
Submitted:
04 December 2023
Posted:
05 December 2023
You are already at the latest version
Abstract
The pine pitch canker pathogen, Fusarium circinatum, is globally regarded as one of the most important threads to commercial pine-based forestry. Although genome sequences of this fungus are available, these remain highly fragmented or structurally ill-defined. Our overall goal was to provide high-quality assemblies for two notable strains of F. circinatum, and to characterize these in terms of coding content, repetitiveness, and the position of telomeres and centromeres. For this purpose, we used Oxford Nanopore Technologies MinION long read sequences, as well as Illumina short sequence reads. By leveraging the genomic synteny inherent to F. circinatum and its close relatives, these sequence reads were assembled to chromosome-level, where contiguous sequences mostly spanned from telomere to telomere. Comparative analyses unveiled remarkable variability in the twelfth and smallest chromosome, which is known to be dispensable. It presented a striking length polymorphism, with one strain lacking substantial portions from the chromosome's distal and proximal regions. These regions, characterized by a lower gene density and G+C content and an increased prevalence of repetitive elements, contrast starkly with the syntenic segments of the chromosome, as well as with the core chromosomes. We propose that these unusual regions might have arisen or expanded due to the presence of transposable elements. Comparison of overall chromosome structure revealed that centromeric elements often underpin intrachromosomal differences between F. circinatum strains, especially at chromosomal breakpoints. This suggested a potential role for centromeres in shaping the chromosomal architecture of F. circinatum and its relatives. The publicly available genome data generated here, together with the detailed metadata provided, represent essential resources for future studies of this important plant pathogen.
Keywords:
dispensable chromosome
; centromere
; telomere
; intrachromosomal translocation
1. Introduction
The era of fungal genomics began in 1996 with sequencing of the baker’s yeast (Saccharomyces cerevisiae) genome [1]. In 2003, the genome of the first filamentous fungus, Neurospora crassa, was sequenced [2]. These two milestones in genome research provided the impetus for subsequent fungal work and by 2010, the genome sequences for more than 100 fungi were available in public databases [3]. Towards the end of 2023, one of the main repositories for fungal genomes, MycoCosm (https://mycocosm.jgi.doe.gov), contained data for more than 2500 species, spanning the fungal tree of life, with thousands more genomes currently being sequenced. The collective availability of these resources has revolutionized our knowledge of the ecology, evolution and overall biology of fungi [4,5,6,7,8,9,10,11]. Indeed, genome sequencing has become a routine part of modern fungal research.
The socioeconomically important genus Fusarium (Phylum Ascomycota; Family Nectriaceae; Order Hypocreales) provides an illustrative example of how technological advances and decreasing costs have driven large scale genome initiatives [4,12,13,14]. Apart from genome sequences being publicly available for numerous Fusarium species, multiple strains of many agriculturally/medically important species have also been sequenced. One such species is the pine pitch canker pathogen, Fusarium circinatum, for which the whole genome sequences of 17 strains are currently available in the database of the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/; accessed November 2023). Together with many other socioeconomically important species, this pathogen forms part of the so-called Fusarium fujikuroi species complex (FFSC) [15]. It can infect more than 60 Pinus species at all growth stages and is globally regarded as one of the most important risks to pine-based forestry enterprises [16]. The pathogen typically causes chlorosis and the development of large resinous cankers at infection points on the trunks and branches of trees, often leading to dieback [17]. In the case of younger plants, especially in seedlings in commercial nurseries, infection by F. circinatum causes wilting and chlorosis of needles due to severe root and root collar disease, culminating in plant mortality [17]. The need for effective control strategies has thus sparked considerable interest in the use of genomic tools for studying the genetics, evolution and general biology of this important pathogen.
Fusarium circinatum was the first eukaryote to have its whole genome sequenced on the African continent [18]. As is the case for most publicly available fungal genomes, this was done using second-generation sequencing technologies. The original assembly was compiled for strain FSP34 using the 454 GS FLX system [18] and later augmented with SOLiDTM mate-pair data, but the resulting assembly remained highly fragmented and incomplete [19]. The same is true for the second F. circinatum strain (KS17) that was sequenced using SOLiD mate-pair data [20]. Despite their limited quality, however, these data were invaluable for improving our knowledge of the pitch canker pathogen, especially in terms of its population dynamics [21] and pathogenesis e.g., [22,23]. Furthermore, by making use of the genetic linkage map for strain FSP34 [24] and the macrosyntenic nature of FFSC genomes [25], contigs comprising the initial genome data for FSP34 and KS17 could be ordered into pseudomolecules corresponding to the twelve chromosomes of F. circinatum [19].
The advent of third-generation technologies enabled the real-time reading of nucleotide sequences at the single molecule level [26], allowing the production of long sequence reads (> 2.27 Mb) [27]. A prominent example of such a long-read system is the portable MinION sequencer from Oxford Nanopore Technologies (ONT), which uses nanopores to sequence a single DNA molecule per pore, bypassing the sequencing-by-synthesis method of traditional sequencers [28]. The high error rate inherent to this system is then accounted by using its long-read output in conjunction with short-read data to assemble highly accurate and exceedingly long, contiguous sequences [27]. In fungi, this approach has allowed for the assembly of chromosome-scale sequences, often spanning from telomere to telomere [29,30,31,32]. Such complete or near complete assemblies thus allow for investigations into the structural and architectural properties of genomes, and the role these play in the biology of fungi, as well as their overall genome evolution [29,33].
Insights into the genome architecture and sub-genomic compartmentalization of F. circinatum have been largely limited by not having access to high-quality chromosome-level assemblies. Although genomes produced using long-read systems are available for a number of F. circinatum strains, these were only ordered into pseudomolecules using those produced for F. circinatum FSP34 [34]. Therefore, the overall goal of this study was to provide complete or near complete, fully annotated, chromosome-level genome assemblies for F. circinatum by focusing specifically on the two strains (FSP34 and KS17) most frequently used in laboratory and computational studies e.g., [20,23,35]. Accordingly, our study had three specific objectives: (i) to assemble the genomes of strains FSP34 and KS17 by making use of both second and third generation sequencing technologies; (ii) to annotate the genomes in terms of gene content, chromosome number, and chromosome identity, as well as their telomeric and centromeric features; and (iii) to compile for both strains a detailed set of relevant biological and ecological metadata (i.e., a description of the attributes of the samples from which the genome data was generated). The latter is increasingly regarded as essential for achieving timely and impactful outcomes in genome-based investigations [36,37]. This study would thus aid future research into the chromosomal architecture as well as the genic content of each genome, delivering essential resources for studying this destructive plant pathogen.
2. Materials and Methods
2.1. Genome Sequencing and Assembly
DNA was extracted from F. circinatum strains FSP34 and KS17 as described previously [38]. These DNAs were then subjected to ONT MinION sequencing, as well as Illumina HiSeq 2500 sequencing of a 550bp paired-end library (Macrogen, Seoul, Korea). The data produced by the two systems were then used in a two-step process to compile the respective genomes into assemblies. First, the ONT MinION reads were trimmed and assembled using Canu v1.7.1 [39], with the ‘correctedErrorRate’ setting adjusted, until assemblies were obtained that displayed the expected macrosynteny known in the FFSC [12,25]. The latter was determined using the LASTZ v1.02.00 plugin [40] of Geneious v 7.1.9 [41]. The second step utilized the quality-filtered Illumina reads (>18 bases) returned by CLC Genomics Workbench v 8.0.1 (CLCBio, Aarhus, Denmark). These reads were indexed and aligned to the Canu assembly using BWA [42] and SAMtools [43]. We then used Pilon v1.22 [44] to correct for the occurrence of sequencing errors inherent to the MinION sequencing platform [28]. Where needed, further scaffolding were performed using LASTZ-based alignments and MUMmer v4.x [45] to order and orient contigs into pseudomolecules and to pinpoint assembly breaks. The latter were indicated in the eventual assemblies with the insertion of 100 ambiguous nucleotides (i.e., 100 Ns).
MUMmer was used to evaluate the occurrence and position of a specific chromosomal translocation known to characterize the genomes of Fusarium species in the American clade of the FFSC [25]. This clade represents one of the so-called biogeographic clades that mainly comprises of species isolated from plant hosts originating in the Americas [46]. The current study only utilized those American clade species for which high-quality genome assemblies were available (i.e., Fusarium pilosicola, Fusarium marasasianum, Fusarium pininemorale, Fusarium sororula, Fusarium fracticaudum and F. temperatum) (Table S1). The chromosomal translocation was also investigated in a number of other F. circinatum strains (FFRA, FSOR, UG10, UG27, CMWF560, CMWF567, CMWF1803, and GL1327) for which suitable genome data were available (Table S1).
2.2. Evaluation of Genome Quality and Completeness
Assembly completeness was estimated using the Benchmarking Universal Single-Copy Orthologs (BUSCO) v3 tool, with the “Sordariomyceta” dataset [47]. We also investigated the level to which improved sequencing technologies allowed for the enhancement of the FSP34 and KS17 assemblies. The analyses included the previous [19] and current assemblies for FSP34 (denoted as FSP34previous and FSP34current), and the previous and current assemblies for KS17 (denoted as KS17previous and KS17current).
2.3. Genome Annotation
We identified the putative positions of telomeres and centromeres for each of the pseudomolecules. Telomeres were identified by the occurrence of telomeric repeats (TTAGGG)ncommonly found in filamentous fungi [49] using a sliding window of 1 000 bp with 500 bp increments. To control for the spurious appearance of this repeat in the genome, telomeric caps were identified as those stretches of DNA containing a higher frequency of the telomeric repeats (i.e., ≥ three times the average telomeric density across each pseudomolecule). The putative positions of centromeres were determined as described previously [50]. Briefly, this involved identifying regions with reduced G+C content and increased frequencies of mutations resembling those caused by repeat-induced point (RIP). We then determined whether the identified regions occurred at comparable locations to those of the known centromeres of F. fujikuroi and F. verticillioides [12]. The latter was achieved by using tBLASTn searches (implemented in CLC Genomics Workbench; E < 1x10-5) to compare the positions of genes flanking the centromeres of F. fujikuroi and F. verticillioides [12] with those flanking the putative centromeric regions of FSP34 and KS17. Nomenclature for centromeric positions followed those originally published in Hereditas [51].
The genes encoded by the FSP34 and KS17 genomes were identified using MAKER v2.31.8 [52]. This pipeline incorporated the gene prediction programs AUGUSTUS v3.2.2 [53], GeneMark ES [54] and SNAP [55]. In addition, predicted protein evidence from F. graminearum and F. verticillioides [4], F. fujikuroi [12], F. mangiferae and F. proliferatum [14], as well as F. circinatum [18], was utilized. The predicted genes were then functionally annotated using Blast2GO [56]. Where relevant, gene ontology (GO) term enrichment was evaluated using two-sided Fisher’s exact tests in Blast2GO (P < 0.05), and summarized with the REVIGO web server [57]. Additionally, the repeat content of the two assemblies analysed using REPET v2.5 pipeline [58,59]. This pipeline allowed both detection and annotation of the repeats and other transposable elements (TEs).
2.4. Compilation of Ecological and Biological Metadata for FSP34 and KS17
To complement the high-quality genomes generated in this study, all relevant source and biological data for the two strains were collected from literature. For this purpose, we focused on their origins in terms of geography, host species, tissue type, as well as the population genetics of their source populations. We also collected information generated using previous laboratory-based experimentation regarding their reproductive biology.
In the current study, we additionally investigated these two strain’s pathogenicity and growth rate. For this purpose, pathogenicity tests using six-month-old Pinus patula were conducted as described before [60]. Mycelial growth of the two strains were evaluated at a range of temperatures (10°C to 35°C, at 5°C intervals) on potato dextrose agar medium (20% w/v PDA [Biolab] and 5% w/v agar [BD Difco]). For this purpose, a 6mm mycelial plug taken from the actively growing margin of a 7-day old PDA culture was placed in the center of a 90 mm Petri plate containing PDA and incubated at the respective temperatures for 7 d in the dark. Replicates of five plates per strain at each temperature was performed. Colony diameters were recorded as the average of two measurements taken along two axes at right angles to each other.
3. Results
3.1.1. Chromosome-Level Assemblies for FSP34 and KS17
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession AYJV00000000 and LQBB00000000 for F. circinatum FSP34 and KS17, respectively. The respective assemblies were 45 020 843 and 44 380 849 nucleotides in length (Table 1). For FSP34, this represented an increase in genome size, with the FSP34previous being 1.02% smaller than the one reported here (i.e., FSP34current). The opposite was observed for KS17, with the highly fragmented KS17previous assembly being 1.04% larger than the KS17current assembly. In terms of G+C content, a substantial difference was also observed between the old and new KS17 assemblies and not those for FSP34. G+C content values of 47.41%, 47% and 47.26% were obtained for the FSP34previous, FSP34current and the KS17current assemblies, respectively, while the KS17previous had a G+C content of 44.69%. This could be explained by the G+C bias of the SOLiD technology used in the KS17previous assembly [61].
Both new assemblies displayed N50 values exceeding 4.3Mb (i.e., contigs ≥4.3Mb in length cover at least 50% of the assembly). This high contiguity in the two assemblies gave rise to FSP34current and KS17current assemblies containing substantially fewer scaffolds (49 and 96 scaffolds, respectively) than the previous assemblies (Table 1). When using these scaffolds to compile each of the 12 known chromosomes for F. circinatum (Table 2), the pseudomolecules for the FSP34current assembly contained twelve-fold fewer scaffolds than was the case for the FSP34previous assembly.
In contrast to the previous assembly, only a small proportion of contigs (representing less than 0.60% of the total genome size) could not be incorporated into the pseudomolecules. Also, for FSP34current, the pseudomolecules were larger in size than was predicted from FSP34previous, with pseudomolecule 12 the only exception (i.e., it was slightly smaller in the FSP34current assembly). The sizes of the pseudomolecules of the FSP34current and KS17current assembly had the same general trend, although pseudomolecule 12 of the KS17current assembly was almost 1.66 times larger due to the presence of distal and proximal regions that were not found in its FSP34 counterpart (Figure 1).
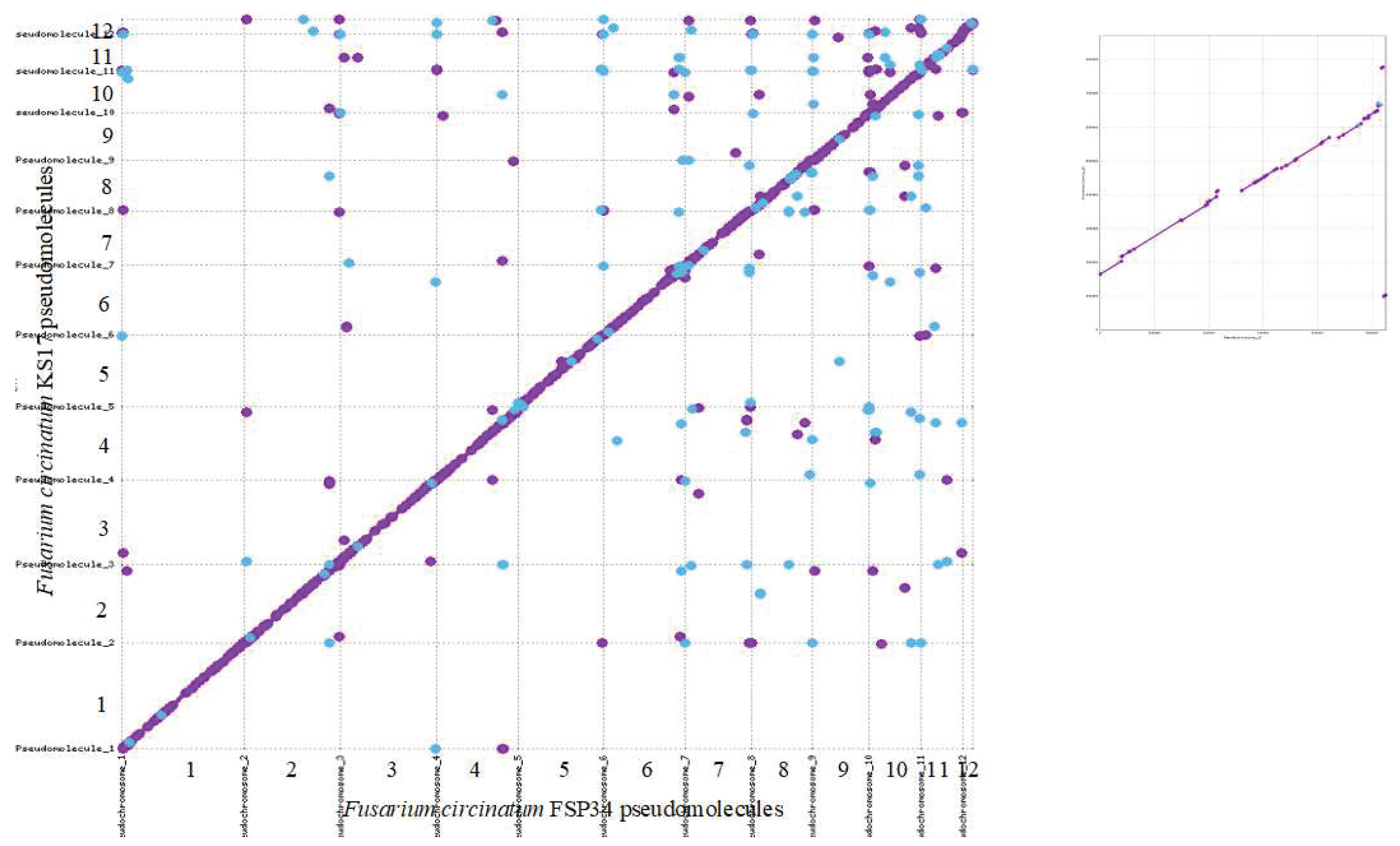
Overall, the FSP34current and KS17current assemblies were highly syntenic, as indicated by the MUMmer analysis (Figure 1). Also, the reciprocal translocation between chromosomes 8 and 11 [25] previously suggested to occur in FSP34previous [19], was found in the new assemblies. Both pseudomolecule 8 and 11 have scaffolds traversing the breakpoint in FSP34current, and the same was true for these pseudomolecules in KS17current.
Figure 1.
Sequence comparison between the set of pseudomolecules compiled for each genome. MUMmer revealed high levels of synteny across the F. circinatum FSP34 and KS17 assemblies generated in this study. A close-up of the comparison between twelfth pseudomolecules from the two genome assemblies is shown in the inset (top right corner). Forward matches are indicated with purple dots and reverse matches with blue dots.
Figure 1.
Sequence comparison between the set of pseudomolecules compiled for each genome. MUMmer revealed high levels of synteny across the F. circinatum FSP34 and KS17 assemblies generated in this study. A close-up of the comparison between twelfth pseudomolecules from the two genome assemblies is shown in the inset (top right corner). Forward matches are indicated with purple dots and reverse matches with blue dots.
3.1.2. Identification of Telomeres and Centromeres
To determine whether the 12 pseudomolecules for each assembly had their telomeric caps, we analyzed the distribution of the eukaryotic telomere repeat motif (Figure 2). The data indicated that several of the pseudomolecules compiled for the two assemblies represented chromosomes that were sequenced end to end. A total of 17 and 18 (out of the expected 24) telomeric caps were identified in the FSP34current and KS17current assemblies, respectively. These were present at both ends of pseudomolecule 7, 10, 11 and 12 of both assemblies, as well as for FSP34’s pseudomolecule 4 and 9 and KS17’s pseudomolecule 1, 3 and 6. This was a substantial improvement over the FSP34previous assembly for which only 7 of the 24 ends had telomere caps, while in the KS17previous assembly telomere annotation was not done due to its highly fragmented nature.
Based on G+C content depletion, combined with comparisons to the structurally annotated chromosomes of closely related species, we predicted the putative locations of centromeric regions in all 12 of the pseudomolecules in each of the two genome assemblies (Figure 2; Table S2 and S3). Analyses of regions corresponding to centromeres showed that these regions were less syntenic, gene poor, G+C-depleted and were richer in repeats and RIP mutations that the rest of the respective pseudomolecules [62,63]. Specifically, all the centromeric regions in the two genome assemblies had a G+C content below 20% (Table S2) compared to the 47% average for the genome-wide G+C content (Table 1). Also, these regions showed a high level of conservation in terms of the genes flaking them, when compared to those neighbouring the centromeres of F. fujikuroi and F. verticillioides (Table S3). The only exceptions were for pseudomolecules 8, 11 and 12, where the observed synteny and collinearity differed substantially from what is known for FFSC species (see below).
Most of the predicted centromeric regions in the assemblies were located in intermediate locations on the pseudomolecules (i.e., submetacentric, metacentric and subtelomeric in pseudomolecules 1, 2 and 3, respectively) [51]. In most instances, similar positions for centromeres were also predicted for corresponding pseudomolecules in the two assemblies, except for pseudomolecules 5, 8, 11 and 12 (Table S2). Pseudomolecule 5 of FSP34current had a metacentric position, but a submetacentric position in KS17current. This was supported by the fact that the conserved gene flanking the proximal side of the centromere, was not detected in the KS17current assembly (Table S3), implying that the shift in centromere location was associated with altered synteny on pseudomolecule 5.
Figure 2.
Schematic overview of the relative positions of centromeres (blue bars) and telomeres (orange bars) identified and/or predicted for each of the 12 pseudomolecules in the FSP34 and KS17 genome assemblies generated in this study (pink bar in FSP34 pseudomolecule 12 is indicative that the centromere and telomere are both positioned distally in close proximity to each other).
Figure 2.
Schematic overview of the relative positions of centromeres (blue bars) and telomeres (orange bars) identified and/or predicted for each of the 12 pseudomolecules in the FSP34 and KS17 genome assemblies generated in this study (pink bar in FSP34 pseudomolecule 12 is indicative that the centromere and telomere are both positioned distally in close proximity to each other).
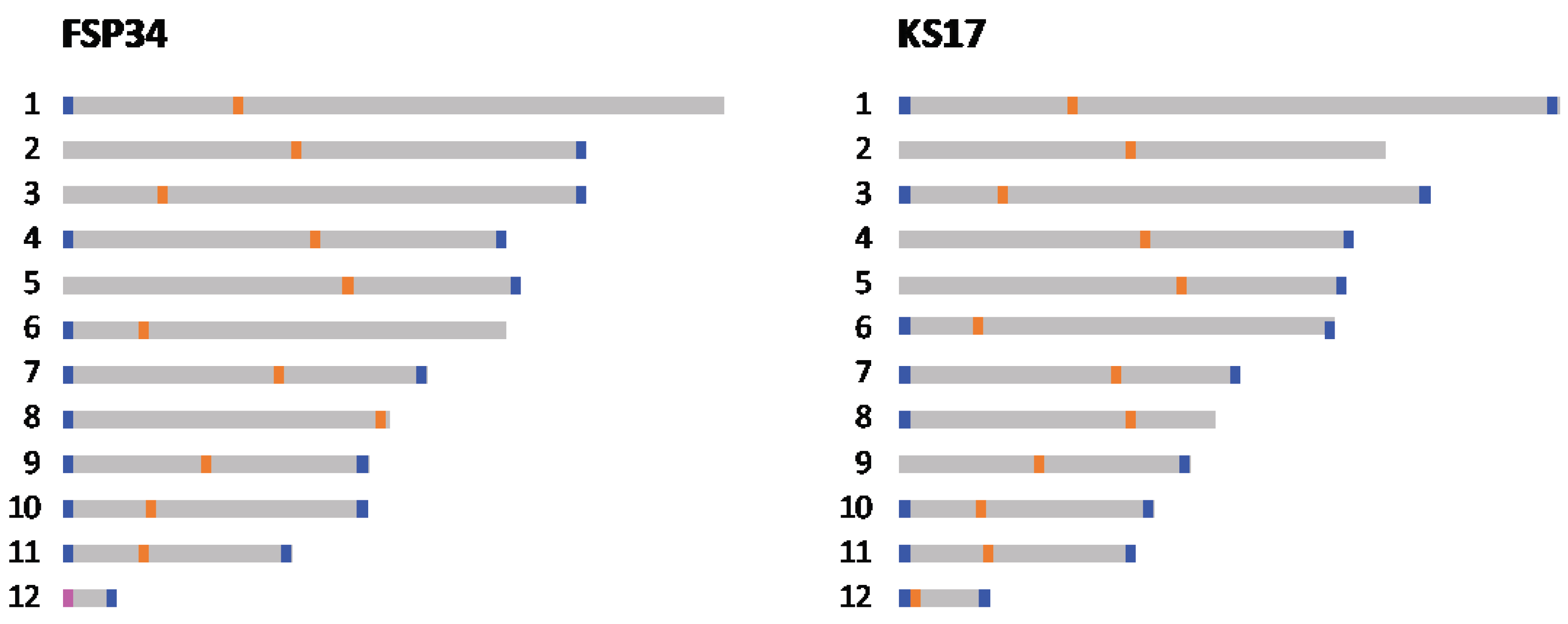
In pseudomolecule 12 of FSP34, the centromere is located distally with a telocentric position (centromere present at the end of a chromosome), while it is acrocentric in KS17. In both of the pseudomolecule 12 assemblies, the corresponding centromeric gene from the distal side of F. fujikuroi/F. verticillioides chromosome 12 (Table S3), is not present. This genomic region also corresponds to the area where the size variation was observed for this chromosome in F. circinatum FSP34 and KS17 (Figure 1, Table 2), suggesting that loss of the distal portion of the chromosome in FSP34 occurred in close proximity to the centromere (see below).
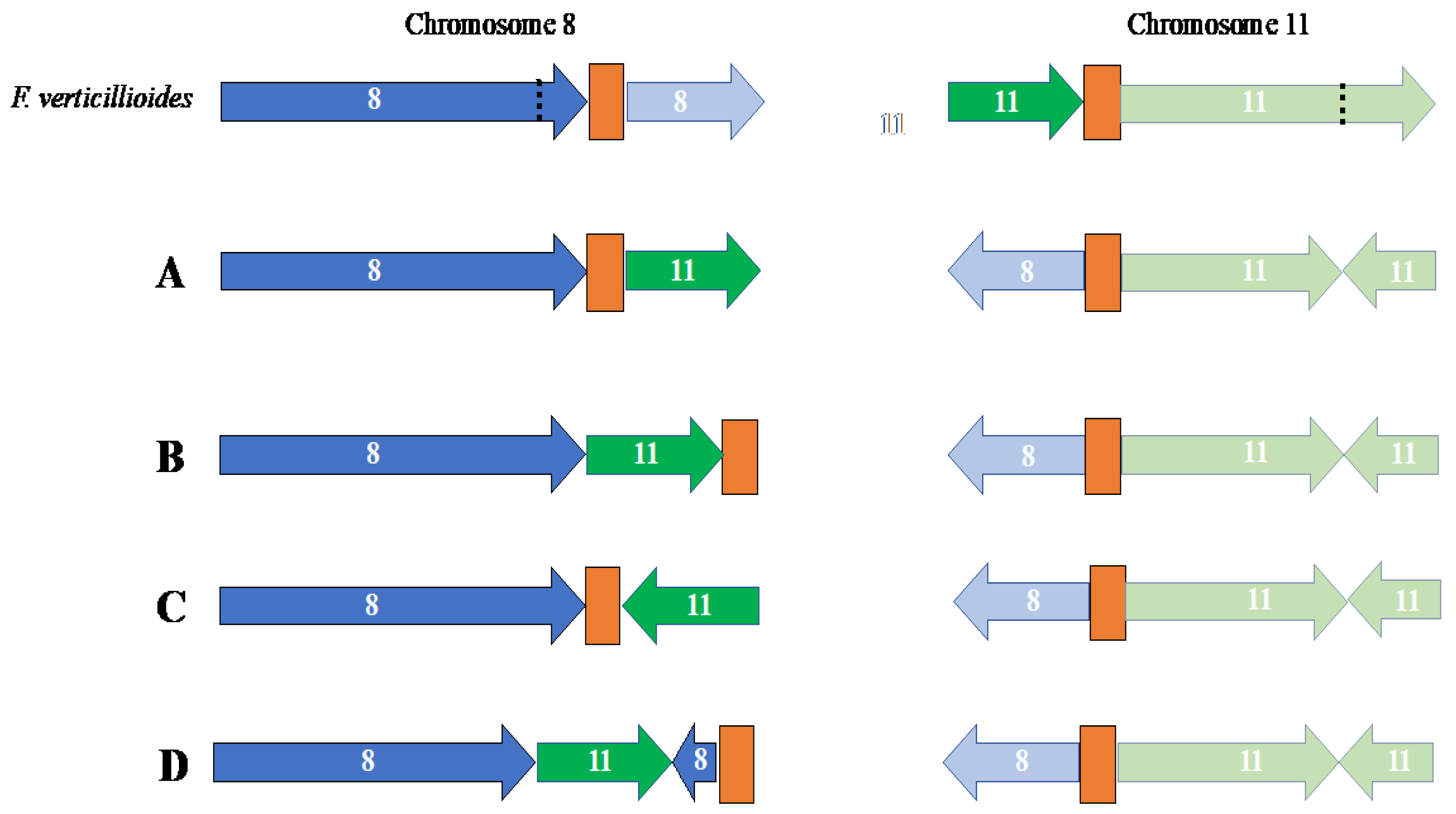
In pseudomolecule 8, the centromeric region is predicted to be telocentric in the FSP34current assembly, compared to being submetacentric in the KS17current assembly (Table S2). In terms of pseudomolecule 11, the predicted centromeric regions were submetacentric for FSP34current and metacentric in KS17current. Conserved genes flanking the distal sides of the centromere for pseudomolecule 8, as well as the proximal sides of pseudomolecule 11, were not syntenous. These findings supported the presence of the reciprocal translocation between these chromosomes, thus showing synteny to centromeric genes from chromosomes 11 and 8, respectively. The reciprocal translocation in chromosome 11 was uniform for F. circinatum FSP34 and KS17, but the centromeric region of chromosome 8 had an internal position in KS17 (see arrangement A in Figure 3). In FSP34 it was located at the distal end of the chromosome in a telocentric position (arrangement B in Figure 3).
Comparison of a diverse set of F. circinatum strains, as well as closely related species from the American clade FFSC species (Table S1) revealed the presence of the reciprocal translocation between chromosome 8 and 11 in all the genomes examined (Figure 3). It had a conserved position in chromosome 11, but the position in chromosome 8 was variable, irrespective of species or strain identity. For example, F. circinatum displayed three iterations of the arrangment oberseved for the translocation on chromosome 8 (arrangements A and B described above, and arrangent C in Figure 3). The third iteration was similar to the one observed in KS17, but the translocated portion of chromosome 11 present on chromosome 8, was inverted (Figure 3C). In addition, one of the arrangements of this translocation was only observed in F. pininemorale, with an additional chromosomal rearrangement observed on chromosome 8 (Figure 3D). In all these arrangements, the breakage for this reciprocal translocation appeared to be associated with the centromeres.
Figure 3.
Schematic representation of the reciprocal translocation between chromosome 8 and 11 in various American clade species of the FFSC (Table S1) relative to F. verticillioides. Portions of chromosomes originating from chromosome 8 in F. verticillioides are indicated in blue, whilst those originating from chromosome 11 in F. verticillioides are indicated in green. The centromeric regions are indicated in orange. Black dotted line represents chromosomal breakpoints not involving centromeric regions. Only chromosomal arrangements larger than 200 000bp are indicated in this representation. Arrangement (A) represents the translocation in F. circinatum strains KS17, FFRA, UG10, UG27, CMWF560, CMWF1803 and GL1327, as well as F. pilosicola, F. temperatum, F. marasasianum and F. sororula. Arrangement (B) represents the translocation in F. circinatum FSP34. Arrangement (C) represents the translocation in F. circinatum strains FSOR and CMWF567, as well as F. fracticaudum, while arrangement (D) represents the one in F. pininemorale.
Figure 3.
Schematic representation of the reciprocal translocation between chromosome 8 and 11 in various American clade species of the FFSC (Table S1) relative to F. verticillioides. Portions of chromosomes originating from chromosome 8 in F. verticillioides are indicated in blue, whilst those originating from chromosome 11 in F. verticillioides are indicated in green. The centromeric regions are indicated in orange. Black dotted line represents chromosomal breakpoints not involving centromeric regions. Only chromosomal arrangements larger than 200 000bp are indicated in this representation. Arrangement (A) represents the translocation in F. circinatum strains KS17, FFRA, UG10, UG27, CMWF560, CMWF1803 and GL1327, as well as F. pilosicola, F. temperatum, F. marasasianum and F. sororula. Arrangement (B) represents the translocation in F. circinatum FSP34. Arrangement (C) represents the translocation in F. circinatum strains FSOR and CMWF567, as well as F. fracticaudum, while arrangement (D) represents the one in F. pininemorale.
3.1.3. Genome Completeness and Gene Content
The genome assemblies generated here were highly complete (i.e., 97.3% for FSP34current and 98.1% for KS17current), containing >3624 of the set of 3725 BUSCO genes used to estimate genome completeness in the class Sordariomycetes (Table 1). This was a substantial improvement over the older genome assemblies for these fungi, particularly for the strain KS17. With a completeness score of 76.2%, the KS17pevious assembly lacked almost 900 of the expected BUSCO genes.
As expected, substantially more genes were identified and annotated by MAKER in the FSP34current assembly than the previous version (Table 1). The new assembly contained >500 more genes, at a slightly higher density of 344.06 genes/Mb (compared to 339.68 genes/Mb in the FSP34previous assembly). Direct comparisons between the new and the older versions of the KS17 assembly could not be made as the annotations for the older assembly utilized software lacking the same capabilities as MAKER [20]. Nevertheless, similar gene densities were estimated for the FSP34current and KS17current assemblies, and this similarity was also generally extended to individual sets of pseudomolecules (results not shown). Overall, pseudomolecule 10 had the highest gene density (i.e., 377.20 genes/Mb for FSP34current and 382.52 genes/Mb for KS17current) while it was lowest on pseudomolecule 12 (i.e., 325.67 genes/Mb for FSP34current and 294.02 genes/Mb KS17current).
Of the genes predicted for the FSP34current assembly, only 642 (4.14%) lacked relevant BLAST hits to any known protein (Table S4). In total, 4 694 (30.30%) and 1204 (7.77%) genes could not be assigned GO or InterPro identifications, respectively. A similar pattern was observed for the KS17current assembly (Table S5), with 626 (4.14%) lacking BLAST hits, 5 072 (33.56%) not assigned GO identifications, and 1 201 (7.95%) not assigned any InterPro identifications. See Tables S4 and S5 for the full lists of functional annotations for the genes predicted in the FSP34current and KS17current assemblies.
Due to the differences observed between pseudomolecule 12 and the remainder of the pseudomolecules in the new assemblies, we compared the gene content using two-sided Fisher’s exact tests on the GO terms. For FSP34, pseudomolecule 12 was significantly (P < 0.05) enriched for GO terms involved in biological functions associated with cellular aromatic compound metabolism, primary metabolism and organic cyclic compound metabolism, as well as GO terms associated with nitrogen compound metabolism, transport and localization (Table S6; Figure S1A). In the case of KS17current, pseudomolecule 12 was also enriched for GO terms involved in cellular amide metabolism, organic substance transport, organelle organization and protein metabolism, as well as GO terms associated with cellular response to stimulus, catabolism and cellular catabolism (Table S7; Figure S1B).
3.1.4. Genome Repetitiveness and TE Content
Improvement in the genome assemblies was evident in their increased repeat and TE content (Table 1; Table S8). FSP34previous had 2.81% of the genome that was characterized as repetitive elements, whilst this increased to 8.75% in FSP34current. The KS17current assembly contained a comparable 8.60% of repetitive elements. When considering only pseudomolecules 1-11 (i.e., those representing core chromosomes), the repetitive content was 4.68-15.15% across the FSP34current core set and 4.80-11.97% for the KS17current core set, with the two assemblies sharing similar content patterns. Although the repeat content of pesudomolecule 12 in the FSP34current assembly was 6.92%, its counterpart in the KS17current assembly was 26.6% (Table 3).
The large difference in repeat content between pseudomolecule 12 in the two assemblies were examined in more detail. This revealed that the additional regions located at the proximal and distal parts of pseudomolecule 12 of KS17current (see inset in Figure 1) accounted for most of the observed increased repetitive content (Table 3). Interestingly, the distal part of the pseudomolecule was also highly depleted in G+C content (31.88%) compared to the average of 41.73% for the entire pseudomolecule. The additional regions at the proximal and distal part of pseudomolecule 12 of KS17current are also extremely gene poor (Table 3).
Both Class I TEs (i.e., retrotransposons) and Class II TEs (i.e., DNA transposons) were identified in the FSP34previous (Table S9), FSP34current (Table S10) and KS17current assemblies (Table S11). FSP34current had Class I TEs, namely TRIM (Terminal Repeat transposons in Miniature), LARD (Large Retrotransposon derivatives), LINE (Long Interspersed Nuclear Elements) and unclassified retrotransposons (non-autonomous retrotransposon), as well as Class II TE TIR (Terminal Inverted Repeats) (Figure 4A; Figure S2). KS17current had LARD, unclassified retrotransposons and TIR transposable elements in very similar densities on each pseudomolecule (Figure 4B; Figure S2). In addition, the frequency of the TEs in FSP34current was LARD > unclassified retrotransposons > TIR > LINE > TRIM, whilst in KS17current it was LARD > TIR > unclassified retrotransposons (Figure S2).
3.1.5. Detailed Source and Biological Information for KS17 and FSP34
For summarizing available information regarding the ecology and biology of F. circinatum strains KS17 and FSP34, we utilized the published literature as well as the findings of our in vitro growth studies and pathogenicity tests. Details regarding their origins, reproductive biology and the population dynamics of their source populations are provided in Table 4. Note, however, that most of the information regarding the origins of FSP34 comes from a study by Gordon et al. [65], although they never explicitly mentioned the strain. Also, strain KS17 was isolated during a study by Steenkamp et al. [66], but it did not form part of their experiments.
Additionally, the results of our pathogenicity tests showed that both KS17 and FSP34 are pathogenic to P. patula seedlings (Supplementarry Figure S4A). Three weeks after inoculation, lesions were observed for the FSP34 and KS17 treatments, respectively, while no lesions developed in the control treatment. FSP34 was, however, significantly more virulent or aggressive, with lesion lengths averaging 15.45 mm compared to the 6.2 mm of KS17 (P < 0.001). Likewise, compared to KS17, FSP34 grew significantly faster at all the temperatures tested, although neither grew ar 35° (Supplmentary Figure S4B).
4. Discussion
By harnessing the power of second and third-generation sequencing technologies [74,75], we determined the whole genome sequences for two important strains of F. circinatum strains (FSP34 and KS17) [18,19,20]. For both, we obtained near complete chromosome-level assemblies, consisting of contiguous sequence mostly spanning entire molecules from telomere to telomere. The two new assemblies emphasized the macrosyntenic nature of the FFSC genomes [12,25] and allowed detailed examination of the previously suggested reciprocal translocation between chromosomes 8 and 11 in species from the American clade of FFSC [19,25,76,77,78]. The data presented here further revealed significant variability in chromosome 12 of F. circinatum.
Comparison of the two F. circinatum strains revealed that their genomes were highly syntenic. This was expected for members of the same species, and it also echoed the high level of conservation of large-scale chromosomal arrangements typically observed across the FFSC; e.g., between F. circinatum and more distantly related FFSC species such as Fusarium verticillioides (causal agent of maize ear rot) and Fusarium fujikuroi (causal agent of bakanae disease in rice seedlings) [12,25]. Our results are also consistent with those of a study exploring intraspecific genome plasticity in F. circinatum, which reported that >90% of a strain’s genome sequence is conserved and “alignable” with that of another strain of the fungus [34]. Nonetheless, structural changes to several chromosomes of F. circinatum were evident with most variation located on the subtelomeric regions of chromosome 1-11, with chromosome 12 being the most variable.
The findings presented here improved our understanding of the origin and evolution of the reciprocal translocation between chromosomes 8 and 11 of species from the American clade of the FFSC. Our high-quality, chromosome-level assemblies confirmed its existence in the two F. circinatum strains examined. Although the translocation was mentioned in previous studies [19,25,76,77,78], all the genome assemblies in question were generated using short-read sequences. However, our use of ONT MinION long-read sequences allowed for the assembly of contiguous sequences across the predicted chromosomal breakpoints. Apart from providing the strongest evidence yet for the existence of this translocation, our data also showed that the region associated with it is highly variable and that this variability is likely associated with centromeres. This was well illustrated by the different arrangements obtained for the translocated region on chromosome 8 relative to the centromeric region (see arrangements A and B in Figure 3). Such centromere-associated “evolvability” of chromosomes has also been reported in other eukaryotes [79] including fungi [80]. Double-stranded breaks occurring in the repeat-rich centromeric DNA of Cryptococcus, for example, have been shown to mediate chromosomal translocations and rearrangements, which are thought to have played a significant role in the evolution of these fungi [81,82]. In the case of F. circinatum and the remainder of the American clade of the FFSC, the emergence of such as centromere-mediated translocation involving chromosomes 8 and 11 would have predated the divergence of extant species and likely occurred in the ancestor of the clade. The fact that the region varies among strains of F. circinatum and among other American clade species, irrespective of their species identities, suggests a continued role for centromeres in shaping genome architecture of these fungi.
We now have a more comprehensive understanding of the repetitive nature of the F. circinatum genome, which is important as these repetitive elements are known to bring about changes and variation in genome architecture [83]. Indeed, the dynamic and plastic nature of fungal genomes [84] is epitomized in chromosome 12 of F. circinatum. This chromosome in KS17 had substantially more TEs compared to FSP34 and the presence of repetitive elements likely enabled formation of the chromosome length polymorphism observed. In particular, this chromosome in KS17 has more Class I TEs than in FSP34, and the activity of retrotransposons are known to give rise to sequence polymorphism and genome expansion [85].
Apart from its repetitive nature, chromosome 12 of F. circinatum also displayed unusual length polymorphism relative to the core chromosomes. Strain KS17’s twelfth chromosome being >1.6x longer than that of FSP34. Similar patterns have been found in other members of the FFSC for chromosome 12, especially F. fracticaudum where this molecule is over 1Mb in length [78]. In the current study, the length polymorphism was found associated with the proximal and distal portion of the KS17 chromosome 12. This was also evident from genomic alignments of chromosome 12 from F. circinatum CMWF1803 (isolated from diseased P. patula branches in Hidalgo, Mexico [86] to those of FSP34 and KS17 (see Figure S3 for details). Interestingly, the “middle” portion of the KS17 chromosome 12 (i.e., the region that is syntenic to the entire chromosome 12 of FSP34), is characterized by gene density, G+C content and repeats content that are similar to those of chromosomes 1-11. Therefore, FSP34 chromosome 12 seems to have either lost the regions enabling variation or could represent an ancestral or fundamental version of chromosome 12. A possible role for centromeric elements in these chromosome length polymorphisms cannot be excluded, as chromosome 12 of FSP34 has a centromere at one of its ends and not a telomere as in KS17.
The biological functions associated with chromosome 12 of F. circinatum remain unclear. In the FFSC, the twelfth chromosome is generally regarded both as dispensable and strain-specific [12,13]. In notable pathogens such as F. oxysporum, such chromosomes have been shown to be involved in pathogenicity, in part due to the small effector proteins they encode [4]. In chromosome 12 of F. circinatum FSP34 and KS17 there was an increase in genes involved in different processes and functions between them, with no clear-cut enrichment of genes specific for chromosome 12. However, elimination of chromosome 12 from the genome of the fungus has been linked to reduced virulence on P. radiata seedlings [87,88]. Clearly, further research is needed for clarifying the evolution and role of this chromosome in F. circinatum.
The improved F. circinatum assemblies provided in this study represents and invaluable resource genomic comparisons in the FFSC. We have shown the benefit of resequencing previous genomic sequence data generated using second generation sequencing technologies, and highlighted various structural chromosomal differences shown using the ONT MinION sequencing technology, in conjunction with Illumina HiSeq for error correction. Studies on intraspecific genetic variation has the capacity to provide understanding into how genetic variation is accumulated within genomes, and to address how this impacts adaptability and variability of fungi [34,89]. Combined with the detailed metadata compiled for the two strains (see Table 4) our high-quality chromosome-level assembly for F. circinatum will undoubtedly facilitate more comprehensive genomic and comparative studies into this destructive plant pathogen.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: REVIGO treemap summarizing GO biological process categories enriched in the accessory chromosome 12 of F. circinatum FSP34 (A) and KS17 (B).; Figure S2: Classes and orders of transposable elements (TEs) identified in FSP34current (A) and KS17current (B), based on the classification of [64].; Figure S3: Sequence comparison of the chromosome length polymorphism (CLP) of chromosome 12.; Table S1: Genomes used for genomic comparisons for the reciprocal translocation between chromosomes 8 and 11.; Table S2: Locations of the putative centromeric regions of Fusarium circinatum FSP34 and KS17.; Table S3: Nearest genes to Fusarium circinatum centromeres and synteny with Fusarium fujikuroi and Fusarium verticillioides.; Table S4: Functional annotation of F. circinatum FSP34 using Blast2GO.; Table S5: Functional annotation of F. circinatum KS17 using Blast2GO.; Table S6: Enrichment analysis of the accessory chromosome 12 of F. circinatum FSP34.; Table S7: Enrichment analysis of the accessory chromosome 12 of F. circinatum KS17.; Table S8: Percentage of each pseudomolecule that contains repetive sequences.; Table S9: Transposable elements for FSP34previous which was detected, annotated and analyzed using the REPET v2.5 pipeline.; Table S10: Transposable elements for FSP34current which was detected, annotated and analyzed using the REPET v2.5 pipeline.; Table S11: Transposable elements for KS17current which was detected, annotated and analyzed using the REPET v2.5 pipeline.
Author Contributions
Conceptualization: L.dV., M.A.vdN., E.T.S. and B.D.W.; Methodology: L.dV., M.A.vdN., E.T.S., B.D.W., Q.C.S., K.S.L. and S.vW.; Formal analysis: L.dV., Q.C.S., K.S.L. and S.vW; Resources: M.A.vdN., B.D.W.; Data curation: L.dV., M.A.vdN. and Q.C.S.; Writing—original draft preparation: L.dV., M.A.vdN. and E.T.S.; Writing—review and editing: L.dV. and M.A.vdN., E.T.S., B.D.W., Q.C.S., K.S.L. and S.vW. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the Department of Science and Innovation (DSI) - National Research Foundation (NRF) Centre of Excellence in Tree Health Biotechnology and SARChI Chair in Fungal Genomics for financial support.
Data Availability Statement
The Whole Genome Shotgun project for Fusarium circinatum has been deposited at DDBJ/ENA/GenBank under the accession AYJV00000000 and LQBB00000000 for F. circinatum FSP34 and KS17, respectively.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The reference genome sequence of Saccharomyces cerevisiae: Then and Now. G3: Genes | Genomes | Genetics 2014, 4, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. Mycocosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Research 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-J.; Van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Sengupta, A.; Ghosh, R.; Agarwal, G.; Tarafdar, A.; Nagavardhini, A.; Pande, S.; Varshney, R.K. Genome wide transcriptome profiling of Fusarium oxysporum f. sp. ciceris conidial germination reveals new insights into infection-related genes. Scientific Reports 2016, 6, 37353. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K. Fungal genome sequencing: Basic biology to biotechnology. Critical Reviews in Biotechnology 2016, 36, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sánchez-García, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nature Communications 2021, 11, 5125. [Google Scholar] [CrossRef]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.; Rooney, A.P.; O'Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annual Review of Microbiology 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A genome-scale phylogeny of the kingdom Fungi. Current Biology 2021, 31, 1653–1665.e1655. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, M.P.A.; Santana, Q.C.; Steenkamp, E.T.; Wingfield, B.D.; Wingfield, M.J. Fungal genomes enhance our understanding of the pathogens affecting trees cultivated in Southern Hemisphere plantations. Southern Forests 2020, 82, 215–232. [Google Scholar] [CrossRef]
- Delulio, G.A.; Guo, L.; Zhang, Y.; Goldberg, J.M. Kinome expansion in the Fusarium oxysporum species complex driven by accessory chromosomes. mSphere 2018, 3, e00231–e00218. [Google Scholar] [CrossRef]
- Wiemann, P.; Sieber, C.M.K.; Von Bargen, K.W.; Studt, L.; Niehaus, E.-M.; Hub, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; Espino, J.J.; et al. Unleashing the cryptic genome: Genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens 2013, 9, e1003475. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, L.; Guo, L.; Dora, S.; Wang, B.; Ye, K.; Sánchez-Rodríguez, C.; Croll, D. A chromosome-scale genome assembly for the Fusarium oxysporum strain Fo5176 to establish a model Arabidopsis-fungal pathosystem. G3: Genes | Genomes | Genetics 2020, 10, 3549–3555. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, E.-M.; Münsterkötter, M.; Proctor, R.H.; Brown, D.W.; Sharon, A.; Idan, Y.; Oren-Young, L.; Sieber, C.M.; Novák, O.; Pĕnčík, A.; et al. Comparative "omics"of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biology and Evolution 2017, 8, 3574–3599. [Google Scholar] [CrossRef] [PubMed]
- Geiser, D.M.; Aoki, T.; Bacon, C.W.; Baker, S.E.; Bhattacharyya, M.K.K.; Brandt, M.E.; Brown, D.W.; Burgess, L.W.; Chulze, S.N.; Coleman, J.J.; et al. One fungus, one name: Defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathology 2013, 103, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Drenkhan, R.; Ganley, B.; Martín-García, J.; Vahalík, P.; Adamson, K.; Adamčíková, K.; Ahumada, R.; Blank, L.; Bragança, H.; Capretti, P.; et al. Global geographic distribution and host range of Fusarium circinatum, the causal agent of pine pitch canker. Forests 2020, 11, 724. [Google Scholar] [CrossRef]
- Wingfield, M.J.; Hammerbacher, A.; Ganley, R.J.; Steenkamp, E.T.; Gordon, T.R.; Wingfield, B.D.; Coutinho, T.A. Pitch canker caused by Fusarium circinatum - a growing threat to pine plantations and forests worldwide. Australasian Plant Pathology 2008, 37, 319–334. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Steenkamp, E.T.; Santana, Q.C.; Coetzee, M.P.A.; Bam, S.; Barnes, I.; Beukes, C.W.; Chan, W.Y.; De Vos, L.; Fourie, G.; et al. First fungal genome sequence from Africa: A preliminary analysis. South African Journal of Science 2012, 108, 1–9. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Liu, M.; Nguyen, H.D.T.; Lane, F.A.; Morgan, S.W.; De Vos, L.; Wilken, P.M.; Duong, T.A.; Aylward, J.; Coetzee, M.P.A. ; et al. Nine draft genome sequences of Claviceps purpurea s. lat., including C. arundinis, C. humidiphila, and C. cf. spartinea, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria euclaypti, and Teratospahaeria destructans. IMA Fungus 2018, 9, 401–418. [CrossRef]
- van Wyk, S.; Wingfield, B.D.; De Vos, L.; Santana, Q.C.; Van der Merwe, N.A.; Steenkamp, E.T. Multiple independent origins for a subtelomeric locus associated with growth rate in Fusarium circinatum. IMA Fungus 2018, 9, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Santana, Q.C.; Coetzee, M.P.A.; Steenkamp, E.T.; Mlonyeni, O.X.; Hammond, G.N.A.; Wingfield, M.J.; Wingfield, B.D. Microsatellite discovery by deep sequencing of enriched genomic libraries. BioTechniques 2009, 46, 217–223. [Google Scholar] [CrossRef]
- Phasha, M.M.; Wingfield, B.D.; Wingfield, M.J.; Coetzee, M.P.A.; Hammerbacher, A.; Steenkamp, E.T. Deciphering the effect of FUB1 disruption on fusaric acid production and pathogenicity in Fusarium circinatum. Fungal Biology 2021. [Google Scholar] [CrossRef] [PubMed]
- Phasha, M.M.; Wingfield, M.J.; Wingfield, B.D.; Coetzee, M.P.A.; Hallen-Adams, H.; Fru, F.; Swalarsk-Parry, B.S.; Yilmaz, N.; Duong, T.A.; Steenkamp, E.T. Ras2 is important for growth and pathogenicity in Fusarium circinatum. Fungal Genetics and Biology 2021, 150, 103541. [Google Scholar] [CrossRef] [PubMed]
- De Vos, L.; Myburg, A.A.; Wingfield, M.J.; Desjardins, A.E.; Gordon, T.R.; Wingfield, B.D. Complete genetic linkage maps from an interspecific cross between Fusarium circinatum and Fusarium subglutinans. Fungal Genetics and Biology 2007, 44, 701–714. [Google Scholar] [CrossRef]
- De Vos, L.; Steenkamp, E.T.; Martin, S.H.; Santana, Q.C.; Fourie, G.; Van der Merwe, N.A.; Wingfield, M.J.; Wingfield, B.D. Genome-wide macrosynteny among Fusarium species within the Gibberella fujikuroi complex revealed by amplified fragment length polymorphisms. PLoS ONE 2014, 9, e114682. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotechnology 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.; Harrison, J.; O'Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomolecular Detection and Quantification 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saud, Z.; Kortsinoglou, A.M.; Kouvelis, V.N.; Butt, T.M. Telomere length de novo assembly of all 7 chromosomes and mitogenome sequencing of the model entomopathogenic fungus, Metarhizium brunneum, by means of a novel assembly pipeline. BMC Genomics 2021, 22, 87. [Google Scholar] [CrossRef]
- Wang, B.; Liang, X.; Gleason, M.L.; Hsiang, T.; Zhang, R.; Sun, G. A chromosome-scale assembly of the smallest Dothideomycete genome reveals a unique genome compaction mechanism in filamentous fungi. BMC Genomics 2020, 21, 321. [Google Scholar] [CrossRef] [PubMed]
- Crestana, G.S.; Taniguti, L.M.; dos Santos, C.P.; Benevenuto, J.; Ceresini, P.C.; Carvalho, G.; Kitajima, J.P.; Monteiro-Vitorello, C.B. Complete chromosome-scale genome sequence resource for Sporisorium panici-leucophaei, the causal agent of sourgrass smut disease. Molecular Plant-Microbe Interactions 2021, 34, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, H.; Jia, Y.; Dong, Q.; Steinberg, C.; Alabouvette, C.; Edel-Hermann, V.; Kistler, H.C.; Ye, K.; Ma, L.-J.; et al. Chromosome-scale genome assembly of Fusarium oxysporum strain Fo47, a fungal endophyte and biocontrol agent. Molecular Plant-Microbe Interactions 2020, 33, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, S.K.; Walston, R.F.; Allen, J.L. Complete, high-quality genomes from long-read metagenomic sequencing of two wolf lichen thalli reveals enigmatic genome architecture. Genomics 2020, 112, 3150–3156. [Google Scholar] [CrossRef] [PubMed]
- Maphosa, M.N.; Steenkamp, E.T.; Kanzi, A.M.; van Wyk, S.; De Vos, L.; Santana, Q.C.; Duong, T.A.; Wingfield, B.D. Intra-species genomic variation in the pine pathogen Fusarium circinatum. Journal of Fungi 2022, 8, 657. [Google Scholar] [CrossRef] [PubMed]
- Swalarsk-Parry, B.S.; Steenkamp, E.T.; van Wyk, S.; Santana, Q.C.; van der Nest, M.A.; Hammerbacher, A.; Wingfield, B.D.; De Vos, L. Identification and characterization of a QTL for growth of Fusarium circinatum on pine-based medium. Journal of Fungi 2022, 8, 1214. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, L.A.; Stajich, J.E. Fungal biodiversity and conservation mycology in light of new technology, big data, and changing attitudes. Current Biology 2021, 31, R1312–R1325. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; LeRoy, N.J.; Khoroshevskyi, O. Challenges to sharing sample metadata in computational genomics. Frontiers in Genetics 2023, 14, 154198. [Google Scholar] [CrossRef] [PubMed]
- Duong, T.A.; De Beer, Z.W.; Wingfield, B.D.; Wingfield, M.J. Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biology 2013, 117, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillipy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Research 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved pairwise alignment of genomic DNA. Pennsylvania State University, Pennsylvania, USA, 2007.
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics Applications Note 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biology 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- O'Donnell, K.; Cigelnik, E.; Nirenberg, H.I. Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia 1998, 90, 465–493. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Molecular Biology and Evolution 2017, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- van Wyk, S. Molecular characterization of the growth rate determining quantitative trait locus in Fusarium circinatum. University of Pretoria, Pretoria, South Africa, 2015.
- Wu, C.; Kim, Y.-S.; Smith, K.M.; Li, W.; Hood, H.M.; Staben, C.; Selker, E.U.; Sachs, M.S.; Farman, M.L. Characterization of chromosome ends in the filamentous fungus Neurospora crassa. Genetics 2009, 181, 1129–1145. [Google Scholar] [CrossRef] [PubMed]
- van Wyk, S.; Harrison, C.H.; Wingfield, B.D.; De Vos, L.; van der Merwe, N.A.; Steenkamp, E.T. The RIPper, a web-based tool for genome-wide quantification of Repeat-Induced Point (RIP) mutations. PeerJ 2019, 7, e7447. [Google Scholar] [CrossRef] [PubMed]
- Levan, A.; Fredga, K.; Sandberg, A.A. Nomenclature for centromeric position on chromosomes. Hereditas 1964, 201–220. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Research 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinformatics 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Flutre, T.; Dupart, E.; Feuillet, C.; Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef] [PubMed]
- Quesneville, H.; Bergman, C.M.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabehere, D. Combined evidence annotation of transposable elements in genome sequences. PLoS Computational Biology 2005, 1, e22. [Google Scholar] [CrossRef] [PubMed]
- De Vos, L.; Van der Nest, M.A.; Van der Merwe, N.A.; Myburg, A.A.; Wingfield, M.J.; Wingfield, B.D. Genetic analysis of growth, morphology and pathogenicity in the F1 progeny of an interspecific cross between Fusarium circinatum and Fusarium subglutinans. Fungal Biology 2011, 115, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Zapatka, M.; Lasitschka, B.; Jones, D.; Northcott, P.; Hutter, B.; Jäger, N.; Kool, M.; Taylor, M.; Lichter, P.; et al. Coverage bias and sensitivity of variant calling for four whole-genome sequencing technologies. PLoS ONE 2013, 8, e66621. [Google Scholar] [CrossRef]
- van Wyk, S.; Wingfield, B.D.; De Vos, L.; van der Merwe, N.A.; Steenkamp, E.T. Genome-wide analysis of Repeat-Induced Point mutations in the Ascomycota. Frontiers in Microbiology 2021, 11, 3625. [Google Scholar] [CrossRef]
- van Wyk, S.; Wingfield, B.D.; van der Merwe, N.A.; De Vos, L.; Maphosa, M.; Potgieter, L.; Steenkamp, E.T. Repeat-induced point mutations (RIP) drives the formation of distinct sub-genomic comparrtments in Fusarium circinatum. Preprints 2020, 2020120697. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.R.; Storer, A.J.; Okamoto, D. Population structure of the pitch canker pathogen, Fusarium subglutinans f. sp. pini, in California. Mycological Research 1996, 100, 850–854. [Google Scholar] [CrossRef]
- Steenkamp, E.T.; Makhari, O.M.; Coutinho, T.A.; Wingfield, B.D.; Wingfield, M.J. Evidence for a new introduction of the pitch canker fungus Fusarium circinatum in South Africa. Plant Pathology 2014, 63, 530–538. [Google Scholar] [CrossRef]
- Aylward, J.; Steenkamp, E.T.; Dreyer, L.L.; Roets, F.; Wingfield, B.D.; Wingfield, M.J. Genome sequences of Knoxdaviesia capensis and K. protea (Fungi: Ascomycota) from Protea trees in South Africa. Standards in Genomic Sciences 2016, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Mashandule, O.M. Population biology of Fusarium cirrcinatum Nirenberg et O’Donnell associated with South African Pinus radiata D. Don plantation trees. PhD, University of Pretoria, Pretoria, South Africa, 2014.
- Steenkamp, E.T.; Coutinho, T.A.; Desjardins, A.E.; Wingfield, B.D.; Marasas, W.F.O.; Wingfield, M.J. Gibberella fujikuroi mating population E is associated with maize and teosinte. Molecular Plant Pathology 2001, 2, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.E.; Plattner, R.D.; Gordon, T.R. Gibberella fujikuroi mating population A and Fusarium subglutinans from teosinte species and maize from Mexico and Central America. Mycological Research 2000, 104, 865–872. [Google Scholar] [CrossRef]
- McCain, A.H.; Koehler, C.S.; Tjosvold, S.A. Pitch canker threatens California pines. California Agriculture 1987, November-December, 22-23.
- Berbegal, M.; Pérez-Sierra, A.; Armengol, J.; Grünwald, N.J. Evidence for multiple introductions and clonality in Spanish populations of Fusarium circinatum. Phytopathology 2013, 103, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Britz, H.; Coutinho, T.A.; Wingfield, B.D.; Marasas, W.F.O.; Wingfield, M.J. Diversity and differentiation in two populations of Gibberella circinata in South Africa. Plant Pathology 2005, 54, 46–52. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends in Genetics 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-generation sequencing and emerging technologies. Seminars in Thrombosis and Homestasis 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, B.D.; Barnes, I.; De Beer, Z.W.; De Vos, L.; Duong, T.A.; Kanzi, A.M.; Naidoo, K.; Nguyen, H.D.T.; Santana, Q.C.; Sayari, M.; et al. Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus 2015, 6, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, B.D.; Berger, D.K.; Steenkamp, E.T.; Lim, H.-J.; Duong, T.A.; Bluhm, B.H.; de Beer, Z.W.; De Vos, L.; Fourie, G.; Naidoo, K.; et al. Draft genome of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens and Ophiostoma ips. IMA Fungus 2017, 8, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, B.D.; Bills, G.F.; Dong, Y.; Huang, W.; Nel, W.J.; Swalarsk-Parry, B.S.; Vaghefi, N.; Wilken, P.M.; An, Z.; de Beer, Z.W. ; et al. Draft genome sequence of Annulohypoxylon stygium, Aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. hyalina, and Morchella septimelata. IMA Fungus 2018, 9, 199–223. [CrossRef]
- Coghlan, A.; Eichler, E.E.; Oliver, S.G.; Paterson, A.H.; Stein, L. Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends in Genetics 2005, 21, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Guin, K.; Sreekumar, L.; Sanyal, K. Implications of the Evolutionary Trajectory of Centromeres in the Fungal Kingdom. Annual Review of Microbiology 2020, 74, 835–853. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yadav, V.; Billmyre, R.B.; Cuomo, C.A.; Nowrousian, M.; Wang, L.; Souciet, J.-L.; Boekhout, T.; Porcel, B.; Wincker, P.; et al. Fungal genome and mating system transitions facilitated by chromosomal translocations involving intercentromeric recombination. PLos Biology 2017, 15, e2002527. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Sun, S.; Coelho, M.A.; Heitmans, J. Centromere scission drives chromosome shuffling and reproductive isolation. Proceedings of the National Academy of Sciences 2020, 117, 7917–7928. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Sharda, S.; Oddi, V.; Nandineni, M.R. The landscape of repetitive elements in the refined genome of chilli anthracnose fungus Collectotrichum truncatum. Frontiers in Microbiology 2018, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H.; Croll, D. The evolving fungal genome. Fungal Biology Reviews 2014, 28, 1–12. [Google Scholar] [CrossRef]
- Depotter, J.R.L. , Ökmen, B., Ebert, M.K., Beckers, J., Kruse, J., Thines, M. and Doehlemann, G. High nucleotide substitution rates associated with retrotransposon proliferation drive dynamic secretome evolution in smut pathogens. Microbiology Spectrum, 2022, 10, e00349–e22. [Google Scholar] [CrossRef]
- Malewski, T.; Matić, S.; Okorski, A.; Borowik, P.; Oszako, T. Annotation of the 12th chromosome of the forest pathogen Fusarium circinatum. Agronomy 2023, 13, 773. [Google Scholar] [CrossRef]
- Slinski, S.L. , Kirkpatrick, S.C. and Gordon, T.R. Inheritance of virulence in Fusarium circinatum, the cause of pitch canker in pines. Plant Pathology 2016, 65, 1292–1296. [Google Scholar] [CrossRef]
- van der Nest, M.A. , Beirn, L.A., Crouch, J.A., Demers, J.E., De Beer, Z.W., De Vos, L., Gordon, T.R., Moncalvo, J.M., Naidoo, K., Sanchez-Ramirez, S. and Roodt, D. Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA fungus, 2014, 5, 472–486. [Google Scholar] [CrossRef]
- Fumero, M.V.; Villani, A.; Susca, A.; Haidukowski, M.; Cimmarusti, M.T.; Toomajian, C.; Leslie, J.F.; Chulze, S.N.; Moretti, A. Fumonisin and beauvericin chemotypes and genotypes of the sister species Fusarium subglutinans and Fusarium temperatum. Appled and Environmental Microbiology 2020, 86, e00133–e00120. [Google Scholar] [CrossRef] [PubMed]
Figure 4.
Classes and orders of transposable elements (TEs) identified in FSP34current (A), KS17current (B) and FSP34previous (C) based on the classification of [64]. The TEs are given as Class I (TRIM - Terminal Repeat transposons in Miniature, LARD - Large Retrotransposon derivatives, LINE - Long Interspersed Nuclear Elements and unclassified - non-autonomous retrotransposon), and Class II (TIR - Terminal Inverted Repeats).
Figure 4.
Classes and orders of transposable elements (TEs) identified in FSP34current (A), KS17current (B) and FSP34previous (C) based on the classification of [64]. The TEs are given as Class I (TRIM - Terminal Repeat transposons in Miniature, LARD - Large Retrotransposon derivatives, LINE - Long Interspersed Nuclear Elements and unclassified - non-autonomous retrotransposon), and Class II (TIR - Terminal Inverted Repeats).
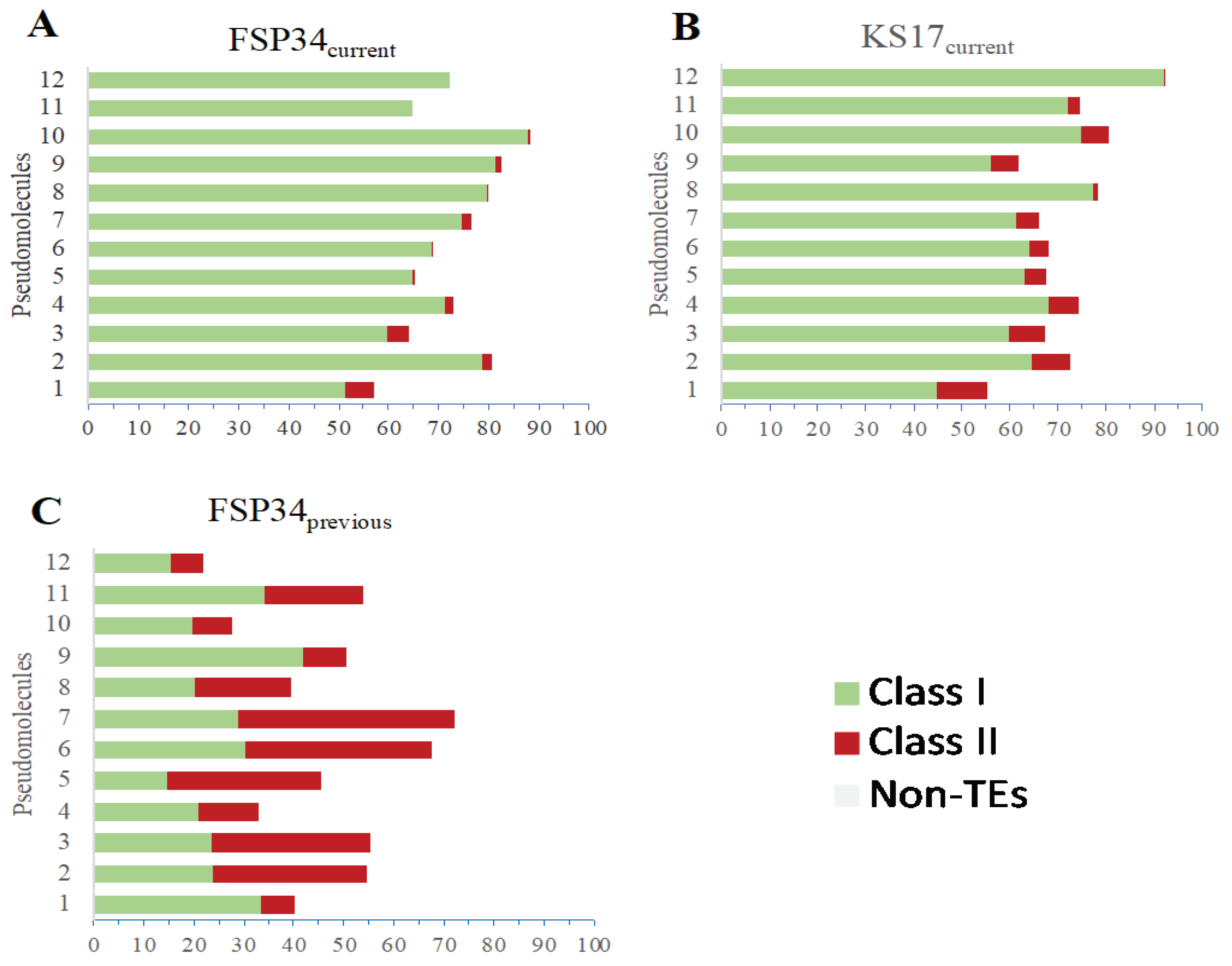

Table 1.
Genome statistics of F. circinatum strains FSP34 and KS17.
| Genome Statistic | FSP34 | KS17 | |||
| Previous 1 | Current | Previous 2 | Current | ||
| Genome size (bp) | 43 932 912 | 45 020 843 | 46 325 048 | 44 380 849 | |
| G+C content (%) | 47.41 | 47.00 | 44.69 | 47.26 | |
| Number of open reading frames | 14 923 3 | 15 490 | 16 502 4 | 15 113 | |
| Gene density (orfs/Mb) | 339.68 | 344.06 | 356.22 | 340.53 | |
| Number of scaffolds | 585 | 49 | 6 033 | 96 | |
| N50 (bp) | 363 633 | 4 313 168 | 95 695 | 4 401 926 | |
| Average scaffold size (bp) | 75 085 | 1 667 439 | 7 679 | 1 431 640 | |
| Unmapped scaffolds (total % of genome) | 418 (3.03%) | 15 (0.60%) | - 5 | 19 (0.78%) | |
| Repeat content 6 | 2.81% | 8.75% | - | 8.60% | |
| Genome completeness 7 | 94.8% | 97.3% | 76.2% | 98.1% | |
1 Wingfield et al. [19]. 2 Van Wyk et al. [20]. 3 Annotated using MAKER [18]. 4 Annotated using WebAUGUSTUS [20]. 5 Scaffolds were never assigned to chromosomes [20]. 6 Determined using REPET v2.5 [58,59]. Assembly of KS17PREVIOUS was too fragmented to determine the repeat content. 7 Based on BUSCO v3.0.2 using the “Sordariomycete” database [47].
Table 2.
Size (base pairs) of the pseudomolecules from F. circinatum strains FSP34 and KS17.
| Pseudomolecule | FSP34 1 | KS17current | |
| Previous | Current | ||
| 1 | 6 190 704 (14) | 6 407 689 (2) | 6 397 914 (8) |
| 2 | 4 773 114 (22) | 5 066 197 (3) | 4 709 326 (5) |
| 3 | 4 756 822 (18) | 5 081 888 (3) | 5 148 568 (3) |
| 4 | 4 140 424 (12) | 4 313 168 (2) | 4 401 926 (5) |
| 5 | 4 399 406 (10) | 4 432 553 (2) | 4 304 443 (7) |
| 6 | 4 053 349 (21) | 4 301 895 (3) | 4 219 930 (9) |
| 7 | 3 472 423 (19) | 3 541 054 (5) | 3 312 103 (8) |
| 8 | 3 024 507 (17) | 3 172 915 (5) | 3 066 990 (9) |
| 9 | 2 773 158 (8) | 2 981 544 (1) | 2 828 005 (4) |
| 10 | 2 371 510 (16) | 2 698 820 (3) | 2 483 521 (7) |
| 11 | 2 122 486 (11) | 2 228 420 (3) | 2 291 537 (5) |
| 12 | 525 791 (6) | 525 065 (1) | 870 680 (3) |
1 The previous FSP34 genome was sequenced by Wingfield et al. [19]. Number of scaffolds used for building pseudomolecules are indicated in brackets.
Table 3.
G+C and repeat content, and gene density of pseudomolecule 12 of FSP34 and KS17.
| Content Estimates | FSP34 Pseudomolecule 12 1 | KS17 Pseudomolecule 12 2 | |||
| Whole Molecule | Distal | Middle | Proximal | ||
| G+C content (%) | 46.36 | 41.73 | 31.88 | 45.18 | 41.48 |
| Gene density (orfs/Mb) | 325.67 | 294.01 | 123.53 | 349.52 | 300.00 |
| Repeats (%) | 6.92 | 26.63 | 59.67 | 12.67 | 33.48 |
1 FSP34previous has pseudomolecule 12 assembled into one scaffold with a telomeric cap at both ends. 2 The “Distal” portion of KS17current pseudomolecule 12 corresponds to the first 170 000bp that has a length polymorphism compared to FSP34current pseudomolecule 12. “Middle” is the portion of KS17current pseudomolecule 12 corresponding to position 170 001-670 679bp displaying synteny to the FSP34current pseudomolecule 12. “Proximal” in KS17current pseudomolecule 12 corresponds to the last 200 000bp which has a length polymorphism in comparison to FSP34current pseudomolecule 12.
Table 4.
Summary of the available metadata for F. circinatum strains FSP34 and KS17.
| Data/Properties | FSP34 | KS17 | References |
| Strain origin | |||
| Collection date | Unknown date between March 1993 and April 1995 | October 2005 | [65,66,67,68] |
| Collector | TR Gordon | OM Mashandula, ET Steenkamp | [65,68] 1 |
| Host plant | Pinus radiata | Pinus radiata | [65] |
| Host tissue | Tissue from the leading edge of a canker on the branch of a mature tree | Diseased root tissue of a nursery seedling | [65] |
| Geographic location | Monterey, California (USA) | Karatara, Western Cape (South Africa) | [65,68] |
| Location description | Exact location is unknown, but collected in a region where mature trees of native Pinus species displayed symptoms of pitch canker | Commercial seedling nursery; was collected during an outbreak of F. circinatum-associated root disease | [65] |
| Reproductive biology | |||
| Mating type | MAT1-1 | MAT1-2 | [68,69] |
| Fertility | Male fertile; mostly female sterile; also capable of mating with sexually compatible strains of Fusarium temperatum to produce fertile hybrid progeny | Male fertile; displays some level of fertility as a female | [24,69,70](unpublished) |
| Source population dynamics | Forms part of a population with limited genetic diversity that propagates asexually | Forms part of a moderately diverse population that propagates mainly asexually | [66,71,72,73] |
| Growth in culture | Grows at 5-30°C, but not at 35°C; grows faster than KS17 at 15-30°C | Grows at 5-30°C, but not at 35°C; grows slower than FSP34 at 15-30°C | [60], this study |
| Pathogenicity | Capable of inducing lesions when inoculated onto the apices of the main stems of P. patula seedlings; more virulent than KS17 | Capable of inducing lesions when inoculated onto the apices of the main stems of P. patula seedlings; less virulent than FSP34 | This study |
1 Fusarium circinatum strain KS17 was isolated at the same time and location, and from the same tissue type as those used in the study by Steenkamp et al. [66] but did not form part of that study.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



