
Submitted:
13 October 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
Plastics streams dominated by polyolefins and PVC demand a design framework that links synthesis to end-of-life reactivity. This work integrates polymerization-derived microstructure with depolymerization mechanisms to guide selective valorization. We synthesize mechanistic and kinetic evidence connecting coordination and radical polymerization (linear HDPE, branched LDPE, stereoregular PP; PVC with backbone C–Cl) to degradation pathways, and evaluate catalytic topologies (Brønsted/Lewis acidity, framework Al siting, micro/mesoporosity), initiators, and termination/quench strategies under relevant process variables (temperature, heating rate, vapor residence time, pressure). The analysis shows that microstructure prescribes reaction manifolds and attainable product slates: strong Brønsted acidity and shape-selective micropores favor C₂–C₄ olefins and BTX, whereas weaker acidity and hierarchical porosity preserve chain length to paraffinic oils/waxes; mesopore enrichment shortens contact times and suppresses secondary cracking; initiators lower onset energies and expand operability; diffusion management and surface passivation mitigate deactivation. For PVC, continuous HCl removal and basic/redox co-catalysts or ionic liquids lower dehydrochlorination temperatures and yield cleaner fractions, making staged dechlorination followed by residue cracking essential. Framing process design as “polymerization → structure → depolymerization” enables predictive yield targeting and energy-lean operation across mixed wastes, providing actionable guidance on catalyst selection, severity and residence-time control, regeneration, and integrated halogen management.
Keywords:
polyolefin pyrolysis
; catalytic pyrolysis
; reaction mechanism
; product selectivity
; vapor residence time
; initiators
; light olefins/BTX
1. Introduction
Diverting polyolefin- and PVC-rich waste away from landfills is no longer just a matter of space and leachate control; it is an opportunity to recover carbon as useful energy carriers and chemicals via thermal and catalytic valorization [1,2,3]. Across these materials, product selectivity is ultimately rooted in mechanism: the chain architectures encoded during polymerization (linear High Density Polyethylene (HDPE), branched Low Density Polyethylene (LDPE), stereoregular Polypropylene (PP), chlorine-bearing Polyvinyl Chloride (PVC) precondition how bonds will break under heat, determining whether the carbon flux ends up as cogeneration-grade gases, C2–C4 olefins/BTX, or paraffinic waxes and oils [1,2,3,4,5]. Strategic catalytic interventions—hydrogenolysis, metathesis, and acid-site-mediated cracking on micro/mesoporous frameworks—translate that mechanistic control into targeted product slates while minimizing waste formation [1,2,3].
For polyolefins, well-resolved kinetic and mechanistic studies converge on random C–C scission, β-scission, hydrogen abstraction, and backbiting as the primary routes that set the baseline gas vs. condensable yields [6,7]. Process severity and heat-flux history strongly modulate light-olefin formation; under high heating rates, pressure and heating rate redistribute diene/alkene/alkane balances, while parametric datasets with advanced analytics clarify how temperature and residence time steer primary volatiles without prolongcracking [8,9]. Mixed virgin/waste PP/LDPE systems exhibit lowered apparent barriers, enabling comparable conversions at milder temperatures—an operational advantage for waste streams [10]. Once acids are introduced, shape selectivity compresses distributions toward gasoline/BTX cuts over HZSM-5, whereas weaker acidity and larger pores, coupled with rapid quenching, preserve longer chains conducive to wax/oil fractions [11]. PP’s tertiary centers accentuate formation of C1–C4 and gasoline-range intermediates at moderate severities; temperature-programmed profiles delineate the window to pivot between off-gas for CHP and liquids for upgrading [12].
PVC requires a sequenced flowsheet. The first stage is controlled dehydrochlorination, governed by zipper-type autocatalysis captured by classical kinetics and supported by evidence for parallel unimolecular and radical pathways; a modern two-step kinetic picture aligns with these fundamentals and explicitly links the structure of the dehydrochlorinated residue to subsequent reactivity [13,14,15]. Selectivity and plant hygiene improve when HCl is continuously removed or scavenged: additives and processing strategies that curtail HCl activity slow the autocatalytic loop and redirect pathways [16]. Metal chlorides/oxides lower the onset temperature for dehydrochlorination and can suppress aromatic growth from polyenes, enabling cleaner gases and residues for downstream conversion [17,18]. When aromatics form, they arise via intramolecular cyclizations of conjugated sequences—mechanistic insight that justifies short residence and the use of mild basic/redox co-catalysts downstream to limit condensation [19]. Halogen–metal interactions matter operationally: Cu-based species markedly perturb HCl/VOC release profiles, underscoring the need for halogen guards and compatible metallurgy [20]. At the molecular scale, defect-assisted chlorine migration and TG-FTIR/EGA diagnostics on cable grades reinforce an analytics-led two-stage design (dechlorination → residue cracking) and help place capture media and catalysts effectively [21,22].
Catalyst microstructure—especially the balance of Brønsted/Lewis acidity, framework Al siting, and pore architecture—governs how polymer-derived intermediates traverse these degradation networks. Hierarchical ZSM-5 families and tuned acidity/alkalinity increase light-olefin and aromatic yields while suppressing coke by mitigating secondary hydrogen transfer and cyclization in tight micropores [23,24,25]. Micropore topology dictates where coke nucleates and grows, linking void geometry to deactivation kinetics and selectivity loss [26]. Precise placement of framework Al can tilt pathways toward primary β-scission over secondary oligomerization, boosting olefin formation from LDPE/PP feeds [27].
Mechanistically, acidity-aware molecular kinetic models and first-principles microkinetics illuminate when monomolecular cracking, bimolecular hydride transfer, or aromatization dominate, and how temperature or acid-site density toggles among these regimes [28,29]. Zeolite-confined β-scission barriers depend jointly on hydrocarbon structure and pore confinement, providing predictive handles to match feed microstructure (branching, unsaturation) with catalyst topology [30].
Operational levers couple with microstructure to steer outcomes. CO2 co-feeds promote dehydrogenation and shift equilibria toward aromatics over mesoporous HZSM-5 and Ga/ZSM-5, reducing co-produced H2 [31]. In PP/PE upgrading, mesopore enrichment shortens contact times and curbs secondary reactions, raising propylene/olefin selectivity without deep cracking [32]. Phase-aware residence-time control and staged/co-feed strategies modulate pool chemistry, enabling concurrent formation of ethylene/propylene and para-xylene on a single catalyst bed when architecture and conditions are co-optimized [33,34].
Stability hinges on diffusion management and acid–site tailoring. Micropore-diffusion control and hierarchical/core–shell architectures delay pore-mouth blockage and lower coke yield; phosphorus modification or external-surface passivation further balances acidity to sustain activity at high conversion [25,35,36]. Pairing mesoporous domains in bifunctional systems limits methane and other light-gas losses during hydrogenolysis/hydrocracking, extending time-on-stream while biasing liquids over gases [37]. Recent demonstrations with HMFI catalysts show that polypropylene can be converted to light olefins even below classical pyrolytic temperatures when carbocation chemistry and external surface sites are leveraged, broadening the operable window for selective, energy-lean upgrading [38]. Figure 1 synthesizes this structure–mechanism–operation logic: starting from monomer supply and polymerization mode (Ziegler–Natta vs. radical) that fix HDPE/LDPE/PP/PVC microstructures, it maps the dominant thermolysis routes (random/β-scission, H-abstraction/backbiting; zipper dehydrochlorination for PVC), the key operating levers (temperature, heating rate, residence time, rapid quench), and the role of optional catalysts/initiators and HCl management (for PVC) in steering outcomes from light gases to paraffinic oils/waxes.
A mechanistic viewpoint converts “heat-and-hope” into design: it identifies which bonds break first, which intermediates dominate, and where to intervene with catalysts (acid strength, metal function, topology), initiators (to set onset and radical populations), and suppressors/rapid quench (to limit secondary cracking and preserve chain length). That same insight maps operating envelopes, such as heating rate ramps and final temperatures that bias gases for cogeneration versus paraffinic waxes/oils suitable for candles and heat storage materials (e.g. paraffins) – Phase Change Materials (PCM) materials, turning landfilled liabilities into controllable product slates with defensible process conditions.
2. Coordination and Radical Polymerization Mechanisms in the Synthesis of HDPE, LDPE, PP, and PVC
2.1. Coordination Polymerization Mechanism of HDPE and PP
Coordination polymerization initiates through the formation of an active metal-alkyl complex at a Group IV transition metal site, typically involving titanium or zirconium as the central metal. This species is generated in situ by reacting a stable precatalyst with an alkylaluminum co-catalyst (e.g., Et3Al, MAO), which simultaneously alkylates the metal and removes ligands to create a vacant coordination. This vacancy allows the olefin to bind via π-complexation, where electron donation and back-donation between the monomer and the metal stabilize the complex [39,40,41]. In titanium-based systems, this interaction facilitates 1,2-insertion of the monomer into the metal–carbon bond, forming the first covalent link in the polymer chain and converting the site into an active center for propagation. Initially, Ti(IV) alkyl cations were proposed as the active species, but recent studies show that Ti(III) alkyls, formed by partial reduction with the co-catalyst, often dominate. These Ti(III) species are more effective due to their unpaired electron, which enhances monomer coordination and insertion [39]. In copolymerizations (e.g., ethylene–propylene), each monomer follows the same insertion pathway, though differences in reactivity lead to a mix of random and block segments [42]. Activation energies for initiation are moderate, ranging from 33 to 67 kJ mol−1, depending on the catalyst type. Homogeneous systems generally show lower barriers, supporting efficient initiation under mild conditions, though high initiator concentrations may still lead to side reactions [43,44,45,46].
In the coordination polymerization of HDPE, chain propagation proceeds via repeated insertion of ethylene monomers into a growing metal–carbon bond at an active center such as Ti–C or Zr–C. This occurs through a classical migratory insertion mechanism, which begins with π-complexation of ethylene to a vacant site on the transition metal, followed by insertion into the metal–alkyl bond [46]. In Ziegler–Natta systems, propagation is highly stereoselective and leads to linear HDPE with low branching. Metallocene catalysts, such as zirconocene complexes, allow more precise control over polymer architecture [47]. Computational studies show that while the first few insertions face slightly increasing energy barriers due to steric hindrance and agostic interactions, these barriers later decrease, stabilizing the propagation process [48].
The coordination of ethylene to the active site involves π-complexation, wherein the ethylene molecule donates electron density to the empty d-orbitals of the titanium center, forming a non-covalent π-complex [39,49]. This coordination orients the monomer for the subsequent insertion step but does not yet form a covalent bond. This step, along with the preceding catalyst activation, is illustrated in Figure 2a–b.
The migratory insertion of the coordinated ethylene into the Ti–C bond marks the beginning of chain growth, also known as chain initiation (Figure 2c) [50]. This step results in the formation of a new Ti–CH2–CH2–R species, where R denotes the rest of the growing polymer chain. Once the first insertion has occurred, the catalyst–polymer complex is fully activated for propagation [51]. Propagation in the coordination polymerization of high-density polyethylene (HDPE) involves the repeated migratory insertion of ethylene monomers into the growing metal–carbon bond, as shown in Figure 2d [52]. Each propagation step requires a fresh coordination of ethylene, followed by insertion into the Ti–C bond at the active site, typically a titanium–alkyl species. This process is highly stereoselective and typically yields linear polyethylene with minimal branching when Ziegler–Natta or metallocene catalysts are used. [39,53]
Chain termination occurs through several competing mechanisms, the most common being β-hydride elimination (BHE), β-hydride transfer to monomer (BHT), and hydrogenolysis (chain transfer to hydrogen). In β-hydride elimination, a β-hydrogen from the growing polymer chain is transferred to the metal, resulting in the formation of a vinyl-terminated polymer and a metal–hydride species (Figure 2e). This pathway becomes more dominant when monomer concentration is low and is generally less favored energetically than BHT [54]. β-Hydride transfer to monomer, shown in Figure 2f, involves the transfer of a β-hydrogen to a nearby ethylene molecule rather than to the metal center, producing a saturated chain end and regenerating the active site. This route is often preferred due to its lower activation energy and ability to maintain polymerization activity, especially under ethylene-rich conditions [55,56]. Finally, chain transfer to hydrogen, or hydrogenation, involves hydrogenolysis of the metal–carbon bond (Figure 2g). This process leads to saturated alkyl-terminated chains and is commonly used industrially to control molecular weight distribution. It is highly selective and depends on hydrogen concentration as well as the structure of the catalyst [57,58]. Finally, chain transfer to hydrogen, or hydrogenation, involves hydrogenolysis of the metal–carbon bond (Figure 2h). This process leads to saturated alkyl-terminated chains and is commonly used industrially to control molecular weight distribution. It is highly selective and depends on hydrogen concentration as well as the structure of the catalyst [58]. The resulting linear polyethylene structure with minimal branching is represented in Figure 2i.
In the coordination polymerization of PP, the process initiates with catalyst activation identical to HDPE, involving the formation of a Ti(III)–ethyl complex on MgCl2 after alkylation by AlEt3 (Figure 3a) [59]. Propylene coordinates to the active site through its C=C double bond, forming a π-complex with specific facial orientation governed by the stereoelectronic environment of the metal center (Figure 3b). This orientation dictates the regio- and stereochemistry of insertion, which occurs via migratory insertion of the CH2 group into the Ti–C bond (Figure 3c) [60]. The resulting growing chain maintains a defined stereoregularity—either isotactic, syndiotactic, or atactic—depending on the symmetry and ligand design of the catalyst system [61]. Electron donors, both internal (e.g., diisobutyl phthalate) and external (e.g., silane-based modifiers), fine-tune this environment by altering the electron density at the titanium center and the coordination geometry of the π-complex [62,63].
The propagation of PP chains continues via successive insertions, each guided by steric and electronic effects that enforce specific tacticity. C2-symmetric zirconocene catalysts are known to promote high isotacticity through chiral induction and enantiomorphic site control [64]. Kinetic studies have shown that the first propylene insertion is stereodetermining, while subsequent insertions propagate the same configuration unless interrupted by chain transfer or site deactivation (Figure 3d–e) [65]. Metallocenes and advanced Ziegler–Natta catalysts can achieve turnover frequencies ranging from 0.1 to 1 monomer per active site per second under optimized conditions [66].
Termination mechanisms in PP polymerization are more diverse due to the additional methyl group on the monomer, which permits β-methyl elimination in addition to β-hydride elimination. In β-methyl elimination, the methyl group is removed from the β-position and transferred to the metal center, forming allylic or branched end-groups [67]. β-Hydride elimination, the more common route, leads to vinyl-terminated PP chains and Ti–H species (Figure 3f), whereas β-hydride transfer to monomer results in a new Ti–alkyl center and a terminally unsaturated chain (Figure 3g) [68,69]. Octahedral zirconium complexes with carefully tuned ligand geometries demonstrate comparable precision: while one catalyst may yield site-controlled isotactic chains, a structurally similar complex may produce syndiotactic polymers under chain-end control, emphasizing the sensitivity of stereochemical outcome to ligand design [70]. The migratory insertion mechanism becomes essential in certain catalyst systems, particularly those based on C2-symmetric {SBI}-type zirconocenes. These catalysts direct monomer coordination to the sterically crowded site, favoring chain migration and high isotacticity. Thermochemical modeling of the first few insertion steps correlates well with experimental pentad distributions, confirming that migratory insertion mechanisms dominate in these systems and are necessary for achieving highly isoselective propagation [71].
Chain transfer to hydrogen is especially significant industrially, where molecular hydrogen is used to produce PP with reduced molecular weight and improved processability (Figure 3h) [57,58]. This pathway involves the cleavage of the metal–carbon bond by hydrogen, forming a saturated polymer and a new Ti–H center, which can reinitiate polymerization or become deactivated depending on the system [72].
Computational studies have further refined these mechanistic pathways, revealing two dominant transition states in β-hydride transfer: TSA, which favors direct metal–hydrogen bonding and is common in less sterically hindered environments, and TSC, more prominent in bulkier catalysts where propagation is sterically hindered and termination is favored [56,73]. The selection between these routes is governed by subtle changes in ligand geometry, donor type, and monomer concentration. These findings demonstrate the crucial role of catalyst design and polymerization environment in determining the final properties of PP.
Overall, coordination polymerization of HDPE and PP follows a fundamentally similar mechanism driven by metal-catalyzed coordination–insertion, but their differences in monomer structure lead to distinct stereochemical pathways, termination behavior, and catalyst requirements. The design of catalyst ligands, use of electron donors, and operational parameters such as hydrogen pressure and monomer concentration together allow precise control of polymer microstructure, molecular weight distribution, and end-group functionality. These parameters must be finely tuned to meet specific industrial requirements, particularly for sustainable and efficient synthesis of polyolefins under modern green chemistry constraints.
2.2. Free Radical Polymerization of LDPE and PVC
The synthesis of low-density polyethylene (LDPE) via high-pressure free radical polymerization is initiated by the thermal decomposition of organic peroxides, which generate reactive radicals capable of initiating chain growth [74,75,76,77]. The efficiency of this initiation step is highly dependent on the peroxide’s molecular structure, decomposition kinetics, and compatibility with process parameters such as temperature, pressure, and residence time [75,78]. Among the commonly used initiators, lauroyl peroxide is frequently applied due to its predictable decomposition behavior [79]. The decomposition pathway of lauroyl peroxide is illustrated in Figure 4a, where homolytic O–O bond cleavage leads to the formation of two acyloxy radicals [79]. These radicals subsequently react with ethylene monomers to form the first macroradical species, as shown in Figure 4b [80].
Initiator structure plays a pivotal role in defining polymerization performance [81]. For instance, bi-functional peroxides produce multiple radical centres upon decomposition, enabling more rapid initiation and increased monomer conversion relative to their mono-functional counterparts [82]. This enhanced efficiency is crucial for maintaining high yields and narrow molecular weight distributions, especially under the extreme conditions characteristic of LDPE production [83]. Meanwhile, tetrafunctional initiators like JWEB50 have been shown to outperform conventional mono-functional initiators in other monomer systems, underscoring the broader utility of multifunctional peroxides in radical polymerizations [84].
The decomposition of organic peroxides typically proceeds via homolytic O–O bond cleavage, producing acyloxy, alkyl, or alkoxy radicals depending on the peroxide type [85]. These radicals differ in reactivity and thermal stability, attributes that can be fine-tuned through steric and electronic modifications to the initiator structure [86]. For example, dialkyl peroxides yield fast initiation with reduced side reactions, while hydroperoxides and ketone peroxides, though reactive, pose safety concerns and are limited to niche applications [76].
Initiator performance evolves with changes in process conditions [87]. Each peroxide displays an optimal temperature range where radical generation is maximized and undesired side reactions are minimized [88]. Beyond this range, excessive decomposition reduces radical efficiency, diminishing polymer yield despite higher initiator consumption [89]. The introduction of composite initiation strategies—such as combining azo and peroxide initiators—has been explored to balance radical reactivity and propagation control, especially in systems where fine-tuning molecular architecture is desirable [76,85,90,91].
Overall, initiation in LDPE polymerization hinges on precise radical generation via carefully selected peroxides [76,85,90]. By optimizing initiator structure, dosing, and thermal behavior in response to reactor conditions, industrial processes can achieve efficient chain initiation with minimal waste and maximum product control [92].
Propagation in the free radical polymerization of LDPE proceeds through the sequential addition of ethylene monomers to growing chain radicals, a process governed by a delicate balance between chain growth, intermolecular chain transfer, and intramolecular branching [93,94,95]. This propagation sequence is shown in Figure 4c–e [93]. One of the dominant branching mechanisms is 1,5-hydrogen backbiting, where a hydrogen atom is abstracted from a methylene unit five carbon atoms upstream, producing a secondary or tertiary radical that enables the formation of short-chain branches—an essential structural feature of LDPE [93]. This reaction is shown in Figure 4f, where the terminal radical folds back and abstracts a hydrogen atom from an internal CH2 group, generating a branched radical that can further propagate (Figure 4g) [79,89,95]. This pathway has a relatively low activation energy −1, making it kinetically favorable under high-pressure industrial conditions [96].
The competing intermolecular chain transfer reactions—particularly those involving hydrogen abstraction by solvent, monomer, or polymer—exhibit higher activation energies (~100 kJ mol−1) and primarily regulate molecular weight [97,98]. All three kinetic processes—propagation, chain transfer, and branching—scale quadratically with ethylene fugacity, reinforcing the role of pressure [99]. Kinetic models have been developed that explicitly account for primary propagation and secondary branching mechanisms [100,101]. These simulate molecular weight and branching distributions by solving recursion equations under realistic reactor conditions [102].
Termination occurs predominantly through bimolecular reactions between macroradicals, with recombination (combination) and disproportionation as the main pathways [97]. These are exemplified in Figure 4h, where two growing macroradicals undergo recombination to form a saturated polymer chain [103]. Chain-length-dependent termination (CLDT) has been confirmed using Pulsed Laser Polymerization (PLP) and Electron Paramagnetic Resonance (EPR) techniques, showing that short radicals terminate faster than longer ones [104,105]. Activation energy for termination is also chain-length dependent: short chains exhibit Ea of 25–39 kJ mol−1, while longer radicals show values of 18–24 kJ mol−1 [106]. The selectivity between termination modes is influenced by viscosity, radical size, and temperature. Higher temperatures and lower viscosities favor disproportionation, while viscous media promote recombination [103].
Termination dynamics are modelled using MWD-based simulations and Monte Carlo approaches that account for diffusion, backbiting, and gel effects [92]. Advanced techniques such as SP-PLP-EPR quantify propagation and backbiting effects with high resolution [107,108].
The free radical polymerization of vinyl chloride (VC) is initiated by the thermal decomposition of azo compounds such as azobisisobutyronitrile (AIBN), which cleaves into two carbon-centered radicals [85,93,103]. This decomposition step is depicted in Figure 5a. The generated radicals add to vinyl chloride monomers, resulting in the formation of primary macroradicals as shown in Figure 5b [109]. Chain propagation proceeds predominantly through head-to-tail monomer addition, forming a linear and stereoregular structure, illustrated in Figure 5c [110]. The propagation step is highly exothermic, with activation energies at 24.9 kJ mol−1, depending on the tacticity and chain environment [111,112].
Intramolecular chain transfer via 1,5-hydrogen backbiting becomes significant as conversion increases, with the activation energy estimation at around 54.3 kJ mol−1 [95,112]. This step is visualized in Figure 5d, where the growing terminal radical abstracts a hydrogen atom from a methylene group within the chain, forming a mid-chain radical (MCR) [113,114,115]. The rate constant for the first propagation step from the branching site (BP1) is several orders of magnitude lower than for terminal propagation, although subsequent propagation steps (BP2, BP3) recover near-terminal reactivity [112,114,116].
At high monomer concentrations, chain transfer to monomer occurs via hydrogen abstraction, creating a new initiating radical and terminating the existing chain [114,117]. Although slower than propagation, this process becomes significant at elevated conversions and influences both molecular weight and the distribution of internal unsaturations [117,118]. Chain transfer to polymer results in long-chain branching and higher polydispersity, especially under bulk polymerization conditions [117].
Internal double bonds are formed through β-chlorine elimination or long-range hydrogen transfer [118]. These unsaturations behave like chain-end groups and contribute to thermal and UV instability of the polymer [119]. Stereoselective backbiting is also known to introduce tacticity-related defects, particularly at lower conversions when radicals exhibit greater conformational flexibility [120].
As polymerization proceeds toward high conversion (>85%), monomer availability in the reaction phase decreases sharply, and radical propagation slows due to increasing viscosity and reduced chain mobility [110,121]. Under these conditions, propagation shifts toward monomolecular reactions, and structural defects such as branching and internal unsaturation increase [121]. Chloroallylic end groups, commonly associated with chain termination, decrease in frequency at these stages, indicating a shift in dominant termination mechanisms [121,122].
Figure 5e depicts termination via recombination, where two macroradicals couple to form a single, saturated chain [123]. Alternatively, Figure 5f shows disproportionation, in which a hydrogen atom is transferred from one radical to another, resulting in one saturated and one unsaturated chain [124]. Figure 5g presents a second variant of disproportionation that yields a saturated macromolecule via a stabilized internal shift [125]. The balance between recombination and disproportionation is influenced by radical size, temperature, and the viscosity of the medium [125,126]. Short-chain radicals favor disproportionation due to higher diffusion rates and greater reactivity, whereas recombination is more prevalent with larger, more hindered radicals [106].
Activation energies for termination processes vary depending on the mechanism and radical chain length. Disproportionation requires 25–39 kJ mol−1, while recombination occurs at 18–24 kJ mol−1 [106]. Chain-length-dependent termination (CLDT) has been confirmed through experiments using pulsed laser polymerization and EPR spectroscopy [106,125]. These findings are relevant for modeling termination kinetics and predicting molecular weight distributions in industrial PVC production [110,124].
Chain transfer appears to play a major role in PVC polymerization, particularly under high conversion and bulk-phase conditions. The observed number of initiator-derived end groups per chain is often below 0.4, which indicates that a single radical may initiate several chains through sequential transfer reactions [124]. This mechanism aligns with industrial observations of lower than expected molecular weights despite moderate initiator concentrations [124]. Terminal unsaturations introduced by disproportionation are critical for stability, as they can act as reactive sites under thermal or UV stress [127].
Accurate modelling of PVC polymerization must consider conversion-dependent changes in phase composition, chain mobility, and radical diffusion [106,128]. Such models have incorporated chain transfer, backbiting, and termination kinetics using detailed mechanistic frameworks [106]. Advanced computational methods like G3(MP2)-RAD and ONIOM have also been applied to estimate propagation constants and structural defect pathways, with results in agreement with experimental observations [94,129]. These approaches reveal that defect formation is not random but governed by steric constraints, energetic preferences, and local radical dynamics [127].
Ultimately, the development of structure–property relationships in PVC depends on precise control of chain propagation, transfer, and termination processes. By adjusting processing conditions such as initiator concentration, temperature, and monomer feed rates, polymer microstructure and product performance can be tuned to meet specific application requirements.
3. Mechanistic Pathways and Kinetic Features of Thermal Degradation in Polyolefins and PVC
3.1. HDPE
The initiation of thermal degradation in HDPE proceeds via homolytic cleavage of carbon–carbon (C–C) bonds, triggered when the system temperature exceeds the bond dissociation energy threshold. This process results in the formation of primary alkyl radicals that mark the onset of degradation, initiating radical chain reactions across the polymer backbone [80,81,82].
Early degradation events are predominantly described by a random scission model, wherein each C–C bond along the linear polymer chain has an equal probability of breaking under thermal stress (Figure 6a). This statistical approach successfully captures observed kinetic behavior and molecular weight distributions during pyrolysis, particularly in the absence of branching or catalytic residues [80,82,83]. However, site-specific initiation mechanisms have also been proposed, emphasizing preferential scission at structurally weaker sites—such as allylic positions or residual catalyst centres—where degradation may commence at lower temperatures [84].
The homolytic scission events result in the generation of highly reactive radicals, which participate in subsequent reactions such as hydrogen abstraction (Figure 6c), vinyl formation, and β-scission (Figure 6b). These radicals create vinyl and vinylidene end groups, with vinyl groups forming at rates significantly higher than the evolution of ethylene in early degradation stages, suggesting that functional group generation dominates over monomer recovery in the initiation phase [86].
Experimental studies have shown that the initiation step is highly sensitive to molecular weight, crystallinity, and thermal history. Lower molecular weight fractions degrade more readily due to enhanced chain mobility and reduced thermal stability, while crystalline regions offer greater resistance to bond rupture [81,86,130]. The onset temperature for random scission typically ranges between 400–440 °C, although the presence of impurities, chain defects, or catalytic residues can lower this threshold by as much as 100 °C [131].
Kinetic modelling of the initiation phase supports a homolytic mechanism with reported activation energies for HDPE degradation ranging from approximately 222 kJ mol−1 to 270 kJ mol−1, depending on the method used (e.g., Kissinger or Ozawa-Flynn-Wall and Vyazovkin isoconversional method) and the polymer architecture [131]. These values reflect the energy requirement for C–C bond cleavage and provide a quantitative foundation for process optimization in thermal recycling.
Although the random scission model remains widely accepted, recent work has highlighted the importance of distinguishing between uniformly random (chain reaction) and structurally biased (attack on β – positioned carbon) initiation. These distinctions are particularly relevant for refining pyrolysis models and improving the accuracy of product yield predictions in both open and closed systems [7,81].
Ultimately, the initiation of HDPE thermal degradation is best described as a complex interplay between random homolytic bond scission and site-specific activation, both are governed by temperature gradient rate and final temperature. Understanding this initiation step is essential for controlling downstream reaction pathways and optimizing the pyrolytic conversion of HDPE into valuable hydrocarbon fractions.
The propagation phase of HDPE thermal degradation is governed by a complex network of free-radical-driven reactions that proceed following the initial homolytic cleavage of C–C bonds. At the beginning (at lower temperatures) primary mechanism is random scission and then with rising temperatures and consequently concentration of β–carbon positions, β–scission becomes dominant mechanism (Figure 6b). Hypothesis can be supported by formation of alkene products at higher temperatures of pyrolysis [132,133].
Intermolecular hydrogen abstraction (Figure 6c) is another key step, where radicals extract hydrogen atoms from neighbouring polymer chains or smaller hydrocarbon molecules, thereby generating new radical sites and further driving the degradation process [86,133]. Intramolecular hydrogen transfer (Figure 6d), commonly referred to as backbiting, plays a critical role in creating secondary radical sites that undergo secondary β-scission (Figure 6e), contributing to the diversity of volatile products [130,134].
Radical isomerization, including alkyl shifts, promotes the rearrangement of radical intermediates to more stable configurations (Figure 6f), thereby altering the final product distribution [133,135]. Secondary cracking of olefins, especially under elevated temperatures and prolonged vapor residence times, further reduces long-chain olefins into lighter hydrocarbons (Figure 6g) [131,136].
Cyclization reactions can occur during degradation, where radicals undergo ring closure to form cyclic hydrocarbons (Figure 6h). These cyclic intermediates can subsequently undergo dehydrogenation and further rearrangements, eventually leading to the formation of aromatics and polycyclic aromatic hydrocarbons (PAHs), especially at temperatures above 600 °C [131,133,135,136].
Vinylidene formation is another important reaction pathway, leading to the development of vinyl-type structures through radical-induced elimination reactions, contributing to the unsaturation of the degradation products [131,132,133]. Minor radical β-scission and recombination equilibria also play a role in controlling the balance between chain fragmentation and radical stability, particularly at elevated temperatures (Figure 6i) [133,137].
Radical-induced disproportionation further contributes to the diversity of degradation products (Figure 6j) by forming stable radicals and smaller hydrocarbons [110]. Additionally, aromatization and PAH formation pathways are significant under severe pyrolytic conditions, as cyclic radicals undergo dehydrogenation and ring rearrangement to form stable aromatic structures [131,132].
Ring opening reactions of cyclic hydrocarbons contribute to the formation of linear chain products, further enhancing the diversity of volatile products [130,131]. Dehydrogenation processes, especially at elevated temperatures, increase the unsaturation level in the resulting hydrocarbons and promote the formation of aromatic compounds [130,136,138].
Experimental studies confirm that the progression of propagation reactions and the final product distribution are highly dependent on the thermal environment and residence time. Low temperatures and short vapor residence times favour a broader spectrum of alkanes, alkenes, and waxes, while higher temperatures and longer residence times predominantly yield light hydrocarbons, monoaromatics, and PAHs [133,135].
Oxidative pathways, although not the focus of the propagation phase under inert pyrolysis conditions, may become significant in processing environments such as extrusion, where oxygen exposure leads to the formation of oxidized volatiles like aldehydes and carboxylic acids [139]. Additives, including flame retardants and catalytic residues, can also modulate the degradation pathway by accelerating or redirecting radical reactions toward desired product distributions or char formation [140,141].
Overall, the propagation phase in HDPE thermal degradation involves a dynamic interplay of β-scission, hydrogen abstraction, backbiting, isomerization, secondary cracking, cyclization, vinylidene formation, and dehydrogenation reactions. These mechanisms collectively dictate the evolution of product species and govern the molecular weight reduction, unsaturation levels, and aromatic content in the final pyrolysis products.
The termination phase in the thermal degradation of high-density polyethylene (HDPE) under vacuum occurs predominantly via radical recombination (Figure 6i), disproportionation (Figure 6j), and crosslinking (Figure 6k). These mechanisms are governed by radical concentrations, melt viscosity, temperature, and the absence of oxygen, which suppresses oxidative pathways and alters radical lifetimes [142].
Radical recombination proceeds through the coupling of two macroradicals to form a stable σ-bond, effectively removing reactive centres and terminating the chain reaction. This process is diffusion-limited in high-viscosity polymer melts, and its efficiency decreases with increasing molecular weight and polymer chain entanglement [143]. Under vacuum conditions, the suppression of oxidative termination further enhances the relevance of recombination. In metallocene-based HDPE, where unsaturation levels are low, recombination appears less prevalent compared to Phillips-type HDPE, which exhibits higher vinyl content and radical reactivity [144,145].
Disproportionation constitutes a parallel radical termination mechanism in which a hydrogen atom is abstracted from one radical by another, yielding a saturated and an unsaturated chain end. The formation of vinyl and vinylidene groups observed in degraded HDPE supports the occurrence of this pathway, particularly under conditions of low chain mobility [142,146]. Disproportionation is more likely to compete with recombination when radical concentrations are high and chain diffusion is limited, conditions typically found at lower temperatures or during early stages of degradation in viscous phases [147].
Crosslinking arises when two macroradicals from separate chains recombine to form inter-chain covalent bonds, producing a three-dimensional network structure. This mechanism is facilitated by the presence of unsaturated sites (e.g., vinyl groups) and occurs more readily in HDPE grades with higher branching or residual catalyst fragments. The addition of radicals to vinyl functionalities has been identified as a key step in promoting crosslinking during pyrolysis [144]. At moderate temperatures, crosslinking can outcompete scission, increasing molecular weight, particularly in the absence of oxygen and under shear-free conditions [147].
The relative dominance of termination pathways depends strongly on thermal conditions. At higher temperatures, chain scission rates increase, lowering molecular weight and favoring the generation of volatiles. In contrast, at lower temperatures and in the absence of oxygen, radical termination via recombination and crosslinking is more prominent [148]. Overall, termination mechanisms in HDPE degradation under vacuum are criticallyinfluenced by viscosity, temperature, and polymer structure. The prevalence of recombination, disproportionation, and crosslinking determines the balance between molecular fragmentation and network formation, thereby shaping the final product distribution and dictating processability in thermal recycling applications.
3.2. LDPE
The thermal degradation of LDPE under vacuum initiates through random homolytic scission of C–C bonds along the polymer backbone (Figure 6a). This unimolecular process occurs without oxidative interference and becomes significant at elevated temperatures ranging between 410 and 475 °C, where the thermal energy exceeds the bond dissociation threshold of saturated carbon chains [149]. The resulting alkyl radicals generated in this initiation step mark the starting point for subsequent β-scission and hydrogen abstraction reactions.
Although often modelled as purely random, the cleavage of C–C bonds in LDPE is not uniformly statistical. Thermochemical studies and experimental analyses suggest a preference for scission at structurally weaker sites, such as allylic positions or tertiary carbons, due to their lower dissociation energies [7,81,150]. These preferential sites enable radical formation at lower activation energies and contribute to the early evolution of unsaturated species such as vinyl and vinylidene groups [151].
Upon C–C bond cleavage, primary and secondary alkyl radicals are formed. The stability of these radicals influences their reactivity, with secondary radicals generally being more thermodynamically favored and more likely to propagate the degradation process [149]. The radicals can also undergo stabilization through resonance delocalization or hydrogen abstraction from neighboring chains, forming longer-lived radical intermediates capable of driving further decomposition [152].
The initiation phase also results in important structural changes within the polymer matrix. These include the formation of double bonds and unsaturated end-groups, an increase in reactive chain ends, and the production of low molecular weight aliphatic fragments such as alkanes and alkenes [153].These effects are consistent with both experimental measurements and kinetic simulations, which have reported early-stage molecular weight reduction and volatile product evolution as primary indicators of radical initiation [154].
Additionally, molecular dynamics and variational transition state theory have shown that each bond dissociation rates increase with chain length up to decane, after which the effect reach plateau. This chain length dependence influences the overall fragmentation rate of the polymer backbone [150].
The propagation phase is primarily governed by β-scission of alkyl radicals (Figure 6b), which cleave adjacent C–C bonds to generate unsaturated chain ends and smaller hydrocarbon fragments [155,156]. Following initial radical formation, backbiting via intermolecular hydrogen abstraction (Figure 6c) frequently occurs, particularly in LDPE due to its branched structure, facilitating proximity between radical sites and hydrogen atoms within the same polymer chain [132,157]. Intramolecular hydrogen transfer (Figure 6d) then repositions the radical along the polymer backbone, preparing it for secondary β-scission (Figure 6e), which further breaks down the chain [156,158].
Radical isomerization, through 1,2- or 1,3-alkyl shifts (Figure 6f), enables migration of radical centres to thermodynamically more stable positions, affecting both chain stability and subsequent degradation steps [159]. Vinylidene groups form via elimination of hydrogen from terminal radicals (Figure 6g), introducing unsaturation into the polymer. Cyclization (Figure 6h), especially in flexible chain segments, results in the formation of cyclic intermediates, which at elevated temperatures undergo aromatization via dehydrogenation (Figure 6i), yielding aromatic compounds such as benzene or substituted aromatics [156].
The olefins generated in the primary degradation steps may undergo secondary cracking (Figure 6j), yielding smaller hydrocarbons such as ethylene, propylene, and butenes, especially at prolonged residence times and elevated temperatures [86,160].
Several additional processes are intertwined with the radical propagation phase. Random chain scission persists throughout degradation, generating fragments of varying molecular weights [160,161]. Depolymerization, characterized by the stepwise loss of monomer units, further reduces the polymer chain length and contributes to low-molecular-weight product formation [162]. Under specific thermal conditions, crosslinking between polymer chains may also occur (Figure 6m) [101].
It has been demonstrated that the degradation rate transitions from zero-order to first-order kinetics as the temperature increases from ~340 °C to above 425 °C, indicating the activation of radical-driven propagation reactions above a critical temperature threshold [155]. The relevance of these radical propagation pathways extends to practical applications such as pyrolytic recycling and the thermal stability of polyethylene-based materials. The formation of cyclic intermediates, aromatics, and PAHs, although more prevalent under severe pyrolytic conditions, can have significant environmental and material performance implications [156,158,160].
In summary, the propagation phase of LDPE thermal degradation under vacuum is defined by a series of interconnected radical processes, including β-scission (Figure 6b,e), hydrogen abstraction (Figure 6c,d), radical isomerization (Figure 6f), vinylidene formation (Figure 6g), cyclization (Figure 6h) and aromatization (Figure 6i).
Termination in the thermal degradation of low-density polyethylene (LDPE) under vacuum proceeds through multiple radical-driven pathways. Radical recombination (Figure 6k) constitutes a key termination route, leading to saturated covalent bonds and loss of radical activity. Disproportionation (Figure 6l) results in one saturated and one unsaturated chain by hydrogen transfer between radicals [162,163,164]. Crosslinking (Figure 6m) occur at high temperatures in the melt phase forming a three-dimensional network, but in minor extent [101,165,166].
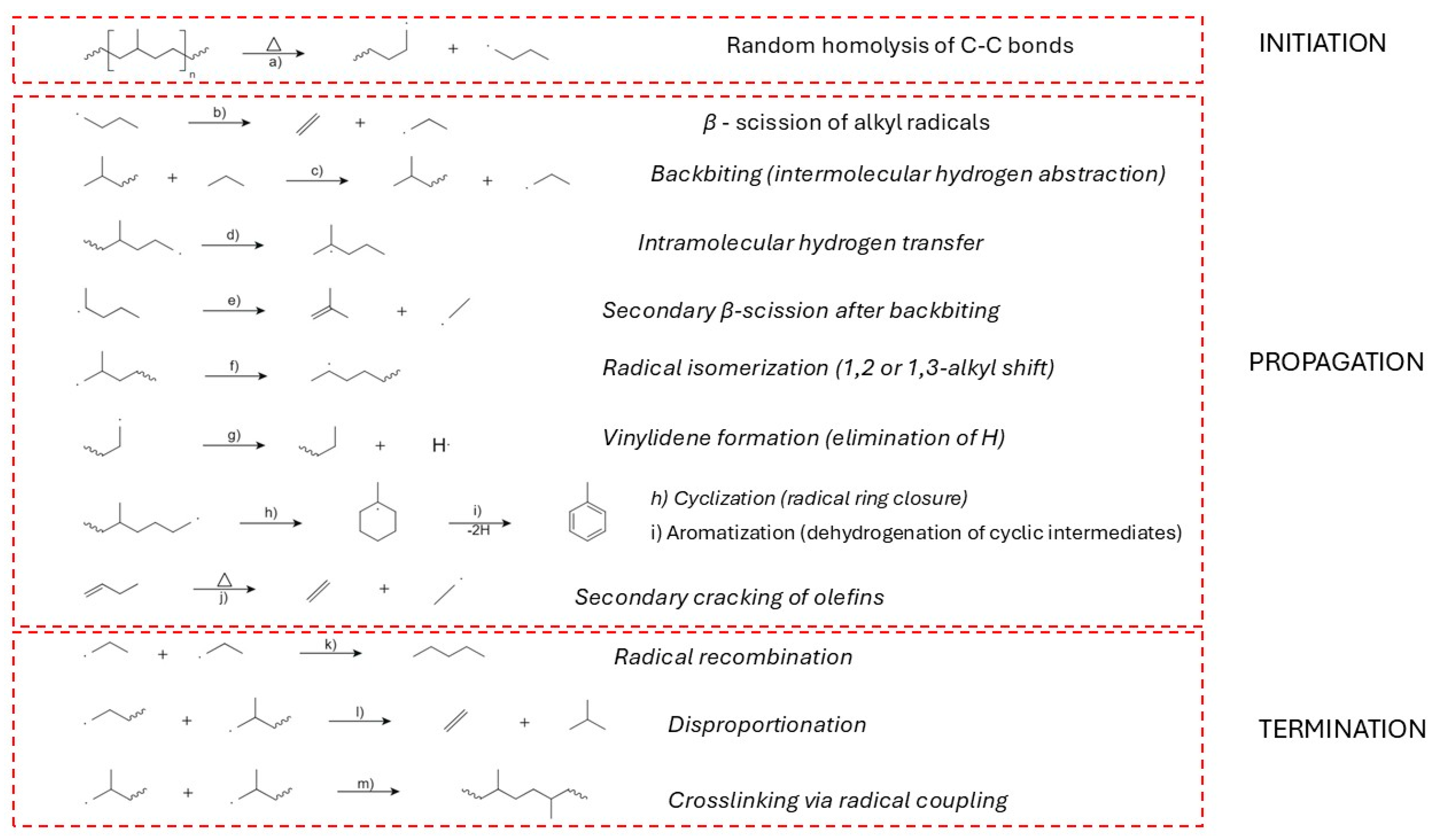
Figure 7.
Thermal degradation mechanism of low-density polyethylene (LDPE): a) Random C–C bond scission (homolysis); b) β-scission of alkyl radicals; c) Backbiting (intermolecular hydrogen abstraction); d) Intramolecular hydrogen transfer; e) Secondary β-scission after backbiting; f) Radical isomerization (1,2 or 1,3-alkyl shift); g) Vinylidene formation (H elimination); h) Cyclization (ring closure); i) Aromatization (dehydrogenation of cyclic intermediates); j) Secondary cracking of olefins; k) Radical recombination; l) Disproportionation; m) Crosslinking via radical coupling.
Figure 7.
Thermal degradation mechanism of low-density polyethylene (LDPE): a) Random C–C bond scission (homolysis); b) β-scission of alkyl radicals; c) Backbiting (intermolecular hydrogen abstraction); d) Intramolecular hydrogen transfer; e) Secondary β-scission after backbiting; f) Radical isomerization (1,2 or 1,3-alkyl shift); g) Vinylidene formation (H elimination); h) Cyclization (ring closure); i) Aromatization (dehydrogenation of cyclic intermediates); j) Secondary cracking of olefins; k) Radical recombination; l) Disproportionation; m) Crosslinking via radical coupling.
3.3. PP
The initiation phase of polypropylene (PP) thermal degradation under vacuum is governed predominantly by random homolytic cleavage of carbon–carbon (C–C) bonds along the polymer backbone (Figure 8a). This unimolecular process leads to the generation of primary, secondary, and particularly tertiary alkyl radicals, which serve as reactive intermediates for subsequent propagation pathways [167,168,169].
The homolytic scission in PP occurs statistically across the polymer chain, but structural factors such as the presence of methyl substituents adjacent to tertiary carbons in isotactic PP significantly influence the selectivity and stability of the resulting radicals. The formation of tertiary-centred radicals is favored both thermodynamically and kinetically due to the stabilization conferred by the polymer’s tacticity and branching pattern [170,171,172].
Computational and experimental analyses confirm that although scission is initially random, the resulting product distribution and radical reactivity are highly non-random. Product analysis from PP pyrolysis shows a high prevalence of branched olefins—e.g., 2,4-dimethyl-1-pentene—which points to a mechanistic preference for radical transfer to tertiary centres and subsequent β-scission at these sites (Figure 8b) [6,170]. This implies that radical transfer, both intra- and intermolecular, plays a central role during initiation, allowing chain mobility and localized reactivity to guide the degradation pathway.
The activation energy for initiation via random backbone cleavage is reported to be approximately 301.3 kJ mol−1, while secondary side-scission processes (e.g., at tertiary radicals) may proceed with significantly lower barriers (~123.9 kJ mol−1) (Figure 8b) [168]. These energy differences support a dual-mechanism model: initial radical formation requires substantial thermal input, while subsequent reactions proceed via more accessible routes. This aligns with PHASE-based kinetic studies showing a sharp two-stage reactivity pattern, where an initial rapid degradation phase is followed by a slower transformation of unsaturated residues and char precursors [173].
Mechanistic modelling of PP pyrolysis, incorporating majority reactions and species, validates the dominance of random C–C homolysis in initiating degradation, especially at elevated temperatures above 380 °C (Figure 8a) [169,170,174]. The transition from weak-link activation at lower temperatures (200–350 °C) to homolysis-dominated initiation at higher temperatures (>380 °C) is well supported by changes in volatile product yields and degradation kinetics [12,175].
Additionally, initiation events are closely linked to hydrogen abstraction processes, which either stabilize radicals or lead to new radical species (Figure 8e). These transfer reactions are facilitated by the presence of labile tertiary hydrogen atoms, which are abundant in isotactic PP, enhancing the likelihood of radical rearrangement and unsaturation formation (Figure 8d) [6,12,171].
Environmental conditions, particularly vacuum and inert atmospheres, affect the onset and rate of initiation. Under vacuum, the absence of oxygen inhibits oxidative side reactions, thereby amplifying the contribution of homolytic scission and suppressing low-temperature degradation pathways [168,174,175]. Moreover, the local polymer morphology influences initiation efficiency, with amorphous regions favoring radical formation due to reduced steric constraints and enhanced chain mobility [174].
Spectroscopic and molecular weight analyses further confirm the initiation process. A measurable decrease in intrinsic viscosity correlates with chain scission extent, and FTIR observations reveal increasing carbonyl and vinyl group concentrations as degradation progresses [172]. These findings indicate that unsaturation and chain end functionalities emerge early during pyrolysis, reflecting the impact of radical generation and migration in the initiation stage.
The propagation phase is driven by a complex series of radical-mediated reactions that evolve from the primary macroradicals formed during initiation. Once generated, these radicals—primarily tertiary in structure due to the methyl-substituted backbone of PP—undergo β-scission, a dominant propagation step that cleaves C–C bonds adjacent to the radical centre (Figure 8b). This process results in the formation of shorter hydrocarbon fragments and new secondary radicals, effectively sustaining the degradation cascade. The β-scission of tertiary radicals is thermodynamically favorable and leads to the formation of unsaturated hydrocarbons such as propylene, 2-methyl-1-pentene, and other branched olefins, which are frequently detected among the pyrolysis products [12,169,170,176,177,178]
Methyl radicals are also formed during this process, either through side-group fragmentation or as secondary products of β-scission events (Figure 8c). These small radicals are highly reactive and play a key role in product diversification and hydrogen abstraction reactions. The system further propagates through backbiting reactions, particularly the 1,5-hydrogen shift, where a hydrogen atom is transferred intramolecularly to a radical site via a six-membered transition state (Figure 8d). This leads to the formation of more stable radicals, often tertiary, which are prone to subsequent β-scission. Backbiting is facilitated by the backbone structure of PP and is especially active near chain ends or in regions with local chain mobility [170,178].
In parallel, intermolecular hydrogen abstraction allows radicals to abstract hydrogen atoms from neighboring polymer chains (Figure 8e). Hydrogen abstraction step generates new radical centres while stabilizing the abstracting radical, thus spreading radical activity throughout the polymer matrix. This mechanism contributes significantly to the degradation process, particularly under high-radical-density conditions typical in vacuum pyrolysis [6,12,171,178]. Alongside hydrogen abstraction, radical isomerization also plays a substantial role in redistribution of radical centres (Figure 8f). This occurs through hydrogen shifts such as 1,2- or 1,3-transfers, enabling the radical to migrate to more thermodynamically stable sites, usually adjacent to tertiary carbon atoms. The resulting rearrangements increase the variety and stability of degradation intermediates and influence the composition of the resulting product pool [169,179].
Terminal double bonds are indicative of end-chain scission and are commonly observed among PP pyrolysis products, especially in the form of monoolefins (Figure 8g) [170]. The formation of allylic radicals, stabilized through resonance with adjacent double bonds, is another critical aspect of propagation. These radicals contribute to the formation of conjugated dienes and persist longer in the radical pool, influencing both the rate and the extent of subsequent decomposition steps [169,180].
Cyclization reactions, although less prevalent, are recognized in PP degradation, particularly under thermal conditions that permit chain folding and intramolecular radical attack (Figure 8h). The formation of cyclic intermediates or products has been supported by the frequent overrepresentation of specific hydrocarbon fragments such as C9 and C15, which align with mechanistically favored ring closure and β-scission events involving cyclic transition states [169]. In some cases, these intermediates can further undergo dehydrogenation to yield aromatic products, although such transformations are secondary in PP due to the saturated nature of its backbone.
The accumulation and interaction of radicals during propagation also enable recombination reactions, particularly at elevated concentrations or at later stages of degradation (Figure 8i). Radical recombination leads to the formation of heavier, more complex hydrocarbons and can serve as a partial termination route, though in vacuum conditions the low-pressure environment favors continued radical propagation over termination. The product distribution observed in PP pyrolysis under vacuum—especially the dominance of C5–C15 olefins—reflects the combined influence of β-scission, backbiting, hydrogen abstraction, isomerization, and occasional cyclization steps [12,177,181].
Overall, the propagation phase of PP thermal degradation under vacuum is governed by an interdependent network of radical reactions. These reactions are modulated by chain structure, stereochemistry, and thermal environment, and they ultimately determine the molecular weight reduction rate and the identity of volatile degradation products. The predominance of specific product series (e.g., hydrocarbons with carbon numbers divisible by three) confirms that propagation is not entirely random but influenced by preferred radical transfer routes and chain segment stabilities. The combination of detailed mechanistic modelling and empirical product analysis provides robust evidence that radical propagation in PP involves multiple pathways [130].
In the thermal degradation of polypropylene (PP) under vacuum, termination reactions play a central role in determining both the product distribution and the final structural characteristics of the degraded polymer. Among these, radical recombination, disproportionation, and crosslinking are the principal termination mechanisms (Figure 8i-k).
Radical recombination involves the coupling of two carbon-centred radicals to form a stable covalent bond, thereby neutralizing both radicals and preventing further propagation (Figure 8i). This mechanism is particularly significant under vacuum conditions. Intermolecular recombination is more likely to occur when radical concentrations are high, such as at elevated temperatures where β-scission and hydrogen abstraction are prevalent. Studies have shown that the recombination of macroradicals becomes more competitive as radical mobility increases, particularly near 400 °C, where an increased yield of higher molecular weight alkanes and alkenes is observed, consistent with recombination pathways [12,95,177]. Intramolecular recombination, though less frequently emphasized, can also occur in the semi-solid or viscous phase of degradation, potentially leading to cyclic structures or contributing to local crosslink formation [12,170].
Disproportionation represents an alternative radical termination route, in which a hydrogen atom is transferred from one radical to another (Figure 8j). This process results in the formation of a saturated molecule and an unsaturated one, typically an alkane and a terminal olefin. Although generally less dominant than recombination, disproportionation becomes relevant at elevated temperatures, where radical mobility and hydrogen transfer rates are enhanced. In PP degradation, this mechanism contributes to the formation of unsaturated products such as vinylidene end-groups and terminal alkenes [6,86]. The competition between recombination and disproportionation is governed by factors including radical structure, steric hindrance, and local concentration gradients. In environments with high radical density but limited diffusion—such as in condensed or semi-solid phases—disproportionation may become kinetically favorable despite its lower statistical likelihood compared to recombination [182].
Crosslinking occurs when radicals from different PP chains combine to form covalent interchain bonds, resulting in the formation of a three-dimensional network (Figure 8k). While PP is less prone to crosslinking than polyethylene due to its branched methyl side groups, crosslinking can still take place under conditions of high radical concentration or in the presence of external energy inputs (e.g., gamma radiation or peroxide initiation) [183]. The resulting crosslinked structures exhibit increased thermal stability and rigidity but may compromise the processability and recyclability of the polymer. Experimental findings confirm that radical-induced crosslinking becomes relevant at high degradation temperatures or under radiation-assisted decomposition, where chain mobility and reactive site proximity support network formation [6,86,167]. However, in vacuum pyrolysis, where volatility and molecular fragmentation dominate, extensive crosslinking is less likely unless degradation occurs in the solid or melt state with restricted chain motion [6].
Altogether, these termination mechanisms act in tandem to regulate the balance between continued radical propagation and product stabilization. Radical recombination is the most direct path to neutralize radicals and dominates at high radical concentrations. Disproportionation introduces unsaturation into the degradation products and contributes to olefin formation, while crosslinking alters the polymer's physical network, influencing thermal and mechanical properties. The relative prevalence of each pathway is dictated by temperature, radical concentration, and molecular mobility—all of which are modulated under vacuum conditions typical of PP pyrolysis.
3.4. PVC
Under vacuum, the thermolysis of PVC is governed by the polarity and relative weakness of the C–Cl bond and by the heterogeneous distribution of labile structural condition that seed chemical change. Cleavage events originate preferentially at allylic and tertiary chlorines, head-to-head linkages, and other polymerization defects where electronic (resonance, hyperconjugation) and steric factors reduce the barrier for C–Cl scission [184,185,186]. A syn-periplanar >CH–CCl< geometry aligns σC-H and σC-Cl orbitals to support concerted 1,2-elimination through a four-center transition state; in tertiary environments six-center concerted pathways are frequently accessible and display substantially lower activation energies (≈ 173.6 kJ mol−1) than regular repeat units (≈ 230.5 kJ mol−1) [187,188]. Quantum-chemical calculations (MNDO/AM1/DFT) and model-compound studies reproduce these energetic trends and rationalize why carefully purified (reprecipitated) PVC shows more uniform kinetics: the average length of initially dehydrochlorinated segments (lavl) typically spans ~4–12, tracking the density and topology of labile sites [189]. Microstructure (tacticity, branching) modulates the population and spatial disposition of tertiary chlorines; temperature and Lewis-acid coordination to chlorine further polarize and weaken C–Cl bonds, advancing onset and rate [186,189,190]. Because evolved HCl is efficiently removed under vacuum, bulk acid does not accumulate, autocatalysis is damped, and molecular (ionic) elimination dominates [184,191].
The mechanistic map captured in Figure 9a–l begins with Figure 9a, which shows random homolysis of C–Cl bonds as a minor but mechanistically relevant entry that creates macroradicals on the backbone together with chlorine radicals (Cl•) [96,97]. The dominant pathway, however, is the autocatalytic zipper dehydrochlorination illustrated in Figure 9b–d: successive eliminations of HCl from adjacent –CH2–CHCl– pairs progressively extend a conjugated polyene segment along the same chain—growth in conjugation length, not in molecular weight [189,193,194]. Two intrinsic, non-radical channels operate alongside the zipper: Figure 9e depicts self-catalyzed dehydrochlorination via chloride (Cl−), an E2-like process in which Cl− is regenerated, and Figure 9f shows a pericyclic/E2-like concerted elimination that proceeds through a six-center transition state when stereogeometry permits [13,15,193,195]. Although ionic routes dominate under vacuum, Figure 9g–i summarize a minor radical loop: homolytic C–Cl rupture forms macroradicals and Cl• (Figure 9g), Cl• abstracts hydrogen from saturated segments to produce HCl and new macroradicals (Figure 9h), and β-elimination on those radicals extends the double bond while regenerating Cl• (Figure 9i) [189,191,194]. As the zipper advances, Figure 9j emphasizes the buildup of polyene sequences (–CH=CH–)n, which act as platforms for secondary propagation processes. Most notably, Figure 9k shows a thermally allowed 6π- electrocyclic ring closure of a triene fragment to cyclohexadiene-like motifs (thermal disrotatory pathway consistent with Woodward–Hoffmann rules), thereby embedding cyclic/aromatic precursors into the backbone [184,193,196]. Figure 9l then highlights interchain radical coupling between polyene segments (crosslinking), which—together with concurrent cis–trans isomerization that biases the π-array toward lower-energy trans configurations—progressively immobilizes and stiffens the material [196,197,198].
Kinetic observations align with this picture. Early conversion is well described by apparent first-order behavior with respect to the concentration of labile motifs, consistent with defect-initiated progress of the zipper [184,193,199]. β-Chloroallyl and polyenyl-chloride sequences are kinetically privileged, accelerating subsequent HCl loss; where HCl removal is locally imperfect, the acid can lower the enthalpy of dehydrochlorination by up to ~110 kJ mol−1 in susceptible fragments, transiently increasing the rate [184,191,195,200]. Polyenyl segments generally eliminate faster than β-chloroallyl motifs, establishing a moving hierarchy of reactivity as conjugation grows [195]. Morphology (crystallinity, plasticizer content) modulates segmental mobility and the diffusion of HCl and Cl−, thereby shaping lavl and the spatial pattern of zipper advance [13,184,189,199]. There is also evidence that cation-radical species formed by interactions of polyenes with HCl can abstract hydrogen and sensitize unreacted sequences, creating localized autocatalytic pockets even when radical concentrations are globally low [199].
As conjugated domains mature, the chemistry tilts toward stabilization pathways summarized in Figure 10m–o. Figure 10m depicts char formation: long polyenes cyclize (via repeated 6π closures), dehydrogenate, and crosslink to yield condensed aromatic frameworks that are carbon-rich, thermally robust, and poorly volatile [12,184,196,201]. Transition-metal additives such as NiO or MoO3 can reinforce this evolution by catalyzing intermolecular chlorine loss and producing NiCl2 in situ, which promotes further unsaturation and raises crosslink density, thereby increasing char yield while suppressing volatiles [201,202,203]. Figure 10n shows radical recombination—bimolecular coupling of macroradicals formed during propagation—to generate new C–C bonds that locally quench reactivity and add crosslinks to the network [184,196,201]. Figure 10o illustrates reversible HCl re-addition to double bonds; under efficient vacuum this route is minor, but where HCl is not fully removed it can transiently regenerate –CH–CH(Cl)– units and interrupt further eliminations, thus moderating the pace of unsaturation [184,196,200,201] [99,104,115]. At elevated temperatures, the late-stage polyene backbone can also fragment, releasing light alkenes and aromatic species; radical cyclization/dehydrogenation of these aromatics leads to polycyclic aromatic hydrocarbons (PAHs), a fraction of which volatilizes while the remainder integrates into the growing char network [204,205].
Taken together, Figure 9 and Figure 10 outline a coherent mechanistic trajectory under vacuum: selective C–Cl scission at structurally weak sites seeds unsaturation; conjugation grows chiefly by zipper dehydrochlorination, aided by self-catalyzed and concerted E2-like eliminations and accompanied by minor radical cycles (Cl•, macroradicals); polyenes then reorganize via 6π electrocyclizations, cis–trans isomerization, and interchain radical coupling; and termination proceeds by aromatization and crosslinking into carbon-rich char, with local radical recombination and small, reversible HCl additions shaping fine details of the residue. This mechanistic synthesis not only rationalizes kinetic signatures (first-order on labile motifs, sensitivity to lavl, morphology, and HCl removal) but also provides levers for stabilization strategies in processing—namely, controlling defect populations and microstructure, enhancing acid scavenging and HCl evacuation, and judiciously using Lewis-acid inhibitors or transition-metal traps to steer the balance between volatiles and char [184,196,206].
4. Tuning Thermal Degradation Pathways with Potential Catalysts and Initiators: Lowering Onset and Enabling Selective Termination
4.1. Polyolefins and PVC Inffluence of Catalyst, Initiators and Termiantion Procedures
4.1.1. Catalyst Effect for HDPE, LDPE, PP and PVC Pyrolysis
Catalytic pyrolysis represents a critical pathway for upgrading plastic waste streams, particularly polyolefins such as HDPE, LDPE, PP and PVC. The introduction of tailored catalysts lowers degradation onset temperatures, accelerates kinetics, and enables control of product selectivity, while in PVC, catalytic strategies focus additionally on dehydrochlorination, polyene stabilization, and HCl management. The following synthesis integrates current literature across these four major polymers, highlighting mechanistic pathways, catalyst classes, and practical challenges.
In the case of HDPE, catalytic pyrolysis consistently lowers decomposition temperatures and improves reaction rates compared to pure thermal cracking. Zeolites dominate catalytic performance, with Hydrogen-form Zeolite Socony Mobil–5 (HZSM-5) and Hydrogen-form Y-type Zeolite (HY) being the most studied. HZSM-5, with its strong Brønsted acidity and shape-selective micropores, enriches light olefin yields such as propylene and butenes, whereas HY, due to its larger pore size, enhances aromatic selectivity [207,208,209]. Adjustment of the SiO2/Al2O3 ratio in HZSM-5 strengthens acidity and has been reported to boost olefin production up to 58 wt% [208]. Mesoporous materials such as Mobil Composition of Matter 41 (MCM-41), Santa Barbara Amorphous-15 (SBA-15), and hierarchical Beta expand diffusion channels, and when acidified, shift product distributions toward branched and aromatic hydrocarbons [210,211]. Metal oxides including MgO, ZnO, and MgₓAlOᵧ are effective in yielding paraffin-rich liquids with superior catalyst stability, with MgₓAlOᵧ particularly suited to mixed-plastic feedstocks [212,213]. Natural clays such as bentonite and kaolin are less active but remain economically favorable alternatives [214]. Transition-metal modifications, exemplified by Ni/Y, lower activation energies and extend catalyst life, though often with reduced initial activity [207,215]. Across catalysts, kinetic studies confirm significant activation energy reductions, enabling accelerated cracking at lower temperatures [216,217]. Catalyst choice steers distribution sharply: HZSM-5 promotes light olefins, Hydrogen-form Ultra-Stable Y-type Zeolite (HUSY) and Hydrogen-form Beta Zeolite (HBEA) increase aromatics and isoparaffins, while thermal pyrolysis favors 1-olefins and n-paraffins [11,209,218,219]. Two-step configurations, where thermal pyrolysis is followed by catalytic upgrading, further enhance selectivity and minimize coking [208]. Persistent challenges include coke deposition and rapid deactivation, particularly in heterogeneous waste feeds [220,221,222], and although certain systems (e.g., ZnCl2, NH4Y) offer marginal benefits, further research is needed on catalyst regeneration and integration with downstream refining [222,223].
The catalytic pyrolysis of LDPE shows similar improvements, with distinct product evolution compared to HDPE. Acidic zeolites, especially ZSM-5, show high selectivity toward light aromatics such as benzene, toluene, and xylenes. Under optimized low-temperature operation, ZSM-5 has achieved 50.6 wt% aromatic yield at 280 °C for 1h with 90.9% selectivity, attributed to micropore confinement and in situ hydrogen transfer [224]. HZSM-5 enhances olefins and aromatics, whereas HUSY favors paraffins in the C4–C8 range [209,225]. Zeolite Beta, when engineered to hierarchical porosity with tuned acidity, has achieved selectivities as high as 88.7% toward lubricating base oil [226], while HY zeolites promote volatile paraffins through disproportionation mechanisms [225]. A key limitation is deactivation by coke; HZSM-5 gradually deactivates until stabilizing after several cycles [227], while LDPE generates more pronounced fouling in HUSY than HDPE [219]. To mitigate diffusion limitations, mesoporous materials are widely used. MCM-41, with large pores and moderate acidity, increases oil yield while reducing secondary cracking [228], and SBA-15 functionalized with sulfated zirconia boosts acid site density and promotes cracking at milder conditions [210]. Hierarchical Beta further enhances aromatic yields through improved molecular transport [229,230]. Sulphates such as Zr(SO4)2 on SBA-15 combine acidity with mesoporosity, providing efficient cracking pathways [154]. Other oxides, including Al2O3 and silica-alumina, reduce onset temperature and activation energy, with silica-alumina lowering Ea from 117.2 kJ mol−1 to 97.3 kJ mol−1 [231]. Carbon-based catalysts, including activated carbons and nanotubes, enable chain adsorption and breakdown, with selectivity tuned by surface functionalities [232]. Mechanistic investigations underscore carbocation intermediates as central, with ZSM-5 favoring dehydroaromatization and HY favoring iso-paraffins [224,233]. In practice, MCM-41 maximizes oil yields (78.4% at 650 °C), while ZSM-5 produces high aromatic content (65.9% at 500 °C) [228]. Silica-alumina provides paraffinic, low-aromatic liquids ideal for clean fuels [232]. Natural zeolites such as clinoptilolite can accelerate decomposition and alter selectivity, though reuse is hindered by coke [234]. Across studies, acidity, pore topology, and catalyst stability emerge as the key determinants of LDPE catalytic pyrolysis efficiency [11,219,235].
For PP, product distributions are strongly shaped by catalyst acidity, structure, and pore dimensions. HZSM-5 enhances formation of C3–C12 hydrocarbons and aromatics such as benzene, toluene, and xylene through its microporous and strongly Brønsted acidic nature, but generally reduces wax yields compared to thermal pyrolysis [236,237]. Other zeolites including HZSM-11 and Y, sometimes combined with metal–organic frameworks (MOFs) like MIL-53 (Cu), further increase aromatic and light-oil selectivity [212,238]. Low-cost natural zeolites (e.g. clinoptilolite) increase paraffinic liquid fractions, with milder acidity and porosity preserving longer hydrocarbons [239,240]. MOF's such as UiO-66, rich in Lewis-acidic zirconia sites, enable selective C–C cleavage while minimizing residue and steering products toward aliphatics rather than aromatics [241]. Metal-doped systems add further flexibility: Ni–Cr supported on natural zeolites boosts gasoline-range hydrocarbons near 450 °C through combined acid and hydrogen transfer/dehydrogenation functions, while cobalt oxides reduce activation energy and yield fuel-grade oils under milder conditions (~430 °C) [242]. Silica-alumina has achieved oil yields up to 91 wt% at 500 °C, outperforming clays such as kaolin or bentonite [243]. Spent FCC catalysts are widely available and cost-effective but tend to produce more gaseous products, limiting wax recovery [244]. Overall, zeolites consistently favor aromatic pathways, while weaker acidity and larger pore catalysts, including MOFs, are better suited for retaining paraffinic waxes under inert or vacuum conditions [245,246].
PVC pyrolysis differs fundamentally, involving initial dehydrochlorination (270–320 °C) to polyenes, followed by secondary scission, aromatization, or char formation [195,196,202,203]. Catalysts serve dual purposes: lowering onset temperatures and managing HCl. Metal chlorides such as ZnCl2 and BaCl2 accelerate dehydrochlorination, leading to early HCl and benzene release below 350 °C and shifting product distributions toward volatiles, especially under hydrogen co-feeding at 400 °C [17]. However, these may also stabilize intermediates, increasing char via metal-organic complexes. Metal hydroxides and oxides such as Ca(OH)2, CaO, Mg(OH)2, and MgO mainly act as HCl traps, reacting to form stable chlorides and producing a cleaner gas phase. Calcium systems outperform magnesium, and hydroxides outperform oxides in HCl uptake [247,248]. Transition metal oxides (MoO3, CuO, Fe2O3, Co3O4) alter degradation trajectories: MoO3 promotes char and reduces aromatics [249]; CuO favors aliphatic hydrocarbons via intermolecular crosslinking; Fe2O3 and ZnO promote deeper cracking, lowering liquids and enriching gases [248,250,251]. These behaviors reflect Lewis acidity and redox functions in stabilizing polyene intermediates. TiO2, ZnO, and SnO2 modulate gas compositions, with TiO2 enhancing aromatics (chlorobenzenes, naphthalenes) and ZnO/SnO2 suppressing aromatics in favor of lighter aliphatic gases at 400–500 °C [248]. More recent work has applied ionic liquids such as [P4444][Cl], achieving up to 98% dehydrochlorination at 80–180 °C in 60 minutes through anion-assisted stabilization of elimination intermediates [252]. Alkaline additives such as CaO and K2CO3 also capture HCl and steer degradation: CaO has reduced HCl emissions below 20% at 1:1.6 additive/PVC ratios, while K2CO3 enhances both neutralization and gas-phase yields [250]. Thus, catalytic design in PVC pyrolysis is a balance between early HCl removal, light-gas maximization, char suppression, and toxic aromatic minimization, typically optimized within 300–500 °C.
Together, these studies confirm that catalyst selection—acidity, pore structure, redox potential, and regeneration possibilities—exerts decisive influence on product selectivity and environmental implications in plastic pyrolysis. Strong Brønsted acidity and micropores favor olefin and aromatic products; mesoporous or hierarchical structures extend diffusion and enhance paraffinic and oil yields; metal functions enable hydrogen transfer, dehydrogenation, or selective dechlorination; and specific additives in PVC serve crucial roles in HCl management to assure HCl free pyrolysis gases. Persistent bottlenecks remain in coke formation, feed impurities, and catalyst regeneration, but advances in hierarchical structures, two-stage processing, and ionic liquid dechlorination strategies point toward increasingly selective and sustainable catalytic pyrolysis.
4.1.2. Initiator Effects for HDPE, LDPE, PP and PVC Pyrolysis
The use of radical and thermal initiators represents a central strategy in controlling polymer pyrolysis under vacuum conditions, where oxygen is excluded and reaction pathways are dominated by radical chemistry [11]. Initiators not only lower the degradation onset temperature but also reduce activation energies and steer selectivity toward valuable products, thereby enhancing the efficiency and scalability of vacuum pyrolysis [253].
For high-density polyethylene (HDPE), pyrolysis in inert conditions usually commences around 400–500 °C with an activation energy close to 227 kJ mol−1. Radical initiators reduce this threshold substantially. Peroxides such as dicumyl peroxide (DCP) or cumene hydroperoxide decompose above 300 °C to form alkoxy radicals, which abstract hydrogens from the polymer chain and initiate β-scission. More remarkable is the action of 2-ethylhexyl nitrate (2-EHN), which decomposes via O–N bond homolysis around 150 °C under oxygen-free conditions, yielding an alkoxy radical (RO·) and a nitrogen dioxide radical (NO2·). The RO· initiates hydrogen abstraction from the HDPE backbone, while NO2· further propagates radical formation or undergoes recombination to N2O4. This process provides an efficient radical source without requiring molecular oxygen, thereby reducing the energy demand of the pyrolytic system [222]. Kinetic studies show that activation energy values may be lowered to ~122 kJ mol−1 in the presence of initiators, with corresponding enthalpy reductions (ΔH ~156–220 kJ mol−1) and Gibbs free energies that remain positive, confirming the endothermic nature of the process [254,255,256,257,258]. Product distribution also improves, with liquid fractions shifting toward gasoline- and diesel-range hydrocarbons, while wax formation is suppressed.
Low-density polyethylene (LDPE) degrades more readily due to its higher branching and less crystalline morphology. Flash vacuum pyrolysis experiments have confirmed that initiators boost the recovery of light olefins, with ethylene comprising more than 60% of the gas fraction, while also enhancing monomer recovery [259,260,261]. Co-feeding initiators with solid catalysts creates a synergistic effect: silica–alumina and ZnO reduce the activation energy, while HZSM-5 channels the radical fragments into BTX aromatics. Oil yields of up to 80% have been reported when radical initiators are combined with mesoporous or acidic catalysts, showing the promisable potential for LDPE valorization [262,263,264,265].
The pyrolysis of polypropylene (PP) is particularly sensitive to radical initiators because of its tertiary carbon centres, which are prone to chain scission. Conventional peroxides efficiently accelerate degradation, with tetra-functional peroxides outperforming di-functional ones due to their higher radical output. Nitroxide-mediated systems (NOR) provide more controlled reductions in molecular weight, limiting runaway fragmentation and preserving tunable rheological properties. Organosulfur compounds act differently: instead of directly producing radicals, they lower the activation energy (~146 kJ mol−1) by stabilizing transition states, making chain scission more accessible. Organo-Sn and macromolecular initiators such as polyglycidyl methacrylate (PGMA) contribute through additional radical release pathways but raise toxicity or stability concerns [213,266,267,268]. Synergistic use of initiators with molecular sieves or cobalt oxides reduces activation barriers to as low as ~64 kJ mol−1, shifting selectivity toward liquid fuels, while Ziegler–Natta catalysts, traditionally employed in polymer synthesis, can be repurposed to promote radical depolymerization at reduced temperatures [242,269]. Overall, radical initiators allow PP pyrolysis to be tuned between light olefin/BTX production and heavier wax/oil fractions depending on initiator chemistry and operating temperature.
The role of initiators in polyvinyl chloride (PVC) degradation is especially critical because of the polymer’s two-step breakdown: initial dehydrochlorination at 270–320 °C and subsequent polyene scission. In vacuum conditions, initiators such as ZnCl2 and BaCl2 accelerate dehydrochlorination by lowering the onset to below 350 °C, facilitating early HCl release and reducing polyene buildup [17]. Alkaline oxides and carbonates, particularly CaO and K2CO3, act as both acid scavengers and initiators, trapping up to 80% of released HCl and thereby preventing chlorine contamination of pyrolysis oils [250]. Zeolitic systems like H-ZSM-5 provide dual functionality: they initiate dehydrochlorination via their strong acidity and simultaneously convert intermediates into olefinic gases, reducing chlorine residues to as low as 20 ppm in secondary oil products [270]. Transition metal oxides, including Fe2O3 and ZnO, further alter PVC pyrolysis pathways through redox and adsorption effects, shifting selectivity toward light aliphatic gases and suppressing toxic aromatics. Recent innovations have explored ionic liquids (e.g., [P4444][Cl]) as initiators, achieving nearly complete dehydrochlorination at temperatures as low as 80–180 °C, thereby opening low-temperature routes for PVC valorization [252].
Taken together, initiators significantly extend the operational flexibility of vacuum pyrolysis. They reduce energy input, accelerate degradation kinetics, and steer selectivity toward fuels or specialty chemicals, while also mitigating undesirable byproducts such as waxes or chlorinated aromatics. When combined with catalysts, initiators enable finely tuned, lower-temperature processes that maximize fuel quality and minimize environmental impacts. Their chemistry is thus central to advancing vacuum pyrolysis into a scalable solution for circular plastic waste management. It is always mandatory to think about recyclability and environmental implications.
4.1.3. Termination Strategies for HDPE, LDPE, PP and PVC Pyrolysis
Termination strategies in the pyrolysis of High-Density Polyethylene (HDPE) aim to inhibit uncontrolled chain scission and radical propagation, thus enhancing selectivity toward desired liquid hydrocarbons and reducing the formation of low-molecular-weight byproducts [134]. One of the most extensively studied approaches involves the use of butylated hydroxytoluene (BHT), which acts as a radical scavenger. When immobilized on hydrophobic silica fillers, BHT exhibits improved radical suppression by reducing its volatility and enhancing interaction with the polymer matrix. This results in significantly lower yields of volatile products and a shift toward higher-molecular-weight hydrocarbons, while hydrophilic silica–BHT systems show diminished effectiveness due to weaker retention and lower scavenging activity [134]. An alternative radical-trapping approach involves the use of fullerene (C60), which stabilizes carbon-centered radicals formed during HDPE decomposition. Its radical scavenging effect increases the activation energy required for degradation, thereby suppressing early-stage decomposition. This suppression slows the degradation rate under inert atmospheres and results in a greater proportion of stable, liquid-phase hydrocarbons while minimizing gaseous products [271].
Catalyst-based termination methods also offer effective control over degradation. Microporous zeolites such as HZSM-5, HY, and USY provide Brønsted acid sites that facilitate targeted cracking and secondary transformations, including isomerization and aromatization [272,273]. Their pore architecture enables shape-selective conversion, leading to an increase in liquid products rich in olefins and aromatics, with HZSM-5 favoring C5–C8 olefins and HY enhancing aromatic hydrocarbon yield, sometimes up to 50% of the liquid fraction [274]. Spent fluid catalytic cracking (FCC) catalysts represent a cost-effective and industrially scalable solution. These catalysts significantly lower the degradation temperature and increase the formation rate of low boiling, olefinic liquids. When applied to mixed polyolefin streams, they improve product uniformity and decrease the molecular weight distribution of the resulting liquids, aligning their composition more closely with gasoline-range hydrocarbons [275,276]. Collectively, termination modulation in HDPE pyrolysis through additives like BHT and fullerene, or via catalytic systems such as zeolites and FCC catalysts, enables suppression of undesired fragmentation pathways and enhances product selectivity [134].
In the case of Low-Density Polyethylene (LDPE), termination strategies play a crucial role in maintaining liquid yields and avoiding excessive scission into light hydrocarbons [239]. Catalytic control is again important, with natural zeolites such as clinoptilolite and Philippine natural zeolite providing cost-effective alternatives that reduce uncondensed gases while improving liquid oil recovery [239]. Rapid quenching of pyrolysis vapors also provides a physical termination mechanism, stabilizing liquid products and suppressing secondary radical-driven fragmentation into smaller volatile molecules [178]. The reusability and regeneration of catalysts, such as HZSM-5 restored at 700 °C in inert atmospheres, further support long-term stability in termination efficiency and minimize coke deposition that otherwise alters product selectivity [236]. These combined strategies for LDPE highlight the importance of both chemical and physical termination in maintaining process control.
Controlling termination in the pyrolysis of Polypropylene (PP) is particularly important to preserve long-chain hydrocarbons such as waxes and mid-distillate fuels, since uncontrolled propagation often leads to over-cracking into lighter gaseous fractions [277,278]. Thermal regulation has been shown to be one of the most effective strategies, with operations in the 450–550 °C range enabling a balance between conversion efficiency and selectivity. For instance, pyrolysis at 500 °C yielded a stable liquid fraction of ~76% within 30 minutes, underscoring the benefit of moderate conditions [279]. Catalytic intervention further shapes the termination process, with acidic zeolites promoting gasoline-range hydrocarbons (C5–C12), while kaolin, activated carbon, and iron-modified catalysts enhance mid-range hydrocarbon selectivity and reduce oxygenated byproducts [32,280]. Optimization of operating variables such as nitrogen flow rate and feed input via statistical design methods has yielded fuel oil fractions above 89%, while minimizing gas formation and suppressing excessive secondary cracking [278,281]. Rapid quenching is also widely recognized as an effective physical termination step, particularly when integrated with catalytic systems [282]. Catalyst regeneration ensures consistent activity across multiple cycles, enabling long-term process stability and controlled termination [283]. Collectively, these strategies underline the multifaceted approaches available for PP termination under inert pyrolysis conditions.
For PVC, termination strategies aim to suppress degradation beyond dehydrochlorination, thereby preventing char formation and improving the recovery of volatile hydrocarbons [250]. Since PVC degradation proceeds via initial HCl release at ~270–320 °C followed by polyene formation and eventual cross-linking, termination focuses on stabilizing these intermediates or halting their progression. Rapid cooling under vacuum effectively quenches intermediates and suppresses polyene cross-linking [284]. A two-step pyrolysis protocol has also proven effective, where dehydrochlorination at ~300–350 °C is followed by high-temperature backbone cracking, decoupling chlorine elimination from subsequent degradation [285]. Hydrogen co-feeding provides radical termination by stabilizing polyene fragments and limiting aromatic or char pathways [286,287]. Alkaline hydroxides such as Ca(OH)2 and Mg(OH)2 neutralize HCl and disrupt radical propagation, further suppressing polyene formation [247]. Stabilizers including organotin compounds and dichlorotin dioxine delay dehydrochlorination, while ionic liquids facilitate both HCl removal and hydrogenation of intermediates [288,289]. Advanced additives such as alkaline earth stearates raise the activation energy for dehydrochlorination, delay degradation onset, and synergistically neutralize HCl when combined with lead stearate [290]. Mechanistically, these strategies target the interruption of free-radical propagation through either chemical scavenging or physical quenching, enabling selective recovery of volatile, non-chlorinated products [251,291].
5. Conclusions
A unified treatment of polymerization and depolymerization is essential because the microstructure encoded at synthesis governs bond strengths, radical stabilities, and elimination geometries, thereby fixing the kinetic pathways and attainable product slates at end-of-life. Linear HDPE, branched LDPE, stereoregular PP with dense tertiary centers, and PVC with backbone C–Cl functionality present fundamentally different degradation manifolds before any reactor variable is selected. Recognizing this “polymerization → structure → depolymerization” causality allows chemical engineers to replace empirical parameter sweeps with mechanism-anchored design of yields—whether the objective is light olefins/BTX for petrochemical integration, paraffinic oils/waxes for materials and fuels, or clean combustible gases for CHP.
For polyolefins, random scission, β-scission, hydrogen abstraction, and backbiting establish the baseline partitioning between gases and condensables. Catalytic topology and acidity then translate that intrinsic chemistry into selective outcomes: strong Brønsted acidity in shape-selective micropores compresses distributions toward C2–C4 olefins and BTX via carbocation chemistry and controlled secondary reactions, whereas weaker acidity and meso/hierarchical porosity preserve longer chains and favor paraffinic oils and waxes. Process variables map directly onto these mechanisms. Temperature and heating rate set the onset and depth of primary cracking; vapor residence time and pressure regulate the extent of secondary scission, oligomerization, and aromatization; rapid quench kinetically traps liquids before over-cracking. Initiators lower activation thresholds and tune radical populations, reducing energy input and expanding the operable window. Diffusion management is co-equal with acidity for stability: hierarchical frameworks, external-surface passivation, and judicious Al siting mitigate pore-mouth blockage, slow coke nucleation, and sustain selectivity at conversion.
PVC imposes a flowsheet constraint that further underscores the value of the coupled perspective. Controlled dehydrochlorination must precede residue cracking to suppress autocatalysis, protect metallurgy, and prevent chlorine carryover. Continuous HCl removal or in-situ scavenging, together with basic/redox co-catalysts or ionic-liquid strategies, lowers the dechlorination temperature, limits polyene cyclization/condensation, and yields cleaner gas and liquid fractions. Only after halogen management is secured should severity and residence time be applied to upgrade the dechlorinated residue, with short contact and rapid quench again decisive for suppressing aromatization and char.
Viewed through this lens, process design becomes a targeted exercise in matching feed microstructure to catalyst function and transport, then selecting operating envelopes that pivot deliberately among off-gas, light olefins/aromatics, and paraffinic liquids. The practical implications are clear: (i) predictive yield control via acidity/topology tuned to polymer architecture; (ii) lower specific energy demand by combining initiators with appropriate catalysts; (iii) longer time-on-stream by engineering diffusion pathways that defer deactivation; and (iv) for PVC, integrated halogen capture that decouples dechlorination from backbone scission. Coupling polymerization-derived structure with depolymerization mechanisms in this way turns heterogeneous plastic waste from a liability into a tunable feedstock, enabling scalable, selective, and energy-lean routes that align product quality, plant operability, and circular-carbon objectives.
Author Contributions
Conceptualization, T.T. and D.K.; methodology, T.T.; software, T.T.; validation, T.T., D.K., D.P and K.R.; formal analysis, T.T., D.P. and K.R. investigation, T.T., D.K., D.P and K.R.; resources, T.T., D.K., D.P and K.R.; data curation, T.T., D.K., D.P.; writing—original draft preparation, T.T., D.K., D.P and K.R.; writing—review and editing, T.T., D.K. and D.P.; visualization, T.T.; supervision, D.K., D.P. and K.R..; project administration, T.T., D.K. and K.R..; funding acquisition, T.T, D.K. and K.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Slovenian Research and Innovation Agency, research programme number P2-0414, Process systems engineering and sustainable developmentand and project number J7-50228, Mining the technosphere for efficient use of resources and improving the state of the environment. and The APC was funded by the Scientific Research Centre Bistra Ptuj.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Sun, J.; Dong, J.; Gao, L.; Zhao, Y.-Q.; Moon, H.; Scott, S.L. Catalytic Upcycling of Polyolefins. Chem. Rev. 2024, 124, 9457–9579. [CrossRef]
- Xu, S.; Tang, J.; Fu, L. Catalytic Strategies for the Upcycling of Polyolefin Plastic Waste. Langmuir 2024, 40, 3984–4000. [CrossRef] [PubMed]
- Faust, K.; Denifl, P.; Hapke, M. Recent Advances in Catalytic Chemical Recycling of Polyolefins. ChemCatChem 2023, 15, e202300310. [CrossRef]
- Lopez, E.C.R. Pyrolysis of Polyvinyl Chloride, Polypropylene, and Polystyrene: Current Research and Future Outlook. Eng. Proc. 2023, 56, 1–9. [CrossRef]
- Svadlenak, S.; Wojcik, S.; Ogunlalu, O.; Vu, M.; Dor, M.; Boudouris, B.W.; Wildenschild, D.; Goulas, K.A. Upcycling of Polyvinyl Chloride to Hydrocarbon Waxes via Dechlorination and Catalytic Hydrogenation. Appl. Catal. B Environ. 2023, 338, 123065. [CrossRef]
- Kruse, T.M.; Wong, H.; Broadbelt, L.J. Mechanistic Modeling of Polymer Pyrolysis : Polypropylene. 2003, 36, 9594–9607. [CrossRef]
- Popov, K. V; Knyazev, V.D. Initial Stages of the Pyrolysis of Polyethylene. J. Phys. Chem. A 2015, 119, 11737–11760. [CrossRef]
- Gascoin, N.; Fau, G.; Gillard, P.; Mangeot, A. Flash Pyrolysis of High Density Polyethylene. 49th AIAA/ASME/SAE/ASEE Jt. Propuls. Conf. 2013, 1 PartF. [CrossRef]
- J V, J.; Perez, B.; Toraman, H. Parametric Study of Polyethylene Primary Decomposition Using a Micropyrolyzer Coupled with Two-Dimensional Gas Chromatography. ACS Sustain. Chem. Eng. 2024, 12. [CrossRef]
- Yan, G.; Jing, X.; Wen, H.; Xiang, S. Thermal Cracking of Virgin and Waste Plastics of PP and LDPE in a Semibatch Reactor under Atmospheric Pressure. Energy & Fuels 2015, 29, 2289–2298. [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Evolution of Products Generated during the Dynamic Pyrolysis of LDPE and HDPE over HZSM5. Energy & Fuels 2008, 22, 2917–2924. [CrossRef]
- Hujuri, U.; Ghoshal, A.; Gumma, S. Temperature-Dependent Pyrolytic Product Evolution Profile for Polypropylene. J. Appl. Polym. Sci. 2011, 119, 2318–2325. [CrossRef]
- Danforth, J.D.; Spiegel, J.; Bloom, J. The Kinetics and Mechanism of the Thermal Dehydrochlorination of Poly(Vinyl Chloride). J. Macromol. Sci. Part A - Chem. 1982, 17, 1107–1127. [CrossRef]
- Nolan, K.; Shapiro, J. Presence of Dual Mechanism in Poly(Vinyl Chloride) Dehydrochlorination. J. Polym. Sci. Polym. Symp. 2007, 55, 201–209. [CrossRef]
- Wu, J.; Papanikolaou, K.G.; Cheng, F.; Addison, B.; Cuthbertson, A.A.; Mavrikakis, M.; Huber, G.W. Kinetic Study of Polyvinyl Chloride Pyrolysis with Characterization of Dehydrochlorinated PVC. ACS Sustain. Chem. Eng. 2024, 12, 7402–7413. [CrossRef]
- Patel, K.; Velazquez, A.; Calderon, H.S.; Brown, G.R. Studies of the Solid-State Thermal Degradation of PVC. I. Autocatalysis by Hydrogen Chloride. J. Appl. Polym. Sci. 1992, 46, 179–187. [CrossRef]
- Cheng, W.-H.; Liang, Y.C. Catalytic Pyrolysis of Polyvinylchloride in the Presence of Metal Chloride. J. Appl. Polym. Sci. 2000, 77, 2464–2471. [CrossRef]
- Ballistreri, A.; Foti, S.; Maravigna, P.; Montaudo, G.; Scamporrino, E. Effect of Metal Oxides on the Evolution of Aromatic Hydrocarbons in the Thermal Decomposition of PVC. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 3101–3110. [CrossRef]
- Lattimer, R.P.; Kroenke, W.J. Mechanisms of Formation of Volatile Aromatic Pyrolyzates from Poly(Vinyl Chloride). J. Appl. Polym. Sci. 1982, 27, 1355–1366. [CrossRef]
- Jafari, A.J.; Donaldson, J.D. Determination of HCl and VOC Emission from Thermal Degradation of PVC in the Absence and Presence of Copper, Copper(II) Oxide and Copper(II) Chloride. J. Chem. 2009, 6, 753835. [CrossRef]
- Meng, H.; Liu, J.; Xia, Y.; Hu, B.; Sun, H.; Li, J.; Lu, Q. Migration and Transformation Mechanism of Cl during Polyvinyl Chloride Pyrolysis: The Role of Structural Defects. Polym. Degrad. Stab. 2024, 224, 110750. [CrossRef]
- Beneš, M.; Milanov, N.; Matuschek, G.; Kettrup, A.; Plaček, V.; Balek, V. Thermal Degradation of PVC Cable Insulation Studied by Simultaneous TG-FTIR and TG-EGA Methods. J. Therm. Anal. Calorim. - J THERM ANAL CALORIM 2004, 78, 621–630. [CrossRef]
- Xu, S.; Sun, Z.; Hou, K.; Wang, G.; Liu, M.; Zhang, Y.; Zhang, C.; Zhang, Z. Quantifying the Modulation of Modified ZSM-5 Acidity/Alkalinity on Olefin Catalytic Pyrolysis to Maximize Light Olefin Selectivity. J. Energy Inst. 2024, 115, 101688. [CrossRef]
- Farah, E.; Demianenko, L.; Engvall, K.; Kantarelis, E. Controlling the Activity and Selectivity of HZSM-5 Catalysts in the Conversion of Biomass-Derived Oxygenates Using Hierarchical Structures: The Effect of Crystalline Size and Intracrystalline Pore Dimensions on Olefins Selectivity and Catalyst Deactivatio. Top. Catal. 2023, 66, 1310–1328. [CrossRef]
- Wang, R.; Gong, Y.; Wang, P.; Zheng, A.; Wang, Z.; Sha, Y.; Jiang, Q.; Xin, M.; Cao, D.; Song, H.; et al. 3D-Printed Monolithic ZSM-5@nano-ZSM-5: Hierarchical Core-Shell Structured Catalysts for Enhanced Cracking of Polyethylene-Derived Pyrolysis Oils. Addit. Manuf. 2024, 79, 103890. [CrossRef]
- Rzepka, P.; Sheptyakov, D.; Wang, C.; van Bokhoven, J.A.; Paunović, V. How Micropore Topology Influences the Structure and Location of Coke in Zeolite Catalysts. ACS Catal. 2024, 14, 5593–5604. [CrossRef]
- Wang, T.; Feng, X.; Lin, D.; Li, Y.; Shang, J.; Zhang, J.; Li, S.; Liu, Y.; Zhao, H.; Ma, Z.; et al. Regulating Framework Aluminum Location towards Boosted Light Olefins Generation in Ex-Situ Catalytic Pyrolysis of Low-Density Polyethylene. Chem. Eng. J. 2024, 485, 149737. [CrossRef]
- Chen, Z.; Lyu, W.; Wang, R.; Li, Y.; Xu, C.; Jiang, G.; Zhang, L. A Molecular Kinetic Model Incorporating Catalyst Acidity for Hydrocarbon Catalytic Cracking. AIChE J. 2023, 69, e18060. [CrossRef]
- Chen, D.; Liu, D.; He, H.; Zhao, L.; Gao, J.; Xu, C. Rational Tuning of Monomolecular, Bimolecular and Aromatization Pathways in the Catalytic Pyrolysis of Hexane on ZSM-5 from a First-Principles-Based Microkinetics Analysis. Fuel 2024, 366, 131368. [CrossRef]
- Ureel, Y.; Alexopoulos, K.; Van Geem, K.M.; Sabbe, M.K. Predicting the Effect of Framework and Hydrocarbon Structure on the Zeolite-Catalyzed Beta-Scission. Catal. Sci. Technol. 2024, 14, 7020–7036. [CrossRef]
- Zhang, J.; Dengguo, L.; Chen, Z.; Wang, X.; Xiong, Q.; Li, J.; Zhang, X.; Oboirien, B.O.; Xu, G. CO2-Assisted Catalytic Pyrolysis of Polyolefins to Aromatics over Mesoporous HZSM-5 and Ga/ZSM-5 Catalysts. ACS Sustain. Chem. Eng. 2024, 12, 13137–13148. [CrossRef]
- He, Q.; Akin, O.; Ureel, Y.; Yazdani, P.; Li, L.; Varghese, R.J.; Geem, K.M. Van Enhancing Catalytic Pyrolysis of Polypropylene Using Mesopore-Modified HZSM-5 Catalysts: Insights and Strategies for Improved Performance. Front. Chem. Eng. 2024, 6. [CrossRef]
- Kim, C.A.; Sahasrabudhe, C.A.; Wang, Y.-Y.; Yappert, R.; Heyden, A.; Huang, W.; Sadow, A.D.; Peters, B. Population Balance Equations for Reactive Separation in Polymer Upcycling. Langmuir 2024, 40, 4096–4107. [CrossRef]
- Wu, Y.; Han, J.; Zhang, W.; Yu, Z.; Wang, K.; Fang, X.; Wei, Y.; Liu, Z. Combined Strategies Enable Highly Selective Light Olefins and Para-Xylene Production on Single Catalyst Bed. J. Am. Chem. Soc. 2024, 146, 8086–8097. [CrossRef]
- Duan, J.; Chen, W.; Wang, C.; Wang, L.; Liu, Z.; Yi, X.; Fang, W.; Wang, H.; Wei, H.; Xu, S.; et al. Coking-Resistant Polyethylene Upcycling Modulated by Zeolite Micropore Diffusion. J. Am. Chem. Soc. 2022, 144, 14269–14277. [CrossRef] [PubMed]
- Eschenbacher, A.; Varghese, R.J.; Delikonstantis, E.; Mynko, O.; Goodarzi, F.; Enemark-Rasmussen, K.; Oenema, J.; Abbas-Abadi, M.S.; Stefanidis, G.D.; Van Geem, K.M. Highly Selective Conversion of Mixed Polyolefins to Valuable Base Chemicals Using Phosphorus-Modified and Steam-Treated Mesoporous HZSM-5 Zeolite with Minimal Carbon Footprint. Appl. Catal. B Environ. 2022, 309, 121251. [CrossRef]
- Tennakoon, A.; Wu, X.; Meirow, M.; Howell, D.; Willmon, J.; Yu, J.; Lamb, J. V.; Delferro, M.; Luijten, E.; Huang, W.; et al. Two Mesoporous Domains Are Better Than One for Catalytic Deconstruction of Polyolefins. J. Am. Chem. Soc. 2023, 145, 17936–17944. [CrossRef] [PubMed]
- Ando, Y.; Miyakage, T.; Anzai, A.; Huang, M.; Ait El Fakir, A.; Toyao, T.; Nakasaka, Y.; Phuekphong, A.; Ogawa, M.; Kolganov, A.A.; et al. Conversion of Polypropylene to Light Olefins by HMFI Catalysts below Pyrolytic Temperature: Catalytic, Spectroscopic, and Theoretical Studies. J. Phys. Chem. C 2025, 129, 1678–1691. [CrossRef]
- Ashuiev, A.; Allouche, F.; Wili, N.; Searles, K.; Klose, D.; Copéret, C.; Jeschke, G. Molecular and Supported Ti(Iii)-Alkyls: Efficient Ethylene Polymerization Driven by the π-Character of Metal-Carbon Bonds and Back Donation from a Singly Occupied Molecular Orbital. Chem. Sci. 2021, 12, 780–792. [CrossRef]
- Nakayama, Y.; Shiono, T. Coordination Polymerization (Olefin and Diene). In Encyclopedia of Polymeric Nanomaterials; Kobayashi, S., Müllen, K., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2021; pp. 1–6 ISBN 978-3-642-36199-9.
- Tran, T. V; Do, L.H. Tunable Modalities in Polyolefin Synthesis via Coordination Insertion Catalysis. Eur. Polym. J. 2021, 142, 110100. [CrossRef]
- Fushimi, M.; Damma, D. Exploring Ti Active Sites in Ziegler-Natta Catalysts through Realistic-Scale Computer Simulations with Universal Neural Network Potential. Mol. Catal. 2024, 565, 114414. [CrossRef]
- Young, M.J.; Ma, C.C.M.; Ting, C. Activation Energy and Transition State Determination of the Olefin Insertion Process of Metallocene Catalysts Using a Semiempirical Molecular Orbital Calculation. Russ. J. Coord. Chem. Khimiya 2002, 28, 25–31. [CrossRef]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Calculating Accurate Barriers for Olefin Insertion and Related Reactions. J. Organomet. Chem. 2015, 775, 39–49. [CrossRef]
- Clementi, E.; Giunchi, G.; Introduction, I. Theoretical Study on a Reaction Pathway of Ziegler-Natta- Type Catalysis. 1978, 68.
- Ortega, D.E.; Matute, R.A.; Toro-Labbé, A. Exploring the Nature of the Energy Barriers on the Mechanism of the Zirconocene-Catalyzed Ethylene Polymerization: A Quantitative Study from Reaction Force Analysis. J. Phys. Chem. C 2020, 124, 8198–8209. [CrossRef]
- Huang, J.; Rempel, G.L. Ziegler-Natta Catalysts for Olefin Polymerization: Mechanistic Insights from Metallocene Systems. Prog. Polym. Sci. 1995, 20, 459–526. [CrossRef]
- Fan, H.; Kang, X.; Dai, S. Stable Ultrahighly Branched Polyethylene Synthesis via Externally Robust Chain-Walking Polymerization. ACS Catal. 2024, 13531–13541. [CrossRef]
- Ashuiev, A.; Humbert, M.; Gajan, D.; Norsic, S.; Searles, K.; Klose, D.; Lesage, A.; Pintacuda, G.; Ashuiev, A.; Humbert, M.; et al. Spectroscopic Signature and Structure of Active Centers in Ziegler-Natta Polymerization Catalysts Revealed by Electron Paramagnetic Resonance To Cite This Version : HAL Id : Hal-03017383 Spectroscopic Signature and Structure of Active Centers in Ziegler-N. 2020. [CrossRef]
- So, L.C.; Faucher, S.; Zhu, S. Synthesis of Low Molecular Weight Polyethylenes and Polyethylene Mimics with Controlled Chain Structures. Prog. Polym. Sci. 2014, 39, 1196–1234. [CrossRef]
- Kissin, Y. V Active Centers in Ziegler–Natta Catalysts: Formation Kinetics and Structure. J. Catal. 2012, 292, 188–200. [CrossRef]
- Shiga, A. Theoretical Study of Ethylene Polymerization on Ziegler–Natta Catalysts and on Metallocene Catalysts. J. Mol. Catal. A Chem. 1999, 146, 325–334. [CrossRef]
- Koltzenburg, S.; Maskos, M.; Nuyken, O. Coordination Polymerization. In Polymer Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 2023; pp. 295–324 ISBN 978-3-662-64929-9.
- Yu, Y.; Fu, Z.; Fan, Z. Chain Transfer Reactions of Propylene Polymerization Catalyzed by AlEt 3 Activated TiCl 4/MgCl 2 Catalyst under Very Low Monomer Addition Rate. J. Mol. Catal. A Chem. 2012, 363–364, 134–139. [CrossRef]
- Margl, P.; Deng, L.; Ziegler, T. A Unified View of Ethylene Polymerization by D0 and D0f(n) Transition Metals. 3. Termination of the Growing Polymer Chain. J. Am. Chem. Soc. 1999, 121, 154–162. [CrossRef]
- Talarico, G.; Budzelaar, P.H.M. A Second Transition State for Chain Transfer to Monomer in Olefin Polymerization Promoted by Group 4 Metal Catalysts. J. Am. Chem. Soc. 2006, 128, 4524–4525. [CrossRef]
- Tsutsui, T.; Kashiwa, N.; Mizuno, A. Effect of Hydrogen on Propene Polymerization with Ethylenebis(1-indenyl)Zirconium Dichloride and Methylalumoxane Catalyst System. Die Makromol. Chemie, Rapid Commun. 1990, 11, 565–570. [CrossRef]
- Kissin, Y. V.; Rishina, L.A.; Vizen, E.I. Hydrogen Effects in Propylene Polymerization Reactions with Titanium-Based Ziegler-Natta Catalysts. II. Mechanism of the Chain-Transfer Reaction. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1899–1911. [CrossRef]
- Guo, Y.; Zhang, Z.; Guo, W.; Khan, A.; Fu, Z.; Xu, J.; Fan, Z. Kinetics and Mechanism of Metallocene-Catalyzed Olefin Polymerization: Comparison of Ethylene, Propylene Homopolymerizations, and Their Copolymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 867–875. [CrossRef]
- Naweephattana, P.; Walaijai, K.; Rungnim, C.; Luanphaisarnnont, T.; Watthanaphanit, A.; Patthamasang, S.; Phiriyawirut, P.; Surawatanawong, P. The Role of Organoaluminum and Electron Donors in Propene Insertion on the Ziegler–Natta Catalyst. Dalt. Trans. 2024, 53, 11050–11059. [CrossRef]
- Kumawat, J.; Gupta, V.K.; Vanka, K. Effect of Donors on the Activation Mechanism in Ziegler-Natta Catalysis: A Computational Study. ChemCatChem 2016, 8, 1809–1818. [CrossRef]
- Kissin, Y. V.; Liu, X.; Pollick, D.J.; Brungard, N.L.; Chang, M. Ziegler-Natta Catalysts for Propylene Polymerization: Chemistry of Reactions Leading to the Formation of Active Centers. J. Mol. Catal. A Chem. 2008, 287, 45–52. [CrossRef]
- Magni, E.; Somorjai, G.A. Ethylene and Propylene Polymerization Catalyzed by a Model Ziegler-Natta Catalyst Prepared by Gas Phase Deposition of Magnesium Chloride and Titanium Chloride Thin Films. Catal. Letters 1995, 35, 205–214. [CrossRef]
- Fushimi, M.; Damma, D. The Role of External Donors in Ziegler-Natta Catalysts through Nudged Elastic Band Simulations on Realistic-Scale Models Employing a Universal Neural Network Potential. J. Phys. Chem. C 2024, 128, 6646–6657. [CrossRef]
- Li, C.; Feng, H.; Liu, H.; Zhuang, Z.; Zhou, J.; Liu, D. Effect of Dioldibenzoate Isomers as Electron Donors on the Performances of Ziegler–Natta Polypropylene Catalysts: Experiments and Calculations. J. Phys. Chem. C 2023, 127, 2294–2302. [CrossRef]
- Tritto, I.; Sacchi, M.C.; Locatelli, P. Ziegler-Natta Polymerization of Propene: Cooperative Effects of Titanium Ligands on the Steric Control of the First Addition of Monomer. Die Makromol. Chemie 1986, 187, 2145–2151. [CrossRef]
- Resconi, L.; Piemontesi, F.; Franciscono, G.; Abis, L.; Fiorani, T. Olefin Polymerization at Bis(Pentamethylcyclopentadienyl)Zirconium and -Hafnium Centers: Chain-Transfer Mechanisms. J. Am. Chem. Soc. 1992, 114, 1025–1032. [CrossRef]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Haspeslagh, L.; Atwood, J.L.; Bott, S.G.; Robinson, K. Metallocene/Polypropylene Structural Relationships: Implications on Polymerization and Stereochemical Control Mechanisms. Makromol. Chemie. Macromol. Symp. 1991, 48–49, 253–295. [CrossRef]
- Busico, V.; Cipullo, R.; Pellecchia, R.; Ronca, S.; Roviello, G.; Talarico, G. Design of Stereoselective Ziegler-Natta Propene Polymerization Catalysts. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15321–15326. [CrossRef] [PubMed]
- Busico, V.; Cipullo, R.; Ronca, S.; Budzelaar, P.H.M. Mimicking Ziegler-Natta Catalysts in Homogeneous Phase, 1: C2-Symmetric Octahedral Zr(IV) Complexes with Tetradentate [ONNO]-Type Ligands. Macromol. Rapid Commun. 2001, 22, 1405–1410. [CrossRef]
- Castro, L.; Therukauff, G.; Vantomme, A.; Welle, A.; Haspeslagh, L.; Brusson, J.M.; Maron, L.; Carpentier, J.F.; Kirillov, E. A Theoretical Outlook on the Stereoselectivity Origins of Isoselective Zirconocene Propylene Polymerization Catalysts. Chem. - A Eur. J. 2018, 24, 10784–10792. [CrossRef]
- Chadwick, J.C.; Heere, J.J.R.; Sudmeijer, O. Factors Influencing Chain Transfer with Monomer and with Hydrogen in Propene Polymerization Using MgCl2-Supported Ziegler-Natta Catalysts. Macromol. Chem. Phys. 2000, 201, 1846–1852. [CrossRef]
- Cavallo, L.; Guerra, G.; Corradini, P. Mechanisms of Propagation and Termination Reactions in Classical Heterogeneous Ziegler-Natta Catalytic Systems: A Nonlocal Density Functional Study. J. Am. Chem. Soc. 1998, 120, 2428–2436. [CrossRef]
- Santoro, O.; Piola, L.; Mc Cabe, K.; Lhost, O.; Den Dauw, K.; Vantomme, A.; Welle, A.; Maron, L.; Carpentier, J.F.; Kirillov, E. Long-Chain Branched Polyethylene via Coordinative Tandem Insertion and Chain-Transfer Polymerization Using Rac-{EBTHI}ZrCl2/MAO/Al-Alkenyl Combinations: An Experimental and Theoretical Study. Macromolecules 2020, 53, 8847–8857. [CrossRef]
- Sifri, R.J.; Padilla-Vélez, O.; Coates, G.W.; Fors, B.P. Controlling the Shape of Molecular Weight Distributions in Coordination Polymerization and Its Impact on Physical Properties. J. Am. Chem. Soc. 2020, 142, 1443–1448. [CrossRef]
- Becker, P.; Buback, M.; Sandmann, J. Initiator Efficiency of Peroxides in High-Pressure Ethene Polymerization. 2002, 2113–2123.
- Davis, T.P. HANDBOOK OF RADICAL; ISBN 047139274X.
- Goller, A.; Obenauf, J.; Kretschmer, W.P.; Kempe, R. The Highly Controlled and Efficient Polymerization of Ethylene. Angew. Chemie - Int. Ed. 2023, 62. [CrossRef]
- Scorah, M.J.; Cosentino, R.; Dhib, R.; Penlidis, A. Experimental Study of a Tetrafunctional Peroxide Initiator: Bulk Free Radical Polymerization of Butyl Acrylate and Vinyl Acetate. Polym. Bull. 2006, 57, 157–167. [CrossRef]
- Koptelov, A.A.; Milekhin, Y.M.; Baranets, Y.N. Simulation of Thermal Decomposition of a Polymer at Random Scissions of C – C Bonds. 2012, 6, 626–633. [CrossRef]
- Poutsma, M.L. Reexamination of the Pyrolysis of Polyethylene : Data Needs , Free-Radical Mechanistic Considerations , and Thermochemical Kinetic Simulation of Initial Product-Forming Pathways. 2003, 8931–8957.
- Yeiser, T.M.; Kline, G.M.; Arnett, R.L.; Stacy, C.J. Kinetics of the Thermal Degradation of Linear Polyethylene Complete Description of Thermal Degradation Would. 1966.
- Ding, W.; Liang, J.; Anderson, L.L. Thermal and Catalytic Degradation of High Density Polyethylene and Commingled Post-Consumer Plastic Waste. 1997, 3820.
- Holmstrom, A.; Sorvik, E.M. NITROGEN ATMOSPHERE OF LOW OXYGEN CONTENT . TYPES OF HIGH-DENSITY POLYETHYLENE. 1976, 53, 33–53.
- Lee, E.J.; Park, H.J.; Kim, S.M.; Lee, K.Y. Effect of Azo and Peroxide Initiators on a Kinetic Study of Methyl Methacrylate Free Radical Polymerization by DSC. Macromol. Res. 2018, 26, 322–331. [CrossRef]
- Pielichowski, K.; Njuguna, J.; Majka, T. Thermal Degradation of Polymers, Copolymers, and Blends. In; 2023; pp. 49–147 ISBN 9780128230237.
- Natta, G. Olefin Polymerization with Ziegler-Natta Catalyst. 1963, 1–6.
- Barta, J. Recent Advances in the Synthesis and Applications of Azo Initiators. 2016, 5133–5145. [CrossRef]
- Scorah, M.J.; Dhib, R.; Penlidis, A. Use of a Novel Tetrafunctional Initiator in the Free Radical Homo- and Copolymerization of Styrene, Methyl Methacylate and α-Methyl Styrene. J. Macromol. Sci. - Pure Appl. Chem. 2005, 42 A, 403–426. [CrossRef]
- Luft, G.; Bitsch, H.; Seidl, H. Effectiveness of Organic Peroxide Initiators in the High-Pressure Polymerization of Ethylene. J. Macromol. Sci. Part A - Chem. 1977, 11, 1089–1112. [CrossRef]
- Myers, T.N. Initiators, Free-Radical. 2001, In Kirk-Ot. [CrossRef]
- Khubi-Arani, Z.; Salami-Kalajahi, M.; Najafi, M.; Roghani-Mamaqani, H.; Haddadi-Asl, V.; Ghafelebashi-Zarand, S.M. Simulation of Styrene Free Radical Polymerization over Bi-Functional Initiators Using Monte Carlo Simulation Method and Comparison with Mono-Functional Initiators. Polym. Sci. - Ser. B 2010, 52, 184–192. [CrossRef]
- Machi, S.; Kise, S.; Hagiwara, M.; Kagiya, T. Mechanisms of Propagation, Transfer, and Short-chain Branching Reactions in the Free-radical Polymerization of Ethylene. J. Polym. Sci. Part A-1 Polym. Chem. 1967, 5, 3115–3128. [CrossRef]
- Izgorodina, E.I.; Coote, M.L. Accurate Ab Initio Prediction of Propagation Rate Coefficients in Free-Radical Polymerization : Acrylonitrile and Vinyl Chloride. 2006, 324, 96–110. [CrossRef]
- Ashfaq, A.; Clochard, M.C.; Coqueret, X.; Dispenza, C.; Driscoll, M.S.; Ulański, P.; Al-Sheikhly, M. Polymerization Reactions and Modifications of Polymers by Ionizing Radiation. Polymers (Basel). 2020, 12, 1–67. [CrossRef]
- Liu, Y.A.; Sharma, N. Free Radical Polymerizations: LDPE and EVA ; 2023; ISBN 9783527843831.
- Mavroudakis, E.; Cuccato, D.; Moscatelli, D. Quantum Mechanical Investigation on Bimolecular Hydrogen Abstractions in Butyl Acrylate-Based Free Radical Polymerization Processes. J. Phys. Chem. A 2014, 118, 1799–1806. [CrossRef]
- Konstantinov, I.; Ewart, S.; Brown, H.; Eddy, C.; Mendenhall, J.; Munjal, S. Accurate Density Functional Theory (DFT) Protocol for Screening and Designing Chain Transfer and Branching Agents for LDPE Systems. Mol. Syst. Des. Eng. 2018, 3, 228–242. [CrossRef]
- Beuermann, S.; Buback, M. Free-Radical Polymerization Under High Pressure. In High Pressure Molecular Science; Winter, R., Jonas, J., Eds.; Springer Netherlands: Dordrecht, 1999; pp. 331–367 ISBN 978-94-011-4669-2.
- Seif, A.; Domingo, L.R.; Ahmadi, T.S. Calculation of the Rate Constants for Hydrogen Abstraction Reactions by Hydroperoxyl Radical from Methanol, and the Investigation of Stability of CH3OH.HO2 Complex. Comput. Theor. Chem. 2020, 1190, 113010. [CrossRef]
- Chabira, S.F.; Sebaa, M.; G’sell, C. Oxidation and Crosslinking Processes during Thermal Aging of Low-Density Polyethylene Films. J. Appl. Polym. Sci. 2012, 124, 5200–5208. [CrossRef]
- Iedema, P.; Wulkow, M.; Hoefsloot, H. Modeling Molecular Weight and Degree of Branching Distribution of Low-Density Polyethylene. Macromolecules 2000, 33. [CrossRef]
- Odian, G. Radical Chain Polymerization. In Principles of Polymerization; John Wiley & Sons, Ltd, 2004; pp. 198–349 ISBN 9780471478751.
- Barner-Kowollik, C.; Russell, G.T. Chain-Length-Dependent Termination in Radical Polymerization: Subtle Revolution in Tackling a Long-Standing Challenge. Prog. Polym. Sci. 2009, 34, 1211–1259. [CrossRef]
- de Kock, J.B.L. Chain-Length Dependent Bimolecular Termination in Free-Radical Polymerization : Theory, Validation and Experimental Application of Novel Model-Independent Methods; 1999; ISBN 90-386-2701-7.
- Alghamdi, M.M.; Russell, G.T. On the Activation Energy of Termination in Radical Polymerization , as Studied at Low Conversion. 2024.
- Barner-kowollik, C.; Buback, M.; Egorov, M.; Fukuda, T.; Goto, A.; Friedrich, O.; Russell, G.T.; Vana, P.; Yamada, B.; Zetterlund, P.B. Critically Evaluated Termination Rate Coefficients for Free-Radical Polymerization : Experimental Methods. 2005, 30, 605–643. [CrossRef]
- Nikitin, A.N.; Hutchinson, R.A. Determination of the Mode of Free Radical Termination from Pulsed Laser Polymerization Experiments. 2007, 29–42. [CrossRef]
- Cauter, K. Van; Speybroeck, V. Van; Waroquier, M. Ab Initio Study of Poly ( Vinyl Chloride ) Propagation Kinetics : Head-to-Head versus Head-to-Tail Additions. 2007, 541–552. [CrossRef]
- Cuccato, D.; Dossi, M.; Moscatelli, D.; Storti, G. A Density Functional Theory Study of Poly ( Vinyl Chloride ) ( PVC ) Free Radical Polymerization. 2011, 100–109. [CrossRef]
- Ibrahim, A.; Ali, Y.; Saad, H.; Amur, I. Kinetics and Mechanism of Bulk Polymerization of Vinyl Chloride in a Polymerization Reactor. J. Eng. Res. 2015, 12, 41–50. [CrossRef]
- De Roo, T.; Wieme, J.; Heynderickx, G.J.; Marin, G.B. Estimation of Intrinsic Rate Coefficients in Vinyl Chloride Suspension Polymerization. Polymer (Guildf). 2005, 46, 8340–8354. [CrossRef]
- Abreu, C.M.R.; Fonseca, A.C.; Rocha, N.M.P.; Guthrie, J.T.; Serra, A.C.; Coelho, J.F.J. Poly(Vinyl Chloride): Current Status and Future Perspectives via Reversible Deactivation Radical Polymerization Methods. Prog. Polym. Sci. 2018, 87, 34–69. [CrossRef]
- Bárkányi, Á.; Németh, S.; Lakatos, B.G. Modelling and Simulation of Suspension Polymerization of Vinyl Chloride via Population Balance Model. Comput. Chem. Eng. 2013, 59, 211–218. [CrossRef]
- Cuccato, D.; Dossi, M.; Moscatelli, D.; Storti, G. Quantum Chemical Investigation of Secondary Reactions in Poly ( Vinyl Chloride ) Free-Radical Polymerization. 2012, 1–16. [CrossRef]
- Дeдoв, A. Modeling the Kinetics of the Extraction of Vinyl Chloride from Polyvinyl Chloride. Fibre Chem. 2012, 44. [CrossRef]
- Starnes, W.H. MECHANISM AND MICROSTRUCTURE IN THE FREE-RADICAL POLYHERIZATION OF VINYL CHLORIDE: HEAD-TO-HEAD ADDITION REVISITED 1 W. H. Starnes, Jr.,. B. 1993, 11, 1–11.
- Wieme, J.; Marin, G.B. Microkinetic Modeling of Structural Properties of Poly ( Vinyl Chloride ). 2009, 7797–7810. [CrossRef]
- Percec, V.; Popov, A. V; Ramirez-Castillo, E.; Weichold, O. Living Radical Polymerization of Vinyl Chloride Initiated with Iodoform and Catalyzed by Nascent Cu0/Tris(2-aminoethyl)Amine or Polyethyleneimine in Water at 25 °C Proceeds by a New Competing Pathways Mechanism. J. Polym. Sci. Part A 2003, 41, 3283–3299. [CrossRef]
- Pauwels, K.F.D.; Agostini, M.; Bruinsma, M.; Vorenkamp, E.J.; Schouten, A.J.; Coote, M.L. Experimental and Theoretical Evaluation of the Reactions Leading to Formation of Internal Double Bonds in Suspension PVC. 2008, 5527–5539.
- Purmova, J.; Pauwels, K.F.D.; Zoelen, W. Van; Vorenkamp, E.J.; Schouten, A.J.; Coote, M.L. New Insight into the Formation of Structural Defects in Poly ( Vinyl Chloride ). 2005, 6352–6366.
- Heuts, J.P.A.; Heuts, J.P.A.; Sudarko; Gilbert, R.G. First-principles Prediction and Interpretation of Propagation and Transfer Rate Coefficients. Macromol. Symp. 1996, 111, 147–157. [CrossRef]
- Buback, M.; Egorov, M.; Gilbert, R.G.; Kaminsky, V.; Olaj, O.F.; Russell, G.T.; Vana, P.; Zifferer, G. Critically Evaluated Termination Rate Coefficients for Free-Radical Polymerization , 1 The Current Situation. 2002, 2570–2582.
- Phillips, E.O.; Pino, P.; Mazzanti, J.; Anderson, A.W.; Ashby, C.E.; Ford, B.M.; Jeselson, M.; Tsvetkova, V.I.; Chirkov, N.M.; Sendel, E.B.; et al. THE MECHANISM OF CHAIN TERMINATION IN THE FREE-RADICAL POLYMERIZATION OF VINYL CHLORIDE BY 14C-LABELLED INITIATORS *. 1961, 1020–1025.
- Buback, M.; Russell, G.T. Detailed Analysis of Termination Kinetics in Radical Polymerization. 2023. [CrossRef]
- Yamin, N.; Tosaka, M.; Yamago, S. Elucidation of the Termination Mechanism of the Radical Polymerization of Isoprene. 2025. [CrossRef]
- Moad, G.; Solomon, D.H. 5 - Termination. In The Chemistry of Radical Polymerization (Second Edition); Moad, G., Solomon, D.H., Eds.; Elsevier Science Ltd: Amsterdam, 2005; pp. 233–278 ISBN 978-0-08-044288-4.
- Dubikhin, V. V; Knerel’man, E.I.; Nazin, G.M.; Prokudin, V.G.; Stashina, G.A.; Shastin, A. V; Shunina, I.G. Solvent and External Pressure Effects on the Ratio of the Cyanoisopropyl Radical Recombination and Disproportionation Rates. Kinet. Catal. 2013, 54, 404–407. [CrossRef]
- Starnes, W. Structural Defects in Poly(Vinyl Chloride) and the Mechanism of Vinyl Chloride Polymerization: Comments on Recent Studies. Procedia Chem. 2012, 4, 1–10. [CrossRef]
- Seifali, M.; Abadi, A. The Effect of Process and Structural Parameters on the Stability , Thermo - Mechanical and Thermal Degradation of Polymers with Hydrocarbon Skeleton Containing PE , PP , PS , PVC , NR , PBR and SBR. J. Therm. Anal. Calorim. 2020. [CrossRef]
- Natesakhawat, S.; Weidman, J.; Garcia, S.; Means, N.C.; Wang, P. Pyrolysis of High-Density Polyethylene: Degradation Behaviors, Kinetics, and Product Characteristics. J. Energy Inst. 2024, 116, 101738. [CrossRef]
- N.A. Slovokhotova, M.A. Magrupov, V.A.K. Thermal Degradation of Polyethylene. 1979, 1974–1979.
- Holmstrom, A. Thermal Degradation of Polyethylene in a Nitrogen Atmosphere of Low Oxygen Content . 11 . Structural Changes Occurring in Low-Density Polyethylene at an Oxygen Content. 1974, 18, 761–778. [CrossRef]
- Kuzema, P.O.; Bolbukh, Y.M.; Tertykh, V.A.; Laguta, I. V. Vacuum Thermal Decomposition of Polyethylene Containing Antioxidant and Hydrophilic/Hydrophobic Silica. J. Therm. Anal. Calorim. 2015, 121, 1167–1180. [CrossRef]
- Tsuchiyat, Y.; Sumit, K. Thermal Decomposition Products of Polyethylene *. 1968, 6, 415–424.
- Gracida-alvarez, U.R.; Mitchell, M.K.; Sacramento-rivero, J.C.; Shonnard, D.R. Effect of Temperature and Vapor Residence Time on the Micropyrolysis Products of Waste High Density Polyethylene. 2018. [CrossRef]
- Cruz, M.; Schoors, L. Van; Benzarti, K.; Colin, X.; Cruz, M.; Schoors, L. Van; Benzarti, K.; Thermo-oxidative, X.C. Thermo-Oxidative Degradation of Additive Free Polyethylene . Part I . Analysis of Chemical Modifications at Molecular and Macromolecular Scales To Cite This Version : HAL Id : Hal-02291131. 2019.
- Rychlý, J.; Rychlá, L. Polyolefins: From Thermal and Oxidative Degradation to Ignition and Burning. In; Springer, Cham, 2016; pp. 285–314.
- Andersson, T.; Stålbom, B.; Wesslén, B. Degradation of Polyethylene during Extrusion. II. Degradation of Low-Density Polyethylene, Linear Low-Density Polyethylene, and High-Density Polyethylene in Film Extrusion. J. Appl. Polym. Sci. 2004, 91, 1525–1537. [CrossRef]
- Bifulco, A.; Gaan, S.; Price, D.; Horrocks, A.R. Thermal Decomposition of Flame Retardant Polymers. In; Informa, 2024; pp. 11–35.
- Chen, X.; Zhuo, J.; Jiao, C. Thermal Degradation Characteristics of Fl Ame Retardant Polylactide Using TG-IR. Polym. Degrad. Stab. 2012, 97, 2143–2147. [CrossRef]
- Vesely, D.; Castro-Diaz, L. Diffusion Controlled Oxidative Degradation of Un-Stabilised Polyethylene. 2016, 4, 1–7. [CrossRef]
- Harlin, A.L.I.; Heino, E. Comparison of Rheological Properties of Cross-Linked and Thermal-Mech Anically Degraded HDPE. 1995, 479–486.
- Sa, T.; Allen, N.S.; Liauw, C.M.; Johnson, B. Effects of Type of Polymerization Catalyst System on the Degradation of Polyethylenes in the Melt State Part 1 : Unstabilized Polyethylenes ( Including Metallocene Types ). [CrossRef]
- Pinheiro, L.A.; Chinelatto, M.A.; Canevarolo, S. V The Role of Chain Scission and Chain Branching in High Density Polyethylene during Thermo-Mechanical Degradation. Polym. Degrad. Stab. 2004, 86, 445–453. [CrossRef]
- del Teso Sánchez, K.; Allen, N.S.; Liauw, C.M.; Johnson, B. Effects of Type of Polymerization Catalyst System on the Degradation of Polyethylenes in the Melt State. Part 1: Unstabilized Polyethylenes (Including Metallocene Types). J. Vinyl Addit. Technol. 2011, 17, 28–39. [CrossRef]
- Rideal, G.R.; Padget, J.. C. The Thermal-Mechanical Degradation of High Density Polyethylene. J. polym. sci., C Polym. symp 1976, 15, 1–15. [CrossRef]
- Srivastava, D.; Kumar, P.; Mathur, G.N. Aging Characteristics of Ternary Blends of Polyethylenes. I. Mater. Manuf. Process. 2001, 16, 419–425. [CrossRef]
- Dickens, B. Thermally Degrading Polyethylene Studied by Means of Factor- Jump Thermogravimetry. 1982, 20, 1065–1087.
- Popov, K. V; Knyazev, V.D. Molecular Dynamics Simulation of C − C Bond Scission in Polyethylene and Linear Alkanes : E Ff Ects of the Condensed Phase. 2014.
- Peterson, J.D.; Vyazovkin, S.; Wight, C.A. Kinetics of the Thermal and Thermo-Oxidative Degradation of Polystyrene, Polyethylene and Poly(Propylene). Macromol. Chem. Phys. 2001, 202, 775–784. [CrossRef]
- Smagala, T.G.; McCoy, B.J. Mechanisms and Approximations in Macromolecular Reactions: Reversible Initiation, Chain Scission, and Hydrogen Abstraction. Ind. Eng. Chem. Res. 2003, 42, 2461–2469. [CrossRef]
- Kiran, E.; Gillham, J.K. Pyrolysis-Molecular Weight Chromatography: A New on-Line System for Analysis of Polymers. II. Thermal Decomposition of Polyolefins: Polyethylene, Polypropylene, Polyisobutylene. J. Appl. Polym. Sci. 1976, 20, 2045–2068. [CrossRef]
- Seeger, M.; Cantow, H.-J. Theory of Thermal Decomposition and Volatilization of Normal Alkanes and Linear Polyethylene. Polym. Bull. 1979, 1, 347–354. [CrossRef]
- David A. Anderson, E.S.; Freeman The Kinetics o f the Thermal Degradation of Polystyrene and Polyethylene. 1961, 253–260.
- Jellinek, H.H.G. Thermal Degradation of Polystyrene and Polyethylene. Part III. J. Polym. Sci 1949, 4, 13–36. [CrossRef]
- Sawaguchi, T.; Ikemura, T.; Seno, M. Thermal Degradation of Polymers in the Melt, 2. Kinetic Approach to the Formation of Volatile Oligomers by Thermal Degradation of Polyisobutylene. Macromol. Chem. Phys. 1996, 197, 215–222. [CrossRef]
- Wedlake, M.D.; Kohl, P.A. Thermal Decomposition Kinetics of Functionalized Polynorbornene. J. Mater. Res. 2002, 17, 632–640. [CrossRef]
- Kumar, G.S.; Kumar, V.R.; Madras, G. Continuous Distribution Kinetics for the Thermal Degradation of LDPE in Solution. J. Appl. Polym. Sci. 2002, 84, 681–690. [CrossRef]
- Pielichowski, K.; Njuguna, J.; Majka, T. Mechanisms of Thermal Degradation of Polymers. In; 2023; pp. 9–11 ISBN 9780128230237.
- Madorsky, S.L. Rates of Thermal Degradation of Polystyrene and Polyethylene in a Vacuum. J. Polym. Sci. 1952, 9, 133–156. [CrossRef]
- Kruse, T.M.; Woo, O.S.; Wong, H.; Khan, S.S.; Broadbelt, L.J. Mechanistic Modeling of Polymer Degradation : A Comprehensive Study of Polystyrene. 2002, 7830–7844.
- Sohma, J. Radical Migration as an Elementary Process in Degradation. 1983, 55, 1595–1601. [CrossRef]
- Timpe, H.-J.; Kronfeld, K.-P. Kinetic Treatment of Termination Processes at Radical Polymerizations. Acta Polym. 1991, 42, 415–419. [CrossRef]
- Thomas, J.K. Fundamental Aspects of the Radiolysis of Solid Polymers: Crosslinking and Degradation. ChemInform 2009, 40. [CrossRef]
- Costa, L.; Bracco, P. Mechanisms of Cross-Linking, Oxidative Degradation, and Stabilization of UHMWPE. In; Elsevier BV, 2016; pp. 467–487.
- Davis, T.E.; Tobias, R.L.; Peterli, E.B. Thermal Degradation of Polypropylene. J. Polym. Sci. 1962, 56, 485–499. [CrossRef]
- Dickens, B. Thermal Degradation Study of Isotactic Polypropylene Using Factor-Jump Thermogravimetry. J. Polym. Sci. Part A 1982, 20, 1169–1183. [CrossRef]
- Ishikawa, T.; Ohkawa, T.; Suzuki, M.; Tsuchiya, T.; Takeda, K. Semiquantitative Analysis of the Thermal Degradation of Polypropylene. J. Appl. Polym. Sci. 2003, 88, 1465–1472. [CrossRef]
- Tsuchiya, Y.; Sumi, K. Thermal Decomposition Products of Polypropylene. J. Polym. Sci. Part A 1969, 7, 1599–1607. [CrossRef]
- Kumar, R.; Madras, G. Thermal Degradation Kinetics of Isotactic and Atactic Polypropylene. J. Appl. Polym. Sci. 2003, 90, 2206–2213. [CrossRef]
- Qian, S.; Igarashi, T.; Nitta, K. Thermal Degradation Behavior of Polypropylene in the Melt State : Molecular Weight Distribution Changes and Chain Scission Mechanism. 2011, 1661–1670. [CrossRef]
- Sidhu, N.; Mastalski, I.; Zolghadr, A.; Patel, B.; Uppili, S.; Go, T.; Maduskar, S.; Wang, Z.; Neurock, M.; Dauenhauer, P.J. On the Intrinsic Reaction Kinetics of Polypropylene Pyrolysis. Matter 2023, 6, 3413–3433. [CrossRef]
- Bresler, S.E.; Os’minskaia, A.T.; Popov, A.G. The Thermal Degradation of Stereoregular Polypropylene. Polym. Sci. U.s.s.r. 1961, 2, 224–227. [CrossRef]
- Fu, Z.; Sun, Q.; Hua, F.; Yang, S.; Ji, Y.; Cheng, Y. A Molecular-Level Kinetic Model for the Primary and Secondary Reactions of Polypropylene Pyrolysis. J. Anal. Appl. Pyrolysis 2023, 175, 106182. [CrossRef]
- Chen, Y.; Wang, B. Effect of Diatomite on the Thermal Degradation Behavior of Polypropylene and Formation of Graphene Products. 2022.
- Rätzsch, M.; Arnold, M.; Borsig, E.; Bucka, H.; Reichelt, N. Radical Reactions on Polypropylene in the Solid State. Prog. Polym. Sci. 2002, 27, 1195–1282. [CrossRef]
- Sawaguchi, T.; Seno, M. On the Stereoisomerization in Thermal Degradation of Isotactic Poly(Propylene). Macromol. Chem. Phys. 1996, 197, 3995–4015. [CrossRef]
- Straznicky, J.I.; Iedema, P.D.; Remerie, K.; Mcauley, K.B. A Deterministic Model to Predict Tacticity Changes During Controlled Degradation of Polypropylene. Chem. Eng. Sci. 2024, 293, 120064. [CrossRef]
- He, P.; Xiao, Y.; Zhang, P.-M.; Zhu, N.; Zhu, X.; Yan, D. In Situ Fourier Transform Infrared Spectroscopic Study of the Thermal Degradation of Isotactic Poly(Propylene). Appl. Spectrosc. 2005, 59, 33–38. [CrossRef]
- Alam, S. Revising the Mechanism of Polyolefin, Degradation and Stabilisation: Insights from Chemiluminescence, Volatiles and Extractables. Manchester Metrop. Univ. 2019.
- Shibryaeva, L. Thermal Oxidation of Polypropylene and Modified Polypropylene - Structure Effects. 2012. [CrossRef]
- Yoshiga, A.; Otaguro, H.; Parra, D.F.; Lima, L.F.C.P.; Lugão, A.B. Controlled Degradation and Crosslinking of Polypropylene Induced by Gamma Radiation and Acetylene. Polym. Bull. 2009, 63, 397–409. [CrossRef]
- Troitskii, B.B.; Troitskaya, L.S.; Myakov, V.N.; Lepaev, A.F. Mechanism of the Thermal Degradation of Poly(Vinyl Chloride). J. Polym. Sci. Polym. Symp. 2007, 42, 1347–1361. [CrossRef]
- Lv, Y.; Liu, J.; Luo, Z.; Wang, H.; Wei, Z. Construction of Chain Segment Structure Models, and Effects on the Initial Stage of the Thermal Degradation of Poly(Vinyl Chloride). RSC Adv. 2017, 7, 37268–37275. [CrossRef]
- Abbås, K.B.; Sörvik, E.M. On the Thermal Degradation of Poly(Vinyl Chloride). IV. Initiation Sites of Dehydrochlorination. J. Appl. Polym. Sci. 1976, 20, 2395–2406. [CrossRef]
- Lukáš, R.; Přádová, O. Thermal Dehydrochlorination of Poly(Vinyl Chloride), 2. Transiently and Permanently Acting Structural Defects. Die Makromol. Chemie 1986, 187, 2111–2122. [CrossRef]
- Wang, Y.; Wang, X.; Liu, L.; Peng, X. Theoretical Study on the Thermal Dehydrochlorination of Model Compounds for Poly(Vinyl Chloride). J. Mol. Struct. THEOCHEM 2009, 896, 34–37. [CrossRef]
- Bacaloglu, R.; Fisch, M.H. Reaction Mechanism of Poly(Vinyl Chloride) Degradation. Molecular Orbital Calculations. J. Vinyl Addit. Technol. 1995, 1, 241–249. [CrossRef]
- Rogestedt, M.; Hjertberg, T. Structure and Degradation of Commercial Poly(Vinyl Chloride) Obtained at Different Temperatures. Macromolecules 1993, 26, 60–64. [CrossRef]
- Troitskii, B.B.; Troitskaya, L.S. Some Aspects of TheThermal Degradation of Poly(Vinyl Chloride). Int. J. Polym. Mater. 1998, 41, 285–324. [CrossRef]
- Hujuri, U.; Ghoshal, A.K.; Gumma, S. Temperature-Dependent Pyrolytic Product Evolution Profile for Polypropylene. 2010. [CrossRef]
- Fisch, M.H.; Bacaloglu, R. Kinetics and Mechanism of the Thermal Degradation of Poly(Vinyl Chloride). J. Vinyl Addit. Technol. 1995, 1, 233–240. [CrossRef]
- Krongauz, V. V Kinetics of Plasticized Poly(Vinyl Chloride) Thermal Degradation, Induction, Autocatalysis, Glass Transition, Diffusion. Chemistryselect 2024, 9. [CrossRef]
- Yanborisov, V.M.; Borisevich, S.S. Quantum-Chemical Modeling of the Mechanism of Autocatalytic Dehydrochlorination of PVC. Theor. Exp. Chem. 2005, 41, 352–358. [CrossRef]
- Chatterjee, N.; Basu, S.; Palit, S.K.; Maiti, M.M. A Reexamination of the Degradation of Polyvinylchloride by Thermal Analysis. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1225–1236. [CrossRef]
- Yanborisov, V.; Borisevich, S. Mechanism of Initiation and Growth of Polyene Sequences during Thermal Degradation of Poly(Vinyl Chloride). Polym. Sci. - Ser. A 2005, 47, 844–854.
- Bettens, T.; Eeckhoudt, J.; Hoffmann, M.; Alonso, M.; Geerlings, P.; Dreuw, A.; Proft, F. Designing Force Probes Based on Reversible 6π-Electrocyclizations in Polyenes Using Quantum Chemical Calculations. J. Org. Chem. 2021, XXXX. [CrossRef]
- Starnes, W.H.; Ge, X. Mechanism of Autocatalysis in the Thermal Dehydrochlorination of Poly(Vinyl Chloride). Macromolecules 2004, 37, 352–359. [CrossRef]
- TÜDŐS, F.; KELEN, T. INVESTIGATION OF THE KINETICS AND MECHANISM OF PVC DEGRADATION. In Macromolecular Chemistry–8; SAARELA, K., Ed.; Butterworth-Heinemann, 1973; pp. 393–412 ISBN 978-0-408-70516-5.
- Ye, L.; Li, T.; Hong, L. Understanding Enhanced Char Formation in the Thermal Decomposition of PVC Resin: Role of Intermolecular Chlorine Loss. Mater. Today Commun. 2021, 26, 102186. [CrossRef]
- Lattimer, R.P.; Kroenke, W.J. 15 - STUDIES OF VOLATILE PYROLYZATE, SMOKE, AND CHAR FORMATION IN POLY(VINYL CHLORIDE). In Analytical Pyrolysis; Voorhees, K.J., Ed.; Butterworth-Heinemann, 1984; pp. 453–473 ISBN 978-0-408-01417-5.
- Brauman, S.K. Char Formation in Polyvinyl Chloride. III. Mechanistic Aspects of Isothermal Degradation of PVC Containing Some Dehydrochlorination/Charring Agents. J. Appl. Polym. Sci. 1981, 26, 353–371. [CrossRef]
- Nagy, T.; Iván, B.; Turcsányi, B.; Kelen, T.; Tüdös, F. Crosslinking, Scission and Benzene Formation during PVC Degradation under Various Conditions. Polym. Bull. 1980, 3, 613–620. [CrossRef]
- Kelen, T. Secondary Processes of Thermal Degradation of PVC. J. Macromol. Sci. Part A 1978, 12, 349–360. [CrossRef]
- Gupta, V.P.; Pierre, L.E. St. Thermal Degradation of Poly(Vinyl Chloride). II. Degradation Mechanism Based on Decomposition Energetics. J. Polym. Sci. Part A 1973, 11, 1841–1850. [CrossRef]
- Coelho, A.; Fonseca, I.M.; Matos, I.; Marques, M.M.; Botelho do Rego, A.M.; Lemos, M.A.N.D.A.; Lemos, F. Catalytic Degradation of Low and High Density Polyethylenes Using Ethylene Polymerization Catalysts: Kinetic Studies Using Simultaneous TG/DSC Analysis. Appl. Catal. A Gen. 2010, 374, 170–179. [CrossRef]
- Artetxe, M.; Lopez, G.; Amutio, M.; Elordi, G.; Bilbao, J.; Olazar, M. Cracking of High Density Polyethylene Pyrolysis Waxes on HZSM-5 Catalysts of Different Acidity. Ind. Eng. Chem. Res. 2013, 52, 10637–10645. [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Thermal and Catalytic Pyrolysis of Polyethylene over HZSM5 and HUSY Zeolites in a Batch Reactor under Dynamic Conditions. Appl. Catal. B Environ. 2009, 86, 78–86. [CrossRef]
- van de Minkelis, J.H.; Hergesell, A.H.; van der Waal, J.C.; Altink, R.M.; Vollmer, I.; Weckhuysen, B.M. Catalytic Pyrolysis of Polyethylene with Microporous and Mesoporous Materials: Assessing Performance and Mechanistic Understanding. ChemSusChem 2025, 18, e202401141. [CrossRef] [PubMed]
- Lee, H.W.; Park, Y.-K. Catalytic Pyrolysis of Polyethylene and Polypropylene over Desilicated Beta and Al-MSU-F. Catalysts 2018, 8, 501. [CrossRef]
- Mousavi, S.A.H.S.; Dehaghani, A.H.S. Catalytic Pyrolysis of Plastic Waste to Gasoline, Jet Fuel and Diesel with Nano MOF Derived-Loaded Y Zeolite: Evaluation of Temperature, Zeolite Crystallization and Catalyst Loading Effects. Energy Convers. Manag. 2024, 299, 117825. [CrossRef]
- Psarreas, A.; Tzoganakis, C.; McManus, N.T.; Penlidis, A. Nitroxide-Mediated Controlled Degradation of Polypropylene. Polym. Eng. Sci. 2007, 47, 2118–2123. [CrossRef]
- Kharitontsev, V.; Tissen, E.; Matveenko, E.; Mikhailov, Y.; Tretyakov, N.; Zagoruiko, A.; Elyshev, A. Estimating the Efficiency of Catalysts for Catalytic Pyrolysis of Polyethylene. Katal. v promyshlennosti 2023, 23, 58–65. [CrossRef]
- Manos, G.; Garforth, A.; Dwyer, J. Catalytic Degradation of High-Density Polyethylene on an Ultrastable-Y Zeolite. Nature of Initial Polymer Reactions, Pattern of Formation of Gas and Liquid Products, and Temperature Effects. Ind. Eng. Chem. Res. 2000, 39, 1203–1208. [CrossRef]
- Coelho, A.; Costa, L.; Marques, M.D.M.; Fonseca, I.; Lemos, M.A.; Lemos, F. Using Simultaneous DSC/TG to Analyze the Kinetics of Polyethylene Degradation-Catalytic Cracking Using HY and HZSM-5 Zeolites. React. Kinet. Mech. Catal. 2010, 99, 5–15. [CrossRef]
- Ivanova, S.R.; Minsker, K.S.; Zaikov, G.E. Catalytic Degradation of Polyolefins. Oxid. Commun. 2002, 25, 325–349. [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Evolution of Products during the Degradation of Polyethylene in a Batch Reactor. J. Anal. Appl. Pyrolysis 2009, 86, 14–21. [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Evolution with the Temperature of the Compounds Obtained in the Catalytic Pyrolysis of Polyethylene over HUSY. Ind. Eng. Chem. Res. 2008, 47, 6896–6903. [CrossRef]
- Mesquita, K.; Pinto, J.; Pacheco, H. Assessment of Performance and Deactivation Resistance of Catalysts in the Pyrolysis of Polyethylene and Post-Consumer Polyolefin Waste. Macromol. React. Eng. 2024, 18. [CrossRef]
- Yang, G.; Peng, P.; Guo, H.; Song, H.; Li, Z. The Catalytic Pyrolysis of Waste Polyolefins by Zeolite-Based Catalysts: A Critical Review on the Structure-Acidity Synergies of Catalysts. Polym. Degrad. Stab. 2024, 222, 110712. [CrossRef]
- Pinto, F.; Costa, P.; Gulyurtlu, I.; Cabrita, I. Pyrolysis of Plastic Wastes. 2. Effect of Catalyst on Product Yield. J. Anal. Appl. Pyrolysis 1999, 51, 57–71. [CrossRef]
- Saha, B.; Vedachalam, S.; Dalai, A.K.; Saxena, S.; Dally, B.; Roberts, W.L. Review on Production of Liquid Fuel from Plastic Wastes through Thermal and Catalytic Degradation. J. Energy Inst. 2024, 114, 101661. [CrossRef]
- Zhang, Z.; Chen, H.; Li, G.; Hu, W.; Niu, B.; Long, D.; Zhang, Y. Highly Selective Upgrading of Polyethylene into Light Aromatics via a Low-Temperature Melting-Catalysis Strategy. ACS Catal. 2024. [CrossRef]
- Hesse, N.D.; Lin, R.; Bonnet, E.; Cooper, J.; White, R.L. In Situ Analysis of Volatiles Obtained from the Catalytic Cracking of Polyethylene. J. Appl. Polym. Sci. 2001, 82, 3118–3125. [CrossRef]
- Liu, Y.; Dai, W.; Zheng, J.; Du, Y.; Wang, Q.; Hedin, N.; Qin, B.; Li, R. Selective and Controllable Cracking of Polyethylene Waste by Beta Zeolites with Different Mesoporosity and Crystallinity. Adv. Sci. 2024. [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Study of the Deactivation Process of HZSM5 Zeolite during Polyethylene Pyrolysis. Appl. Catal. A-general 2007, 333, 57–66. [CrossRef]
- Liu, T.; Li, Y.; Zhou, Y.; Deng, S.; Zhang, H. Efficient Pyrolysis of Low-Density Polyethylene for Regulatable Oil and Gas Products by ZSM-5, HY and MCM-41 Catalysts. Catalysts 2023, 13, 1–13. [CrossRef]
- Kang, J.; Kim, J.Y.; Sung, S.; Lee, Y.; Gu, S.; Choi, J.W.; Yoo, C.J.; Suh, D.J.; Choi, J.; Ha, J.M. Chemical Upcycling of PVC-Containing Plastic Wastes by Thermal Degradation and Catalysis in a Chlorine-Rich Environment. Environ. Pollut. 2024, 342, 123074. [CrossRef]
- Tan, J.Z.; Ortega, M.; Miller, S.A.; Hullfish, C.W.; Kim, H.; Kim, S.; Hu, W.; Hu, J.; Lercher, J.A.; Koel, B.E.; et al. Catalytic Consequences of Hierarchical Pore Architectures within MFI and FAU Zeolites for Polyethylene Conversion. ACS Catal. 2024. [CrossRef]
- Upare, D.P.; Lee, C.W.; Lee, D.K.; Kang, Y.S. Effect of Acidity of Solid Acid Catalysts during Non-Oxidative Thermal Decomposition of LDPE. Carbon Lett. 2024. [CrossRef]
- Rahman, M.; Mondal, B.K.; Ahmed, N.; Hossain, M.D. Catalytic Pyrolysis of Waste High-Density (HDPE) and Low-Density Polyethylene (LDPE) to Produce Liquid Hydrocarbon Using Silica-Alumina Catalyst. J. Bangladesh Acad. Sci. 2023. [CrossRef]
- Pyra, K.; Tarach, K.A.; Janiszewska, E.; Majda, D.; Góra-Marek, K. Evaluation of the Textural Parameters of Zeolite Beta in LDPE Catalytic Degradation: Thermogravimetric Analysis Coupled with FTIR Operando Studies. Molecules 2020, 25, 926. [CrossRef]
- TOMASZEWSKA, K.; KalŁuzna-CZAPLIŃSKA, J.; JOZWIAK, W. Thermo-Catalytic Degradation of Low Density Polyethylene over Clinoptilolite - The Effect of Carbon Residue Deposition. Polimery/Polymers 2010, 55, 222–226. [CrossRef]
- Okonsky, S.T.; Krishna, J.V.J.; Toraman, H.E. Catalytic Co-Pyrolysis of LDPE and PET with HZSM-5{,} H-Beta{,} and HY: Experiments and Kinetic Modelling. React. Chem. Eng. 2022, 7, 2175–2191. [CrossRef]
- Inayat, A.; Inayat, A.; Schwieger, W.; Klemencova, K.; Lestinsky, P. Chemical Recycling of Waste Polypropylene via Thermo-catalytic Pyrolysis over HZSM-5 Catalysts. Chem. Eng. Technol. 2023, 46, 1289–1297. [CrossRef]
- Bozkurt, O.D.; Toraman, H.E. Conversion of Polypropylene into Light Hydrocarbons and Aromatics by Metal Exchanged Zeolite Catalysts. Langmuir 2024. [CrossRef]
- Lim, S.Y.; Fu, S.; Lee, J. Enhancement of Light Hydrocarbon Production from Polypropylene Waste by HZSM-11-Catalyzed Pyrolysis. 2024. [CrossRef]
- Bautista, A.S.; Rivera, K.N.O.; Suratos, T.A.K.M.; Dimaano, M. Conversion of Polypropylene (PP) Plastic Waste to Liquid Oil through Catalytic Pyrolysis Using Philippine Natural Zeolite. 2024. [CrossRef]
- Irawan, A.; Kurniawan, T.; Nurkholifah, N.; Melina, M.; Nandiyanto, A.B.D.; Firdaus, M.A.; Alwan, H.; Bindar, Y. Pyrolysis of Polyolefins into Chemicals Using Low-Cost Natural Zeolites. Waste and Biomass Valorization 2023, 14, 1705–1719. [CrossRef]
- Heng, J.Z.X.; Tan, T.T.Y.; Li, X.; Loh, W.W.; Chen, Y.; Xing, Z.; Lim, Z.; Ong, J.L.Y.; Lin, K.S.; Nishiyama, Y.; et al. Pyrolytic Depolymerization of Polyolefins Catalysed by Zirconium-Based UiO-66 Metal–Organic Frameworks. Angew. Chemie Int. Ed. 2024, 63, e202408718. [CrossRef]
- Nisar, J.; Farid, R.; Ali, G.; Muhammad, F.; Shah, A.; Farooqi, Z.H.; Shah, F. Kinetics and Fuel Properties of the Oil Obtained from the Pyrolysis of Polypropylene over Cobalt Oxide. Clean. Chem. Eng. 2022, 4, 100083. [CrossRef]
- Pal, R.K.; Tiwari, A.C. Comparative Study of Catalytic Pyrolysis of Waste Polypropylene(PP) Using Silica Alumina, Kaolin Clay and Calcium Bentonite Catalyst. J. Emerg. Technol. Innov. Res. 2020.
- Aisien, Felix Aibuedefe; Aisien, E.T. Liquid Fluids from Thermo-Catalytic Degradation of Waste Low-Density Polyethylene Using Spent Fcc Catalyst. Detritus 2022, 75–83. [CrossRef]
- Akhtar, M.N.; Riaz, S.; Ahmad, N.; Jaseer, E.A. Pioneering Aromatic Generation from Plastic Waste via Catalytic Thermolysis: A Minireview. Energy and Fuels 2024, 38, 11363–11390. [CrossRef]
- Miandad, R.; Rehan, M.; Barakat, M.A.; Aburiazaiza, A.S.; Khan, H.; Ismail, I.M.I.; Dhavamani, J.; Gardy, J.; Hassanpour, A.; Nizami, A.S. Catalytic Pyrolysis of Plastic Waste: Moving toward Pyrolysis Based Biorefineries. Front. Energy Res. 2019, 7, 1–17. [CrossRef]
- Wootthikanokkhan, J.; Jaturapiree, A.; Meeyoo, V. Effect of Metal Compounds and Experimental Conditions on Distribution of Products from PVC Pyrolysis. J. Polym. Environ. 2003, 11, 1–6. [CrossRef]
- Iida, T.; Nakanishi, M.; Goto, K. Investigations on Poly(Vinyl Chloride) - 1. Evolution of Aromatics on Pyrolysis of Poly(Vinyl Chloride) and Its Mechanism. J Polym Sci Part A-1 Polym Chem 1974, 12, 737–749. [CrossRef]
- Lattimer, R.P.; Kroenke, W.J. The Functional Role of Molybdenum Trioxide as a Smoke Retarder Additive in Rigid Poly(Vinyl Chloride). J. Appl. Polym. Sci. 1981, 26, 1191–1210. [CrossRef]
- Loong, G.K.M.; Okada, K.; Morishige, N.; Konakahara, N.; Yokota, M.; Tanoue, K. Investigation of the Uptake and Catalytic Effect of Calcium and Potassium-Based Additives under Low-Temperature Pyrolysis of Polyvinyl Chloride. Environ. Prog. \& Sustain. Energy 2024, 43, e14352. [CrossRef]
- Fedorov, A.A.; Chekryshkin, Y.S.; Rudometova, O. V.; Vnutskikh, Z.A. Application of Inorganic Compounds at the Thermal Processing of Polyvinylchloride. Russ. J. Appl. Chem. 2008, 81, 1673–1685. [CrossRef]
- Glas, D.; Hulsbosch, J.; Dubois, P.; Binnemans, K.; Vos, D. De End-of-Life Treatment of Poly(Vinyl Chloride) and Chlorinated Polyethylene by Dehydrochlorination in Ionic Liquids. ChemSusChem 2014, 7, 610–617. [CrossRef]
- Triacca, V.J.; Gloor, P.E.; Zhu, S.; Hrymak, A.N.; Hamielec, A.E. Free Radical Degradation of Polypropylene : Random Chain Scission. Polym. Eng. Sci. 1993, 33, 445–454. [CrossRef]
- Kurt Dr. Rauer, P.D. Degradation of Polyethylene by Means of Agents Generating Free Radicals. Eur. Pat. Off. 1987.
- Uebe, J.; Zukauskaite, A.; Kryzevicius, Z.; Vanagiene, G. Use of 2-Ethylhexyl Nitrate for the Slow Pyrolysis of Plastic Waste. Processes 2022, 10. [CrossRef]
- Costa, C.S.; Fernandes, A.; Muñoz, M.; Ribeiro, M.R.; Silva, J.M. Analyzing HDPE Thermal and Catalytic Degradation in Hydrogen Atmosphere: A Model-Free Approach to the Activation Energy. Catalysts 2024. [CrossRef]
- Coelho, A.; Costa, L.; Marques, M.M.; Fonseca, I.; Lemos, M.A.N.D.A.; Lemos, F. The Effect of ZSM-5 Zeolite Acidity on the Catalytic Degradation of High-Density Polyethylene Using Simultaneous DSC/TG Analysis. Appl. Catal. A-general 2012, 413, 183–191. [CrossRef]
- Koç, A. Thermal Pyrolysis of Waste Disposable Plastic Syringes and Pyrolysis Thermodynamics. Adv. Chem. Eng. Sci. 2022, 12, 96–113. [CrossRef]
- Karaduman, A.; Koçak, M.Ç.; Bilgesü, A.Y. Flash Vacuum Pyrolysis of Low Density Polyethylene in a Free-Fall Reactor. Polym. Plast. Technol. Eng. 2003, 42, 181–191. [CrossRef]
- Kayacan, İ.; Doğan, Ö.M. Pyrolysis of Low and High Density Polyethylene. Part I: Non-Isothermal Pyrolysis Kinetics. Energy Sources Part A-recovery Util. Environ. Eff. 2008, 30, 385–391. [CrossRef]
- Khaghanikavkani, E.; Farid, M. Thermal Pyrolysis of Polyethylene: Kinetic Study. Energy Sci. Technol. 2011, 2, 1–10. [CrossRef]
- Audisio, G.; Bertini, F.; Beltrame, P.L.; Carniti, P. Catalytic Degradation of Polyolefins. Macromol. Symp. 1992, 57, 191–209. [CrossRef]
- Anene, F.; Fredriksen, B.; Sætre, K.A.; Tokheim, L.-A. Experimental Study of Thermal and Catalytic Pyrolysis of Plastic Waste Components. 2018. [CrossRef]
- Xu, D.; Huang, G.; Guo, L.; Chen, Y.; Ding, C.; Ding, C.; Liu, C. Enhancement of Catalytic Combustion and Thermolysis for Treating Polyethylene Plastic Waste. 2021, 1–17. [CrossRef]
- Rajan, K.P.; Mustafa, I.; Gopanna, A.; Thomas, S.P. Catalytic Pyrolysis of Waste Low-Density Polyethylene (LDPE) Carry Bags to Fuels: Experimental and Exergy Analyses. 2023. [CrossRef]
- Scorah, M.J.; Zhu, S.; Psarreas, A.; McManus, N.T.; Dhib, R.; Tzoganakis, C.; Penlidis, A. Peroxide-controlled Degradation of Polypropylene Using a Tetra-functional Initiator. Polym. Eng. Sci. 2009, 49, 1760–1766. [CrossRef]
- Masai, Y.; Kiyotsukuri, T. Effects of Additves on the Thermal Decomposition of Polypropylene. Sen-i Gakkaishi 1991, 47, 37–43. [CrossRef]
- Mizutani, Y.; Yamamoto, K.; Matsuoka, S.; Hisano, S. Studies on Reactions of Polypropylene. VIII. The Thermal Degradation of Polypropylene Accelerated by Polyglycidyl Methacrylate. Bull. Chem. Soc. Jpn. 1967, 40, 1526–1530. [CrossRef]
- Zorriqueta, I.J. Pyrolysis of Polypropylene by Ziegler-Natta Catalysts. 2006.
- Hu, Y.; Li, M.; Zhou, N.; Yuan, H.; Guo, Q.; Jiao, L.; Ma, Z. Catalytic Stepwise Pyrolysis for Dechlorination and Chemical Recycling of PVC-Containing Mixed Plastic Wastes: Influence of Temperature, Heating Rate, and Catalyst. Sci. Total Environ. 2024, 908, 168344. [CrossRef]
- Zhao, L.; Cao, Z.; Fang, Z.; Guo, Z. Influence of Fullerene on the Kinetics of Thermal and Thermo-Oxidative Degradation of High-Density Polyethylene by Capturing Free Radicals. J. Therm. Anal. Calorim. 2013, 114, 1287–1294. [CrossRef]
- Abbas, A.S.; Saber, M.G. Thermal and Catalytic Degradation Kinetics of High-Density Polyethylene Over NaX Nano-Zeolite. J. Chem. Pet. Eng. 2016, 17, 33–43.
- Zaggout, F.R.; Mughari, A.R. Al; Garforth, A. Catalytic Degradation of High Density Polyethylene Using Zeolites. J. Environ. Sci. Heal. Part A-toxic/hazardous Subst. Environ. Eng. 2001, 36, 163–175. [CrossRef]
- Anggoro, D.D. Optimization of Catalytic Degradation of Plastic to Aromatics Over HY Zeolite. 2005.
- Lee, K.-H.; Jeon, S.-G.; Kim, K.-H.; Noh, N.-S.; Shin, D.-H.; Seo, Y.-H.; Yee, J.-J.; Kim, G.-T. Thermal and Catalytic Degradation of Waste High-Density Polyethylene (HDPE) Using Spent FCC Catalyst. Korean J. Chem. Eng. 2003, 20, 693–697. [CrossRef]
- Lee, K.-H.; Shin, D.-H. Catalytic Degradation of Waste Polyolefinic Polymers Using Spent FCC Catalyst with Various Experimental Variables. Korean J. Chem. Eng. 2003, 20, 89–92. [CrossRef]
- Kumar, R.; Sadhukhan, A.K.; Gupta, P.; Singh, R.K.; Ruj, B. Recovery of Enhanced Gasoline-Range Fuel from Catalytic Pyrolysis of Waste Polypropylene: Effect of Heating Rate, Temperature, and Catalyst on Reaction Kinetics, Products Yield, and Compositions. Process Saf. Environ. Prot. 2024, 188, 793–806. [CrossRef]
- Prabha, B.; Ramesh, D.; Sriramajayam, S.; Uma, D. Optimization of Pyrolysis Process Parameters for Fuel Oil Production from the Thermal Recycling of Waste Polypropylene Grocery Bags Using the Box–Behnken Design. Recycling 2024, 9. [CrossRef]
- Aremanda, R.B.; Singh, R.K. Conversion of Waste Polypropylene Disposable Cups into Liquid Fuels by Thermal and Catalytic Pyrolysis Using Activated Carbon. Sustinere J. Environ. Sustain. 2022, 6, 79–91. [CrossRef]
- Fu, H.; Li, X.; Shao, S.; Cai, Y. Jet Fuel Range Hydrocarbon Generation from Catalytic Pyrolysis of Lignin and Polypropylene with Iron-Modified Activated Carbon. J. Anal. Appl. Pyrolysis 2024. [CrossRef]
- Faisal, F.; Rasul, M.; Chowdhury, A.A.; Jahirul, M.I. Optimisation of Process Parameters to Maximise the Oil Yield from Pyrolysis of Mixed Waste Plastics. Sustainability 2024. [CrossRef]
- Jaydev, S.D.; Martín, A.J.; Pérez-Ramírez, J. Direct Conversion of Polypropylene into Liquid Hydrocarbons on Carbon-Supported Platinum Catalysts. ChemSusChem 2021. [CrossRef]
- Al-Zaidi, B.Y.; Almukhtar, R.; Hamawand, I. Optimization of Polypropylene Waste Recycling Products as Alternative Fuels through Non-Catalytic Thermal and Catalytic Hydrocracking Using Fresh and Spent Pt/Al2O3 and NiMo/Al2O3 Catalysts. Energies 2023, 16, 4871. [CrossRef]
- Suzuki, K.; Tadauchi, M. Pyrolytically Decomposing Waste Plastic, e.g. PVC 1994.
- Castro, A.; Carneiro, C.; Vilarinho, C.; Soares, D.; Maçães, C.; Sousa, C.; Castro, F. Study of a Two Steps Process for the Valorization of PVC-Containing Wastes. Waste and Biomass Valorization 2013, 4, 55–63. [CrossRef]
- O’Rourke, G.; Stalpaert, M.; Skorynina, A.A.; Bugaev, A.L.; Janssens, K.; Emelen, L. Van; Lemmens, V.; Colemonts, C.M.C.J.; Sakellariou, D.; Vos, D. De Catalytic Tandem Dehydrochlorination–Hydrogenation of PVC towards Valorisation of Chlorinated Plastic Waste. Chem. Sci. 2023, 14, 4401–4412. [CrossRef]
- Scalfani, V.; Ezendu, S.; Ryoo, D.; Shinde, P.S.; Anderson, J.L.; Szilvási, T.; Rupar, P.A.; Bara, J.E. PVC Modification through Sequential Dehydrochlorination–Hydrogenation Reaction Cycles Facilitated via Fractionation by Green Solvents. ACS Appl. Polym. Mater. 2024. [CrossRef]
- Figge, K.; Findeiß, W. Untersuchungen Zum Mechanismus Der PVC-Stabilisierung Mit Organozinn-Verbindungen. Angew. Makromol. Chemie 1975, 47, 141–179. [CrossRef]
- Ahmad, Z.; ur Rehman, H.; Ali, S.; Sarwar, M.I. Thermal Degradation of Poly (Vinyl Chloride)-Stabilization Effect of Dichlorotin Dioxine. Int. J. Polym. Mater. 2000, 46, 547–559. [CrossRef]
- Mahmood, F.; Qadeer, R. Effects of Alkaline Earth Metal Stearates on the Dehydrochlorination of Poly(Vinyl Chloride). J. Therm. Anal. 1994, 42, 1167–1173. [CrossRef]
- Zhang, B.; Yan, X.Y.; Shibata, K.; Uda, T.; Tada, M.; Hirasawa, M. Thermogravimetric-Mass Spectrometric Analysis of the Reactions between Oxide (ZnO, Fe2O3 or ZnFe2O4) and Polyvinyl Chloride under Inert Atmosphere. Mater. Trans. JIM 2000, 41, 1342–1350. [CrossRef]
Figure 1.
Overview of the pathway from raw materials to pyrolysis for HDPE, LDPE, PP, and PVC. Monomer supply (ethylene, propylene; VCM) and polymerization mode (coordination vs. radical) set chain microstructures (branching, tertiary C, C–Cl) that precondition thermal behavior. At end-of-life, fractions not suited for mechanical recycling are routed to pyrolysis. Dominant degradation modes are random/β-scission (HDPE), random scission with H-abstraction/backbiting (LDPE), β-scission (PP), and zipper dehydrochlorination to HCl and a polyene residue (PVC). Operating variables (temperature, heating rate, residence time, rapid quench) and optional catalysts/initiators (e.g., zeolites, metals, peroxides) tune product windows from gases to oils/waxes; for PVC, staged dechlorination with HCl capture precedes cracking of the residue.
Figure 1.
Overview of the pathway from raw materials to pyrolysis for HDPE, LDPE, PP, and PVC. Monomer supply (ethylene, propylene; VCM) and polymerization mode (coordination vs. radical) set chain microstructures (branching, tertiary C, C–Cl) that precondition thermal behavior. At end-of-life, fractions not suited for mechanical recycling are routed to pyrolysis. Dominant degradation modes are random/β-scission (HDPE), random scission with H-abstraction/backbiting (LDPE), β-scission (PP), and zipper dehydrochlorination to HCl and a polyene residue (PVC). Operating variables (temperature, heating rate, residence time, rapid quench) and optional catalysts/initiators (e.g., zeolites, metals, peroxides) tune product windows from gases to oils/waxes; for PVC, staged dechlorination with HCl capture precedes cracking of the residue.
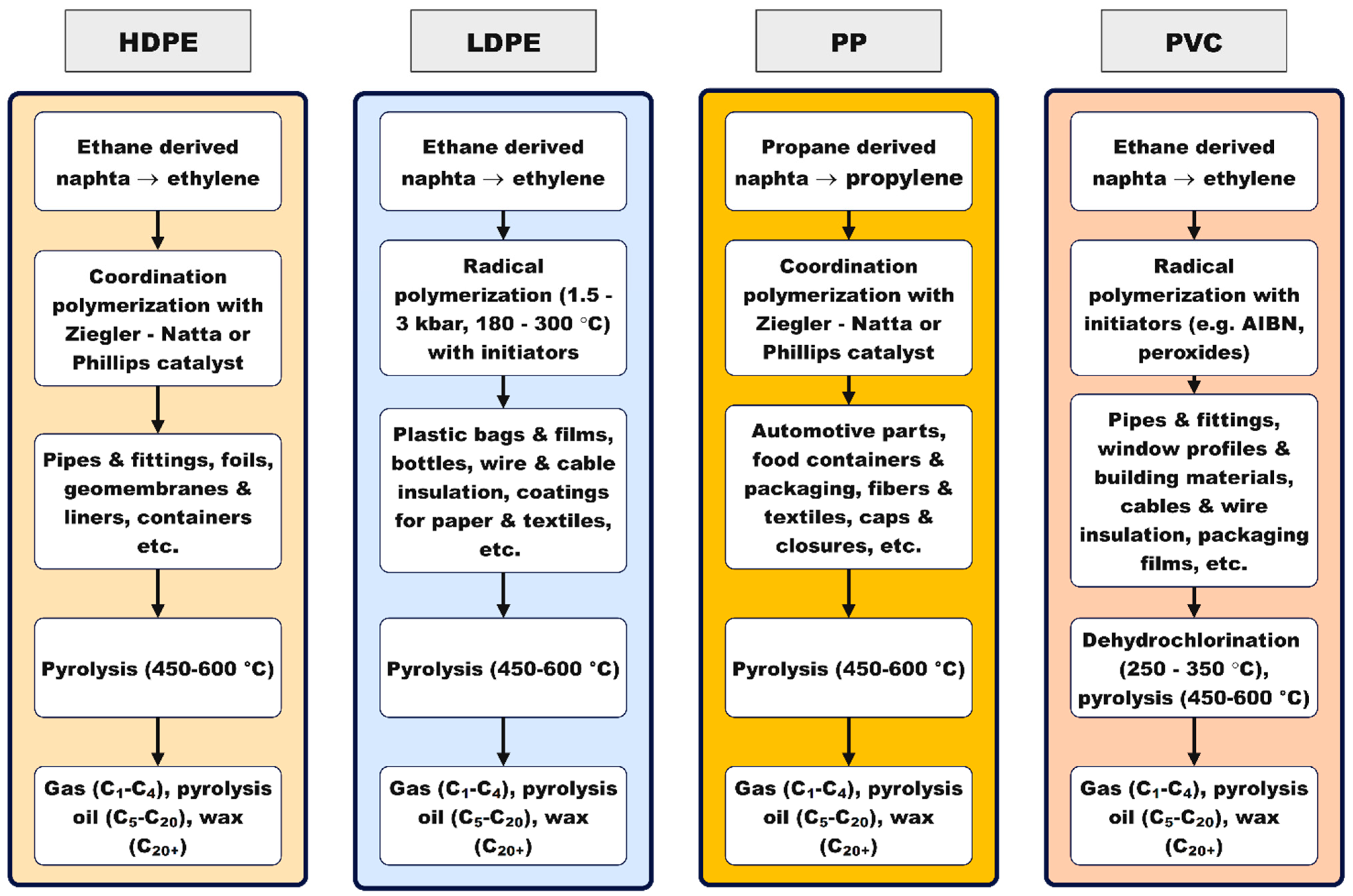
Figure 2.
Proposed reaction mechanism for the coordination polymerization of ethylene to high-density polyethylene (HDPE) using a TiCl4/Al(C2H5)3 Ziegler–Natta catalytic system. (a) Activation of the TiCl4 catalyst by triethylaluminum, forming an active titanium–ethyl species, (b) π-coordination of ethylene to the Ti3+ center, (c) migratory insertion of ethylene into the Ti–C bond, extending the chain by two carbon atoms, (d) coordination of another ethylene molecule to the titanium center, (e) repeated migratory insertions of ethylene during chain propagation, (f) chain termination by β-hydride elimination forming a terminal double bond, (g) chain transfer to monomer via β-hydride transfer, producing a saturated chain and regenerating a Ti–ethyl site, (h) chain transfer to hydrogen (hydrogenation), terminating the chain with a fully saturated alkyl end, and (i) formation of the final linear HDPE chain structure, which is highly crystalline due to minimal branching.
Figure 2.
Proposed reaction mechanism for the coordination polymerization of ethylene to high-density polyethylene (HDPE) using a TiCl4/Al(C2H5)3 Ziegler–Natta catalytic system. (a) Activation of the TiCl4 catalyst by triethylaluminum, forming an active titanium–ethyl species, (b) π-coordination of ethylene to the Ti3+ center, (c) migratory insertion of ethylene into the Ti–C bond, extending the chain by two carbon atoms, (d) coordination of another ethylene molecule to the titanium center, (e) repeated migratory insertions of ethylene during chain propagation, (f) chain termination by β-hydride elimination forming a terminal double bond, (g) chain transfer to monomer via β-hydride transfer, producing a saturated chain and regenerating a Ti–ethyl site, (h) chain transfer to hydrogen (hydrogenation), terminating the chain with a fully saturated alkyl end, and (i) formation of the final linear HDPE chain structure, which is highly crystalline due to minimal branching.
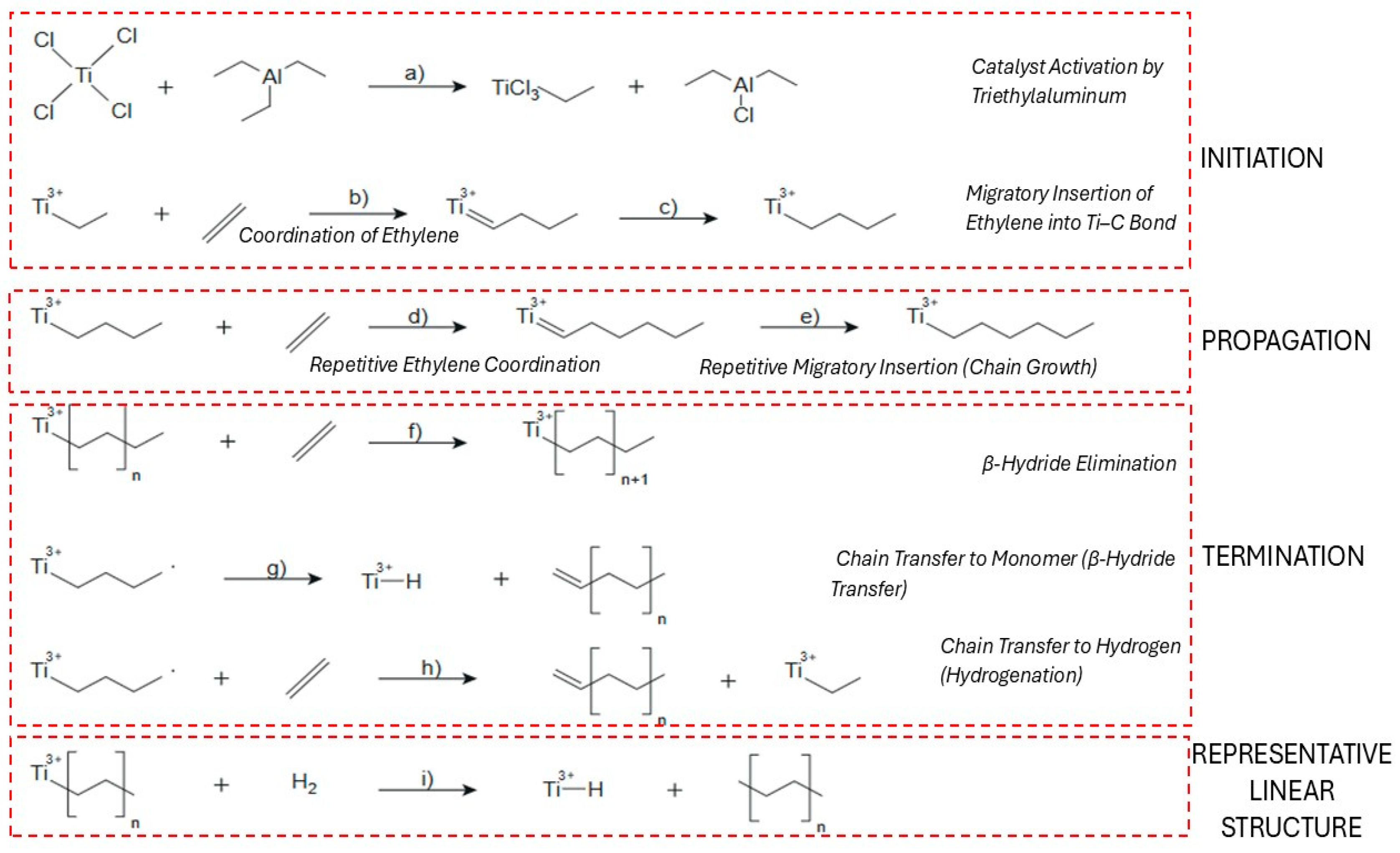
Figure 3.
Reaction mechanism pathway of synthesis of PP using Ziegler - Natta catalyst TiCl4. a) present the catalyst activation or reduction of Ti(IV) to Ti (III) and alkylization of the transition metal center, formation of the Ti3+ -ethyl complex; (b) monomer coordination or π complex formation; (c) migratory insertion or chain initiation; (d) chain propagation (repeated migratory insertions); (e) chain termination – β hydride elimination; (f) chain termination – β – hydride transfer to monomer (g) chain termination – hydrogenation or chain transfer to hydrogen.
Figure 3.
Reaction mechanism pathway of synthesis of PP using Ziegler - Natta catalyst TiCl4. a) present the catalyst activation or reduction of Ti(IV) to Ti (III) and alkylization of the transition metal center, formation of the Ti3+ -ethyl complex; (b) monomer coordination or π complex formation; (c) migratory insertion or chain initiation; (d) chain propagation (repeated migratory insertions); (e) chain termination – β hydride elimination; (f) chain termination – β – hydride transfer to monomer (g) chain termination – hydrogenation or chain transfer to hydrogen.
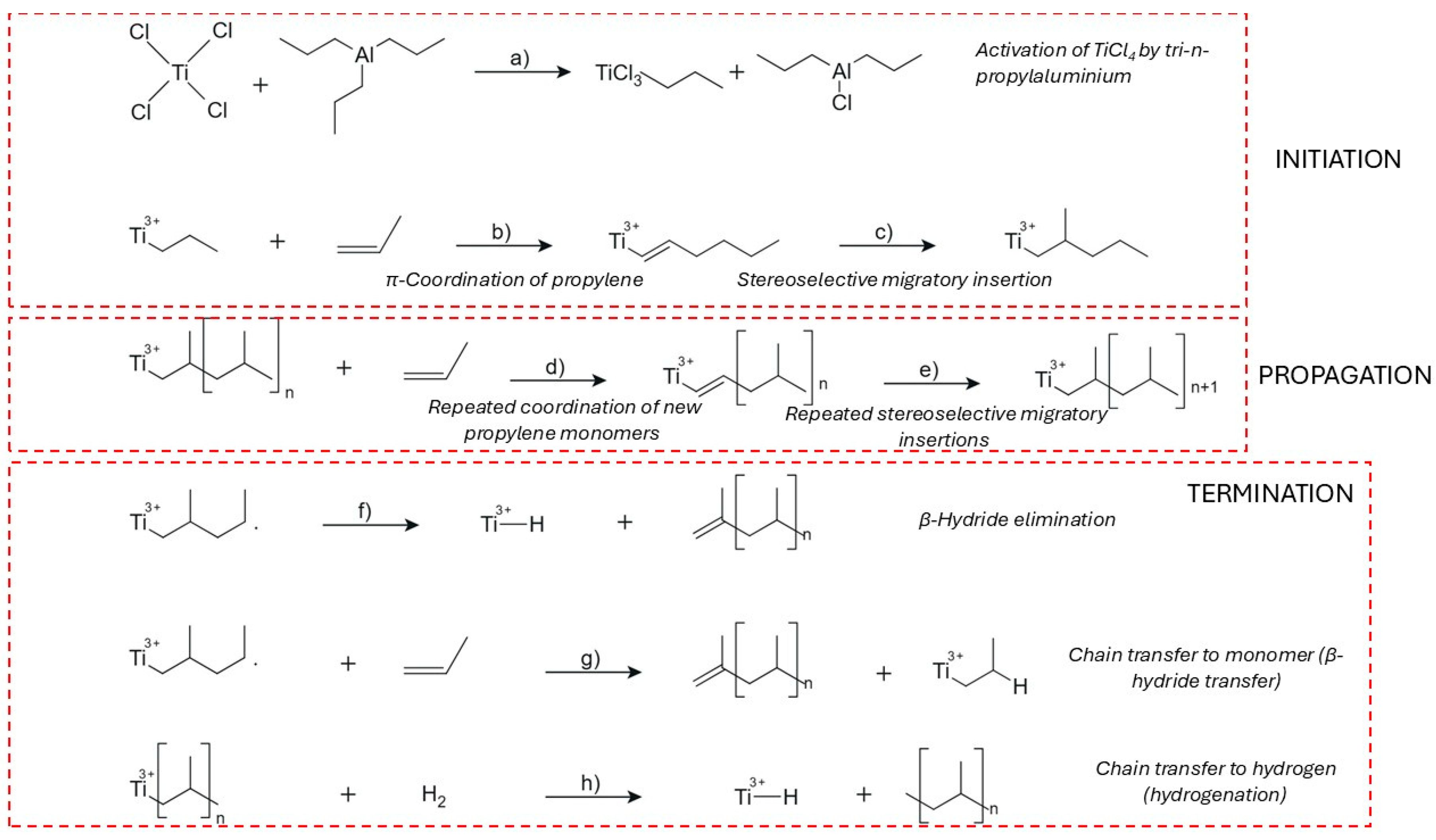
Figure 4.
Proposed free-radical polymerization mechanism for the formation of low-density polyethylene (LDPE) using lauroyl peroxide as initiator. (a) Decomposition of lauroyl peroxide yields two primary radicals, (b) radical addition to ethylene initiates chain growth, (c) propagation by sequential ethylene additions, (d) continued linear propagation via repetitive monomer insertions, (e) extended chain growth under high-pressure conditions, (f) intramolecular hydrogen transfer (1,5-backbiting) forms a secondary internal radical, (g) branch formation via ethylene addition at the side-chain radical site, (g2) branch elongation through continued propagation from the newly formed branch point, and (h) termination by recombination of two radical chain ends.
Figure 4.
Proposed free-radical polymerization mechanism for the formation of low-density polyethylene (LDPE) using lauroyl peroxide as initiator. (a) Decomposition of lauroyl peroxide yields two primary radicals, (b) radical addition to ethylene initiates chain growth, (c) propagation by sequential ethylene additions, (d) continued linear propagation via repetitive monomer insertions, (e) extended chain growth under high-pressure conditions, (f) intramolecular hydrogen transfer (1,5-backbiting) forms a secondary internal radical, (g) branch formation via ethylene addition at the side-chain radical site, (g2) branch elongation through continued propagation from the newly formed branch point, and (h) termination by recombination of two radical chain ends.
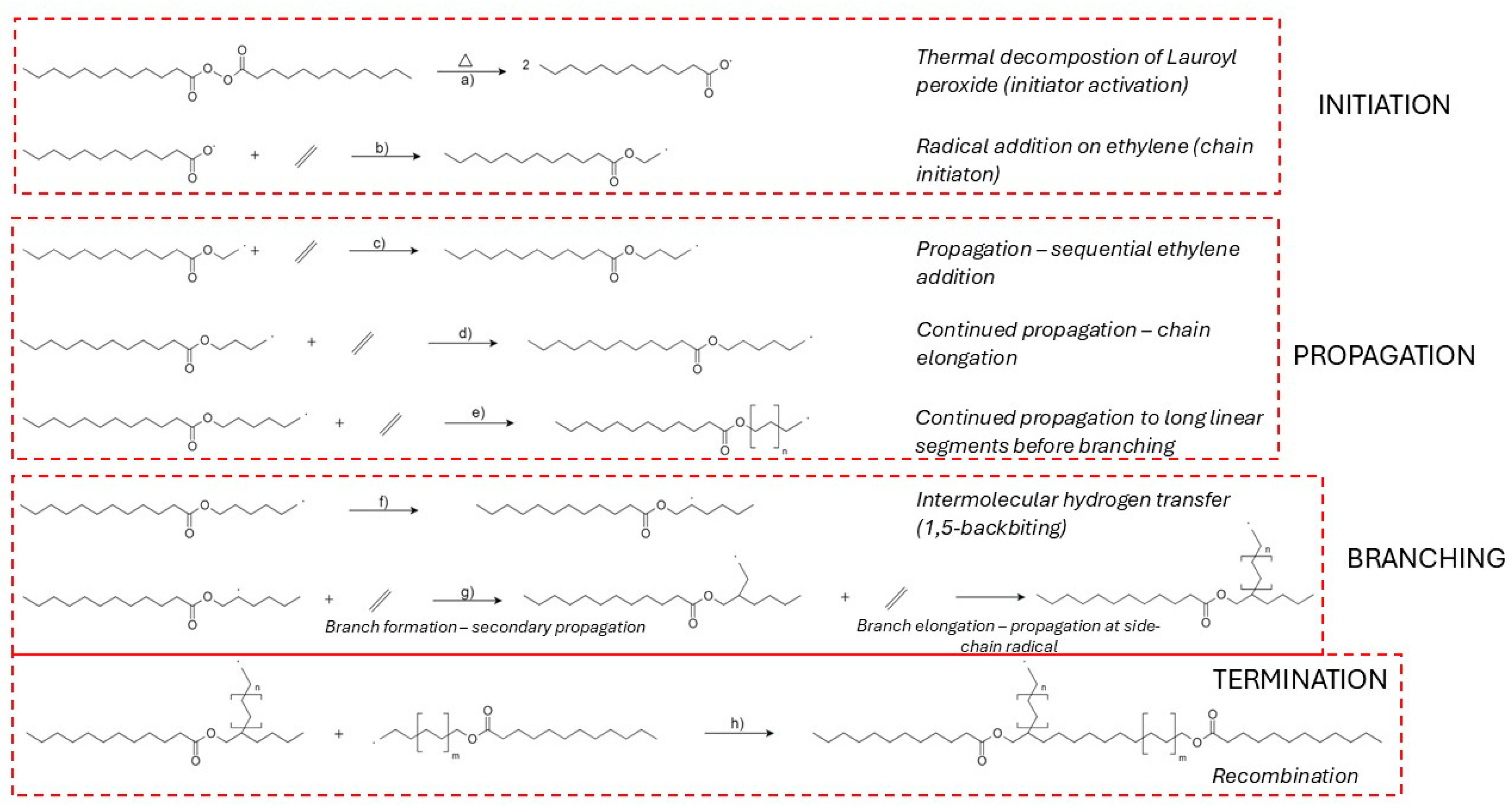
Figure 5.
Proposed mechanism of free-radical polymerization of vinyl chloride (VC) using azobisisobutyronitrile (AIBN) as initiator. (a) Thermal decomposition of AIBN generates two 2-cyano-2-propyl radicals and nitrogen gas. (b) The primary radical adds to vinyl chloride, forming an initiating macroradical. (c) Propagation occurs via head-to-tail addition of successive vinyl chloride monomers, yielding a stereoregular –CH2–CHCl– polymer backbone. (d) Intramolecular chain transfer through 1,5-hydrogen backbiting forms a mid-chain radical, initiating branching. (e) Termination by recombination, where two macroradicals couple to form a saturated chain. (f) Termination by disproportionation via hydrogen atom transfer, producing one saturated and one unsaturated PVC chain. (g) An alternative disproportionation route, involving an internal hydrogen shift, yielding a saturated polymer end-group.
Figure 5.
Proposed mechanism of free-radical polymerization of vinyl chloride (VC) using azobisisobutyronitrile (AIBN) as initiator. (a) Thermal decomposition of AIBN generates two 2-cyano-2-propyl radicals and nitrogen gas. (b) The primary radical adds to vinyl chloride, forming an initiating macroradical. (c) Propagation occurs via head-to-tail addition of successive vinyl chloride monomers, yielding a stereoregular –CH2–CHCl– polymer backbone. (d) Intramolecular chain transfer through 1,5-hydrogen backbiting forms a mid-chain radical, initiating branching. (e) Termination by recombination, where two macroradicals couple to form a saturated chain. (f) Termination by disproportionation via hydrogen atom transfer, producing one saturated and one unsaturated PVC chain. (g) An alternative disproportionation route, involving an internal hydrogen shift, yielding a saturated polymer end-group.
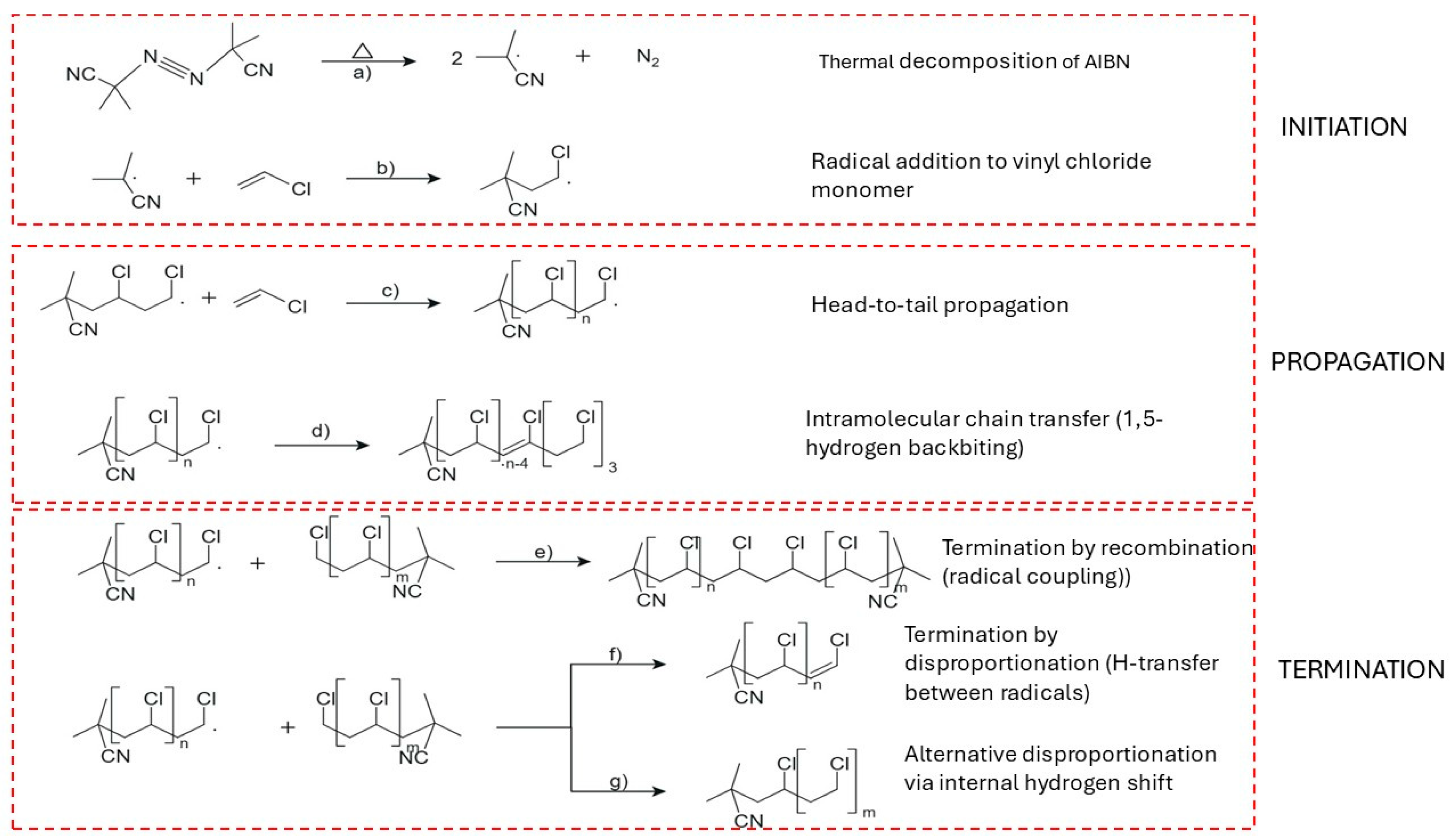
Figure 6.
Thermal degradation mechanism of high-density polyethylene (HDPE): a) Initiation by random C–C bond homolysis; b) β-scission of primary/secondary alkyl radicals; c) Intermolecular hydrogen abstraction; d) Intramolecular hydrogen transfer (1,5-backbiting); e) Secondary β-scission following backbiting; f) Radical isomerization via alkyl shift; g) Secondary cracking of olefins; h) Cyclization into cyclic radicals; i) Radical recombination forming saturated chains; j) Disproportionation yielding unsaturated end groups; k) Crosslinking via radical coupling between chains.
Figure 6.
Thermal degradation mechanism of high-density polyethylene (HDPE): a) Initiation by random C–C bond homolysis; b) β-scission of primary/secondary alkyl radicals; c) Intermolecular hydrogen abstraction; d) Intramolecular hydrogen transfer (1,5-backbiting); e) Secondary β-scission following backbiting; f) Radical isomerization via alkyl shift; g) Secondary cracking of olefins; h) Cyclization into cyclic radicals; i) Radical recombination forming saturated chains; j) Disproportionation yielding unsaturated end groups; k) Crosslinking via radical coupling between chains.
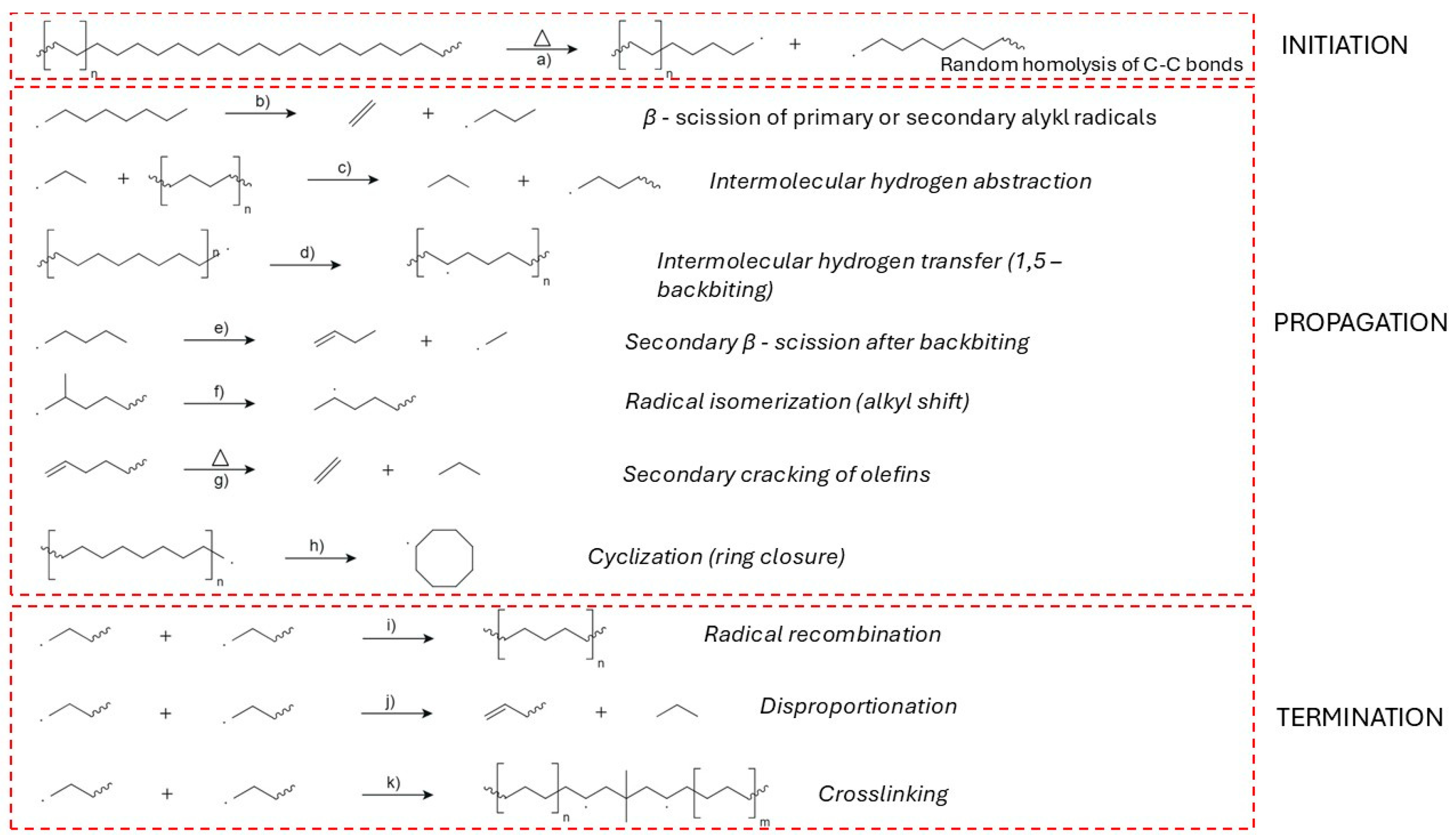
Figure 8.
Reaction mechanism of the thermal degradation of polypropylene (PP). a) Random homolysis of C–C bonds; b) β-scission of tertiary radicals; c) methyl radical formation; d) backbiting (1,5-hydrogen shift); e) intermolecular hydrogen abstraction; f) radical isomerization (1,2/1,3-alkyl shifts); g) vinylidene formation (terminal hydrogen elimination); h) cyclization via radical-induced ring closure; i) radical recombination forming saturated chains; j) disproportionation yielding one saturated and one unsaturated chain; k) crosslinking through radical coupling between polymer chains.
Figure 8.
Reaction mechanism of the thermal degradation of polypropylene (PP). a) Random homolysis of C–C bonds; b) β-scission of tertiary radicals; c) methyl radical formation; d) backbiting (1,5-hydrogen shift); e) intermolecular hydrogen abstraction; f) radical isomerization (1,2/1,3-alkyl shifts); g) vinylidene formation (terminal hydrogen elimination); h) cyclization via radical-induced ring closure; i) radical recombination forming saturated chains; j) disproportionation yielding one saturated and one unsaturated chain; k) crosslinking through radical coupling between polymer chains.
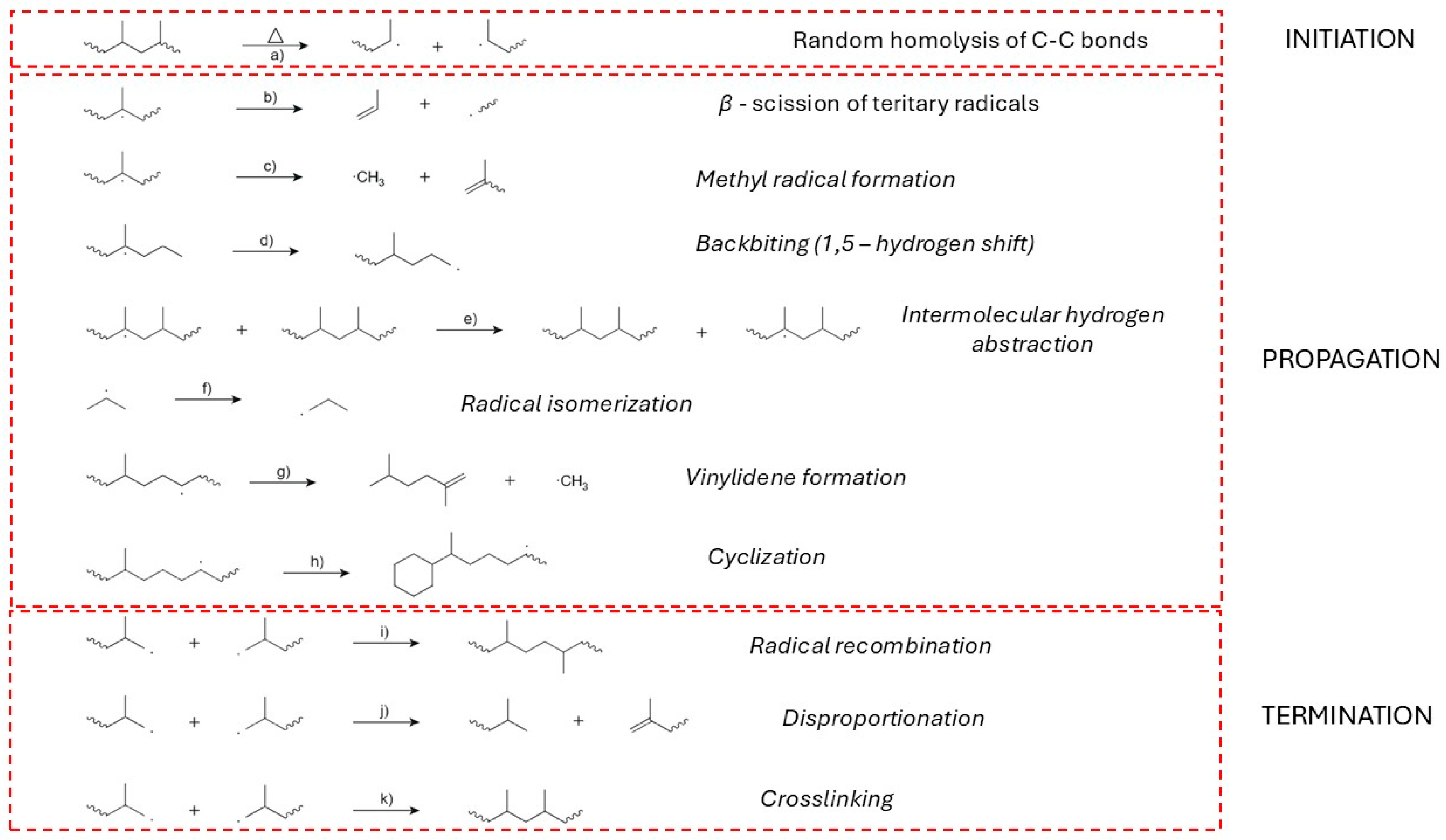
Figure 9.
Initiation and propagation steps in the thermal degradation of PVC. The sequence includes (a) random homolysis of C–Cl bonds, which generates initiation sites, (b) autocatalytic dehydrochlorination via the zipper mechanism, (c) extension of the conjugated segment in autocatalytic dehydrochlorination, (d) further extension of conjugation along the chain, (e) self-catalyzed dehydrochlorination via chloride ions, (f) pericyclic/E2-like concerted elimination, (g) the minor radical pathway, (h) propagation through hydrogen abstraction by chlorine radicals, (i) elimination steps extending the double bond with regeneration of Cl radicals, (j) polyene sequence formation with growth of conjugated domains, (k) polyene transformations through secondary propagation including 6π electrocyclic ring closure (cyclization), and (l) crosslinking between polyene chains via radical coupling.
Figure 9.
Initiation and propagation steps in the thermal degradation of PVC. The sequence includes (a) random homolysis of C–Cl bonds, which generates initiation sites, (b) autocatalytic dehydrochlorination via the zipper mechanism, (c) extension of the conjugated segment in autocatalytic dehydrochlorination, (d) further extension of conjugation along the chain, (e) self-catalyzed dehydrochlorination via chloride ions, (f) pericyclic/E2-like concerted elimination, (g) the minor radical pathway, (h) propagation through hydrogen abstraction by chlorine radicals, (i) elimination steps extending the double bond with regeneration of Cl radicals, (j) polyene sequence formation with growth of conjugated domains, (k) polyene transformations through secondary propagation including 6π electrocyclic ring closure (cyclization), and (l) crosslinking between polyene chains via radical coupling.
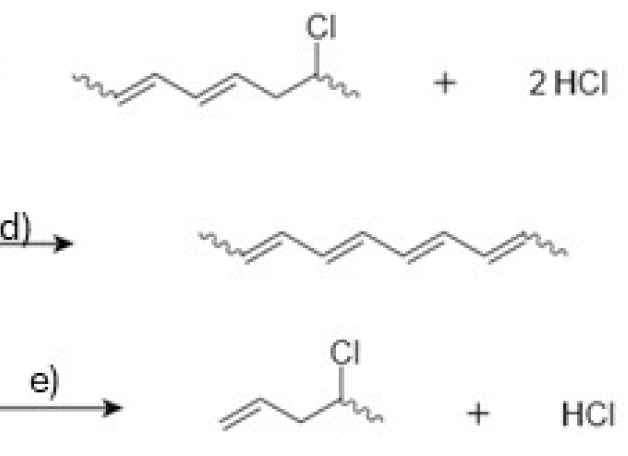
Figure 10.
Termination steps in the thermal degradation of PVC. The processes comprise (m) char formation through the development of cross-linked aromatic structures, (n) radical recombination leading to local quenching of chain reactivity, and (o) reversible re-addition of HCl to unsaturated sites. These termination pathways highlight how conjugated polyenes evolve toward stable aromatic char residues, with small fractions undergoing reversible reactions or quenching events.
Figure 10.
Termination steps in the thermal degradation of PVC. The processes comprise (m) char formation through the development of cross-linked aromatic structures, (n) radical recombination leading to local quenching of chain reactivity, and (o) reversible re-addition of HCl to unsaturated sites. These termination pathways highlight how conjugated polyenes evolve toward stable aromatic char residues, with small fractions undergoing reversible reactions or quenching events.
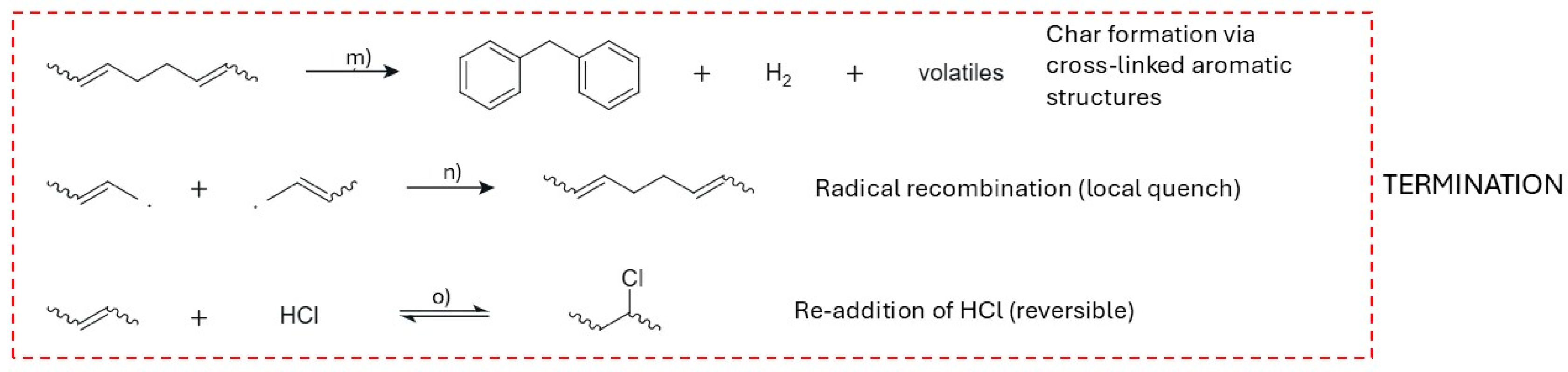
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



