Submitted:
04 August 2025
Posted:
06 August 2025
You are already at the latest version
Abstract
Cancer-associated fibroblasts (CAFs) are crucial regulators of the tumor microenvironment (TME), promoting cancer progression, immune suppression, and therapy resistance. Single-cell transcriptomics has identified at least five distinct CAF subtypes—myofibroblastic (myCAFs), inflammatory (iCAFs), antigen-presenting (apCAFs), metabolic (meCAFs), and vascular/developmental (vCAFs/dCAFs)—each with unique localization, signaling, and functions. While CAFs are well studied in epithelial cancers, their roles in sarcomas are less understood, despite the shared mesenchymal origin of tumor and stromal cells. This overlap blurs the line between malignant and non-malignant fibroblasts, raising fundamental questions about the identity of CAFs in mesenchymal tumors. In this narrative review, we explore the heterogeneity and plasticity of CAFs across solid tumors, focusing on their role in immune evasion, epithelial-to-mesenchymal transition (EMT), and resistance to chemotherapy, targeted therapy, and immunotherapy. We highlight emerging evidence on CAF-like cells in sarcomas and their contribution to tumor invasion, immune exclusion, and metastatic niche formation. We also assess new strategies to target or reprogram CAFs and suggest that CAF profiling may serve as a potential biomarker for patient stratification. Understanding CAF biology across various tumor types, including those with dense stroma and immunologically cold sarcomas, is crucial for developing more effective, personalized cancer treatments.
Keywords:
cancer-associated fibroblasts
; tumor microenvironment
; sarcoma
; fibroblast heterogeneity
; CAF-plasticity
; mesenchymal tumors
; stromal remodeling
; tumor-stroma interaction
; immune modulation
; 3D tumor models
1. Introduction
In normal tissues, fibroblasts play a vital structural and regulatory role, maintaining the extracellular matrix (ECM) and supporting tissue homeostasis [1]. They are responsible for producing ECM proteins such as collagen and fibronectin, and contribute to tissue repair after injury [2,3]. Under physiological conditions, fibroblasts stay quiescent but become temporarily activated during wound healing, adopting a contractile, myofibroblast-like phenotype that promotes matrix remodeling and secretes growth factors [4].
In the tumor microenvironment (TME), however, this activation becomes chronic and dysregulated [5]. Extended exposure to inflammatory signals, hypoxia, and tumor-derived cytokines reprograms normal fibroblasts into cancer-associated fibroblasts (CAFs)—a diverse group of persistently active cells that support tumor growth [6,7]. Unlike temporary wound-healing fibroblasts, CAFs remain in an active state, continuously releasing pro-tumor factors, reshaping the extracellular matrix (ECM), promoting angiogenesis, and influencing immune responses to promote the growth, invasion, and immune evasion of cancer cells [8,9,10].
CAFs exhibit remarkable phenotypic plasticity and can originate from various sources, including resident fibroblasts, bone marrow–derived mesenchymal stem cells, pericytes, adipocytes, and even epithelial or endothelial cells through epithelial-to-mesenchymal transition (EMT) or endothelial-to-mesenchymal transition (EndMT) [11,12,13,14,15]. This diversity enhances their functional heterogeneity, as shown in recent single-cell RNA sequencing studies.
This review offers a comprehensive overview of CAF heterogeneity, function, and clinical significance across solid tumors, with particular emphasis on their understudied roles in sarcomas. We investigate the origins of CAFs, their subtype-specific functions, and their roles in immune evasion, ECM remodeling, and therapeutic resistance. Because sarcomas originate from mesenchymal cells, the distinction between tumor cells and CAFs becomes unclear, raising important biological and clinical questions. We also review emerging strategies to target or reprogram CAFs and consider how understanding their context-dependent roles could enhance the effectiveness of cancer treatment.
2. Data Collection
This narrative review was developed following a comprehensive literature search conducted using PubMed and Google Scholar, covering publications from January 2000 to May 2025. The search strategy aimed to identify studies related to the heterogeneity, biological functions, and clinical implications of cancer-associated fibroblasts (CAFs) across both epithelial and mesenchymal tumors. We included original research articles, clinical studies, review papers, and preclinical investigations (both in vitro and in vivo), with a focus on mechanistic insights, stromal–immune interactions, and therapeutic resistance.
To ensure broad coverage, we used Boolean operators to combine various relevant keywords and phrases, including: 'cancer-associated fibroblasts” OR “CAFs” OR “fibroblast heterogeneity” OR “myofibroblastic CAFs” OR “inflammatory CAFs” OR “antigen-presenting CAFs” OR “CAF subtypes” OR “CAF reprogramming” OR “tumor-promoting CAFs” OR “tumor-suppressive CAFs” OR “CAF plasticity” OR “CAF immune evasion” OR “tumor stroma” OR “tumor microenvironment” OR “TME” OR “stromal remodeling” OR “extracellular matrix” OR “TGF-β” OR “CXCL12” OR “IL-6” OR “immune exclusion” OR “immune checkpoint resistance” OR “CAF-mediated resistance” OR “ferroptosis and CAFs” OR “CAF biomarkers” OR “spatial transcriptomics CAF” OR “single-cell RNA sequencing CAF” OR “sarcomas and CAFs” OR “fibroblastic sarcoma” OR “fibromatosis” OR “desmoid tumor” OR “Ewing sarcoma CAF” OR “LRRC15” OR “CAF-targeted therapy” OR “FAP” OR “ABBV-085” OR “CAF exosomes” OR “CAF and CSC” OR “CAF and angiogenesis” OR “CAF in PDAC” OR “CAF in breast cancer” OR “CAF in NSCLC” OR “CAF in soft tissue sarcoma”.
We also searched for MeSH terms related to “fibroblasts,” “carcinoma,” “sarcoma,” “tumor microenvironment,” “immunotherapy,” “chemoresistance,” and “angiogenesis.” Citation tracking was conducted to identify additional relevant studies not found in the initial search. Only articles published in English were reviewed in full.
Articles were included if they (1) described phenotypic or functional CAF subtypes, (2) explored CAF–immune cell interactions, (3) addressed CAF contributions to therapy resistance, or (4) investigated CAFs in sarcomas and their distinction from tumor cells. Exclusion criteria included studies unrelated to CAFs, papers focused solely on non-oncologic fibroblast biology, and non-English publications.
3. Subtypes of Cancer-Associated Fibroblasts (CAFs)
The traditional view of CAFs as a uniform group has changed significantly over the past decade. Advances in single-cell RNA sequencing and spatial transcriptomics have revealed that CAFs exhibit considerable diversity in both appearance and function. Researchers have identified at least five main CAF subtypes, each playing different roles in tumor biology based on their location, surface markers, secretory profiles, and interactions with other parts of the TME [16,17].
3.1. Myofibroblastic CAFs (myCAFs)
Myofibroblastic cancer-associated fibroblasts (myCAFs) are a specific subset of CAFs characterized by their high expression of α-smooth muscle actin (α-SMA) and their spatial localization near tumor cells [18,19,20]. These cells exhibit a contractile phenotype like that of wound-healing myofibroblasts, playing a crucial role in remodeling the ECM [4,21]. By secreting collagen, fibronectin, and lysyl oxidase, myCAFs contribute to the desmoplastic reaction, which increases tissue stiffness and forms physical barriers that support tumor invasion and hinder the delivery of drugs [22,23]. Importantly, this ECM remodeling is not only structural; it also influences mechanotransduction pathways in cancer cells, such as Yes-Associated Protein and Transcriptional Co-Activator with PDZ-binding motif (YAP/TAZ) signaling, which promotes proliferation, migration, and therapy resistance [24,25].
Despite their protumorigenic roles, myCAFs have also been associated with tumor suppression in certain situations [26,27]. In genetically engineered mouse models of pancreatic ductal adenocarcinoma (PDAC), removing α-SMA+ myoCAFs resulted in decreased survival, increased immunosuppressive infiltrates, such as regulatory T cells, and tumors that were less differentiated and more aggressive [28,29]. This paradox highlights the adaptability of myCAFs and underscores their ability to both promote and inhibit tumor growth depending on the molecular and spatial context.
Recent single-cell transcriptomic studies have revealed that myCAFs are not a single, uniform group, but rather include subclusters with distinct gene expression patterns. For example, in breast cancer, different subsets of ECM-myCAFs and Transforming Growth Factor Beta (TGFβ)-myCAFs have been identified; both help tumors evade the immune system and resist immunotherapy by decreasing CD8-positive T cells (CD8+ T) cell infiltration and increasing TGF-β signaling [30,31]. Additionally, myCAFs may facilitate the development of immune-excluded tumor types, which is a common feature of resistance to immune checkpoint blockade therapies [32,33].
3.2. Inflammatory CAFs (iCAFs)
Inflammatory cancer-associated fibroblasts (iCAFs) are a phenotypically and functionally distinct subset of cancer-associated fibroblasts. Low levels of α-SMA and high secretion of pro-inflammatory cytokines such as Interleukin-6 (IL-6), Interleukin-11 (IL-11), C-X-C motif chemokine ligand 1 (CXCL1), SDF-1, Stromal cell-derived factor 1 (CXCL12), and Leukemia Inhibitory Factor (LIF) characterize them [34,35]. Unlike contractile myCAFs, iCAFs are usually located farther from tumor cells and are transcriptionally activated by interleukin-1 (IL-1) through the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathway [36]. Conversely, TGF-β signaling suppresses this phenotype and promotes the differentiation of myCAFs [30,37]. This reciprocal signaling dependence highlights the dynamic plasticity of CAF states, allowing transitions between iCAF and myCAF identities in response to microenvironmental cues [38].
Functionally, iCAFs play a key role in creating an immunosuppressive TME. The cytokines secreted by iCAFs can attract myeloid-derived suppressor cells (MDSCs) and direct tumor-associated macrophages (TAMs) toward an M2-like phenotype, which then suppresses cytotoxic T cell activity and promotes immune evasion [39,40]. IL-6, in particular, is a powerful driver of Signal Transducer and Activator of Transcription 3 (STAT3) activation in both immune and tumor cells, supporting chronic inflammation, resistance to cell death, and the preservation of cancer stem cell traits [41,42] Additionally, CXCL12 secretion by iCAFs creates a physical and chemotactic barrier that prevents CD8+ T cells from reaching the tumor core, aiding resistance to immune checkpoint inhibitors [43,44].
Recent single-cell RNA sequencing (ssRNA-Seq) studies in pancreatic and breast cancers have further refined the iCAF phenotype, distinguishing subclusters based on differences in cytokine expression profiles and spatial distribution [45]. In some models, iCAFs have also been linked to triggering EMT through paracrine TGF-β and IL-6 signaling, encouraging invasion and metastatic potential [46].
3.3. Antigen-Presenting CAFs (apCAFs)
Antigen-presenting CAFs (apCAFs) are a distinct and functionally complex subtype of CAFs characterized by their expression of Major Histocompatibility Complex (MHC) class II molecules and components of the antigen-processing machinery [47]. Notably, they lack classical co-stimulatory molecules [48]. Initially identified in pancreatic ductal adenocarcinoma (PDAC) through ssRNA-Seq, apCAFs were shown to interact with Cluster of Differentiation 4 positive T lymphocytes (CD4+ T-cells) via MHC–II–dependent mechanisms, potentially presenting antigens and influencing T-cell responses without full activation due to the absence of co-stimulation [48,49].
Recent studies have expanded the significance of apCAFs beyond PDAC, identifying them in lung and gastric cancers. In gastric cancer, apCAFs were found to be enriched in tumor regions near tertiary lymphoid structures (TLS), indicating a role in supporting local immune cell activation and organization [50]. Spatial transcriptomics and immunohistochemistry analyses have confirmed their colocalization with T- and B-cell clusters, and their abundance is associated with improved responses to immune checkpoint blockade [50]. Mechanistically, apCAFs have been shown to enhance the activation and killing ability of CD4+ T-cells, as well as promote M1-like macrophage polarization, thereby creating a positive feedback loop that boosts anti-tumor immunity [50,51]. However, the duality of the apCAF function remains an area of investigation. In specific settings, the absence of co-stimulatory molecules may lead to T-cell anergy or the differentiation of regulatory T-cells (Tregs), potentially suppressing immunity [52,53].
3.4. Metabolic CAFs (meCAFs)
Metabolically active cancer-associated fibroblasts (meCAFs) constitute a distinct subset of CAFs characterized by their pivotal role in promoting tumor metabolism through reprogramming [54]. Unlike myCAFs or iCAFs, meCAFs are primarily identified by their increased expression of glycolytic enzymes, glucose transporters (GLUT1), lipid metabolism enzymes, and amino acid transporters, indicating their active involvement in modifying the tumor’s metabolic environment [5].
CAFs play a key role in the reverse Warburg effect. The Warburg effect describes how cancer cells prefer aerobic glycolysis over oxidative phosphorylation, leading to increased glucose intake and lactate production even in the presence of oxygen [55]. This metabolic shift supports rapid growth and creates an acidic microenvironment that affects CAF activation and function [55]. These metabolites are taken up by cancer cells and used in the tricarboxylic acid (TCA) cycle to produce energy through oxidative phosphorylation (OXPHOS) and promote tumor growth [56]. As a result, CAFs in breast and lung tumors show higher levels of glycolytic enzymes, including lactate dehydrogenase (LDH), pyruvate kinase M2 (PKM2), and the lactate transporter Monocarboxylate Transporter 4 (MCT4([57,58]. Lactate functions as an energy shuttle between stromal and cancer cells, like its role in the brain and heart [59,60,61]. This reverse Warburg-like metabolic interaction enhances tumor growth, invasiveness, and resistance to therapy [62]. Importantly, meCAFs help support cancer cell growth in nutrient-poor or hypoxic tumor areas by supplying alternative energy sources and buffering the acidic microenvironment [63,64].
Mechanistically, the development of meCAF phenotypes is driven by paracrine signals, such as IL-6, TGF-β, and tumor-derived exosomal miRNAs (e.g., miR-105), which activate the Myelocytomatosis oncogene (MYC) and Ataxia Telangiectasia Mutated (ATM) signaling pathways in fibroblasts, thereby causing metabolic shifts [65]. In breast cancer, high meCAF activity has been linked to resistance to chemotherapy and immune checkpoint blockade, due to both metabolic support and the promotion of an immunosuppressive microenvironment through lactic acid–induced T-cell dysfunction and macrophage polarization [66,67].
3.5. Vascular and Developmental CAFs (vCAFs and dCAFs)
Among the recently identified subsets of cancer-associated fibroblasts, vascular cancer-associated fibroblasts (vCAFs) and developmental cancer-associated fibroblasts (dCAFs) are specialized groups with distinct transcriptional profiles and locations within the tumor microenvironment. vCAFs originate from perivascular stromal cells and are found near tumor blood vessels [68]. These cells exhibit high levels of pro-angiogenic mediators such as ascular Endothelial Growth Factor A (VEGFA), Angiopoietin-like 4 (ANGPTL4), and IL-6, along with matrix-remodeling enzymes like MMP9, which aid endothelial cell movement and new vessel formation [69]. Functionally, vCAFs play a key role in stabilizing abnormal tumor blood vessels, enhancing blood flow, and supplying nutrients, ultimately supporting rapid tumor growth and contributing to therapy resistance [70].
3.6. Tumor-Promoting VERSUS Tumor-Suppressive Functions of CAFs
CAFs show a functional split within the TME, acting either as promoters or suppressors of tumor growth [71]. This dual role results from the wide variety and adaptability of these cells, which are affected by spatial location, tumor type, and ongoing cellular interactions. Most studies have historically focused on the tumor-promoting activities of CAFs, which include facilitating cancer cell invasion, immune evasion, angiogenesis, and therapeutic resistance.
Tumor-promoting CAFs (pCAFs) actively support carcinogenesis through various mechanisms. They remodel the ECM by producing structural proteins such as collagen and fibronectin, along with matrix metalloproteinases (MMPs), which degrade ECM components, increase tissue stiffness, and facilitate the creation of invasion pathways for tumor cells [72]. The production of MMPs enables CAFs to promote further invasion of cancer cells, making these enzymes viable therapeutic targets [72]. In lung cancer, ECM remodeling and the secretion of growth factors by CAFs contribute to increased tissue stiffness, which enhances the adhesion of metastatic cancer cells to the tumor endothelium, thereby exacerbating metastatic progression [72]. Additionally, pCAFs secrete a variety of cytokines and chemokines, including interleukin-6 (IL-6), CXCL12, and transforming growth factor-beta (TGF-β), which inhibit cytotoxic T-lymphocyte activity and attract immunosuppressive cells such as regulatory T-cells (Tregs), TAMs, and MDSCs [73,74]. These CAFs also promote angiogenesis by releasing vascular endothelial growth factor (VEGF) and supplying metabolic substrates, such as lactate and glutamine, to cancer cells, thereby supporting their metabolic reprogramming and growth [75]. Moreover, CAFs contribute to resistance against chemotherapy and immunotherapy by strengthening physical barriers within the tumor stroma and maintaining an immunosuppressive environment [76]. For instance, in pancreatic ductal adenocarcinoma, CAF-secreted stromal cell-derived factor 1 (SDF-1) increases Special AT-rich Sequence-Binding Protein 1 (SATB-1) expression in tumor cells, thereby contributing to gemcitabine resistance [77].
In contrast, a subset of CAFs has tumor-suppressive properties. These tumor-restraining CAFs (rCAFs) can act as physical barriers, thereby limiting tumor spread and maintaining tissue architecture [9,79]. Recent studies have demonstrated that specific subsets of CAFs, particularly those producing Cluster of Differentiation 9 (CD9)-positive exosomes, can inhibit melanoma cell proliferation and are associated with improved long-term survival in patients [80]. In lung cancer, Cluster of Differentiation 200 (CD200)-expressing CAFs have been shown to increase tumor cell sensitivity to targeted treatments, such as Epidermal Growth Factor Receptor (EGFR) inhibitors, and this effect disappears when CD200 is absent [81]. Other evidence suggests that factors secreted by CAFs, including Insulin-like Growth Factor (IGF) and IGF-binding proteins, may enhance the response to drugs in non-small cell lung cancer [82,83]. In pancreatic cancer, removing specific myCAF populations has been associated with increased immunosuppression and faster tumor growth, suggesting that these cells may have a protective role [84]. Additionally, antigen-presenting CAFs (apCAFs) may help activate immune responses against tumors [84]. These findings highlight the complexity of CAF biology and emphasize the need for further research into their context-dependent anti-tumor roles. Cancer-associated fibroblasts (CAFs) have dynamic and evolving roles in tumor progression. They adapt their phenotype and functions as the TME changes during different stages of cancer development.
4.0. Crosstalk with Cancer Stem Cells (CSCs)
CAFs also play a crucial role in maintaining cancer stem cells (CSCs), a subpopulation of tumor cells characterized by their ability to self-renew and exhibit pluripotency [1]. They are responsible for producing ECM proteins such as collagen and fibronectin, and contribute to tissue repair after injury [2,3]. Under physiological conditions, fibroblasts remain quiescent but become temporarily activated during wound healing, adopting a contractile, myofibroblast-like phenotype that is associated with a high metastatic potential [85,86]. The interaction between CAF and cancer stem cells (CSC) is facilitated through a complex network of secreted factors, signaling molecules, and extracellular vesicles that work together to enhance the stem-like qualities of cancer cells [87].
IL-6 secreted by CAFs activates the Janus Kinases/Signal Transducer and Activator of Transcription 3 (JAK/STAT3) signaling pathway in tumor cells, thereby enhancing stemness, EMT, and resistance to apoptosis [88,89]. Additionally, CAFs secrete Wingless/Integrated signaling proteins (WNT) ligands and TGF-β, both of which are essential for maintaining the CSC phenotype [90]. WNT signaling promotes β-catenin activation, while TGF-β facilitates EMT and niche adaptation, fostering metastatic ability [90,91]. Beyond soluble factors, CAFs also communicate with CSCs through the release of exosomes [92,93]. These extracellular vesicles can carry various bioactive molecules, including microRNAs (miRNAs), metabolites, and proteins that promote stemness and therapeutic resistance [92,93]. For example, CAF-derived exosomes enriched with miR-21 or miR-146a have been shown to enhance CSC traits and resistance to chemotherapeutic drugs [94,95]
Additionally, the close physical proximity between CAFs and CSCs in the tumor microenvironment supports the notion that CAFs play a crucial role in the CSC niche [96]. Through direct cell-to-cell contact and remodeling of the ECM, CAFs contribute to creating a microenvironment that protects CSCs from immune surveillance and the stress caused by treatments, thereby promoting tumor recurrence and metastasis [97].
5. CAFs in Sarcomas
Sarcomas are a biologically and clinically diverse group of cancers that arise from mesenchymal tissues, such as bone, muscle, fat, cartilage, and connective tissue [98]. This origin from mesenchymal tissue clearly distinguishes sarcomas from carcinomas, which originate from epithelial cells [99]. As a result, the role and understanding of CAFs in sarcomas are less clearly defined and may differ significantly from their roles in epithelial tumors. Therefore, comprehending how CAFs influence the tumor microenvironment in sarcomas is crucial and may necessitate unique conceptual and experimental approaches.
Figure 1.
Distinct Origins and Functional Heterogeneity of Cancer-Associated Fibroblasts in Carcinomas and Sarcomas.
Figure 1.
Distinct Origins and Functional Heterogeneity of Cancer-Associated Fibroblasts in Carcinomas and Sarcomas.
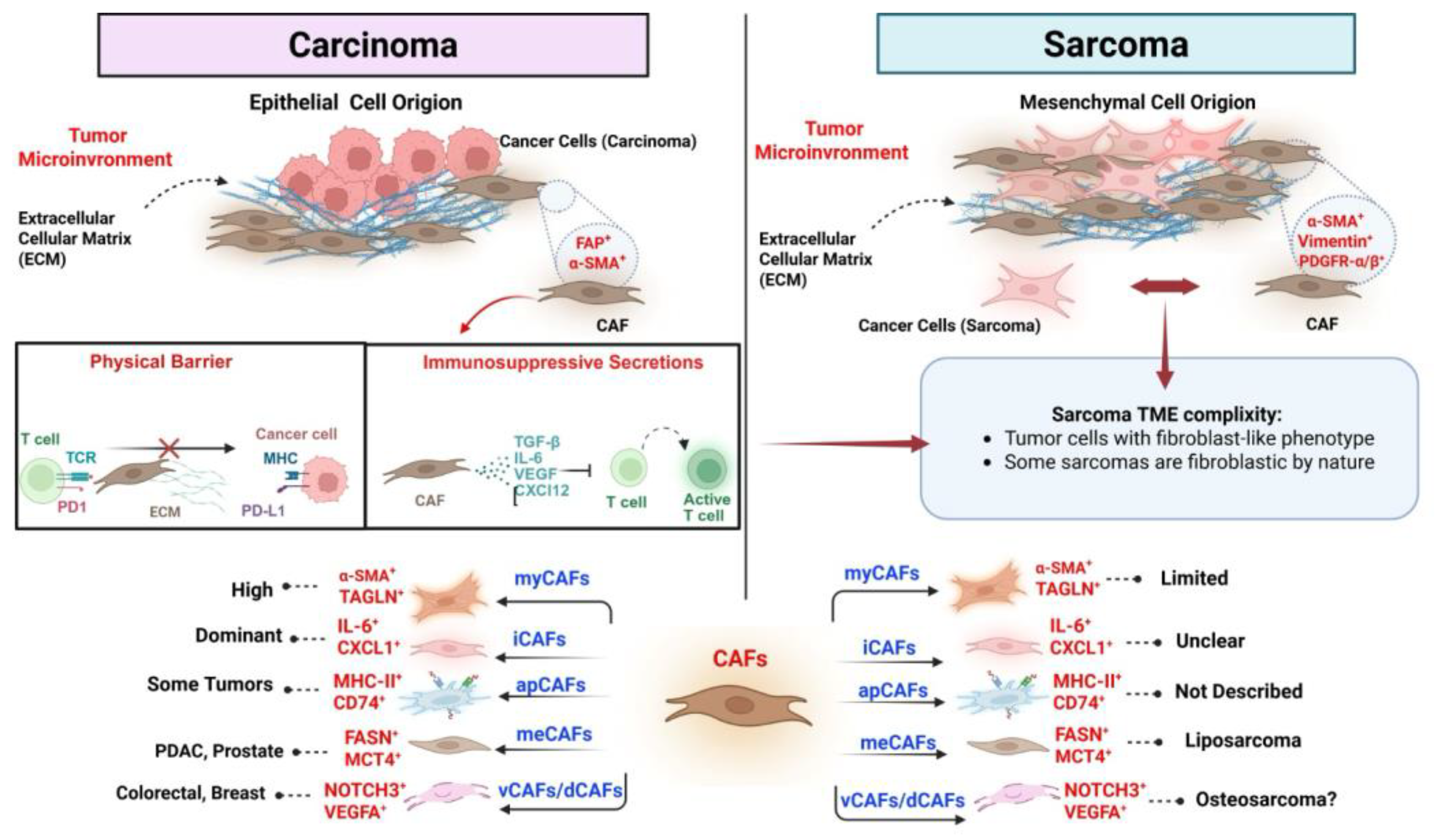
This figure compares the tumor microenvironment (TME) and cancer-associated fibroblast (CAF) characteristics in carcinomas versus sarcomas, highlighting both their origins and immunomodulatory functions. On the left side, carcinomas are depicted as tumors of epithelial cell origin, where CAFs—typically marked by expression of FAP and α-SMA—play a pivotal role in shaping the TME. These fibroblasts contribute to immune evasion through two primary mechanisms: creating a dense extracellular matrix (ECM) that forms a physical barrier to T-cell infiltration, and releasing immunosuppressive cytokines, such as TGF-β, IL-6, VEGF, and CXCL12, which inhibit T-cell activation. Carcinoma-associated CAFs exhibit substantial heterogeneity and can be classified into subtypes such as myCAFs, iCAFs, apCAFs, meCAFs, and vCAFs/dCAFs, each defined by distinct molecular profiles and functional roles. In contrast, sarcomas arise from mesenchymal cells and exhibit a more complex interplay with the surrounding stroma. Some sarcoma tumor cells exhibit fibroblast-like features, blurring the distinction between malignant and stromal compartments. Sarcoma-associated CAFs also express markers such as α-SMA, vimentin, and PDGFR-α/β; however, the functional roles and prevalence of specific CAF subtypes remain less clearly defined. For example, myCAFs appear to be limited in sarcomas; the presence of iCAFs is still unclear, and apCAFs have not yet been described in this tumor type. Specific CAF subsets—such as those expressing FASN and MCT4—have been associated with sarcoma subtypes, including liposarcoma. In contrast, vCAFs/dCAFs (characterized by NOTCH3 and VEGFA) may play a role in osteosarcoma, although this remains speculative.
6. Ambiguity in CAF Identity
In carcinomas, CAFs are typically reactive, non-malignant stromal cells that are activated by signals from the tumor [100]. They promote tumor progression through immunosuppression, remodeling of the ECM, and facilitating angiogenesis [101]. In these contexts, CAFs are usually distinguishable from tumor cells based on lineage markers and histological features. In contrast, the identity of CAFs in sarcomas remains much less defined. Many sarcomas, particularly those with fibroblastic or myofibroblastic differentiation, such as fibrosarcoma and desmoid-type fibromatosis, are composed of tumor cells that themselves express typical CAF markers, including α-SMA, fibroblast activation protein (FAP), platelet-derived growth factor receptors (PDGFR-α/β), and vimentin [101,102,103,104]. This phenotypic similarity complicates the distinction between malignant tumor cells and non-neoplastic fibroblasts within the tumor microenvironment. One study investigated the role of transcriptional heterogeneity in Ewing sarcoma (EwS) and its contribution to phenotypic diversity within tumors. Although EwS is genetically and histologically uniform, its tumor cells show varying degrees of mesenchymal differentiation at the transcriptional level [105]. Using single cell proteogenomic sequencing of EwS cell lines and integrating this data with patient tumor transcriptomic data, the researchers identified distinct subpopulations of tumor cells, particularly a subset characterized by high expression of Cluster of Differentiation 73 (CD73) [105]. CD73+ EwS cells exhibited features typically associated with CAFs, including upregulation of ECM-related genes, increased migratory capacity, and reduced activity of the EWS: FLI1 fusion oncoprotein [105]. These cells were observed to produce large amounts of ECM proteins and were therefore termed “CAF-like tumor cells.” Spatial profiling revealed that these cells are heterogeneously distributed across tumors, often clustering in invasive fronts and peri-necrotic areas [105]. The findings demonstrate that EwS tumor cells can acquire CAF-like characteristics and contribute to the remodeling of the tumor microenvironment from within. This challenges the conventional notion that ECM production in tumors is solely the function of stromal fibroblasts, highlighting an intrinsic mechanism by which tumor cells themselves modulate their microenvironment.
7. Functional Contributions of Stromal CAFs
Despite the ambiguity surrounding cellular identity, accumulating evidence suggests that authentic non-malignant CAF populations exist in sarcomas and play functional roles in tumor progression, immune modulation, and therapy resistance. These CAFs may originate from resident fibroblasts, pericytes, or bone marrow–derived mesenchymal stem cells (MSCs), like their counterparts in epithelial tumors [11]. A scRNA-seq study of tumors in osteosarcoma has explored the TME of primary, recurrent, and metastatic osteosarcoma (OS).The analysis revealed that CAFs are more abundant in recurrent OS and are strongly enriched in the EMT pathway [106]. Notably, CAFs in recurrent tumors showed high expression of lysyl oxidase (LOX), an enzyme implicated in extracellular matrix remodeling and EMT induction. Functional experiments demonstrated that LOX plays a central role in regulating CAF activity, promoting macrophage polarization, and reshaping the immune microenvironment in OS [106].
Furthermore, pharmacological inhibition of LOX significantly reduced tumor cell migration and enhanced apoptosis, both in vitro and in vivo. The findings highlight a novel mechanism by which CAFs contribute to OS progression through Oxidation-mediated (OX-mediated) EMT and immune modulation [106]. Targeting LOX in CAFs may offer a promising strategy for remodeling the TME and improving outcomes in recurrent osteosarcoma. While the specific CAF subtypes, known as iCAFs, myCAFs, and apCAFs, have been well-characterized in epithelial cancers, such as pancreatic cancer, their classification in sarcomas remains under investigation, and similar but not identical subpopulations have been reported.
Several studies have highlighted key molecular pathways through which CAFs facilitate tumor invasion. A study found that Collagen Type VI Alpha 1 Chain (COL6A1) is commonly upregulated in OS, especially in lung metastases, and is associated with poor prognosis [107]. This upregulation is driven by c-Jun binding to E1A Binding Protein p300 (p300), which increases histone H3 lysine 27 (H3K27) acetylation at the COL6A1 promoter [107]. COL6A1 promotes OS cell migration and invasion by interacting with Suppressor of Cytokine Signaling 5 (SOCS5) to suppress STAT1 expression and activation via ubiquitination and proteasomal degradation [107]. Additionally, COL6A1 is packaged into osteosarcoma-derived exosomes, which convert normal fibroblasts into cancer-associated fibroblasts (CAFs) that secrete IL-6 and Interleukin-8 (IL-8) [107]. These activated CAFs enhance the invasion and migration of osteosarcoma cells through the TGF-β/COL6A1 signaling pathway [107]. Overall, COL6A1 promotes OS metastasis by both suppressing STAT1 in tumor cells and activating CAFs [107].
Another study identified a specific group of glycolytic CAFs (glyCAFs) in soft tissue sarcomas, characterized by high GLUT1-dependent glycolysis and the expression of CD73 and CD90 [108]. These glyCAFs produce C-X-C motif chemokine ligand 16 (CXCL16), which traps cytotoxic CD8+ T cells at the tumor margins, restricting their infiltration and promoting an immunosuppressive environment [108]. Inhibiting glycolysis decreased glyCAF accumulation and CXCL16 secretion, improved T-cell infiltration, and enhanced chemotherapy response [108]. This study highlights the role of the metabolic state of CAFs in sarcomas in contributing to therapy resistance and immune evasion. These CAFs depend on GLUT1-mediated glycolysis to produce CXCL16, forming a barrier to CD8+ T-cell infiltration [108]. Targeting their metabolism restored T-cell infiltration and worked synergistically with chemotherapy, revealing a promising strategy to overcome immune exclusion.
Another study demonstrated that reprogramming CAFs, rather than depleting them, may be therapeutically advantageous. Their nanocomposite hydrogel delivered a NADPH oxidase 4 (Nox4) inhibitor followed by doxorubicin, reprogramming the stromal environment and enhancing anti– Programmed Cell Death Protein 1 (PD–1) responses in osteosarcoma models [109]. An analysis of 133 soft-tissue sarcoma cases showed that high expression of CAF markers Fibroblast Activation Protein (FAP), Cluster of Differentiation 10 (CD10), and podoplanin, both within tumors and at their margins, was linked to significantly worse disease-free, metastasis-free, and local recurrence-free survival [110]. These findings underscore the prognostic importance of CAF marker expression in soft-tissue sarcomas and support targeting the CAF compartment as a potential therapeutic approach.
In contrast to other sarcomas, rhabdomyosarcoma (RMS) appears largely CAF-independent. Rhabdomyosarcoma (RMS) seems to rely less on stromal fibroblasts. In comparative analyses using 2D and 3D culture systems and mouse xenografts, RMS cells exhibited minimal interaction with fibroblasts, tended to lose their spheroid structure on stromal layers, and showed low CAF infiltration in vivo [111]. These findings indicate that RMS cells directly remodel their ECM and depend less on CAF support for tumor expansion [111]. Analysis of Leucine-Rich Repeat Containing 15 (LRRC15) expression across 711 soft-tissue sarcoma (STS) cases revealed that, unlike in epithelial tumors, LRRC15 is present not only in stromal cells but also in tumor cells in various STS subtypes [112]. Elevated LRRC15 expression was associated with higher tumor grade and poorer prognosis [113]. Preclinical data demonstrated that targeting LRRC15 with the antibody-drug conjugate ABBV-085 produced substantial anti-tumor effects in LRRC15-positive Soft Tissue Sarcoma (STS) models [112]. These findings highlight LRRC15 as a potential biomarker and therapeutic target in STS, supporting ongoing clinical evaluation of ABBV-085.
8. Clinical Implications
CAFs are increasingly recognized as pivotal players in shaping the TME and influencing therapeutic outcomes. Their roles in promoting tumor progression, immunosuppression, and resistance to multiple treatment modalities, particularly immunotherapy, make them attractive but complex clinical targets. Emerging therapeutic strategies seek to either eliminate CAFs, reprogram their phenotype, or disrupt their interactions with cancer and immune cells. These approaches must carefully navigate the heterogeneity and context dependence. For instance, the depletion of α-SMA+ myofibroblastic CAFs in pancreatic cancer models paradoxically accelerated tumor progression, reduced overall survival, and increased immunosuppressive cell infiltration [113]. These findings underscore the risk of indiscriminate CAF ablation and have shifted therapeutic interest toward selective modulation or reprogramming of specific CAF subtypes.
Reprogramming strategies aim to convert tumor-promoting CAFs into quiescent or even tumor-restraining phenotypes. This alternative therapeutic approach focuses on transforming activated CAFs into less harmful fibroblasts or into CAF subpopulations with tumor-suppressive functions rather than eliminating them [114]. One promising agent in this context is all-trans retinoic acid (ATRA), a metabolite of vitamin A known to promote cellular differentiation and immune modulation [115]. ATRA has been shown to restore quiescence in activated fibroblasts, thereby limiting their tumor-supporting behavior in several epithelial tumor models [115].
Beyond its effect on CAFs, ATRA also targets Myeloid-Derived Suppressor Cells (MDSCs), a key immunosuppressive population within the tumor microenvironment [116]. When combined with immune checkpoint inhibitors, ATRA has been shown in preclinical models of mesothelioma, fibrosarcoma, and non-small cell lung cancer (NSCLC) to reduce MDSC accumulation and promote an interferon-driven, CD8+ T-cell-enriched tumor milieu, ultimately enhancing the response to anti-PD-1 therapy [116]. These dual actions make ATRA a compelling agent for reshaping the tumor microenvironment and improving immunotherapeutic efficacy. This effect is also supported by Phase II trials in melanoma that combine ATRA with ipilimumab or pembrolizumab [117]. Another strategy to limit CAF activity is to restore their quiescent state using retinoic acid (a metabolite of vitamin A) or vitamin D compounds. Vitamin A or D deficiency is linked to CAF activation, so reintroducing these pathways may reverse this effect [118,119]. In pancreatic cancer models, ATRA treatment induces CAF quiescence, thereby reducing tumor cell proliferation and increasing apoptosis [120]. Similarly, the vitamin D analog calcipotriol reprograms the stroma to a quiescent state, thereby improving drug delivery and enhancing the efficacy of chemotherapy [121]. Another study found that Minichromosome Maintenance Complex Component 2 (MCM2) is upregulated in liposarcoma tissues and cells, promoting a CAF-like phenotype characterized by increased expression of FAP, α-SMA, and elevated secretion of IL-6, IL-8, and TGF-β [122]. These MCM2-activated CAF-like cells enhanced liposarcoma proliferation, migration, and invasion. While doxorubicin alone had little effect, combining MCM2 knockdown with doxorubicin suppressed proliferation and induced apoptosis [122]. In vivo, silencing MCM2 reduced tumor growth [122]. Overall, MCM2 drives CAF-like activation and contributes to liposarcoma progression and chemoresistance, suggesting it as a potential therapeutic target [122]. In osteosarcoma, for instance, CAF-derived exosomal microRNA-1228 (miR-1228) promotes tumor invasiveness by suppressing SCAI in cancer cells [123]. Interruption of this axis reduces metastatic potential and may restore chemosensitivity [123]. Similarly, glycolytic CAFs (glyCAFs) in soft tissue sarcomas create an immunosuppressive niche by producing CXCL16, which establishes a physical and chemotactic barrier to CD8+ T cell infiltration [123]. Targeting glycolysis in these CAFs restored immune cell access and synergized with chemotherapy, offering a new angle for combinatorial therapy.
8.1. CAF Subtypes as Predictive Biomarkers
Beyond their role as therapeutic targets, CAFs and their molecular signatures are emerging as potential predictive biomarkers. Single-cell transcriptomic analyses have identified subtypes of CAFs that correlate with immune exclusion, desmoplasia, and poor responses to immunotherapy. For instance, in lung adenocarcinoma, CAF-based gene expression profiles have been shown to predict resistance to immune checkpoint blockade [124]. Research has shown that iCAFs, characterized by a secretory and inflammatory phenotype, are often located distal to tumor cells. In contrast, myCAFs, which express contractile and ECM-remodeling genes, are found closer to tumors [125]. Gene ontology analyses highlight that both iCAF and myCAF populations are heavily involved in extracellular matrix production and remodeling. Importantly, dense and stiff ECM—often generated by myCAFs—serves as both a physical barrier and a biochemical inhibitor to CD8+ T-cell infiltration, contributing to immune exclusion [126]. Additionally, ECM-rich myCAF signatures have been correlated with increased T-cell dysfunction, as indicated by the elevated expression of checkpoint molecules, such as PD-1 and CTLA-4, on tumor-infiltrating lymphocytes [72].
8.2. CAFs and Therapeutic Resistance
CAFs play a crucial role in promoting resistance to cancer treatments by supporting the survival of tumor cells under therapeutic stress. CAFs also remodel the ECM, creating a dense, fibrotic microenvironment that impedes drug penetration and promotes tumor cell invasion [127,128,129]. Additionally, CAFs facilitate immune evasion by attracting immunosuppressive cells (e.g., MDSCs, Tregs) and preventing the infiltration of cytotoxic T cells, further supporting tumor persistence [130,131,132]. Overall, these complex interactions position CAFs as key orchestrators of treatment resistance, making them potential targets for therapy to improve clinical outcomes. These features enable tumors to evade cytotoxic effects and adapt to the pressures of treatment.
8.2.1. Chemotherapy Resistance
CAFs are increasingly recognized as central mediators of chemotherapy resistance in various solid tumors. Through a combination of paracrine signaling, extracellular vesicle (EV) release, and metabolic reprogramming, CAFs reshape the tumor microenvironment to shield cancer cells from drug-induced cytotoxicity [133,134]. A consistent finding across malignancies is the role of CAF-derived cytokines, particularly interleukin-6 (IL-6) [135]. IL-6 secretion activates the STAT3 signaling pathway in tumor cells, promoting survival, proliferation, and resistance to Deoxyribonucleic Acid (DNA)-damaging agents, such as cisplatin [136]. In esophageal squamous cell carcinoma, IL-6 collaborates with exosomal miR-21 to induce monocytic myeloid-derived suppressor cells (M-MDSCs), which further contribute to immunosuppression and chemoresistance [137]. Inhibiting both IL-6 and miR-21 partially reversed resistance, highlighting the therapeutic relevance of this axis [138].
Another mechanism by which CAFs promote chemoresistance is through the transfer of non-coding RNAs via exosomes [139]. In bladder cancer, exosomal Long Intergenic Non-Protein Coding RNA 355 (LINC00355) derived from CAFs was shown to sponge microRNA-34b-5p (miR-34b-5p), consequently upregulating ATP-Binding Cassette Subfamily B Member 1 (ABCB1), a key drug efflux transporter [140]. This mechanism directly reduced cisplatin sensitivity in tumor cells and was reversed upon knockdown of LINC00355 or overexpression of miR-34b-5p [140]. Similarly, in ovarian and oral cancers, CAF-secreted midkine (MK) was found to upregulate the long non-coding RNA Antisense Noncoding RNA in the INK4 Locus (ANRIL) in cancer cells [141]. ANRIL modulates apoptosis regulators and efflux transporters, ultimately reducing the efficacy of cisplatin [141]. These findings demonstrate that Long Non-Coding RNA (lncRNA)-mediated crosstalk between CAFs and tumor cells significantly contributes to therapy resistance.
In addition to transcriptional changes, CAFs also rewire the metabolism of cancer cells to favor survival under chemotherapeutic stress. In ovarian cancer, CAF-derived Holliday Junction Recognition Protein (HJURP) enhances glutamine metabolism and tricarboxylic acid (TCA) cycle activity, facilitating resistance to doxorubicin [142]. This metabolic adaptation not only sustains energy production but also buffers redox imbalances caused by chemotherapy. Importantly, co-culture of tumor cells with CAF-conditioned media results in elevated HJURP expression and increased IC50 values, highlighting the functional relevance of this stromal–tumor metabolic axis [142].
8.2.2. CAFs and Resistance to Immunotherapy
CAFs have emerged as crucial mediators of resistance to immune checkpoint blockade (ICB) across various tumor types. By secreting immunosuppressive cytokines (e.g., TGF-β, IL-6, CXCL12), expressing immune checkpoint ligands Programmed Death-Ligand 1/Programmed Death-Ligand 2 (PD-L1/PD-L2), and producing metabolic modulators such as Indoleamine 2,3-Dioxygenase (IDO) and Arginase 2 (ARG2), CAFs shape the TME into an immune-excluded, T-cell-dysfunctional landscape [72,74,143]. Studies have demonstrated that targeting this axis—through the inhibition of C-X-C Chemokine Receptor Type 4 (CXCR4) or TGF-β signaling—can remodel the tumor stroma, enhance CD8+ T cell infiltration, and improve the effectiveness of immune checkpoint blockade [144,145]. Recent studies have improved our understanding of CAF heterogeneity in this context. For instance, subsets such as iCAFs and myCAFs are typically immunosuppressive, whereas apCAFs may support anti-tumor responses [146,147]. Clinical correlations are emerging; Microfibril-Associated Glycoprotein 2 (MFAP2) + CAFs in gastric cancer have been shown to impair responses to both chemotherapy and immunotherapy through macrophage migration inhibitory factor (MIF)–mediated immune modulation [148]. Similarly, desmoplastic CAFs expressing Decorin (DCN) were found to indicate Immune Checkpoint Blockade (ICB)-resistant phenotypes in metastatic gastric cancer, correlating with low PD-L1 and interferon signaling [149]. Mechanistically, CAFs can induce resistance through direct interactions with immune cells or by remodeling the extracellular matrix. Endo180 (MRC2), a receptor enriched on myCAFs, has been shown to suppress CD8+ T cell infiltration and limit the efficacy of αPD-1/αCTLA-4 in murine models and melanoma patients [150]. In PDAC, CAF-secreted Tenascin C (TNC) promotes epithelial-to-mesenchymal transition (EMT) and immune exclusion through integrin αV/β3 signaling, identifying a novel stromal checkpoint amenable to therapeutic targeting [151]. Emerging resistance mechanisms are also evident in cellular therapies. In multiple myeloma, CAFs within the bone marrow niche decreased Chimeric Antigen Receptor T-cell therapy (CAR-T) efficacy by impairing cytotoxic T cell function [152]. Dual targeting of CAFs and tumor cells restored the response, indicating the need to design CAR-T constructs that address stromal barriers.
Finally, CAF-driven resistance extends beyond immune exclusion. Recent studies suggest that CAFs may also foster resistance by inhibiting ferroptosis in tumor cells—a type of regulated cell death that can enhance anti-tumor immune responses through the release of danger-associated molecular patterns (DAMPs) and the promotion of immune activation [153,154]. For instance, in gastrointestinal tumors, the overexpression of ANO1 facilitates CAF recruitment via TGF-β signaling, resulting in the formation of an immunosuppressive niche that resists anti-PD-1 therapy [155]. Anoctamin-1, (ANO1) has also been linked to the modulation of lipid metabolism and iron homeostasis, which may contribute to ferroptosis resistance [155]. Inhibition of ANO1 reverses both CAF accumulation and ferroptosis suppression, presenting a therapeutic opportunity to enhance immunotherapy responses by targeting CAF-related metabolic and signaling pathways [155].
Figure 2.
CAF-mediated mechanisms of resistance to chemotherapy and immunotherapy in the tumor microenvironment.
Figure 2.
CAF-mediated mechanisms of resistance to chemotherapy and immunotherapy in the tumor microenvironment.
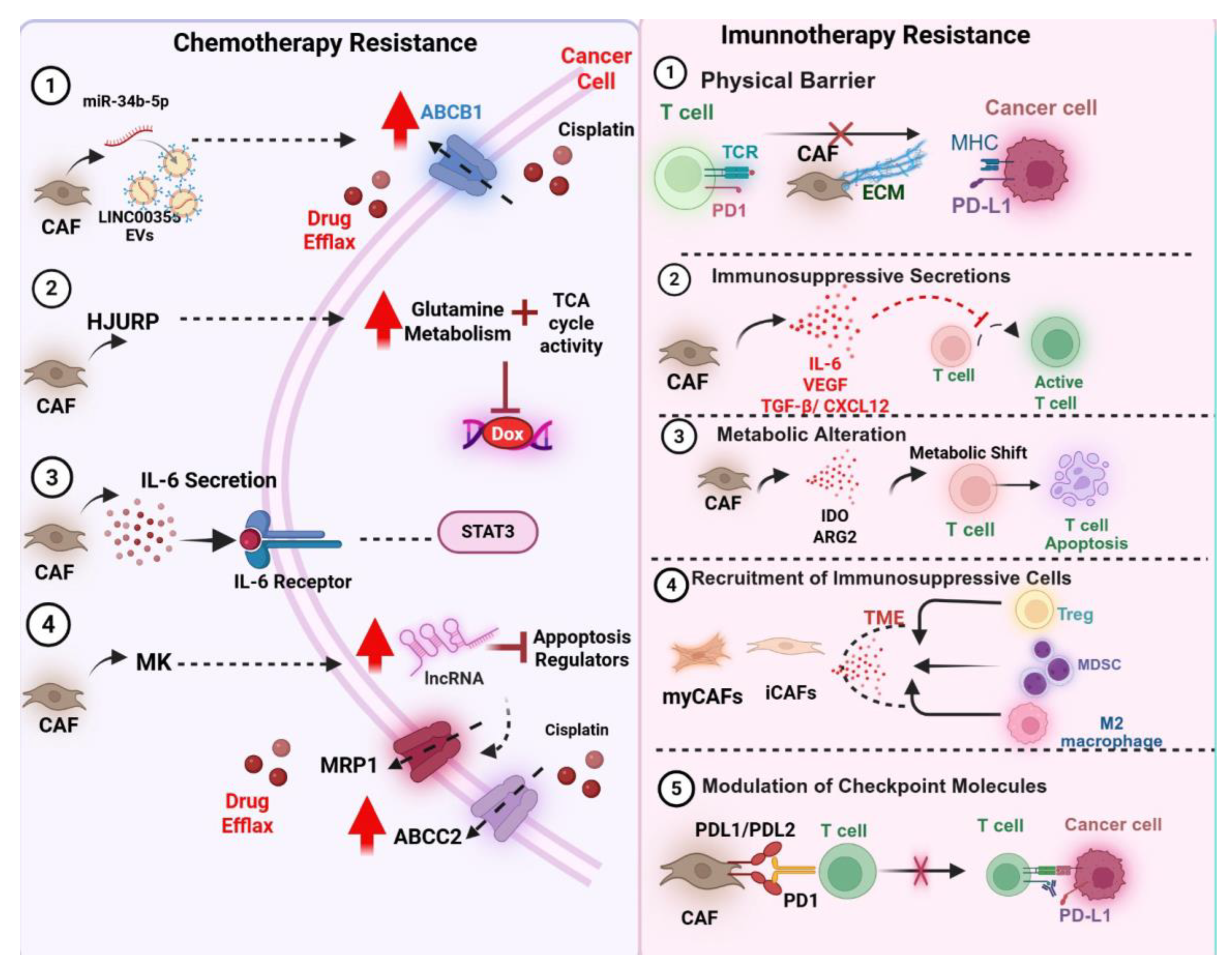
This figure illustrates the diverse mechanisms by which cancer-associated fibroblasts (CAFs) contribute to therapeutic resistance in cancer, highlighting both chemotherapy (left panel) and immunotherapy (right panel) pathways. On the left, several CAF-driven mechanisms that promote chemotherapy resistance are shown. CAFs can transfer long non-coding RNAs (e.g., LINC00355) and microRNAs (e.g., miR-34b-5p) via extracellular vesicles, leading to upregulation of drug efflux transporters such as ABCB1, MRP1, and ABCC2 in cancer cells, thereby reducing intracellular drug accumulation and efficacy. In parallel, CAF-derived factors, such as HJURP, enhance glutamine metabolism and increase tricarboxylic acid (TCA) cycle activity, thereby conferring resistance to agents like doxorubicin. Additionally, IL-6 secretion by CAFs activates the IL-6/STAT3 signaling pathway in tumor cells, promoting survival and reducing apoptotic response to chemotherapy. Other CAF-secreted mediators, such as midkine (MK), modulate apoptosis regulators through long non-coding RNAs, further reinforcing resistance.
8.2.3. CAFs and Resistance to Endocrine and Targeted Therapies
CAFs play a complex role in mediating resistance to endocrine and targeted therapies across different cancer types. In hepatocellular carcinoma (HCC), researchers developed murine and human 3D co-culture models of liver tumor organoids with CAFs to investigate their interactions and treatment response [156]. CAFs promoted tumor organoid growth through direct contact and paracrine signaling, while cancer cells influenced the physiology of CAFs. In xenograft models, co-transplantation with CAFs has been shown to enhance tumor growth [156]. Additionally, the presence of CAFs or their conditioned medium reduced the effectiveness of sorafenib, regorafenib, and 5-Fluorouracil (5-FU), underscoring the role of CAFs in fostering liver cancer growth and therapy resistance [156]. Also in HCC, another study found that fibronectin extra domain A (FN-EDA), derived from cancer-associated fibroblasts, plays a key role in promoting sorafenib resistance in hepatocellular carcinoma (HCC) [157]. FN-EDA activates the Toll-Like Receptor 4/Nuclear Factor kappa-light-chain-enhancer of activated B cells (TLR4/NF-κB) pathway in HCC cells, leading to increased expression of Serine Hydroxymethyltransferase 1(SHMT1), a crucial enzyme in one-carbon metabolism, which helps cancer cells counter sorafenib-induced oxidative stress [157]. In non-small cell lung cancer (NSCLC), cancer-associated fibroblasts (CAFs) drive resistance to EGFR-TKIs through multiple mechanisms. Yi et al. demonstrated that CAF-secreted HGF and IGF-1 activate the Mesenchymal-Epithelial Transition factor/Insulin-like Growth Factor 1 Receptor (c-MET/IGF-1R) signaling pathway, inducing Annexin A2 (ANXA2) expression and EMT, which mediates gefitinib resistance [158]. Zhang et al. described a subset of Collagen Triple Helix Repeat Containing (CTHRC1+) CAFs that sustain EGFR-TKI resistance through metabolic reprogramming and histone acetylation [159]. Clinically, high stromal ANXA2 and CAF density were correlated with poor progression-free survival [160]. Podoplanin-positive CAFs have been shown to promote primary resistance in tumors by sustaining persistent ERK pathway activation, which supports the survival and proliferation of tumor cells despite therapy [9]. Meanwhile, C-X-C motif chemokine ligand 14(CXCL14)-positive CAFs contribute to cancer progression by enhancing epithelial-mesenchymal transition (EMT) and promoting angiogenesis, facilitating increased tumor invasiveness and vascularization [161]. In breast cancer, one study identified a subset of Cluster of Differentiation 63 (CD63+) CAFs in ER-positive breast cancer that are enriched in tumors resistant to CDK4/6 inhibitors (CDK4/6i) [162]. These CAFs promote resistance by releasing exosomes containing miR-20, which targets and downregulates Retinoblastoma 1 (RB1) in cancer cells, thereby reducing the sensitivity to Cyclin-Dependent Kinases 4 and 6 (CDK4/6) inhibitors, using cyclic Arg-Gly-Asp peptide (cRGD)-miR-20 sponge nanoparticles to neutralize miR-20 restored drug sensitivity in vitro and in vivo, suggesting that targeting CD63+ CAFs and their exosomal miR-20 could improve CDK4/6i efficacy in breast cancer [162]. Another study identified a subset of CD63+ CAFs in Estrogen Receptor Alpha (Erα)-positive breast cancer that contributes to tamoxifen resistance [163]. These CAFs secrete exosomes enriched with miR-22, which targets ERα and Phosphatase and Tensin Homolog (PTEN) in cancer cells, promoting drug resistance [163]. Splicing Factor, Arginine/Serine-Rich 1 (SFRS1) mediates packaging of miR-22 into exosomes, while CD63 supports CAF phenotype via STAT3 activation [163]. Blocking CD63+ CAFs with neutralizing antibodies or miR-22 sponge nanoparticles restored tamoxifen sensitivity, highlighting CD63+ CAFs as potential therapeutic targets to overcome resistance [163].
Also in breast cancer, CAFs influence responses to anti-Human Epidermal Growth Factor Receptor 2 (HER2) therapies (e.g., trastuzumab) by increasing cancer stem cell populations and activating IL–6–mediated Signal Transducer and Activator of Transcription 3/Phosphoinositide 3-Kinase (STAT3/PI3K) pathways [164]. Additionally, low stromal Extracellular Signal-Regulated Kinase (ERK) phosphorylation predicted a poor response to tamoxifen, and the loss of caveolin-1 in cancer-associated fibroblasts (CAFs) was associated with worse outcomes in hormonal therapy [165]. In prostate cancer, CAF-secreted C-C Motif Chemokine Ligand 5 (CCL5) increases Androgen Receptor (AR) and PD-L1 expression, promoting resistance to enzalutamide [166], and CTHRC1+ myofibroblast-like CAFs modulate androgen receptor signaling through the Cellular Communication Network Factor 2/Caveolin-1/Androgen Receptor (CCN2/CAV1/AR) pathway, further contributing to anti-androgen resistance [167]. In ovarian cancer, CAFs mediate resistance to anti-angiogenic therapy through sustained pro-angiogenic signaling and expression of immune checkpoints [168]. Similarly, in head and neck squamous cell carcinoma (HNSCC), CAFs promote resistance to EGFR inhibitors through the secretion of Matrix Metalloproteinases (MMPs) and Amphiregulin (AREG)-mediated receptor stabilization [169]. Neuroendocrine tumors (NETs) show resistance to everolimus due to CAF-induced STAT3 activation, which promotes proliferation and drug resistance [170].
Figure 3.
CAF-mediated mechanisms of resistance to endocrine and targeted therapies in cancer.
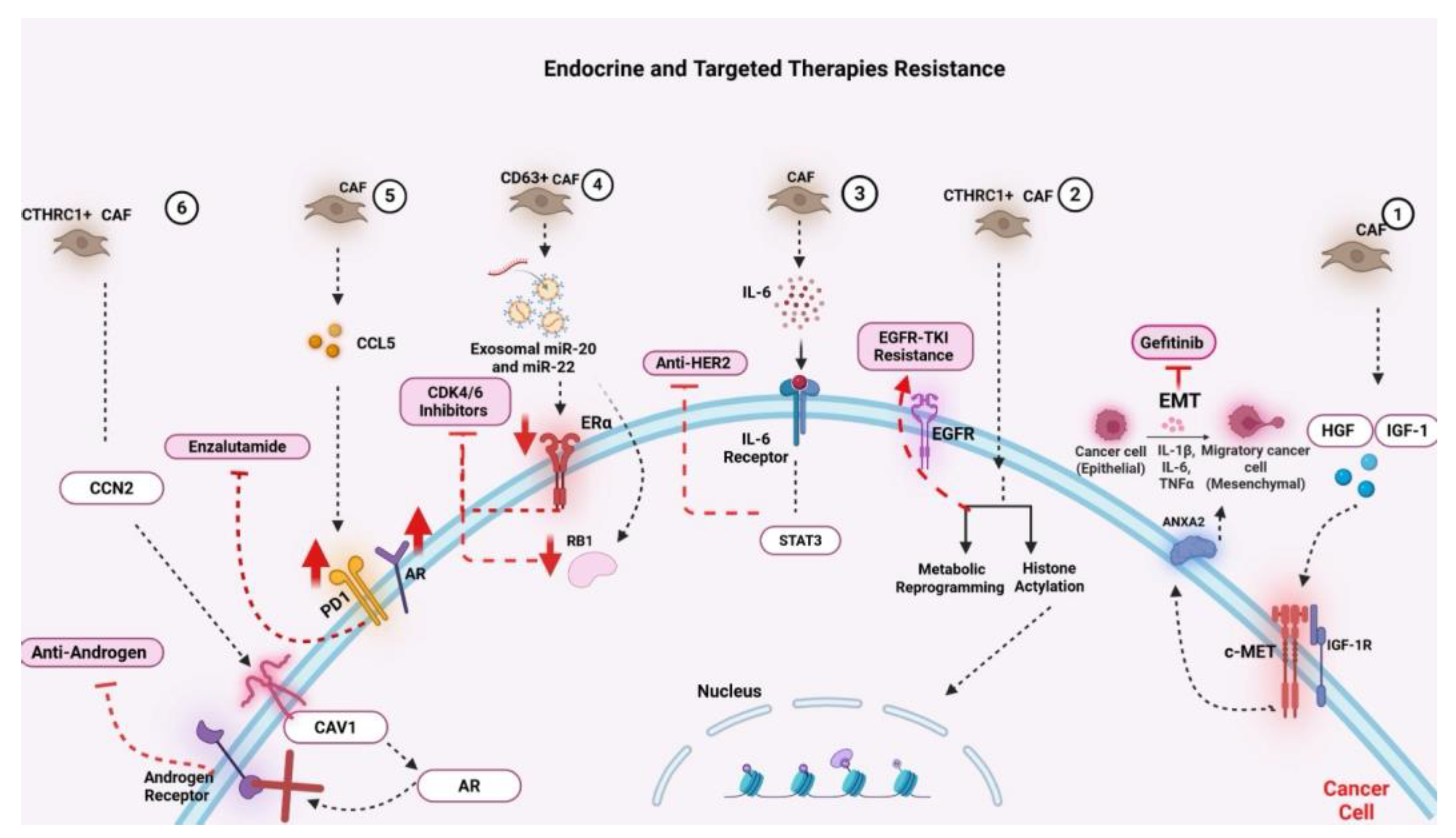
This figure illustrates how cancer-associated fibroblasts (CAFs) contribute to resistance against various endocrine and targeted therapies through diverse molecular and cellular mechanisms within the tumor microenvironment. On the far right, CAF-secreted growth factors, such as hepatocyte growth factor (HGF) and insulin-like growth factor-1 (IGF-1), activate the c-MET and IGF-1R signaling pathways in tumor cells, promoting proliferation, survival, and therapeutic evasion. CAF-induced epithelial-to-mesenchymal transition (EMT), driven by pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α) and facilitated by ANXA2, underlies resistance to EGFR inhibitors, such as gefitinib. Progressing leftward, CAFs also induce resistance to EGFR tyrosine kinase inhibitors (TKIs) by upregulating EGFR expression, promoting metabolic reprogramming, and enhancing histone acetylation in cancer cells. In parallel, IL-6 secreted by CAFs activates the IL-6 receptor/STAT3 signaling axis, which confers resistance to anti-HER2 therapies. Further resistance to CDK4/6 inhibitors is mediated by CD63+ CAFs through the delivery of exosomal miR-20 and miR-22, leading to estrogen receptor alpha (ERα) downregulation and retinoblastoma (RB1) inactivation, undermining cell-cycle control. Another subset of CAFs enhances endocrine resistance by secreting CCL5, which activates downstream signals that reinforce resistance to anti-estrogen therapy. In the context of prostate cancer, CAF-driven expression of caveolin-1 (CAV1) and PD-L1 enhances androgen receptor (AR) signaling and immune evasion, ultimately contributing to resistance against anti-androgens such as enzalutamide. Additionally, CTHRC1+ CAFs upregulate CCN2 expression, further driving resistance through activation of the AR pathway.
9. Challenges and Future Directions
Despite the growing interest in CAF-targeted therapies, clinical translation remains limited due to several unresolved challenges. A significant obstacle is the absence of specific, universal CAF markers that distinguish pro-tumorigenic from tumor-restraining subsets, which increases the risk of off-target effects and disrupts regular tissue repair [87]. Additionally, the inherent plasticity of CAFs allows for dynamic phenotype switching under therapeutic pressure, raising the possibility that targeting one subtype may lead to the compensatory activation of others, thereby preserving tumor-supportive functions [9,97]. These challenges are particularly evident in mesenchymal tumors, such as sarcomas, where the distinction between malignant tumor cells and CAFs is often unclear due to overlapping lineage markers [171]. This complicates efforts to design stromal-specific interventions and highlights the need for improved lineage-tracing and spatial profiling techniques in these tumors. Future research should focus on developing subtype-specific and context-dependent cancer-associated fibroblast (CAF) therapies, guided by single-cell RNA sequencing, spatial transcriptomics, and epigenetic profiling. These technologies may facilitate the identification of actionable CAF subpopulations that drive resistance to treatment or immune exclusion. Longitudinal studies exploring the temporal evolution of CAF phenotypes during chemotherapy, radiotherapy, and immunotherapy will be essential for understanding how CAFs adapt throughout treatment.
Simultaneously, organotypic 3D culture models and patient-derived tumor-stroma co-cultures can act as physiologically relevant systems to assess the effectiveness of CAF-modulating agents. These models may aid in unraveling the reciprocal interactions between cancer-associated fibroblasts (CAFs), cancer stem cells, immune populations, and endothelial cells, which are critical relationships that impact tumor relapse and metastasis. Additionally, incorporating CAF profiling into the design of clinical trials could improve patient stratification, especially in stromally dense or immune-excluded tumors like pancreatic cancer, soft tissue sarcomas, and desmoplastic gastric cancer. Stromal gene signatures may function not only as prognostic markers but also as predictive tools for combinatorial therapies.
10. Conclusions
Cancer-associated fibroblasts (CAFs) are dynamic and multifaceted components of the tumor microenvironment. Their ability to support tumor growth, suppress anti-tumor immunity, and mediate therapy resistance positions them as key therapeutic targets, while also making them complex, context-dependent regulators. Notably, CAFs can also inhibit tumor progression under certain conditions, underscoring the need for precise, subtype-specific targeting strategies rather than indiscriminate depletion.
This review highlights the diversity, adaptability, and dual roles of CAFs in both epithelial and mesenchymal tumors. While the biology of CAFs in carcinomas has been widely studied, our understanding of CAF-like populations in sarcomas remains limited, further complicated by the overlapping lineage between malignant and stromal cells. Bridging this knowledge gap is crucial for advancing therapeutic strategies in these aggressive and often treatment-resistant cancers.
Future efforts should focus on refining CAF classification, identifying functional subtypes, and integrating stromal profiling into clinical decision-making. Combining CAF-targeted interventions with immunotherapy, chemotherapy, or anti-angiogenic agents may yield synergistic benefits, particularly in stromally dense and immunologically "cold" tumors.
Ultimately, unlocking the therapeutic potential of CAF modulation will require context-aware, personalized approaches informed by a comprehensive understanding of the tumor microenvironment's spatial and temporal dynamics.
Author Contributions
Conceptualization, O.B. and I.C.; investigation, O.B. and I.C.; writing—original draft preparation, O.B.; writing—review and editing, O.B., I.C., and G.B.-S. Supervision, G.B.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. Research in our Lab is currently funded by the Israel Cancer Association grant number (20240075) and the Roche disease Project in Breast Cancer.
Institutional Review Board Statement
Not applicable. This study is a narrative literature review and did not involve human or animal subjects.
Informed Consent Statement
Not applicable. This review did not involve human participants or identifiable patient data.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing does not apply to this article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare that they have no conflicts of interest. The funders had no role in the design of the study, in the collection, analysis, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
CAF – Cancer-Associated Fibroblast
myCAF – Myofibroblastic Cancer-Associated Fibroblast
iCAF – Inflammatory Cancer-Associated Fibroblast
apCAF – Antigen-Presenting Cancer-Associated Fibroblast
meCAF – Metabolic Cancer-Associated Fibroblast
vCAF – Vascular Cancer-Associated Fibroblast
dCAF – Developmental Cancer-Associated Fibroblast
rCAF – Tumor-Restraining Cancer-Associated Fibroblast
pCAF – Tumor-Promoting Cancer-Associated Fibroblast
CSCs – Cancer Stem Cells
ECM – Extracellular Matrix
EMT – Epithelial-to-Mesenchymal Transition
EndMT – Endothelial-to-Mesenchymal Transition
TGF-β – Transforming Growth Factor Beta
IL-6 – Interleukin 6
IL-10 – Interleukin 10
CXCL12 – C-X-C Motif Chemokine Ligand 12
CXCL1 – C-X-C Motif Chemokine Ligand 1
CXCL16 – C-X-C Motif Chemokine Ligand 16
JAK/STAT – Janus Kinase/Signal Transducer and Activator of Transcription
NF-κB – Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells
PDGF – Platelet-Derived Growth Factor
VEGF – Vascular Endothelial Growth Factor
WNT – Wingless/Integrated Signaling Pathway
HGF – Hepatocyte Growth Factor
IGF-1 – Insulin-Like Growth Factor 1
PD-L1/PD-L2 – Programmed Death Ligand 1/2
TLR4 – Toll-Like Receptor 4
ATM – Ataxia Telangiectasia Mutated (signaling kinase)
IDO – Indoleamine 2,3-Dioxygenase
ARG2 – Arginase 2
α-SMA – Alpha Smooth Muscle Actin
FAP – Fibroblast Activation Protein
CD4+ T cells – Cluster of Differentiation 4 Positive T Lymphocytes
CD8+ T cells – Cluster of Differentiation 8 Positive T Lymphocytes
Tregs – Regulatory T Cells
TAMs – Tumor-Associated Macrophages
MDSCs – Myeloid-Derived Suppressor Cells
MHC – Major Histocompatibility Complex
TLS – Tertiary Lymphoid Structures
DCN – Decorin
CTHRC1 – Collagen Triple Helix Repeat Containing 1
CD63 – Cluster of Differentiation 63
CD10 – Cluster of Differentiation 10
PDGFR – Platelet-Derived Growth Factor Receptor
SDF-1 – Stromal-Derived Factor 1
CXCR4 – C-X-C Chemokine Receptor Type 4
scRNA-seq – Single-Cell RNA Sequencing
TME – Tumor Microenvironment
TIME – Tumor Immune Microenvironment
3D – Three-Dimensional
EVs – Extracellular Vesicles
PDAC – Pancreatic Ductal Adenocarcinoma
NSCLC – Non-Small Cell Lung Cancer
HCC – Hepatocellular Carcinoma
RMS – Rhabdomyosarcoma
UPS – Undifferentiated Pleomorphic Sarcoma
STS – Soft Tissue Sarcoma
EwS – Ewing Sarcoma
HNSCC – Head and Neck Squamous Cell Carcinoma
NETs – Neuroendocrine Tumors
MHC-II – Major Histocompatibility Complex Class II
ZEB1 – Zinc Finger E-Box Binding Homeobox 1
TBX2 – T-Box Transcription Factor 2
SOX9 – SRY-Box Transcription Factor 9
COL6A1 – Collagen Type VI Alpha 1 Chain
HJURP – Holliday Junction Recognition Protein
MCM2 – Minichromosome Maintenance Complex Component 2
ANXA2 – Annexin A2
AR – Androgen Receptor
CXCL14 – C-X-C Motif Chemokine Ligand 14
ABCB1 – ATP-Binding Cassette Sub-Family B Member 1
ABCC2 – ATP-Binding Cassette Sub-Family C Member 2
MRP1 – Multidrug Resistance-associated Protein 1
miRNA/miR – MicroRNA
e.g., miR-105, miR-21, miR-146a, miR-1228, miR-4717-5p
lncRNA – Long Non-Coding RNA
e.g., ANRIL, LINC00355
ICB – Immune Checkpoint Blockade
CAR-T – Chimeric Antigen Receptor T-Cell Therapy
TKIs – Tyrosine Kinase Inhibitors
ADC – Antibody–Drug Conjugate
MMAE – Monomethyl Auristatin E
OXPHOS – Oxidative Phosphorylation
TCA Cycle – Tricarboxylic Acid Cycle (Krebs Cycle)
References
- Plikus M, V. , Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar]
- Diller, R.B.; Tabor, A.J. The Role of the Extracellular Matrix (ECM) in Wound Healing: A Review. Biomimetics 2022, 7, 87. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Y.; Li, D.; Wei, J.; Chen, K.; Zhang, E.; Liu, G.; Chu, X.; Liu, X.; Liu, W.; et al. Cancer associated fibroblasts and metabolic reprogramming: unraveling the intricate crosstalk in tumor evolution. J. Hematol. Oncol. 2024, 17, 1–32. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef]
- Kim, I.; Choi, S.; Yoo, S.; Lee, M.; Kim, I.-S. Cancer-Associated Fibroblasts in the Hypoxic Tumor Microenvironment. Cancers 2022, 14, 3321. [Google Scholar] [CrossRef]
- Lan, X.; Li, W.; Zhao, K.; Wang, J.; Li, S.; Zhao, H. Revisiting the role of cancer-associated fibroblasts in tumor microenvironment. Front. Immunol. 2025, 16, 1582532. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, Y.; Yang, J.; Li, Z.; Liu, F.; Wang, A.; Gao, Z.; Wu, C.; Yin, H. Overcoming cancer treatment resistance: Unraveling the role of cancer-associated fibroblasts. J. Natl. Cancer Cent. 2025, 5, 237–251. [Google Scholar] [CrossRef]
- Chhabra, Y.; Weeraratna, A.T. Fibroblasts in cancer: Unity in heterogeneity. Cell 2023, 186, 1580–1609. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1–TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef]
- Raffaghello, L.; Vacca, A.; Pistoia, V.; Ribatti, D. Cancer associated fibroblasts in hematological malignancies. Oncotarget 2014, 6, 2589–2603. [Google Scholar] [CrossRef]
- Fotsitzoudis C, Koulouridi A, Messaritakis I, Konstantinidis T, Gouvas N, Tsiaoussis J, et al. Cancer-Associated Fibroblasts: The Origin, Biological Characteristics and Role in Cancer-A—A Glance on Colorectal Cancer. Cancers (Basel). 2022, 14, 4394.
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Liu, Y.; Sinjab, A.; Min, J.; Han, G.; Paradiso, F.; Zhang, Y.; Wang, R.; Pei, G.; Dai, Y.; Liu, Y.; et al. Conserved spatial subtypes and cellular neighborhoods of cancer-associated fibroblasts revealed by single-cell spatial multi-omics. Cancer Cell 2025, 43, 905–924.e6. [Google Scholar] [CrossRef]
- Huang, J.; Tsang, W.-Y.; Li, Z.-H.; Guan, X.-Y. The Origin, Differentiation, and Functions of Cancer-Associated Fibroblasts in Gastrointestinal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 503–511. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Peiffer, R.; Boumahd, Y.; Gullo, C.; Crake, R.; Letellier, E.; Bellahcène, A.; Peulen, O. Cancer-Associated Fibroblast Diversity Shapes Tumor Metabolism in Pancreatic Cancer. Cancers 2022, 15, 61. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb. Perspect. Biol. 2022, 15, a041231. [Google Scholar] [CrossRef]
- Mancini, A.; Gentile, M.T.; Pentimalli, F.; Cortellino, S.; Grieco, M.; Giordano, A. Multiple aspects of matrix stiffness in cancer progression. Front. Oncol. 2024, 14, 1406644. [Google Scholar] [CrossRef]
- Sato, H.; Hara, T.; Meng, S.; Tsuji, Y.; Arao, Y.; Saito, Y.; Sasaki, K.; Kobayashi, S.; Doki, Y.; Eguchi, H.; et al. Multifaced roles of desmoplastic reaction and fibrosis in pancreatic cancer progression: Current understanding and future directions. Cancer Sci. 2023, 114, 3487–3495. [Google Scholar] [CrossRef]
- Piccolo, S.; Panciera, T.; Contessotto, P.; Cordenonsi, M. YAP/TAZ as master regulators in cancer: modulation, function and therapeutic approaches. Nat. Cancer 2022, 4, 9–26. [Google Scholar] [CrossRef]
- Lee, J.J.; Ng, K.Y.; Bakhtiar, A. Extracellular matrix: unlocking new avenues in cancer treatment. Biomark. Res. 2025, 13, 1–25. [Google Scholar] [CrossRef]
- Kwon, J.Y.; Vera, R.E.; Fernandez-Zapico, M.E. The multi-faceted roles of cancer-associated fibroblasts in pancreatic cancer. Cell. Signal. 2025, 127, 111584. [Google Scholar] [CrossRef]
- Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Saúde-Conde, R.; Öztürk, A.A.; Stosic, K.; Senar, O.A.; Navez, J.; Bouchart, C.; Arsenijevic, T.; Flamen, P.; Van Laethem, J.-L. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma or a Metaphor for Heterogeneity: From Single-Cell Analysis to Whole-Body Imaging. Biomedicines 2024, 12, 591. [Google Scholar] [CrossRef]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-β Mediated Immune Evasion in Cancer—Spotlight on Cancer-Associated Fibroblasts. Cancers 2020, 12, 3650. [Google Scholar] [CrossRef]
- Freeman, P.; Mielgo, A. Cancer-Associated Fibroblast Mediated Inhibition of CD8+ Cytotoxic T Cell Accumulation in Tumours: Mechanisms and Therapeutic Opportunities. Cancers 2020, 12, 2687. [Google Scholar] [CrossRef]
- Hou, W. Role of TGFβ-activated cancer-associated fibroblasts in the resistance to checkpoint blockade immunotherapy. Front. Oncol. 2025, 15, 1602452. [Google Scholar] [CrossRef]
- Clifton, G.T.; Rothenberg, M.; Ascierto, P.A.; Begley, G.; Cecchini, M.; Eder, J.P.; Ghiringhelli, F.; Italiano, A.; Kochetkova, M.; Li, R.; et al. Developing a definition of immune exclusion in cancer: results of a modified Delphi workshop. J. Immunother. Cancer 2023, 11, e006773. [Google Scholar] [CrossRef]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Xue, V.W.; Chung, J.Y.-F.; Córdoba, C.A.G.; Cheung, A.H.-K.; Kang, W.; Lam, E.W.-F.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. Transforming Growth Factor-β: A Multifunctional Regulator of Cancer Immunity. Cancers 2020, 12, 3099. [Google Scholar] [CrossRef]
- Xia Z, De Wever O. The plasticity of cancer-associated fibroblasts. Trends Cancer. 2025.
- Cha, Y.J.; Koo, J.S. Role of Tumor-Associated Myeloid Cells in Breast Cancer. Cells 2020, 9, 1785. [Google Scholar] [CrossRef]
- Shakiba, M.; Tuveson, D.A. Macrophages and fibroblasts as regulators of the immune response in pancreatic cancer. Nat. Immunol. 2025. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021, 33, 127–148. [Google Scholar]
- Amer, H.; Flanagan, K.L.; Kampan, N.C.; Itsiopoulos, C.; Scott, C.L.; Kartikasari, A.E.R.; Plebanski, M. Interleukin-6 Is a Crucial Factor in Shaping the Inflammatory Tumor Microenvironment in Ovarian Cancer and Determining Its Hot or Cold Nature with Diagnostic and Prognostic Utilities. Cancers 2025, 17, 1691. [Google Scholar] [CrossRef]
- Bruni, S.; Mercogliano, M.F.; Mauro, F.L.; Russo, R.I.C.; Schillaci, R. Cancer immune exclusion: breaking the barricade for a successful immunotherapy. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef]
- Zhang X, Zhu R, Yu D, Wang J, Yan Y, Xu K. Single-cell RNA sequencing to explore cancer-associated fibroblasts heterogeneity: “Single” vision for “heterogeneous” environment. Cell Prolif. 2024, 57.
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Song, J.; Wei, R.; Liu, C.; Zhao, Z.; Liu, X.; Wang, Y.; Liu, F.; Liu, X. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat. Commun. 2025, 16, 1–16. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Kerdidani, D.; Aerakis, E.; Verrou, K.-M.; Angelidis, I.; Douka, K.; Maniou, M.-A.; Stamoulis, P.; Goudevenou, K.; Prados, A.; Tzaferis, C.; et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef]
- Caligiuri, G.; Tuveson, D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell 2023, 41, 434–449. [Google Scholar] [CrossRef]
- Song, J.; Wei, R.; Liu, C.; Zhao, Z.; Liu, X.; Wang, Y.; Liu, F.; Liu, X. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat. Commun. 2025, 16, 1–16. [Google Scholar] [CrossRef]
- Wei SC, Sharma R, Anang NAAS, Levine JH, Zhao Y, Mancuso JJ, et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity. 2019, 50, 1084–1098.e10. [Google Scholar]
- Hünig, T.; Beyersdorf, N.; Kerkau, T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. ImmunoTargets Ther. 2015, ume 4, 111–122. [Google Scholar] [CrossRef]
- Li, Z.; Sun, C.; Qin, Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11, 8322–8336. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Lee, M.; Yoon, J.-H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef]
- van Hall G, Stømstad M, Rasmussen P, Jans Ø, Zaar M, Gam C, et al. Blood Lactate is an Important Energy Source for the Human Brain. Journal of Cerebral Blood Flow & Metabolism. 2009, 29, 1121–1129.
- Medina, J.M.; Tabernero, A. Lactate utilization by brain cells and its role in CNS development. J. Neurosci. Res. 2004, 79, 2–10. [Google Scholar] [CrossRef]
- Bartelds, B.; Knoester, H.; Beaufort-Krol, G.C.M.; Smid, G.B.; Takens, J.; Zijlstra, W.G.; Heymans, H.S.A.; Kuipers, J.R.G. Myocardial Lactate Metabolism in Fetal and Newborn Lambs. Circulation 1999, 99, 1892–1897. [Google Scholar] [CrossRef]
- Wu, D.; Zhuo, L.; Wang, X. Metabolic reprogramming of carcinoma-associated fibroblasts and its impact on metabolic heterogeneity of tumors. Semin. Cell Dev. Biol. 2017, 64, 125–131. [Google Scholar] [CrossRef]
- Kay, E.J.; Zanivan, S. The tumor microenvironment is an ecosystem sustained by metabolic interactions. Cell Rep. 2025, 44, 115432. [Google Scholar] [CrossRef]
- Antonio MJ, Zhang C, Le A. Different Tumor Microenvironments Lead to Different Metabolic Phenotypes. In 2021. p. 137–47.
- Ruksha, T.; Palkina, N. Role of exosomes in transforming growth factor-β-mediated cancer cell plasticity and drug resistance. Explor. Target. Anti-tumor Ther. 2025, 6, 1002322. [Google Scholar] [CrossRef]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef]
- Yan, X.; Xie, Y.; Yang, F.; Hua, Y.; Zeng, T.; Sun, C.; Yang, M.; Huang, X.; Wu, H.; Fu, Z.; et al. Comprehensive description of the current breast cancer microenvironment advancements via single-cell analysis. J. Exp. Clin. Cancer Res. 2021, 40, 1–15. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Lorenc, P.; Sikorska, A.; Molenda, S.; Guzniczak, N.; Dams-Kozlowska, H.; Florczak, A. Physiological and tumor-associated angiogenesis: Key factors and therapy targeting VEGF/VEGFR pathway. Biomed. Pharmacother. 2024, 180, 117585. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef]
- Zhang, H.; Yue, X.; Chen, Z.; Liu, C.; Wu, W.; Zhang, N.; Liu, Z.; Yang, L.; Jiang, Q.; Cheng, Q.; et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol. Cancer 2023, 22, 1–50. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, Y.; Yang, J.; Li, Z.; Liu, F.; Wang, A.; Gao, Z.; Wu, C.; Yin, H. Overcoming cancer treatment resistance: Unraveling the role of cancer-associated fibroblasts. J. Natl. Cancer Cent. 2025, 5, 237–251. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer 2021, 20, 1–30. [Google Scholar] [CrossRef]
- Wanandi, S.I.; Ningsih, S.S.; Asikin, H.; Hosea, R.; Neolaka, G.M.G. Metabolic Interplay between Tumour Cells and Cancer-Associated Fibroblasts (CAFs) under Hypoxia versus Normoxia. Malays. J. Med Sci. 2018, 25, 7–16. [Google Scholar] [CrossRef]
- Feng, B.; Wu, J.; Shen, B.; Jiang, F.; Feng, J. Cancer-associated fibroblasts and resistance to anticancer therapies: status, mechanisms, and countermeasures. Cancer Cell Int. 2022, 22, 1–15. [Google Scholar] [CrossRef]
- Wei L, Ye H, Li G, Lu Y, Zhou Q, Zheng S, et al. Correction: Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2021, 12, 232.
- Barbazan, J.; Pérez-González, C.; Gómez-González, M.; Dedenon, M.; Richon, S.; Latorre, E.; Serra, M.; Mariani, P.; Descroix, S.; Sens, P.; et al. Cancer-associated fibroblasts actively compress cancer cells and modulate mechanotransduction. Nat. Commun. 2023, 14, 1–17. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Sun, L.; Yuan, Y.; Zhu, Y. Potential mechanisms of cancer-associated fibroblasts in therapeutic resistance. Biomed. Pharmacother. 2023, 166, 115425. [Google Scholar] [CrossRef]
- Fujii, N.; Yashiro, M.; Hatano, T.; Fujikawa, H.; Motomura, H. CD9-positive Exosomes Derived from Cancer-associated Fibroblasts Might Inhibit the Proliferation of Malignant Melanoma Cells. Anticancer. Res. 2022, 43, 25–33. [Google Scholar] [CrossRef]
- Ishibashi, M.; Neri, S.; Hashimoto, H.; Miyashita, T.; Yoshida, T.; Nakamura, Y.; Udagawa, H.; Kirita, K.; Matsumoto, S.; Umemura, S.; et al. CD200-positive cancer associated fibroblasts augment the sensitivity of Epidermal Growth Factor Receptor mutation-positive lung adenocarcinomas to EGFR Tyrosine kinase inhibitors. Sci. Rep. 2017, 7, 46662. [Google Scholar] [CrossRef]
- Papavassiliou KA, Sofianidi AA, Cholidou K, Papavassiliou AG. The IGF Signalling Axis in Lung Cancer: Clinical Significance and Therapeutic Challenges. J Cell Mol Med. 2025, 29.
- Hanley, C.J.; Thomas, G.J. Targeting cancer associated fibroblasts to enhance immunotherapy: emerging strategies and future perspectives. Oncotarget 2021, 12, 1427–1433. [Google Scholar] [CrossRef]
- Song, J.; Wei, R.; Liu, C.; Zhao, Z.; Liu, X.; Wang, Y.; Liu, F.; Liu, X. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat. Commun. 2025, 16, 1–16. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Razi, S.; Haghparast, A.; Khameneh, S.C.; Sadrabadi, A.E.; Aziziyan, F.; Bakhtiyari, M.; Nabi-Afjadi, M.; Tarhriz, V.; Jalili, A.; Zalpoor, H. The role of tumor microenvironment on cancer stem cell fate in solid tumors. Cell Commun. Signal. 2023, 21, 1–23. [Google Scholar] [CrossRef]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: a versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev. 2024, 43, 1095–1116. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef]
- Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017, 8, 20741–20750.
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X.-J. Transforming Growth Factor-β Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Huang, T.-X.; Guan, X.-Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. 2019, 9, 1889–1904.
- Yang, J.; Teng, Y. Harnessing cancer stem cell-derived exosomes to improve cancer therapy. J. Exp. Clin. Cancer Res. 2023, 42, 1–15. [Google Scholar] [CrossRef]
- Li, Q.; He, G.; Yu, Y.; Li, X.; Peng, X.; Yang, L. Exosome crosstalk between cancer stem cells and tumor microenvironment: cancer progression and therapeutic strategies. Stem Cell Res. Ther. 2024, 15, 1–19. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Najafgholian, S.; Salehi, R.; Goli, M.; Ranjbar, M.; Nickho, H.; Javanmard, S.H.; Ferns, G.A.; Manian, M. The role of cancer-associated fibroblasts and exosomal miRNAs-mediated intercellular communication in the tumor microenvironment and the biology of carcinogenesis: a systematic review. Cell Death Discov. 2024, 10, 1–20. [Google Scholar] [CrossRef]
- Liu, G.; Liu, J.; Li, S.; Zhang, Y.; Ren, H. Exosome-Mediated Chemoresistance in Cancers: Mechanisms, Therapeutic Implications, and Future Directions. Biomolecules 2025, 15, 685. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Jia, H.; Chen, X.; Zhang, L.; Chen, M. Cancer associated fibroblasts in cancer development and therapy. J. Hematol. Oncol. 2025, 18, 1–31. [Google Scholar] [CrossRef]
- Popovich JR, KSGD; et al. Sarcoma. Sarcoma. 2023 Aug 14.
- Muse ME SKCJ. Physiology, Epithelialization. 2023 Sep 4.
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Muchlińska, A.; Nagel, A.; Popęda, M.; Szade, J.; Niemira, M.; Zieliński, J.; Skokowski, J.; Bednarz-Knoll, N.; Żaczek, A.J. Alpha-smooth muscle actin-positive cancer-associated fibroblasts secreting osteopontin promote growth of luminal breast cancer. Cell. Mol. Biol. Lett. 2022, 27, 1–14. [Google Scholar] [CrossRef]
- Mori, Y.; Novruzov, E.; Schmitt, D.; Cardinale, J.; Watabe, T.; Choyke, P.L.; Alavi, A.; Haberkorn, U.; Giesel, F.L. Clinical applications of fibroblast activation protein inhibitor positron emission tomography (FAPI-PET). npj Imaging 2024, 2, 1–21. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Mishima, Y.; Hayashibe, K.; Sasase, A. α-Smooth Muscle Actin Expression in Tumor and Stromal Cells of Benign and Malignant Human Pigment Cell Tumors. J. Investig. Dermatol. 1992, 98, 116–120. [Google Scholar] [CrossRef]
- Wrenn, E.D.; Apfelbaum, A.A.; Rudzinski, E.R.; Deng, X.; Jiang, W.; Sud, S.; Van Noord, R.A.; Newman, E.A.; Garcia, N.M.; Miyaki, A.; et al. Cancer-Associated Fibroblast-Like Tumor Cells Remodel the Ewing Sarcoma Tumor Microenvironment. Clin. Cancer Res. 2023, 29, 5140–5154. [Google Scholar] [CrossRef]
- Huang, X.; Wang, L.; Guo, H.; Zhang, W.; Shao, Z. Single-cell transcriptomics reveals the regulative roles of cancer associated fibroblasts in tumor immune microenvironment of recurrent osteosarcoma. Theranostics 2022, 12, 5877–5887. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Yang, X.; Lu, W.; Chen, Y.; Lin, Y.; Wang, J.; Lin, S.; Yun, J.-P. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts: Erratum. Theranostics 2022, 12, 4604–4605. [Google Scholar] [CrossRef]
- Broz, M.T.; Ko, E.Y.; Ishaya, K.; Xiao, J.; De Simone, M.; Hoi, X.P.; Piras, R.; Gala, B.; Tessaro, F.H.G.; Karlstaedt, A.; et al. Metabolic targeting of cancer associated fibroblasts overcomes T-cell exclusion and chemoresistance in soft-tissue sarcomas. Nat. Commun. 2024, 15, 1–18. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Wei, R.; Zhang, J.; Zhu, J.; Wang, W.; Wang, Z.; Wupur, Z.; Li, Y.; Meng, H. Synergistic Chemoimmunotherapy Augmentation via Sequential Nanocomposite Hydrogel-Mediated Reprogramming of Cancer-Associated Fibroblasts in Osteosarcoma. Adv. Mater. 2023, 36, e2309591. [Google Scholar] [CrossRef]
- Umakoshi, M.; Kudo-Asabe, Y.; Tsuchie, H.; Li, Z.; Koyama, K.; Miyabe, K.; Yoshida, M.; Nagasawa, H.; Nanjo, H.; Okada, K.; et al. Prognostic value of CAF marker expression in the intratumoral and marginal areas of soft tissue sarcoma. Pathobiology 2024, 92, 1–1. [Google Scholar] [CrossRef]
- D’aGostino, S.; Tombolan, L.; Saggioro, M.; Frasson, C.; Rampazzo, E.; Pellegrini, S.; Favaretto, F.; Biz, C.; Ruggieri, P.; Gamba, P.; et al. Rhabdomyosarcoma Cells Produce Their Own Extracellular Matrix With Minimal Involvement of Cancer-Associated Fibroblasts: A Preliminary Study. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Perret, R.; Huang, Y.; Courgeon, F.; Gokhale, P.C.; Laroche-Clary, A.; Eschle, B.K.; Velasco, V.; Le Loarer, F.; Algeo, M.-P.; et al. LRRC15 Targeting in Soft-Tissue Sarcomas: Biological and Clinical Implications. Cancers 2020, 12, 757. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Sunami, Y.; Böker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697. [Google Scholar] [CrossRef]
- Lavudi, K.; Nuguri, S.M.; Olverson, Z.; Dhanabalan, A.K.; Patnaik, S.; Kokkanti, R.R. Targeting the retinoic acid signaling pathway as a modern precision therapy against cancers. Front. Cell Dev. Biol. 2023, 11, 1254612. [Google Scholar] [CrossRef]
- Tilsed, C.M.; Casey, T.H.; de Jong, E.; Bosco, A.; Zemek, R.M.; Salmons, J.; Wan, G.; Millward, M.J.; Nowak, A.K.; Lake, R.A.; et al. Retinoic Acid Induces an IFN-Driven Inflammatory Tumour Microenvironment, Sensitizing to Immune Checkpoint Therapy. Front. Oncol. 2022, 12, 849793. [Google Scholar] [CrossRef]
- Tobin, R.P.; Cogswell, D.T.; Cates, V.M.; Davis, D.M.; Borgers, J.S.; Van Gulick, R.J.; Katsnelson, E.; Couts, K.L.; Jordan, K.R.; Gao, D.; et al. Targeting MDSC Differentiation Using ATRA: A Phase I/II Clinical Trial Combining Pembrolizumab and All-Trans Retinoic Acid for Metastatic Melanoma. Clin. Cancer Res. 2022, 29, 1209–1219. [Google Scholar] [CrossRef]
- Łabędź, N.; Anisiewicz, A.; Stachowicz-Suhs, M.; Banach, J.; Kłopotowska, D.; Maciejczyk, A.; Gazińska, P.; Piotrowska, A.; Dzięgiel, P.; Matkowski, R.; et al. Dual effect of vitamin D3 on breast cancer-associated fibroblasts. BMC Cancer 2024, 24, 1–25. [Google Scholar] [CrossRef]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Moro, C.F.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 1–11. [Google Scholar] [CrossRef]
- Kim, S.-I.; Chaurasiya, S.; Sivanandam, V.; Kang, S.; Park, A.K.; Lu, J.; Yang, A.; Zhang, Z.; Bagdasarian, I.A.; Woo, Y.; et al. Priming stroma with a vitamin D analog to optimize viroimmunotherapy for pancreatic cancer. Mol. Ther. - Oncolytics 2022, 24, 864–872. [Google Scholar] [CrossRef]
- Bai, C.; Li, S.; Tan, Z.; Fan, Z. Targeting MCM2 activates cancer-associated fibroblasts-like phenotype and affects chemo-resistance of liposarcoma cells against doxorubicin. Anti-Cancer Drugs 2024, 35, 883–892. [Google Scholar] [CrossRef]
- Wang, J.-W.; Wu, X.-F.; Gu, X.-J.; Jiang, X.-H. Exosomal miR-1228 From Cancer-Associated Fibroblasts Promotes Cell Migration and Invasion of Osteosarcoma by Directly Targeting SCAI. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2019, 27, 979–986. [Google Scholar] [CrossRef]
- Moutafi, M.; Martinez-Morilla, S.; Divakar, P.; Vathiotis, I.; Gavrielatou, N.; Aung, T.N.; Yaghoobi, V.; Fernandez, A.I.; Zugazagoitia, J.; Herbst, R.S.; et al. Discovery of Biomarkers of Resistance to Immune Checkpoint Blockade in NSCLC Using High-Plex Digital Spatial Profiling. J. Thorac. Oncol. 2022, 17, 991–1001. [Google Scholar] [CrossRef]
- Grout, J.A.; Sirven, P.; Leader, A.M.; Maskey, S.; Hector, E.; Puisieux, I.; Steffan, F.; Cheng, E.; Tung, N.; Maurin, M.; et al. Spatial Positioning and Matrix Programs of Cancer-Associated Fibroblasts Promote T-cell Exclusion in Human Lung Tumors. Cancer Discov. 2022, 12, 2606–2625. [Google Scholar] [CrossRef]
- Minini, M.; Fouassier, L. Cancer-Associated Fibroblasts and Extracellular Matrix: Therapeutical Strategies for Modulating the Cholangiocarcinoma Microenvironment. Curr. Oncol. 2023, 30, 4185–4196. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Ma, W.; Wu, J.; Wu, Q.; Sun, C. Paracrine signaling in cancer-associated fibroblasts: central regulators of the tumor immune microenvironment. J. Transl. Med. 2025, 23, 1–22. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol. Cancer 2021, 20, 1–30. [Google Scholar] [CrossRef]
- Zhang C, Fei Y, Wang H, Hu S, Liu C, Hu R, et al. Front Pharmacol. 2023, 14.
- Koppensteiner, L.; Mathieson, L.; O’cOnnor, R.A.; Akram, A.R. Cancer Associated Fibroblasts - An Impediment to Effective Anti-Cancer T Cell Immunity. Front. Immunol. 2022, 13, 887380. [Google Scholar] [CrossRef]
- Liao, T.; Chen, X.; Qiu, F.; Zhang, X.; Wu, F.; Zhao, Z.; Xu, M.; Chen, M.; Shen, J.-W.; Shen, Q.; et al. Regulation of cancer-associated fibroblasts for enhanced cancer immunotherapy using advanced functional nanomedicines: an updated review. J. Nanobiotechnology 2025, 23, 1–34. [Google Scholar] [CrossRef]
- Peng, Z.; Tong, Z.; Ren, Z.; Ye, M.; Hu, K. Cancer-associated fibroblasts and its derived exosomes: a new perspective for reshaping the tumor microenvironment. Mol. Med. 2023, 29, 1–24. [Google Scholar] [CrossRef]
- Shoucair, I.; Mello, F.W.; Jabalee, J.; Maleki, S.; Garnis, C. The Role of Cancer-Associated Fibroblasts and Extracellular Vesicles in Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6837. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Song Z, Ren D, Xu X, Wang Y. Molecular cross-talk of IL-6 in tumors and new progress in combined therapy. Thorac Cancer. 2018, 9, 669–675.
- Zhao, Q.; Huang, L.; Qin, G.; Qiao, Y.; Ren, F.; Shen, C.; Wang, S.; Liu, S.; Lian, J.; Wang, D.; et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021, 518, 35–48. [Google Scholar] [CrossRef]
- Johnson, D.E.; O'KEefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Zhu, S.; Mao, J.; Zhang, X.; Wang, P.; Zhou, Y.; Tong, J.; Peng, H.; Yang, B.; Fu, Q. CAF-derived exosomal lncRNA FAL1 promotes chemoresistance to oxaliplatin by regulating autophagy in colorectal cancer. Dig. Liver Dis. 2023, 56, 330–342. [Google Scholar] [CrossRef]
- Luo, G.; Zhang, Y.; Wu, Z.; Zhang, L.; Liang, C.; Chen, X. Exosomal LINC00355 derived from cancer-associated fibroblasts promotes bladder cancer cell resistance to cisplatin by regulating miR-34b-5p/ABCB1 axis. Acta Biochim. et Biophys. Sin. 2021, 53, 558–566. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, L.; Li, Y.; Ren, J.; Shi, G.; Wang, Y.; Zhao, S.; Ni, Y.; Hou, Y. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci. Rep. 2017, 7, 16231–16231. [Google Scholar] [CrossRef]
- Lan, Y.; Xu, H.; Jin, L. HJURP Derived from Cancer-Associated Fibroblasts Promotes Glutamine Metabolism to Induce Resistance to Doxorubicin in Ovarian Cancer. Tohoku J. Exp. Med. 2024, 264, 31–39. [Google Scholar] [CrossRef]
- Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021, 302, 259–272.
- Yang Y, Li J, Lei W, Wang H, Ni Y, Liu Y, et al. CXCL12-CXCR4/CXCR7 Axis in Cancer: from Mechanisms to Clinical Applications. Int J Biol Sci. 2023, 19, 3341–3359.
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Morgan, A.; Griffin, M.; Kameni, L.; Wan, D.C.; Longaker, M.T.; Norton, J.A. Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Biology 2023, 12, 1044. [Google Scholar] [CrossRef]
- Wei, R.; Song, J.; Liu, X.; Huo, S.; Liu, C.; Liu, X. Immunosuppressive MFAP2+ cancer associated fibroblasts conferred unfavorable prognosis and therapeutic resistance in gastric cancer. Cell. Oncol. 2023, 47, 55–68. [Google Scholar] [CrossRef]
- Kim, K.T.; Lee, M.H.; Shin, S.-J.; Cho, I.; Kuk, J.C.; Yun, J.; Choi, Y.Y. Decorin as a key marker of desmoplastic cancer-associated fibroblasts mediating first-line immune checkpoint blockade resistance in metastatic gastric cancer. Gastric Cancer 2024, 28, 12–26. [Google Scholar] [CrossRef]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef]
- Furuhashi, S.; Morita, Y.; Matsumoto, A.; Ida, S.; Muraki, R.; Kitajima, R.; Takeda, M.; Kikuchi, H.; Hiramatsu, Y.; Takeuchi, H. Tenascin C in pancreatic cancer-associated fibroblasts enhances epithelial mesenchymal transition and is associated with resistance to immune checkpoint inhibitor. 2023, 13, 5641–+.
- Sakemura, R.; Hefazi, M.; Siegler, E.L.; Cox, M.J.; Larson, D.P.; Hansen, M.J.; Roman, C.M.; Schick, K.J.; Can, I.; Tapper, E.E.; et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood 2022, 139, 3708–3721. [Google Scholar] [CrossRef]
- Li J, Cao F, Yin H liang, Huang Z jian, Lin Z tao, Mao N, et al. Ferroptosis: past, present, and future. Cell Death Dis. 2020, 11, 88.
- Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022, 7, 196.
- Jiang, F.; Jia, K.; Chen, Y.; Ji, C.; Chong, X.; Li, Z.; Zhao, F.; Bai, Y.; Ge, S.; Gao, J.; et al. ANO1-Mediated Inhibition of Cancer Ferroptosis Confers Immunotherapeutic Resistance through Recruiting Cancer-Associated Fibroblasts. Adv. Sci. 2023, 10, e2300881. [Google Scholar] [CrossRef]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, Y.; Wang, Y.; Zhao, X.; Zi, R.; Hao, J.; Ding, Q.; Jiang, H.; Wang, X.; Lu, F.; et al. Cancer-associated fibroblasts derived fibronectin extra domain A promotes sorafenib resistance in hepatocellular carcinoma cells by activating SHMT1. Genes Dis. 2024, 11, 101330. [Google Scholar] [CrossRef]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, W.; Xu, H.; Xu, J.; Li, J.; Liu, X.; Lu, X.; Dai, J.; Jiang, Y.; Wang, W.; et al. Cancer-associated fibroblasts promote EGFR-TKI resistance via the CTHRC1/glycolysis/H3K18la positive feedback loop. Oncogene 2025, 44, 1400–1414. [Google Scholar] [CrossRef]
- Ibrahim FMk, Helal DS, Ali DA, Abd-Ellatif RN, Elkady AM, Sharshar R, et al. Prognostic role of annexin A2 and cancer-associated fibroblasts in advanced non-small cell lung cancer: Implication in epithelial-mesenchymal transition and gefitinib resistance. Pathol Res Pract. 2023, 241, 154293.
- Xu W, Yang H, Xu K, Zhu A, Hall SRR, Jia Y, et al. Transitional CXCL14 + cancer-associated fibroblasts enhance tumour metastasis and confer resistance to EGFR-TKIs, revealing therapeutic vulnerability to filgotinib in lung adenocarcinoma. Clin Transl Med. 2025, 15.
- Sun J, Du R, Li X, Liu C, Wang D, He X, et al. CD63+ cancer-associated fibroblasts confer CDK4/6 inhibitor resistance to breast cancer cells by exosomal miR-20. Cancer Lett. 2024, 588, 216747.
- Gao Y, Li X, Zeng C, Liu C, Hao Q, Li W, et al. CD63 + Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Advanced Science. 2020, 7.
- Mao, Y.; Zhang, Y.; Qu, Q.; Zhao, M.; Lou, Y.; Liu, J.; Huang, O.; Chen, X.; Wu, J.; Shen, K. Cancer-associated fibroblasts induce trastuzumab resistance in HER2 positive breast cancer cells. Mol. Biosyst. 2015, 11, 1029–1040. [Google Scholar] [CrossRef]
- Busch, S.; Rydén, L.; Stål, O.; Jirström, K.; Landberg, G.; Aboussekhra, A. Low ERK Phosphorylation in Cancer-Associated Fibroblasts Is Associated with Tamoxifen Resistance in Pre-Menopausal Breast Cancer. PLOS ONE 2012, 7, e45669. [Google Scholar] [CrossRef]
- Xiong, Z.; Yu, S.-L.; Xie, Z.-X.; Zhuang, R.-L.; Peng, S.-R.; Wang, Q.; Gao, Z.; Li, B.-H.; Xie, J.-J.; Huang, H.; et al. Cancer-associated fibroblasts promote enzalutamide resistance and PD-L1 expression in prostate cancer through CCL5-CCR5 paracrine axis. iScience 2024, 27, 109674. [Google Scholar] [CrossRef]
- Liang H qi, He Q huan, Wei Q ju, Mo Q zhou, Yang G lin, Wei F ye, et al. CTHRC1 expresses in cancer-associated fibroblasts and is associated with resistance to anti-androgen therapy in prostate cancer. Genes Genomics. 2025, 47, 541–557.
- Sulaiman, R.; De, P.; Aske, J.C.; Lin, X.; Dale, A.; Koirala, N.; Gaster, K.; Espaillat, L.R.; Starks, D.; Dey, N. Patient-Derived Primary Cancer-Associated Fibroblasts Mediate Resistance to Anti-Angiogenic Drug in Ovarian Cancers. Biomedicines 2023, 11, 112. [Google Scholar] [CrossRef]
- Cooper, J.B.; Cohen, E.E.W. Mechanisms of resistance to EGFR inhibitors in head and neck cancer. Head Neck 2009, 31, 1086–1094. [Google Scholar] [CrossRef]
- Amin, T.; Viol, F.; Krause, J.; Fahl, M.; Eggers, C.; Awwad, F.; Schmidt, B.; Benten, D.; Ungefroren, H.; Fraune, C.; et al. Cancer-Associated Fibroblasts Induce Proliferation and Therapeutic Resistance to Everolimus in Neuroendocrine Tumors through STAT3 Activation. Neuroendocrinology 2022, 113, 501–518. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



