
Submitted:
18 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Senecavirus A (SVA) is an emerging threat to swine populations due to its potential to cause vesicular lesions, which are difficult to differentiate from other vesicular diseases of swine such as foot and mouth disease (FMD), requiring significant resources for differential diagnosis. The first Taiwanese isolate of SVA was identified in 2006, although the first clinical case was not reported until 2012. The genetic characteristics and seroprevalence of SVA in Taiwan remain unclear. This study aimed to assess the seroprevalence and genetic diversity of SVA in nurserys and finishers on Taiwanese pig farms. Phylogenetic analysis of seven Taiwanese SVA isolates revealed clustering into groups I and II. The 2006 and 2012 isolates shared 95.5% and 95.7% identity, respectively, with an early USA strain (MT360258), while more recent strains collected between 2018 and 2022 exhibited 95.7–98.8% identity with a 2020 USA strain (MZ733977). Serological analysis of 300 farms showed significantly higher herd-level seroprevalence in nurserys (53%) than finishers (6.7%). Furthermore, comparative analysis of nine known B cell epitopes showed high sequence conservation across Taiwanese and global strains. These findings provide important baseline data on the genetic diversity and seroprevalence of SVA in Taiwan and support the development of improved surveillance strategies for this emerging swine pathogen.
Keywords:
senecavirus A
; seroprevalence
; phylogenetic analysis
; Taiwan
; vesicular disease
1. Introduction
Senecavirus A (SVA), also known as Seneca Valley virus, is a non-enveloped, single-stranded positive-sense RNA virus belonging to the family Picornaviridae [1]. It is the sole member of the genus Senecavirus. The SVA genome is approximately 7.2 kb in length and encodes a polyprotein that is processed into four structural proteins (VP1, VP2, VP3, and VP4) and eight non-structural proteins. SVA was first identified in 2002 as a contaminant in cell culture media and considered a harmless virus [2,3].
Since its initial discovery, retrospective analyses have revealed that SVA had been silently circulating in North American swine populations as early as the late 1980s. The virus was sporadically detected in various clinical samples between 1988 and 2005, though its pathogenic role was unclear at the time. A 2007 case in Canada, where vesicular lesions were observed in pigs transported to the United States, raised suspicions about SVA as a potential vesicular disease agent. Similarly, an isolated case in Indiana, USA, in 2012, also linked SVA to vesicular symptoms. However, it was not until large-scale outbreaks in Brazil (2014) and the United States (2015) that SVA was recognized as an emerging pathogen responsible for vesicular disease in swine. The virus was soon identified in several other countries, including the People’s Republic of China (PROC), Colombia, and Thailand, demonstrating its potential for global spread [3,5,6,8,9,10]. These outbreaks, particularly in Brazil, were associated not only with vesicular lesions in pigs but also with epidemic transient neonatal losses (ETNL), characterized by high mortality in neonatal piglets.
In the USA, SVA has circulated in swine populations for over 30 years, with serological studies confirming its persistence[1]. The clinical signs of SVA infection, including lethargy, anorexia, fever, lameness, and the presence of vesicles on the snout, oral cavity, and coronary band, are indistinguishable from those of other vesicular diseases, including FMD, swine vesicular disease (SVD), vesicular stomatitis (VS), and vesicular exanthema of swine (VES)[4,8,9].
Molecular epidemiology studies have reclassified SVA strains into distinct phylogenetic clades, with more recent strains displaying increased pathogenicity compared to historical isolates. The widespread circulation of SVA and its ability to induce lesions indistinguishable from those caused by foot and mouth disease virus (FMDV) pose significant challenges to veterinary diagnostics and disease control. In non-FMD-endemic regions such as Taiwan, where strict surveillance measures are in place to maintain FMD-free status, the presence of SVA as a vesicular disease agent highlights the importance of continued monitoring and investigation into its epidemiology and genetic characteristics.
SVA was first isolated from blood-contaminated effluent collected from a truck used to transport pig carcasses to a rendering plant in 2006. However, the first clinical detection of SVA in Taiwan occurred in 2012, when a contracted veterinarian at a farrow-to-finish swine farm in Hualien County reported suspected vesicular lesions on the coronary bands of finisher swine to the Local Animal Disease Inspection Authority (LADIA). Samples were collected by veterinarians from LADIA and submitted to the Veterinary Research Institute (VRI) under the Ministry of Agriculture in Taiwan for testing. All samples tested negative for FMD, SVD, VS, and VES, but SVA was identified. Subsequently, SVA was isolated from two clinical cases of pigs with vesicular lesions in Taiwan. In 2018, a veterinary meat inspector at an abattoir in Tainan City also observed vesicular lesions in swine in lairage and reported them to the Animal and Plant Health Inspection Agency. In 2020, the owner of a farrow-to-finish swine farm in Tainan City also reported vesicular lesions in finisher swine to the LADIA. Samples from both cases were collected by veterinarians of LADIA and sent to the VRI for testing for FMD, SVD, VS, VES, and SVA. SVA was isolated from these samples, while agents of other vesicular diseases were not detected and these diseases ruled out. Although SVA has not been associated with significant economic losses to date, its emergence as a vesicular disease pathogen that results in clinical signs almost identical to FMD highlights the need for enhanced surveillance and understanding of its epidemiology[4].
Among vesicular diseases of swine, foot and mouth disease (FMD) is prominent due to its highly contagious nature and devastating economic consequences, posing significant threats to the swine industry[10]. After more than 23 years of concerted effort, Taiwan achieved FMD-free status without vaccination for the Taiwan, Penghu, and Matsu regions in 2020. To maintain this status, all vesicular disease cases in swine must be treated as suspected FMD until confirmed otherwise through laboratory testing[11]. This underscores the critical importance of understanding the epidemiology of the emerging vesicular disease pathogen, SVA, in Taiwan.
However, there is limited information on the prevalence and genetic characteristics of SVA in Taiwanese swine population. Establishing baseline epidemiological and phylogenetic data for SVA is essential for improving understanding of its infection dynamics and assessing its potential impact on the swine industry[12].
To address these gaps, this study was designed to: investigate the phylogenetic and epidemiological characteristics of SVA in Taiwan by determining the animal level and farm level seroprevalence of SVA in swine across the country; and characterize the genetic profile of Taiwanese SVA isolates. By comparing these isolates with those from other countries, this study seeks to provide critical insights of the epidemiology of SVA in Taiwan and support the development of effective surveillance and control strategies.
2. Materials and Methods
2.1. Sample Collection
Samples archived from previous investigations of cases of vesicular disease were also included in this study for sequencing and phylogenetic analysis. These samples had all been confirmed, at the time of collection, to be negative for FMD, SVD, VS and VES. These archived samples included vesicular epithelial samples collected from cases of vesicular lesions in finisher pigs aged 20–26 weeks in Taiwan in 2012, 2018, and 2020. Additionally, a 2006 strain (ID: HC061119) was isolated from blood-contaminated effluent collected from a truck used for transporting pig carcasses to a rendering plant. Two SVA isolates from 2021 and 2022 were also obtained from oral swab samples collected from swine that had initially tested positive for FMDV NSP antibodies in a separate FMD surveillance program. These FMD test results were subsequently confirmed as false positives based on negative results in virus neutralization tests (VNT) and nucleic acid detection for FMDV. Specifically, one strain from 2021 (ID: 1102577) was isolated from oral swabs collected from 20–26-week-old pigs in a farm in Tainan City, and one strain from 2022 (ID: 1111570) was isolated from oral swabs collected from a 20-week-old pig in a farm in Pingtung County. Additionally, another strain from 2022 (ID: 1111388) was isolated from lymph node tissue collected from a dead 8-week-old pig from a farm in Pingtung County. According to records from the veterinarian of local animal disease inspection agency, this pig had exhibited agonal respiration (gasping) prior to death. Testing conducted at the VRI identified PRRSV and SVA from the collected tissues, while FMDV and other major swine diseases were ruled out.
2.2. RNA Extraction and cDNA Synthesis
Oral swab samples were placed in 3 mL of Minimum Essential Media (MEM; Thermo Fisher Scientific, Waltham, MA, USA) immediately after collection. At the laboratory the cotton swab was pressed against the tube wall to extract the liquid and the samples centrifuged at 3,000 rpm and the supernatant collected. To prepare a 10% (w/v) homogenate, vesicular epithelial samples were homogenized and mixed with 3 mL of MEM supplemented with 5% fetal calf serum and 1% antibiotics. The homogenates were centrifuged at 3,000 rpm, and the supernatant collected. The supernatants derived from swabs and tissue homogenates were subjected to automated nucleic acid extraction using the TANBead Nucleic Acid Extraction Kit (Taiwan Advanced Nanotech Inc., Taoyuan City, Taiwan). The nucleic acid extract was eluted with 100 μL elution buffer according to the manufacturer’s instructions. cDNA was synthesized using the LightCycler® 480 system (Roche Diagnostics GmbH, Mannheim, Germany) in a 20 µL reaction mixture containing 4 µL of LightCycler® 480 Probes Master (2×; Roche Diagnostics, Mannheim, Germany), 0.1 µL of AMV Reverse Transcriptase (10 U/µL; Promega Corporation, Madison, WI, USA), 1 µL each of forward and reverse primers (20 µM), 0.5 µL of probe (10 µM), 3 µL of RNA template, and 10.4 µL of DEPC-treated water (Thermo Fisher Scientific, Waltham, MA, USA). The thermal program was as follows: 50°C for 30 min (1 cycle), 95°C for 3 min (1 cycle), followed by 45 cycles of 95°C for 15 s and 60°C for 1 min, and a final step at 40°C for 30 s. The synthesized cDNA was stored at −80°C until use.
2.3. Amplification of the SVA Genome and Sequencing
To amplify the SVA genome, we designed seven primer pairs based on the sequence of strain USA/KS15-031348/2015 (MN233025), as detailed in Table 1. PCR conditions were optimized to amplify the complete genome with the Phusion Green Hot Start II High Fidelity DNA Polymerase kit (Thermo Fisher Scientific).
PCR products were purified from agarose gels followed by Sanger’s sequencing using the BigDye Terminator V3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with the same primers as in the PCR. The sequencing reaction was prepared according to the manufacturer's instructions. The PCR reactions were performed in a GeneAmp PCR System 2700 Thermal cycler (Applied Biosystems, Foster City, CA, USA) with the following cycling parameters: 96°C initial denaturation for 1 min, followed by 45 cycles of 96°C denaturation for 10 sec, 50°C annealing for 10 sec, and 60°C elongation for 4 min. The sequencing products were analyzed using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). Each nucleotide position was confirmed by sequencing at least two times.
2.4. Sequence Identity and Phylogenetic Analysis
The full length sequences and 4 structural proteins(VP1-4) of the 7 isolated SVA strains from this study were aligned with 18 reference sequences available in GenBank (https://www.ncbi.nlm.nih.gov/genbank/), as well as 12 additional sequences provided by the Agricultural Research Service (ARS), United States Department of Agriculture (USDA).
A phylogenetic tree was constructed using the Maximum Likelihood method in MEGA XI software (Pennsylvania State University, PA, USA) with a substitution model and 1,000 bootstrap replicates to determine the genetic relationships between the Taiwanese SVA strains and those from other countries.
2.5. Epitope Sequencec Analysis
In addition, amino acid sequence comparisons of known B cell epitopes were conducted by MEGA XI software. A total of nine previously identified linear B cell epitopes within VP1, VP2, and VP3 regions were analyzed based on published literature[13,14,15]. The selected epitopes included 21GELAAP26 within VP1; 12DRVITQT18, 71WTKAVK76, 98GGAFTA103, 150KSLQELN156, 177SLGTYYR183, 248YKEGAT253 and 266SPYFNGL272 within VP2; 192GWFSLHKLTK201 within VP3. Aligned amino acid sequences from seven Taiwanese isolates and 30 representative global SVA strains were analyzed to identify potential amino acid substitutions within these epitope regions.
2.6. Serological Surveillance
Serum samples were collected from commercial swine farms in Taiwan across 19 administrative divisions (counties/cities) of five regions: northern, central, southern, eastern, and the offshore islands regions. Three administrative divisions—Keelung City, Taipei City, and Lienchiang County—were excluded due to the absence of representative swine populations. The sample size was determined using the online Epitools sample size calculator (https://epitools.ausvet.com.au/) assuming an expected herd level seroprevalence of 7.4% as reported in a 2022 study conducted on SVA seroprevalence in swine older than 20 weeks of age in the USA [16], with a 5% precision and a 95% confidence interval. Based on a 2020 survey by the Ministry of Agriculture, Taiwan there were a total of 6,497 swine farms in Taiwan. Using this number as the population of farms at risk, at least 106 swine farms were required to be sampled, however a total of 300 farms were subsequently sampled to increase representativeness and reliability of the study. Serum samples were collected from at least 14 swine on each selected farm, a number calculated to be 95% confident of detecting at least three seropositive animals assuming a within-herd animal level seroprevalence of 20%. If there were fewer than 14 swine on the selected farm, all swine on that farm were sampled. The selected swine were monitored longitudinally, with two serum samples collected. The first sample set of samples was taken during the nursery stage (6-12 weeks), and the second set collected from the same pigs during the finisher stage (20-26 weeks) stages. This sampling design allowed for the detection of neutralizing antibody changes between the two stages. Serum samples were heat-inactivated at 56 °C for 30 minutes and stored at -20°C for subsequent analysis.
2.7. Anti-SVA Neutralizing Anitibody Assay
Antibodies to SVA were detected using the VNT assay. Serial two-fold dilutions of serum were carried out and mixed with 100 TCID50 of the W107-0691 Taiwanese strain. After incubation at 37°C for 1 hour, then added the BHK-21 cells and inoculated at 37°C for 48 hours. Neutralizing titers were defined as the highest serum dilution that completely inhibited the cytopathic effect. Samples with neutralization antibody titters ≥1:64 were classified as positive[17]. Farms were classified as seropositive for SVA if farm had over than two anti-SVA VNT-positive pigs of the same stage.
2.8. Statistical Analysis
To compare the herd level seroprevalence of SVA among different groups, pairwise comparisons were conducted using GraphPad Prism 10.4.1 (GraphPad Software, LLC, San Diego, CA, USA ). Odds ratios (ORs) and their 95% confidence intervals (CIs) were also calculated to compare the seroprevalence between the regions. The OR calculations followed the methodology described in Statistical Methods in Epidemiology, Monographs in Epidemiology and Biostatistics, Vol. 12, Oxford University Press, by Kahn HA and Sempos CT (1989). The SVA seroprevalence in administrative divisions was displayed using maps created with QGIS 3.28.7 (QGIS.org, an Open Source Geospatial Foundation Project headquartered in Beaverton, OR, USA).
3. Results
3.1. Phylogenetic Analysis of Full-Length Sequence in Taiwanese SVA Strains
3.1.1. Full-Length Sequence Analysis
Seven Taiwanese strains of SVA obtained from previous sampling investigations and the FMD surveillance program were sequenced and analyzed in this study. Based on full-length sequences analysis, nucleotide and amino acids sequence identities between 7 Taiwanese isolates ranged from 92.2% to 99.6% and 96.0% to 99.0%. It showed the highest nucleotide and amino acids sequence identities to the recent USA strain SVA-USA-OH-NADC4-2020 (MZ733977) with identities ranging from 92.4% to 98.8% and 95.7% to 98.4%, respectively.
The full-length sequence phylogenetic tree revealed two groups among the Taiwanese isolates. The early isolates HC061119 (2006) and HC121223 (2012), which clustered phylogenetically with the prototype strain SVV-001, were classified into group Ia. The other five contemporary Taiwanese isolates collected after 2018, were classified into group II and clustered with USA strains from 2020 (Figure 1a). The mean nucleotide divergence (p-distance) within each group was 2.90% for group Ia, 1.57% for group Ib, and 3.27% for group II. The divergence between group Ia and Ib was 7.51%, and between group Ib and II was 6.60%, supporting their classification into distinct phylogenetic groups. HC061119-PT-2006 and HC121223 exhibited 95.1%–95.7% nucleotide and 96.7%–97.3% amino acid sequence identity with other members of group Ia. The five contemporary Taiwanese isolates obtained after 2018 showed the highest nucleotide and amino acid sequence identities to the recent USA strain SVA-USA-OH-NADC4-2020 (MZ733977), with identities ranging from 95.7% to 98.8% and 97.2% to 98.4%, respectively.
3.1.2. Structural Protein Sequence Analysis
Maximum likelihood phylogenetic trees were constructed using partial sequences of the VP1, VP2, VP3, and VP4 genes (Figures 1b–e). Across the VP1, VP2, and VP3 trees, three phylogenetic groups were consistently observed, and the bootstrap values of most nodes exceeding 70%. These groups showed topological consistency with the grouping patterns observed in the full-length sequence phylogenetic analysis result. Historical Taiwanese strains (HC061119 and HC121223) clustered within group Ia, alongside the prototype strain SVV-001. In contrast, Taiwanese isolates collected after 2018 (W107-0691, W109-05470, 1102577, 1111570, and 1111388) together with USA contemporary strains clustered within group II, indicating a shift toward contemporary virus lineages. The bootstrap support values of VP4 were generally lower, and clear separation into three groups was not observed.
3.1.3. Epitope Sequence Analysis
Amino acid sequence alignment of the nine linear B cell epitopes across seven Taiwanese and 30 representative global SVA strains revealed a high degree of conservation. Seven out of nine epitopes were completely conserved across all analyzed strains, including ²¹GELAAP²⁶ in VP1; ⁹⁸GGAFTA¹⁰³, ¹⁵⁰KSLQELN¹⁵⁶, ¹⁷⁷SLGTYYR¹⁸³, ²⁴⁸YKEGAT²⁵³, and ²⁶⁶SPYFNGL²⁷² in VP2; and ¹⁹²GWFSLHKLTK²⁰¹ in VP3. Two minor amino acid variations were observed in the VP2 ¹²DRVITQT¹⁸ epitope, three strains—MT360258, HC061119, and KY486158—contained a threonine (T) instead of isoleucine (I) at position 15, resulting in the sequence DRVTTQT. All other strains, including Taiwanese isolates, carried the DRVITQT variant. In the VP2 ⁷¹WTKAVK⁷⁶ epitope, one Taiwanese strain (1111570) exhibited a unique substitution of glycine (G) for tryptophan (W) at position 71, resulting in WGKAVK. No other substitutions were detected in the remaining epitope regions among the analyzed strains.
3.2. Seroprevalence of SVA in Nursery and Finishing Stages
3.2.1. Herd and Animal Level Seroprevalence in Nursery Swine
A total of 300 herds in nursery stage were sampled and assayed. The results revealed that 159 herds were classified as seropositive and herd level seroprevalence of 53.0%; 95% CI, 47.2–58.8. Overall, the herd level seroprevalence in nursery stage differed significantly across the five regions (P = 0.0001, χ² = 22.88; df = 4,1). The herd seroprevalence of nursery stage was highest in the southern region (65.7%, 95% CI, 57.0–73.7), followed by the central region (47.2%, 95% CI, 38.1–56.4). The eastern region had a similar herd level seroprevalence of 37.5% (95% CI, 8.5–75.5) in nursery stage to the northern region (37.0%, 95% CI, 19.4–57.6). No nursery swine from herds located on offshore islands were seropositive (95% CI, 0.0-45.9) (Table 2). The herd level seroprevalence of nursery stage in the southern region was significantly higher than that of the northern region (OR 3.3; 95% CI, 1.4–7.7, P < 0.01). All other regional herd level seroprevalences of nursery stage were not significantly different (Table 2 and Figure 2a).
Overall there was a significant difference in the herd seroprevalence of nurserys between the 19 counties/cities (P < 0.0001, χ² = 53.01; df = 18,1). Notably, Pingtung County had the highest herd level seroprevalence in nursery stage (82.1%, 95% CI: 69.6–91.1), which was significantly higher than that in several other counties/cities, including Penghu County (P = 0.0399), Kinmen County (P = 0.0001), Taitung County (P = 0.0021), Changhua County (P = 0.0031),Tainan City (P = 0.0003), Changhua County (P < 0.0001), Yunlin County (P = 0.0017), Miaoli County (P = 0.0053), Taichung City (P = 0.0007), Taoyuan City (P = 0.0200), and Yilan County (P = 0.0002).
Of the 4,508 nursery pigs sampled, 1,634 were seropositive, with an animal level seroprevalence of 36.2% (95% CI, 34.8–37.7). The animal level seroprevalence for nurserys also varied between regions (P < 0.0001, χ² = 222.78; df = 4,1): southern region 46.3% (95% CI, 44.1–48.6), central region 31.4% (95% CI, 29.3–33.6), eastern region 27.5% (95% CI, 19.7–36.4), northern region 24.0% (95% CI, 19.9–28.4), and offshore islands 0.0% (95% CI, 0.0–2.4) (Table 2). The animal level seroprevalence of nursery stage in the central region was significantly higher than in the northern region (OR: 1.5, 95% CI, 1.1-1.9, P = 0.0033), and the southern region also showed significantly higher odds than the northern region (OR: 2.7, 95% CI, 2.2-3.5, P < 0.0001). Although no positive samples were detected in swine from offshore islands, the comparison with the northern region still yielded a statistically significant result on a Chi-square test (P < 0.0001); however OR could not be calculated for this group (Table 2).
A county level comparison of animal level seroprevalence in nursery stage also showed an overall significant difference among the 19 counties/cities sampled (P < 0.0001, χ² = 90.56; df = 18,1). Pingtung County exhibited the highest animal level seroprevalence in nursery stage (57.5%, 95% CI: 54.1–60.9), which was significantly higher than that observed in several other counties/cities, including Penghu County (P < 0.0001), Kinmen County (P < 0.0001), Taitung County (P < 0.0001), Chiayi City (P < 0.0001), Chiayi County (P < 0.0001), Kaohsiung City (P = 0.0082), Tainan City (P < 0.0001), Changhua County (P < 0.0001), Yunlin County (P < 0.0001), Miaoli County (P < 0.0001), Nantou County (P < 0.0001), Taichung City (P < 0.0001), Hsinchu County (P < 0.0001), Hsinchu City (P < 0.0001), Taoyuan City (P < 0.0001), and Yilan County (P < 0.0001).
3.1.2. Herd and Animal Level Seroprevalence in Finisher Swine
Of the 300 herds of finisher stage sampled and assayed. The results revealed only 20 herds were classified as seropositive and herd level seroprevalence of 6.7%; 95% CI, 4.1–10.1. The highest herd level seroprevalence for finishers was observed in the central region (8.9%; 95% CI, 4.5–15.4, followed by the northern region (7.4%, 95% CI, 0.9–24.3) and southern region (5.2%; 95% CI, 2.1–10.5). No finisher herds were classified as seropositive in the eastern region or offshore islands (Table 3 and Figure 2b).
The herd level seroprevalence of finisher stage was similar between the five regions (P = 0.62, χ² = 2.64; df = 4,1). Similarly, no significant differences were observed in the herd level seroprevalences of finisher stage among the 19 counties/cities (P = 0.13, χ² = 24.68; df = 18,1). Nevertheless, Miaoli County exhibited the highest herd level seroprevalence in finisher stag (37.5%, 95% CI: 8.5–75.5), which was significantly higher than that in several other counties/cities, including Kaohsiung City (P = 0.0359), Pingtung County (P = 0.0222), Tainan City (P = 0.0457), and Yunlin County (P = 0.0209).
A total of 4,145 finisher swine were tested, and 192 pigs were classified as seropositive (animal level seroprevalence of 4.6%; 95% CI, 4.0–5.3). The animal level seroprevalence for finisher swine varied between the five regions (P = 0.0004, χ² = 20.65; df = 4,1) with the highest animal level seroprevalence of finisher stage in the central region (6.1%; 95% CI, 5.0–7.4) compared to the southern region (4.2%; 95% CI, 3.3–5.2), northern region (2.6%; 95% CI, 1.3–4.8), eastern region (0.9%; 95% CI, 0.0–4.7), and offshore islands (0.8%; 95% CI, 0.0–4.2) (Table 2). The animal level seroprevalence of finisher stage in the central region was significantly higher than that of the offshore islands (OR 8.47; 95% CI, 1.17–61.23, P = 0.0055). All other regional animal level seroprevalences in finisher stage were not significantly different (Table 3).
Overall the animal level seroprevalence of finishers was significantly different (P < 0.0001, χ² = 486.63; df = 18,1) between the 19 counties/cities. Miaoli County had the highest animal level seroprevalence in finisher stage(17.65%, 95% CI, 11.27–25.70), which was significantly higher than that observed in several other counties/cities, including Penghu County (P = 0.0080), Kinmen County (P < 0.0001), Hualien County (P = 0.0003), Taitung County (P = 0.0026), Chiayi City (P = 0.0142), Chiayi County (P = 0.0007), Kaohsiung City (P < 0.0001), Pingtung County (P < 0.0001), Tainan City (P < 0.0001), Changhua County (P < 0.0001), Yunlin County (P < 0.0001), Nantou County (P = 0.0013), Taichung City (P < 0.0001), Hsinchu County (P < 0.0001), Hsinchu City (P = 0.0080), New Taipei City (P = 0.0011), and Yilan County (P < 0.0001).
3.1.3. Comparison of Seroprevalence Between Nursery and Finisher Stages
The herd level seroprevalence of nursery pigs (53.0%; 95% CI, 47.2–58.8) was significantly higher than that of finishers (6.7%; 95% CI, 4.1-10.1, P < 0.0001). Similarly, the animal level seroprevalence of nursery pigs was significantly higher than that of finishers (36.2%; 95% CI, 34.8–37.7% for nursery pigs vs. 4.6%; 95% CI, 4.0–5.3% for finishers, P < 0.0001).
To further explore the relationship between seroprevalence in the nursery and finisher stages, the results were classified to four patterns: (1) Pattern I, which were seropositive in both the nursery and finisher stages (n = 14); (2) Pattern II, which were seropositive in the nursery stage but seronegative in the finisher stage (n = 145); (3) Pattern III, which were seronegative in the nursery stage but seropositive in the finisher stage (n = 6); and (4) Pattern IV, which were seronegative in both stages (n = 135). There was no significant difference in the overall herd level seroprevalence between nursery pigs and finishers among all patterns (P = 0.11, χ² = 2.49; df = 1; OR 2.17, 95% CI, 0.81–5.82). Notably, RRT-PCR testing was conducted among the six herds classified as Pattern 3, and three of these herds tested positive for SVA RNA.
4. Discussion
The phylogenetic analysis of SVA isolates from Taiwan revealed distinct clustering patterns corresponding to group Ia and II, consistent with studies by Joshi et al. (2020). HC061119 and HC121223, belonging to group Ia, showed high nucleotide identity (95.5% and 95.7%) with US-SVV-001-P3-2002, a strain with close phylogenetic relationship to the earliest characterized SVA viruses in the USA[5]. This suggested that the Taiwanese historical strains may have originated from an early lineage of SVA. Furthermore, the five more recent Taiwanese strains (2018–2022) exhibited higher nucleotide (95.7–98.8%) and amino acid (97.2%–98.4%) sequence identity with SVA-USA-TN-NADC5-2020 (MZ733977), indicating the potential influence of international animal transport on viral evolution. Similar clustering of recent isolates within group II has been observed in other countries, emphasizing the global spread and diversification of SVA. A study by Buckley et al. (2021) further supported the evolutionary trends of SVA strains, reporting genetic differences between the SVV 001/2002 strain and other contemporary SVA strains. Their study revealed that SVV 001/2002 belonged to an independent evolutionary clade, while the other strains clustered within a separate clade[18]. These findings aligned with our analysis of Taiwanese SVA strains, which also demonstrated genomic evolution of SVA. To further delineate the genetic relationships among SVA strains, we constructed phylogenetic trees based on the complete genome and the four structural protein-coding regions: VP4, VP2, VP3, and VP1. The clustering patterns derived from nucleotide sequences analyses of VP1-VP3 were largely consistent with those from the full-length sequences phylogenetic tree, reinforcing the robustness of the observed phylogenetic groupings. Conversely, VP4 exhibited lower resolution with inconsistent clustering, likely due to the shorter sequence length and limited variability of the VP4 region. Therefore, VP4-based phylogenetic analysis should be interpreted with caution and regarded as supplementary to the more informative VP1–VP3-based results. These structural protein nucleotide sequence level comparisons provide additional evidence for the genetic divergence between the older Taiwanese strains (e.g., SVA-HC121223-HL-2012 in group Ia) and contemporary isolates, supporting the hypothesis of multiple introduction events or independent evolutionary events. Joshi et al. (2020) analyzed the full-length sequences of historical and contemporary SVA strains and reported a 6.32% genetic divergence between them. Their phylogenetic analysis revealed that most SVA strains cluster based on their geographic origins, suggesting independent evolution in different regions. However, some contemporary strains from PROC and Colombia clustered with strains isolated from the USA, indicating possible cross-regional transmission of SVA strains[5]. Houston et al. (2020) provided a comprehensive review of the global distribution and evolutionary trends of SVA. They noted that historical SVA strains were primarily detected in North America, whereas contemporary SVA strains have spread globally, including the Americas and Asia. Furthermore, they highlighted the genetic divergence between historical and contemporary SVA strains, which aligns with the findings of Joshi et al. (2020). Previous reviews supported our observations on the global dissemination and genetic evolution of SVA, reinforcing the complexity of its evolutionary dynamics and transmission patterns[3]. Furthermore, Saeng-chuto et al. (2018) reported that the SVA strains detected in Thailand were closely related to Canadian strains, suggesting that international swine transport is a contributing factor in the spread of SVA[19]. The clustering of contemporary Taiwanese SVA isolates within group II highlights potential regional transmission patterns and viral evolution. Understanding these evolutionary dynamics is crucial for developing targeted control measures.
Moreover, analysis of linear B cell epitopes revealed high sequence conservation across Taiwanese and global strains, supporting their potential utility in broad-range diagnostic assays and immunogen design. Notably, most epitopes remained unchanged despite phylogenetic divergence, indicating antigenic stability. The few observed amino acid substitutions were rare and limited to individual strains, suggesting minimal impact on epitope-based recognition. These findings reinforce the suitability of conserved epitopes as reliable molecular targets. Interestingly, a substitution observed within the VP2 ¹²DRVITQT¹⁸ epitope was only observed in early North American isolates, including the first reported USA strain MT360258 (2002), NC011349 (2008), and one Canadian strain KY486158 (2015). This variation was absent in all other strains, including recent global and Taiwanese isolates. Such a pattern suggests that the T15I substitution may represent a variation from early SVA lineages rather than a feature of ongoing antigenic drift. Its disappearance in contemporary strains might reflect evolutionary constraints or selection pressure favoring the conserved DRVITQT motif. Similarly, a unique W71G substitution in the VP2 ⁷¹WTKAVK⁷⁶ epitope was observed in only one recent Taiwanese strain (1111570), with no comparable variation detected in other strains. The high conservation of this epitope in contemporary strains further supports its potential for serological assay development.
As an emerging vesicular disease pathogen in swine, SVA has spread across major swine-producing regions in the Americas and Asia [8], with recent cases now reported in Europe. Notably, during a bilateral video conference on SVA research between VRI and the Pirbright Institute in the United Kingdom in 2024, it was highlighted that SVA cases have been detected in the UK, further emphasizing its expanding geographic distribution (UK Government News, 2024). Despite its growing geographic distribution in the swine industry, information on the animal and herd level prevalence of SVA is scant. One of the major challenges in studying the epidemiology of SVA is its tendency to result in subclinical infections, which may be unlikely to be detected without active surveillance or laboratory confirmation [5,16,20]. Therefore, establishing baseline prevalence data is crucial for enhancing our understanding of the virus’s occurrence within pig populations and its national geographic distribution.
Our study revealed distinct differences in the seroprevalence of SVA between nursery and finisher swine, both at the herd and animal levels. The herd level seroprevalence for nursery swine was 53.0% (95% CI, 47.2–58.8), while for finisher swine, it was significantly lower at 6.7% (95% CI, 4.1–10.1, P<0.0001). Similarly, the animal level seroprevalence in nursery swine was 36.2% (95% CI, 34.8–37.7), which was significantly higher (P < 0.0001) than the 4.6% (95% CI, 4.0–5.3) detected in finisher swine. However, herds were not significantly likely to seroconvert or become seronegative between the two sampling time points. Among the six herds classified as late seroconversion—those seronegative at the nursery stage but seropositive at the finisher stage—RRT-PCR testing was performed, and viral RNA was detected in three of them. This indicates that active or recent infection likely occurred between the two sampling points. The detection of SVA RNA in these herds provides additional support for the interpretation that late seroconversion reflects recent exposure post-weanings. It also underscores the dynamic nature of SVA transmission and highlights the value of integrating serological and molecular surveillance to capture the complete picture of virus circulation within herds.
The seropositive pigs in the nursery stage that turned seronegative in the finisher stage suggest possible shifts in immunity. Which are likely driven by maternal antibody transfer followed by subsequent viral exposure to the environment. These findings provide valuable insight into the immune status of pigs across different stages of growth and support the hypothesis that maternal immunity plays a key role in the immunity patterns observed in Taiwanese swine, as reported in other international studies ([21,22]). The observed reduction in seroprevalence with age in our study differs from the findings reported by Houston et al. (2019), where the herd level seroprevalence in nursery-finisher pigs (6–26 weeks) and sows (>26weeks) in the USA were 42.7% and 75.8%, respectively. Houston et al. noted in their discussion that while pigs around six weeks of age were included in their study, the presence of maternal antibodies in the youngest pigs could not be completely ruled out. This grouping approach and the potential influence of maternal immunity may have contributed to the higher overall seroprevalence reported in their study compared to our stratified analysis of different growth stages [22]. In contrast, the seroprevalence of 7.4% in finisher pigs (20 weeks or older) reported by Preis et al. (2022) aligns closely with the low seroprevalence observed in finisher pigs in our study. Our study found a higher seroprevalence in nursery pigs compared to finisher pigs, which is consistent with Houston et al.'s findings of a higher prevalence in nursery-finisher pigs compared to the lower prevalence reported by Preis et al. in finisher pigs. Additionally, Preis et al. reported a herd level seroprevalence of 17.3% in breeding pigs, which was higher than that observed in finisher pigs[23]. The stark contrast between the high sow seroprevalence reported by Houston et al. (2016) (75.8%) and the much lower rate observed by Preis et al. (2022) (17.3%) may indicate a declining trend in SVA circulation in the USA swine population over time. Notably, Houston’s study was conducted in 2016, shortly after the peak of SVA outbreaks in 2015, whereas Preis’s study was conducted during 2018–2019, when viral transmission may have diminished. This temporal difference underscores the importance of longitudinal surveillance in tracking the epidemiological dynamics of emerging vesicular diseases. In addition to temporal factors, methodological differences may also account for some of the variation in seroprevalence estimates. Preis et al. used immunofluorescence assay (IFA) for serological detection, while Houston et al. employed both ELISA and IFA with parallel interpretation, potentially increasing assay sensitivity. In contrast, our study used VNT, which, while more time and resource-intensive, is recommended by WOAH as the gold standard for antibody detection in other vesicular disease of swine caused by Picornaviridae viruses, such as foot and mouth disease, due to its high specificity and reliability.
SVA transmission can occur through both direct contact with infected pigs and indirect contact with contaminated environments, transportation vehicles, or personnel. Breeding herds typically have higher stocking densities compared to nursery-finisher herds, which may facilitate more efficient viral transmission, further exacerbating the spread of SVA in these environments[24]. Furthermore, sows could experience immune suppression during their reproductive cycles, which may increase their susceptibility to SVA infections. This immune suppression in sows could contribute to the higher SVA prevalence reported in these animals[25], as the compromised immune response makes them more likely to virus shedding and subsequent transmission to their piglets. This, in conjunction with persistent viral shedding, may contribute to the higher seroprevalence in sows compared to nursery-finisher pigs [25]. It was demonstrated that piglets born to SVA-infected sows acquire maternal antibodies, which can persist for up to 98 days post-birth[21]. As sows are usually kept in commercial piggeries for several years they are more likely to be exposed to infection. The persistent nature of SVA infections complicates control efforts, as sows have been shown to shed the virus for up to 60 days post-infection. In contrast, when pigs are moved to nursery-finisher pens after weaning, they are housed in larger spaces with lower stocking density, reducing their potential exposure to the virus. Additionally, the faster turnover rate of nursery-finisher pigs—typically slaughtered around six months—further reduces their exposure time. Maternal immunity[22] likely contributes to the higher seroprevalence observed in nursery swine compared to finishers, which aligns with the findings of this study.
Our study also highlights significant regional variations in SVA seroprevalence. The herd level seroprevalence in nursery swine was significantly higher in the southern region compared to the northern region (OR 3.3; 95% CI, 1.4–7.7, P < 0.01). Also, the animal level seroprevalence in the central region was 1.5 times higher than that in the northern region (OR 1.5; 95% CI, 1.1–1.9, P = 0.0033), while the southern region exhibited even higher odds of seropositivity (OR 2.7; 95% CI, 2.2–3.5, P < 0.0001). For finisher swine, the highest herd level seroprevalence was observed in the central region, where the animal level seroprevalence was significantly higher than that of offshore islands (OR 8.5; 95% CI, 1.2–61.2, P = 0.0055). This regional difference may suggest that environmental or management factors, such as swine density, and farm-to-farm contacts, may play a role in SVA spread. Swine farming in Taiwan is primarily concentrated in the central and southern regions, which could contribute to the higher seroprevalence observed in these areas. Previous studies have highlighted the importance of regional differences and management factors in influencing the prevalence of swine diseases[11,16]. The close contact among pigs during mixing at markets or transportation significantly increases the risk of SVA transmission. Studies from North Carolina in USA have demonstrated that the circulation of SVA is prevalent in secondary markets (slaughterhouses that purchased lower quality pigs and cull sow markets[8]). This highlighted that direct or indirect close contact between pigs, whether through mixing at markets or in densely stocked environments, facilitates the spread of the virus. Interestingly, no seropositive farms were detected from offshore islands in both the nursery and finisher stages. This might be a result of their geographical isolation.
Although SVA infection typically does not cause systemic disease, its vesicular lesions are clinically indistinguishable from those caused by high-consequence diseases such as FMD. The time and resources required for differential diagnosis are substantial, emphasizing the importance of studies like this in providing essential baseline data and informing the development of improved surveillance and diagnostic strategies.
5. Conclusions
This study confirmed the widespread presence of SVA in Taiwanese swine herds, with both herd level and animal level seroprevalence significantly higher in nursery swine compared to finisher swine. While this suggests that SVA is likely spreading within swine populations, the specific mechanisms of transmission were not directly investigated in this study, underscoring the need for further research to elucidate its pathways and dynamics. Although SVA infection does not typically result in clinical disease, its vesicular lesions are similar to those of notifiable diseases such as FMD. The differentiation of these conditions requires time and resources, emphasizing the importance of understanding the epidemiology of SVA. This study provides critical baseline data to support targeted surveillance and control measures, which are essential for reducing the diagnostic burden and economic costs associated with vesicular disease outbreaks in swine populations.
The observed regional differences in herd seroprevalence highlight the central and southern regions as potential hotspots for SVA transmission. Phylogenetic analyses revealed that historical Taiwanese SVA strains are closely related to early USA strains, whereas Taiwanese strains from 2018–2022 show a closer relationship to contemporary USA strains. Additionally, the high conservation of key B cell epitopes among Taiwanese and global SVA strains suggests that currently available or future serological assays and immunogen-based interventions may be applicable. These findings enhance our understanding of the epidemiology and molecular evolution of SVA, offering valuable insights for the development of effective strategies to mitigate its impact on the swine industry.
Supplementary Materials
None.
Author Contributions
Conceptualization, C.-J. Pan, K.-J. Tsai, and Y.-L. Huang; Methodology, C.-J. Pan, Y.-L. Huang, K.-J. Tsai, and I. D. Robertson; Formal Analysis: C.-J. Pan, Y.-L. Huang and I. D. Robertson; Supervision, Y.-L. Huang, K.-J. Tsai, M.-C. Deng and I. D. Robertson; Investigation, C.-J. Pan; Resources, N.-N. Lin, and K. Lager; Data Curation, C.-J. Pan; Writing—Original Draft, C.-J. Pan; Writing—Review & Editing, Y.-L. Huang, K.-J. Tsai, and I. D. Robertson; Project Administration: J.-C. Chang, and M.-C. Deng; Funding Acquisition, M.-C. Deng. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Agriculture, Taiwan, under grant number 111AS-5.1.1-HI-H7.
Institutional Review Board Statement
Ethical review and approval were waived for this study as the animal specimens used were collected as part of national Taiwanese routine surveillance programs and in compliance with national animal health protocols. Serum samples were obtained from swine under the classical swine fever surveillance program. All samples were collected by veterinarians from the local animal disease inspection authority. Vesicular epithelium samples were collected following the guidelines outlined in the WOAH Manual, Chapter 3.1.8: Foot and Mouth Disease (Infection with Foot and Mouth Disease Virus). No additional procedures were performed on the animals solely for the purpose of this research study.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding authors upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Houston, E.; Giménez-Lirola, L.G.; Magtoto, R.; Mora-Díaz, J.C.; Baum, D.; Piñeyro, P.E. Seroprevalence of Senecavirus A in Sows and Nursery-Finisher Pigs in Major Swine Producing-States in the United States. Prev. Vet. Med. 2019, 165, 1–7. [CrossRef]
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete Genome Sequence Analysis of Seneca Valley Virus-001, a Novel Oncolytic Picornavirus. J. Gen. Virol. 2008, 89, 1265–1275. [CrossRef]
- Houston, E.; Temeeyasen, G.; Piñeyro, P.E. Comprehensive Review on Immunopathogenesis, Diagnostic and Epidemiology of Senecavirus A. Virus Res. 2020, 286, 198038. [CrossRef]
- Arzt, J.; Bertram, M.R.; Vu, L.T.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Palinski, R.; Stenfeldt, C.; Fish, I.H.; Hoang, B.H.; et al. First Detection and Genome Sequence of Senecavirus A in Vietnam. Microbiol. Resour. Announc. 2019, 8, e01247-18. [CrossRef]
- Joshi, L.R.; Mohr, K.A.; Gava, D.; Kutish, G.; Buysse, A.S.; Vannucci, F.A.; Piñeyro, P.E.; Crossley, B.M.; Schiltz, J.J.; Jenkins-Moore, M.; et al. Genetic Diversity and Evolution of the Emerging Picornavirus Senecavirus A. J. Gen. Virol. 2020, 101, 175–187. [CrossRef]
- Leme, R.A.; Zotti, E.; Alcântara, B.K.; Oliveira, M. V.; Freitas, L.A.; Alfieri, A.F.; Alfieri, A.A. Senecavirus A: An Emerging Vesicular Infection in Brazilian Pig Herds. Transbound. Emerg. Dis. 2015, 62, 603–611. [CrossRef]
- Sun, D.; Vannucci, F.; Knutson, T.P.; Corzo, C.; Marthaler, D.G. Emergence and Whole-Genome Sequence of Senecavirus A in Colombia. Transbound. Emerg. Dis. 2017, 64, 1346–1349. [CrossRef]
- Hause, B.M.; Myers, O.; Duff, J.; Hesse, R.A. Senecavirus A in Pigs, United States, 2015. Emerg. Infect. Dis. 2016, 22, 1323–1325. [CrossRef]
- Joshi, L.R.; Fernandes, M.H.V.; Clement, T.; Lawson, S.; Pillatzki, A.; Resende, T.P.; Vannucci, F.A.; Kutish, G.F.; Nelson, E.A.; Diel, D.G. Pathogenesis of Senecavirus a Infection in Finishing Pigs. J. Gen. Virol. 2016, 97, 3267–3279. [CrossRef]
- Paton, D.J.; Gubbins, S.; King, D.P. Understanding the Transmission of Foot-and-Mouth Disease Virus at Different Scales. Curr. Opin. Virol. 2018, 28, 85–91. [CrossRef]
- Segalés, J.; Barcellos, D.; Alfieri, A.; Burrough, E.; Marthaler, D. Senecavirus A: An Emerging Pathogen Causing Vesicular Disease and Mortality in Pigs? Vet. Pathol. 2017, 54, 11–21. [CrossRef]
- Leme, R.A.; Alfieri, A.F.; Alfieri, A.A. Update on Senecavirus Infection in Pigs. Viruses 2017, 9, 170. [CrossRef]
- Fan, H.; Zhu, H.; Li, S.; Shi, M.; Zhou, E.; Wang, X.; Jiang, P.; Bai, J. Identification of Linear B Cell Epitopes on VP1 and VP2 Proteins of Senecavirus A (SVA) Using Monoclonal Antibodies. Vet. Microbiol. 2020, 247, 108753,. [CrossRef]
- Chen, M.; Chen, L.; Wang, J.; Mou, C.; Chen, Z. Identification of a B-Cell Epitope in the Vp3 Protein of Senecavirus a. Viruses 2021, 13, 2300. [CrossRef]
- Zhou, H.; Sun, M.; Su, S.; Meng, L.; Yang, W.; Yang, L.; Shi, X.; Li, X.; Wang, H.; Ma, H.; et al. Identification of a Linear B-Cell Epitope on the “Puff” Loop of the Senecavirus A VP2 Protein Involved in Receptor Binding. Front. Microbiol. 2024, 15, 1387309. [CrossRef]
- Preis, G.; Sanhueza, J.M.; Vilalta, C.; Vannucci, F.A.; Culhane, M.R., Corzo, C.A. Senecavirus A Seroprevalence and Risk Factors in United States Pig Farms. Front. Vet. Sci. 2022, 9, 1011975. [CrossRef]
- Goolia, M.; Vannucci, F.; Yang, M.; Patnayak, D.; Babiuk, S.; Nfon, C.K. Validation of a Competitive ELISA and a Virus Neutralization Test for the Detection and Confirmation of Antibodies to Senecavirus A in Swine Sera. J. Vet. Diagn. Invest. 2017, 29, 250–253. [CrossRef]
- Buckley, A.C.; Michael, D.D.; Faaberg, K.S.; Guo, B.; Yoon, K.J.; Lager, K.M. Comparison of Historical and Contemporary Isolates of Senecavirus A. Vet. Microbiol. 2021, 253, 108946. [CrossRef]
- Saeng-chuto, K.; Rodtian, P.; Temeeyasen, G.; Wegner, M.; Nilubol, D. The First Detection of Senecavirus A in Pigs in Thailand, 2016. Transbound. Emerg. Dis. 2018, 65, 285–288. [CrossRef]
- Gimenez-Lirola, L.G.; Rademacher, C.; Linhares, D.; Harmon, K.; Rotolo, M.; Sun, Y.; Baum, D.H.; Zimmerman, J.; Piñeyro, P. Serological and Molecular Detection of Senecavirus a Associated with an Outbreak of Swine Idiopathic Vesicular Disease and Neonatal Mortality. J. Clin. Microbiol. 2016, 54, 2082–2089. [CrossRef]
- Yang, F.; Zhu, Z.; Liu, H.; Cao, W.; Zhang, W.; Wei, T.; Zheng, M.; Zhang, K.; Tian, H.; Zeng, Q.; et al. Evaluation of Antibody Response in Sows after Vaccination with Senecavirus a Vaccine and the Effect of Maternal Antibody Transfer on Antibody Dynamics in Offspring. Vaccines (Basel) 2021, 9, 1066. [CrossRef]
- Houston, E.; Giménez-Lirola, L.G.; Magtoto, R.; Mora-Díaz, J.C.; Baum, D.; Piñeyro, P.E. Seroprevalence of Senecavirus A in Sows and Nursery-Finisher Pigs in Major Swine Producing-States in the United States. Prev. Vet. Med. 2019, 165, 1–7. [CrossRef]
- Preis, G.; Sanhueza, J.M.; Vilalta, C.; Vannucci, F.A.; Culhane, M.R.; Corzo, C.A. Senecavirus A Seroprevalence and Risk Factors in United States Pig Farms. Front. Vet. Sci. 2022, 9, 1011975. [CrossRef]
- Dee, S.; Brands, L.; Edler, R.; Schelkopf, A.; Nerem, J.; Spronk, G.; Kikuti, M.; Corzo, C.A. Further Evidence That Science-Based Biosecurity Provides Sustainable Prevention of Porcine Reproductive and Respiratory Syndrome Virus Infection and Improved Productivity in Swine Breeding Herds. Animals 2024, 14, 2530. [CrossRef]
- Maggioli, M.F.; Fernandes, M.H. V.; Joshi, L.R.; Sharma, B.; Tweet, M.M.; Noll, J.C.G.; Bauermann, F. V.; Diel, D.G. Persistent Infection and Transmission of Senecavirus A from Carrier Sows to Contact Piglets. J. Virol. 2019, 93, e00819-19. [CrossRef]
Figure 1.
Maximum likelihood phylogenetic trees of Senecavirus A strains constructed using MEGA XI with 1,000 bootstrap replicates based on (a) full-length sequence, (b) VP1, (c) VP2, (d) VP3, and (e) VP4 sequences. Bootstrap values above 70% are shown at the nodes. Solid circles indicate SVA strains from Taiwan identified in this study. Solid squares represent strains closely related to the Taiwanese viruses based on full-length sequence phylogenetic analysis. The corresponding GenBank accession numbers for each Taiwanese strain are: HC061119 (PV002715), HC121223 (PV002716), W107-0691 (PV002717), W109-0547 (PV002718), 1102577 (PV002719), 1111388 (PV002713), and 1111570 (PV002714).
Figure 1.
Maximum likelihood phylogenetic trees of Senecavirus A strains constructed using MEGA XI with 1,000 bootstrap replicates based on (a) full-length sequence, (b) VP1, (c) VP2, (d) VP3, and (e) VP4 sequences. Bootstrap values above 70% are shown at the nodes. Solid circles indicate SVA strains from Taiwan identified in this study. Solid squares represent strains closely related to the Taiwanese viruses based on full-length sequence phylogenetic analysis. The corresponding GenBank accession numbers for each Taiwanese strain are: HC061119 (PV002715), HC121223 (PV002716), W107-0691 (PV002717), W109-0547 (PV002718), 1102577 (PV002719), 1111388 (PV002713), and 1111570 (PV002714).
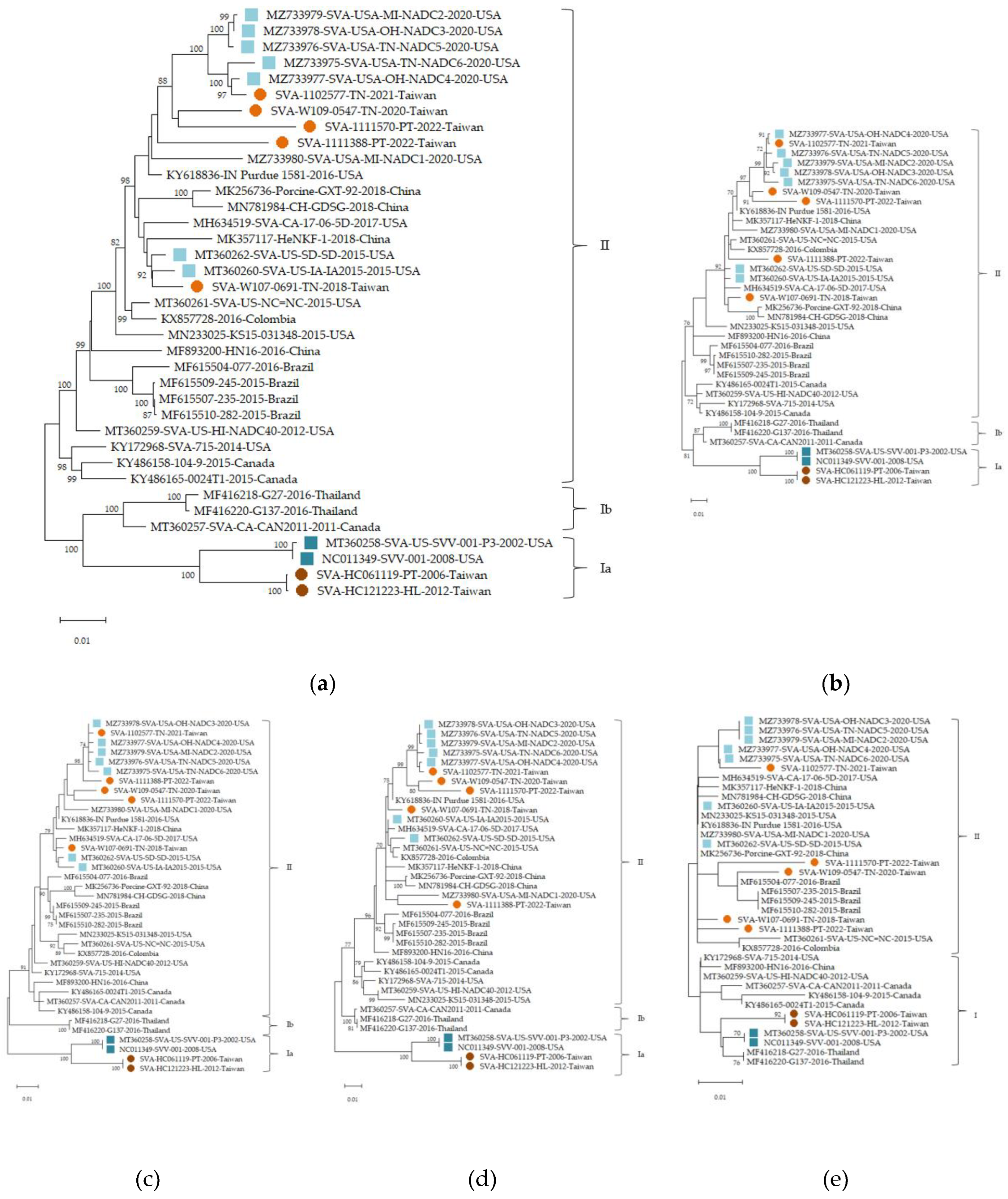
Figure 2.
Geographical distribution of SVA herd level seroprevalence across counties and cities in Taiwan during the nursery stage (a) and the finisher stage (b). The map shows the seroprevalence of Senecavirus A (SVA) across different counties in Taiwan. Each county is colored according to its county/city level seroprevalence (%), highlighting areas with high and low infection rates.
Figure 2.
Geographical distribution of SVA herd level seroprevalence across counties and cities in Taiwan during the nursery stage (a) and the finisher stage (b). The map shows the seroprevalence of Senecavirus A (SVA) across different counties in Taiwan. Each county is colored according to its county/city level seroprevalence (%), highlighting areas with high and low infection rates.
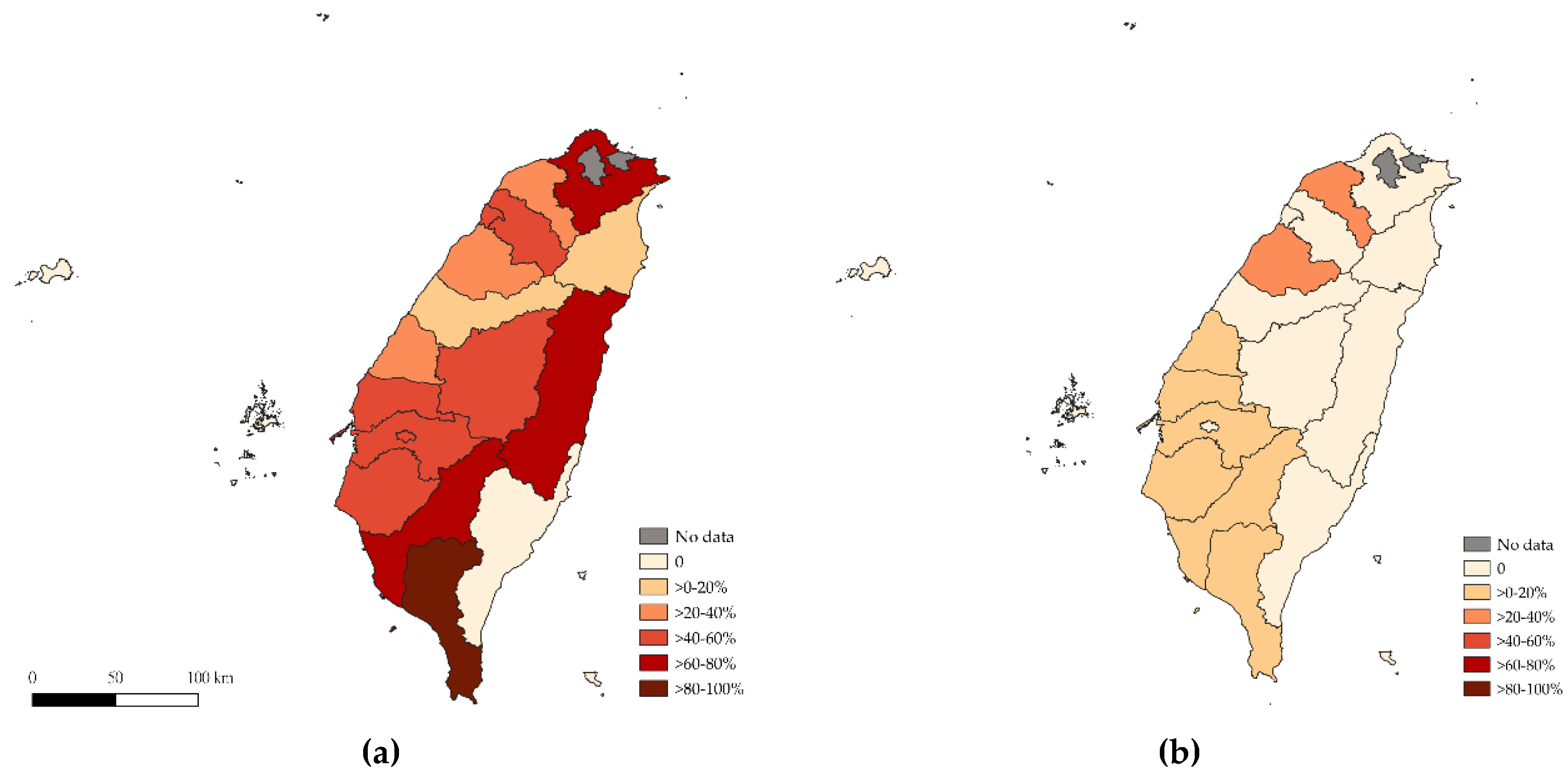

Table 1.
Primers used for full-length genome sequencing of Senecavirus (SVA) in this study.
| Primer | Sequence (5′ -3′ ) | Location of MN233025(nt) |
| SVA-1F | ATGCCCAGTCCTTCCTTTCC | 18-782 |
| SVA-1R | CGAATCGTAAACACCATTGTTCACC | 18-782 |
| SVA-2F | TACTGCCTGATAGGGCGAC | 608-1408 |
| SVA-2R | CCGTTGAGGCCTCCCT | 608-1408 |
| SVA-3F | GCCATCGACAGGTGGTACA | 1290-2497 |
| SVA-3R | TAGTCACTGGGCGAGATGTAG | 1290-2497 |
| SVA-4F | ATGGCAAGAGGGAAATTCCT | 2343-3821 |
| SVA-4R | TGGAGGAGGCGGTTCTAC | 2343-3821 |
| SVA-5F | CGCTATCTAACCAAGCTTCAG | 3689-5198 |
| SVA-5R | GTTAGGCTGTTGCATTTCCAT | 3689-5198 |
| SVA-6F | AAGTACTTCTCTGGCTCTGATACA | 5049-6434 |
| SVA-6R | AGGATGGGATTGAAACTTGG | 5049-6434 |
| SVA-7F | CTACTCTGATCATGTCTTCCAAAC | 6272-7295 |
| SVA-7R | TTTTTTTTTTTTTTTTCCCTTTTCTGTCCC | 6272-7295 |
Table 2.
Herd level seroprevalence of SVA in swine at the nursery stage across different regions of Taiwan.
Table 2.
Herd level seroprevalence of SVA in swine at the nursery stage across different regions of Taiwan.
| Regions | Number of test-positive farms/Total number tested |
Herd level seroprevalence (95% CI) |
OR (95% CI) |
p-Value | Number of test-positive animals/Total number tested |
Animal level seroprevalence (95% CI) |
OR (95% CI) |
p-Value |
| Northern | 10/27 | 37.0%(19.4-57.6) | 1.0 | - | 97/405 | 24.0%(19.9-28.4) | 1.0 | - |
| Central | 58/123 | 47.2%(38.1-56.4) | 1.5(0.6-3.6) | 0.3969 | 573/1,824 | 31.4%(29.3-33.6) | 1.5(1.1-1.9) | 0.0033* |
| Southern | 88/134 | 65.7%(57.0 -73.7) | 3.3(1.4-7.7) | 0.0087 * | 931/2,009 | 46.3%(44.1-48.6) | 2.7(2.2-3.5) | <0.0001* |
| Eastern | 3/8 | 37.5%(8.5-75.5) | 1.0(0.2-5.2) | 1 | 33/120 | 27.5%(19.7-36.4) | 1.2(0.8-1.9) | 0.4702 |
| Offshore islands | 0/8 | 0.0(0.0-36.9) | - | 0.0734 | 0/150 | 0.0%(0.0-2.4) | - | <0.0001* |
| Total | 159/300 | 53.0%(47.2-58.8) | - | - | 1,634/4,508 | 36.2%(34.8-37.7) | - | - |
* Indicates a statistically significant difference (P < 0.05) compared to the reference regions, which is the northern region for herd level and animal level seroprevalence.
Table 3.
Herd level seroprevalence of SVA in swine at the finisher stage across different regions of Taiwan.
Table 3.
Herd level seroprevalence of SVA in swine at the finisher stage across different regions of Taiwan.
| Regions | Number of test-positive farms/Total number tested |
Herd level seroprevalence (95% CI) |
OR (95% CI) |
p-Value | Number of test-positive animals/Total number tested |
Animal level seroprevalence (95% CI) |
OR (95% CI) |
p-Value |
| Northern | 2/27 | 7.4%(0.9-24.3) | 1.45(0.3-7.4) | 0.6473 | 10/380 | 2.6%(1.3-4.8) | 3.5(0.5-27.7) | 0.304 |
| Central | 11/123 | 8.9%(4.6-15.4) | 1.78(0.7-4.8) | 0.3286 | 102/1667 | 6.1%(5.0-7.4) | 8.5 (1.2-61.2) | 0.0055* |
| Southern | 7/134 | 5.2%(2.1-10.5) | 1.0 | - | 78/1852 | 4.2%(3.3-5.2) | 5.7(0.8-41.4) | 0.0596 |
| Eastern | 0/8 | 0(0.0-36.9) | - | 1 | 1/115 | 0.9%(0.0-4.7) | 1.1 (0.1-18.4) | 1 |
| Offshore islands | 0/8 | 0(0.0-36.9) | - | 1 | 1/131 | 0.8%(0.0-4.2) | 1.0 | - |
| Total | 20/300 | 6.7%(4.1-10.1%) | - | - | 192/4145 | 4.6%(4.0-5.3%) | - | - |
* Indicates a statistically significant difference (P < 0.05) compared to the reference regions, which is the southern region for herd level seroprevalence and the offshore islands for animal level seroprevalence.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



