
Submitted:
16 October 2025
Posted:
17 October 2025
You are already at the latest version
Abstract
We investigate the defect-dependent electronic structure and gas-sensing potential of cubic α-CsPbI₃ using first-principles density functional theory and nonadiabatic molecular dy-namics simulations. Among the intrinsic defects, interstitials, vacancies, antisites, and switches studied, the IPb and PbI antisite defects exhibit transition energy levels near the middle of the band gap, thus functioning as deep traps. Short-term adsorption of ammo-nia selectively modifies the electronic structure, coordinating with Pb at PbI sites and Cs at IPb sites, significantly altering recombination pathways. Detailed analysis reveals that NH3 reduces anharmonicity at IPb defects, enabling enhanced recombination at elevated tem-peratures, while trap-assisted recombination dominates at room temperature. Other ana-lytes, including CH3NH2 and NO2, show negligible impact on the band gap or recombina-tion dynamics, highlighting the selectivity of NH3 interactions. Ab initio nonadiabatic mo-lecular dynamics simulations at 300 K and 600 K further demonstrate tempera-ture-dependent modulation of carrier lifetimes, with NH3 accelerating recombination at ambient conditions and suppressing certain pathways at higher temperatures. These findings suggest that α-CsPbI₃ can serve as a selective and sensitive ammonia sensor over a broad temperature range and offer insights for ammonia detection under industrially relevant conditions.
Keywords:
perovskites
; defect chemistry
; ammonia sensor
; DFT
; α-CsPbI3
1. Introduction
Given the current stability challenges [1,2,3,4,5,6] presented by the lead-based perovskite solar cells (PSC), it may seem counterintuitive to propose to use such metastable materials as gas sensors. However, it should be noted that while the stability of lead-based perovskite materials is a concern, they also demonstrate a propensity for self-healing in some unusual conditions, which appears to be reproducible at standard operating conditions if the interfaces and grains are appropriately passivated by a stabilizing species. The exact conditions required to stabilize these materials remain under active investigation. Nevertheless, just as they have shown remarkable improvements in power conversion efficiency (PCE) in photovoltaics [7], they have also demonstrated great promise as gas sensors. Previously, we investigated the exceptional properties of an FAPbCl3-based resistor-type sensor, which demonstrated both high selectivity and sensitivity towards ammonia, a combination that is known to be quite challenging to find in chemical gas sensors [8,9]. DFT calculations led to the conclusion that this is due to the unique properties of the material associated with the defect chemistry, where certain deep-trap defects can be temporarily saturated by ammonia gas, thus decreasing the electrical resistance of the material. Because of size and polarity, only the ammonia molecule could properly fit into the defect and saturate the dangling bond. We termed this effect the “lock-and-key” mechanism akin to substrate-enzyme interactions in cellular biology. According to Shockley-Read-Hall (SRH) theory [10,11], such deep traps may serve as nonradiative recombination centers; however, due to the long carrier lifetime of the material, it becomes important to account for electron-phonon coupling [12].
A key challenge in sensor design is balancing material reactivity towards analytes with the risk of degradation, which is often accelerated at the higher temperatures required by many industrial process streams. In chemical process design, it is often crucial to have sensors as close to the reaction stream as possible to minimize dead-time between the actuation and response, which, for some fast processes, indicates the need for a high-temperature sensor. This has led to the proliferation of the ABO3 perovskites for the sensing of combustion products such as COx and NOx.[13,14,15,16,17,18,19]
Black-phase CsPbI3 under certain conditions exhibits superior stability relative to its organic-inorganic hybrid perovskite (OIHP) counterparts due to the lack of an organic A-site cation. These cations contribute some instability to the material and provide additional avenues of degradation [20,21]. However, it should also be noted that under some exotic conditions, these organic cations can induce self-healing effects [22]. For CsPbI3, the photoactive black cubic (α) phase is stable in pure form above temperatures of around 573 K to 633 K [23,24], tending to degrade at lower temperatures as a result of Cs being too small to occupy the interstitial space between octahedra, which is quantifiable by the low Goldschmidt tolerance factor of 0.81 [25]. This can be ameliorated by the incorporation of larger A-site cations [25] or smaller X-site halogens [26,27], lowering the stable temperature range while creating other stability issues related to phase segregation [28,29,30]. This high-temperature stability of all-inorganic α-CsPbI3 indicates that it can be useful for gas sensing in harsh environments such as those encountered in conventional ammonia production.
In the present work, the defect-dependent electronic structure of α-CsPbI3 is characterized with and without several gas analytes. Gas analytes were selected based on industrial relevance, with a size restriction to allow for diffusion throughout the large interstitial spaces between the BX6 octahedra, created by the large A-site cation. Studied gas molecules include CO, CO2, NO, NO2, H2, CH3NH2 (as a representative volatile organic compound (VOC)), and NH3. Among all of the analyte’s ammonia exhibits a significant influence on the perovskite’s electronic structure, which may be exploited as the operating mechanism in a novel chemical sensor. We note that in the case of a reactor-based sensor, the selectivity of the sensor is likely of less relevance than sensitivity; thus, we focus on the most promising analyte: NH3.
One pair of frequently encountered surface-based defects (labeled using Kröger–Vink[31] notation) in black phase perovskites are the antisite defects IPb and PbI, [32,33] both of which may be related to the destructive Pb2+/Pb0 and I−/I3− surface-based redox chemistry [34]. Recent DFT calculations have demonstrated that within bulk, the PbI defect becomes favorable for n-type doped conditions; however, the most dominant defects in bulk are shallow [35], indicating that our proposed sensor mechanism will be surface and grain-boundary-based. This agrees with the experiment, which typically shows surface-based recombination (τ1) to be an order of magnitude faster than bulk-based recombination (τ2) [36].
In the present work, we calculated the nonradiative dynamics of the deep IPb and PbI defects, as well as other defects, in α-CsPbI3 and compared them to the pristine material as well as with their NH3-absorbed counterparts. NH3 is then selected for further exploration due to the global significance of the energy-intensive Haber-Bosch process in the production of fertilizer. This was done with the express purpose of investigating whether this material may be an effective photoluminescence (PL) –based detector of NH3. Ammonia’s interaction with the deep traps may enable carrier recombination and thus is predicted to allow α-CsPbI3 to function as an ammonia sensor.
2. Theoretical Methods
For all the DFT calculations performed, we utilized the Vienna Ab initio Simulation Package (VASP) [37]. Initial structural optimizations employed the Perdew-Burke-Ernzerhof (PBE) [38] generalized gradient approximation (GGA) functional and projector augmented-wave (PAW) pseudopotentials [39] and Grimme D3 dispersion corrections [40] were applied throughout. Both the atomic positions and lattice constants of the cubic α-CsPbI3 unit cell were relaxed; a 4×4×4 supercell was used to mitigate interaction of virtual images of the defects. This large supercell allows sufficient sampling of k-space with a single Γ point, particularly as for an even-numbered cubic perovskite cell, the band-gap folds to the Γ-point for bandgaps at the Γ, R, M, or X point. Each defect was introduced to the cell and relaxed, after which band structures were characterized by single-point (SP) calculations using the Strongly Constrained and Appropriately Normed (SCAN) [41,42] mGGA functional. The results of our calculations were analyzed with VASPKIT [43] for band-unfolding according to the methodology proposed by Ku et al [44]. The defects considered are interstitials, vacancies, antisites (mostly neglected in perovskite literature until relatively recently) and ‘switches,’ defined as pairs of mutually compensating antisites. The cubic lattice vectors were fixed for the subsequent step. Defect formation energy (DFE) as a function of the Fermi level was calculated by the equation below:
where is the DFE of defect in charge state , is the energy of the pristine crystal, and refer to the number and chemical potential of the species removed from or added to the crystal to form a defect , is the Fermi level relative to the top of the valence band, , and is the first-order Makov-Payne correction [45]:
where is the Madelung constant, is the dielectric constant, and is the characteristic length of the cell. Defects with transition energy levels (TELs) near mid-gap were selected for further study, with the lattice vectors allowed to vary.
With the deep traps identified, analytes of interest were introduced to each, and the cells were re-relaxed using PBE, and their electronic structures were re-characterized with SCAN SP calculations and unfolded with VASPKIT. The results were used to identify the analyte with the strongest interaction, which motivated further excited-state study using the NONRAD package, developed for the study of nonradiative recombination dynamics [46,47]. Because NONRAD requires identical lattices for both charge states, and each defect has a set of three consecutive charges, the middle charge state’s lattice was used. NONRAD employs a one-dimensional approximation of phonon modes, which has shown to be sufficiently accurate [48]. The generalized configuration coordinate is defined as:
where is an index running over the atoms in the supercell, is each atom’s mass, is the Cartesian coordinate associated with the mid-gap state of the atom for the transition being considered, and the same but for the state corresponding to the fully geometrically-relaxed VBM state or electronically-excited geometrically-relaxed CBM state (with h+ and e- carriers). The energy of these states along the paths is shifted relative to the energy of its associated minimum, and also by the carrier energies, i.e., the band gap magnitude for the CBM + h+ + e- state, the associated TEL for the mid-gap states, and zero in the case of the VBM state.
To elucidate the effect of the NH3 addition to α-CsPbI3, we performed ab initio molecular dynamics (AIMD) calculations. Equilibration was performed for 2000 fs in the canonical ensemble (NVT). Velocity rescaling was applied to maintain the target temperature during equilibration. We chose two temperature regimes: 300 K and 600 K. The latter is stable, while the former may be stabilized through novel passivation strategies [49]. Production runs were then performed for 10 ps each in the microcanonical ensemble (NVE), allowing the temperature to vary throughout the trajectory. A 1 fs time step was utilized for all AIMD runs, and in this case, we used 3 x 3 x 3 superslabs to ensure that the calculations were tractable to perform.
To run the nonadiabatic molecular dynamics (NAMD) analysis, we cut the last 2 ps of the run and prepare them as snapshots. These snapshots were calculated using SCF convergence with a tightened criterion of 1x10-6 eV at the R-point, which is where the direct band gap is located for an odd-numbered α-CsPbI3 supercell. To enable quick evaluation of the nonadiabatic coupling (NAC) elements, we used the Concentric Approximation Nonadiabatic Coupling (CA-NAC) code, which was specifically written to be compatible with the PAW formalism in VASP [50]. The NACs, eigenvalues, and dephasing time generated by CA-NAC were then used to perform semiclassical decoherence-induced surface hopping (DiSH) simulations [51,52] using the parallelized Hefei-NAMD program [53,54]. The NAMD simulations were performed with 1000 independent trajectories and 100 stochastic samples. Each trajectory was allowed to evolve for 10 ns with a timestep of 1.0 fs. In all cases, an extra 10 bands were included above and below the band edges for the DiSH simulations. This approach was used to determine the nonradiative recombination lifetimes (nr) of charge carriers for the pristine, NH3·Pristine, IPb , NH3·IPb , PbI and NH3·PbI systems. For the pristine and NH3·Pristine systems, nr was extracted by fitting the decaying CBM population from the NAMD simulation to an exponential decay function:
For the other four cases, there were defects inside of the bandgap, and thus three-term (in case of NH3·PbI and NH3·IPb) and four-term (in case of PbI and IPb) differential equation models were required to fit them. In the case of the 4-State Ladder Model (CBM → D1 → D2 → VBM) the following equations were solved:
In the three-term case, where , , and represent the carrier populations in the respective states, whereas the k1, k2, and k3 constants are the rate constants corresponding to CBM-to-D1 trapping, D1 → D2 relaxation, and final D2 → VBM recombination, respectively. In the case of the 5-State Ladder Model (CBM → D1 → D2 → D3 →VBM), the equations were the same as the equations above, but with equation (5) replaced with the following two equations:
For simplicity, the three constants (k1-k3) of the former model and the four constants (k1-k4) of the latter model represent sequential trapping and recombination rates along the ladder pathway. Since the intermediate ladder steps relax much faster than the final transition, the effective nonradiative recombination lifetime was determined by the slowest step of the ladder. Optical properties were calculated using a summation over states approach [55]; see our previous works for theoretical details [56,57,58,59].
3. Results and Discussions
3.1. Defect Characterization and Band Structure Analysis
Of the defects surveyed (interstitials, vacancies, antisites, and switches, or pairs of superimposed mutually compensating antisites where adjacent atoms swap sites), only PbI and IPb exhibited TELs within the band gap, (+3/+2) and (+2/+1) for PbI and (0/-1) and (-1/-2) for IPb. Both defects also have lower energy two-carrier charge transitions [(+1/-1) for IPb and (+3/+1) for PbI], but the low probability of double carrier capture kinetically limits these transitions. Figure 1 shows the geometries of the middle charges for each defect case. For PbI+2, the I forming the corners of the adjacent PbI6 octahedra both rotate to bond with the extra Pb, whereas for IPb–1 the smaller (in the sense of ionic radius) antisite I has relatively minimal impact on the local geometry, which is not surprising considering iodine’s ability to form chains [57].
Figure 2 shows the unfolded band diagrams [44] of pristine α-CsPbI3 (Figure 2a), as well as the antisite PbI+2 (Figure 2b) and IPb–1 (Figure 2c) defects. The pristine band structure matches that of the expected unit cell, with a direct band gap at the R-symmetric point (see Ku et al [44] for the convoluting effect increasing supercell size has on the resulting band structure; the direct band gap should, in theory, allow for both easier carrier excitation/combination through the lack of need for kinetically hindering phonon absorption/emission in order to obey conservation of momentum. This gap is lower than the one which is experimentally observed, as expected for a mGGA functional. The disorder introduced by the defects is readily apparent in the band diagrams of the two defects in the splitting of energy levels as compared to the discrete levels in the pristine case, particularly for PbI+2 for which the higher disorder results in a more continuous valence band. Both defects have energy levels which oscillate along the M-Γ-R path, corresponding to high dispersion, but which are flat along the Γ-X path. Flatter bands indicate higher effective electron masses and reduced kinetic energy,[58] making it harder for electrons in these states to escape.
3.2. Gas Absorption Effects
Next, analytes were introduced to study the effects on the band structure in search of sensing opportunities. It was previously unknown why α-CsPbI3 has such low recombination rates, even though it has thermodynamically viable deep trap states. This behavior likely arises from the soft, anharmonic nature of the perovskite lattice: carriers persist for nanoseconds if allowed sufficient time to absorb multiple phonons [59]. This suggests that significant geometric contortions and energy barriers must be overcome to complete one or more of the charge transitions.[60] To search for analytes which could alter this delicate dynamic, band structures were examined for strong trapping potential. NH3 showed the greatest enhancement of recombination rates, indicated by the mid-gap band levels. In contrast to the gas-less cases of Figure 2b and 2c, Figure 3 shows the relaxed geometries of the two deep traps with NH3, along with the associated band structures.
First, by comparing Figure 3 with Figure 2, one can see that NH3 has little effect on the band gap of defective CsPbI3. This is likely due to the coordination of NH3 with Cs, which contributes little to the structure of the valence and conduction bands as seen by its contribution (red) in Figure 4a. As is typical for perovskites, the X-site antibonding halogen orbitals (with a small amount of Pb(s)) dominate the valence band while the B-site lead metal dominates the conduction band [56] Figure 4b compares the total density of states (DOS) of pristine CsPbI3 with that of IPb with and without NH3; when comparing pristine CsPbI3 with IPb the introduction of the trap state below the conduction band can be seen, its notable that the addition of NH3 increases the DOS near the valence band maximum (VBM) and decreases it near the conduction band minimum (CBM).
In contrast to NH3, most other analytes (Figures S1-S4 for PbI and Figures S5-S9 for IPb) reduced trapping potential or had no effect on the band gap of pristine and defective CsPbI3. For example, see Figure 5a, where NO2·PbI results in flat energy levels within the bands near the edges. While CH3NH2·PbI’s band diagram looks very similar to that of NH3·PbI (as shown in Figure 5b) for the case of CH3NH2·IPb, in Figure 5c, it resembles NO2·PbI with its absence of band gap TEL trap states. This indicates that, despite the chemically similar R–NH2 functional group, CH3NH2 is unlikely to increase nonradiative recombination rate as NH3 does, a positive indication for the selectivity of a sensor with this operating mechanism. According to SRH theory, it is likely that the perovskite + gas system would continue to have conductivity similar to its pristine counterpart when exposed to these gases.
With NH3 identified as a potential enabler of the recombination centers PbI and IPb, the energetics along the charge transition path must be characterized. The resulting paths are shown in Figure 6. Note that these were calculated with PBE rather than SCAN, and thus the magnitude of the energy shifts corresponds to the PBE calculations (i.e., a band gap shift of 1.48 eV and respective shifts for each TEL based on the point of intersection for the charge states as calculated by eq. 1 using the PBE values). Most significantly, we note the lower difference in ∆Q between charge states than previously reported for the case of (gasless) δ-CsPbI3.[60] This is likely due to the NH3 absorbing some of the donated charge and mitigating consequent geometry shifts. For the anionic defects (Figure 6b and 6d), the IPb ( – 1/ – 2) transition is almost barrier-less for carrier capture from CBM to mid-gap state, but a sharper energy increase is observed for both charge states in the −Q direction, resulting in a |∆Q| > 20 amu½ Å shift required for hole capture. Notably, for the IPb (0/ – 1) transition ∆Q between the geometries associated with each state is very small, but due to very similar relatively large contortions for both e− and h+ capture are still required, however, at much lower energy barriers. In contrast, in the case of the cationic defects, the results from the calculations along the PbI (+3/+2) path again show easy e− capture followed by more difficult h+ capture, although for this case it appears only a mild ∆Q and ∆E are required, attributable to the lower for PbI+2 relative to PbI+3.
For PbI(+2/+1), there are large barriers present. The origin of this anharmonic scaling of E vs. Q is a consequence of the significant change in the coordination environment of the antisite Pb from +1 to +2/+3; at +1 the antisite Pb remains in the displaced I’s position, while at +2/+3 the extra h+ on the Pb induces stronger bonding with nearby I, pulling away from the location the displaced I would occupy in the pristine lattice. This was previously described in Zhang et al.’s work, meaning this defect’s behavior matches their work qualitatively, although the geometry shift is mitigated with the presence of the NH3.[60] Returning to IPb(0/ – 1/ – 2), the NH3 almost completely heals the anharmonicity; the reason can be seen in Figure 3b. The NH3 displaces one of the corner I’s (foreground) in what would make up part of a PbI6 octahedron in the pristine lattice and coordinates with the Cs via the lone electron pair on the N. Additional e− are apparently delocalized, which minimizes their effect on the geometry around the defect. This makes the local energy environment of the potential energy surface (PES) behave more like a harmonic oscillator, which is particularly visible in the case of the PES for NH3·IPb(0/ – 1) (Figure 6d), where the two curves resemble parabolas even far from their minima. This near-constant curvature results in smaller perturbations needed to overcome the finite geometric and energy barriers between the states. Although NH3·IPb(–1/–2) does retain some degree of anharmonicity, it is greatly reduced as compared to the steeply accelerating curvature in the −Q direction of e.g. PbI(+2/+1), which strengthens the case for NH3 enabling IPb to function as a recombination center, especially at the higher temperatures relevant to sensor operation.
3.3. Recombination Times via Nonadiabatic Molecular Dynamics
The results of our NAMD calculations are presented in Table 1 (300 K) and 2 (600 K). By comparing these tables, we see that at 300 K, NH₃ shortens the intrinsic recombination time relative to pristine (492 vs. 797 ns), while at 600 K the trend inverts, with NH₃:Pristine exhibiting a slightly longer lifetime than pristine (557 vs. 352 ns). A similar inversion of temperature dependence is observed in antisite systems: for IPb and PbI recombination slows at higher temperature (23 → 28 ns and 1 → 0 ns, respectively), whereas in their NH₃-complexed counterparts recombination instead accelerates (30 → 22 ns for NH₃:IPb and 0 → 1 ns for NH₃:PbI). This consistent inversion suggests that NH₃ perturbs the recombination landscape differently from bare defects, suppressing certain scattering pathways at elevated temperature while leaving trap-assisted channels dominant at room temperature.

Table 1.
Calculated band gap (Eg), root-mean-square nonadiabatic coupling (NAC) magnitude, pure dephasing time, and recombination time (τNR) for the pristine, NH3·Pristine, IPb , NH3·IPb , PbI, and NH3·PbI, systems. Simulations at 300 K.
Table 1.
Calculated band gap (Eg), root-mean-square nonadiabatic coupling (NAC) magnitude, pure dephasing time, and recombination time (τNR) for the pristine, NH3·Pristine, IPb , NH3·IPb , PbI, and NH3·PbI, systems. Simulations at 300 K.
| System | Transition | Eg (eV) | NACs (meV) | Dephasing Time (ps) | Τ (ns) |
| Pristine | VBM-CBM | 1.68±0.04 | 0.77 | 16.49 | 797 |
| NH3·Pristine | VBM-CBM | 1.64±0.06 | 0.52 | 11.08 | 492 |
| IPb | VBM-D3 | 1.74±0.04 | 21.27 | 24.29 | 23 |
| D2-D3 | 0.49 | 2.65 | |||
| D2-D1 | 63.62 | 2.66 | |||
| D1-CBM | 42.81 | 42.66 | |||
| NH3·IPb | VBM-D2 | 1.70±0.04 | 30.60 | 27.71 | 30 |
| D2-D1 | 0.28 | 3.12 | |||
| D1-CBM | 82.06 | 3.17 | |||
| PbI | VBM-D3 | 2.23±0.08 | 1.43 | 5.57 | 1 |
| D2-D3 | 2.40 | 6.98 | |||
| D2-D1 | 40.24 | 17.43 | |||
| D1-CBM | 44.38 | 81.69 | |||
| NH3·PbI | VBM-D2 | 2.24±0.09 | 4.09 | 5.29 | 0 |
| D2-D1 | 4.29 | 6.55 | |||
| D1-CBM | 57.00 | 54.11 |
Table 2.
Calculated band gap (Eg), root-mean-square nonadiabatic coupling (NAC) magnitude, pure dephasing time, and recombination time (τNR) for the pristine, NH3·Pristine, IPb, NH3·IPb, PbI, and NH3·PbI, systems. Simulations at 600 K.
Table 2.
Calculated band gap (Eg), root-mean-square nonadiabatic coupling (NAC) magnitude, pure dephasing time, and recombination time (τNR) for the pristine, NH3·Pristine, IPb, NH3·IPb, PbI, and NH3·PbI, systems. Simulations at 600 K.
| System | Transition | Eg (eV) | NACs (meV) | Dephasing Time (ps) | Τ (ns) |
| Pristine | VBM-CBM | 1.68±0.04 | 0.77 | 16.49 | 797 |
| NH3·Pristine | VBM-CBM | 1.64±0.06 | 0.52 | 11.08 | 492 |
| IPb | VBM-D3 | 1.74±0.04 | 21.27 | 24.29 | 23 |
| D2-D3 | 0.49 | 2.65 | |||
| D2-D1 | 63.62 | 2.66 | |||
| D1-CBM | 42.81 | 42.66 | |||
| NH3·IPb | VBM-D2 | 1.70±0.04 | 30.60 | 27.71 | 30 |
| D2-D1 | 0.28 | 3.12 | |||
| D1-CBM | 82.06 | 3.17 | |||
| PbI | VBM-D3 | 2.23±0.08 | 1.43 | 5.57 | 1 |
| D2-D3 | 2.40 | 6.98 | |||
| D2-D1 | 40.24 | 17.43 | |||
| D1-CBM | 44.38 | 81.69 | |||
| NH3·PbI | VBM-D2 | 2.24±0.09 | 4.09 | 5.29 | 0 |
| D2-D1 | 4.29 | 6.55 | |||
| D1-CBM | 57.00 | 54.11 |
3.4. Ammonia-Driven Phase Change and Photoluminescent Detection Pathways
For the gas-less defects, a large supercell reveals that the cubic symmetry is preserved beyond the immediately adjacent unit cells (Figure 7a). While Figure 1a highlights the locally broken symmetry, Figure 7a demonstrates that the distortions do not propagate. In contrast, NH3 interacts strongly with the Pb atom at the center of the PbI6 polyhedron, inducing rotations throughout the 4x4x4 supercell and driving a α → δ phase change.
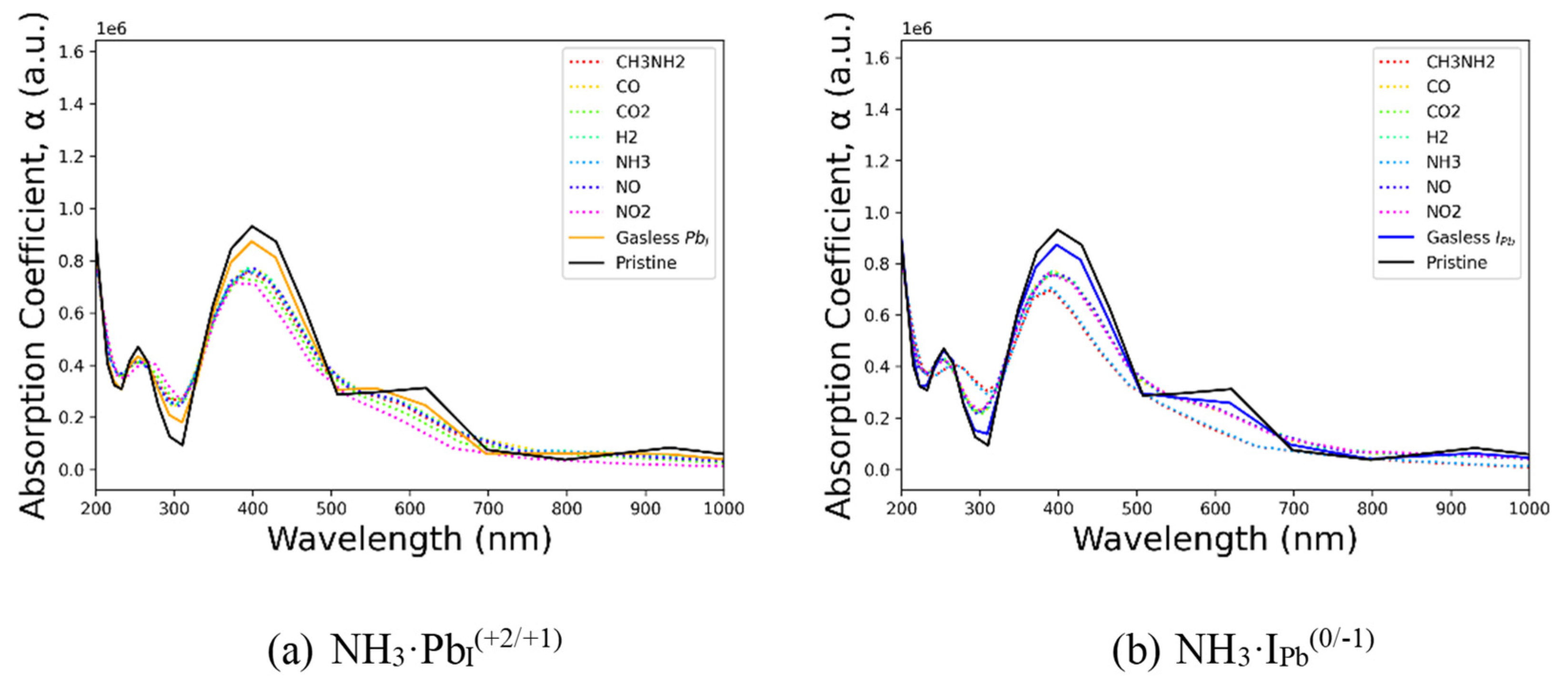
To evaluate photoluminescent sensing potential, optoelectronic calculations were also conducted and are shown in Figure 8, implying again the stronger interaction of NH3 with IPb. The presence of either defect significantly lowers the absorption coefficient, as seen by comparing the solid lines in each subfigure. In Figure 8a, the cationic defect interacts most strongly with CO2 and NO2 and least with CO, NO, and H2. R–NH2 compounds fall between these extremes. In Figure 8b, the effect is more binary: analytes without an amine group follow a trend of moderate absorption reduction, while CH3NH2 and NH3 show increased absorption. This suggests that a light-absorption-based sensor could effectively distinguish amines from non-amines but may not be suitable for differentiating NH3 from other small amine-containing compounds. A tandem chemo-optical resistor sensor could be particularly effective, combining spectral differences with the increased resistivity caused by gas interactions with deep carrier traps.
4. Conclusions
The point defect physics and electronic structure of CsPbI3 were analyzed, confirming previous reports that the IPb and PbI antisite defects are the only potential nonradiative recombination centers. Additional calculations incorporating gas analytes revealed that NH3 significantly influences the trap state energy levels by coordinating with the metal cations (Pb for PbI, Cs for IPb). Since prior studies suggest that anharmonicity can hinder recombination, we examined the energetics of the recombination processes, revealing that in the case of IPb the presence of NH3 on the site reduces the anharmonicity and may allow it to function as a recombination center at higher temperatures. This was explored using nonadiabatic molecular dynamics, revealing potential for ammonia sensing at conditions relevant to industrially relevant processes. Phase change inducement was noted as an alternative sensing mechanism, and optoelectronic calculations were conducted, indicating that light absorption may be sensitive to amines. Taken together, this work contributes a novel band-driven approach to studying gas-solid interactions while also demonstrating an NH3 sensor ripe for industrial applications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1-S4 include the unfolded band structures involving the addition of CO, CO2, H2, and NO2 analytes to the CsPbI3 supercell with a PbI defect. Figures S4-S8 show the unfolded band structure with the addition of CO, CO2, H2, NO, and NO2 to the CsPbI3 supercell with an IPb defect.
Author Contributions
Conceptualization, S.N. and L.G.; methodology, S. N., L. G., O.V.P.; validation, S. N., L. G., Y.D., B. R. R.; formal analysis, S. N., L. G., Y.D., B. R. R.; writing—original draft preparation, S. N., L. G., Y.D., B. R. R., S. W.; writing—review and editing, S. N., L. G., Y.D., B. R. R., S. W.; supervision, Y.D., B. R. R., S. W.; funding acquisition, Y.D., B. R. R., S. W. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
Data will be made available on request.
Acknowledgements
S.N. thanks the Louisiana Board of Regents for supporting his research through the LBR Graduate Fellowship. O.V.P. acknowledges support from the Department of Energy, grant DE-SC0014429. Y.D. was supported in part by the National Energy Technology Laboratory (NETL) Research and Innovation Center’s Sensors, Controls & Other Novel Concepts Program (MYRP #1025037). B. R. R. was supported by the US National Science Foundation under grant number OIA-1946231 and the Louisiana Board of Regents for the Louisiana Materials Design Alliance (LAMDA). S. W. acknowledges support from the National Science Foundation (OIA 2418390). All authors thank the Louisiana Optical Network Infrastructure (LONI) for the computational infrastructure used to complete this project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ni, Z.; Jiao, H.; Fei, C.; Gu, H.; Xu, S.; Yu, Z.; Yang, G.; Deng, Y.; Jiang, Q.; Liu, Y.; Yan, Y.; Huang, J. Evolution of Defects during the Degradation of Metal Halide Perovskite Solar Cells under Reverse Bias and Illumination. Nat Energy 2022, 7 (1), 65–73. [CrossRef]
- Zhu, H.; Teale, S.; Lintangpradipto, M. N.; Mahesh, S.; Chen, B.; McGehee, M. D.; Sargent, E. H.; Bakr, O. M. Long-Term Operating Stability in Perovskite Photovoltaics. Nat Rev Mater 2023, 8 (9), 569–586. [CrossRef]
- Khan, M. R.; Schwenzer, J. A.; Lehr, J.; Paetzold, U. W.; Lemmer, U. Emergence of Deep Traps in Long-Term Thermally Stressed CH3NH3PbI3 Perovskite Revealed by Thermally Stimulated Currents. Journal of Physical Chemistry Letters 2022, 13 (2), 552–558. [CrossRef]
- Bera, S.; Saha, A.; Mondal, S.; Biswas, A.; Mallick, S.; Chatterjee, R.; Roy, S. Review of Defect Engineering in Perovskites for Photovoltaic Application. Mater. Adv. 2022, 3 (13), 5234–5247. [CrossRef]
- Zhuang, J.; Wang, J.; Yan, F. Review on Chemical Stability of Lead Halide Perovskite Solar Cells. Nano-Micro Lett. 2023, 15 (1), 84. [CrossRef]
- Wang, R.; Mujahid, M.; Duan, Y.; Wang, Z.-K.; Xue, J.; Yang, Y. A Review of Perovskites Solar Cell Stability. Advanced Functional Materials 2019, 29 (47), 1808843. [CrossRef]
- Best Research-Cell Efficiency Chart | Photovoltaic Research | NREL. https://www.nrel.gov/pv/cell-efficiency.html (accessed 2023-11-13).
- Parfenov, A. A.; Yamilova, O. R.; Gutsev, L. G.; Sagdullina, D. K.; Novikov, A. V.; Ramachandran, B. R.; Stevenson, K. J.; Aldoshin, S. M.; Troshin, P. A. Highly Sensitive and Selective Ammonia Gas Sensor Based on FAPbCl3 Lead Halide Perovskites. Journal of Materials Chemistry C 2021, 9 (7), 2561–2568. [CrossRef]
- Nations, S.; Gutsev, L.; Bala Ramachandran, ab; Aldoshin, S.; Duan, Y.; Wang, S. First-Principles Study of the Defect-Activity and Optical Properties of FAPbCl3. Materials Advances 2022, 2022, 3897. [CrossRef]
- Shockley, W.; Read, W. T. Statistics of the Recombinations of Holes and Electrons. Physical Review 1952, 87 (5), 835–842. [CrossRef]
- Alkauskas, A.; Dreyer, C. E.; Lyons, J. L.; Van de Walle, C. G. Role of Excited States in Shockley-Read-Hall Recombination in Wide-Band-Gap Semiconductors. Phys. Rev. B 2016, 93 (20), 201304. [CrossRef]
- Yamada, Y.; Kanemitsu, Y. Electron-Phonon Interactions in Halide Perovskites. NPG Asia Mater 2022, 14 (1), 1–15. [CrossRef]
- Wu, Y. N.; Saidi, W. A.; Wuenschell, J. K.; Tadano, T.; Ohodnicki, P.; Chorpening, B.; Duan, Y. Anharmonicity Explains Temperature Renormalization Effects of the Band Gap in SrTiO3. Journal of Physical Chemistry Letters 2020, 11 (7), 2518–2523. [CrossRef]
- Duan, Y.; Ohodnicki, P.; Chorpening, B.; Hackett, G. Electronic Structural, Optical and Phonon Lattice Dynamical Properties of Pure- and La-Doped SrTiO3: An Ab Initio Thermodynamics Study. Journal of Solid State Chemistry 2017, 256 (August), 239–251. [CrossRef]
- Jia, T.; Zeng, Z.; Lin, H. Q.; Duan, Y.; Ohodnicki, P. First-Principles Study on the Electronic, Optical and Thermodynamic Properties of ABO3 (A = La,Sr, B = Fe,Co) Perovskites. RSC Advances 2017, 7 (62), 38798–38804. [CrossRef]
- Qi, H.; Lee, Y. L.; Yang, T.; Li, W.; Li, W.; Ma, L.; Hu, S.; Duan, Y.; Hackett, G. A.; Liu, X. Positive Effects of H2O on the Hydrogen Oxidation Reaction on Sr2Fe1.5Mo0.5O6-δ-Based Perovskite Anodes for Solid Oxide Fuel Cells. ACS Catalysis 2020, 10 (10), 5567–5578. [CrossRef]
- Jia, T.; Zeng, Z.; Zhang, X.; Ohodnicki, P.; Chorpening, B.; Hackett, G.; Lekse, J.; Duan, Y. The Influence of Oxygen Vacancy on the Electronic and Optical Properties of ABO3-: δ (A = La, Sr, B = Fe, Co) Perovskites. Physical Chemistry Chemical Physics 2019, 21 (36), 20454–20462. [CrossRef]
- Jia, T.; Ohodnicki, P.; Chorpening, B.; Lekse, J.; Hackett, G.; Duan, Y. Theoretical Study of the Optical and Thermodynamic Properties of La: XSr1- xCo1- yFeyO3- δ (x / y = 0.25, 0.5, 0.75) Perovskites. Physical Chemistry Chemical Physics 2019, 21 (47), 26117–26122. [CrossRef]
- Fan, Z.; Sun, K.; Wang, J. Perovskites for Photovoltaics: A Combined Review of Organic-Inorganic Halide Perovskites and Ferroelectric Oxide Perovskites. Journal of Materials Chemistry A 2015, 3 (37), 18809–18828. [CrossRef]
- Ouedraogo, N. A. N.; Chen, Y.; Xiao, Y. Y.; Meng, Q.; Han, C. B.; Yan, H.; Zhang, Y. Stability of All-Inorganic Perovskite Solar Cells. Nano Energy 2020, 67, 104249. [CrossRef]
- Wang, B.; Novendra, N.; Navrotsky, A. Energetics, Structures, and Phase Transitions of Cubic and Orthorhombic Cesium Lead Iodide (CsPbI3) Polymorphs. Journal of the American Chemical Society 2019, 141 (37), 14501–14504. [CrossRef]
- Boldyreva, A. G.; Frolova, L. A.; Zhidkov, I. S.; Gutsev, L. G.; Kurmaev, E. Z.; Ramachandran, B. R.; Petrov, V. G.; Stevenson, K. J.; Aldoshin, S. M.; Troshin, P. A. Unravelling the Material Composition Effects on the Gamma Ray Stability of Lead Halide Perovskite Solar Cells: MAPbI3 Breaks the Records. Journal of Physical Chemistry Letters 2020, 11 (7), 2630–2636. [CrossRef]
- Wang, Q.; Zheng, X.; Deng, Y.; Zhao, J.; Chen, Z.; Huang, J. Stabilizing the α-Phase of CsPbI3 Perovskite by Sulfobetaine Zwitterions in One-Step Spin-Coating Films. Joule 2017, 1 (2), 371–382. [CrossRef]
- Masi, S.; Gualdrón-Reyes, A. F.; Mora-Seró, I. Stabilization of Black Perovskite Phase in FAPbI3and CsPbI3. ACS Energy Letters 2020, 5 (6), 1974–1985. [CrossRef]
- Jena, A. K.; Kulkarni, A.; Sanehira, Y.; Ikegami, M.; Miyasaka, T. Stabilization of α-CsPbI3 in Ambient Room Temperature Conditions by Incorporating Eu into CsPbI3. Chemistry of Materials 2018, 30 (19), 6668–6674. [CrossRef]
- Dastidar, S.; Egger, D. A.; Tan, L. Z.; Cromer, S. B.; Dillon, A. D.; Liu, S.; Kronik, L.; Rappe, A. M.; Fafarman, A. T. High Chloride Doping Levels Stabilize the Perovskite Phase of Cesium Lead Iodide. Nano Lett. 2016, 16 (6), 3563–3570. [CrossRef]
- Sutton, R. J.; Eperon, G. E.; Miranda, L.; Parrott, E. S.; Kamino, B. A.; Patel, J. B.; Hörantner, M. T.; Johnston, M. B.; Haghighirad, A. A.; Moore, D. T.; Snaith, H. J. Bandgap-Tunable Cesium Lead Halide Perovskites with High Thermal Stability for Efficient Solar Cells. Advanced Energy Materials 2016, 6 (8), 1502458. [CrossRef]
- Liu, L.; Lu, J.; Wang, H.; Cui, Z.; Giorgi, G.; Bai, Y.; Chen, Q. A-Site Phase Segregation in Mixed Cation Perovskite. Materials Reports: Energy 2021, 1 (4), 100064. [CrossRef]
- DuBose, J. T.; Kamat, P. V. Hole Trapping in Halide Perovskites Induces Phase Segregation. Acc. Mater. Res. 2022, 3 (7), 761–771. [CrossRef]
- Motti, S. G.; Patel, J. B.; Oliver, R. D. J.; Snaith, H. J.; Johnston, M. B.; Herz, L. M. Phase Segregation in Mixed-Halide Perovskites Affects Charge-Carrier Dynamics While Preserving Mobility. Nat Commun 2021, 12 (1), 6955. [CrossRef]
- Kröger, F. A.; Vink, H. J. Relations between the Concentrations of Imperfections in Crystalline Solids. Solid State Physics - Advances in Research and Applications 1956, 3 (C), 307–435. [CrossRef]
- Chen, B.; Rudd, P. N.; Yang, S.; Yuan, Y.; Huang, J. Imperfections and Their Passivation in Halide Perovskite Solar Cells. Chem. Soc. Rev. 2019, 48 (14), 3842–3867. [CrossRef]
- Wang, R.; Xue, J.; Wang, K.-L.; Wang, Z.-K.; Luo, Y.; Fenning, D.; Xu, G.; Nuryyeva, S.; Huang, T.; Zhao, Y.; Yang, J. L.; Zhu, J.; Wang, M.; Tan, S.; Yavuz, I.; Houk, K. N.; Yang, Y. Constructive Molecular Configurations for Surface-Defect Passivation of Perovskite Photovoltaics. Science 2019, 366 (6472), 1509–1513. [CrossRef]
- Frolova, L. A.; Luchkin, S. Y.; Lekina, Y.; Gutsev, L. G.; Tsarev, S. A.; Zhidkov, I. S.; Kurmaev, E. Z.; Shen, Z. X.; Stevenson, K. J.; Aldoshin, S. M.; Troshin, P. A. Reversible Pb2+/Pb0 and I−/I3− Redox Chemistry Drives the Light-Induced Phase Segregation in All-Inorganic Mixed Halide Perovskites. Advanced Energy Materials 2021, 11 (12), 2002934.
- Xue, H.; Vicent-Luna, J. M.; Tao, S.; Brocks, G. Compound Defects in Halide Perovskites: A First-Principles Study of CsPbI3. J. Phys. Chem. C 2023, 127 (2), 1189–1197. [CrossRef]
- Yang, Z.-L.; Zhang, Z.-Y.; Fan, W.-L.; Hu, C.; Zhang, L.; Qi, J.-J. High-Performance g-C3N4 Added Carbon-Based Perovskite Solar Cells Insulated by Al2O3 Layer. Solar Energy 2019, 193, 859–865. [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van De Walle, C. G. First-Principles Calculations for Point Defects in Solids. Reviews of Modern Physics 2014, 86 (1), 253–305. [CrossRef]
- Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Physical Review Letters 1996, 77 (18), 3865–3868. [CrossRef]
- Joubert, D.; Kresse, G. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Physical Review B - Condensed Matter and Materials Physics 1999, 59 (3), 1758–1775. [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. Journal of Chemical Physics 2010, 132 (15), 24103. [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Physical Review Letters 2015, 115 (3), 036402. [CrossRef]
- Sun, J.; Remsing, R. C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; Klein, M. L.; Perdew, J. P. Accurate First-Principles Structures and Energies of Diversely Bonded Systems from an Efficient Density Functional. Nature Chemistry 2016, 8 (9), 831–836. [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A User-Friendly Interface Facilitating High-Throughput Computing and Analysis Using VASP Code. Computer Physics Communications 2021, 267, 108033. [CrossRef]
- Ku, W.; Berlijn, T.; Lee, C. C. Unfolding First-Principles Band Structures. Physical Review Letters 2010, 104 (21), 216401. [CrossRef]
- Makov, G.; Payne, M. C. Periodic Boundary Conditions in Ab Initio Calculations. Phys. Rev. B 1995, 51 (7), 4014–4022. [CrossRef]
- Alkauskas, A.; Yan, Q.; Van De Walle, C. G. First-Principles Theory of Nonradiative Carrier Capture via Multiphonon Emission. Physical Review B 2014, 90 (7), 075202. [CrossRef]
- Turiansky, M. E.; Alkauskas, A.; Engel, M.; Kresse, G.; Wickramaratne, D.; Shen, J.-X.; Dreyer, C. E.; Van de Walle, C. G. Nonrad: Computing Nonradiative Capture Coefficients from First Principles. Computer Physics Communications 2021, 267, 108056. [CrossRef]
- Alkauskas, A.; Yan, Q.; Van De Walle, C. G. First-Principles Theory of Nonradiative Carrier Capture via Multiphonon Emission. Physical Review B - Condensed Matter and Materials Physics 2014, 90 (7). [CrossRef]
- Wang, H.; Li, B.; Liu, F.; Zhan, W.; Feng, M.; Guo, J.; Wang, S.; Liang, Y.; Fan, Y.; Chen, Y.; Miao, Y.; Zhao, Y. 2D Capping Layer Passivation toward Inorganic CsPbI3 Perovskite Minimodule. Advanced Functional Materials 2025, 35 (35), 2423397. [CrossRef]
- Chu, W.; Prezhdo, O. V. Concentric Approximation for Fast and Accurate Numerical Evaluation of Nonadiabatic Coupling with Projector Augmented-Wave Pseudopotentials. J. Phys. Chem. Lett. 2021, 12 (12), 3082–3089. [CrossRef]
- Jaeger, H. M.; Fischer, S.; Prezhdo, O. V. Decoherence-Induced Surface Hopping. The Journal of Chemical Physics 2012, 137 (22), 22A545. [CrossRef]
- Liu, D.; Wang, B.; Vasenko, A. S.; Prezhdo, O. V. Decoherence Ensures Convergence of Non-Adiabatic Molecular Dynamics with Number of States. The Journal of Chemical Physics 2024, 161 (6), 064104. [CrossRef]
- Jaeger, H. M.; Fischer, S.; Prezhdo, O. V. Decoherence-Induced Surface Hopping. The Journal of Chemical Physics 2012, 137 (22), 22A545. [CrossRef]
- Zheng, Q.; Chu, W.; Zhao, C.; Zhang, L.; Guo, H.; Wang, Y.; Jiang, X.; Zhao, J. Ab Initio Nonadiabatic Molecular Dynamics Investigations on the Excited Carriers in Condensed Matter Systems. WIREs Computational Molecular Science 2019, 9 (6), e1411. [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear Optical Properties in the Projector-Augmented Wave Methodology. Physical Review B - Condensed Matter and Materials Physics 2006, 73 (4), 045112. [CrossRef]
- Nations, S.; Jia, T.; Wang, S.; Duan, Y. Electronic and Optical Properties of Orthorhombic (CH3NH3)BX3(B = Sn, Pb; X = F, Cl, Br, I) Perovskites: A First-Principles Investigation. RSC Advances 2021, 11 (36), 22264–22272. [CrossRef]
- Svensson, P. H.; Kloo, L. Synthesis, Structure, and Bonding in Polyiodide and Metal Iodide−Iodine Systems. Chem. Rev. 2003, 103 (5), 1649–1684. [CrossRef]
- Liu, Z.; Liu, F.; Wu, Y.-S. Exotic Electronic States in the World of Flat Bands: From Theory to Material. Chinese Phys. B 2014, 23 (7), 077308. [CrossRef]
- Pokryshkin, N. S.; Mantsevich, V. N.; Timoshenko, V. Y. Anti-Stokes Photoluminescence in Halide Perovskite Nanocrystals: From Understanding the Mechanism towards Application in Fully Solid-State Optical Cooling. Nanomaterials 2023, 13 (12), 1833. [CrossRef]
- Zhang, X.; Turiansky, M. E.; Van de Walle, C. G. Correctly Assessing Defect Tolerance in Halide Perovskites. The Journal of Physical Chemistry C 2020, 124 (11), 6022–6027. [CrossRef]
Figure 1.
Defect cases found to have TELs in the band gap functioning as deep traps: (a) Pb in place of an I with +2 charge; (b) I in place of a Pb with -1 charge. Cs is teal, Pb is gray, and I is purple.
Figure 1.
Defect cases found to have TELs in the band gap functioning as deep traps: (a) Pb in place of an I with +2 charge; (b) I in place of a Pb with -1 charge. Cs is teal, Pb is gray, and I is purple.
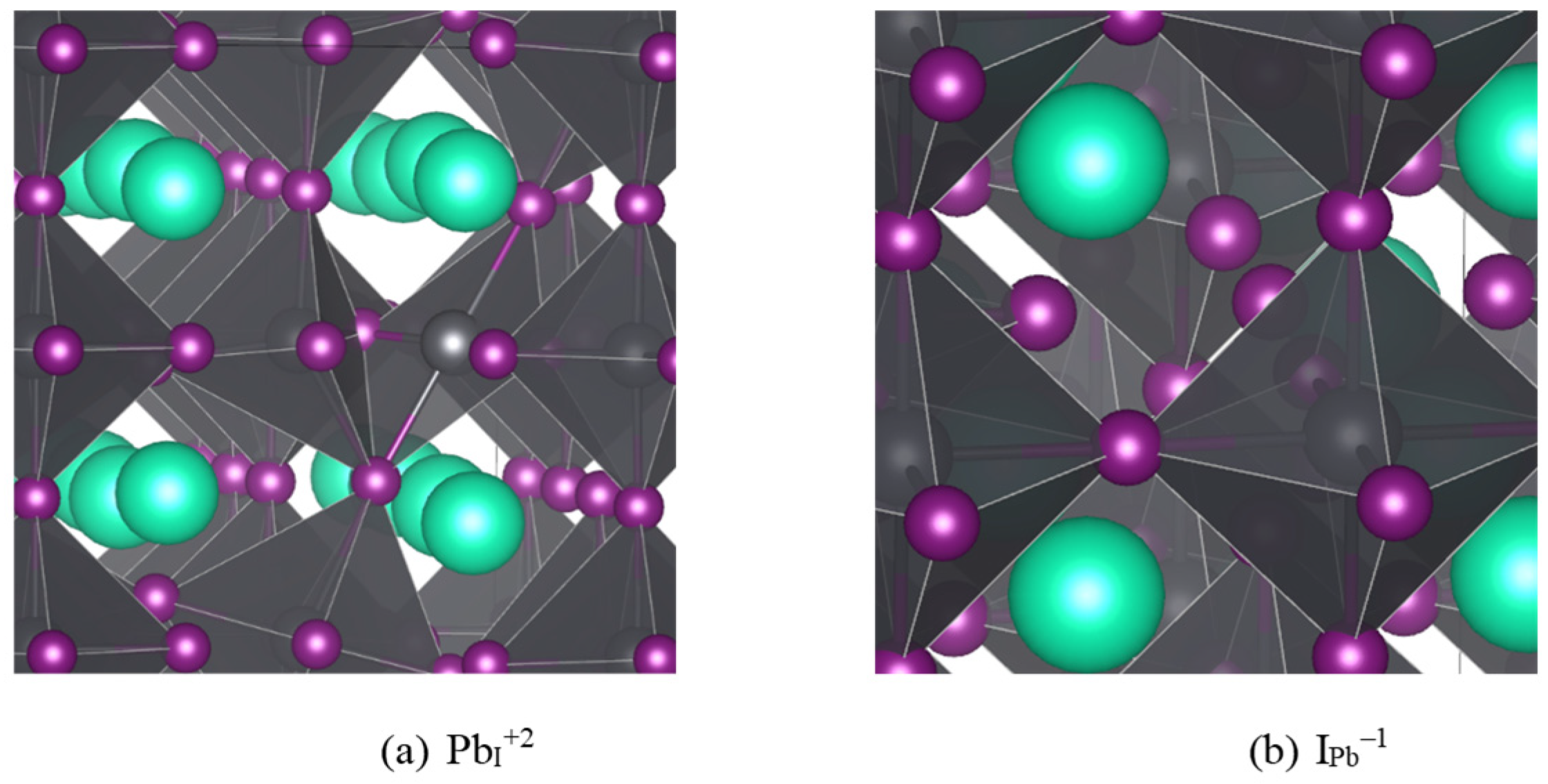
Figure 2.
Band diagrams for pristine CsPbI3 and defect cases found to have TELs levels inside the band gap, functioning as deep traps: (a) Pristine α-CsPbI3; (b) Pb in place of an I with +2 charge; (c) I in place of a Pb with -1 charge.
Figure 2.
Band diagrams for pristine CsPbI3 and defect cases found to have TELs levels inside the band gap, functioning as deep traps: (a) Pristine α-CsPbI3; (b) Pb in place of an I with +2 charge; (c) I in place of a Pb with -1 charge.
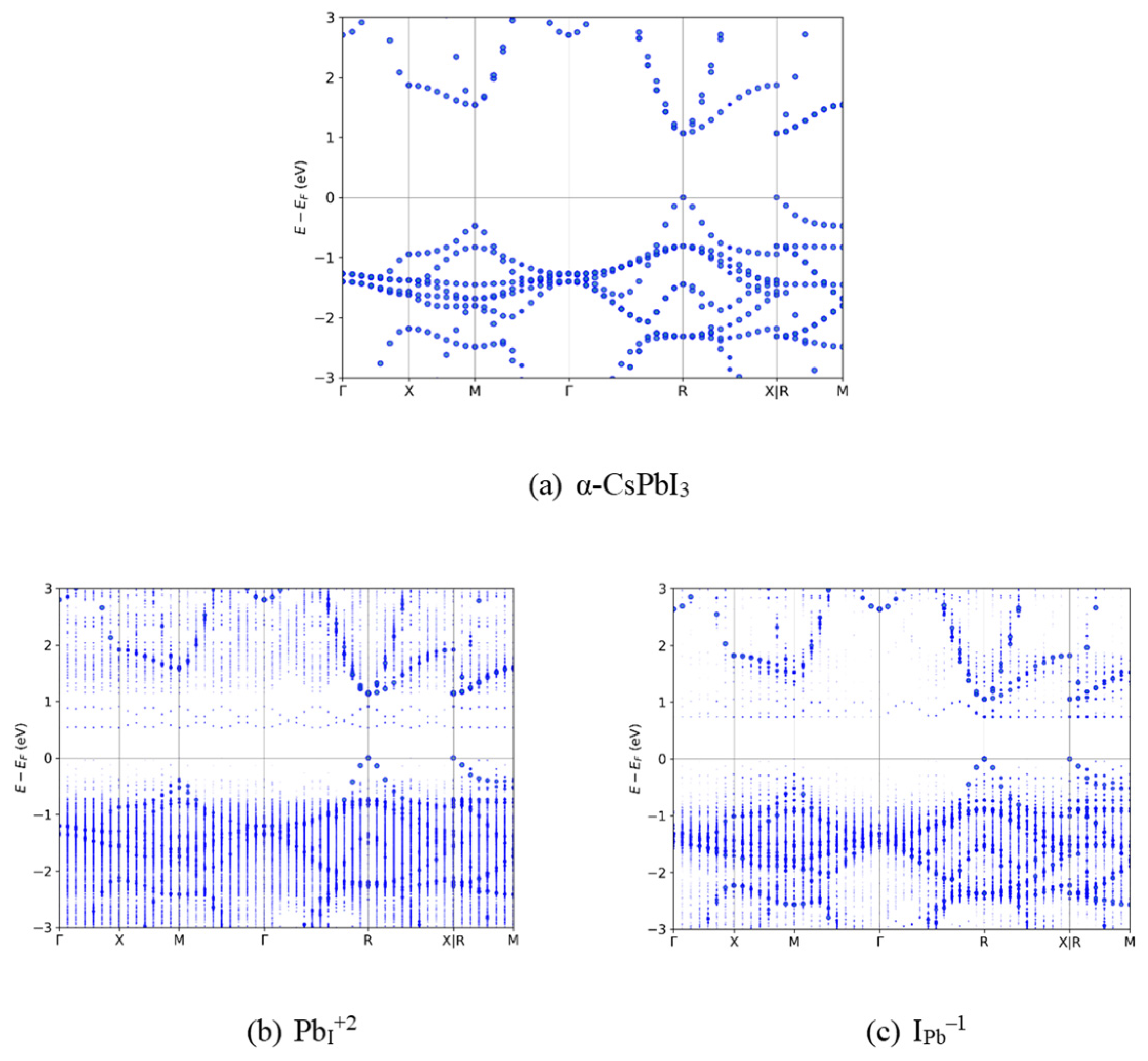
Figure 3.
Geometries and band diagrams for ammonia on trap defect sites: Figure 3a and 3c: Pb in place of an I; Figure 3b and 3d: I in place of a Pb. Cs is teal, Pb is dark gray, I is purple, N is light gray, and H is pink. Band figures are shown with the highest magnitude charge case to illustrate energy levels which are not present in more neutral cases.
Figure 3.
Geometries and band diagrams for ammonia on trap defect sites: Figure 3a and 3c: Pb in place of an I; Figure 3b and 3d: I in place of a Pb. Cs is teal, Pb is dark gray, I is purple, N is light gray, and H is pink. Band figures are shown with the highest magnitude charge case to illustrate energy levels which are not present in more neutral cases.
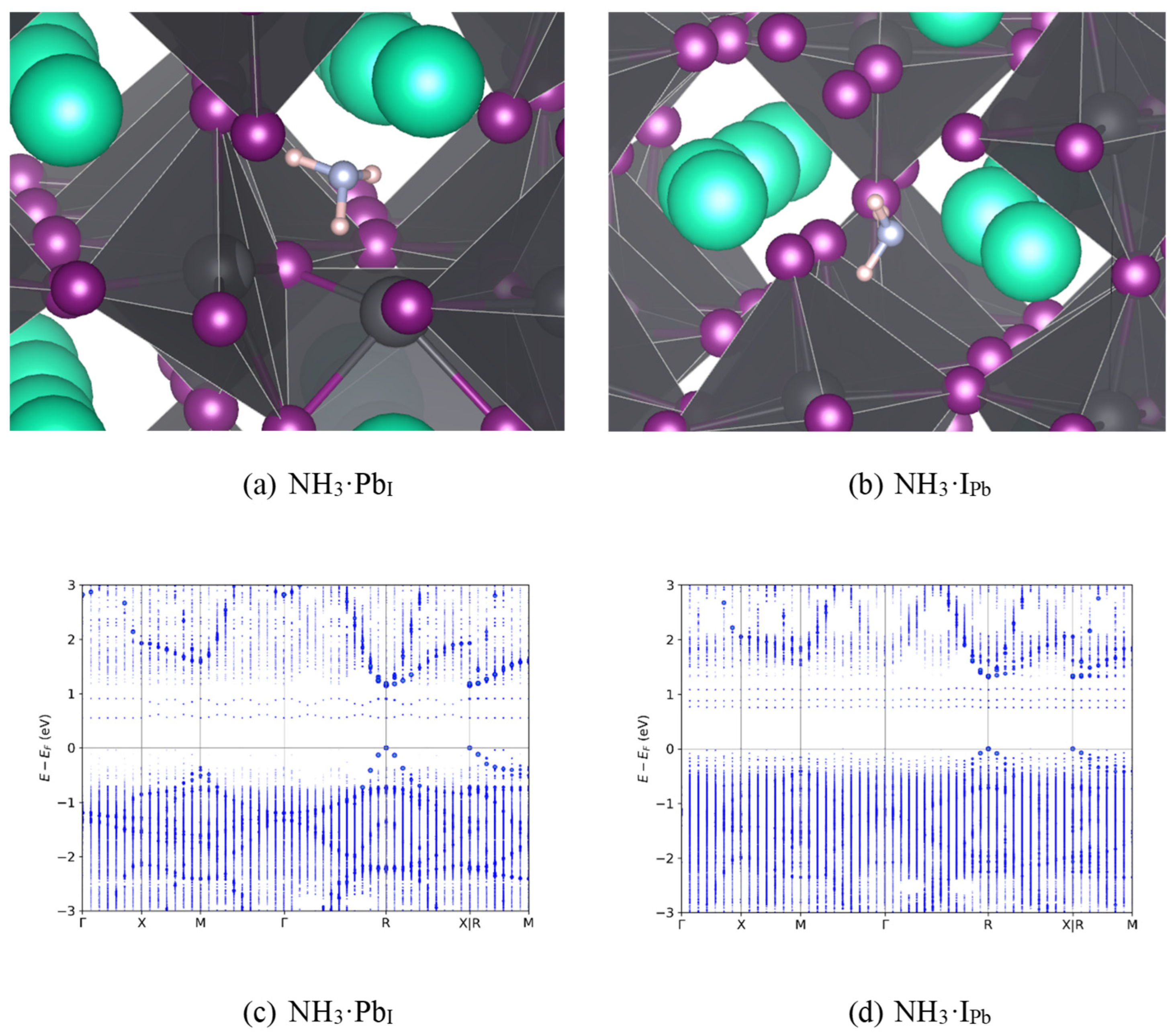
Figure 4.
The calculated density of states. (a) The pristine perovskite with partial density of states for Cs (red), I (green), and Pb (purple); (b) the total density of states for gasless IPb (blue), NH3·IPb (orange), and pristine (green).
Figure 4.
The calculated density of states. (a) The pristine perovskite with partial density of states for Cs (red), I (green), and Pb (purple); (b) the total density of states for gasless IPb (blue), NH3·IPb (orange), and pristine (green).
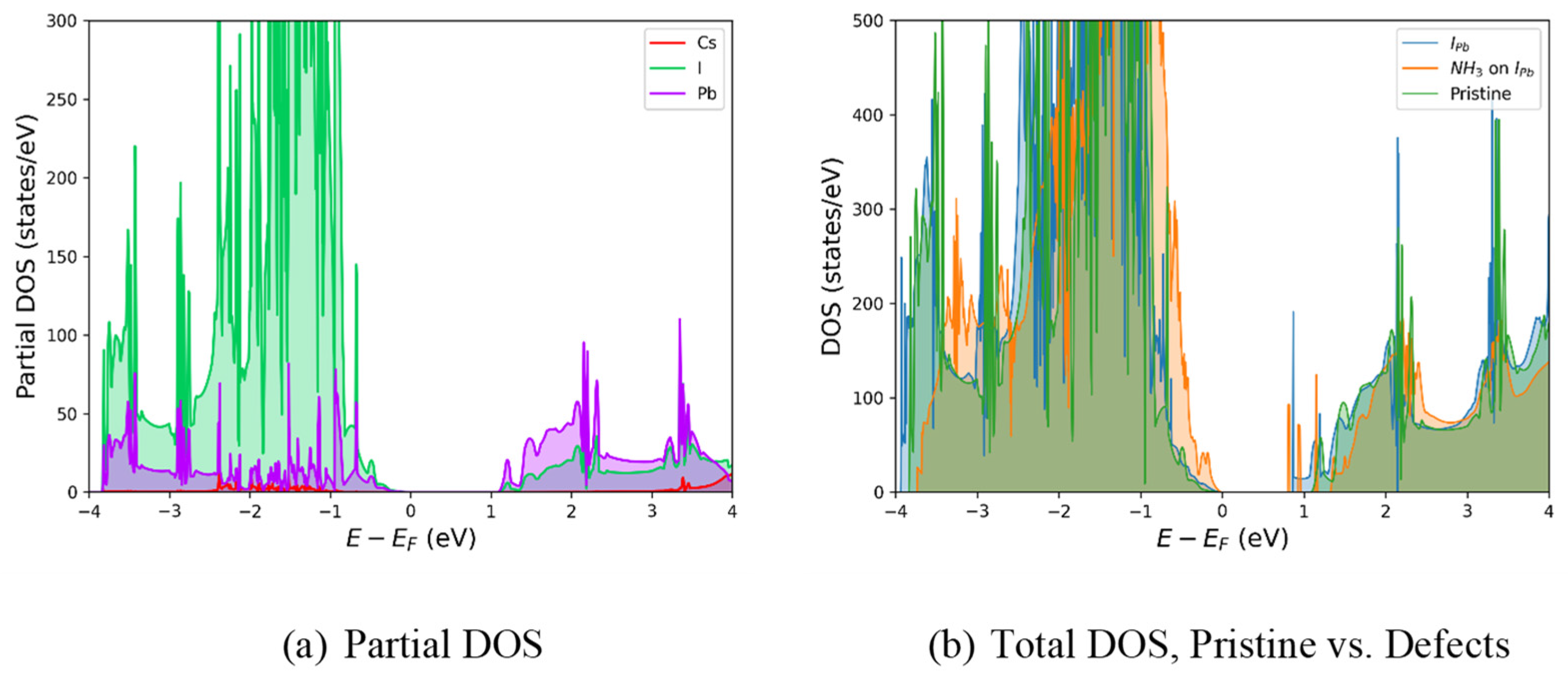
Figure 5.
Exemplary band diagrams for alternative, non-NH3 analytes. (a) NO2 on PbI, (b) CH3NH2 on PbI and (c) CH3NH2 on IPb.
Figure 5.
Exemplary band diagrams for alternative, non-NH3 analytes. (a) NO2 on PbI, (b) CH3NH2 on PbI and (c) CH3NH2 on IPb.
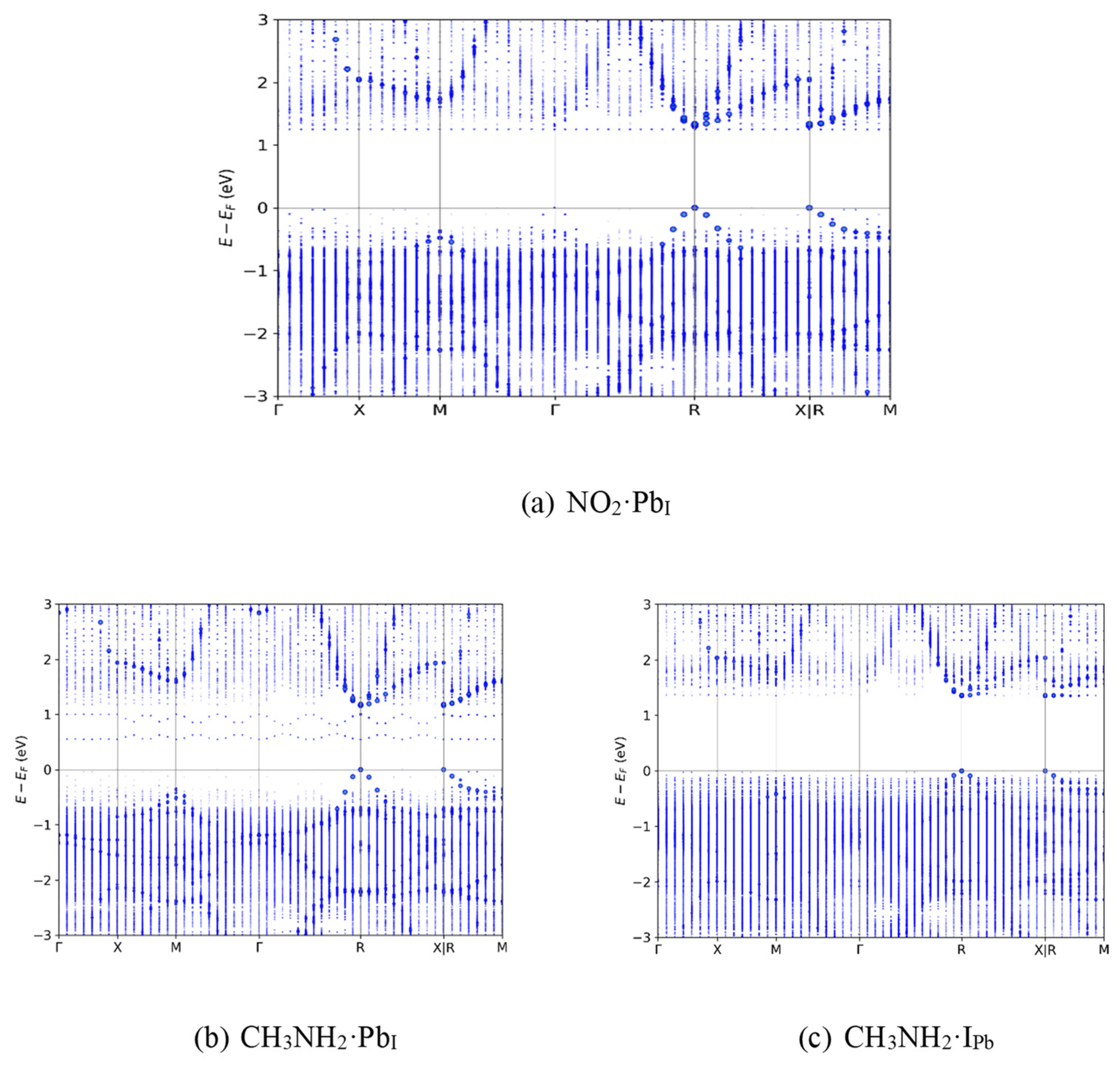
Figure 6.
Nonradiative recombination processes calculated with PBE for the case of NH3 on each of the two deep traps present for CsPbI3: (a) and (c): Pb in place of an I; (b) and (d): I in place of a Pb.
Figure 6.
Nonradiative recombination processes calculated with PBE for the case of NH3 on each of the two deep traps present for CsPbI3: (a) and (c): Pb in place of an I; (b) and (d): I in place of a Pb.
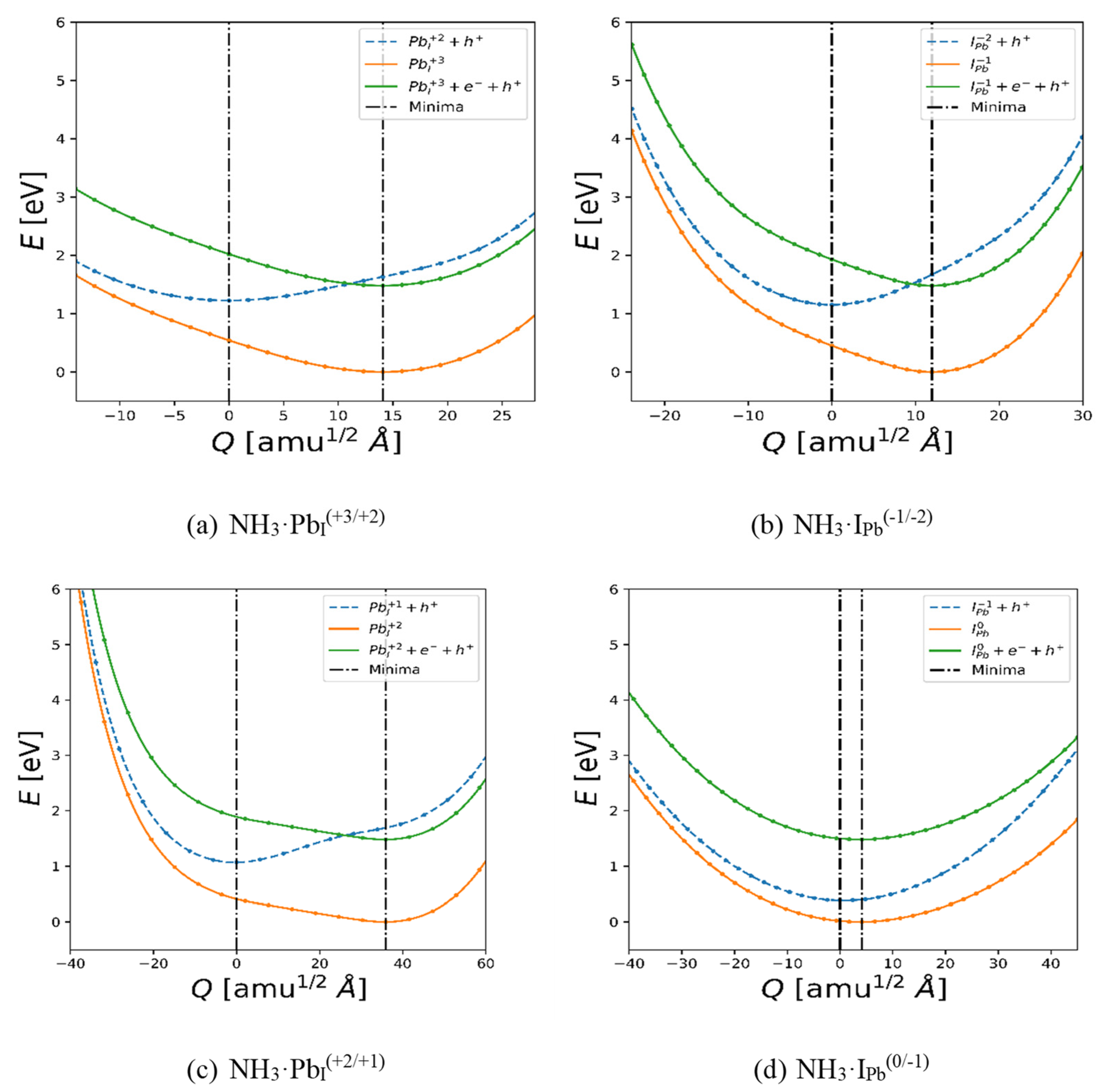
Figure 7.
Geometries for the PbI+2 (a) without and (b) with NH3. Cs is teal, Pb is gray and I is purple.
Figure 7.
Geometries for the PbI+2 (a) without and (b) with NH3. Cs is teal, Pb is gray and I is purple.
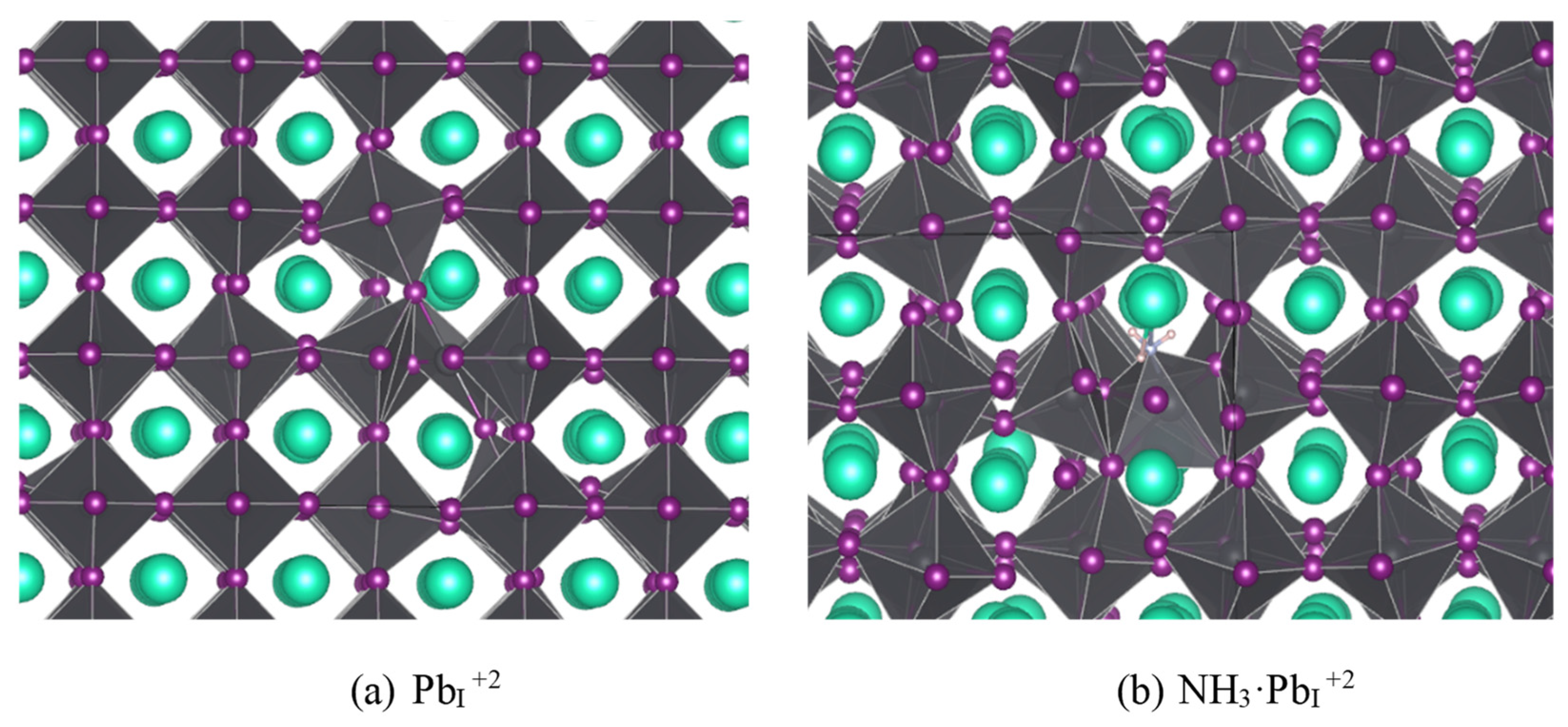
Figure 8.
Absorption spectra for CsPbI3 deep traps with various gas analytes on (a) PbI and (b) IPb.
Figure 8.
Absorption spectra for CsPbI3 deep traps with various gas analytes on (a) PbI and (b) IPb.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



