deposition. Thus, inflammatory molecules function as biochemical amplifiers, transforming normally transient PS-exposed membranes into persistent catalytic platforms that drive thromboinflammation.
Cellular Senescence
Senescent cells accumulate in ageing phenotypes, but are also implicated in both noncommunicable and communicable diseases. For instance, cardiovascular disease, diabetes, and autoimmune conditions exhibit higher levels of cellular senescence [91,92,93,94]. Some viruses, such as SARS-CoV-2, and other pathogens are capable of inducing cellular senescence in a range of cell types [95,96]. SARS-CoV-2 is known to induce endothelial [97] and leukocyte senescence [98,99]. The senescent phenotype is characterized by an increase and dysregulation in pro-inflammatory molecules such as cytokines and chemokines, oxidative stress, dysregulation of growth factors, and procoagulant factors, such as VWF and TF [97,100], specifically in haematological and vascular cells. Additionally, senescent cells and processes are also linked to amyloidogenesis [101,102], and there are also indications that amyloid proteins and aggregates can induce cellular senescence [103,104].
Hence, senescent processes can also induce thrombosis and implicate immune function. Senescent endothelial cells exhibit impaired nitric oxide production, increased reactive oxygen species, and loss of glycocalyx integrity, all of which predispose to endothelial barrier disruption and platelet adhesion [105,106]. Furthermore, senescent cells often upregulate TF [107], thereby promoting a procoagulant membrane phenotype similar to that seen in apoptosis or eryptosis. The increase in procoagulant factors such as vWF and TF by senescent endothelial and other cells can exacerbate fibrin formation at prothrombotic surfaces in blood and at the vascular wall.
Importantly, the plasma membranes of senescent cells undergo biophysical remodeling, including altered lipid composition, membrane stiffening, and redistribution of key proteins, contributing to impaired endocytosis, antigen presentation, and coagulation regulation [108,109,110]. These changes are compounded by loss of membrane lipid asymmetry and surface exposure of PS and annexins in some cases, as well as damage-associated molecular patterns (DAMPs) [107,111], creating an environment primed for immune recognition, macrophage recruitment, and coagulation cascade activation. The persistence of senescent cells is particularly exacerbated in conditions where immune dysfunction is prominent, resulting in a decreased ability to recognize and remove senescent cells. Thus, cellular senescence represents a third major pathway – alongside apoptosis and necrosis – through which membrane alterations promote prothrombotic transformation of vascular and blood cell surfaces.
Decaying Cells Can Break Down into Smaller and Smaller Fragments and Micropartices/Microvesicles That Become Prothrombotic Seeding Areas
As cells decay, their fragments or the resulting microparticles [112,113] still carry these prothrombotic signals [114,115] and can accumulate fibrinogen/fibrin and inflammatory molecules. Such circulating “micro” clots might not immediately be in high enough quantities or concentrations to initiate a true clot. However, these smaller entities may be able to associated with fibrin networks. It is e.g., known that microparticles (that are also prothrombotic entities) can accumulate and build into fibrin networks [116,117,118]. Zubairova et al. in 2015 showed that microparticles accelerate fibrin polymerisation and support formation of more dense fibrin clots that resist fibrinolysis. Such microparticles drive faster thrombin generation, impact thrombin-mediated kinetic effects of fibrin formation, and impacts fibrin structure and properties. Fibrin clots formed in the presence of microparticles, contain 0.1–0.5-μm size granular and CD61-positive material on fibres, suggesting that platelet-derived microparticles attach to fibrin [116] (see Figure 2).
Microclots Are Not ‘Just’ Microparticles
Although microparticles can bind to fibrin(ogen) [116,117,119], and there is evidence that microclots can contain microparticles, we would stress that they are not synonymous for a variety of reasons:
- Number. Microparticles of the above size range can be present in plasma in very large numbers, values quoted ranging from 8.106 to 4.109 /mL 120; ~3.106 /mL has been stated just for platelet-derived microparticles [131]. Orozco and coworkers found ~108 microparticles /mL [132], Albert and co-workers over 106 /mL [133] and Chandler and coworkers, numbers from 3.106 to 108 /mL [120]. By contrast, microclots greater than 1 μm in equivalent diameter are commonly present in numbers with a median below 1000 /mL [129] and a maximum value around 6.105 /mL, even in pathological conditions.
- Composition. The composition of microparticles simply reflects the composition of the cells from which they originate, and these cellular origins typically include platelets [117,134,135]. erythrocytes [136], leukocytes [137,138], and endothelial cells [138,139,140,141]. Unsurprisingly, their origin affects their thrombotic potential [117,142,143,144] as well as reflecting the diseases with which they are associated [145,146]. We note too the possibility that nanoplastics may also contribute to a pathological microparticle burden [147,148] as they themselves are amyloidogenic [149]. The same issues pertain for similarly sized particulate matter ingested via air pollution (e.g., [150,151]). Importantly, because these items are essentially insoluble they too can contribute to the blockage of the microcirculation that underpins so much of the pathology of fibrinaloid microclots. However, microparticles themselves are not specifically enriched in fibrin(ogen) albeit that they can bind it. We note that by contrast the fibrinaloid microclots are dominated by fibrin(ogen) subunits [152,153,154] and are significantly enriched in amyloidogenic proteins [155,156].
- Causality. It would seem that lipid microvesicles will bind fibrin(ogen) in microparticles but that actual clotting in the microclots traps other things, including microparticles. Consequently the order of adding fibrin in the two structures is opposite.
Biochemical Characteristics of Molecules That Can Associate with Decaying Membranes or Act as Prothrombotic Seeding Areas
Many pro-inflammatory molecules that have previously been found to accumulate on procoagulant cellular membrane debris have an inherent amyloidogenicity; for a review see [155]. Any protein’s amyloidogenicity can be determined by using predictive bioinformatics tools such as AmyloGram [157]. Such molecules may also themselves interact with circulating fibrinogen and plasma proteins when added to healthy plasma. Examples of such interactions are SAA, complement, as well as interleukins [62,158,159].
As mentioned in previous paragraphs, SAA is an abundant inflammatory molecule and has numerous receptors on cell membranes [63,64,66,70,87,160]. SAA and fibrinogen share receptors, suggesting that co-binding, or adjacent receptor binding to the receptors left on the debris occurs. This provides evidence that these seeding areas are not only prothrombotic but also amyloidogenic. We have found SAA to be significantly upregulated in various inflammatory diseases [161,162,163,164]. Proteomic analyses show that unusually amyloidogenic proteins are enriched in such clots, probably by actually being incorporated into cross-beta elements of the developing fibrils [155,156].
In purified fibrin(ogen) systems, defined amyloid fibrils from SARS-CoV-2 spike peptides exert segment-specific effects on clot dynamics and lysis [165]. Westman and coworkers showed that fibrils from spike601 (aa601–620) delayed thrombin-driven fibrin formation by sequestering fibrinogen without impairing subsequent fibrinolysis, whereas fibrils from spike685 (aa685–701; just C-terminal to the furin site) promoted denser clot architecture and yielded plasmin-resistant residues in a dose-dependent manner [165]. Fluorescence co-localization and TEM indicated that spike685 fibrils co-assemble with fibrin into granular/fibrous aggregates that persist after tPA/plasmin treatment, consistent with impaired fibrinolysis [166].
In the case of fibrinogen, the fibrinogen α-chain subunit, and specifically its αC domain, is often considered to be the most amyloidogenic [167,168], on the grounds that hereditary mutations in the fibrinogen Aα (FGA) gene can lead to Fibrinogen Alpha Chain (AFib) amyloidosis [169,170].
These findings provide a concrete biochemical mechanism by which spike-derived amyloid, particularly spike685, could seed fibrinolysis-resistant (micro) clots relevant to specifically long COVID pathophysiology. While the data derive from controlled in vitro assays mostly do not model whole-blood shear [171] or cellular clearance, they identify an aggregation-prone spike segment with clear potential to bias clot structure toward persistent, prothrombotic states [165].
A Healthy Circulation Avoids Uncontrolled SAA– or Fibrinogen–Receptor Binding
There are various mechanisms whereby receptor binding does not randomly activate in the presence of possible ligands like SAA and fibrinogen. In healthy circulation, random or uncontrolled binding of SAA or fibrinogen to their receptors is prevented by several protective mechanisms. Platelet integrins such as αIIbβ3 remain inactive until specifically triggered by inside-out signaling, ensuring that fibrinogen cannot bind under basal conditions [17]. The intact endothelial glycocalyx, together with continuous release of nitric oxide (NO) and prostacyclin (PGI2), enforces an anti-adhesive and anti-thrombotic vascular surface [172]. Circulating acute-phase proteins such as SAA are normally sequestered in high-density lipoprotein (HDL) complexes, limiting their free availability for receptor engagement [173,174]. Finally, maintenance of membrane asymmetry by flippases keeps PS restricted to the inner leaflet, thereby preventing exposure of the negatively charged surfaces that otherwise act as procoagulant platforms [17]. See Table 2, Table 3 and Table 4 for summaries.
The Heterogeneity of Complex Hydrophobic, Amyloid-Containing Biological Structures, Including Lipofuscin, Atherosclerotic Plaques, and Fibrinaloid Microclot Complexes and Macroclots
Most simple biological structures are more or less homogeneous, as evolution has selected them to maintain specific functions and hence structures, although we recognise the ability of various ‘metamorphic’ [156] or ‘fold-switching’ [184,185,186,187] proteins to adopt multiple stable conformations In particular, prions and other proteins can adopt stable, amyloid states that often self-organise into insoluble fibrils of fairly similar diameters in the range 10-20nm governed by the length of the cross-beta motif that characterises such amyloid.
Many classical amyloidoses have been described [188,189]. For the present context, however, we emphasize our finding [166] that fibrinogen can interact with amyloidogenic inflammatory molecules either directly in circulation or at sites where fibrinogen receptors are expressed. These interactions give rise to insoluble, misfolded, amyloidogenic protein complexes that can be observed in both whole blood and platelet-poor plasma and they stain positively with the fluorogenic dyes like ThT. We have termed these misfolded and amyloidogenic heterogenous deposits, fibrinaloid microclot complexes. Moreover, the clotting cascade, culminating in the conversion of prothrombin to thrombin, can be triggered, resulting in true fibrin complexes that have also adopted an anomalous, misfolded and potentially amyloidogenic structure.
Cross-seeding of amyloidogenic proteins leads to fibrils containing a very large variety of different proteins [155]. The hydrophobic patches of amyloids also bind strongly to lipids, something important to their cytotoxicity [190,191,192]. The presence of metals such as iron that can catalyse the Fenton reaction [193,194] is also likely since many of the proteins are highly oxidised (see also for atherosclerotic plaques [195,196,197]).
Another set of structures that also contain lipids, amyloid proteins and DNA in a very heterogeneous layout is represented by lipofuscin [198,199,200,201], a complex pigment found in the lysosomes of ageing cells.,[202,203,204].
Yet another complex set of structures involving lipids and amyloidogenic proteins (as well as other materials, not least unliganded iron ions that catalyse oxidations [193]), is represented by atherosclerotic plaques [202,203,204,205], that are obviously highly important in the generation of myocardial infarctions and other vascular problems. Amyloid(ogenic) proteins that they are known to contain include alpha1-antitrypsin [206,207], serum amyloid A [208,209,210], apolipoprotein A1 [211,212,213] and amyloid-beta [214,215]. Strikingly the first three of these have also been found in fibrinaloid microclots [62,152,154], consistent with their appearance in and contributions to a variety of thrombotic diseases [216] and the general biophysics underlying cross-seeding [155,156]. The same is true for the kinds of heterogeneous plaque found in Alzheimer’s dementia [217].
Finally, we recently established that the macroclots isolated from individuals following an ischaemic stroke are also amyloid in nature [218], with the heterogeneity being especially evident from the comparison of brightfield and fluorescence images [219]. Consequently, characterisation of this kind of heterogeneity becomes important if we are to understand its significance.
Characterizing (Heterogenous) Prothrombotic Complexes
Characterization of these (heterogenous) proinflammatory circulating complexes in the form of “micro”clots remains challenging. We have been using Thioflavin T (ThT), which is a benzothiazole dye that remains non-fluorescent when free in solution because its two aromatic rings can rotate freely around a central single bond. Fluorescence occurs when ThT binds to a surface that restricts its rotational freedom, locking the dye into a planar conformation. This mechanism underlies its widespread use as an amyloid probe: aligned within the repetitive grooves of cross-β sheet fibrils, ThT becomes immobilized and emits a strong fluorescent signal [220,221,222] However, the same mechanism also means that ThT can interact with other repetitive or rigid hydrophobic surfaces, potentially including lipid assemblies [223], nucleic acids [224] and damaged or misfolded proteins [225,226,227].
In complex biological environments, ThT fluorescence can therefore also indicate the presence of structured hydrophobic binding sites. We have previously shown that we can induce ThT binding to purified fibrinogen after addition of SAA [62,161], LPS, LTA, gingipains [228,229,230] and also spike protein [231,232]. SAA is, of course, a known amyloidogenic molecule [160,233,234] and is upregulated in the circulation in various inflammatory diseases [161,162,235]. The SARS-CoV-2 spike protein also has amyloidogenic potential and its spike protein (especially in the presence of LPS or amylin) [236] may form aggregates, and Amytracker probes show small aggregates [237,238,239].
In summary, ThT fluorescence thus arises when its rotational freedom is restricted, most strongly when the dye intercalates into the repetitive grooves of cross-β sheet amyloid fibrils. While ThT is known to also bind certain non-amyloid surfaces, in complex biological systems such as plasma or damaged cell membranes, its signal is most informative when it highlights regions where amyloidogenic proteins may accumulate. Proteins like fibrinogen, SAA, or other aggregation-prone inflammatory molecules can adopt β-sheet–rich conformations upon interaction with perturbed membranes, creating surfaces that both restrict ThT rotation and report amyloidogenic potential. Thus, even though ThT can bind to hydrophobic structures in general, in the prothrombotic context a ThT signal may suggest the presence of interactions between proteins with amyloidogenic potential and damaged membrane surfaces or such amyloidogenic inflammatory molecules interacting with fibrin(ogen). Both these scenarios provide evidence for the presence of complexes with amyloid-like properties in inflammatory and post-viral diseases.
Summarising Our Thoughts on the Various Phenotypes of Circulating Prothrombotic Complexes (We Termed Fibrinaloid Microclot Complexes (FMCs)) May Drive Thrombo-Inflammation on β-Sheet Rich and Amyloidogenic Surfaces
We suggest that circulating prothrombotic complexes (or fibrinaloid microclot complexes (FMCs)), drive thrombo-inflammation can be classified into several distinct phenotypic forms (see Figure 3, Figure 4, Figure 5 and Figure 6).
Our suggested phenotypes are:
- o Cell-derived debris
These are characterized by dying cells or large fragments of damaged or apoptotic endothelial cells, erythrocytes, platelets, and leukocytes. These cellular debris retain intact membrane patches with exposed phosphatidylserine and clustered membrane proteins, serving as initial scaffolds for clotting factor binding. Furthermore, proteins in decaying membranes become β-sheet rich, as they unfold.
- o Subcellular vesicles and microparticles
Apoptotic bodies and microparticles (0.1–1 μm), derived from shedding or fragmentation of the parent cell membranes are rich in membrane proteins, PS patches, and receptors; capable of binding fibrinogen, prothrombinase, and annexins.
- o Nucleoprotein immune complexes
Neutrophil extracellular traps (NETs) are DNA–protein assemblies composed primarily of extracellular chromatin, histones, and neutrophil enzymes such as myeloperoxidase (MPO). These structures can act as nucleation sites for fibrin(ogen), recruiting additional inflammatory and coagulation factors to form prothrombotic complexes. As shown in Figure 6, NETs can associate with pre-existing amyloidogenic seeding surfaces or independently promote microclot formation. NETs are frequently observed in inflammatory conditions including COVID-19, Long COVID [240,241], and cancer, where they contribute to persistent vascular inflammation and immunothrombosis. Recent findings demonstrate that microclots in Long COVID frequently co-localize with NETs, visualized using THT, myeloperoxidase (MPO), and Hoechst staining in PPP. This association likely stabilizes microclots, impairs fibrinolysis, and amplifies endothelial injury, positioning NET-associated microclots as key drivers of immunothrombosis and potential diagnostic targets [240].
- o Plasma protein aggregates associated with amyloidogenic inflammatory molecules
Fibrin(ogen) that associate with SAA, VWF and other inflammatory circulating proteins, such as spike protein, can co-aggregate or associate with fibrin(ogen) and thus can result in amyloid-like conformations. The rational for this statement is because types of proteins or peptides are either prone to cross-β sheet formation, or a amyloid molecule themselves (e.g., SAA).
Together, these phenotypic forms, represent a pathological mode of alteration that is distinct from canonical thrombin-driven clotting, which normally yields fibrin clots that are susceptible to fibrinolysis and generate breakdown products such as D-dimer. These immune-thrombotic complexes are rather circulating signaling entities that show significant ThT-binding capacity and that are resistant to classical fibrinolytic processes due to their composition. We therefore suggest that these structures act as prothrombotic seeding areas that recruit fibrinogen, fibrin, and inflammatory molecules, forming the initial scaffolds of “micro” clots. At this stage, they may not yet contain sufficient material to develop into a fully formed thrombus, but they can be detected using markers for fibrin(ogen) or associated inflammatory molecules. The kinetics of their formation, and how their composition evolves over time, remain largely unexplored. Among circulating amyloidogenic proteins, SAA is one of the most prominent and shares several receptors with fibrinogen, suggesting a mechanistic link between inflammation, amyloid formation, and early thrombotic signaling.
Thus in conclusion, we suggest that Insoluble protein complexes, arising from inflammatory molecules, interactions with fibrinogen, or deposits of cellular prothrombotic debris, can act as scaffolds. We refer to these early pathological deposits as circulating “prothombotic microclot complexes”, reflecting their role as triggers of downstream thrombotic events. These complexes form catalytic surfaces for thrombin generation. In the presence of prothrombin, thrombin, or other activators of the coagulation cascade, these complexes have the potential to initiate bona fide and “true” clot formation either alone or on damage cells of the vasculature. We therefore collectively term these precursor prothrombotic and misfolded circulating complexes, ThT-positive fibrinaloid microclot complexes (or FMCs).
Author Contributions
Conceptualization, DBK & EP; Formal Analysis, DBK & EP; Resources, DBK & EP; Writing and Original Draft Preparation, DBK; Writing – Review & Editing, DBK & EP; Visualization, DBK & EP; Funding Acquisition, DBK & EP; Editing of manuscript, ART; Microscopy, CV; Cellular Scenenscence, JMN.
Funding
DBK thanks the Balvi Foundation (grant 18) and the Novo Nordisk Foundation for funding (grant NNF20CC0035580). EP thanks PolyBio Research Foundation, Kanro Research Foundation and Balvi Research Foundation for funding. JMN thanks Kanro Research Foundation. The content and findings reported and illustrated are the sole deduction, view and responsibility of the researchers and do not reflect the official position and sentiments of the funders. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of Interest
EP is a named inventor on a patent disclosing the use of fluorescence microscopy in Long COVID.
References
- Kaiser, R. , Gold, C., and Stark, K. (2025). Recent Advances in Immunothrombosis and Thromboinflammation. Thromb Haemost. [CrossRef]
- Engelmann, B. , and Massberg, S. (2013). Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13, 34-45. [CrossRef]
- Almskog, L.M. , and Ågren, A. (2025). Thromboinflammation vs. immunothrombosis: strategies for overcoming anticoagulant resistance in COVID-19 and other hyperinflammatory diseases. Is ROTEM helpful or not? Front Immunol 16, 1599639. [CrossRef]
- Che, X. , Ranjan, A., Guo, C., Zhang, K., Goldsmith, R., Levine, S., Moneghetti, K.J., Zhai, Y., Ge, L., Mishra, N., et al. (2025). Heightened innate immunity may trigger chronic inflammation, fatigue and post-exertional malaise in ME/CFS. npj Metabolic Health and Disease 3, 34. [CrossRef]
- Smith, S.A. , Travers, R.J., and Morrissey, J.H. (2015). How it all starts: Initiation of the clotting cascade. Crit Rev Biochem Mol Biol 50, 326-336. [CrossRef]
- Zhang, Y. , Chen, X., Gueydan, C., and Han, J. (2018). Plasma membrane changes during programmed cell deaths. Cell Research 28, 9-21. [CrossRef]
- Wang, J. , Yu, C., Zhuang, J., Qi, W., Jiang, J., Liu, X., Zhao, W., Cao, Y., Wu, H., Qi, J., and Zhao, R.C. (2022). The role of phosphatidylserine on the membrane in immunity and blood coagulation. Biomarker Research 10, 4. [CrossRef]
- Zha, D. , Wang, S., Monaghan-Nichols, P., Qian, Y., Sampath, V., and Fu, M. (2023). Mechanisms of Endothelial Cell Membrane Repair: Progress and Perspectives. Cells 12. [CrossRef]
- Dejana, E. , Tournier-Lasserve, E., and Weinstein, B.M. (2009). The Control of Vascular Integrity by Endothelial Cell Junctions: Molecular Basis and Pathological Implications. Developmental Cell 16, 209-221. [CrossRef]
- Zwaal, R.F., and Schroit, A.J. (1997). Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89, 1121–1132. [CrossRef]
- Kell, D.B. (2021). The Transporter-Mediated Cellular Uptake and Efflux of Pharmaceutical Drugs and Biotechnology Products: How and Why Phospholipid Bilayer Transport Is Negligible in Real Biomembranes. Molecules 26. [CrossRef]
- Dupuy, A.D., and Engelman, D.M. (2008). Protein area occupancy at the center of the red blood cell membrane. Proceedings of the National Academy of Sciences 105, 2848–2852. [CrossRef] [PubMed]
- Jeon, J.-H. , Javanainen, M., Martinez-Seara, H., Metzler, R., and Vattulainen, I. (2016). Protein Crowding in Lipid Bilayers Gives Rise to Non-Gaussian Anomalous Lateral Diffusion of Phospholipids and Proteins. Physical Review X 6, 021006. [CrossRef]
- Alberts, B. , Heald, R., Johnson, A., Morgan, D., Raff, M., Roberts, K., & Walter, P. (2022). Molecular Biology of the Cell. 7th Edition (Garland Science). https://wwnorton.com/books/9780393884821.
- Aman, J. , and Margadant, C. (2023). Integrin-Dependent Cell-Matrix Adhesion in Endothelial Health and Disease. Circ Res 132, 355-378. [CrossRef]
- Čopič, A. , Dieudonné, T., and Lenoir, G. (2023). Phosphatidylserine transport in cell life and death. Curr Opin Cell Biol 83, 102192. [CrossRef]
- Nagata, S. , Suzuki, J., Segawa, K., and Fujii, T. (2016). Exposure of phosphatidylserine on the cell surface. Cell Death & Differentiation 23, 952-961. [CrossRef]
- Dal Col, J. , Lamberti, M.J., Nigro, A., Casolaro, V., Fratta, E., Steffan, A., and Montico, B. (2022). Phospholipid scramblase 1: a protein with multiple functions via multiple molecular interactors. Cell Communication and Signaling 20, 78. [CrossRef]
- Millington-Burgess, S.L. , and Harper, M.T. (2022). Maintaining flippase activity in procoagulant platelets is a novel approach to reducing thrombin generation. J Thromb Haemost 20, 989-995. [CrossRef]
- Filep, J.G. (2023). Two to tango: endothelial cell TMEM16 scramblases drive coagulation and thrombosis. J Clin Invest 133. [CrossRef]
- Tkachenko, A. , Alfhili, M.A., Alsughayyir, J., Attanzio, A., Al Mamun Bhuyan, A., Bukowska, B., Cilla, A., Quintanar-Escorza, M.A., Föller, M., Havranek, O., et al. (2025). Current understanding of eryptosis: mechanisms, physiological functions, role in disease, pharmacological applications, and nomenclature recommendations. Cell Death & Disease 16, 467. [CrossRef]
- Lentz, B.R. (2003). Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Progress in Lipid Research 42, 423-438. [CrossRef]
- Mizuguchi, C. , Nakamura, M., Kurimitsu, N., Ohgita, T., Nishitsuji, K., Baba, T., Shigenaga, A., Shimanouchi, T., Okuhira, K., Otaka, A., and Saito, H. (2018). Effect of Phosphatidylserine and Cholesterol on Membrane-mediated Fibril Formation by the N-terminal Amyloidogenic Fragment of Apolipoprotein A-I. Scientific Reports 8, 5497. [CrossRef]
- Clarke, R.J. (2023). Electrostatic switch mechanisms of membrane protein trafficking and regulation. Biophys Rev 15, 1967-1985. [CrossRef]
- Calianese, D.C. , and Birge, R.B. (2020). Biology of phosphatidylserine (PS): basic physiology and implications in immunology, infectious disease, and cancer. Cell Communication and Signaling 18, 41. [CrossRef]
- Hipp, M.S. , Kasturi, P., and Hartl, F.U. (2019). The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20, 421-435. [CrossRef]
- Koklic, T. , Majumder, R., Weinreb, G.E., and Lentz, B.R. (2009). Factor XA binding to phosphatidylserine-containing membranes produces an inactive membrane-bound dimer. Biophys J 97, 2232-2241. [CrossRef]
- Carman, C.V. , Nikova, D.N., Sakurai, Y., Shi, J., Novakovic, V.A., Rasmussen, J.T., Lam, W.A., and Gilbert, G.E. (2023). Membrane curvature and PS localize coagulation proteins to filopodia and retraction fibers of endothelial cells. Blood Adv 7, 60-72. [CrossRef]
- Banjade, S. , and Rosen, M.K. (2014). Phase transitions of multivalent proteins can promote clustering of membrane receptors. Elife 3. [CrossRef]
- Li, L. , Hu, J., Różycki, B., Ji, J., and Song, F. (2022). Interplay of receptor-ligand binding and lipid domain formation during cell adhesion. Front Mol Biosci 9, 1019477. [CrossRef]
- Lee, S.H. , Meng, X.W., Flatten, K.S., Loegering, D.A., and Kaufmann, S.H. (2013). Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death & Differentiation 20, 64-76. [CrossRef]
- Shin, H.W. , and Takatsu, H. (2020). Phosphatidylserine exposure in living cells. Crit Rev Biochem Mol Biol 55, 166-178. [CrossRef]
- Birge, R.B. , Boeltz, S., Kumar, S., Carlson, J., Wanderley, J., Calianese, D., Barcinski, M., Brekken, R.A., Huang, X., Hutchins, J.T., et al. (2016). Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ 23, 962-978. [CrossRef]
- Reddy, E.C. , and Rand, M.L. (2020). Procoagulant Phosphatidylserine-Exposing Platelets in vitro and in vivo. Front Cardiovasc Med 7, 15. [CrossRef]
- Chu, Y. , Guo, H., Zhang, Y., and Qiao, R. (2021). Procoagulant platelets: Generation, characteristics, and therapeutic target. J Clin Lab Anal 35, e23750. [CrossRef]
- Bourguignon, A. , Tasneem, S., and Hayward, C.P.M. (2022). Update on platelet procoagulant mechanisms in health and in bleeding disorders. Int J Lab Hematol 44 Suppl 1, 89-100. [CrossRef]
- Park, S. , and Park, J.K. (2024). Back to basics: the coagulation pathway. Blood Res 59, 35. [CrossRef]
- Segawa, K. , and Nagata, S. (2015). An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends in Cell Biology 25, 639-650. [CrossRef]
- Tschirhart, B.J. , Lu, X., Gomes, J., Chandrabalan, A., Bell, G., Hess, D.A., Xing, G., Ling, H., Burger, D., and Feng, Q. (2023). Annexin A5 Inhibits Endothelial Inflammation Induced by Lipopolysaccharide-Activated Platelets and Microvesicles via Phosphatidylserine Binding. Pharmaceuticals 16, 837.
- Muller, M.P. , Wang, Y., Morrissey, J.H., and Tajkhorshid, E. (2017). Lipid specificity of the membrane binding domain of coagulation factor X. Journal of Thrombosis and Haemostasis 15, 2005-2016. [CrossRef]
- Wan, P. , Choksi, S., Park, Y.J., Chen, X., Yan, J., Foroutannejad, S., Liu, Z., Chen, J., Lake, R., Liu, C., and Liu, Z.G. (2025). Soluble tissue factor generated by necroptosis-triggered shedding is responsible for thrombosis. Cell Res. [CrossRef]
- Ansari, S.A. , Pendurthi, U.R., Sen, P., and Rao, L.V. (2016). The Role of Putative Phosphatidylserine-Interactive Residues of Tissue Factor on Its Coagulant Activity at the Cell Surface. PLoS One 11, e0158377. [CrossRef]
- Spronk, H.M. , ten Cate, H., and van der Meijden, P.E. (2014). Differential roles of tissue factor and phosphatidylserine in activation of coagulation. Thromb Res 133 Suppl 1, S54-56. [CrossRef]
- El Masri, R. , Crétinon, Y., Gout, E., and Vivès, R.R. (2020). HS and Inflammation: A Potential Playground for the Sulfs? Front Immunol 11, 570. [CrossRef]
- Farrugia, B.L. , Lord, M.S., Melrose, J., and Whitelock, J.M. (2018). The Role of Heparan Sulfate in Inflammation, and the Development of Biomimetics as Anti-Inflammatory Strategies. J Histochem Cytochem 66, 321-336. [CrossRef]
- Cripps, J.G. , Crespo, F.A., Romanovskis, P., Spatola, A.F., and Fernández-Botrán, R. (2005). Modulation of acute inflammation by targeting glycosaminoglycan–cytokine interactions. International Immunopharmacology 5, 1622-1632. [CrossRef]
- Sobczak, A.I.S., Pitt, S.J., and Stewart, A.J. (2018). Glycosaminoglycan Neutralization in Coagulation Control. Arteriosclerosis, Thrombosis, and Vascular Biology 38, 1258–1270. [CrossRef] [PubMed]
- Milusev, A. , Despont, A., Shaw, J., Rieben, R., and Sorvillo, N. (2023). Inflammatory stimuli induce shedding of heparan sulfate from arterial but not venous porcine endothelial cells leading to differential proinflammatory and procoagulant responses. Scientific Reports 13, 4483. [CrossRef]
- Lever, R. , Smailbegovic, A., and Page, C. (2001). Role of glycosaminoglycans in inflammation. InflammoPharmacology 9, 165-169. [CrossRef]
- Lietha, D. , and Izard, T. (2020). Roles of Membrane Domains in Integrin-Mediated Cell Adhesion. Int J Mol Sci 21. [CrossRef]
- Morrissey, J.H. , Davis-Harrison, R.L., Tavoosi, N., Ke, K., Pureza, V., Boettcher, J.M., Clay, M.C., Rienstra, C.M., Ohkubo, Y.Z., Pogorelov, T.V., and Tajkhorshid, E. (2010). Protein-phospholipid interactions in blood clotting. Thromb Res 125 Suppl 1, S23-25. [CrossRef]
- Medfisch, S.M. , Muehl, E.M., Morrissey, J.H., and Bailey, R.C. (2020). Phosphatidylethanolamine-phosphatidylserine binding synergy of seven coagulation factors revealed using Nanodisc arrays on silicon photonic sensors. Sci Rep 10, 17407. [CrossRef]
- Schreuder, M. , Reitsma, P.H., and Bos, M.H.A. (2019). Blood coagulation factor Va’s key interactive residues and regions for prothrombinase assembly and prothrombin binding. J Thromb Haemost 17, 1229-1239. [CrossRef]
- Bradford, H.N. , Orcutt, S.J., and Krishnaswamy, S. (2013). Membrane binding by prothrombin mediates its constrained presentation to prothrombinase for cleavage. J Biol Chem 288, 27789-27800. [CrossRef]
- Shim, J.Y. , Lee, C.J., Wu, S., and Pedersen, L.G. (2015). A model for the unique role of factor Va A2 domain extension in the human ternary thrombin-generating complex. Biophys Chem 199, 46-50. [CrossRef]
- Weisel, J.W. , and Litvinov, R.I. (2013). Mechanisms of fibrin polymerization and clinical implications. Blood 121, 1712-1719. [CrossRef]
- Kattula, S. , Byrnes, J.R., and Wolberg, A.S. (2017). Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler Thromb Vasc Biol 37, e13-e21. [CrossRef]
- Wang, Y. , Carrim, N., and Ni, H. (2015). Fibronectin orchestrates thrombosis and hemostasis. Oncotarget 6, 19350-19351. [CrossRef]
- Cho, J. , and Mosher, D.F. (2006). Enhancement of thrombogenesis by plasma fibronectin cross-linked to fibrin and assembled in platelet thrombi. Blood 107, 3555-3563. [CrossRef]
- Ni, H. , Yuen, P.S., Papalia, J.M., Trevithick, J.E., Sakai, T., Fässler, R., Hynes, R.O., and Wagner, D.D. (2003). Plasma fibronectin promotes thrombus growth and stability in injured arterioles. Proc Natl Acad Sci U S A 100, 2415-2419. [CrossRef]
- Wirth, F. , Lubosch, A., Hamelmann, S., and Nakchbandi, I.A. (2020). Fibronectin and Its Receptors in Hematopoiesis. Cells 9. [CrossRef]
- Page, M.J. , Thomson, G.J.A., Nunes, J.M., Engelbrecht, A.M., Nell, T.A., de Villiers, W.J.S., de Beer, M.C., Engelbrecht, L., Kell, D.B., and Pretorius, E. (2019). Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci Rep 9, 3102. [CrossRef]
- Papareddy, P. , and Herwald, H. (2025). From immune activation to disease progression: Unraveling the complex role of Serum Amyloid A proteins. Cytokine & Growth Factor Reviews 83, 77-84. [CrossRef]
- Abouelasrar Salama, S. , Gouwy, M., Van Damme, J., and Struyf, S. (2021). The turning away of serum amyloid A biological activities and receptor usage. Immunology 163, 115-127. [CrossRef]
- Kam, P.C. , and Egan, M.K. (2002). Platelet glycoprotein IIb/IIIa antagonists: pharmacology and clinical developments. Anesthesiology 96, 1237-1249. [CrossRef]
- Urieli-Shoval, S. , Shubinsky, G., Linke, R.P., Fridkin, M., Tabi, I., and Matzner, Y. (2002). Adhesion of human platelets to serum amyloid A. Blood 99, 1224-1229. [CrossRef]
- Kahner, B.N. , Kato, H., Banno, A., Ginsberg, M.H., Shattil, S.J., and Ye, F. (2012). Kindlins, integrin activation and the regulation of talin recruitment to αIIbβ3. PLoS One 7, e34056. [CrossRef]
- Ma, Y.Q., Qin, J., and Plow, E.F. (2007). Platelet integrin αIIbβ3: activation mechanisms. Journal of Thrombosis and Haemostasis 5, 1345-1352. [CrossRef]
- Ellis, C. , Ward, N.L., Rice, M., Ball, N.J., Walle, P., Najdek, C., Kilinc, D., Lambert, J.C., Chapuis, J., and Goult, B.T. (2024). The structure of an amyloid precursor protein/talin complex indicates a mechanical basis of Alzheimer’s disease. Open Biol 14, 240185. [CrossRef]
- Gouwy, M. , De Buck, M., Abouelasrar Salama, S., Vandooren, J., Knoops, S., Pörtner, N., Vanbrabant, L., Berghmans, N., Opdenakker, G., Proost, P., et al. (2018). Matrix Metalloproteinase-9-Generated COOH-, but Not NH(2)-Terminal Fragments of Serum Amyloid A1 Retain Potentiating Activity in Neutrophil Migration to CXCL8, With Loss of Direct Chemotactic and Cytokine-Inducing Capacity. Front Immunol 9, 1081. [CrossRef]
- De Buck, M. , Berghmans, N., Pörtner, N., Vanbrabant, L., Cockx, M., Struyf, S., Opdenakker, G., Proost, P., Van Damme, J., and Gouwy, M. (2015). Serum amyloid A1α induces paracrine IL-8/CXCL8 via TLR2 and directly synergizes with this chemokine via CXCR2 and formyl peptide receptor 2 to recruit neutrophils. J Leukoc Biol 98, 1049-1060. [CrossRef]
- Lind, S. , Sundqvist, M., Holmdahl, R., Dahlgren, C., Forsman, H., and Olofsson, P. (2019). Functional and signaling characterization of the neutrophil FPR2 selective agonist Act-389949. Biochem Pharmacol 166, 163-173. [CrossRef]
- Jayaraman, S. , Urdaneta, A., Bullitt, E., Fändrich, M., and Gursky, O. (2023). Lipid clearance and amyloid formation by serum amyloid A: exploring the links between beneficial and pathologic actions of an enigmatic protein. J Lipid Res 64, 100429. [CrossRef]
- Huang, J. , Li, X., Shi, X., Zhu, M., Wang, J., Huang, S., Huang, X., Wang, H., Li, L., Deng, H., et al. (2019). Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting. J Hematol Oncol 12, 26. [CrossRef]
- Zou, J. , Swieringa, F., de Laat, B., de Groot, P.G., Roest, M., and Heemskerk, J.W.M. (2022). Reversible Platelet Integrin αIIbβ3 Activation and Thrombus Instability. International Journal of Molecular Sciences 23, 12512.
- Al-Yafeai, Z., Pearson, B.H., Peretik, J.M., Cockerham, E.D., Reeves, K.A., Bhattarai, U., Wang, D., Petrich, B.G., and Orr, A.W. (2021). Integrin affinity modulation critically regulates atherogenic endothelial activation in vitro and in vivo. Matrix Biology 96, 87-103. [CrossRef]
- van Buul, J.D. , van Rijssel, J., van Alphen, F.P., van Stalborch, A.M., Mul, E.P., and Hordijk, P.L. (2010). ICAM-1 clustering on endothelial cells recruits VCAM-1. J Biomed Biotechnol 2010, 120328. [CrossRef]
- Suehiro, K. , Gailit, J., and Plow, E.F. (1997). Fibrinogen Is a Ligand for Integrin α5β1 on Endothelial Cells*. Journal of Biological Chemistry 272, 5360-5366. [CrossRef]
- Mosesson, M.W. (2005). Fibrinogen and fibrin structure and functions. J Thromb Haemost 3, 1894-1904. [CrossRef]
- Semenov, A.N. , Lugovtsov, A.E., Shirshin, E.A., Yakimov, B.P., Ermolinskiy, P.B., Bikmulina, P.Y., Kudryavtsev, D.S., Timashev, P.S., Muravyov, A.V., Wagner, C., et al. (2020). Assessment of Fibrinogen Macromolecules Interaction with Red Blood Cells Membrane by Means of Laser Aggregometry, Flow Cytometry, and Optical Tweezers Combined with Microfluidics. Biomolecules 10. [CrossRef]
- Moon, B. , Yang, S., Moon, H., Lee, J., and Park, D. (2023). After cell death: the molecular machinery of efferocytosis. Experimental & Molecular Medicine 55, 1644-1651. [CrossRef]
- Arandjelovic, S. , and Ravichandran, K.S. (2015). Phagocytosis of apoptotic cells in homeostasis. Nature Immunology 16, 907-917. [CrossRef]
- Fadeel, B. , Xue, D., and Kagan, V. (2010). Programmed cell clearance: molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem Biophys Res Commun 396, 7-10. [CrossRef]
- Hari-Dass, R. , Shah, C., Meyer, D.J., and Raynes, J.G. (2005). Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem 280, 18562-18567. [CrossRef]
- Bryckaert, M. , Rosa, J.P., Denis, C.V., and Lenting, P.J. (2015). Of von Willebrand factor and platelets. Cell Mol Life Sci 72, 307-326. [CrossRef]
- Jayaraman, S. , Gantz, D.L., Haupt, C., and Gursky, O. (2017). Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc Natl Acad Sci U S A 114, E6507-e6515. [CrossRef]
- Nady, A. , Reichheld, S.E., and Sharpe, S. (2024). Structural studies of a serum amyloid A octamer that is primed to scaffold lipid nanodiscs. Protein Sci 33, e4983. [CrossRef]
- Westermark, G.T. , Fändrich, M., Lundmark, K., and Westermark, P. (2018). Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission. Cold Spring Harb Perspect Med 8. [CrossRef]
- van Genderen, H.O. , Kenis, H., Hofstra, L., Narula, J., and Reutelingsperger, C.P.M. (2008). Extracellular annexin A5: Functions of phosphatidylserine-binding and two-dimensional crystallization. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1783, 953-963. [CrossRef]
- Leventis, P.A. , and Grinstein, S. (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 39, 407-427. [CrossRef]
- Mehdizadeh, M. , Aguilar, M., Thorin, E., Ferbeyre, G., and Nattel, S. (2022). The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nature Reviews Cardiology 19, 250-264. [CrossRef]
- Tedeschi, V. , Paldino, G., Kunkl, M., Paroli, M., Sorrentino, R., Tuosto, L., and Fiorillo, M.T. (2022). CD8+ T Cell Senescence: Lights and Shadows in Viral Infections, Autoimmune Disorders and Cancer. International Journal of Molecular Sciences 23, 3374.
- Narasimhan, A., Flores, R.R., Robbins, P.D., and Niedernhofer, L.J. (2021). Role of cellular senescence in type II diabetes. Endocrinology 162, bqab136.
- Kell, L. , Simon, A.K., Alsaleh, G., and Cox, L.S. (2023). The central role of DNA damage in immunosenescence. Frontiers in Aging Volume 4 - 2023. [CrossRef]
- Gioia, U. , Tavella, S., Martínez-Orellana, P., Cicio, G., Colliva, A., Ceccon, M., Cabrini, M., Henriques, A.C., Fumagalli, V., and Paldino, A. (2023). SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nature Cell Biology 25, 550-564.
- Yang, L. , Kim, T.W., Han, Y., Nair, M.S., Harschnitz, O., Zhu, J., Wang, P., Koo, S.Y., Lacko, L.A., and Chandar, V. (2024). SARS-CoV-2 infection causes dopaminergic neuron senescence. Cell Stem Cell 31, 196-211. e196.
- Nunes, M. , Kell, L., Slaghekke, A., Wüst, R., Fielding, B., Kell, D., and Pretorius, E. (2025). Virus-Induced Endothelial Senescence as a Cause and Driving Factor for ME/CFS and Long COVID: Mediated by a Dysfunctional Immune System.
- Lin, Y. , Postma, D., Steeneken, L., Melo dos Santos, L., Kirkland, J., Espindola-Netto, J., Tchkonia, T., Borghesan, M., Bouma, H., and Demaria, M. (2023). Circulating monocytes expressing senescence-associated features are enriched in COVID-19 patients with severe disease. Aging Cell 22, e14011.
- Berentschot, J.C. , Drexhage, H.A., Aynekulu Mersha, D.G., Wijkhuijs, A.J., GeurtsvanKessel, C.H., Koopmans, M.P., Voermans, J.J., Hendriks, R.W., Nagtzaam, N.M., and de Bie, M. (2023). Immunological profiling in long COVID: overall low grade inflammation and T-lymphocyte senescence and increased monocyte activation correlating with increasing fatigue severity. Frontiers in immunology 14, 1254899.
- Ohtani, N. (2022). The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflammation and regeneration 42, 11.
- Suelves, N. , Saleki, S., Ibrahim, T., Palomares, D., Moonen, S., Koper, M.J., Vrancx, C., Vadukul, D.M., Papadopoulos, N., Viceconte, N., et al. (2023). Senescence-related impairment of autophagy induces toxic intraneuronal amyloid-β accumulation in a mouse model of amyloid pathology. Acta Neuropathologica Communications 11, 82. [CrossRef]
- Liu, J. , Yi, C., Qi, J., Cui, X., Yuan, X., Deng, W., Chen, M., and Xu, H. (2025). Senescence Alters Antimicrobial Peptide Expression and Induces Amyloid-β Production in Retinal Pigment Epithelial Cells. Aging cell 24, e70161.
- Ungerleider, K. , Beck, J.A., Lissa, D., Joruiz, S., Horikawa, I., and Harris, C.C. (2022). Δ133p53α protects human astrocytes from amyloid-beta induced senescence and neurotoxicity. Neuroscience 498, 190-202.
- Flanary, B.E., Sammons, N.W., Nguyen, C., Walker, D., and Streit, W.J. (2007). Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation research 10, 61-74.
- Sun, L. , Wang, L., Ye, K.X., Wang, S., Zhang, R., Juan, Z., Feng, L., and Min, S. (2023). Endothelial glycocalyx in aging and age-related diseases. Aging and disease 14, 1606.
- McConnell, M.J., Kostallari, E., Ibrahim, S.H., and Iwakiri, Y. (2023). The evolving role of liver sinusoidal endothelial cells in liver health and disease. Hepatology 78, 649-669.
- Seki, M., Arashiki, N., Takakuwa, Y., Nitta, K., and Nakamura, F. (2020). Reduction in flippase activity contributes to surface presentation of phosphatidylserine in human senescent erythrocytes. Journal of Cellular and Molecular Medicine 24, 13991-14000.
- Wi, J.H. , Heo, C.H., Gwak, H., Jung, C., and Kim, S.Y. (2021). Probing Physical Properties of the Cellular Membrane in Senescent Cells by Fluorescence Imaging. J Phys Chem B 125, 10182-10194. [CrossRef]
- Picos, A. , Seoane, N., Campos-Toimil, M., and Viña, D. (2025). Vascular senescence and aging: mechanisms, clinical implications, and therapeutic prospects. Biogerontology 26, 118. [CrossRef]
- Bochenek, M.L. , Schütz, E., and Schäfer, K. (2016). Endothelial cell senescence and thrombosis: Ageing clots. Thrombosis research 147, 36-45.
- Dasgupta, S.K., Argaiz, E.R., Mercado, J.E.C., Maul, H.O.E., Garza, J., Enriquez, A.B., Abdel-Monem, H., Prakasam, A., Andreeff, M., and Thiagarajan, P. (2010). Platelet senescence and phosphatidylserine exposure. Transfusion 50, 2167-2175.
- Curtis, A.M. , Edelberg, J., Jonas, R., Rogers, W.T., Moore, J.S., Syed, W., and Mohler, E.R. (2013). Endothelial microparticles: Sophisticated vesicles modulating vascular function. Vascular Medicine 18, 204-214. [CrossRef]
- Deng, F. , Wang, S., and Zhang, L. (2017). Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of circulatory hypoxia-related diseases: a literature review. J Cell Mol Med 21, 1698-1710. [CrossRef]
- Haghbin, M. , Sotoodeh Jahromi, A., Hashemi Tayer, A., and Ghasemi Nejad, Z. (2025). The Potential Clinical Relevance of Procoagulant Microparticles as Biomarkers of Blood Coagulation in Breast Cancer: A Systematic Review. Asian Pac J Cancer Prev 26, 23-32. [CrossRef]
- Aung, H.H. , Tung, J.-P., Dean, M.M., Flower, R.L., and Pecheniuk, N.M. (2017). Procoagulant role of microparticles in routine storage of packed red blood cells: potential risk for prothrombotic post-transfusion complications. Pathology 49, 62-69. [CrossRef]
- Zubairova, L.D. , Nabiullina, R.M., Nagaswami, C., Zuev, Y.F., Mustafin, I.G., Litvinov, R.I., and Weisel, J.W. (2015). Circulating Microparticles Alter Formation, Structure and Properties of Fibrin Clots. Scientific Reports 5, 17611. [CrossRef]
- Aleman, M.M. , Gardiner, C., Harrison, P., and Wolberg, A.S. (2011). Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. Journal of Thrombosis and Haemostasis 9, 2251-2261. [CrossRef]
- Owens, A.P. , 3rd, and Mackman, N. (2011). Microparticles in hemostasis and thrombosis. Circ Res 108, 1284-1297. [CrossRef]
- Merten, M. , Pakala, R., Thiagarajan, P., and Benedict, C.R. (1999). Platelet microparticles promote platelet interaction with subendothelial matrix in a glycoprotein IIb/IIIa-dependent mechanism. Circulation 99, 2577-2582. [CrossRef]
- Chandler, W.L. , Yeung, W., and Tait, J.F. (2011). A new microparticle size calibration standard for use in measuring smaller microparticles using a new flow cytometer. Journal of Thrombosis and Haemostasis 9, 1216-1224. [CrossRef]
- Zwicker, J.I. , Lacroix, R., Dignat-George, F., Furie, B.C., and Furie, B. (2012). Measurement of platelet microparticles. Methods Mol Biol 788, 127-139. [CrossRef]
- Bergaglio, T. , Synhaivska, O., and Nirmalraj, P.N. (2023). Digital holo-tomographic 3D maps of COVID-19 microclots in blood to assess disease severity. bioRxiv, 2023.2009.2012.557318. [CrossRef]
- Reid, V.L., and Webster, N.R. (2012). Role of microparticles in sepsis. British Journal of Anaesthesia 109, 503-513. [CrossRef]
- Burnouf, T. , Chou, M.-L., Goubran, H., Cognasse, F., Garraud, O., and Seghatchian, J. (2015). An overview of the role of microparticles/microvesicles in blood components: Are they clinically beneficial or harmful? Transfusion and Apheresis Science 53, 137-145. [CrossRef]
- Braga-Lagache, S. , Buchs, N., Iacovache, M.-I., Zuber, B., Jackson, C.B., and Heller, M. (2016). Robust Label-free, Quantitative Profiling of Circulating Plasma Microparticle (MP) Associated Proteins*. Molecular & Cellular Proteomics 15, 3640-3652. [CrossRef]
- Zwicker, J.I. , Trenor, C.C., Furie, B.C., and Furie, B. (2011). Tissue Factor–Bearing Microparticles and Thrombus Formation. Arteriosclerosis, Thrombosis, and Vascular Biology 31, 728-733. [CrossRef]
- Nieri, D. , Neri, T., Petrini, S., Vagaggini, B., Paggiaro, P., and Celi, A. Cell-derived microparticles and the lung. European Respiratory Review 25, 266-277. [CrossRef]
- Kalluri, R., and LeBleu, V.S. (2020). The biology, function, and biomedical applications of exosomes. Science 367, eaau6977. [CrossRef]
- Turner, S. , Laubscher, G.J., Khan, M.A., Kell, D.B., and Pretorius, E. (2023). Accelerating discovery: A novel flow cytometric method for detecting fibrin(ogen) amyloid microclots using long COVID as a model. Heliyon 9. [CrossRef]
- Kell, D.B. , Laubscher, G.J., and Pretorius, E. (2022). A central role for amyloid fibrin microclots in long COVID/PASC: origins and therapeutic implications. Biochemical Journal 479, 537-559. [CrossRef]
- Cointe, S. , Judicone, C., Robert, S., Mooberry, M.J., Poncelet, P., Wauben, M., Nieuwland, R., Key, N.S., Dignat-George, F., and Lacroix, R. (2017). Standardization of microparticle enumeration across different flow cytometry platforms: results of a multicenter collaborative workshop. Journal of Thrombosis and Haemostasis 15, 187-193. [CrossRef]
- Orozco, A.F. , and Lewis, D.E. (2010). Flow cytometric analysis of circulating microparticles in plasma. Cytometry A 77, 502-514. [CrossRef]
- Albert, V. , Subramanian, A., and Pati, H.P. (2018). Correlation of Circulating Microparticles with Coagulation and Fibrinolysis Is Healthy Individuals. Blood 132, 4975-4975. [CrossRef]
- Burnier, L. , Fontana, P., Kwak, B.R., and Angelillo-Scherrer, A. (2009). Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost 101, 439-451.
- Rank, A. , Nieuwland, R., Delker, R., Köhler, A., Toth, B., Pihusch, V., Wilkowski, R., and Pihusch, R. (2010). Cellular origin of platelet-derived microparticles in vivo. Thrombosis Research 126, e255-e259. [CrossRef]
- Westerman, M. , and Porter, J.B. (2016). Red blood cell-derived microparticles: An overview. Blood Cells, Molecules, and Diseases 59, 134-139. [CrossRef]
- Angelillo-Scherrer, A. , Weber, C., and Mause, S. (2012). Leukocyte-Derived Microparticles in Vascular Homeostasis. Circulation Research 110, 356-369. [CrossRef]
- Lacroix, R. , Plawinski, L., Robert, S., Doeuvre, L., Sabatier, F., Martinez de Lizarrondo, S., Mezzapesa, A., Anfosso, F., Leroyer, A.S., Poullin, P., et al. (2012). Leukocyte- and endothelial-derived microparticles: a circulating source for fibrinolysis. Haematologica 97, 1864-1872. [CrossRef]
- Chironi, G.N. , Boulanger, C.M., Simon, A., Dignat-George, F., Freyssinet, J.-M., and Tedgui, A. (2009). Endothelial microparticles in diseases. Cell and Tissue Research 335, 143-151. [CrossRef]
- Dignat-George, F., and Boulanger, C.M. (2011). The Many Faces of Endothelial Microparticles. Arteriosclerosis, Thrombosis, and Vascular Biology 31, 27-33. [CrossRef]
- Horstman, L.L. , Jy, W., Jimenez, J.J., and Ahn, Y.S. (2004). Endothelial microparticles as markers of endothelial dysfunction. FBL 9, 1118-1135. [CrossRef]
- Mege, D. , Crescence, L., Ouaissi, M., Sielezneff, I., Guieu, R., Dignat-George, F., Dubois, C., and Panicot-Dubois, L. (2017). Fibrin-bearing microparticles: marker of thrombo-embolic events in pancreatic and colorectal cancers. Oncotarget 8.
- Chou, J. , Mackman, N., Merrill-Skoloff, G., Pedersen, B., Furie, B.C., and Furie, B. (2004). Hematopoietic cell-derived microparticle tissue factor contributes to fibrin formation during thrombus propagation. Blood 104, 3190-3197. [CrossRef]
- Agouti, I., Cointe, S., Robert, S., Judicone, C., Loundou, A., Driss, F., Brisson, A., Steschenko, D., Rose, C., Pondarré, C., et al. (2015). Platelet and not erythrocyte microparticles are procoagulant in transfused thalassaemia major patients. British Journal of Haematology 171, 615-624. [CrossRef]
- Nomura, S. , Ozaki, Y., and Ikeda, Y. (2008). Function and role of microparticles in various clinical settings. Thrombosis Research 123, 8-23. [CrossRef]
- Nomura, S. , Inami, N., Shouzu, A., Urase, F., and Maeda, Y. (2009). Correlation and association between plasma platelet-, monocyte- and endothelial cell-derived microparticles in hypertensive patients with type 2 diabetes mellitus. Platelets 20, 406-414. [CrossRef]
- Rajendran, D. , and Chandrasekaran, N. (2023). Journey of micronanoplastics with blood components. RSC Advances 13, 31435-31459. [CrossRef]
- Kopatz, V. , Wen, K., Kovács, T., Keimowitz, A.S., Pichler, V., Widder, J., Vethaak, A.D., Hollóczki, O., and Kenner, L. (2023). Micro- and Nanoplastics Breach the Blood–Brain Barrier (BBB): Biomolecular Corona’s Role Revealed. Nanomaterials 13. [CrossRef]
- Bashirova, N. , Schölzel, F., Hornig, D., Scheidt, H.A., Krueger, M., Salvan, G., Huster, D., Matysik, J., and Alia, A. (2025). The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation. Molecules 30, 1432.
- Zhang, Q. , Wang, Y., Xiao, Q., Geng, G., Davis, S.J., Liu, X., Yang, J., Liu, J., Huang, W., He, C., et al. (2025). Long-range PM2.5 pollution and health impacts from the 2023 Canadian wildfires. Nature 645, 672-678. [CrossRef]
- Zhang, X., Liu, H., Wu, X., Jia, L., Gadhave, K., Wang, L., Zhang, K., Li, H., Chen, R., Kumbhar, R., et al. (2025). Lewy body dementia promotion by air pollutants. Science 389, eadu4132. [CrossRef]
- Kruger, A. , Vlok, M., Turner, S., Venter, C., Laubscher, G.J., Kell, D.B., and Pretorius, E. (2022). Proteomics of fibrin amyloid microclots in Long COVID/ Post-Acute Sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system. Cardiovasc Diabetol 21, 190. [CrossRef]
- Schofield, J. , Abrams, S.T., Jenkins, R., Lane, S., Wang, G., and Toh, C.H. (2024). Amyloid-Fibrinogen Aggregates (“Microclots”) Predict Risks of Disseminated Intravascular Coagulation and Mortality. Blood Adv. [CrossRef]
- Pretorius, E. , Vlok, M., Venter, C., Bezuidenhout, J.A., Laubscher, G.J., Steenkamp, J., and Kell, D.B. (2021). Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol 20, 172. [CrossRef]
- Kell, D.B. , and Pretorius, E. (2024). Proteomic Evidence for Amyloidogenic Cross-Seeding in Fibrinaloid Microclots. International Journal of Molecular Sciences 25, 10809.
- Kell, D.B. , and Pretorius, E. (2025). The Proteome Content of Blood Clots Observed Under Different Conditions: Successful Role in Predicting Clot Amyloid(ogenicity). Molecules 30, 668.
- Burdukiewicz, M. , Sobczyk, P., Rödiger, S., Duda-Madej, A., Mackiewicz, P., and Kotulska, M. (2017). Amyloidogenic motifs revealed by n-gram analysis. Sci Rep 7, 12961. [CrossRef]
- Bester, J. , Matshailwe, C., and Pretorius, E. (2018). Simultaneous presence of hypercoagulation and increased clot lysis time due to IL-1β, IL-6 and IL-8. Cytokine 110, 237-242. [CrossRef]
- Bester, J. , and Pretorius, E. (2016). Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep 6, 32188. [CrossRef]
- Sack, G.H. (2020). Serum Amyloid A (SAA) Proteins. Subcell Biochem 94, 421-436. [CrossRef]
- Bezuidenhout, J.A. , Venter, C., Roberts, T.J., Tarr, G., Kell, D.B., and Pretorius, E. (2020). Detection of Citrullinated Fibrin in Plasma Clots of Rheumatoid Arthritis Patients and Its Relation to Altered Structural Clot Properties, Disease-Related Inflammation and Prothrombotic Tendency. Front Immunol 11, 577523. [CrossRef]
- Turner, S. , Naidoo, C.A., Usher, T.J., Kruger, A., Venter, C., Laubscher, G.J., Khan, M.A., Kell, D.B., and Pretorius, E. (2023). Increased Levels of Inflammatory and Endothelial Biomarkers in Blood of Long COVID Patients Point to Thrombotic Endothelialitis. Semin Thromb Hemost. [CrossRef]
- Visser, M.J.E. , Venter, C., Roberts, T.J., Tarr, G., and Pretorius, E. (2021). Psoriatic disease is associated with systemic inflammation, endothelial activation, and altered haemostatic function. Sci Rep 11, 13043. [CrossRef]
- Randeria, S.N. , Thomson, G.J.A., Nell, T.A., Roberts, T., and Pretorius, E. (2019). Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc Diabetol 18, 72. [CrossRef]
- Westman, H. , Hammarström, P., and Nyström, S. (2025). SARS-CoV-2 spike protein amyloid fibrils impair fibrin formation and fibrinolysis. bioRxiv, 2025.2006.2030.661938. [CrossRef]
- Kell, D.B. , and Pretorius, E. (2017). Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Progress in Biophysics and Molecular Biology 123, 16-41. [CrossRef]
- Soria, J. , Mirshahi, S., Mirshahi, S.Q., Varin, R., Pritchard, L.L., Soria, C., and Mirshahi, M. (2019). Fibrinogen αC domain: Its importance in physiopathology. Research and Practice in Thrombosis and Haemostasis 3, 173-183. [CrossRef]
- Chapman, J. , and Dogan, A. (2019). Fibrinogen alpha amyloidosis: insights from proteomics. Expert Review of Proteomics 16, 783-793. [CrossRef]
- Yazaki, M. , Yoshinaga, T., Sekijima, Y., Kametani, F., and Okumura, N. (2018). Hereditary Fibrinogen Aα-Chain Amyloidosis in Asia: Clinical and Molecular Characteristics. International Journal of Molecular Sciences 19, 320.
- Jin, S. , Shen, Z., Li, J., Lin, P., Xu, X., Ding, X., and Liu, H. (2021). Fibrinogen A Alpha-Chain Amyloidosis Associated With a Novel Variant in a Chinese Family. Kidney Int Rep 6, 2726-2730. [CrossRef]
- Grixti, J.M. , Theron, C.W., Salcedo-Sora, J.E., Pretorius, E. and Kell, D.B. (2024). Automated microscopic measurement of fibrinaloid microclots and their degradation by nattokinase, the main natto protease. Journal of Experimental and Clinical Application of Chinese Medicine https://ojs.exploverpub.com/index.php/jecacm/article/view/201.
- Reitsma, S. , Slaaf, D.W., Vink, H., van Zandvoort, M.A., and oude Egbrink, M.G. (2007). The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch 454, 345-359. [CrossRef]
- Artl, A. , Marsche, G., Lestavel, S., Sattler, W., and Malle, E. (2000). Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol 20, 763-772. [CrossRef]
- Kisilevsky, R. , and Manley, P.N. (2012). Acute-phase serum amyloid A: perspectives on its physiological and pathological roles. Amyloid 19, 5-14. [CrossRef]
- Shattil, S.J. , and Newman, P.J. (2004). Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood 104, 1606-1615. [CrossRef]
- Radomski, M.W. , Palmer, R.M., and Moncada, S. (1987). Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 2, 1057-1058. [CrossRef]
- Uhlar, C.M. , and Whitehead, A.S. (1999). Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265, 501-523. [CrossRef]
- Shattil, S.J. , Kim, C., and Ginsberg, M.H. (2010). The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol 11, 288-300. [CrossRef]
- Van Lenten, B.J. , Hama, S.Y., de Beer, F.C., Stafforini, D.M., McIntyre, T.M., Prescott, S.M., La Du, B.N., Fogelman, A.M., and Navab, M. (1995). Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest 96, 2758-2767. [CrossRef]
- de Beer, M.C., Yuan, T., Kindy, M.S., Asztalos, B.F., Roheim, P.S., and de Beer, F.C. (1995). Characterization of constitutive human serum amyloid A protein (SAA4) as an apolipoprotein. J Lipid Res 36. 526–534.
- Frame, N.M. , and Gursky, O. (2016). Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett 590, 866-879. [CrossRef]
- Webb, N.R. (2021). High-Density Lipoproteins and Serum Amyloid A (SAA). Curr Atheroscler Rep 23, 7. [CrossRef]
- Gursky, O. (2020). Structural Basis for Vital Function and Malfunction of Serum Amyloid A: an Acute-Phase Protein that Wears Hydrophobicity on Its Sleeve. Curr Atheroscler Rep 22, 69. [CrossRef]
- Ashe, K.H. , and Aguzzi, A. (2013). Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 7, 55-59. [CrossRef]
- Frontzek, K. , Bardelli, M., Senatore, A., Henzi, A., Reimann, R.R., Bedir, S., Marino, M., Hussain, R., Jurt, S., Meisl, G., et al. (2022). A conformational switch controlling the toxicity of the prion protein. Nature Structural & Molecular Biology 29, 831-840. [CrossRef]
- Porter, L.L. , and Looger, L.L. (2018). Extant fold-switching proteins are widespread. Proc Natl Acad Sci U S A 115, 5968-5973. [CrossRef]
- Retamal-Farfán, I. , González-Higueras, J., Galaz-Davison, P., Rivera, M., and Ramírez-Sarmiento, C.A. (2023). Exploring the structural acrobatics of fold-switching proteins using simplified structure-based models. Biophys Rev 15, 787-799. [CrossRef]
- Wechalekar, A.D. , Gillmore, J.D., and Hawkins, P.N. (2016). Systemic amyloidosis. Lancet 387, 2641-2654. [CrossRef]
- Cuddy, S.A.M. , and Falk, R.H. (2020). Amyloidosis as a Systemic Disease in Context. Can J Cardiol 36, 396-407. [CrossRef]
- Sanderson, J.M. (2022). The association of lipids with amyloid fibrils. Journal of Biological Chemistry 298, 102108. [CrossRef]
- Kurouski, D. (2023). Elucidating the Role of Lipids in the Aggregation of Amyloidogenic Proteins. Accounts of Chemical Research 56, 2898-2906. [CrossRef]
- Hellstrand, E. , Nowacka, A., Topgaard, D., Linse, S., and Sparr, E. (2013). Membrane lipid co-aggregation with α-synuclein fibrils. PLoS One 8, e77235. [CrossRef]
- Kell, D.B. (2009). Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Medical Genomics 2, 2. [CrossRef]
- Kell, D.B. , and Pretorius, E. (2018). No effects without causes: the Iron Dysregulation and Dormant Microbes hypothesis for chronic, inflammatory diseases. Biol Rev Camb Philos Soc 93, 1518-1557. [CrossRef]
- Cornelissen, A. , Guo, L., Sakamoto, A., Virmani, R., and Finn, A.V. (2019). New insights into the role of iron in inflammation and atherosclerosis. eBioMedicine 47, 598-606. [CrossRef]
- Sharkey-Toppen, T.P. , Tewari, A.K., and Raman, S.V. (2014). Iron and Atherosclerosis: Nailing Down a Novel Target with Magnetic Resonance. Journal of Cardiovascular Translational Research 7, 533-542. [CrossRef]
- Kopriva, D. , Kisheev, A., Meena, D., Pelle, S., Karnitsky, M., Lavoie, A., and Buttigieg, J. (2015). The Nature of Iron Deposits Differs between Symptomatic and Asymptomatic Carotid Atherosclerotic Plaques. PLoS One 10, e0143138. [CrossRef]
- Kun, A. , González-Camacho, F., Hernández, S., Moreno-García, A., Calero, O., and Calero, M. (2018). Characterization of Amyloid-β Plaques and Autofluorescent Lipofuscin Aggregates in Alzheimer’s Disease Brain: A Confocal Microscopy Approach. Methods Mol Biol 1779, 497-512. [CrossRef]
- Lowman, R.L. , and Yampolsky, L.Y. (2023). Lipofuscin, amyloids, and lipid peroxidation as potential markers of aging in Daphnia. Biogerontology 24, 541-553. [CrossRef]
- Ng, K.-P. , Gugiu, B., Renganathan, K., Davies, M.W., Gu, X., Crabb, J.S., Kim, S.R., Różanowska, M.B., Bonilha, V.L., Rayborn, M.E., et al. (2008). Retinal Pigment Epithelium Lipofuscin Proteomics. Molecular & Cellular Proteomics 7, 1397-1405. [CrossRef]
- Ottis, P. , Koppe, K., Onisko, B., Dynin, I., Arzberger, T., Kretzschmar, H., Requena, J.R., Silva, C.J., Huston, J.P., and Korth, C. (2012). Human and rat brain lipofuscin proteome. Proteomics 12, 2445-2454. [CrossRef]
- Babaniamansour, P. , Mohammadi, M., Babaniamansour, S., and Aliniagerdroudbari, E. (2020). The Relation between Atherosclerosis Plaque Composition and Plaque Rupture. Journal of Medical Signals & Sensors 10, 267-273. [CrossRef]
- He, Z. , Luo, J., Lv, M., Li, Q., Ke, W., Niu, X., and Zhang, Z. (2023). Characteristics and evaluation of atherosclerotic plaques: an overview of state-of-the-art techniques. Front Neurol 14, 1159288. [CrossRef]
- Tomey, M.I. , Narula, J., and Kovacic, J.C. (2014). Advances in the Understanding of Plaque Composition and Treatment Options: Year in Review. Journal of the American College of Cardiology 63, 1604-1616. [CrossRef]
- Howlett, G.J. , and Moore, K.J. (2006). Untangling the role of amyloid in atherosclerosis. Curr Opin Lipidol 17, 541-547. [CrossRef]
- Dichtl, W. , Moraga, F., Ares, M.P., Crisby, M., Nilsson, J., Lindgren, S., and Janciauskiene, S. (2000). The carboxyl-terminal fragment of alpha1-antitrypsin is present in atherosclerotic plaques and regulates inflammatory transcription factors in primary human monocytes. Mol Cell Biol Res Commun 4, 50-61. [CrossRef]
- von zur Muhlen, C. , Schiffer, E., Sackmann, C., Zurbig, P., Neudorfer, I., Zirlik, A., Htun, N., Iphofer, A., Jansch, L., Mischak, H., et al. (2012). Urine proteome analysis reflects atherosclerotic disease in an ApoE-/- mouse model and allows the discovery of new candidate biomarkers in mouse and human atherosclerosis. Mol Cell Proteomics 11, M111 013847. [CrossRef]
- Dong, Z. , Wu, T., Qin, W., An, C., Wang, Z., Zhang, M., Zhang, Y., Zhang, C., and An, F. (2011). Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med 17, 1357-1364. [CrossRef]
- Getz, G.S. , Krishack, P.A., and Reardon, C.A. (2016). Serum amyloid A and atherosclerosis. Curr Opin Lipidol 27, 531-535. [CrossRef]
- Shridas, P. , and Tannock, L.R. (2019). Role of serum amyloid A in atherosclerosis. Curr Opin Lipidol 30, 320-325. [CrossRef]
- Westermark, P. , Mucchiano, G., Marthin, T., Johnson, K.H., and Sletten, K. (1995). Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am J Pathol 147, 1186-1192.
- Mucchiano, G.I. , Jonasson, L., Haggqvist, B., Einarsson, E., and Westermark, P. (2001). Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am J Clin Pathol 115, 298-303. [CrossRef]
- Obici, L. , Franceschini, G., Calabresi, L., Giorgetti, S., Stoppini, M., Merlini, G., and Bellotti, V. (2006). Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 13, 191-205. [CrossRef]
- Williams, B. (2015). Amyloid beta and cardiovascular disease: intriguing questions indeed. J Am Coll Cardiol 65, 917-919. [CrossRef]
- Gagno, G. , Ferro, F., Fluca, A.L., Janjusevic, M., Rossi, M., Sinagra, G., Beltrami, A.P., Moretti, R., and Aleksova, A. (2020). From Brain to Heart: Possible Role of Amyloid-beta in Ischemic Heart Disease and Ischemia-Reperfusion Injury. Int J Mol Sci 21, 9655. [CrossRef]
- Kell, D.B. , Salcedo-Sora, J.E., and Pretorius, E. (2025). Amyloidogenic potential of plaque and thrombus proteomes and of fold-switching metamorphic proteins. Preprints, 2025081049. [CrossRef]
- Rajendran, L. , Honsho, M., Zahn, T.R., Keller, P., Geiger, K.D., Verkade, P., and Simons, K. (2006). Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences 103, 11172-11177. [CrossRef]
- Grixti, J.M. , Chandran, A., Pretorius, J.H., Walker, M., Sekhar, A., Pretorius, E., and Kell, D.B. (2025). Amyloid Presence in Acute Ischemic Stroke Thrombi: Observational Evidence for Fibrinolytic Resistance. Stroke. [CrossRef]
- Grixti, J.M. , Chandran, A., Pretorius, J.-H., Walker, M., Sekhar, A., Pretorius, E., and Kell, D.B. (2024). The clots removed from ischaemic stroke patients by mechanical thrombectomy are amyloid in nature. medRxiv, 2024.2011.2001.24316555. [CrossRef]
- Biancalana, M. , and Koide, S. (2010). Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta 1804, 1405-1412. [CrossRef]
- Gade Malmos, K. , Blancas-Mejia, L.M., Weber, B., Buchner, J., Ramirez-Alvarado, M., Naiki, H., and Otzen, D. (2017). ThT 101: a primer on the use of thioflavin T to investigate amyloid formation. Amyloid 24, 1-16. [CrossRef]
- Xue, C. , Lin, T.Y., Chang, D., and Guo, Z. (2017). Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R Soc Open Sci 4, 160696. [CrossRef]
- Das, S. , Purkayastha, P. (2017). Selective Binding of Thioflavin T in Sequence-Exchanged Single Strand DNA Oligomers and Further Interaction with Phospholipid Membranes. Chemistry Select 2, 5000.
- Hanczyc, P. , Rajchel-Mieldzioć, P., Feng, B., and Fita, P. (2021). Identification of Thioflavin T Binding Modes to DNA: A Structure-Specific Molecular Probe for Lasing Applications. The Journal of Physical Chemistry Letters 12, 5436-5442. [CrossRef]
- Schlein, M. (2017). Insulin Formulation Characterization—the Thioflavin T Assays. The AAPS Journal 19, 397-408. [CrossRef]
- Harada, A. , Azakami, H., and Kato, A. (2008). Amyloid Fibril Formation of Hen Lysozyme Depends on the Instability of the C-Helix (88-99). Bioscience, Biotechnology, and Biochemistry 72, 1523-1530. [CrossRef]
- Chen, Y.-H. , Tseng, C.-P., How, S.-C., Lo, C.-H., Chou, W.-L., and Wang, S.S.S. (2016). Amyloid fibrillogenesis of lysozyme is suppressed by a food additive brilliant blue FCF. Colloids and Surfaces B: Biointerfaces 142, 351-359. [CrossRef]
- Pretorius, E. , Mbotwe, S., Bester, J., Robinson, C.J., and Kell, D.B. (2016). Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J R Soc Interface 13. [CrossRef]
- Pretorius, E. , Page, M.J., Hendricks, L., Nkosi, N.B., Benson, S.R., and Kell, D.B. (2018). Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: assessment with novel Amytracker™ stains. J R Soc Interface 15. [CrossRef]
- Nunes, J.M. , Fillis, T., Page, M.J., Venter, C., Lancry, O., Kell, D.B., Windberger, U., and Pretorius, E. (2020). Gingipain R1 and Lipopolysaccharide From Porphyromonas gingivalis Have Major Effects on Blood Clot Morphology and Mechanics. Front Immunol 11, 1551. [CrossRef]
- Grobbelaar, L.M. , Venter, C., Vlok, M., Ngoepe, M., Laubscher, G.J., Lourens, P.J., Steenkamp, J., Kell, D.B., and Pretorius, E. (2021). SARS-CoV-2 spike protein S1 induces fibrin(ogen) resistant to fibrinolysis: implications for microclot formation in COVID-19. Biosci Rep 41. [CrossRef]
- Grobbelaar, L.M. , Kruger, A., Venter, C., Burger, E.M., Laubscher, G.J., Maponga, T.G., Kotze, M.J., Kwaan, H.C., Miller, J.B., Fulkerson, D., et al. (2022). Relative Hypercoagulopathy of the SARS-CoV-2 Beta and Delta Variants when Compared to the Less Severe Omicron Variants Is Related to TEG Parameters, the Extent of Fibrin Amyloid Microclots, and the Severity of Clinical Illness. Semin Thromb Hemost 48, 858-868. [CrossRef]
- Frame, N.M. , and Gursky, O. (2017). Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. Amyloid 24, 13-14. [CrossRef]
- Bilog, M. , Vedad, J., Capadona, C., Profit, A.A., and Desamero, R.Z.B. (2024). Key charged residues influence the amyloidogenic propensity of the helix-1 region of serum amyloid A. Biochimica et Biophysica Acta (BBA) - General Subjects 1868, 130690. [CrossRef]
- Zinellu, A. , Paliogiannis, P., Carru, C., and Mangoni, A.A. (2021). Serum amyloid A concentrations, COVID-19 severity and mortality: An updated systematic review and meta-analysis. Int J Infect Dis 105, 668-674. [CrossRef]
- Bilog, M. , Cersosimo, J., Vigil, I., Desamero, R.Z.B., and Profit, A.A. (2024). Effect of a SARS-CoV-2 Protein Fragment on the Amyloidogenic Propensity of Human Islet Amyloid Polypeptide. ACS Chem Neurosci 15, 4431-4440. [CrossRef]
- Petrlova, J. , Samsudin, F., Bond, P.J., and Schmidtchen, A. (2022). SARS-CoV-2 spike protein aggregation is triggered by bacterial lipopolysaccharide. FEBS Lett 596, 2566-2575. [CrossRef]
- Petruk, G. , Puthia, M., Petrlova, J., Samsudin, F., Strömdahl, A.C., Cerps, S., Uller, L., Kjellström, S., Bond, P.J., and Schmidtchen, A.A. (2020). SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J Mol Cell Biol 12, 916-932. [CrossRef]
- Nyström, S. , and Hammarström, P. (2022). Amyloidogenesis of SARS-CoV-2 Spike Protein. J Am Chem Soc 144, 8945-8950. [CrossRef]
- Thierry, A.R., Usher, T., Sanchez, C., Turner, S., Venter, C., Ha, T., Pastor, B., Waters, M., Thompson, A., Mirandola, A., Pisareva, E., Prevostel, C., Laubscher, GJ., Kell, D.B., Pretorius, E. (2025). Circulating microclots are structurally associated with Neutrophil Extracellular Traps and their amounts are strongly elevated in long COVID patients. Journal of Medical Virology 97. e70613. [CrossRef]
- Pisareva, E. , Badiou, S., Mihalovičová, L., Mirandola, A., Pastor, B., Kudriavtsev, A., Berger, M., Roubille, C., Fesler, P., Klouche, K., et al. (2023). Persistence of neutrophil extracellular traps and anticardiolipin auto-antibodies in post-acute phase COVID-19 patients. J Med Virol 95, e28209. [CrossRef]
Figure 1.
Membranes in health and disease. A) Biochemical composition of membranes [11] (~50:50 lipids versus proteins by area [12]). B) The structure of a healthy cell membrane. In a physiologically intact cell, the lipid bilayer maintains asymmetry between inner and outer leaflets, and integral proteins are properly folded and distributed. This arrangement preserves selective permeability, electrochemical gradients, and homeostatic signaling. (Protein/lipid distribution not to scale). C) Early membrane damage to form a prothrombotic surface. Oxidative stress, inflammatory mediators, or mechanical injury disrupt lipid asymmetry, expose phosphatidylserine and other negatively charged lipids, and induce clustering or unfolding of membrane proteins. These changes provide catalytic sites for coagulation reactions, contributing to early thrombo-inflammatory signaling. D) Late stage membrane changes. Progressive damage leads to loss of membrane integrity, protein unfolding, and the release of microvesicles and cellular debris. These fragments expose negatively charged and amyloidogenic surfaces that bind inflammatory molecules, thereby serving as circulating procoagulant platforms and amplifying thrombo-inflammatory cascades. Created in https://BioRender.com.
Figure 1.
Membranes in health and disease. A) Biochemical composition of membranes [11] (~50:50 lipids versus proteins by area [12]). B) The structure of a healthy cell membrane. In a physiologically intact cell, the lipid bilayer maintains asymmetry between inner and outer leaflets, and integral proteins are properly folded and distributed. This arrangement preserves selective permeability, electrochemical gradients, and homeostatic signaling. (Protein/lipid distribution not to scale). C) Early membrane damage to form a prothrombotic surface. Oxidative stress, inflammatory mediators, or mechanical injury disrupt lipid asymmetry, expose phosphatidylserine and other negatively charged lipids, and induce clustering or unfolding of membrane proteins. These changes provide catalytic sites for coagulation reactions, contributing to early thrombo-inflammatory signaling. D) Late stage membrane changes. Progressive damage leads to loss of membrane integrity, protein unfolding, and the release of microvesicles and cellular debris. These fragments expose negatively charged and amyloidogenic surfaces that bind inflammatory molecules, thereby serving as circulating procoagulant platforms and amplifying thrombo-inflammatory cascades. Created in https://BioRender.com.
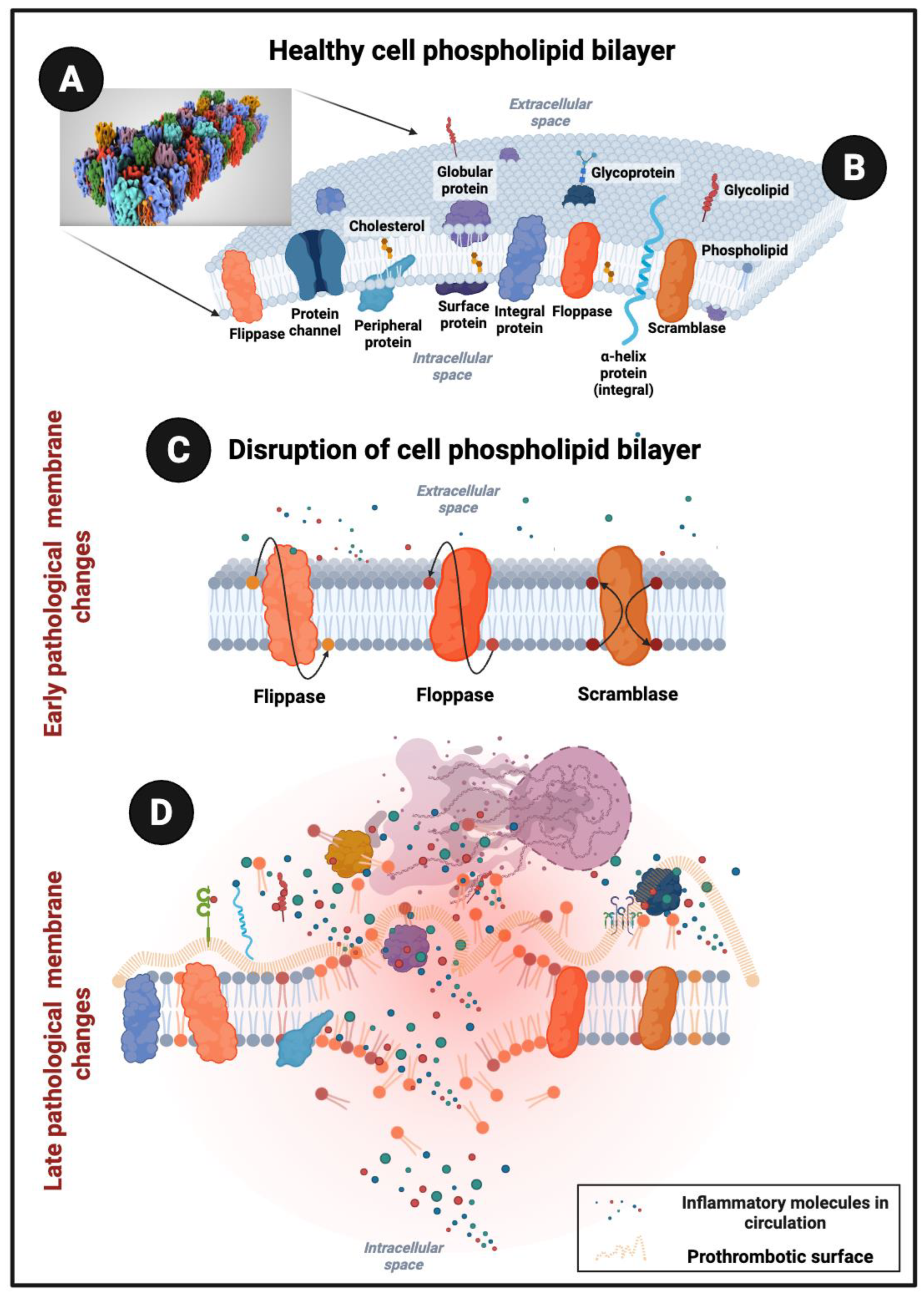
Figure 2.
A) Resting (non-activated) platelet with numerous low-affinity receptors on its surface. B) The complexity of platelet membrane receptors during platelet activation where platelets can form complexes with other activated platelets, white blood cells and damaged endothelial cells. C) The formation of a procoagulant and amyloidogenic surface to seed and fibrinogen and other pro-clotting molecules. Progressive membrane injury in platelets, leukocytes, and endothelial cells exposes phosphatidylserine and denatured or misfolded proteins, generating surfaces capable of binding fibrinogen, fibronectin, and serum amyloid A (SAA). These damaged membranes, enriched in amyloidogenic and negatively charged domains, act as catalytic scaffolds that seed procoagular surfaces. Created in https://BioRender.com.
Figure 2.
A) Resting (non-activated) platelet with numerous low-affinity receptors on its surface. B) The complexity of platelet membrane receptors during platelet activation where platelets can form complexes with other activated platelets, white blood cells and damaged endothelial cells. C) The formation of a procoagulant and amyloidogenic surface to seed and fibrinogen and other pro-clotting molecules. Progressive membrane injury in platelets, leukocytes, and endothelial cells exposes phosphatidylserine and denatured or misfolded proteins, generating surfaces capable of binding fibrinogen, fibronectin, and serum amyloid A (SAA). These damaged membranes, enriched in amyloidogenic and negatively charged domains, act as catalytic scaffolds that seed procoagular surfaces. Created in https://BioRender.com.
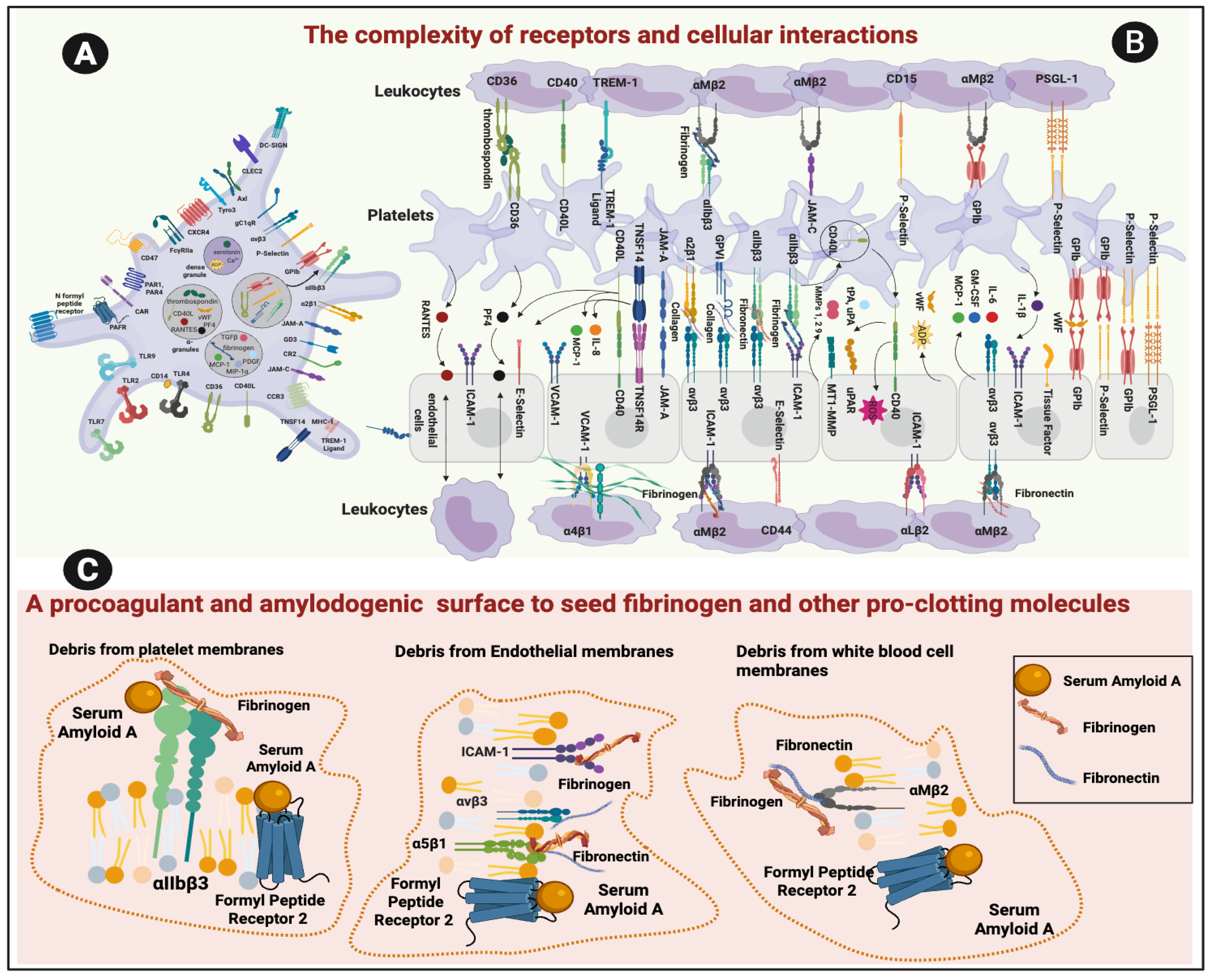
Figure 3.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the cellular debris pathway. (1) Healthy and damaged cells: In healthy membranes, phosphatidylserine (PS) is internalized and integrin αIIbβ3 (GPIIb/IIIa) remains in a low-affinity state. Upon injury, PS externalizes (“PS flip”), receptors activate, and membranes become prothrombotic and immune-silent. Progressive damage generates debris that binds inflammatory molecules.(2) Prothrombotic fragments and microparticles: Exposed receptors interact with serum amyloid A (SAA) and fibrin(ogen), forming β-sheet–rich, amyloidogenic scaffolds that seed fibrinaloid microclots.(3) Confocal microscopy: (A–B) Whole blood: red blood cells and platelets stained with cell mask (red); amyloid signal detected with Thioflavin T ((ThT) green)). (C–D) Platelet-poor plasma (PPP): membrane debris or fibrin(ogen) α-chain antibody (pink) co-localizing with ThT-positive microclot complexes. Created in https://BioRender.com.
Figure 3.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the cellular debris pathway. (1) Healthy and damaged cells: In healthy membranes, phosphatidylserine (PS) is internalized and integrin αIIbβ3 (GPIIb/IIIa) remains in a low-affinity state. Upon injury, PS externalizes (“PS flip”), receptors activate, and membranes become prothrombotic and immune-silent. Progressive damage generates debris that binds inflammatory molecules.(2) Prothrombotic fragments and microparticles: Exposed receptors interact with serum amyloid A (SAA) and fibrin(ogen), forming β-sheet–rich, amyloidogenic scaffolds that seed fibrinaloid microclots.(3) Confocal microscopy: (A–B) Whole blood: red blood cells and platelets stained with cell mask (red); amyloid signal detected with Thioflavin T ((ThT) green)). (C–D) Platelet-poor plasma (PPP): membrane debris or fibrin(ogen) α-chain antibody (pink) co-localizing with ThT-positive microclot complexes. Created in https://BioRender.com.
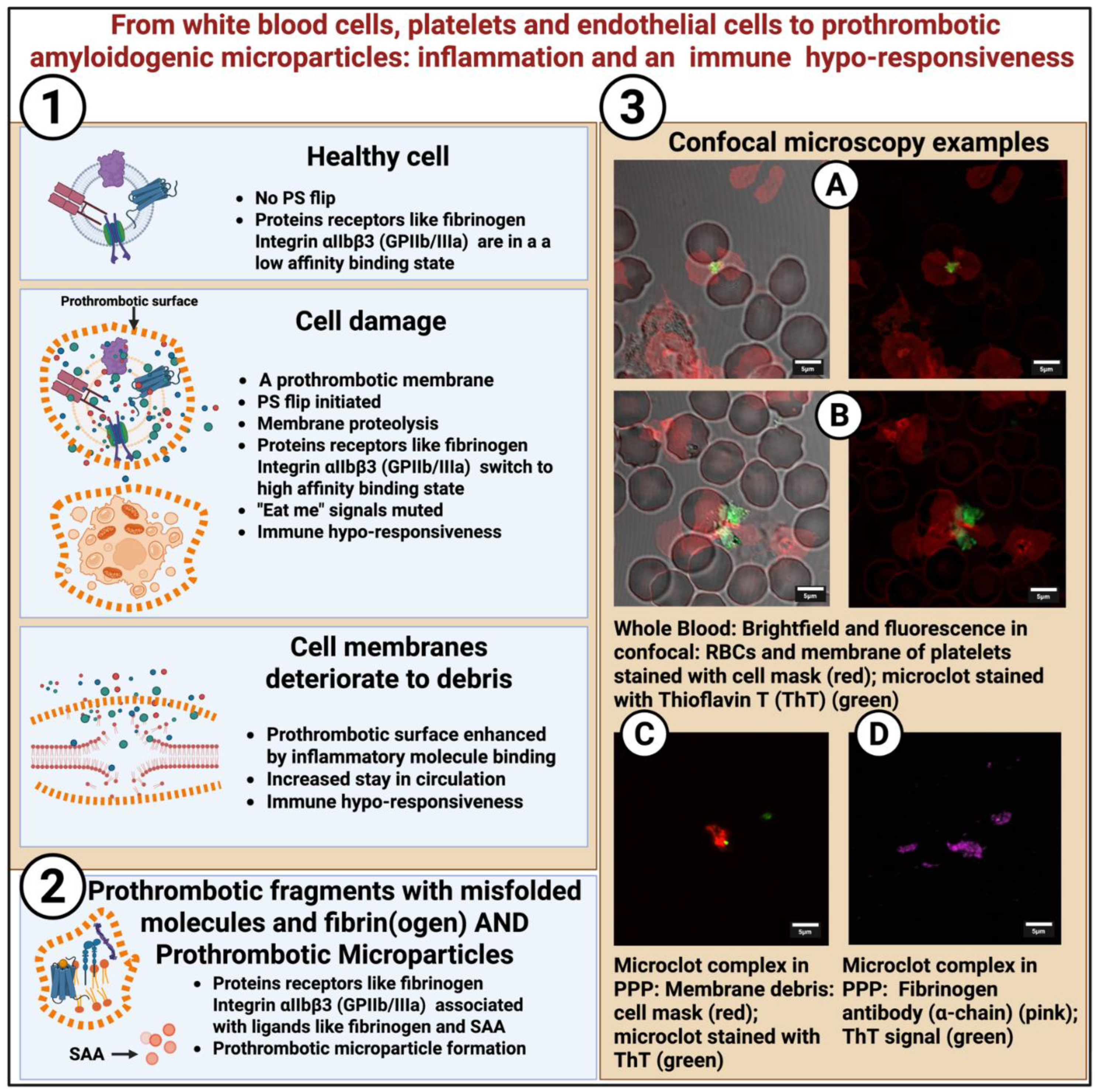
Figure 4.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the fibrinogen and inflammatory molecules pathway. (A) Conventional fibrin formation: Thrombin converts fibrinogen into fibrin monomers that polymerize into α-helical fibrin fibers, forming organized, degradable clots. (B) Pathological polymerization: Inflammatory molecules bind fibrinogen, inducing structural misfolding and β-sheet formation. These aberrant fibrin(ogen) complexes (“fibrinaloid” structures) are visualized with Thioflavin T ((ThT) green)) and fluorescent fibrinogen α-chain antibody (red) in purified fibrinogen and platelet-poor plasma (PPP).(C) Amyloidogenic inflammatory molecules: Circulating inflammagens such as spike protein, lipopolysaccharide (LPS), and serum amyloid A (SAA) are intrinsically amyloidogenic and can bind fibrinogen to form heterogeneous prothrombotic deposits. Created in https://BioRender.com.
Figure 4.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the fibrinogen and inflammatory molecules pathway. (A) Conventional fibrin formation: Thrombin converts fibrinogen into fibrin monomers that polymerize into α-helical fibrin fibers, forming organized, degradable clots. (B) Pathological polymerization: Inflammatory molecules bind fibrinogen, inducing structural misfolding and β-sheet formation. These aberrant fibrin(ogen) complexes (“fibrinaloid” structures) are visualized with Thioflavin T ((ThT) green)) and fluorescent fibrinogen α-chain antibody (red) in purified fibrinogen and platelet-poor plasma (PPP).(C) Amyloidogenic inflammatory molecules: Circulating inflammagens such as spike protein, lipopolysaccharide (LPS), and serum amyloid A (SAA) are intrinsically amyloidogenic and can bind fibrinogen to form heterogeneous prothrombotic deposits. Created in https://BioRender.com.
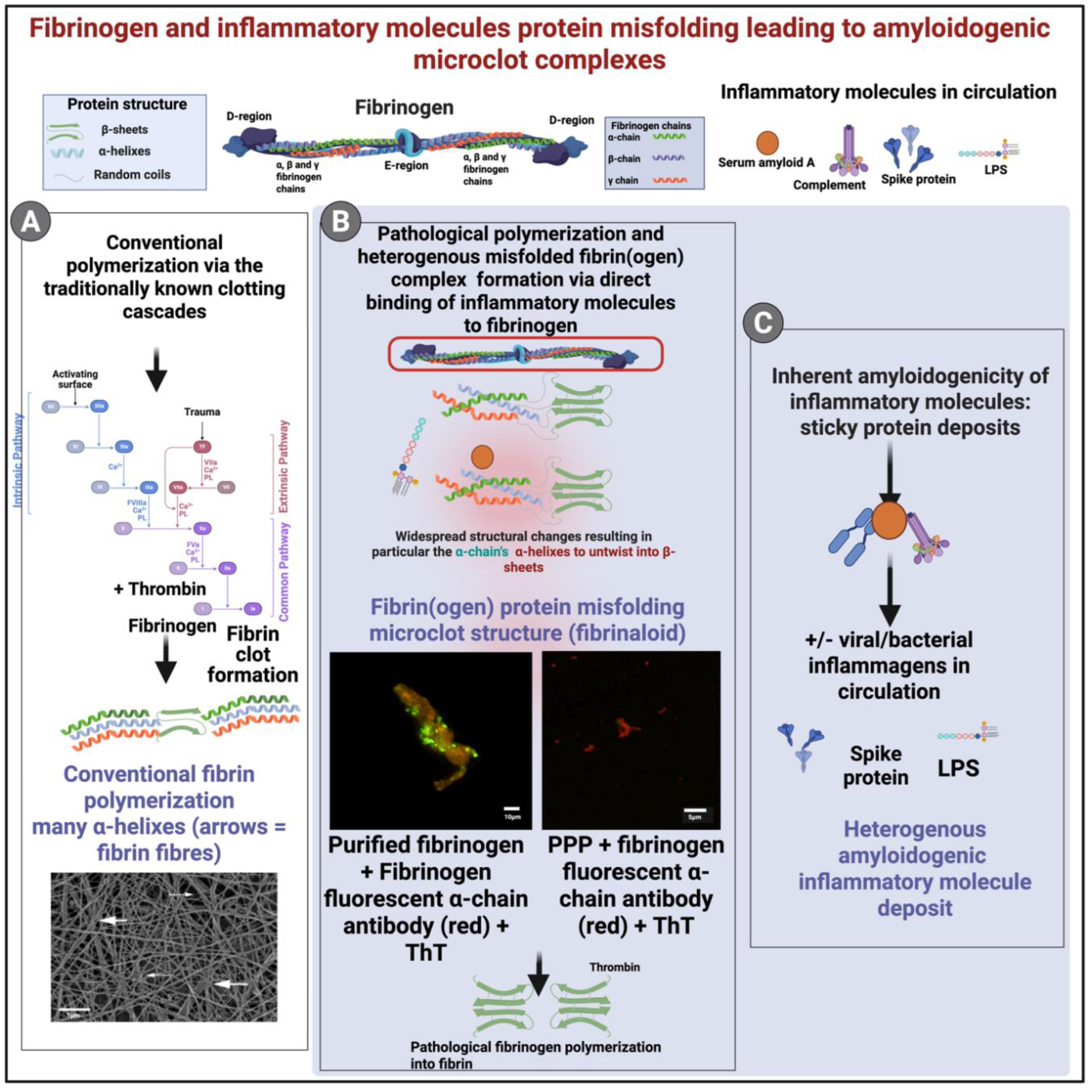
Figure 5.
The roadmap to circulating prothrombotic complexes (microclots) (and/or microparticles) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the seeding onto functional cells of the vasculature pathway (example given: seeding onto red blood cells: scanning electron micrographs of whole blood).
Figure 5.
The roadmap to circulating prothrombotic complexes (microclots) (and/or microparticles) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces via the seeding onto functional cells of the vasculature pathway (example given: seeding onto red blood cells: scanning electron micrographs of whole blood).
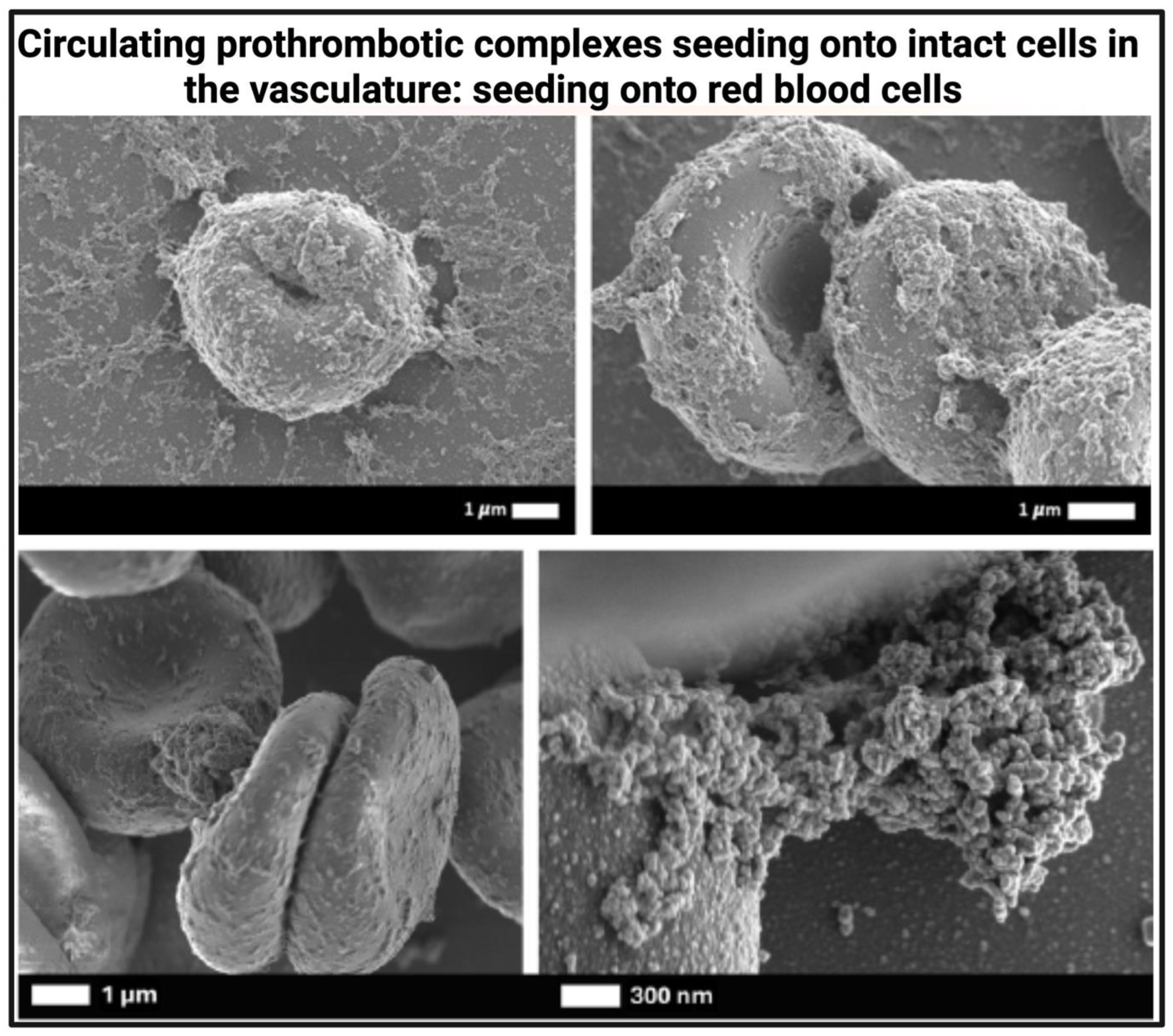
Figure 6.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces. From microclot complexes to anomalous fibrin dense matted deposits or “true” clots. (1–3) Progressive stages of pathological complex formation, beginning with misfolded fibrin(ogen) bound to amyloidogenic inflammatory molecules, evolving to mixed inflammatory deposits and misfolded cellular debris with prothrombotic potential. (4–6) FMCs are associated with neutrophil extracellular traps (NETs) and immune complexes (ThT+/MPO+/DNA+) and can interact with functional blood cells, including red blood cells and platelets, as shown by fluorescence and scanning electron microscopy. (See micrograph at (5) taken from [240]. (7) These insoluble protein complexes, arising from inflammatory molecules or fibrinogen interactions, act as scaffolds for thrombin generation and may seed further amyloid or fibrin deposition. In the presence of prothrombin or thrombin, they can initiate bona fide clot formation on vascular or cellular surfaces.Collectively, these Thioflavin T (ThT)-positive misfolded circulating complexes are termed fibrinaloid microclot complexes (FMCs). Created in https://BioRender.com.
Figure 6.
The roadmap to circulating prothrombotic complexes (microclots) that may drive thrombo-inflammation on β-sheet rich and amyloidogenic surfaces. From microclot complexes to anomalous fibrin dense matted deposits or “true” clots. (1–3) Progressive stages of pathological complex formation, beginning with misfolded fibrin(ogen) bound to amyloidogenic inflammatory molecules, evolving to mixed inflammatory deposits and misfolded cellular debris with prothrombotic potential. (4–6) FMCs are associated with neutrophil extracellular traps (NETs) and immune complexes (ThT+/MPO+/DNA+) and can interact with functional blood cells, including red blood cells and platelets, as shown by fluorescence and scanning electron microscopy. (See micrograph at (5) taken from [240]. (7) These insoluble protein complexes, arising from inflammatory molecules or fibrinogen interactions, act as scaffolds for thrombin generation and may seed further amyloid or fibrin deposition. In the presence of prothrombin or thrombin, they can initiate bona fide clot formation on vascular or cellular surfaces.Collectively, these Thioflavin T (ThT)-positive misfolded circulating complexes are termed fibrinaloid microclot complexes (FMCs). Created in https://BioRender.com.
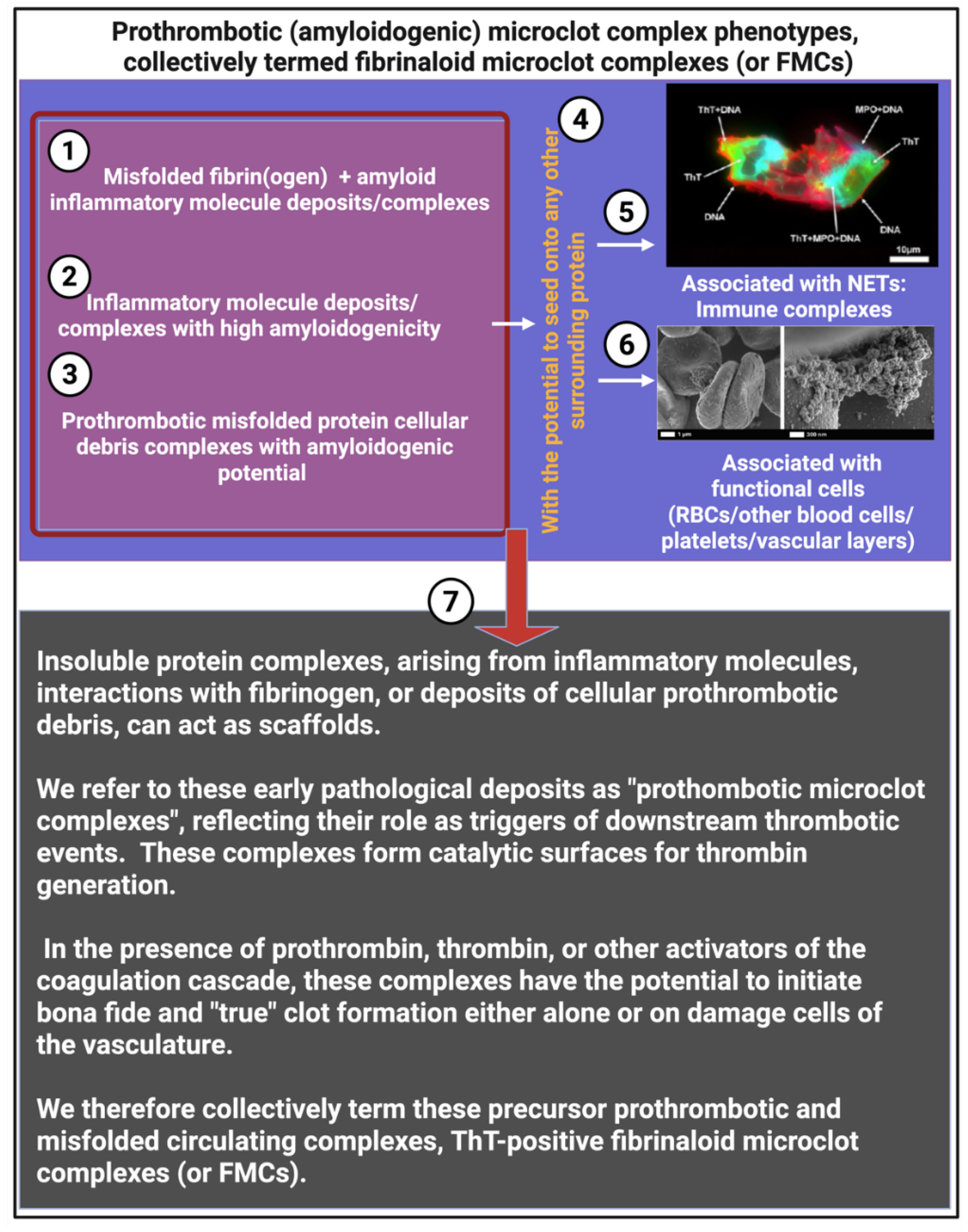

Table 1.
Fibrinogen receptors and their associated cell types.
| Cell type | Main fibrinogen-binding receptors | Comments |
| Platelets | Integrin αIIbβ3 (GPIIb/IIIa) [74] FPR2 [64] |
The dominant fibrinogen receptor αIIbβ3 essential for platelet aggregation and thrombus formation. SAA also binds to this αIIbβ3 receptor [64]. Platelets also have FPR2 receptors. αIIbβ3 is preferentially and highly expressed on resting platelets with 60,000–80,000 copies per cell [75]. |
| Endothelial cells | Integrins αvβ3 and α5β1 [15,76] ICAM1 [77] FPR2 |
Mediate fibrinogen binding, endothelial adhesion, angiogenesis, and leukocyte interactions Fibrinogen receptors contribute to clot stability at vessel walls α5β1 on endothelial cells, atheroprone flow plus oxidized lipoproteins increases the high-affinity conformation of α5β1, making ECs more adhesive and proinflammatory [76] α5β1 binds fibrin and fibronectin [78] FPR2 binds SAA [64] |
| Leukocytes (esp. neutrophils, monocytes, macrophages) | Integrin αMβ2 [79] (Mac-1, CD11b/CD18) FPR2 |
Binds fibrinogen, supports leukocyte adhesion, migration, and immunothrombosis. FRP2 important in linking inflammation to coagulation [64] |
| Erythrocytes | Less well-defined but perhaps GPIIbIIIa [80] |
Red cells can bind fibrinogen under inflammatory/prothrombotic conditions, but possibly not via a dedicated high-affinity receptor like platelets Not finally confirmed but GPIIbIIIa can possibly serve as fibrinogen binding sites. The presence of the GPIIbIIIa inhibitors reduces the amount of adsorbed fibrinogen, leading to a decrease in the hydrodynamic stability of RBC aggregates. |
Table 2.
Regulatory processes to prevent receptor activation.
| Regulatory Layer | Mechanism | Effect on SAA / Fibrinogen / Receptors | Key References |
| Receptor conformation | Platelet integrin αIIbβ3 is kept in a low-affinity state until “inside-out” signaling (e.g., via thrombin, ADP) activates it | Prevents fibrinogen and SAA from binding under resting conditions | [175] |
| Endothelial anti-adhesive surface | Endothelial glycocalyx (heparan sulfates, proteoglycans) blocks receptor access; NO and PGI2 secretion suppress platelet activation | Prevents SAA and fibrinogen interaction with receptors on healthy endothelium and platelets | [172,176] |
| Plasma binding partners | SAA is mostly HDL-bound in health; fibrinogen requires thrombin cleavage to reveal αIIbβ3 binding sites | Circulating ligands are “shielded” from receptor engagement | [177] |
| Membrane lipid asymmetry | Phosphatidylserine (PS) is restricted to inner leaflet by flippases | Prevents formation of procoagulant binding sites for clotting factors and SAA | [17] |
Table 3.
Protective mechanisms preventing receptor–ligand interactions in healthy circulation and their alteration during inflammation.
Table 3.
Protective mechanisms preventing receptor–ligand interactions in healthy circulation and their alteration during inflammation.
| Protective mechanism (healthy circulation) | Change during inflammation/disease |
| Integrins (e.g., αIIbβ3) inactive → low-affinity state maintained until platelet inside-out signaling activates them [175,178] | Platelet activation by thrombin, ADP, thromboxane A2, or cytokines triggers conformational change of αIIbβ3 → high-affinity fibrinogen and SAA binding [178] |
| Endothelial glycocalyx + NO/PGI2 enforce anti-adhesion, maintain vascular quiescence [172] | Glycocalyx degraded by ROS, proteases, and inflammatory enzymes; reduced NO/PGI2 signaling → adhesion molecules and receptors exposed (Lipowsky, 2012; Schmidt et al., 2020) |
| SAA sequestered in HDL complexes under baseline conditions, minimal free circulating SAA [179] | Acute-phase response: SAA upregulated 1000-fold and dissociates from HDL [180] Large pool of free can then potentially SAA binds receptors and fibrinogen. |
| PS restricted to inner leaflet by flippases maintains lipid asymmetry, no catalytic surface for coagulation [17,22] | In apoptosis/activation: scramblase activation + flippase inhibition; PS externalization, generating negatively charged catalytic surfaces for factor binding [17] |
Table 4.
Safety mechanisms of SAA in health and disease.
| Protective partner (health) | Mechanism of “safety” | Status in health (SAA levels <5 mg/L) | What changes in inflammation (SAA up to 1000 mg/L) | References |
| HDL (apoA-I containing particles) | Major carrier, prevents SAA functioning as an inflammatory molecule | SAA tightly bound to HDL; free SAA negligible | HDL composition altered; apoA displaced by SAA (“acute-phase HDL”) with reduced protective function; free SAA increases | [181,182] |
| Apolipoproteins | ApoA stabilize HDL, reducing SAA exposure | ApoA is abundant; maintain HDL’s anti-amyloid function | ApoA levels fall; displaced by SAA, exposure of hydrophobic domains; ApoE overwhelmed. | [177] |
| Lipid components (phospholipids, cholesterol esters) | SAA intercalates into HDL phospholipid monolayer, hydrophobic residues shielded | Normal lipid ratios keep SAA soluble within HDL | Acute-phase HDL lipid ratios altered; SAA less shielded and more aggregation-prone | [181,183] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



