Submitted:
29 September 2025
Posted:
30 September 2025
You are already at the latest version
Abstract
Background: Porcine reproductive and respiratory syndrome virus type 2 (PRRSV-2) remains a significant threat to swine production globally, including Japan. While the genetic diversity of PRRSV-2 has been reported previously, the potential association with modified live vaccines (MLV) is not well understood. This study aimed to characterize PRRSV-2 strains currently circulating in Japan and assess possible links with MLV. Methods: A total of 1,190-nucleotide open reading frame 5 sequences of PRRSV-2 were collected across Japan between 2020 and 2023, and phylogenetic analyses were performed to classify genetic clusters. Additionally, correlations between cluster distribution and MLV usage were examined, using sequences detected in the Kanto region. Results: Phylogenetic analysis revealed that 48.5% of the sequences belonged to Cluster III, with a median nucleotide identity of 90.5% to the Japanese reference strain EDRD-1. Notably, the sequence identity between the strains detected in this study and EDRD-1 was significantly lower than that of strains identified in 1992–1993 (p < 0.05). In the Kanto region, Cluster I and II variants, which exhibited high sequence homology to MLV strains, were exclusively detected on farms with a history of MLV usage. Furthermore, Cluster IV displayed substantial genetic divergence, suggesting it comprises a heterogeneous group of distinct lineages. Conclusions: These findings demonstrated the temporal changes in the genetic diversity of Cluster III and provided clear evidence that MLV usage directly influences PRRSV-2 cluster distribution, with Clusters I and II likely representing vaccine-origin viruses. The marked heterogeneity of Cluster IV also highlights the limitations of the current cluster-based classification.
Keywords:
porcine reproductive and respiratory syndrome virus 2
; PRRSV-2
; phylogenetic analysis
; modified live vaccine
; MLV
1. Introduction
Porcine reproductive and respiratory syndrome (PRRS) caused by PRRS virus (PRRSV) is a contagious disease causing reproductive failure in sows and respiratory distress in piglets [1]. First identified in the late 1980s in the U.S. and Europe [2], PRRSV has since led to significant economic losses globally, including an estimated 664 million USD annually in the U.S. and 28.3 billion JPY in Japan [3,4]. PRRSV, a single-stranded RNA virus belonging to Betaarterivirus genus, Arteriviridae family, consists of two species: PRRSV-1 (European type, alternately Betaarterivirus suid 1) and PRRSV-2 (North American type, alternately Betaarterivirus suid 2) [5]. The virus exhibits frequent mutations, particularly in the open reading frame (ORF) 5 gene, which encodes glycoprotein (GP) 5—a key membrane protein involved in host cell binding and virus neutralization [6,7]. ORF5 sequence analysis is widely used to classify PRRSV strains and study their genetic evolution [8-12].
The classification system for PRRSV-2, which prevails in Japan, has evolved over time. Initially, phylogenetic typing of PRRSV-2 employed restriction fragment length polymorphism (RFLP) typing based on restriction enzyme (MluI, HincII, SacII) cleavage patterns of the DNA fragment coding for the ORF5 gene in 1998 to discriminate RespPRRSV/Ingelvac PRRS MLV strains (RFLP 2-5-2) from wild-type strains [13]. Although this technique was widely used in North America, it presented limitations in assessing the genetic similarity of different strains [9,14]. Subsequently, a sequence-based phylogeny of ORF5 was established in 2010 [15], enabling a more detailed understanding of the genetic divergence of PRRSV-2. Through this approach, PRRSV-2 has been classified into 11 lineages and 21 sub-lineages [9]. This lineage taxonomy has gained wide acceptance internationally, and global data revealed that lineage distribution exhibits regional variation, with predominant lineages replaced over time [16,17]. On the other hand, in Japan, the classification of PRRSV-2 was reported by Yoshii et al. in 2005 to form five clusters [18], which subsequently became the standard system for the genotyping of PRRSV-2 in Japan. Historical analyses reveal that only Cluster I and III were detected in 1992-1993 [18], whereas Cluster II and V, in addition to Cluster I and III, were identified in 2000-2001 [19]. Furthermore, comparative sequence analyses of isolates collected between 1992 and 2008 demonstrated a progressive increase in nucleotide diversity within Cluster III, indicating ongoing genomic evolution over time [19]. Notably, Cluster IV was detected for the first time in Japan in 2008 [19]. More recent data indicate that cluster II predominated among the sequences detected in 2018-2020, followed by Cluster IV, while Cluster III, which had previously dominated, has exhibited a decline [20].
Control strategies for PRRS include a variety of approaches, namely early detection and isolation, and hygiene control, with vaccination representing one of the most crucial and widely implemented interventions worldwide [5,21,22]. The commercial PRRSV vaccines are dominated by two types, modified live vaccine (MLV) and killed vaccine, with MLV being more extensively used [5,23]. In the Japanese market, MLV products include Ingelvac PRRS® MLV, Fostera® PRRS, and Unistrain® PRRS from three different pharmaceutical companies. Ingelvac PRRS® MLV was launched in 1998, and Fostera® PRRS was launched in 2018. The active ingredient of the vaccines corresponds to Cluster I and II of PRRSV-2, respectively. Notably, only Unistrain® PRRS uses PRRSV-1 as a vaccine strain and was launched in 2023.
The extensive genetic variability of PRRSV represents one of the main challenges to the effective control of the virus [16,24,25]. Persistent PRRSV gene changes over time have been reported in PRRSV-endemic areas, and the accumulation of these changes is expected to impair the effectiveness of vaccination-based disease control strategies as they produce genetic and antigenic diversity [16,25]. Therefore, a comprehensive phylogenetic analysis of PRRSV is critical for understanding the evolutionary dynamics of the virus and guiding the development of more effective vaccination and control measures.
Given these considerations, this study firstly aimed to analyze the genetic divergence of PRRSV-2 detected in Japan during 2020-2023 and to elucidate temporal changes in the genetic diversity of PRRSV in the country by comparing these contemporary sequences with those published in previous studies. Secondly, the proportions of the farms using MLV were compared between clusters to assess the relationship between MLV use and the distribution of clusters to which prevalent strains belong.
2. Materials and Methods
2.1. Study 1.. Description of Genetic Variability of PRRSV-2 Detected in Japan During 2020–2023
2.1.1. Study 1.1: Broad Regional Analysis (2020-2023)
2.1.1.1. Sample Collection
Between 2020 and 2023, samples were collected from 69 farms across 18 prefectures as part of health screening and disease investigation. A total of 1,190 sequences were obtained from 1,136 serum samples, 37 oral fluid samples, and 17 organ samples.
2.1.1.2. Sample Analysis
All laboratory procedures—including RNA extraction, qualitative and quantitative reverse transcription polymerase chain reaction (RT-PCR/RT-qPCR) targeting PRRSV-2, and sequencing of the ORF5 region—were performed by SMC Co., Ltd. (Kanagawa, Japan) or Nippon Institute for Biological Science (NIBS) (Toyo, Japan). Sequences were aligned in MUSCLE using default settings. Phylogenetic analyses were conducted using the Neighbor-joining method, following protocols established in previous studies [18-20], and implemented in MEGA 11 software [26]. The phylogenetic analyses were conducted using previously published strain sequences and strains isolated by NIBS between 1997 and 2019. Table 1 shows strains that were isolated domestically, while Table 2 shows strains that were isolated overseas [18-20]. Sequences were clustered based on phylogenetic analysis following a cluster classification scheme described in previous reports [18-20].
2.1.2. Study 1.2: Regional Analysis of the Kanto Region (2020-2023)
2.1.2.1. Sample Collection
Study 1.2 focused on sequences derived from samples collected in the Kanto region, which represent a subset of the nationwide dataset described in Study 1.1. Specifically, 576 sequences were obtained from samples that tested positive for PRRSV-2 by RT-PCR or RT-qPCR, out of a total of 1,670 samples submitted for health screening or disease investigation, comprising 1,275 serum samples, 380 oral fluid samples, and 15 organ samples. These samples were collected from 34 farms across four prefectures in the Kanto region: Chiba (18 farms), Ibaraki (10 farms), Tochigi (5 farms), and Kanagawa (1 farm).
2.1.2.2. Sample Analysis
A total of 1,670 samples were subjected to RT-qPCR or RT-PCR targeting PRRSV-2. For each RT-PCR-positive batch, one representative sample was selected for sequence analysis. All laboratory procedures—including RNA extraction, RT-PCR/RT-qPCR, and sequencing of the ORF5 region—were performed by SMC Co., Ltd. or NIBS, following the same protocols as in Study 1.1. All 576 sequences obtained in Study 1.2 were already included in the dataset analyzed in Study 1.1. and had been classified according to its established cluster framework.
2.1.3. Statistical Analysis
A descriptive epizootiological analysis was carried out to investigate the geographical and production-stage distribution of PRRSV-2 clusters. The Mann-Whitney U test was used to compare the sequence identity of strains belonging to the same cluster between this study and previous studies.
2.2. Study 2. Assessment of the Relationship Between MLV Use and the Distribution of PRRSV-2 Clusters of Prevalent Strains
2.2.1. Sample Collection and Sequencing
To assess the relationship between the use of MLV and the distribution of prevalent strains, a subset of 576 sequences derived from the Kanto region in study 1.2 was analyzed. In addition, information regarding the use of MLV was collected from all participating farms.
2.2.2. Statistical Analysis
Chi-square tests were used to compare the proportions of farms using MLV among the clusters, including only farms with known MLV usage status. As four clusters, I–IV, except Cluster V, were identified, pairwise comparisons of proportions were subsequently conducted using the pairwise.prop.test function in R version 4.3.3. [27] and RStudio Cloud [28].
3. Results
3.1. Study 1: Description of Genetic Variability of PRRSV-2 Detected in Japan During 2020–2023 and Comparison of Sequence Homology with Previous Reports
3.1.1. Broad Regional Phylogenetic Analysis and Cluster Identification (Study 1.1)
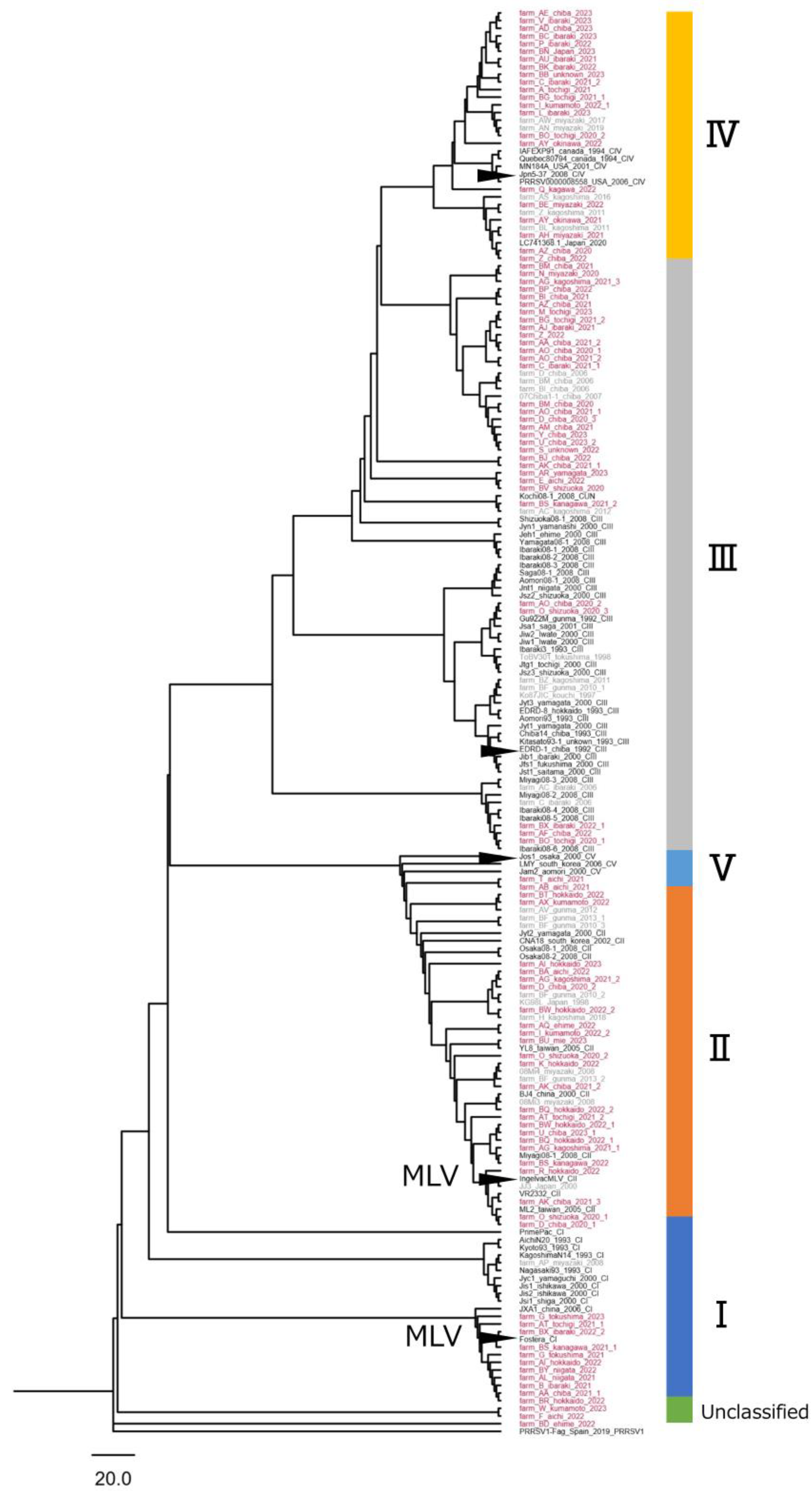
PRRSV-2 ORF5 gene sequences collected between 2020 and 2023 were classified into five clusters, I–V, based on reference sequences reported in previous studies [18-20]. Sequences that did not correspond to any established cluster were designated as Unclassified (Fig. 1). Cluster I included numerous sequences closely related to the MLV vaccine strain, Fostera® PRRS. Similarly, Cluster II contained a large number of sequences, many of which exhibited close homology to the MLV vaccine strain, Ingelvac® PRRS MLV. Cluster III showed marked genetic divergence; sequences detected between 2020 and 2023 primarily occupied phylogenetic positions distinct from those identified between 1992 and 2008. The proportions of PRRSV-2 ORF5 gene sequences assigned to each cluster were calculated based on the total number of sequences obtained (n = 1,190). Specifically, 13.3% for Cluster I, 18.6% for Cluster II, 48.5% for Cluster III, 18.4% for Cluster IV, 0.2% for Cluster V, and 1.0% for Unclassified. Regarding the distribution of clusters at the pig farm level, 53 farms (76.8%) harbored PRRSV-2 strains belonging to only one cluster, 15 farms (21.7%) contained those belonging to two clusters, and 1 farm (1.4%) hosted those classified into three clusters.
Figure 1.
Phylogenetic tree and cluster classification of PRRSV strains detected between 1992 and 2023 based on reference sequences. Clusters are color-coded as follows: Cluster I (blue), Cluster II (orange), Cluster III (gray), Cluster IV (yellow), Cluster V (light blue) and Unclassified (green). Black labels indicate the reference strains, gray labels indicate strains collected between 1997 and 2019 at NIBS, red labels denote strains analyzed in the present study. Arrowheads indicate the reference strains used for the homology analysis.
Figure 1.
Phylogenetic tree and cluster classification of PRRSV strains detected between 1992 and 2023 based on reference sequences. Clusters are color-coded as follows: Cluster I (blue), Cluster II (orange), Cluster III (gray), Cluster IV (yellow), Cluster V (light blue) and Unclassified (green). Black labels indicate the reference strains, gray labels indicate strains collected between 1997 and 2019 at NIBS, red labels denote strains analyzed in the present study. Arrowheads indicate the reference strains used for the homology analysis.
3.1.2. Descriptive Epizootiology of Distribution of Clusters Across Regions in Japan (Study 1.1)
In Figure 2, the regional distributions of PRRSV-2 clusters in Japan are shown. Marked differences in cluster distribution were observed among the six regions: Hokkaido, Tohoku/Hokuriku, Kanto, Tokai, Shikoku, and Kyushu. For example, only two clusters were detected in Hokkaido (Clusters I and II) and Tohoku/Hokuriku (Clusters I and III), while several clusters were detected in Kanto (Clusters I, II, III, IV), Tokai (Clusters II, III, V, Unclassified), Shikoku (Clusters I, II, IV, Unclassified) and Kyushu (Clusters II, III, IV, Unclassified). The predominant clusters by region were as follows: Cluster I in Tohoku and Shikoku (94.7% and 45.5%), Cluster II in Hokkaido (70.6%), and Cluster III in Kanto, Tokai, and Kyushu (46.7%, 63.8%, and 59.8%), respectively (Fig. 2).
3.1.3. Sequence Homology to the Reference Strains (Study 1.1)
Homology analysis revealed distinct sequence similarities among the PRRSV-2 clusters identified in this study. PRRSV-2 sequences belonging to Cluster I showed the highest homology with that of the reference strain, Fostera® PRRS, with a median of 99.2% and a homology range of 95.2–100%. The 25th percentile for Cluster I was 98.3%, and the 75th percentile was 99.6%. PRRSV-2 sequences belonging to Cluster II exhibited homology with that of the reference strain, Ingelvac® PRRS MLV, ranging from 88.1–100%, with a median of 99.6%, a 25th percentile of 98.8%, and a 75th percentile of 99.7%. On the other hand, PRRSV-2 sequences in Cluster III, compared with the sequence of EDRD-1, had a broader homology range (85.3–96.7%), with a median of 90.5%, a 25th percentile of 87.4%, and a 75th percentile of 91.9%. PRRSV-2 sequences in Cluster IV, when compared with the sequence of Jpn5-37, showed a homology range of 79.5–95.3%, with a median of 84.9%, a 25th percentile of 84.2%, and a 75th percentile of 85.2%. Meanwhile, PRRSV-2 sequences in Cluster V, compared with the sequence of Jos1, had a homology range of 82.7–88.5%, with a median of 85.6%, a 25th percentile of 84.1%, and a 75th percentile of 87.0% (Table 3). Notably, Cluster I sequences in this study showed significantly higher homology to Fostera® PRRS compared with Cluster I sequences detected during 1992–2008 (92.5% [18,19], p < 0.05). Conversely, Cluster III sequences showed significantly lower homology to EDRD-1 than those detected during 1992–1993 (95.7% [18], p < 0.05).
3.2 Study 1.2: Regional Analysis of PRRSV-2 in the Kanto Region (2020-2023)
3.2.1. PRRSV-2 Detection Rates Across Different Production Stages (Study 1.2)
PRRSV-2 was detected by RT-PCR across all developmental stages of swine production on farms in the Kanto region. The lowest positivity rate of PRRSV-2 (PCR-positive samples/total test samples) was observed in sows (28.1%) and the highest in fetuses (90.0%) (Table 4).
3.2.2. Distribution of PRRSV-2 Clusters Across Production Stages (Study 1.2)
Although multiple PRRSV-2 sequences in different clusters were detected across most production stages, only PRRSV-2 sequences belonging to Clusters III and IV were detected in gilts and sows. In contrast, those belonging to Clusters I and IV were detected in aborted fetuses (Table 5). No PRRSV-2 sequences belonging to Cluster V were detected in the Kanto region.
3.3. Study 2: Assessment of the Relationship Between MLV Use and Distribution of PRRSV-2 Clusters of Prevalent Strains in the Kanto Region
A chi-square test showed that the proportions of pig farms using MLV differed significantly among the four identified clusters (x2 = 255.9, df = 3, p < 0.001). All farms from which PRRSV-2 sequences belonging to Clusters I and II were detected had a history of MLV use (100%, Table 6). Pairwise comparisons of proportions showed significant differences in MLV usage between the following cluster pairs: Clusters I and III (p < 0.001), Clusters I and IV (p < 0.001), Clusters II and III (p < 0.001), Clusters II and IV (p < 0.001), and Clusters III and IV (p < 0.001) (Table 6). The proportion of farms using MLV was the third highest in Cluster III (79.5%) and the lowest in Cluster IV (19.1%).
4. Discussion
This study performed a comprehensive molecular phylogenetic analysis of 1190 PRRSV-2 sequences obtained from 69 farms across Japan over a four-year period (2020–2023). The findings provide valuable insights into the genetic variability of contemporary PRRSV-2 strains circulating in the country. Comparative analysis with previously published PRRSV data and this study’s results revealed that Cluster III has undergone substantial genetic differentiation over time, resulting in notable sequence divergence from EDRD-1, the domestic reference strain, when compared with strains isolated in 1992-1993 [18]. This finding suggests that Cluster III has persisted in the country for an extended period, gradually accumulating genetic changes. Furthermore, analysis of PRRSV from farms in the Kanto region revealed that Clusters I to IV were detected frequently on farms with a history of MLV use, whereas PRRSV-2 belonging to only Clusters III and IV were detected on farms without such a history. Statistically significant differences in cluster detection rates were observed between farms with and without MLV use for each cluster. This observation strongly suggests that the use of MLV may have contributed to changes in the distribution of PRRSV-2 clusters in the region.
This study confirmed that Cluster III remains the most predominant PRRSV-2 cluster in Japan at the moment and appears to have accumulated genetic variation over time. These findings are consistent with the report by Iseki et al., who identified Cluster III between 2007 and 2008 as the most frequently detected one [19]. However, our findings differ from those of Kyutoku et al., who found that Clusters II and IV were dominant in Japan between 2018 and 2020 [20]. This discrepancy may be attributable to differences in the geographic distributions of the farms sampled, as PRRSV genetic lineages have been shown to vary depending on regional and farm-specific control strategies [9,29]. The observed pattern of genetic variation in Cluster III corresponds with the findings of Iseki et al., who reported a significant reduction in the sequence homology between Cluster III strains from 1992 to 1993 and those from 2007 to 2008 [19]. In contrast, Kyutoku et al. reported no significant changes in the sequence identity among Cluster III strains detected between 2018 and 2020 [20]. Based on the present findings, we hypothesize that genetic variation of PRRSV-2 in Cluster III occurs gradually rather than rapidly, becoming apparent only over scales of approximately a decade. These observations therefore suggest that PRRSV-2 belonging to Cluster III is undergoing slow but steady genetic diversification over extended periods by repeating the multiplications in pigs.
The results of the present study showed that Cluster IV exhibited a low mean sequence homology of 84.7% with its reference strain, Jpn5-37, suggesting a substantial genetic divergence between the strain identified in 2008 and the Cluster IV strains detected in this study. This divergence was also evident in our phylogenetic tree analysis, which showed considerable branch expansion within Cluster IV and the presence of multiple lineages with high genetic diversity. A particularly notable finding was the identification of Cluster IV sequences from a pig farm in Okinawa that were classified as sublineage 1.5 of Lineage 1 (L1A in the lineage taxonomy [30]). Further phylogenetic, RFLP patterns, and glycosylation site analyses revealed that these sequences were analogous to the NADC34 strain, which has caused large-scale outbreaks in the U.S. and Asia since 2017 [30]. These L1A sequences showed relatively low homology (86.1–87.6%) to the reference strain used in this study, but high homology (90.9–92.9%) to the NADC34 strain [30]. This suggests that certain Cluster IV lineages may have origins distinct from the historical Japanese reference strain. Kyutoku et al. also reported a low average homology of approximately 85% between Cluster IV and Jpn5-37 in sequences detected between 2018 and 2020 [20]. In addition, they identified more than 99% homology between a strain detected in Austria (AY875854) and one in Denmark (AF095515), suggesting a probable non-Japanese origin for some Cluster IV members [20]. These findings suggest that Cluster IV is comprised of a heterogeneous group of genetically distinct lineages, rather than a single evolutionary lineage. Further, this heterogeneity highlights a limitation of cluster-based classification in fully capturing the diversity of these genetic lineages. We therefore propose that the classification of PRRSV should incorporate a combined approach, integrating detailed sequence-based phylogenetic analysis and lineage-level classification, in addition to traditional cluster classification.
Analysis of trends in Cluster I revealed that the strains detected in this study and those reported prior to the introduction of Fostera® PRRS formed distinct groupings on the phylogenetic tree. Their homologies with the MLV strain showed significant differences. Kyutoku et al. similarly analyzed sequence homology to MLV strains using isolates detected between 2018 and 2020 [20]. They found that strains detected after 2019 had significantly higher homology to MLV strains than those detected in 2018 and attributed this shift to the impact of Fostera® PRRS, which was introduced in 2018. Based on these results, we conclude that the detection rate of Cluster I field strains, previously distributed mainly in western Japan, has decreased in recent years. This decline likely reflects selective pressure exerted by vaccine-induced immunity—particularly against field strains closely related to the MLV strain—as well as improved sanitary management practices on pig farms, and other factors that contribute to the reduction of field strain circulation.
Our results provide compelling evidence that MLV usage patterns influence the genetic and phylogenetic distribution of PRRSV within farms. In the Kanto region, Clusters I and II were detected exclusively on the farms with a history of MLV use. Notably, many of the strains within these clusters exhibited high sequence homology to MLV strains, with mean values of 98% for both clusters. These findings suggest a strong link between MLV use and the epizootiological dynamics of Clusters I and II. This observation is consistent with the report by Kyutoku et al., who found that PRRSV Clusters I and II sequences detected between 2018 and 2020 in Japan were highly homologous to MLV strains used in Japan [20]. Similar trends have been observed in other countries, where studies have shown that the detection rate of field strains with more than 98% homology to MLV strains and decreased detection rates of strains with less than 95% homology following the introduction of MLV to the market [9,31]. Paploski et al. also noted that PRRSV genetic lineages may vary selectively according to the immune status of swine populations [17], suggesting that immunological differences between farms with and without a history of MLV use may contribute to the observed differences in the prevalence of Clusters III and IV. Taken together, our findings, which include the exclusive detection of Clusters I and II on vaccinated farms and their high sequence homology to vaccine strains, indicate that these clusters likely represent viruses of vaccine origin. The observed association between cluster distributions and farm-level MLV use history, combined with high levels of sequence homology, raise the possibility that the MLV strains persist within farms and undergo genetic changes over time. These dynamics have lasting effects on PRRSV genetic variability and the composition of prevalent strains within farms. This has important implications for the efficacy of vaccination programs and highlights the need for careful selection of vaccine strains and the potential development of new control strategies.
An important observation in this study was that although multiple PRRSV-2 clusters were detected in piglets, growers, and finishers, RT-PCR positivity was low in sows, and Clusters I and II were not detected in this age group. Several biological factors may explain this result. First, resistance to PRRSV infection is known to increase with age, and sows have physiologically robust immune defenses compared with younger pigs [32]. Second, sow herds on PRRS-positive farms are likely to have experienced repeated infections, resulting in acquired immunity to circulating field strains [33]. Third, on farms using MLV, sows are typically vaccinated at regular intervals, which may enhance specific and robust immune responses against Clusters I and II, thereby reducing the likelihood of viral replication and detection in this group [34,35]. The combined effects of these factors may have led to the lower detection rates for Clusters I and II in sows. However, this interpretation remains speculative, and additional investigations such as longitudinal sampling of individual animals and integration with serological tests are needed to better understand the dynamics of the virus in sow herds.
This study has several methodological limitations. First, sampling bias may have influenced the results due to the limited number of farms surveyed and the unequal number of sequences obtained from each farm. This sampling bias may have disproportionately reflected the characteristics of specific regions or farm management systems, thereby limiting the generalizability of the results. Second, the analysis in this study was based solely on ORF5 sequences. Although ORF5 is commonly used for PRRSV classification, more comprehensive, whole-genome sequencing may yield more comprehensive and potentially divergent results. Third, the validity of treating populations as single clusters remains questionable, particularly for populations that exhibit substantial internal variability, such as Clusters III and IV. These limitations constrain the interpretation and generalization of the results and highlight challenges that should be addressed in future research. Subsequent studies would benefit from sampling more farms using statistically appropriate sample sizes, incorporating whole-genome sequencing to capture PRRSV genetic diversity more comprehensively, and adopting alternative taxonomic frameworks, such as lineage classification. Furthermore, adopting a longitudinal sampling design would enable the monitoring of within-farm viral dynamics over time and provide a more accurate assessment of the long-term impact of vaccine use.
5. Conclusions
This study highlighted the genetic diversity and evolutionary dynamics of PRRSV-2 in Japan, confirming Cluster III as the predominant cluster exhibiting gradual changes in genetic variation over time. The use of MLV appears to significantly influence cluster distribution, with Clusters I and II primarily detected on farms that have a history of MLV use. Additionally, Cluster IV exhibited substantial genetic divergence, suggesting the presence of multiple distinct sub-clusters within this group. Despite methodological limitations, this study emphasized the importance of continued molecular surveillance and the refinement of classification frameworks. Future research should incorporate whole-genome sequence analysis and longitudinal sampling to more comprehensively characterize PRRSV evolution and vaccine-associated dynamics, thereby contributing to the development of improved disease control strategies.
Author Contributions
Conceptualization, Y.Y. and O.T.; Methodology, O.T., R.M., and K.M.; Formal Analysis, Y.Y.; Investigation, Y.Y.; Data Curation, Y.Y., R.N., R.T., and N.T.; Writing – Original Draft Preparation, Y.Y.; Writing – Review & Editing, O.T., A.K., and K.M.; Visualization, A.K.; Supervision, R.M., and K.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work did not receive support from any organization except for the regular institutional budget.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained verbally from all farmers who submitted samples used for analysis in this study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We would like to express our sincere gratitude to the members of the farm who kindly provided samples for this study, as well as to the Sales Department of Nisseiken Co., Ltd. and the staff of the Nippon Institute for Biological Science for their generous support.
Conflicts of Interest
All authors declare no conflict of interest. However, Y.Y., N.T., and R.N. are employees of Nisseiken Co., Ltd.
Abbreviations
The following abbreviations are used in this manuscript:
| GP | glycoprotein |
| MLV | modified live vaccine |
| NIBS | Nippon Institute of Biological Science |
| ORF | open reading frame |
| PRRS | porcine reproductive and respiratory syndrome |
| PRRSV | porcine reproductive and respiratory syndrome virus |
| RFLP | restriction fragment length polymorphism |
| RT-PCR | qualitative reverse transcription polymerase chain reaction |
| RT-qPCR | quantitative reverse transcription polymerase chain reaction |
References
- Fiers, J.; Cay, A.B.; Maes, D.; Tignon, M. A Comprehensive Review on Porcine Reproductive and Respiratory Syndrome Virus with Emphasis on Immunity. Vaccines 2024, 12, 942. [Google Scholar] [CrossRef]
- Ruedas-Torres, I.; Rodríguez-Gómez, I.M.; Sánchez-Carvajal, J.M.; Larenas-Muñoz, F.; Pallarés, F.J.; Carrasco, L.; Gómez-Laguna, J. The jigsaw of PRRSV virulence. Vet. Microbiol. 2021, 260, 109168. [Google Scholar] [CrossRef]
- Yamane, I.; Kure, K.; Ishikawa, H.; Tagagi, M.; Miyazaki, A.; Suzuki, T.; Shibahara, T.; Kubo, M.; Kobayashi, H.; Kokuho, T.; et al. Evaluation of the economical losses due to the outbreaks of porcine reproductive and respiratory syndrome. (A questionnaire-based epidemiological survey and estimation of the total economical losses in Japan). Proc. Jpn. Pig Vet. Soc. 2009, 55, 33–37. (In Japanese) [Google Scholar]
- Holtkamp, D.; Kliebenstein, J.; Neumann, E.; Zimmerman, J.; Rotto, H.; Yoder, T.; Wang, C.; Yeske, P.; Mowrer, C.; Haley, C. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on U.S. pork producers. J. Swine Health Prod. 2013, 21, 72–84. [Google Scholar] [CrossRef]
- Chae, C. Commercial PRRS Modified-Live Virus Vaccines. Vaccines 2021, 9, 185. [Google Scholar] [CrossRef]
- Luo, Q.; Zheng, Y.; Zhang, H.; Yang, Z.; Sha, H.; Kong, W.; Zhao, M.; Wang, N. Research Progress on Glycoprotein 5 of Porcine Reproductive and Respiratory Syndrome Virus. Animals: an Open Access Journal from MDPI 2023, 13, 813. [Google Scholar] [CrossRef] [PubMed]
- Kim, W. I. , Kim, J. J., Cha, S. H., Wu, W. H., Cooper, V., Evans, R., Choi, E. J., & Yoon, K. J. Significance of genetic variation of PRRSV ORF5 in virus neutralization and molecular determinants corresponding to cross neutralization among PRRS viruses. Vet. Microbiol. 2013, 162, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Bálint, Á.; Molnár, T.; Kecskeméti, S.; Kulcsar, G.; Soós, T.; Szabo, P.; Kaszab, E.; Fornyos, K.; Zádori, Z.; Bányai, K.; et al. Genetic Variability of PRRSV Vaccine Strains Used in the National Eradication Programme, Hungary. Vaccines 2021, 9, 849. [Google Scholar] [CrossRef] [PubMed]
- Yim-Im, W.; Anderson, T.K.; Paploski, I.A.D.; VanderWaal, K.; Gauger, P.; Krueger, K.; Shi, M.; Main, R.; Zhang, J. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol. Spectr. 2023, 11, e0291623. [Google Scholar] [CrossRef]
- Jian, Y.; Lu, C.; Shi, Y.; Kong, X.; Song, J.; Wang, J. Genetic evolution analysis of PRRSV ORF5 gene in five provinces of Northern China in 2024. BMC Vet. Res. 2025, 21, 4679. [Google Scholar] [CrossRef]
- Zeller, M.A.; Chang, J.; Trevisan, G.; Main, R.G.; Gauger, P.C.; Zhang, J. Rapid PRRSV-2 ORF5-based lineage classification using Nextclade. Front. Vet. Sci. 2024, 11, 1419340. [Google Scholar] [CrossRef]
- Chandra, S.; Cezar, G.; Rupasinghe, K.; Magalhães, E.; Silva, G.; Almeida, M.; Crim, B.; Burrough, E.; Gauger, P.; Madson, D.; et al. Harnessing sequencing data for porcine reproductive and respiratory syndrome virus (PRRSV): tracking genetic evolution dynamics and emerging sequences in US swine industry. Front. Vet. Sci. 2025, 12, 1571020. [Google Scholar] [CrossRef] [PubMed]
- Wesley, R.D.; Mengeling, W.L.; Lager, K.M.; Clouser, D.F.; Landgraf, J.G.; Frey, M.L. Differentiation of a Porcine Reproductive and Respiratory Syndrome Virus Vaccine Strain from North American Field Strains by Restriction Fragment Length Polymorphism Analysis of ORF 5. J. Vet. Diagn. Invest. 1998, 10, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, G.; Sharma, A.; Gauger, P.; Harmon, K.M.; Zhang, J.; Main, R.; Zeller, M.; Linhares, L.C.M.; Linhares, D.C.L. PRRSV2 genetic diversity defined by RFLP patterns in the United States from 2007 to 2019. J. Vet. Diagn. Invest. 2021, 33, 920–931. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.Y.; Hon, C.C.; Hui, R.K.H.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C.C. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Paploski, I.A.D.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder, D.C.; VanderWaal, K. Temporal Dynamics of Co-circulating Lineages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10, 2486. [Google Scholar] [CrossRef]
- Paploski, I.A.D.; Pamornchainavakul, N.; Makau, D.N.; Rovira, A.; Corzo, C.A.; Schroeder, D.C.; Cheeran, M.C.J.; Doeschl-Wilson, A.; Kao, R.R.; Lycett, S.; et al. Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States. Vaccines 2021, 9, 608. [Google Scholar] [CrossRef]
- Yoshii, M.; Kaku, Y.; Murakami, Y.; Shimizu, M.; Kato, K.; Ikeda, H. Genetic variation and geographic distribution of porcine reproductive and respiratory syndrome virus in Japan. Arch. Virol. 2005, 150, 2313–2324. [Google Scholar] [CrossRef]
- Iseki, H.; Takagi, M.; Miyazaki, A.; Katsuda, K.; Mikami, O.; Tsunemitsu, H. Genetic analysis of ORF5 in porcine reproductive and respiratory syndrome virus in Japan. Microbiol. Immunol. 2011, 55, 211–216. [Google Scholar] [CrossRef]
- Kyutoku, F.; Yokoyama, T.; Sugiura, K. Genetic Diversity and Epidemic Types of Porcine Reproductive and Respiratory Syndrome (PRRS) Virus in Japan from 2018 to 2020. Epidemiologia 2022, 3, 285–296. [Google Scholar] [CrossRef]
- Charerntantanakul, W. Porcine reproductive and respiratory syndrome virus vaccines: Immunogenicity, efficacy and safety aspects. World J. Virol. 2012, 1, 23. [Google Scholar] [CrossRef]
- Cho, J.G.; Dee, S.A. Porcine reproductive and respiratory syndrome virus. Theriogenology 2006, 66, 655–662. [Google Scholar] [CrossRef]
- Li, J.; Miller, L.C.; Sang, Y. Current Status of Vaccines for Porcine Reproductive and Respiratory Syndrome: Interferon Response, Immunological Overview, and Future Prospects. Vaccines 2024, 12, 606. [Google Scholar] [CrossRef]
- Montaner-Tarbes, S.; del Portillo, H.A.; Montoya, M.; Fraile, L. Key gaps in the knowledge of the porcine respiratory reproductive syndrome virus (PRRSV). Front. Vet. Sci. 2019, 6, 38. [Google Scholar] [CrossRef]
- Chen, N.; Xiao, Y.; Ye, M.; Li, X.; Li, S.; Xie, N.; Wei, Y.; Wang, J.; Zhu, J. High genetic diversity of Chinese porcine reproductive and respiratory syndrome viruses from 2016 to 2019. Res. Vet. Sci. 2020, 131, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, 2024. https://www.R-project.org/.
- Posit Team. RStudio: Integrated Development Environment for R. Posit Software, PBC, Boston, MA, 2024. https://www.posit.co/.
- Shen, Y.F.; Arruda, A.G.; Koscielny, M.P.; Cheng, T.Y. Contrasting PRRSV temporal lineage patterns at the individual farm, production system, and regional levels in Ohio and neighboring states from 2017 to 2021. Prev. Vet. Med. 2024, 226, 106186. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, Y.; Taira, O.; Omori, T.; Tsutsumi, N.; Sugiura, K. First detection of NADC34-like porcine reproductive and respiratory syndrome virus strains in Japan. J. Vet. Med. Sci. 2025, 87, 110–114. [Google Scholar] [CrossRef]
- Kikuti, M.; Sanhueza, J.; Vilalta, C.; Paploski, I.A.D.; VanderWaal, K.; Corzo, C.A. Porcine reproductive and respiratory syndrome virus 2 (PRRSV-2) genetic diversity and occurrence of wild type and vaccine-like strains in the United States swine industry. PLoS One 2021, 16, e0259531. [Google Scholar] [CrossRef]
- Klinge, K.L.; Vaughn, E.M.; Roof, M.B.; Bautista, E.M.; Murtaugh, M.P. Age-dependent resistance to Porcine reproductive and respiratory syndrome virus replication in swine. Virol. J. 2009, 6, 177. [Google Scholar] [CrossRef]
- Plaza-Soriano, Á.; Martínez-Lobo, F.J.; Garza-Moreno, L.; Castillo-Pérez, J.; Caballero, E.; Castro, J.M.; Simarro, I.; Prieto, C. Determination of the frequency of individuals with broadly cross-reactive neutralizing antibodies against PRRSV in the sow population under field conditions. Porcine Health Manag. 2024, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Lebret, A.; Berton, P.; Normand, V.; Messager, I.; Robert, N.; Bouchet, F.; Brissonnier, M.; Boulbria, G. PRRSV detection by qPCR in processing fluids and serum samples collected in a positive stable breeding herd following mass vaccination of sows with a modified live vaccine. Porcine Health Manag. 2021, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Matamoros, A.; Camprodon, A.; Maldonado, J.; Pedrazuela, R.; Miranda, J. Safety and long-lasting immunity of the combined administration of a modified-live virus vaccine against porcine reproductive and respiratory syndrome virus 1 and an inactivated vaccine against porcine parvovirus and Erysipelothrix rhusiopathiae in breeding pigs. Porcine Health Manag. 2019, 5, 11. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Percentage distribution of PRRSV-2 clusters across different regions of Japan, based on strains detected between 2020 and 2023. For each region shown on the map of Japan, two pie charts are presented: the distribution of clusters based on the number of farms (left) and the distribution based on the number of sequences (right). The number at the center of each pie chart represents the total count, indicating the total number of farms (left) and the total number of identified sequences (right). Clusters I to V and Unclassified strains are shown as follows: Cluster I (blue), Cluster II (orange), Cluster III (gray), Cluster IV (yellow), Cluster V (light blue), and Unclassified (green). Duplication was allowed in counting the number of farms from which multiple-cluster strains were detected. Samples from six farms of unknown location and the obtained sequences derived from them were excluded from the analysis.
Figure 2.
Percentage distribution of PRRSV-2 clusters across different regions of Japan, based on strains detected between 2020 and 2023. For each region shown on the map of Japan, two pie charts are presented: the distribution of clusters based on the number of farms (left) and the distribution based on the number of sequences (right). The number at the center of each pie chart represents the total count, indicating the total number of farms (left) and the total number of identified sequences (right). Clusters I to V and Unclassified strains are shown as follows: Cluster I (blue), Cluster II (orange), Cluster III (gray), Cluster IV (yellow), Cluster V (light blue), and Unclassified (green). Duplication was allowed in counting the number of farms from which multiple-cluster strains were detected. Samples from six farms of unknown location and the obtained sequences derived from them were excluded from the analysis.
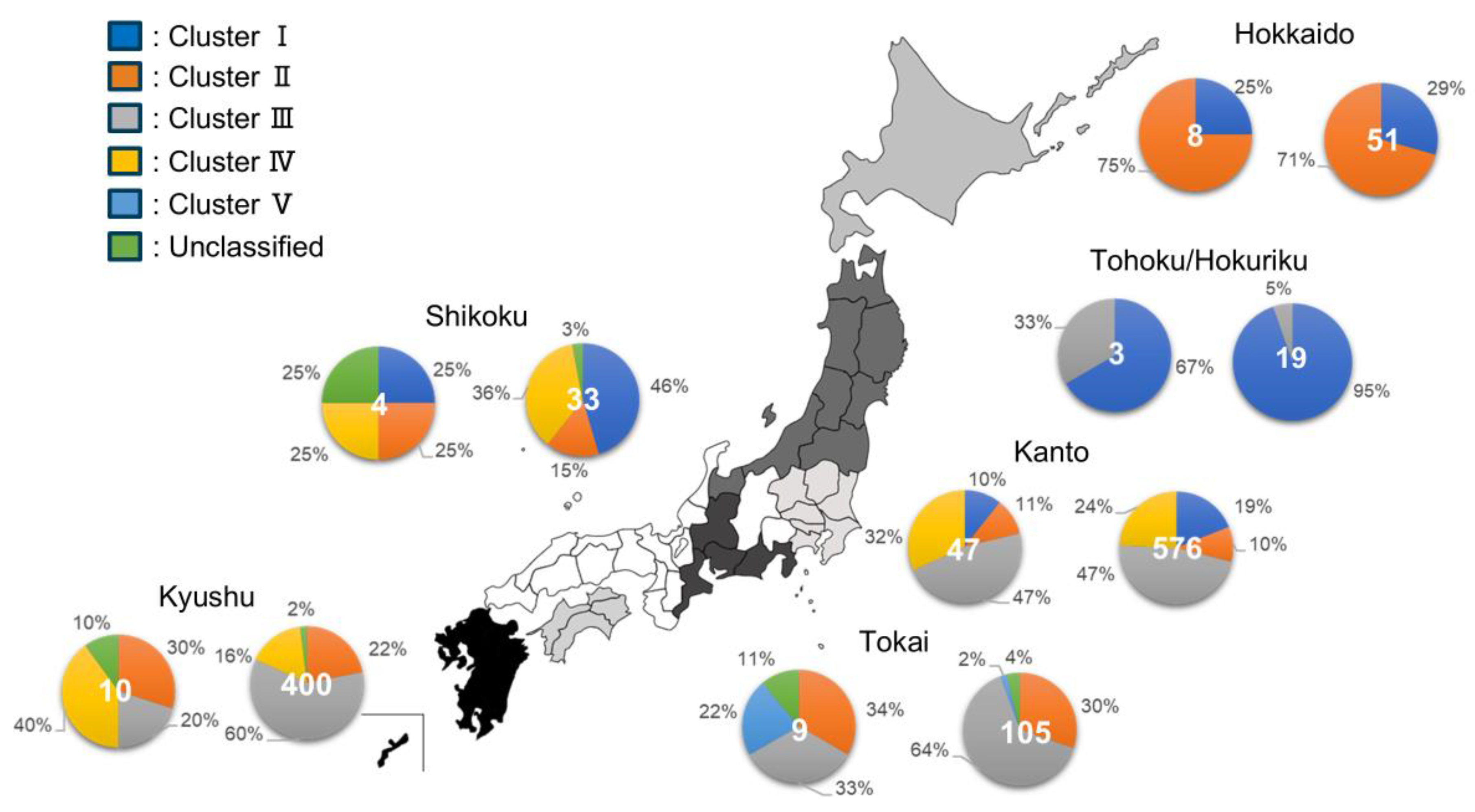

Table 1.
Reference sequences of Japanese-origin PRRSV-2 strains used for phylogenetic analysis and their cluster classification.
Table 1.
Reference sequences of Japanese-origin PRRSV-2 strains used for phylogenetic analysis and their cluster classification.
| Phylogenetic cluster |
Year of isolation |
Prefecture | Name of isolate | Accession no. |
|---|---|---|---|---|
| I | 1993 | Aichi | Aichi N20 | AB175715 |
| I | 1993 | Kyoto | Kyoto 93 | AB175724 |
| I | 1993 | Nagasaki | Nagasaki 93 | AB175725 |
| I | 1993 | Kagoshima | Kagoshima N14 | AB175723 |
| I | 2000 | Ishikawa | Jis1 | AB175694 |
| I | 2000 | Ishikawa | Jis2 | AB175695 |
| I | 2000 | Shiga | Jsi1 | AB175701 |
| I | 2000 | Yamaguchi | Jyc1 | AB175709 |
| II | 2000 | Aomori | Jam2 | AB175690 |
| II | 2000 | Yamagata | Jyt2 | AB175713 |
| II | 2007–2008** | Miyagi | Miyagi08-1 | AB546104 |
| II | 2007–2008** | Osaka | Osaka08-1 | AB546120 |
| II | 2007–2008** | Osaka | Osaka08-2 | AB546121 |
| III | 1992 | Chiba | EDRD-1* | D45852 |
| III | 1992 | Gunma | Gu922M | AB175721 |
| III | 1993 | Hokkaido | EDRD-8 | AB175720 |
| III | 1993 | Aomori | Aomori93 | AB175716 |
| III | 1993 | Ibaraki | Ibaraki3 | AB175722 |
| III | 1993 | Chiba | Chiba 14 | AB175717 |
| III | 1993 | Tokyo | Kitasato 93-1 | AB023782 |
| III | 2000 | Iwate | Jiw1 | AB175696 |
| III | 2000 | Iwate | Jiw2 | AB175697 |
| III | 2000 | Yamagata | Jyt1 | AB175712 |
| III | 2000 | Yamagata | Jyt3 | AB175714 |
| III | 2000 | Niigata | Jnt1 | AB175698 |
| III | 2000 | Ibaraki | Jib1 | AB175693 |
| III | 2000 | Tochigi | Jtg1 | AB175708 |
| III | 2000 | Saitama | Jst1 | AB175702 |
| III | 2000 | Yamanashi | Jyn1 | AB175710 |
| III | 2000 | Shizuoka | Jsz2 | AB175704 |
| III | 2000 | Ehime | Jeh1 | AB175691 |
| III | 2001 | Saga | Jsa1 | AB175700 |
| III | 2007–2008** | Aomori | Aomori08-1 | AB546102 |
| III | 2007–2008** | Miyagi | Miyagi08-2 | AB546105 |
| III | 2007–2008** | Miyagi | Miyagi08-3 | AB546106 |
| III | 2007–2008** | Yamagata | Yamagata08-1 | AB546108 |
| III | 2007–2008** | Ibaraki | Ibaraki08-1 | AB546109 |
| III | 2007–2008** | Ibaraki | Ibaraki08-2 | AB546110 |
| III | 2007–2008** | Ibaraki | Ibaraki08-3 | AB546111 |
| III | 2007–2008** | Ibaraki | Ibaraki08-4 | AB546112 |
| III | 2007–2008** | Ibaraki | Ibaraki08-5 | AB546113 |
| III | 2007–2008** | Ibaraki | Ibaraki08-6 | AB546114 |
| III | 2007–2008** | Shizuoka | Shizuoka08-1 | AB546118 |
| III | 2007–2008** | Kochi | Kochi08-1 | AB546123 |
| IV | 2008 | Jpn5-37* | - | AB546125 |
| IV | 2020 | 6145-1L | - | LC741368 |
| III | 2007–2008** | Saga | Saga08-1 | AB546124 |
| V | 2000 | Osaka | Jos1* | AB175699 |
| * Reference strains used for homology analysis in this study | ||||
| **The exact obtained year is not provided as the annotation | ||||
Table 2.
Reference sequences of non-Japanese-origin PRRSV strains used for phylogenetic analysis and their cluster classification.
Table 2.
Reference sequences of non-Japanese-origin PRRSV strains used for phylogenetic analysis and their cluster classification.
| Phylogenetic cluster |
Year of isolation | Country/ Region |
Name of isolate | Accession no. |
|---|---|---|---|---|
| I | 2006 | China | JXA1 | EF112445 |
| I | 2019 | USA | Fostera® PRRS* | Fostera® PRRS* |
| II | 1992 | USA | VR2332 | EF536003 |
| II | 1997 | USA | Ingelvac® PRRS MLV* | AF020048 |
| II | 2000 | China | BJ-4 | AF331831 |
| II | 2002 | South Korea | CNA18 | DQ473472 |
| II | 2005 | Taiwan | ML2 | EU273672 |
| II | 2006 | Taiwan | YL8 | EU273700 |
| IV | 1994 | Canada | IAF-EXP91 | L40898 |
| IV | 1994 | Canada | Quebec 807/94 | Z82995 |
| IV | 2001 | USA | MN184A | DQ176019 |
| IV | 2006 | USA | PRRSV0000008558 | EU758599 |
| V | 2006 | South Korea | LMY | DQ473474 |
| PRRSV-1 | 2019 | Spain | PRRSV1-Fag | MZ318699 |
| * Reference strains used for homology analysis in this study | ||||
| ** Year of submission to GenBank | ||||
Table 3.
Nucleotide sequence homology of PRRSV-2 ORF5 genes obtained between 2020 and 2023, compared with reference strains.
Table 3.
Nucleotide sequence homology of PRRSV-2 ORF5 genes obtained between 2020 and 2023, compared with reference strains.
| Cluster | Reference strain |
No. of Obtained sequences |
Homology distribution (%) |
Mean (%) |
Median (%) |
25th percentile (%) |
75th percentile (%) |
|---|---|---|---|---|---|---|---|
| I | Fostera® PRRS | 158 | 95.2-100 | 98.9 | 99.2 | 98.3 | 99.6 |
| II | Ingelvac®PRRS MLV | 221 | 88.1-100 | 98.9 | 99.6 | 98.8 | 99.7 |
| III | EDRD-1 | 577 | 85.3-96.7 | 90.0 | 90.5 | 87.4 | 91.9 |
| IV | Jpn5-37 | 219 | 79.5-95.3 | 84.7 | 84.9 | 84.2 | 85.2 |
| V | Jos1 | 2 | 82.7-88.5 | 85.6 | 85.6 | 84.1 | 87.0 |
| Unclassified | 13 | ||||||
| Total | 1190 |
Table 4.
Positivity rates of PRRSV-2 RT-PCR by production stage on pig farms in the Kanto region.
| Pig production stage | |||||||
| Gilt | Sow | Fetus | Piglet | Grower | Finisher | Unknown | |
| No. of positive samples / No. of samples |
35/61 | 70/249 | 9/10 | 49/116 | 601/927 | 85/262 | 27/45 |
| Positivity rate (%) |
57.4% | 28.1% | 90.0% | 42.2% | 64.8% | 32.4% | 60.0% |
Table 5.
Distribution of PRRSV-2 phylogenetic clusters by production stage on farms in the Kanto region.
Table 5.
Distribution of PRRSV-2 phylogenetic clusters by production stage on farms in the Kanto region.
| Pig production stage | ||||||||||||||
| Cluster | Gilt (%) (n/N) |
Sow (%) (n/N) |
Fetus (%) (n/N) |
Piglet (%) (n/N) |
Grower (%) (n/N) |
Finisher (%) (n/N) |
Unknown (%) (n/N) |
|||||||
| I | 0.0% | (0/7) | 0.0% | (0/23) | 33.3% | (1/3) | 3.6% | (1/28) | 17.9% | (82/459) | 50.0% | (17/34) | 36.4% | (8/22) |
| II | 0.0% | (0/7) | 0.0% | (0/23) | 0.0% | (0/3) | 10.7% | (3/28) | 11.5% | (53/459) | 2.9% | (1/34) | 4.6% | (1/22) |
| III | 28.6% | (2/7) | 17.4% | (4/23) | 0.0% | (0/3) | 35.7% | (10/28) | 52.3% | (240/459) | 11.8% | (4/34) | 40.9% | (9/22) |
| IV | 71.4% | (5/7) | 82.6% | (19/23) | 66.7% | (2/3) | 50.0% | (14/28) | 18.3% | (84/459) | 35.3% | (12/34) | 18.2% | (4/22) |
| V | 0.0% | (0/0) | 0.0% | (0/0) | 0.0% | (0/0) | 0.0% | (0/0) | 0.0% | (0/0) | 0.0% | (0/0) | 0.0% | (0/0) |
| *Values are expressed as percentages. N indicates the total number of sequences obtained for each production stage, and n denotes the number of sequences classified into each phylogenetic cluster within the corresponding stage. | ||||||||||||||
Table 6.
Comparisons of MLV vaccine usage among farms by the PRRSV-2 cluster in the Kanto region.
| Cluster | Sequences from farms using MLV | Sequences from farms not using MLV | Sequences from farms with unknown MLV status | Total |
| I | 109 | 0 | 0 | 109 |
| II | 58 | 0 | 0 | 58 |
| III | 213 | 55 | 1 | 269 |
| IV | 26 | 110 | 4 | 140 |
| Total | 406 | 165 | 5 | 576 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



