Submitted:
05 August 2025
Posted:
20 August 2025
You are already at the latest version
Abstract
The airway epithelial barrier (AEB) is a dynamic interface that maintains respiratory homeostasis. Complex networks of epithelial cells, intercellular junctions, and immune constituents support the structural and functional integrity of the AEB. This review synthesizes how the respiratory exposome components disrupt AEB physiology by compromising junctional integrity, triggering oxidative stress, and inducing inflammation. The review further analyzes how these perturbations lead to maladaptive responses in chronic respiratory diseases (CRDs) and the effectiveness of emerging biologics targeting epithelial-derived alarmins in treating CRDs. By integrating exposome science with epithelial physiology, we provide a unified framework for understanding environmental impacts on airway health.
Keywords:
airway epithelial barrier
; respiratory exposome
; chronic respiratory diseases
; precision medicine
1. Introduction
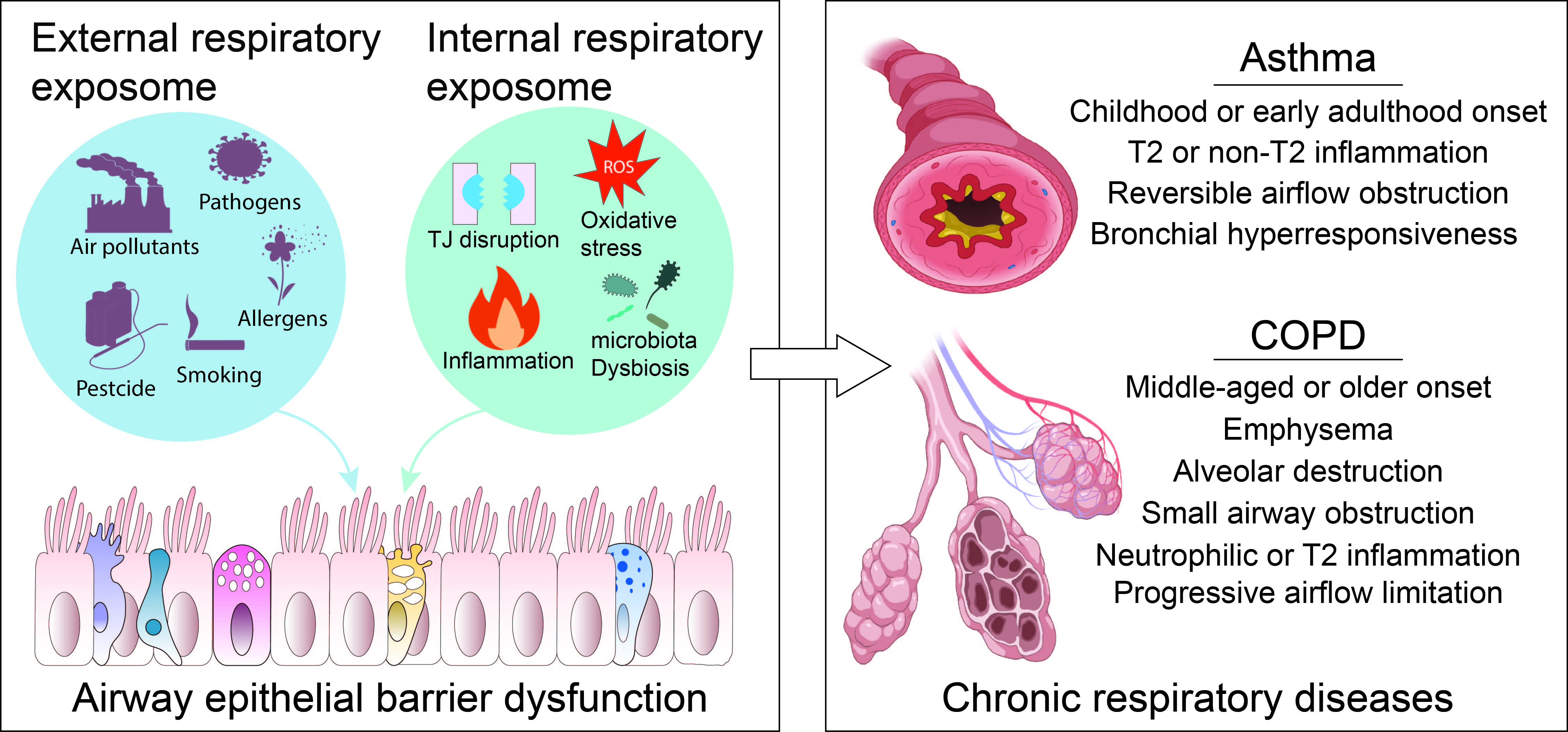
Chronic respiratory diseases (CRDs), such as asthma, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), and cystic fibrosis (CF), represent a significant and growing global health challenge (GBD Chronic Respiratory Disease Collaborators, 2020). With a frequency of 454.6 million cases and almost 4 million fatalities annually, CRDs were the third most common cause of death globally in 2019. This represents increases of 28.5% and 39.8%, respectively, in total fatalities and prevalence when compared to 1990, although the age-standardized rates decrease by 41.7% and 16.9%, respectively, during this period (GBD Chronic Respiratory Disease Collaborators, 2023). Asthma is the most prevalent chronic respiratory disease, whereas COPD is the leading cause of death among CRDs (H. Feng et al., 2025).
Both genetic and environmental factors play a significant role in the occurrence of CRDs (Sayers et al., 2024), whereas new insights into how environmental factors contribute to the development of CRDs are emerging. With industrialization, urbanization, and modernization, the respiratory system is increasingly exposed to a broader range of harmful substances. As a wider array of risk factors is being uncovered, the demand for thorough analysis of these environmental influences continues to grow. The term “exposome” was introduced by Christopher Wild, a British cancer epidemiologist, in 2005. It serves as a vital concept to emphasize the need for balancing research between genetic drivers of health and the non-genetic factors that affect us throughout our lives (Wild, 2005). Adverse environmental exposure is a significant cause of the development of CRDs. To explore the connection between the exposome and CRDs, we define the respiratory exposome as the sum of all environmental elements to which the airway is exposed. Then, we examine how these factors set off the CRDs.
The airway is divided into the upper airway and the lower airway. The upper airway includes the nasal cavity, the pharynx, and the larynx. The lower airway is dominated by the tracheobronchial tree structure, which encompasses the trachea, bronchi, and bronchioles. The airway can be divided into the transducing zone (from the nasal cavity to the bronchioles) and the respiratory zone (the alveoli) (Davis and Wypych, 2021). The forefront of this system lies the epithelium, a continuous cellular lining that extends from the trachea to the alveoli, forming a critical interface between the external and internal environments. The cellular organization and the appended molecular apparatus of this interface constitute a barrier against the external environment, and the integrity and functionality of this barrier are paramount, serving as a primary defense against inhaled pathogens and environmental insults, thereby maintaining overall respiratory health (Ganesan et al., 2013; Knight and Holgate, 2003). Disruptions in epithelial structure or function, however, can initiate a cascade of pathological events, leading to both localized lung diseases and systemic manifestations, a testament to the epithelium’s far-reaching influence.
The review will dissect the delicate and complex structure of the airway epithelial barrier (AEB), including the highly heterogeneous cellular components, and the intercellular junctions that regulate the permeability of the barrier. It will also discuss the respiratory exposome factors that cause pulmonary epithelium injury and the mechanisms that underlie the related diseases, such as asthma and COPD. Finally, we will discuss how respiratory exposome research is implicated in precision medicine for CRDs.
2. AEB Structure
2.1. Airway Epithelial Cells (AECs) Landscape
In human and mouse airways, there are multiple types of epithelial cells, including ciliated cells, goblet cells, club cells, Tuft cells, ionocytes, neuroendocrine (NE) cells, mucous cell, serous cells, myoepithelial cells, and basal cells (Hogan and Tata, 2019) (Figure 1A). The initial identification and classification of AECs relied heavily on traditional histological techniques, primarily various forms of microscopy and staining protocols, which lead to the discovery of the ciliated cells, the goblet cells, and the club cells. However, the advent of single-cell RNA sequencing (scRNA-seq) has revolutionized the discovery of rare and functionally unique cell types, such as Tuft cells, ionocytes, and neuroendocrine (NE) cells (Travaglini et al., 2020).
Although the airway epithelium was first mentioned in 1600, it wasn’t until 1834 that the presence of cilia was documented. Later research has shown that the ciliated cells are essential for pushing mucus along the airways (Widdicombe, 2019a). Ciliated cells are equipped with hairlike projections called cilia, and drive mucociliary clearance by moving mucus and trapped particles out of the airways using their motile cilia. The airway cilia also express Chemosensory receptors. They sense the bitter compounds and trigger intracellular signals that increase the ciliary beat frequency (Shah et al., 2009).
Goblet cells, characterized by their distinct morphology resembling a drinking goblet, serve as primary secretory cells within the superficial epithelium of the large airways, where they synthesize and secrete mucin glycoproteins that form essential components of the protective airway surface fluid (Cortez and Schultz-Cherry, 2021).
Club cells, initially described by Rudolph Albert von Kölliker in 1881 (Blackburn et al., 2023), are non-ciliated, non-mucous secretory cells that display a characteristic cuboidal shape with a distinctive bulging surface facing the bronchiolar lumen. These cells actively participate in the biotransformation of numerous harmful compounds introduced through inhaled air, including furan, hydrocarbons, naphthalene, ozone, and various components of tobacco smoke, thereby serving a detoxification function (Rokicki et al., 2016). They secrete multiple proteins, including the well-characterized CC16 protein, which demonstrates significant anti-inflammatory and immunomodulatory properties (Almuntashiri et al., 2020). Club cells can transdifferentiate into ciliated cells, a process that involves a critical regulatory mechanism through the inhibition of Notch signaling pathways, and plays a key role in airway epithelial regeneration and repair after injury (Bray and Bigas, 2025). Club cells also serve as precursors for goblet cells, and the differentiation process from club cells to goblet cells requires the expression of several transcription factors, notably SPDEF, which can be influenced by environmental stimuli, such as allergens and inflammatory cytokines (e.g., IL-13) (Cumplido-Laso et al., 2023).
Tuft cells, another rare but distinctive lineage of epithelial cells, were first discovered in rats in 1956 (Rhodin and Dalhamn, 1956) and in the human trachea in 1959 (J. Rhodin, 1959). Tuft cells have a bottle-like shape and are named for their distinctive apical “tuft-like” microvilli extending into the luminal space of the airway. These cells are chemosensory sentinels and play a role in mediating type II inflammation by producing IL-25 and cysteinyl leukotrienes (Barr et al., 2022).
The existence of pulmonary ionocytes was first suggested nearly three decades ago by Engelhardt and colleagues while surveying airway epithelial cells with cystic fibrosis (CF) transmembrane conductance regulator (CFTR), a chloride channel mutated in CF (Engelhardt et al., 1992). CF is a muco-obstructive lung disease characterized by pulmonary ionocytes that express high levels of CFTR and other ion channels and transporters, such as epithelial sodium channel (ENaC), V-ATPase proton pump, barttin/Cl- channels, and anion exchanger protein 2 (AE2), forming a sophisticated molecular machinery for ion transport regulation (Lei et al., 2023; Okuda and Gentzsch, 2024). One of the most significant functions of these cells is the regulation of airway surface liquid (ASL) pH through a complex interplay of ion transport mechanisms (Luan et al., 2024; Vila-Gonzalez et al., 2024). Although rare ionocytes exhibit the highest levels of CFTR expression, club cells comprise the majority of CFTR-expressing cells, albeit with lower levels per individual cell. While ionocytes regulate ASL pH, club cells modulate ASL volumes and hydration, both of which are linked to airway mucus dysfunctions and need to be targeted in CF therapy (Vila-Gonzalez et al., 2024).
The pulmonary neuroendocrine cells (PNECs) were discovered by Fröhlich, who termed them ‘helle zellen’ or ‘bright cells’ in German, as being responsible for detecting harmful agents (Thakur et al., 2024). This cell type displays a distinct architectural organization within the airways, appearing either as solitary cells or forming specialized clusters known as neuroepithelial bodies (NEBs). The sophisticated innervation of NEBs, involving both afferent and efferent nerve fibers, establishes a complex neural network that enables rapid communication between the airway environment and the central nervous system. The neural architecture, along with the expression of specialized molecular machinery, renders PNECs capable of detecting both chemical and mechanical environmental changes (Noguchi et al., 2020).
Airway basal cells, anchored to the basal lamina via hemidesmosomes, are a sophisticated population of stem cells. Basal cells are remarkably regenerative and can differentiate into virtually every type of cell in a pseudostratified epithelium under steady-state conditions and after acute injury within the airways (Bray and Bigas, 2025). Beyond their stem cell functions, basal cells emerge as crucial players in pulmonary host defense and immune regulation. When exposed to Haemophilus influenzae, these cells demonstrate their immunological prowess by upregulating antimicrobial proteins, such as RNase7, and producing pro-inflammatory cytokines, including IL-6 and IL-8 (Ruysseveldt et al., 2021).
Mucous cells, serous cells, and myoepithelial cells constitute the airway submucosal glands (SMGs). The earliest mentions of airway glands can be traced back to 1602, but it was not until the end of the 19th century that the major histological features of airway submucosal glands were described (Widdicombe, 2019b). Mucous cells are primarily responsible for mucin production, and serous cells focus on the secretion of antimicrobial proteins and watery fluids (Di and Mou, 2024). In most parts of the SMG acini, the strategic intermingling of serous and mucous cells suggests a coordinated effort in producing mucus with optimal properties. While serous cells occasionally form clusters at the distal part of an acinus, areas solely populated by mucous cells are not typically observed (Yu et al., 2022). Myoepithelial cells are found enveloping the SMG acini, situated around the secretory units of exocrine glands, and between the basal lamina and secretory cells. Myoepithelial cells are less exposed to environmental hazards and represent a stem cell reservoir that can repair severely injured epithelium by differentiating into other types of epithelial cells (Duval et al., 2018). During injury, the SMGs’ basal myoepithelial cells can self-renew and differentiate into mucous cells and serous cells, and they can also differentiate back into basal cells (Lynch et al., 2018).
Two types of epithelial cells contribute to the alveoli architecture: the Type 1 alveolar epithelial cells (AEC1 or AT1) and the Type 2 alveolar epithelial cells (AEC2 or AT2). AT1 exhibits a distinctive squamous morphology, characterized by its thin, flattened structure, and emerges as the predominant cellular constituent, covering an impressive 96% of the internal surface of each alveolus. AT2 cuboidal morphology and specialized secretory capabilities. AT1 demonstrates three primary functions: facilitating gas exchange, maintaining ion and fluid balance within the alveoli, and engaging in sophisticated communication with AT2 to coordinate surfactant secretion in response to mechanical stimuli. AT2 produces and secretes pulmonary surfactant, expressing immunomodulatory proteins necessary for host defense, facilitating transepithelial water movement, and serving as progenitor cells capable of self-renewal and regenerating the AT2 (Joo et al., 2024). In human respiratory bronchioles, a new type of secretory cell, which expresses SCGB1A1 and SCGB3A2, is called the respiratory airway secretory cell (RASC). RASCs are a lung progenitor population that can unidirectionally differentiate into AT2, a process controlled by Notch and Wnt signaling (Basil et al., 2022).
2.1. Intercellular Junction Complexes
The lung is a complex organ characterized by an intricate network of epithelial cells that perform vital functions in gas exchange and maintaining lung integrity. Among the structural components of these epithelial cells are four primary types of intercellular junctions: tight junctions (TJs), adherens junctions (AJs), desmosomes, and gap Junctions (GJs) (Figure 1B) (Fu et al., 2022; Kageyama et al., 2024).
TJs, also known as zonula occludens, are multiprotein complexes that seal the paracellular space and function as a selective diffusion barrier, regulating the paracellular passage of ions, water, and various molecules across tissues. They are localized in the apicolateral regions of pulmonary epithelial cells and have a “fence function,” which prevents the mixing of membrane proteins and lipids between the apical and basolateral domains of the cell membrane. This helps maintain cellular polarity, which is essential for the proper functioning of epithelial cells (Farquhar and Palade, 1963). AJs, or zonula adherens, are multiprotein architectures predominantly found in the apical region of epithelial cells and are closely associated with TJs (Naser et al., 2023; Wittekindt, 2017). AJs are essential adhesive contacts between neighboring epithelial cells, initiating cell-cell contacts and mediating their maturation and maintenance. They link the actin cytoskeleton of one cell to that of an adjacent cell, enabling groups of cells to function as robust structural units (Campas et al., 2024). Desmosomes, also known as macula adherens, are localized at the midpoints of neighboring epithelial cells (Zimmer and Kowalczyk, 2024). It provides strong adhesion between cells, preventing them from tearing apart. They mechanically integrate adjacent cells by coupling adhesive interactions mediated by desmosomal cadherins (desmogleins and desmocollins) to the intermediate filament network (Perl et al., 2024). Gap junctions are membrane channels that facilitate direct chemical and electrical communication between adjacent cells, allowing the passage of small molecules, ions, and second messengers. GJs facilitate direct electrical coupling and metabolic exchange between cells, enabling rapid and coordinated cellular responses (Jagielnicki et al., 2024).
TJs are the primary intercellular junctions responsible for regulating the intercellular permeability and the barrier function of the airway epithelium (Otani and Furuse, 2020). TJs are referred to as zonula occludens because they form a thin, continuous circumferential belt that seals the intercellular space. In freeze-fracture electron microscopy (FFEM) micrographs, AJs are shown as a circumferential network of TJ strands or fibrils, which correspond to the focal sites of intimate apposition of outer plasma membrane leaflets in the transmission electron microscopy (TEM) images. The TJ strands are now defined as being formed by Claudin family proteins. The claudin family in mammals comprises 27 protein members, which share a structural topology of tetraspan membrane segments, a large extracellular loop containing a consensus sequence motif, and a second, shorter extracellular loop (Gunzel and Yu, 2013).
The TJ complex contains other essential components, such as TJ-associated MARVEL proteins (TAMPs) and junctional adhesion molecules (JAMs). The TAMP family includes Occludin, Tricellulin, and MarvelD3 proteins. Occludin and Tricellulin were shown to jointly contribute to TJ strand branching point formation and epithelial barrier maintenance, but marvelD3 was not associated with the morphology of TJ strands and barrier function in MDCK II (Saito et al., 2022). Occludin is a ~65 kDa integral membrane protein. Like Claudin, it also has four transmembrane domains and two extracellular regions. Evidence showed copolymerization of Occludin with Claudin(s) for proper stabilization of TJ strands (Cummins, 2012; Furuse et al., 1998). JAMs, including JAM-A, -B, and C, belong to the immunoglobulin (Ig) superfamily and are found to be localized in TJs. JAM-1 plays a role in maintaining the integrity of TJ morphology and barrier function (Otani et al., 2019).
Beneath the membrane of TJs, there is a plaque constituted by scaffold proteins, including ZO-1, ZO-2, ZO-3, MUPP1, MAGI-1, etc. (Gonzalez-Mariscal et al., 2003). These proteins contain specialized domains, including PDZ, Src homology 3 (SH3), and guanylate kinase domains, which facilitate protein-protein interactions and signal transduction (Godbole et al., 2022). ZO-1 undergoes liquid–liquid phase separation (LLPS) during TJ formation, which is regulated by multimerization, multivalent interactions, mechanical force, and dephosphorylation of ZO-1. Several additional TJ scaffolding and adaptor proteins are predicted to undergo LLPS based on the presence of intrinsically disordered domains (Sun et al., 2022). ZO-1 contains an actin-binding region that interacts with branched actin filaments so that it connects the TJ transmembrane proteins to a branched actin filament network (Citi et al., 2024).
TJs and AJs are intimately connected spatially and functionally, and are collectively referred to as the apical junctional complexes (AJCs) (Jang, 2014; Naser et al., 2023). Both TJs and AJs form continuous circumferential belts that encircle the apical regions of epithelial cells, working together to integrate individual cells into a cohesive tissue and establish barrier function. The AJ is positioned immediately below the TJ and provides the structural integrity necessary for cell-cell adhesion, a prerequisite for the formation and function of TJs. While AJs maintain close cell apposition, they cannot form a seal, a function provided by TJs. AJs are located immediately below TJs and are associated with E-cadherin (CDH1) through α-catenin/β-catenin heterodimers (Arbore et al., 2022), which in turn connect to actin filaments (F-actins). Nectins are also crucial components of AJs. They are transmembrane proteins that, along with cadherins, form the adhesive domain of the cells. Their ectodomains facilitate adhesion, while their cytosolic regions connect the adhesive contact to the F-actin cytoskeleton via the adaptors Afadin and PLEKHA7 (Troyanovsky, 2023). The F-actins, combined with Myosin-2 proteins, form a continuous circumferential actomyosin cytoskeleton belt beneath the cytoplasmic membrane, which plays a fundamental role in the architecture and assembly of AJs. The actomyosin belt connects to the AJs through a network of branched actin filaments, which act as a linker and force transducer to ZO-1 and TJ transmembrane proteins (Figure 1C).
Structural biology has contributed a lot to the understanding of the architecture of the AJCs (Figure 1D). Crystal structure of mouse Claudin-15 (mCldn15) showed that the four transmembrane (TM1 to TM4) segments form a typical left-handed four-helix bundle, and large portions of the two extracellular segments form a prominent β-sheet structure. The two extracellular segments are referred to as extracellular strand 1 and 2 (ECS1 and ECS2). At the immediate downstream of ECS1, there is a short extracellular helix (ECH). In the crystal lattice, the protomers of mCldn15 constitute side-by-side interactions and form a linear polymer. The linear arrangement is maintained by a conserved hydrophobic residue (Met68) that protrudes from the ECH in one protomer and fits into a hydrophobic pocket comprising TM3 and ECS2 (Phe146, Phe147, and Leu158) of the adjacent protomer. It might present the linearly arranged TJs strands observed in FFEM. Claudins facilitate cell adhesion by head-to-head interactions between claudins in adjoining cell membranes. This could be mediated by the two variable loop regions (V1 and V2) located at ECS1 and ECS2, respectively (Suzuki et al., 2014).
JAMs, including JAM-A, -B, and C, belong to the immunoglobulin (Ig) superfamily and are found to be localized in TJs. JAM-1 plays a role in maintaining the integrity of TJ morphology and barrier function [52]. In the crystal structure of the soluble extracellular part of JAM-A, two protomers form a U-shaped dimer via the interactions between the N-terminal Ig domains from each protomer. This structure provided a model of homophilic interactions of JAM-A to explain its adhesive function (Kostrewa et al., 2001; Otani et al., 2019).
In the crystal structure, C-cadherins have five extracellular cadherin-like domains (EC1 to EC5), where the membrane-distal end (EC1 domain) is approximately perpendicular to the membrane-proximal end (EC5 domain). EC1 of two cadherin molecules from the juxtaposed cells form the “strand dimer” interface. A twofold symmetric exchange of the N-terminal beta strands between the EC1 domains of adjoining molecules defines this interaction. This strand dimer is oriented to mediate the adhesion between cadherins presented from adjacent cells, implying its role in trans interactions. The C-cadherin crystal structure also reveals a distinct “cis-oriented” interface that is proposed to mediate interactions between cadherin molecules on the same cell surface. These molecules are arranged in a parallel, front-to-back fashion, forming a continuous line of molecules. This type of cis interaction is believed to be necessary for cadherin adhesive function, likely by increasing avidity through molecular clustering (Boggon et al., 2002).
Dimerization is a fundamental step in mediating Nectin adhesive functions, both on the surface of the same cell and between neighboring cells. Nectin molecules first dimerize in cis on the surface of the same cell. This cis-dimerization is a prerequisite for subsequent trans-dimerization. The crystal structure of the extracellular region of nectin-1 revealed that it forms a V-shaped homophilic dimer through its first immunoglobulin (Ig)-like domain, representing a cis-dimer. Following cis-dimer formation, these cis-dimers then undergo trans-dimerization between neighboring cells (Narita et al., 2011).
3. AEB Functions
3.1. Physical Barrier
The airway epithelium barrier represents a sophisticated biological interface that serves as the primary defense mechanism against environmental challenges while maintaining essential respiratory functions. As a physical barrier, the airway epithelium produces mucus primarily through goblet cells and submucosal glands, creating a protective layer that traps inhaled pathogens, particles, and allergens. This mucus contains mucins, high-molecular-weight glycoproteins such as MUC5AC and MUC5B, which form a viscoelastic barrier essential for airway protection. Tight junctions and adherens junctions at the apicolateral border of epithelial cells form a selective physical barrier that controls paracellular permeability. These junctions prevent the penetration of inhaled pathogens and environmental toxins into deeper tissues while maintaining cell polarity and structural integrity (Carlier et al., 2021). The airway epithelium is lined with ciliated cells, each bearing approximately 200 to 300 motile cilia that beat at frequencies of 20–25 Hz. This coordinated beating propels the mucus, along with trapped particles and microbes, away from the lungs toward the throat for expulsion. This mucociliary escalator is a critical defense mechanism that works synergistically with mucus production to maintain airway cleanliness and prevent infection. Respiratory cilia beat continuously in a coordinated metachronous fashion, with a rapid forward power stroke followed by a slower recovery stroke, generating effective mucus transport along the airway surfaces. This complex ultrastructure of respiratory cilia enables their essential function in airway defense by efficiently moving mucus and trapped particles out of the respiratory tract.
Respiratory cilia are microtubule-based organelles approximately 6–7 μm long and 0.2-0.3 μm in diameter that extend from the apical membranes of airway ciliated cells (Horton et al., 2025; Kuek and Lee, 2020). They are motile cilia, and the core structure of each cilium is the axoneme, which features a “9 + 2” microtubule arrangement: nine outer doublet microtubule protofilaments arranged in a ring surrounding two central single microtubules. The axoneme arises from basal bodies located just beneath the cell membrane on the apical surface of AECs. Basal bodies anchor the cilium and initiate its growth. They consist of triplet microtubules that transition into the doublets of the axoneme (Greenan et al., 2020).
The fundamental structural units of the ciliary axoneme are dynein-decorated doublet microtubules (DMTs), the microtubule-based molecular machine that powers the rhythmic beating of motile cilia. Each DMT is composed of two distinct parts: a complete A-tubule and a partial B-tubule. The A- and B-tubules are made of 13 and 10 protofilaments that spread along the extension direction of the DMT, respectively. The protofilaments consist of ⍺- and β-tubulin isoforms. The external surface of the DMT is patterned by a 96-nm repeating unit. This repeat is defined by the regular positioning of key axonemal complexes, including outer dynein arms (ODAs), inner dynein arms (IDAs), radial spokes (RSs), and the nexin–dynein regulatory complex (N-DRC). These complexes attach to the A-tubule of the DMT or link adjacent DMTs. The N-DRC links neighboring DMTs, the ODAs project from the A microtubule, and the RSs project from the A-tubule. The luminal surfaces of DMTs are patterned by microtubule inner proteins (MIPs), which have an overall 48 nm periodic architecture. Of the 29 MIPs identified in bovine DMTs, four tektins and a tektin-interacting protein, TEKTIP1 (C19orf71), form a bundle of helical tektin filaments within the A-tubule lumen. The tektin bundle likely stabilizes DMTs to withstand waveform-dependent mechanical forces (Gui et al., 2021).
ODAs are motor proteins that use ATP hydrolysis to generate mechanical force for ciliary beating. These dynein arms cause sliding between adjacent microtubule doublets, resulting in ciliary motion that is necessary to propel mucus (Walton et al., 2023). Respiratory cilia perform efficient mucus transport along the airway surfaces by beating constantly in a coordinated metachronous fashion, with a fast forward power stroke followed by a delayed recovery stroke (Jing et al., 2017).
3.2. Chemical Barrier
The pulmonary epithelium isn’t merely a passive physical barrier; it actively secretes a variety of substances that contribute to the chemical defense of the lungs. These secretions form a complex milieu that neutralizes harmful agents and maintains a healthy environment in the airway. The AECs produce a variety of antimicrobial proteins and peptides (AMPPs), including human β-defensins (HBDs), cathelicidins (hCAP18/LL-37), PLUNC (palate, lung, and nasal epithelium clone) family proteins, lactoferrin, Lysozymes, secretory leukocyte protease inhibitor (SLPI), surfactant proteins (SP-A and SP-D). These AMPPs exhibit antimicrobial activity against viral, fungal, bacterial, or protozoan pathogens, playing a pivotal role in host defense (Di et al., 2024). The epithelial cells also produce antioxidants, which combat oxidative stress caused by inhaled pollutants and inflammatory processes (Huff et al., 2019).
3.3. Immunological Barrier
The airway epithelium is an immunological barrier. It is equipped with an array of pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and Nod-like receptors (NLRs) (2021). These receptors act as sentinels, constantly surveilling the airways for the presence of pathogen-associated molecular patterns (PAMPs) from invading microorganisms and damage-associated molecular patterns (DAMPs) released from injured or stressed cells. Upon detecting these danger signals, epithelial cells initiate a cascade of intracellular signaling pathways, leading to the production and release of various inflammatory mediators, including cytokines and chemokines (2022). These mediators, in turn, recruit and activate immune cells, orchestrating a coordinated immune response that eliminates the threat and restores tissue homeostasis. This early response is crucial for modulating subsequent immune activity (Burgoyne et al., 2021).
The crosstalk between resident and recruited immune cells is crucial for maintaining immune homeostasis in the airways (Hewitt and Lloyd, 2021). Airway macrophages, especially alveolar macrophages (AMs), are the primary immune cells in the lung at steady state, primarily detecting and reacting to inhaled pollutants and pathogens, and thus play a role in causing both protective and pro-inflammatory reactions (Y. Feng et al., 2025). AECs maintain AMs in a quiescent state by expressing anti-inflammatory membrane proteins such as CD200. They significantly inhibit lipopolysaccharide (LPS)-induced pro-inflammatory responses in macrophages and increase the secretion of TGF-β and IL-10 from macrophages even without LPS stimulation. This suggests that AECs induce tolerance in macrophages (Bissonnette et al., 2020).
Pulmonary DCs, located beneath the AECs, are essential for initiating effective immune responses to harmful pathogens while maintaining tolerance against harmless antigens (Yoshida et al., 2025). Under homeostatic conditions, AECs secrete factors such as retinoic acid (RA), Transforming Growth Factor-beta (TGF-β), and granulocyte macrophage colony-stimulating factor (GM-CSF), which render DCs tolerogenic and help prevent inflammation in the airways. RA, in particular, is crucial for maintaining mucosal tolerance by acting on DCs to induce Foxp3+ regulatory T cells (Conrad et al., 2025). On the other hand, AECs enhance the immune surveillance capacity of myeloid DCs (mDCs) through upregulation of PRRs and downstream signaling molecules in mDCs, enabling them to react to danger signals and enhance leukocyte recruitment (Agrawal et al., 2017).
Resident T cells, particularly intraepithelial γδ T cells, play pivotal roles in maintaining homeostasis in barrier tissues, including the airway. Epithelial resident γδ T cells are essential mediators of homeostasis through the regulation of the AMPPs. Through crosstalk with neighboring epithelial cells, these T cells can also detect the presence of invading bacteria. Besides, γδ T cells are involved in epithelial tissue repair (Witherden and Havran, 2013). Distinct immune cell populations, including resident memory B cells (BRM) and T cells (TRM), can be identified in the human nasal and nasopharyngeal swab samples. These cells were stable over time for more than one year in healthy subjects. In subjects with SARS-CoV-2 breakthrough infections, local virus-specific BRM, plasma cells, germinal center B cells, and virus-specific memory CD4+ TRM, and CD8+ TRM were identified, with age-dependent upper airway immunological shifts observed (Ramirez et al., 2024). Therefore, the airway epithelium serves as a storage pool for resident adaptive immune cells, which can respond rapidly to reencountered pathogens.
4. The Respiratory Exposome
As previously discussed, the airway is constantly exposed to a complex mixture of environmental factors, collectively referred to as the respiratory exposome. Christopher Wild, who coined the concept of exposome, divided the exposome into three distinct and complementary research domains, which include general external (social-economic factors, the urban environment and climate factors, etc.); specific external (an individual’s direct regional environment, including exposure to chemicals, diet, physical activity, tobacco smoking and infections); and internal (referring to the internal biological processes, such as oxidative stress, inflammation, epigenetic changes, metabolism and the internal microbiome) (Vermeulen et al., 2020; Wild, 2012, 2005). Accordingly, the external respiratory exposome can be classified into the general external respiratory exposome (encompassing general external environmental factors associated with respiratory health) and the specific external respiratory exposome (comprising local external environmental components that impact the respiratory health of particular individuals). The internal respiratory exposome encompasses the entirety of internal biological processes triggered by the external respiratory exposome within the respiratory system. Given the importance of the airway epithelial barrier in the development of CRDs, the endogenous cellular and molecular processes occurring in the airway epithelium will be discussed here.
Examples of general external respiratory exposome components include climate conditions (He et al., 2023), urban/rural environment (Reyfman et al., 2021), built environment (Bole et al., 2024), housing conditions (Kovesi et al., 2022), and green space (Nieuwenhuijsen et al., 2024). The specific external exposome components encompass ambient and indoor pollutants, pesticides, detergents, macro- and nano-plastics, pathogens, and allergens. Many of these agents have been reported to disrupt the airway epithelial barrier (Table 1). The most discussed biological processes that are associated with AEB damage include TJ disruption, ciliary clearance movement abnormalities, oxidative stress, inflammation, microbiome dysbiosis, etc. These pathophysiological processes typically initiate the CRD development and belong to the internal respiratory exposome domain (Figure 2).
5. The AEB Damage Involved in the Development of CRDs
5.1. Asthma
Asthma is characterized by reversible airflow restriction, airway inflammation, and bronchial hyperresponsiveness. AEB dysfunction is a significant contributor to the development of asthma (Heijink et al., 2020). Environmental triggers of asthma include primarily inhaled dust mites, molds, pollen, smoke, and other airborne particulates (Pijnenburg and Nantanda, 2021). The detrimental exposures, especially during the vulnerable early life stage (Rackley and Stripp, 2012), cause structural compromise in the AEB, including disruption of AJC and detachment of ciliated cells, which in turn increases mucosal permeability, allowing more allergens and pathogens to traverse the epithelium into the submucosa, where they encounter immune cells (Calven et al., 2020; Gon and Hashimoto, 2018). Most asthma patients display an aberrant epithelial barrier, with remarkable loss of E-cadherin and claudin-18 that constitute AJCs (Heijink et al., 2007; Sweerus et al., 2017).
The epithelial barrier disruption in asthma is associated with evaluated oxidative stress due to various environmental factors (Table 1). The excess reactive oxygen species (ROS) produced in oxidative stress activates signaling pathways such as NF-κB and Src-family kinases, leading to downregulation and structural disorganization of junction proteins like ZO-1, Occludins, Claudins, and E-cadherin, and thus directly impairs the airway epithelial barrier by damaging AJC proteins via downregulating, decreasing transepithelial electrical resistance, and increasing permeability. Cytokines induced by oxidative stress, such as TNF-α and IL-1β, further promote junctional disruption and inflammation (Zhang et al., 2025).
The epithelial cells also respond to damaging environmental factors by releasing epithelial cytokines known as alarmins, including thymic stromal lymphopoietin (TSLP), interleukin (IL)-25, and IL-33. A newly discovered alarmin is TNF-like cytokine 1A (TL1A), a member of the TNF cytokine superfamily that is expressed by epithelial cells and binds to a trimeric receptor, DR3 (death receptor 3), expressed on a broad spectrum of immune and non-immune cells (Varricchi et al., 2025). These alarmins act on dendritic cells (DCs), group 2 innate lymphoid cells (ILC2s), and T cells, promoting the recruitment and activation of Th2 cells and other immune cells that produce Th2 cytokines such as IL-4, IL-5, and IL-13. These cytokines amplify eosinophilic inflammation, airway hyperresponsiveness, mucus hypersecretion, and airway remodeling, which are hallmarks of asthma. In asthma, ILC2s are particularly abundant in airway tissues and produce large quantities of IL-5 and IL-13 in response to alarmins, playing a key role in early and augmenting type 2 responses in the airway. Therefore, Th2 cells and ILC2s are the primary regulators of type 2 inflammation (Kato and Kita, 2025).
The Th2 cytokine-driven eosinophils release cytotoxic granule proteins, leukotrienes, and reactive oxygen species that contribute to tissue damage, airway remodeling, and mucus production (McBrien and Menzies-Gow, 2017). The Th2 cytokines also drive B cells to produce allergen-specific IgE, which binds with high affinity to FcεRI on mast cells and basophils. Allergen crosslinking of IgE bound to FcεRI triggers degranulation, releasing histamine, leukotrienes, prostaglandins, and cytokines that cause bronchoconstriction and mucus secretion (Owen, 2007).
IL-4, IL-13, TNF-α, and inflammatory mediators released by sensitized airway smooth muscle (ASM) contribute to airway hyperresponsiveness (AHR), characterized by an exaggerated narrowing response of the airways to stimuli. The AEB disruption exposes the airway smooth muscle to inhaled irritants and reduces epithelial-derived relaxing factors, thereby enhancing ASM contraction and AH [118]. Th2 cytokines IL-13 and IL-14 induce mucin gene expression (particularly MUC5AC) in goblet cells and cause mucus hypersecretion, contributing to airway obstruction, cough, and airway hyperresponsiveness, with increased MUC5AC secretion playing a dominant role (Evans et al., 2009).
In asthma, chronic airway inflammation triggers repeated tissue injury and repair, resulting in structural changes within the airways, a phenomenon known as airway remodeling. This is associated with a molecular process known as Epithelial-Mesenchymal Transition (EMT). The AECs stimulated with pollutants or allergens release transforming growth factor-beta (TGF-β) and epidermal growth factor (EGF). These factors promote EMT and perpetuate airway inflammation. Eosinophils, inflammatory cells prominent in asthma, also induce EMT by secreting TGF-β1, aggravating airway fibrosis and remodeling. EMT-related mechanisms are pivotal in the development of persistent airway remodeling in asthma, which limits respiratory function and exacerbates disease severity (Zhang et al., 2025).
ABE disruption also leads to alteration of the airway microbiome and microbiota dysbiosis. The airway harbors a consortium of microorganisms collectively known as the airway microbiome. The epithelial hypothesis posits that damage to the epithelial barrier allows microbiota typically located on the surface of the epithelium to translocate to the deeper layers beneath the epithelial cells. It stimulates the immune system, contributing to inflammatory processes. The epithelial damaged tissue is also characterized by microbial dysbiosis and transepithelial translocation of commensal microbes, as well as further colonization by opportunistic pathogens (Akdis, 2021). Firmicutes and Bacteroidetes are the predominant phyla in healthy lung microbiota, with Prevotella, Veillonella, and Streptococcus being the most common genera. The airway microbiota was found to differ in patients with asthma. Increased Proteobacteria were observed in asthmatic patients, and non-Proteobacteria taxa, such as Porphyromonas, Fusobacterium, and Sphingomonadaceae, were also elevated. A differential mycobiome was also evident according to asthma endotypes, with Fusarium, Cladosporium, and Aspergillus specifically enriched in T2-high asthma (refer to the following sections) (Yi et al., 2022).
5.2. COPD
COPD is one of the leading contributors to global mortality. It is a progressive condition of chronic bronchitis, small airway obstruction, and emphysema that represents a leading cause of death worldwide (Rao et al., 2020). It is characterized by the presence of persistent respiratory symptoms, structural abnormalities within the pulmonary system, and compromised lung function, which may manifest individually or in combination (Celli and Agusti, 2018). The primary risk factors of COPD are tobacco smoking, and smoke from biomass fuel used for cooking and heating are also risk factors (Agusti and Hogg, 2019; Christenson et al., 2022). In an umbrella review, smoking, ambient air pollution including nitrogen dioxide, a low BMI, indoor biomass burning, childhood asthma, and occupational dust exposure were identified as the exposome risk factors for COPD (Holtjer et al., 2023).
COPD bronchial epithelium showed lower Ciliary beat frequency (CBF) and higher dyskinesia index (DI), compared to healthy controls. It also exhibits a significant loss of ciliated cells, accompanied by shorter cilia and ultrastructural defects in the axoneme. Studies have consistently confirmed the presence of these defects across airway regions, from the nasal epithelium to the bronchioles, in patients with COPD and smokers. Furthermore, gene expression related to cilia structure and function, such as dynein arm proteins (DNAH5, DNAH9, DNAH11) and infraglabellar transport proteins (IFT43, IFT57, IFT144, IFT172), is reduced in COPD epithelial cells, exacerbating the dysfunction [127]. Additionally, bronchial epithelial cells from COPD patients exhibit an impaired ability to differentiate into ciliated cells, resulting in a sustained deficiency of functional cilia in the airway epithelium (Hedstrom et al., 2021; Hessel et al., 2014). These combined changes compromise mucociliary clearance, contributing to mucus retention, chronic infections, and airway inflammation characteristic of COPD (Thomas et al., 2021).
In COPD, chronic inhalation of harmful pollutants such as cigarette smoke (CS) and biomass smoke generates ROS and promotes oxidative stress in AECs. This oxidative burden leads to inflammation, the secretion of proinflammatory cytokines, altered epigenetic modifications, and premature epithelial cell senescence, resulting in loss of epithelial integrity and promoting barrier dysfunction (Ortiz-Quintero et al., 2022).
CS extract downregulates multiple TJ and AJ proteins and causes airway epithelial barrier damage (Aghapour et al., 2018). Woodsmoke contains many of the same toxic compounds as CS, including polycyclic aromatic hydrocarbons, carbon monoxide, and free radicals, which may contribute to the breakdown of alveolar structure and function through a p44/42 MAPK-dependent pathway, with chronic exposure leading to the development and/or exacerbation of respiratory pathologies (Lee et al., 2021). Toxic exposures disrupt these intercellular junctions. PM2.5 led to the polarization of macrophages to the M2 subtype, characterized by the high expression of MMP12 via the IL-4/STAT6 pathway. In addition, PM2.5-exposed macrophages reduced the level of E-cadherin in alveolar epithelial cells, resulting in alveolar epithelial barrier dysfunction and excessive extracellular matrix (ECM) degradation, which ultimately led to COPD progression (Guo et al., 2024).
Abnormal epithelial remodeling in smokers and patients with COPD involves the EMT, a process in which epithelial cells lose their characteristic polarity, cell-cell adhesion, and attachment to the basal membrane. This transition is characterized by the downregulation of epithelial markers, such as E-cadherin and tight junction proteins (e.g., ZO-1), alongside the upregulation of mesenchymal markers, including vimentin, N-cadherin, fibronectin, and transcription factors like Snail, Slug, and Twist. Such molecular and morphological changes contribute to airway remodeling, fibrosis, and obstruction, which are commonly observed in COPD (Su et al., 2022). The EMT process in COPD drives pathological tissue remodeling by promoting extracellular matrix deposition and facilitating epithelial barrier dysfunction, which leads to persistent inflammation and impaired repair mechanisms. Overall, EMT is a critical pathological mechanism linking chronic cigarette smoke exposure to airway fibrosis, remodeling, and the increased risk of lung cancer in COPD (Yang and Shaykhiev, 2017).
6. CRD Precision Medicine by Targeting Airway Epithelial Barrier
Precision medicine aims to tailor therapies based on individual genetic, biomarker, phenotypic, or psychosocial factors (Jameson and Longo, 2015). CRDs, including asthma and COPD, are highly heterogeneous diseases. However, until recently, individual patients were still diagnosed based on clinical presentation and associated lung function abnormalities and managed with a similar regimen. This results in suboptimized treatment, undesirable effects, or unnecessary reverse effects. As early as 2017, the European Respiratory Society convened a research seminar to discuss the application of precision medicine in airway diseases. The seminar questioned the traditional, physiology-based classification system that labels the airway diseases as asthma, COPD, etc. Instead, it advocated for relying on the biological network determined by the exposome and the genetic background, and classified airway diseases into endotypes and clinical phenotypes that describe differences between individuals as they relate to clinically meaningful outcomes, and identify biomarkers that can be used as treatable traits (Agusti et al., 2017).
A clinical phenotype refers to any measurable or observable trait related to a disease’s manifestation in a patient. It is how a disease presents itself clinically, including symptoms, signs, and measurable biological markers (Chung and Adcock, 2013). Endotypes describe distinct pathophysiologic mechanisms at a cellular and molecular level. Precision medicine is used to describe therapy targeted at patient endotypes (Kuruvilla et al., 2019).
Asthma endotypes can be broadly categorized as type 2 (T2) high or non-T2 (Kuruvilla et al., 2019) (Ray et al., 2020). The T2 high asthma is driven by Type 2 cytokines (IL-4, IL-5, and IL-13). The three key biomarkers for T2 asthma are blood eosinophils, FeNO, and IgE [141]. AECs respond to the damaging stimuli by releasing alarmins (TSLP, IL-25, IL-33), which activate ILC2s, the potent producers of IL-5 and IL-13, and propagate early type 2 immune responses upon the alarmin stimulation. Alarmins and activated DCs also induce Th2 cells to secrete IL-4, IL-5, and IL-13, and these type 2 cytokines are associated with high IgE antibody titers and eosinophilia. A cardinal mark of T2 high asthma is the large number of eosinophils recruited to the inflammatory site, further amplifying T2 inflammation and driving goblet cell metaplasia or basement membrane thickening, which leads to tissue remodeling (Lu et al., 2021).
The non-T2 asthma endotype is characterized by the absence of type 2 (T2) inflammation markers and typically involves neutrophilic or paucigranulocytic airway inflammation. It does not exhibit elevated eosinophils or the classical T2-high cytokine profile commonly seen in allergic asthma, and often shows poor responsiveness to corticosteroid therapy. This endotype includes subtypes defined by increased neutrophil presence (neutrophilic asthma) or normal levels of both neutrophils and eosinophils (paucigranulocytic asthma). The underlying molecular mechanisms involve pathways such as the activation of Th1 and Th17 cells, as well as inflammasomes like NLRP3, which contribute to corticosteroid resistance and persistent airway inflammation. Biomarkers for non-T2 asthma are less well-defined but may include sputum neutrophil counts and markers such as IL-6 and MMP-9. This endotype generally represents a heterogeneous group with variable clinical courses and limited targeted treatment options currently available (Adrish and Akuthota, 2023).
To date, the most well-characterized endotype of COPD is α1-antitrypsin (AAT) deficiency (AATD), which is commonly associated with early-onset lower-lobe emphysema and is supported by robust biomarkers at both the genetic and protein levels, thereby enabling targeted replacement therapy (Chapman et al., 2015; Ghosh et al., 2022). AAT is an endogenous serine protease inhibitor. In susceptible individuals—particularly those with a genetic deficiency of AAT—chronic inhalational exposure leads to dysregulated proteolytic activity, predominantly by neutrophil elastase (NE), resulting in unchecked degradation of the elastic matrix within the lung parenchyma (Gooptu et al., 2009). Recently, natural products have been used to restore AAT activity and slow disease progression in AATD patients (Sun et al., 2025).
In approximately 10–40% of adults with COPD, there is evidence of type 2 (T2)–mediated airway inflammation. This is commonly measured by counting eosinophils in the airways or blood. Elevated eosinophils mark the best-studied COPD endotype and are associated with a greater risk of exacerbations (Bhatt et al., 2023). Since T2 inflammation is present in asthma (50-70% of asthma patients), COPD, and certain forms of interstitial lung diseases, targeting this endotype can enhance clinical outcomes in responsive individuals while reducing unnecessary side effects in those less likely to benefit from specific therapies (Beech et al., 2024; Howell et al., 2023; Pellicano et al., 2023).
Because many patients with T2-high asthma do not respond to approved IgE- or T2 cytokine-targeting therapies, the trend in precision medicine has shifted toward targeting the AEB (Akenroye et al., 2025). The airway epithelium barrier continuously monitors and mounts appropriate responses to environmental exposures, and is also a primary initiating site for CRD, including not only asthma but also COPD. In both diseases, the epithelial barrier exhibits functional and structural abnormalities, including the increased release of epithelial alarmins, such as TSLP and IL-33, in response to environmental triggers like viruses, allergens, pollutants, and cigarette smoke. This heightened alarm release contributes to the initiation and exacerbation of airway inflammation and disease progression, as well as the potential of using alarmin singletons as a precision therapy target (Calderon et al., 2023).
Several biologicals targeting IL-33/ST2 (the IL-33 receptor) axis have demonstrated effectiveness in clinical trials. In Phase II asthma trials, itepekimab, an anti-33 monoclonal antibody, significantly reduced the loss of asthma control events and improved lung function compared to placebo, although its efficacy was moderate (Wechsler et al., 2021). For COPD, phase 2a and 3 clinical studies showed that itepekimab reduced exacerbation rates and improved lung function, particularly in former smokers with moderate-to-severe disease. A notable clinical trial reported a 51% lower adjusted annualized exacerbation rate in this subgroup (Rabe et al., 2021). Astegolimab, an anti-ST2 antibody, significantly reduces the annualized asthma exacerbation rate (AER) in a broad population of patients with severe asthma, including both eosinophil-high (T2) and eosinophil-low (non-T2) subgroups. In clinical trials, at the highest tested dose of 490 mg, astegolimab reduced AER by 43% overall and by 54% specifically in patients with eosinophil-low disease, indicating a strong effect in the non-T2 subgroup (Kelsen et al., 2021). In COPD patients, astegolimab did not significantly reduce exacerbations. However, it improves health status and quality of life measures (Yousuf et al., 2022). Tozorakimab is a novel human anti-IL-33 monoclonal antibody with a dual mechanism of action, inhibiting both reduced IL-33 (IL-33red) and oxidized IL-33 (IL-33ox) forms, thereby blocking IL-33 receptor ST2- and RAGE/EGFR-mediated pathways. Preclinical data show that tozorakimab has extremely high affinity and fast association rate with IL-33red, superior even to the natural decoy receptor ST2, suggesting potent neutralization capabilities (England et al., 2023). Phase 1 clinical evaluation demonstrated that tozorakimab is well-tolerated, with no safety concerns, in healthy subjects and patients with mild COPD, as indicated by biomarker analyses showing reduced inflammation. A Phase 2a COPD trial for tozorakimab demonstrated positive efficacy signals in a subgroup of COPD patients, although full results have yet to be reported (Singh et al., 2025).
Similarly, clinical trials also demonstrate the efficacy of anti-TSLP antibodies. Tezepelumab, a monoclonal antibody targeting TSLP, has shown strong efficacy in asthma by reducing exacerbations and improving lung function in patients with severe, uncontrolled asthma (Salvati et al., 2021). Furthermore, tezepelumab effectively reduces exacerbations across both T2 high (>150/µL blood eosinophils) and T2 low phenotypes of asthma (Agache et al., 2025; Corren et al., 2023). In COPD, although direct large-scale trial data are limited in the documents provided, tezepelumab is recognized for its potential to modulate upstream inflammatory pathways and is currently under investigation in clinical trials for COPD (Gauvreau et al., 2023).
7. Conclusive Remarks and Perspectives
The AEB plays a central role in maintaining respiratory homeostasis and defending against environmental insults, but is also associated with CRDs. The respiratory exposome’s impact on the AEB is a key driver in the onset and progression of CRDs, as it promotes barrier disruption, inflammation, structural remodeling, and susceptibility to infection. Precision medicine approaches that incorporate respiratory exposome knowledge with molecular and clinical data offer a promising avenue to personalize prevention and treatment strategies focused on restoring epithelial barrier function and modulating immune responses, ultimately improving outcomes for patients with CRDs.
The advancement of exposome methodology is opening a new window to systematically measure the characteristics of exposure to both external and internal environments at both individual and population levels. For example, satellites equipped with optical, infrared, microwave, synthetic aperture radar (SAR), and hyperspectral imagers can capture data across different wavelengths and detect pollution in air, water, and soil. Geographical Information Technologies (GIT), such as the Global Positioning System (GPS), can be used to monitor individuals’ geographic locations and potential pollution in the surrounding area. Personal, portable sensors implemented in various wearables have been developed to detect a wide range of environmental factors, such as air pollution, noise, temperature, and green space, as well as health responses, including blood pressure, heart rate, lung function, emotional status, and physical activity levels (Turner et al., 2017). The smartphone-based geospatial tools and sensors provide real-time, high-resolution data on individual exposure to multiple environmental factors (Wan et al., 2025). For respiratory exposome assessment, several portable spirometers are available for measuring FEV1, FVC, and PEF, including Spirotel and Piko-1 (Hu, 2025).
To determine the internal exposome, omics technologies such as transcriptomics, proteomics, metabolomics, epigenomics, and microbiomics analysis provide unprecedented breadth and high-throughput capabilities for comprehensively profiling molecular changes. However, molecular techniques that in-depth analyze a few targeted individual biological molecules—such as specific proteins, genes, and metabolites—and their interactions remain essential for precisely elucidating molecular pathways and mechanisms (Hu, 2025). For the respiratory exposome, biological samples derived from the airway epithelium are particularly valuable for detecting omics and molecular mechanisms. These samples can be obtained from primary nasal, tracheal, and bronchial epithelial cells, which are collected using cytology brushes or bronchoscopes (Fawcett et al., 2023; Garner et al., 2024), or from in vitro culture systems such as air-liquid interface (Lee et al., 2023) (Baldassi et al., 2021), airway organoids (Matkovic Leko et al., 2023), 3D-printed airway models (Lee et al., 2024), or airway-on-chip (Gao et al., 2024).
In addition to the technologies above, high-resolution mass spectrometry (HRMS) offers approaches to simultaneously measure a vast number of exogenous and endogenous compounds, offering a resolution of both external and internal chemical exposomes (Liu et al., 2020). The profile of the external exposome defines the expotype, while the internal exposome defines the endotype. The ultimate avenue of precision medicine would be combining the expotype and the endotype (Figure 3). The term “expotype” refers to a specific subset or collection of exposures to which the investigated individuals are exposed. It primarily focuses on specific external exposures accumulated over a given time and space (Martin-Sanchez et al., 2021).
Current precision medicine distinguishes and treats CRD patients using only a limited number of biomarkers, such as those related to inflammation. Many other pathogenetic molecular processes haven’t been adequately explored for their potential in defining the endotype or used as therapeutic targets, and the expotype hasn’t been widely considered in disease management. These defects may be the reasons for the unsatisfying treatment outcomes. Filling the gap between respiratory exposome research and clinical therapeutic practice may hold promise for improving respiratory health management and precision medicine.
References
- Adrish, M., and P. Akuthota. 2023. Approach to non-type 2 asthma. Respir Med 216: 107327. [Google Scholar] [CrossRef]
- Agache, I., I. M. Adcock, F. Baraldi, K. F. Chung, I. Eguiluz-Gracia, S. L. Johnston, M. Jutel, P. Nair, A. Papi, C. Porsbjerg, O. S. Usmani, D. A. Meyers, M. Zemelka-Wiacek, and E. R. Bleecker. 2025. Personalised therapeutic approaches for asthma. J Allergy Clin Immunol. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, M., P. Raee, S. J. Moghaddam, P. S. Hiemstra, and I. H. Heijink. 2018. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am J Respir Cell Mol Biol 58: 157–169. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S., R. Srivastava, F. Rahmatpanah, C. Madiraju, L. BenMohamed, and A. Agrawal. 2017. Airway epithelial cells enhance the immunogenicity of human myeloid dendritic cells under steady state. Clin Exp Immunol 189: 279–289. [Google Scholar] [CrossRef]
- Agusti, A., and J. C. Hogg. 2019. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N Engl J Med 381: 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A., M. Bafadhel, R. Beasley, E. H. Bel, R. Faner, P. G. Gibson, R. Louis, V. M. McDonald, P. J. Sterk, M. Thomas, C. Vogelmeier, and I. D. Pavord. 2017. on behalf of all participants in the, seminar Precision medicine in airway diseases: Moving to clinical practice. Eur Respir J 50. [Google Scholar] [CrossRef]
- Akdis, C. A. 2021. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol 21: 739–751. [Google Scholar] [CrossRef]
- Akenroye, A., J. A. Boyce, and H. Kita. 2025. Targeting alarmins in asthma: From bench to clinic. J Allergy Clin Immunol 155: 1133–1148. [Google Scholar] [CrossRef]
- Almuntashiri, S., Y. Zhu, Y. Han, X. Wang, P. R. Somanath, and D. Zhang. 2020. Club Cell Secreted Protein CC16: Potential Applications in Prognosis and Therapy for Pulmonary Diseases. J Clin Med 9. [Google Scholar] [CrossRef]
- Alvarez, F., G. D. De Melo, F. Larrous, L. Kergoat, B. Boëda, V. Michel, D. Seilhean, M. Tichit, D. Hing, D. Hardy, E. Kornobis, H. Bourhy, N. Wolff, and C. Caillet-Saguy. 2025. The SARS-CoV-2 envelope PDZ binding motif acts as a virulence factor disrupting host’s epithelial cell–cell junctions. Cell. Mol. Biol. Lett. 30. [Google Scholar] [CrossRef]
- Arbore, C., M. Sergides, L. Gardini, G. Bianchi, A. V. Kashchuk, I. Pertici, P. Bianco, F. S. Pavone, and M. Capitanio. 2022. alpha-catenin switches between a slip and an asymmetric catch bond with F-actin to cooperatively regulate cell junction fluidity. Nat Commun 13: 1146. [Google Scholar] [CrossRef]
- Baldassi, D., B. Gabold, and O. Merkel. 2021. Air-liquid interface cultures of the healthy and diseased human respiratory tract: Promises, challenges and future directions. Adv Nanobiomed Res 1. [Google Scholar] [CrossRef] [PubMed]
- Barbier, E., J. Carpentier, O. Simonin, P. Gosset, A. Platel, M. Happillon, L. Y. Alleman, E. Perdrix, V. Riffault, T. Chassat, J.-M. Lo Guidice, S. Anthérieu, and G. Garçon. 2023. Oxidative stress and inflammation induced by air pollution-derived PM2. 5 persist in the lungs of mice after cessation of their sub-chronic exposure. Environ. Int. 181: 108248. [Google Scholar] [CrossRef] [PubMed]
- Barr, J., M. E. Gentile, S. Lee, M. E. Kotas, M. Fernanda de Mello Costa, N. P. Holcomb, A. Jaquish, G. Palashikar, M. Soewignjo, M. McDaniel, I. Matsumoto, R. Margolskee, J. Von Moltke, N. A. Cohen, X. Sun, and A. E. Vaughan. 2022. Injury-induced pulmonary tuft cells are heterogenous, arise independent of key Type 2 cytokines, and are dispensable for dysplastic repair. Elife 11. [Google Scholar] [CrossRef]
- Basil, M. C., F. L. Cardenas-Diaz, J. J. Kathiriya, M. P. Morley, J. Carl, A. N. Brumwell, J. Katzen, K. J. Slovik, A. Babu, S. Zhou, M. M. Kremp, K. B. McCauley, S. Li, J. D. Planer, S. S. Hussain, X. Liu, R. Windmueller, Y. Ying, K. M. Stewart, M. Oyster, J. D. Christie, J. M. Diamond, J. F. Engelhardt, E. Cantu, S. M. Rowe, D. N. Kotton, H. A. Chapman, and E. E. Morrisey. 2022. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 604: 120–126. [Google Scholar] [CrossRef] [PubMed]
- Beech, A., A. Higham, S. Booth, V. Tejwani, F. Trinkmann, and D. Singh. 2024. Type 2 inflammation in COPD: Is it just asthma? Breathe Sheff 20: 230229. [Google Scholar] [CrossRef]
- Bhatt, S. P., A. Agusti, M. Bafadhel, S. A. Christenson, J. Bon, G. C. Donaldson, D. D. Sin, J. A. Wedzicha, and F. J. Martinez. 2023. Phenotypes, Etiotypes, and Endotypes of Exacerbations of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 208: 1026–1041. [Google Scholar] [CrossRef]
- Bissonnette, E. Y., J. F. Lauzon-Joset, J. S. Debley, and S. F. Ziegler. 2020. Cross-Talk Between Alveolar Macrophages and Lung Epithelial Cells is Essential to Maintain Lung Homeostasis. Front Immunol 11: 583042. [Google Scholar] [CrossRef]
- Blackburn, J. B., N. F. Li, N. W. Bartlett, and B. W. Richmond. 2023. An update in club cell biology and its potential relevance to chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 324: L652–L665. [Google Scholar] [CrossRef]
- Boggon, T. J., J. Murray, S. Chappuis-Flament, E. Wong, B. M. Gumbiner, and L. Shapiro. 2002. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296: 1308–1313. [Google Scholar] [CrossRef]
- Bole, A., A. Bernstein, M. J. White, A. Bole, S. J. Balk, L. G. Byron, G. M. Huerta-Montañez, P. J. Landrigan, S. M. Marcus, A. L. Nerlinger, L. H. Patel, R. Philipsborn, A. D. Woolf, L. Zajac, K. A. Gray, J. Briskin, N. G. DeNicola, M. Karwowski, M. H. Ward, and COUNCIL ON ENVIRONMENTAL HEALTH AND CLIMATE CHANGE. 2024. SECTION ON MINORITY HEALTH, EQUITY, AND INCLUSION The Built Environment and Pediatric Health. Pediatrics 153. [Google Scholar] [CrossRef] [PubMed]
- Bray, S. J., and A. Bigas. 2025. Modes of Notch signalling in development and disease. Nat. Rev. Mol. Cell Biol. 26: 522–537. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R. A., A. J. Fisher, and L. A. Borthwick. 2021. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells 10. [Google Scholar] [CrossRef] [PubMed]
- Calderon, A. A., C. Dimond, D. F. Choy, R. Pappu, M. A. Grimbaldeston, D. Mohan, and K. F. Chung. 2023. Targeting interleukin-33 and thymic stromal lymphopoietin pathways for novel pulmonary therapeutics in asthma and COPD. Eur Respir Rev 32. [Google Scholar] [CrossRef]
- Calven, J., E. Ax, and M. Radinger. 2020. The Airway Epithelium-A Central Player in Asthma Pathogenesis. Int J Mol Sci 21. [Google Scholar] [CrossRef]
- Campas, O., I. Noordstra, and A. S. Yap. 2024. Adherens junctions as molecular regulators of emergent tissue mechanics. Nat Rev Mol Cell Biol 25: 252–269. [Google Scholar] [CrossRef]
- Carlier, F. M., C. de Fays, and C. Pilette. 2021. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front Physiol 12: 691227. [Google Scholar] [CrossRef]
- Celli, B. R., and A. Agusti. 2018. COPD: Time to improve its taxonomy? ERJ Open Res 4. [Google Scholar] [CrossRef]
- Chapman, K. R., J. G. Burdon, E. Piitulainen, R. A. Sandhaus, N. Seersholm, J. M. Stocks, B. C. Stoel, L. Huang, Z. Yao, J. M. Edelman, N. G. McElvaney, and Rapid Trial Study Group. 2015. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): A randomised, double-blind, placebo-controlled trial. Lancet 386: 360–368. [Google Scholar] [CrossRef]
- Christenson, S. A., B. M. Smith, M. Bafadhel, and N. Putcha. 2022. Chronic obstructive pulmonary disease. Lancet 399: 2227–2242. [Google Scholar] [CrossRef]
- Chung, K. F., and I. M. Adcock. 2013. How variability in clinical phenotypes should guide research into disease mechanisms in asthma. Ann Am Thorac Soc 10: S109–17. [Google Scholar] [CrossRef] [PubMed]
- Citi, S., M. Fromm, M. Furuse, L. Gonzalez-Mariscal, A. Nusrat, S. Tsukita, and J. R. Turner. 2024. A short guide to the tight junction. J Cell Sci 137. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M. L., G. Barrientos, X. Cai, S. Mukherjee, M. Das, E. Stephen-Victor, and H. Harb. 2025. Regulatory T cells and their role in allergic disease. Allergy 80: 77–93. [Google Scholar] [CrossRef]
- Corren, J., A. Menzies-Gow, G. Chupp, E. Israel, S. Korn, B. Cook, C. S. Ambrose, A. Hellqvist, S. L. Roseti, N. A. Molfino, J. P. Llanos, N. Martin, K. Bowen, J. M. Griffiths, J. R. Parnes, and G. Colice. 2023. Efficacy of Tezepelumab in Severe, Uncontrolled Asthma: Pooled Analysis of the PATHWAY and NAVIGATOR Clinical Trials. Am J Respir Crit Care Med 208: 13–24. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V., and S. Schultz-Cherry. 2021. The role of goblet cells in viral pathogenesis. FEBS J 288: 7060–7072. [Google Scholar] [CrossRef]
- Cummins, P. M. 2012. Occludin: One protein, many forms. Mol Cell Biol 32: 242–250. [Google Scholar] [CrossRef]
- Cumplido-Laso, G., D. A. Benitez, S. Mulero-Navarro, and J. M. Carvajal-Gonzalez. 2023. Transcriptional Regulation of Airway Epithelial Cell Differentiation: Insights into the Notch Pathway and Beyond. Int J Mol Sci 24. [Google Scholar] [CrossRef]
- Davis, J. D., and T. P. Wypych. 2021. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol 14: 978–990. [Google Scholar] [CrossRef]
- Di, Y. P., and H. Mou. 2024. Airway Serous Cells: A Comparative Study of Spatial Distribution and Abundance among Species. J Respir Biol Transl Med 1. [Google Scholar] [CrossRef]
- Di, Y. P., J. M. Kuhn, and M. L. Mangoni. 2024. Lung antimicrobial proteins and peptides: From host defense to therapeutic strategies. Physiol Rev 104: 1643–1677. [Google Scholar] [CrossRef]
- Duval, C., M. Watanabe, and G. Donati. 2018. Buried myoepithelial stem cells as a reservoir for repairing the exposed airway epithelium. Stem Cell Investig 5: 45. [Google Scholar] [CrossRef]
- Engelhardt, J. F., J. R. Yankaskas, S. A. Ernst, Y. Yang, C. R. Marino, R. C. Boucher, J. A. Cohn, and J. M. Wilson. 1992. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2: 240–248. [Google Scholar] [CrossRef]
- England, E., D. G. Rees, I. C. Scott, S. Carmen, D. T. Y. Chan, C. E. Chaillan Huntington, K. F. Houslay, T. Erngren, M. Penney, J. B. Majithiya, L. Rapley, D. A. Sims, C. Hollins, E. C. Hinchy, M. D. Strain, B. P. Kemp, D. J. Corkill, R. D. May, K. A. Vousden, R. J. Butler, T. Mustelin, T. J. Vaughan, D. C. Lowe, C. Colley, and E. S. Cohen. 2023. Tozorakimab (MEDI3506): An anti-IL-33 antibody that inhibits IL-33 signalling via ST2 and RAGE/EGFR to reduce inflammation and epithelial dysfunction. Sci Rep 13: 9825. [Google Scholar] [CrossRef]
- Evans, C. M., K. Kim, M. J. Tuvim, and B. F. Dickey. 2009. Mucus hypersecretion in asthma: Causes and effects. Curr Opin Pulm Med 15: 4–11. [Google Scholar] [CrossRef]
- Farquhar, M. G., and G. E. Palade. 1963. Junctional complexes in various epithelia. J Cell Biol 17: 375–412. [Google Scholar] [CrossRef]
- Fawcett, L. K., N. Turgutoglu, K. M. Allan, Y. Belessis, J. Widger, A. Jaffe, and S. A. Waters. 2023. Comparing Cytology Brushes for Optimal Human Nasal Epithelial Cell Collection: Implications for Airway Disease Diagnosis and Research. J Med 13. [Google Scholar] [CrossRef] [PubMed]
- Feng, H., Z. Li, and R. Zheng. 2025. The global burden of chronic respiratory diseases attributable to tobacco from 1990 to 2021: A global burden of disease study 2021. BMC Public Health 25: 456. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y., X. Tang, H. Fu, X. Fan, J. Wei, J. Liu, H. Wang, H. Bi, Z. Chen, X. Wei, and Y. Zheng. 2025. Mechanistic insights into carbon black-activated AKT/TMEM175 cascade impairing macrophage-epithelial cross-talk and airway epithelial proliferation. Env. Pollut 372: 126076. [Google Scholar] [CrossRef] [PubMed]
- Fu, R., X. Jiang, G. Li, Y. Zhu, and H. Zhang. 2022. Junctional complexes in epithelial cells: Sentinels for extracellular insults and intracellular homeostasis. FEBS J 289: 7314–7333. [Google Scholar] [CrossRef]
- Furuse, M., H. Sasaki, K. Fujimoto, and S. Tsukita. 1998. A single gene product, claudin-1 or-2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143: 391–401. [Google Scholar] [CrossRef]
- Ganesan, S., A. T. Comstock, and U. S. Sajjan. 2013. Barrier function of airway tract epithelium. Tissue Barriers 1: e24997. [Google Scholar] [CrossRef] [PubMed]
- Gao, N., A. Raduka, and F. Rezaee. 2022. Respiratory syncytial virus disrupts the airway epithelial barrier by decreasing cortactin and destabilizing F-actin. J Cell Sci 135. [Google Scholar] [CrossRef] [PubMed]
- Gao, W., K. R. Kanagarajah, E. Graham, K. Soon, T. Veres, T. J. Moraes, C. E. Bear, R. A. Veldhuizen, A. P. Wong, and A. Günther. 2024. Collagen Tubular Airway-on-Chip for Extended Epithelial Culture and Investigation of Ventilation Dynamics. Small 20. [Google Scholar] [CrossRef] [PubMed]
- Garner, J. L., P. L. Shah, F. Herth, and D. J. Slebos. 2024. ERJ Advances: Interventional bronchoscopy. Eur Respir J 64. [Google Scholar] [CrossRef]
- Gauvreau, G. M., J. M. Hohlfeld, J. M. FitzGerald, L. P. Boulet, D. W. Cockcroft, B. E. Davis, S. Korn, O. Kornmann, R. Leigh, I. Mayers, H. Watz, S. S. Grant, M. Jain, M. Cabanski, P. E. Pertel, I. Jones, J. R. Lecot, H. Cao, and P. M. O’Byrne. 2023. Inhaled anti-TSLP antibody fragment, ecleralimab, blocks responses to allergen in mild asthma. Eur Respir J 61. [Google Scholar] [CrossRef]
- GBD Chronic Respiratory Disease Collaborators. 2020. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: A systematic analysis for the Global Burden of Disease Study. (2017). Lancet Respir Med 8: 585–596. [Google Scholar] [CrossRef]
- GBD Chronic Respiratory Disease Collaborators. 2023. Global burden of chronic respiratory diseases and risk factors, 1990-2019: An update from the Global Burden of Disease Study 2019. EClinicalMedicine 59: 101936. [Google Scholar] [CrossRef]
- Gerayeli, F. V., H. Y. Park, S. Milne, X. Li, C. X. Yang, J. Tuong, R. L. Eddy, S. M. Vahedi, E. Guinto, C. Y. Cheung, J. S. W. Yang, C. Gilchrist, D. Yehia, T. Stach, H. Dang, C. Leung, T. Shaipanich, J. Leipsic, G. J. Koelwyn, J. M. Leung, and D. D. Sin. 2024. Single-cell sequencing reveals cellular landscape alterations in the airway mucosa of patients with pulmonary long COVID. Eur. Respir. J. 64: 2301947. [Google Scholar] [CrossRef]
- Ghosh, A. J., B. D. Hobbs, M. Moll, A. Saferali, A. Boueiz, J. H. Yun, F. Sciurba, L. Barwick, A. H. Limper, K. Flaherty, G. Criner, K. K. Brown, R. Wise, F. J. Martinez, D. Lomas, P. J. Castaldi, V. J. Carey, D. L. DeMeo, M. H. Cho, E. K. Silverman, C. P. Hersh, and COPDGene Investigators. 2022. Alpha-1 Antitrypsin MZ Heterozygosity Is an Endotype of Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 205: 313–323. [Google Scholar] [CrossRef]
- Godbole, N. M., A. A. Chowdhury, N. Chataut, and S. Awasthi. 2022. Tight Junctions, the Epithelial Barrier, and Toll-like Receptor-4 During Lung Injury. Inflammation 45: 2142–2162. [Google Scholar] [CrossRef]
- Gon, Y., and S. Hashimoto. 2018. Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergol Int 67: 12–17. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, L., A. Betanzos, P. Nava, and B. E. Jaramillo. 2003. Tight junction proteins. Prog Biophys Mol Biol 81: 1–44. [Google Scholar] [CrossRef] [PubMed]
- Gooptu, B., U. I. Ekeowa, and D. A. Lomas. 2009. Mechanisms of emphysema in alpha1-antitrypsin deficiency: Molecular and cellular insights. Eur Respir J 34: 475–488. [Google Scholar] [CrossRef] [PubMed]
- Greenan, G. A., R. D. Vale, and D. A. Agard. 2020. Electron cryotomography of intact motile cilia defines the basal body to axoneme transition. J Cell Biol 219. [Google Scholar] [CrossRef]
- Gui, M., H. Farley, P. Anujan, J. R. Anderson, D. W. Maxwell, J. B. Whitchurch, J. J. Botsch, T. Qiu, S. Meleppattu, S. K. Singh, Q. Zhang, J. Thompson, J. S. Lucas, C. D. Bingle, D. P. Norris, S. Roy, and A. Brown. 2021. De novo identification of mammalian ciliary motility proteins using cryo-EM. Cell 184: 5791–5806 e19. [Google Scholar] [CrossRef]
- Gunzel, D., and A. S. Yu. 2013. Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569. [Google Scholar] [CrossRef]
- Guo, X., S. Yang, H. Zhu, F. Liu, K. Li, G. Li, Y. Lin, H. Yu, W. Qiu, H. Xu, Q. Liu, X. Xie, Y. Sun, P. Zheng, B. Chen, Z. Liu, X. Yuan, S. Peng, X. Bi, J. Yang, N. Y. Shao, and J. Dai. 2024. Involvement of M2 macrophages polarization in PM2. 5-induced COPD by upregulating MMP12 via IL4/STAT6 pathway. Ecotoxicol Env. Saf 283: 116793. [Google Scholar] [CrossRef]
- He, Y., W. J. Liu, N. Jia, S. Richardson, and C. Huang. 2023. Viral respiratory infections in a rapidly changing climate: The need to prepare for the next pandemic. eBioMedicine 93: 104593. [Google Scholar] [CrossRef]
- Hedstrom, U., L. Oberg, O. Vaarala, G. Dellgren, M. Silverborn, L. Bjermer, G. Westergren-Thorsson, O. Hallgren, and X. Zhou. 2021. Impaired Differentiation of Chronic Obstructive Pulmonary Disease Bronchial Epithelial Cells Grown on Bronchial Scaffolds. Am J Respir Cell Mol Biol 65: 201–213. [Google Scholar] [CrossRef]
- Heijink, I. H., P. M. Kies, H. F. Kauffman, D. S. Postma, A. J. van Oosterhout, and E. Vellenga. 2007. Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J Immunol 178: 7678–7685. [Google Scholar] [CrossRef]
- Heijink, I. H., V. N. S. Kuchibhotla, M. P. Roffel, T. Maes, D. A. Knight, I. Sayers, and M. C. Nawijn. 2020. Epithelial cell dysfunction, a major driver of asthma development. Allergy 75: 1902–1917. [Google Scholar] [CrossRef]
- Hessel, J., J. Heldrich, J. Fuller, M. R. Staudt, S. Radisch, C. Hollmann, B. G. Harvey, R. J. Kaner, J. Salit, J. Yee-Levin, S. Sridhar, S. Pillai, H. Hilton, G. Wolff, H. Bitter, S. Visvanathan, J. Fine, C. S. Stevenson, R. G. Crystal, and A. E. Tilley. 2014. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS ONE 9: e85453. [Google Scholar] [CrossRef]
- Hewitt, R. J., and C. M. Lloyd. 2021. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 21: 347–362. [Google Scholar] [CrossRef]
- Hoagland, D. A., P. Rodríguez-Morales, A. O. Mann, A. Y. Baez Vazquez, S. Yu, A. Lai, H. Kane, S. M. Dang, Y. Lin, L. Thorens, S. Begum, M. A. Castro, S. D. Pope, J. Lim, S. Li, X. Zhang, M. O. Li, C. F. Kim, R. Jackson, R. Medzhitov, and R. A. Franklin. 2025. Macrophage-derived oncostatin M repairs the lung epithelial barrier during inflammatory damage. Science 389: 169–175. [Google Scholar] [CrossRef]
- Hogan, B., and P. R. Tata. 2019. Cellular organization and biology of the respiratory system. Nat Cell Biol. [Google Scholar] [CrossRef] [PubMed]
- Holtjer, J. C. S., L. D. Bloemsma, R. Beijers, M. E. B. Cornelissen, B. Hilvering, L. Houweling, R. C. H. Vermeulen, G. S. Downward, A. H. Maitland-Van der Zee, and P. O. consortium. 2023. Identifying risk factors for COPD and adult-onset asthma: An umbrella review. Eur Respir Rev 32. [Google Scholar] [CrossRef] [PubMed]
- Horton, K., P. A. C. Wing, C. L. Jackson, C. J. McCormick, M. P. Carroll, and J. S. Lucas. 2025. Interplay between respiratory viruses and cilia in the airways. Eur Respir Rev 34. [Google Scholar] [CrossRef] [PubMed]
- Howell, I., A. Howell, and I. D. Pavord. 2023. Type 2 inflammation and biological therapies in asthma: Targeted medicine taking flight. J Exp Med 220. [Google Scholar] [CrossRef]
- Hu, X. 2025. The lung exposome: Accelerating precision respiratory health. The Lung: pp. 629–645. [Google Scholar]
- Huff, R. D., C. Carlsten, and J. A. Hirota. 2019. An update on immunologic mechanisms in the respiratory mucosa in response to air pollutants. J Allergy Clin Immunol 143: 1989–2001. [Google Scholar] [CrossRef]
- Jacquet, A. 2011. Interactions of airway epithelium with protease allergens in the allergic response. Clin Exp Allergy 41: 305–311. [Google Scholar] [CrossRef]
- Jagielnicki, M., I. Kucharska, B. C. Bennett, A. L. Harris, and M. Yeager. 2024. Connexin Gap Junction Channels and Hemichannels: Insights from High-Resolution Structures. Biol. Basel 13. [Google Scholar] [CrossRef]
- Jameson, J. L., and D. L. Longo. 2015. Precision medicine--personalized, problematic, and promising. N Engl J Med 372: 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Jang, A. S. 2014. The apical junctional complex in respiratory diseases. Chonnam Med J 50: 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jia, J., J. Xia, R. Zhang, Y. Bai, S. Liu, M. Dan, T. Li, T. Yan, L. Chen, S. Gong, P. Niu, and T. Chen. 2019. Investigation of the impact of PM2. 5 on the ciliary motion of human nasal epithelial cells. Chemosphere 233: 309–318. [Google Scholar] [CrossRef] [PubMed]
- Jing, J. C., J. J. Chen, L. Chou, B. J. F. Wong, and Z. Chen. 2017. Visualization and Detection of Ciliary Beating Pattern and Frequency in the Upper Airway using Phase Resolved Doppler Optical Coherence Tomography. Sci Rep 7: 8522. [Google Scholar] [CrossRef]
- Joo, H., S. Min, and S. W. Cho. 2024. Advanced lung organoids for respiratory system and pulmonary disease modeling. J Tissue Eng 15: 20417314241232502. [Google Scholar] [CrossRef]
- Kageyama, T., T. Ito, S. Tanaka, and H. Nakajima. 2024. Physiological and immunological barriers in the lung. Semin Immunopathol 45: 533–547. [Google Scholar] [CrossRef]
- Kato, A., and H. Kita. 2025. The immunology of asthma and chronic rhinosinusitis. Nat Rev Immunol. [Google Scholar] [CrossRef]
- Kelsen, S. G., I. O. Agache, W. Soong, E. Israel, G. L. Chupp, D. S. Cheung, W. Theess, X. Yang, T. L. Staton, D. F. Choy, A. Fong, A. Dash, M. Dolton, R. Pappu, and C. E. Brightling. 2021. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J Allergy Clin Immunol 148: 790–798. [Google Scholar] [CrossRef]
- Kim, K. A., J. H. Jung, Y. S. Choi, and S. T. Kim. 2024. Ginsenoside Re protects rhinovirus-induced disruption of tight junction through inhibition of ROS-mediated phosphatases inactivation in human nasal epithelial cells. Heliyon 10: e27688. [Google Scholar] [CrossRef]
- Knight, D. A., and S. T. Holgate. 2003. The airway epithelium: Structural and functional properties in health and disease. Respirology 8: 432–446. [Google Scholar] [CrossRef] [PubMed]
- Koch, C. M., A. D. Prigge, L. Setar, K. R. Anekalla, H. C. Do-Umehara, H. Abdala-Valencia, Y. Politanska, A. Shukla, J. Chavez, G. R. Hahn, and B. M. Coates. 2022. Cilia-related gene signature in the nasal mucosa correlates with disease severity and outcomes in critical respiratory syncytial virus bronchiolitis. Front Immunol 13: 924792. [Google Scholar] [CrossRef] [PubMed]
- Kostrewa, D., M. Brockhaus, A. D’Arcy, G. E. Dale, P. Nelboeck, G. Schmid, F. Mueller, G. Bazzoni, E. Dejana, T. Bartfai, F. K. Winkler, and M. Hennig. 2001. X-ray structure of junctional adhesion molecule: Structural basis for homophilic adhesion via a novel dimerization motif. EMBO J 20: 4391–4398. [Google Scholar] [CrossRef] [PubMed]
- Kovesi, T., G. Mallach, Y. Schreiber, M. McKay, G. Lawlor, N. Barrowman, A. Tsampalieros, R. Kulka, A. Root, L. Kelly, M. Kirlew, and J. D. Miller. 2022. Housing conditions and respiratory morbidity in Indigenous children in remote communities in Northwestern Ontario, Canada. CMAJ 194: E80–E88. [Google Scholar] [CrossRef]
- Kuek, L. E., and R. J. Lee. 2020. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol.-Lung Cell. Mol. Physiol. 319: L603–L619. [Google Scholar] [CrossRef]
- Kuruvilla, M. E., F. E. Lee, and G. B. Lee. 2019. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin Rev Allergy Immunol 56: 219–233. [Google Scholar] [CrossRef]
- Lee, D. C., H. Choi, J.-M. Oh, Junuk Lee, Joohyung Lee, H. Y. Lee, and J. Y. Kang. 2020. Urban particulate matter regulates tight junction proteins by inducing oxidative stress via the Akt signal pathway in human nasal epithelial cells. Toxicol. Lett. 333: 33–41. [Google Scholar] [CrossRef]
- Lee, P. H., S. Park, Y. G. Lee, S. M. Choi, M. H. An, and A. S. Jang. 2021. The Impact of Environmental Pollutants on Barrier Dysfunction in Respiratory Disease. Allergy Asthma Immunol Res 13: 850–862. [Google Scholar] [CrossRef]
- Lee, R. E., B. Reidel, M. R. Nelson, J. K. Macdonald, M. Kesimer, and S. H. Randell. 2023. Air-Liquid interface cultures to model drug delivery through the mucociliary epithelial barrier. Adv. Drug Deliv. Rev. 198: 114866. [Google Scholar] [CrossRef]
- Lee, Y., M. K. Lee, H.-R. Lee, B. Kim, M. Kim, and S. Jung. 2024. 3D-printed airway model as a platform for SARS-CoV-2 infection and antiviral drug testing. Biomaterials 311: 122689. [Google Scholar] [CrossRef]
- Lei, L., S. Traore, G. S. Romano Ibarra, P. H. Karp, T. Rehman, D. K. Meyerholz, J. Zabner, D. A. Stoltz, P. L. Sinn, M. J. Welsh, P. B. McCray, and I. M. Thornell. 2023. CFTR-rich ionocytes mediate chloride absorption across airway epithelia. J Clin Invest 133. [Google Scholar] [CrossRef]
- Lima, C., M. A. P. Falcao, J. G. S. Rosa, G. R. Disner, and M. Lopes-Ferreira. 2022. Pesticides and Their Impairing Effects on Epithelial Barrier Integrity, Dysbiosis, Disruption of the AhR Signaling Pathway and Development of Immune-Mediated Inflammatory Diseases. Int J Mol Sci 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, X., X. Zhao, X. Li, S. Lv, R. Ma, Y. Qi, A. Abulikemu, H. Duan, C. Guo, Y. Li, and Z. Sun. 2020. PM(2. 5) triggered apoptosis in lung epithelial cells through the mitochondrial apoptotic way mediated by a ROS-DRP1-mitochondrial fission axis. J Hazard Mater 397: 122608. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y., Y. Huang, J. Li, J. Huang, L. Zhang, J. Feng, J. Li, Q. Xia, Q. Zhao, L. Huang, S. Jiang, and S. Su. 2021. Eosinophil extracellular traps drive asthma progression through neuro-immune signals. Nat Cell Biol 23: 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Luan, X., N. Henao Romero, V. A. Campanucci, Y. Le, J. Mustofa, J. S. Tam, and J. P. Ianowski. 2024. Pulmonary Ionocytes Regulate Airway Surface Liquid pH in Primary Human Bronchial Epithelial Cells. Am J Respir Crit Care Med 210: 788–800. [Google Scholar] [CrossRef]
- Lynch, T. J., P. J. Anderson, P. G. Rotti, S. R. Tyler, A. K. Crooke, S. H. Choi, D. T. Montoro, C. L. Silverman, W. Shahin, R. Zhao, C. W. Jensen-Cody, A. Adamcakova-Dodd, T. I. A. Evans, W. Xie, Y. Zhang, H. Mou, B. P. Herring, P. S. Thorne, J. Rajagopal, C. Yeaman, K. R. Parekh, and J. F. Engelhardt. 2018. Submucosal Gland Myoepithelial Cells Are Reserve Stem Cells That Can Regenerate Mouse Tracheal Epithelium. Cell Stem Cell 22: 653–667 e5. [Google Scholar] [CrossRef]
- Martin-Sanchez, F., M. Atienza-Maderuelo, G. Lopez-Campos, and P. Collado. 2021. Use of informatics to characterise the exposome of COVID-19. BMJ Health Care Inf 28. [Google Scholar] [CrossRef]
- Matkovic Leko, I., R. T. Schneider, T. A. Thimraj, N. Schrode, D. Beitler, H.-Y. Liu, K. Beaumont, Y.-W. Chen, and H.-W. Snoeck. 2023. A distal lung organoid model to study interstitial lung disease, viral infection and human lung development. Nat. Protoc. 18: 2283–2312. [Google Scholar] [CrossRef]
- McBrien, C. N., and A. Menzies-Gow. 2017. The Biology of Eosinophils and Their Role in Asthma. Front Med Lausanne 4: 93. [Google Scholar] [CrossRef]
- Montgomery, M. T., S. P. Sajuthi, S.-H. Cho, J. L. Everman, C. L. Rios, K. C. Goldfarbmuren, N. D. Jackson, B. Saef, M. Cromie, C. Eng, V. Medina, J. R. Elhawary, S. S. Oh, J. Rodriguez-Santana, E. K. Vladar, E. G. Burchard, and M. A. Seibold. 2020. Genome-Wide Analysis Reveals Mucociliary Remodeling of the Nasal Airway Epithelium Induced by Urban PM2. 5. Am. J. Respir. Cell Mol. Biol. 63: 172–184. [Google Scholar] [CrossRef]
- Moratin, H., J. Lang, M. S. Picker, A. Rossi, C. Wilhelm, A. von Fournier, M. Stoth, M. Goncalves, N. Kleinsasser, S. Hackenberg, A. Scherzad, and T. J. Meyer. 2025. The Impact of NO(2) on Epithelial Barrier Integrity of a Primary Cell-Based Air-Liquid Interface Model of the Nasal Respiratory Epithelium. J Appl Toxicol 45: 482–491. [Google Scholar] [CrossRef]
- Narita, H., Y. Yamamoto, M. Suzuki, N. Miyazaki, A. Yoshida, K. Kawai, K. Iwasaki, A. Nakagawa, Y. Takai, and T. Sakisaka. 2011. Crystal Structure of the cis-Dimer of Nectin-1: Implications for the architecture of cell-cell junctions. J Biol Chem 286: 12659–12669. [Google Scholar] [CrossRef]
- Naser, A. N., Q. Lu, and Y. H. Chen. 2023. Trans-Compartmental Regulation of Tight Junction Barrier Function. Tissue Barriers 11: 2133880. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuijsen, M., A. De Nazelle, J. Garcia-Aymerich, H. Khreis, and B. Hoffmann. 2024. Shaping urban environments to improve respiratory health: Recommendations for research, planning, and policy. Lancet Respir. Med. 12: 247–254. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K., K. A. Brune, N. Putcha, P. Mandke, W. K. O’Neal, D. Shade, V. Srivastava, M. Wang, H. Lam, S. S. An, M. B. Drummond, N. N. Hansel, D. N. Robinson, and V. K. Sidhaye. 2017. Cigarette smoke disrupts monolayer integrity by altering epithelial cell-cell adhesion and cortical tension. Am. J. Physiol. Lung Cell. Mol. Physiol. 313: L581–L591. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M., K. T. Furukawa, and M. Morimoto. 2020. Pulmonary neuroendocrine cells: Physiology, tissue homeostasis and disease. Model Mech 13. [Google Scholar] [CrossRef]
- Okuda, K., and M. Gentzsch. 2024. Pulmonary Ionocytes: What Are They Transporting and Which Way? Am J Respir Crit Care Med 210: 705–707. [Google Scholar] [CrossRef]
- Onufer, A. P., J. C. Mell, L. Cort, A. Rao, N. V. Mdluli, and A. J. Carey. 2025. Influenza virus-induced type I interferons disrupt alveolar epithelial repair and tight junction integrity in the developing lung. Mucosal Immunol. 18: 607–619. [Google Scholar] [CrossRef]
- Ortiz-Quintero, B., I. Martinez-Espinosa, and R. Perez-Padilla. 2022. Mechanisms of Lung Damage and Development of COPD Due to Household Biomass-Smoke Exposure: Inflammation, Oxidative Stress, MicroRNAs, and Gene Polymorphisms. Cells 12. [Google Scholar] [CrossRef]
- Otani, T., and M. Furuse. 2020. Tight Junction Structure and Function Revisited. Trends Cell Biol 30: 805–817. [Google Scholar] [CrossRef]
- Otani, T., T. P. Nguyen, S. Tokuda, K. Sugihara, T. Sugawara, K. Furuse, T. Miura, K. Ebnet, and M. Furuse. 2019. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J Cell Biol 218: 3372–3396. [Google Scholar] [CrossRef]
- Owen, C. E. 2007. Immunoglobulin E: Role in asthma and allergic disease: Lessons from the clinic. Pharmacol Ther 113: 121–133. [Google Scholar] [CrossRef]
- Pellicano, C., L. Vantaggio, A. Colalillo, K. Pocino, V. Basile, M. Marino, U. Basile, and E. Rosato. 2023. Type 2 cytokines and scleroderma interstitial lung disease. Clin Exp Med 23: 3517–3525. [Google Scholar] [CrossRef]
- Perl, A. L., J. L. Pokorny, and K. J. Green. 2024. Desmosomes at a glance. J Cell Sci 137. [Google Scholar] [CrossRef] [PubMed]
- Pijnenburg, M. W., and R. Nantanda. 2021. Rising and falling prevalence of asthma symptoms. Lancet 398: 1542–1543. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K. F., B. R. Celli, M. E. Wechsler, R. M. Abdulai, X. Luo, M. M. Boomsma, H. Staudinger, J. E. Horowitz, A. Baras, M. A. Ferreira, M. K. Ruddy, M. C. Nivens, N. Amin, D. M. Weinreich, G. D. Yancopoulos, and H. Goulaouic. 2021. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: A genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med 9: 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Rackley, C. R., and B. R. Stripp. 2012. Building and maintaining the epithelium of the lung. J Clin Invest 122: 2724–2730. [Google Scholar] [CrossRef]
- Ramirez, S. I., F. Faraji, L. B. Hills, P. G. Lopez, B. Goodwin, H. D. Stacey, H. J. Sutton, K. M. Hastie, E. O. Saphire, H. J. Kim, S. Mashoof, C. H. Yan, A. S. DeConde, G. Levi, and S. Crotty. 2024. Immunological memory diversity in the human upper airway. Nature 632: 630–636. [Google Scholar] [CrossRef]
- Rao, W., S. Wang, M. Duleba, S. Niroula, K. Goller, J. Xie, R. Mahalingam, R. Neupane, A. A. Liew, M. Vincent, K. Okuda, W. K. O’Neal, R. C. Boucher, B. F. Dickey, M. E. Wechsler, O. Ibrahim, J. F. Engelhardt, T. C. J. Mertens, W. Wang, S. S. K. Jyothula, C. P. Crum, H. Karmouty-Quintana, K. R. Parekh, M. L. Metersky, F. D. McKeon, and W. Xian. 2020. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 181: 848–864 e18. [Google Scholar] [CrossRef]
- Ray, A., M. Camiolo, A. Fitzpatrick, M. Gauthier, and S. E. Wenzel. 2020. Are We Meeting the Promise of Endotypes and Precision Medicine in Asthma? Physiol Rev 100: 983–1017. [Google Scholar] [CrossRef]
- Reyfman, P. A., B. Khuder, J. B. Pierce, D. A. Meza, M. R. Carnethon, R. Kalhan, and S. S. Khan. 2021. Urban–Rural Mortality Disparities from Chronic Lower Respiratory Diseases in the United States, 1999–2019. Am. J. Respir. Crit. Care Med. 203: 1435–1437. [Google Scholar] [CrossRef]
- Rhodin, J. 1959. LXVII ultrastructure of the tracheal ciliated mucosa in rat and man. Ann. Otol. Rhinol. Laryngol. 68: 964–974. [Google Scholar] [CrossRef]
- Rhodin, J., and T. Dalhamn. 1956. Electron microscopy of the tracheal ciliated mucosa in rat. Z Zellforsch Mikrosk Anat 44: 345–412. [Google Scholar] [CrossRef] [PubMed]
- Rokicki, W., M. Rokicki, J. Wojtacha, and A. Dzeljijli. 2016. The role and importance of club cells (Clara cells) in the pathogenesis of some respiratory diseases. Kardiochir Torakochirurgia Pol 13: 26–30. [Google Scholar] [CrossRef] [PubMed]
- Russo, R. C., D. Togbe, I. Couillin, N. Segueni, L. Han, V. F. J. Quesniaux, T. Stoeger, and B. Ryffel. 2025. Ozone-induced lung injury and inflammation: Pathways and therapeutic targets for pulmonary diseases caused by air pollutants. Environ. Int. 198: 109391. [Google Scholar] [CrossRef] [PubMed]
- Ruysseveldt, E., K. Martens, and B. Steelant. 2021. Airway Basal Cells, Protectors of Epithelial Walls in Health and Respiratory Diseases. Front Allergy 2: 787128. [Google Scholar] [CrossRef]
- Saito, A. C., C. Endo, Y. Fukazawa, T. Higashi, and H. Chiba. 2022. Effects of TAMP family on the tight junction strand network and barrier function in epithelial cells. Ann N Acad Sci 1517: 234–250. [Google Scholar] [CrossRef]
- Sajjan, U., Q. Wang, Y. Zhao, D. C. Gruenert, and M. B. Hershenson. 2008. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med 178: 1271–1281. [Google Scholar] [CrossRef]
- Salvati, L., L. Maggi, F. Annunziato, and L. Cosmi. 2021. Thymic stromal lymphopoietin and alarmins as possible therapeutical targets for asthma. Curr Opin Allergy Clin Immunol 21: 590–596. [Google Scholar] [CrossRef]
- Sayers, I., C. John, J. Chen, and I. P. Hall. 2024. Genetics of chronic respiratory disease. Nat Rev Genet 25: 534–547. [Google Scholar] [CrossRef]
- Schreiner, T., L. Allnoch, G. Beythien, K. Marek, K. Becker, D. Schaudien, S. Stanelle-Bertram, B. Schaumburg, N. Mounogou Kouassi, S. Beck, M. Zickler, G. Gabriel, W. Baumgartner, F. Armando, and M. Ciurkiewicz. 2022. SARS-CoV-2 Infection Dysregulates Cilia and Basal Cell Homeostasis in the Respiratory Epithelium of Hamsters. Int J Mol Sci 23. [Google Scholar] [CrossRef] [PubMed]
- Shah, A. S., Y. Ben-Shahar, T. O. Moninger, J. N. Kline, and M. J. Welsh. 2009. Motile cilia of human airway epithelia are chemosensory. Science 325: 1131–1134. [Google Scholar] [CrossRef] [PubMed]
- Short, K. R., J. Kasper, S. van der Aa, A. C. Andeweg, F. Zaaraoui-Boutahar, M. Goeijenbier, M. Richard, S. Herold, C. Becker, D. P. Scott, R. W. Limpens, A. J. Koster, M. Barcena, R. A. Fouchier, C. J. Kirkpatrick, and T. Kuiken. 2016. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur Respir J 47: 954–966. [Google Scholar] [CrossRef] [PubMed]
- Singh, D., P. Guller, F. Reid, S. Doffman, U. Seppala, I. Psallidas, R. Moate, R. Smith, J. Kiraga, E. Jimenez, D. Brooks, A. Kelly, L. H. Nordenmark, M. Waqas Sadiq, L. Mateos Caballero, C. Kell, M. G. Belvisi, and H. Pandya. 2025. A phase 2a trial of the IL-33 mAb tozorakimab in patients with COPD: FRONTIER-4. Eur Respir J. [Google Scholar] [CrossRef]
- Smallcombe, C. C., D. T. Linfield, T. J. Harford, V. Bokun, A. I. Ivanov, G. Piedimonte, and F. Rezaee. 2019. Disruption of the airway epithelial barrier in a murine model of respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol 316: L358–L368. [Google Scholar] [CrossRef]
- Srisomboon, Y., K. Iijima, M. Colwell, P. J. Maniak, M. Macchietto, C. Faulk, H. Kita, and S. M. O’Grady. 2023. Allergen-induced DNA release by the airway epithelium amplifies type 2 immunity. J Allergy Clin Immunol 151: 494–508 e6. [Google Scholar] [CrossRef]
- Su, X., W. Wu, Z. Zhu, X. Lin, and Y. Zeng. 2022. The effects of epithelial-mesenchymal transitions in COPD induced by cigarette smoke: An update. Respir Res 23: 225. [Google Scholar] [CrossRef]
- Sun, D., I. LuValle-Burke, K. Pombo-Garcia, and A. Honigmann. 2022. Biomolecular condensates in epithelial junctions. Curr Opin Cell Biol 77: 102089. [Google Scholar] [CrossRef]
- Sun, S., C. Wang, J. Hu, P. Zhao, X. Wang, and W. E. Balch. 2025. Spatial covariance reveals isothiocyanate natural products adjust redox stress to restore function in alpha-1-antitrypsin deficiency. Cell Rep Med 6: 101917. [Google Scholar] [CrossRef]
- Suzuki, H., T. Nishizawa, K. Tani, Y. Yamazaki, A. Tamura, R. Ishitani, N. Dohmae, S. Tsukita, O. Nureki, and Y. Fujiyoshi. 2014. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science 344: 304–307. [Google Scholar] [CrossRef]
- Sweerus, K., M. Lachowicz-Scroggins, E. Gordon, M. LaFemina, X. Huang, M. Parikh, C. Kanegai, J. V. Fahy, and J. A. Frank. 2017. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J Allergy Clin Immunol 139: 72–81 e1. [Google Scholar] [CrossRef]
- Taylor-Blair, H. C., A. C. W. Siu, A. Haysom-McDowell, S. Kokkinis, A. Bani Saeid, D. K. Chellappan, B. G. G. Oliver, K. R. Paudel, G. De Rubis, and K. Dua. 2024. The impact of airborne particulate matter-based pollution on the cellular and molecular mechanisms in chronic obstructive pulmonary disease (COPD). Sci Total Env. 954: 176413. [Google Scholar] [CrossRef]
- Thakur, A., S. Mei, N. Zhang, K. Zhang, B. Taslakjian, J. Lian, S. Wu, B. Chen, J. Solway, and H. J. Chen. 2024. Pulmonary neuroendocrine cells: Crucial players in respiratory function and airway-nerve communication. Front Neurosci 18: 1438188. [Google Scholar] [CrossRef]
- Thomas, B., M. S. Koh, C. O’Callaghan, J. C. Allen, A. Rutman, R. A. Hirst, J. Connolly, S. Y. Low, O. Thun How, L. Chian Min, W. T. Lim, L. Lin Ean Oon, Q. He, O. H. Teoh, and T. S. Lapperre. 2021. Dysfunctional Bronchial Cilia Are a Feature of Chronic Obstructive Pulmonary Disease (COPD). COPD 18: 657–663. [Google Scholar] [CrossRef]
- Travaglini, K. J., A. N. Nabhan, L. Penland, R. Sinha, A. Gillich, R. V. Sit, S. Chang, S. D. Conley, Y. Mori, J. Seita, G. J. Berry, J. B. Shrager, R. J. Metzger, C. S. Kuo, N. Neff, I. L. Weissman, S. R. Quake, and M. A. Krasnow. 2020. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587: 619–625. [Google Scholar] [CrossRef] [PubMed]
- Troyanovsky, S. M. 2023. Adherens junction: The ensemble of specialized cadherin clusters. Trends Cell Biol 33: 374–387. [Google Scholar] [CrossRef] [PubMed]
- Turner, M. C., M. Nieuwenhuijsen, K. Anderson, D. Balshaw, Y. Cui, G. Dunton, J. A. Hoppin, P. Koutrakis, and M. Jerrett. 2017. Assessing the Exposome with External Measures: Commentary on the State of the Science and Research Recommendations. Annu Rev Public Health 38: 215–239. [Google Scholar] [CrossRef]
- Varricchi, G., R. Poto, G. Criscuolo, C. Strisciuglio, P. Nair, and G. Marone. 2025. TL1A, a novel alarmin in airway, intestinal, and autoimmune disorders. J. Allergy Clin. Immunol. 155: 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, R., E. L. Schymanski, A. L. Barabasi, and G. W. Miller. 2020. The exposome and health: Where chemistry meets biology. Science 367: 392–396. [Google Scholar] [CrossRef]
- Vila-Gonzalez, M., L. Pinte, R. Fradique, E. Causa, H. Kool, M. Rodrat, C. M. Morell, M. Al-Thani, L. Porter, W. Guo, R. Maeshima, S. L. Hart, F. McCaughan, A. Granata, D. N. Sheppard, R. A. Floto, E. L. Rawlins, P. Cicuta, and L. Vallier. 2024. In vitro platform to model the function of ionocytes in the human airway epithelium. Respir Res 25: 180. [Google Scholar] [CrossRef]
- Walton, T., M. Gui, S. Velkova, M. R. Fassad, R. A. Hirst, E. Haarman, C. O’Callaghan, M. Bottier, T. Burgoyne, H. M. Mitchison, and A. Brown. 2023. Axonemal structures reveal mechanoregulatory and disease mechanisms. Nature 618: 625–633. [Google Scholar] [CrossRef]
- Wan, M., E. M. Simonin, M. M. Johnson, X. Zhang, X. Lin, P. Gao, C. J. Patel, A. Yousuf, M. P. Snyder, X. Hong, X. Wang, V. Sampath, and K. C. Nadeau. 2025. Exposomics: A review of methodologies, applications, and future directions in molecular medicine. EMBO Mol Med 17: 599–608. [Google Scholar] [CrossRef]
- Wang, M., G. Tan, A. Eljaszewicz, Y. Meng, P. Wawrzyniak, S. Acharya, C. Altunbulakli, P. Westermann, A. Dreher, L. Yan, C. Wang, M. Akdis, L. Zhang, K. C. Nadeau, and C. A. Akdis. 2019. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol 143: 1892–1903. [Google Scholar] [CrossRef]
- Wechsler, M. E., M. K. Ruddy, I. D. Pavord, E. Israel, K. F. Rabe, L. B. Ford, J. F. Maspero, R. M. Abdulai, C. C. Hu, R. Martincova, A. Jessel, M. C. Nivens, N. Amin, D. M. Weinreich, G. D. Yancopoulos, and H. Goulaouic. 2021. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N Engl J Med 385: 1656–1668. [Google Scholar] [CrossRef]
- Widdicombe, J. H. 2019a. Early studies on the surface epithelium of mammalian airways. Am J Physiol Lung Cell Mol Physiol 317: L486–L495. [Google Scholar] [CrossRef]
- Widdicombe, J. H. 2019b. Early studies of airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 316: L990–L998. [Google Scholar] [CrossRef] [PubMed]
- Wild, C. P. 2005. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomark. Prev 14: 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Wild, C. P. 2012. The exposome: From concept to utility. Int J Epidemiol 41: 24–32. [Google Scholar] [CrossRef] [PubMed]
- Witherden, D. A., and W. L. Havran. 2013. Cross-talk between intraepithelial gammadelta T cells and epithelial cells. J Leukoc Biol 94: 69–76. [Google Scholar] [CrossRef]
- Wittekindt, O. H. 2017. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch 469: 135–147. [Google Scholar] [CrossRef]
- Yang, J., and R. Shaykhiev. 2017. Reply: Epithelial-Mesenchymal Transition: A Necessary New Therapeutic Target in Chronic Obstructive Pulmonary Disease? Am J Respir Crit Care Med 196: 394–395. [Google Scholar] [CrossRef]
- Yi, X., J. Gao, and Z. Wang. 2022. The human lung microbiome-A hidden link between microbes and human health and diseases. Imeta 1: e33. [Google Scholar] [CrossRef]
- Yoshida, M., R. Arzili, and M. Z. Nikolic. 2025. Immune-epithelial cell interactions in lung development, homeostasis and disease. Int J Biochem Cell Biol 178: 106703. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, A. J., S. Mohammed, L. Carr, M. Yavari Ramsheh, C. Micieli, V. Mistry, K. Haldar, A. Wright, P. Novotny, S. Parker, S. Glover, J. Finch, N. Quann, C. L. Brookes, R. Hobson, W. Ibrahim, R. J. Russell, C. John, M. A. Grimbaldeston, D. F. Choy, D. Cheung, M. Steiner, N. J. Greening, and C. E. Brightling. 2022. Astegolimab, an anti-ST2, in chronic obstructive pulmonary disease (COPD-ST2OP): A phase 2a, placebo-controlled trial. Lancet Respir Med 10: 469–477. [Google Scholar] [CrossRef]
- Yu, W., T. O. Moninger, A. L. Thurman, Y. Xie, A. Jain, K. Zarei, L. S. Powers, A. A. Pezzulo, D. A. Stoltz, and M. J. Welsh. 2022. Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs. Proc Natl Acad Sci U A 119. [Google Scholar] [CrossRef]
- Zhang, B., X. Feng, L. Tian, B. Xiao, L. Hou, B. Mo, and D. Yao. 2025. Epithelial-mesenchymal transition in asthma: Its role and underlying regulatory mechanisms. Front Immunol 16: 1519998. [Google Scholar] [CrossRef]
- Zimmer, S. E., and A. P. Kowalczyk. 2024. The desmosome as a dynamic membrane domain. Curr. Opin. Cell Biol. 90: 102403. [Google Scholar] [CrossRef]
Figure 1.
The structures of the airway epithelial barrier. A. The cell components of the pulmonary epithelial barrier. The airway epithelium contains basal cells, ciliated cells, club cells, PNECs, ionocytes, and Tuft cells. Basal cells can differentiate into the other six types of cells and self-renew. The airway SMG is composed of myoepithelial cells, mucous cells, and serous cells. Myoepithelial cells can differentiate into mucous cells and serous cells. It can self-renew and differentiate into basal cells. The Alveoli are composed of AT1 and AT2. AT2 can differentiate into AT1, and they can self-renew. B. The four types of intercellular junctions between AECs. C. The structural model of TACs. The TJs and AJs together form the TACs. The membrane proteins in TJs include claudins, occludins, and JAMs. The cytoplasmic tails of these proteins are connected to the branched actin filament network. The AJ membrane proteins include nectins and cadherins. The intracellular domains of these proteins are linked to the actomyosin belts, which are constituted of actin filaments and myosins. D. The three-dimensional structures of the AJC proteins. The examples of these structures shown here include claudin-15 (PDB ID: 7KP4), JAMs (PDB: 1nbq), Occludin (AlphaFold Protein Structure Database: AF-Q16625-F1-v4), Nectins (PDB ID: 3ALP), and Cadherins (PDB ID: IL3W).
Figure 1.
The structures of the airway epithelial barrier. A. The cell components of the pulmonary epithelial barrier. The airway epithelium contains basal cells, ciliated cells, club cells, PNECs, ionocytes, and Tuft cells. Basal cells can differentiate into the other six types of cells and self-renew. The airway SMG is composed of myoepithelial cells, mucous cells, and serous cells. Myoepithelial cells can differentiate into mucous cells and serous cells. It can self-renew and differentiate into basal cells. The Alveoli are composed of AT1 and AT2. AT2 can differentiate into AT1, and they can self-renew. B. The four types of intercellular junctions between AECs. C. The structural model of TACs. The TJs and AJs together form the TACs. The membrane proteins in TJs include claudins, occludins, and JAMs. The cytoplasmic tails of these proteins are connected to the branched actin filament network. The AJ membrane proteins include nectins and cadherins. The intracellular domains of these proteins are linked to the actomyosin belts, which are constituted of actin filaments and myosins. D. The three-dimensional structures of the AJC proteins. The examples of these structures shown here include claudin-15 (PDB ID: 7KP4), JAMs (PDB: 1nbq), Occludin (AlphaFold Protein Structure Database: AF-Q16625-F1-v4), Nectins (PDB ID: 3ALP), and Cadherins (PDB ID: IL3W).
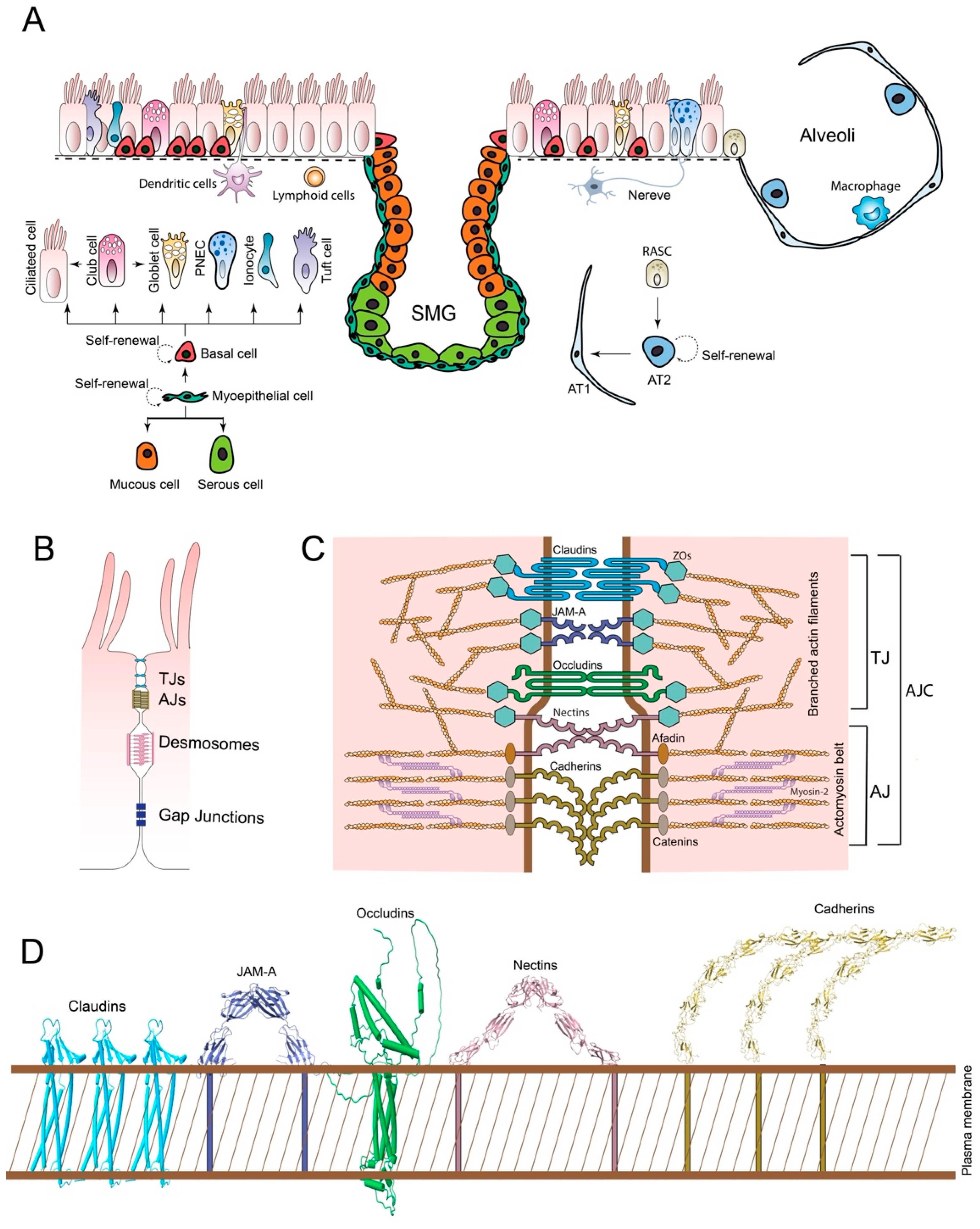
Figure 2.
The respiratory exposome. The respiratory exposome is divided into three domains: the general external respiratory exposome, the specific external respiratory exposome, and the internal respiratory exposome. Some representative components for the three domains of the respiratory exposome are shown.
Figure 2.
The respiratory exposome. The respiratory exposome is divided into three domains: the general external respiratory exposome, the specific external respiratory exposome, and the internal respiratory exposome. Some representative components for the three domains of the respiratory exposome are shown.
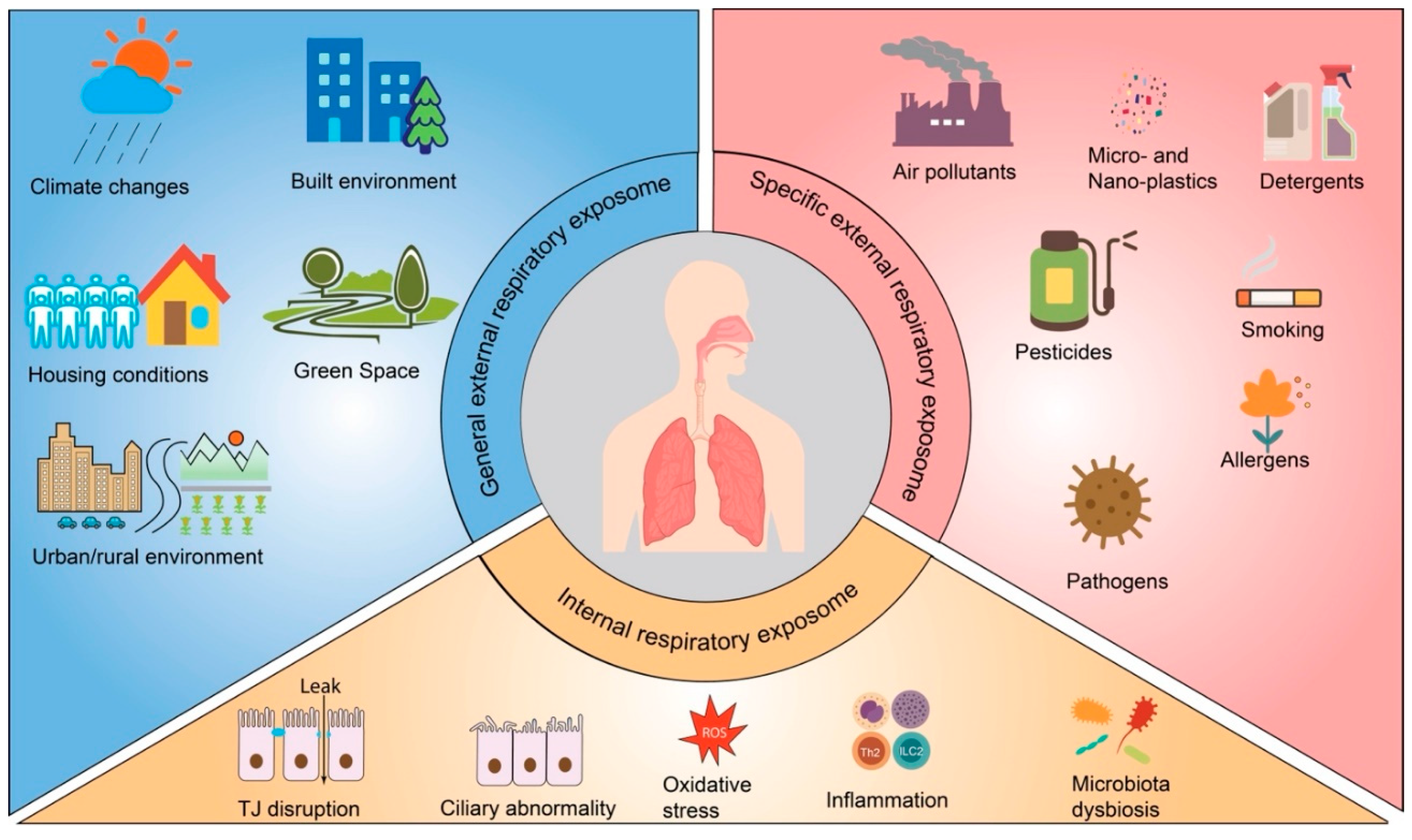
Figure 3.
Framework for integrating external and internal exposome data using computational models to derive expotypes and endotypes for precision respiratory medicine. Remote sensors, local environmental monitoring stations, or personal portable devices collect the external exposome data. In contrast, the internal exposome data are acquired using single- or multiple-omics technologies or molecular analysis (not shown here). Through a series of data processing and computational modeling methods, the expotype of the individual patients can be defined from the external exposome, and the endotype can be defined from the internal exposome. Both the expotype and the endotype can serve as references for upgrading the precision medicine strategy.
Figure 3.
Framework for integrating external and internal exposome data using computational models to derive expotypes and endotypes for precision respiratory medicine. Remote sensors, local environmental monitoring stations, or personal portable devices collect the external exposome data. In contrast, the internal exposome data are acquired using single- or multiple-omics technologies or molecular analysis (not shown here). Through a series of data processing and computational modeling methods, the expotype of the individual patients can be defined from the external exposome, and the endotype can be defined from the internal exposome. Both the expotype and the endotype can serve as references for upgrading the precision medicine strategy.
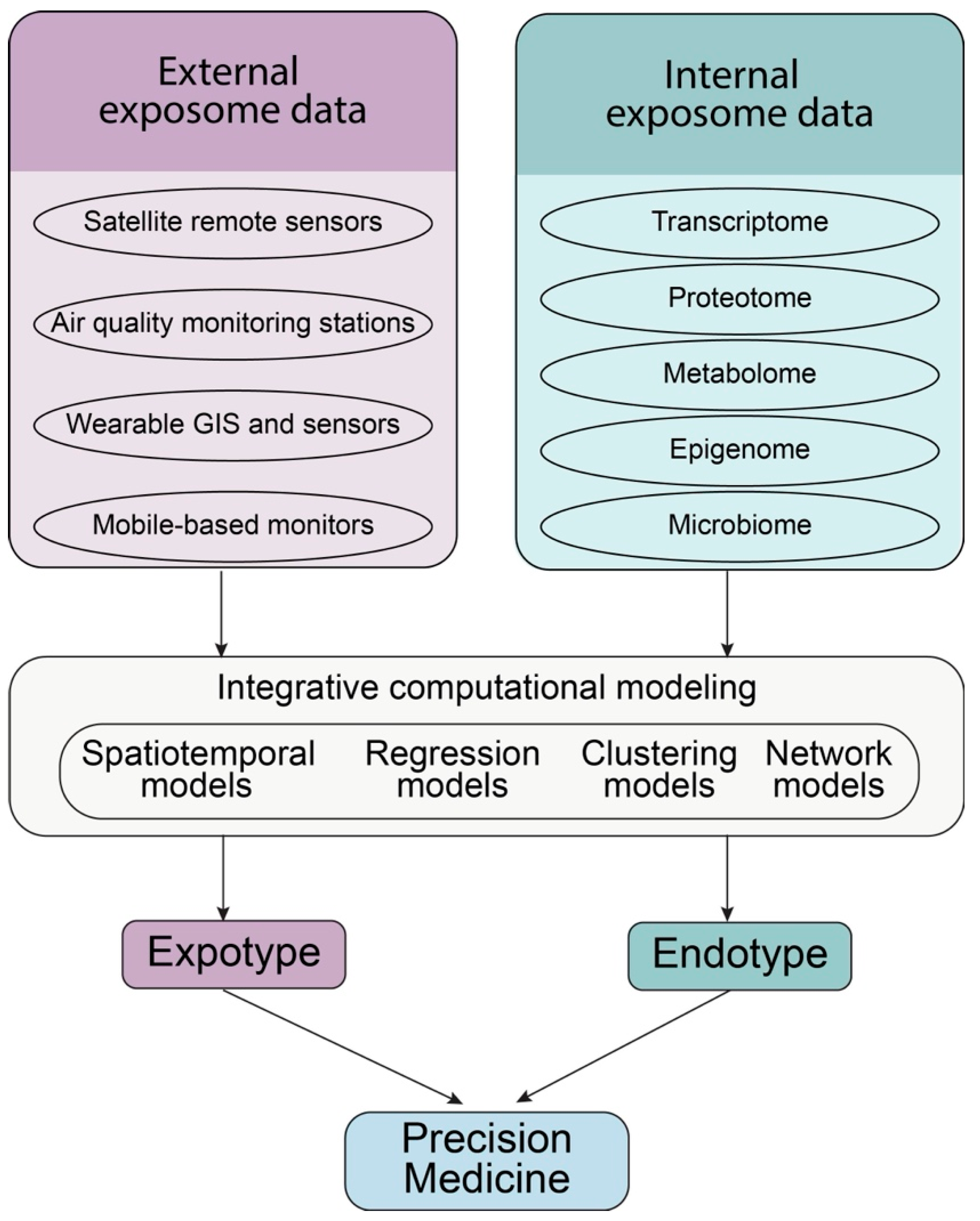

Table 1.
The specific external respiratory exposome factors that disrupt AEB integrity.
| Exposure factors | Mechanisms |
| Air pollution | |
| Particle matters (PMs) | Induce oxidative stress and inflammation (Barbier et al., 2023). |
| Lead to airway remodeling through oxidative stress, inflammation, and altered epithelial plasticity (Taylor-Blair et al., 2024). | |
| Cause mucociliary dysfunction by activating mucus secretory expression gene (Montgomery et al., 2020) and impacting the ciliary motion (Jia et al., 2019). | |
| Downregulate the level of Occludin, Claudin-1, E-cadherin, and ZO-1 (Lee et al., 2020). | |
| Ozone | Induced respiratory epithelial cell death, oxidative stress, inflammation, and barrier damage (Russo et al., 2025). |
| Nitrogen dioxide (NO2) | Decrease TJ protein expression and induce inflammation (Moratin et al., 2025). |
| Tobacco smoke | Reduce E-cadherin and ß-catenin expression and further destabilize cell adhesion by reducing the tension between epithelial cells via increasing actin polymer levels (Nishida et al., 2017). |
| Pesticides | Induce oxidative stress, and alter or disrupt of apical cell-cell junctions via decrease in the expression of proteins like E-cadherin, β-catenin, Occludin, and ZOs (Lima et al., 2022). |
| Laundry detergents | Disrupt epithelial barrier function with decreased transepithelial electrical resistance, increased paracellular flux, and irregular tight TJ structure (Wang et al., 2019). |
| Allergens | Induce mitochondrial or nuclear DNA release and nuclear DNA fragmentation in human bronchial epithelial cells (Srisomboon et al., 2023). |
| Directly digest Occludin and zonula occludens-1 (ZO-1) in airway epithelium (Jacquet, 2011). | |
| Viruses | |
| Respiratory syncytial virus (RSV) | Disrupt AJC by decrease the expression of ZO-1, Occludin, Claudin-1, Cleaves extracellular fragments of E-cadherin, depolymerizes F-actin and decreases cortactin, an actin-binding protein crucial for barrier stability (Gao et al., 2022; Smallcombe et al., 2019). |
| Lead to cilia loss and impaired mucociliary clearance (Koch et al., 2022). | |
| Influenza A viruses (IAV) | Causes significant damage to the alveolar epithelial barrier, leading to loss of tight junction integrity primarily through reduction or loss of tight junction proteins such as claudin-4 (Short et al., 2016). |
| Induce production of IFN-I, disrupting alveolar epithelial repair and tight junction integrity (Onufer et al., 2025), and causing the AEC death (Hoagland et al., 2025). | |
| Coronaviruses (CoV) | SARS-CoV-2 Disrupts TJ by the viral E protein-ZO-1 interaction (Alvarez et al., 2025). |
| SARS-CoV-2 infection led to cilia loss in hamsters (Schreiner et al., 2022). | |
| In bronchoscopy samples from long COVID patients, genes related to AEB dysfunction and mucus production were up-regulated (Gerayeli et al., 2024) | |
| Rhinovirus (RV) | Generate ROS and decrease Claudin, Occludin, E-cadherin and ZO-1 (Kim et al., 2024; Sajjan et al., 2008). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



