
Submitted:
16 October 2025
Posted:
17 October 2025
You are already at the latest version
Abstract
Although dry eye disease (DED) is common with age, the cellular and metabolic changes in the lacrimal gland (LG) that contribute to this condition are not fully un-derstood. Here, we applied fluorescence lifetime imaging microscopy (FLIM) to ex-amine metabolic alterations in LG tissue from aged (20-22 months) female C57BL/6J mice, a model of age-related DED, versus young (~3 months) C57BL/6J mice. Phasor analysis of NAD(P)H fluorescence revealed a shift in aged LG toward more glycolytic metabolism and reduced oxidative phosphorylation. We recently identified a novel subpopulation of F4/80-enriched multinucleated macrophages rich in lipids and lipid metabolizing enzymes in aged female mice. Using FLIM combined with immuno-labeling enabled isolation of the metabolic signature of these macrophages, confirming their increased NADPH oxidase 2 (NOX2) activity, an enzyme which generates reactive oxygen species which are characteristically expressed in M1-type macrophages. In-creased phosphorylation of P47phox, associated with NOX2 activation, was also ob-served in these macrophages, supporting their classification as M1-like cells. FLIM thus provides a valuable tool both to capture metabolic changes in the LG overall, and in defining metabolic features of specific cell populations that may be important in diseases such as DED.
Keywords:
fluorescence life time imaging microscopy (FLIM)
; lacrimal gland
; ageing
; metabolism
; macrophages
1. Introduction
One in six people in the world will be aged 60 or over by 2030, while the population is expected to increase from 1 billion in 2020 to 1.4 billion by 2030 and 2.1 billion by 2050 [1]. Many chronic disorders, such as heart disease, cancer, obesity, and mental disorders are increasing in prevalence in parallel with aging of the population [2,3]. While studies support the role of prevention through lifestyle modification in many chronic diseases [4], such research is not available to support preventive strategies to avoid a chronic disease of the eye: dry eye disease (DED). DED affects 18.6 percent of individuals aged >75-years compared to only 2.7 percent of individuals aged 18 to 34 years; due to the aging population, a 60 percent increase in DED patient numbers is expected by 2030 [5,6,7]. With this impending increase in patient numbers, it is essential that we develop a deeper understanding of age-related DED.
The lacrimal gland (LG) is an exocrine gland that plays a pivotal role in producing the aqueous layer of the tear film, which in turn provides moisture, lubrication, and nutrients to the ocular surface [8,9]. Age-related changes in the LG such as lymphocytic [10] and fatty infiltration [11], fibrosis [11], volumen [12], and decreased tear secretion [13,14,15] may contribute to age-related DED. C57BL/6J mice exhibit comparable age-related DED symptoms by 6-9 months of age including development of corneal surface irregularity, corneal barrier disruption (corneal staining), and altered tear composition, compared to 8-week mice in both sexes [16,17,18]. Moreover, the aged C57BL/6J mouse LG exhibits increased lymphocytic infiltration and formation of ectopic lymphoid structures comparable to those seen in human LG [17,19]. Thus, the aged C57BL/6J mouse models age-related changes in the LG associated with DED.
Macrophages, part of the innate immune system, play critical roles in tissue development, homeostasis, and repair of damaged tissue [20]. These phagocytic cells have been traditionally categorized into M1 (classically-activated, pro-inflammatory) and M2 (alternatively-activated, anti-inflammatory) types. Differentiation into M1 versus M2 populations in vitro is driven by stimuli including lipopolysaccharides (LPS) and/or IFN-g or IL-4 and/or IL-13 respectively [21,22]. Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase 2 (NOX2) is a key enzyme responsible for generation of superoxide in host defense that is abundant in pro-inflammatory M1 macrophages [23,24]. NOX2 activation involves phosphorylation of p47phox by phosphokinase C, initiating its translocation with several other cytosolic protein subunits to gp91phox, the transmembrane catalytic subunit [25]. Once activated, NOX2 oxidizes NADPH to NADP+ to produce superoxide which is rapidly converted to H2O2, leading to a pro-inflammatory response by M1 macrophages [26]. The relationship between NOX2 activity and macrophage function is a key determinant in macrophage differentiation and indicative of the status of the immune system.
Depending on location, macrophages can be highly specialized in their function and phenotype, with some implicated in lipid clearance. Foamy macrophages accumulate lipids and cholesterol esters and are hallmarks of early atherosclerotic plaques [27]. In areas of high lipid concentration, foam cells can aggregate into multinucleated giant cells named Touton giant cells [28,29]. Multinucleated macrophages are also reported in different tissues [30,31,32]. We recently identified a foamy multinucleated macrophage population that accumulates in the aged female mouse LG and that is enriched in lipid metabolizing enzymes [33]. Other properties of these macrophages including their status as M1- or M2-like are so far unclear.
Here, we have applied fluorescence lifetime imaging microscopy (FLIM) to compare the metabolic status of young and aged LG. NAD(P)H is an intrinsic cellular fluorescent marker, and its fluorescence decay time (lifetime) can be used as a general indicator of cellular energy metabolism. A shorter fluorescent decay lifetime for NAD(P)H is correlated with a lower percentage of bound NAD(P)H and suggestive of higher free NAD(P)H concentration, suggesting a shift towards utilization of glycolysis for energy production. A longer fluorescent decay lifetime for NAD(P)H is correlated with a higher percentage of NAD(P)H bound to enzymes, suggesting more metabolic activity and the utilization of oxidative phosphorylation (OxPhos) to produce ATP [34,35,36,37,38,39,40]. Signals for NAD(P)H associated with individual metabolic enzymes can also be identified within the phasor. FLIM phasor analysis is a powerful tool for interacting with live-cell imaging data and for extracting meaning from metabolic imaging.
Our FLIM analysis reveals a general shift towards glycolytic metabolism with age in whole LG sections imaged ex vivo. FLIM analysis of NOX2 activity in vitro and in immunoprecipitates from LPS-activated M1 macrophages reveal specific signatures that can be mapped to the foamy multinucleate macrophages in the aged LG that are consistent with the increased activity of NADPH oxidase 2 (NOX2), suggesting that these foamy macrophages have M1-like properties. This study illustrates for the first time, the power of FLIM to non-invasively probe the global metabolic landscape of the LG and to identify changes in metabolism in specific cell populations potentially linked to developing pathology.
2. Materials and Methods
2.1. Mice
DED severity and prevalence is greater in females than in males [20], thus we used female mice in the studies. Female C57BL/6J (B6) mice were received from the NIH/National Institute of Aging, or purchased from the Jackson Laboratory (Bar Harbor, ME) (strain #00664) and aged for up to 2 years. For all experiments, young mice are female C57BL/6J mice aged ~3 months and old mice are female C57BL/6J mice aged 20-22 months. Following compliance with the animal use policies approved by the University of Southern California, we used intraperitoneal injection of 100 mg/kg ketamine + 10 mg/kg xylazine for mouse anesthesia and cervical dislocation for euthanasia. After euthanasia, the LG was recovered and processed immediately or stored at -80°C for future experiments.
2.2. Materials and Reagents
Goat anti-mouse NOX2 polyclonal antibody (cat# PA5-142646), donkey anti-rat Alexa-Fluor® (AF) 488 secondary antibody (cat# A-21208), rat anti-mouse F4/80-AF561 primary antibody (cat# 505-4801-82), donkey anti-goat AF568 secondary antibody (cat# A-11057), donkey anti-rabbit AF594 secondary antibody (cat# A-21207), DAPI (cat# D-1306), phalloidin-AF647 (cat# A22287), Acridine Orange (cat # A1301), goat anti-mouse NOX2 Polyclonal Antibody (cat# PA5-142646), 10% Tris-Glycine protein gels (cat# XP00105BOX), iBlot 2 Nitrocellulose transfer stacks (cat # IB23001), and Protein G beads (cat# 10003D) were obtained from ThermoFisher (Waltham, MA). SiR-DNA (cat# CY-SC007) was from Cytoskeleton, Inc (Denver, CO). Protein G Plus-Agarose Suspension (cat# IP04-1.5 mL), IGEPAL CA-630 (cat# 18896-50 mL), the RAW264.7 mouse macrophage cell line, normal Mouse IgG (cat# 12-371), tetramethylethylenediamine (cat# T9281), ammonium persulfate (cat# A3678), lipopolysaccharide (cat # L2880-25MG), and Tris buffered saline with Tween 20 (TBS-T), pH 8.0 (cat# T9039) were from Sigma Aldrich (St. Louis, MO). Rabbit anti-mouse gp91phox primary antibody (cat# ab310337) was from Abcam (Cambridge, United Kingdom). Acrylamide (cat# 1610144) was from Biorad (Hercules, CA). Rabbit anti-mouse phospho-p47phox antibody (cat# PA5-36863), goat anti-mouse p47phox antibody (cat# PA1-9073), and rat anti-mouse F/480 (cat# MA1-91124) were from Invitrogen (Grand Island, NY). Donkey anti-goat IR680 (cat# 926-68074) and total protein stain kit (cat # 926-11016) were purchased from LiCor (Lincoln, NE). Laemmli SDS sample buffer (6x) (cat# SAB03-02) were from Bioland Scientific (Paramount, CA). Protease/phosphatase Inhibitor (cat # 5872s) was from Cell Signaling Technologies (Danvers, MA). Glyceraldehyde-3-phosphate dehydrogenase (cat # 9001-50-7), NADH (cat# 606-68-8), and NADPH (cat# 2646-71-1) were purchased from MilliporeSigma (Burlington MA). All other chemicals were obtained at high purity from standard suppliers.
2.3. Immunoprecipitation of NOX2 (gp91phox) from RAW 264.7 Cells
Mouse RAW 264.7 cells, a macrophage cell line, were seeded in DMEM/F-12 complete media with 10% FBS in 100 mm plates and incubated for 3 h to settle into wells. Cells were either exposed to LPS (100 ng/mL) or complete media (DMEM/F-12 containing 10% fetal bovine serum, FBS) for 24 hours. At this time, the cell media was removed, and lysis medium was added. The lysis medium consisted of sterile-filtered 20 mM Tris HCl, pH 8.0, 140 mM NaCl, 1% IGEPAL® CA-630, containing 1 mM EDTA, to which a protease/phosphatase inhibitor cocktail was added immediately before use. Using a cell scraper, the cells were collected and homogenized in microfuge tubes with Fisherbrand pellet pestles (cat#12-141-363). After centrifuging lysates at 100 rcf for 5 min at 4 °C, the supernatant was collected. Protein G beads were washed three times with lysis buffer before incubation with samples and antibodies. Samples were precleared with mouse IgG and Protein G beads for 1 h, before incubation overnight with rabbit anti-mouse NOX2 antibody at 4 °C on a tube rotator. Then, Protein G beads with NOX2 antibody were added to the sample and incubated for 1 h at 4 °C on a tube rotator. Beads were washed 5 times and aliquots were imaged by FLIM with 2 mM NADPH as below. At least 7 fields were acquired from samples from LPS-treated cells, untreated cells, and NADPH only.
The remainder of the samples on beads were used for Western Blotting. Samples were eluted from the beads with reducing dye and b-mercaptoethanol (6x, 9% V/V) at 95°C for 5 min. Each sample (including beads) was loaded onto precast 10% Tris-Glycine gels and resolved by SDS-PAGE at 120 V at 4°C for 2 h. Proteins on gels were transferred to nitrocellulose membranes using an iBlot™ 2 gel transfer machine. After blocking for 1 h with blocking buffer at room temperature with shaking, membranes were washed 3 times for 5 min with TBS-T, then incubated with goat anti-mouse gp91phox antibody in blocking buffer at 4°C overnight. After 3 washes, 5 min each with TBS-T, membranes were incubated with donkey anti-Goat IR 680 (1:4000) in blocking buffer at room temperature for 1 h. After another 3 washes for 5 min each with TBS-T, membranes were imaged with a LiCor® Odyssey Fc machine. Signal quantification used Image Studio Version 5.2. Controls included blots processed without exposure to primary antibody.
2.4. Histology
Right exorbital LG and ocular adnexa from old mice were excised, fixed in 10% formalin, paraffin-embedded, and cut into 5-µm sections using a microtome (Microm HM 340E, Thermo Fisher Scientific Waltham, MA). Histological sections were stained with H&E [41]. Digital images were visualized and acquired using a light microscope (Eclipse E400; Nikon, Tokyo, Japan).
2.5. Ex Vivo, Organotypic Metabolic Imaging of Live Young and Old LG Sections
LG were removed from C57BL/6J mice (n = 3/group) at young and old ages and briefly placed at -20°C on dry ice. 10x Tris-buffered Saline (TBS), consisting of 200 mM Tris, pH 7.5, with 1.5 M NaCl, was diluted with deionized water and 40% acrylamide to produce a solution of 15% acrylamide in 1x TBS. 10% ammonium persulfate and tetramethylethylenediamine were added to the mixture and poured into a 35mm cell culture dish. The LG was dipped multiple times into the liquid to produce maximum exposure and minimum bubbles. After polymerization of the acrylamide gel around the LG, it was cut in a trapezoidal shape to accommodate optimal cutting with a Leica vibratome. 100 mm sections were cut at 25% 50% and 75% depth in the LG and each section was imaged. Each section was put on a slide with a coverslip and imaged on the Leica SP8 DIVE FALCON inverted microscope with a 25x objective (0.95 Numerical Aperture) water immersion lens. The tunable multiphoton excitation laser was set at 740 nm wavelength, 0.5mW power at the stage (13% of 2.03W peak power), 2.66 μs pixel dwell time, and fluorescence emission was collected from 425-450 nm, and the z-stack image of the whole LG section was acquired. The FLIM decay measurements from each pixel were transformed using FAST-Fourier transformation resulting in cosine and sine terms plotted in a FLIM phasor. The images were then analyzed with the Leica LAS-X FLIM/FCS module version 4.05.
2.6. Imaging of Macrophages on Culture Dishes with/Without LPS
Mouse RAW 264.7 cells were cultured and seeded into 35mm plastic bottomed culture dishes suitable for fluorescence imaging from Ibidi (cat# 81156) with half of the dishes treated with LPS in the same manner as with the IP experiment described above. After 24 h of 100 ng/mL LPS treatment, control and LPS-treated cells were FLIM imaged and analyzed as described previously.
2.7. FLIM Imaging of old LG Sections Labeled to Identify Macrophages
LG from 3 old mice were embedded in acrylamide as above and similarly sectioned (25%, 50%, and 75% depths of the LG). Sections were submerged in a TBS solution containing a 1:20 dilution of rat anti-mouse F4/80 antibody conjugated to AF561 and a 1:200 dilution of acridine orange for 1 h in an incubator at 37°C with gentle shaking. Sections were washed with TBS 3 times, for 10 min in the incubator with gentle shaking. These sections were then placed on a slide with a coverslip and imaged on the Leica SP8 DIVE FALCON inverted microscope using the 63x water immersion objective (1.2 NA). Samples were imaged using the 740 nm excitation wavelength for NADPH signal, 515 nm (2% laser power) for acridine orange and 594 nm (6% laser power) for sub-optimal excitation of AF561 while minimizing bleed through of acridine orange into the F4/80-AF561 detector. Z-stack images were obtained for regions of the LG enriched in the F4/80 stain. At least 5 to 7 images were taken of each section for increased signal of the same z plane. Images were then analyzed with the Leica LAS-X program.
2.8. Immunofluorescence of Phospho-p47 (Pp47), p47, and F4/80
LG from young and old mice were fixed in 4% paraformaldehyde and 4% sucrose in PBS for 3 h at room temperature, then incubated overnight in a 30% sucrose PBS solution at 4°C. LG were embedded in O.C.T. the following day, frozen on dry ice, and stored at –80 °C. Frozen O.C.T. blocks were sectioned at 5 µm thickness and mounted on superfrost plus microscope slides. While submerged in sterile phosphate-buffered saline (PBS), the slides were photobleached to eliminate autofluorescence from lipofuscin using one of two different methods: using an LED desk lamp (Rotary LED Light Model Q1) in a 4 °C cold room for 7 days as published [33] or with a TiYo Autofluorescence Quenching System (Bulldog Bio, Portsmouth, NH) for one 2 h cycle in accord with the manufacturer’s recommended protocol. After photobleaching, slides were quenched with ammonium chloride in PBS for 15 min at room temperature with shaking between each step. The slides were then blocked with 5% BSA in 0.3% Triton X-100 for 3 h at room temperature. The sections were then incubated with primary antibodies as follows: rabbit anti-mouse to p47 (1:100), goat anti-mouse phospho-p47 (1:100), or rat anti-mouse F/480 (1:100) in blocking buffer overnight at 4°C. On the second day, slides were washed with PBS 3 times, for 15 min each at room temperature with gentle shaking. The sections were then incubated with secondary antibodies as follows: donkey anti-rat AF488 (1:200, for F4/80), donkey anti-rabbit AF594 (1:200, for p47) or donkey anti-goat AF 568 (1:200 for pP47). DAPI and Phalloidin AF647 (1:200) were added concurrently with secondary antibodies and incubated for 1 h at 37°C. After incubation with secondary antibodies, slides were washed again with PBS 3 times, then mounted with ProLong antifade mounting medium and a glass coverslip and were left to dry overnight. Images were acquired with a Zeiss LSM 800 with Airyscan using a 63x oil objective (NA 1.4). All images were subjected to equivalent image processing with brightness and contrast of the proteins of interest being conserved using the QuPath program version 0.4.2.
RAW 264.7 cells were seeded in a 12-well plate on 20 mm glass coverslips in DMEM/F-12 complete media with 10% FBS for at least 4 h. The cells were exposed without or with 100 ng/mL LPS for 24 h. Then cells on coverslips were fixed and permeabilized with -20 °C acetone/methanol (1:1) for 20 min. After the cells were blocked with 1% BSA in PBS, they were processed as described above for LG sections starting with incubation with primary antibodies. Immunofluorescence was detected using a Zeiss LSM 800 microscope equipped with AiryScan as above.
2.9. Bulk RNA Sequencing and Data Analysis
Our previously published bulk RNA sequencing data from young and aged LGs was used. The original description and data analysis methods can be found in [17]. The dataset for the published study is publicly available in the GEO repository (Access ID GSE224596).
2.10. Statistical Considerations
Phasor analysis is performed on a pixel-by-pixel basis, and then regions of interest (ROI) selected either in the phasor or image are then analyzed within the image or phasor, respectively. The power of this method is in analysis of all images in an experiment together under one rubric for both standardization and an unbiased approach. Our FLIM pixel dwell time is 2.66 μs and the Spectra Physics Insight X3 laser pulses at 80 MHz which means each pixel in an image is sampled about 213 times per scan. Further, we accumulate 10 images per tile or z-section, therefore each pixel is sampled 2130 times per condition/replicate and the average lifetime for each pixel is plotted in the phasor. We choose a laser intensity and frame accumulations necessary to collect a minimum of 10 photons per pixel for an accurate measurement in the tissue or cells we intend to analyze.
3. Results
3.1. The FLIM Phasor
Phasor analysis begins with pixel-by-pixel Fourier transformation of FLIM decay time measurements into real (G) and imaginary (S) components which are plotted, resulting in a FLIM fingerprint of the sample. In cells and tissues, there are many enzymes to which NAD(P)H can bind, each having a particular NAD(P)H lifetime and phasor position. Thus, changes in cellular metabolism involving differential utilization of enzymes binding NAD(P)H can provide a snapshot of the cell’s metabolic status that reflects its physiology/pathophysiology.
The phasor is a 2D representation of a 3D histogram with a rainbow lookup table which serves as a heat map of pixel density. The FLIM phasor semi-circle (Supplemental Figure S1A) demarcates where single-component exponential decays are found. The phasor semi-circle is a continuous plot from instantaneous to infinite lifetimes with dilation of lifetimes in the clockwise direction and constricting in the counter-clockwise direction. Any pixels residing within the semi-circle are mixtures of decays. The location is a linear combination of components falling on a chord in the semi-circle (Supplemental Figure S1B). A pixel’s position along the chord is a ratiometric analysis of lifetime contributions. For instance, Figure 1A depicts the phasor representing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity when mixed with different amounts of NADH. 100% unbound NADH (0:0 Bound/Total ratio) has a lifetime of 0.4 ns and exhibits a single exponential decay on the semi-circle at the blue tip of the rainbow cone (Figure 1B). The chord associated with GAPDH binding to NADH can be delineated by extending a line from 100% unbound NADH through the centroid of a phasor distribution associated with intermediate NADH binding (Figure 1C) and to the point on the arc associated with NADH saturation (Figure 1D), The rainbow scale bar reflects the fraction of NADH bound (Figure 1A) and can be used to pseudo-color the images for visualization of major changes.
3.2. FLIM Signal from Young and Old LG Imaged Ex Vivo
Live LG from young versus old mice were sectioned and imaged by FLIM. In samples from aged LG, an intense fluorescent signal arising from accumulated lipofuscin that we have previously reported in these aged female mice [33,42] initially overpowered the subtle differences in NAD(P)H signal, as lipofuscin and NADH have overlapping emission spectra. By thresholding out the brightest pixels corresponding to lipofuscin, we were able to highlight differences in NAD(P)H FLIM signals between samples.
The remaining fluorescent signal was relatively photon-limited and therefore needed de-noising and amplification of signal-to-noise ratio (SNR) by applying Leica’s Complex Wavelet Filter [42]. Using this approach, the FLIM phasor distribution of young LG was clearly distinct from that of old LG (Figure 2).
LG of young mice had greater OxPhos FLIM signature and a smooth, gradient-like distribution of metabolic states relative to LG of old mice (Figure 2A). While the old mice showed some regions with similar distributions to young mice, in other regions the metabolic signal was patchy and irregular. With the same rainbow scale bar applied to both phasors, we observed higher OxPhos signal (red colored) on the young versus the old (Figure 3B, top and bottom). The higher OxPhos areas in the young LG are distributed in the outer edges throughout the 3 mice samples, while the old LG show the higher OxPhos areas in the interior regions. The distribution of regions of lower OxPhos (blue colored) are evident in the old LG (Figure 2B) in both the whole tissue section as well as the magnified (top and bottom row respectively) images.
The old LG tissues show an accumulation of lipofuscin which shows up in the phasor as a very short lifetime species that pulls the metabolic signal in the lipofuscin-confounded pixels away from the purely metabolic axis shown in Supplemental Figure S1, stretching in a line from the majority NADH-bound GAPDH to the 100% unbound NADH. This secondary axis we designate as the Lipofuscin-Metabolic Axis (LMA) (Supplemental Figure S2) which stretches in a linear fashion from the metabolic signal’s center of mass to the phasor location associated with lipofuscin. Because these pixels fall on the LMA and there are three components, it has been shown that the third component, lipofuscin, can be deconvolved and separated such that the metabolic signal can be accurately analyzed [43].
3.3. Aged LG Exhibit Increased Expression of NOX2 Subunits
A principal finding in a previous study of the aged female LG was the presence of multinucleate, lipid-laden macrophages that were F4/80 positive. Figure 3 shows the phenotype of these macrophages in both H&E labeled LG sections (Figure 3A) and photobleached LG sections processed for immunofluorescence (Figure 3B). Beyond this phenotype, little is known about the properties of these macrophages that accumulate with age in the LG. Bulk RNASeq provided one indication that these macrophages might have M1-like properties, since increased gene expression was detected of the subunits of the NOX2 enzyme (Table 1) that is a marker of M1-type macrophages. We decided to test this hypothesis using FLIM imaging of the multinucleate macrophages ex vivo.
3.4. NOX2 FLIM Signal from Activated Macrophages and Immunoprecipitates Analyzed with the FLIM Phasor
In order to use FLIM to determine whether the novel multinucleate lipid-enriched macrophages that accumulate with age (Figure 3) in LG were M1-like and enriched in NOX2, as suggested by bulk RNASeq (Table 1), we first needed to obtain a FLIM signature for NOX2. We utilized a macrophage cell line, which when induced to the M1 phenotype increases NOX2 activity, to visualize NAD(P)H lifetime in situ. Figure 4A shows the FLIM signature of RAW 264.7 cells with and without activation to the M1-state by stimulation with LPS. Application of the same rainbow lookup table to the phasor pixels obtained from these two cell populations revealed a significant red shift in the activated M1 macrophages (right column).
To see if this induced signal might represent activated NOX2, we evaluated the NOX2 FLIM signal in vitro. NOX2 was immunoprecipitated from lysates of non-stimulated (LPS) or stimulated (LPS+) RAW 264.7 cells using a NOX2 (gp91phox) antibody. Successful immunoprecipitation was confirmed by Western blotting using an additional NOX2 antibody raised in a different host animal (Figure 4C). The band for NOX2 runs near 55-65 kD and was stronger than the signal obtained with secondary antibody alone. The presence of background signal in samples with secondary antibody alone may be due to the remnants of mouse IgG (heavy chain ~50 kDa) used for preclearing which slightly overlaps with the heterogeneous glycosylated NOX2 signal. IP beads from LPS-activated or non-activated macrophages were then evaluated by FLIM in the presence of NADPH (Figure 4B). A large background autofluorescent signal, verified with free beads alone, arises from the Protein A+G agarose IP beads, and was noted in both phasor plots (arrow). For an unbiased approach to our cell metabolism analysis, we stretched a rainbow color scale bar from the experimental NADPH-bound NOX2 localization in the IP, to the 100% free NADPH signal at 4 ns lifetime and on the circle (Figure 4B, left column phasor) that we designate as the NOX2 metabolic axis (Nx2-axis). The Nx2-axis in both angle and extent was applied to all the macrophage cell line FLIM-phasor images. Beyond this specific signal, it is obvious that there is a difference in imaging. There is only red signal associated with samples on beads from the LPS+ samples. As activated NOX2 should bind more to NADPH, the signal emitted by this enzyme would be expected to be shifted towards the top left of the Nx2-axis and to have more red and orange than non-LPS treated cells, indicating that the NOX2 from LPS-activated cells has more bound NADPH.
3.5. Multinucleated Macrophages with Distinct FLIM Signal in Old LG
We previously identified a unique, multinucleated, foamy macrophage population i old LG from female mice [33]. To identify the location of the multinucleated macrophages in the LG sections to study their metabolism, we used acridine orange and AF 564 conjugated F4/80 antibodies in the live sections from old mouse LG. We used the single photon FLIM signal of both acridine orange and AF 564 to identify the pixels associated with both multinucleated and F4/80-positive cells which define these macrophages in the LG [33]. Figure 5A (left column) confirms the presence of multinucleated macrophages in the old LG outside of the acinar cell clusters, while F4/80 defines the cellular boundary of the macrophages. We then masked out the metabolic signal from acini by selecting those pixels in the phasor as black in the images, thus enabling our focus on the localization and metabolic differences of the macrophages without the distraction of signal from the surrounding tissue. The metabolic signal that arises from those F4/80 positive pixels represents the macrophage metabolic signatures. Figure 5A (right column) shows the F4/80-positive pixels highlighted in the rainbow scale using the phasor-pixel selection shown in the corresponding phasor. As a reminder, metabolic signal from macrophages in old LG tissues is confounded with lipofuscin signal that dominates due to abundance of the molecule and its higher quantum efficiency versus NAD(P)H. These metabolic signatures are convolved with approximately 3 times the amount of lipofuscin signal as compared to the NAD(P)H signal as shown by the 3/4 distance shift along the LMA toward the known phasor localization of pure lipofuscin. This technique not only reveals the dynamic range of metabolism in these cells but also highlights the higher metabolic signature originating from regions close to the cytoplasmic membrane of the soma and filopodia (arrowheads).
Using these phasor selections in a feed-forward manner we could highlight the metabolic signal in unlabeled young and old LGs (Figure 5B). This technique enabled us to see the distribution and proportion of pixels, ostensibly activated macrophages, that are also filled with lipofuscin.
3.6. LPS-Activated Macrophages and Foamy Multinucleated Macrophages in Old LG Show Increased Expression of Phosphorylated p47phox
As further validation that some of the metabolic FLIM signal from macrophages in vitro and ex vivo in the live LG sections was associated with activation of NOX2, we evaluated its distribution by immunofluorescence. Exposure of RAW 264.7 cells to 100 ng/mL LPS for 24 h in vitro generates the classically activated M1 phenotype which includes activation of NOX2 [44,45], as confirmed in Figure 4A. NOX2 activation involves phosphorylation of p47phox (pP47phox) and its translocation, with other subunits to gp91phox, the transmembrane catalytic subunit[25]. Figure 6A and B reveal a marked increase in p47 and pP47 by immunofluorescence in LPS-stimulated macrophages, including detection of both p47 and pP47 within puncta associated with the plasma membrane in accord with NOX2 activation.
LG sections were photobleached to remove the significant autofluorescent lipofuscin signal associated with aging that we previously reported [33], prior to labeling with antibodies to p47phox and pP47phox. In old LG, we detected multinucleated macrophages labeled with F4/80 (Figure 7A, B). These multinucleated macrophages contained both p47phox (p47) and pP47phox, whereas the occasional single nucleated macrophages in the young LG labeled with F4/80 primarily located between acinar cells did not label strongly for the pP47phox. This suggests that the activated transmembrane NOX2, which should be bound to the pP47, is concentrated in the foamy multinucleated macrophages in the old LG.
4. Discussion
Authors Oxidative stress due to diminished cellular ability to neutralize free radicals including reactive oxygen species (ROS) generated by metabolic processes is a hallmark of aging. In conjunction with inflammation, it is considered to be a principal driving factor in many age-related diseases. Oxidative stress can induce a shift in cellular energy metabolism from oxidative phosphorylation to glycolysis, in part as an adaptive mechanism to restore redox balance through increased availability of NADPH which can neutralize ROS. Mediators of the oxidative stress-induced shift towards glycolysis include hypoxia inducible factor 1 (HIF-1), which is known to be activated as a protective response to prevent acinar cell death in DED [46]. In the comparison between the FLIM phasors of live young and old mouse LG sections, we were able to clearly observe that the young LG exhibited characteristics of a higher OxPhos profile versus the old LG, suggesting that the old LG are less metabolically active. This is also evidenced by the buildup of lipofuscin in the aged LG which is clearly present in the phasor as a long tail (Figure 5A, blue circle). These findings are in accord with prior publications indicating the increase in lipofuscin accumulation in mouse tissues as early as 8 months [15,47]. Increased oxidative stress has previously been correlated with aging and development of DED in the LG in both humans and mice utilizing different technologies [18,48,49]. In fact, activation of Nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of antioxidant defenses, has recently been proposed as a therapeutic strategy to treat DED [18]. Although a role for oxidative stress in aging and development of DED has been proposed, the current study is the first demonstration of such an effect using FLIM. In addition to studying the aging process, this technique has potential to rapidly screen the ability of various agents to effectively reduce oxidative stress in the LG.
While it is true that the FLIM profile is illustrative of general oxidative stress and increased glycolytic metabolism in old LG tissue, some of the whole tissue phasor data may be misleading if we do not take into consideration the high levels of accumulation of lipofuscin in macrophages that has been previously reported with aging in the LG [15,33,47] and elsewhere [50] in aged mice. Perhaps this is not surprising as macrophages are abundant in lysosomes, which are the primary site for lipofuscin deposition. This strong pull of the lipofuscin signal toward the shorter lifetime components creates a false sense that glycolytic metabolism is predominant, which is not the case specifically for these multinucleate foamy macrophages in aged LG. Therefore, we use the LMA rather than a usual metabolic axis and because lipofuscin has a single location in the phasor, we can deconvolve out the metabolic signal from these pixels. We did this experimentally in Figure 5 by shifting the dynamic range of FLIM analysis toward the lipofuscin signal, enabling us to tease out the activated NOX2 signal from all the other metabolic signals within the lipofuscin-filled macrophage.
The multinucleated macrophages that we previously characterized in the old LG [33] are F4/80-positive and enriched in lipid metabolizing enzymes [33], but their phenotype (M1 versus M2) was not characterized. However, other foamy macrophages have been previously suggested to contribute to tissue pathology. Both in atherosclerosis and non-alcoholic fatty liver disease, diseases associated with lipid deposition, the M1 type macrophage seem to exacerbate the disease[51,52,53,54]. The apparent increase in NOX2 activity in foamy multinucleate macrophages in the LG would likewise suggest a pro-inflammatory phenotype that may contribute to DED pathology. Although FLIM imaging suggests increased NOX2 in F4/80 positive multinucleate foamy macrophages, other approaches may be useful in determining whether this phenotype is universal for all macrophages in the LG, or whether this population has additional heterogeneity.
Here we present a characterization of aged LG from mice exhibiting DED having a decreased OxPhos metabolic profile with a discrete lipofuscin content. Building upon our prior study of multinucleate macrophages that contain lipid metabolizing enzymes and cholesterol transporters and are enriched in F4/80, we have identified these cells to have an activated NOX2 profile, which suggests their classically activated, pro-inflammatory phenotype. This observation by FLIM is consistent with other data showing increased expression of NOX2 components in the LG by RNASeq and by the increased pP47phox signal in multinucleate macrophages in aged LG. Further studies will explore the modulation of these macrophages in order to treat or prevent age-related DED.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Diagram of the FLIM phasor semi-circle, Figure S2: Phasor map of NOX2 metabolic axis (Nx2-axis) and lipofuscin-metabolic axis (LMA).
Author Contributions
Conceptualization, M.C, J.J., M.C.E, C.S.d.P. and S.H.A.; methodology, M.C, and J.J. C.T, A.R.M. and C.S.d.P; formal analysis M.C, J.J and , C.S.d.P..; writing—original draft preparation, M.C, J.J. and S.H.A; writing—review and editing, All.; visualization, M.C, J.J. and C.T and A.R.M.; funding acquisition, C.S.d.P and S.H.A. All authors have read and agreed to the published version of the manuscript.
Funding
Supported by the following grants from the National Eye Institute, National Institutes of Health: R01EY030447 to C.S.d.P., R01EY011386 to S.H.A, and R01EY026635 to S.H.A. Researh was also supported by the following Core grants: P30EY002520 to the Department of Ophthalmology at Baylor College of Medicine, P30EY029220 to the Department of Ophthalmology at USC Keck School of Medicine; and P30EY021725 to Baylor College of Medicine). Further support was provided by unrestricted grants from Research to Prevent Blindness to the Department of Ophthalmology at Baylor College of Medicine and the Roski Eye Institute, Department of Ophthalmology, USC Keck School of Medicine. The Hamill Foundation, The Sid Richardson Foundation, and P30 Cancer Center Support Grant (NCI-CA125123) which supports the Human Tissue Acquisition and Pathology Core at Baylor College of Medicine also supported the project. C.S.d.P holds the Caroline F. Elles Endowed Professorship, which provides salary support.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
M. Choi, None; J. Junge, none; C. Toscano, None; A.R. Mulay, none; M.C. Edman, None; C.S. de Paiva, None; S.F. Hamm-Alvarez, None.
Abbreviations
The following abbreviations are used in this manuscript:
| B6 | C57BL/6 |
| DED | Dry Eye Disease |
| FLIM | Fluorescence Lifetime Imaging Microscopy |
| IFN | Interferon |
| LG | Lacrimal Gland |
| LPS | lipopolysaccharides |
| LMA | lipofuscin-metabolic axis |
| NAPDH | Nicotinamide Adenine Dinucleotide Phosphate |
| NOX2 | NADPH oxidase 2 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| Nx2-axis | NOX2 metabolic axis |
| OxPhos | Oxidative phosphorylation |
| TBS | Tris Buffered Saline |
References
- World Health Organization. Aging and health. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on May 25).
- Collaborators, U.S.B.o.D.; Mokdad, A.H.; Ballestros, K.; Echko, M.; Glenn, S.; Olsen, H.E.; Mullany, E.; Lee, A.; Khan, A.R.; Ahmadi, A.; et al. The State of US Health, 1990-2016: Burden of Diseases, Injuries, and Risk Factors Among US States. JAMA 2018, 319, 1444–1472. [Google Scholar] [CrossRef] [PubMed]
- Benavidez, G.A.; Zahnd, W.E.; Hung, P.; Eberth, J.M. Chronic Disease Prevalence in the US: Sociodemographic and Geographic Variations by Zip Code Tabulation Area. Prev Chronic Dis 2024, 21, E14. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Preventing Chronic Diseases: What You Can Do Now. Available online: https://www.cdc.gov/chronic-disease/prevention/index.html (accessed on May 25).
- Papas, E.B. The global prevalence of dry eye disease: A Bayesian view. Ophthalmic Physiol Opt 2021, 41, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Schaumberg, D.A.; Dana, R.; Buring, J.E.; Sullivan, D.A. Prevalence of dry eye disease among US men: estimates from the Physicians' Health Studies. Arch Ophthalmol 2009, 127, 763–768. [Google Scholar] [CrossRef]
- Schaumberg, D.A.; Sullivan, D.A.; Buring, J.E.; Dana, M.R. Prevalence of dry eye syndrome among US women. Am J Ophthalmol 2003, 136, 318–326. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; Stern, M.E. Biological functions of tear film. Exp Eye Res 2020, 197, 108115. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, S.Z.; Koh, S.K.; Chen, L.; Vaz, C.; Tanavde, V.; Li, X.R.; Beuerman, R.W. In-depth analysis of the human tear proteome. Journal of proteomics 2012, 75, 3877–3885. [Google Scholar] [CrossRef]
- Nasu, M.; Matsubara, O.; Yamamoto, H. Post-mortem prevalence of lymphocytic infiltration of the lacrymal gland: a comparative study in autoimmune and non-autoimmune diseases. J Pathol 1984, 143, 11–15. [Google Scholar] [CrossRef]
- Obata, H.; Yamamoto, S.; Horiuchi, H.; Machinami, R. Histopathologic study of human lacrimal gland. Statistical analysis with special reference to aging. Ophthalmology 1995, 102, 678–686. [Google Scholar] [CrossRef]
- Bukhari, A.A.; Basheer, N.A.; Joharjy, H.I. Age, gender, and interracial variability of normal lacrimal gland volume using MRI. Ophthalmic Plast Reconstr Surg 2014, 30, 388–391. [Google Scholar] [CrossRef]
- Roen, J.L.; Stasior, O.G.; Jakobiec, F.A. Aging changes in the human lacrimal gland: role of the ducts. The CLAO journal : official publication of the Contact Lens Association of Ophthalmologists, Inc 1985, 11, 237–242. [Google Scholar] [PubMed]
- Schein, O.D.; Tielsch, J.M.; Munoz, B.; Bandeen-Roche, K.; West, S. Relation between signs and symptoms of dry eye in the elderly. A population-based perspective. Ophthalmology 1997, 104, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Alves, M.; Rios, J.D.; Dartt, D.A. The aging lacrimal gland: changes in structure and function. The ocular surface 2008, 6, 162–174. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Volpe, E.A.; Zhang, X.; Darlington, G.J.; Li, D.Q.; Pflugfelder, S.C.; de Paiva, C.S. Ocular surface disease and dacryoadenitis in aging C57BL/6 mice. Am J Pathol 2014, 184, 631–643. [Google Scholar] [CrossRef]
- Galletti, J.G.; Scholand, K.K.; Trujillo-Vargas, C.M.; Yu, Z.; Mauduit, O.; Delcroix, V.; Makarenkova, H.P.; de Paiva, C.S. Ectopic lymphoid structures in the aged lacrimal glands. Clin Immunol 2023, 248, 109251. [Google Scholar] [CrossRef]
- de Souza, R.G.; Yu, Z.; Hernandez, H.; Trujillo-Vargas, C.M.; Lee, A.; Mauk, K.E.; Cai, J.; Alves, M.R.; de Paiva, C.S. Modulation of Oxidative Stress and Inflammation in the Aged Lacrimal Gland. Am J Pathol 2021, 191, 294–308. [Google Scholar] [CrossRef]
- Trujillo-Vargas, C.M.; Mauk, K.E.; Hernandez, H.; de Souza, R.G.; Yu, Z.; Galletti, J.G.; Dietrich, J.; Paulsen, F.; de Paiva, C.S. Immune phenotype of the CD4(+) T cells in the aged lymphoid organs and lacrimal glands. Geroscience 2022, 44, 2105–2128. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. Journal of cellular physiology 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Frontiers in immunology 2019, 10, 792. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Dinauer, M.C. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood 2019, 133, 2130–2139. [Google Scholar] [CrossRef]
- Alfonso-Garcia, A.; Smith, T.D.; Datta, R.; Luu, T.U.; Gratton, E.; Potma, E.O.; Liu, W.F. Label-free identification of macrophage phenotype by fluorescence lifetime imaging microscopy. J Biomed Opt 2016, 21, 46005. [Google Scholar] [CrossRef] [PubMed]
- Nocella, C.; D'Amico, A.; Cammisotto, V.; Bartimoccia, S.; Castellani, V.; Loffredo, L.; Marini, L.; Ferrara, G.; Testa, M.; Motta, G.; et al. Structure, Activation, and Regulation of NOX2: At the Crossroad between the Innate Immunity and Oxidative Stress-Mediated Pathologies. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.G. NADPH Oxidases Are Essential for Macrophage Differentiation. The Journal of biological chemistry 2016, 291, 20030–20041. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, M.; Aerts, J. The Unique Phenotype of Lipid-Laden Macrophages. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Gupta, G.; Athanikar, S.B.; Pai, V.V.; Naveen, K.N. Giant cells in dermatology. Indian J Dermatol 2014, 59, 481–484. [Google Scholar] [CrossRef]
- Aterman, K.; Remmele, W.; Smith, M. Karl Touton and his "xanthelasmatic giant cell." A selective review of multinucleated giant cells. Am J Dermatopathol 1988, 10, 257–269. [Google Scholar] [CrossRef]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin Immunol 2008, 20, 86–100. [Google Scholar] [CrossRef]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9. [Google Scholar] [CrossRef]
- Pagan, A.J.; Ramakrishnan, L. The Formation and Function of Granulomas. Annual review of immunology 2018, 36, 639–665. [Google Scholar] [CrossRef]
- Choi, M.; Toscano, C.; Edman, M.C.; de Paiva, C.S.; Hamm-Alvarez, S.F. The Aging Lacrimal Gland of Female C57BL/6J Mice Exhibits Multinucleate Macrophage Infiltration Associated With Lipid Dysregulation. Invest Ophthalmol Vis Sci 2024, 65, 1. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.W.; Arnesano, C.; Harutyunyan, N.; Khuu, T.; Martinez, J.C.; Pollack, H.A.; Koos, D.S.; Lee, T.C.; Fraser, S.E.; Moats, R.A.; et al. Structural and Functional Characterization of Human Stem-Cell-Derived Retinal Organoids by Live Imaging. Invest Ophthalmol Vis Sci 2017, 58, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Colyer, R.A.; Lee, C.; Gratton, E. A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc Res Tech 2008, 71, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical journal 2008, 94, L14–16. [Google Scholar] [CrossRef]
- Kolenc, O.I.; Quinn, K.P. Evaluating Cell Metabolism Through Autofluorescence Imaging of NAD(P)H and FAD. Antioxid Redox Signal 2018. [Google Scholar] [CrossRef]
- Stringari, C.; Edwards, R.A.; Pate, K.T.; Waterman, M.L.; Donovan, P.J.; Gratton, E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci Rep 2012, 2, 568. [Google Scholar] [CrossRef]
- Stringari, C.; Nourse, J.L.; Flanagan, L.A.; Gratton, E. Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PloS one 2012, 7, e48014. [Google Scholar] [CrossRef]
- Wright, B.K.; Andrews, L.M.; Jones, M.R.; Stringari, C.; Digman, M.A.; Gratton, E. Phasor-FLIM analysis of NADH distribution and localization in the nucleus of live progenitor myoblast cells. Microsc Res Tech 2012, 75, 1717–1722. [Google Scholar] [CrossRef]
- Henriksson, J.T.; de Paiva, C.S.; Farley, W.; Pflugfelder, S.C.; Burns, A.R.; Bergmanson, J.P. Morphologic Alterations of the Palpebral Conjunctival Epithelium in a Dry Eye Model. Cornea 2012. [Google Scholar] [CrossRef]
- Wang, P.; Hecht, F.; Ossato, G.; Tille, S.; Fraser, S.E.; Junge, J.A. Complex wavelet filter improves FLIM phasors for photon starved imaging experiments. Biomed Opt Express 2021, 12, 3463–3473. [Google Scholar] [CrossRef]
- Ranjit, S.; Datta, R.; Dvornikov, A.; Gratton, E. Multicomponent Analysis of Phasor Plot in a Single Pixel to Calculate Changes of Metabolic Trajectory in Biological Systems. J Phys Chem A 2019, 123, 9865–9873. [Google Scholar] [CrossRef]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Chen, T.L.; Chen, R.M. Lipopolysaccharide triggers macrophage activation of inflammatory cytokine expression, chemotaxis, phagocytosis, and oxidative ability via a toll-like receptor 4-dependent pathway: validated by RNA interference. Toxicol Lett 2009, 191, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Ji, Y.W.; Lee, S.M.; Shim, J.; Noh, H.; Yeo, A.; Park, C.; Park, M.S.; Chang, E.J.; Lee, H.K. Activation of HIF-1alpha (hypoxia inducible factor-1alpha) prevents dry eye-induced acinar cell death in the lacrimal gland. Cell death & disease 2014, 5, e1309. [Google Scholar] [CrossRef]
- Rios, J.D.; Horikawa, Y.; Chen, L.L.; Kublin, C.L.; Hodges, R.R.; Dartt, D.A.; Zoukhri, D. Age-dependent alterations in mouse exorbital lacrimal gland structure, innervation and secretory response. Exp Eye Res 2005, 80, 477–491. [Google Scholar] [CrossRef]
- Damato, B.E.; Allan, D.; Murray, S.B.; Lee, W.R. Senile atrophy of the human lacrimal gland: the contribution of chronic inflammatory disease. Br J Ophthalmol 1984, 68, 674–680. [Google Scholar] [CrossRef]
- Sacca, S.C.; Cutolo, C.A.; Ferrari, D.; Corazza, P.; Traverso, C.E. The Eye, Oxidative Damage and Polyunsaturated Fatty Acids. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Vida, C.; de Toda, I.M.; Cruces, J.; Garrido, A.; Gonzalez-Sanchez, M.; De la Fuente, M. Role of macrophages in age-related oxidative stress and lipofuscin accumulation in mice. Redox Biol 2017, 12, 423–437. [Google Scholar] [CrossRef]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2016, 63, 1310–1324. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep 2017, 7, 44612. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phasor approach to FLIM analysis and trends in the phasor as percentage of NAD(P)H-enzyme binding changes. Each image is a snapshot of metabolic activity where fluorescence decays of NAD(P)H are recorded for each pixel in an image. These fluorescence decay curves are transformed using FAST Fourier algorithms as Cosine and Sine functions whose coefficients are plotted into the metabolic phasor as G and S coordinates (G, S) respectively, where each image-pixel has a corresponding pixel mapped in the phasor plot. The location of each phasor-pixel relates to both the percentage of NAD(P)H bound to enzyme and reflects the combination of enzymes in a life-tissue sample. (A) This roadmap for the metabolic phasor shows how analysis of NAD(P)H-binding percentages is performed and applied to real data of NAD(P)H-bound percentages to Glycerol-3-phosphate dehydrogenase (G3PDH) for the combined phasors for images B-D. The rainbow look-up table is the colorimetric scale bar used to reflect percentages of bound NADH in this analysis. (B) Pure NADH in PBS has 0% bound, (C) higher concentrations of NADH versus G3PDH has variable binding percentages of 30-95% bound, and (D) excess G3PDH where 98% of NADH is bound.
Figure 1.
Phasor approach to FLIM analysis and trends in the phasor as percentage of NAD(P)H-enzyme binding changes. Each image is a snapshot of metabolic activity where fluorescence decays of NAD(P)H are recorded for each pixel in an image. These fluorescence decay curves are transformed using FAST Fourier algorithms as Cosine and Sine functions whose coefficients are plotted into the metabolic phasor as G and S coordinates (G, S) respectively, where each image-pixel has a corresponding pixel mapped in the phasor plot. The location of each phasor-pixel relates to both the percentage of NAD(P)H bound to enzyme and reflects the combination of enzymes in a life-tissue sample. (A) This roadmap for the metabolic phasor shows how analysis of NAD(P)H-binding percentages is performed and applied to real data of NAD(P)H-bound percentages to Glycerol-3-phosphate dehydrogenase (G3PDH) for the combined phasors for images B-D. The rainbow look-up table is the colorimetric scale bar used to reflect percentages of bound NADH in this analysis. (B) Pure NADH in PBS has 0% bound, (C) higher concentrations of NADH versus G3PDH has variable binding percentages of 30-95% bound, and (D) excess G3PDH where 98% of NADH is bound.
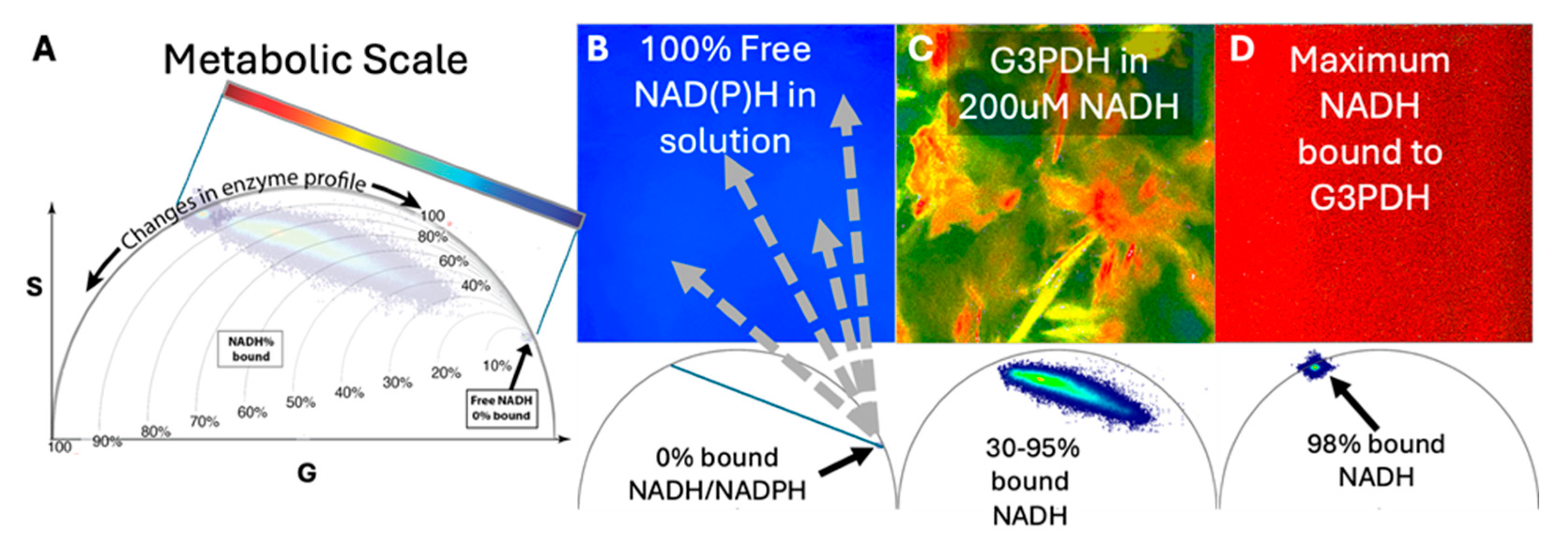
Figure 2.
Metabolic FLIM imaging of young versus old LG. (A) The metabolic phasors show both the differences of metabolism in old versus young LG as well as the well-documented prevalence of lipofuscin in aged LGs displayed in the distribution’s tail pointing to the lower right of the phasor. These lipofuscin-associated phasor-pixels have been masked out in black so they do not appear in the corresponding old LG images, otherwise the rainbow look-up table are consistent between the two phasors for direct comparison between young and old LGs. (B) The young LG shows a more regular distribution of metabolic signals with higher OxPhos on the outer side of the acini, while the old LG have an uneven metabolic distribution across the tissue and seem to show less OxPphos signal. However, we cannot rule out that this metabolic shift is due in part to lipofuscin signal and potentially contributes to a likely false glycolytic shift. Images at 5X magnification of each highlighted area (white box) are shown below images of the whole tissue section. Scale bars: 500 µm and 100 µm (Insets), respectively. Phasors are comprised of 3 layers (25%, 50%, and 75% depth) in n=3 LG/group, thus 9 image composites per phasor.
Figure 2.
Metabolic FLIM imaging of young versus old LG. (A) The metabolic phasors show both the differences of metabolism in old versus young LG as well as the well-documented prevalence of lipofuscin in aged LGs displayed in the distribution’s tail pointing to the lower right of the phasor. These lipofuscin-associated phasor-pixels have been masked out in black so they do not appear in the corresponding old LG images, otherwise the rainbow look-up table are consistent between the two phasors for direct comparison between young and old LGs. (B) The young LG shows a more regular distribution of metabolic signals with higher OxPhos on the outer side of the acini, while the old LG have an uneven metabolic distribution across the tissue and seem to show less OxPphos signal. However, we cannot rule out that this metabolic shift is due in part to lipofuscin signal and potentially contributes to a likely false glycolytic shift. Images at 5X magnification of each highlighted area (white box) are shown below images of the whole tissue section. Scale bars: 500 µm and 100 µm (Insets), respectively. Phasors are comprised of 3 layers (25%, 50%, and 75% depth) in n=3 LG/group, thus 9 image composites per phasor.
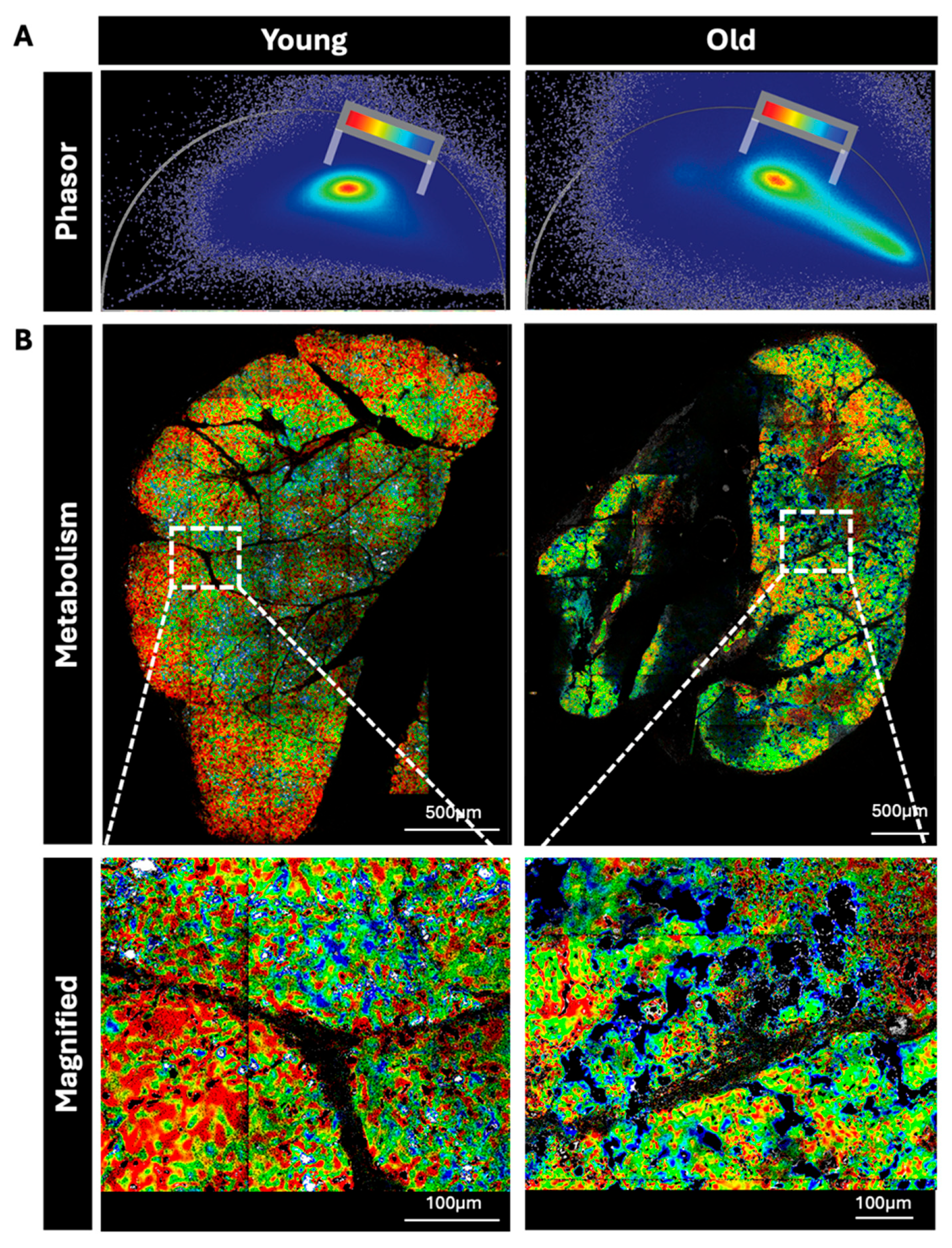
Figure 3.
Identification of multinucleate macrophages in old LG sections. A) Immunofluorescence labeling of photobleached sections from old LG shows F4/80 positive (green) multinucleate macrophages (white arrows). A macrophage with a single nucleus also labeled with F4/80 is also shown in the same panel (white arrowhead) and *, luminal regions. Scale Bar= 10 µm B) H&E staining of sections from old LG reveals multinucleate macrophages. The apparent boundaries of these macrophages in tissue is marked in black. Scale Bar= 50 µm.
Figure 3.
Identification of multinucleate macrophages in old LG sections. A) Immunofluorescence labeling of photobleached sections from old LG shows F4/80 positive (green) multinucleate macrophages (white arrows). A macrophage with a single nucleus also labeled with F4/80 is also shown in the same panel (white arrowhead) and *, luminal regions. Scale Bar= 10 µm B) H&E staining of sections from old LG reveals multinucleate macrophages. The apparent boundaries of these macrophages in tissue is marked in black. Scale Bar= 50 µm.
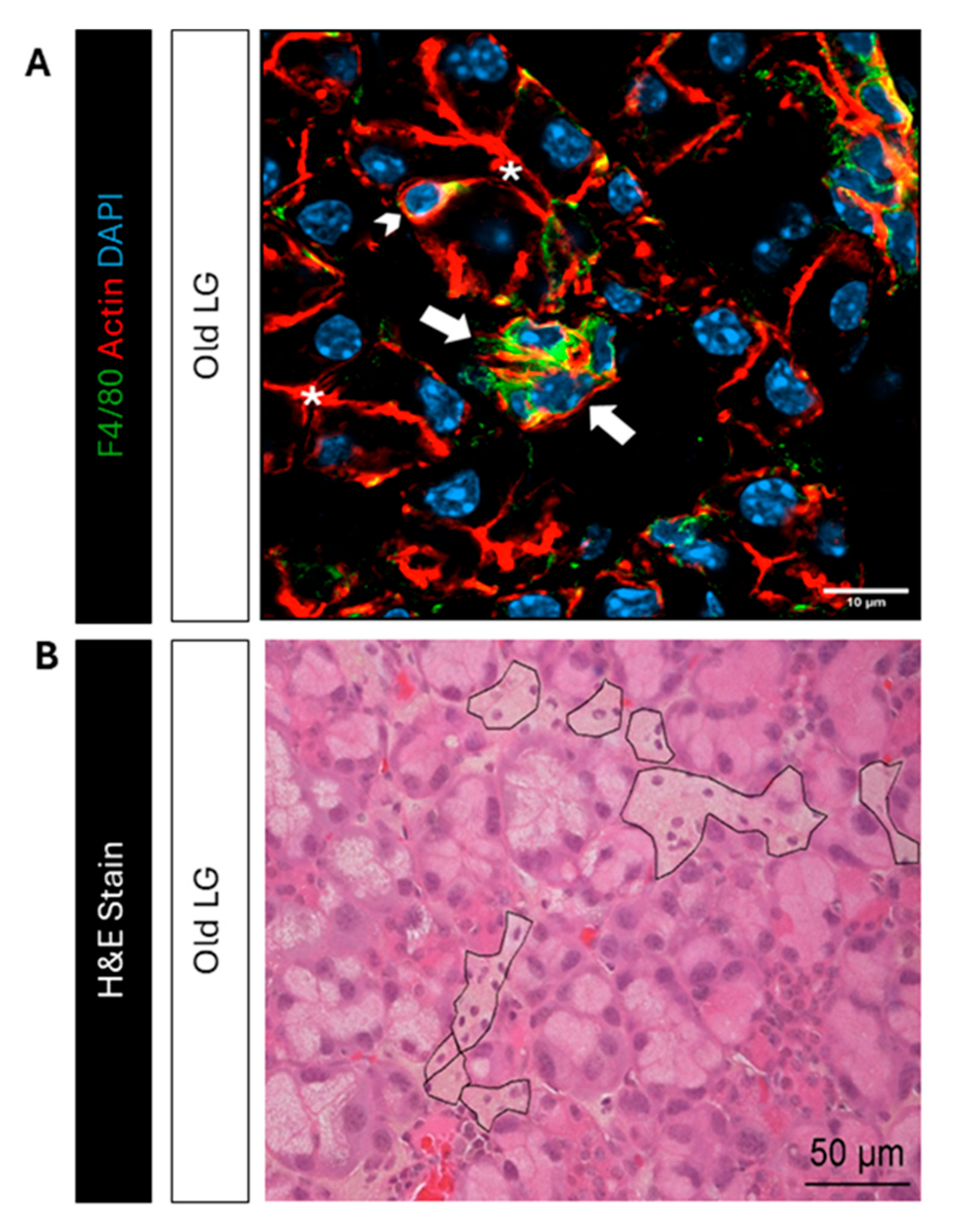
Figure 4.
Immunoprecipitation of NOX2 with FLIM imaging and Western blot analyses. (A) Macrophages in culture were imaged for metabolic FLIM before and after treatment with LPS, exhibiting an appropriate and expected shift in the metabolic phasor (B) IP beads from untreated versus LPS-treated macrophages show a difference in signal, the side distribution (marked in white arrows) not included in the colorimetric analysis arises from background autofluorescence of the beads. (C) Western blotting of NOX2. The first 10 lanes of each blot show the washes from the beads from unstimulated (-) and stimulated (+) lysates. In the first few washes, cross-reactive material was eluted from beads containing both samples, which was not apparent in the last few washes. The last two lanes are the beads loaded with LPS- and LPS+ samples. The left membrane was exposed to the goat anti-mouse NOX2 antibody plus the anti-goat IR680 antibody, while the right membrane was only exposed to the anti-goat IR680 antibody.
Figure 4.
Immunoprecipitation of NOX2 with FLIM imaging and Western blot analyses. (A) Macrophages in culture were imaged for metabolic FLIM before and after treatment with LPS, exhibiting an appropriate and expected shift in the metabolic phasor (B) IP beads from untreated versus LPS-treated macrophages show a difference in signal, the side distribution (marked in white arrows) not included in the colorimetric analysis arises from background autofluorescence of the beads. (C) Western blotting of NOX2. The first 10 lanes of each blot show the washes from the beads from unstimulated (-) and stimulated (+) lysates. In the first few washes, cross-reactive material was eluted from beads containing both samples, which was not apparent in the last few washes. The last two lanes are the beads loaded with LPS- and LPS+ samples. The left membrane was exposed to the goat anti-mouse NOX2 antibody plus the anti-goat IR680 antibody, while the right membrane was only exposed to the anti-goat IR680 antibody.
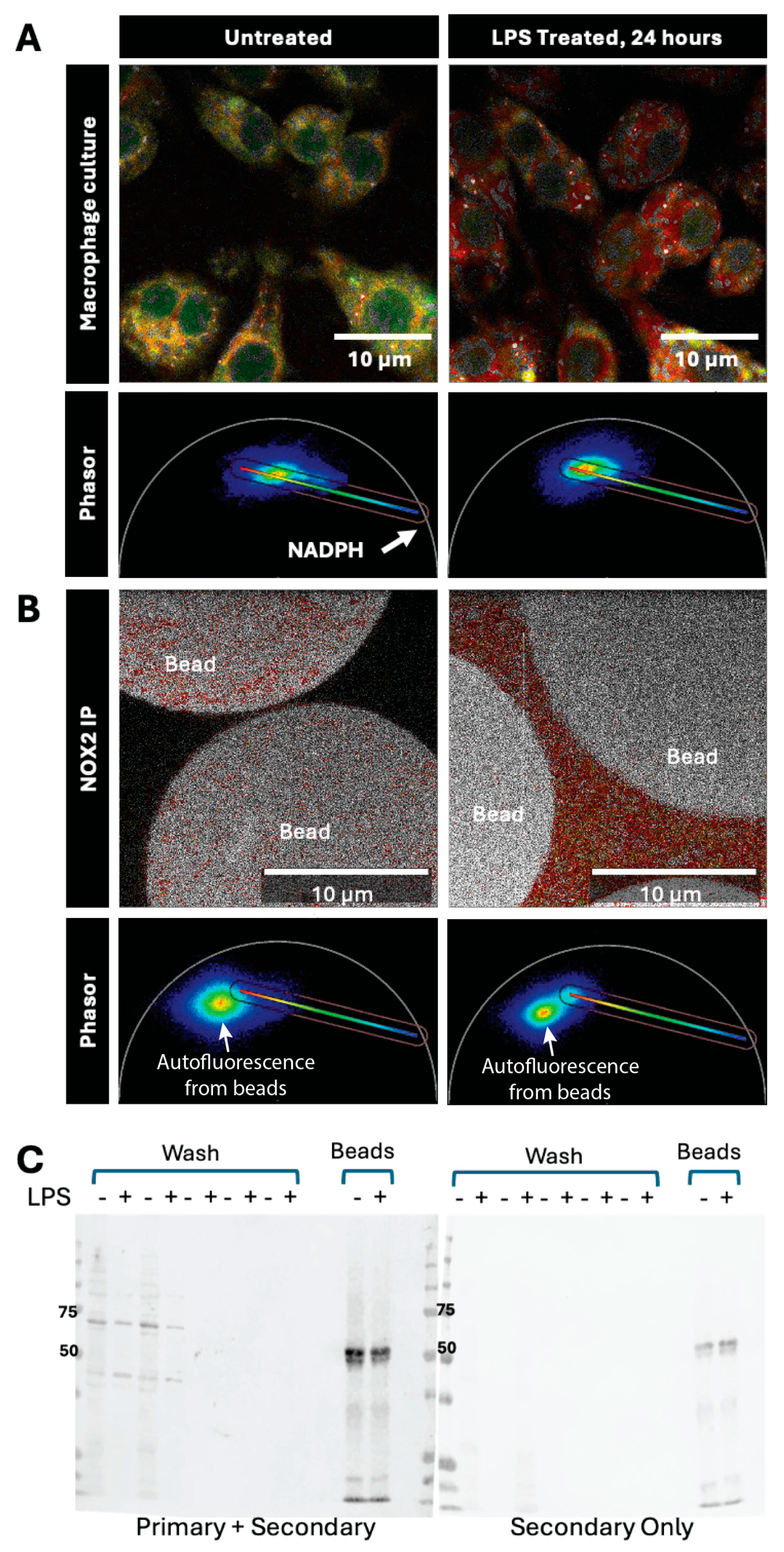
Figure 5.
FLIM imaging of fluorophores and metabolism in multinucleate macrophages in live-stained old LG. (A) 2 images of the same old LG are shown with different signals in the respective columns. The left column shows single-photon FLIM analysis of acridine orange (green) and F4/80 (magenta) labeling highlighted from phasor-selected pixels, while the right column shows metabolic signal arising from F4/80 positive pixels with the colorimetric scale bar shown in the phasor applied. The blue pixels appear confounded with lipofuscin (blue) but the metabolic signal is apparent in the green to red pixels. Higher metabolic signature originates from regions close to the cytoplasmic membrane of the soma and filopodia (arrowheads). Scale bar: 20 µm. (B) The left column shows the young LG data while the right column shows old LG data. A magnified section of each LG is shown in the following row. Both phasor schemes are the same as the one used in the right column of panel A. The majority of metabolic signal is masked out with black in the magnified images while it is not masked out in whole LG images.
Figure 5.
FLIM imaging of fluorophores and metabolism in multinucleate macrophages in live-stained old LG. (A) 2 images of the same old LG are shown with different signals in the respective columns. The left column shows single-photon FLIM analysis of acridine orange (green) and F4/80 (magenta) labeling highlighted from phasor-selected pixels, while the right column shows metabolic signal arising from F4/80 positive pixels with the colorimetric scale bar shown in the phasor applied. The blue pixels appear confounded with lipofuscin (blue) but the metabolic signal is apparent in the green to red pixels. Higher metabolic signature originates from regions close to the cytoplasmic membrane of the soma and filopodia (arrowheads). Scale bar: 20 µm. (B) The left column shows the young LG data while the right column shows old LG data. A magnified section of each LG is shown in the following row. Both phasor schemes are the same as the one used in the right column of panel A. The majority of metabolic signal is masked out with black in the magnified images while it is not masked out in whole LG images.
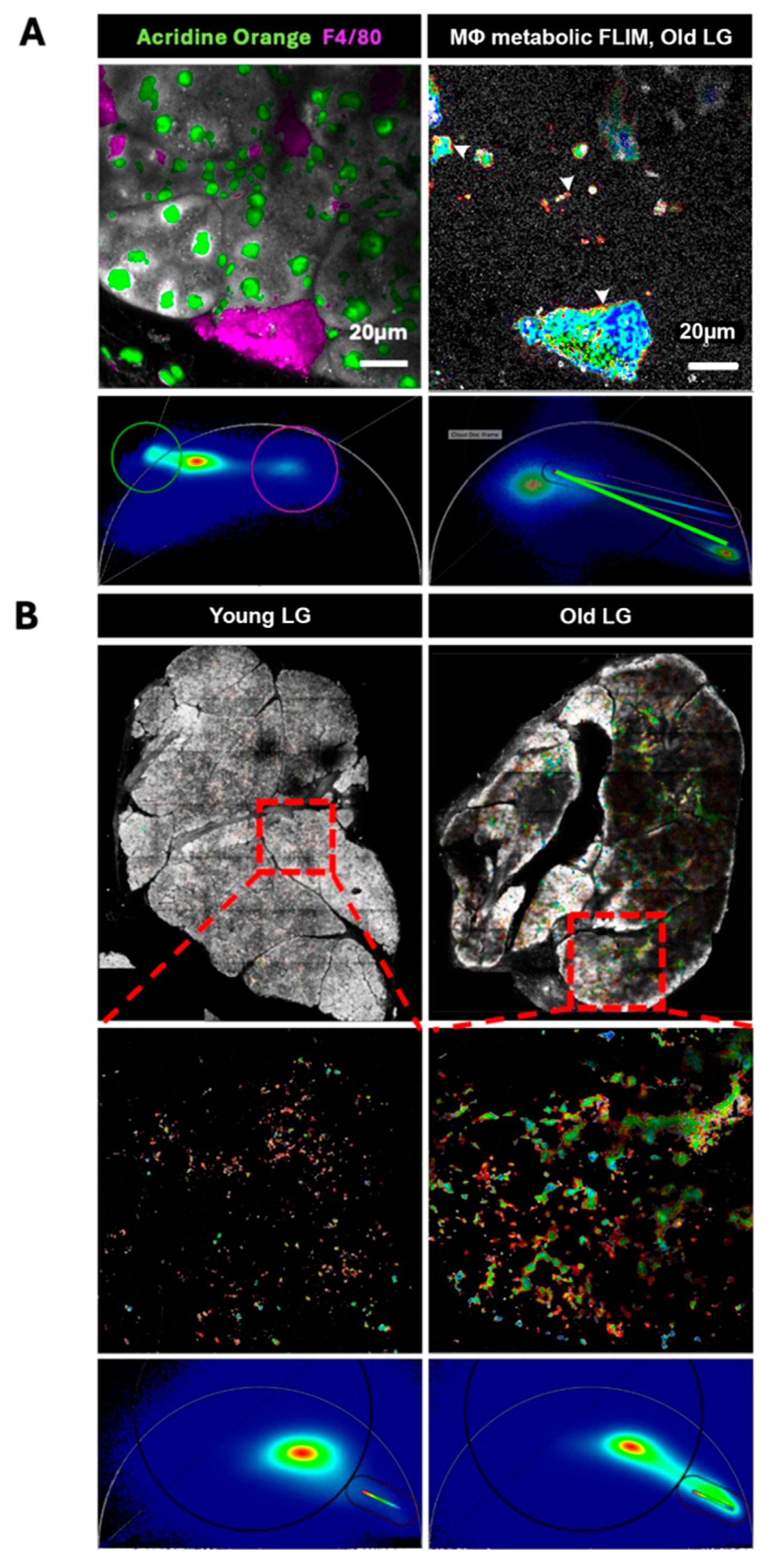
Figure 6.
Immunofluorescence labeling reveals that LPS-stimulated RAW 264.7 cells have higher P47phox and pP47phox. (A) RAW 264.7 cells express P47phox. LPS stimulation results in higher signal intensity and accumulation of immunofluorescence associated with P47phox (green) in puncta on the membrane. Scale bar: 20 µm (B) While there is residual pP47phox (green) signal in non-stimulated macrophages, LPS-stimulated macrophages show a much greater immunofluorescence signal, also localized to puncta (green) on the plasma membrane. Green arrows indicate the accumulations of pP47phox in LPS-stimulated cells. Scale bar: 20 µm.
Figure 6.
Immunofluorescence labeling reveals that LPS-stimulated RAW 264.7 cells have higher P47phox and pP47phox. (A) RAW 264.7 cells express P47phox. LPS stimulation results in higher signal intensity and accumulation of immunofluorescence associated with P47phox (green) in puncta on the membrane. Scale bar: 20 µm (B) While there is residual pP47phox (green) signal in non-stimulated macrophages, LPS-stimulated macrophages show a much greater immunofluorescence signal, also localized to puncta (green) on the plasma membrane. Green arrows indicate the accumulations of pP47phox in LPS-stimulated cells. Scale bar: 20 µm.
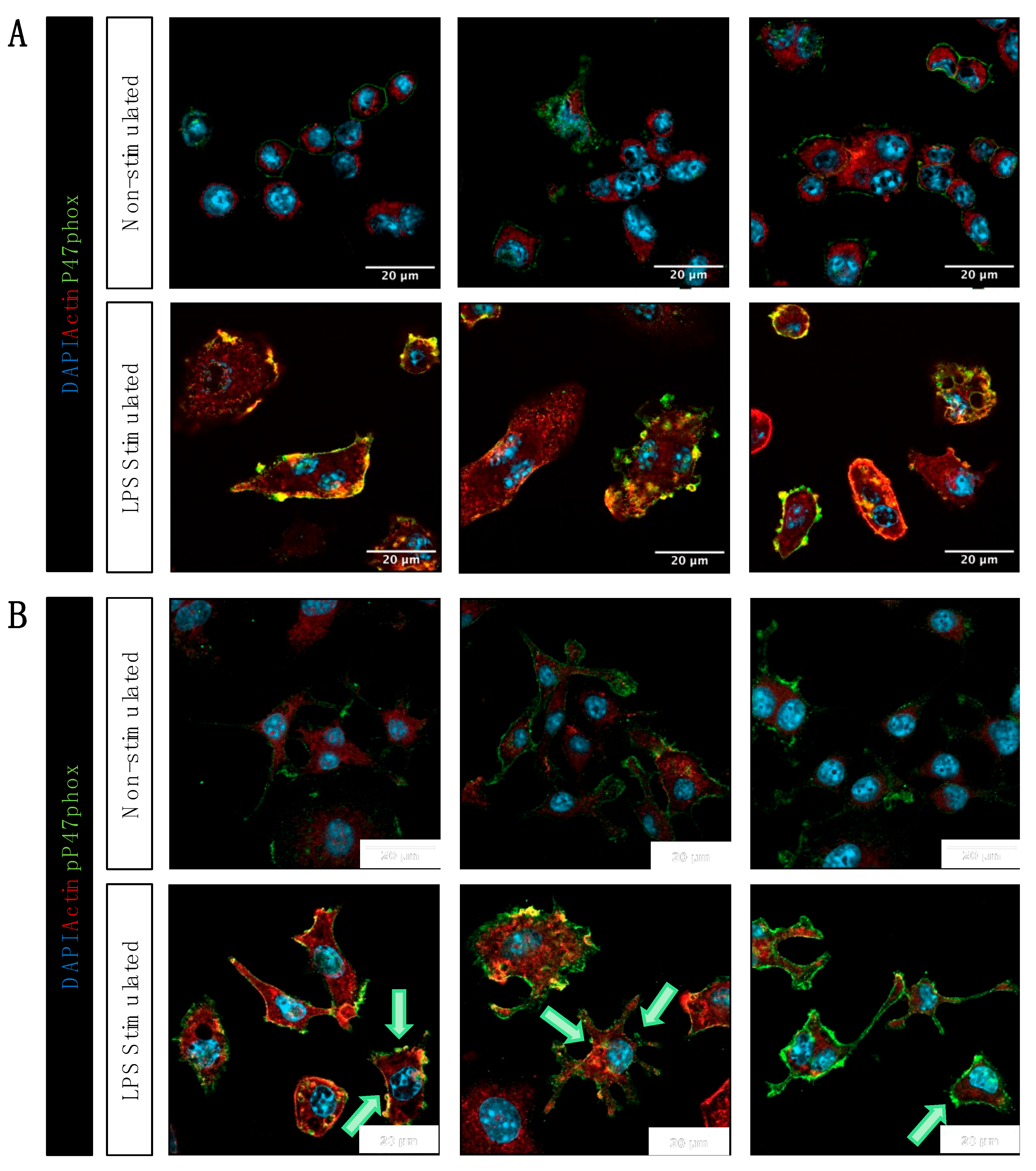
Figure 7.
Immunofluorescence labeling of shows increased phospho-P47phox in multinucleate macrophages in old LG. (A) Young LG have fewer macrophages (magenta) while multinucleated macrophages are detected in the old LG. P47 labeling (green) is primarily detected in multinucleated macrophages in old LG. Scale bar: 20 µm (B) Only macrophages in old LG (magenta) show pP47phox signal (green) while none is detectable in macrophages in young LG. Scale bar: 500 µm. Results were obtained from n=3 mice/group. Images in columns labeled Mouse 1, Mouse 2, and Mouse 3 are from different mice at each age. Mouse 1, 2, and 3 are not necessarily the same across different panels or figures. Dashed lines outline groups of acinar cells in each image.
Figure 7.
Immunofluorescence labeling of shows increased phospho-P47phox in multinucleate macrophages in old LG. (A) Young LG have fewer macrophages (magenta) while multinucleated macrophages are detected in the old LG. P47 labeling (green) is primarily detected in multinucleated macrophages in old LG. Scale bar: 20 µm (B) Only macrophages in old LG (magenta) show pP47phox signal (green) while none is detectable in macrophages in young LG. Scale bar: 500 µm. Results were obtained from n=3 mice/group. Images in columns labeled Mouse 1, Mouse 2, and Mouse 3 are from different mice at each age. Mouse 1, 2, and 3 are not necessarily the same across different panels or figures. Dashed lines outline groups of acinar cells in each image.
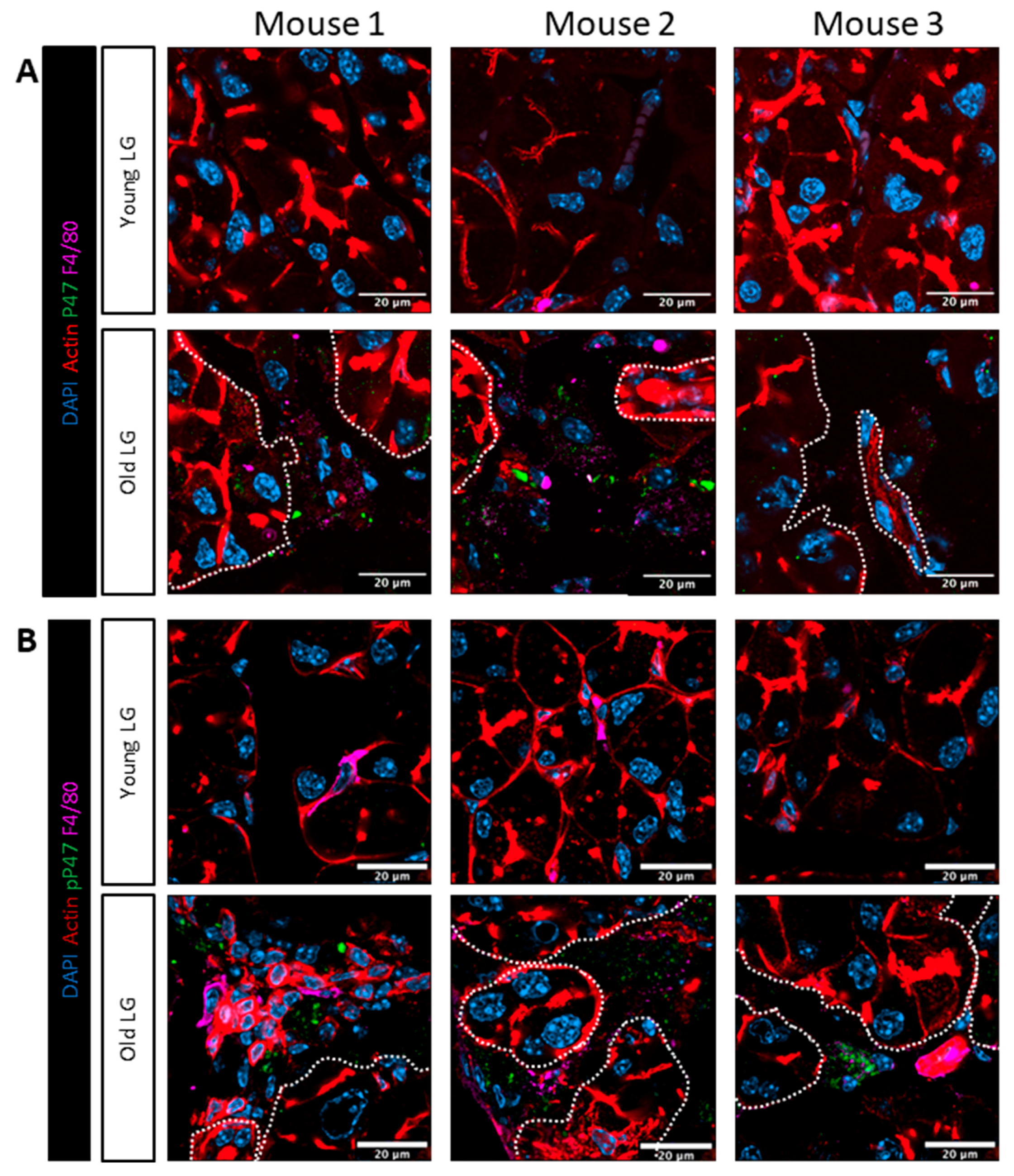

Table 1.
Gene expression of NOX2 isoforms in aged LGs showing log2fold change compared to young LGs.
Table 1.
Gene expression of NOX2 isoforms in aged LGs showing log2fold change compared to young LGs.
| Subunit | Gene | Log2 FoldChange | P adjusted |
|---|---|---|---|
| gp91-phox | Cybb | 1.26 | 2.53 E-09 |
| P22-phox | Cyba | 1.18 | 4.37 E-07 |
| P47-phox | Ncf1 | 1.60 | 7.67 E-07 |
| P67-phox | Ncf2 | 1.09 | 0.001 |
| P40-phox | Ncf4 | 1.64 | 2.4 E-05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



