
Submitted:
29 August 2025
Posted:
01 September 2025
You are already at the latest version
Abstract
We present a detailed first-principles investigation of the vibrational and spectroscopic properties of four layered boron nitride (BN) polymorphs–e-BN (AA), h-BN (AA′), r-BN (ABC), and b-BN (AB). Using density functional perturbation theory with van der Waals corrections, we calculate phonon frequencies and Raman and infrared (IR) modes at the Γ point to identify distinct stacking-dependent spectral signatures. Our results reveal that r-BN exhibits the strongest combined Raman and IR response, dominated by a high-frequency E mode at 1399.9 cm−1, while e-BN is characterized by a sharp, isolated E′ mode at 1402.4 cm−1. In h-BN, the characteristic E2g Raman mode at 1396.7 cm−1 is accompanied by a unique mid-frequency A2u IR mode at 704.4 cm−1. The b-BN polytype shows its most distinctive IR feature at 728.3 cm−1 (A2′′), together with a high-frequency E′ Raman/IR mode at 1409.5 cm−1. These spectroscopic fingerprints provide a reliable means to distinguish BN polytypes and directly support recent experimental efforts to determine stacking order via vibrational spectroscopy. This study establishes a robust theoretical framework for interpreting Raman and IR spectra of BN polymorphs and guiding the characterization of layered sp2-bonded materials.
Keywords:
boron nitride
; polytypes
; first-principles calculations
; vibrational properties
1. Introduction
Hexagonal boron nitride (h-BN) is a prototypical layered material with a honeycomb lattice structure analogous to graphite, composed of strongly bonded in-plane B–N atoms ( hybridization) and weakly interacting interlayer planes. This structural anisotropy imparts h-BN with a wide band gap, exceptional thermal and chemical stability, and a rich vibrational spectrum, positioning it as a vital material in diverse applications including deep ultraviolet optoelectronics, two-dimensional (2D) heterostructures, and quantum light emission [1,2]. One of the most intriguing and technologically relevant aspects of h-BN is its tendency to form various stacking sequences, or polytypes, due to the nearly degenerate interlayer energy landscape. These polytypes–notably AA′ (h-BN), ABC (r-BN), AA (e-BN), and AB (b-BN)–differ only in how their layers are arranged along the out-of-plane direction, yet exhibit markedly distinct electronic, vibrational, and optical properties [2,3,4,5,6]. The co-existence of these polytypes within single crystals leads to structural disorder, interfaces, and stacking faults, which can affect charge transport, phonon lifetimes, and emission characteristics [3,7].
Despite extensive research, the thermodynamic and vibrational stability of boron nitride (BN) polymorphs remains a topic of active investigation and debate. While cubic BN (c-BN) is the ground state under high pressure, recent high-level calculations and experiments suggest that h-BN is thermodynamically stabilized at ambient conditions through entropic contributions and many-body van der Waals (vdW) interactions [8,9]. In particular, the subtle energy differences between the low-energy phases (such as h-BN and r-BN) are on the order of tens of meV per formula unit–a scale that demands theoretical methods beyond conventional density functional theory (DFT) to resolve [8,9,10]. Including nonlocal correlation effects is thus essential for capturing the relative stability and interlayer coupling in these materials. Stacking order is not merely a structural detail but a critical determinant of the electronic structure and vibrational response. For instance, AA′ stacking (common in h-BN) is predicted to be the lowest energy configuration among the layered forms [11]. In contrast, ABC stacking (r-BN) yields distinct optical and electronic properties, including a redshifted conduction band minimum and richer infrared (IR) and Raman activity [3,4,6]. Additionally, b-BN and e-BN represent higher-symmetry and more idealized stackings (AB and AA, respectively), which, while less studied experimentally, serve as critical structural models to explore the influence of interlayer registry and symmetry on physical properties [5].
Experimental studies using cathodoluminescence, Raman scattering, X-ray absorption, UV photoluminescence, and IR spectroscopy have revealed that polytypism dramatically influences the optoelectronic response of BN [2,3,7,12]. Notably, recent photoluminescence studies have succeeded in distinguishing BN polytypes through subtle spectral shifts and emission signatures [12,13]. However, interpreting these data is complicated by the frequent co-existence of polytypes, nanoscale disorder, and temperature-induced transformations [8,9]. In this context, vibrational spectroscopy-particularly Raman and IR techniques-has emerged as a powerful diagnostic tool for identifying BN polytypes, owing to its sensitivity to symmetry, stacking order, and interlayer coupling.
To support this approach, computational modeling plays a pivotal role in interpreting vibrational spectra and guiding polytype-specific material characterization. In this work, we present a comprehensive first-principles investigation of the vibrational and dielectric properties of four BN polymorphs–e-BN (AA), h-BN (AA′), r-BN (ABC), and b-BN (AB). We use density functional perturbation theory (DFPT) with and without vdW corrections to examine the stacking-dependent evolution of phonon frequencies, Raman and infrared activities, and dielectric properties. Our results provide detailed spectroscopic fingerprints-including irreducible representations and intensities-that enable clear identification of BN polytypes. Understanding these intricate relationships is crucial for the fundamental science of 2D materials and for engineering BN-based optoelectronic and quantum devices with targeted performance. The findings presented here aim to bridge this gap and contribute to a more predictive understanding of polytype-dependent vibrational behavior in layered BN.
2. Computational Details
We employed DFT, as implemented in the Quantum ESPRESSO (QE) package, to investigate the structural, electronic, and vibrational properties of different BN phases. The calculations were initiated using the experimental unit cell, with explicitly defined lattice parameters in Cartesian coordinates. A plane-wave energy cutoff of 80 Ry was chosen to ensure numerical accuracy, following a systematic convergence analysis. The exchange-correlation interactions were treated using the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA). Brillouin zone sampling was performed using a -centered Monkhorst-Pack k point mesh to ensure accurate integration. Given the layered nature of BN, vdW corrections were incorporated using the DFT-D approach [14]. The convergence criteria were set to Ry for total energy and Bohr for atomic forces. Projector-augmented wave (PAW) pseudopotentials were employed for electron-ion interactions, providing a reliable description of core-valence interactions while maintaining computational efficiency. The cohesive energy per atom, , was calculated as where and are the total energies of isolated boron and nitrogen atoms, respectively; and are the numbers of B and N atoms in the BN unit cell; is the total energy of the BN polymorph; and is the total number of atoms. The atomic energies were computed in large cubic cells to eliminate spurious interactions.
3. Results
3.1. Crystal Structures for Different Polymorphs of BN
The atomic structures of four layered BN polymorphs–e-BN (AA), h-BN (AA′), r-BN (ABC), and b-BN (AB)–are illustrated in Figure 1a–d. These polymorphs differ in stacking sequences, symmetry groups, and interlayer arrangements. Structural parameters, cohesive energies (), and electronic band gaps () are summarized in Table 1. The stacking order is a key factor controlling the interlayer spacing and stability of these -bonded systems.
e-BN (AA) crystallizes in the non-centrosymmetric space group (No. 187) and features direct AA stacking, where boron and nitrogen atoms are aligned vertically across layers (Figure 1a). This configuration leads to weak interlayer bonding in the absence of vdW corrections. Inclusion of vdW interactions significantly contracts the c-axis from 5.06 to 3.38 Å, as shown in Table 1, improving agreement with values reported by Gil et al. [2]. No experimental data exist for this stacking, but theoretical values are consistent across methods.
h-BN (AA′) is the most commonly observed polymorph, adopting the (No. 194) space group, with alternating B and N atoms stacked in a staggered bilayer configuration (Figure 1b). The calculated lattice constants with vdW corrections ( Å, Å) agree well with both theoretical and experimental values reported in Table 1 of Gil et al. [2] and Ahmed et al. [15], where Å and Å are cited as experimental averages. This stacking is energetically favored and widely accepted as the most stable -BN structure under ambient conditions.
r-BN (ABC) belongs to the rhombohedral space group (No. 166) and exhibits a three-layer ABC stacking sequence (Figure 1c). The calculated interlayer distance ( Å with vdW) aligns reasonably with experimental reports ( Å) in Table 1 of Gil et al. [2]. Our calculations also show a slightly larger cohesive energy compared to h-BN (7.207 vs. 7.205 eV), consistent with the finding by Nikaido et al. [9] that r-BN and h-BN are nearly degenerate in energy, although h-BN remains thermodynamically most stable at 0 K.
b-BN (AB) is a less commonly studied polymorph with symmetry, characterized by a two-layer AB stacking, also called Bernal stacking (Figure 1d). The optimized lattice parameters with vdW corrections ( Å, Å) fall within the range of those reported for -BN systems in Gil et al. [2]. The relative stability of b-BN is comparable to r-BN in our calculations, and it exhibits a direct band gap, suggesting unique electronic properties discussed in Sec. Section 3.2.
Across all polymorphs, we observe a strong sensitivity of the c-axis lattice parameter to vdW corrections, while the in-plane lattice constant a remains nearly invariant. This agrees with trends reported in both Gil et al. [2] and Ahmed et al. [15], whose comprehensive tables include both theoretical and experimental lattice data. The cohesive energies calculated here suggest a delicate balance in stability among BN polytypes, with h-BN slightly preferred, in line with diffusion Monte Carlo results by Nikaido et al. [9]. These results highlight the essential role of interlayer stacking in modulating structural and energetic properties of layered BN and provide a consistent theoretical framework in support of experimental data. Overall, the structural polymorphism in -bonded BN results in a delicate balance between symmetry, interlayer stacking, and vdW interactions, all of which are crucial for understanding the phase stability and physical properties of BN-based materials.
3.2. Electronic Band Structure
The electronic band structures of the four studied BN polymorphs were calculated with and without the vdW correction. The results are shown in Figure 2, while a summary of the band gaps’ details is presented in Table 1. All polymorphs exhibit wide band gaps in the range of 3.94 to 4.63 eV, consistent with the semiconducting nature of -bonded BN. The inclusion of vdW interactions significantly affects the out-of-plane lattice constants and, consequently, the electronic band structure, underscoring the importance of capturing weak interlayer forces in layered materials. Across the studied BN polymorphs, the electronic band structure reveals predominantly indirect band gaps, except for b-BN, which exhibits a direct transition at the K point. Specifically, e-BN, h-BN, and r-BN show indirect band gaps ranging from 3.94 to 4.63 eV, typically between the K and or K and M points. Among them, e-BN has the widest gap (4.63 eV), while r-BN has the narrowest (3.94 eV), highlighting the influence of stacking geometry and interlayer coupling. The reduced gap in r-BN, a rhombohedral phase, can be attributed to enhanced interlayer orbital overlap due to its three-layer ABC stacking, which modifies the conduction band minimum. In contrast, b-BN exhibits a direct band gap (K–K), a property desirable for optoelectronic applications relying on vertical transitions. This distinction underscores the sensitivity of the electronic structure to interlayer interactions and symmetry, and emphasizes the potential of stacking-engineered BN for tailored band gap applications.
Comparison with earlier work by Ahmed et al. [15] shows qualitative agreement in the trends of band gap variations across BN polymorphs. Their full-potential LAPW calculations using the Engel–Vosko GGA functional yielded a gap of 4.18 eV for h-BN and 4.21 eV for r-BN, closely matching our PBE+vdW results. However, Ahmed et al. emphasize the importance of using GGA-EV to better align with experimental values, suggesting a pathway for further refinement. Additional insight is provided by Olovsson and Magnuson [3], who investigated h-, r-, and turbostratic BN using X-ray absorption near-edge structure (XANES) spectroscopy. Their DFT+core-hole simulations revealed distinctive and features sensitive to stacking order. The observed shift of the onset in r-BN relative to h-BN correlates with our finding of a narrower band gap in the rhombohedral structure. Moreover, their turbostratic BN models exhibited an average band gap of approximately 3.86 eV, further supporting the notion that stacking disorder can be exploited to engineer BN’s electronic properties.
Lastly, our results show that interlayer stacking and vdW interactions are critical determinants of the electronic properties of BN polymorphs. The transition between indirect and direct band gaps, combined with variations in gap magnitude, offers opportunities for targeted design of BN-based materials in optoelectronics, UV photonics, and quantum applications.
3.3. Phonon Frequencies
The vibrational properties of layered BN polymorphs are susceptible to weak interlayer forces and stacking configurations. To accurately capture these effects, we computed phonon frequencies at the point of the Brillouin zone using DFPT with and without vdW corrections, as implemented in QE. The refined phonon spectra–including full irreducible representation labeling and explicit identification of silent (S), infrared (I), and Raman (R) active modes–are summarized in Table 2. All optical phonons are included, providing a comprehensive mode-by-mode comparison across BN polymorphs. Including vdW interactions significantly impacts phonon frequencies, particularly in polymorphs with strong interlayer coupling such as h-BN and r-BN. Consistent with prior studies [2,9], vdW corrections soften out-of-plane optical modes and bring theoretical spectra closer to experimental IR and Raman observations. The detailed mode analysis reveals prominent shifts upon vdW inclusion, especially for low-frequency modes below 200 cm and high-frequency optical branches near 1350 cm. For example, in h-BN, the out-of-plane infrared-active mode shifts from 781.1 cm (no vdW) to 723 cm (with vdW), while the silent mode also softens notably. Similarly, r-BN exhibits rich vibrational behavior with multiple modes showing dual IR and Raman activity (labeled as I+R), especially between 730–800 cm, where vdW corrections shift the modes by 20–40 cm. In e-BN, the phonon spectrum reflects the high symmetry and absence of staggered stacking. We observe fewer distinct branches and minimal splitting between optical modes. The dominant IR-active and modes remain near 780 cm and 1343 cm, respectively, with vdW corrections introducing only moderate shifts. Notably, silent modes, such as those transforming as in analogous systems, are absent here due to symmetry constraints. Conversely, b-BN shows a broader vibrational landscape. Low-frequency IR-active modes shift from 55 cm (no vdW) to 180 cm (with vdW), reflecting stronger interlayer coupling. Intermediate-frequency and modes also emerge around 730–800 cm, with rich IR and Raman activity reflecting the lower symmetry of the AB stack. The high-frequency Raman-active modes near 1350 cm remain prominent and shift modestly upon vdW inclusion.
These results directly connect to the subsequent analysis of Raman and IR intensities (Figuers Figure 3 and Figure 4), where stacking-dependent features, such as the rich mid-frequency IR activity of r-BN and the sharper Raman peaks of e-BN, mirror the phonon characteristics detailed here. Overall, this detailed vibrational analysis highlights the critical role of vdW interactions and stacking order in shaping the phonon spectra of layered BN polymorphs. The combination of full irreducible representation labeling and activity classification provides a robust framework for both theoretical interpretation and experimental verification of BN polytypes.
3.4. Raman and Infrared Intensities
To obtain accurate IR and Raman intensities, pseudopotentials compatible with the linear and nonlinear response formalism in QE are essential. In particular, norm-conserving (NC) pseudopotentials within the local density approximation (LDA) enable reliable Raman intensity calculations, as they provide the second-order response properties, such as polarizability derivatives, required for this task. We therefore reoptimized the lattice parameters and atomic positions of each BN polymorph using optimized NC Vanderbilt pseudopotentials (ONCVPs) from the PseudoDojo project [16], with all calculations performed in LDA. The resulting LDA-ONCVP phonon frequencies (with vdW corrections; Supporting Information Table S1) follow the same mode ordering and show close quantitative agreement, within expected functional- and pseudopotential-dependent shifts, with the PBE + DFT-D results in Table 2. This comparison highlights both the robustness of the trends across computational setups and the importance of using consistent pseudopotentials and exchange-correlation functionals when modeling spectroscopic properties of layered materials.
Figure 3 and Figure 4 display the Raman and IR intensity spectra at the point. In both panels, the peaks labeled with their symmetry assignments directly correspond to the fingerprint modes listed in Table 3. Complete symmetry-resolved listings are provided in the Supporting Information (Tables S1 and S2). For e-BN, the most prominent signal is the high-frequency in-plane mode at 1402.4 cm, which is both Raman- and IR-active (R ≈ 200.54 a.u.; IR ≈ 27.32 a.u.). In h-BN, the signature Raman feature is the mode at 1396.7 cm (R ≈ 333.68 a.u.), while the mid-frequency mode at 704.4 cm (IR ≈ 10.50 a.u.) is unique to this polytype. In r-BN, a strong coincident Raman/IR high-frequency peak occurs at 1399.9 cm (R ≈ 510.66 a.u.; IR ≈ 80.66 a.u.), with a secondary fingerprint provided by the mid-frequency mode at 725.7 cm (R ≈ 32.17 a.u.; IR ≈ 12.27 a.u.). Finally, for b-BN, the mode at 1409.5 cm (R ≈ 46.69 a.u.; IR ≈ 6.46 a.u.) defines the high-frequency response, while the mode at 728.3 cm (IR ≈ 7.95 a.u.) marks its mid-frequency fingerprint.
This combined analysis of spectra and symmetry-resolved intensities enables direct matching between experimental Raman/IR data and computed fingerprints, providing a practical route for BN polytype identification.
4. Summary
We have carried out a comprehensive first-principles investigation of the structural, vibrational, and spectroscopic properties of four layered BN polymorphs–e-BN (AA), h-BN (AA′), r-BN (ABC), and b-BN (AB)–with emphasis on vdW interactions and stacking-dependent fingerprints. Comparison of calculations with and without vdW corrections demonstrates that dispersion forces are essential for accurately describing relative stability, lattice parameters, and phonon spectra. Including vdW interactions brings lattice constants and electronic band gaps into closer agreement with experiment and resolves subtle stacking-dependent vibrational features. All polymorphs are dynamically stable, as indicated by the absence of imaginary phonon modes. Symmetry-resolved Raman and IR intensity calculations identify distinct spectroscopic fingerprints: r-BN exhibits a strong coincident Raman/IR E mode at 1399.9 cm complemented by mid-frequency activity; e-BN is characterized by a sharp, isolated Raman/IR mode at 1402.4 cm; h-BN displays the characteristic Raman mode at 1396.7 cm and a unique IR-active mode at 704.4 cm; and b-BN is distinguished by mid-frequency IR activity at 728.3 cm alongside a high-frequency Raman/IR mode at 1409.5 cm. This validated computational framework enables reliable experimental identification of BN polytypes and can be directly extended to other layered -bonded materials for advanced optoelectronic and quantum applications.
Author Contributions
Conceptualization, P.M. and N.G.S.; methodology, P.M.; validation, P.M.; investigation, P.M. and N.G.S.; writing and original draft preparation, P.M.; writing, review, and editing, N.G.S.; visualization, P.M.; supervision, N.G.S. All authors reviewed the manuscript.
Acknowledgments
The use of supercomputers at the Interdisciplinary Centre for Mathematical and Computational Modelling (ICM) at the University of Warsaw is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tan, T.; Jiang, X.; Wang, C.; Yao, B.; Zhang, H. 2D Material Optoelectronics for Information Functional Device Applications: Status and Challenges. Advanced Science 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Gil, B.; Desrat, W.; Rousseau, A.; Elias, C.; Valvin, P.; Moret, M.; Li, J.; Janzen, E.; Edgar, J.H.; Cassabois, G. Polytypes of sp2-Bonded Boron Nitride. Crystals 2022, 12, 782. [Google Scholar] [CrossRef]
- Olovsson, W.; Magnuson, M. Rhombohedral and Turbostratic Boron Nitride Polytypes Investigated by X-ray Absorption Spectroscopy. The Journal of Physical Chemistry C 2022, 126, 21101–21108. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.P.; Shen, Z.X. Structural and electronic properties ofh-BN. Physical Review B 2003, 68. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Pham, T.; Dogan, M.; Oh, S.; Shevitski, B.; Schumm, G.; Liu, S.; Ercius, P.; Aloni, S.; Cohen, M.L.; et al. Alternative stacking sequences in hexagonal boron nitride. 2D Materials 2019, 6, 021006. [Google Scholar] [CrossRef]
- Novotný, M.; Dubecký, M.; Karlický, F. Toward accurate modeling of structure and energetics of bulk hexagonal boron nitride. Journal of Computational Chemistry 2023, 45, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Ordin, S.V.; Sharupin, B.N.; Fedorov, M.I. Normal lattice vibrations and the crystal structure of anisotropic modifications of boron nitride. Semiconductors 1998, 32, 924–932. [Google Scholar] [CrossRef]
- Cazorla, C.; Gould, T. Polymorphism of bulk boron nitride. Science Advances 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, Y.; Ichibha, T.; Hongo, K.; Reboredo, F.A.; Kumar, K.C.H.; Mahadevan, P.; Maezono, R.; Nakano, K. Diffusion Monte Carlo Study on Relative Stabilities of Boron Nitride Polymorphs. The Journal of Physical Chemistry C 2022, 126, 6000–6007. [Google Scholar] [CrossRef]
- Korona, T.; Chojecki, M. Exploring point defects in hexagonal boron-nitrogen monolayers. International Journal of Quantum Chemistry 2019, 119, e25925. [Google Scholar] [CrossRef]
- Constantinescu, G.; Kuc, A.; Heine, T. Stacking in Bulk and Bilayer Hexagonal Boron Nitride. Physical Review Letters 2013, 111. [Google Scholar] [CrossRef] [PubMed]
- Iwański, J.; Korona, K.P.; Tokarczyk, M.; Kowalski, G.; Dąbrowska, A.K.; Tatarczak, P.; Rogala, I.; Bilska, M.; Wójcik, M.; Kret, S.; et al. Revealing polytypism in 2D boron nitride with UV photoluminescence. npj 2D Materials and Applications 2024, 8. [Google Scholar] [CrossRef]
- Korona, K.P.; Binder, J.; Dąbrowska, A.K.; Iwański, J.; Reszka, A.; Korona, T.; Tokarczyk, M.; Stępniewski, R.; Wysmołek, A. Growth temperature induced changes of luminescence in epitaxial BN: from colour centres to donor–acceptor recombination. Nanoscale 2023, 15, 9864–9877. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Casarin, M.; Forrer, D.; Pavone, M.; Sambi, M.; Vittadini, A. Role and effective treatment of dispersive forces in materials: Polyethylene and graphite crystals as test cases. Journal of Computational Chemistry 2009, 30, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; e Aleem, F.; Hashemifar, S.J.; Akbarzadeh, H. First principles study of structural and electronic properties of different phases of boron nitride. Physica B: Condensed Matter 2007, 400, 297–306. [Google Scholar] [CrossRef]
- van Setten, M.; Giantomassi, M.; Bousquet, E.; Verstraete, M.; Hamann, D.; Gonze, X.; Rignanese, G.M. The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table. Computer Physics Communications 2018, 226, 39–54. [Google Scholar] [CrossRef]
Figure 1.
Crystal structures of BN polymorphs (green = B, gray = N): (a) e-BN [ (187)], (b) h-BN [ (194)], (c) r-BN [ (166)], and (d) b-BN [ (194)].
Figure 1.
Crystal structures of BN polymorphs (green = B, gray = N): (a) e-BN [ (187)], (b) h-BN [ (194)], (c) r-BN [ (166)], and (d) b-BN [ (194)].
Figure 2.
Electronic band structures of different BN polymorphs with and without vdW corrections. Black solid lines indicate results without vdW, and red dashed lines include vdW corrections.
Figure 2.
Electronic band structures of different BN polymorphs with and without vdW corrections. Black solid lines indicate results without vdW, and red dashed lines include vdW corrections.
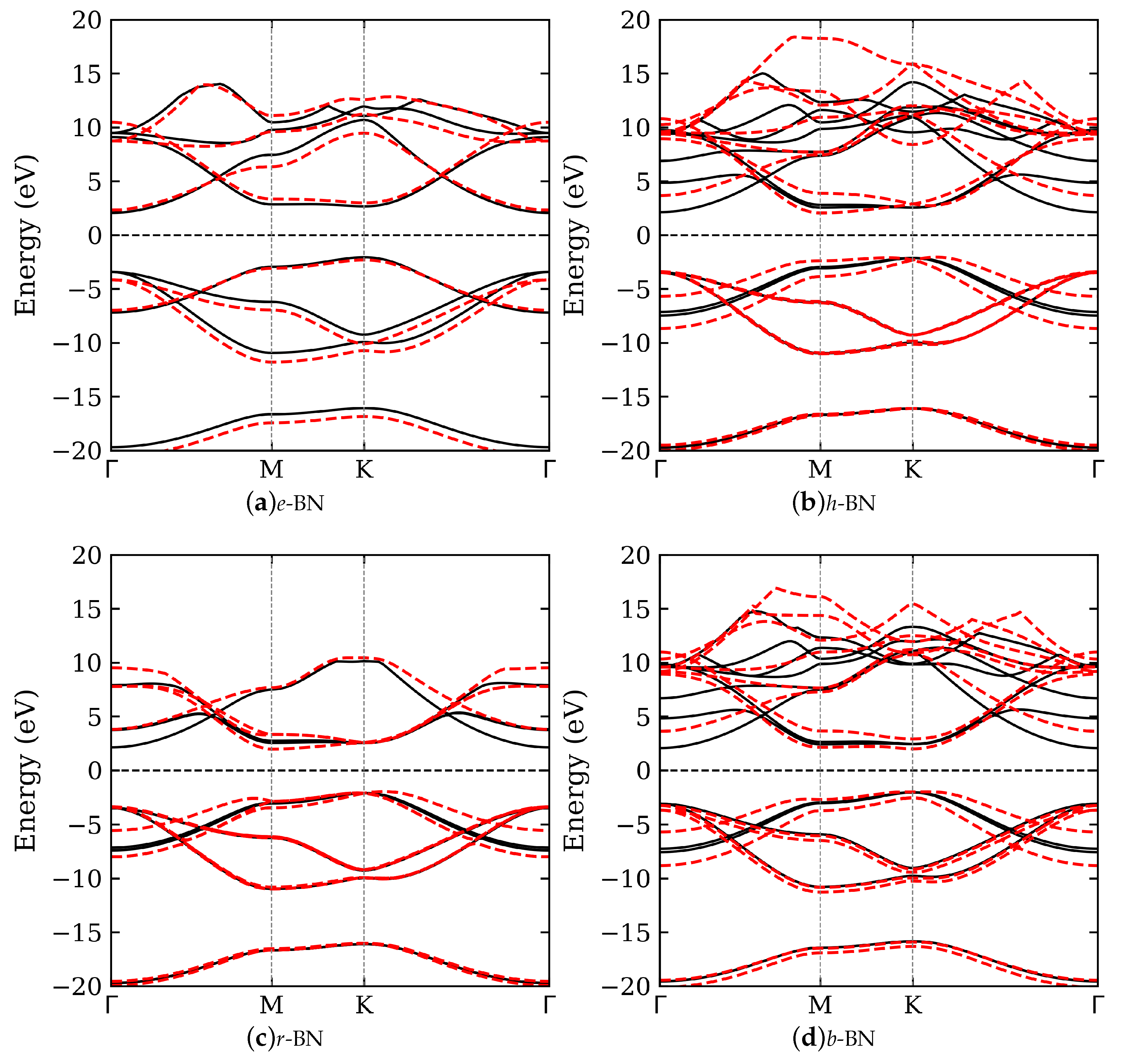
Figure 3.
Computed Raman intensity spectra at the point for the four BN polymorphs.
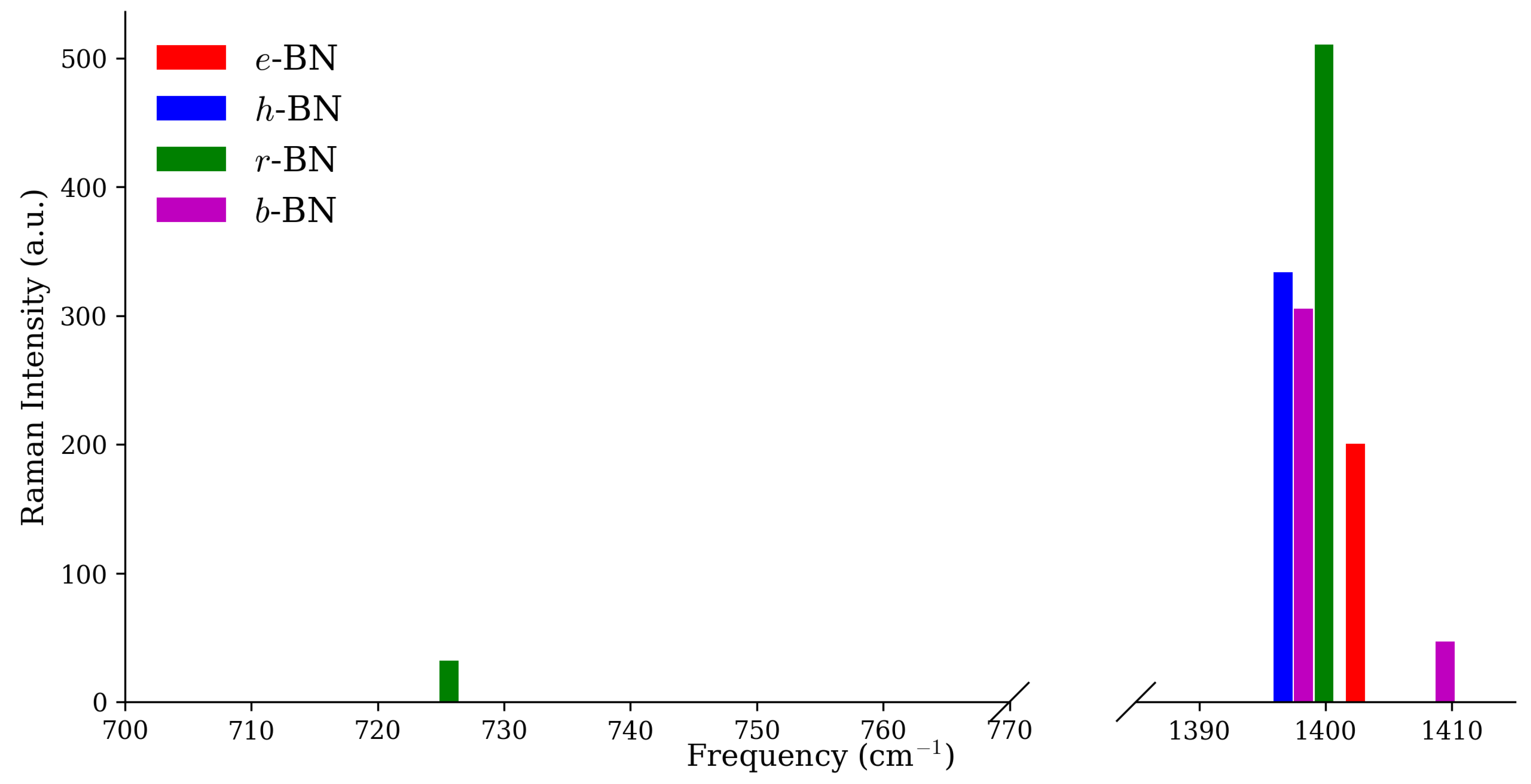
Figure 4.
Computed infrared (IR) intensity spectra at the point for the four BN polymorphs.
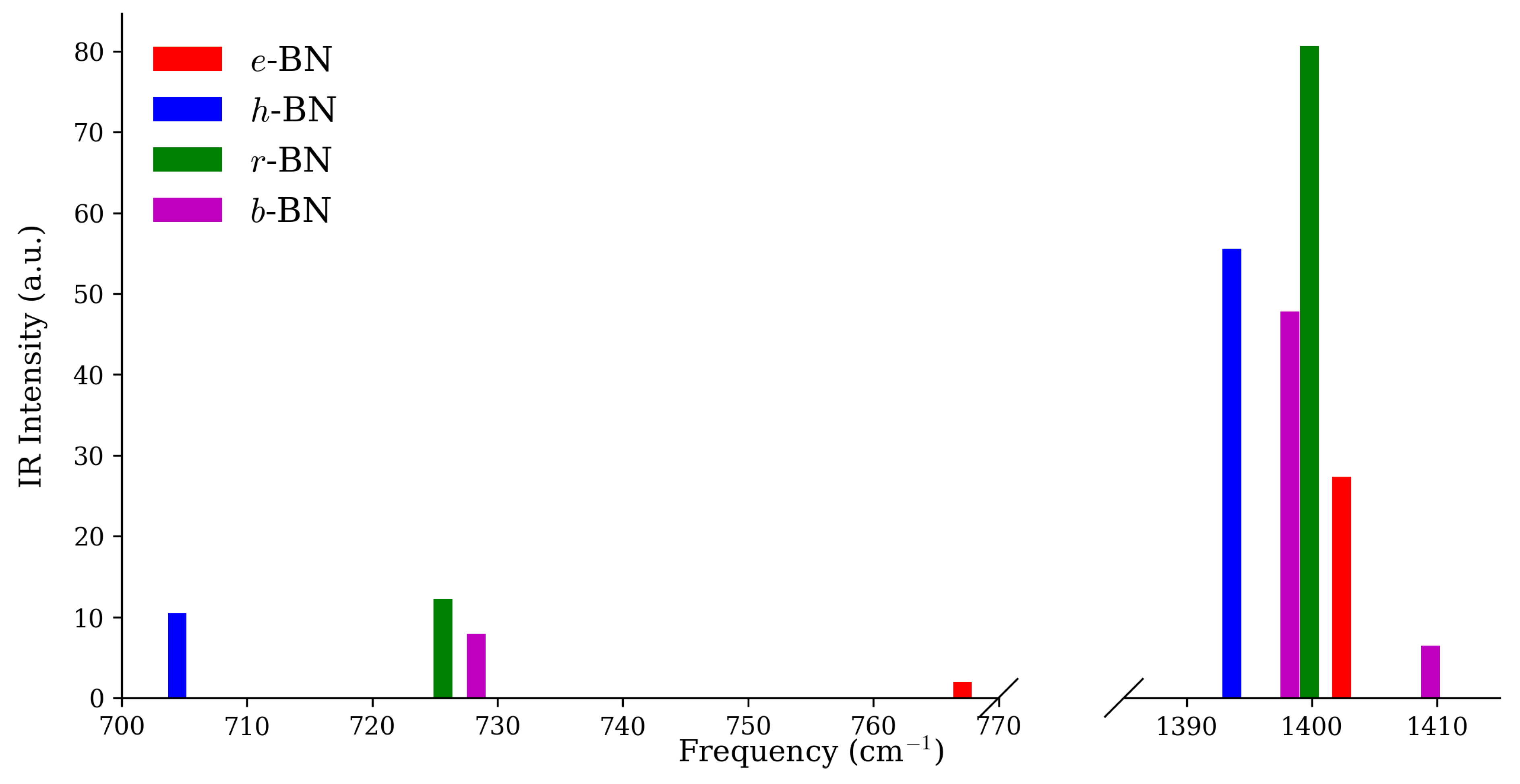

Table 1.
Comparison of structural parameters, cohesive energy (), and electronic band gap () for layered BN polymorphs with and without vdW corrections.
Table 1.
Comparison of structural parameters, cohesive energy (), and electronic band gap () for layered BN polymorphs with and without vdW corrections.
| Method | a [Å] | c [Å] | [eV] | [eV] |
|---|---|---|---|---|
| e-BN (AA) | ||||
| without vdW | 2.514 | 5.059 | 7.064 | 4.12 (indirect, K–) |
| with vdW | 2.511 | 3.383 | 7.181 | 4.63 (indirect, K–) |
| literature | 2.476 [2] | 3.476 [2] | – | – |
| h-BN (AA′) | ||||
| without vdW | 2.515 | 9.136 | 7.065 | 4.25 (indirect, K–) |
| with vdW | 2.512 | 6.179 | 7.205 | 4.10 (indirect, K–M) |
| literature | 2.478 [2] | 6.354 [2] | 7.055 [15] | 4.25 [3] |
| r-BN (ABC) | ||||
| without vdW | 2.515 | 13.743 | 7.065 | 4.24 (indirect, K–) |
| with vdW | 2.511 | 9.168 | 7.207 | 3.94 (indirect, K–M) |
| literature | 2.476 [2] | 9.679 [2] | – | 4.21 [3] |
| b-BN (AB) | ||||
| without vdW | 2.514 | 9.229 | 7.065 | 4.11 (indirect, K–) |
| with vdW | 2.511 | 6.117 | 7.207 | 3.98 (direct, K–K) |
| literature | 2.477 [2] | 6.319 [2] | – | – |
Table 2.
Phonon frequencies (in cm) at the point for each BN polymorph, calculated using DFPT with and without vdW corrections. Each entry lists the frequency shift from no-vdW to vdW, followed by the irreducible representation and mode activity: infrared (I), Raman (R), both (I+R), or silent (S). Only optical phonons are included, with vdW-corrected values shown second in each pair.
Table 2.
Phonon frequencies (in cm) at the point for each BN polymorph, calculated using DFPT with and without vdW corrections. Each entry lists the frequency shift from no-vdW to vdW, followed by the irreducible representation and mode activity: infrared (I), Raman (R), both (I+R), or silent (S). Only optical phonons are included, with vdW-corrected values shown second in each pair.
| e-BN | h-BN | r-BN | b-BN | ||||
|---|---|---|---|---|---|---|---|
| Freq. (no→vdW) | Mode | Freq. (no→vdW) | Mode | Freq. (no→vdW) | Mode | Freq. (no→vdW) | Mode |
| 783 → 757.2 | (I) | 0 → 39.6 | (R) | 0 → 37.3 | E (I+R) | 0 → 48.5 | (I+R) |
| 1343.5 → 1352.5 | (I+R) | 52.9 → 182.8 | (S) | 0 → 39.3 | E (I+R) | 55.2 → 180.0 | (I) |
| 781.1 → 723.0 | (I) | 0 → 150.2 | (I+R) | 781.6 → 732.0 | (I) | ||
| 803.0 → 792.1 | (S) | 49.2 → 159.2 | (I+R) | 803.0 → 793.9 | (I) | ||
| 1341.6 → 1349.3 | (I) | 778.5 → 730.6 | (I+R) | 1343.6 → 1351.9 | (I+R) | ||
| 1341.6 → 1350.3 | (R) | 799.2 → 795.7 | (I+R) | 1343.6 → 1358.7 | (I+R) | ||
| 801.4 → 797.8 | (I+R) | ||||||
| 1343.2 → 1353.1 | E (I+R) | ||||||
| 1343.2 → 1356.4 | E (I+R) | ||||||
| 1343.3 → 1356.4 | E (I+R) | ||||||
Table 3.
Strongest Raman (R) modes and most distinctive infrared (IR) modes for each BN polymorph at the point, selected for their value in polytype identification. Frequencies are in cm and intensities in arbitrary units (a.u.). Irreducible representations follow the symmetry assignments in Supporting Information Table S1. Full symmetry-resolved listings are also provided in the Supporting Information (Tables S1 and S2).
Table 3.
Strongest Raman (R) modes and most distinctive infrared (IR) modes for each BN polymorph at the point, selected for their value in polytype identification. Frequencies are in cm and intensities in arbitrary units (a.u.). Irreducible representations follow the symmetry assignments in Supporting Information Table S1. Full symmetry-resolved listings are also provided in the Supporting Information (Tables S1 and S2).
| Polymorph | Raman (cm) | Mode (R) | Intensity (R) [a.u.] | IR (cm) | Mode (IR) | Intensity (IR) [a.u.] |
|---|---|---|---|---|---|---|
| e-BN | 1402.4 | 200.54 | 1402.4 | 27.32 | ||
| h-BN(2) | 1396.7 | 333.68 | 704.4 | 10.50 | ||
| r-BN(3) | 1399.9 | E | 510.66 | 1399.9 | E | 80.66 |
| b-BN(4) | 1409.5 | 46.69 | 728.3 | 7.95 |
Spectroscopic fingerprints:(1) Sharp, isolated Raman/IR peak at 1402 cm−1. (2) Unique mid-frequency IR mode at 704 cm−1.
(3) Strong coincident Raman/IR peak at 1399.9 cm−1 plus concurrent mid-frequency A1 mode at 725.7 cm−1. (4) Mid-frequency
IR activity at 728 cm−1 with high-frequency Raman/IR peak at 1409.5 cm−1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



