
Submitted:
21 August 2025
Posted:
22 August 2025
You are already at the latest version
Abstract
Assembling high-quality fungal genomes, specifically telomere-to-telomere (T2T) gapless assemblies, often necessitates the integration of multiple sequencing platforms. This requirement poses a limitation on the number of fungal genomes that can feasibly be generated within a single project. Here, we demonstrate that haplotype-aware error correction (HERRO) of Oxford Nanopore simplex reads enables the generation of high-quality assemblies from a single sequencing platform. We present an automated Snakemake workflow that, without manual intervention, produced gapless genome assemblies for industrially relevant strains: Neurospora intermedia NRRL 2884, Trichoderma asperellum TA1, and Aspergillus oryzae CBS 466.91, each achieving complete BUSCO scores exceeding 98 %. Among these, only the T. asperellum assembly yielded a fully telomere-to-telomere gapless genome, while the N. intermedia and A. oryzae assemblies were gapless but near-telomere-to-telomere. Manual curation was required for the mitochondrial genome assembly of N. intermedia.
Keywords:
telomere-to-telomere
; nanopore sequencing
; long read sequencing
; high molecular weight DNA
; fungal genomes
; snakemake
1. Introduction
The first whole-genome sequence of a filamentous fungus was reported in 2003 [1], a milestone that required substantial resources. Advances in genome sequencing technologies now allow such projects to be completed routinely within weeks, depending on the volume and type of data required. High-throughput short-read sequencing with Illumina can be performed rapidly, whereas projects requiring high molecular weight (HMW) DNA for long-read sequencing generally take longer. Nevertheless, both approaches are considerably faster and more cost-effective than the methods available in 2003 [2]. The reduction in sequencing time and cost is largely attributable to the capacity of second- and third-generation sequencing platforms to generate gigabases of data per run [3,4]. With the advent of third-generation sequencing, it has become increasingly feasible to assemble genomes to telomere-to-telomere (T2T) completeness [5]. Achieving such assemblies, however, often necessitates combining multiple complementary sequencing platforms, for example, long-read data from PacBio and/or Oxford Nanopore for assembly, short-read Illumina data for polishing, and long-range data such as Hi-C for scaffolding [6,7,8]. For Nanopore sequencing, polishing with high-quality short reads has historically been standard practice due to its higher error rate compared to PacBio [9]. The accuracy of Nanopore reads can be improved substantially, from an average Phred quality score of Q20 to Q30, through duplex sequencing, enabling the resolution of complex genomes into high-quality assemblies [10]. Accuracy can also be enhanced through pre-processing methods such as haplotype-aware error correction (HERRO), which has recently been applied to produce T2T human genome assemblies from Nanopore simplex data alone [11], as well as four T2T assemblies of Colletotrichum lini strains, a pathogenic fungus of flax [12]. T2T assemblies allow the resolution of long repetitive regions, such as telomeres and centromeres [13], and can resolve into fully gap-free assemblies in which no contigs are joined post-assembly (i.e., no ambiguous “N” gaps) [5,14]. The absence of scaffolding gaps improves gene prediction by preventing artificial fragmentation of coding sequences (CDS). This is particularly important for pathogenicity and toxicity studies, as biosynthetic gene clusters (BGCs) can span up to 220 Kb [15,16] and may be missed or fragmented in assemblies of low contiguity [17]. Because BGCs are predominantly located in subtelomeric regions [17] which were difficult to resolve with older sequencing and assembly approaches, their identification benefits greatly from T2T assemblies. Resolving BGCs, including polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), terpene synthases, and ribosomally synthesized post-translationally modified peptides (RiPPs), is critical for novel drug discovery [18,19], a process now accelerated by the unprecedented pace of fungal genome sequencing. Similarly, the identification of carbohydrate-active enzymes (CAZymes) is advancing rapidly, supporting research into lignocellulosic biomass degradation [20]. Species of Neurospora, Trichoderma, and Aspergillus are among the most widely studied filamentous fungi [21]. In industrial contexts, Trichoderma and Aspergillus dominate patents for the production of organic acids (notably citric acid) and proteins [21], while N. intermedia and N. sitophila are recognized for their role in producing red oncom, a traditional Indonesian fermented food [22]. High-quality fungal genomes are essential for species identification [23], understanding fungal plant virulence [24], enabling drug discovery [18], and supporting the development of industrial production strains [25].
In this study, we present: (i) the first near-T2T gapless genome of Neurospora intermedia NRRL 2884; (ii) a near-T2T gapless genome of Aspergillus oryzae CBS 466.91; (iii) a fully gapless T2T genome of Trichoderma asperellum TA1; and (iv) a Snakemake workflow for generating fungal gapless near-T2T genomes exclusively from Oxford Nanopore simplex data.
2. Materials and Methods
2.1. DNA Extraction and Sequencing
Neurospora sitophila (NRRL 2884) was acquired from ATCC® (36935™), and was isolated from Oncom, Indonesia. Aspergillus oryzae CBS 466.91 was acquired from the WI-KNAW culture collection in the Netherlands, and originally isolated in Osaka, Japan. Trichoderma asperellum TA1 was isolated from field soil at a farm belonging to University of Copenhagen, Denmark, and initially morphologically identified as Trichoderma harzianum but renamed as T. asperellum based on ITS sequencing (unpublished results). All isolates were stored at -80 °C either as agar plugs or conidia in 15 % glycerol. The acquired strain of Neurospora sitophila will be denominated as Neurospora intermedia NRRL 2884 as it was incorrectly identified when acquired from the strain collection in 2022. Revisiting the collection in 2025, it is stated that “The DNA sequences indicate that this strain is phylogenetically closer to N. intermedia than N. sitophila" which also is confirmed in this study. The strains were grown on yeast extract peptone glucose (YPG) agar plates (Glucose 25 g/L, Peptone 20 g/L, Yeast extract 10 g/L, Agar 20 g/L) for 5 days at 30 °C. Subsequently, 10 plugs (3 mm ø) were prepared from each plate and used for inoculation of 100 ml liquid medium in 250 ml Erlenmeyer flasks. N. intermedia NRRL 2884 and A. oryzae CBS 466.91 were grown for 2 and 5 days, respectively in YPG medium at 30 °C and T. asperellum TA1 was grown for 2 days at 25 °C in yeast extract sucrose (YES) medium (Sucrose 150 g/L, Yeast extract 20 g/L, 0.5 g/L, 1 mL YES trace solution ( 16 g/L, 5 g/L), pH 6.5) with agitation of 150 rpm. Mycelium was harvested by pouring into autoclaved double layered Mira cloth (merckmillipore) and washed with autoclaved water. The harvested mycelium was frozen in liquid , then lyophilized (Scanvac Coolsafe Freeze Dryer), and ground with a mortar and pestle. HMW DNA from N. intermedia NRRL 2884 and A. oryzae CBS 466.91 was extracted with the phenol-chloroform method and purified, as described in [26]. HMW DNA extraction from T. asperellum TA1 was done with the NucleoBond®HMW (DNA Macherey-Nagel™) kit according to the manufacturer’s protocol (Enzymatic lysis) and further purified via the isopropanol precipitation method described in [26]. All samples were subjected to short read elimination via the Circulomics Short Read Eliminator XS kit (Circulomics). DNA purity was evaluated with NanoDrop One and Qubit 4. DNA length was determined on an Agilent TapeStation 4150 with Genomic DNA ScreenTapes. Library prep and barcoding was done according to the manufacturers protocol with the Native Barcoding Kit 24 V14, which was loaded on a PromethION Flow Cell (R10.4.1). Sequencing was done on a PromethION 24 with MinKNOW and run for 72 hours with five other fungi, not part of this study, the samples were live basecalled with Super accurate basecalling Guppy (v7.1.4) [27] on a local machine, trimmed, and de-multiplexed in pod5 files for later basecalling with the Dorado [28].
2.2. Snakemake Workflow for High Quality Assembly
All computation was done on a high-performance computing (HPC) cluster running Slurm. The assembly pipeline was run in a custom snakemake (v7.18.2) [29] workflow. A general overview is visualized in Figure 1. Basecalling was performed with Dorado basecaller (v0.9.1) [28] using the sup model (v5.0.0) from the de-multiplexed pod5 files. SAMtools (v1.21) was used to convert Bam files to fastq files [30]. Porchop_abi (v0.5.0) [31] was used to remove fragmented adapters and ligators. Chopper (v0.9.0) [32] was used to filter reads based on length and quality, >Q10 and >10 Kb as the input for Read error correction, >Q20 and >20 Kb as the input for Flye (v2.9.3) [33], >Q10 and >50 Kb as the ultralong reads as one of the inputs for Hifiasm (v0.24.0) [34]. Flye (v2.9.3), was used to make draft assemblies for mitochondrial genome extraction for the assembly graph input of GetOrganelle (v1.7.7.1) [35]. Before Read error correction Rasusa (v2.1.0) [36] was used to limit the reads to a maximum coverage of 125x for a genome of 40 Mb. HERRO [11] was run as implemented in the Dorado workflow for error correction of reads with Dorado correct (v0.9.1). Seqtk (v1.4.0) [37] was employed to convert fasta files from HERRO to fastq. Error corrected reads were then filtered based on the lengths >10 Kb, >15 Kb, >20 Kb, >25 Kb, and >30 Kb with seqkit (v2.9.0) [38], these 5 different read bins were then used, together with the ultralong reads, to assemble draft genomes with Hifiasm (v0.24.0) [34]. Draft assemblies from Hifiasm (v0.24.0), were converted from fga files to fa files with a bash command. From these five draft assemblies obtained for each fungus, the draft assembly with the highest contiguity for each fungus was identified with a bash command and selected for polishing. The draft genome selected for polishing was then aligned with the bam file from the basecalling step with Dorado aligner (v0.9.1), sorted and indexed with SAMtools (v1.21), this was then used for polishing with Dorado polish (v0.9.1). The final assemblies were assessed with BUSCO (v5.7.1) [39] using the fungi_odb10 (01/08/24) dataset, run in genome mode. The full workflow and dependencies are available at https://github.com/TerpmikaelAAU/T2T_fungal_genome_pipeline. All other computation methods were run with either bash or python3.
2.3. Phylogenetic Determination with Universal Fungal Core Genes
Phylogeny was determined with the UFCG (v1.0.6) pipeline with default settings [40]. All reference genomes were downloaded from NCBI for their respective genus (24/03/2025) with NCBI Datasets (v16.2.0) [41]. For the Neurospora phylogeny, the four HQ (high quality) assemblies of N. sitophila [42] were also included as there was no N. sitophila genomes available in the NCBI database. Phylogenetic tree with Genealogical Sorting Index (GSI) values was visualized in iTOL (v7.2.1) [43]. For T. asperellum TA1, initial phylogeny was not sufficient to determine the specific species, therefore the closest neighbors were used in a second run on the UFCG pipeline.
2.4. Whole-Genome Alignment
2.5. Gene prediction and functional annotation
Ab initio gene prediction and subsequent functional annotations were done with the Funannotate (v1.8.17) pipeline [48]. Including antiSMASH 7.0 (v7.1) [49] and InterProScan (v5.73-104.0) [50,51]. Telomeres were identified via tidk (v0.2.63) [52] by searching with the experimental validated telomere sequences in Telobase (17/04/25) [53]. Functional genes and telomeres were visualized with pyCirclize (v1.9.0).
2.6. Validation of Workflow
To validate the snakemake workflow, the basecalled reads of Colletotrichum lini 394-2 [12] was downloaded from the Sequence Read Archive. The workflow for these reads was started after the Porchop_abi step, no polishing was done, and the assembly quality was validated with the BUSCO (v5.3.2) glomerellales_odb10 (2024/01/08) dataset run with default parameters in genome mode.
3. Results
3.1. Snakemake Workflow Output and Assembly Stats
The raw basecalled reads for the three different fungi were of high depth and quality with the average Phred score > Q20 for all the samples and a N50 of > 10 Kb. After the first filtering step to prepare the reads for error correction, the coverage was halved for N. intermedia NRRL 2884 and A. oryzae CBS 466.91, and for T. asperellum TA1 25 % was lost. After this, the samples displayed N50 > 20 Kb. Some reads were lost after error correction and as the output of the Dorado error correction is in the fasta format, the quality scores are lost. Sub-sampling reads for the highest contiguity draft genome resulted in a much lower coverage for N. intermedia NRRL 2884 compared to A. oryzae CBS 466.91 and T. asperellum TA1. The ultra-long non corrected reads had a coverage between 7.5 and 9.4. (Table 1). The percentage of reads >Q30 was 85 % or more in all samples after basecalling, however most of these were below 10 Kb and not included in further analysis. We observed that by not limiting the error correction step to reads below 50 Kb, higher contiguous draft assemblies were produced.
The BUSCO assessment of all genomes resulted in complete BUSCO scores > 98 % for all samples and a genome size in congruences with what has been observed in the literature [54,55] (Table 2). All assemblies resolved into genomes with the same count of contigs as observed chromosomes in each fungus, without the mitochondrial genome. However, this was not the case for the N. intermedia NRRL 2884 assembly as it had a mitochondrial contig with a size of 258 Kb, and no circular consensus of the mitochondrial genome could be assembled by Getorganelle in the snakemake workflow. Manually rerunning Getorganelle on the output graph from Hifiasm did not produce a circular consensus either. In contrast, mitochondrial genomes were assembled automatically with Flye and Getorganelle for T. asperellum TA1 and A. oryzae CBS 466.91.
3.2. Phylogenetic Determination
To determine/verify the taxonomic classification of the N. intermedia NRRL 2884 strain, it was aligned with the Universal Fungal Core Genes (UFCG) from UFCG. The produced tree showed that instead of being grouped in the sitophila clade it was grouped with Neurospora crassa and closest related to N. intermedia as shown on Figure 2.
With prior ITS classification done on T. asperellum TA1 the taxonomical tree was also produced for verification/determination of the taxonomical classification. This was however not possible as the T. asperellum TA1 assembly clustered close to T. asperellum and T. asperelloides as shown on Figure 3, and therefore there was no clear delineation of this fungus.
To achieve a higher resolution of taxonomical classification the genomes available in the NCBI database of T. asperellum, T. asperelloides and T. yunnanense were used for the alignment. Here a clearer classification was observed of the T. asperellum TA1 assembly, being that of the T. asperellum species as shown on Figure 4. Our phylogenetic analysis thereby supports the initial classification by ITS sequencing. Moreover the T. asperellum TA1 is in a distinct clade of T. asperellum relatively far removed from the type strain T. asperellum CBS 433.97.
With A. oryzae being very closely related to A. flavus known to produce aflatoxin [56], clear taxonomic classification is imperative. In the alignment of all NCBI GenBank reference genomes of Aspergillus, the A. oryzae CBS 466.91 assembly produced in this study was neatly clustered with the reference strain of A. oryzae RIB40 as shown on Figure 5.
3.3. Whole Genome Alignment with High Quality Closest Neighbor
Whole genome alignment was made between N. intermedia NRRL 2884 and NCBI GenBank reference genome N. intermedia HJ2022-06 to identify potential chromosomal rearrangements. No rearrangements were observed between these two species, however the higher contiguity of our assembly was evident as several of the smaller contigs of strain HJ2022-06 were mapped to different parts of whole chromosomes (Figure 6). The large 258 Kb mitochondrial genome assembled for N. intermedia NRRL 2884 mapped to the smaller 87 Kb mitochondrial genome of scaffold 11 from N. intermedia HJ2022-06. Aligning these two mitochondria with blastn showed a query cover of 98 % and an identity of 99.98 %. By rerunning filtering with new parameters >Q25, >10 Kb and using the assembly graph from Flye for the mitochondrial genome assembly with Getorgannelle a mitochondrial genome of 56 Kb was achieved. This was in much closer congruence with the HQ mitochondrial genome in N. crassa OR74 of 64.8 Kb. An alignment of these two with blastn had a query cover of 94 % and an identity of 99.92 %.
Whole genome alignment of T. asperellum TA1 and T. asperellum TCTP001 again show how some of the smaller contigs in T. asperellum TCTP001 are part of the fully assembled chromosomes in our assembly. However, there is a high number of chromosomal rearrangements either originating from the assembly method, or from chromosomal rearrangements events, as shown on Figure 7. The mitochondrial genomes shared high similarity as T. asperellum TA1 had a size of 29.7 Kb resembling the T. asperellum TCTP001 mitochondrial genome of 28.5 Kb, and alignment with blastn gave a query cover of 96 % and a percent identity of 99.79 %.
Some rearrangements were observed between A. oryzae CBS 466.91 and A. oryzae RIB40, with some of them being small parts of the genome and others being 1 Mb parts of the chromosomes, aligned to different chromosomes between the two strains, as seen on Figure 8. The three unplaced scaffolds from A. oryzae RIB40 each mapped to different chromosomes in A. oryzae CBS 466.91, 9 mapped to chromosome 2, 10 mapped to chromosome 4, and 11 mapped to chromosome 7.
The mitochondrial genome of A. oryzae CBS 466.91 and A. oryzae RIB40 were similar in size spanning 29.1 Kb and 29.2 Kb, respectively. Aligning the two mitochondrial genomes with blastn resulted in a percent identity 100 % and a query coverage of 99.93 %
3.4. Predicted Genes and Telomeres
Funannotate gene prediction and annotation identified 8,790 genes in N. intermedia NRRL 2884, including 345 CAZymes, 8 PKSs, 5 NRPSs/NRPSs-like genes, 3 terpene synthases, and 4 RiPP clusters (Table 3). In comparison, T. asperellum TA1 had 9,805 genes and A. oryzae CBS 466.91 had 13,042 genes, both showing a higher amount of BGCs (Table 3).
N. intermedia NRRL 2884 had identifiable telomeres on all chromosomes, except one telomere in chromosome 2 and a clear lack of CDS in chromosomes 1, 5, 6, and 7 which indicates that the centromeric regions are assembled correctly [57] as illustrated on Figure 9.
Telomeres were identified on all chromosomes of T. asperellum TA1; however, in this assembly, the centromeric regions are less clearly defined than in N. intermedia NRRL 2884 (Figure 10).
A. oryzae CBS 466.91 contained telomeres on all chromosomes except chromosome 8 (Figure 11). Despite the lack of telomeres, chromosome 8 is interpreted as a real chromosome as it aligns with chromosome 7 in A. oryzae RIB40 and retains annotated functional genes.
3.5. Workflow Validation
The workflow presented in this study was tested with the reads of Colletotrichum lini 394-2. The two assemblies yielded identical BUSCO scores, with the only difference being a slightly smaller genome size in the assembly generated using the method described in this study (Table 4). In this case, the mitochondrial genome was produced during the initial Hifasm assembly rather than by GetOrganelle. These results further demonstrate that the approach presented here can generate fungal T2T assemblies solely from Nanopore simplex sequencing data, without the need for complementary sequencing from other platforms.
4. Discussion
The basecalling, filtering, read error correction, draft assembly, polishing, and mitochondrial genome recovery were performed in a near-fully automated manner by the newly developed Snakemake workflow. This approach yielded a near-T2T gapless genome for Neurospora intermedia NRRL 2884 and Aspergillus oryzae CBS 466.91, and a fully gapless T2T genome for Trichoderma asperellum TA1, all generated exclusively from Nanopore simplex sequencing data.
There is a pressing need to produce higher-quality fungal genomes, as 17,789 fungal genomes are currently available in the NCBI database, of which 91.6 % are assembled only to the scaffold or contig level [58]. The ability to assemble fungal genomes to a gapless chromosome level, or to chromosome level, with a single, relatively inexpensive sequencing approach holds significant potential for increasing the overall quality of fungal genome assemblies in the future.
The workflow was validated by assembling the genome of Colletotrichum lini 394-2 [12], yielding a genome of comparable quality to that reported by the original authors. To our knowledge, this remains the only published study describing T2T fungal genome assemblies generated exclusively from Nanopore simplex data. In our implementation, the mitochondrial genome was assembled directly from the provided dataset, whereas Sigova et al. [12] incorporated the mitochondrial genome from a previous assembly because mitochondrial reads had been lost during the error-correction process, which was also observed in the present study for T. asperellum TA1 and A. oryzae CBS 466.91.
The mitochondrial genome of N. intermedia NRRL 2884 was initially assembled with corrected reads by Hifasm as a 258 Kb genome, many times larger than a typical Neurospora mitochondrial genome [59], and no contiguous mitochondrial genome was recovered in the initial GetOrganelle assembly. To address this anomaly, the filtering parameters for uncorrected reads in the Flye assembly were modified, resulting in an assembly graph from which GetOrganelle could recover a mitochondrial genome much more consistent with that of a typical fungal mitochondrial genome. In contrast, the mitochondrial genomes of T. asperellum TA1 and A. oryzae CBS 466.91 were successfully assembled by GetOrganelle without such adjustments. For C. lini 394-2, mitochondrial reads were not lost during the error correction step and were correctly assembled by Hifasm. These findings highlight that mitochondrial genome assembly using this approach can be complex and may require further methodological refinement.
Accurate phylogenetic determination is often necessary to confirm or revise prior taxonomic classifications. While whole-genome alignments between species provide a more robust alternative to single-gene approaches such as UFCG [40] or BUSCO [39], such analyses impose substantial computational demands, particularly when handling large numbers of genomes [60]. Consequently, the use of multiple single-copy orthologs, as implemented in UFCG, remains the most practical strategy for phylogenetic analysis of newly assembled high-quality genomes. As observed in Trichoderma spp., greater resolution may be required for accurate species-level determination, likely due to the low synteny within the genus [61].
High-molecular-weight (HMW) DNA extraction is critical for next-generation sequencing. In this study, the phenol–chloroform extraction method successfully produced HMW DNA of suitable quality for N. intermedia NRRL 2884 and A. oryzae CBS 466.91, but not for T. asperellum TA1. For the latter, a commercially available kit employing enzymatic lysis followed by cetyltrimethylammonium bromide (CTAB) purification was used, yielding HMW DNA of sufficient quality for sequencing.
Using Snakemake, we provide tools for the rapid and accurate sequencing and assembly of complex fungal genomes with the aim of enabling future sequencing efforts to proceed more efficiently and at a faster pace.
Author Contributions
Conceptualization, Mikael Terp; Investigation, Mikael Terp and Mark Nyitrai; Software, Mikael Terp; Writing – original draft, Mikael Terp; Writing – review & editing, Mikael Terp, Mette Lübeck, Teis Esben Sondergaard, Christian Rusbjerg-Weberskov and Mark Nyitrai. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been carried out in the frames of the NNF project: HYDROPHOBINS: Specialized Proteins for Food (HFBfood). Contract no. NNF22OC0079146. The authors are grateful to the Novo Nordisk Foundation for financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequencing data and genomes produced in this study can be found in the NCBI database under the BioProject accession number: PRJNA1302126. The snakemake workflow and other code used in this study can be found at: https://github.com/TerpmikaelAAU/T2T_fungal_genome_pipeline
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BGCs | Biosynthetic gene clusters |
| BUSCO | Benchmarking Universal Single-Copy Orthologs |
| CAZymes | Carbohydrate-Active Enzymes |
| CTAB | Cetyltrimethylammonium bromide |
| GSI | Genealogical Sorting Index |
| CDS | Coding sequence |
| HERRO | Haplotype-aware error correction |
| HMW | High molecular weight DNA |
| HPC | High-performance computing |
| HQ | High quality |
| NRPSs | Nonribosomal peptide synthetases |
| PKSs | Polyketide synthases |
| RiPPs | Post-translationally modified peptides |
| T2T | Telomere to telomere |
| Tidk | Telomere identification toolkit |
| UFCG | Universal Fungal Core Genes |
| YES | Yeast extract sucrose |
| YPG | Yeast extract Peptone Glucose |
References
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Schwarze, K.; Buchanan, J.; Fermont, J.M.; Dreau, H.; Tilley, M.W.; Taylor, J.M.; Antoniou, P.; Knight, S.J.; Camps, C.; Pentony, M.M.; et al. The complete costs of genome sequencing: a microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genetics in Medicine 2020, 22, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Genome assembly in the telomere-to-telomere era. Nature Reviews Genetics 2024, 25, 658–670. [Google Scholar] [CrossRef]
- Wang, H.; Yao, G.; Chen, W.; Ayhan, D.H.; Wang, X.; Sun, J.; Yi, S.; Meng, T.; Chen, S.; Geng, X.; A gap-free genome assembly of Fusarium oxysporum f., sp.; et al. conglutinans, a vascular wilt pathogen. Scientific Data 2024, 11. [Google Scholar] [CrossRef]
- Li, Z.; Yang, J.; Ji, X.; Liu, J.; Yin, C.; Bhadauria, V.; Zhao, W.; Peng, Y.L. First telomere-to-telomere gapless assembly of the rice blast fungus Pyricularia oryzae. Scientific Data 2024, 11. [Google Scholar] [CrossRef]
- Yao, G.; Chen, W.; Sun, J.; Wang, X.; Wang, H.; Meng, T.; Zhang, L.; Guo, L. Gapless genome assembly of Fusarium verticillioides, a filamentous fungus threatening plant and human health. Scientific Data 2023, 10. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biology 2020, 21. [Google Scholar] [CrossRef]
- Koren, S.; Bao, Z.; Guarracino, A.; Ou, S.; Goodwin, S.; Jenike, K.M.; Lucas, J.; McNulty, B.; Park, J.; Rautiainen, M.; et al. Gapless assembly of complete human and plant chromosomes using only nanopore sequencing. Genome Research 2024, 34, 1919–1930. [Google Scholar] [CrossRef]
- Stanojevic, D.; Lin, D.; Nurk, S.; Florez De Sessions, P.; Sikic, M. Telomere-to-Telomere Phased Genome Assembly Using HERRO-Corrected Simplex Nanopore Reads. bioRxiv 2024. [Google Scholar] [CrossRef]
- Sigova, E.A.; Dvorianinova, E.M.; Arkhipov, A.A.; Rozhmina, T.A.; Kudryavtseva, L.P.; Kaplun, A.M.; Bodrov, Y.V.; Pavlova, V.A.; Borkhert, E.V.; Zhernova, D.A.; et al. Nanopore Data-Driven T2T Genome Assemblies of Colletotrichum lini Strains. Journal of Fungi 2024, 10, 874. [Google Scholar] [CrossRef] [PubMed]
- Cechova, M. Probably Correct: Rescuing Repeats with Short and Long Reads. Genes 2020, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.K.; Bouvagnet, P. Assembling and gap filling of unordered genome sequences through gene checking. Genome Biology 2001, 2, preprint0008.1. [Google Scholar] [CrossRef]
- Khaldi, N.; Seifuddin, F.T.; Turner, G.; Haft, D.; Nierman, W.C.; Wolfe, K.H.; Fedorova, N.D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genetics and Biology 2010, 47, 736–741. [Google Scholar] [CrossRef]
- Robey, M.T.; Caesar, L.K.; Drott, M.T.; Keller, N.P.; Kelleher, N.L. An interpreted atlas of biosynthetic gene clusters from 1, 000 fungal genomes. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef]
- Zhang, X.; Leahy, I.; Collemare, J.; Seidl, M.F. Genomic Localization Bias of Secondary Metabolite Gene Clusters and Association with Histone Modifications in Aspergillus. Genome Biology and Evolution 2024, 16. [Google Scholar] [CrossRef]
- Greco, C.; Keller, N.P.; Rokas, A. Unearthing fungal chemodiversity and prospects for drug discovery. Current Opinion in Microbiology 2019, 51, 22–29. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, H.; Liu, Y.; Hong, Y.; Yang, H.; Zhou, C.; Tao, L. Computational advances in biosynthetic gene cluster discovery and prediction. Biotechnology Advances 2025, 79, 108532. [Google Scholar] [CrossRef]
- Lange, L.; Barrett, K.; Meyer, A.S. New Method for Identifying Fungal Kingdom Enzyme Hotspots from Genome Sequences. Journal of Fungi 2021, 7, 207. [Google Scholar] [CrossRef]
- Füting, P.; Barthel, L.; Cairns, T.C.; Briesen, H.; Schmideder, S. Filamentous fungal applications in biotechnology: a combined bibliometric and patentometric assessment. Fungal Biology and Biotechnology 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, C.H.; Nuraida, L.; Nuramalia, D.R.; Hardanti, S.; Świąder, K. Oncom: A Nutritive Functional Fermented Food Made from Food Process Solid Residue. Applied Sciences 2024, 14, 10702. [Google Scholar] [CrossRef]
- Qi, G.; Hao, L.; Xin, T.; Gan, Y.; Lou, Q.; Xu, W.; Song, J. Analysis of Whole-Genome facilitates rapid and precise identification of fungal species. Frontiers in Microbiology 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, H.P.; Gottschalk, C.; Cooper, B.; Bukowski, M.R.; Yang, R.; Gaskins, V.L.; Luciano-Rosario, D.; Fonseca, J.M.; Jurick, W.M. Omics-Based Comparison of Fungal Virulence Genes, Biosynthetic Gene Clusters, and Small Molecules in Penicillium expansum and Penicillium chrysogenum. Journal of Fungi 2024, 11, 14. [Google Scholar] [CrossRef]
- Salazar-Cerezo, S.; de Vries, R.P.; Garrigues, S. Strategies for the Development of Industrial Fungal Producing Strains. Journal of Fungi 2023, 9, 834. [Google Scholar] [CrossRef]
- Petersen, C.; Sørensen, T.; Westphal, K.R.; Fechete, L.I.; Sondergaard, T.E.; Sørensen, J.L.; Nielsen, K.L. High molecular weight DNA extraction methods lead to high quality filamentous ascomycete fungal genome assemblies using Oxford Nanopore sequencing. Microbial Genomics 2022, 8. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biology 2019, 20. [Google Scholar] [CrossRef]
- Nanoporetech. GitHub - dorado. https://github.com/nanoporetech/dorado, 2022. [Accessed 14-08-2025].
- Köster, J.; Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bonenfant, Q.; Noé, L.; Touzet, H. Porechop_ABI: discovering unknown adapters in Oxford Nanopore Technology sequencing reads for downstream trimming. Bioinformatics Advances 2022, 3. [Google Scholar] [CrossRef]
- De Coster, W.; Rademakers, R. NanoPack2: population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Hall, M. Rasusa: Randomly subsample sequencing reads to a specified coverage. Journal of Open Source Software 2022, 7, 3941. [Google Scholar] [CrossRef]
- lh3. GitHub - lh3/seqtk: Toolkit for processing sequences in FASTA/Q formats — github.com. https://github.com/lh3/seqtk, 2016. [Accessed 21-03-2025].
- Shen, W.; Sipos, B.; Zhao, L. SeqKit2: A Swiss army knife for sequence and alignment processing. iMeta 2024, 3. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Kim, D.; Gilchrist, C.L.M.; Chun, J.; Steinegger, M. UFCG: database of universal fungal core genes and pipeline for genome-wide phylogenetic analysis of fungi. Nucleic Acids Research 2022, 51, D777–D784. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Cox, E.; Holmes, J.B.; Anderson, W.R.; Falk, R.; Hem, V.; Tsuchiya, M.T.N.; Schuler, G.D.; Zhang, X.; Torcivia, J.; et al. Exploring and retrieving sequence and metadata for species across the tree of life with NCBI Datasets. Scientific Data 2024, 11. [Google Scholar] [CrossRef]
- Svedberg, J.; Vogan, A.A.; Rhoades, N.A.; Sarmarajeewa, D.; Jacobson, D.J.; Lascoux, M.; Hammond, T.M.; Johannesson, H. An introgressed gene causes meiotic drive inNeurospora sitophila. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Research 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biology 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y. pyCirclize: Circular visualization in Python. https://github.com/moshi4/pyCirclize, 2022. [Accessed 08-04-2025].
- Krzywinski, M.; Schein, J.; Birol, İ.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Research 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: a better web interface. Nucleic Acids Research 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Palmer, J.M.; Stajich, J. Funannotate v1.8.1: Eukaryotic genome annotation, 2020. [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Research 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Apweiler, R. GitHub - ebi-pf-team/interproscan: Genome-scale protein function classification — github.com. https://github.com/ebi-pf-team/interproscan, 2001. [Accessed 08-04-2025].
- Zdobnov, E.M.; Apweiler, R. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef]
- Brown, M.R.; Manuel Gonzalez de La Rosa, P.; Blaxter, M. tidk: a toolkit to rapidly identify telomeric repeats from genomic datasets. Bioinformatics 2025, 41. [Google Scholar] [CrossRef]
- Lyčka, M.; Bubeník, M.; Závodník, M.; Peska, V.; Fajkus, P.; Demko, M.; Fajkus, J.; Fojtová, M. TeloBase: a community-curated database of telomere sequences across the tree of life. Nucleic Acids Research 2023, 52, D311–D321. [Google Scholar] [CrossRef]
- Schalamun, M.; Schmoll, M. Trichoderma – genomes and genomics as treasure troves for research towards biology, biotechnology and agriculture. Frontiers in Fungal Biology 2022, 3. [Google Scholar] [CrossRef]
- Wieloch, W. Chromosome visualisation in filamentous fungi. Journal of Microbiological Methods 2006, 67, 1–8. [Google Scholar] [CrossRef]
- Han, D.M.; Baek, J.H.; Choi, D.G.; Jeon, M.S.; Eyun, S.i.; Jeon, C.O. Comparative pangenome analysis of Aspergillus flavus and Aspergillus oryzae reveals their phylogenetic, genomic, and metabolic homogeneity. Food Microbiology 2024, 119, 104435. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. What makes a centromere? Experimental Cell Research 2020, 389, 111895. [Google Scholar] [CrossRef] [PubMed]
- Zaccaron, A.Z.; Stergiopoulos, I. The dynamics of fungal genome organization and its impact on host adaptation and antifungal resistance. Journal of Genetics and Genomics 2025, 52, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.; Pratas, D.; Videira, A.; Pereira, F. Revisiting the Neurospora crassa mitochondrial genome. Letters in Applied Microbiology 2021, 73, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Buckler, E.S.; Stitzer, M.C. New whole-genome alignment tools are needed for tapping into plant diversity. Trends in Plant Science 2024, 29, 355–369. [Google Scholar] [CrossRef]
- Li, W.C.; Lin, T.C.; Chen, C.L.; Liu, H.C.; Lin, H.N.; Chao, J.L.; Hsieh, C.H.; Ni, H.F.; Chen, R.S.; Wang, T.F. Complete Genome Sequences and Genome-Wide Characterization of Trichoderma Biocontrol Agents Provide New Insights into their Evolution and Variation in Genome Organization, Sexual Development, and Fungal-Plant Interactions. Microbiology Spectrum 2021, 9. [Google Scholar] [CrossRef]
Figure 1.
Overview of Snakemake workflow.
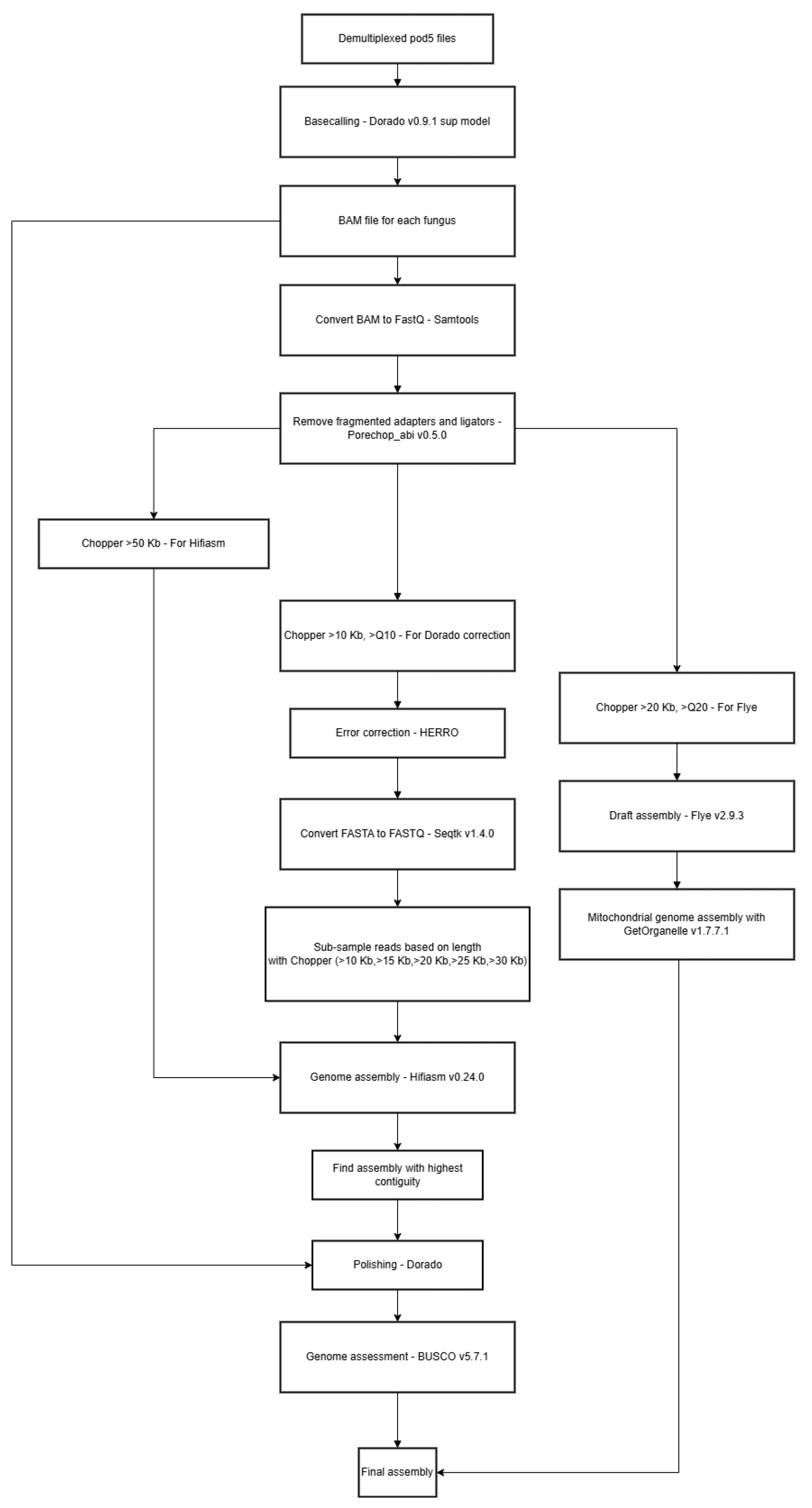
Figure 2.
Phylogenetic tree of NCBI GenBank reference genomes of the Neurospora genus, with the Neurospora intermedia NRRL 2884 assembly produced in this study, clustering with N. intermedia HJ2022-06, highlighted in green, and not N. sitophila as it was first identified and acquired as from the strain collection. Genealogical Sorting Index (GSI) values shown on branches.
Figure 2.
Phylogenetic tree of NCBI GenBank reference genomes of the Neurospora genus, with the Neurospora intermedia NRRL 2884 assembly produced in this study, clustering with N. intermedia HJ2022-06, highlighted in green, and not N. sitophila as it was first identified and acquired as from the strain collection. Genealogical Sorting Index (GSI) values shown on branches.
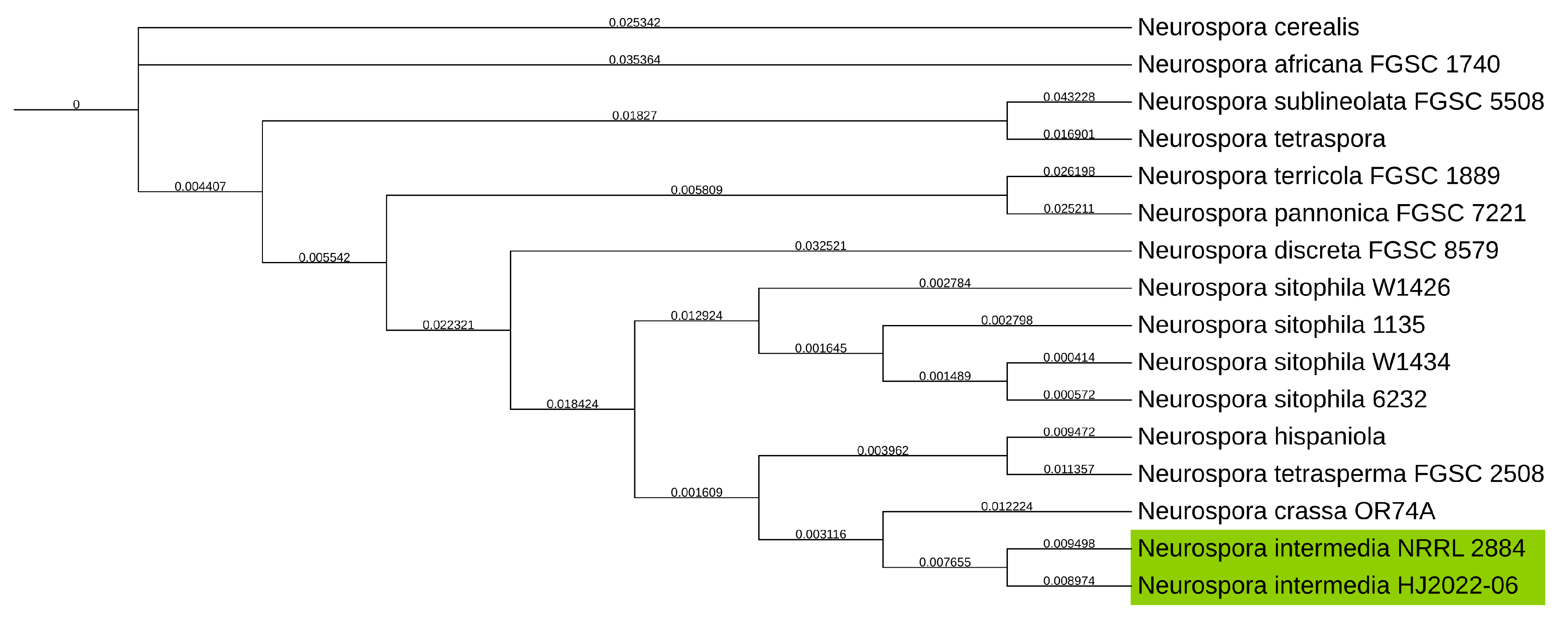
Figure 3.
Phylogenetic tree of NCBI GenBank reference genomes of the Trichoderma genus, with the T. asperellum TA1 assembly from this study clustering close to T. asperelloides, T. yunnanense, and T. asperellum. T. asperellum TA1 assembly produced in this study highlighted in green. GSI values shown on branches.
Figure 3.
Phylogenetic tree of NCBI GenBank reference genomes of the Trichoderma genus, with the T. asperellum TA1 assembly from this study clustering close to T. asperelloides, T. yunnanense, and T. asperellum. T. asperellum TA1 assembly produced in this study highlighted in green. GSI values shown on branches.
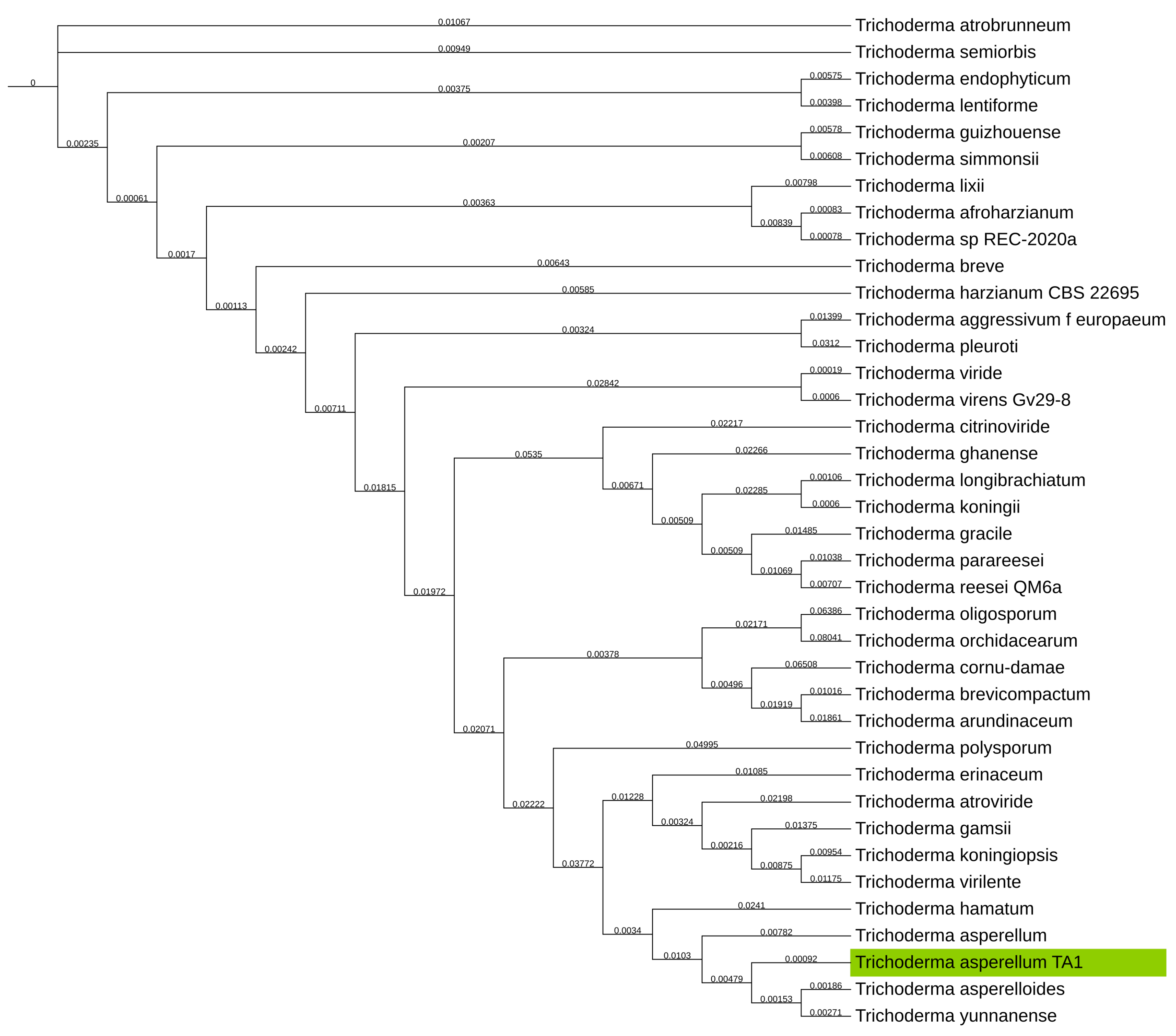
Figure 4.
Phylogenetic tree of NCBI genomes of T. asperelloides, T. yunnanense and T. asperellum, for a higher resolution taxonomical classification. With the T. asperellum TA1 assembly produced in this study clustering closest to T. asperellum highlighted in green. GSI values shown on branches. NCBI GenBank reference genomes are marked with *. T. asperellum type strain is marked with T.
Figure 4.
Phylogenetic tree of NCBI genomes of T. asperelloides, T. yunnanense and T. asperellum, for a higher resolution taxonomical classification. With the T. asperellum TA1 assembly produced in this study clustering closest to T. asperellum highlighted in green. GSI values shown on branches. NCBI GenBank reference genomes are marked with *. T. asperellum type strain is marked with T.
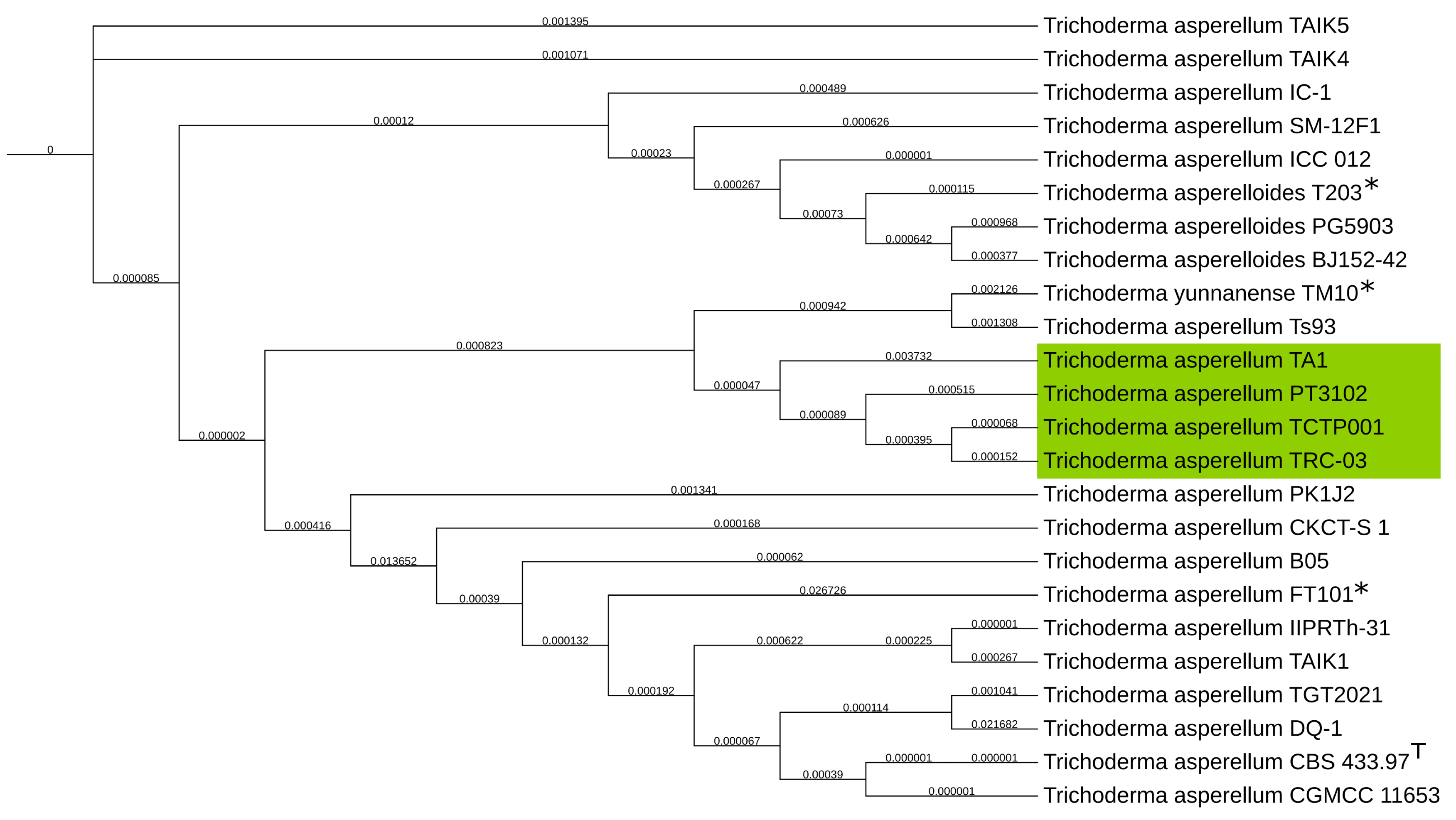
Figure 5.
Phylogenetic tree of NCBI GenBank reference genomes of the Aspergillus genus showing the clustering of the A. oryzae CBS 466.91 genome assembled in this study clustering with A. oryzae RIB40 highlighted in green. GSI values shown on branches.
Figure 5.
Phylogenetic tree of NCBI GenBank reference genomes of the Aspergillus genus showing the clustering of the A. oryzae CBS 466.91 genome assembled in this study clustering with A. oryzae RIB40 highlighted in green. GSI values shown on branches.
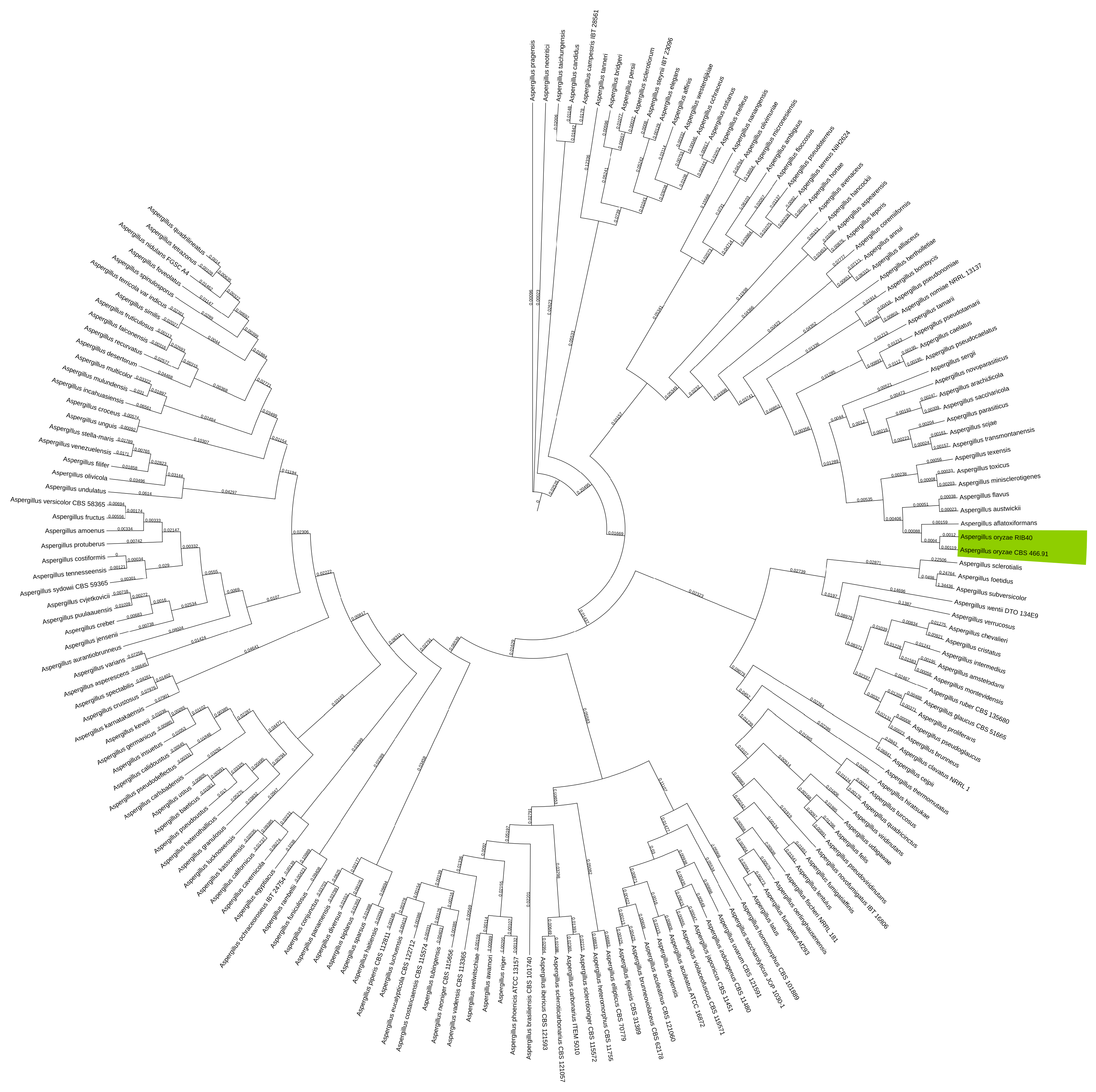
Figure 6.
Whole-genome alignment of N. intermedia NRRL 2884 and the closest related HQ genome N. intermedia HJ2022-06 in the NCBI GenBank database. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
Figure 6.
Whole-genome alignment of N. intermedia NRRL 2884 and the closest related HQ genome N. intermedia HJ2022-06 in the NCBI GenBank database. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
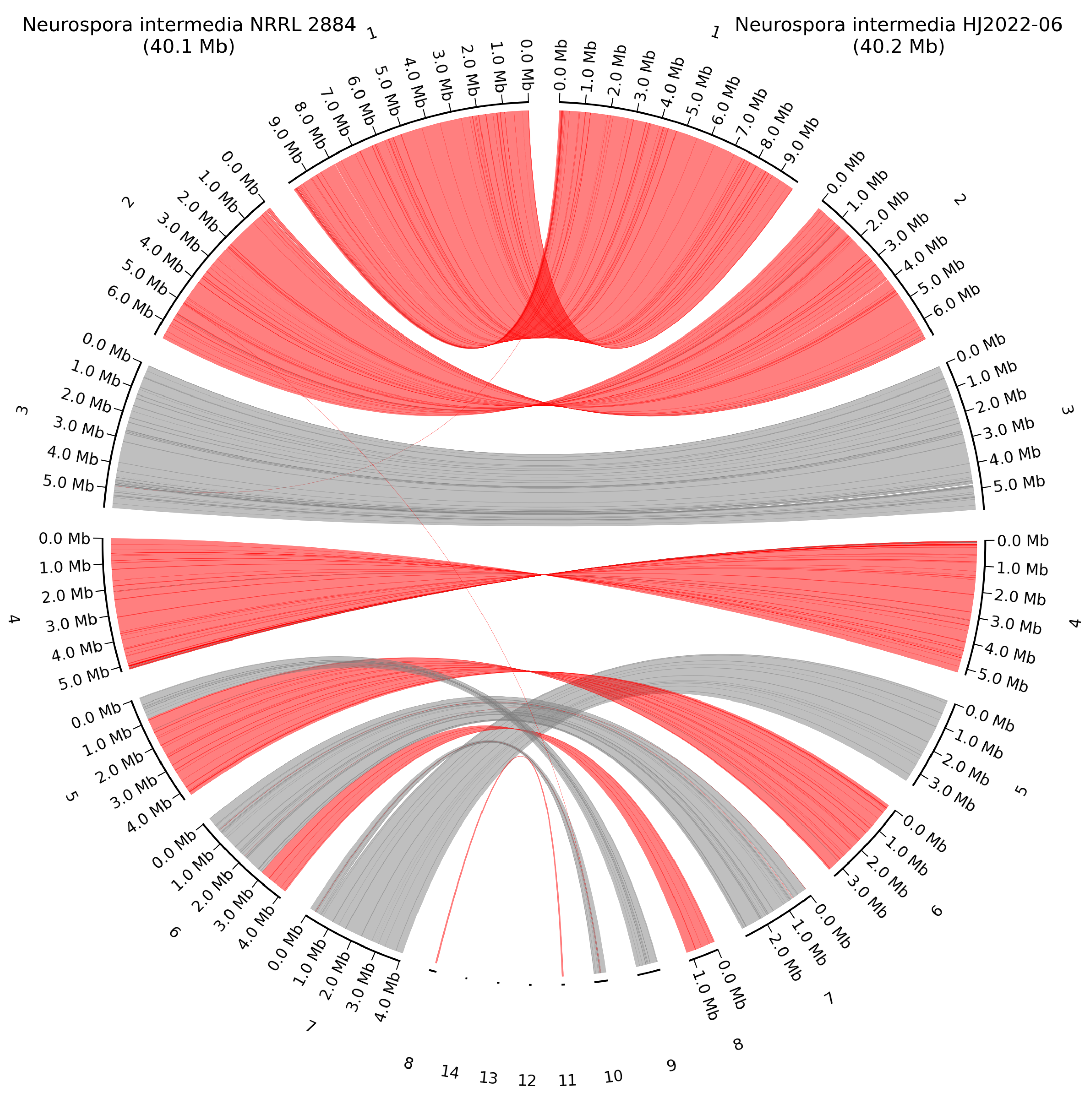
Figure 7.
Whole-genome alignment of T. asperellum TA1 and the closest related HQ genome T. asperellum TCTP001. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
Figure 7.
Whole-genome alignment of T. asperellum TA1 and the closest related HQ genome T. asperellum TCTP001. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
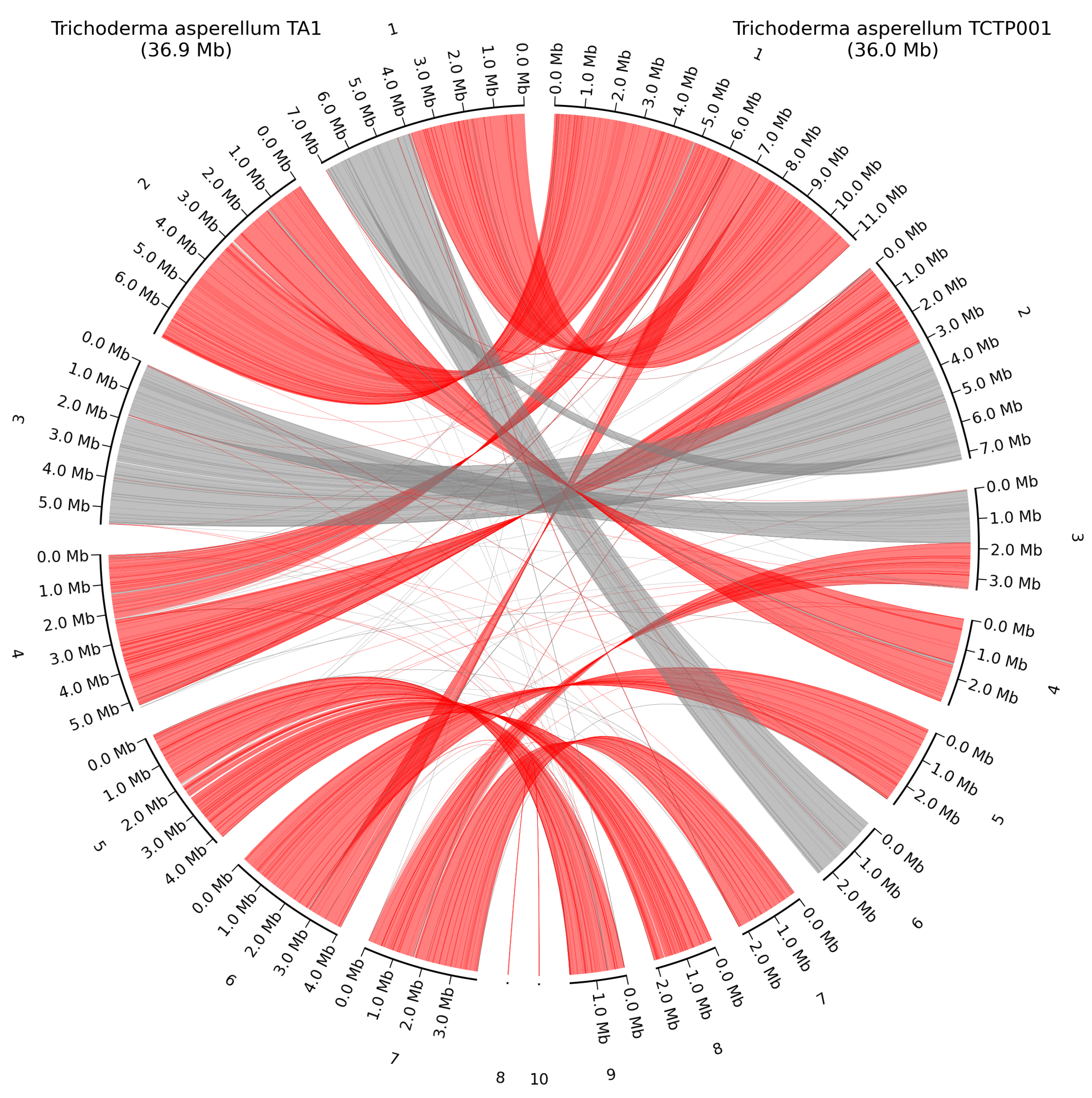
Figure 8.
Whole-genome alignment of A. oryzae CBS 466.91 and the closest related HQ genome A. oryzae RIB40. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
Figure 8.
Whole-genome alignment of A. oryzae CBS 466.91 and the closest related HQ genome A. oryzae RIB40. Grey lines show direct alignment, red lines show reverse complement alignment. Numbering is from the largest contig to the smallest.
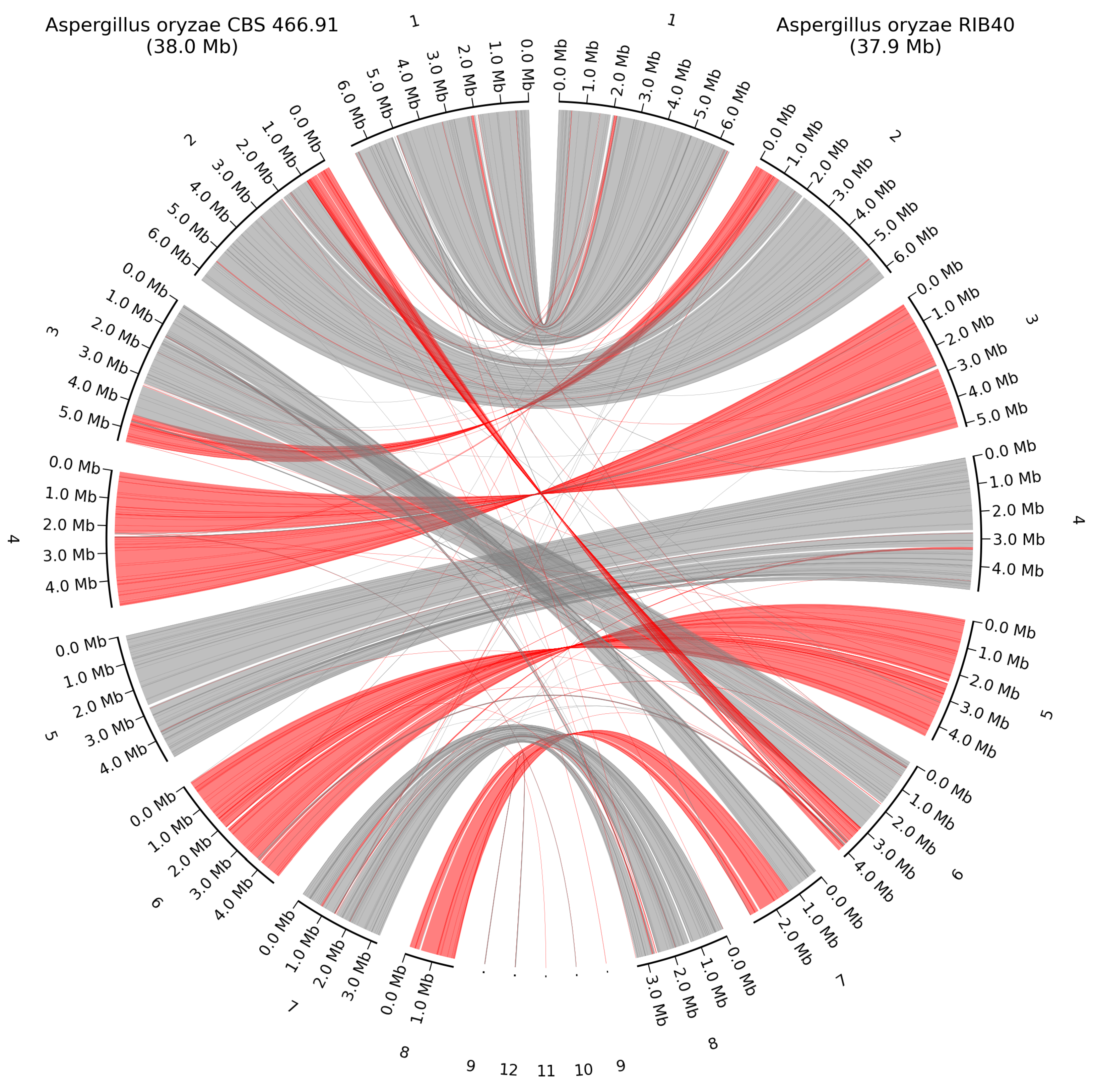
Figure 9.
Overview of predicted annotation in N. intermedia NRRL 2884. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward Coding sequence (CDS) in red, Reverse CDS in blue, tRNAs in pink, biosynthetic gene clusters (BGCs) in green, carbohydrate-active enzymes (Cazymes) genes in purple.
Figure 9.
Overview of predicted annotation in N. intermedia NRRL 2884. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward Coding sequence (CDS) in red, Reverse CDS in blue, tRNAs in pink, biosynthetic gene clusters (BGCs) in green, carbohydrate-active enzymes (Cazymes) genes in purple.
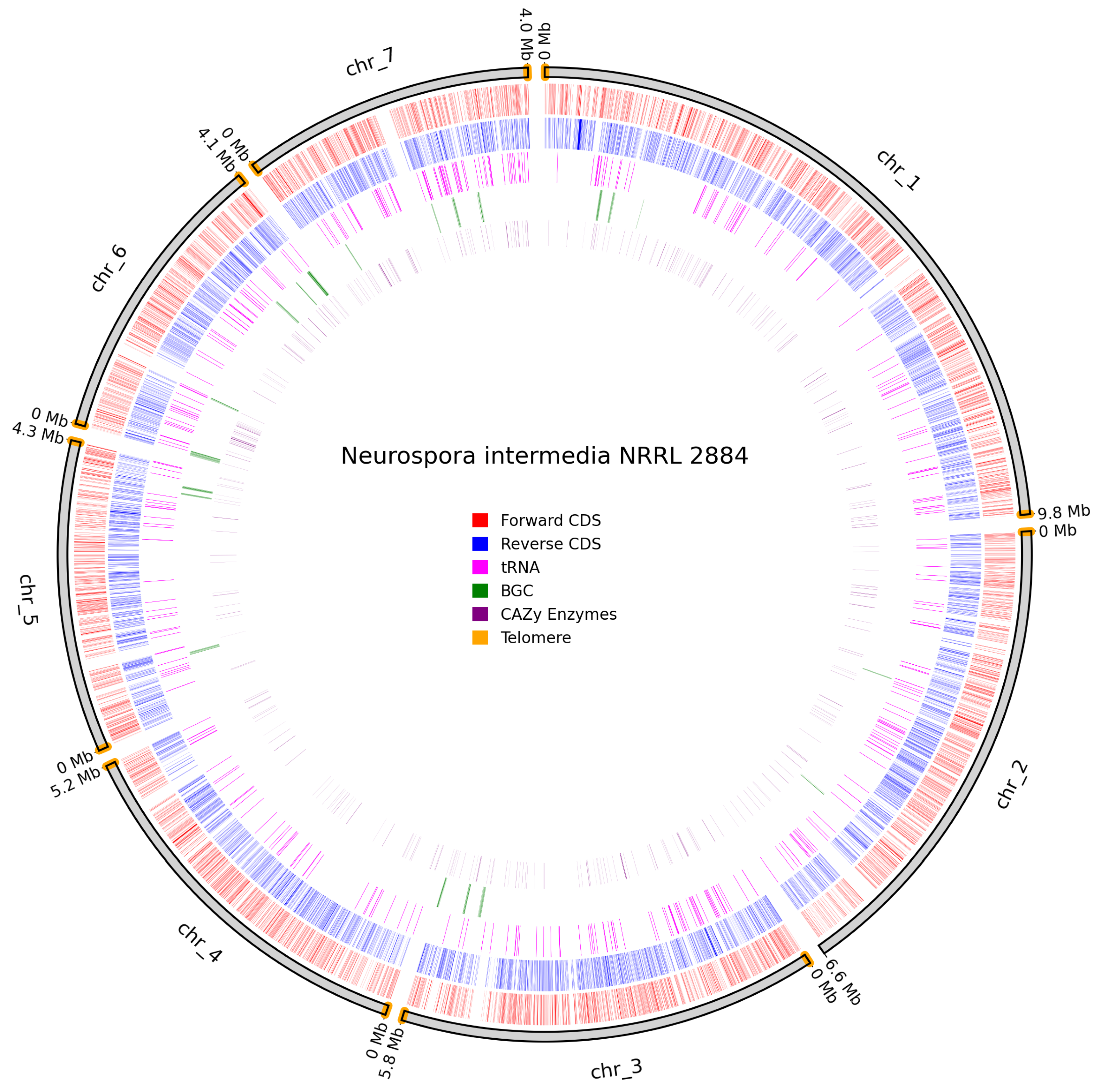
Figure 10.
Overview of predicted annotation in T. asperellum TA1. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward CDS in red, Reverse CDS in blue, tRNAs in pink, BGCs in green, Cazy genes in purple.
Figure 10.
Overview of predicted annotation in T. asperellum TA1. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward CDS in red, Reverse CDS in blue, tRNAs in pink, BGCs in green, Cazy genes in purple.
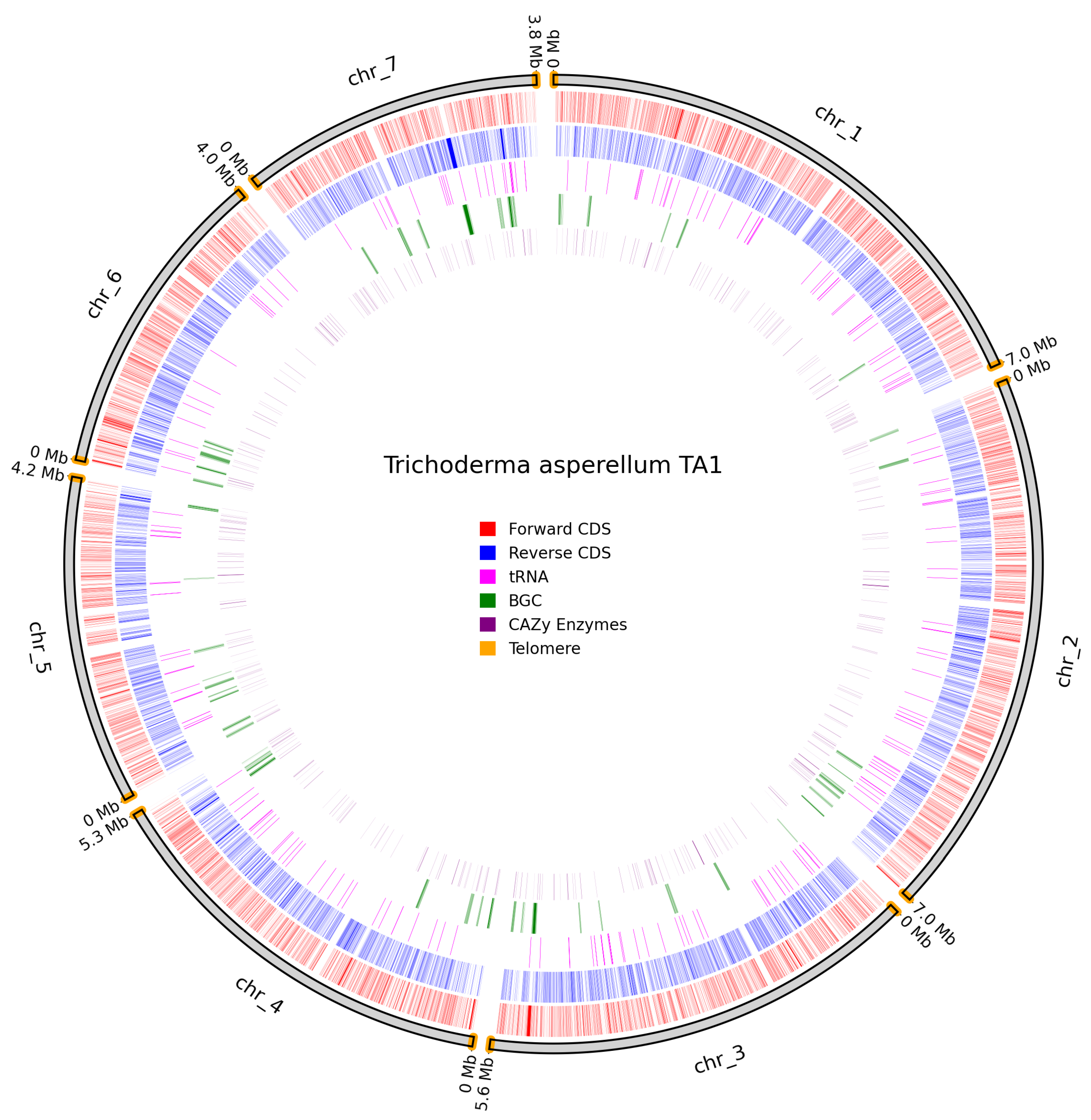
Figure 11.
Overview of predicted annotation in A. oryzae CBS 466.91. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward CDS in red, Reverse CDS in blue, tRNAs in pink, BGCs in green, Cazy genes in purple.
Figure 11.
Overview of predicted annotation in A. oryzae CBS 466.91. Chromosome naming scheme on the outer track with chromosome size in Mb. Identified telomeres are coloured in orange. Forward CDS in red, Reverse CDS in blue, tRNAs in pink, BGCs in green, Cazy genes in purple.
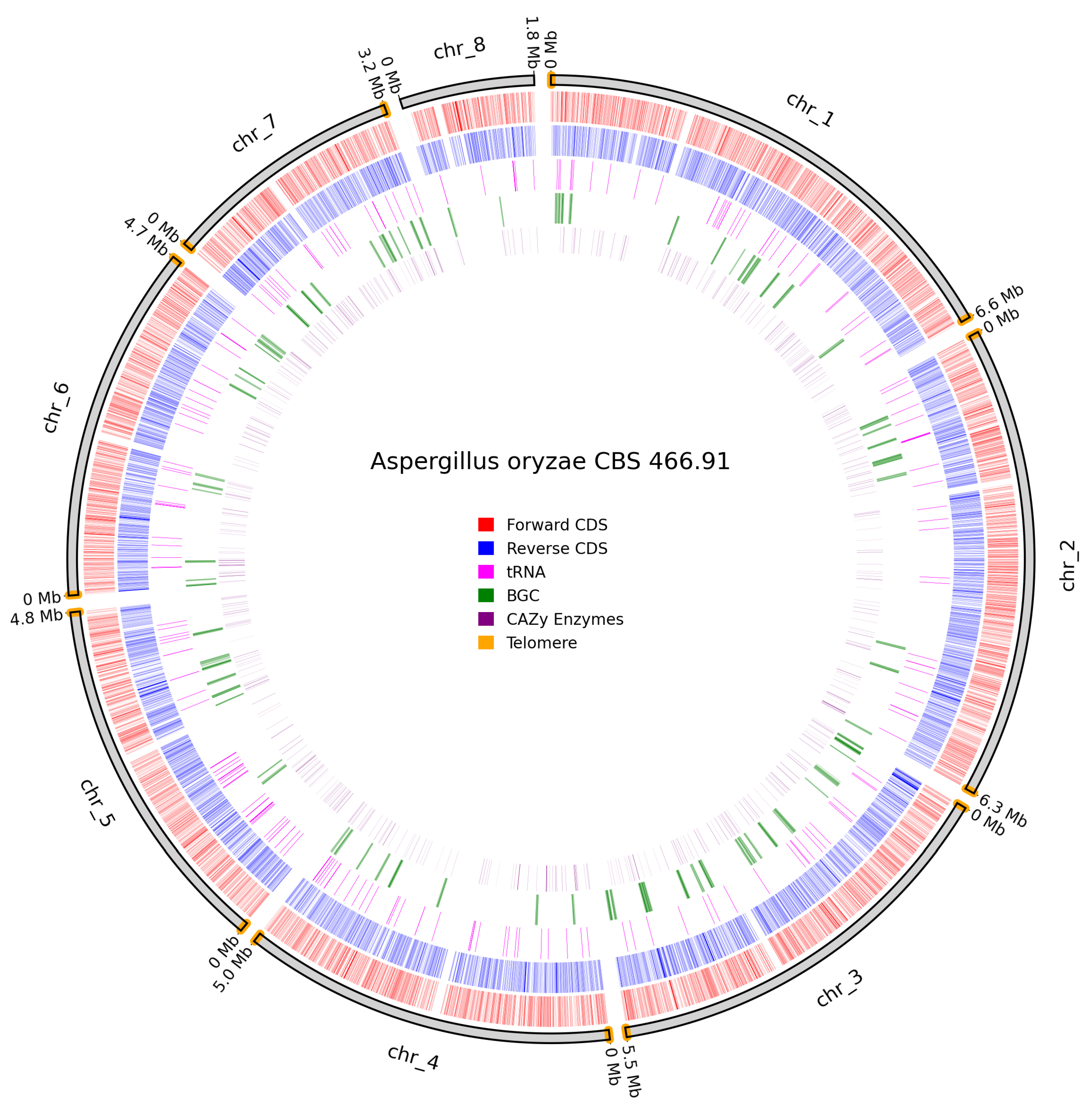

Table 1.
Read statistics for the snakemake workflow basecalling, filtering and error correction results. Coverage was calculated based on the final genome assembly. Average quality was not calculated after error correction and the subsequent subsampling as there is no intrinsic quality score after error correction.
Table 1.
Read statistics for the snakemake workflow basecalling, filtering and error correction results. Coverage was calculated based on the final genome assembly. Average quality was not calculated after error correction and the subsequent subsampling as there is no intrinsic quality score after error correction.
| Genus | Method | Coverage [X] | N50 [Kb] | Average Quality |
|---|---|---|---|---|
| Neurospora | Raw-Basecalled | 194.56 | 11.375 | 20.31 |
| Neurospora | Pre-Correction | 104.67 | 21.338 | 20.58 |
| Neurospora | Corrected | 94.05 | 20.317 | n/a |
| Neurospora | Contiguity | 33.68 | 34.920 | n/a |
| Neurospora | Ultra-long | 7.53 | 59.306 | 20.14 |
| Neurospora | Flye | 42.53 | 30.951 | 23.34 |
| Trichoderma | Raw-Basecalled | 66.34 | 22.114 | 20.35 |
| Trichoderma | Pre-Correction | 49.06 | 29.752 | 20.56 |
| Trichoderma | Corrected | 44.78 | 27.294 | n/a |
| Trichoderma | Contiguity | 43.60 | 27.993 | n/a |
| Trichoderma | Ultra-long | 9.37 | 61.816 | 20.27 |
| Trichoderma | Flye | 26.45 | 37.005 | 23.45 |
| Aspergillus | Raw-Basecalled | 238.93 | 11.980 | 20.94 |
| Aspergillus | Pre-Correction | 136.76 | 19.340 | 21.13 |
| Aspergillus | Corrected | 118.29 | 18.200 | n/a |
| Aspergillus | Contiguity | 114.65 | 18.633 | n/a |
| Aspergillus | Ultra-long | 7.60 | 59.144 | 20.40 |
| Aspergillus | Flye | 51.27 | 29.417 | 23.74 |
Table 2.
BUSCO results. C (complete BUSCO), S (Single copy BUSCO), D (Duplicated BUSCO), F (Fragmented BUSCO), M (Missing BUSCO), DB (BUSCO dataset).
Table 2.
BUSCO results. C (complete BUSCO), S (Single copy BUSCO), D (Duplicated BUSCO), F (Fragmented BUSCO), M (Missing BUSCO), DB (BUSCO dataset).
| Genus | Contigs | Size [Mb] | C [%] | S [%] | D [%] | F [%] | M [%] | n | DB |
|---|---|---|---|---|---|---|---|---|---|
| Neurospora | 7(+1)1 | 40 | 99.4 | 99.1 | 0.3 | 0.3 | 0.3 | 758 | fungi_odb10 |
| Trichoderma | 7 | 37 | 98.8 | 98.5 | 0.3 | 0.3 | 0.9 | 758 | fungi_odb10 |
| Aspergillus | 8 | 38 | 98.7 | 98.3 | 0.4 | 0.5 | 0.8 | 758 | fungi_odb10 |
1The extra contig was a wrongly assembled mitochondrial genome
Table 3.
Genes and BGCs predicted for the fungal genomes. statistics.
| Strain | Genes | CAZymes | PKSs | NRPSs/NRPSs-like | Terpene synthases | RiPPs |
|---|---|---|---|---|---|---|
| N. intermedia NRRL 2884 | 8790 | 345 | 8 | 5 | 3 | 4 |
| T. asperellum TA1 | 9805 | 401 | 17 | 18 | 10 | 3 |
| A. oryzae CBS 466.91 | 13042 | 579 | 28 | 32 | 12 | 6 |
Table 4.
BUSCO results. C (complete BUSCO), S (Single copy BUSCO), D (Duplicated BUSCO), F (Fragmented BUSCO), M (Missing BUSCO), DB (BUSCO dataset).
Table 4.
BUSCO results. C (complete BUSCO), S (Single copy BUSCO), D (Duplicated BUSCO), F (Fragmented BUSCO), M (Missing BUSCO), DB (BUSCO dataset).
| Sample | Contigs | Size [Mb] | C [%] | S [%] | D [%] | F [%] | M [%] | DB |
|---|---|---|---|---|---|---|---|---|
| C. lini 394-2 1 | 13 | 53.56 | 96.8 | 96.6 | 0.2 | 0.6 | 2.6 | glomerellales_odb10 |
| C. lini 394-2 | 13 | 53.69 | 96.8 | 96.6 | 0.2 | 0.6 | 2.6 | glomerellales_odb10 |
1 Assembled in this study
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



