Submitted:
17 September 2025
Posted:
18 September 2025
Read the latest preprint version here
Abstract
Pro-fibrotic mediators derived from tubular cells have a central role in he development of kidney fibrosis. Our previous studies revealed that GEF-H1 (ARHGEF2)/RhoA signalling is a crucial regulator of tubular mediator release. Fibrotic stimuli elevate GEF-H1 expression and activity, but the molecular mechanisms controlling tubular GEF-H1 activity during fibrotic reprogramming are incompletely explored. Here we used immunoprecipitation and proximity ligation assay to show that GEF-H1 interacts with Myotonic Dystrophy Kinase-related Cdc42-binding kinase (MRCK)α in porcine and human tubular cells. Using GEF-H1 mutants we mapped the interacting domain to the N-terminus of GEF-H1 and showed the requirement for an intact DH domain. MRCKα silencing elevated GEF-H1 activity, induced GEF-H1-dependent RhoA activation and augmented stress fibre formation and phospho-cofilin levels. Interestingly, TNFα or TGFβ1 addition rapidly increased binding between GEF-H1 and MRCKα, suggesting a negative feedback role. Indeed, the effect of TNFα or TGFβ1 on GEF-H1 activation was augmented in the absence of MRCKα. Using an mRNA array, we found that MRCKα depletion elevated basal and TGFβ1-induced expression of key fibrosis-related genes. MRCKα silencing also promoted nuclear translocation of the profibrotic transcriptional co-activator Myocardin- related Transcription Factor (MRTF). Depletion of MRTF-A and B prevented the increase in ACTA-2 (smooth muscle actin) and transgelin (TAGLN), key markers of fibrotic reprogramming, induced by MRCKα-silencing and TGFβ1 treatment. Taken together, we identified MRCKα as a new suppressor of GEF-H1/RhoA/MRTF signaling and tubular fibrotic gene expression. Cytokines augment binding between the two proteins, thereby mitigating GEF-H1 activation in a negative feedback cycle. These effects could be crucial for preventing RhoA overactivation.
Keywords:
kidney fibrosis
; tubular cells
; RhoA signaling
; guanine nucleotide exchange factor
; ARHGEF2
; myocardin-related transcription factor
; fibrogenic mediators
; myotonic dystrophy-related Cdc42-binding kinase α
1. Introduction
Chronic kidney disease (CKD), a common consequence of diabetes and high blood pressure, is characterized by the gradual loss of kidney functions. The underlying pathology is hallmarked by chronic inflammation and tubulointerstitial fibrosis, i.e., excessive deposition of extracellular matrix (ECM) that destroys functional tissue [1]. The number of patients with CKD and kidney fibrosis is rapidly increasing, leading to a serious public health problem [2]. However, our understanding of the molecular pathogenesis of fibrosis remains incomplete. Activated fibroblasts and myofibroblasts that overproduce ECM, are key pathogenic executors in fibrosis. Importantly, accumulating evidence from many studies now also reveals the importance of cues derived from surrounding cells, including the tubular epithelium, in activation of these ECM producing cells (reviewed in [3,4,5]). Epithelial-mesenchymal crosstalk is mediated by factors released from tubular cells undergoing a pro-fibrotic phenotypic reprogramming, or partial epithelial-mesenchymal transition, leading to a pro-fibrotic epithelial phenotype (PEP) [6]. This process can be induced by injury and cytokines, including TNFα and TGFβ1 [7,8,9,10]). TGFβ1 is one of the most important inducers of fibrogenesis (e.g., [11]). We and others have highlighted the significance of RhoA signaling in tubular pro-fibrotic reprogramming [6,12,13,14]). Our data revealed early RhoA activation in tubular cells in a murine model of kidney injury and fibrosis [6] and following stimulation by inflammatory mediators [12,15]. Myocardin-related Transcription Factors (MRTF-A and B) are key co-activators of serum response factor (SRF) that act downstream from RhoA (reviewed in [16,17]). MRTFs shuttle between the cytosol and the nucleus, controlled by the polymerization state of actin. RhoA-induced F-actin polymerization induces their nuclear accumulation, where the MRTF/SRF complex binds to CARG domains in the promoters of fibrosis-related target genes and initiates their transcription [18,19]. Key MRTF-dependent tubule-derived fibrogenic mediators include Cellular communication network factor 2 (CCN2), also known as Connective Tissue Growth Factor (CTGF) and TGFβ1 itself [6].
RhoA dysregulation during inflammation and injury repair contributes to a vicious cycle leading to maladaptive repair and fibrosis [17,20]. Rho family proteins are controlled by a large network of guanine nucleotide exchange factors (GEF) that promote their activation, and GTPase activating proteins (GAP) that enhance inactivation [21,22]. We have identified GEF-H1 (ARHGEF2), as a key activator of RhoA in kidney tubular cells [15]. GEF-H1 is a Dbl family GEF highly expressed in epithelial cells [23,24]. Its dysregulation was associated with various pathological conditions, including cancer, organ fibrosis and kidney disease [13,25,26,27,28]. We have recently shown that GEF-H1 expression is elevated in a mouse kidney fibrosis model and in tubular cells stimulated by pro-fibrotic cytokines [12]. Thus, accumulating evidence points to a central role of GEF-H1 in fibrogenesis. GEF-H1 is controlled by interacting proteins that localize it to the microtubules and intercellular junctions, and by phosphorylation (e.g., [29,30,31,32,33]). It is activated in response to an array of stimuli, including various inflammatory and fibrogenic input, such as TGFβ1, TNFα and mechanical stress [15,28,34,35]. However, fibrosis-relevant control of GEF-H1 activity remains incompletely understood. In search for new regulators, in this study we identified Myotonic dystrophy-related Cdc42-binding kinase α (MRCKα/CDC42BPA), as an interactor of GEF-H1. MRCK belongs to the AGC (PKA, PKG and PKC) kinase family [36,37,38,39]. It has three isoforms, closely related to Rho kinase. MRCK family kinases control epithelial apico-basal polarization, cell migration and tissue remodeling [38]. However, their role in organ fibrosis or kidney disease remains unexplored.
In this study we show that MRCKα inhibits GEF-H1/RhoA signaling, thereby reducing cytoskeleton remodelling, MRTF nuclear translocation and MRTF-dependent fibrotic gene expression. Overall, our findings establish MRCKα as a new suppressor of the GEF-H1/RhoA/MRTF axis, with important implications for fibrogenesis.
2. Materials and Methods
2.1. Reagents and Antibodies
TGFβ1 was from MedChem Express (Monmouth Junction, NJ, USA, Cat#HY-P7118) or R&D Systems (Minneapolis, MN, USA, Cat#240-B-002). Tumor Necrosis Factor α (TNFα) was purchased from MedChem Express (Cat# HY-P7058). The following GEF-H1 antibodies were used: for immunoprecipitation: Cat# NBP2-21577, RRID: AB_3265603 (Novus, Centennial, CO, USA); for proximity ligation assay, mouse anti-GEF-H1 (Thermo Fisher Scientific, Waltham, MA, USA) Cat# MA5-27803, RRID:AB_2735201) and for western blotting: Cat# 4076, RRID: AB_2060032 (Cell Signaling Technologies, Danvers, Massachusetts, USA). MRCKα antibodies used were as follows: for immunoprecipitation and western blotting Cat# A302-694A, RRID:AB_10750425 (Thermo Fisher Scientific); and rabbit anti-MRCKα for proximity ligation assay: Cat#GTX10259 RRID:AB_1240609 (GeneTex Inc., Irvine, CA, USA). The following antibodies were from Cell Signaling Technology: FLAG (Cat# 8146, RRID: AB_10950495), RhoA (Cat# 2117S, RRID: AB_10693922), Phospho-Myosin Light Chain 2 (Thr18/Ser19) (E2J8F0) (Cat# 95777, RRID:AB_3677547), p-cofilin (Ser3) (Cat# 3313, RRID: AB_2080597), cofilin (Cat# 5175, RRID: AB_10622000), MRTF B (Cat# 14613, RRID: AB_2798539). Other antibodies used were: GFP (Cat# 66002-1-Ig, RRID: AB_11182611) and MRTF-A (Cat# 21166-1-AP, RRID:AB_2878822) from Proteintech (Rosemont, IL, USA); GAPDH (Cat# 39-8600, RRID: AB_2533438) from Thermo Fisher Scientific and α-smooth muscle actin (SMA) (Cat# ab150301, RRID:AB_3675461) from Abcam (Cambridge, MA). Secondary antibodies used for Western blotting were from Cell Signaling Technology: HRP-linked anti-rabbit IgG (Cat# 7074, RRID: AB_2099233) and HRP-linked anti-mouse IgG, (Cat#7076).
2.2. Cell Culture and Treatment
LLC-PK1, a kidney tubule epithelial cell line (male) was from the European Collection of Authenticated Cell Cultures (Wiltshire, UK), (ECACC Cat# 86121112, RRID: CVCL_0391). This cell line was used in our previous studies, e.g., ([12,40]). Human Embryonic kidney cells-293 (HEK-293) (female) (Cat# CRL-1573, RRID: CVCL_0045) was from the American Type Culture Collection (Manassas, VA, USA). Tissue culture media and reagents for culturing LLC-PK1 and HEK cells were from Thermo Fisher/Life Technologies. Both cell lines were maintained in a low glucose DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in an atmosphere containing 5% CO2. Unless otherwise stated, cells were serum depleted overnight before experiments. Human hTERT-immortalized renal proximal tubule epithelial cells (RPTEC/TERT1) (male) were from The American Type Culture Collection (CRL-4031). These cells were cultured in Minimum Essential Medium Eagle (Cat# M4526, Millipore/Sigma-Aldrich, Burlington, ON, Canada), supplemented with RPTEC Complete supplement (Cat# MTOXRCSUP) and Gentamicin (G1397). RPTECs were incubated in culture medium without the supplement overnight prior to experiments.
For cytokine treatment, cells were grown to 100% confluence, serum/supplement depleted as indicated, and TNFα (20 ng/ml) or TGFβ1 (10 ng/mL) was added for the indicated times.
2.3. Plasmid and Short Interfering RNA (siRNA) Transfection
FLAG-tagged GEF-H1 plasmids (WT and N-terminal GEF-H1 (1-443) in pCMV-Tag 2B and C-terminal GEF-H1 (444-985) in pCMV-Tag 2C vector) were a kind gift from Dr. H. Miki (Nishikyō-ku, Kyoto, Japan) [32]. GFP-GEF-H1-WT or GFP- GEF-H1-ΔC constructs were a kind gift from Dr. M. Kohno (Nagayo, Nagasaki, Japan). These constructs were generated as described in [41] by cloning into a pEGFP-C1 expression vector. GFP-GEF-H1-DH mutant was a kind gift from Dr. M. Bokoch (The Scripps Research Institute, La Jolla, CA). This mutant was prepared by site-directed mutagenesis to generate a Tyr to Ala mutation at residue 393 in the conserved QRITKY sequence in the DH domain of GEF-H1 [29]. MRCKα (CDC42BPA) cloned into the pReceiver-M56 vector containing an mCherry tag was purchased from GeneCopoeia (Maryland, Mid-Atlantic, USA Cat#EX-T0588-M56).
For immunoprecipitation experiments HEK-293 cells were grown in 10 cm dishes to 70-80% confluence and transfected with 4 µg of FLAG- or GFP-tagged GEF-H1 constructs using Fugene 6 transfection reagent (Cat# E2691, Promega Madison, WI, USA) following the manufacturer’s instructions. Cells were used 48 h post-transfection.
For siRNA-mediated silencing oligonucleotides were purchased from Dharmacon/Horizon Discovery (Lafayette, CO, USA). The non-related control siRNA (NR siRNA) was purchased from Dharmacon/Horizon Discovery (Cat nr D-001810-01-50) or MedChemExpress (Cat#HY-150150). Table 1 lists the siRNA sequences used in LLC-PK1 cells. For silencing proteins in the human RPTECs, the following predesigned siRNAs were used: ON-TARGETplus Human CDC42BPA (8476) siRNA (MRCK) cat# J-003814-14 and J-003814-15 (Horizon Discovery) and human ARHGEF2 siRNA (9181) cat #J-009883-06 and J-009883-07.
Cells were transfected using 100nM siRNA using Lipofectamine RNAiMAX transfection reagent (Cat#13778075, Thermo Fisher Scientific) following the manufacturer’s instructions. For MRTF A and B silencing, 50nM of each siRNA was used. Unless otherwise indicated, the experiments were performed 48 hours after transfection. For co-silencing GEF-H1 and MRCKα, the cells were sequentially transfected, as follows. First, they were transfected with GEF-H1 siRNA, and 24 hours or 6 hours later (as indicated in the legends), they were transfected with MRCKα siRNA for an additional 24h. Downregulation was routinely verified using Western blotting.
2.4. Immunoprecipitation (IP)
LLC-PK1 and HEK-293 cells were grown in 10 cm dishes. Where indicated, cells were transfected as described above. Cells were grown to 100% confluency, washed with ice-cold PBS and lysed with ice-cold NP-40 lysis buffer (150 mM NaCl, 30mM HEPES, 1% NP40, 0.25% Sodium deoxycholate, PH 7.5) containing1 mM Na3VO4 (Cat#0758, New England Biolabs, Whitby, ON, Canada), 1 mM phenylmethylsulphonyl fluoride (Cat# PMS444, Bioshop Burlington, ON Canada), protease and phosphatase inhibitors (cOmplete Mini, EDTA-free (cat# 4693159001) and PhosSTOP (cat# 4906845001) from Roche Diagnostics, Basel, Switzerland). Lysates were centrifuged at 12,000 rpm for 5 mins at 4°C. An aliquot of the supernatant was retained as input sample and the remaining supernatant was used for pre-clearing. For precipitating endogenous proteins, Pierce™ Protein A/G Agarose beads (Cat#PI20421, Thermo Fisher Scientific) were blocked by incubating with 2% bovine serum albumin (BSA) in NP40 lysis buffer for 1 hour at 4°C in a rotator, followed by washes with NP40 lysis buffer. The supernatants from the cell lysates were precleared for 1 hour at 4°C in a rotator using the A/G Agarose beads, then incubated with 3 µg of GEF-H1, or MRCKα antibody (1h), followed by incubation with blocked Pierce™ Protein A/G Agarose beads (3h).
For precipitating FLAG- and GFP-tagged GEF-H1, lysates were precleared using ChromoTek Binding Control Agarose Beads (Cat# bab, Proteintech), and immunoprecipitation was performed using ChromoTek DYKDDDDK Fab-Trap® Agarose (Cat#ffa, RRID:AB_2894836) or ChromoTek GFP-Trap® Agarose (cat#:gta, RRID:AB_2631357) from Proteintech, respectively, for 3 hours at 4°C using a rotator.
For all IP, following indicated times, beads were washed with ice-cold NP-40 lysis buffer three times, resuspended and boiled in Laemmli Sample Buffer (Bio-Rad, Hercules, CA, USA, Cat# 1610737) and analysed by Western blotting.
For Mass spectrometry, cells were grown in 10 cm dishes, lysed as above, and lysates from 3 plates/sample were used for each condition. The IP was performed using the GEF-H1 antibody as above. Mass Spectrometry was performed by the Sparc Biocentre at The Hospital for Sick Children, Toronto, ON, Canada.
2.5. Western Blotting
Western blotting was done as in our previous studies (e.g., [12]). Briefly, following the indicated treatments, cells were washed, lysed with ice-cold lysis buffer (100 mM NaCl, 30 mM HEPES pH7.5, 20 mM NaF, 1 mM EGTA, 1% Triton X-100) supplemented with 1 mM Na3VO4, 1 mM phenylmethylsulphonyl fluoride, and protease and phosphatase inhibitors, as for the NP40 lysis buffer used for IP. Lysates were centrifuged and protein concentration determined using bicinchoninic acid assay (Thermo Fisher Scientific/ Pierce Biotechnology). Protein was separated by SDS polyacrylamide gel electrophoresis and transferring to nitrocellulose membrane using standard protocols. The blots were blocked in Tris-buffered saline with Tween (TBST) containing 3% BSA for 1h, then incubated with the indicated primary antibodies overnight at 4°C, followed by washes, incubation with the corresponding peroxidase-conjugated secondary antibodies and visualization with the enhanced chemiluminescence (ECL) method (Cat#1705060, Bio-Rad, Hercules, CA, USA). For multiple probing of the same blot, the blot was either stripped and re-probed or the membrane was cut horizontally and probed with different primary antibodies. ECL signals were captured using a BioRad ChemiDoc Imaging system. Densitometry was performed using ImageLab. Values were normalized using GAPDH as loading control, and expressed as fold change from control, taken as unity.
2.6. GEF-H1 and RhoA Activation Assay
Active GEF-H1 and RhoA (GTP-bound) were captured from cell lysates using GST-RhoA(G17A) or GST-RhoA-binding domain (RBD), amino acids 7–89 of Rhotekin, respectively as described in [15,42]. RhoA(G17A) cannot bind nucleotide and therefore has high affinity for activated GEFs [43]. Confluent LLC-PK1 cells were lysed with ice-cold assay buffer (100 mM NaCl, 50 mM Tris base (pH 7.6), 20 mM NaF, 10 mM MgCl2, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS) containing 1 mM Na3VO4 and cOmplete Mini, EDTA-free protease inhibitor. After centrifugation, an aliquot of the supernatant was retained to sample input (total GEF-H1 or RhoA). The remaining supernatants were incubated with 20-25 µg of GST-RhoA(G17A) or GST-RBD beads (45 min, at 4°C), followed by extensive washes. Total and captured (active) proteins were analyzed by Western blotting and quantified by densitometry, as previously described ([12]). Precipitated (active) GEF-H1 or RhoA for each condition was normalized using the corresponding total protein, and the fold change relative to control was calculated.
2.7. Immunofluorescence Microscopy
LLC-PK1 cells grown on coverslips were transfected and treated as indicated. Cells were then fixed with 4% paraformaldehyde, washed with PBS, permeabilized and quenched with 0.1% Triton-PBS containing 100mM glycine. Following washing, coverslips were blocked with 3% BSA in PBS, then incubated with primary antibody (1:100) for 1 hour. Next, coverslips were washed and incubated with Alexa Fluor® 488-conjugated anti-rabbit IgG (H+L), F(ab’)2 Fragment secondary antibody (1:1000, Cell Signaling Technology, Cat# 4413, RRID: AB_10694110) and 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) (1:1000) (Cat# 10236276001, Roche Diagnostics). For F-actin staining, fixed, quenched and permeabilized cells were incubated with Alexa Fluor® 488 Phalloidin (Cat# 8878, Cell Signaling Technology) for 30 mins along with DAPI. The slides were visualized either using a Zeiss Widefield Microscope (63x objective), or a WaveFX spinning-disk confocal microscope (Quorum Technologies, Guelph, Canada) with an ORCA-flash4.0 digital camera with Gen II sCMOS image sensor. For confocal images, maximum intensity projection pictures were generated from Z-stack using the Metamorph software. All parameters for image acquisition were kept constant. Control and treatment groups were processed in parallel at the same time under the same conditions.
2.8. Proximity Ligation Assay (PLA)
RPTEC/TERT1 cells grown on coverslips were fixed, washed and permeabilized as described in 2.7. The PLA assay was performed using the Duolink In Situ Orange kit (cat#DUO92102, Millipore/Sigma-Aldrich), as recommended by the manufacturer. Briefly, cells were blocked with the Duolink Blocking Solution (1h, room temperature). Primary antibodies (mouse anti-GEF-H1 (1:50) and rabbit MRCKα (1:500)) were diluted in the antibody diluent and incubated in a humidified chamber overnight at 4°C. Following washing, the PLA probes (Duolink In Situ PLA Probe Anti-Mouse MINUS and Anti-Rabbit PLUS, 1:5 dilution in antibody diluent) were added for 1h in a pre-heated humidified chamber at 37°C. Ligation was performed using Duolink Ligase (1:40 in ligation buffer, 30 min) at 37°C. The amplification reaction was performed using Duolink Polymerase (1:80, 100 min, 37°C). Cells were mounted with Duolink In Situ Mounting Medium containing DAPI and viewed using a WaveFX spinning-disk confocal microscope (Quorum Technologies, Guelph, Canada) with an ORCA-flash4.0 digital camera with Gen II sCMOS image sensor. Maximum intensity projection pictures were generated from Z-stack using the Metamorph software. The PLA signal was quantified by counting the puncta in each microscopic field and dividing it by the number of nuclei. For each experiment 3-5 randomly selected fields/coverslips were counted.
2.9. RT2 Profiler Fibrosis Array
An RT2 Profiler™ Pig Fibrosis PCR Array was used to determine levels of 84 fibrosis-related genes (Cat# 330231, GeneGlobe ID: PASS-120Z, Qiagen (Montreal, QC, Canada). LLC-PK1 cells were transfected with 100 nM NR or MRCKα siRNA. 24 hours post-transfection, the medium was replaced with serum-free DMEM medium followed by treatment with 10 ng/ml TGFβ1 for 24 hours. RNA was extracted using the RNeasy Mini Kit (Cat# 74104, Qiagen) and 1.5µg of RNA was used for gDNA elimination and cDNA synthesis using the RT2 First strand kit (Cat# 330401, Qiagen). A the stock solution for each tested condition was prepared using RT2 SYBR Green ROX qPCR Mastermix (Cat# 330523, Qiagen), plated in the RT2 Profiler™ PCR Array and PCR was performed using a QuantStudio™ 7 Flex Real-Time PCR System (Thermo Fisher Scientific). Relative gene expression was calculated using the ∆∆Ct method, and fold changes from control expressed. Fold changes were inputted into the R Studio Statistical software (version 4.4.2, R core team 2023, and Heatmap R package version 1.0.12, “pheatmap” plugin) (https://CRAN.R-project.org/package=pheatmap) . Heatmaps show Z scores (indicating the number of standard deviations a data point is from the mean) of genes across treatment groups. Fold change of selected genes upregulated by MRCKα silencing in the presence or absence of TGFβ1 was also calculated and graphed.
2.10. RNA Extraction and RT-PCR
LLC-PK1 cells were transfected with the indicated siRNAs. Where indicated, 24 hours post-transfection, the medium was replaced with serum-free DMEM medium and treated with 10 ng/mL of TGFβ1 for 24 hours. RNA was extracted using the RNeasy Mini Kit (Qiagen) and 1µg of RNA was converted to cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad, Cat# 1708890) according to the manufacturer’s instruction. SYBR green-based real-time PCR was performed using QuantStudio™ 7 Flex Real-Time PCR System. Peptidylprolyl isomerase A (PPIA) was used as the reference gene. Relative gene expression was calculated using the ∆∆Ct method. Table 2 lists the qPCR Primer sequences used in this study.
2.11. Statistical Analysis
Western Blot and immunofluorescent pictures shown are representatives of at least three independent experiments. The graphs were generated using GraphPad Prism software (version 10.2.3) and the data are presented as means ±S.D. of the number of independent experiments indicated in the figure legends (n). Statistical analysis was performed using GraphPad Prism. Data normalized to the control, taken as unity, were analysed using one-sample t-test, with 1 as the hypothetical value, and significance was depicted using * symbols. Non-normalized samples were compared using unpaired t-test or one-way ANOVA, as indicated in the legends. Significance was denoted using #. Significance indicated on the figures is as follows: one symbol (# or *) p<0.05; two symbols (## or **) p<0.01; three symbols (### or ***) p<0.001; four symbols (#### or ****) p<0.0001.
3. Results
3.1. Association Between MRCKα and the N-Terminus of GEF-H1
In our previous studies we identified GEF-H1 as a master regulator of RhoA signalling and fibrotic reprogramming in tubular cells (e.g., [12,13]). To find interactors that may regulate GEF-H1 in this context, we immunoprecipitated endogenous GEF-H1 from LLC-PK1 cells and submitted samples for Mass Spectrometry analysis. From this screen, MRCKα (CDC42BPA) was identified as one of the highest-ranked interactors of GEF-H1. To confirm this interaction, we immunoprecipitated endogenous GEF-H1 from LLC-PK1 cells and verified co-precipitation of MRCKα using western blotting (Figure 1A). Conversely, precipitation of endogenous MRCKα yielded well-detectable co-precipitation of GEF-H1, providing a double-sided verification of the interaction between the two proteins. Omitting the primary antibody during the IP resulted in no precipitated MRCKα or GEF-H1 (Figure 1A).
To further verify the association between GEF-H1 and MRCKα in tubular cells, we performed in situ proximity ligation assays (PLA) in human proximal tubule cells (RTPEC/hTERT). We used the Duolink PLA kit to visualize proximity of the specific antibodies against GEF-H1 and MRCKα. The presence of discrete red puncta indicates that the proteins are within <40nm (Figure 1B). The signal depended on the presence of the primary antibodies, and it was significantly reduced in cells transfecetd with an MRCKα siRNA. Taken together, these data verify in situ interaction between MRCKα and GEF-H1.
We next aimed at identifying the GEF-H1 segment required for binding to MRCKα. To this end, we used truncated mutants of GEF-H1, as summarized in Figure 1C. GEF-H1 contains a zinc finger-like motif (C1), a Dbl-homologous domain (DH), a Pleckstrin homology domain (PH) and a coiled-coil (CC) region [24]. The full-length (WT) or truncated FLAG- or GFP-tagged GEF-H1 constructs were transfected into HEK cells and IP-ed through their respective tags. As shown in Figure 1D and E, MRCKα strongly co-precipitated with both FLAG-tagged and GFP-tagged full length GEF-H1. Truncated mutants containing only the N-terminal portion (GEF-H1 1-443 and GEF-H1ΔC) also bound MRCKα (Figure 1C and D). In contrast, the C-terminal portion (GEF-H1 444-985) failed to precipitate MRCKα (Figure 1C). Since the N-terminal region contains the DH-domain, that is responsible for the GEF activity, we wondered if this domain was required for binding to MRCKα. We used a DH-domain mutant containing a Tyr to Ala mutation at residue 393, which renders it inactive [29]. Interestingly, this GEF-H1Y393A mutant did not bind to MRCKα (Figure 1E), suggesting a key role for an intact DH domain in mediating association of the two proteins.
3.2. MRCKα Inhibits GEF-H1-Mediated RhoA Activation
Having verified the interaction between MRCKα and GEF-H1, we next asked if MRCKα influenced GEF-H1 activity. We silenced MRCKα in LLC-PK1 cells (Figure 2A) or in human RPTEC/hTERT cells (Figure 2B), using two different MRCKα-specific siRNAs for each cell line. All siRNAs potently reduced MRCKα expression. GEF-H1 activity was detected using affinity precipitation with GST-RhoA(G17A), a mutant with high affinity for activated GEFs, as in our earlier studies (e.g., [15]). Silencing MRCKα resulted in a significant augmentation of GEF-H1 activity in both cell lines (Figure 2A, B). The two siRNAs yielded similar results in both cell lines, verifying that this effect was indeed due to MRCKα depletion. These data strongly suggest that in resting cells, MRCKα is an inhibitor of GEF-H1.
We next asked whether MRCKα silencing-induced GEF-H1 activation was sufficient to promote RhoA activation. The GST-RBD affinity precipitation assay revealed that MRCKα silencing indeed significantly increased RhoA activity (Figure 2C). Importantly, co-silencing GEF-H1 prevented RhoA activation induced by MRCKα downregulation, indicating that RhoA activation was mediated by GEF-H1.
3.3. MRCKα Silencing Augments GEF-H1 Dependent Stress Fiber Formation and Cofilin Phosphorylation
We next explored the effects of MRCKα on actin remodelling in tubular cells. MRCKα was silenced in LLC-PK1 cells with or without co-silencing of GEF-H1. Visualizing F-actin using fluorescent rhodamine revealed that MRCKα silencing induced a general increase in F-actin, with an elevated abundance of stress fibres and stronger peripheral staining (Figure 3A). The cells also appeared more elongated follwing MRCKα silencing. Co-silencing of GEF-H1 potently reduced this effect, showing that in resting cells MRCKα is an inhibitor of GEF-H1-dependent actin stress fibre formation. Efficient silencing was verified using Western blotting (Figure 3B). MRCK can directly phosphorylate myosin light chain (MLC) [44]. However, it could also have an indirect effect, through RhoA/Rho kinase, a pathway that is also a key regulator of pMLC[45]. To test how MRCKα silencing affects pMLC, we stained pMLC in cells transfected with MRCKα siRNA (Figure 3C). We found that pMLC was augmented in cells where MRCKα was silenced. Co-silencing GEF-H1 indeed reduced this effect, showing that MRCKα depletion enhances MLC phosphorylation through GEF-H1/RhoA/Rho kinase (Figure 3C). Another mechanism whereby RhoA could augment stress fibers is through Rho kinase dependent activation of LIM domain kinase (LIMK), that in turn phosphorylates and inactivates the F-actin severing protein cofilin. However, MRCK itself was shown to promote LIMK-dependent cofilin phosphorylation [46]. Therefore, we next assessed the overall effect of MRCKα silencing on cofilin phosphorylation. As shown on Figure 3D MRCKα depletion significantly increased phospho-cofilin levels, indicating that in tubular cells MRCKα is a suppressor of cofilin phosphorylation and actin polymerization.
3.4. MRCKα Exerts a Negative Feed-Back on GEF-H1 Activation
GEF-H1 is known to be activated by an array of inputs. To further understand the role of MRCKα in GEF-H1 regulation, we asked whether GEF-H1-activating inputs affect the interaction between MRCKα and GEF-H1. We repeated the PLA in cells with or without stimulation with TNFα or TGFβ1, two known activators of GEF-H1 (Figure 4A). Interestingly, we found that both of these stimuli significantly increased the number of puncta indicating enhanced interaction between the two proteins. This lead us to hypothesize that in addition to inhibiting basal GEF-H1, MRCKα might also exert a negative feedback that blunts stimulus-induced GEF-H1 activation. To test this assumption, we compared the magnitude of stimulus-induced GEF-H1 activation with or without silencing MRCKα in LLC-PK1 cells. In accordance with previously published data, both TNFα and TGFβ1 induced a well detectable activation of GEF-H1 (Figure 4B). As shown earlier (Figure 2), MRCKα silencing augmented basal GEF-H1 activity. Importantly, both stimuli induced significantly stronger GEF-H1 activation in the absence of MRCKα, supporting the existence of a negative feed-back loop exerted by MRCKα.
To substantiate this conclusion, we asked whether MRCKα overexpression can affect GEF-H1 activation. To this end, we overexpressed an mCherry-tagged MRCKα in LLC-PK1 cells along with an HA-tagged GEF-H1. This allowed us to follow activation of HA-GEF-H1 exclusively in cells that also overexpressed MRCKα. Activity of HA-GEF-H1 was followed using the RhoA(G17A) precipitation assay. As shown on Figure 4C, HA-tagged GEF-H1 was strongly activated by TGFβ1. In contrast, when MRCKα was also expressed along with HA-GEF-H1, TGFβ1-induced GEF-H1 activation was abolished. Taken together, these data substantiate that MRCKα can suppress both basal and stimulus-induced GEF-H1 activation.
3.5. MRCKα Silencing Elevates Expression of Fibrosis-Related Genes
Studies from us and others have implicated GEF-H1/RhoA signalling in pro-fibrotic epithelial reprogramming [6,25,26]. Since our above-described findings implicated MRCKα as an inhibitor of GEF-H1, we next asked if MRCKα also controlled expression of key pro-fibrotic genes in tubular cells. To address this, we used a Qiagen RT2 Profiler™ Pig Fibrosis PCR Array allowing assessment of the expression of 84 genes. LLC-PK1 cells were transfected with control (NR) or MRCKα-specific siRNA followed by treatment with or without TGFβ1. As shown on Figure 5, we noted changes in several genes upon MRCKα silencing. Figure 5A depicts results from the array expressed as Z scores, revealing difference from the mean calculated for each gene using all treatment groups. These data show that MRCKα depletion by itself altered expression of several genes, and also modulated the effects of TGFβ1. Figure 5B depicts genes that were at least 1.5 fold elevated upon MRCKα silencing, including CCN2/CTGF, endothelin-1 (EDN1) and SMAD7. Further, we observed a consistent increasing trend in TGF superfamily members TGFβ1 and 2 (TGFB1, TGFB2), and inhibin β (INHBE); ECM regulating proteins TIMP1 and 2, and matrix metalloproteinase 14 (MMP14); as well as Tissue-type plasminogen activator (PLAT). For these genes the fold changes varied in a gene-dependent manner between 1.3 and 10-fold. Importantly, many of the uregulated genes have been implicated as contributors to fibrosis. Since TGFβ1 is a key pro-fibrotic cytokine that is known to induce many of these genes, we also asked if MRCKα silencing altered the effects of TGFβ1. Figure 5C depicts genes that were at least 1.5 fold upregulated by TGFβ1 when MRCKα was silenced compared to TGFβ1 treatment in the control. The genes significantly upregulated included the apoptosis inhibitor BCL2; the ECM protein Collagen Type 1 α chain (COL1A2); integrin linked protein kinase (ILK) that mediates focal adhesion signalling; Latent Transforming Growth Factor Beta Binding Protein, a regulator of TGFβ access; TIMP1; and ACTA2 (α-smooth muscle actin), a marker of myofibroblast activation. Several other genes were consistently, upregulated, albeit the effect in this series did not reach significance. These include genes that were also upregulated by MRCKα silencing without TGFβ1 addition: EDN1, TGFB2 , as well TIMP3; the transcription factor CCAAT Enhancer Binding Protein Beta (CEPBP); angiotensinogen (AGT); and hepatocyte growth factor (HGF). Taken together, these experiments revealed that MRCKα controls the expression of a variey of fibrosis-related genes.
One of the strongly upregulated genes in these data sets was ACTA2, that encodes α-smooth muscle actin (α-SMA), a hallmark of the myofibroblast phenotype [47,48]. MRCKα silencing augmented the effect of TGFβ1 on ACTA2 by 4-10-fold in the three experiments (Figure 5C and D). In fact, the combined treatment of MRCKα silencing and TGFβ1 elevated ACTA2 close to a 100-fold compared to the control (Figure 5D). Western Blotting to detect α-SMA protein verified that the effect of TGFβ1 was strongly augmented by MRCKα depletion (Figure 5E). Thus, MRCKα suppresses the basal expression of a variety of genes involved in fibrosis and mitigates TGFβ1-induced gene expression.
3.6. MRCKα Silencing Promotes MRTF-Dependent Gene Transcription
MRTF-A and B are crucial fibrogenic transcription factors activated by RhoA signalling. In fact, several of the genes we found to be augmented by MRCKα silencing are known MRTF targets, suggesting that MRCKα might regulate MRTF. To test this assumption, we silenced MRCKα in LLC-PK1 cells, and explored effects on MRTF in the absence or presence of TGFβ1. Immunofluorescent staining visualizing MRTF-A (Figure 6A) revealed that in control cells MRTF-A was cytosolic or partially nuclear.
MRCKα depletion by itself augmented nuclear presence of MRTF-A. Thus, MRCKα indeed acts as a suppressor of MRTF nuclear accumulation. To explore the functional relevance of MRTF regulation by MRCKα, we depleted this protein with and without co-silencing of the two MRTF isoforms, MRTF-A and B. Figure 6B verifies successful downregulation of all three proteins. We selected typical MRTF-dependent fibrosis related genes, transgelin (TAGLN), α-SMA/ACTA2 and CCN2/CTGF, as in our earlier studies [6,14,19,49]. RT-qPCR analysis verified that TAGLN mRNA levels were augmented by MRCKα depletion alone (Figure 6C, left). Moreover, it also elevated levels of TAGLN in TGFβ1-treated cells (Figure 6C right graph). MRTF-A and B silencing strongly and significantly reduced this effect. Similarly, the combined effect of MRCKα and TGFβ1 on ACTA2 was prevented when MRTF-A and B were silenced (Figure 6D). Finally, we also explored effects on CCN2 mRNA. RT-qPCR analysis verified findings from the fibrosis array (Figure 5A-C), showing that MRCKα silencing significantly elevated the levels of CCN2 both in the absence and presence of TGFβ1 (Figure 6E). MRTF-A and B silencing showed a string trend towards reducing the effect, although the change did not reach significance, likely due to the fact that CCN2 is regulated by multiple transcription factors. Overall, our data strongly support an anti-fibrogenic role for MRCKα through suppressing of MRTF-dependent gene transcription.
4. Discussion
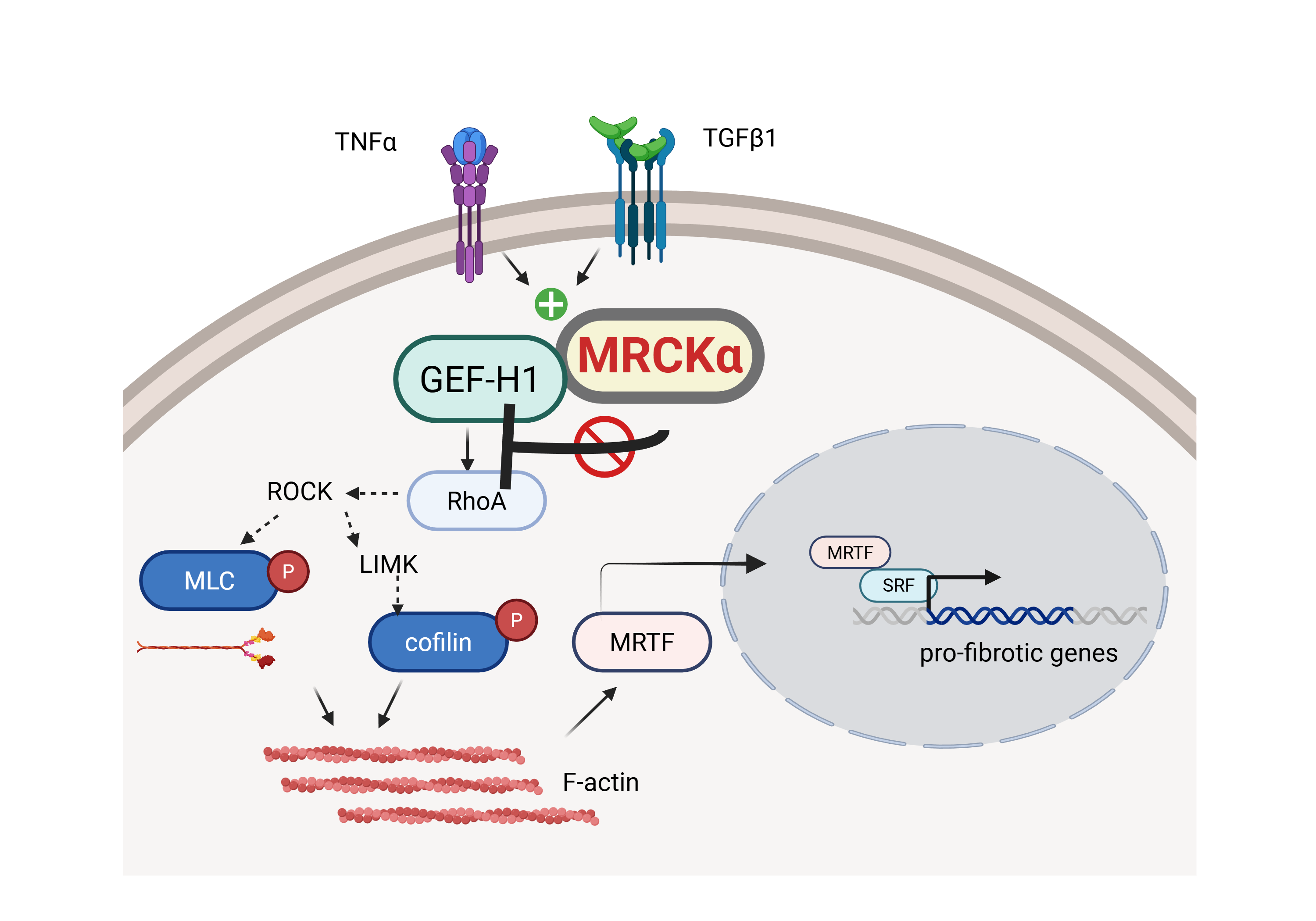
The key finding of this study is the demonstration that MRCKα interacts with and inhibits GEF-H1. We showed that MRCKα depletion augmented both basal and stimulus-induced GEF-H1 activity, promoted RhoA and MRTF activation and augmented TGFβ1-induced expression of several fibrosis-related genes. Taken together, our study identifies MRCKα as a new suppressor of the pro-fibrotic epithelial phenotype switch.
GEF-H1 is a ubiquitously expressed exchange factor for RhoA, RhoB and Rac, with vital roles in cytoskeletal organization, motility, epithelial polarization and permeability, as well as gene expression, including pro-fibrotic epithelial reprogramming (reviewed in [23,24]). Fine regulation of this protein is crucial for normal cellular functions, including epithelial homeostasis. Accordingly, GEF-H1 dysregulation was associated with various diseases. For example, GEF-H1-dependent signalling was reported to be upregulated in various cancers (e.g., [50,51,52,53,54]). Studies from us and others also implicated GEF-H1 in heart, kidney and eye disease and organ fibrosis [12,25,27,28]. These diverse physiological and pathological functions are fine-tuned in a complex and incompletely understood manner. Resting GEF-H1 activity is suppressed by binding to intercellular junction proteins and/or the microtubules [13,29,30,31,55,56,57]. Multiple phosphorylation sites in GEF-H1, that are targeted by a variety of kinases are also crucial for tight spatiotemporal control [23]. For example, ERK1/2 is an activating kinase that was implicated in mediating effects of an array of stimuli (e.g., [15,41]). Interestingly, however, the majority of kinases were shown to interact with GEF-H1 appear to be inhibitory. Examples include the PAK family [31,58], Protein kinase A [59], MARK2/Par1b [32,60] and cell cycle associated kinases such as Aurora A [61]. Thus, kinase-dependent suppression is a key mechanism of GEF-H1 activity control.
Our current study identifies MRCKα as a new interactor and inhibitor of GEF-H1. Using co-precipitation and in situ proximity ligation we verified interaction between MRCKα and GEF-H1 in kidney tubular cells. This finding is in line with a previous study that found these proteins in complex with BepC, an effector protein of the bacterium Bartonella [62]. Moreover, GEF-H1 and MRCKα co-precipitated even in the absence of BepC. However, the functional relevance of this interaction remained unclear, as MRCKα knockout did not affect BepC-induced cytoskeleton remodelling. In the current study we provide further details of the interaction between these proteins. First, we mapped the MRCK binding site to the N-terminal portion of GEF-H1. Second, we showed that the interaction required an intact GEF-H1 DH domain, since a point mutation of a conserved site within the DH domain (Y393A) completely abolished the binding. The GEF-H1 DH domain physically interact with RhoA, and harbors the GEF activity [24]. The Y393A mutant was shown to prevent interaction between GEF-H1 and RhoA [24]. Thus, one possibility is that MRCKα binding might mask the site for the GEF-H1-RhoA interaction, resulting in inhibition of the effect of GEF-H1 on RhoA. MRCKα may also be in a tripartite complex with GEF-H1 and RhoA, affecting RhoA activation. Alternatively, MRCKα may phosphorylate GEF-H1, leading to its suppression. Although in LLC-PK1 cells we have not been able to detect consistent changes in GEF-H1 phosphorylation upon MRCKα silencing, this may be due to techjnical limitations. Thus, further studies are warranted to define the exact mechanism, which may include MRCK-induced inhibitory GEF-H1 phosphorylation and/or hindrance of RhoA association or activation by GEF-H1.
Surprisingly, TNFα or TGFβ1, two cytokines known to activate GEF-H1 [15,28], increased the PLA signal, indicating that a larger pool of MRCKα and GEF-H1 were in close proximity after stimulation. This strongly suggests a negative feed-back mechanism, that may limit GEF-H1 activity and prevent its overactivation. This conclusion is supported by our data showing that MRCKα silencing augments not only basal, but also stimulus-induced GEF-H1 activation, and overexpression of a tagged MRCKα can mitigate TGFβ1-induced GEF-H1 activation.
Members of the MRCK family were identified as interactors of the Rho family small GTPase Cdc42 [39,44]. However, the exact role of Cdc42 in MRCK regulation remains unclear. Overexpression of active Cdc42 or silencing endogenous Cdc42 did not result in MRCKα activity changes [63,64]. Instead, Cdc42 binding likely controls MRCK localization, as Cdc42 downregulation was found to cause loss of lamellipodial MRCK in Hela cells [64]. On the other hand, expression of a kinase-dead MRCKα was shown to block some downstream effects of active Cdc42 [44]. Localized crosstalk between Rho family proteins has been documented in many systems, and MRCKα may represent a focal point for such interplay. Our finding that MRCKα is an inhibitor of GEF-H1/RhoA reveals an additional layer of complexity whereby Cdc42 might affect localized RhoA activity. Cdc42-dependent recruitment of MRCKα may lead to local inhibition of GEF-H1/RhoA signalling, for example in the lamellipodium of migrating cells, or during the development of epithelial polarity. Such crosstalk could also fine-tune perijunctional actomyosin contractility [65]. Interestingly, GEF-H1 appears to integrate input from Rac and Cdc42 through PAK family kinases, that are also effectors of these small GTPases. PAK1 was shown to phosphorylate and inactivate GEF-H1, at least in part by regulating its microtubule binding [31]. Further, in tubular cells we showed that TNFα-induced Rac activation promoted ERK-dependent stimulation of GEF-H1 towards RhoA [33], and GEF-H1 phosphorylation status can determine which small GTPAse (e.g Rac or RhoA) is activated downstream [33]. Together with the current study, these data further highlight the complext network of Rhio family regulation.
One key effect of MRCKα is the regulation of acto-myosin contractility. MRCKs can directly phosphorylate MLC, and can also control cofilin, a ubiquitous actin severing protein, through activating LIMK [46,66]. LIMK phosphorylates, and thereby inactivates cofilin, leading to increased actin polymerization [67]. Both LIMK and MLC activity, however, are also controlled by RhoA/Rho kinase, and the effects of MRCKα on through these pathways appear to be opposite. Thus, the overall effect on pMLC, cofilin and F-actin of MRCKα in a given cell likely depends on the balance of the direct activating and indirect inactivating (through RhoA/Rho kinase) effects. We found that in tubular cells, downregulating MRCKα increased pMLC. It also induced cofilin phosphorylation and augmented F-actin levels through GEF-H1. This latter effects is similar to what was reported in breast cancer cells, where MRCKα knockout was also found to augment cofilin phosphorylation [68]. Overall the cytoskeletal effecst we found in tubualr cells are in line with a suppressor role for MRCKα on RhoA-dependent cytoskeleotn remodelling and support the notion that MRCKα might also reduce MRTF avtivation, that is downstrem from F-actin.
GEF-H1 and RhoA are also key regulators of injury-induced epithelial reprogramming in tubular cells, and promote the production of pro-fibrotic mediators [6]. Reprogrammed epithelial cells release various mediators to activate mesenchymal cells and enhance ECM production, a key step in fibrosis. Indeed, our data show that MRCK silencing augments the production of the matricellular signaling protein CCN2 (CTGF), a key profibrotic mediator. MRCKα silencing by itself also augments other fibrosis-related genes, and promotes the effects of TGFβ1. We also showed that MRCKα altered MRTF nuclear translocation, and MRTF-dependent gene transcription (e.g., ACTA2 and TNGLN) [47]. However, the overall effect of MRCK is complex in this respect. While MRCKα silencing exerted a general positive effect on many genes we tested, a subset of genes showed reduced expression upon MRCKα silencing. Thus MRCKα may be a positive regulator of these . Further, MRCK silencing also significantly augmented SMAD7, that exerts a negative feed-back in TGFβ signalling and is considered anti-fibrotic [69]. The comprehensive effect of MRCKα on TGFβ1 signalling itself warrants further studies. Overall, future studies involving animal models should reveal whether MRCKα expression or activity are affected by fibrogenesis, and explore its overall role in fibrogenesis.
5. Conclusions
This study identified a new negative regulatory mechanism controlling GEF-H1/RhoA signalling under basal and cytokine-stimulated conditions. The MRCKα-GEF-H1 interaction modulates actin remodelling in tubular cells. Accordingly, MRCKα is a novel regulator of MRTF signalling, that plays a crucial role in fibrogenic tubular reprogramming. Taken together, our study identified MRCKα as a new suppressor of fibrosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Full blots used in the figures.
Author Contributions
Conceptualization, V.S..S, K.S and A.K.; methodology, V.S.S., Q.D., B.W., S.V.; formal analysis, V.S.S., Q.D., B.W., S.V.; investigation, V.S.S., Q.D., B.W., S.V.; resources, K.S.; data curation, V.S.S.; writing—original draft preparation, V.S.S and K.S.; writing—review and editing, K.S and A.K.; visualization, V.S.S, B.W., S.V. and K.S.; supervision, K.S.; project administration, V.S.S.; funding acquisition, K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the CANADIAN INSTITUTES OF HEALTH RESEARCH (CIHR) project grants to KS (PJT-180624 and PJT 180482), and a grant from the KIDNEY FOUNDATION OF CANADA (grant reference: 25KHRG-1416869). VSS was supported by a Keenan Post-Doctoral Scholarship from the St. Michael’s Foundation. KS is supported by a Keenan Professorship in Translational Medicine from the St. Michael’s Foundation.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Acknowledgments
The authors thank the Keenan Research Center Core facility scientists for their help with the experiments described in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CTGF | Connective tissue growth factor |
| CCN2 | Cellular Communication Network Factor 2 |
| GEF | Guanine Nucleotide Exchange Factor |
| CKD | Chronic kidney disease |
| ECM | Extracellular matrix |
| LIMK | LIM domain kinase |
| MRCK | Myotonic Dystrophy Kinase-related Cdc42-binding kinase |
| MRTF | Myocardin-Related Transcription Factor |
| NR | non-related |
| PLA | Proximity Ligation Assay |
| SMA | Smooth muscle actin |
| TAGLN | Transgelin |
| TGFβ1 | Transforming Growth Factor β1 |
| TNFα | Tumor Necrosis Factor-α |
References
- Panizo, S.; Martinez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martin-Carro, B.; Fernandez-Martin, J.L.; Naves-Diaz, M.; Carrillo-Lopez, N.; Cannata-Andia, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int J Mol Sci 2021, 22. [CrossRef]
- Ammirati, A.L. Chronic Kidney Disease. Rev Assoc Med Bras (1992) 2020, 66Suppl 1, s03–s09. [CrossRef]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct Target Ther 2023, 8, 129. [CrossRef]
- Li, L.; Fu, H.; Liu, Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat Rev Nephrol 2022, 18, 545–557. [CrossRef]
- Schiessl, I.M. The Role of Tubule-Interstitial Crosstalk in Renal Injury and Recovery. Semin Nephrol 2020, 40, 216–231. [CrossRef]
- Bialik, J.F.; Ding, M.; Speight, P.; Dan, Q.; Miranda, M.Z.; Di Ciano-Oliveira, C.; Kofler, M.M.; Rotstein, O.D.; Pedersen, S.F.; Szaszi, K.; et al. Profibrotic epithelial phenotype: a central role for MRTF and TAZ. Sci Rep 2019, 9, 4323. [CrossRef]
- Baker, M.L.; Cantley, L.G. Adding insult to injury: the spectrum of tubulointerstitial responses in acute kidney injury. J Clin Invest 2025, 135. [CrossRef]
- Li, Z.L.; Li, X.Y.; Zhou, Y.; Wang, B.; Lv, L.L.; Liu, B.C. Renal tubular epithelial cells response to injury in acute kidney injury. EBioMedicine 2024, 107, 105294. [CrossRef]
- Quaggin, S.E.; Kapus, A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int 2011, 80, 41–50, doi:ki201177 [pii] . [CrossRef]
- Chatterjee, A.; Tumarin, J.; Prabhakar, S. Cellular cross-talk drives mesenchymal transdifferentiation in diabetic kidney disease. Front Med (Lausanne) 2024, 11, 1499473. [CrossRef]
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-beta as a driver of fibrosis: physiological roles and therapeutic opportunities. J Pathol 2021, 254, 358–373. [CrossRef]
- Venugopal, S.; Dan, Q.; Sri Theivakadadcham, V.S.; Wu, B.; Kofler, M.; Layne, M.D.; Connelly, K.A.; Rzepka, M.F.; Friedberg, M.K.; Kapus, A.; et al. Regulation of the RhoA exchange factor GEF-H1 by profibrotic stimuli through a positive feedback loop involving RhoA, MRTF, and Sp1. Am J Physiol Cell Physiol 2024, 327, C387–C402. [CrossRef]
- Dan, Q.; Shi, Y.; Rabani, R.; Venugopal, S.; Xiao, J.; Anwer, S.; Ding, M.; Speight, P.; Pan, W.; Alexander, R.T.; et al. Claudin-2 suppresses GEF-H1, RHOA, and MRTF thereby impacting proliferation and profibrotic phenotype of tubular cells. J Biol Chem 2019, 294, 15446–15465. [CrossRef]
- Fan, L.; Sebe, A.; Peterfi, Z.; Masszi, A.; Thirone, A.C.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szaszi, K.; Mucsi, I.; et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol Biol Cell 2007, 18, 1083–1097. [CrossRef]
- Kakiashvili, E.; Speight, P.; Waheed, F.; Seth, R.; Lodyga, M.; Tanimura, S.; Kohno, M.; Rotstein, O.D.; Kapus, A.; Szaszi, K. GEF-H1 Mediates Tumor Necrosis Factor-{alpha}-induced Rho Activation and Myosin Phosphorylation: role in the regulation of tubular paracellular permeability. J Biol Chem 2009, 284, 11454–11466. [CrossRef]
- Miranda, M.Z.; Lichner, Z.; Szaszi, K.; Kapus, A. MRTF: Basic Biology and Role in Kidney Disease. Int J Mol Sci 2021, 22. [CrossRef]
- Small, E.M. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res 2012, 5, 794–804. [CrossRef]
- Sun, Q.; Chen, G.; Streb, J.W.; Long, X.; Yang, Y.; Stoeckert, C.J., Jr.; Miano, J.M. Defining the mammalian CArGome. Genome Res 2006, 16, 197–207, doi:gr.4108706 [pii] . [CrossRef]
- Lichner, Z.; Ding, M.; Khare, T.; Dan, Q.; Benitez, R.; Praszner, M.; Song, X.; Saleeb, R.; Hinz, B.; Pei, Y.; et al. Myocardin-Related Transcription Factor Mediates Epithelial Fibrogenesis in Polycystic Kidney Disease. Cells 2024, 13. [CrossRef]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol Lett 2020, 219, 15–26. [CrossRef]
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10. [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev 2013, 93, 269–309. [CrossRef]
- Joo, E.; Olson, M.F. Regulation and functions of the RhoA regulatory guanine nucleotide exchange factor GEF-H1. Small GTPases 2021, 12, 358–371. [CrossRef]
- Birkenfeld, J.; Nalbant, P.; Yoon, S.H.; Bokoch, G.M. Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF: is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol 2008, 18, 210–219. [CrossRef]
- Hu, Q.; Lai, J.; Chen, H.; Cai, Y.; Yue, Z.; Lin, H.; Sun, L. Reducing GEF-H1 Expression Inhibits Renal Cyst Formation, Inflammation, and Fibrosis via RhoA Signaling in Nephronophthisis. Int J Mol Sci 2023, 24. [CrossRef]
- Mills, C.; Hemkemeyer, S.A.; Alimajstorovic, Z.; Bowers, C.; Eskandarpour, M.; Greenwood, J.; Calder, V.; Chan, A.W.E.; Gane, P.J.; Selwood, D.L.; et al. Therapeutic Validation of GEF-H1 Using a De Novo Designed Inhibitor in Models of Retinal Disease. Cells 2022, 11. [CrossRef]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci Rep 2016, 6, 39383. [CrossRef]
- Tsapara, A.; Luthert, P.; Greenwood, J.; Hill, C.S.; Matter, K.; Balda, M.S. The RhoA activator GEF-H1/Lfc is a transforming growth factor-beta target gene and effector that regulates alpha-smooth muscle actin expression and cell migration. Mol Biol Cell 2010, 21, 860–870. [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 2002, 4, 294–301. [CrossRef]
- Aijaz, S.; D’Atri, F.; Citi, S.; Balda, M.S.; Matter, K. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev Cell 2005, 8, 777–786. [CrossRef]
- Zenke, F.T.; Krendel, M.; DerMardirossian, C.; King, C.C.; Bohl, B.P.; Bokoch, G.M. p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J Biol Chem 2004, 279, 18392–18400. [CrossRef]
- Yoshimura, Y.; Miki, H. Dynamic regulation of GEF-H1 localization at microtubules by Par1b/MARK2. Biochem Biophys Res Commun 2011, 408, 322–328, doi:S0006-291X(11)00606-1 [pii] . [CrossRef]
- Waheed, F.; Dan, Q.; Amoozadeh, Y.; Zhang, Y.; Tanimura, S.; Speight, P.; Kapus, A.; Szaszi, K. Central role of the exchange factor GEF-H1 in TNF-alpha-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol Biol Cell 2013, 24, 1068–1082. [CrossRef]
- Birukova, A.A.; Fu, P.; Xing, J.; Yakubov, B.; Cokic, I.; Birukov, K.G. Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am J Physiol Lung Cell Mol Physiol 2010, 298, L837–848, doi:ajplung.00263.2009 [pii] . [CrossRef]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol 2011, 13, 722–727. [CrossRef]
- Kale, V.P.; Hengst, J.A.; Desai, D.H.; Amin, S.G.; Yun, J.K. The regulatory roles of ROCK and MRCK kinases in the plasticity of cancer cell migration. Cancer Lett 2015, 361, 185–196. [CrossRef]
- Zhao, Z.; Manser, E. Myotonic dystrophy kinase-related Cdc42-binding kinases (MRCK), the ROCK-like effectors of Cdc42 and Rac1. Small GTPases 2015, 6, 81–88. [CrossRef]
- Zihni, C. MRCK: a master regulator of tissue remodeling or another ‘ROCK’ in the epithelial block? Tissue Barriers 2021, 9, 1916380. [CrossRef]
- Unbekandt, M.; Olson, M.F. The actin-myosin regulatory MRCK kinases: regulation, biological functions and associations with human cancer. J Mol Med (Berl) 2014, 92, 217–225. [CrossRef]
- Amoozadeh, Y.; Anwer, S.; Dan, Q.; Venugopal, S.; Shi, Y.; Branchard, E.; Liedtke, E.; Ailenberg, M.; Rotstein, O.D.; Kapus, A.; et al. Cell confluence regulates claudin-2 expression: possible role for ZO-1 and Rac. Am J Physiol Cell Physiol 2018, 314, C366–C378. [CrossRef]
- Fujishiro, S.H.; Tanimura, S.; Mure, S.; Kashimoto, Y.; Watanabe, K.; Kohno, M. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem Biophys Res Commun 2008, 368, 162–167. [CrossRef]
- Waheed, F.; Speight, P.; Dan, Q.; Garcia-Mata, R.; Szaszi, K. Affinity precipitation of active Rho-GEFs using a GST-tagged mutant Rho protein (GST-RhoA(G17A)) from epithelial cell lysates. J Vis Exp 2012. [CrossRef]
- Garcia-Mata, R.; Wennerberg, K.; Arthur, W.T.; Noren, N.K.; Ellerbroek, S.M.; Burridge, K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 2006, 406, 425–437.
- Leung, T.; Chen, X.Q.; Tan, I.; Manser, E.; Lim, L. Myotonic dystrophy kinase-related Cdc42-binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol Cell Biol 1998, 18, 130–140. [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2011, 67, 545–554. [CrossRef]
- Sumi, T.; Matsumoto, K.; Shibuya, A.; Nakamura, T. Activation of LIM kinases by myotonic dystrophy kinase-related Cdc42-binding kinase alpha. J Biol Chem 2001, 276, 23092–23096. [CrossRef]
- Masszi, A.; Di Ciano, C.; Sirokmany, G.; Arthur, W.T.; Rotstein, O.D.; Wang, J.; McCulloch, C.A.; Rosivall, L.; Mucsi, I.; Kapus, A. Central role for Rho in TGF-beta1-induced alpha-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol 2003, 284, F911–924. [CrossRef]
- Younesi, F.S.; Son, D.O.; Firmino, J.; Hinz, B. Myofibroblast Markers and Microscopy Detection Methods in Cell Culture and Histology. Methods Mol Biol 2021, 2299, 17–47. [CrossRef]
- Masszi, A.; Speight, P.; Charbonney, E.; Lodyga, M.; Nakano, H.; Szaszi, K.; Kapus, A. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol 2010, 188, 383–399. [CrossRef]
- Nakao, Y.; Nakagawa, S.; Yamashita, Y.I.; Umezaki, N.; Okamoto, Y.; Ogata, Y.; Yasuda-Yoshihara, N.; Itoyama, R.; Yusa, T.; Yamashita, K.; et al. High ARHGEF2 (GEF-H1) Expression is Associated with Poor Prognosis Via Cell Cycle Regulation in Patients with Pancreatic Cancer. Ann Surg Oncol 2021. [CrossRef]
- Lieb, W.S.; Lungu, C.; Tamas, R.; Berreth, H.; Rathert, P.; Storz, P.; Olayioye, M.A.; Hausser, A. The GEF-H1/PKD3 signaling pathway promotes the maintenance of triple-negative breast cancer stem cells. Int J Cancer 2020, 146, 3423–3434. [CrossRef]
- Cao, J.; Yang, T.; Tang, D.; Zhou, F.; Qian, Y.; Zou, X. Increased expression of GEF-H1 promotes colon cancer progression by RhoA signaling. Pathol Res Pract 2019, 215, 1012–1019. [CrossRef]
- Kent, O.A.; Sandi, M.J.; Rottapel, R. Co-dependency between KRAS addiction and ARHGEF2 promotes an adaptive escape from MAPK pathway inhibition. Small GTPases 2019, 10, 441–448. [CrossRef]
- Shi, J.; Guo, B.; Zhang, Y.; Hui, Q.; Chang, P.; Tao, K. Guanine nucleotide exchange factor H1 can be a new biomarker of melanoma. Biologics 2016, 10, 89–98. [CrossRef]
- Guillemot, L.; Paschoud, S.; Jond, L.; Foglia, A.; Citi, S. Paracingulin regulates the activity of Rac1 and RhoA GTPases by recruiting Tiam1 and GEF-H1 to epithelial junctions. Mol Biol Cell 2008, 19, 4442–4453. [CrossRef]
- Raya-Sandino, A.; Castillo-Kauil, A.; Dominguez-Calderon, A.; Alarcon, L.; Flores-Benitez, D.; Cuellar-Perez, F.; Lopez-Bayghen, B.; Chavez-Munguia, B.; Vazquez-Prado, J.; Gonzalez-Mariscal, L. Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim Biophys Acta 2017, 1864, 1714–1733. [CrossRef]
- Azoitei, M.L.; Noh, J.; Marston, D.J.; Roudot, P.; Marshall, C.B.; Daugird, T.A.; Lisanza, S.L.; Sandi, M.J.; Ikura, M.; Sondek, J.; et al. Spatiotemporal dynamics of GEF-H1 activation controlled by microtubule- and Src-mediated pathways. J Cell Biol 2019, 218, 3077–3097. [CrossRef]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J Cell Sci 2005, 118, 1861–1872.
- Meiri, D.; Greeve, M.A.; Brunet, A.; Finan, D.; Wells, C.D.; LaRose, J.; Rottapel, R. Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol Cell Biol 2009, 29, 5963–5973. [CrossRef]
- Yamahashi, Y.; Saito, Y.; Murata-Kamiya, N.; Hatakeyama, M. Polarity-regulating Kinase Partitioning-defective 1b (PAR1b) Phosphorylates Guanine Nucleotide Exchange Factor H1 (GEF-H1) to Regulate RhoA-dependent Actin Cytoskeletal Reorganization. J Biol Chem 2011, 286, 44576–44584. [CrossRef]
- Birkenfeld, J.; Nalbant, P.; Bohl, B.P.; Pertz, O.; Hahn, K.M.; Bokoch, G.M. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell 2007, 12, 699–712. [CrossRef]
- Marlaire, S.; Dehio, C. Bartonella effector protein C mediates actin stress fiber formation via recruitment of GEF-H1 to the plasma membrane. PLoS Pathog 2021, 17, e1008548. [CrossRef]
- Tan, I.; Seow, K.T.; Lim, L.; Leung, T. Intermolecular and intramolecular interactions regulate catalytic activity of myotonic dystrophy kinase-related Cdc42-binding kinase alpha. Mol Cell Biol 2001, 21, 2767–2778. [CrossRef]
- Tan, I.; Yong, J.; Dong, J.M.; Lim, L.; Leung, T. A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 2008, 135, 123–136. [CrossRef]
- Zihni, C.; Terry, S.J. RhoGTPase signalling at epithelial tight junctions: Bridging the GAP between polarity and cancer. Int J Biochem Cell Biol 2015, 64, 120–125. [CrossRef]
- Lee, I.C.J.; Leung, T.; Tan, I. Adaptor protein LRAP25 mediates myotonic dystrophy kinase-related Cdc42-binding kinase (MRCK) regulation of LIMK1 protein in lamellipodial F-actin dynamics. J Biol Chem 2014, 289, 26989–27003. [CrossRef]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechere, L. LIM kinases: cofilin and beyond. Oncotarget 2017, 8, 41749–41763. [CrossRef]
- Kwa, M.Q.; Brandao, R.; Phung, T.H.; Ge, J.; Scieri, G.; Brakebusch, C. MRCKalpha Is Dispensable for Breast Cancer Development in the MMTV-PyMT Model. Cells 2021, 10. [CrossRef]
- Gifford, C.C.; Tang, J.; Costello, A.; Khakoo, N.S.; Nguyen, T.Q.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Negative regulators of TGF-beta1 signaling in renal fibrosis; pathological mechanisms and novel therapeutic opportunities. Clin Sci (Lond) 2021, 135, 275–303. [CrossRef]
Figure 1.
MRCKα interacts with the N-terminus of GEF-H1. (A) Endogenous GEF-H1 or MRCKα were immunoprecipitated from LLC-PK1 cells and co-immunoprecipitated proteins were detected by Western Blotting (representative blot of n = 3). “No Ab” indicates IP without primary antibody, with beads alone. (B) RPTEC/hTERT cells grown on coverslips were transfected with control (non-related, NR) siRNA, or an MRCKα-specific siRNA, as indicated. PLA was performed using GEF-H1 and MRCKα-specific antibodies, as described in the Methods. Where indicated, the primary antibodies were omitted. The nuclei were counterstained using DAPI. The red puncta in 3-5 visual fields/coverslips were counted and divided by the number of nuclei. The graph shows mean+/-SD of 10 fields from 2 independent experiments (*** p<0.001, unpaired t-test, n=10). (C) FLAG- and GFP-tagged GEF-H1 constructs used. Domains of GEF-H1 include C1: zinc finger-like motif; DH: Dbl-homologous domain; PH: pleckstrin homology domain; Coiled: coiled-coil region. (D,E) FLAG- (D) or GFP- (E)-tagged GEF-H1 constructs were transfected in HEK-293 cells and immunoprecipitated through the corresponding tags. The co-immunoprecipitation of MRCKα was visualized by Western blotting (representative blots from n=3).
Figure 1.
MRCKα interacts with the N-terminus of GEF-H1. (A) Endogenous GEF-H1 or MRCKα were immunoprecipitated from LLC-PK1 cells and co-immunoprecipitated proteins were detected by Western Blotting (representative blot of n = 3). “No Ab” indicates IP without primary antibody, with beads alone. (B) RPTEC/hTERT cells grown on coverslips were transfected with control (non-related, NR) siRNA, or an MRCKα-specific siRNA, as indicated. PLA was performed using GEF-H1 and MRCKα-specific antibodies, as described in the Methods. Where indicated, the primary antibodies were omitted. The nuclei were counterstained using DAPI. The red puncta in 3-5 visual fields/coverslips were counted and divided by the number of nuclei. The graph shows mean+/-SD of 10 fields from 2 independent experiments (*** p<0.001, unpaired t-test, n=10). (C) FLAG- and GFP-tagged GEF-H1 constructs used. Domains of GEF-H1 include C1: zinc finger-like motif; DH: Dbl-homologous domain; PH: pleckstrin homology domain; Coiled: coiled-coil region. (D,E) FLAG- (D) or GFP- (E)-tagged GEF-H1 constructs were transfected in HEK-293 cells and immunoprecipitated through the corresponding tags. The co-immunoprecipitation of MRCKα was visualized by Western blotting (representative blots from n=3).
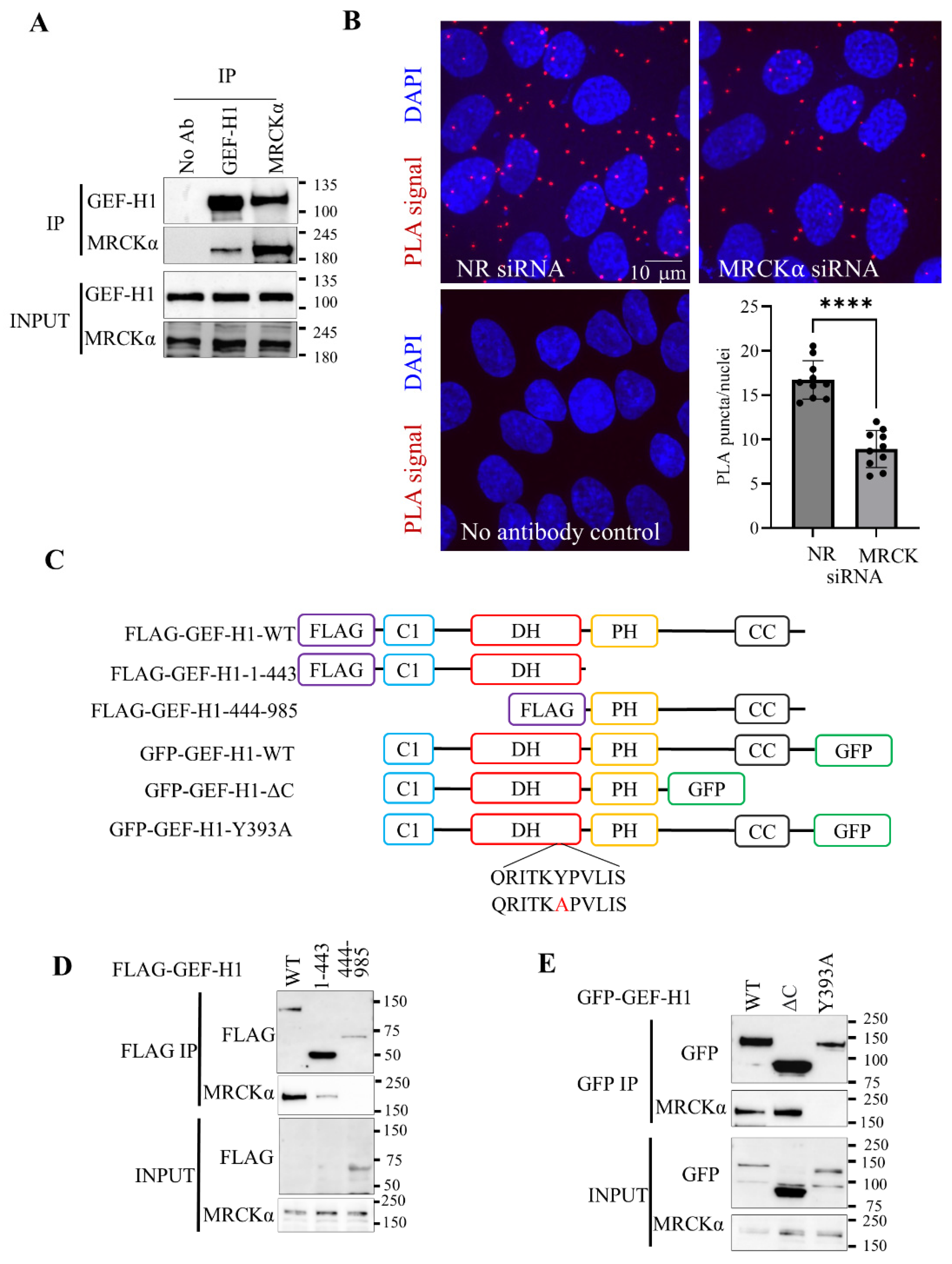
Figure 2.
MRCKα suppresses GEF-H1 and RhoA activation. (A,B) LLC-PK1 (A) or RPTEC/hTERT cells (B) were transfected with NR siRNA, or two different MRCKα-specific siRNAs (100nM) for 48 hours. The cells were lysed and active GEF-H1 was captured using GST-RhoA(G17A) beads. Precipitated (active) and total GEF-H1 were detected by Western blotting and quantified by densitometry. The graphs show combined results obtained with the two siRNAs. Active GEF-H1 was normalized by the corresponding total GEF-H1 and expressed as fold change from control (taken as 1) (n=6 (3/siRNA), *p < 0.05, one sample t test vs. 1). (C) LLC-PK1 cells were transfected with NR, MRCKα or GEF-H1-specific siRNAs or their combination, as indicated. Active RhoA was captured using GST–RBD-coupled beads, and precipitated (active) and total RhoA, as well as GEF-H1 and MRCKα in the total cell lysates were detected by Western blotting. Active RhoA was normalized to the corresponding total RhoA, and expressed as fold change from control (taken as 1). (n=5, *p< 0.05, one sample t-test; # p< 0.05, one way ANOVA vs the indicated condition).
Figure 2.
MRCKα suppresses GEF-H1 and RhoA activation. (A,B) LLC-PK1 (A) or RPTEC/hTERT cells (B) were transfected with NR siRNA, or two different MRCKα-specific siRNAs (100nM) for 48 hours. The cells were lysed and active GEF-H1 was captured using GST-RhoA(G17A) beads. Precipitated (active) and total GEF-H1 were detected by Western blotting and quantified by densitometry. The graphs show combined results obtained with the two siRNAs. Active GEF-H1 was normalized by the corresponding total GEF-H1 and expressed as fold change from control (taken as 1) (n=6 (3/siRNA), *p < 0.05, one sample t test vs. 1). (C) LLC-PK1 cells were transfected with NR, MRCKα or GEF-H1-specific siRNAs or their combination, as indicated. Active RhoA was captured using GST–RBD-coupled beads, and precipitated (active) and total RhoA, as well as GEF-H1 and MRCKα in the total cell lysates were detected by Western blotting. Active RhoA was normalized to the corresponding total RhoA, and expressed as fold change from control (taken as 1). (n=5, *p< 0.05, one sample t-test; # p< 0.05, one way ANOVA vs the indicated condition).
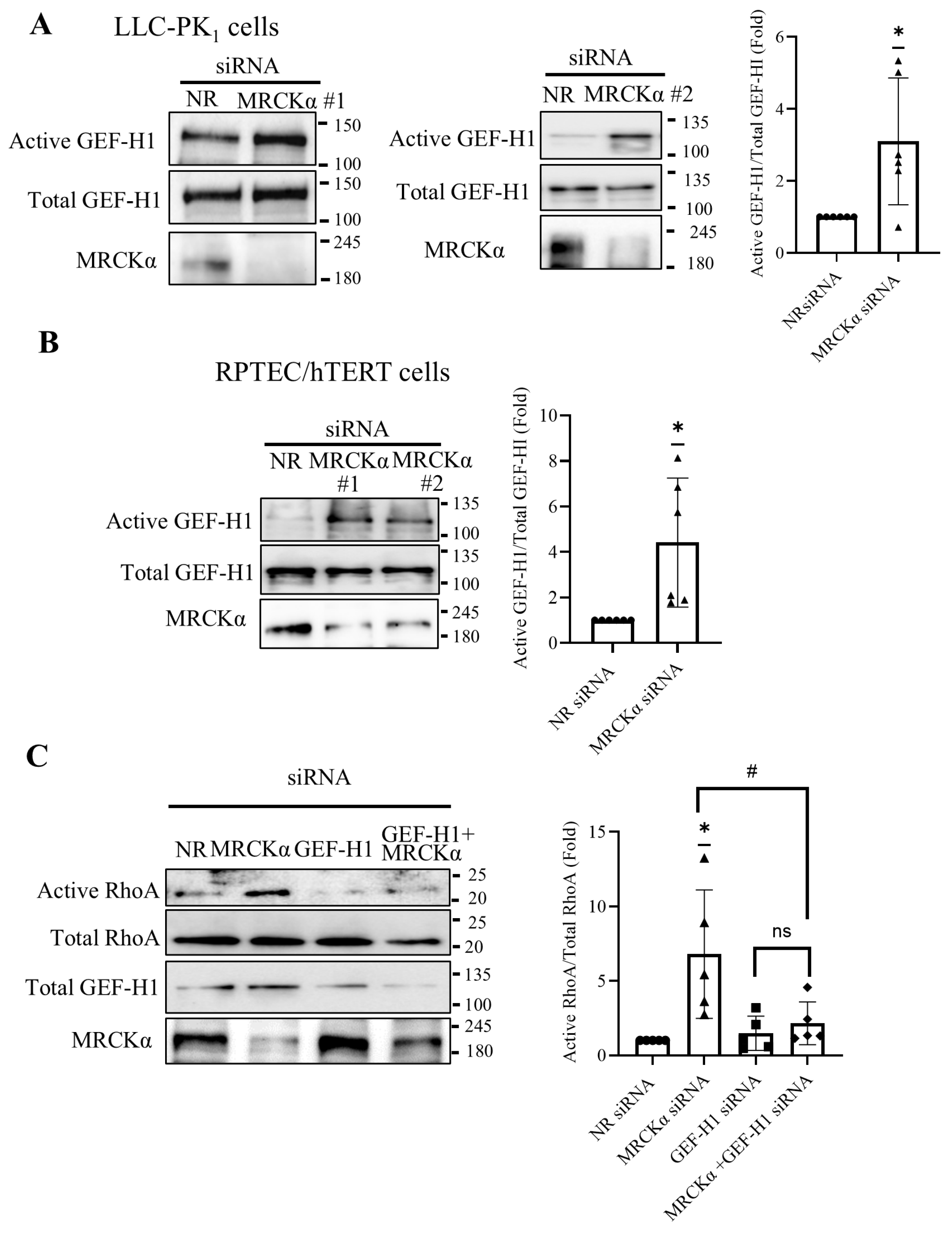
Figure 3.
MRCKα suppresses phospho-cofilin and reduces GEF-H1-dependent actin stress fibers. LLC-PK1 cells were transfected with GEF-H1 siRNA and 6 hours or 24 hours later with MRCKα siRNA (24h or 42h total). (A) cells were fixed, permeabilized, and F-actin was visualized using Alexa Fluor® 488 Phalloidin and nuclei counterstained using DAPI. Pictures were taken using a Zeiss Widefield Microscope (63x objective). The scale bar represents 10µm. (B). GEF-H1 and MRCKα silencing was verified by Western Blotting. (C). Cells were transfected as indicated, fixed, permeabilized, and phospho-Ser18/Thr19 MLC and nuclei were stained. Pictures were taken using a confocal microscope. Maximum intensity projection pictures generated from Z-stack are show. The scale bar corresponds to 10 μm and applies to all images. (D). Phosphorylated and total cofilin were quantified using Western Blotting. Densitometry values show data for the 48h silencing of phospho-cofilin, normalized using the corresponding total cofilin, and expressed as fold change from control (taken as 1) (n=6, **p < 0.01, one sample t-test vs. 1).
Figure 3.
MRCKα suppresses phospho-cofilin and reduces GEF-H1-dependent actin stress fibers. LLC-PK1 cells were transfected with GEF-H1 siRNA and 6 hours or 24 hours later with MRCKα siRNA (24h or 42h total). (A) cells were fixed, permeabilized, and F-actin was visualized using Alexa Fluor® 488 Phalloidin and nuclei counterstained using DAPI. Pictures were taken using a Zeiss Widefield Microscope (63x objective). The scale bar represents 10µm. (B). GEF-H1 and MRCKα silencing was verified by Western Blotting. (C). Cells were transfected as indicated, fixed, permeabilized, and phospho-Ser18/Thr19 MLC and nuclei were stained. Pictures were taken using a confocal microscope. Maximum intensity projection pictures generated from Z-stack are show. The scale bar corresponds to 10 μm and applies to all images. (D). Phosphorylated and total cofilin were quantified using Western Blotting. Densitometry values show data for the 48h silencing of phospho-cofilin, normalized using the corresponding total cofilin, and expressed as fold change from control (taken as 1) (n=6, **p < 0.01, one sample t-test vs. 1).
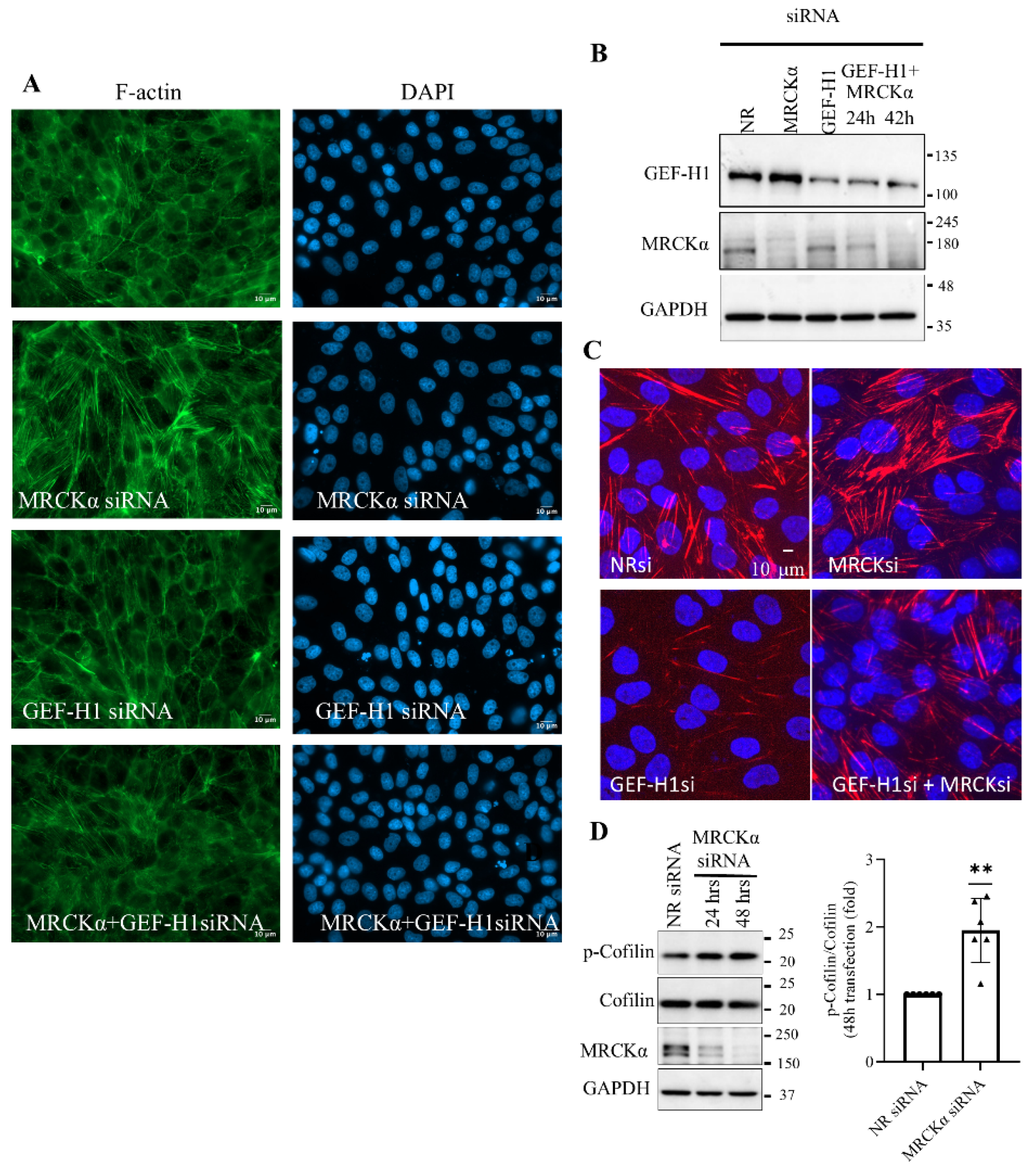
Figure 4.
MRCKα blunts cytokine-induced GEF-H1 activation. (A). RPTECs were stimulated by TNFα (20 mg/ml) or TGFβ1 (10 ng/ml) for 15 min, and PLA was performed and quantified as in Figure 1 (n=6 independet experiments for control and TNF treatment, n=3 for TGFβ1 treatment, 3 fields/experiment) **** p<0.0001, unpaired t-test. (B). LLC-PK1 cells were transfected with control (NR) or MRCKα-specific siRNAs. Cells were stimulated with TNFα or TGFβ1 for 15 min and active GEF-H1 precipitated and quantified as in Figure 2 (n=4-5, (n=5, *p< 0.05, **p< 0.01, ***p< 0.001, one sample t-test vs 1; ## p< 0.01, one way ANOVA vs the indicated condition). (C). LLC-PK1 cells were transfected with HA-tagged GEF-H1 with or without mCherry-tagged-MRCKα, and 48 h later the cells were stimulated with TGFβ1(10 min). GEF-H1 activation was measured as in A, and the precipitation of active HA-GEF-H1 was detected by Western blotting (representative of n=3).
Figure 4.
MRCKα blunts cytokine-induced GEF-H1 activation. (A). RPTECs were stimulated by TNFα (20 mg/ml) or TGFβ1 (10 ng/ml) for 15 min, and PLA was performed and quantified as in Figure 1 (n=6 independet experiments for control and TNF treatment, n=3 for TGFβ1 treatment, 3 fields/experiment) **** p<0.0001, unpaired t-test. (B). LLC-PK1 cells were transfected with control (NR) or MRCKα-specific siRNAs. Cells were stimulated with TNFα or TGFβ1 for 15 min and active GEF-H1 precipitated and quantified as in Figure 2 (n=4-5, (n=5, *p< 0.05, **p< 0.01, ***p< 0.001, one sample t-test vs 1; ## p< 0.01, one way ANOVA vs the indicated condition). (C). LLC-PK1 cells were transfected with HA-tagged GEF-H1 with or without mCherry-tagged-MRCKα, and 48 h later the cells were stimulated with TGFβ1(10 min). GEF-H1 activation was measured as in A, and the precipitation of active HA-GEF-H1 was detected by Western blotting (representative of n=3).
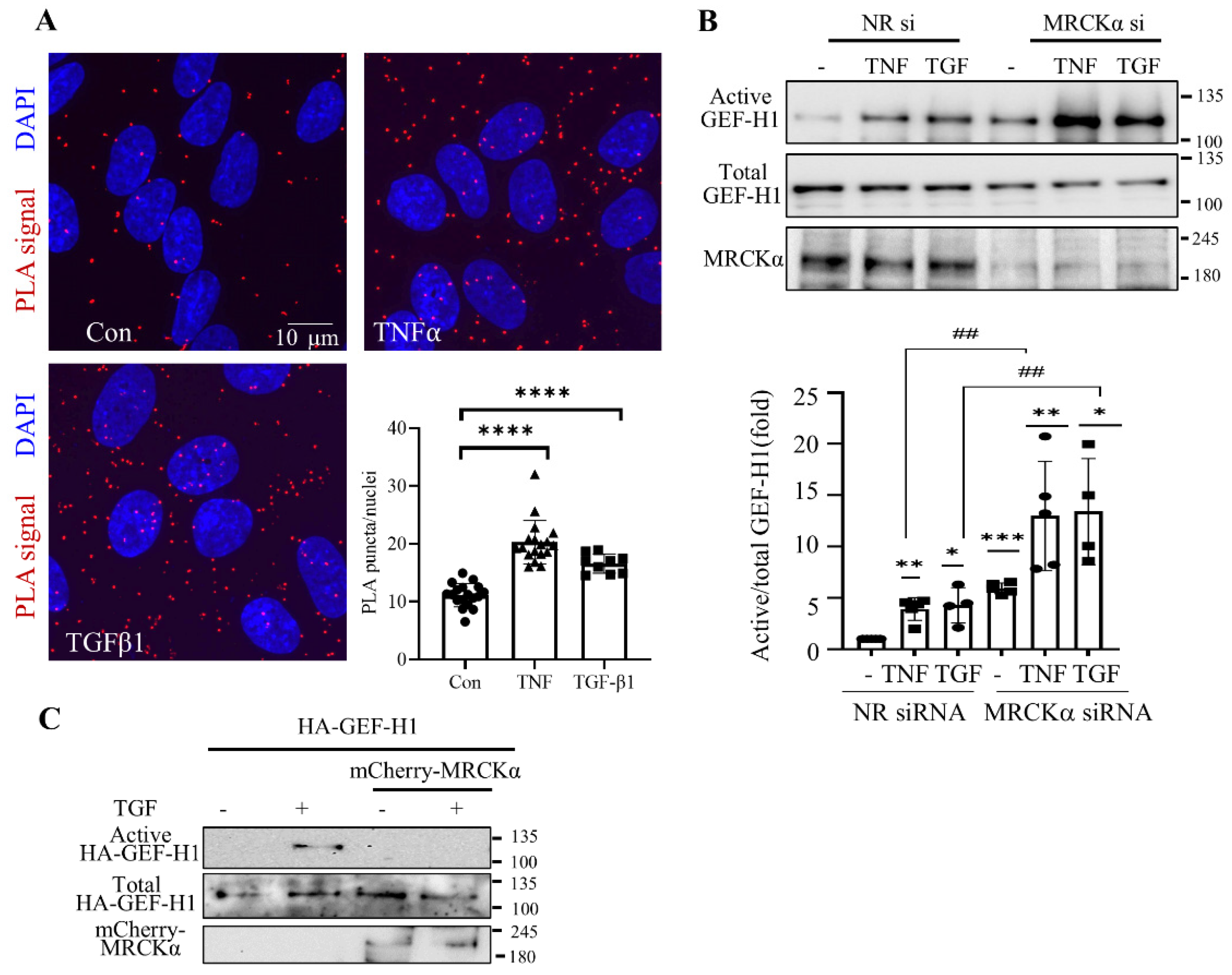
Figure 5.
MRCKα suppresses fibrosis-associated genes. (A). LLC-PK1 cells were transfected with NR or MRCKα specific siRNA for 48 hours. Twenty-four hours post-transfection the medium was replaced with serum-free DMEM supplemented with 10 ng/mL TGFβ1 for 24 hours, as indicated. The samples were analyzed using a Qiagen RT2 Profiler™ Pig Fibrosis gene PCR array. The heatmap shows Z scores, calculated using fold changes of the relative gene expression. (B) Genes that are upregulated in the array by at least 1.5-fold upon MRCKα silencing compared to the control (taken as 1). (C) Genes that are upregulated in the array by at least 1.5-fold by TGFβ1 in MRCKα siRNA transfecetd cells vs NR transfected cells taken as 1 (fold change). In B and C, n=3, *p < 0.05, **p < 0.01 one sample t-test vs1). (D). Data from the fibrosis array for the ACTA2 mRNA expressed as fold change from control. (n=3, *p < 0.05, **p < 0.01, one sample t-test vs. 1; #### p<0.0001, one-way Anova vs the indicated condition). (E) LLC-PK1 cells were transfected with the indicated siRNAs, and 4 hours later the medium was changed to serum-free DMEM with or without TGFβ1 (10 ng/ml) for 48 hours. αSMA, MRCKα and GAPDH were detected by Western blotting (n=3, *p < 0.05, **p < 0.01, one sample t-test vs. 1; #### p<0.0001, one-way Anova vs the indicated condition). The MRCKα specific band is indicated by the arrow.
Figure 5.
MRCKα suppresses fibrosis-associated genes. (A). LLC-PK1 cells were transfected with NR or MRCKα specific siRNA for 48 hours. Twenty-four hours post-transfection the medium was replaced with serum-free DMEM supplemented with 10 ng/mL TGFβ1 for 24 hours, as indicated. The samples were analyzed using a Qiagen RT2 Profiler™ Pig Fibrosis gene PCR array. The heatmap shows Z scores, calculated using fold changes of the relative gene expression. (B) Genes that are upregulated in the array by at least 1.5-fold upon MRCKα silencing compared to the control (taken as 1). (C) Genes that are upregulated in the array by at least 1.5-fold by TGFβ1 in MRCKα siRNA transfecetd cells vs NR transfected cells taken as 1 (fold change). In B and C, n=3, *p < 0.05, **p < 0.01 one sample t-test vs1). (D). Data from the fibrosis array for the ACTA2 mRNA expressed as fold change from control. (n=3, *p < 0.05, **p < 0.01, one sample t-test vs. 1; #### p<0.0001, one-way Anova vs the indicated condition). (E) LLC-PK1 cells were transfected with the indicated siRNAs, and 4 hours later the medium was changed to serum-free DMEM with or without TGFβ1 (10 ng/ml) for 48 hours. αSMA, MRCKα and GAPDH were detected by Western blotting (n=3, *p < 0.05, **p < 0.01, one sample t-test vs. 1; #### p<0.0001, one-way Anova vs the indicated condition). The MRCKα specific band is indicated by the arrow.
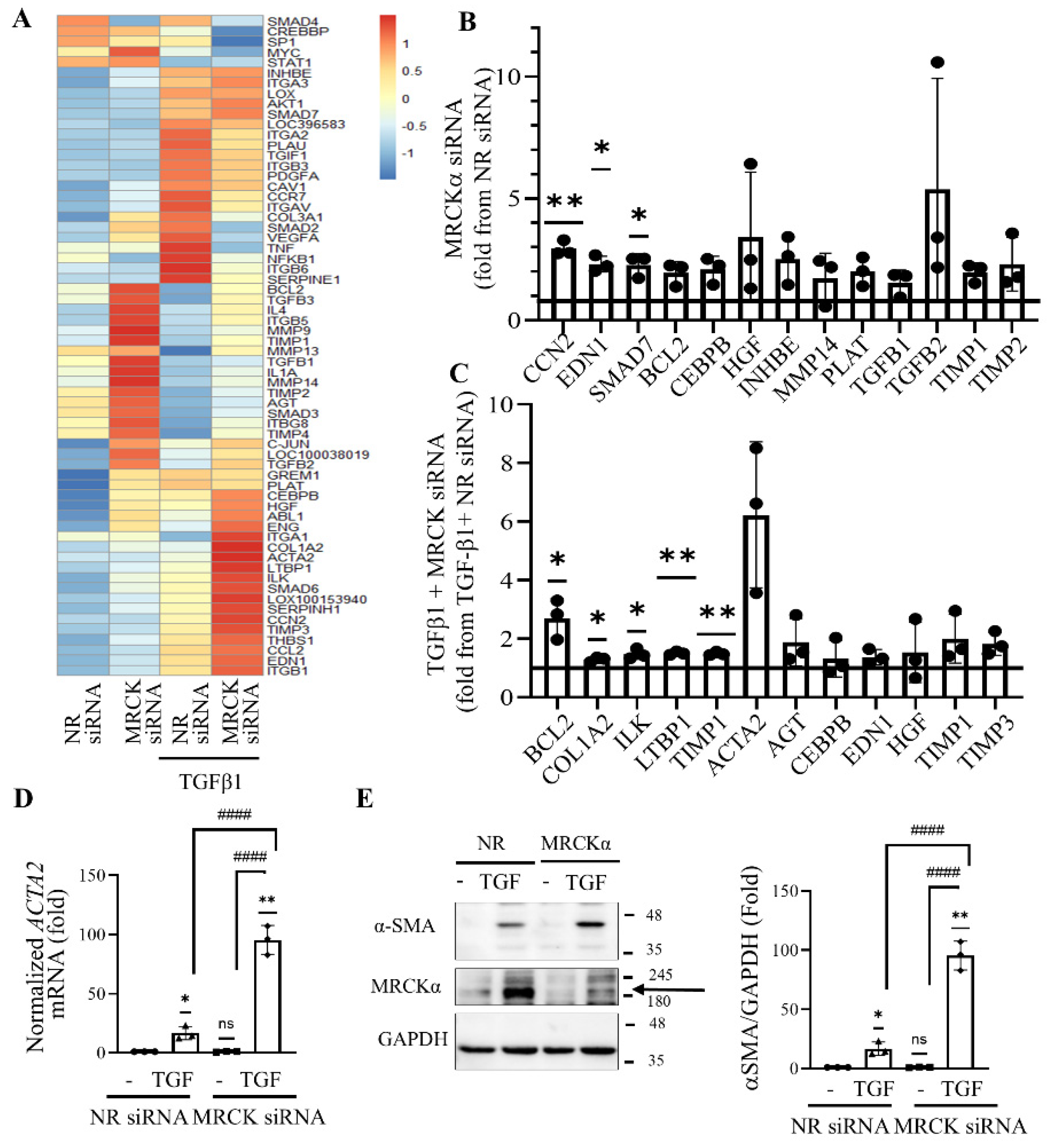
Figure 6.
MRCKα regulates MRTF-A nuclear translocation and MRTF-dependent gene transcription. (A). LLC-PK1 cells grown on coverslips were transfected with the indicated siRNAs, and 32 h later the medium was changed to serum-free DMEM for 16 h. Cells were treated with or without TGFβ1 (10 ng/ml, 30 mins). MRTF-A was visualized by immunofluorescence using an Alexa Fluor 555-labelled secondary antibody. Nuclei were stained using DAPI. Pictures were taken using a Zeiss Widefield Microscope (63x objective). The scale bar represents 10µm. (B). LLC-PK1 cells were transfected and treated with TGFβ1 (48h), and the indicated siRNAs, and proteins detected using the indicated antibodies. The arrow points to MRCKα. (C-E). LLC-PK1 cells were transfected with the indicated siRNAs, and where indicated, treated with TGFβ1 for 24 h. RT-PCR was performed to measure TAGLN (C), ACTA2 (D) and CCN2 (E) mRNA as described in the Methods, using PPIA as the reference gene. Changes were expressed as fold change from NR siRNA transfected samples taken as 1 (n=5-7, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one sample t-test vs. 1; # p<0.05, ## p<0.01, ### p<0.001, #### p<0.0001, one-way Anova vs the indicated condition).
Figure 6.
MRCKα regulates MRTF-A nuclear translocation and MRTF-dependent gene transcription. (A). LLC-PK1 cells grown on coverslips were transfected with the indicated siRNAs, and 32 h later the medium was changed to serum-free DMEM for 16 h. Cells were treated with or without TGFβ1 (10 ng/ml, 30 mins). MRTF-A was visualized by immunofluorescence using an Alexa Fluor 555-labelled secondary antibody. Nuclei were stained using DAPI. Pictures were taken using a Zeiss Widefield Microscope (63x objective). The scale bar represents 10µm. (B). LLC-PK1 cells were transfected and treated with TGFβ1 (48h), and the indicated siRNAs, and proteins detected using the indicated antibodies. The arrow points to MRCKα. (C-E). LLC-PK1 cells were transfected with the indicated siRNAs, and where indicated, treated with TGFβ1 for 24 h. RT-PCR was performed to measure TAGLN (C), ACTA2 (D) and CCN2 (E) mRNA as described in the Methods, using PPIA as the reference gene. Changes were expressed as fold change from NR siRNA transfected samples taken as 1 (n=5-7, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one sample t-test vs. 1; # p<0.05, ## p<0.01, ### p<0.001, #### p<0.0001, one-way Anova vs the indicated condition).
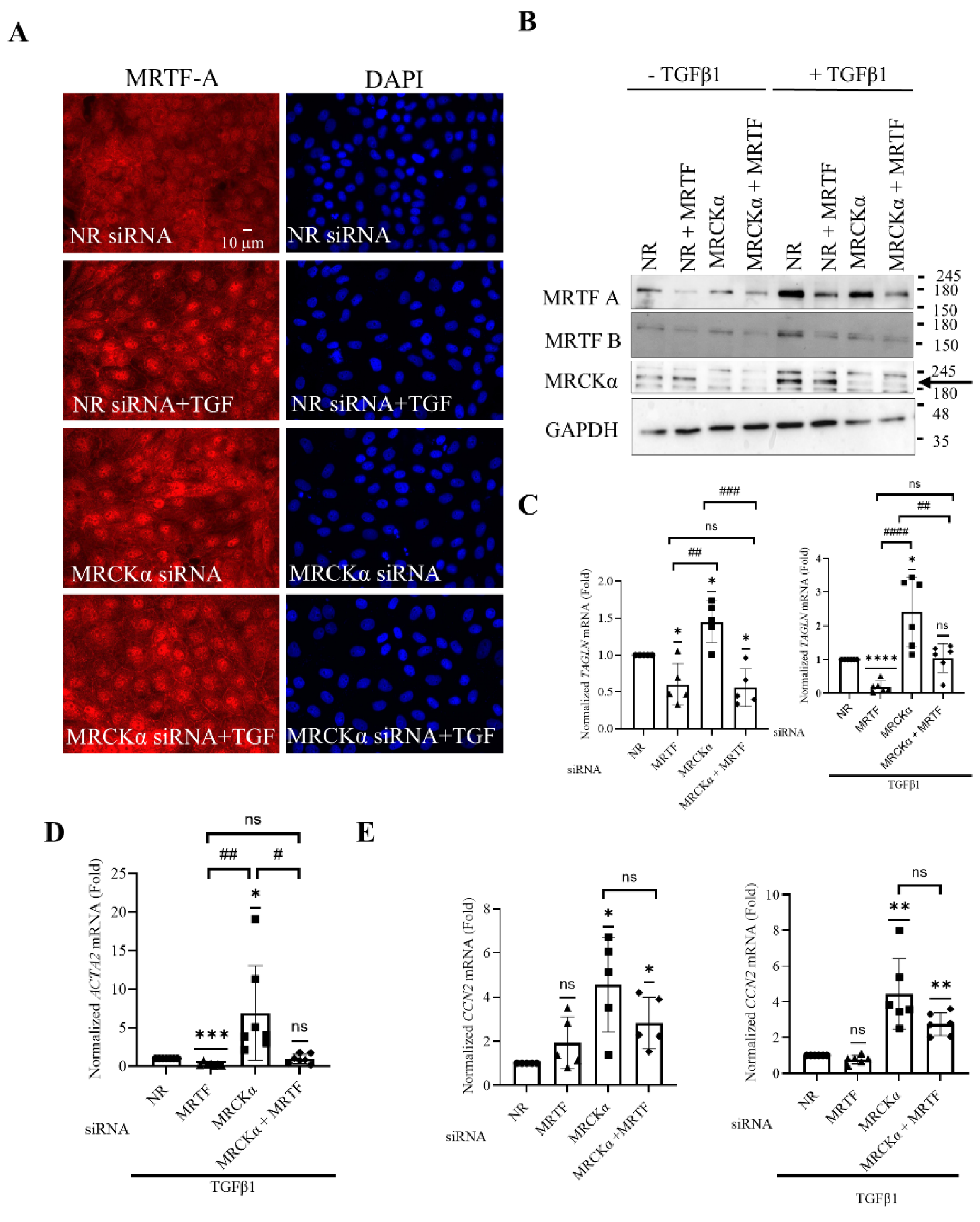

Table 1.
Short interfering RNA sequences against porcine proteins (LLC-PK1 cells).
| siRNA | sequence |
|---|---|
| Porcine MRCKα #1 | GGG AAA UGA AGA AGG GUU AUU |
| Porcine MRCKα #2 | AGU UAG AAG AAG AGG UAA AUU |
| Porcine MRTF-A | CCA AGG AGC UGA AGC CAA A |
| Porcine MRTF-B | CGA CAA ACA CCG UAG CAA A |
Table 2.
Primers for detecting porcine mRNA used in the study.
| siRNA | sequence |
|---|---|
| CCN2/CTGF F | GTG AAG ACA TAC CGG GCT AAG |
| CCN2/ CTGF R | GAC ACT TGA ACT CCA CAG GAA |
| TAGLN E3 F | GAG CAG GTG GCT CAG TTC TT |
| TAGLN E3 R | CCA CGG TAG TGT CCA TCA TTC |
| ACTA2 F | CGTCCTAGACATCAGGGGGT |
| ACTA2 R | GGGGCAACACGAAGCTCATT |
| PPIA F | CGG GTC CTG GCA TCT TGT |
| PPIA R | TGG CAG TGC AAA TGA AAA ACT G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



