
Submitted:
01 September 2025
Posted:
03 September 2025
You are already at the latest version
Abstract
Ticks are important vectors of pathogens affecting humans and animals, posing a serious threat to health. For the first time, we studied the metagenomic profile of the microbial composition of Hyalomma scupense and Hyalomma asiaticum ticks in Kazakhstan. For microbiome analysis, 94 adult H. asiaticum and H. scupense ticks collected from cattle in Kazakhstan in 2023 were randomly selected. 16S rRNA gene sequencing was performed using the Ion Torrent NGS platform. Taxonomic classification carried out in the BV-BRC platform with the Kraken2 database. Metagenomic analysis revealed 26 bacterial genera, including both pathogenic and symbiotic taxa. In H. scupense, the dominant groups were Francisella (89.0%), Staphylococcus (76.0%), and Candidatus Midichloria (61.0%), while in H. asiaticum they were Francisella (99.0% and 95.0%) and Helcococcus (65.0%). In male H. scupense, the proportion of Francisella reached 89%, whereas in females it varied from 2% to 28%. In H. asiaticum, Helcococcus accounted for 65% in males compared to 11% in females. This is the first report on the metagenomic profile of the microbiota of H. scupense and H. asiaticum in Kazakhstan. The detection of pathogens indicates a risk of their transmission to humans and animals and highlights the need to develop new tick control strategies.
Keywords:
tick
; H. scupense
; H. asiaticum
; microbiome
; bacteria
; sequencing
; 16S rRNA
* Correspondents: sultankul70@mail.ru (K.T.S.); omb65@mail.ru (O.M.B.)
1. Introduction
Ticks are widely distributed across the globe, and tick infestations pose a significant threat to livestock due to their capacity to transmit tick-borne pathogens (TBPs) and cause various diseases. Ticks are considered the second most important vectors of human diseases worldwide after mosquitoes, yet they are the primary vectors of pathogenic diseases in both domestic and wild animals [1,2]. In addition to pathogens, ticks also harbor a diverse array of symbiotic and commensal microorganisms [3,4].
Currently, 896 tick species have been recorded worldwide, of which 27 species belong to the genus Hyalomma [5,6]. The tick fauna of Kazakhstan comprises over 30 ixodid tick species, including 8 species of the genus Hyalomma. The natural conditions of Kazakhstan are favorable for the habitation of various tick species, among which H. scupense and H. asiaticum are prominent. In the desert landscapes of Kazakhstan, H. asiaticum predominates, whereas H. scupense is more common in semi-desert and low-mountain steppe regions. Ticks from the southern region of Kazakhstan are recognized as vectors and reservoirs of multiple pathogens, causing Q fever, tick-borne spotted fevers, arboviruses, and piroplasmoses [8,9,10,11,12].
Current data analysis reveals that the metagenomic profile of bacterial communities in H. scupense and H. asiaticum ticks inhabiting the southern and southeastern regions of Kazakhstan remains insufficiently studied.
Furthermore, the potential applications and prospects of metagenomic approaches in the diagnosis and epidemiological surveillance of infectious diseases have not yet been considered. Until recently, most studies focused primarily on the identification of tick-borne pathogens and their epidemiology using traditional methods [9,10,11,12]. Advancements in metagenomics have fundamentally transformed the ability to characterize the taxonomic composition of microbial ecosystems [13]. Recent metagenomic projects have revealed that the diversity of life is far greater and more complex than previously imagined through classical methods that rely on visually observable biodiversity [14,15,16,17].
To date, there are no available data on the microbiome of the most widespread and significant Hyalomma tick species in Kazakhstan. Therefore, our study aims to describe the microbial diversity and richness of the microbiota associated with H. scupense and H. asiaticum ticks. In this research, 16S rRNA metagenomics was employed to investigate bacterial communities in ticks collected from cattle in the southern and southeastern regions of Kazakhstan. The findings of this work may contribute to disease risk assessment for livestock and human populations in these areas and serve as a foundation for developing targeted control strategies.
2. Materials and Methods
2.1 Tick Sampling
Tick collection was conducted in April - September 2023 in the Kyzylorda, Zhambyl, Turkestan, and Zhetysu regions of Kazakhstan. All ticks were collected from cattle. The collection was performed in accordance with the permit issued by the Committee for Veterinary Control and Supervision of the Ministry of Agriculture of the Republic of Kazakhstan and with the consent of the animal owners. During tick collection, personnel adhered to strict safety measures, including wearing protective suits with sealed collars and cuffs, and regularly performing self- and mutual inspections to detect any crawling or attached ticks.
Ticks were removed using blunt forceps from the inner thigh, udder, scrotum, neck, and axillary regions of the animals. Live ticks were placed into plastic tubes with screw caps. To maintain humidity, a leaf of a cereal plant was usually added to each tube. Prior to analysis, ticks were kept alive in a cool place or refrigerated in standard tubes with plant material. Detailed labels were attached to all collected samples. Each arthropod was identified using an Altami PC0745 stereomicroscope (RS0745, Altami, Saint Petersburg, Russia). A total of 1260 ticks were collected. For subsequent microbiome analysis, 94 adult H. asiaticum and H. scupense ticks were randomly selected from cattle across various locations in the southern and southeastern regions of Kazakhstan (Table 1). Female tick samples were pooled in groups of 10 based on location, species, and sex. Two separate pools of male ticks were created, consisting of 6 and 8 individuals respectively. Additional information, including collection sites and tick counts, is provided in Table 1.
Geographic distribution of H. scupense and H. asiaticum ticks visualized using cartographic analysis performed in QGIS software (Figure 1).
2.2. Molecular-Genetic Identification of Ticks
Tick specimens were initially identified using morphological methods [18], using an Altami stereomicroscope (RS0745, Altami, Saint Petersburg, Russia). The morphological identifications were subsequently confirmed by molecular genetic analysis targeting the mitochondrial cytochrome c oxidase subunit 1 gene (COX1) [19].
A fragment of the COX1 gene (820 bp) was amplified by polymerase chain reaction (PCR) for molecular identification using the following primers: Cox1F: 5’-GGAACAATATATTTAATTTTTGG-3’ and Cox1R: 5’-ATCTATCCCTACTGTAAATATATG-3’. The amplification conditions were as follows: initial denaturation at 94 °C for 2 minutes; followed by 35 cycles of denaturation at 94 °C for 30 seconds, annealing at 54 °C for 45 seconds, and extension at 72 °C for 1 minute; with a final extension step at 72 °C for 10 minutes.
PCR products of the COX1 gene were subjected to nucleotide sequencing on an Applied Biosystems 3130 automated DNA sequencer (ABI, 3130, City, State, USA) using the Bigdye Terminator V3.1 loop sequencing kit (Applied Biosystems, Inc., Vilnius, Lithuania) for gene sequencing using an Applied Biosystems 3130 genetic analyzer (HITACHI, Tokyo, Japan).
The obtained nucleotide sequences were analyzed using the Sequencher v. 4.5 program (Gene Codes Corporation, Ann Arbor, MI, USA). The nucleotide sequence was aligned using the Mega 7.0 computer program complex. A set of nucleotide sequences from the international GenBank database of the National Center for Biotechnology Information (NCBI) was used to construct a phylogenetic tree. Phylogenetic analysis of the sequences was performed using the Mega 11.0 program.
For the analysis, COX1 gene sequences of ticks obtained from GenBank were used (accession numbers: MW498400, JQ737073, OR533789, NC053941, MN907845, MN821375, MN964348, MN907831, OM743222.
2.3. DNA Extraction
Ticks, previously sterilized with 70% ethanol, were homogenized using a mechanical homogenizer in centrifuge tubes containing 500 µL of chilled sterile phosphate-buffered saline (PBS, 1X). The homogenized samples were then centrifuged at 12,000 × g for 10 minutes at 4 °C, and the supernatant was collected. Total DNA was extracted from the supernatant using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The purity of the extracted DNA was assessed by agarose gel electrophoresis, and DNA samples were stored at −80 °C until further use.
2.4. Library Preparation
DNA concentration from the microbial community was measured using the Qubit™ dsDNA HS (High Sensitivity) Assay Kit (Life Technologies, Carlsbad, CA, USA). Library preparation was carried out using the Ion 16S™ Metagenomics Kit (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, 12 µL of DNA was mixed with 15 µL of Environmental Master Mix. Subsequently, 3 µL of each 10× 16S Primer Set was added: one tube with primers targeting regions V2-4-8 (Pool 1) and another with primers targeting V3-6,7-9 (Pool 2). Samples were subjected to PCR under the following thermal cycling conditions: initial denaturation at 95 °C for 10 minutes; followed by 25 cycles of 95 °C for 30 seconds, 58 °C for 30 seconds, and 72 °C for 30 seconds; with a final extension at 72 °C for 7 minutes. Amplification products were purified using AMPure XP beads (Beckman Coulter, Brea, CА, USA) and eluted in nuclease-free water. The concentrations of PCR products from Pool 1 and Pool 2 were assessed by agarose gel electrophoresis and subsequently combined.
End repair was performed by adding 20 µL of 5× End Repair Buffer and 1 µL of End Repair Enzyme to each sample, followed by incubation at room temperature for 20 minutes. The pooled amplicons were purified again using AMPure XP beads and eluted in Low TE buffer. Ligation and nick repair were performed using 10× Ligase Buffer, Ion P1 Adaptor, Ion Xpress™ Barcodes, dNTP Mix, DNA Ligase, Nick Repair Polymerase, nuclease-free water, and sample DNA. The thermal protocol included incubation at 25 °C for 15 minutes followed by 72 °C for 5 minutes. Adapter-ligated and nick-repaired DNA was again purified using AMPure XP beads and eluted in Low TE buffer.
Library amplification was performed using the Ion Plus Fragment Library Kit (Thermo Fisher Scientific, Carlsbad, USA) under the following PCR conditions: 95 °C for 5 minutes; followed by 7 cycles of 95 °C for 15 seconds, 58 °C for 15 seconds, and 70 °C for 1 minute; and a final extension at 70 °C for 1 minute. The amplified library was purified using AMPure XP beads and eluted in Low TE buffer. The optimal library concentration for template preparation was quantified by qPCR on a QuantStudio™ 5 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the Ion Universal Library Quantitation Kit (Thermo Fisher Scientific, Waltham, MA, USA). Each library was normalized to a concentration of 30 pM, and equal volumes of each library were pooled for downstream processing.
2.5. Sequencing
Libraries were prepared for sequencing using the Ion Chef Instrument and the Ion 510™ & 520™ & Ion 530™ Kit–Chef (hermo Fisher Scientific, Waltham, MA, USA). Chips were then loaded onto the Ion GeneStudio S5 System (Ion Torrent platform) (Thermo Fisher Scien-tific, Marsiling Industrial Estate, Woodlands, Singapore) along with Ion S5 Sequencing Kit reagents (Thermo Fisher Scientific, Waltham, MA, USA) and sequenced at the laboratory. Samples in this study were sequenced on Ion 530 chips using 400bp sequencing size.
2.6. Taxonomic Classification
Taxonomic analysis of the bacterial community was performed by high-throughput sequencing of the hypervariable region V2-4-8 and V3-6,7-9 of the 16S rRNA gene on the Ion Torrent platform using next-generation sequencing technology. The obtained data were taxonomically classified on the platform of The Bacterial and Viral Bioinformatics Research Center (BV-BRC) using the standard Kraken2 database.
Taxonomic analysis of the bacterial community was performed by high-throughput sequencing of the hypervariable V2-4-8 и V3-6,7-9 regions of the 16S rRNA gene using the Ion Torrent next-generation sequencing platform. The obtained sequences were taxonomically classified on the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) platform using the standard Kraken2 database.
2.7. Statistical Data Analysis
Alpha diversity parameters of microbial communities were assessed by calculating the Shannon-Wiener diversity index [20], Simpson’s dominance index [21], and Margalef’s richness index [22].
Beta diversity analysis to compare the taxonomic composition of the microbiomes of H. scupense and H. asiaticum ticks was performed using the Bray-Curtis dissimilarity index [23]. To quantify the similarity of bacterial community species composition between H. scupense and H. asiaticum, the Jaccard index was applied [24]. Table 2 presents the formulas used for the quantitative estimation of alpha and beta diversity parameters.
All analyses were performed and processed using the R programming environment (https://www.r-project.org/).
PCoA Diagram Construction. To analyze the geographical distribution of bacteria associated with H. scupense and H. asiaticum, Principal Coordinates Analysis (PCoA) was employed. Data visualization was carried out using the Python programming language.
Analysis of Bacterial Composition and Similarity. To assess the similarity of bacterial communities in ticks, the Jaccard similarity index was used. This index reflects the proportion of shared bacterial taxa between sample pairs.
Data visualization was also conducted in Python. Hierarchical clustering analysis was applied to identify natural groupings of bacteria and tick samples based on their microbiome profiles. The clustering method used was agglomerative hierarchical clustering with the nearest neighbor (single linkage) algorithm.
3. Results
3.1. Tick Collection and Identification
Tick samples randomly selected for microbiome analysis were identified to the species level using a combination of morphological and molecular data.
A total of 94 adult ticks, initially morphologically identified and grouped into 10 pools, were subjected to PCR amplification targeting a fragment of the cytochrome c oxidase subunit I (COX1) gene, followed by sequencing of PCR products from positive samples. Phylogenetic analysis of the COX1 gene sequences confirmed the identification of the ticks as H. scupense and H. asiaticum, fully confirming the results of the morphological examination.
The COX1 gene sequences of H. scupense obtained in this study have been deposited in GenBank under the following accession numbers: PQ560690 (H. scupense Kyzylorda_Zhalagash1_KZ), PQ560881 (H. scupense Kyzylorda_Kazaly_KZ), PQ573248 (H. scupense Kyzylorda_Zhalagash2_KZ), PQ569618 (H. scupense Turkestan_Otyrar_KZ), and PQ870262 (H. scupense Zhambyl_Shu_KZ).
The COX1 gene sequences of H. asiaticum have been deposited in GenBank under accession numbers: PQ569438 (H. asiaticum Zhambyl_Zhanatas1_KZ), PQ569449 (H. asiaticum Zhambyl_Zhanatas2_KZ), PQ560954 (H. asiaticum Zhetysu_Zharkent_KZ), PQ560955 (H. asiaticum Zhetysu_Chulakai_KZ), and PQ578371 (H. asiaticum Zhambyl_Ryskulov_KZ) (Figure 2).
3.2. Assessment of the Bacterial Diversity Profile Based on 16S rRNA Gene Sequencing
All 10 tick pools were analyzed for the presence of tick-borne bacterial agents using 16S rRNA gene sequencing data processed on the Ion Torrent next-generation sequencing platform. Metagenomic analysis of H. scupense ticks revealed the presence of bacterial genera including Corynebacterium, Bifidobacterium, Staphylococcus, Candidatus Midichloria, Mannheimia, Francisella, Coxiella, Pseudomonas, Acinetobacter, Stenotrophomonas, Rickettsia, Lachnoclostridium, Clostridium sensu stricto 3, Atopostipes, and Sphingobacterium. In H. asiaticum ticks, bacterial genera identified included Escherichia-Shigella, Francisella, Candidatus Midichloria, Mannheimia, Fusobacterium, Parvimonas, Helcococcus, Campylobacter, Porphyromonas, Staphylococcus, Trueperella, Pseudomonas, Acinetobacter, Erwinia, Clostridium sensu stricto 3, Streptococcus, Atopostipes, Solibacillus, Bacillus, and Corynebacterium (Figure 3).
At the genus level, the bacterial community composition in H. scupense was dominated by Francisella (89.0%), Staphylococcus (76.0%), and Candidatus Midichloria (61.0%), whereas in H. asiaticum, a higher relative abundance of Francisella (99.0% and 95.0%) and Helcococcus (65.0%) was observed.
The results indicate that the microbiome of Kazakhstani ticks varies in composition and microbial diversity depending on the geographic region.
From a geographic perspective, H. scupense and H. asiaticum ticks collected from the Zhambyl region exhibited significantly higher prevalence of Francisella (99% in Zhambyl_Zhanatas2, 95% in Zhambyl_Zhanatas1, and 89% in Zhambyl_Shu) compared to other locations.
H. scupense ticks from southern regions of Kazakhstan (Turkestan, Zhambyl, and Kyzylorda regions) showed a higher relative abundance of Candidatus Midichloria (61% in Kyzylorda_Zhalagash1), whereas this microorganism was not detected in H. asiaticum or H. scupense from the Zhetysu region.
H. scupense from the Turkestan region exhibited elevated relative abundances of several bacterial genera, including Coxiella (43.0%), Pseudomonas (11.0%), Acinetobacter (2.0%), Stenotrophomonas (3.0%), Rickettsia (5.0%), Lachnoclostridium (2.0%), Clostridium sensu stricto 3 (3.0%), Atopostipes (2.0%), Corynebacterium (9.0%), and Sphingobacterium (2.0%) (Figure 3D).
H. asiaticum from the Zhambyl region harbored more abundant bacterial genera such as Pseudomonas (3.0%), Acinetobacter (14.0%), Erwinia (32.0%), Clostridium sensu stricto 3 (2.0%), Staphylococcus (7.0%), Streptococcus (2.0%), Atopostipes (2.0%), Solibacillus (3.0%), Bacillus (7.0%), and Corynebacterium (7.0%) (Figure 3K).
3.3. Bacterial Microbiome Diversity in H. scupense and H. asiaticum
Alpha diversity of bacterial communities associated with H. scupense and H. asiaticum ticks is determined by the Shannon-Wiener, Margalef, and Simpson indices (Figure 4).
The bacterial abundance distribution curves for H. scupense (Figure 4A) and H. asiaticum (Figure 4B) show that a small number of bacterial genera (top ranks) carry the greatest abundance, while the majority of taxa occur at lower abundance levels. The broken shape of the curve suggests the presence of dominant bacteria with a gradual decline in abundance among other taxa. Notably, H. asiaticum demonstrates a more pronounced dominance of a single bacterial genus, whereas H. scupense exhibits a more even distribution among several top-ranked genera.
Shannon-Wiener index (Figure 4 C): the median for H. scupense was 0.17 (interquartile ranges (IQR) 0.17 - 0.33), for H. asiaticum - 0.21 (IQR 0.14 - 0.29), which reflects the high uniformity of communities in H. asiaticum.
Margalef index (Figure 4 D): for H. scupense the median is 0.52 (IQR 0.52 - 1.29), for H. asiaticum the median is - 0.78 (IQR 0.26 - 1.57), which indicates species richness in H. asiaticum.
Simpson index (Figure 4 E): median values were 0.003 (IQR 0.003 - 0.013) for H. scupense and 0.010 (IQR 0.001 - 0.020) for H. asiaticum, confirming the high diversity in H. asiaticum.
3.4. Microbial Variations Depending on Geographic Origin
We constructed distribution diagrams and cladograms of microbial communities, demonstrating differences in the bacterial composition of H. scupense and H. asiaticum ticks according to their geographic origin. To assess differences in bacterial communities between regions, principal coordinate analysis (PCoA) was performed. Coordinates were calculated based on the Bray-Curtis distance matrix.
The microbiome of H. asiaticum showed no significant differences between regions (p-value = 0.299, p > 0.05, ANOVA test), suggesting a similar bacterial composition across different locations. This indicates that H. asiaticum harbors a relatively stable microbiota independent of geographic location.
For H. scupense, differences between regions were statistically significant, with a p-value of 0.023 (p < 0.05) according to the ANOVA test. The microbiome of H. scupense significantly varies among regions, which is supported by the visual analysis of the PCoA plot.
A statistically significant difference in bacterial composition was observed between the Kyzylorda and Zhetysu regions for H. scupense (p = 0.016). This may indicate that environmental or host-related factors differ between these regions. For example, the climate in the Kyzylorda region is more arid and located further south, whereas the Zhetysu region is more humid and situated to the east, potentially leading to distinct microbial community structures (Figure 5).
3.5. Beta diversity of the Bacterial Community in the Microbiome of H. scupense and H. asiaticum Ticks
The Jaccard index was used to assess the similarity in bacterial community composition between H. scupense and H. asiaticum, reflecting the proportion of shared bacterial taxa between sample pairs.
Hierarchical clustering analysis was applied to identify natural groups of bacteria within the tick microbiomes (Figure 6).
Light yellow shades correspond to low similarity (Jaccard index ≤ 0.50), while dark green shades indicate high similarity (Jaccard index > 0.50). The heatmap was generated using the Python environnt. The clustering method applied was agglomerative nearest neighbor clustering.
The constructed heatmap with hierarchical clustering revealed patterns in the distribution of bacterial composition among ticks. Statistically significant differences between groups (p < 0.001, t-test) indicate that bacterial communities of different tick species form distinguishable clusters.
Some bacterial genera, such as Mannheimia, Staphylococcus, Pseudomonas, Acinetobacter, Clostridium sensu stricto 3, and Atopostipes, are present in both samples with equal proportions (0.5), indicating partial overlap in bacterial composition. The genera Corynebacterium (0.75 and 0.25) and Francisella (0.43 and 0.57) occur in both samples but at different frequencies. The similarity level of bacterial communities between H. scupense and H. asiaticum ticks based on the Jaccard index is only 30.8%, with only 8 shared bacterial genera identified.
Certain bacteria, including Coxiella, Candidatus Midichloria, Stenotrophomonas, Rickettsia, Lachnoclostridium, Sphingobacterium, and Bifidobacterium, are found exclusively in H. scupense, indicating a species-specific bacterial composition. Conversely, genera such as Fusobacterium, Escherichia-Shigella, Parvimonas, Helcococcus, Campylobacter, Porphyromonas, Trueperella, Erwinia, Streptococcus, Solibacillus, and Bacillus are unique to H. asiaticum, reflecting bacterial diversity specific to this tick species.
Thus, the analysis demonstrates that the bacterial communities of the two tick species comprise both shared and unique taxa, suggesting differences in their ecology or potential interactions with pathogens.
4. Discussion
Tick-borne pathogens remain poorly understood, despite their important medical and veterinary significance. In many countries, including Kazakhstan and neighboring regions of Central Asia, data on the composition and distribution of microorganisms associated with ticks of the genus Hyalomma are extremely limited. This creates significant gaps in understanding the epidemiological role of the most common tick species of the genus Hyalomma. The lack of systematic studies prevents the identification of the spectrum of pathogens circulating in natural foci and does not allow a full assessment of their potential impact on human and animal health.
According to the scientific literature, a number of studies of microbial populations in ticks of the genera Ixodes, Rhipicephalus, Haemaphysalis, Dermacentor and Amblyomma have been conducted to date. [25,26,27].
In Kazakhstan, there is very little data on the composition of microbial communities in the most common species - H. scupense and H. asiaticum. Studying their microbiota and symbionts is important for understanding the bacterial communities of ticks. Such research can help to elucidate their ability to transmit pathogens to vertebrate hosts.
Next generation sequencing of the V2-4-8 and V3-6,7-9 variable regions of the bacterial 16S rRNA gene was performed using H. scupense and H. asiaticum samples using the Ion Torrent platform.
We found that the most dominant group in H. scupense were Francisella (89.0%), Staphylococcus (76.0%) and Candidatus Midichloria (61.0%). And in H. аsiaticum, the most dominant bacteria were Francisella (99.0% and 95.0%), Helcococcus (65.0%).
In the bacterial microbiota of Kazakhstan ticks H. scupense and H. asiaticum, the symbiotic/pathogenic bacterium Francisella predominates in the microbiome. According to literature data, Francisella predominates in the microbiome of H. excavatum and H. marginatum [28].
Recent studies confirm the widespread distribution of Francisella in other representatives of the genus Hyalomma. Francisella-like endosymbionts are transmitted vertically [29] and constitute the main components of the Hyalomma microbiome worldwide [17]. Thus, they have been identified in H. anatolicum in Pakistan, H. lusitanicum in Spain, H. aegyptium in Turkey, H. dromedarii in the Middle East, and also in H. asiaticum in China. [30,31,32,33].
These results demonstrate that the Francisella symbiont/pathogen plays a key role in shaping the microbial communities of Hyalomma ticks and likely has a significant impact on its vector competence. Bacterial endosymbionts may influence tick physiology and reproductive capacity, as well as the ability of ticks to transmit transmitted pathogens, and finally may interact with tick hosts, which may have veterinary and zoonotic implications, in particular for Francisella and Rickettsia bacteria [34]. Staphylococcus and Helcococcus were the most common pathogens in H. scupense and H. asiaticum, respectively, indicating the possible presence of these microorganisms in animal populations parasitized by these tick species.
Identification of Helcococcus spp. is challenging due to the slow growth of these organisms [35]. In our study, Helcococcus was identified using 16S rRNA gene sequencing. Members of the genus Helcococcus are known to cause mastitis and urocystitis in animals [36]. Different species of Staphylococci vary in their virulence, which determines different levels of health threat. Staphylococcus probably enters the tick's body from the environment and persists throughout its life cycle [37].
Their exact role is still unknown, but pathogenic species such as Staphylococcus lentus and Staphylococcus saprophyticus, which were previously found in Hyalomma ticks [ 38], may be the reason why staphylococci are frequently detected in these ticks. In our study, Staphylococcus accounted for 7.0% of the microbiota in H. asiaticum and 76% in H. scupense, confirming the prevalence of this genus of bacteria in these tick species. Candidatus Midichloria mitochondrii is a widespread endosymbiont of ticks [39]. This bacterium plays a critical role in the growth and development of the host, providing it with additional sources of ATP and B vitamins, which contributes to an increase in the adaptive capabilities and general biology of ticks [40]. Previously, Candidatus Midichloria mitochondrii was detected in H. anatolicum in Xinjiang, China [41]. In our study, this endosymbiont was detected only in H. scupense.
Blood feeding is known to have a strong impact on tick microbial diversity, composition, and species richness [42]. Ticks acquire more microorganisms, including pathogens, from hosts during blood feeding. More diverse and larger numbers of vertebrate hosts result in a more diverse microbiota [43].
Comparative analysis of the microbial communities associated with H. scupense and H. asiaticum revealed significant differences in their bacterial composition. A total of 15 bacterial genera were identified in H. scupense, while 19 genera were identified in H. asiaticum. Eight genera of Corynebacterium, Staphylococcus, Mannheimia, Francisella, Pseudomonas, Acinetobacter, Clostridium sensu stricto 3, and Atopostipes (30.8%) were common to both species, forming a conserved component of the microbiota. At the same time, 69.2% of the bacterial composition is accounted for by species-specific taxa: 7 unique genera in H. scupense (26.9% of the total), including Bifidobacterium, Candidatus Midichloria, Coxiella, Stenotrophomonas, Rickettsia, Lachnoclostridium, and Sphingobacterium and 11 unique genera in H. asiaticum (42.3%), including Escherichia-Shigella, Fusobacterium, Parvimonas, Helcococcus, Campylobacter, Porphyromonas, Trueperella, Erwinia, Streptococcus, Solibacillus, and Bacillus.
Despite the presence of a common bacterial base, a significant proportion of the bacterial composition differs between H. scupense and H. asiaticum. The data obtained highlight both the species-specificity of microbial associations and the potential influence of environmental [44,45], geographic [46] and other factors on the formation of microbial communities in ticks.
Our study revealed a difference in the microbiota composition between the sexes of H. scupense and H. asiaticum ticks. In male H. scupense, a higher prevalence of the endosymbiont Francisella was found, which constituted the dominant part of the microbiome - 89% in samples from Zhambyl_Shu. At the same time, in females, the proportion of Francisella was significantly lower: 13% - Kyzylorda_Zhalagash 1, 28% Kyzylorda_Zhalagash 2 and only 2% - Turkestan_Otyrar. This difference may indicate sexual characteristics in the formation of microbial communities and the role of endosymbionts in H. scupense, which requires further study.
H. asiaticum exhibits significant sex differences in the microbiota composition. In males from the Zhetysu_Chulakai region, the proportion of Helcococcus was 65%, while in females from Zhetysu_Zharkent it was only 11%. This indicates a potential sex specificity in the formation of the bacterial community. According to the literature, Helcococcus was not so widely detected in H. truncatum collected from cattle; it was present with a relative frequency of about 4.7% [47].
Males and females may have different contacts with the host, habitat, and food, which may determine differences in the microbiota, including the predominance of Helcococcus in males.
Our results expand our understanding of the biodiversity and microbiota ecology of Hyalomma in Central Asia and highlight the need for further research to understand the contribution of endosymbionts and pathogens to the vector competence of these ticks. These results are particularly important for Central Asian countries, where Hyalomma ticks are widespread and play an important role in the transmission of zoonotic infections. The lack of data on the microbiota of local tick populations hinders understanding of epidemiological risks in the region. Our results fill this gap and provide a basis for the development of preventive and diagnostic strategies aimed at reducing the threat to human and animal health in Kazakhstan and neighboring Central Asian countries.
5. Conclusions
Our study characterized the microbial communities of H. scupense and H. asiaticum from the southern and southeastern regions of Kazakhstan for the first time using next-generation sequencing.
H. scupense and H. asiaticum ticks contained 15 and 19 genera of bacteria, respectively, with only eight (30.8%) being common. 69.2% of the bacterial composition consisted of species-specific taxa, with 26.9% for H. scupense and 42.3% for H. asiaticum. The dominance of Francisella in both species confirms its key role in the microbiome of the genus Hyalomma, while the identified sex differences - the predominance of Francisella in male H. scupense and Helcococcus in male H. asiaticum - indicate an additional level of regulation of microbial associations. These results expand our understanding of the biodiversity and microbiota ecology of Hyalomma in Central Asia and highlight the need for further research aimed at understanding the contribution of endosymbionts and pathogens to the vector competence of these ticks.
Author Contributions
Conceptualization, K.T.S. and O.M.B.; Methodology, N.S.K. and G.O.Sh.; Software, T.O.A.; Validation, N.S.K., M.D.A. and S.B.Zh.; Formal Analysis, O.V.Ch. M.R.A.; Investigation, G.O.Sh.; Resources, G.O.Sh.; Data Curation, K.T.S.; Writing - Original Draft Prepa-ration, K.T.S.; Writing – Review & Editing, K.T.S. and O.M.B.; Visualization, M.D.A., G.O.Sh. and N.S.K.; Supervision, O.V.Ch and N.S.K.; Project Administration, K.T.S. and T.O.A.; Funding Acquisition, K.T.S.
Funding
This research was funded by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan (Grant No. АР19677632).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of the Research Institute for Biological Safety Problems of the Ministry of Health of the Republic of Kazakhstan (permit number: №10/14-11-22, 14 November 2022).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Stachurski F, Moutailler S, Boissier J. Etude des tiques et maladies transmises en milieu insulaire méditerranéen; l ‘exemple du cheptel bovin corse Ticks and tick-borne pathogens on a Mediterranean island environment: the Corsican cattle. 2016, 335–338.
- Parola, P.; Raoult, D. Ticks and tickborne bacterial diseases in humans: An emerging infectious threat. Clin. Infect. Dis. 2001, 32, 897–928. [Google Scholar] [CrossRef]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasit. Vectors. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Wu-Chuang, A.; Hodzic, A.; Mateos-Hernandez, L.; Estrada-Pena, A.; Obregon, D.; Cabezas-Cruz, A. Current debates and advances in tick microbiome research. Curr. Res. Parasitol. Vector Borne Dis. 2021, 1, 100036. [Google Scholar] [CrossRef] [PubMed]
- Guglielmone, A.A.; Robbins, R.G.; Apanaskevich, D.A.; Petney, T.N.; Estrada-Peña, A.; Horak, I.G.; Shao, R.; Barker, S. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida) of the world: a list of valid species names. Zootaxa. 2010, 2528, 1–28. [Google Scholar] [CrossRef]
- Sonenshine, D.E.; Lane, R.; Nicholson, W. Ticks (Ixodida). In: Mullen, G.; Durden, L., Eds. Medical and Veterinary Entomology; Academia: Orlando. 2002; pp. 517–558.
- Sayakova, Z.Z.; Abdybekova, A.M.; Zhaksylykova, A.A.; Kenessary, S.A.; Berdiakhmetkyzy, S.; Kulemin, M.V. Distribution of Hyalomma asiaticum Schulze et Schlottke, 1929 (Acari, Ixodidae) ticks in southern Kazakhstan. Science and education. 2024, 2024. 3, 76. [Google Scholar]
- Kulemin, M.V.; Rapoport, L.P.; Vasilenko, A.V.; Kobeshova, Zh.B.; Shokputov, T.M.; Saylaubekuly, R.; Atovullaeva, L.M. Ixodid ticks of farm animals in Southern Kazakhstan: fauna structure, abundance, epizootological significance. Parazitologiya. 2020, 54, 25–31. [Google Scholar]
- Perfilyeva, Y.V.; Shapiyeva, Z.Zh.; Ostapchuk, Y.O.; Berdygulova, Z.A.; Bissenbay, A.O.; Kulemin, M.V.; Ismagulova, G.A.; Skiba, Y.A.; Sayakova, Z.Z.; Mamadaliyev, S.M.; Maltseva, E.R.; Dmitrovskiy, A.M. Tick-borne pathogens and their vectors in Kazakhstan. Ticks and Tick-Borne Dis. 2020, 11, 101498. [Google Scholar] [CrossRef]
- Sultankulova, K.T.; Shynybekova, G.O.; Kozhabergenov, N.S.; Mukhami, N.N.; Chervyakova, O.V.; Burashev, Y.D.; Zakarya, K.D.; Nakhanov, A.K.; Barakbayev, K.B.; Orynbayev, M.B. The prevalence and genetic variants of the CCHF virus circulating among ticks in the southern regions of Kazakhstan. Pathogens. 2022, 11, 841. [Google Scholar] [CrossRef]
- Sultankulova, K.T.; Shynybekova, G.O.; Issabek, A.U.; Mukhami, N.N.; Melisbek, A.M.; Chervyakova, O.V.; Kozhabergenov, N.S.; Barmak, S.M.; Bopi, A.K.; Omarova, Z.D.; Alibekova, D.A.; Argimbayeva, T.U.; Namet, A.M.; Zuban, I.A.; Orynbayev, M.B. The prevalence of pathogens among ticks collected from livestock in Kazakhstan. Pathogens. 2022, 11, 1206. [Google Scholar] [CrossRef]
- Orynbayev, M.B.; Rystaeva, R.A.; Omarova, Z.D.; Kerimbaev, A.A.; Sarsenbaeva, G.Zh.; Kopeev, S.K.; Nakhanov, A.K.; Strochkov, V.M.; Sultankulova, K. Isolation of Coxiella burnetii from ticks in Kazakhstan. Biosafety Biotechnol. 2020, 1, 62–67. [Google Scholar]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Kratou, M.; Maitre, A.; Abuin-Denis, L.; Selmi, R.; Belkahia, H.; Alanazi, A.D.; Gattan, H.; Al-Ahmadi, B.M.; Shater, A.F.; Mateos-Hernández, L.; Obregón, D.; Messadi, L.; Cabezas-Cruz, A.; Ben Said, M. Microbial community variations in adult Hyalomma dromedarii ticks from Saudi Arabia and Tunisia. Front. Microbiol. 2025, 16, 1543560. [Google Scholar] [CrossRef]
- Aguilar-Díaz, H.; Quiroz-Castañeda, R.E.; Cobaxin-Cárdenas, M.; Salinas-Estrella, E.; Amaro-Estrada, I. Advances in the study of the tick cattle microbiota and the influence on vectorial capacity. Front. Vet. Sci. 2021, 8, 710352. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Ruiz, P. The tick microbiome: the “other bacterial players” in tick biocontrol. Microorganisms. 2024, 12, 2451. [Google Scholar] [CrossRef] [PubMed]
- Masri, M.T.A.; Al-Deeb, M.A. A systematic review of the microbiome of Hyalomma Koch, 1844 ticks using next-generation sequencing of the 16S rRNA gene. Vet. World. 2025, 18, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Apanaskevich, D.A.; Horak, I.G. The genus Hyalomma. XI. Redescription of all parasitic stages of H. (Euhyalomma) asiaticum and notes on its biology. Exp. Appl. Acarol. 2010, 52, 207–220. [Google Scholar] [CrossRef]
- Lv, J.; Wu, S.; Zhang, Y. Assessment of four DNA fragments (COI, 16S rDNA, ITS2, 12S rDNA) for species identification of the Ixodida (Acari: Ixodida). Parasit. Vectors. 2014, 7, 93. [Google Scholar] [CrossRef]
- Thukral, A.K.; Bhardwaj, R.; Kumar, V.; Sharma, A. New indices regarding the dominance and diversity of communities, derived from sample variance and standard deviation. Heliyon. 2019, 5, e02606. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature. 1949, 168, 668. [Google Scholar] [CrossRef]
- Margalef, D.R. Information theory in ecology. Gen. Syst. 1958, 3, 36–71. [Google Scholar]
- Krebs, C.J. Ecological Methodology; Menlo Park: California. 1999.
- Pesenko, Yu.A. Principles and methods of quantitative analysis in faunistic studies; Moscow. 1982.
- Varela-Stokes, A.S.; Park, S.H.; Kim, S.A.; Ricke, S.C. Microbial communities in North American ixodid ticks of veterinary and medical importance. 2017, 4, 1–7. 4.
- René-Martellet, M.; Minard, G.; Massot, R.; Van, V.T.; Moro, C.V.; Chabanne, L.; et al. Bacterial microbiota associated with Rhipicephalus sanguineus ticks from France, Senegal and Arizona. 2017, 1–10.
- Lim, F.S.; Khoo, J.J.; Tan, K.K.; Zainal, N.; Loong, S.K.; Khor, C.S.; et al. Bacterial communities in Haemaphysalis, Dermacentor and Amblyomma ticks collected from wild boar in Malaysia. Ticks Tick-Borne Dis. 2020, 11.
- Keskin, A.; Bursali, A.; Snow, D.E.; Dowd, S.E.; Tekin, S. Assessment of bacterial diversity in Hyalomma. Exp. Appl. Acarol. 2017. [Google Scholar]
- Scoles, G.A. Phylogenetic analysis of the Francisella-like endosymbionts of Dermacentor ticks. J. Med. Entomol. 2004, 41, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.; Budachetri, K.; Mukherjee, N.; Williams, J.; Kausar, A.; Hassan, M.J.; et al. A study of ticks and tick-borne livestock pathogens in Pakistan. 2017, 1–17.
- Ghafar, A.; Cabezas-Cruz, A.; Galon, C.; Obregon, D.; Gasser, R.B.; Moutailler, S.; et al. Bovine ticks harbour a diverse array of microorganisms in Pakistan. Parasit. Vectors. 2020, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kratou, M.; Maitre, A.; Abuin-Denis, L. ; Selmi, R; Belkahia, H. ; Alanazi, A.D.; et al. Microbial community variations in adult Hyalomma dromedarii ticks from Saudi Arabia and Tunisia. Front. Microbiol. 2025, 16, 1543560. [Google Scholar]
- Duan, L.; Yang, X.; Zhang, L. The differences in microbial communities and tick-borne pathogens between Dermacentor marginatus and Hyalomma asiaticum collected from Northwestern China. BMC Infect. Dis. 2025, 25, 1019. [Google Scholar] [CrossRef]
- Benyedem, H.; Lekired, A.; Mhadhbi, M.; Dhibi, M.; Romdhane, R.; Chaari, S.; Rekik, M.; Ouzari, H.I.; Hajji, T.; Darghouth, M.A. First insights into the microbiome of Tunisian Hyalomma ticks with a focus on H. scupense. PLoS ONE. 2022, 17, e0269823. [Google Scholar] [CrossRef]
- Collins, M.D.; Falsen, E.; Brownlee, K.; Lawson, P.A. Helcococcus sueciensis sp. nov., isolated from a human wound. Int. J. Syst. Evol. Microbiol. 2004, 54, 1557–1560. [Google Scholar] [CrossRef]
- Collins, M.D.; Falsen, E.; Foster, G.; Monasterio, L.R.; Dominguez, L.; Fernandez-Garayzabal, J.F. Helcococcus ovis sp. nov., a Gram-positive organism from sheep. Int. J. Syst. Bacteriol. 1999, 49, 1429–1432. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhou, X.J.; Chen, K.L.; Masoudi, A.; Liu, J.Z.; Zhang, Y.K. Bacterial microbiota analysis demonstrates that ticks acquire bacteria from habitat and host blood meal. Exp. Appl. Acarol. 2021, 87, 81–95. [Google Scholar]
- Li, C.H.; Cao, J.; Zhou, Y.Z.; Zhang, H.S.; Gong, H.Y.; Zhou, J.L. The midgut bacterial flora of laboratory-reared ticks (Haemaphysalis longicornis, Hyalomma asiaticum, Rhipicephalus haemaphysaloides). J. Integr. Agric. 2014, 13, 1766–1771. [Google Scholar] [CrossRef]
- Beninati, T.; Lo, N.; Sacchi, L.; Genchi, C.; Noda, H.; Bandi, C. A novel alpha-Proteobacterium resides in mitochondria of ovarian cells of Ixodes ricinus. Appl. Environ. Microbiol. 2004, 70, 2596–2602. [Google Scholar] [CrossRef]
- Olivieri, E.; Epis, S.; Castelli, M. Tissue tropism and metabolic pathways of Midichloria mitochondrii in Ixodes ricinus. Ticks and Tick-Borne Dis. 2019, 10, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Hu, E.; Gan, L.; Yang, D.; Wu, J.; Gao, S.; Tuo, X.; Bayin, C.G.; Hu, Z.; Guo, Q. Candidatus Midichloria mitochondrii can be vertically transmitted in Hyalomma anatolicum. Exp. Parasitol. 2024, 265, 108828. [Google Scholar] [CrossRef] [PubMed]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017, 11, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Pollet, T.; Sprong, H.; Lejal, E.; Krawczyk, A.I.; Moutailler, S.; Cosson, J.F.; et al. Scale affects identification and distribution of microbial communities in ticks. Parasit. Vectors. 2020, 1–13. [Google Scholar] [CrossRef]
- Aivelo, T.; Norberg, A.; Tschirren, B. Bacterial microbiota composition of Ixodes ricinus: environmental variation, tick characteristics and microbial interactions. 2019, 1–25.
- Trout Fryxell, R.T.; DeBruyn, J.M. The microbiome of Ehrlichia-infected and uninfected lone star ticks (Amblyomma americanum). PLoS ONE. 2016, 11, e0168994. [Google Scholar]
- van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; et al. Variation in the microbiota of Ixodes ticks with geography, species, and sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef]
- Chigwada, A.D.; Mapholi, N.O.; Ogola, H.J.O.; Mbizeni, S.; Masebe, T.M. Pathogenic and endosymbiotic bacteria and associated antibiotic resistance biomarkers in Amblyomma and Hyalomma ticks infesting Nguni cattle. Pathogens. 2022, 11, 432. [Google Scholar] [CrossRef]
Figure 1.
Visualization of the geographic distribution of H. scupense and H. asiaticum ticks using cartographic analysis in QGIS.  - Kyzylorda_Zhalagash1_H. scupense, Kyzylorda_Zhalagash2_H. scupense, Kyzylorda_Kazaly_H. scupense, Zhambyl_Shu_H. scupense, Turkestan_Otyrar_H. scupense.
- Kyzylorda_Zhalagash1_H. scupense, Kyzylorda_Zhalagash2_H. scupense, Kyzylorda_Kazaly_H. scupense, Zhambyl_Shu_H. scupense, Turkestan_Otyrar_H. scupense.  - Zhambyl_Zhanatas1_H. asiaticum, Zhambyl_Zhanatas2_H. asiaticum, Zhambyl_Ryskulov_H. asiaticum, Zhetysu_Zharkent_H. asiaticum, Zhetysu_Chulakai_H. asiaticum
- Zhambyl_Zhanatas1_H. asiaticum, Zhambyl_Zhanatas2_H. asiaticum, Zhambyl_Ryskulov_H. asiaticum, Zhetysu_Zharkent_H. asiaticum, Zhetysu_Chulakai_H. asiaticum
- Kyzylorda_Zhalagash1_H. scupense, Kyzylorda_Zhalagash2_H. scupense, Kyzylorda_Kazaly_H. scupense, Zhambyl_Shu_H. scupense, Turkestan_Otyrar_H. scupense. - Zhambyl_Zhanatas1_H. asiaticum, Zhambyl_Zhanatas2_H. asiaticum, Zhambyl_Ryskulov_H. asiaticum, Zhetysu_Zharkent_H. asiaticum, Zhetysu_Chulakai_H. asiaticum
Figure 1.
Visualization of the geographic distribution of H. scupense and H. asiaticum ticks using cartographic analysis in QGIS. - Kyzylorda_Zhalagash1_H. scupense, Kyzylorda_Zhalagash2_H. scupense, Kyzylorda_Kazaly_H. scupense, Zhambyl_Shu_H. scupense, Turkestan_Otyrar_H. scupense. - Zhambyl_Zhanatas1_H. asiaticum, Zhambyl_Zhanatas2_H. asiaticum, Zhambyl_Ryskulov_H. asiaticum, Zhetysu_Zharkent_H. asiaticum, Zhetysu_Chulakai_H. asiaticum
- Kyzylorda_Zhalagash1_H. scupense, Kyzylorda_Zhalagash2_H. scupense, Kyzylorda_Kazaly_H. scupense, Zhambyl_Shu_H. scupense, Turkestan_Otyrar_H. scupense. - Zhambyl_Zhanatas1_H. asiaticum, Zhambyl_Zhanatas2_H. asiaticum, Zhambyl_Ryskulov_H. asiaticum, Zhetysu_Zharkent_H. asiaticum, Zhetysu_Chulakai_H. asiaticum
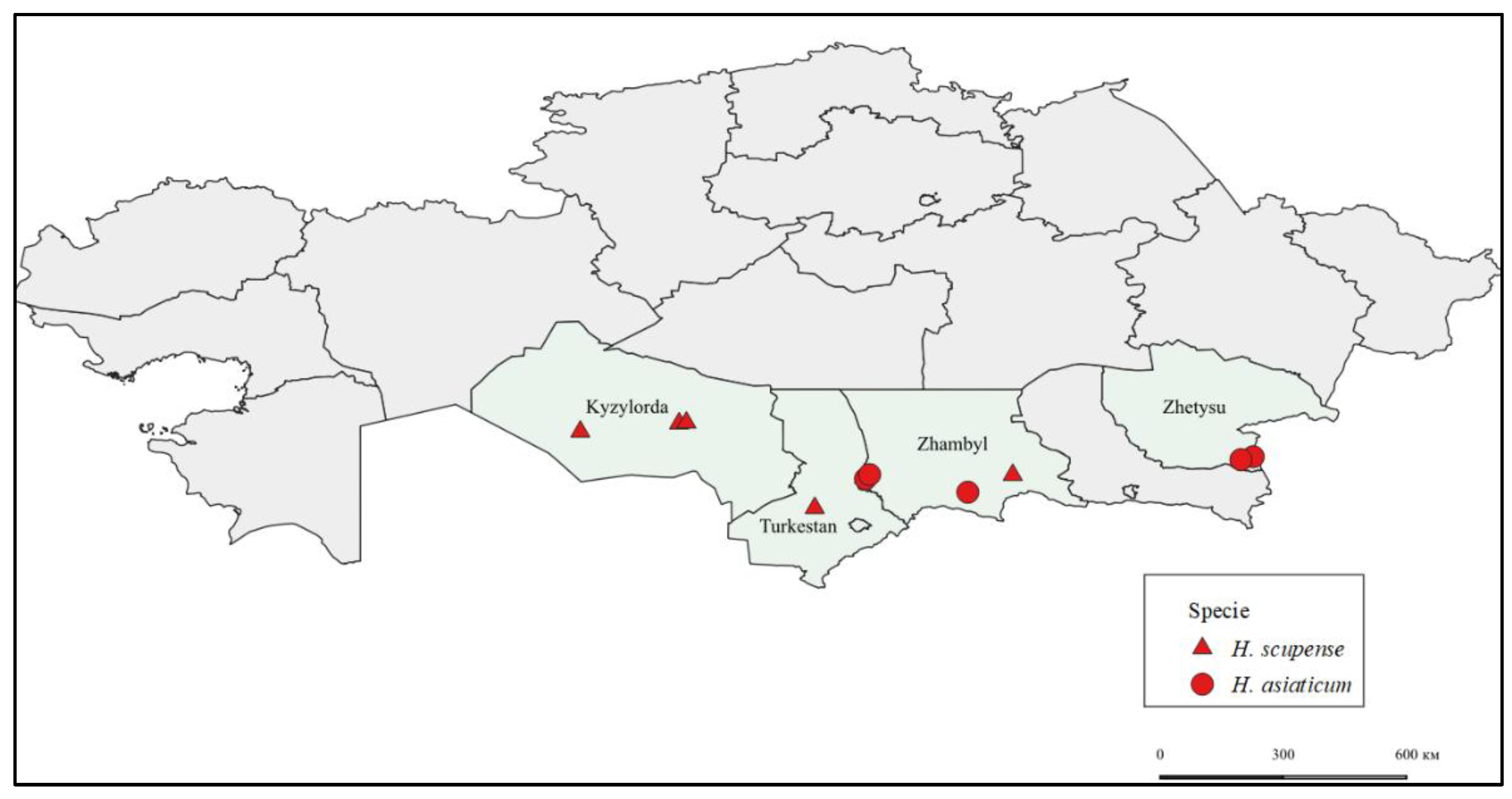
Figure 2.
Phylogenetic analysis of the cytochrome c oxidase subunit I (COX1) gene of H. scupense and H. asiaticum ticks.  - Hyalomma scupense identified in this study based on the COX1 gene.
- Hyalomma scupense identified in this study based on the COX1 gene.  - Hyalomma asiaticum identified in this study based on the COX1 gene.
- Hyalomma asiaticum identified in this study based on the COX1 gene.
- Hyalomma scupense identified in this study based on the COX1 gene. - Hyalomma asiaticum identified in this study based on the COX1 gene.
Figure 2.
Phylogenetic analysis of the cytochrome c oxidase subunit I (COX1) gene of H. scupense and H. asiaticum ticks. - Hyalomma scupense identified in this study based on the COX1 gene. - Hyalomma asiaticum identified in this study based on the COX1 gene.
- Hyalomma scupense identified in this study based on the COX1 gene. - Hyalomma asiaticum identified in this study based on the COX1 gene.
Figure 3.
Metagenomic profiles of the prevalence of 26 major bacterial genera detected in H. scupense and H. asiaticum tick samples using the Ion GeneStudio S5 System.
Figure 3.
Metagenomic profiles of the prevalence of 26 major bacterial genera detected in H. scupense and H. asiaticum tick samples using the Ion GeneStudio S5 System.

Figure 4.
Assessment of alpha diversity and variation of bacterial communities in H. scupense and H. asiaticum ticks. Rank and abundance diagrams of bacterial communities in H. scupense (A) and H. asiaticum (B). Shannon-Wiener index (C), Margalef index (D), and Simpson index (E). The line inside each boxplot indicates the median value.
Figure 4.
Assessment of alpha diversity and variation of bacterial communities in H. scupense and H. asiaticum ticks. Rank and abundance diagrams of bacterial communities in H. scupense (A) and H. asiaticum (B). Shannon-Wiener index (C), Margalef index (D), and Simpson index (E). The line inside each boxplot indicates the median value.
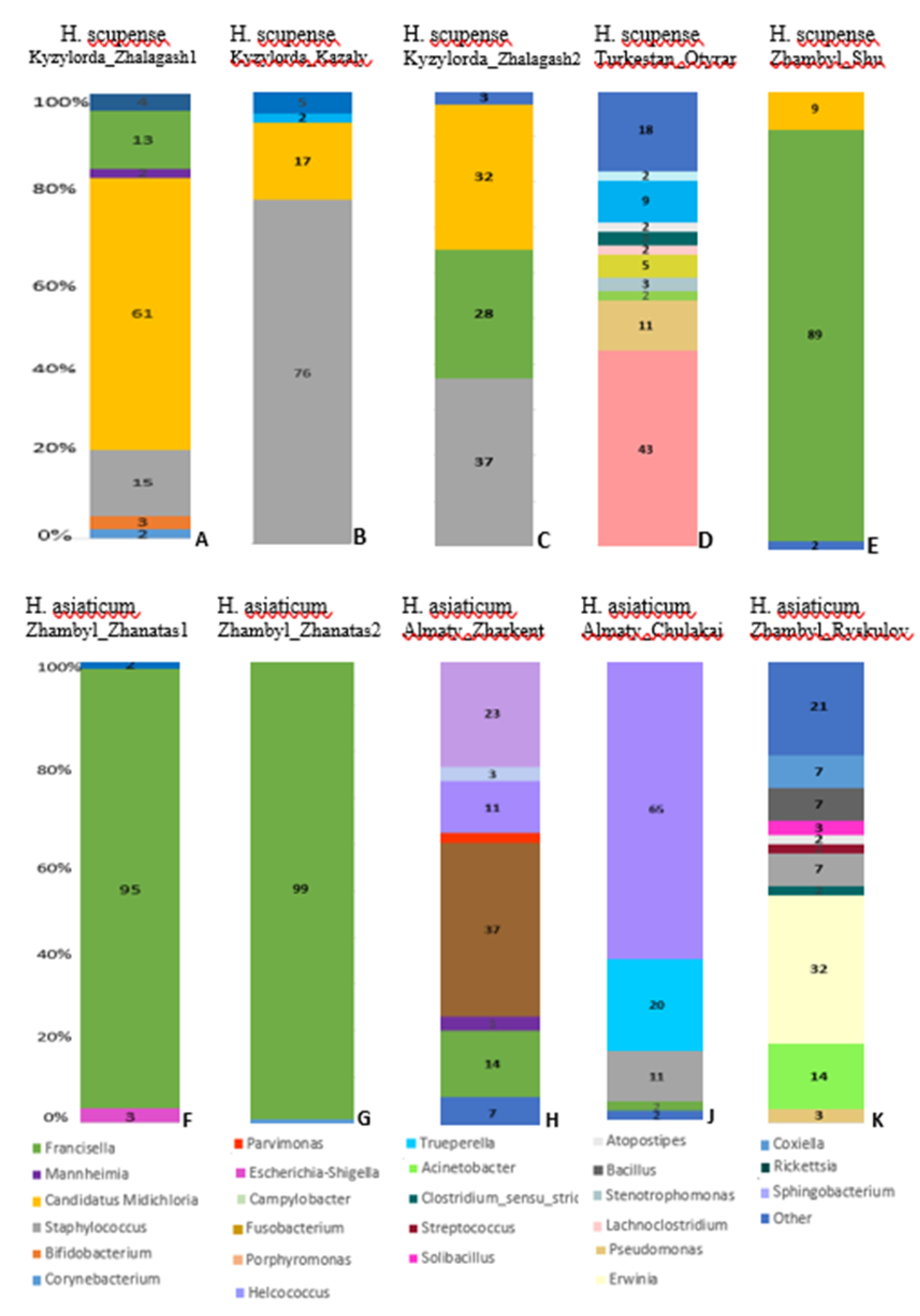
Figure 5.
Geographic distribution of bacteria carried by H. scupense and H. asiaticum ticks based on Principal Coordinates Analysis (PCoA). Scatter plots visualize the data with PCoA1 and PCoA2 axes. Each point on the graphs corresponds to tick samples of H. asiaticum (A) and H. scupense (B), with point colors indicating their regional origin. Microbial community cladograms for H. scupense (C) and H. asiaticum (D) illustrate bacterial composition differences according to regions of Kazakhstan: Kyzylorda, Turkestan, Zhambyl and Zhetysu.
Figure 5.
Geographic distribution of bacteria carried by H. scupense and H. asiaticum ticks based on Principal Coordinates Analysis (PCoA). Scatter plots visualize the data with PCoA1 and PCoA2 axes. Each point on the graphs corresponds to tick samples of H. asiaticum (A) and H. scupense (B), with point colors indicating their regional origin. Microbial community cladograms for H. scupense (C) and H. asiaticum (D) illustrate bacterial composition differences according to regions of Kazakhstan: Kyzylorda, Turkestan, Zhambyl and Zhetysu.
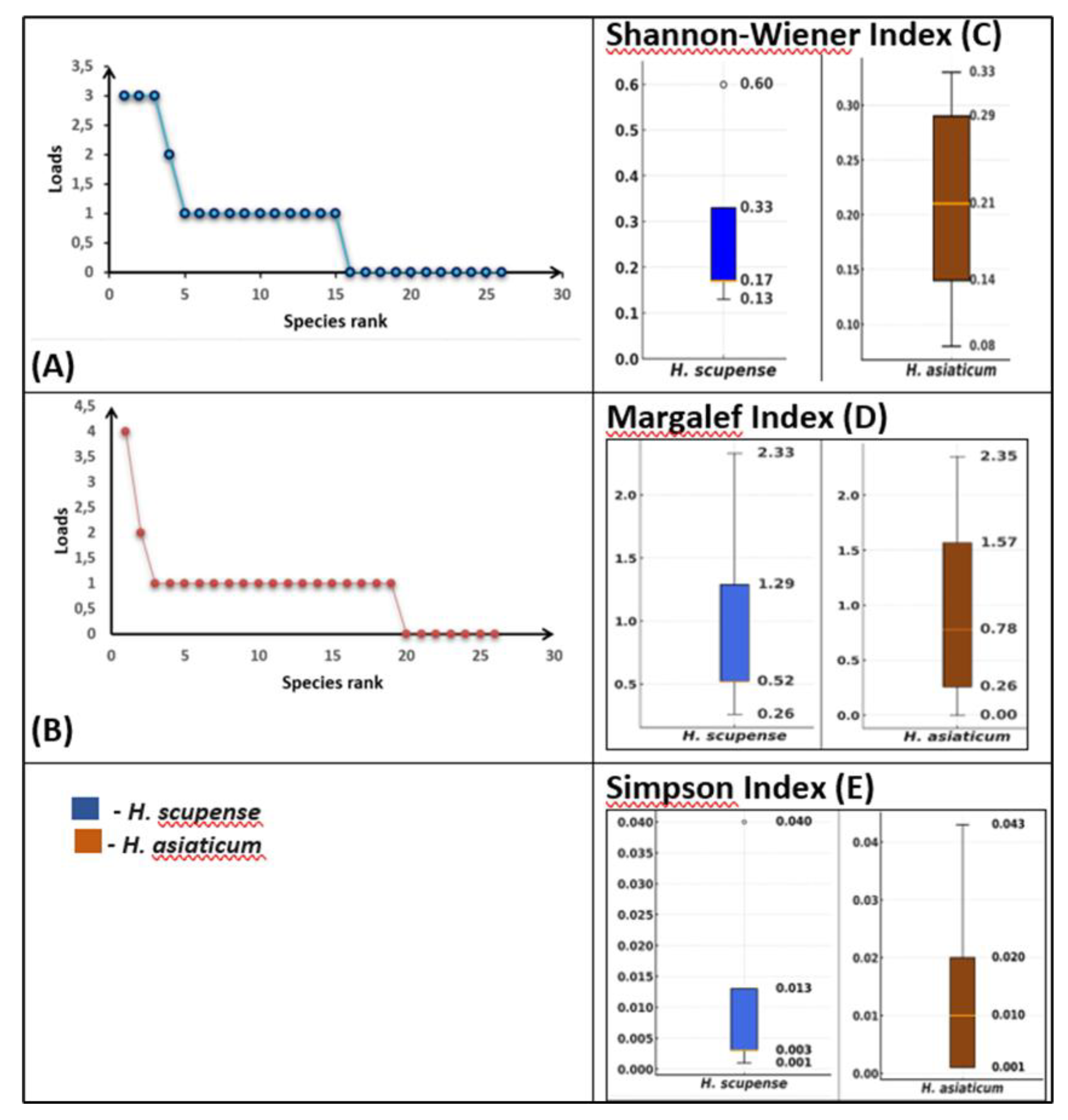
Figure 6.
Heatmap of the relative abundance of bacteria present in H. scupense and H. asiaticum ticks.
Figure 6.
Heatmap of the relative abundance of bacteria present in H. scupense and H. asiaticum ticks.
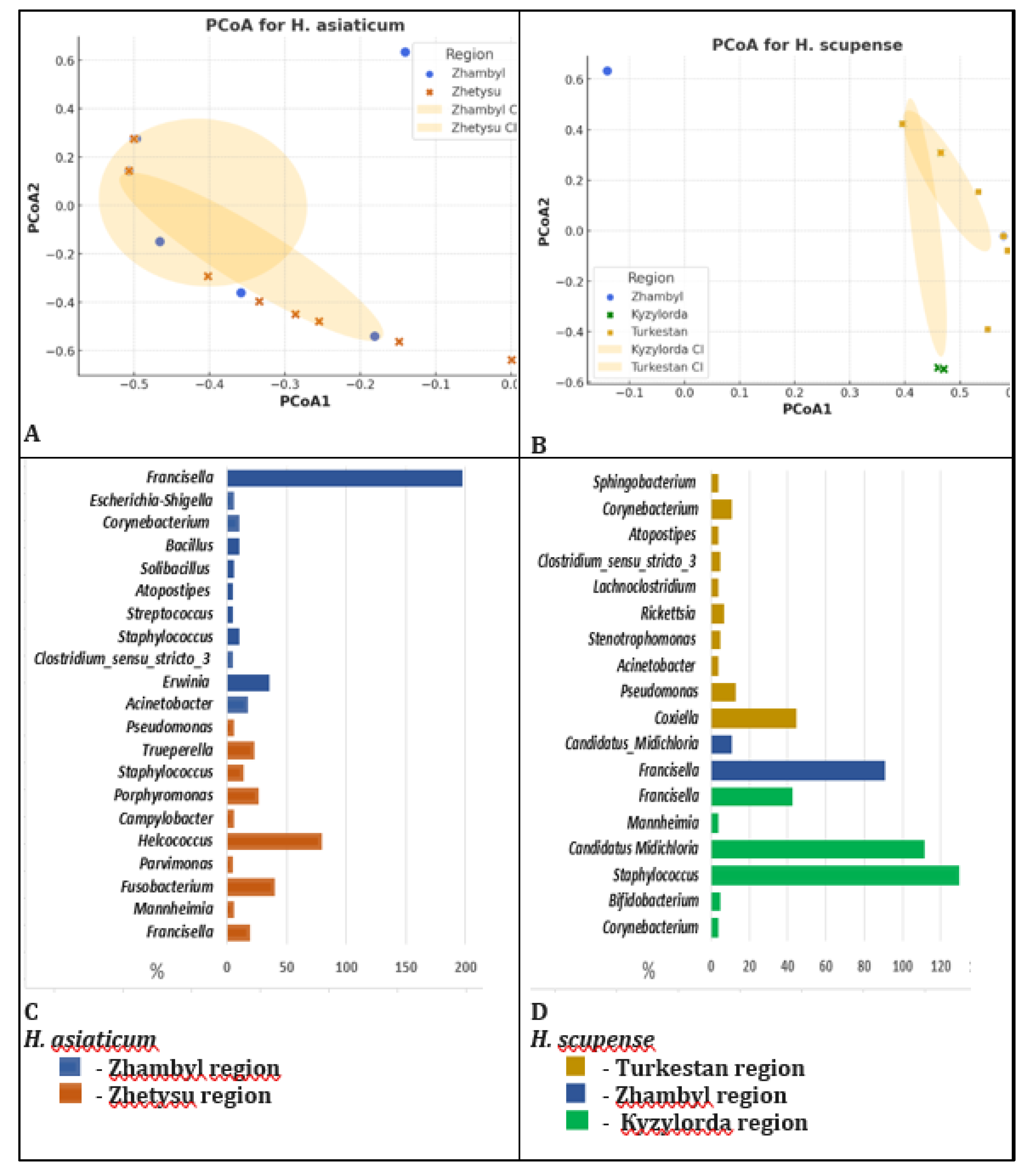

Table 1.
Tick sampling information from southern and southeastern regions of Kazakhstan.
| Sample Collection Site | Coordinates | No. of Ticks | Tick Species | Sex | Host | Genbank Accession No | |
|---|---|---|---|---|---|---|---|
| 1 | Kyzylorda_Zhalagash1 | 45°04′55″ 64°40′51″ |
10 | H. scupense | ♀ | Cattle | PQ560690 |
| 2 | Zhambyl_Zhanatas1 | 43°34′00″ 69°45′00″ |
10 | H. asiaticum | ♀ | Cattle | PQ569438 |
| 3 | Kyzylorda_Kazaly | 44°51′00″ 61°59′24″ |
10 | H. scupense | ♀ | Cattle | PQ560881 |
| 4 | Kyzylorda_Zhalagash2 | 45°04′55″ 64°40′51″ |
10 | H. scupense | ♀ | Cattle | PQ573248 |
| 5 | Zhambyl_Zhanatas2 | 43°34′00″ 69°45′00″ |
10 | H. asiaticum | ♀ | Cattle | PQ569449 |
| 6 | Zhambyl_Shu | 43°40′32″ 73°45′40″ |
8 | H. scupense | ♂ | Cattle | PQ571834 |
| 7 | Zhetysu_Zharkent |
44°10′00″ 80°00′00″ |
10 | H. asiaticum | ♀ | Cattle | PQ560954 |
| 8 | Zhetysu _Chulakai |
44°05′15″ 79°58′00″ |
6 | H. asiaticum | ♂ | Cattle | PQ560955 |
| 9 | Zhambyl_Ryskulov | 43°12′00″ 72°32′00″ |
10 | H. asiaticum | ♀ | Cattle | PQ578371 |
| 10 | Turkestan_Otyrar | 42°46′36″ 68°22′09″ |
10 | H. scupense | ♀ | Cattle | PQ569618 |
Table 2.
Indices for quantitative assessment of alpha and beta diversity parameters.
| Index Name | Formula | Diversity Level |
|---|---|---|
| Shannon–Wiener diversity index | 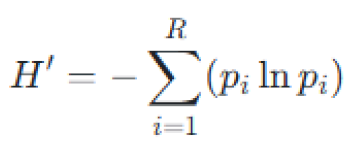 |
Alpha diversity |
| Margalef richness index |
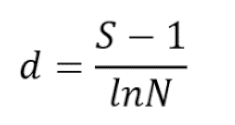 |
Alpha diversity |
| Simpson’s dominance index | 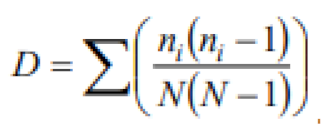 |
Alpha diversity |
| Bray–Curtis dissimilarity index | 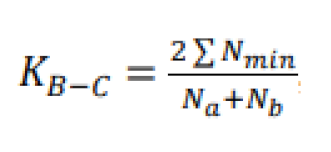 |
Beta diversity |
| Jaccard similarity index | 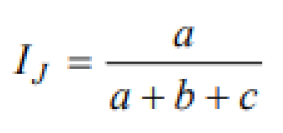 |
Beta diversity |
| Note: Shannon-Wiener index (H'): where pipi is the proportion of individuals belonging to the ii-th species, RR is the total number of species, and ln denotes the natural logarithm. Margalef’s index (d): where SS is species richness (total number of species), and NN is the sample size (total number of individuals in the community). Simpson’s index (D): where nini is the number of individuals of the ii-th species, and NN is the total number of individuals in the sample. Bray-Curtis dissimilarity index (BC): where NaNa is the total abundance (sum of counts) in the first community, NbNb is the total abundance in the second community, and Nmin represents the sum of the lesser abundances for each taxon shared between the two communities. Jaccard similarity index (J): where aa is the number of species common to both lists, bb is the number of species present only in the first list, and cc is the number of species present only in the second list. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



