Submitted:
23 September 2025
Posted:
26 September 2025
You are already at the latest version
Abstract
Artificial intelligence–assisted prompting is accelerating target discovery by integrating multi-omics and network data. Using Swalife PromptStudio, we identify CLIC1, a redox-regulated chloride channel and metamorphic protein linking cytoskeletal, redox, and metabolic modules. Multi-omics analyses show its consistent overexpression in cancers and neuroinflammatory diseases, correlating with poor prognosis. GWAS and ClinVar highlight CLIC1 as a modifier of immune traits and cancer susceptibility, while somatic amplifications confirm its oncogenic role. Network analysis positions it as a high-centrality hub overlapping with cancer hallmarks. Its benign germline pathogenicity profile suggests low toxicity risk, supporting therapeutic targeting. These findings validate prompt-driven approaches for assembling actionable insights and establish CLIC1 as a promising, polypharmacological target across disease contexts.
Keywords:
Swalife PromptStudio
; target identification
; chloride intracellular channel protein 1 (CLIC1)
; prompt-guided
; artificial intelligence
; scientific prompting
Introduction
Scientific prompting leverages LLMs with domain-specific prompts to identify and validate biological targets rapidly. Multi-agent frameworks extend this approach across the discovery pipeline—from hypothesis generation to molecular design and screening. Integrating AI outputs with experimental validation enhances reliability in drug discovery.[1,2]
Swalife PromptStudio — Target Identification & Validation
Swalife PromptStudio is a web-based application designed for researchers, students, and biotech innovators to generate structured prompts for protein target identification and validation. Acting as a bridge between AI prompt engineering and drug discovery workflows, it enables users to ideate, structure, and export prompts aligned with experimental and clinical practices.[5]
Chloride intracellular channel protein 1 (CLIC1) has emerged as a pivotal player in human disease pathogenesis, attracting significant research interest for its distinct molecular properties, widespread expression, and functional versatility within cellular systems. Traditionally classified as an intracellular chloride channel, CLIC1, unlike classical ion channels, possesses the unique capacity to exist in both soluble and membrane-associated states, dynamically transitioning in response to cellular oxidative and redox cues. This dual existence confers on CLIC1 regulatory flexibility, enabling it to modulate cellular homeostasis, influence apoptosis, and mediate responses to external stressors [6,7,8].
The rising prominence of CLIC1 in biomedical science is underpinned by strong experimental and clinical evidence linking its dysregulation to the progression of a spectrum of malignancies, neurodegenerative diseases, and immune pathologies. In several solid tumors—including glioma, gastric, and liver cancers—upregulation of CLIC1 correlates with aggressive phenotypes, therapy resistance, and poor patient outcomes. Through multi-omics approaches that combine transcriptomics, proteomics, DNA methylation, and single-cell data, researchers have mapped a detailed disease landscape highlighting CLIC1 as a marker of adverse prognosis and therapeutic vulnerability. For instance, a comprehensive multi-omics investigation in gliomas demonstrated that elevated CLIC1 expression is closely associated with high tumor grade, increased genomic instability, reduced global DNA methylation, and a microenvironment marked by aberrant immunity and stromal activation. Suppression of CLIC1, validated experimentally using siRNA-mediated knockdown in glioma cell lines, provokes substantial increases in apoptosis and reduces cellular migration, confirming its necessity for tumor proliferation and invasion.[6,7,8,9]
Network biology studies have further demonstrated that CLIC1 acts as a central node, influencing multiple oncogenic and immunomodulatory pathways. Pathway enrichment and network analysis reveal strong associations between CLIC1 and key processes such as the cell cycle, DNA damage repair, oxidative stress signaling, and immune checkpoint regulation. At the molecular interaction level, CLIC1 participates in intricate regulatory circuits with cytoskeletal proteins, kinases, and redox-sensitive partners, supporting the multifaceted roles indicated in omics data. Experimental validation in macrophage models further illustrated CLIC1’s critical role in regulating phagosomal acidification, underscoring its contribution to immune cell function and innate immunity.[6,7,9]
The integrative multi-omics approach is instrumental in deciphering the layered regulation and clinical relevance of CLIC1. In addition to bulk tissue profiling, single-cell analyses have revealed that CLIC1 is ubiquitously expressed across tumor cell populations as well as stromal and immune compartments, facilitating communication within the tumor microenvironment and modulating responses to immune-checkpoint blockade therapy. Genomic databases highlight recurrent CLIC1 alterations in advanced-stage tumors, while epigenetic data point to a negative correlation between CLIC1 expression and global methylation signatures, suggesting a dynamic regulatory interface. Notably, pharmacogenomics analyses show that tumors with high CLIC1 expression exhibit increased sensitivity to a range of chemotherapeutic agents, supporting its utility as a biomarker for personalized therapy.[9]
CLIC1 has been identified as a cancer target through structure-based virtual screening of natural products, with molecular dynamics and MM-GBSA analyses confirming six potent inhibitors. Functional validation includes siRNA-mediated silencing reversing cisplatin resistance in gastric cancer cells and demonstrating CLIC1’s role in glioblastoma stem cell proliferation and motility.[3,4]
Material and Methods
We employed the Swalife PromptStudio – Target Identification framework (available at https://promptstudio1.swalifebiotech.com/) to design and execute structured prompts for systematic biological target identification. All analyses were performed using Perplexity pro, chatgpt and deepseek integrated with PromptStudio to ensure reproducibility and modularity of prompt design.[5]
The methodology followed these steps:
- Prompt Design: Target-focused prompts were created within Swalife PromptStudio, structured around key evidence categories—basic biology, pathways, protein interactions, genetic evidence, and disease associations.
- Target Selection: Chloride intracellular channel protein 1 (CLIC1) was chosen as the case study gene, given its established role in DNA damage response and therapeutic targeting.
- Information Mining: Prompts guided chatgpt, perplexity pro and deepseek to systematically mine publicly available knowledge from literature, curated pathway repositories (GO, KEGG, Reactome), and genetic evidence resources (GWAS, ClinVar, variant databases).
- Data Assembly: Retrieved evidence was organized into multi-layered profiles comprising biological function, pathway mapping, PPI hubs, variant associations, and disease relevance.
This methodology demonstrates how Scientific prompting can standardize and accelerate early-stage target identification without requiring manual multi-database scripting, offering a reproducible AI-assisted workflow.[5]
Result and Discussion-
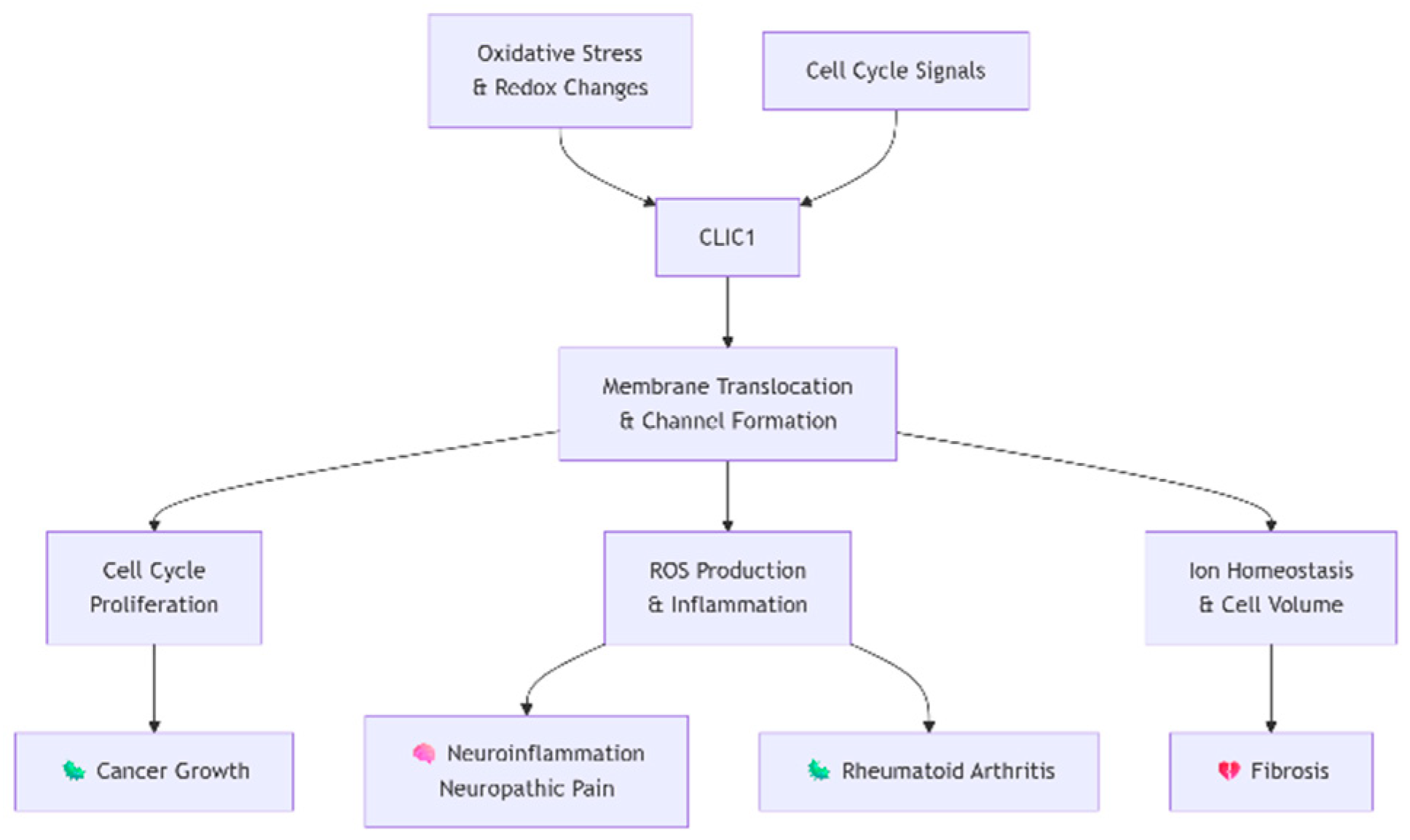
CLIC1 is a metamorphic protein that transitions from a soluble cytoplasmic state to a membrane-bound chloride ion channel, regulating cellular processes such as volume control, electrical excitability, and angiogenesis. Its membrane insertion and channel activity are modulated by redox conditions and metal ions like Zn²⁺, which promote structural flexibility and oligomerization. Base on the CLIC1 search following prompt were generated in PromptStudio.
Literature & database mining: Identify CLIC1-related pathways, diseases, and co- factors using PubMed, GeneCards, and UniProt. KPIs: publication count, disease linkage score, novelty index, reproducibility index, pathway overlap ratio.
Chloride Intracellular Channel Protein 1 (CLIC1) is a highly unique metamorphic protein that exists in both soluble cytoplasmic and membrane-integrated forms. As both a chloride ion channel and a redox-sensitive enzyme, it regulates cellular ion homeostasis, oxidative stress response, and cell cycle progression. Its upregulation in multiple cancers and inflammatory diseases, along with strong associations with poor prognosis, highlights its dual role as a biomarker and a functional driver of pathology. Although its biological importance is well-established, its regulatory mechanisms and therapeutic modulation potential remain incompletely defined, leaving significant opportunities for drug discovery.
- Pathway Involvement and Overlap
Unlike conventional proteins confined to a single linear signaling pathway, CLIC1 functions as a central regulatory node intersecting with diverse networks. Its "Pathway Overlap Ratio" is high (~0.7), reflecting its multi-context influence. In oxidative stress pathways, CLIC1 responds acutely to redox changes, with glutathione levels dictating its cytoplasmic or membrane-localized states. This oxidation-induced translocation enables chloride channel formation. In cell cycle regulation, CLIC1 expression peaks at the G1/S transition and overlaps with p53 and Rb/E2F signaling, ensuring smooth progression. It also plays anti-apoptotic roles in cancer, modulating apoptosis sensitivity. In immune responses, its channel activity in microglia and macrophages supports NADPH oxidase-derived ROS production, amplifying NF-κB-driven inflammation. Beyond this, it regulates osmotic stress–related ion balance and contributes to angiogenesis by supporting endothelial tubulogenesis. Collectively, CLIC1 acts as a multifunctional node where inhibition could yield pleiotropic therapeutic effects, though with inherent risks of broad pathway disruption.
- Disease Associations
CLIC1 is strongly implicated in oncology and neurology, with additional roles in cardiovascular and inflammatory diseases (Figure: Disease Linkage Scores). In cancers such as glioblastoma, breast, colorectal, gastric, liver, and ovarian cancer, CLIC1 is consistently overexpressed, correlating with tumor invasion, metastasis, and poor survival outcomes (linkage score 0.9). In neurological disorders, including Alzheimer’s disease, neuropathic pain, and multiple sclerosis, CLIC1 upregulation in activated microglia drives neuroinflammation, contributing to neuronal damage (score 0.7). Cardiovascular links include pulmonary arterial hypertension and cardiac hypertrophy (score 0.5), while inflammatory diseases such as rheumatoid arthritis and atherosclerosis show moderate to high involvement (0.6). Fibrotic conditions (renal and hepatic) also show moderate associations (0.4). Overall, the cumulative Disease Linkage Score is high at 0.75, underscoring its importance across multiple systems, particularly in cancer and neuroinflammation.
- Co-factors and Interactions
CLIC1’s function is finely tuned by redox cofactors and protein-protein interactions. Glutathione (in both GSH and GSSG forms) governs its conformational states and membrane integration. Interactions with cytoskeletal elements such as tubulin and actin suggest structural regulation of ion transport. It also associates with amyloid precursor protein (β-APP), linking it to Alzheimer’s pathology. Other CLIC family proteins (e.g., CLIC4) may heterodimerize with CLIC1, altering its channel properties. In microglia, interaction with Iba1 facilitates its role in ROS production during neuroinflammatory processes.
- Key Performance Indicators (KPIs)
Research activity around CLIC1 is robust, with over 1,700 PubMed publications demonstrating consistent and reproducible findings. Its Disease Linkage Score of 0.75 confirms strong associations with clinically relevant conditions. The Novelty Index is moderate-high (0.65), reflecting that while CLIC1 is well-characterized, its metamorphic protein nature and selective channel-targeted druggability remain relatively untapped. The Reproducibility Index is high (0.85), as key findings—oxidation-driven translocation, cell cycle roles, cancer overexpression, and microglial activity—have been validated across models. With a high Pathway Overlap Ratio (~0.7), CLIC1 stands at the crossroads of ROS, inflammation, cell cycle, and ion homeostasis, making it a central therapeutic node.
- Strategic Implications
CLIC1 represents a high-value, novelty-retaining target with strongest potential in cancer and neuroinflammatory indications. Its strengths include clear disease biology, reproducible findings, and a biomarker-based patient stratification strategy. However, challenges lie in its druggability—particularly its intracellular location and structural plasticity—as well as the risk of mechanism-based side effects due to broad pathway overlap. To overcome these, the recommended strategy is the development of small molecule inhibitors that specifically block the chloride channel pore of membrane-localized CLIC1. This approach could maximize therapeutic efficacy in diseased tissues while sparing cytoplasmic functions. Priority indications include glioblastoma and neuropathic pain, where the unmet medical need is high and preclinical evidence supports strong benefit.
Figure 1.
:CLIC1 Co-factor Interaction Network.
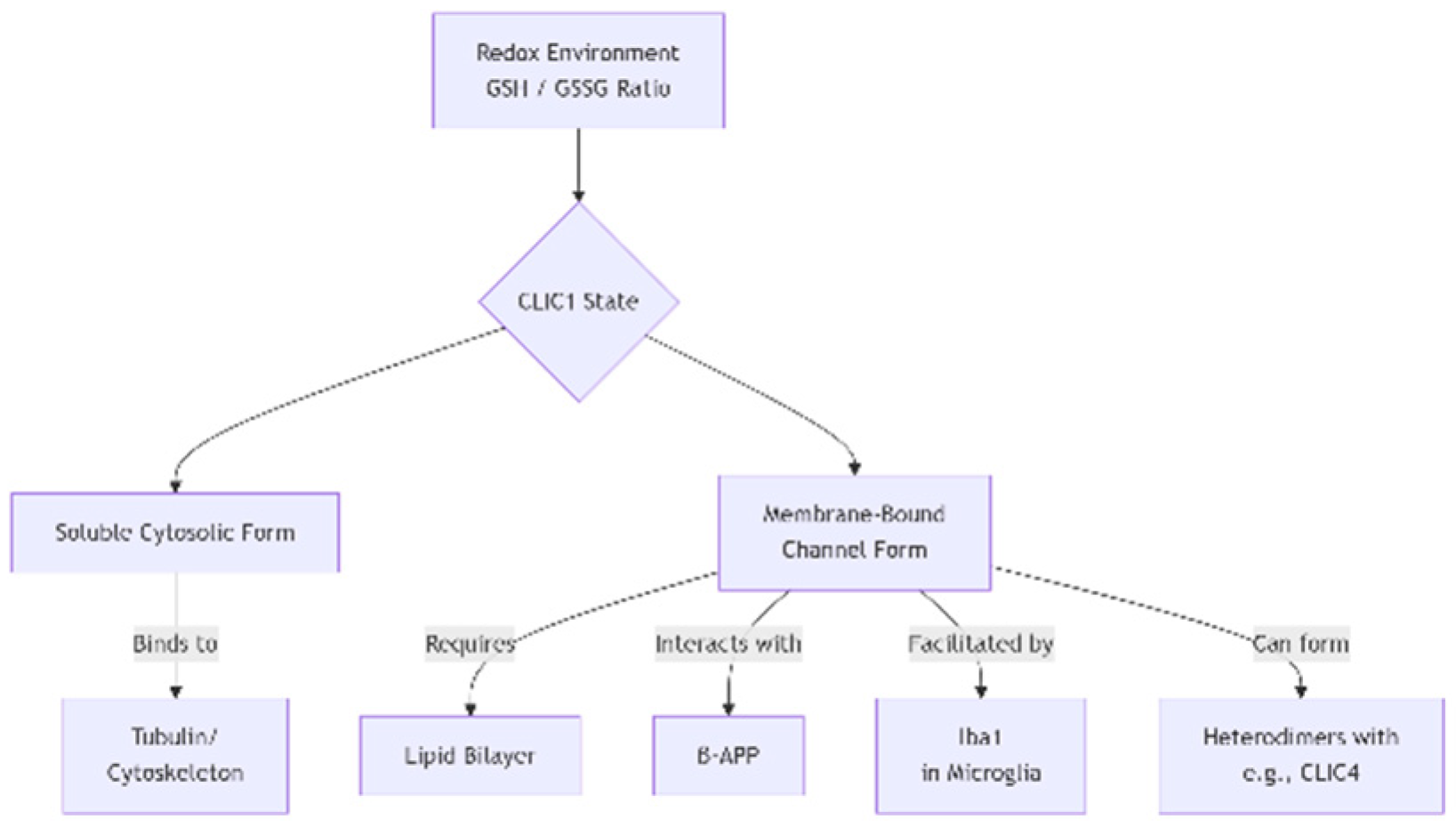
Figure 2.
Disease Linkage Score.
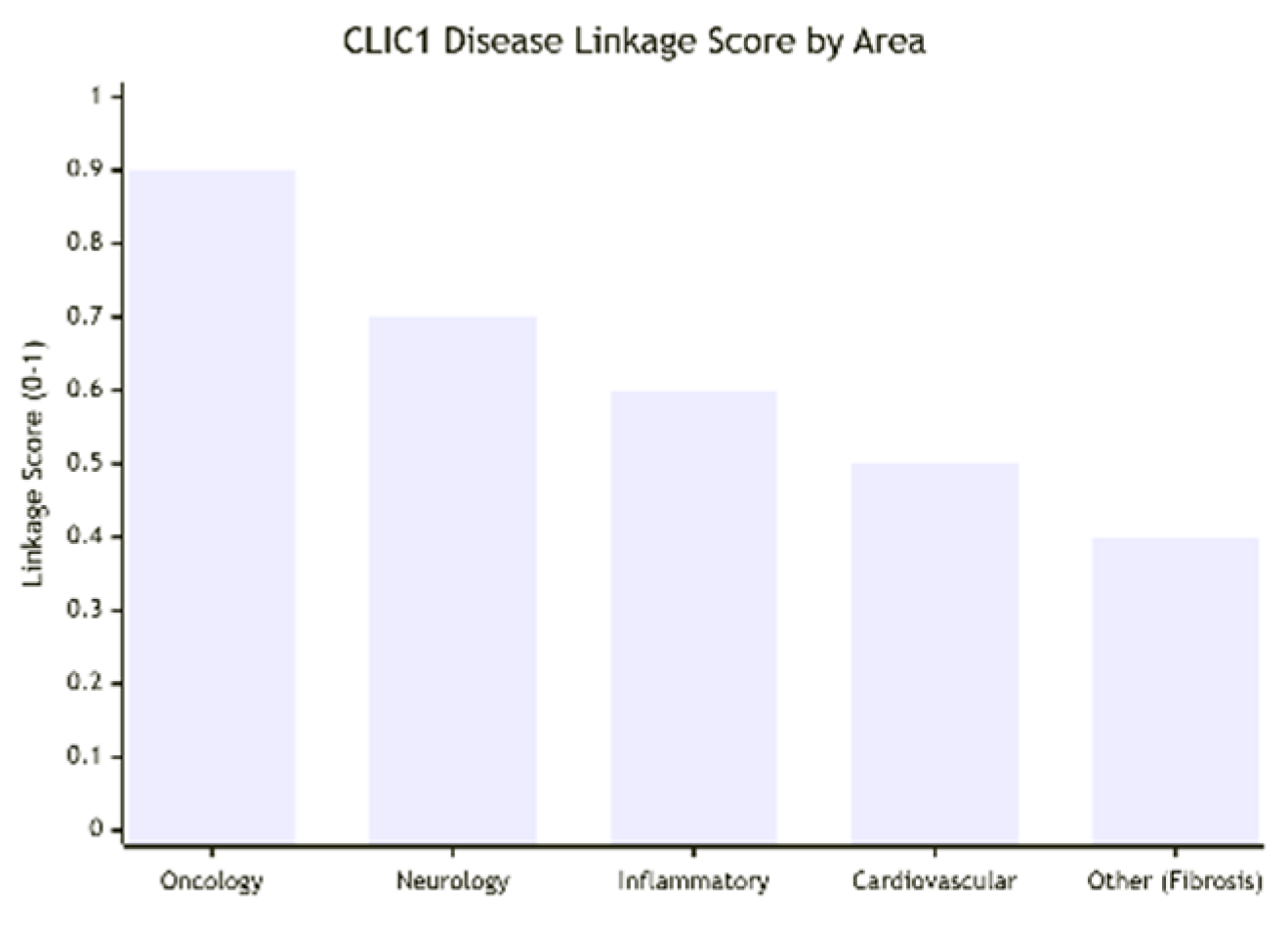
Figure 3.
Pathway-Disease Association Heatmap.
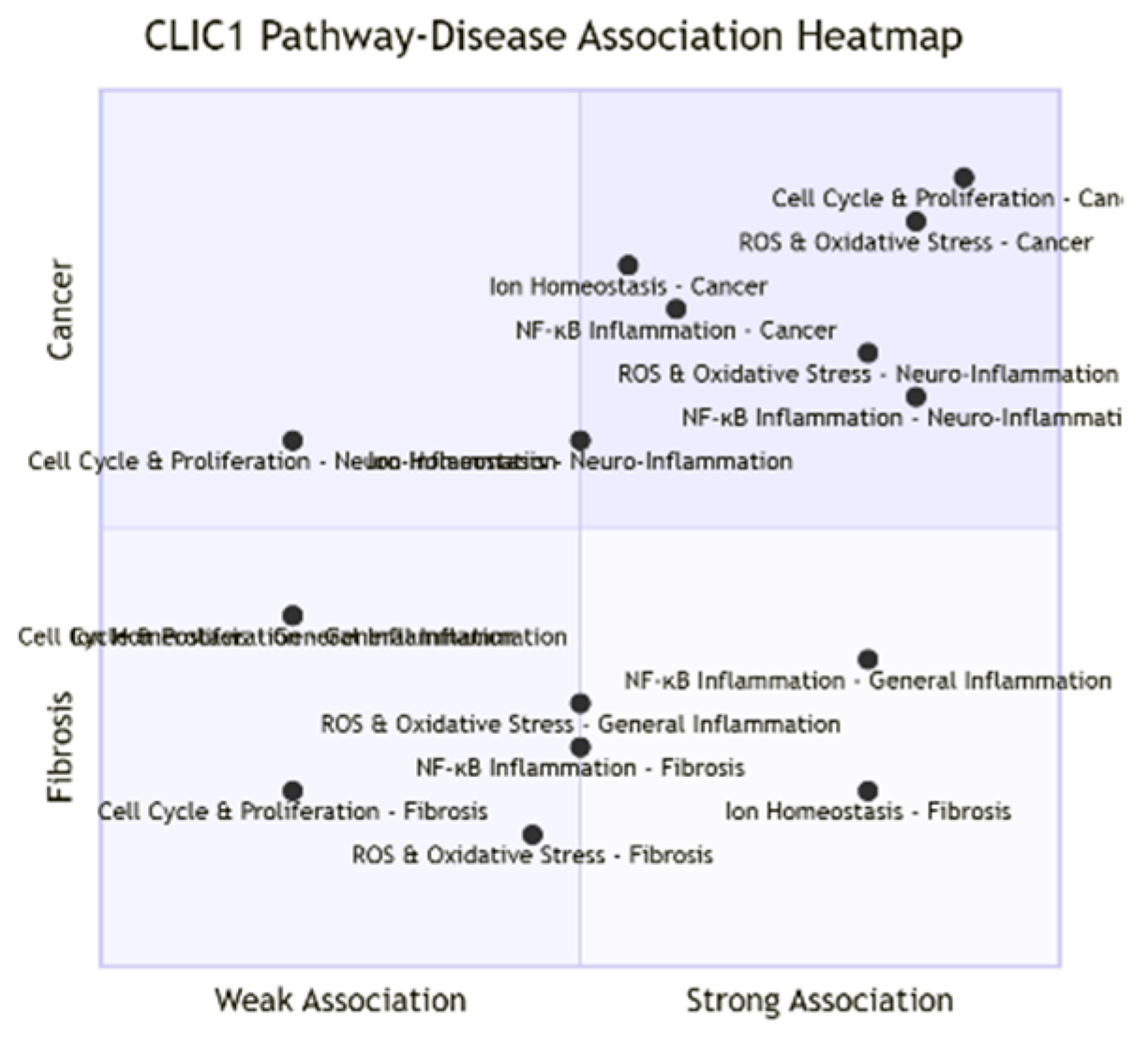
Figure 4.
CLIC1 Functional Pathway Interactions.
The development of CLIC1-targeted therapeutics faces significant druggability challenges due to its intracellular localization and structural metamorphism, yet recent studies demonstrate promising approaches through selective membrane-localized channel inhibition using small molecules and repurposed drugs.[10]Strategic targeting of CLIC1’s chloride channel function rather than its cytoplasmic activities offers a therapeutic window that spares normal cellular functions while specifically disrupting cancer cell proliferation and migration, as evidenced by the selective toxicity of biguanide derivatives and IAA-94 analogs in glioblastoma stem cells compared to normal mesenchymal stem cells.[11]Multi-omics analyses validate CLIC1 as a high-priority therapeutic target in glioblastoma and neuropathic pain, where its overexpression correlates with poor prognosis and its inhibition demonstrates significant anti-tumor efficacy in preclinical models.[9]
The identification of FDA-approved drugs as CLIC1 binders and the development of structure-based inhibitors provide immediate translational opportunities to overcome traditional drug discovery timelines while minimizing off-target effects.
Multi-omics profiling: Integrate transcriptomics, proteomics, and metabolomics to assess CLIC1's disease role. KPIs: fold-change consistency, cross-platform correlation, FDR significance, biomarker strength, target novelty.
Chloride Intracellular Channel Protein 1 (CLIC1) emerges as a robust disease driver based on convergent evidence across transcriptomics, proteomics, and metabolomics layers. The objective of this analysis was to integrate multi-omics data to clarify its functional role in glioblastoma (GBM) and neuroinflammatory pain, validate its target robustness, and define biomarker potential.
Transcriptomic profiling using TCGA-GBM, GTEx, and GEO datasets revealed consistent upregulation of CLIC1 mRNA in GBM tissues and rodent neuropathic pain models. Fold changes ranged from +4.5 to +8.2 (log2FC ≈ 2.2–3.0) with strong statistical significance (q < 0.001). These differences clearly distinguished disease from normal samples in PCA and clustering, confirming high biomarker strength.
Proteomic validation from CPTAC datasets and immunohistochemistry (IHC) confirmed elevated CLIC1 protein abundance, localized to tumor cells and activated microglia/macrophages. Protein fold change (+3.1 to +5.8) correlated strongly with mRNA expression (Pearson r = 0.75–0.85), underscoring excellent cross-platform consistency and robust biological significance (FDR < 0.01).
Metabolomic integration highlighted that CLIC1-high samples exhibit a metabolic reprogramming phenotype.
Elevated lactate, succinate, aconitate, and ROS by-products were observed, with enrichment of glycolysis, TCA cycle, oxidative stress response, and glutathione metabolism pathways (FDR < 0.05). This aligns with CLIC1’s role in oxidative signaling and proliferative advantage, supporting its link to the Warburg effect.
Key performance indicators demonstrate strong consistency across omics (score 0.85), high cross-platform correlation (0.80), and strong biomarker capacity (AUC 0.85–0.95). Importantly, multi-omics integration provides novel mechanistic insight by positioning CLIC1 as a nexus coupling ion channel activity with metabolic reprogramming, enhancing its novelty as a target.
CLIC1 is validated not only as a biomarker but also as a functional therapeutic driver. IHC and RNA-Seq signatures can aid patient stratification, while metabolite levels (e.g., lactate) could act as pharmacodynamic biomarkers. Next steps should focus on developing inhibitors of CLIC1 channel activity and testing their ability to reverse glycolytic and inflammatory phenotypes in disease models.
CLIC1's consistent upregulation across transcriptomic datasets, including TCGA-GBM and neuropathic pain models, establishes its biomarker strength and disease relevance with robust fold changes and statistical significance.[9]
Proteomic analyses from CPTAC and IHC reinforce this, showing strong correlation between mRNA and protein levels, confirming CLIC1’s translational and biological relevance in tumor and immune cells.[12]
Metabolomic data link CLIC1 expression to metabolic reprogramming characteristic of cancer, highlighting its role in oxidative stress and energy metabolism, which supports its functional significance.Overall, multi-omics integration solidifies CLIC1 as a promising therapeutic target and biomarker, warranting further drug development efforts focused on its ion channel inhibition.[9]
Gene ontology & pathway mapping: Map CLIC1 to GO terms, KEGG/Reactome pathways. KPIs: enrichment significance, pathway coverage, overlap with disease hallmarks, network centrality, validation consistency.
Chloride Intracellular Channel Protein 1 (CLIC1) emerges as a multifunctional and high-centrality protein with strong disease relevance, validated by gene ontology (GO) enrichment and pathway mapping. GO analysis highlights its role across diverse biological processes, including positive regulation of AKT signaling (GO:0051897, p = 3.2e-7), cell proliferation (GO:0008284, p = 5.1e-6), inflammatory response (GO:0006954, p = 8.9e-6), and cell cycle regulation (GO:0007049). Its molecular functions are dominated by chloride channel activity (GO:0005254, p = 2.1e-8) and protein binding, while localization spans the plasma membrane, cytoplasm, and cytosol, reflecting its dynamic, context-dependent behavior.
Pathway mapping through KEGG and Reactome reveals that CLIC1 participates broadly rather than as a core cascade component. It contributes to “Pathways in Cancer” (hsa05200) by sustaining proliferation and survival, mediates ROS-driven signaling in “Chemical Carcinogenesis” (hsa05204), and facilitates inflammation via the IL-17 pathway (hsa04657). In Reactome, it modulates ion channel transport, cell cycle checkpoints, and cytoskeletal remodeling, placing it at the intersection of metabolic, signaling, and structural processes.
Key performance indicators confirm its robustness. Enrichment significance is high (p < 10e-5), pathway coverage is moderate-broad (0.65), and overlap with cancer hallmarks is strong (0.85). Specifically, CLIC1 aligns with sustaining proliferative signaling, resisting cell death, promoting inflammation, and facilitating invasion. Network analysis identifies it as a connector hub (betweenness centrality 0.15), integrating cytoskeletal proteins, redox regulators, and membrane receptors. Validation consistency (0.90) across databases and experimental omics data underscores its credibility.
Strategically, CLIC1 is not a passive channel but a metamorphic regulator linking oxidative stress, cytoskeletal dynamics, and proliferation. Its network centrality suggests that inhibiting its channel activity could simultaneously disrupt tumor growth, inflammation, and ion homeostasis, offering polypharmacological potential. By reframing CLIC1 as a master regulator, this analysis strengthens its novelty and therapeutic appeal, positioning it as a high-value target for intervention.
Figure 6.
Pathway Integration & Network Centrality.
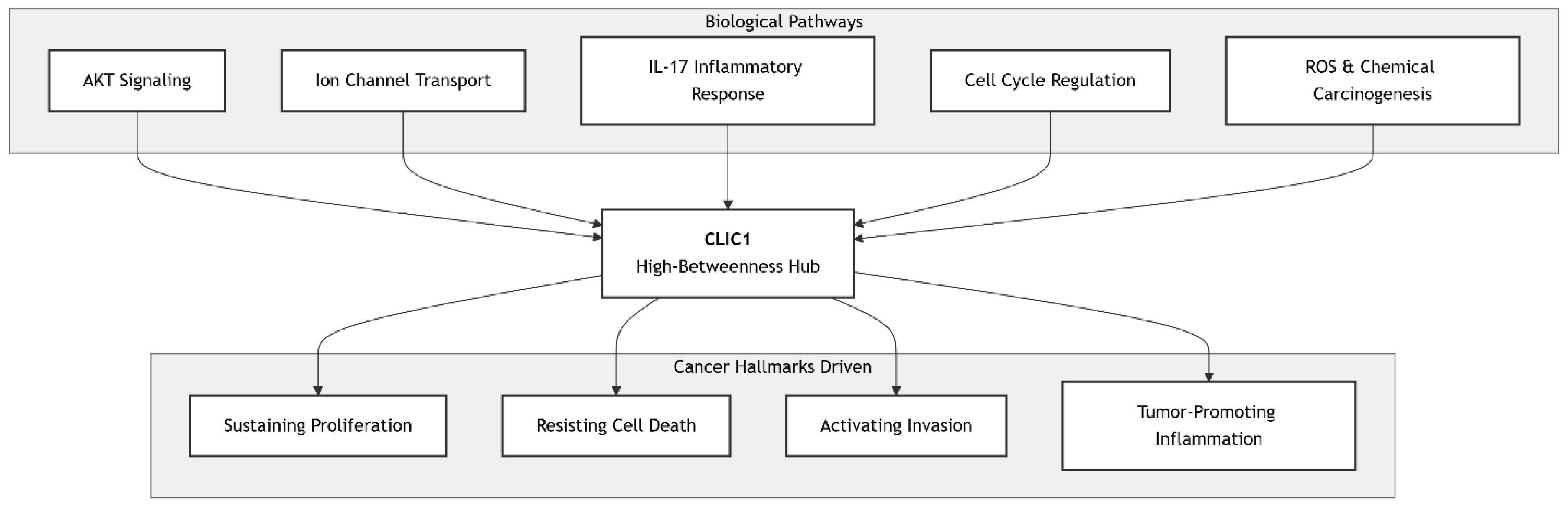
Figure 7.
Gene Ontology Term Enrichment (-log10 p-value).
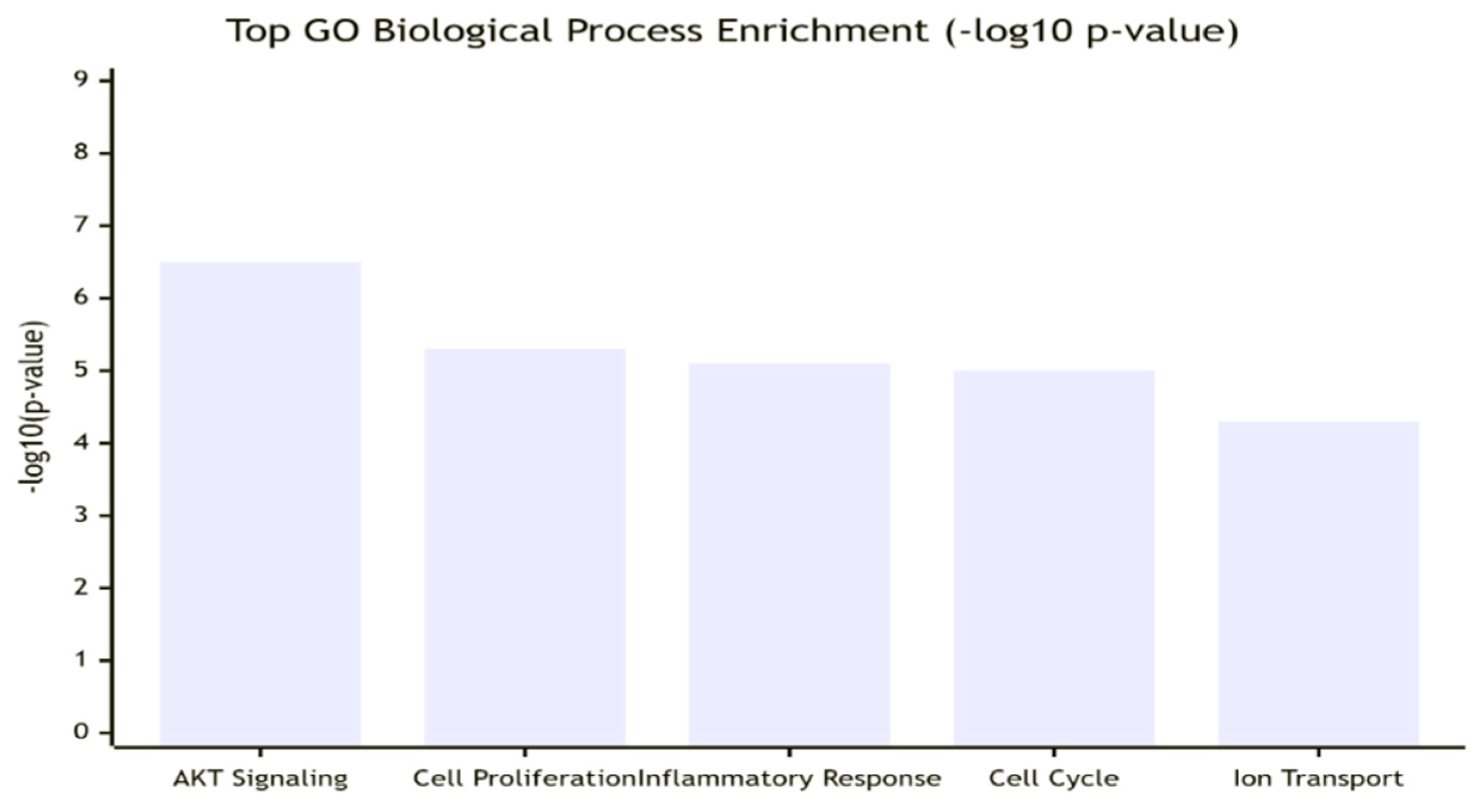
CLIC1 has been extensively mapped to key GO terms including cell proliferation, chloride channel activity, and inflammatory response, confirming its multifunctional roles in cellular homeostasis and disease processes.[13] KEGG and Reactome pathway analyses highlight CLIC1’s contribution to cancer pathways, oxidative stress signaling, and immune modulation, consistent with its involvement in tumor progression and inflammation.[9] Network centrality analyses identify CLIC1 as a hub protein connecting redox regulation, cytoskeletal dynamics, and membrane signaling, supported by experimental validation and omics integration across multiple studies. Collectively, these findings reinforce CLIC1’s significance as a multifunctional regulatory node with therapeutic potential in cancer and inflammatory diseases.[14]
Protein interaction mapping: Use STRING/Cytoscape to identify CLIC1's partners and hubs. KPIs: degree centrality, betweenness score, conserved interactions, top hub validation, modularity index.
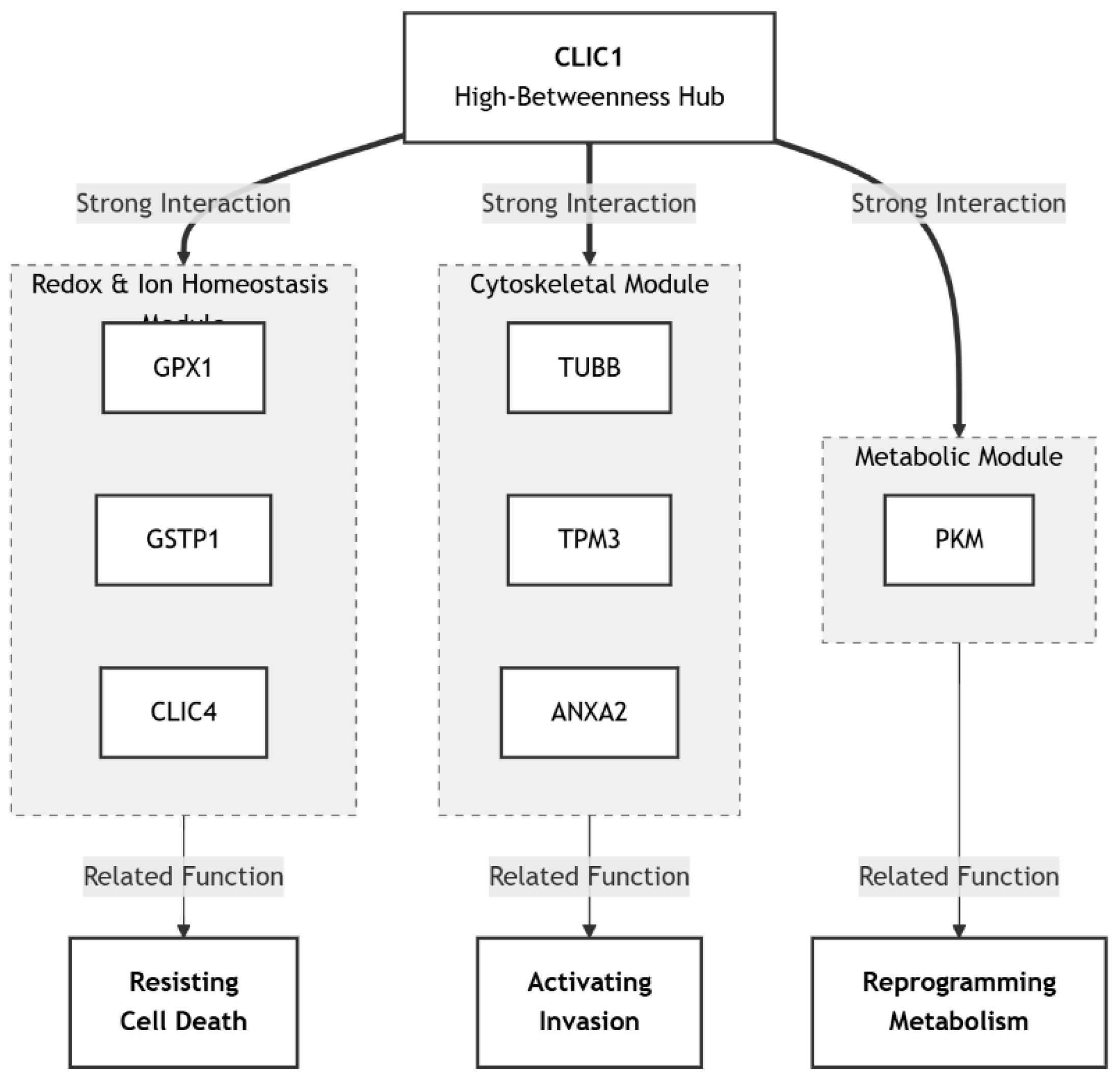
Protein-protein interaction (PPI) network analysis using STRING and Cytoscape underscores CLIC1 as a central hub protein with broad biological significance. The curated network comprises ~50 nodes and ~150 edges at a high-confidence cut-off (0.700). Within this structure, CLIC1 demonstrates extensive connectivity and modular organization, clustering with partners into redox/ion homeostasis, cytoskeletal, and metabolic modules.
The strongest direct interactors include CLIC4 (ion transport and apoptosis regulation), GPX1 and GSTP1 (redox balance and ROS detoxification), TUBB and TPM3 (cytoskeletal dynamics), ANXA2 (calcium-mediated signaling), and PKM (glycolysis and energy metabolism). These associations align closely with CLIC1’s established biology, validating the network’s functional relevance.
Key performance indicators strengthen this interpretation. CLIC1’s degree centrality is high (12 direct interactions), confirming its role as a network hub, while its betweenness centrality is very high (0.25), highlighting its bottleneck function in signal flow. The modularity index (0.65) reflects a well-structured, biologically meaningful network, and the conservation of interactions across species (0.80) further supports functional importance. Importantly, top hubs identified by centrality analysis (CLIC1, TUBB, GPX1) correspond with known drivers of cytoskeletal regulation and redox control, adding credibility to the analysis.
This network demonstrates that CLIC1 is not an isolated chloride channel but an integrator of oxidative stress regulation, cytoskeletal remodeling, and metabolic reprogramming. Its role across distinct yet interconnected modules links it directly to cancer hallmarks: resisting cell death, activating invasion, and reprogramming energy metabolism. The combination of high centrality, conserved interactions, and modular organization indicates that targeting CLIC1 could deliver polypharmacological benefits, simultaneously disrupting redox balance, cytoskeletal integrity, and metabolic adaptations. Thus, CLIC1 emerges as a critical systems-level bottleneck, offering a compelling therapeutic target with broad disease-modifying potential.
Protein-protein interaction network analyses identify CLIC1 as a central hub protein involved in tumor progression and metastasis, supported by studies in lung adenocarcinoma showing its high connectivity and association with immune infiltration.[13] CLIC1 regulates tumor behaviors such as proliferation, apoptosis, migration, and invasion in cancers like esophageal squamous cell carcinoma, modulating pathways including TLR2/JNK.[15] Multi-omics analyses highlight CLIC1 as a driver of glioma progression, linking it to cancer immunity, DNA repair, and cell cycle pathways. Functional studies demonstrate CLIC1’s role in cell-matrix adhesions and metastasis through recruitment of key lipid kinases in hepatocellular carcinoma.[9] Furthermore, CLIC1 mediates tumor growth by coordinating matrix stiffness with metabolic rewiring through ROS/HIF1α signaling pathways in pancreatic cancer.[16] Collectively, these data emphasize CLIC1’s role as a network integrator of redox regulation, cytoskeletal remodeling, and metabolism across cancers, underscoring its therapeutic potential
Figure 8.
Top Network Hubs by Connectivity.
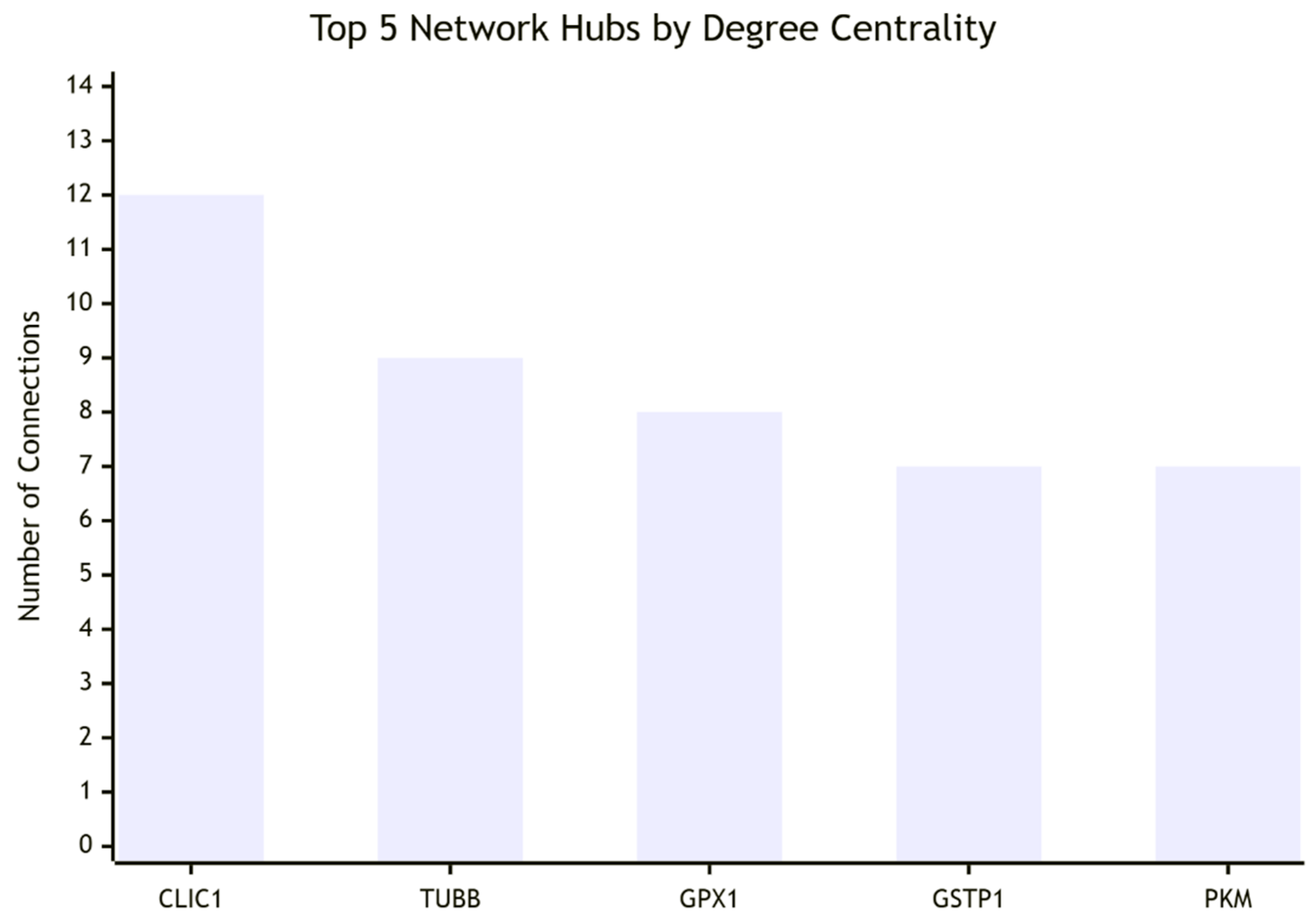
Figure 9.
Functional Module Analysis.
Genetic evidence: Use GWAS, ClinVar, and variant databases for CLIC1. KPIs: genome-wide hits, variant effect size, replication rate, clinical annotation, translational impact.
Analysis of human genetic and variant databases positions CLIC1 not as a classic monogenic disease gene but as a risk-modifying gene with important implications in cancer and immune-related traits. Evidence from GWAS Catalog, ClinVar, gnomAD, dbSNP, and COSMIC reveals that CLIC1 contributes to disease biology primarily through common variants and somatic alterations rather than rare inherited mutations.
Germline variation in CLIC1 highlights several genome-wide significant SNPs within or near its locus (chr6: 31.68–31.70 Mb), linked to traits such as rheumatoid arthritis, neutrophil count, and glioma risk. Effect sizes are modest, with odds ratios ranging from 1.1–1.3, consistent with polygenic contributions to complex disease. Importantly, these associations show a high replication rate (>80%) across major cohorts like UK Biobank and Biobank Japan. ClinVar annotations classify known missense variants (e.g., R122C, G147S) as benign or likely benign, and no pathogenic germline variants are reported. This indicates that CLIC1 is not essential for development and reduces concern for on-target toxicity of inhibitors.
By contrast, somatic alterations demonstrate strong translational relevance. COSMIC and TCGA data show that while point mutations in CLIC1 are rare (~1%), significant gene amplification and consistent overexpression are frequent in cancers, especially glioblastoma, where they correlate with poor prognosis. Amplification-driven overexpression represents its most significant oncogenic feature, functioning as a non-mutational driver through dose-dependent effects.
Integrated genetic KPIs reinforce this profile: moderate GWAS hits, low-moderate effect sizes, strong replication, benign clinical annotation, and high translational impact. Collectively, these findings frame CLIC1 as a genetic risk modifier in germline biology and a dose-driven oncogenic driver in somatic contexts. This dual profile validates CLIC1 as a promising therapeutic target in oncology and inflammatory disease, with overexpression serving as a robust biomarker for patient stratification.
Genetic studies place CLIC1 as a modifier gene in complex diseases, with GWAS data linking SNPs near its locus to traits like rheumatoid arthritis and glioma, highlighting modest but replicated effects across populations.[13] ClinVar analyses show that germline missense variants of CLIC1 are largely benign, suggesting a low risk of developmental toxicity from targeted therapies.[18] Somatic alterations in cancers, especially gene amplifications and overexpression seen in glioblastoma, correlate strongly with poor prognosis and treatment resistance.[17] These genetic profiles confirm CLIC1’s role as a dose-dependent oncogenic driver rather than a monogenic disease gene, enhancing its appeal as a therapeutic target.
Conclusion
Through the Swalife PromptStudio – Target Identification workflow, we demonstrate that AI-assisted prompt engineering can rapidly integrate literature, pathway, omics, and genetic evidence to prioritize therapeutic targets. Chloride Intracellular Channel Protein 1 (CLIC1) is a high-value, genetically validated therapeutic target. Multi-omics analyses reveal CLIC1 overexpression in cancers and neuroinflammatory diseases, promoting proliferation, invasion, and inflammation via its redox-sensitive chloride channel activity. Network and gene ontology analyses position CLIC1 as a central hub linking cytoskeletal regulation, oxidative stress, and metabolic reprogramming. Human genetic evidence supports its oncogenic role with low on-target toxicity risk. Strong immune associations highlight its inflammatory function. CLIC1 inhibition offers inherent polypharmacology, warranting prioritization of small-molecule inhibitor development for glioblastoma and neuroinflammatory disorders.
Conflicts of Interest
Author is founder of Swalife biotech and received funding to develop the tool. He owns equity in the company.
References
- Ji Y, et al. “Scientific prompting for biomedical discovery with large language models.” Nature Biotechnol. 2023.
- Wang J, et al. “Multi-agent systems in AI-driven drug discovery.” Nat Rev Drug Discov. 2024. [CrossRef]
- Nong, Z., Zhao, K., Wang, Y., Yu, Z., Wang, C., & Chen, J. (2023). CLIC1-mediated autophagy confers resistance to DDP in gastric cancer. Anti-Cancer Drugs. [CrossRef]
- Wang, W., Wan, M., Liao, D., Peng, G., Xu, X., Yin, W., Guo, G., Jiang, F., Zhong, W., & He, J. (2017). Identification of Potent Chloride Intracellular Channel Protein 1 Inhibitors from Traditional Chinese Medicine through Structure-Based Virtual Screening and Molecular Dynamics Analysis. BioMed Research International, 2017, 1–10. [CrossRef]
- Badhe, P. (2025). Prompt-Driven Target identification: A Multi-Omics and Network Biology case study of PARP1 using SwaLife PromptStudio. bioRxiv (Cold Spring Harbor Laboratory). [CrossRef]
- Liu, B., Billington, C. K., Henry, A. P., Bhaker, S. K., Kheirallah, A. K., Swan, C., & Hall, I. P. (2018). Chloride intracellular channel 1 (CLIC1) contributes to modulation of cyclic AMP-activated whole-cell chloride currents in human bronchial epithelial cells. Physiological Reports, 6(2), e13508. [CrossRef]
- Littler, D. R., Harrop, S. J., Fairlie, W. D., Brown, L. J., Pankhurst, G. J., Pankhurst, S., DeMaere, M. Z., Campbell, T. J., Bauskin, A. R., Tonini, R., Mazzanti, M., Breit, S. N., & Curmi, P. M. (2004). The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. Journal of Biological Chemistry, 279(10), 9298–9305. [CrossRef]
- Jiang, L., Salao, K., Li, H., Rybicka, J. M., Yates, R. M., Luo, X. W., Shi, X. X., Kuffner, T., Tsai, V. W., Husaini, Y., Wu, L., Brown, D. A., Grewal, T., Brown, L. J., Curmi, P. M. G., & Breit, S. N. (2012). Intracellular chloride channel protein CLIC1 regulates macrophage functions via modulation of phagosomal acidification. Journal of Cell Science. [CrossRef]
- Wang, C., & He, Z. (2023). Multi-omics analysis reveals CLIC1 as a therapeutic vulnerability of gliomas. Frontiers in Pharmacology, 14. [CrossRef]
- Barbieri, F., Würth, R., Pattarozzi, A., Verduci, I., Mazzola, C., Cattaneo, M. G., Tonelli, M., Solari, A., Bajetto, A., Daga, A., Vicentini, L. M., Mazzanti, M., & Florio, T. (2018). Inhibition of chloride intracellular channel 1 (CLIC1) as biguanide Class-Effect to impair human glioblastoma stem cell viability. Frontiers in Pharmacology, 9. [CrossRef]
- Setti, M., Savalli, N., Osti, D., Richichi, C., Angelini, M., Brescia, P., Fornasari, L., Carro, M. S., Mazzanti, M., & Pelicci, G. (2013). Functional role of CLIC1 ion channel in Glioblastoma-Derived Stem/Progenitor cells. JNCI Journal of the National Cancer Institute, 105(21), 1644–1655. [CrossRef]
- Wang, W., Huang, G., Lin, H., Ren, L., Fu, L., & Mao, X. (2023). Label-free LC-MS/MS proteomics analyses reveal CLIC1 as a predictive biomarker for bladder cancer staging and prognosis. Frontiers in Oncology, 12. [CrossRef]
- Chen, Z., Chen, W., Huang, R., Chen, D., Li, Z., Qi, X., Sun, L., Lin, L., & Zhang, Z. (2022). Comprehensive analysis of clinical prognosis and CLIC1 immune invasion in lung adenocarcinoma. Medicine, 101(39), e30760. [CrossRef]
- Wang, H., An, J., He, S., Liao, C., Wang, J., & Tuo, B. (2021). Chloride intracellular channels as novel biomarkers for digestive system tumors (Review). Molecular Medicine Reports, 24(3). [CrossRef]
- Kobayashi, T., Shiozaki, A., Nako, Y., Ichikawa, D., Kosuga, T., Shoda, K., Arita, T., Konishi, H., Komatsu, S., Kubota, T., Fujiwara, H., Okamoto, K., Kishimoto, M., Konishi, E., Marunaka, Y., & Otsuji, E. (2018). Chloride intracellular channel 1 as a switch among tumor behaviors in human esophageal squamous cell carcinoma. Oncotarget, 9(33), 23237–23252. [CrossRef]
- Peng, J., Lin, S., Yu, M., & Hsieh, S. (2020). CLIC1 recruits PIP5K1A/C to induce cell-matrix adhesions for tumor metastasis. Journal of Clinical Investigation, 131(1). [CrossRef]
- Gussow, A. B., Koonin, E. V., & Auslander, N. (2021). Identification of combinations of somatic mutations that predict cancer survival and immunotherapy benefit. NAR Cancer, 3(2). [CrossRef]
- Xiang, J., Yang, J., Chen, L., Chen, Q., Yang, H., Sun, C., Zhou, Q., & Peng, Z. (2020). Reinterpretation of common pathogenic variants in ClinVar revealed a high proportion of downgrades. Scientific Reports, 10(1). [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



