
Submitted:
15 May 2023
Posted:
16 May 2023
You are already at the latest version
Abstract
Prunus tenella is a rare and precious relict plant in China. It is an important genetic resource for almond improvement and an indispensable material in ecological protection and landscaping. However, the research of molecular breeding and genetic evolution has been severely restricted, due to the lack of genome information. In this investigation, we created a chromosome-level genomic pattern of P. tenella, 231Mb in length with a contig N50 of 18.1 Mb by Hi-C techniques and high-accuracy PacBio HiFi sequencing. The present assembly predicted 32088 protein-coding genes, and an examination of the genome assembly indicated that 94.7% among all assembled transcripts were alignable to the genome assembly; most (97.24%) were functionally annotated. By phylogenomic genome comparison, we found that P. tenella is an ancient group that diverged approximately 13.4 million years ago (Mya) from 13 additional closely related species and about 6.5 Mya from the cultivated almond. Collinearity analysis revealed that P. tenella is highly syntenic and has high sequence conservation with almond and peach. However, this species also exhibit many presence/absence variants. Moreover, a large inversion at the 7,588 kb position of chromosome 5 was observed, which may have a significant association with phenotypic traits. Lastly, population genetic structure analysis in eight different populations indicated a high genetic differentiation among the natural distribution of P. tenella. This high-quality genome assembly provides critical clues and comprehensive information for the systematic evolution, genetic characteristics, and functional gene research of P. tenella. Moreover, it provides a valuable genomic resource for in-depth study in protecting, developing, and utilizing P. tenella germplasm resources.
Keywords:
Prunus tenella
; genome
; assembly
; almond
; population genetic structure
1. Introduction
Wild almond (Prunus tenella) is one of the oldest relict plants left from the Tertiary Miocene epoch and is mainly distributed in China and Kazakhstan[1]. In China, wild almond is an endangered and rare species with economic, scientific, and cultural importance and is scattered only in the northern mountainous areas of Xinjiang province[2]. It is considered to be at danger of extinction due to natural and human-caused disturbances, and as a result is on the list of nationally significant protected wild plants.
As a relatively wild species of cultivated almond, wild almond has adapted to extremely harsh environments, displaying impressive cold and drought tolerance[3]. For example, it can grow normally in the hills and mountains in Tacheng and Altay at extremely low temperatures reaching -35° C. Additionally, wild almond possesses exceptional agronomic traits and unique genomic characteristics, making them highly valuable for diverse applications in various fields, such as food processing and medicine[2]. Additionally, these characteristics provide precious genetic materials for studying the adaptive evolution of the wild almond genome and allow for the genetic improvement of cultivated almond, thus improving the resistance of cultivated almond to biotic and abiotic stresses and improving yield.
With the increased attention to wild resources, many desirable traits have been mined and applied to agricultural varieties[4,5,6,7,8]. Moreover, many breakthrough varieties have also benefited from the discovery of wild-breeding genetic resources[9,10,11,12], especially for wheat, rice, soybean, and other economically important food crops[13,14,15,16]. Together, these lines of evidence have shown that introducing wild gene resources has greatly improved disease resistance, insect resistance, and the growth of cultivated varieties[17,18,19,20,21].
In P. tenella, some genes such as self-incompatibility-related genes SBPI, Petcullin1, SFB, SSK1, S-RNas, and the cold resistance-related gene AlsCBF1-A were identified by homologous cloning technology[22,23,24]. Chloroplast DNA (cp DNA), ISSR, and SSR markers are only a few of the molecular markers that were applied for examining population structure and diversity in the past[25,26]. Yet, a thorough knowledge of the economically significant genetic features has been severely hampered by a paucity of genomic resources. More research into the evolutionary adaptations and the development process of distinct features is required before the genetic characteristics of wild almonds can be properly analyzed.
Obtaining high-quality reference genome sequences is the key to revealing allelic variation, genetic relationship, and evolutionary history[27,28,29,30,31]. The present paper describes a highly accurate chromosome-scale standard genome of P. tenella assembled from scratch using Hi-C technique and lengthy PacBio SMRT reads. Additionally, the genetic variation and geographical differentiation of 130 individual plants from 8 wild natural populations were assessed using genome-wide high-resolution molecular markers, which provided important basis for advancing our understanding of origin, formation, and geographical distribution profile of P. tenella.
2. Results
2.1. De novo assembly of the wild almond genome
After quality assessment (GC distribution statistics, quality value Q20, Q30 assessment) and filtering, 29.55 Gb clean data were obtained for genome size assessment and assembly. The sequencing depth totaled approximately 137.00×, with a GC content of approximately 38.15%. Additionally, the proportion of Q20 was greater than 97.19%, and the proportion of Q30 was greater than 91.81%. Based on kmer distribution analysis, the sample genome size was estimated at around 215.65 Mb, consisting of 35.76% repeat sequences and 0.47% heterozygosity. A kmer distribution of k = 21 was observed (Supplementary Figure S1).
Using PacBio SMRT single molecule sequencing, 34.6 GB high-accuracy HiFi reads were obtained for de novo genome assembly. The average length of ccs produced was more than 1,765 bp, and the longest was 34,337 bp. After assembly and deduplication, a contig level assembly of 231 M sequences was obtained, a contig-level assembly of 231 M sequences was obtained, comprising 3,073 contigs (longest = 35.9 Mb) with a 18.1 Mb contig N50 (Table 1). After sequencing quality control, 36.05 Gb Hi-C Fastq clean data were obtained by 3D de novo assembly with the proportion of Q30 was more than 93.10% and the effect rate of 32.5%. Finally, 231 Mb of sequences were anchored onto 8 pseudochromosomes (Figure 2), covering 479 scaffolds (longest = 44.8 Mb, scaffold N50 = 25.6 Mb) (Table 1). A 89.7% mounting rattion have been estimated (Table 2). The BUSCO results showed that more than 2009 (94.7%) genes could be compared to the lineal homologous database, of which 62.5% were single-copy, and 32.7% were duplication (Table 3).

Figure 1.
Morphological characteristics of the Prunus tenella. A) Flower, B) Fruit, and C) Seed.
2.2. Functional annonations, gene prediction, and repetitive sequences
To further describe the wild almond genome, we categorized all sequences that recur using a combination of de novo and homology-based methods. We estimated that transposable elements made up 28.97% of the genome, with TIRs making up to 7.56% and non-TIRs making up to 3.04%. Table 4 shows that only long terminal repeat (LTR) retrotransposons were found. The present assembly projected a total of 32088 protein-coding genes, of which 97.24% had at least one public database functional annotation (Table 5).
2.3. Synteny analysis, and genome evolution and phylogeny
We utilized 10,184 gene families from related species, including, P. avium, P. armeniaca, P. kansuensis, P. ferganensis, P. dulcis, P. davidiana, P. humilus, P. mandshurica, P. mira, P. mume, P. persica, P. salicina, P. sibirica. Among them, 8,674 were common to all species, while 288 were unique to wild almonds (Figure 3).
The period of divergence was then approximated by comparing the protein sequences from every single-copy gene families common to those species, and the resulting phylogenetic trees were created. Bootstrap values greater than 90% provided strong evidence for the association between the species (Figure 4). Indeed, all seven species of the peach genus are on the same branch; among them, wild almond is an ancient group that diverged approximately 13.4 million years ago (Mya). Furthermore, wild almond almost diverged about 6.5 Mya from the cultivated almond. A total of 2679 genes involved in expansion and contraction were identified by gene family analysis, among which 1581 genes in 105 gene families were significantly expanded, and 26 genes in 23 gene families were significantly contracted. Together, these data indicate that the wild almond gene family has undergone a significant contraction (2256 genes) relative to other species (486-1613 genes) in the peach genus, which may be related to natural selection.
Next, synteny analysis was performed to further understand genes’ position relationship on homologous chromosomes and the variation of genome structure. Coding gene and genome-wide collinearity analysis showed a highly linear relationship between wild almond, cultivated almond, and Peach. Meanwhile, 1,540,264 SNPs, 105,280 deletions, and 155,863 insertions (including absence/presence variations) sequences were identified when compared with almond (Supplementary Table S1). Compared with Peach, the 1,574,620 SNP, 106,957 deletions, and 161,747 insertions (including absence/presence variations) sequences were identified (Supplementary Table S2). These structural variations are the main source of genomic variation and may have a significant association with phenotypic traits. In addition, a large inversion(7588kb) was identified on chromosome 5, which is valuable for further understanding gene regulation and epigenetic inheritance in wild almond (Figure 5).
2.4. Population genetic structure and genetic diversity analysis
Using the Illumina HiSeq X Ten platform, 512.5 Gb of clean PE150 paired-end data (approximately 15 average sequencing depth) was generated from 130 separate specimens gathered from the native area in Xinjiang Province, revealing the genetic variants in wild almond across different geographic populations. Based on the genetic distances derived from the genotypes at all the SNP sites of the eight subpopulations in the three areas, a maximum-likelihood and neighbor-joining (Figure 6A) phylogenetic tree was created using the SNPs/genotypes. Some individuals within the subgroups were grouped in other subgroups, but overall, the groupings from the three locations exhibited strong genetic isolation and constituted distinct groups within the phylogenetic tree. The phylogenetic tree's conclusion was confirmed by principal component analysis. Tacheng, Yuoli, and Yumin samples clustered together in a distinct subgroup of the PCA (Figure 6B).
Structure analysis results also showed that the 130 samples were mainly from three ancestral populations, consistent with their distribution areas. The samples from the Tacheng are a pure population, while the TuoLi and Yumin samples are hybridized populations with slight levels of admixture (K = 3; Figure 6C). Genetic diversity analysis showed that Expected_heterozygous_number, Observed_allele_number, Nei_diversity_index, and Shnnon_Wiener_index were 0.17-0.29, 1.31-1.49, 0.18-0.30 and 0.25-0.43, respectively (Supplementary Table S3). Compared with other populations, the genetic diversity of the three populations in Yumin was relatively high, while the Tacheng population was relatively low.
2.5. Genome-wide selection signatures analysis of differentiation
To understand the genetic differentiation among populations, we conducted selective sweep analyses and calculated all pairs of Fst values between 8 sampling populations in three different regions. The results showed that the genetic differentiation between Tacheng and Yumin populations was 0.29-0.32. The genetic differentiation between Tacheng and Tuoli was 0.28-0.3, and the genetic differentiation between Tuoli and Yumin was 0.21-0.27. The inter-population genetic differentiation within the region was low, with the differentiation coefficient falling between 0.05 and 0.1 in the four sample populations of Tuoli and between 0.05 and 0.09 in the three sample populations of Yumin. These results indicate that there had been strong genetic differentiation and geographical variation among different geographical regions of wild almond, which may be due to the obstruction of gene flow caused by geographical isolation.
3. Discussion
Through the use of various sequencing methods, we assembled the first complete reference genome for wild almond in this work. The Prunus genus has several commercially and ecologically significant species in forestry and agriculture, and these data are essential for learning more about wild almonds and the genus as a whole. These findings will also aid in the development of genome-enabled wild almond breeding initiatives. Last but not least, wild almond's status as a relict species makes it a useful model for studying the genetic basis of population formation, evolution, and adaptation to environmental effects under conditions of geographic isolation.
Given the high quality of our wild almond genome assembly, high-depth PacBio long-read, and whole genome re-sequencing data, we now have a comprehensive understanding of the genome of the wild almond. Single-copy, multi-copy, and species-specific gene families were obtained, the evolutionary status was inferred, and the genome’s evolutionary history was traced, laying the foundation for further exploration and research. Additionally, we found many variation sites, including SNPs, insertions, deletions, and inversion. Many of these variants may be associated with phenotypic traits, which will help understand the phylogenetic evolution of wild almond. Moreover, these various sites can be used as important molecular markers for germplasm identification, genetic analysis, functional gene extraction, and assisted breeding.
Through the analysis, we also found some different characteristics of the wild almond genome relative to other species of the same genus. Compared with other tree species, wild almond had relatively more endemic gene families(288), while cultivated almond and peach had relatively few, only 31-37, which may be related to the earlier differentiation time of wild almond. More gene families were contracted in wild almond, which might be related to natural selection caused by the extreme natural environment. The evolutionary status inferred by the phylogenetic tree shows that the wild almond is an independent branch, most closely related to cultivated almond. Importantly, this observation indicates that wild almond has the potential utilization value of providing genetic resources for cultivated almond, and this lays the foundation for further exploration and research.
Population genetic structure can be used to analyze the evolutionary dynamics of a population by describing gene transmission, gene frequency change, and genotype distribution[32,33,34,35]. Based on the SNP information derived from the whole-genome re-sequencing data, thousands of single SNP markers can be used for the fine-scale description of genetic structure[36,37,38]. The results showed that compared with the Tacheng (Nei’s = 0.18; Ho = 0.16)and Tuoli(Nei’s = 0.23-0.26;Ho = 0.16-0.22)populations, the Yumin variety(Nei’s = 0.26-0.3;Ho = 0.17-0.22)has relatively high genetic diversity. This observation is consistent with the study on genetic diversity using chloroplast sequences[4]. Additionally, the results indicated a high genetic differentiation among the natural distribution of wild almond, as the pairwise genetic differentiation (Fst) in a different region is 0.23-0.32, especially within the Tacheng and Yumin group where Fst reached 0.29 to 0.32, values much higher than wright’s high differentiation coefficient[39,40,41]. However, there is little differentiation between subgroups within the Tuoli and Yumin group (0.05-0.1). These results suggest that geographical isolation is an important factor affecting the genetic evolution of wild almonds. This higher differentiation may result from long-term natural selection without gene flow.
In summary, we assembled the first chromosome-level genome of P. tenella and assessed the genetic variation and geographical differentiation of 8 natural populations, which laid a solid foundation for further research on genetic improvement and formation mechanism of important characters in the future.
4. Materials and Methods
4.1. Utilized materials
P. tenella sample materials used for genome assembly were obtained from the germplasm conservation nursery of Xinjiang Academy of Forestry Sciences, Xinjiang, China (Figure 1). Fresh leaves were utilized for Hi-C library development, PacBio HiFi sequencing, and Illumina sequencing. To aid in genome assembly and annotation, fruit, leaf, root, and stem tissues were taken for RNA-seq study.
The fresh leaves used for whole genome re-sequencing were collected from Yumin County, Tuoli County, and Tacheng City, Xinjiang, China. A total of 8 wild almond populations were collected, including 3 from Yumin County, 4 from Tuoli County, and 1 from Tacheng City. Approximately 15-18 samples were collected from each population. In addition, 7 cultivated almond samples were collected for population evolution analysis.
4.2. Genome sequencing and transcriptome sequencing
The experiments were carried out in accordance with Illumina's recommended methodology. The ultrasonic shock was used to physically fragment the qualifying genomic DNA into fragments (350 bp), and then end restoration, adding A, an adapter, and target fragment picking and PCR were used to generate the tiny fragment sequencing library. By using bridge PCR, the library was transferred to the sequencing chip. An Illumina sequencer performed double-ended 150 bp (PE 150) library sequencing.
DNA capture and purification, cyclization, end repair, endonuclease digestion, cell cross-linking, and on-machine sequencing were all necessary steps for HI-C sequencing to be completed. The mRNA was utilized to synthesize full-length cDNA with the help of the SMARTerTM PCR cDNA Synthesis Kit, which was then used to generate sequencing libraries. Using the PacBio system, we sequenced the whole transcriptome.
Library sequencing, library quality testing, library creation, and sample quality testing were all carried out as per Illumina's recommended approach for re-sequencing a variety of population samples. In order to prepare the DNA for sequencing, it was first physically fragmented (using ultrasonic waves), then purified, the ends were mended, the 3' end was augmented with A, and the sequencing joint was linked. Finally, agarose gel electrophoresis was used to determine the optimal fragment size, and PCR amplification was carried out to form the sequencing library.
Transcriptome sequencing of the stem, root, leaf, and fruit tissues was performed on the NovaSeq 6000 platform.
4.3. Assurance of Sequencing Data Quality
Low-quality sequences and duplicated readings in the sequencing data were removed using stringent filtering algorithms that were optimized for the particular platform utilized to assure data integrity and accuracy. Filtering criteria included the following actions for Illumina Hi-Seq data: Firstly, polyG tails were removed. Secondly, paired reads less than 100bp in length were discarded. Thirdly, read pairs containing more than 10% of bases that are the same as the next base were removed. Fourthly, read pairs with over 50% low-quality bases (quality score less than 10) were discarded. The last step was to clean the data of read pairings with a typical quality rating below 20. The Hi-C sequencing results went through a comparable filtering procedure as Illumina Hi-Seq Data before being processed in 3D. With the default settings of the pbccs pipeline, subreads from the PacBio HiFi long readings were filtered and corrected immediately. Approximately 2,000 PacBio HiFi (CCS) reads were randomly selected from the sequencing data and compared with the NT library to evaluate whether the sequencing data contained contamination.
4.4. Heterozygosity and genome size estimation
Heterozygosity and genome size were analyzed before HiFi library construction and sequencing. From the Illumina data, Jellyfish v.2.2.10[42] examined frequency distributions of quality-filtered short fragments (21-mers). Then, based on Jellyfish’s results, genome escope22 was used for genome analysis. This strategy obtained genomic information of P. tenella (Supplementary Figure 1), like heterozygosity, genome size, and proportion of repeat sequences.
4.5. Genome assembly
Following correction and filtering, HiFi CCS reading could be used in the de novo assembly using hifiasm (v0.14-r312) with default parameters. Purge haplotigs was used to remove redundant haploids[43]. In 2017, Dudchenko et al[44]. used the 3D de novo assembly (3D-DNA) software for scaffolding the haploid contigs. The Hi-C readings could be aligned within the draft genome 3D-DNA and Juicebox v1.9.8 was used for the candidate assembly. Assembly Tools (JBAT) [45] was utilized for reviewing the candidate assembly and corrected artificially. The eudicotyledons_odb10 database have been employed in conjunction with BUSCO v3.0.2 (Benchmarking Universal Single-Copy Orthologs)[46] algorithm for assessing genome integrality and gene annotation. A combination of the BWA-MEM method and HISAT2 (v2.1.0)[47] was utilized for mapping small reads filteration obtained by Illumina and the assembled transcripts to the assembly.
4.6. Repetitive element annotations
To annotate the TEs or transposable elements[48], the EDTA genome annotation pipeline was utilized. TEs include retrotransposons and DNA transposons. RepeatModeler was used to identify DNA transposons, including long interspersed nuclear elements (LINEs) of the terminal inverted repeats (TIRs) and retrotransposons, and long tandem repeats (LTRs), as well as helitrons found in DNA transposons. To do this, we used Repbase and RepeatMasker (v4.0.7) and Repbase with the optimal settings to generate a de novo repeat library for repeat sequence identification[49,50].
4.7. Functional annonations and gene prediction
The StringTie (v1.3.5) and HISAT2 (v2.1.0) pipeline was approached for maping the RNA-seq data within the fruits, leavers, stems, and roots were mapped to the genome. Gene prediction together with de novo transcripts assembly were conducted through Trinity [51]. PASA (v2.4.1) pipeline transdecoder4 have also been applied to annotate the transcripts-relevant coding regions[52]. Exonerate v2.2.0 carried out homolog predictions. GlimmerHMM (v3.0.4) and the protein sequences of Prunus dulcis, Prunus mira, Prunus persica, Prunus armeniaca, Prunus mume, and Prunus salicina could also be mapped to the genome [53]. For de novo gene speculation, genes from the PASA results were trained by AUGUSTUS (v3.3.3) and SNAP[54,55]. In order to combine the gene models, EVidenceModeler (v1.1.1) was approached[56]. The predicted protein sequences have been compared to the EuKaryotic Orthologous Groups (KOG), Nr databases, Pfam, SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) to infer possible functions for the protein-coding genes.
4.8. Phylogenetic and gene family analysis
Thirteen closely related species were selected for phylogenetic and gene family analysis along with P.tenella. Additionally, P.avium was selected as the outgroup. The genome database for Rosaceae (www.rosaceae.org), and the NCBI database (www.ncbi.nlm.nih.gov) was used for getting the protein sequences of these species. Alignment quality was ensured by excluding sequences with lengths < 100 bp. OrthoFinder (v2.5.2) have been deployed for identifying single-copy homologous genes and classifying families, using the settings "-M msa -S diamond -T raxml-ng"[57]. RaxML[58] have been approached for estimating and evaluating the phylogenetic connection tree of 14 species using 100 bootstrap repetitions. Time to diverge was computed using PAML's MCMC tree[59]. CAFE (v3.1) have been approached for examining relevant growth patterns and gene families-related declines described by Han et al[60]. By counting the number of ancestral gene families on each branch of the phylogenetic tree, we were able to determine the rate at which gene family sizes shrank or grew. Cafetutorial_clade_and_size_filter.py was used to filter gene families characterized by very high variations in gene copy numbers in an effort to decrease prediction mistakes. Exact data on the contraction and expansion gene families of 14 species was utilized using the script cafetutorial_report_analysis.py, and this data was then analyzed. For selected gene families, we used Fisher's exact test to analyze GO functional enrichment.
4.9. Whole-genome synteny analysis
Almond and peach were selected for whole genome replication (WGD) analysis. Fourfold synonymous (degenerative) third-codon transversion (4DTv) values and synonymous mutation distributions for each synonymous site (Ks) were calculated to analyze the genome replication events. YN substitution model was used to calculate the 4DTv rates based on fourfold degenerate sites. KaKs_Calculator (v2.0)[61] with default parameters was used to calculate Ks values. The minimap2 software was used for genome-wide comparison, and syri software was used to identify collinear regions between the two genomes, structural rearrangements (inversion, translocation, and duplication), local variations (SNP, indel, and CNV), and unaligned regions. The nucmer (4.0.0beta2) program in MUMmer4[62] was used to determine whether similar gene pairs on chromosomal were adjacent in different species.
4.10. Single-nucleotide polymorphism (SNP) calling
Trimmomatic v0.36 was used to eliminate adaptors and low-quality sequences during the preprocessing phase. Every sample's clean reads have been planned using Burrows-Wheeler Aligner to the wild almond standard genome. Next, Picard (http://broad insti tute. github.io/picard/) have been employed for identifying and aligning PCR duplicated sample findings. SNP sites in re-sequencing people from diverse geographical regions were identified using GATK v4 (Genome Analysis Toolkit) for SNP recalling. Each genome's VCF files were generated using variant calling with GATK Hap-lotypeCaller, and then the VCF files for all 137 genomes were combined to create a single VCF file. just SNPs that had a Hardy-Weinberg Equilibrium < 0.001, Minor allele frequency > 0.05, and genotype missing rate 10% for each were kept for further study, narrowing the analysis down to just biallelic variation sites.
4.11. Phylogenetic analysis
A phylogenetic tree was generated using the distance matrix produced by MEGA-CC software (MEGAX)[63] and 1000 bootstrap repetitions to assess the phylogenetic connection of various individuals in order to study the evolutionary links between different populations. In addition, the SMARTPCA application included in the EIGENSOFT software (https://github. com/chrch ang/eigen soft) was utilized to carry out principal component analysis (PCA) and ascertain the subpopulations' clustering status [64].
4.12. Population genetic structure and genetic diversity analysis
In order to learn about the genetic makeup of populations, including their variety, structure, and differentiation, Nucleotide diversity was assessed by dividing each population into 10 kb chunks and analyzing a 100 kb window[65]. Using a Bayesian-based strategy, the K-values (the hypothesized number of populations) ranged between 1 – 10 in ADMIXTURE[66]. The optimal K-value was determined using cross-validation statistics across five separate studies. Bar graphs of the Q matrix for each K-value were made with the aid of the R package Pophelper (http://royfrancis.github.io/pophelper). Fixation index (FST) and nucleotide diversity ratios (π) were computed using VCF methods to identify genomic areas possibly experiencing natural selection sweeps throughout the adaptation process.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Table S1. Variation type Statistics (Prunus dulcis vs Prunus tenella). Supplementary Table S2. Variation type Statistics ( Prunus persica vs Prunus tenella). Supplementary Table S3. Genetic diversity parameters of different populations. Supplementary Figure S1. The kmer distribution of k=21.
Author Contributions
Gaopu Zhu and Han Zhao conceived and coordinated the research. Han Zhao and Hongwei Han investigated and collected the samples. Yue Qin and Zhaoshan Wang assembled and analyzed the data. Yue Qin wrote the manuscript, which has critically revised by Fangdong Li and Gaopu Zhu. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the “National Key R&D Program of China (2022YFD2200400)” and the “Key R&D Program of Xinjiang Uygur Autonomous Region (2022292937)”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data availability: The whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation, under accession number GWHCBGA00000000 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh.The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, J.; Zeng, B.; Luo, S. P.; Li,H.L.; and Madaniyati,W. Protection and propagation ofAmygdalus ledebouriana Schleche in China. Xinjiang AgriculturalSciences. 2006, 43, 61-62.
- Yin, L. K.; Tan, L. X.;Wang, B. Rare endangered endemic higherplants in Xinjiang of China. Urumqi: Xinjiang Science & TechnologyPublishing House. 2006.
- Zhong, H. X.; Lu, C. S.; Luo, S. P.;Li, J. The study of cold resistancetest of dormancy branches and buds of Amygdalus ledebourianaSchleche in Xinjiang. Xinjiang Agricultural Sciences. 2016, 53, 120-125.
- Perazzolli, M., Malacarne, G., Baldo, A., Righetti, L., Bailey, A., Fontana, P., Velasco, R., Malnoy, M., Characterization of resistance gene analogues (RGAs) in apple (Malus × domestica Borkh.) and their evolutionary history of the Rosaceae family. PLoS One. 2014, Feb 5;9(2):e83844. PMID: 24505246; PMCID: PMC3914791. [CrossRef]
- Vinceti, B., Elias, M., Azimov, R., Turdieva, M., Aaliev, S., Bobokalonov, F., Butkov, E., Kaparova, E., Mukhsimov, N., Shamuradova, S., Turgunbaev, K., Azizova, N., & Loo, J. Home gardens of Central Asia: Reservoirs of diversity of fruit and nut tree species. PloS one. 2022, 17(7), e0271398. [CrossRef]
- Singh, R. K., Singh, C., Ambika, Chandana, B. S., Mahto, R. K., Patial, R., Gupta, A., Gahlaut, V., Gayacharan, Hamwieh, A., Upadhyaya, H. D., & Kumar, R. Exploring Chickpea Germplasm Diversity for Broadening the Genetic Base Utilizing Genomic Resourses. Frontiers in genetics. 2022, 13, 905771. [CrossRef]
- Kumar, S., Jacob, S. R., Mir, R. R., Vikas, V. K., Kulwal, P., Chandra, T., Kaur, S., Kumar, U., Kumar, S., Sharma, S., Singh, R., Prasad, S., Singh, A. M., Singh, A. K., Kumari, J., Saharan, M. S., Bhardwaj, S. C., Prasad, M., Kalia, S., & Singh, K. Indian Wheat Genomics Initiative for Harnessing the Potential of Wheat Germplasm Resources for Breeding Disease-Resistant, Nutrient-Dense, and Climate-Resilient Cultivars. Frontiers in genetics. 2022, 13, 834366. [CrossRef]
- Kefale, H., & Wang, L. Discovering favorable genes, QTLs, and genotypes as a genetic resource for sesame (Sesamum indicum L.) improvement. Frontiers in genetics. 2022,13, 1002182. [CrossRef]
- García-Gómez, B. E., Salazar, J. A., Nicolás-Almansa, M., Razi, M., Rubio, M., Ruiz, D., & Martínez-Gómez, P. Molecular Bases of Fruit Quality in Prunus Species: An Integrated Genomic, Transcriptomic, and Metabolic Review with a Breeding Perspective. International journal of molecular sciences. 2020, 22(1), 333. [CrossRef]
- Filip, E., Woronko, K., Stępień, E., & Czarniecka, N. An Overview of Factors Affecting the Functional Quality of Common Wheat (Triticum aestivum L.). International journal of molecular sciences. 2023, 24(8), 7524. [CrossRef]
- Li, Z., Xue, Y., Zhou, H., Li, Y., Usman, B., Jiao, X., Wang, X., Liu, F., Qin, B., Li, R., & Qiu, Y. High-resolution mapping and breeding application of a novel brown planthopper resistance gene derived from wild rice (Oryza. rufipogon Griff). Rice (New York, N.Y.). 2019,12(1), 41. [CrossRef]
- Mamidi, S., Healey, A., Huang, P., Grimwood, J., Jenkins, J., Barry, K., Sreedasyam, A., Lovell, J. T., Feldman, M., Wu, J., Yu, Y., Chen, C., Johnson, J., Sakakibara, H., Kiba, T., Sakurai, T., Tavares, R., Nusinow, D. A., Baxter, I., Schmutz, J., Brutnell, T. P., Kellogg, E. A. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat Biotechnol, 2020, 38,1203-1210.
- Prahalada, G. D., Shivakumar, N., Lohithaswa, H. C., Sidde Gowda, D. K., Ramkumar, G., Kim, S. R., Ramachandra, C., Hittalmani, S., Mohapatra, T., & Jena, K. K. Identification and fine mapping of a new gene, BPH31 conferring resistance to brown planthopper biotype 4 of India to improve rice, Oryza sativa L. Rice (New York, N.Y.). 2017, 10(1), 41. [CrossRef]
- Laugerotte, J., Baumann, U., & Sourdille, P. Genetic control of compatibility in crosses between wheat and its wild or cultivated relatives. Plant biotechnology journal. 2022, 20(5), 812-832. [CrossRef]
- Sharma, S., Schulthess, A. W., Bassi, F. M., Badaeva, E. D., Neumann, K., Graner, A., Özkan, H., Werner, P., Knüpffer, H., & Kilian, B. Introducing Beneficial Alleles from Plant Genetic Resources into the Wheat Germplasm. Biology. 2021, 10(10), 982. [CrossRef]
- Aleem, M., Aleem, S., Sharif, I., Aleem, M., Shahzad, R., Khan, M. I., Batool, A., Sarwar, G., Farooq, J., Iqbal, A., Jan, B. L., Kaushik, P., Feng, X., Bhat, J. A., & Ahmad, P. Whole-Genome Identification of APX and CAT Gene Families in Cultivated and Wild Soybeans and Their Regulatory Function in Plant Development and Stress Response. Antioxidants (Basel, Switzerland). 2022, 11(8), 1626. [CrossRef]
- Mk, A.; Shw, B.; and Ss, A. Wheat wild germplasm: a hidden treasure. Wild Germplasm for Genetic Improvement in Crop Plants.2011, 2021, 55-63.
- Yumurtaci, A. Utilization of wild relatives of wheat, barley, maize and oat in developing abiotic and biotic stress tolerant new varieties. Emirates Journal of Food & Agriculture. 2015, 27.
- Haus, M. J.; Pierz, L. D.; Jacobs, J. L.; Wiersma, A. T.; and Cichy, K. E. Preliminary evaluation of wild bean (phaseolus spp.) germplasm for resistance to fusarium cuneirostrum and fusarium oxysporum. Crop Science. 2021, 3.
- Rostad, H. E.; Reen, R. A.; Mumford, M. H.; Zwart, R.; and Thompson, J. P. Resistance to root-lesion nematode pratylenchus neglectus identified in a new collection of two wild chickpea species (cicer reticulatum and c. echinospermum) from turkey. Plant Pathology. 2022, 5, 71.
- Jeff, E.; Olumide, S.T.; Bruce,D.; Andre, H.; Julianne, A.; Olufemi, A. Resistance in wild macadamia germplasm to phytophthora cinnamomi and phytophthora multivora. Annals of Applied Biology. 2021, 178.
- Wang, B.; Yu, Z.F.; Zeng, B.; Xia, J.H.;Ma,X.X. Self-incompatibility Gene Cullin1 Cloning and Bioinformatics Analysis of Wild Almond in Xinjiang. Chinese Agricultural Science Bulletin. 2017, 33, 63-68.
- Zeng, B.; Liu, N.N.; Xia, J.H.; Liu, M.W.; Wang,J.Y.; Wang, B. Molecular Cloning and Bioinformatics Analysis of SFB Genes Controlling Self-incompatibility in Xinjiang Wild Almond (Prunus tenella Batsch.). Chinese Agricultural Science Bulletin. 2017, 33, 22-30.
- Yu, Z. F.; Wang, B.; Zeng, B.; Wang, J.Y. Cloning and sequence analysisof self - incompatibility gene SBPI of wild almond in Xinjiang.Molecular Plant Breeding. 2018, 16, 6955-6960.
- Zeng, B.; Li,J.; Luo, S. P.; Cheng, Y. J. Identification of Genetic Relationship of Amygdalus Plants by SSR. Xinjiang Agricultural Sciences. 2009, 46, 18-22.
- Lu, Z.J.; Li, J.; Omir, S.T.; Zeng.B.; Luo, S.P. ISSR analysis for genetic diversity of Amygdalus ledebouriana germplasmfrom Xinjiang,China. Journal of Fruit Science. 2010, 27, 918-923.
- Chen, D. X.; Pan, Y.; Wang, Y.; Cui, Y. Z.; & Li, L. Y. The chromosome-level reference genome of coptis chinensis provides insights into genomic evolution and berberine biosynthesis. Horticultural Research. 2021, 8, 11.
- Rush, D. W.; and Epstein, E. Breeding and selection for salt tolerance by the incorporation of wild germplasm into a domestic tomato. Journal American Society for Horticultural Science. 1981, 106, 699-704.
- D'Amico-Willman, K. M.; Ouma, W. Z.; Meulia, T.; Sideli, G. M.; Gradziel, T. M.; and Fresnedo-Ramírez, J. Whole-genome sequence and methylome profiling of the almond ( prunus dulcis [mill.] d.a.webb) cultivar ‘nonpareil’. G3 Genes|Genomes|Genetics. 2022,12(2022)jkac065.
- Liu, J. F.; Wei, H.; Zhang, X.; and Wang, D. Chromosome-level genome assembly and hazelomics database construction provides insights into unsaturated fatty acid synthesis and cold resistance in hazelnut (corylus heterophylla). Frontiers in Plant Science. 2021, 12, 766548.
- Verde, I.; Abbott, A. G.; Scalabrin, S.; Jung, S.; and Rokhsar, D. S. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution-Supplementary Information.Nature Genetics. 2014, 45, 486-495.
- Suguiyama, V. F., Vasconcelos, L. A. B., Rossi, M. M., Biondo, C., & de Setta, N. The population genetic structure approach adds new insights into the evolution of plant LTR retrotransposon lineages. PloS one. 2019, 14(5), e0214542. [CrossRef]
- Ikeda, H. Decades-long phylogeographic issues: complex historical processes and ecological factors on genetic structure of alpine plants in the Japanese Archipelago. Journal of plant research. 2022, 135(2), 191-201. [CrossRef]
- Mori, G. M., Zucchi, M. I., & Souza, A. P. Multiple-geographic-scale genetic structure of two mangrove tree species: the roles of mating system, hybridization, limited dispersal and extrinsic factors. PloS one.2015, 10(2), e0118710. [CrossRef]
- Nishio, S., Takada, N., Terakami, S., Takeuchi, Y., Kimura, M. K., Isoda, K., Saito, T., & Iketani, H. Genetic structure analysis of cultivated and wild chestnut populations reveals gene flow from cultivars to natural stands. Scientific reports. 2021, 11(1), 240. [CrossRef]
- Deb, S., Della Lucia, M. C., Ravi, S., Bertoldo, G., & Stevanato, P. (2023). Transcriptome-Assisted SNP Marker Discovery for Phytophthora infestans Resistance in Solanum lycopersicum L. International journal of molecular sciences, 24(7), 6798. [CrossRef]
- Bali, S., Robinson, B. R., Sathuvalli, V., Bamberg, J., & Goyer, A. Single Nucleotide Polymorphism (SNP) markers associated with high folate content in wild potato species. PloS one. 2018,13(2), e0193415. [CrossRef]
- Roncallo, P. F., Beaufort, V., Larsen, A. O., Dreisigacker, S., & Echenique, V. Genetic diversity and linkage disequilibrium using SNP (KASP) and AFLP markers in a worldwide durum wheat (Triticum turgidum L. var durum) collection. PloS one. 2019,14(6), e0218562. [CrossRef]
- Castilla, A. R., Méndez-Vigo, B., Marcer, A., Martínez-Minaya, J., Conesa, D., Picó, F. X., & Alonso-Blanco, C. Ecological, genetic and evolutionary drivers of regional genetic differentiation in Arabidopsis thaliana. BMC evolutionary biology. 2020, 20(1), 71. [CrossRef]
- Oh, A., & Oh, B. U. Genetic differentiation that is exceptionally high and unexpectedly sensitive to geographic distance in the absence of gene flow: Insights from the genus Eranthis in East Asian regions. Ecology and evolution. 2022,12(6), e9007. [CrossRef]
- Santangelo, J. S., Johnson, M. T. J., & Ness, R. W. Modern spandrels: the roles of genetic drift, gene flow and natural selection in the evolution of parallel clines. Proceedings. Biological sciences. 2018, 285(1878), 20180230. [CrossRef]
- Marcais,G.; and Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011, 27, 764-770. [CrossRef]
- Roach,M. J.; Schmidt, S. A.; and Borneman,A. R. Purge Haplotigs:allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 2018, 19, 460. [CrossRef]
- Dudchenko, O.; Batra, S. S.; Omer, A. D.; Nyquist, S. K.; Hoeger, M.; Durand, N.C. De novo assembly of the Aedes aegypti genome using Hi-C yieldschromosome-length scaffolds.Science. 2017, 356, 92-95. [CrossRef]
- Durand, N.; Robinson, J.; Shamim, S.; Aiden, E. L. Juicebox provides a visualization system for Hi-C contactmaps with unlimited zoom. Cell Syst. 2016, 3, 99-101. [CrossRef]
- Simão, F. A.; Waterhouse, R. M.; Loannidis, P.; Kriventseva, E. V.; and Zdobnov,E.M. BUSCO: assessing genome assembly and annotation completenesswith single-copy orthologs. Bioinformatics. 2015, 31, 3210-3212. [CrossRef]
- Kim, D.; Langmead, B.; and Salzberg, S. L. HISAT: a fast spliced alignerwith low memory requirements.Nat. Methods. 2015, 12, 357-360. [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Doreen, W.; Thomas, P.; Ning, J.; Candice, N. H.; Hufford, M. B. Benchmarking transposable element annotation methods for creationofa streamlined,comprehensive pipeline. Cold Spring Harbor Laboratory. 2019, 1.
- Bao, W.; Kojima, K. K.; and Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015, 6, 11. [CrossRef]
- Tempel, S. Using and Understanding RepeatMasker. Methods Mol. Biol. 2012, 859, 29-51. [CrossRef]
- Grabherr, M. G.; Haas, B. J.; Yassour, M.; Levin, J. Z.; Thompson, D. A.; Amit,I. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644-652. di: 10.1038/nbt1883.
- Haas, B. J.; Delcher, A. L.; Mount, S. M.; Wortman, J. R.; Smith, J.; Hannick, L. I.; Rama, M.; Ronning, C. M.; Rusch, D. B.; Town, C. D. Improving the Arabidopsis genome annotation using maximaltranscript alignment assemblies.Nucleic Acids Res. 2003, 31, 5654-5666. [CrossRef]
- Majoros, W.; Pertea, M.; and Salzberg, S. TigrScan and GlimmerHMM: twoopen source ab initio eukaryotic gene-finders. Bioinformatics. 2004, 20, 2878-2879. [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; and Morgenstern, B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34,W435-W439. [CrossRef]
- Johnson, A. D.; Handsaker, R. E.; Pulit, S. L.; Nizzari, M. M.; O’donnell, C.J.; De Bakker, P.I. SNAP: a web-based tool for identification andannotation of proxy SNPs using HapMap. Bioinformatics. 2008, 24, 2938-2939. [CrossRef]
- Haas, B. J.; Salzberg, S. L.; Zhu, W.; Pertea, M.; Allen, J. E.; Orvis, J.; White, O.; Buell, C. R.; Wortman, J. R. Automated eukaryotic gene structure annotation using evidence modeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [CrossRef]
- Emms, D. M.; Kelly, S. OrthoFinder: phylogenetic orthology inferencefor comparative genomics. Genome Biol. 2019, 20, 238. [CrossRef]
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis andpost-analysis of large phylogenies. Bioinformatics. 2014, 30, 1312-1313. [CrossRef]
- Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Bio.Evol. 2007, 24,1586-1591. [CrossRef]
- Han,M. V.; Thomas,G.W.C.; Lugo-Martinez, J.; and Hahn,M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol. Evol. 2013, 30, 1987-1997. di: 10.1093/molbev/mst100.
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; and Yu, J. KaKs_Calculator 2.0:a toolkit incorporating gamma-series methods and sliding window strategiesGenomics Proteomics Bioinform. 2010, 8, 77-80. [CrossRef]
- Marais,G.; Delcher, A. L.; Phillippy, A. M.; Coston, R.; and Zimin, A. MUMmer4: a fast and versatile genome alignment system. PLOS Comput. Biol. 2018, 14, e1005944. [CrossRef]
- Sudhir, K.; Glen, S.; Michael, L.; Christina, K.; and Koichiro, T. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Molecular biology and evolution. 2018, 35, 1547-1549.
- Alkes, L. P.; Nick, J. P.; Robert, M.P.; Michael,E.W.; Nanch,A.S.; and David. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 2006, 38, 904-909.
- Vilella, A.J.; Blanco-Garcia, A.; Hutter, S.; Rozas, J. VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data.Bioinformatics. 2005, 21, 2791-3.
- Alexander, D. H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome research. 2009, 19, 1655-1664.
Figure 2.
Hi–C assisted assembly of Prunus tenella genome pseudomolecules. A. high-resolution (100 kb) Hi-C interaction heat map among eight chromosomes. B. Chromosome characteristics of the Prunus genome. Following the outermost ring are the following items: Pseudochromosomal visualization of gene density, GC content, repeat content, SNP density, and gene collinearity from a genome assembly.
Figure 2.
Hi–C assisted assembly of Prunus tenella genome pseudomolecules. A. high-resolution (100 kb) Hi-C interaction heat map among eight chromosomes. B. Chromosome characteristics of the Prunus genome. Following the outermost ring are the following items: Pseudochromosomal visualization of gene density, GC content, repeat content, SNP density, and gene collinearity from a genome assembly.
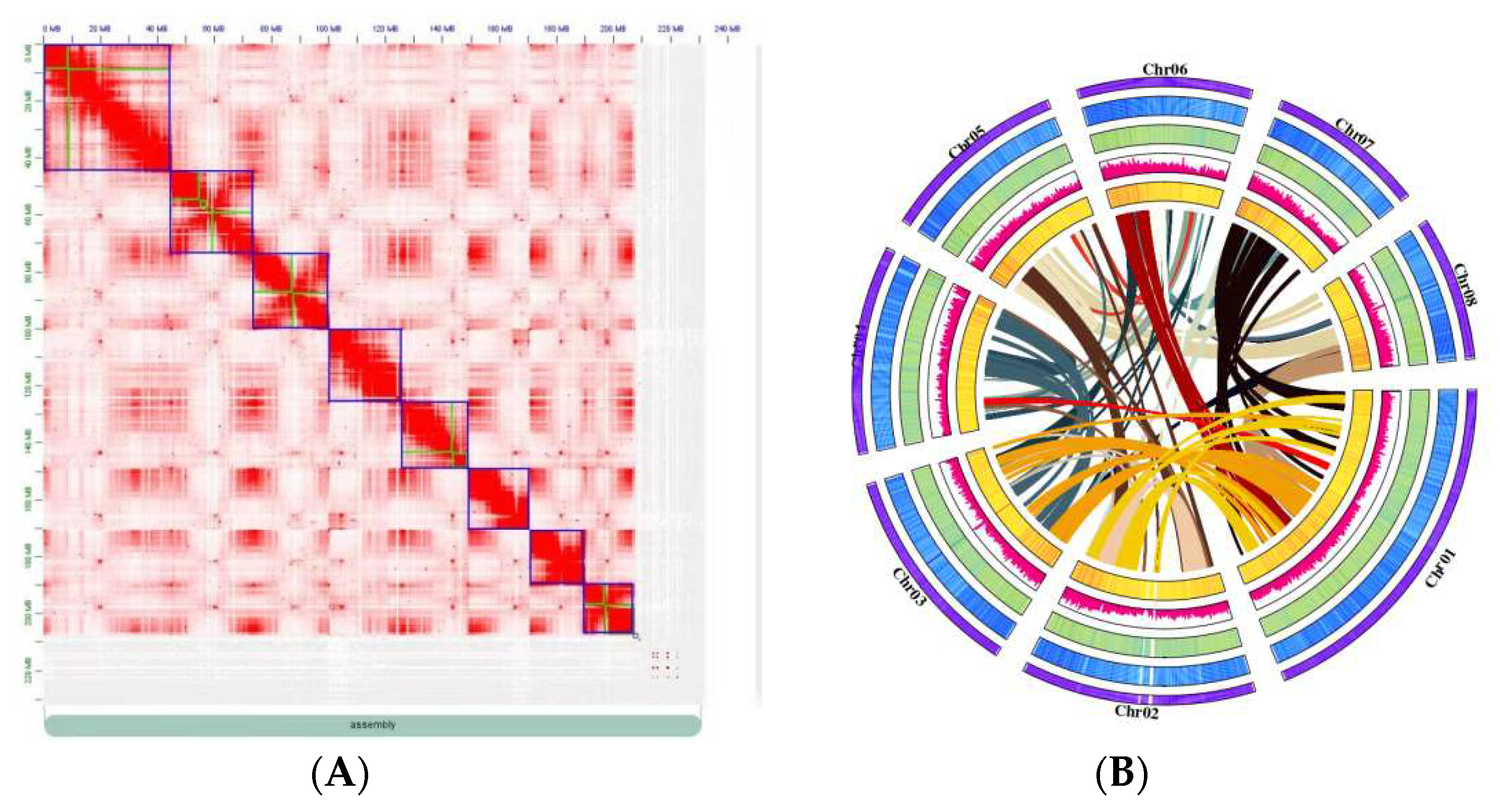
Figure 3.
Number of homologous genes in species and gene family clustering. A) Homologous gene statistics. B) Gene family clustering petal map. .
Figure 3.
Number of homologous genes in species and gene family clustering. A) Homologous gene statistics. B) Gene family clustering petal map. .
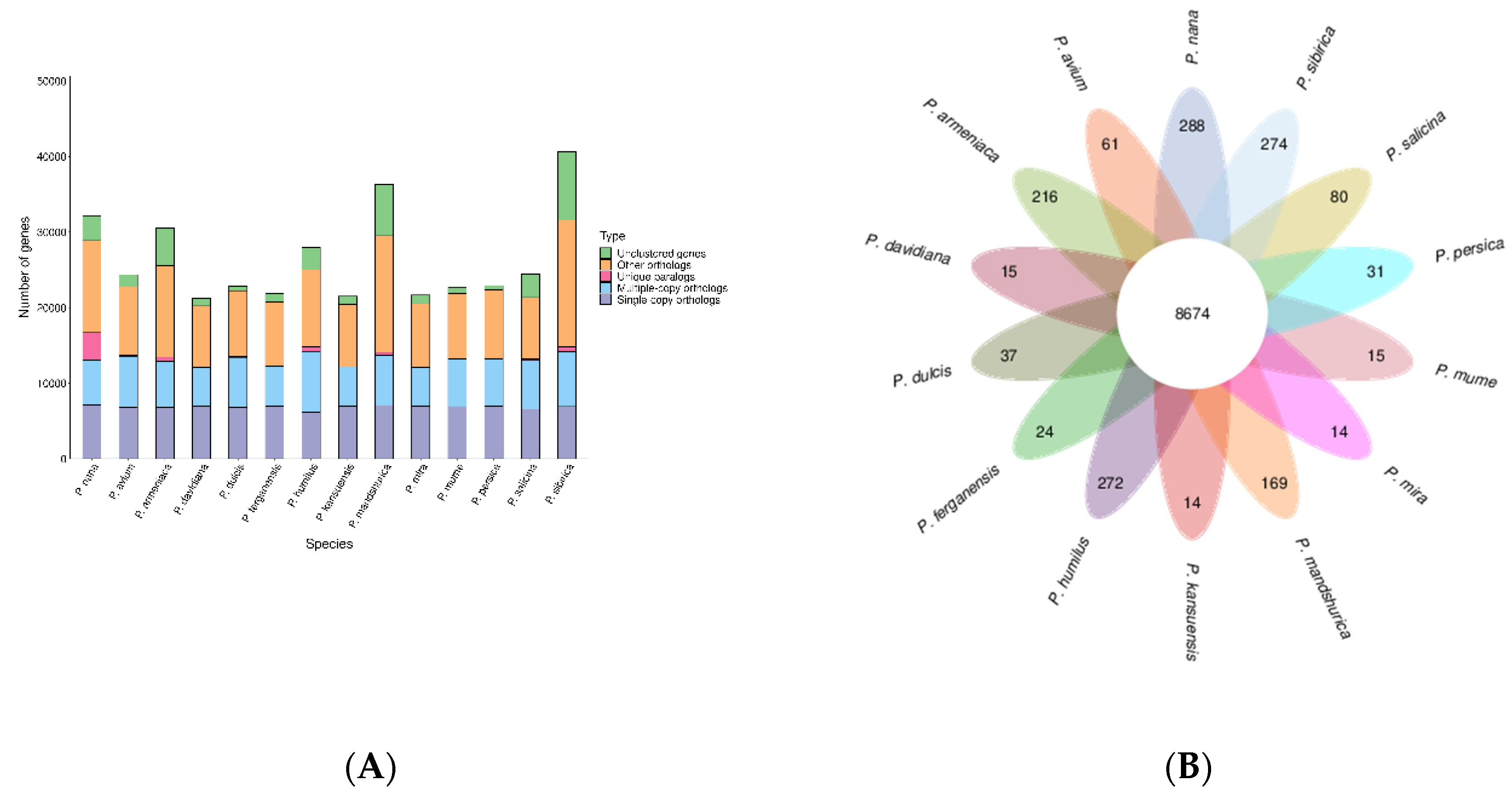
Figure 4.
Divergence time and phylogenetic correlation among species. The percentage of conserved (blue), contracted (red), and expanded (green) gene families among all gene families in the 14 species is shown as a pie chart.
Figure 4.
Divergence time and phylogenetic correlation among species. The percentage of conserved (blue), contracted (red), and expanded (green) gene families among all gene families in the 14 species is shown as a pie chart.
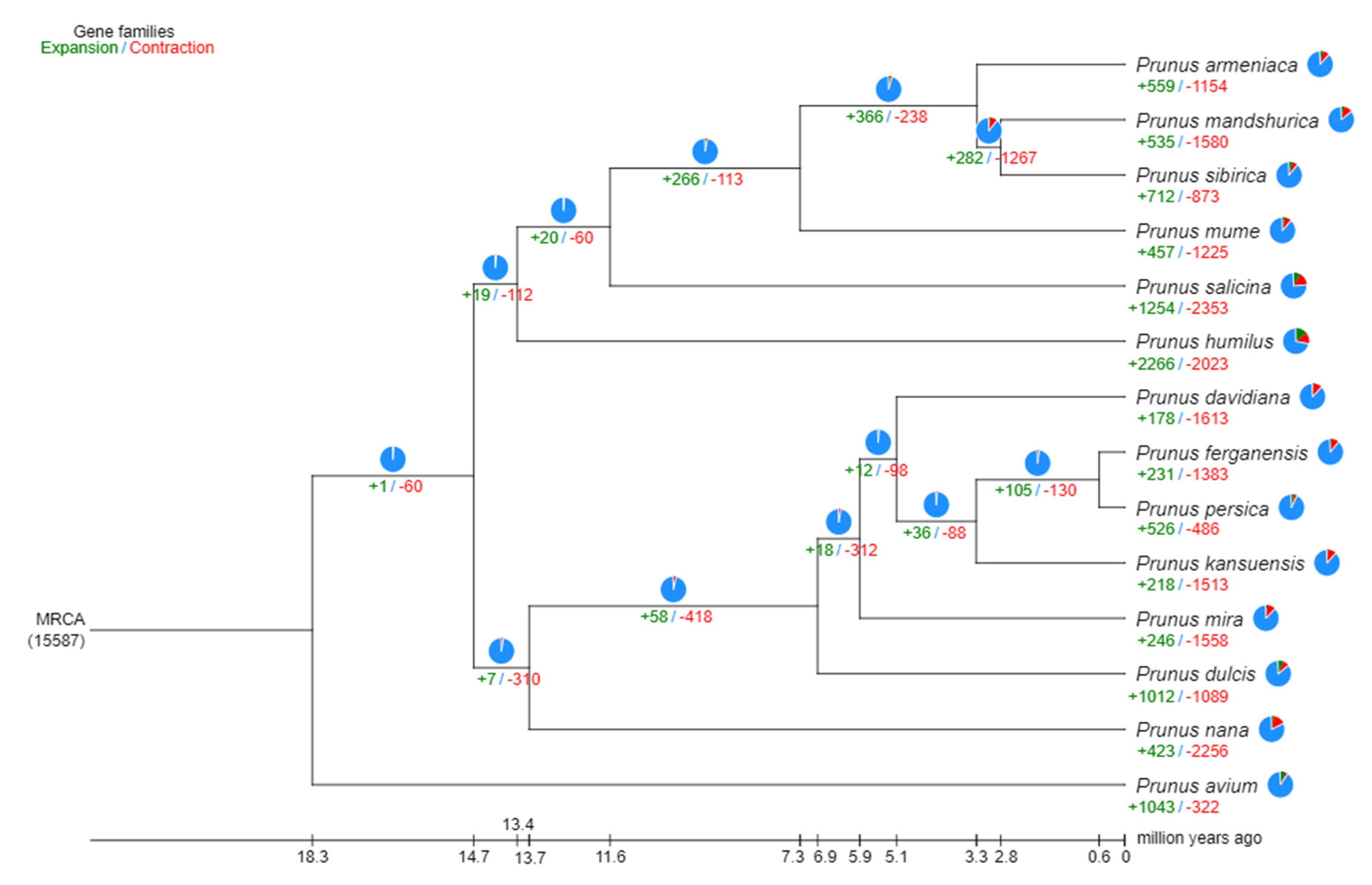
Figure 5.
Genome structure map. The grey area is the collinear relationship area, yellow is the inversion area, green is the translocation area, and blue is the area where reduplication occurs.
Figure 5.
Genome structure map. The grey area is the collinear relationship area, yellow is the inversion area, green is the translocation area, and blue is the area where reduplication occurs.
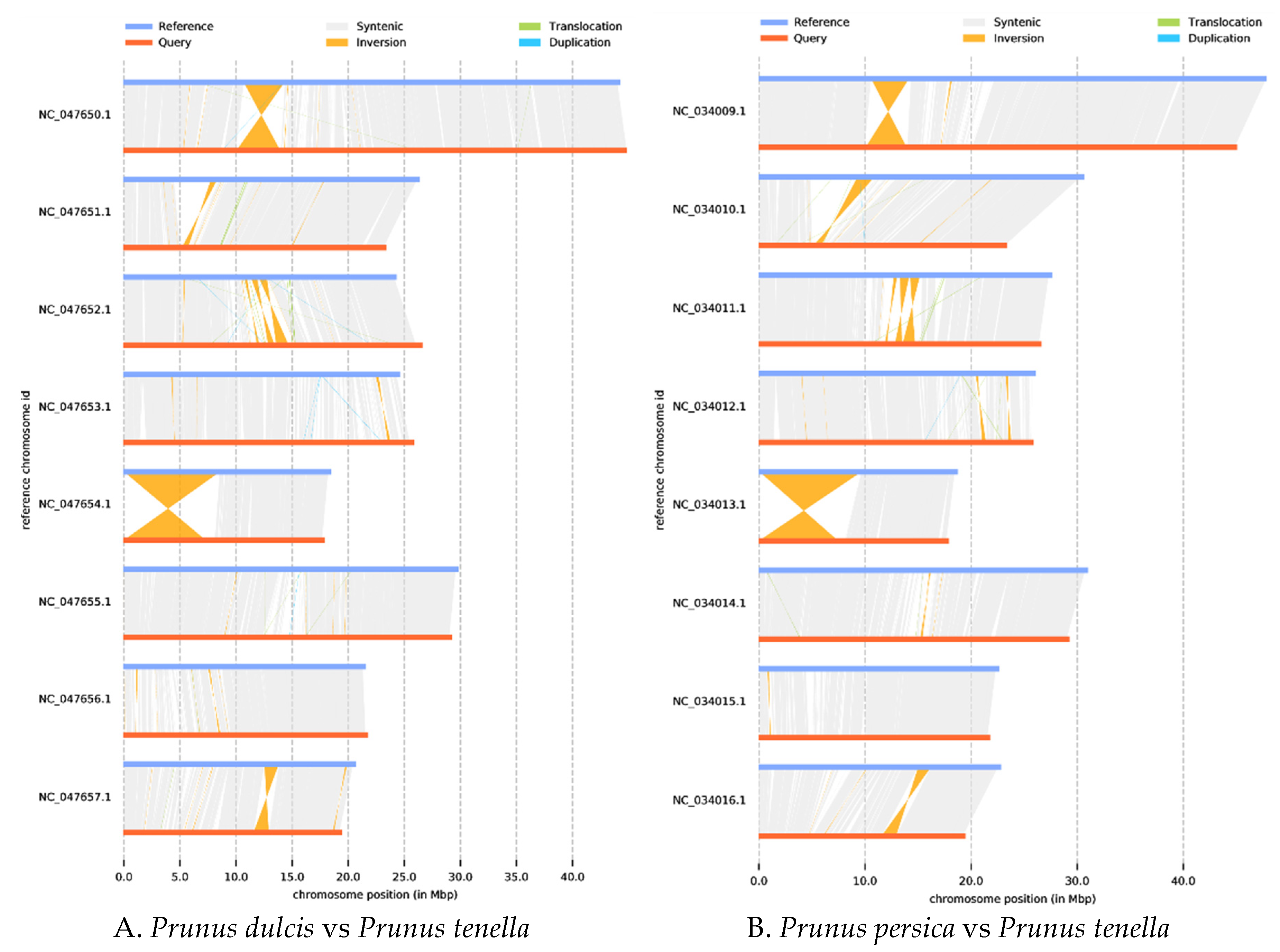
Figure 6.
Phylogeny and population structure of different populations. (A) phylogenetic tree; (B) PCA three-dimensional cluster diagram of samples; (C) Admixture sample clustering results corresponding to K values (3-8).
Figure 6.
Phylogeny and population structure of different populations. (A) phylogenetic tree; (B) PCA three-dimensional cluster diagram of samples; (C) Admixture sample clustering results corresponding to K values (3-8).
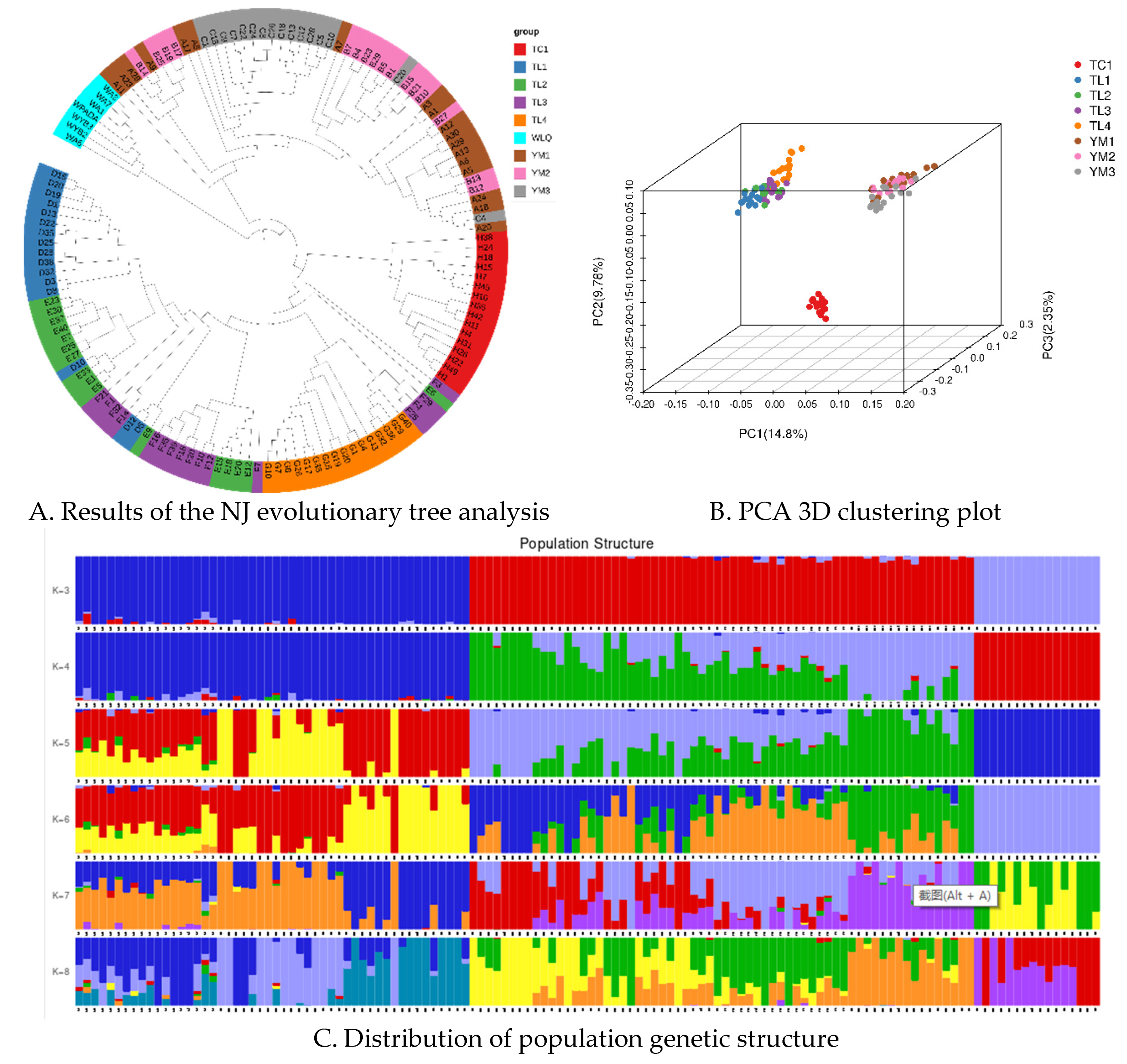
Figure 7.
Schematic of selection signatures between populations. A.)Selective sweep analysis of Tacheng Vs. Tuoli; B.) Selective sweep analysis of Tacheng Vs. Yumin; C.) Selective sweep analysis of Tuoli Vs. Yumin; The ratio of π in the abscissa and Fst in the ordinate corresponding to the frequency distribution diagram above and the frequency distribution diagram on the right, respectively. The dot plot in the middle represents the corresponding Fst and π ratios in different windows. The blue and red areas at the top are the top 5% of the region selected by π, the green area at the right is the top 5% of the region selected by Fst, and the blue and red areas in the middle are the intersection of Fst and π, which are the candidate sites.
Figure 7.
Schematic of selection signatures between populations. A.)Selective sweep analysis of Tacheng Vs. Tuoli; B.) Selective sweep analysis of Tacheng Vs. Yumin; C.) Selective sweep analysis of Tuoli Vs. Yumin; The ratio of π in the abscissa and Fst in the ordinate corresponding to the frequency distribution diagram above and the frequency distribution diagram on the right, respectively. The dot plot in the middle represents the corresponding Fst and π ratios in different windows. The blue and red areas at the top are the top 5% of the region selected by π, the green area at the right is the top 5% of the region selected by Fst, and the blue and red areas in the middle are the intersection of Fst and π, which are the candidate sites.
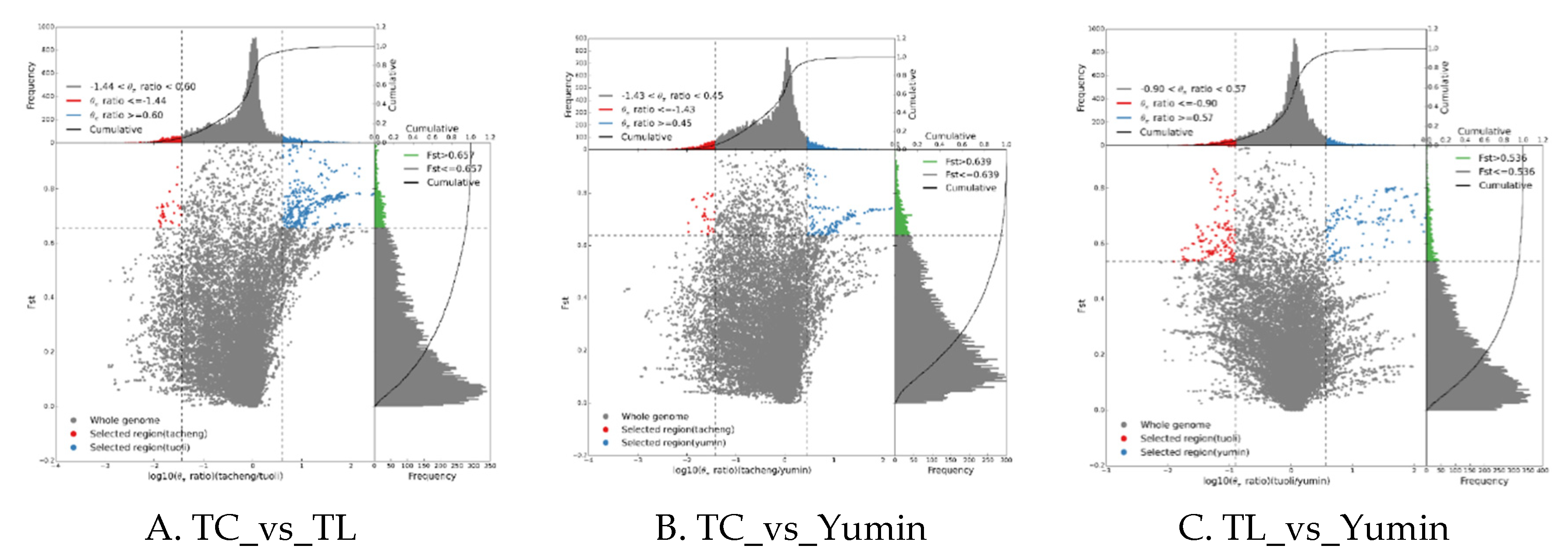

Table 1.
Statistical findings of the assembled Prunus tenella genome.
| Term | Contig number | Contig size (bp) | Scaffold number | Scaffold size (bp) |
| N90 | 5 | 270515 | 9 | 1233122 |
| N80 | 12 | 8938573 | 7 | 19226261 |
| N70 | 11 | 10302541 | 6 | 21538263 |
| N60 | 15 | 14010243 | 5 | 23141640 |
| N50 | 1 | 18100976 | 4 | 25637364 |
| Max length (bp) | 35886393 | 44825466 | ||
| Total size (bp) | 231191648 | 231208648 | ||
| Total number | 513 | 479 | ||
| Average length | 450665.98 | 482690.29 | ||
| Number >= 10kb | 513 | 479 |
Table 2.
Statistical findings of chromosomal level assembly of Prunus tenella.
| Chr ID | Length (bp) |
| Chr1 | 44825466 |
| Chr2 | 29024987 |
| Chr3 | 26387986 |
| Chr4 | 25637364 |
| Chr5 | 23141640 |
| Chr6 | 21538263 |
| Chr7 | 19226261 |
| Chr8 | 17664456 |
| Total chromosome level contig length | 207446423 |
| Total contig length | 231208648 |
| Chromosome length/Total length | 89.7% |
Table 3.
Findings of Prunus tenella genome intgerality assessment by BUSCO.
| Library | eudicotyledons_odb10 |
| Fragmented BUSCOs (F) | 48 |
| Missing BUSCOs (M) | 64 |
| Complete and duplicated BUSCOs (D) | 42 |
| Complete and single-copy BUSCOs (S) | 1967 |
| Complete BUSCOs (C) | 2009 |
| Total BUSCO groups searched | 2121 |
| Summary | 94.7% |
Table 4.
Statistical findings of repeated sequences TE annotations in Prunus tenella genome.
| Class | Length (bp) | Type | Sub-Class | (%) |
| retrotransposons | 8454829 | Ty1/Copia | LTR | 3.66% |
| 18247107 | Ty3/Gypsy | 7.89% | ||
| 15756928 | unknown | 6.82% | ||
| - | LINE | Non-LTR | - | |
| - | unknown | - | ||
| DNA transposons | 1845596 | CACTA | TIR | 0.80% |
| 7818414 | Mutator | 3.38% | ||
| 4181524 | PIF/Harbinger | 1.81% | ||
| 278071 | Tc1/Mariner | 0.12% | ||
| 3345229 | hAT | 1.45% | ||
| 7038992 | helitron | Non-TIR | 3.04% | |
| Total | 66966690 | 28.97% |
Table 5.
Protein-coding genes-related functional annotations in P. tenella genome.
| Database | Gene numbers | (%) |
| GO | 10761 | 33.54 |
| KEGG | 11435 | 35.64 |
| KOG | 19251 | 59.99 |
| Swissprot | 19449 | 60.61 |
| Pfam annotation | 22815 | 71.1 |
| Nr annotation | 31202 | 97.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



