
Submitted:
07 August 2025
Posted:
07 August 2025
You are already at the latest version
Abstract
Obesity-induced insulin resistance and type 2 diabetes mellitus (T2DM) represent complex systemic disorders marked by chronic inflammation, oxidative stress, mito-chondrial dysfunction, and endoplasmic reticulum (ER) stress. These pathophysiologi-cal processes disrupt insulin signaling and β-cell function, leading to impaired glucose homeostasis across multiple organs. Conventional therapies often target isolated pathways, overlooking the intricate molecular crosstalk and organelle-level disturb-ances driving disease progression.
Citrus-derived polyphenols—including hesperidin, naringenin, nobiletin, and tange-retin have emerged as promising agents capable of orchestrating a multi-targeted “metabolic reprogramming.” These compounds modulate key signaling pathways, in-cluding AMPK, PI3K/Akt, NF-κB, and Nrf2, thereby enhancing insulin sensitivity, re-ducing pro-inflammatory cytokine expression, and restoring redox balance. Further-more, they improve mitochondrial biogenesis, stabilize membrane potential, and alle-viate ER stress by modulating the unfolded protein response (UPR), thus supporting cellular energy homeostasis and protein folding capacity.
Evidence from preclinical studies and select clinical trials suggests that citrus poly-phenols can significantly improve glycemic control, reduce oxidative and inflamma-tory markers, and preserve β-cell function. Their pleiotropic actions across molecular and organ-level targets position them as integrative metabolic modulators. This review presents a systems-level synthesis of how citrus polyphenols rewire metabolic signal-ing networks and organelle resilience, offering a holistic therapeutic strategy to miti-gate the root causes of obesity-induced insulin resistance.
Keywords:
citrus polyphenols
; metabolic reprogramming
; insulin resistance
; mitochondrial dysfunction
; endoplasmic reticulum stress
; AMPK signaling
; inflammatory cytokines
; type 2 diabetes mellitus (T2DM)
1. Introduction
It begins quietly, an expanding waistline, a subtle shift in blood sugar levels, a creeping fatigue. But behind these seemingly mundane signs brews a global biological crisis: the unstoppable rise of obesity-linked type 2 diabetes mellitus (T2DM). This twin epidemic, now termed “diabesity,” has grown into a full-blown metabolic pandemic [1,2]. As of 2023, more than 650 million adults are classified as obese, and over 500 million people live with diabetes, with projections pointing toward 700 million cases by 2045 [3,4,5,6]. Numbers, yes, but behind them lies a silent molecular war.
At the heart of this storm is a cascade of metabolic miscommunications. As adipose tissue expands, it not only stores excess energy but also morphs into a pro-inflammatory endocrine organ. Hypertrophied adipocytes release a torrent of cytokines such as TNF-α, IL-6, and MCP-1—recruiting macrophages and igniting a chronic inflammatory loop [7,8,9,10]. The result? The activation of NF-κB, JNK, and SOCS3, key molecular saboteurs that degrade insulin signaling, disrupt glucose uptake, and sow the seeds of insulin resistance across the liver, muscle, and pancreas [10,11,12,13]. But inflammation is only the beginning. With each calorie surplus, cells are flooded with free fatty acids, triggering lipotoxicity, ceramide accumulation, and a collapse in mitochondrial efficiency [14,15]. Overloaded mitochondria generate reactive oxygen species (ROS), feeding a cycle of oxidative damage that mutates proteins, disrupts redox balance, and disables insulin receptors [16,17]. Meanwhile, the endoplasmic reticulum (ER) tasked with protein folding buckles under metabolic pressure. UPR sensors, such as PERK, IRE1, and ATF6, are activated, prompting β-cells to undergo apoptosis and further exacerbating metabolic dysfunction in the body [18,19].
Despite our arsenal of pharmacological tools such as metformin, insulin, and GLP-1 agonists, there remains no cure for diabesity, only containment. These agents address symptoms but leave the root networks of dysfunction largely untouched [20]. This limitation has propelled research toward integrative, multi-targeted interventions derived from nature’s pharmacopeia.
Among these, citrus polyphenols have emerged as compelling candidates. Found in oranges, grapefruits, and tangerines, flavonoids like hesperidin, naringenin, nobiletin, and tangeretin extend far beyond antioxidant function. They operate as molecular systems modulators capable of metabolic reprogramming [21]. These compounds activate AMPK, stimulate PI3K/Akt, inhibit NF-κB, and restore insulin sensitivity in the liver, adipose tissue, and skeletal muscle [21,22,23,24,25]. They modulate SIRT1–PGC1α pathways, stabilize mitochondrial membrane potential, and reduce ER stress through unfolded protein response signaling [21]. In doing so, they mitigate hyperglycemia, improve lipid metabolism, and prevent β-cell failure in preclinical models [25].
This review unpacks how citrus polyphenols intervene at key metabolic junctions, recalibrating molecular circuits, rejuvenating organelle function, and restoring systemic homeostasis. As our understanding deepens, these nature-derived compounds may offer more than dietary support; they may serve as network-based therapeutics in the fight against diabesity.
2. Pathophysiology of Obesity-Induced Diabetes
2.1. Lipotoxicity and Free Fatty Acid-Mediated Insulin Resistance
As the storm of chronic inflammation rages through expanding adipose tissue, another silent disruptor spreads through the bloodstream, free fatty acids (FFAs). In the obese state, basal lipolysis is persistently elevated, turning fat depots into active endocrine disruptors. These circulating FFAs are eagerly taken up by insulin-sensitive tissues, including the liver and skeletal muscle, where they undergo incomplete oxidation or are converted into lipotoxic intermediates such as diacylglycerols (DAGs) and ceramides [26,27].
These molecules act not merely as metabolic byproducts, but as intracellular saboteurs. By activating serine/threonine kinases such as c-Jun N-terminal kinase (JNK) and protein kinase C (PKC), they interfere with insulin signaling at its core, through the inhibitory serine phosphorylation of insulin receptor substrates (IRS1/2). The downstream result is twofold: in skeletal muscle, glucose uptake is impaired; in the liver, glucose production remains inappropriately elevated [28,29,30,31]. These defects form the biochemical bedrock of systemic insulin resistance. Yet, this mechanism is not universally expressed. A paradox remains: not all individuals with obesity develop T2DM. Beneath this variability lies a complex interplay of mitochondrial oxidative capacity, lipid partitioning, and intracellular buffering efficiency. Some individuals appear to be equipped with metabolic machinery capable of neutralizing lipotoxicity [32,33,34,35]. This variability suggests the urgent need for stratified approaches, where mitochondrial phenotype, not body mass index, guides therapeutic strategy.
In the liver, lipotoxicity takes on another form: hepatic insulin resistance. This defect impairs the suppression of gluconeogenic enzymes under insulin stimulation, resulting in fasting and postprandial hyperglycemia. Lipid-laden hepatocytes, struggling under the weight of FFA influx, experience impaired AKT activation and PKC-mediated insulin receptor dysfunction [36,37,38]. Meanwhile, Kupffer cells, the liver’s resident macrophages, release inflammatory cytokines that compound the damage and amplify insulin resistance [39]. Still, one critical question lingers: is hepatic steatosis the cause or merely a companion of insulin resistance? Although these conditions often coexist, emerging evidence suggests that hepatic inflammation and lipotoxicity, not simple steatosis, are the more potent drivers of metabolic impairment [40,41,42]. As such, targeting inflammatory and lipid-signaling pathways within the liver may offer greater therapeutic promise than attempts to merely de-fat the organ.
2.2. Oxidative Stress, Mitochondrial Dysfunction, and ER Disruption
As lipotoxic intermediates sabotage insulin signaling from the surface, a deeper disturbance brews within the cell—oxidative stress. In obesity, the ectopic deposition of lipids in liver and skeletal muscle overburdens cellular respiration, particularly within the mitochondria. The result is an overproduction of reactive oxygen species (ROS), driven not only by mitochondrial electron transport chain leakage but also by upregulated NADPH oxidase (NOX) activity [43,44,45].
These ROS act as rogue messengers, damaging proteins, lipids, and DNA, and activating redox-sensitive inflammatory pathways, such as NF-κB and JNK, which further disrupt insulin receptor signaling [46,47]. Moreover, elevated oxidative stress impairs mitochondrial ATP production, undermines metabolic flexibility, and sets the stage for mitochondrial dysfunction—a central node in the pathogenesis of insulin resistance [48,49]. Yet, ROS are not universally destructive. At physiological levels, they function as essential signaling molecules, fine-tuning insulin sensitivity and energy flux. It is only when their production surpasses the buffering capacity of antioxidant systems that they become pathological. This duality makes ROS modulation a therapeutic tightrope—too much suppression may blunt necessary signaling, while too little invites metabolic collapse [49,50].
But the damage doesn’t end at the mitochondria. Just beyond lies another vulnerable organelle, the endoplasmic reticulum (ER), which is tasked with protein folding, calcium storage, and lipid biosynthesis. In obesity, ER homeostasis is frequently disrupted, as excess nutrients and oxidative signals trigger the unfolded protein response (UPR). Key sensors: PERK, IRE1, and ATF6, become chronically activated, leading to maladaptive signaling, impaired insulin receptor trafficking, and even β-cell apoptosis [50,51,52].
Compounding the crisis, ER stress and mitochondrial dysfunction do not act in isolation. Instead, they engage in bidirectional cross-talk, where ROS produced by mitochondrial overload exacerbate ER stress, and ER stress in turn destabilizes mitochondrial dynamics and calcium homeostasis [53,54]. This self-reinforcing loop amplifies cellular distress, propelling the insulin-resistant phenotype forward.
Thus, in the obese insulin-resistant state, the combined dysfunction of mitochondria and ER forms a molecular sinkhole, collapsing the delicate architecture of intracellular communication and energy balance. Targeting this axis, mitochondrial redox tuning, and restoration of ER homeostasis may be essential for effective metabolic reprogramming.
2.3. β-Cell Compensation and Failure
As insulin resistance deepens in peripheral tissues, the metabolic burden shifts to the pancreas. For a time, the β-cells rise to the occasion. In early obesity, these endocrine sentinels respond to rising glycemic demand with compensatory hypersecretion of insulin, working overtime to preserve normoglycemia [55,56].
But this heroism comes at a cost. With chronic exposure to hyperglycemia and elevated FFAs, a biochemical storm known as glucolipotoxicity, β-cells begin to falter. Initially, their function deteriorates: insulin granule synthesis declines, secretory machinery loses fidelity, and glucose-stimulated insulin secretion becomes blunted [57,58].
Over time, this dysfunction becomes irreversible. A convergence of molecular insults, oxidative stress, ER stress, amyloid deposition, and islet-localized inflammation drives β-cells toward apoptosis and loss of mass [59]. Mitochondrial inefficiency further impairs insulin granule exocytosis, while pro-inflammatory cytokines such as IL-1β and IFN-γ infiltrate islets and exacerbate cell death [60]. Eventually, the tipping point is reached: the remaining β-cell pool is no longer sufficient to compensate. Glucose levels rise unchecked, and what began as a compensatory adaptation gives way to overt T2DM [61]. Yet the fate of the β-cell may not be entirely sealed. Some dysfunction appears reversible, at least in early stages, raising hope for therapeutic intervention. GLP-1 receptor agonists, for example, have been shown to enhance β-cell function, promote insulin gene expression, and even reduce apoptosis. However, restoring β-cell mass, not just improving function, remains a challenging task [62,63].
To this end, regenerative strategies such as β-cell neogenesis, trans-differentiation, and reprogramming of other pancreatic cells are under active exploration. While preclinical data offer tantalizing glimpses of success, translation into durable human therapies is still in its infancy [64]. Thus, the story of the β-cell in diabesity is one of early heroism, gradual unraveling, and an uncertain future. Preserving its function and perhaps, one day, restoring its mass may hold the key to altering the trajectory of metabolic disease.
2.4. Adipose Tissue Dysfunction and Immune Activation
As systemic insulin resistance develops, one of its earliest instigators lies buried in the expansion of fat depots. In the setting of chronic overnutrition, adipocytes enlarge through hypertrophy, outpacing their oxygen supply and creating localized hypoxia. This cellular stress activates hypoxia-inducible factors (HIFs) and initiates an inflammatory cascade [65,66].
Stressed adipocytes begin secreting danger signals, including chemokines such as monocyte chemoattractant protein-1 (MCP-1), which draw immune cells into the adipose microenvironment. There, monocytes differentiate into classically activated (M1) macrophages, forming crown-like structures around dying adipocytes and releasing a storm of pro-inflammatory cytokines: TNF-α, IL-6, and IL-1β [65]. These cytokines interfere with insulin receptor signaling by inhibiting the serine phosphorylation of IRS1, ultimately contributing to systemic insulin resistance [67]. At the same time, levels of adiponectin decline—an insulin-sensitizing adipokine with potent anti-inflammatory and vascular-protective properties. This drop further tips the balance toward a pro-inflammatory adipose phenotype, perpetuating metabolic dysfunction [68].
But this immune-metabolic dialogue is far from uniform. Depot-specific differences—between visceral, subcutaneous, and brown adipose tissue—shape the inflammatory landscape, as do the plastic and dynamic phenotypes of resident immune cells. In some depots, regulatory T cells, eosinophils, and alternatively activated (M2) macrophages attempt to counterbalance inflammation, though their roles remain incompletely understood [65]. As this immunological remodeling unfolds, translational researchers face a challenge: current animal models and cell lines oversimplify the dynamic complexity of human adipose tissue. The precise orchestration of immune-adipocyte interactions during obesity progression and weight loss remains a frontier of investigation, requiring refined model systems and longitudinal tissue profiling [69,70,71].
Thus, the immune-adipocyte axis stands as a central pillar of metabolic inflammation, a battlefield where energy sensing, immune signaling, and hormonal control collide.
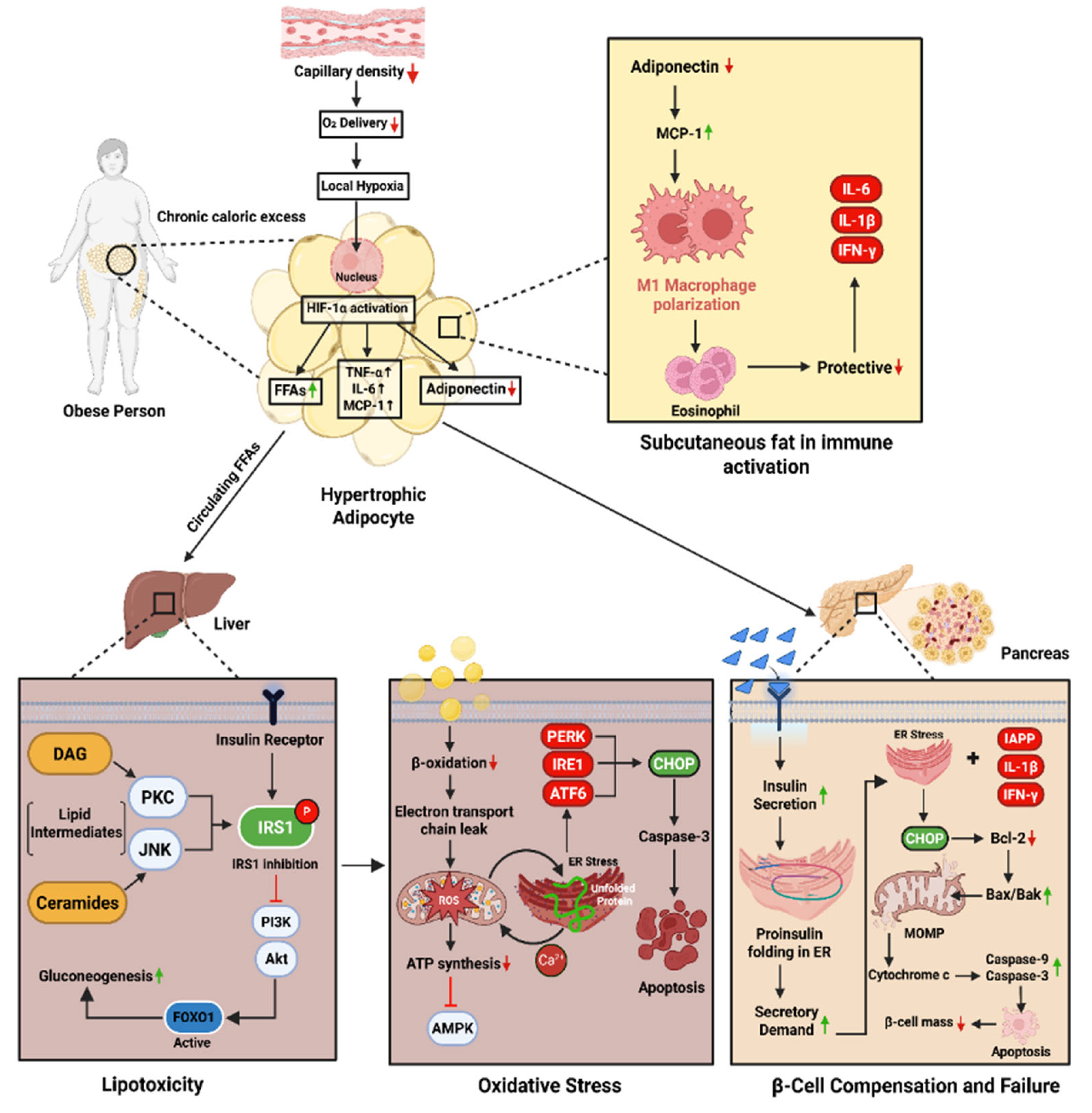
Figure 1.
Schematic representation of the mechanisms linking obesity to β-cell failure and T2DM. Chronic caloric excess induces adipocyte hypertrophy and local hypoxia, activating HIF-1α and promoting the release of FFAs, TNF-α, IL-6, and MCP-1, while reducing adiponectin. This triggers M1 macrophage polarization and inflammation in adipose tissue. Circulating FFAs and lipid intermediates reach the liver, activating PKC/JNK pathways and impairing IRS1–PI3K–Akt signaling, resulting in increased gluconeogenesis. Concurrently, impaired β-oxidation and mitochondrial dysfunction lead to ROS accumulation, ER stress, and apoptosis. In the pancreas, inflammatory and ER stress signals impair insulin synthesis and activate mitochondrial apoptotic pathways, reducing β-cell mass and insulin secretion—contributing to hyperglycemia and T2DM.
Figure 1.
Schematic representation of the mechanisms linking obesity to β-cell failure and T2DM. Chronic caloric excess induces adipocyte hypertrophy and local hypoxia, activating HIF-1α and promoting the release of FFAs, TNF-α, IL-6, and MCP-1, while reducing adiponectin. This triggers M1 macrophage polarization and inflammation in adipose tissue. Circulating FFAs and lipid intermediates reach the liver, activating PKC/JNK pathways and impairing IRS1–PI3K–Akt signaling, resulting in increased gluconeogenesis. Concurrently, impaired β-oxidation and mitochondrial dysfunction lead to ROS accumulation, ER stress, and apoptosis. In the pancreas, inflammatory and ER stress signals impair insulin synthesis and activate mitochondrial apoptotic pathways, reducing β-cell mass and insulin secretion—contributing to hyperglycemia and T2DM.
3. Citrus Polyphenols in Metabolic Reprogramming
3.1. Sour Yet Sweet Salvation: How Citrus Polyphenols Rewire Diabetic Metabolism
Until now, the story has unfolded as a progressive unraveling of metabolic order, characterized by lipotoxicity, oxidative stress, mitochondrial dysfunction, inflammatory infiltration, and β-cell failure [15,72,73]. But nature, too, offers its counterstrike. Quietly nestled among citrus fruits, a class of polyphenolic compounds has shown a surprising capacity to engage the metabolic network, not as blunt tools, but as precise modulators capable of reprogramming disordered pathways [21].
Among these, hesperidin, naringenin, quercetin, tangeretin, kaempferol, and rutin stand out not for acting in isolation, but for their ability to orchestrate multi-system metabolic restoration.
Hesperidin, a major flavanone in oranges, initiates its metabolic rescue by activating AMPK the cell’s energy sensor. This activation restores PI3K/Akt signaling, enabling the translocation of GLUT4 in skeletal muscle and adipose tissue, thereby reviving insulin responsiveness [21,74,75,76]. Simultaneously, hesperidin attenuates ROS accumulation and boosts endogenous antioxidants like SOD, GPx, and catalase, relieving oxidative stress and restoring mitochondrial integrity key to breaking the self-perpetuating cycle of insulin resistance [[75]. Quercetin, abundant in citrus and other plants, reinforces this network-level modulation. It not only improves glycemic control via PI3K/Akt and IRS1 restoration, but also blocks NF-κB-mediated inflammation, decreasing expression of TNF-α and IL-6—two upstream drivers of adipose and hepatic insulin resistance [76,77]. Quercetin’s ability to modulate macrophage polarization further reshapes the immune–metabolic interface in adipose tissue [78,79].
Then comes naringenin, the aglycone of naringin, which enters the liver and shifts metabolic flux toward fatty acid oxidation. It activates PPARα, downregulates SREBP-1c, and inhibits JNK signaling, thereby reducing lipogenesis, inflammation, and insulin resistance in hepatocytes [21,80,81]. In skeletal muscle, it reinforces AMPK signaling, improving glucose uptake and restoring oxidative capacity [82,83].
Tangeretin, sourced from citrus peels, contributes by modulating adipokine profiles—notably restoring adiponectin levels, which enhances insulin sensitivity through both AMPK activation and Pparγ modulation [84,85]. It also suppresses oxidative stress markers in insulin-sensitive tissues, reinforcing mitochondrial health and preserving metabolic signaling fidelity [86]. Kaempferol, while less abundant, wields a unique role in suppressing hepatic gluconeogenesis. By activating AMPK, it downregulates PEPCK and G6Pase, two key enzymes responsible for excess hepatic glucose output in insulin-resistant states [87,88,89,90]. Through this mechanism, it reprograms hepatic energy flow and corrects fasting hyperglycemia [89,90]. Rutin, structurally similar to quercetin, amplifies this ensemble response by restoring mitochondrial biogenesis, enhancing SIRT1–PGC1α signaling, and mitigating oxidative stress, particularly in the pancreas and liver [89].
Together, these compounds do not simply oppose insulin resistance—they rewire the system. Through synergistic targeting of AMPK, PI3K/Akt, NF-κB, PPARs, SIRT1, and UPR components, citrus polyphenols restore cellular energy sensing, reduce inflammatory signaling, and regenerate insulin sensitivity across liver, muscle, adipose, and pancreas. Their strength lies not in singular action, but in their ability to act in concert, modulating interlinked metabolic pathways at the core of diabesity. Thus, citrus polyphenols represent more than nutraceuticals. They are biochemical architects, reshaping the very systems that obesity and hypernutrition seek to dismantle.
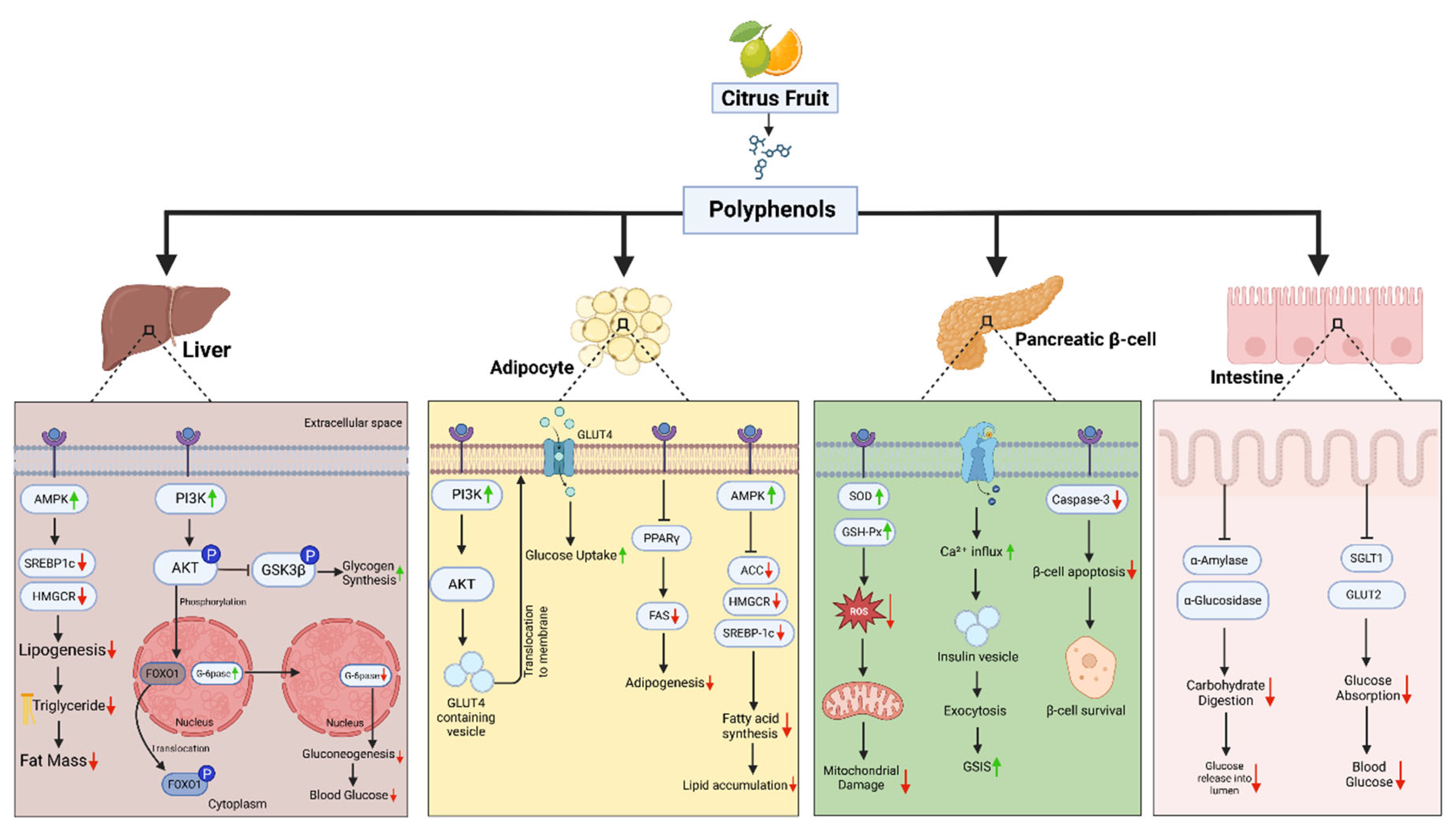
Figure 2.
Multi-Organ Effects of Citrus Fruit Polyphenols in Obesity-Induced Diabetes. This schematic summarizes the metabolic effects of citrus-derived polyphenols across four key insulin-sensitive tissues—liver, adipose tissue, pancreatic β-cells, and the intestine—in the context of obesity and type 2 diabetes. In the liver, polyphenols activate AMPK and PI3K/AKT signaling, suppressing SREBP1c and HMGCR, thereby reducing lipogenesis and triglyceride accumulation. They also inhibit gluconeogenic enzymes (G6Pase, FOXO1), leading to lower hepatic glucose output. In adipocytes, polyphenols enhance PI3K–AKT–GLUT4 translocation, improving glucose uptake. They suppress PPARγ, FAS, and SREBP1c, reducing adipogenesis, fatty acid synthesis, and lipid accumulation. In pancreatic β-cells, citrus polyphenols upregulate antioxidant enzymes (SOD, GSH-Px), reduce ROS, inhibit caspase-3, and promote Ca²⁺ influx, thereby enhancing β-cell survival, insulin exocytosis, and glucose-stimulated insulin secretion (GSIS). In the intestine, citrus polyphenols inhibit α-amylase and α-glucosidase, thereby reducing carbohydrate digestion, and downregulate SGLT1 and GLUT2, which decreases glucose absorption and postprandial glycemia.
Figure 2.
Multi-Organ Effects of Citrus Fruit Polyphenols in Obesity-Induced Diabetes. This schematic summarizes the metabolic effects of citrus-derived polyphenols across four key insulin-sensitive tissues—liver, adipose tissue, pancreatic β-cells, and the intestine—in the context of obesity and type 2 diabetes. In the liver, polyphenols activate AMPK and PI3K/AKT signaling, suppressing SREBP1c and HMGCR, thereby reducing lipogenesis and triglyceride accumulation. They also inhibit gluconeogenic enzymes (G6Pase, FOXO1), leading to lower hepatic glucose output. In adipocytes, polyphenols enhance PI3K–AKT–GLUT4 translocation, improving glucose uptake. They suppress PPARγ, FAS, and SREBP1c, reducing adipogenesis, fatty acid synthesis, and lipid accumulation. In pancreatic β-cells, citrus polyphenols upregulate antioxidant enzymes (SOD, GSH-Px), reduce ROS, inhibit caspase-3, and promote Ca²⁺ influx, thereby enhancing β-cell survival, insulin exocytosis, and glucose-stimulated insulin secretion (GSIS). In the intestine, citrus polyphenols inhibit α-amylase and α-glucosidase, thereby reducing carbohydrate digestion, and downregulate SGLT1 and GLUT2, which decreases glucose absorption and postprandial glycemia.
3.2. Citrus Polyphenols and Inflammatory Reprogramming in Diabesity
In the evolving landscape of diabesity, inflammation emerges not as a byproduct but as a driving force—a molecular amplifier that sustains insulin resistance and disrupts metabolic homeostasis. The epicenter of this inflammatory storm lies within adipose tissue, where immune–metabolic crosstalk distorts insulin signaling at every level.
TNF-α, first identified as a pivotal factor in obese adipose tissue by Hotamisligil et al. [91], sets this cascade in motion [92,93]. It activates JNK and IKKβ, which phosphorylate IRS1 at serine residues, blocking downstream PI3K/Akt signaling and arresting GLUT4 translocation, hallmarks of insulin resistance [94,95,96]. Simultaneously, NF-κB, a master inflammatory transcription factor, translocates to the nucleus, driving the expression of cytokines such as IL-6, MCP-1, and iNOS, which reinforce metabolic dysfunction and immune infiltration [7,97,98]. Amid this inflammatory gridlock, citrus polyphenols have emerged not merely as suppressors of inflammation but as metabolic circuit breakers, capable of disconnecting inflammatory feedback loops and restoring intracellular communication [21].
Naringin, hesperidin, and quercetin have all been shown to suppress NF-κB nuclear translocation, thereby silencing pro-inflammatory transcriptional programs at their source. In murine and cellular models, these polyphenols reduce the expression of COX-2, iNOS, and inflammatory cytokines, thereby reshaping the cytokine environment from a pro-inflammatory to a homeostatic one [99,100]. Further upstream, they interfere with the AGE-RAGE signaling pathway, a critical amplifier of chronic inflammation and oxidative stress in metabolic tissues. Naringin and hesperidin downregulate RAGE expression and reduce AGE accumulation, blunting ROS generation and downstream cytokine bursts [101,102,103]. Importantly, these molecular insights are not confined to the bench. Clinical data reveal that citrus bioflavonoid complexes significantly lower plasma levels of IL-6 and TNF-α, as well as biomarkers of DNA oxidative damage, such as 8-OHdG, suggesting a systemic reprogramming of inflammatory tone in human subjects [104]. This anti-inflammatory reprogramming does not merely reduce cytokines; it restores the integrity of insulin signaling. With NF-κB silenced and JNK/IKKβ activity reduced, IRS1 phosphorylation normalizes, Akt activation is restored, and glucose uptake resumes. This is metabolic recalibration—not via single-point intervention, but through network-level remodeling of the immune–metabolic interface.
Thus, citrus polyphenols act as molecular disruptors of inflammatory inertia, targeting not only the messengers of inflammation but the architecture that sustains it. Their ability to realign immune and metabolic signaling pathways elevates them from anti-inflammatory agents to true agents of metabolic reprogramming.
3.3. Role of Citrus in Mitochondrial Health and Endoplasmic Reticulum Stress: Restoring Protein Homeostasis
In the battle against metabolic disease, restoring energy balance and cellular homeostasis is no longer just a downstream goal, it is a frontline strategy. As insulin signaling falters and inflammation mounts, the cell’s two central organelles for metabolic coordination—the mitochondria and the endoplasmic reticulum (ER), emerge as critical nodes of dysfunction [21]. Their deterioration underlies the systemic collapse in energy sensing, protein folding, and glucose regulation seen in obesity-induced T2DM.
At the heart of this collapse lies mitochondrial dysfunction, driven by nutrient overload, lipotoxicity, and oxidative stress. In metabolically active tissues—especially liver, adipose, and muscle—excess free fatty acids impair mitochondrial respiration, increase reactive oxygen species (ROS), and destabilize membrane potential, culminating in reduced ATP synthesis, and suppression of insulin-stimulated glucose uptake [105,106].
Here, citrus polyphenols—particularly naringenin, hesperidin, and tangeretin—intervene as metabolic stabilizers. Their actions go beyond antioxidant defense. By activating the Nrf2–ARE pathway, they enhance endogenous antioxidant enzyme expression (SOD, catalase, GPx) and limit mitochondrial ROS accumulation [107,108]. More importantly, these polyphenols stimulate PGC-1α, NRF1, and TFAM, transcriptional regulators of mitochondrial biogenesis and energy renewal, reprogramming the cell toward oxidative resilience [107].
Naringenin, in particular, has been shown to preserve mitochondrial membrane potential, boost ATP output, and regulate PINK1/Parkin-mediated mitophagy, thereby eliminating dysfunctional mitochondria and sustaining metabolic flux in hepatocytes and myocytes under lipotoxic conditions[109,110,111]. Meanwhile, hesperidin enhances mitochondrial efficiency and prevents apoptotic signaling by reducing cytochrome c release and caspase activation, thereby preserving cell viability in the face of metabolic overload [112].
Yet mitochondria are not isolated actors. Their function is tightly intertwined with that of the endoplasmic reticulum (ER)—an organelle responsible for protein folding, lipid biosynthesis, and calcium signaling [113]. Under chronic nutrient stress, the ER accumulates misfolded proteins, triggering unfolded protein response (UPR) pathways. If unresolved, this stress spills over into apoptosis, fueling insulin resistance and β-cell failure [71,114,115,116].
Once again, citrus flavonoids offer a path to restoration. Compounds like naringenin and hesperidin selectively attenuate the PERK–eIF2α, IRE1–XBP1, and ATF6 arms of the UPR, preventing the maladaptive amplification of ER stress. In high-fat-diet models, naringenin reduces CHOP expression, a pro-apoptotic UPR marker, and restores adaptive ER signaling, thereby rescuing β-cell and hepatocyte function [114,117,118].
These polyphenols also enhance GRP78/BiP expression, boosting chaperone capacity and facilitating correct protein folding under stress conditions [113,119]. Additionally, by modulating ER-mitochondrial crosstalk, citrus compounds support coordinated calcium transfer, optimize oxidative metabolism, and prevent apoptosis—a level of control that aligns precisely with systemic metabolic reprogramming [120,121,122].
Thus, citrus polyphenols do not merely suppress damage; they restore the intracellular architecture upon which metabolic homeostasis depends. By recalibrating mitochondrial bioenergetics, enhancing mitophagy, stabilizing ER folding capacity, and reinforcing cross-organelle signaling, they initiate a cellular response that rewires the failing metabolic circuitry of diabesity from within.
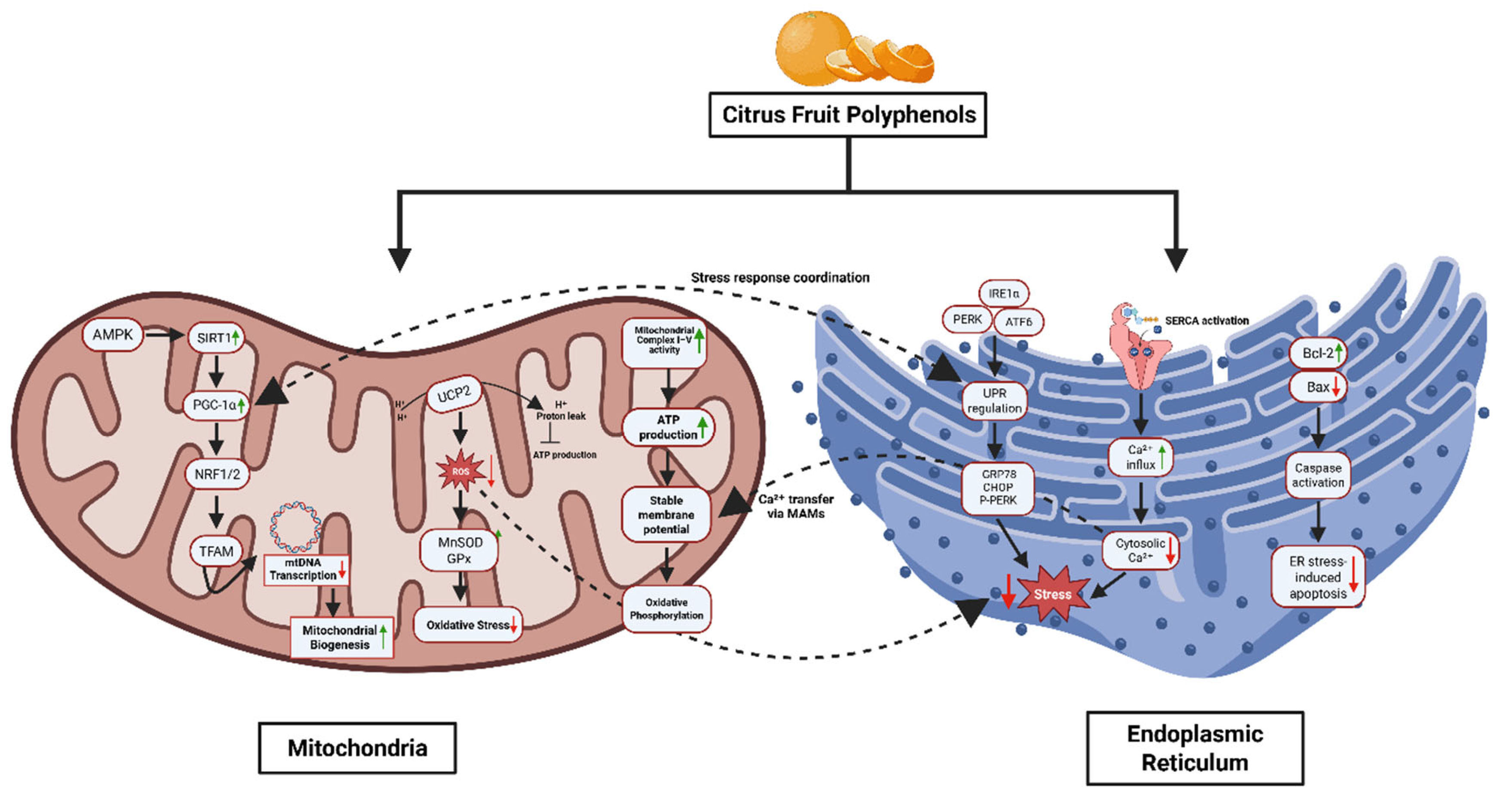
Figure 3.
Citrus Polyphenols Regulate Mitochondrial and Endoplasmic Reticulum (ER) Homeostasis Under Metabolic Stress. This diagram illustrates the coordinated protective effects of citrus fruit polyphenols on mitochondrial and ER function. In mitochondria, polyphenols activate the AMPK–SIRT1–PGC-1α pathway, enhancing mitochondrial biogenesis via NRF1/2 and TFAM, and reducing ROS through upregulation of MnSOD and GPx. They stabilize the mitochondrial membrane potential, support ATP production, and improve complex I/IV activity while reducing proton leak and oxidative stress. In the ER, citrus polyphenols alleviate ER stress by modulating UPR pathways (PERK, IRE1α, ATF6), reducing expression of stress markers (CHOP, p-PERK), enhancing SERCA-mediated Ca2+ influx, and promoting ER–mitochondria Ca2+ transfer. They also inhibit apoptosis by increasing Bcl-2 and suppressing Bax and caspase activation. Together, these actions maintain cellular homeostasis and prevent metabolic dysfunction.
Figure 3.
Citrus Polyphenols Regulate Mitochondrial and Endoplasmic Reticulum (ER) Homeostasis Under Metabolic Stress. This diagram illustrates the coordinated protective effects of citrus fruit polyphenols on mitochondrial and ER function. In mitochondria, polyphenols activate the AMPK–SIRT1–PGC-1α pathway, enhancing mitochondrial biogenesis via NRF1/2 and TFAM, and reducing ROS through upregulation of MnSOD and GPx. They stabilize the mitochondrial membrane potential, support ATP production, and improve complex I/IV activity while reducing proton leak and oxidative stress. In the ER, citrus polyphenols alleviate ER stress by modulating UPR pathways (PERK, IRE1α, ATF6), reducing expression of stress markers (CHOP, p-PERK), enhancing SERCA-mediated Ca2+ influx, and promoting ER–mitochondria Ca2+ transfer. They also inhibit apoptosis by increasing Bcl-2 and suppressing Bax and caspase activation. Together, these actions maintain cellular homeostasis and prevent metabolic dysfunction.
3.4. Free radicals, Oxidative Stress, and Citrus Polyphenols: A Natural Line of defense
As the metabolic storm deepens, a new biochemical disruptor takes center stage: oxidative stress. While reactive oxygen species (ROS) serve as transient messengers under physiological conditions, chronic metabolic overload transforms them into agents of cellular sabotage, eroding the signaling pathways that maintain metabolic equilibrium.
In obesity-induced diabetes, the overproduction of ROS—particularly superoxide (O₂⁻), hydrogen peroxide (H₂O₂), and hydroxyl radicals (•OH)—disrupts insulin receptor integrity, impairs glucose uptake, and accelerates β-cell dysfunction [45,123,124]. These free radicals are not isolated threats; they are embedded within a vicious cycle where lipotoxicity, inflammation, and mitochondrial inefficiency reinforce oxidative damage, amplifying insulin resistance across tissues [125].
The collapse of redox homeostasis is especially destructive in insulin-sensitive cells like hepatocytes, myocytes, and adipocytes, where antioxidant defense systems are often overwhelmed [126,127]. The antioxidant enzymatic triad—superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx)—is essential for neutralizing ROS. However, in metabolic disease, these systems are either underexpressed or dysfunctional, allowing oxidative stress to accumulate unchecked [123,124].
At this critical juncture, citrus polyphenols emerge as a dual-action metabolic defense—scavengers of free radicals and regulators of redox-responsive gene expression. Unlike synthetic antioxidants that act in isolation, these natural compounds integrate into the metabolic network, restoring antioxidant capacity from within [128,129]. For example, supplementation with Citrus unshiu peel extract has been shown to significantly increase hepatic levels of SOD, CAT, GPx, and glutathione (GSH) in high-fat-diet-fed mice, leading to reductions in lipid peroxidation and improvement in insulin sensitivity [130]. This restoration of redox balance helps normalize insulin signaling cascades and suppress stress-induced kinase activation, such as JNK and IKKβ, which are known inhibitors of insulin receptor substrates [128,131].
The effect is not compound-specific but rather synergistic. Flavonoids such as naringenin, hesperidin, and quercetin, abundant in citrus species like Citrus paradisi, Citrus sinensis, and Citrus maxima, not only scavenge ROS directly but also activate the Nrf2–ARE pathway—a master regulator of endogenous antioxidant gene expression [129,132,133]. Activation of Nrf2 leads to increased transcription of SOD, GPx, and CAT, reinforcing intracellular defenses at the transcriptional level [133].
Notably, extracts of red pummelo (Citrus maxima) demonstrated 20–40% higher free radical scavenging capacity than vitamin C or synthetic antioxidants like BHA, underlining the superior efficacy of these phytochemical complexes in redox modulation [134]. Thus, oxidative stress is not merely a consequence of metabolic dysfunction; it is a pathogenic amplifier of insulin resistance and systemic energy imbalance. By disrupting this oxidative-inflammation loop, citrus polyphenols do more than clean up molecular debris; they restore the redox environment required for metabolic precision. In doing so, they enable cells to re-establish insulin responsiveness, protect β-cell integrity, and suppress the molecular triggers of inflammation. This metabolic recalibration through antioxidant programming firmly positions citrus-derived polyphenols as frontline defenders in the systems-level war against diabesity.

Table 1.
Citrus Polyphenols and Metabolic Reprogramming (Animal/Cell Studies).
| Compound | Model / System | Metabolic Mechanism(s) | Source |
| Neohesperidin | HFD-fed mice | ↑ AMPK–PGC-1α → mitochondrial biogenesis, steatosis reduction | [135] |
| Nobiletin | HFD-fed mice | ↑ FA oxidation, energy expenditure; AMPK-independent | [136] |
| Nobiletin | Hepatocytes | Restores Bmal1 → ↑ lipid/OXPHOS metabolism | [137] |
| Nobiletin | Insulin-resistant mice | ↓ VLDL secretion; improves lipid/glucose metabolism | [138] |
| Nobiletin | HepG2 cells | ↑ PGC1α, CPT1, UCP2 → β-oxidation | [138] |
| Nobiletin | ob/ob mice | ↑ GLUT4, ↑ Akt phosphorylation → improved insulin sensitivity | [139] |
| Naringenin | MCD or HFD mice | ↑ AMPK → autophagy, ↑ mitochondrial biogenesis | [140] |
| Naringenin | Hepatocytes/mice | ↑ AMPK, ↑ ATF3 → metabolic inflammation reduction | [141] |
| Naringin | HFD-fed mice | ↑ AMPK → ↓ SREBP-1c/FAS, ↑ redox balance | [142] |
| Naringin | Fructose-fed rats | ↑ Nrf2/HO-1 → antioxidant response; ↓ ChREBP/SREBP-1c | [132] |
| Naringin | HFD mice | ↑ TFEB → lipophagy → ↓ hepatic lipid droplets | [143] |
| Hesperidin | LO2 hepatocytes (HG) | ↑ ATP, restores ΔΨm via AKT/GSK3β | [144] |
| Hesperidin | Hyperlipidemic rats | ↑ SOD, catalase; preserves mitochondrial enzymes | [145] |
| Hesperidin | Neurons (hyperglycemia) | Improves ATP/redox; ↓ mitochondrial dysfunction | [146] |
| Hesperetin | Aging mice | ↑ Cisd2 expression → metabolic health maintenance | [147] |
| Limonene | Mice model | ↑ mitochondrial respiration, ↓ ROS | [148,149] |
| Eriocitrin | HFD rats | ↑ mitochondrial biogenesis, ↓ steatosis | [150] |
| Sudachitin | C57BL/6J, db/db mice | ↑ β-oxidation, ↑ mitochondrial biogenesis | [151] |
| Tangeretin | Diabetic rats | ↑ GLUT4, antioxidant enzymes | [152] |
| Naringenin | NAFLD mice | ↓ NLRP3/NF-κB, ↓ IL-1β → metabolic reprogramming | [153] |
| Naringenin | NAFLD mice (metabolomics) | Gut microbiota modulation → improved host metabolism | [154] |
| Naringenin | Muscle cells | ↑ p-AMPK → ↑ glucose uptake, ↑ mitochondria | [83] |
| Naringin | Hepatocytes, HFD mice | AMPK–IRS1–MAPK pathway → improved insulin signaling | [127] |
| Naringenin NP | MASLD mice | ↑ PPAR, lipid oxidation, gut microbiota shift | [155] |
| Naringenin | Mice (aerobic fitness) | ↑ oxidative fibers, ↑ aerobic metabolism | [156] |
| Naringin | KK-A(y) mice | ↑ AMPK → ↓ glucose/lipids, ↑ insulin sensitivity | [157] |
| Neohesperidin | DIO mice, HepG2 cells | ↑ FGF21, ↑ AMPK → improved lipid regulation | [158] |
| Hesperidin | MASLD mice | ↓ insulin resistance, ↓ oxidative stress | [159] |
| Nobiletin | HepG2 cells | ↑ AMPK, ↓ lipogenesis | [160] |
Figure 4.
Citrus Fruit Polyphenols Mitigate Oxidative Stress in the Liver. This schematic depicts how citrus polyphenols exert antioxidant and anti-inflammatory effects to protect hepatic tissue from oxidative damage. On the left, citrus polyphenols directly scavenge reactive oxygen species (ROS), DPPH⁺, and NO radicals via hydrogen atom donation, preventing lipid peroxidation and radical propagation. Simultaneously, they activate the Keap1–Nrf2 pathway, promoting Nrf2 release and nuclear translocation. Nrf2 then binds to antioxidant response elements (ARE), upregulating endogenous antioxidants such as SOD, CAT, GPx, and HO-1. This enhances hepatic ROS detoxification and reduces oxidative stress markers like malondialdehyde (MDA) and 8-hydroxy-2′-deoxyguanosine (8-OHdG). On the right, citrus polyphenols inhibit xanthine oxidase as well as COX-1/COX-2 and LOX enzymes, decreasing prostaglandin and leukotriene synthesis and limiting inflammatory cell infiltration. Together, these mechanisms mitigate oxidative DNA damage and inflammation, preventing chronic liver injury.
Figure 4.
Citrus Fruit Polyphenols Mitigate Oxidative Stress in the Liver. This schematic depicts how citrus polyphenols exert antioxidant and anti-inflammatory effects to protect hepatic tissue from oxidative damage. On the left, citrus polyphenols directly scavenge reactive oxygen species (ROS), DPPH⁺, and NO radicals via hydrogen atom donation, preventing lipid peroxidation and radical propagation. Simultaneously, they activate the Keap1–Nrf2 pathway, promoting Nrf2 release and nuclear translocation. Nrf2 then binds to antioxidant response elements (ARE), upregulating endogenous antioxidants such as SOD, CAT, GPx, and HO-1. This enhances hepatic ROS detoxification and reduces oxidative stress markers like malondialdehyde (MDA) and 8-hydroxy-2′-deoxyguanosine (8-OHdG). On the right, citrus polyphenols inhibit xanthine oxidase as well as COX-1/COX-2 and LOX enzymes, decreasing prostaglandin and leukotriene synthesis and limiting inflammatory cell infiltration. Together, these mechanisms mitigate oxidative DNA damage and inflammation, preventing chronic liver injury.

3.5. Translating Mechanisms to Humans: Clinical Evidence of Citrus Polyphenol-Driven Metabolic Reprogramming
Having followed citrus polyphenols through cellular landscapes—where they extinguish oxidative stress, rewire mitochondrial signaling, restore protein-folding homeostasis, and silence inflammatory circuits—we arrive at the final test: do these molecular shifts translate to human physiology?
Clinical trials now affirm what systems biology predicted: the pleiotropic actions of citrus-derived polyphenols—hesperidin, naringin, neohesperidin, and bergamot polyphenolic fraction (BPF)—are not confined to preclinical promise. In carefully controlled human studies, these compounds demonstrate a system-level recalibration of metabolic function, validating their role as natural agents of metabolic reprogramming [134,147].
In a double-blind, randomized clinical trial, 500 mg/day of hesperidin administered over three weeks significantly reduced TNF-α and VCAM-1, thereby restoring endothelial function—a proxy for systemic inflammatory tone and insulin sensitivity [22]. Importantly, these effects are not isolated to the vasculature but echo upstream changes in NF-κB signaling and insulin receptor substrate activity, consistent with earlier mechanistic studies.
Naringin, too, has moved from bench to bedside. Oral supplementation in patients with metabolic syndrome resulted in reductions in total cholesterol, LDL-C, and triglycerides, while increasing HDL-C, an indirect yet essential component of insulin signaling restoration [161,162]. These lipid profile improvements align with its known PPARγ activation and inhibition of JNK signaling, bridging preclinical mechanisms with clinical outcome [25,163].
In a dietary intervention study, individuals with T2DM who followed a Mediterranean diet enriched with citrus fruits experienced a reduction in HbA1c from 7.1% to 6.8% over 12 weeks, indicating an improvement in long-term glycemic control through enhanced insulin sensitivity and restored glucose uptake mechanisms [164].
Perhaps most compelling is the clinical validation of bergamot polyphenolic fraction (BPF)—a mixture rich in neoeriocitrin, naringin, and neohesperidin. In hyperlipidemic and prediabetic subjects, BPF not only lowered LDL-C and improved glucose metabolism, but achieved these results with efficacy comparable to statin therapy, yet without pharmacological side effects [165,166].
These studies converge on a single, powerful theme: citrus polyphenols act at the systems level, engaging molecular targets already mapped in preclinical models—AMPK, NF-κB, PI3K/Akt, PPARs—to bring about clinically meaningful metabolic restoration. Their ability to reduce inflammatory biomarkers, improve insulin action, recalibrate lipid metabolism, and preserve vascular function confirms their role not just as dietary supplements, but as translational agents in the therapeutic modulation of diabesity. And so, from the molecular to the metabolic, from mitochondria to macrosystems, citrus polyphenols close the loop: a natural, multifaceted, clinically validated reprogramming of the diseased metabolic network.
Table 2.
Clinical Studies of Citrus Polyphenols on Metabolic Reprogramming.
| Compound(s) | Source | Reported Outcome | Mechanism of Action | Source |
| Neoeriocitrin, Naringin, Neohesperidin | Citrus bergamia (Bergamot) | ↓ Liver fat content, ↓ body weight (vs. placebo) | Enhances bile flow; antioxidant activity reduces oxidative stress | [167] |
| Hesperidin, Naringin, Neohesperidin | Various citrus fruits | ↑ Endothelial function (↑ FMD, vascular tone) | Bile secretion support; antioxidant effects improve vascular inflammation and nitric oxide availability | [168] |
| Hesperidin→Hesperetin; SCFAs | Citrus fruit extracts | Modulates gut microbiota → ↑ beneficial bacteria → systemic inflammation ↓ | Hesperidin metabolized to hesperetin → SCFA production → improved endothelial protection and anti-inflammatory response | [169] |
| Flavones, Flavanones, Oleuropein | Citrus + olive | ↓ Cardiovascular risk biomarkers; improved metabolic-inflammatory profile | Antioxidant activity; inhibits neuro-immune-inflammatory pathways (e.g., cytokines, NF-κB) | [170] |
| Hesperidin, Naringin, Oleuropein | Citrus fruits + olive leaves | ↓ LDL oxidation; ↓ pro-inflammatory cytokines (e.g., TNF-α, IL-6) | Free radical scavenging; anti-inflammatory cytokine modulation | [171] |
| Hesperidin | Orange juice (Citrus sinensis) | ↓ BMI, ↓ waist circumference, ↓ IL-1, IL-6, TNF-α | Inhibits pro-inflammatory cytokine release; protects cells (e.g., keratinocytes, endothelial) from oxidative damage | [172] |
| Hesperidin (1 g/day + lifestyle) | RCT in NAFLD patients | ↓ Liver fat, ALT, weight; improved lipids | NF-κB inhibition; ↓ TNF-α, hs-CRP | [173] |
| Hesperidin (meta-analysis) | RCTs in metabolic subjects (n=525) | ↓ TG, TC, LDL (in BMI>30) | ↓ TNF-α, ↓ IL-6 at high dose | [174] |
| Orange juice (flavonoids) | 4-week RCT in MASLD (n=62) | ↓ Liver steatosis (FibroScan) | ↓ GGT; modest inflammatory effects | [175] |
| Flavonoid-enriched orange juice | RCT in metabolic patients | ↑ antioxidant status; improved glycemic trend | ↓ CRP, ↓ endothelial inflammation | [176] |
| Hesperidin | Vascular study (metabolic syndrome) | ↑ FMD, endothelial function | ↑ NO; ↓ IL-6, TNF-α | [22] |
| Eriomin® (Eriocitrin) | Crossover RCT (n=103) | ↓ FBG, HOMA-IR; ↑ GLP-1, adiponectin | ↓ IL-6, TNF-α, hs-CRP | [177] |
| Polyphenols incl. naringenin | Meta-analysis in NAFLD | ↓ BMI, ALT, AST, TG | ↓ TNF-α (across multiple flavonoids) | [178] |
4. Conclusions & Future Direction
In the orchestra of cellular life, metabolic reprogramming is the conductor’s baton, guiding the tempo of nutrient sensing, mitochondrial energy flow, insulin action, and inflammatory resolution. But in obesity-induced diabetes, the rhythm descends into disarray. The once-synchronized network of metabolic signals becomes a cacophony of oxidative stress, unfolded proteins, inflammatory feedback, and lipotoxic derailment.
Yet, amid this dysfunction, a new score emerges, one not composed in laboratories but grown in groves of citrus, where nature has refined its symphony of healing.
Throughout this review, we followed citrus-derived polyphenols as they stepped into this metabolic dissonance, not as singular antioxidants or hypoglycemic agents, but as systemic modulators orchestrating a return to cellular harmony. From naringenin’s mitochondrial recalibration to hesperidin’s anti-inflammatory tuning, these flavonoids reactivated dormant pathways, rewired dysfunctional circuits, and restored homeostatic balance at every biological level—liver, adipose, muscle, pancreas, and beyond.
Their strength lies not in pharmacological force, but in biological fluency. Citrus polyphenols do not attack the system—they speak its language, modulating master nodes like AMPK, PPARγ, Nrf2, and NF-κB, enabling tissues to heal from within. This systems-level approach is what places them at the forefront of a new therapeutic paradigm: one where nutritional networks, not isolated molecules, reshape disease trajectories.
Clinical studies now echo these findings. From reductions in HbA1c and inflammatory markers to improvements in lipid profiles and insulin sensitivity, the early human evidence is promising. These are not miracle cures—but metabolic nudges that push the body back toward its innate balance, especially when paired with lifestyle realignment.
As we close this chapter, we leave not with finality but with purpose. The story of citrus polyphenols in metabolic reprogramming is still unfolding. While this review focused on their core impact on insulin resistance, oxidative stress, mitochondrial-ER integrity, and inflammatory remodeling, much remains to be explored.
For now, we end where we began: with complexity, with nature, and with a hopeful question. If food is information, and polyphenols are its syntax, then perhaps within the citrus grove lies not just nutrition, but a code for reprogramming metabolism itself.
And we’ve only just started decoding it.
Acknowledgments
We utilized AI-based tools, to assist with grammar and language refinement in this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Allocca, S., et al., Endocrine and Metabolic Mechanisms Linking Obesity to Type 2 Diabetes: Implications for Targeted Therapy. Healthcare (Basel), 2025. 13(12). [CrossRef]
- Kyrou, I., et al., Clinical problems caused by obesity. 2015.
- Saeedi, P., et al., Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract, 2019. 157: p. 107843. [CrossRef]
- Ong, K.L., et al., Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet, 2023. 402(10397): p. 203-234.
- Kocatepe, D., D.C. Büyükkol, and K.N. Hinislioğlu, Obesity Prevalence in World and Türkiye. Northern Journal of Health Sciences, 2025. 1(1): p. 26-32.
- Shimu, S.J., A 10-Year Retrospective Quantitative Analysis of The CDC Database: Examining the Prevalence of Depression in the Us Adult Urban Population. [CrossRef]
- Zatterale, F., et al., Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Frontiers in physiology, 2020. 10: p. 1607. [CrossRef]
- Makki, K., P. Froguel, and I. Wolowczuk, Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. International Scholarly Research Notices, 2013. 2013(1): p. 139239. [CrossRef]
- Li, X., et al., Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Frontiers in Immunology, 2023. 14: p. 1153915. [CrossRef]
- Gkrinia, E.M.M. and A. Belančić, The Mechanisms of Chronic Inflammation in Obesity and Potential Therapeutic Strategies: A Narrative Review. Current Issues in Molecular Biology, 2025. 47(5): p. 357. [CrossRef]
- Gudise, V. and B. Chowdhury, Molecular mechanisms and the vital roles of resistin, TLR 4, and NF-κB in treating type 2 diabetic complications. Beni-Suef University Journal of Basic and Applied Sciences, 2020. 9(1): p. 54.
- Kim, J.K., Endothelial nuclear factor κB in obesity and aging: is endothelial nuclear factor κB a master regulator of inflammation and insulin resistance? 2012, Lippincott Williams & Wilkins Hagerstown, MD. p. 1081-1083.
- Chen, L., et al., Mechanisms linking inflammation to insulin resistance. International journal of endocrinology, 2015. 2015(1): p. 508409. [CrossRef]
- Sergi, D., et al., Ceramides as the molecular link between impaired lipid metabolism, saturated fatty acid intake and insulin resistance: are all saturated fatty acids to be blamed for ceramide-mediated lipotoxicity? Nutrition Research Reviews, 2024: p. 1-11.
- Römer, A., T. Linn, and S.F. Petry, Lipotoxic impairment of mitochondrial function in β-cells: A review. Antioxidants, 2021. 10(2): p. 293. [CrossRef]
- Corkey, B.E., Reactive oxygen species: role in obesity and mitochondrial energy efficiency. Philosophical Transactions of the Royal Society B, 2023. 378(1885): p. 20220210. [CrossRef]
- Apostolova, N., et al., Mitochondrial dysfunction and mitophagy in type 2 diabetes: pathophysiology and therapeutic targets. Antioxidants & Redox Signaling, 2023. 39(4-6): p. 278-320. [CrossRef]
- Patel, S. and A. Majumdar, Endoplasmic Reticulum Stress and Unfolded Protein Response in Metabolic Syndrome, in Biochemical Mechanisms for Metabolic Syndrome. 2024, Springer. p. 203-222.
- Ghosh, R., K. Colon-Negron, and F.R. Papa, Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Molecular metabolism, 2019. 27: p. S60-S68. [CrossRef]
- Aufi, S.S.A. and K. Hasnin, Diabesity An Emerging Epidemic and Advances in Treatment. Barind Medical College Journal, 2024. 10(1): p. 29-34.
- Mohib, M., et al., Beneficial role of citrus fruit polyphenols against hepatic dysfunctions: a review. Journal of dietary supplements, 2018. 15(2): p. 223-250. [CrossRef]
- Rizza, S., et al., Citrus polyphenol hesperidin stimulates production of nitric oxide in endothelial cells while improving endothelial function and reducing inflammatory markers in patients with metabolic syndrome. The Journal of Clinical Endocrinology & Metabolism, 2011. 96(5): p. E782-E792. [CrossRef]
- Gandhi, G.R., et al., Citrus flavonoids as promising phytochemicals targeting diabetes and related complications: A systematic review of in vitro and in vivo studies. Nutrients, 2020. 12(10): p. 2907. [CrossRef]
- Zygmunt, K., et al., Naringenin, a citrus flavonoid, increases muscle cell glucose uptake via AMPK. Biochemical and biophysical research communications, 2010. 398(2): p. 178-183.
- Den Hartogh, D.J. and E. Tsiani, Antidiabetic properties of naringenin: A citrus fruit polyphenol. Biomolecules, 2019. 9(3): p. 99. [CrossRef]
- Hammerschmidt, P. and J.C. Brüning, Contribution of specific ceramides to obesity-associated metabolic diseases. Cellular and Molecular Life Sciences, 2022. 79(8): p. 395. [CrossRef]
- Sears, B. and M. Perry, The role of fatty acids in insulin resistance. Lipids in health and disease, 2015. 14(1): p. 121. [CrossRef]
- Yung, J.H.M. and A. Giacca, Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells, 2020. 9(3): p. 706. [CrossRef]
- Kim, G.-T., et al., Hepatic expression of the serine palmitoyltransferase subunit Sptlc2 reduces lipid droplets in the liver by activating VLDL secretion. Journal of lipid and atherosclerosis, 2020. 9(2): p. 291. [CrossRef]
- Elkanawati, R.Y., S.A. Sumiwi, and J. Levita, Impact of lipids on insulin resistance: insights from human and animal studies. Drug design, development and therapy, 2024: p. 3337-3360. [CrossRef]
- Sokolowska, E. and A. Blachnio-Zabielska, The role of ceramides in insulin resistance. Frontiers in Endocrinology, 2019. 10: p. 577. [CrossRef]
- Schrauwen, P. and M.K. Hesselink, Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes, 2004. 53(6): p. 1412-1417.
- Dubé, J.J., et al., Exercise-induced alterations in intramyocellular lipids and insulin resistance: the athlete's paradox revisited. American Journal of Physiology-Endocrinology and Metabolism, 2008. 294(5): p. E882-E888.
- Goodpaster, B.H. and L.M. Sparks, Metabolic flexibility in health and disease. Cell metabolism, 2017. 25(5): p. 1027-1036. [CrossRef]
- Mohib, M.M., et al., Eplerenone, a mineralocorticoid receptor inhibitor, reduces cirrhosis associated changes of hepatocyte glucose and lipid metabolism. Cell Communication and Signaling, 2024. 22(1): p. 614.
- Cusi, K., Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology, 2012. 142(4): p. 711-725. e6. [CrossRef]
- Qin, W. and J. Weng, Hepatocyte NLRP3 interacts with PKCε to drive hepatic insulin resistance and steatosis. Science Bulletin, 2023. 68(13): p. 1413-1429. [CrossRef]
- Mota, M., et al., Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism, 2016. 65(8): p. 1049-1061.
- Huang, W., et al., Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes, 2010. 59(2): p. 347-57. [CrossRef]
- Ziolkowska, S., et al., The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int J Mol Sci, 2021. 22(20). [CrossRef]
- Gruben, N., et al., Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta, 2014. 1842(11): p. 2329-2343.
- Chen, Z., et al., A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis, 2017. 16(1): p. 203.
- Ly, L.D., et al., Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med, 2017. 49(2): p. e291. [CrossRef]
- Morawietz, H., et al., Cross-Talk of NADPH Oxidases and Inflammation in Obesity. Antioxidants (Basel), 2023. 12(8).
- Vilas-Boas, E.A., et al., Lipotoxicity and β-Cell Failure in Type 2 Diabetes: Oxidative Stress Linked to NADPH Oxidase and ER Stress. Cells, 2021. 10(12). [CrossRef]
- Rains, J.L. and S.K. Jain, Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med, 2011. 50(5): p. 567-75.
- Manna, P. and S.K. Jain, Obesity, Oxidative Stress, Adipose Tissue Dysfunction, and the Associated Health Risks: Causes and Therapeutic Strategies. Metab Syndr Relat Disord, 2015. 13(10): p. 423-44.
- Bhatti, J.S., G.K. Bhatti, and P.H. Reddy, Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis, 2017. 1863(5): p. 1066-1077. [CrossRef]
- Cojocaru, K.A., et al., Mitochondrial Dysfunction, Oxidative Stress, and Therapeutic Strategies in Diabetes, Obesity, and Cardiovascular Disease. Antioxidants (Basel), 2023. 12(3).
- Argaev-Frenkel, L. and T. Rosenzweig, Redox Balance in Type 2 Diabetes: Therapeutic Potential and the Challenge of Antioxidant-Based Therapy. Antioxidants (Basel), 2023. 12(5).
- Shrestha, N., et al., Pathological β-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front Endocrinol (Lausanne), 2021. 12: p. 650158. [CrossRef]
- Kim, G., et al., Endoplasmic Reticulum Stress and Its Impact on Adipogenesis: Molecular Mechanisms Implicated. Nutrients, 2023. 15(24).
- Bhattarai, K.R., et al., The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med, 2021. 53(2): p. 151-167. [CrossRef]
- Veluthakal, R., et al., Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int J Mol Sci, 2024. 25(3).
- Benito-Vicente, A., et al., Molecular mechanisms of lipotoxicity-induced pancreatic β-cell dysfunction. Int Rev Cell Mol Biol, 2021. 359: p. 357-402.
- Biondi, G., et al., Adipose Tissue Secretion Pattern Influences β-Cell Wellness in the Transition from Obesity to Type 2 Diabetes. Int J Mol Sci, 2022. 23(10).
- Ježek, P., et al., Fatty Acid-Stimulated Insulin Secretion vs. Lipotoxicity. Molecules, 2018. 23(6).
- Kim, J.W. and K.H. Yoon, Glucolipotoxicity in Pancreatic β-Cells. Diabetes Metab J, 2011. 35(5): p. 444-50.
- Dalle, S., A. Abderrahmani, and E. Renard, Pharmacological inhibitors of β-cell dysfunction and death as therapeutics for diabetes. Front Endocrinol (Lausanne), 2023. 14: p. 1076343. [CrossRef]
- Brozzi, F. and D.L. Eizirik, ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci, 2016. 121(2): p. 133-9. [CrossRef]
- Lee, J.H. and J. Lee, Endoplasmic Reticulum (ER) Stress and Its Role in Pancreatic β-Cell Dysfunction and Senescence in Type 2 Diabetes. Int J Mol Sci, 2022. 23(9).
- Yang, L., et al., Puerarin Protects Pancreatic β-Cells in Obese Diabetic Mice via Activation of GLP-1R Signaling. Mol Endocrinol, 2016. 30(3): p. 361-71. [CrossRef]
- Yusta, B., et al., GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab, 2006. 4(5): p. 391-406. [CrossRef]
- Li, Y., et al., Recent advances in pancreatic α-cell transdifferentiation for diabetes therapy. Front Immunol, 2025. 16: p. 1551372.
- Choe, S.S., et al., Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front Endocrinol (Lausanne), 2016. 7: p. 30. [CrossRef]
- Huynh, P.M., F. Wang, and Y.A. An, Hypoxia signaling in the adipose tissue. J Mol Cell Biol, 2025. 16(8). [CrossRef]
- Mirabelli, M., et al., Hypoxia in Human Obesity: New Insights from Inflammation towards Insulin Resistance-A Narrative Review. Int J Mol Sci, 2024. 25(18). [CrossRef]
- Begum, M., et al., Adiponectin: a promising target for the treatment of diabetes and its complications. Life, 2023. 13(11): p. 2213.
- Corvera, S., Cellular Heterogeneity in Adipose Tissues. Annu Rev Physiol, 2021. 83: p. 257-278.
- Massier, L., et al., An integrated single cell and spatial transcriptomic map of human white adipose tissue. Nat Commun, 2023. 14(1): p. 1438.
- Sultana, A., et al., Bangladeshi parents’ knowledge and awareness about cervical cancer and willingness to vaccinate female family members against human papilloma virus: a cross sectional study. Int J Community Med Public Health, 2023. 10(10): p. 3446-3453.
- Khin, P.P., J.H. Lee, and H.-S. Jun, Pancreatic beta-cell dysfunction in type 2 diabetes. European Journal of Inflammation, 2023. 21: p. 1721727X231154152. [CrossRef]
- Chen, B., et al., Lipotoxicity: A New Perspective in Type 2 Diabetes Mellitus. Diabetes, Metabolic Syndrome and Obesity, 2025: p. 1223-1237.
- Sandoval, V., et al., Metabolic impact of flavonoids consumption in obesity: from central to peripheral. Nutrients, 2020. 12(8): p. 2393.
- Mirzaei, A., et al., Promising influences of hesperidin and hesperetin against diabetes and its complications: a systematic review of molecular, cellular, and metabolic effects. Excli j, 2023. 22: p. 1235-1263.
- Russo, B., et al., Flavonoids and insulin-resistance: from molecular evidences to clinical trials. International journal of molecular sciences, 2019. 20(9): p. 2061. [CrossRef]
- Yi, H., et al., The therapeutic effects and mechanisms of quercetin on metabolic diseases: pharmacological data and clinical evidence. Oxidative medicine and cellular longevity, 2021. 2021(1): p. 6678662. [CrossRef]
- Dong, J., et al., Quercetin reduces obesity-associated ATM infiltration and inflammation in mice: a mechanism including AMPKα1/SIRT1. J Lipid Res, 2014. 55(3): p. 363-74.
- Tsai, C.F., et al., Regulatory Effects of Quercetin on M1/M2 Macrophage Polarization and Oxidative/Antioxidative Balance. Nutrients, 2021. 14(1).
- Kohjima, M., et al., SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. International journal of molecular medicine, 2008. 21(4): p. 507-511. [CrossRef]
- Lee, J., et al., GLP-1 receptor agonist and non-alcoholic fatty liver disease. Diabetes & metabolism journal, 2012. 36(4): p. 262.
- Li, S., et al., Naringenin improves insulin sensitivity in gestational diabetes mellitus mice through AMPK. Nutrition & diabetes, 2019. 9(1): p. 28. [CrossRef]
- Zygmunt, K., et al., Naringenin, a citrus flavonoid, increases muscle cell glucose uptake via AMPK. Biochem Biophys Res Commun, 2010. 398(2): p. 178-83. [CrossRef]
- Nery, M., et al., Physiological effects of tangeretin and heptamethoxyflavone on obese C57BL/6J mice fed a high-fat diet and analyses of the metabolites originating from these two polymethoxylated flavones. Food Science & Nutrition, 2021. 9(4): p. 1997-2009.
- La Spina, M., et al., Browning effects of a chronic pterostilbene supplementation in mice fed a high-fat diet. International journal of molecular sciences, 2019. 20(21): p. 5377.
- Chen, Q., et al., Tangeretin prevents obesity by modulating systemic inflammation, fat browning, and gut microbiota in high-fat diet-induced obese C57BL/6 mice. The Journal of Nutritional Biochemistry, 2022. 101: p. 108943.
- Gao, J., et al., The combination of cinnamaldehyde and kaempferol ameliorates glucose and lipid metabolism disorders by enhancing lipid metabolism via AMPK activation. Journal of Functional Foods, 2021. 83: p. 104556. [CrossRef]
- Park, J.-e., J. Yoo, and J.-s. Han, HM-Chromanone Alleviates Hyperglycemia by Activating AMPK and PI3K/AKT Pathways in Mice Fed a High-Fat Diet. Nutrients, 2024. 16(22): p. 3972. [CrossRef]
- Chang, W.-T., et al., Rutin and gallic acid regulates mitochondrial functions via the SIRT1 pathway in C2C12 myotubes. Antioxidants, 2021. 10(2): p. 286.
- Alkhalidy, H., et al., The flavonoid kaempferol ameliorates streptozotocin-induced diabetes by suppressing hepatic glucose production. Molecules, 2018. 23(9): p. 2338. [CrossRef]
- Hotamisligil, G.S., N.S. Shargill, and B.M. Spiegelman, Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science, 1993. 259(5091): p. 87-91.
- Hotamisligil, G.S., et al., Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest, 1995. 95(5): p. 2409-15. [CrossRef]
- Shimu, S.J. and S. Islam, Gender Differences in Drug Addiction: Neurobiological, Social, and Psychological Perspectives in Women–A Systematic Review. Journal of Primeasia, 2025. 6(1): p. 1-13.
- Gao, Z., et al., Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem, 2002. 277(50): p. 48115-21.
- Kanety, H., et al., Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J Biol Chem, 1995. 270(40): p. 23780-4.
- Martínez Báez, A., et al., Phosphorylation codes in IRS-1 and IRS-2 are associated with the activation/inhibition of insulin canonical signaling pathways. Current issues in molecular biology, 2024. 46(1): p. 634-649.
- Baker, R.G., M.S. Hayden, and S. Ghosh, NF-κB, inflammation, and metabolic disease. Cell Metab, 2011. 13(1): p. 11-22.
- Kanda, H., et al., MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of clinical investigation, 2006. 116(6): p. 1494-1505. [CrossRef]
- Morshedzadeh, N., et al., A narrative review on the role of hesperidin on metabolic parameters, liver enzymes, and inflammatory markers in nonalcoholic fatty liver disease. Food Science & Nutrition, 2023. 11(12): p. 7523-7533.
- Alam, M.A., et al., Effect of citrus flavonoids, naringin and naringenin, on metabolic syndrome and their mechanisms of action. Advances in nutrition, 2014. 5(4): p. 404-417. [CrossRef]
- Shen, C.-Y., et al., The development of maillard reaction, and advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with AGE-related diseases. Molecules, 2020. 25(23): p. 5591.
- Syed, A.A., et al., Naringin ameliorates type 2 diabetes mellitus-induced steatohepatitis by inhibiting RAGE/NF-κB mediated mitochondrial apoptosis. Life sciences, 2020. 257: p. 118118.
- Cepas, V., et al., Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants (Basel), 2020. 9(2).
- Al-Aubaidy, H.A., et al., Twelve-week mediterranean diet intervention increases citrus bioflavonoid levels and reduces inflammation in people with type 2 diabetes mellitus. Nutrients, 2021. 13(4): p. 1133.
- Passaro, A., et al., The complex interplay between oxinflammation, mitochondrial dysfunction and lipotoxicity: Focus on their role in the pathogenesis of skeletal muscle insulin resistance and modulation by dietary fatty acids. Advances in Redox Research, 2024. 11: p. 100100. [CrossRef]
- Cavaliere, G., et al., From obesity-induced low-grade inflammation to lipotoxicity and mitochondrial dysfunction: altered multi-crosstalk between adipose tissue and metabolically active organs. Antioxidants, 2023. 12(6): p. 1172.
- Kicinska, A. and W. Jarmuszkiewicz, Flavonoids and mitochondria: activation of cytoprotective pathways? Molecules, 2020. 25(13): p. 3060. [CrossRef]
- Armani, A., et al., Nutraceuticals in Brown Adipose Tissue Activation. Cells, 2022. 11(24).
- Zong, Y., et al., Mitochondrial dysfunction: mechanisms and advances in therapy. Signal transduction and targeted therapy, 2024. 9(1): p. 124. [CrossRef]
- Muoio, D.M. and P.D. Neufer, Lipid-induced mitochondrial stress and insulin action in muscle. Cell metabolism, 2012. 15(5): p. 595-605.
- Narendra, D.P., et al., PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS biology, 2010. 8(1): p. e1000298.
- Lemos, G.d.O., R.S. Torrinhas, and D.L. Waitzberg, Nutrients, physical activity, and mitochondrial dysfunction in the setting of metabolic syndrome. Nutrients, 2023. 15(5): p. 1217.
- Ouyang, Y.B., et al., Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion, 2011. 11(2): p. 279-86.
- Chen, X., et al., Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal transduction and targeted therapy, 2023. 8(1): p. 352.
- Alotaibi, G. and A. Alkhammash, Pharmacological landscape of endoplasmic reticulum stress: Uncovering therapeutic avenues for metabolic diseases. European Journal of Pharmacology, 2025: p. 177509. [CrossRef]
- Lenghel, A., et al., What is the sweetest UPR flavor for the β-cell? That is the question. Frontiers in Endocrinology, 2021. 11: p. 614123.
- Lee, J.-H. and J. Lee, Endoplasmic reticulum (ER) stress and its role in pancreatic β-cell dysfunction and senescence in type 2 diabetes. International journal of molecular sciences, 2022. 23(9): p. 4843.
- Zhao, T., J. Du, and H. Zeng, Interplay between endoplasmic reticulum stress and non-coding RNAs in cancer. Journal of Hematology & Oncology, 2020. 13(1): p. 163.
- Amodio, G., et al., Structural and functional significance of the endoplasmic reticulum unfolded protein response transducers and chaperones at the mitochondria–ER contacts: a cancer perspective. Frontiers in Cell and Developmental Biology, 2021. 9: p. 641194. [CrossRef]
- Park, S., et al., Naringenin induces mitochondria-mediated apoptosis and endoplasmic reticulum stress by regulating MAPK and AKT signal transduction pathways in endometriosis cells. MHR: Basic science of reproductive medicine, 2017. 23(12): p. 842-854. [CrossRef]
- Li, X., et al., Effects of hesperidin on mitochondrial function, mitochondria-associated endoplasmic reticulum membranes and IP3R–MCU calcium axis in the intestine of piglets exposed to deoxynivalenol. Food & Function, 2024. 15(12): p. 6459-6474.
- Gou, F., et al., Hesperidin alleviated intestinal barrier injury, mitochondrial dysfunction, and disorder of endoplasmic reticulum mitochondria contact sites under oxidative stress. Journal of Agricultural and Food Chemistry, 2024. 72(29): p. 16276-16286. [CrossRef]
- Khoi, C.-S., T.-Y. Lin, and C.-K. Chiang, Targeting Insulin Resistance, Reactive Oxygen Species, Inflammation, Programmed Cell Death, ER Stress, and Mitochondrial Dysfunction for the Therapeutic Prevention of Free Fatty Acid-Induced Vascular Endothelial Lipotoxicity. Antioxidants, 2024. 13(12): p. 1486. [CrossRef]
- Karunakaran, U. and K.G. Park, A systematic review of oxidative stress and safety of antioxidants in diabetes: focus on islets and their defense. Diabetes Metab J, 2013. 37(2): p. 106-12.
- Sergi, D., et al., Mitochondrial (dys) function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Frontiers in physiology, 2019. 10: p. 449821. [CrossRef]
- González, P., et al., Hyperglycemia and oxidative stress: an integral, updated and critical overview of their metabolic interconnections. International journal of molecular sciences, 2023. 24(11): p. 9352.
- Pu, P., et al., Naringin ameliorates metabolic syndrome by activating AMP-activated protein kinase in mice fed a high-fat diet. Arch Biochem Biophys, 2012. 518(1): p. 61-70.
- Ali, A.M., et al., Antidiabetic Potency, Antioxidant Effects, and Mode of Actions of Citrus reticulata Fruit Peel Hydroethanolic Extract, Hesperidin, and Quercetin in Nicotinamide/Streptozotocin-Induced Wistar Diabetic Rats. Oxid Med Cell Longev, 2020. 2020: p. 1730492.
- Korac, B., et al., Redox changes in obesity, metabolic syndrome, and diabetes. Redox Biol, 2021. 42: p. 101887.
- Park, H.J., et al., Citrus unshiu peel extract ameliorates hyperglycemia and hepatic steatosis by altering inflammation and hepatic glucose- and lipid-regulating enzymes in db/db mice. J Nutr Biochem, 2013. 24(2): p. 419-27. [CrossRef]
- Kim, B.M., B.O. Cho, and S.I. Jang, Anti-obesity effects of Diospyros lotus leaf extract in mice with high-fat diet-induced obesity. Int J Mol Med, 2019. 43(1): p. 603-613.
- Pengnet, S., et al., Naringin attenuates fructose-induced NAFLD progression in rats through reducing endogenous triglyceride synthesis and activating the Nrf2/HO-1 pathway. Front Pharmacol, 2022. 13: p. 1049818.
- T, L.S., et al., Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants (Basel), 2020. 9(10).
- Tsai, H.L., S.K. Chang, and S.J. Chang, Antioxidant content and free radical scavenging ability of fresh red pummelo [Citrus grandis (L.) Osbeck] juice and freeze-dried products. J Agric Food Chem, 2007. 55(8): p. 2867-72.
- Wang, S.W., et al., Neohesperidin enhances PGC-1α-mediated mitochondrial biogenesis and alleviates hepatic steatosis in high fat diet fed mice. Nutr Diabetes, 2020. 10(1): p. 27. [CrossRef]
- Morrow, N.M., et al., The citrus flavonoid nobiletin confers protection from metabolic dysregulation in high-fat-fed mice independent of AMPK. J Lipid Res, 2020. 61(3): p. 387-402.
- Lu, Z., et al., Liver-Specific Bmal1 Depletion Reverses the Beneficial Effects of Nobiletin on Liver Cholesterol Homeostasis in Mice Fed with High-Fat Diet. Nutrients, 2023. 15(11). [CrossRef]
- Mulvihill, E.E., et al., Nobiletin attenuates VLDL overproduction, dyslipidemia, and atherosclerosis in mice with diet-induced insulin resistance. Diabetes, 2011. 60(5): p. 1446-57.
- Lee, Y.S., et al., Nobiletin improves hyperglycemia and insulin resistance in obese diabetic ob/ob mice. Biochem Pharmacol, 2010. 79(11): p. 1674-83. [CrossRef]
- Yang, Y., et al., Naringenin Attenuates Non-Alcoholic Fatty Liver Disease by Enhancing Energy Expenditure and Regulating Autophagy via AMPK. Front Pharmacol, 2021. 12: p. 687095.
- Liu, X., et al., The citrus flavonoid naringenin confers protection in a murine endotoxaemia model through AMPK-ATF3-dependent negative regulation of the TLR4 signalling pathway. Sci Rep, 2016. 6: p. 39735. [CrossRef]
- Sarkar, S., S. Ghosh, and M. Biswas, Naringin ameliorates high-fat diet-induced hepatotoxicity and dyslipidemia in experimental rat model via modulation of anti-oxidant enzymes, AMPK and SERBP-1c signaling pathways. Toxicol Rep, 2025. 14: p. 102062. [CrossRef]
- Guan, L., et al., Naringin Protects against Non-Alcoholic Fatty Liver Disease by Promoting Autophagic Flux and Lipophagy. Mol Nutr Food Res, 2024. 68(3): p. e2200812.
- Tian, M., et al., Hesperidin alleviates insulin resistance by improving HG-induced oxidative stress and mitochondrial dysfunction by restoring miR-149. Diabetol Metab Syndr, 2021. 13(1): p. 50.
- Estruel-Amades, S., et al., Protective Effect of Hesperidin on the Oxidative Stress Induced by an Exhausting Exercise in Intensively Trained Rats. Nutrients, 2019. 11(4). [CrossRef]
- Morshedzadeh, N., et al., A narrative review on the role of hesperidin on metabolic parameters, liver enzymes, and inflammatory markers in nonalcoholic fatty liver disease. Food Sci Nutr, 2023. 11(12): p. 7523-7533.
- Yeh, C.H., et al., Hesperetin promotes longevity and delays aging via activation of Cisd2 in naturally aged mice. J Biomed Sci, 2022. 29(1): p. 53. [CrossRef]
- Durço, A.O., et al., d-Limonene Ameliorates Myocardial Infarction Injury by Reducing Reactive Oxygen Species and Cell Apoptosis in a Murine Model. J Nat Prod, 2019. 82(11): p. 3010-3019.
- Valerii, M.C., et al., Effect of a Fiber D-Limonene-Enriched Food Supplement on Intestinal Microbiota and Metabolic Parameters of Mice on a High-Fat Diet. Pharmaceutics, 2021. 13(11).
- Hiramitsu, M., et al., Eriocitrin ameliorates diet-induced hepatic steatosis with activation of mitochondrial biogenesis. Sci Rep, 2014. 4: p. 3708. [CrossRef]
- Tsutsumi, R., et al., Sudachitin, a polymethoxylated flavone, improves glucose and lipid metabolism by increasing mitochondrial biogenesis in skeletal muscle. Nutr Metab (Lond), 2014. 11: p. 32.
- Sundaram, R., P. Shanthi, and P. Sachdanandam, Effect of tangeretin, a polymethoxylated flavone on glucose metabolism in streptozotocin-induced diabetic rats. Phytomedicine, 2014. 21(6): p. 793-9. [CrossRef]
- Wang, Q., et al., Naringenin attenuates non-alcoholic fatty liver disease by down-regulating the NLRP3/NF-κB pathway in mice. Br J Pharmacol, 2020. 177(8): p. 1806-1821.
- Wu, L., et al., Naringenin Promotes Gastrointestinal Motility in Mice by Impacting the SCF/c-Kit Pathway and Gut Microbiota. Foods, 2024. 13(16).
- Dong, L., et al., Naringenin cationic lipid-modified nanoparticles mitigate MASLD progression by modulating lipid homeostasis and gut microbiota. J Nanobiotechnology, 2025. 23(1): p. 168. [CrossRef]
- Lv, Z., et al., Naringenin improves muscle endurance via activation of the Sp1-ERRγ transcriptional axis. Cell Rep, 2023. 42(11): p. 113288.
- Jia, S., et al., Hypoglycemic and hypolipidemic effects of neohesperidin derived from Citrus aurantium L. in diabetic KK-A(y) mice. Food Funct, 2015. 6(3): p. 878-86.
- Wu, H., et al., Neohesperidin Exerts Lipid-Regulating Effects in vitro and in vivo via Fibroblast Growth Factor 21 and AMP-Activated Protein Kinase/Sirtuin Type 1/Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1α Signaling Axis. Pharmacology, 2017. 100(3-4): p. 115-126.
- Jamal, A., et al., Reduced Insulin Resistance and Oxidative Stress in a Mouse Model of Metabolic Syndrome following Twelve Weeks of Citrus Bioflavonoid Hesperidin Supplementation: A Dose-Response Study. Biomolecules, 2024. 14(6). [CrossRef]
- Yuk, T., et al., Nobiletin Inhibits Hepatic Lipogenesis via Activation of AMP-Activated Protein Kinase. Evid Based Complement Alternat Med, 2018. 2018: p. 7420265.
- Namkhah, Z., et al., Does naringenin supplementation improve lipid profile, severity of hepatic steatosis and probability of liver fibrosis in overweight/obese patients with NAFLD? A randomised, double-blind, placebo-controlled, clinical trial. Int J Clin Pract, 2021. 75(11): p. e14852. [CrossRef]
- Naeini, F., et al., Effects of naringenin supplementation in overweight/obese patients with non-alcoholic fatty liver disease: study protocol for a randomized double-blind clinical trial. Trials, 2021. 22(1): p. 801. [CrossRef]
- Goldwasser, J., et al., Transcriptional regulation of human and rat hepatic lipid metabolism by the grapefruit flavonoid naringenin: role of PPARalpha, PPARgamma and LXRalpha. PLoS One, 2010. 5(8): p. e12399.
- Sleiman, D., M.R. Al-Badri, and S.T. Azar, Effect of mediterranean diet in diabetes control and cardiovascular risk modification: a systematic review. Front Public Health, 2015. 3: p. 69. [CrossRef]
- Carpenito, M., et al., Unveiling the Power of Bergamot: Beyond Lipid-Lowering Effects. Nutrients, 2025. 17(11). [CrossRef]
- Fogacci, F., et al., Metabolic and vascular effect of a new standardized bergamot phytocomplex: a three-arm, placebocontrolled, double-blind clinical trial. Arch Med Sci, 2023. 19(5): p. 1228-1235. [CrossRef]
- Ferro, Y., et al., Citrus Bergamia and Cynara Cardunculus Reduce Serum Uric Acid in Individuals with Non-Alcoholic Fatty Liver Disease. Medicina (Kaunas), 2022. 58(12).
- Jalili, F., et al., The effects of citrus flavonoids supplementation on endothelial function: A systematic review and dose-response meta-analysis of randomized clinical trials. Phytother Res, 2024. 38(6): p. 2847-2859.
- Sost, M.M., et al., A Citrus Fruit Extract High in Polyphenols Beneficially Modulates the Gut Microbiota of Healthy Human Volunteers in a Validated In Vitro Model of the Colon. Nutrients, 2021. 13(11). [CrossRef]
- Sánchez Macarro, M., et al., Effect of a Combination of Citrus Flavones and Flavanones and Olive Polyphenols for the Reduction of Cardiovascular Disease Risk: An Exploratory Randomized, Double-Blind, Placebo-Controlled Study in Healthy Subjects. Nutrients, 2020. 12(5).
- Victoria-Montesinos, D., et al., Effect of Dietary Supplementation with a Natural Extract of Sclerocarya birrea on Glycemic Metabolism in Subjects with Prediabetes: A Randomized Double-Blind Placebo-Controlled Study. Nutrients, 2021. 13(6). [CrossRef]
- Morand, C., et al., Hesperidin contributes to the vascular protective effects of orange juice: a randomized crossover study in healthy volunteers. Am J Clin Nutr, 2011. 93(1): p. 73-80. [CrossRef]
- Yari, Z., et al., The effect of hesperidin supplementation on metabolic profiles in patients with metabolic syndrome: a randomized, double-blind, placebo-controlled clinical trial. Eur J Nutr, 2020. 59(6): p. 2569-2577.
- Khorasanian, A.S., et al., The effects of hesperidin supplementation on cardiovascular risk factors in adults: a systematic review and dose-response meta-analysis. Front Nutr, 2023. 10: p. 1177708. [CrossRef]
- Notarnicola, M., et al., Daily Orange Consumption Reduces Hepatic Steatosis Prevalence in Patients with Metabolic Dysfunction-Associated Steatotic Liver Disease: Exploratory Outcomes of a Randomized Clinical Trial. Nutrients, 2024. 16(18).
- Navajas-Porras, B., et al., Effects of a flavonoid-enriched orange juice on antioxidant capacity, lipid profile, and inflammation in obese patients: A randomized placebo-controlled trial. Food Res Int, 2025. 217: p. 116759. [CrossRef]
- Cesar, T.B., F.M.M. Ramos, and C.B. Ribeiro, Nutraceutical Eriocitrin (Eriomin) Reduces Hyperglycemia by Increasing Glucagon-Like Peptide 1 and Downregulates Systemic Inflammation: A Crossover-Randomized Clinical Trial. J Med Food, 2022. 25(11): p. 1050-1058. [CrossRef]
- Wang, X.C., L. Song, and X.H. Wang, Efficacy of dietary polyphenol supplement in patients with non-alcoholic fatty liver disease: a network meta-analysis. Front Nutr, 2025. 12: p. 1582861.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



