
Submitted:
15 May 2025
Posted:
15 May 2025
You are already at the latest version
Abstract
Cardiovascular disease remains a leading cause of morbidity and mortality worldwide, frequently arising from platelet hyperactivation and subsequent thrombus formation. Although conventional antiplatelet therapies are available, challenges such as drug resistance and bleeding complications require the development of novel agents. In this study, dihydrogeodin (DHG) was isolated from Fennellia flavipes and evaluated using platelets derived from Sprague-Dawley rats. Platelet aggregation induced by collagen, adenosine diphosphate, or thrombin was assessed by light transmission aggregometry; DHG significantly reduced aggregation in a dose-dependent manner. Further assays demonstrated that DHG suppressed intracellular calcium mobilization, adenosine triphosphate release, and integrin αIIbβ3-dependent fibrinogen binding, thereby impairing clot retraction. Western blot analysis revealed that DHG reduced the phosphorylation of mitogen-activated protein kinases (ERK, JNK, p38) and PI3K/Akt, indicating inhibition across multiple platelet signaling pathways. Additionally, SwissADME-assisted pharmacokinetics predicted favorable properties without violations of the Lipinski (Pfizer) filter, Muegge (Bayer) filter, Ghose filter, Veber filter, and Egan filter, and network pharmacology revealed inhibition of calcium and MAPK pathways. These results highlight the potential of DHG as a novel antiplatelet agent with broad-spectrum activity and promising drug-like characteristics. Further studies are warranted to assess its therapeutic window, safety profile, and potential for synergistic use with existing antiplatelet drugs.
Keywords:
dihydrogeodin
; Fennellia flavipes
; platelet aggregation
; MAPK and PI3K/Akt pathways
; marine natural products
1. Introduction
Cardiovascular disease encompasses a broad spectrum of conditions, including atherosclerosis, coronary heart disease, hypertension, and stroke [1]. A key contributor to cardiovascular disease pathophysiology is platelet hyperactivation, which facilitates atherosclerotic plaque development, plaque rupture, and thrombus formation [2]. Under physiological conditions, platelets play a critical role in maintaining hemostasis [3]; however, when excessively activated, they can form occlusive thrombi that impair blood flow, potentially resulting in myocardial infarction, stroke, or other ischemic complications [4]. Accordingly, pharmacological inhibition of platelet function remains a well-established approach for both the treatment and prevention of cardiovascular disease [5,6,7,8]. While synthetic antiplatelet agents have been instrumental in managing cardiovascular diseases, their use is often accompanied by significant adverse effects. For instance, clopidogrel has been associated with rare but severe hematological disorders such as aplastic anemia and thrombotic thrombocytopenic purpura . Similarly, long-term aspirin therapy is linked to gastrointestinal complications, including ulcer formation and an increased risk of bleeding [9]. Considering this, the ethnomedical approach could be a promising strategy for preventing cardiovascular disease and its complications [6].
Platelets contain dense (δ) and alpha (α) granules. δ granules store adenosine diphosphate (ADP), adenosine triphosphate (ATP), Ca2+, and serotonin, whereas α granules contain fibronectin, fibrinogen, von Willebrand factor (vWF), and P-selectin [6,10]. Multiple signaling pathways control granule secretion, including Src family kinases (early platelet activation), intracellular Ca2+ mobilization, the cAMP-VASP-PKA pathway, protein kinase C (PKC) activation, the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) cascades, and RhoA-regulated cytoskeletal reorganization [10]. The increase in intracellular Ca2+ is essential for platelet function, initiating downstream signaling that promotes aggregation and thrombus formation [11]. Platelet activation also depends on integrin αIIbβ3, which undergoes conformational changes during inside-out signaling and binds fibrinogen, a critical step in the formation of stable thrombi [12]. Fibronectin enhances aggregation by interacting with integrins and recruiting signaling molecules to the cytoplasmic tails [13]. Additionally, Rho GTPases and Rho kinases (ROCKs) regulate cytoskeletal remodeling through myosin light chain phosphorylation, influencing platelet morphology, motility, and clot retraction [14]. Notably, treatment with natural products has been shown to reduce Ca2+ mobilization, ATP and serotonin release, fibrinogen binding, fibronectin adhesion, and clot retraction, indicating that such compounds exert antiplatelet effects through the inhibition of granule secretion and integrin αIIbβ3 signaling [12,15]. Despite the effectiveness of current antiplatelet agents such as aspirin and clopidogrel, these drugs are frequently associated with adverse effects and complications [16,17]. Aspirin, for instance, may cause severe gastric ulcers and prolonged bleeding, whereas clopidogrel has been linked to aplastic anemia and thrombocytopenic purpura [6]. Moreover, a substantial proportion of patients exhibit resistance to these therapies [18,19]. Factors contributing to aspirin resistance include genetic polymorphisms in cyclooxygenase-1 (COX-1) and other thromboxane-related genes, upregulated thromboxane biosynthesis from non-platelet sources, elevated platelet turnover, and interindividual variability in drug absorption [5]. These limitations highlight the pressing need for safer and more efficacious antiplatelet agents.
Natural products have historically been a rich source of bioactive molecules [20,21,22]. Numerous plant-derived and microbial metabolites demonstrate potent antiplatelet activity by targeting key enzymes or receptors involved in platelet aggregation [20,23,24,25]. These multitarget mechanisms reflect the broader therapeutic potential of natural compounds, which often exert bidirectional regulatory effects on physiological processes and tend to produce fewer side effects. Several marine fungi have previously been reported to possess bioactive natural products and significantly beneficial antiplatelet activities; for example, Epicoccum sorghinum metabolites inhibit platelet aggregation and inflammation [26], and Aspergillus species have been reported to exhibit antiplatelet as well as anticancer properties [27]. Moreover, marine fungal lipid bioactives display anti-inflammatory and antithrombotic effects, underscoring their promise as sources of novel antiplatelet agents [28].
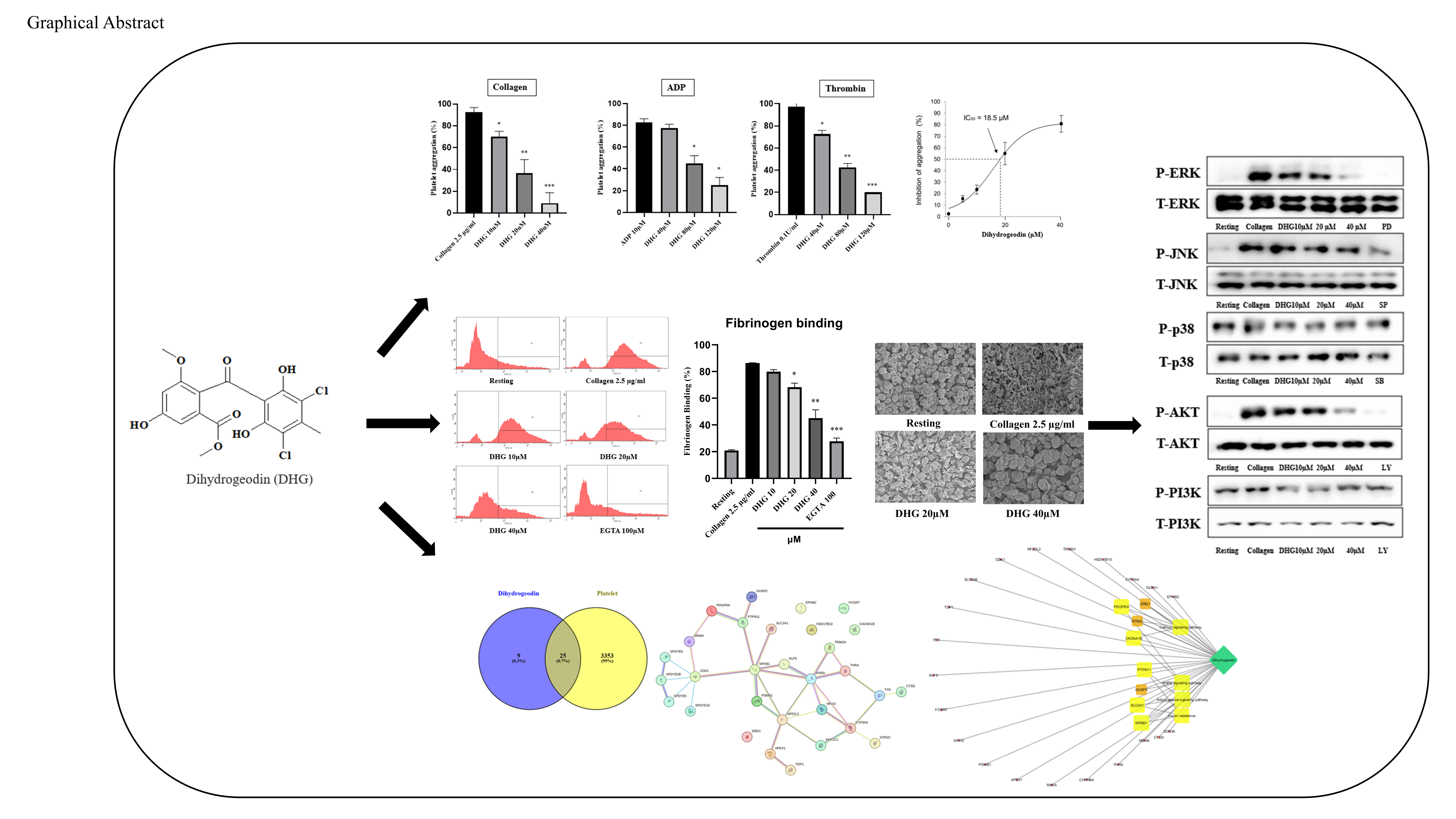
Among microbial sources, Aspergillus species are well known for producing a wide variety of secondary metabolites with therapeutic potential, including antiviral xanthones, diphenyl ether and butenolide derivatives with α-glucosidase inhibitory activity, and benzophenones with antimicrobial properties [29]. Chlorinated diphenyl ethers, in particular, serve as chemical markers of Aspergillus spp. and are generally believed to originate from anthraquinone precursors such as emodin, via intermediates such as sulochrin and grisandienes (e.g., geodin) [29]. The genus Fennellia (family Trichocomaceae), which includes F. flavipes and F. nivea, has been identified as the teleomorph of Aspergillus flavipes [30]. Although A. flavipes has been extensively studied for its production of cytochalasins, alkaloids, polyketides, butenolides, meroterpenoids, diphenyl ethers, benzophenones, and xanthones [31], some of which exhibit cytotoxic, antibacterial, or glucosidase-inhibiting properties, relatively little is known about the bioactive compounds specifically produced by F. flavipes [31]. This study began to address this gap by identifying several notable metabolites from F. flavipes, including a newly characterized dimeric 1,3-dihydroisobenzofuran and five previously known compounds, such as dihydrogeodin (DHG) [31,32]. In the present study, we isolated DHG from the culture broth of F. flavipes and evaluated its in vitro antiplatelet activity. Our findings demonstrate that DHG functions as a potent inhibitor of platelet aggregation, highlighting the potential of F. flavipes as a source of novel antiplatelet agents and pharmacologically active natural products (Graphical abstract).
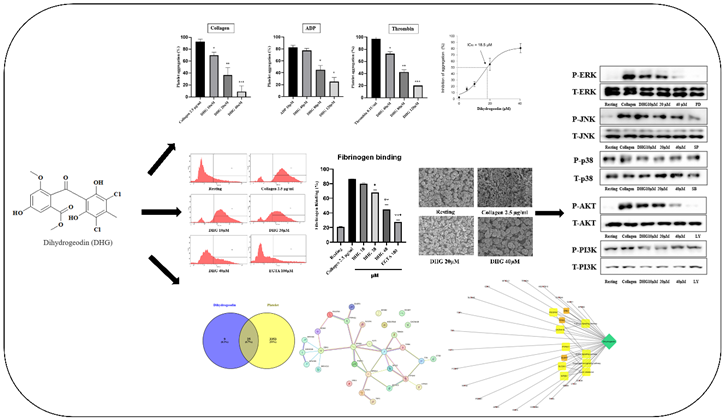
2. Results
2.1. Structural Elucidation
2.2. DHG Inhibits Agonist-Induced Platelet Aggregation
Platelet aggregation can be induced by various agonists, each activating distinct signaling pathways. Collagen initiates platelet adhesion and aggregation at sites of vascular injury [34], ADP promotes platelet activation through P2Y receptors [35], and thrombin induces robust platelet responses via protease-activated receptors [36]. To assess the antiplatelet potential of DHG, we evaluated platelet aggregation induced by each of these three agonists. Figure 1A presents the chemical structure of DHG. As shown in Figure 1B–D, DHG significantly inhibited platelet aggregation induced by collagen, ADP, and thrombin in a dose-dependent manner. Notably, DHG exerted its strongest inhibitory effect on collagen-induced aggregation. In collagen-stimulated platelets (Figure 1B), DHG at 10 µM significantly reduced aggregation compared with collagen alone (*P < 0.05) and completely inhibited aggregation at 40 µM (***P < 0.001). In contrast, ADP-induced aggregation (Figure 1C) required a concentration of 80 μM to achieve significant reduction (*P < 0.05), whereas thrombin-induced aggregation (Figure 1D) was significantly inhibited at 40 µM (*P < 0.05). These findings indicate that DHG more strongly targets the collagen-mediated pathway of platelet activation, while also exhibiting broader inhibitory effects against ADP- and thrombin-induced activation, highlighting its potential as a novel therapeutic agent for the prevention of platelet-mediated thrombotic events. The IC₅₀ of DHG for collagen-induced platelet aggregation was calculated to be 18.5 µM (Figure 1E). No potential cytotoxicity was observed at the indicated concentration (Supplementary Figure S7).
2.3. DHG Inhibits ATP Release and [Ca2+]i Mobilization
To determine whether DHG affects dense granule secretion, we measured ATP release from platelets stimulated with collagen (2.5 µg/mL). As shown in Figure 2 (left panel), collagen substantially increased ATP release compared with resting platelets, whereas DHG at 10, 20, and 40 μM significantly reduced ATP secretion in a dose-dependent manner (*P < 0.05, **P < 0.01, ***P < 0.001 versus collagen-stimulated controls). In parallel, intracellular Ca2+ mobilization, a key event in platelet activation, was assessed using Fura-2/AM-loaded platelets. Collagen stimulation induced a substantial increase in Ca2+ levels relative to resting platelets (Figure 2, right panel). DHG at 10, 20, and 40 μM significantly attenuated this collagen-induced Ca2+ mobilization (**P < 0.01, ***P < 0.001 versus collagen-stimulated controls). These findings suggest that DHG suppresses both dense granule secretion and intracellular calcium signaling, providing further mechanistic evidence for its potent antiplatelet activity.
2.4. DHG Downregulates Inside-Out and Outside-in Signaling and Prevents Platelet Shape Change
Inside-out signaling in platelets is primarily mediated by the activation of integrin αIIbβ3, which promotes fibrinogen binding and supports platelet aggregation [37]. As shown in Figure 3 (left), collagen stimulation (2.5 µg/mL) substantially increased the proportion of platelets binding fibrinogen compared with resting conditions. Pretreatment with DHG at 10, 20, and 40 μM significantly reduced fibrinogen binding in a dose-dependent manner (*P < 0.05, **P < 0.01, ***P < 0.001 versus collagen-stimulated controls). Notably, DHG at 40 μM nearly abolished fibrinogen binding, producing an effect comparable to that of EGTA (100 μM), which chelates calcium and prevents integrin activation [38]. These results indicate that DHG potently inhibits the inside-out signaling pathway leading to αIIbβ3 activation, thereby blocking fibrinogen binding and platelet aggregation.
After integrin activation and aggregation, outside-in signaling via αIIbβ3 coordinates platelet spreading, cytoskeletal reorganization, and clot retraction, a critical process for thrombus stabilization [12]. To assess whether DHG interferes with outside-in signaling, we performed a clot retraction assay using thrombin (1 U/mL) as the stimulus (Figure 4). In the absence of DHG, clot retraction was substantial, resulting in a compact thrombus (Figure 4A). In contrast, treatment with DHG at 20 and 40 μM significantly impaired clot retraction (*P < 0.05, **P < 0.01, ***P < 0.001 versus thrombin alone), yielding larger, less compact, and heavier clots (Figure 4B). The degree of inhibition was comparable to that observed with Y-27632 (10 μM), a known ROCK inhibitor, suggesting that DHG interferes with Rho-dependent signaling pathways. These findings demonstrate that DHG disrupts both inside-out and outside-in signaling in platelets. Further scanning electron microscopy (SEM) was performed to visualize the platelet shape change (Figure 4C). Collagen-induced platelets show clear fibrin meshwork formation which was significantly prevented after treatment with the indicated concentration of DHG.
2.5. DHG Attenuates MAPK and PI3K/Akt Phosphorylation
To determine whether DHG modulates the MAPK and PI3K/Akt pathways, both of which are essential for platelet activation [5], platelets were pretreated with DHG (10, 20, or 40 μM) and subsequently stimulated with collagen (2.5 µg/mL). Phosphorylation levels of extracellular signal-regulated kinase (ERK), Janus kinase (JNK), p38, PI3K, and Akt were assessed by Western blotting (Figure 5). Collagen stimulation substantially increased the phosphorylation of these proteins compared with resting platelets. In contrast, DHG reduced phosphorylation in a dose-dependent manner, whereas total protein levels (T-ERK, T-JNK, T-p38, T-PI3K, T-Akt) remained largely unchanged. Notably, DHG at 40 µM reduced phospho-ERK, phospho-JNK, phospho-PI3K, and phospho-Akt levels to near-basal values, indicating a strong inhibitory effect on MAPK and PI3K/Akt signaling. These results suggest that suppression of these key pathways contributes to the antiplatelet mechanism of DHG.
2.6. Pharmacokinetics, Drug-Likeliness Analysis
To assess the drug-likeness and pharmacokinetic potential of DHG, we utilized SwissADME to predict its physicochemical and pharmacokinetic properties. The analysis revealed that DHG complies with Lipinski’s rule of five, suggesting good oral bioavailability. Additionally, DHG demonstrated favorable lipophilicity (LogP), solubility, and gastrointestinal (GI) absorption, supporting its potential for systemic administration. No violations of drug-likeness criteria were predicted, reinforcing its suitability as a bioactive compound. The pharmacokinetic profile also suggested a low probability of BBB permeability, which may reduce the risk of central nervous system side effects. DHG was also predicted to be a non-substrate for P-glycoprotein (P-gp) efflux transporters, potentially enhancing its bioavailability. However, predicted interactions with cytochrome P450 (CYP) isoforms warrant consideration in future metabolism studies (Figure 6A and B).
2.7. Network Pharmacology
To explore the potential mechanism of DHG in platelet modulation, a network pharmacological approach was employed. A Venn diagram (Figure 7A) was constructed to identify overlapping targets between DHG and platelet activation-related genes, revealing 25 common targets, suggesting a potential pharmacological link between the compound and platelet function. A protein–protein interaction (PPI) network of these overlapping targets was constructed using the STRING database (Figure 7B). Notably, NFKB1, PDGFRA, RXRA, and PTPN11 emerged as key hub nodes, indicating their potential central roles in mediating the effects of DHG. The network shows that these targets are functionally interconnected and may be critical in signal transduction processes related to platelet activity. Further, a compound–target–pathway network (Figure 7C) demonstrated that DHG targets are significantly enriched in several signaling pathways. These include the calcium signaling pathway, MAPK signaling pathway, adipocytokine signaling pathway, and insulin resistance pathways. Notably, these pathways are not only relevant to cardiovascular diseases but also play crucial roles in platelet activation and aggregation, highlighting the therapeutic potential of DHG in thrombosis or related disorders.
3. Discussion
Natural products have attracted considerable interest due to their multitarget activities and relatively low incidence of adverse effects [39]. Recent studies have identified marine-derived compounds as promising sources of antiplatelet agents [40,41,42,43]. Similarly, various herbal medicines and their purified constituents have demonstrated potent antiplatelet activity [6,44,45]. For example, in our recently published study, ginsenoside Rg5 regulates GPVI signaling pathways by inhibiting collagen-induced platelet aggregation. The cryptotanshinone from Salvia miltiorrhiza Bunge exerts P2Y12-independent antiplatelet effects by downregulating the PI3K/Akt, MAPK, and STAT3 pathways [46]. Tryptanthrin from Isatidis Radix interferes with both inside-out and outside-in αIIbβ3 signaling, thus preventing stable thrombus formation [45]. Additionally, dieckol from Eisenia bicyclis inhibits platelet aggregation by suppressing granule secretion, integrin αIIbβ3 function, and the RhoA/ROCK pathway [12]. Similarly, in our study, DHG attenuated integrin αIIbβ3–mediated fibrinogen binding and clot retraction, as well as the phosphorylation of MAPK (ERK, JNK, p38) and PI3K/Akt, underscoring its multifaceted mode of action.
Platelet activation depends on a coordinated network of signaling events triggered by vascular injury [47]. Collagen, ADP, and thrombin activate distinct platelet receptors, GPVI, P2Y12, and protease-activated receptors, respectively, to initiate granule secretion, integrin αIIbβ3 activation, and cytoskeletal reorganization [48]. DHG significantly inhibited platelet aggregation induced by collagen, ADP, and thrombin in a dose-dependent manner, indicating broad inhibitory activity across multiple platelet activation pathways (Figure 1). In addition to its inhibition of aggregation, DHG substantially reduced intracellular Ca2+ mobilization and ATP release in response to collagen stimulation (Figure 2). Given the critical role of Ca2+ in platelet granule secretion, these results suggest that DHG disrupts upstream signaling events essential for platelet activation and may prevent thrombus formation at an early stage. DHG significantly reduced fibrinogen binding to platelets, indicating strong inhibition of αIIbβ3 activation. By blocking this step, DHG prevents the formation of stable platelet plugs (Figure 3). Furthermore, DHG attenuated human thrombin-induced clot retraction, mirroring the effects of established ROCK inhibitors and also prevented collagen-induced platelet shape change (Figure 4). DHG also reduced the phosphorylation of key signaling proteins in a dose-dependent manner without significantly altering total protein levels (Figure 5). Additionally, DHG satisfies Lipinski’s rule of five, suggesting good oral bioavailability. It also demonstrated favorable lipophilicity, solubility, and gastrointestinal absorption, along with a low probability of blood-brain barrier permeability (Figure 6A and B). Venn diagram showing the intersection between DHG-predicted targets and genes associated with platelet activation (Figure 7A). Among the 34 DHG-related genes and 3,353 platelet activation-related genes, 25 common genes were identified, representing potential targets through which DHG may influence platelet function. Figure 7B presents a protein–protein interaction (PPI) network constructed from the 25 overlapping genes using the STRING database. The network reveals a tightly connected hub centered around key nodes such as NFKB1, CDK1, RXRA, PDGFRA, and PTPN11, indicating their crucial roles in mediating the interaction between DHG and platelet-related signaling. These core targets are functionally linked through multiple biological processes, suggesting DHG may exert multi-targeted modulation. Further, Figure 7C shows the compound–target–pathway network constructed using Cytoscape. DHG is connected to several targets, including NFKB1, PDGFRA, DRD1, PTPN11, and HTR2C, which are further linked to relevant pathways such as the MAPK signaling pathway, calcium signaling pathway, adipocytokine signaling pathway, and insulin resistance. These enriched pathways have established roles in platelet function, inflammation, and cardiovascular regulation, supporting the potential of DHG to modulate platelet activity through multiple signaling cascades. Taken together, these pharmacological properties support DHG as a promising candidate for antithrombotic drug development.
Unlike many conventional antiplatelet agents, which typically target a single receptor or enzyme (e.g., P2Y12 for clopidogrel or COX-1 for aspirin), DHG appears to modulate multiple pathways integral to platelet function. Our results demonstrate that DHG inhibits platelet aggregation while reducing ATP release, intracellular Ca2+ mobilization, αIIbβ3 integrin activation, clot retraction, and activation of the MAPK and PI3K/Akt pathways (see schematic in Figure 8). Collectively, these findings suggest a broad-spectrum antiplatelet mechanism, similar to that of other recently reported multitarget natural compounds such as ginsenoside Rg5 [15] cryptotanshinone [46], dieckol [12], and tryptanthrin [45], which also inhibit platelet activation by modulating multiple signaling cascades. However, in vivo studies are necessary to further elucidate the pharmacological effects of DHG in animal models or human platelets. Although the in vitro findings strongly support its antiplatelet efficacy, translation of these results to in vivo systems requires additional investigation. Therefore, future studies using animal models of arterial thrombosis (e.g., FeCl3-induced vascular injury) or venous thrombosis are essential to establish its therapeutic potential.
4. Materials and Methods
4.1. Reagents
Collagen, ADP, and thrombin were obtained from Chrono-Log Co. (Netherlands). Human plasma-derived thrombin and glutaraldehyde were purchased from Sigma-Aldrich. ATP assay kits were acquired from Cayman Chemical (Ann Arbor, MI, USA). Paraformaldehyde, Fura 2-AM, and Alexa Fluor 488-conjugated fibrinogen were obtained from Invitrogen (Eugene, OR, USA). Avertin was prepared as the anesthetic by dissolving 2,2,2-tribromoethanol (Sigma-Aldrich, catalog no. T48402-5G) in 2-methyl-2-butanol (Sigma-Aldrich, catalog no. 152463). All antibodies for western blotting were purchased from Cell Signaling Technology (USA).
4.2. Fungal Material and Fermentation
A fungal strain of F. flavipes (FU00000759) was obtained from the National Marine Biodiversity Institute of Korea (MABIK). This strain was isolated from a tidal flat in Incheon, located on the Western Coast of Korea. The strain was cultured in twenty 5-L flasks, each containing 1 L of potato dextrose broth, at 27°C for 4 weeks under stationary conditions.
4.3. Extraction, Isolation, and Structure Determination
The extraction, isolation, and structure determination of the active compounds from F. flavipes were performed as described in our recent publication [31] and supplementary material section 1.1. The 1H and 13C NMR spectra matched those known for dihydrogeodin [31,32], and complete data is provided in supplementary material Fig. S1-S5.
4.4. Experimental Animals
Seven-week-old male Sprague-Dawley rats weighing 240–260 g were used for in vitro platelet experiments. The animals were acclimated in an environmentally controlled room maintained at approximately 23 ± 2°C and 50% ± 10% humidity, with a 12-h light/dark cycle. All animal procedures were performed in accordance with institutional guidelines and approved by the Animal Care Committee of the College of Veterinary Medicine, Kyungpook National University, Daegu, South Korea (Permit no KNU 2022-0083).
4.5. Light Transmission Aggregometry and Scanning Electron Microscopy (SEM)
Blood was collected from Sprague-Dawley rats via cardiac puncture using a syringe containing acid-citrate-dextrose to isolate purified platelets [6]. The collected blood was transferred into round-bottom tubes containing Tyrode’s buffer and acid-citrate-dextrose at a 1:4 (v/v) ratio. Washed platelets were separated from whole blood by centrifugation at 170 × g for 7 min, followed by a second centrifugation at 350 × g for 10 min. The platelet suspension was adjusted to a concentration of 3 × 108 cells/mL for aggregometry analysis. Platelets were incubated for 1 min with DHG (10, 20, or 40 μM) or with DMSO as a control in the presence of 1 mM CaCl2. Aggregation was then induced by the addition of agonists (collagen at 2.5 μg/mL, ADP at 10 μM, or thrombin at 0.1 U/mL) for 5 min. This method enabled precise quantification of the platelet response under defined experimental conditions, providing valuable insights into the effects of dihydrogeodin on platelet function. Platelets incubated with DHG and agonist (collagen) were fixed in 0.5% paraformaldehyde and 0.5% osmium tetroxide, dehydrated, and freeze-dried at –55 °C before being mounted for field-emission scanning electron microscopy (Hitachi SU8220) to assess ultrastructural shape changes.
4.6. ATP Release Assay, [Ca2+]i Mobilization Assay, and Fibrinogen Binding Assay
Platelets were preincubated with dihydrogeodin and then stimulated with collagen to induce activation. Subsequently, ATP release, intracellular calcium mobilization, and fibrinogen binding were assessed as previously described [6]. ATP secretion was measured in the supernatant using an ATP assay kit to evaluate platelet function. Intracellular calcium concentration ([Ca2+]i) was determined in Fura-2/AM-loaded platelets using the following formula: 224 nM × (F – Fmin) / (Fmax – F). For the fibrinogen binding assay, platelets were stained with an anti-fibrinogen antibody, and activation levels were evaluated by flow cytometry after collagen stimulation in the presence or absence of dihydrogeodin.
4.7. Clot Retraction
Platelet-rich plasma (PRP, 250 μL) was incubated for 2 min with DHG or Y-27632 (a ROCK inhibitor). A final volume of 1 mL was achieved by adding red blood cells (5 μL) and Tyrode’s buffer. Thrombin (1 U/mL) was then added, and clot retraction was assessed at room temperature. Thrombin-induced clots were weighed to compare clot retraction among treatment groups.
4.8. Western Blotting
After preincubation with DHG and stimulation with collagen, lysis buffer was added to initiate lysis, and protein concentrations were quantified. Whole platelet proteins were isolated for subsequent analysis. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked and incubated overnight with primary antibodies, followed by incubation with secondary antibodies for 3 h. The membranes were then washed three times and visualized using enhanced chemiluminescence. This procedure enabled evaluation of changes in protein expression induced by dihydrogeodin treatment.
4.9. Pharmacokinetics, Drug-Likeliness Analysis, and Network Pharmacology
SwissADME, a freely accessible web-based tool, was used to evaluate the pharmacokinetic properties and drug-likeness of DHG, as previously reported [49]. Briefly, the canonical SMILES notation for DHG was retrieved from PubChem (PC-ID 612831 https://pubchem.ncbi.nlm.nih.gov/compound/Dihydrogeodin) and entered into the SwissADME platform (http://www.swissadme.ch/). The resulting output files and images were directly imported from the website. The BOILED-Egg (Brain Or Intestinal Estimated permeation predictive) model provides a rapid and straightforward method for assessing human intestinal absorption (HIA) and blood-brain barrier (BBB) permeation by calculating the lipophilicity and polarity of molecules, followed by generation of a water partition coefficient (WLOGP) versus topological polar surface area (tPSA) plot. For network pharmacology, the potential target proteins of DHG were predicted using the SuperPred webserver, and the results were downloaded as CSV files. Corresponding UniProt IDs for Homo sapiens were retrieved and used for protein mapping via the STRING database to identify protein names (preferred names) associated with DHG. To identify disease-related targets, the keyword “platelet activation” was searched in the GeneCards database, and relevant human gene targets were extracted. The predicted DHG target genes (compound-related genes) and platelet activation-related genes (disease target genes) were compared using Venny 2.1. This analysis identified overlapping targets (common genes) between DHG and platelet activation. These overlapping targets were input into the STRING database (organism: Homo sapiens) to generate a protein–protein interaction (PPI) network and assess potential biological connections. KEGG pathway enrichment analysis was also performed through STRING to identify key signaling pathways associated with the overlapping genes. Finally, the network of DHG, overlapping target genes, and enriched pathways was constructed and visualized using Cytoscape software (version 3.7.2).
4.10. Statistical Analysis
Data were analyzed by one-way analysis of variance (ANOVA), followed by post hoc Dunnett’s test (SAS Institute, Inc., Cary, NC, USA) to determine statistical significance. Results are presented as mean ± standard deviation (SD). A P-value of 0.05 or less was considered statistically significant.
5. Conclusion
Overall, the findings of this study position dihydrogeodin as a promising natural antiplatelet agent capable of multifaceted inhibition of platelet function. By targeting central signaling pathways (MAPK and PI3K/Akt) and integrin αIIbβ3 activation, DHG interferes with both the early and late stages of platelet activation. Coupled with its favorable in silico drug-likeness profile, these results support DHG as a strong candidate for future pharmacological development. Rigorous in vivo studies and detailed mechanistic investigations will be required to fully characterize its therapeutic efficacy and safety in the prevention or treatment of thrombotic diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Abdul Wahab Akram, Dae-Cheol Choi, Bong-Sik Yun, Man Hee Rhee; methodology, Abdul Wahab Akram, Dae-Cheol Choi, Bong-Sik Yun, Man Hee Rhee; software, Abdul Wahab Akram, Bong-Sik Yun, Man Hee Rhee; validation, Bong-Sik Yun, Man Hee Rhee; formal analysis, Abdul Wahab Akram; investigation, Man Hee Rhee; resources, Dae-Cheol Choi, Bong-Sik Yun, Man Hee Rhee; data curation, Abdul Wahab Akram, Dae-Cheol Choi; writing—original draft preparation, Abdul Wahab Akram, Dae-Cheol Choi, Hyung-Kyu Chae , Sung Dae Kim, Dongmi Kwak; writing—review and editing, Abdul Wahab Akram, Dae-Cheol Choi, Hyung-Kyu Chae , Sung Dae Kim, Dongmi Kwak, Bong-Sik Yun, Man Hee Rhee; visualization, Bong-Sik Yun, Man Hee Rhee; supervision, Bong-Sik Yun, Man Hee Rhee; project administration, Bong-Sik Yun, Man Hee Rhee; funding acquisition, Bong-Sik Yun, Man Hee Rhee. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
All animal procedures were performed in accordance with institutional guidelines and approved by the Animal Care Committee of the College of Veterinary Medicine, Kyungpook National University, Daegu, South Korea (Permit no. KNU-2015-60).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary material; further inquiries can be directed to the corresponding authors.
Acknowledgments and Funding
The authors are grateful to Professor Man Hee Rhee for his continuous supervision and support. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2022R1A2C1012963). We are thankful to the National Research Foundation of Korea (NRF) for the grants.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lockhart, P.B.; Sun, Y.P. Diseases of the cardiovascular system. Burket's Oral Medicine 2021, Hoboken (NJ): John Wiley & Sons 505-552.
- Caffrey, C.; Leamy, A.; O’sullivan, E.; Zabetakis, I.; Lordan, R.; Nasopoulou, C. Cardiovascular Diseases and Marine Oils: A Focus on Omega-3 Polyunsaturated Fatty Acids and Polar Lipids. Mar. Drugs 2023, 21, 549. [Google Scholar] [CrossRef] [PubMed]
- Scridon, A. Platelets and Their Role in Hemostasis and Thrombosis—From Physiology to Pathophysiology and Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 12772. [Google Scholar] [CrossRef]
- Młynarska, E.; Czarnik, W.; Fularski, P.; Hajdys, J.; Majchrowicz, G.; Stabrawa, M.; Rysz, J.; Franczyk, B. From Atherosclerotic Plaque to Myocardial Infarction—The Leading Cause of Coronary Artery Occlusion. Int. J. Mol. Sci. 2024, 25, 7295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, D.; Tan, P.; Xian, B.; Jiang, H.; Wu, Q.; Huang, X.; Zhang, P.; Xiao, X.; Pei, J. Mechanism of platelet activation and potential therapeutic effects of natural drugs. Phytomedicine 2022, 108, 154463. [Google Scholar] [CrossRef]
- Akram, A.W.; Saba, E.; Rhee, M.H. Antiplatelet and antithrombotic activities of Lespedeza cuneata via Pharmacological inhibition of integrin αIIbβ3, MAPK, and PI3K/AKT pathways and FeCl3-induced murine thrombosis. Evidence-Based Complementary and Alternative Medicine 2024, 2024, 9927160. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, F.-Q.; Zhang, Q.; Wang, F.-Q.; Hu, Y.-J.; Xia, Z.-N. Natural products for antithrombosis. Evidence-Based Complementary and Alternative Medicine 2015, 2015, 876426. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y.; Yang, S.; Yu, Z.; Xing, Y.; Wu, M. Mechanism and Potential Target of Blood-Activating Chinese Botanical Drugs Combined With Anti-Platelet Drugs: Prevention and Treatment of Atherosclerotic Cardiovascular Diseases. Front. Pharmacol. 2022, 13, 811422. [Google Scholar] [CrossRef]
- Krishnan, K.; Nguyen, T.N.; Appleton, J.P.; Law, Z.K.; Caulfied, M.; Cabrera, C.P.; Lenthall, R.; Hewson, D.; England, T.; McConachie, N.; et al. Antiplatelet Resistance: A Review of Concepts, Mechanisms, and Implications for Management in Acute Ischemic Stroke and Transient Ischemic Attack. Stroke: Vasc. Interv. Neurol. 2023, 3. [Google Scholar] [CrossRef]
- Mussbacher, M.; Kral-Pointner, J.B.; Salzmann, M.; Schrottmaier, W.C.; Assinger, A. Mechanisms of hemostasis: Contributions of platelets, coagulation factors, and the vessel wall. In Fundamentals of Vascular Biology; Springer: 2024; pp. 167-203.
- Shehwar, D.; Barki, S.; Aliotta, A.; Calderara, D.B.; Veuthey, L.; Portela, C.P.; Alberio, L.; Alam, M.R. Platelets and mitochondria: the calcium connection. Mol. Biol. Rep. 2025, 52, 1–9. [Google Scholar] [CrossRef]
- Irfan, M.; Kwon, T.-H.; Kwon, H.-W.; Rhee, M.H. Pharmacological actions of dieckol on modulation of platelet functions and thrombus formation via integrin αIIbβ3 and cAMP signaling. Pharmacol. Res. 2022, 177, 106088. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Calderwood, D.A. Organization, dynamics and mechanoregulation of integrin-mediated cell–ECM adhesions. Nature Reviews Molecular Cell Biology 2023, 24, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Ngo, A.T.P.; Parra-Izquierdo, I.; Aslan, J.E.; McCarty, O.J.T. Rho GTPase regulation of reactive oxygen species generation and signalling in platelet function and disease. Small GTPases 2021, 12, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Akram, A.W.; Shin, J.-H.; Batmunkh, U.; Saba, E.; Kang, Y.-M.; Jung, S.; Han, J.E.; Kim, S.D.; Kwak, D.; Kwon, H.-W.; et al. Ginsenoside Rg5 inhibits platelet aggregation by regulating GPVI signaling pathways and ferric chloride-induced thrombosis. J. Ginseng Res. 2025. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Z.; Shen, B.; Chen, C.; Ding, X.; Song, H. Effects of aspirin on the gastrointestinal tract: Pros vs. cons. Oncology Letters 2020, 20, 2567–2578. [Google Scholar] [CrossRef]
- Bebars, A.; Alhazmi, A.; Aldajan, M.; Alhujeri, N.; Alhaidari, R. Gastrointestinal side effects of antiplatelet drugs (Aspirin and Clopidogrel) in Medina, Saudi Arabia. Int. J. Med. Dev. Ctries. 2021, 210–216. [Google Scholar] [CrossRef]
- Ferreira, M.; Freitas-Silva, M.; Assis, J.; Pinto, R.; Nunes, J.P.; Medeiros, R. The Emergent Phenomenon of Aspirin Resistance: Insights from Genetic Association Studies. Pharmacogenomics 2020, 21, 125–140. [Google Scholar] [CrossRef]
- Hidayat, R.; Nabilah, R.A.; Rasyid, A.; Harris, S.; Harahap, A.R.; Herqutanto, H.; Louisa, M.; Listyaningsih, E.; Rambe, A.S.; Loho, T. Clopidogrel Resistance Among Ischemic Stroke Patients and Its Risk Factors in Indonesia. Acta Medica Acad. 2022, 51, 29–34. [Google Scholar] [CrossRef]
- Chaachouay, N.; Zidane, L. Plant-Derived Natural Products: A Source for Drug Discovery and Development. Drugs Drug Candidates 2024, 3, 184–207. [Google Scholar] [CrossRef]
- Aware, C.B.; Patil, D.N.; Suryawanshi, S.S.; Mali, P.R.; Rane, M.R.; Gurav, R.G.; Jadhav, J.P. Natural bioactive products as promising therapeutics: A review of natural product-based drug development. South Afr. J. Bot. 2022, 151, 512–528. [Google Scholar] [CrossRef]
- Use of Natural Feed Additives as a Remedy for Diseases in Veterinary Medicine. Int. J. Veter- Sci. [CrossRef]
- Wang, H.; He, D.; Duan, L.; Lv, L.; Gao, Q.; Wang, Y.; Yang, S.; Lv, Z. In Vivo Anticoagulant and Antithrombic Activity of Depolymerized Glycosaminoglycan from Apostichopus japonicus and Dynamic Effect–Exposure Relationship in Rat Plasma. Mar. Drugs 2022, 20, 631. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, C.; Xia, Q.; Li, P.; Liu, K.; Zhang, Y. Brevianamide F Exerts Antithrombotic Effects by Modulating the MAPK Signaling Pathway and Coagulation Cascade. Mar. Drugs 2024, 22, 439. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, G.; Li, S.; Li, P.; Zhang, J.; Yin, R.; Yuan, L.; Gao, N.; Zhao, J. Fucosylated Glycosaminoglycan Oligosaccharide HS14, Derived from Sea Cucumbers, Is a Novel Inhibitor of Platelet Toll-like Receptor 2. Mar. Drugs 2025, 23, 110. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Chang, C.-C.; Tsai, Y.-H.; El-Shazly, M.; Wu, C.-C.; Wang, S.-W.; Hwang, T.-L.; Wei, C.-K.; Hohmann, J.; Yang, Z.-J. Anti-inflammatory, antiplatelet aggregation, and antiangiogenesis polyketides from Epicoccum sorghinum: toward an understating of its biological activities and potential applications. ACS omega 2020, 5, 11092–11099. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lin, X.; Fu, J.; Zhang, J.; Tang, W.; He, Z. A Novel Bi-Functional Fibrinolytic Enzyme with Anticoagulant and Thrombolytic Activities from a Marine-Derived Fungus Aspergillus versicolor ZLH-1. Mar. Drugs 2022, 20, 356. [Google Scholar] [CrossRef]
- Papikinou, M.-A.; Pavlidis, K.; Cholidis, P.; Kranas, D.; Adamantidi, T.; Anastasiadou, C.; Tsoupras, A. Marine Fungi Bioactives with Anti-Inflammatory, Antithrombotic and Antioxidant Health-Promoting Properties Against Inflammation-Related Chronic Diseases. Mar. Drugs 2024, 22, 520. [Google Scholar] [CrossRef]
- Ji, Y.B.; Chen, W.J.; Shan, T.Z.; Sun, B.Y.; Yan, P.C.; Jiang, W. Antibacterial diphenyl ether, benzophenone and xanthone derivatives from Aspergillus flavipes. Chemistry & Biodiversity 2020, 17, e1900640. [Google Scholar]
- Peterson, S.W. Phylogenetic analysis of Aspergillus species using DNA sequences from four loci. Mycologia 2008, 100, 205–226. [Google Scholar] [CrossRef]
- Choi, D.-C.; Ki, D.-W.; Kim, J.-Y.; Lee, I.-K.; Yun, B.-S. New dimeric 1,3-dihydroisobenzofuran from culture broth of Fennellia flavipes. Phytochem. Lett. 2025, 65, 141–144. [Google Scholar] [CrossRef]
- Hamed, A.; Ismail, M.; El-Metwally, M.M.; Frese, M.; Stammler, H.G.; Sewald, N.; Shaaban, M. X-ray, structural assignment and molecular docking study of dihydrogeodin from Aspergillus Terreus TM8. Nat. Prod. Res. 2018, 33, 117–121. [Google Scholar] [CrossRef]
- Sato, S.; Okusa, N.; Ogawa, A.; Ikenoue, T.; Seki, T.; Tsuji, T. Identification and Preliminary SAR Studies of (+)-Geodin as a Glucose Uptake Stimulator for Rat Adipocytes. J. Antibiot. 2005, 58, 583–589. [Google Scholar] [CrossRef]
- Lemmens, T.; Luo, Q.; Wielders, S.; Scheijen, J.; Al-Nasiry, S.; Koenen, R.; Wenzel, P.; Cosemans, J. Platelet collagen receptors and their role in modulating platelet adhesion patterns and activation on alternatively processed collagen substrates. Thromb. Res. 2024, 244, 109201. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C.; Hechler, B. Platelet Purinergic Receptors in Thrombosis and Inflammation. Hamostaseologie 2020, 40, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wu, J.; Roest, M.; Heemskerk, J.W.M. Long-term platelet priming after glycoprotein VI stimulation in comparison to Protease-Activating Receptor (PAR) stimulation. PLOS ONE 2021, 16, e0247425. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Lin, K.-C.; Hsia, C.-W.; Hsia, C.-H.; Chen, T.-Y.; Bhavan, P.S.; Sheu, J.-R.; Hou, S.-M. The Antithrombotic Agent Pterostilbene Interferes with Integrin αIIbβ3-Mediated Inside-Out and Outside-In Signals in Human Platelets. Int. J. Mol. Sci. 2021, 22, 3643. [Google Scholar] [CrossRef]
- Xiang, B.; Zhang, G.; Zhang, Y.; Wu, C.; Joshi, S.; Morris, A.J.; Ware, J.; Smyth, S.S.; Whiteheart, S.W.; Li, Z. Calcium Ion Chelation Preserves Platelet Function During Cold Storage. Arter. Thromb. Vasc. Biol. 2020, 41, 234–249. [Google Scholar] [CrossRef]
- Lichota, A.; Szewczyk, E.M.; Gwozdzinski, K. Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds. Int. J. Mol. Sci. 2020, 21, 7975. [Google Scholar] [CrossRef]
- Pan, N.; Li, Z.-C.; Li, Z.-H.; Chen, S.-H.; Jiang, M.-H.; Yang, H.-Y.; Liu, Y.-S.; Hu, R.; Zeng, Y.-W.; Dai, L.-H.; et al. Antiplatelet and Antithrombotic Effects of Isaridin E Isolated from the Marine-Derived Fungus via Downregulating the PI3K/Akt Signaling Pathway. Mar. Drugs 2021, 20, 23. [Google Scholar] [CrossRef]
- Vasconcelos, A.A.; Sucupira, I.D.; Guedes, A.L.; Queiroz, I.N.; Frattani, F.S.; Fonseca, R.J.; Pomin, V.H. Anticoagulant and Antithrombotic Properties of Three Structurally Correlated Sea Urchin Sulfated Glycans and Their Low-Molecular-Weight Derivatives. Mar. Drugs 2018, 16, 304. [Google Scholar] [CrossRef]
- Cao, S.; He, X.; Qin, L.; He, M.; Yang, Y.; Liu, Z.; Mao, W. Anticoagulant and Antithrombotic Properties in Vitro and in Vivo of a Novel Sulfated Polysaccharide from Marine Green Alga Monostroma nitidum. Mar. Drugs 2019, 17, 247. [Google Scholar] [CrossRef]
- Moura, L.D.A.; de Almeida, A.C.M.; Domingos, T.F.S.; Ortiz-Ramirez, F.; Cavalcanti, D.N.; Teixeira, V.L.; Fuly, A.L. Antiplatelet and Anticoagulant Effects of Diterpenes Isolated from the Marine Alga, Dictyota menstrualis. Mar. Drugs 2014, 12, 2471–2484. [Google Scholar] [CrossRef]
- Gholkar, A.A.; Nikam, Y.P.; Zambare, K.K.; Reddy, K.V.; Ghorpade, A.D. Potential Anticoagulant Herbal Plants: A Review. Asian J. Res. Pharm. Sci. 2020, 10, 51. [Google Scholar] [CrossRef]
- Gao, H.; Huang, J.; Huang, X.; Lin, X.; Li, X.; Deng, H.; Zhou, Y.; Wu, L.; Xi, X.; Jin, J.; et al. Tryptanthrin impairs platelet function and thrombus formation by reducing Gp1bα expression. Eur. J. Pharmacol. 2025, 991, 177332. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, R.; Hua, C.; Wu, M.; Yuan, Y.; Zhang, L.; Guo, F.; Liu, J.; Yang, Z.; Liu, G. P2Y12 receptor-independent antiplatelet mechanism of cryptotanshinone: Network pharmacology and experimental validation of multi-target signaling pathways. J. Ethnopharmacol. 2025, 341, 119321. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Platelet-released extracellular vesicles: the effects of thrombin activation. Cell. Mol. Life Sci. 2022, 79, 1–16. [Google Scholar] [CrossRef]
- Zou, J.; Swieringa, F.; de Laat, B.; de Groot, P.G.; Roest, M.; Heemskerk, J.W.M. Reversible platelet integrin αIIbβ3 activation and thrombus instability. International Journal of Molecular Sciences 2022, 23, 12512. [Google Scholar] [CrossRef]
- Quah, Y.; Lee, Y.Y.; Lee, S.-J.; Kim, S.D.; Rhee, M.H.; Park, S.-C. In silico investigation of Panax ginseng lead compounds against COVID-19 associated platelet activation and thromboembolism. J. Ginseng Res. 2022, 47, 283–290. [Google Scholar] [CrossRef]
Figure 1.
Dihydrogeodin (DHG) inhibits agonist-induced platelet aggregation. Washed platelets were incubated for 1 min with DHG in the presence of 1 mM CaCl2. Aggregation was initiated by the addition of agonists. (A) Chemical structure of DHG. (B) Effect of DHG on collagen-induced platelet aggregation. (C) Effect of DHG on ADP-induced platelet aggregation. (D) Effect of DHG on thrombin-induced platelet aggregation. (E) IC₅₀ of DHG for collagen-induced platelet aggregation. Data are expressed as mean ± SD (n = 3). Statistical significance was determined by one-way ANOVA followed by a post hoc Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 versus agonist.
Figure 1.
Dihydrogeodin (DHG) inhibits agonist-induced platelet aggregation. Washed platelets were incubated for 1 min with DHG in the presence of 1 mM CaCl2. Aggregation was initiated by the addition of agonists. (A) Chemical structure of DHG. (B) Effect of DHG on collagen-induced platelet aggregation. (C) Effect of DHG on ADP-induced platelet aggregation. (D) Effect of DHG on thrombin-induced platelet aggregation. (E) IC₅₀ of DHG for collagen-induced platelet aggregation. Data are expressed as mean ± SD (n = 3). Statistical significance was determined by one-way ANOVA followed by a post hoc Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 versus agonist.
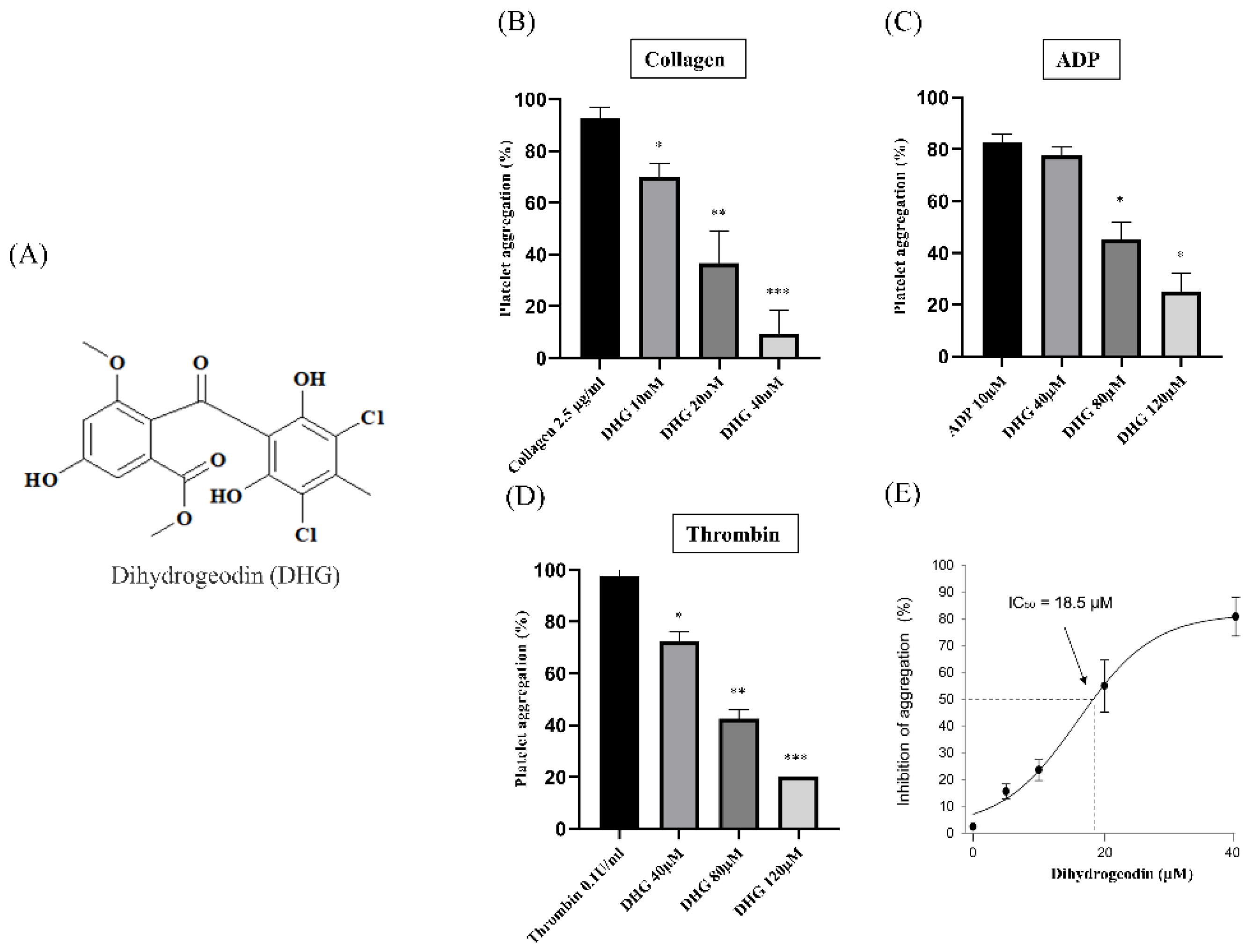
Figure 2.
DHG inhibits platelet granule release. (A) Platelets were preincubated with DHG, stimulated with collagen, and the supernatant was analyzed for ATP using a standard ATP assay kit. (B) Intracellular Ca2+ levels were assessed using Fura-2/AM-loaded platelets. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
Figure 2.
DHG inhibits platelet granule release. (A) Platelets were preincubated with DHG, stimulated with collagen, and the supernatant was analyzed for ATP using a standard ATP assay kit. (B) Intracellular Ca2+ levels were assessed using Fura-2/AM-loaded platelets. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
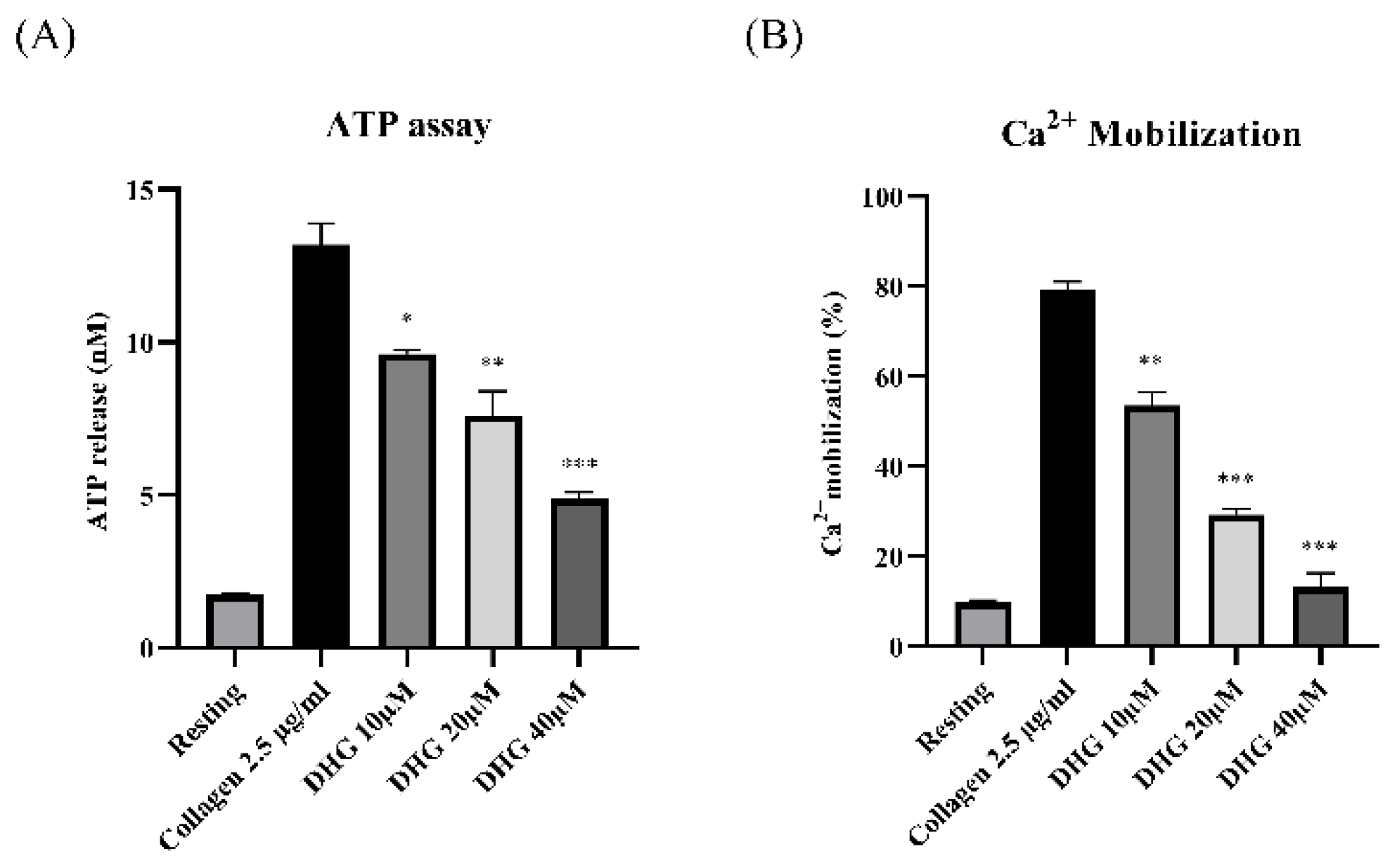
Figure 3.
DHG inhibits integrin αIIbβ3–mediated inside-out signaling pathway. (Left) Flow cytometry histograms showing fibrinogen binding in resting and collagen-stimulated platelets (2.5 μg/mL) in the presence or absence of DHG (10, 20, 40 μM) or EGTA (100 μM). (Right) Quantification of fibrinogen binding. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
Figure 3.
DHG inhibits integrin αIIbβ3–mediated inside-out signaling pathway. (Left) Flow cytometry histograms showing fibrinogen binding in resting and collagen-stimulated platelets (2.5 μg/mL) in the presence or absence of DHG (10, 20, 40 μM) or EGTA (100 μM). (Right) Quantification of fibrinogen binding. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
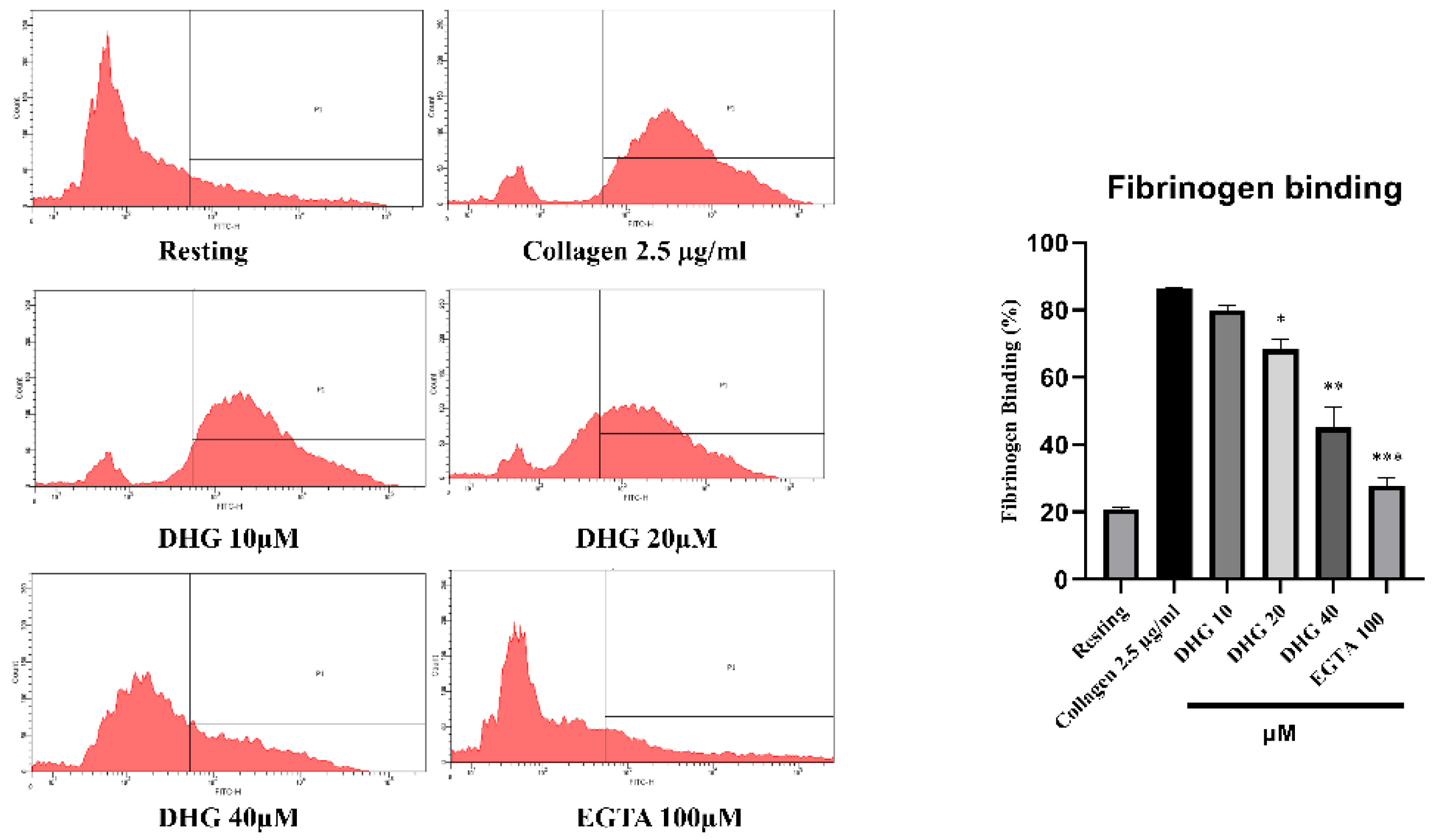
Figure 4.
DHG inhibits clot retraction. Platelet-rich plasma (PRP) was incubated for 2 min with DHG or Y-27632. Human thrombin (1 U/mL) was then added, and (A) clot retraction and clot shape change were assessed at room temperature for 90 min. (B) The clot weight was assessed *P < 0.05, **P < 0.01, ***P < 0.001 versus thrombin alone. (C) Platelet shape change was visualized with scanning electron microscopy (SEM). SEM images of the platelet shape revealed a change in morphology from discoid to a rounded shape containing filopodia upon activation with collagen, which was prevented by treatment with DHG.
Figure 4.
DHG inhibits clot retraction. Platelet-rich plasma (PRP) was incubated for 2 min with DHG or Y-27632. Human thrombin (1 U/mL) was then added, and (A) clot retraction and clot shape change were assessed at room temperature for 90 min. (B) The clot weight was assessed *P < 0.05, **P < 0.01, ***P < 0.001 versus thrombin alone. (C) Platelet shape change was visualized with scanning electron microscopy (SEM). SEM images of the platelet shape revealed a change in morphology from discoid to a rounded shape containing filopodia upon activation with collagen, which was prevented by treatment with DHG.
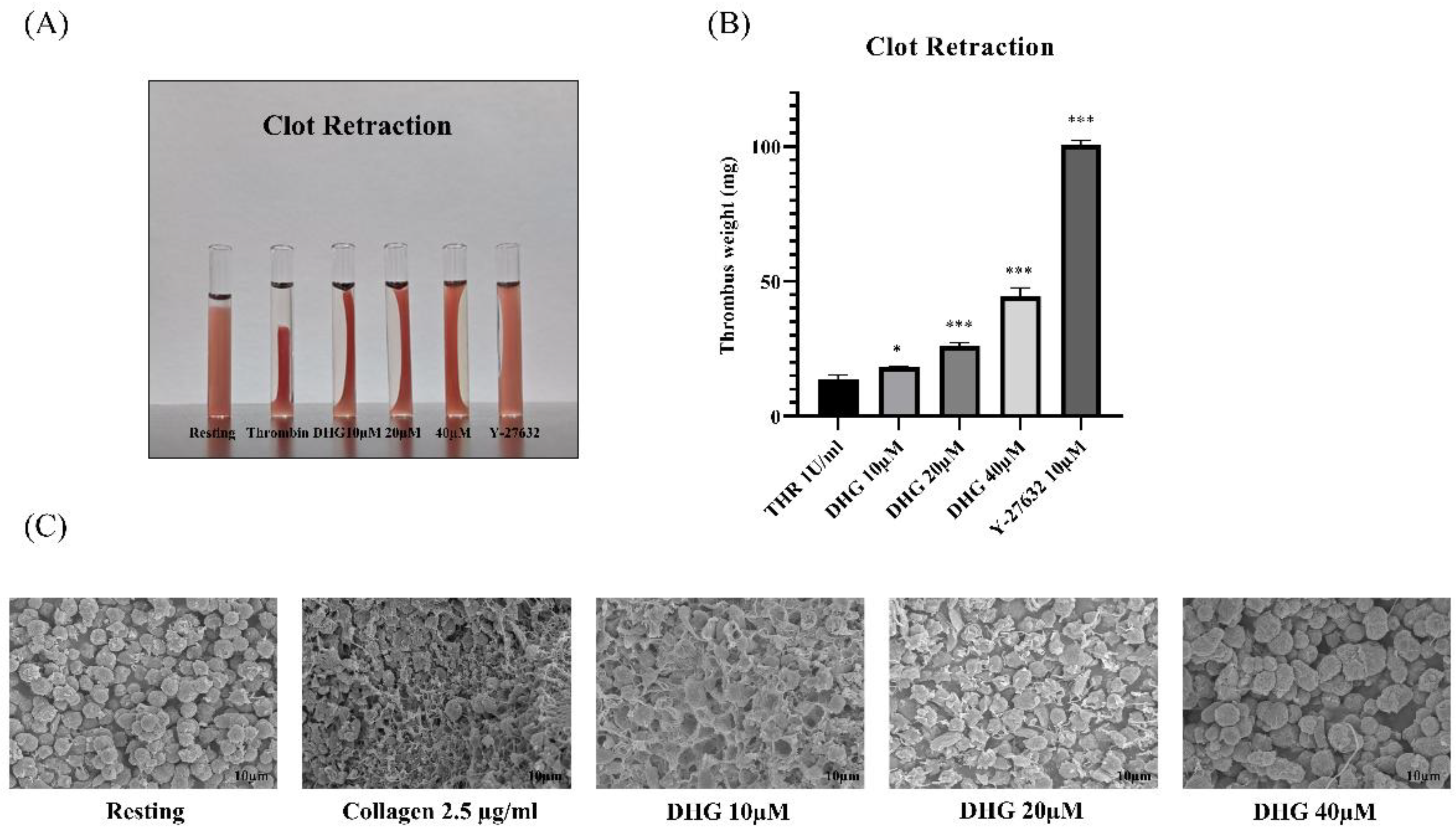
Figure 5.
DHG inhibits protein phosphorylation. Platelets were preincubated with DHG and stimulated with collagen. Aggregation was halted by the addition of lysis buffer, after which protein concentrations were measured. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and probed with specific antibodies after membrane blocking. Visualization was performed using enhanced chemiluminescence. Representative Western blots (left panels) and corresponding quantification (right panels) show the phosphorylation levels of MAPK (ERK, JNK, p38) and PI3K/Akt in platelets pretreated with DHG (10, 20, 40 μM) or a reference inhibitor. Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA followed by a post hoc Dunnett's test *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
Figure 5.
DHG inhibits protein phosphorylation. Platelets were preincubated with DHG and stimulated with collagen. Aggregation was halted by the addition of lysis buffer, after which protein concentrations were measured. Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and probed with specific antibodies after membrane blocking. Visualization was performed using enhanced chemiluminescence. Representative Western blots (left panels) and corresponding quantification (right panels) show the phosphorylation levels of MAPK (ERK, JNK, p38) and PI3K/Akt in platelets pretreated with DHG (10, 20, 40 μM) or a reference inhibitor. Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA followed by a post hoc Dunnett's test *P < 0.05, **P < 0.01, ***P < 0.001 versus collagen alone.
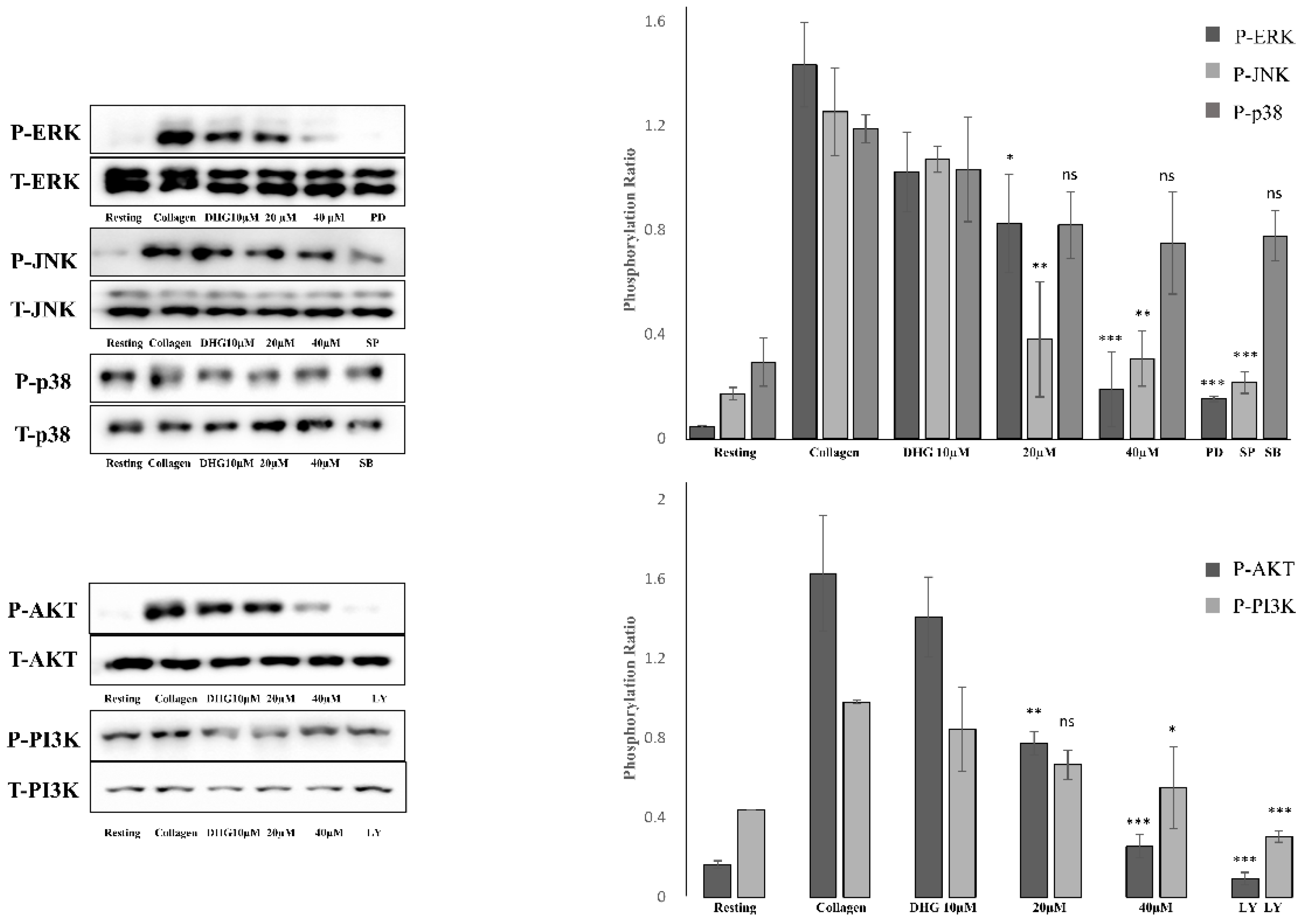
Figure 6.
SwissADME analysis of DHG. The canonical SMILES for DHG were retrieved from PubChem and entered into SwissADME. Output files and visualizations were directly from the website. (A) The BOILED-Egg model evaluated human intestinal absorption (HIA) and blood-brain barrier (BBB) permeability based on lipophilicity and polarity, generating a WLOGP versus tPSA plot. (B) SwissADME predictions showed that DHG complies with Lipinski’s rule of five, indicating good oral bioavailability.
Figure 6.
SwissADME analysis of DHG. The canonical SMILES for DHG were retrieved from PubChem and entered into SwissADME. Output files and visualizations were directly from the website. (A) The BOILED-Egg model evaluated human intestinal absorption (HIA) and blood-brain barrier (BBB) permeability based on lipophilicity and polarity, generating a WLOGP versus tPSA plot. (B) SwissADME predictions showed that DHG complies with Lipinski’s rule of five, indicating good oral bioavailability.
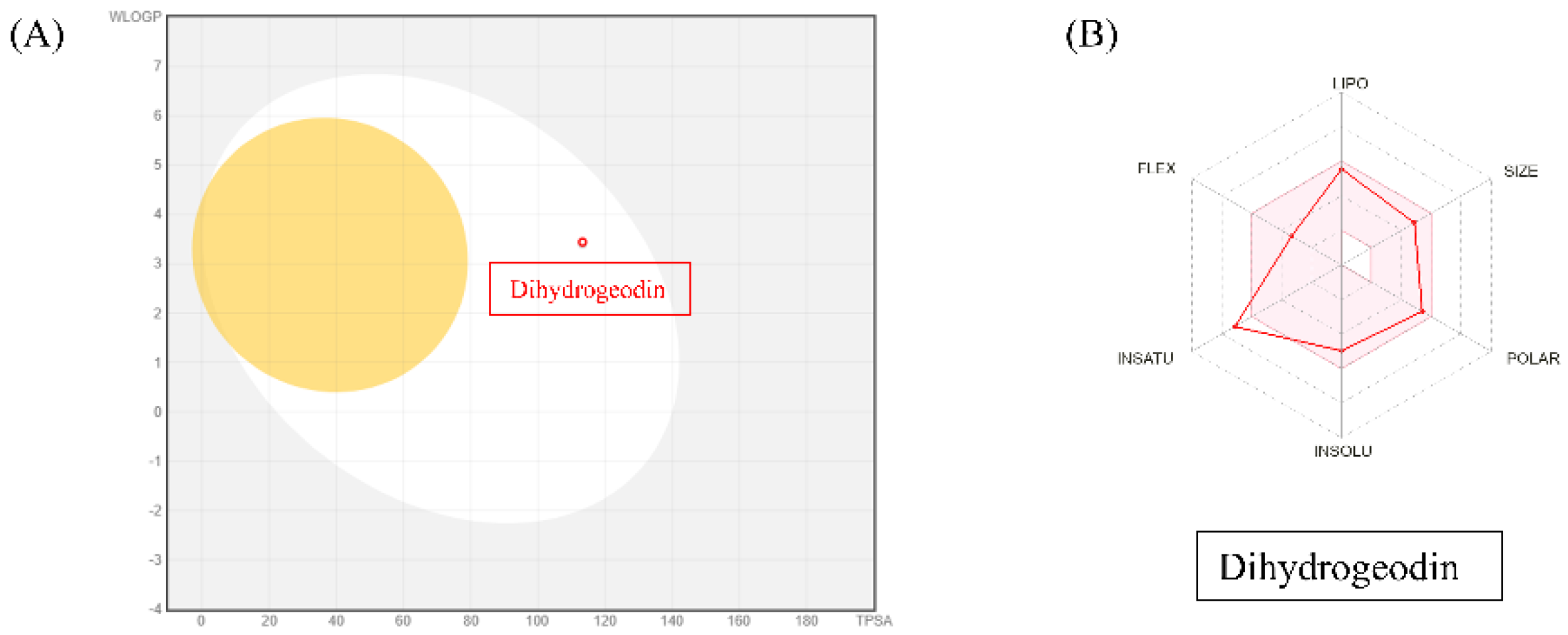
Figure 7.
Network pharmacological assessment of DHG. (A) Venn diagram showing the intersection between predicted targets of DHG and platelet-related genes, where 25 overlapping genes were identified. (B) Protein–protein interaction (PPI) network of the 25 common targets constructed using the STRING database. (C) Compound–target–pathway network illustrating the association between DHG, its core targets, and enriched KEGG pathways.
Figure 7.
Network pharmacological assessment of DHG. (A) Venn diagram showing the intersection between predicted targets of DHG and platelet-related genes, where 25 overlapping genes were identified. (B) Protein–protein interaction (PPI) network of the 25 common targets constructed using the STRING database. (C) Compound–target–pathway network illustrating the association between DHG, its core targets, and enriched KEGG pathways.
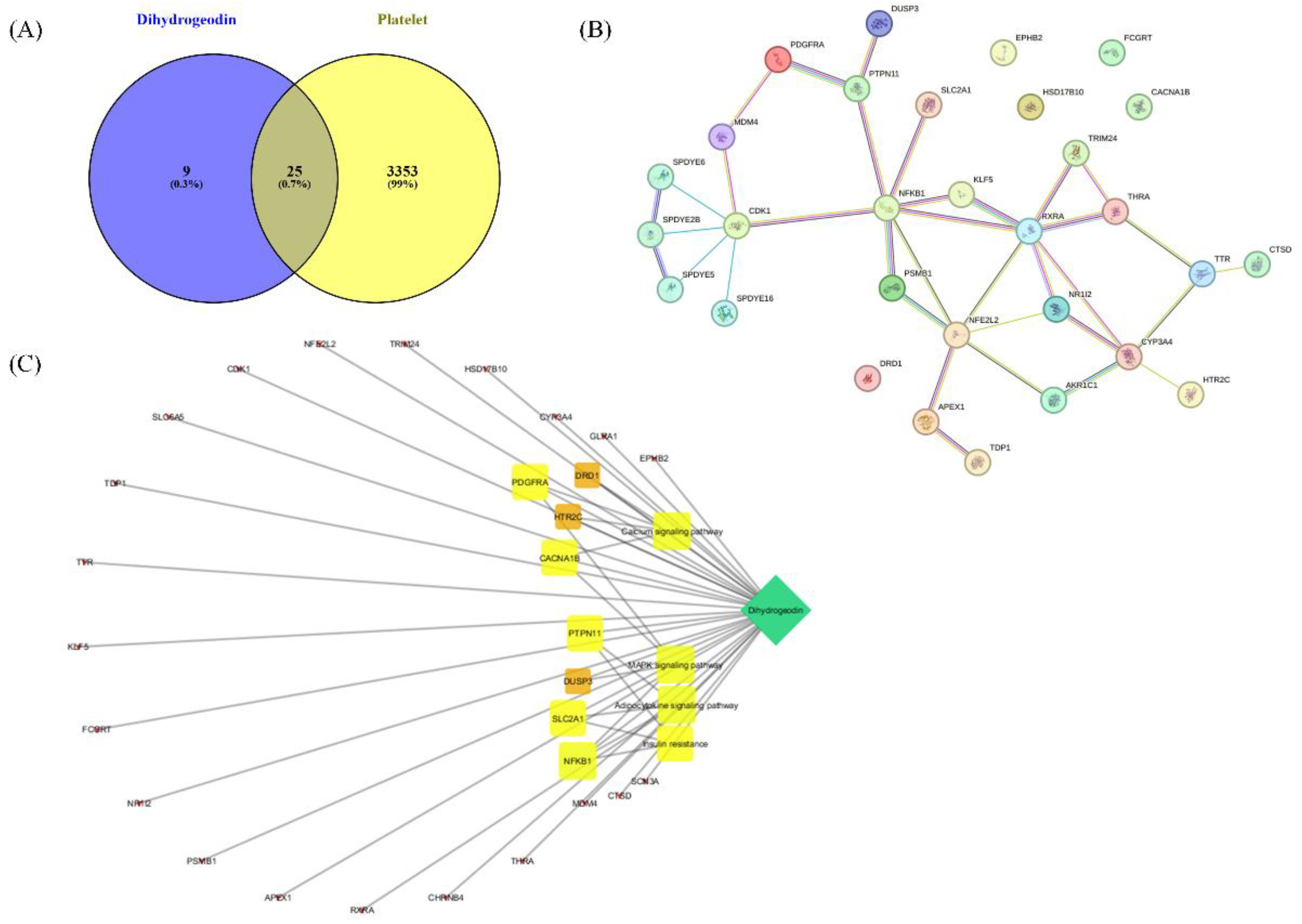
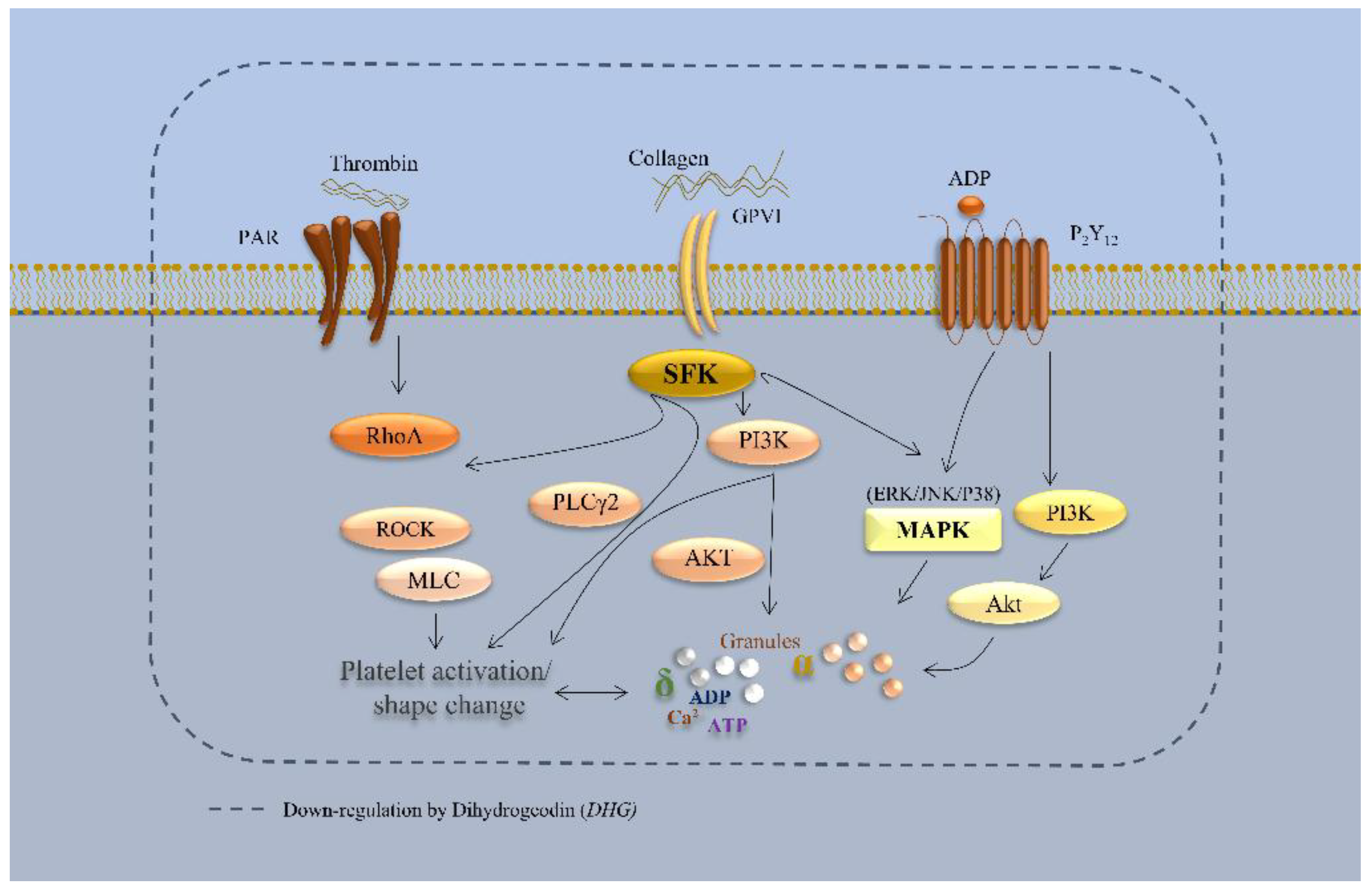
Figure 8.
Schematic representation of DHG-mediated inhibition of platelet signaling pathways. DHG inhibits platelet aggregation while reducing ATP release, intracellular Ca2+ mobilization, αIIbβ3 integrin activation, clot retraction, and activation of the MAPK and PI3K/Akt pathways.
Figure 8.
Schematic representation of DHG-mediated inhibition of platelet signaling pathways. DHG inhibits platelet aggregation while reducing ATP release, intracellular Ca2+ mobilization, αIIbβ3 integrin activation, clot retraction, and activation of the MAPK and PI3K/Akt pathways.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



