
Submitted:
28 August 2025
Posted:
29 August 2025
You are already at the latest version
Abstract
The urgent need for novel therapeutic strategies in triple-negative breast cancer (TNBC)—characterized by absent ER, PR, and HER2 expression—stems from its association with a paucity of effective treatments and an adverse prognosis. This study identifies TNFRSF12A as a key gene specifically overexpressed in TNBC versus other subtypes. Validation with clinical specimens confirmed its exclusive upregulation in TNBC tissues, correlating significantly with worse patient outcomes. Functional enrichment analysis (STRING/DAVID) indicated TNFRSF12A's primary involvement in pathways positively regulating cell migration, angiogenesis, and hypoxia response. Immune infiltration profiling (TIMER/TISCH2) revealed selective enrichment of TNFRSF12A in cancer-associated fibroblasts (CAFs). Its expression showed a significant positive correlation with the CAF marker FAP (ρ = 0.304) and CAF infiltration levels, but inverse correlations with CD8⁺ T-cell (r=-0.165) and B-cell (r=-0.164) infiltration. Regarding chemoresistance, elevated TNFRSF12A expression significantly reduced sensitivity to docetaxel. Molecular docking simulations further verified direct binding between TNFRSF12A and docetaxel, mediated by hydrophobic interactions and hydrogen bonds. To elucidate the underlying molecular mechanisms, cellular experiments revealed that TNFRSF12A knockdown resulted in:1) significantly compromised angiogenic capacity in HUVEC tube formation assays (P< 0.01); 2) markedly augmented cytotoxicity of T cells against tumor cells (P< 0.05); and 3) reduced cellular sensitivity to docetaxel, as evidenced by significantly elevated IC₅₀ values in CCK-8 assays (P< 0.01). In summary, this study systematically elucidates how TNFRSF12A propels TNBC malignant progression by remodeling the tumor immune microenvironment and promoting angiogenesis. Concurrently, we reveal a TNFRSF12A-mediated chemosensitizing effect towards docetaxel. Therefore, these results are crucial for improving the targeting of TNFRSF12A and developing precise combination treatment regimens to improve outcomes for patients with TNBC.
Keywords:
tumor immune microenvironment
; triple-negative breast cancer
; TNFRSF12A
; angiogenesis
1. Introduction
Globally, triple-negative breast cancer (TNBC) imposes a substantial health burden and ranks among the primary causes of cancer death in women due to its aggressive clinical behavior, heightened propensity for local recurrence, and elevated rates of distant metastasis [1]. A key defining feature of TNBC, setting it apart from other breast cancer subtypes, is the lack of expression of three fundamental receptors [2,3]. Currently, the lack of robust TNBC-specific diagnostic and prognostic biomarkers contributes to suboptimal responses to conventional therapies, universally poor clinical outcomes, and eventual tumor recurrence or metastasis in the majority of patients [4,5,6]. Consequently, there is a critical need to identify novel therapeutic targets for TNBC to improve patient prognosis.
While the role of the immune system in tumorigenesis and progression has been extensively documented, immunotherapy has only recently emerged as a transformative breakthrough in oncology, demonstrating remarkable antitumor efficacy and the potential for durable remission or cure [7]. Preclinical studies demonstrate compelling evidence of a significant synergistic interaction between chemotherapeutic agents and immune system functionality in TNBC, thus cooperatively potentiating anti-tumor immune responses [8,9]. The expression levels of particular immunological markers also show significant predictive value for chemotherapy benefit magnitude in TNBC [10]. However, the immunomodulatory mechanisms underlying specific therapeutic agents and their clinical translational potential remain incompletely characterized, warranting further investigation [11]. By delineating the molecular interplay of pivotal immune-associated genes in the immunologically suppressed niche of TNBC, this study endeavors to characterize immune-regulatory mechanisms operative therein. thereby providing a theoretical foundation for developing precision-based therapeutic approaches targeting the TME.
Tumor necrosis factor receptor superfamily member 12A (TNFRSF12A; commonly known as Fn14), constitutes a prominent constituent within the tumor necrosis factor receptor superfamily (TNFRSF) [12,13]. As a type I transmembrane glycoprotein, TNFRSF12A structurally harbors characteristic cysteine-rich domains (CRDs). TWEAK/TNFSF12 binding to the Fn14 ectodomain activates complex downstream signaling cascades [13]. The protein encoded by this gene exerts crucial functions across diverse physiological and pathological processes; it holds a central position, particularly in tissue damage repair, inflammatory responses, and the regulation of cellular homeostasis [14,15]. Accumulating research has progressively unveiled the pathogenic mechanisms of TNFRSF12A in diverse diseases, particularly malignancies and fibroinflammatory disorders [16,17,18,19], establishing it as a promising therapeutic target for disease diagnosis and intervention.
Leveraging breast cancer datasets from the GEO database, this study identified TNFRSF12A as a pivotal research target through differential gene expression analysis and Venn intersection screening with immune-related gene sets. Analytical findings revealed: Significantly elevated TNFRSF12A expression in TNBC tissues, positively correlated with detrimental clinical outcomes—shortened overall survival (OS; HR = 1.89, (1.28–2.81), p = 0.0012), reduced recurrence-free survival (RFS; HR = 1.47, (1.17–1.84), p = 0.00077), and worsened distant metastasis-free survival (DMFS; HR = 1.86, (1.35–2.57), p = 0.00011). Gene set enrichment analysis implicated this gene in tumor-associated neovascularization regulation, with experimental validation demonstrating markedly impaired vascular formation capacity following TNFRSF12A knockdown. Further immune infiltration assessment established an inverse correlation between heightened TNFRSF12A expression level and infiltration level of CD8⁺ T cells and B lymphocytes. In vitro cytotoxicity assays revealed that genetic ablation of TNFRSF12A augmented T-cell-mediated tumor cell lysis. Single-cell transcriptomic profiling revealed selective enrichment of TNFRSF12A expression in cancer-associated fibroblasts (CAFs), with its expression levels positively correlating with fibroblast infiltration density in the tumor microenvironment (McCluster algorithm: Cor=0.316, P =2.15 ×10⁻⁵; TIDE algorithm: Cor =0.329, P=9.12 × 10⁻⁶). Pharmacological susceptibility analysis coupled with experimental validation confirmed significantly increased sensitivity to docetaxel in high TNFRSF12A-expressing cohorts (validated by CCK-8 assay, P<0.01). Molecular docking simulations identified potential binding sites between TNFRSF12A protein and docetaxel (binding energy < -6.7 kcal/mol). Collectively, these findings establish TNFRSF12A as a promising therapeutic target for TNBC with clinical potential for docetaxel combination regimens.
2. Materials and Methods
2.1. Computational Breast Cancer Data Analysis
R provides a freely accessible computational environment for statistical analysis, data visualization, and data mining. We employed the limma package within this ecosystem to curate and analyze breast cancer datasets sourced from GEO repositories.
2.2. Patient Tissue Samples
Paired tumor and adjacent normal tissues from nineteen treatment-naïve triple-negative breast cancer (TNBC) patients were procured from Xiangya Hospital, Central South University (Changsha, China). All specimens underwent formalin-fixed paraffin-embedding (FFPE) without prior exposure to chemotherapy, radiotherapy, or immunotherapy. This study protocol received ethics approval from the Institutional Review Board of Xiangya Hospital.
2.3. Isolation of RNA from Tissue Samples
Following dewaxing with xylene, total RNA was extracted from formalin-fixed paraffin-embedded (FFPE) triple-negative breast cancer and normal tissue specimens using the AmoyDx® FFPE RNA Isolation Kit (AmoyDx, Xiamen, China) per manufacturer’s protocols.
2.4. Gene Expression Analysis
UALCAN (https://ualcan.path.uab.edu/) is an interactive web portal enabling multi-omics analytical capabilities leveraging The Cancer Genome Atlas (TCGA) database. Its functionalities encompass transcriptomic differential expression profiling, survival outcome assessment, correlation analysis, protein-level expression comparisons, and non-coding RNA (miRNA/lncRNA) investigations. The platform further facilitates visualization of analytical outputs derived from TCGA oncogenomic datasets. We employed this resource to interrogate TNFRSF12A expression patterns in triple-negative breast carcinoma while concurrently retrieving genes exhibiting expression correlation with TNFRSF12A across breast cancer subtypes.
The HPA constitutes (https://www.proteinatlas.rg) a comprehensive proteomic knowledgebase dedicated to exhaustive characterization of human gene and protein expression patterns. This resource integrates immunohistochemistry (IHC), immunofluorescence, and high-throughput antibody generation technologies, encompassing spatial distribution data for >24,000 human proteins across normal tissues, neoplastic specimens, cell lines, and hematopoietic cells. Leveraging the platform’s IHC repository, we extracted expression profiles of TNFRSF12A in breast carcinoma tissues relative to normal counterparts.
2.5. Prognostic Analysis
Employing the KM Plotter platform (https://kmplot.com/analysis/)—an established online resource for survival analysis integrating data from GEO and TCGA repositories—we examined the prognostic impact of TNFRSF12A in triple-negative breast cancer patients. This comprehensive repository enables biomarker discovery and validation by establishing associations between gene expression and survival outcomes across >30,000 samples encompassing 21 malignancies.
2.6. Exploring the Function of TNFRSR12A
STRING (https://cn.string-db.rg/) constitutes a protein-protein interaction (PPI) network resource integrating evidence from public repositories and curated literature. It aggregates computationally processed data from multiple primary sources—including UniProt, KEGG, NCBI, and Gene Ontology—to generate a unified PPI network framework. We leveraged this platform to delineate putative interactors of TNFRSF12A and performed functional enrichment profiling of these candidate binding partners using its analytical framework.
The DAVID bioinformatics resource (https://david.ncifcrf.gov/) provides an integrated analytical platform that synthesizes biological data and computational tools. This system delivers comprehensive functional annotation for extensive gene/protein inventories (spanning hundreds to thousands of identifiers), enabling extraction of actionable biological insights from omics-scale datasets. Leveraging DAVID’s analytical infrastructure, we conducted functional annotation profiling of genes exhibiting expression correlation with TNFRSF12A.
The BioLadder platform (https://www.bioladder.cn/web/) provides an integrated bioinformatics environment for computational analysis and analytical visualization. Utilizing this resource, we visualized enrichment analysis outputs, specifically prioritizing the top-ranked statistically significant enrichment terms (p < 0.05) for graphical representation.
2.7. Tumor Microenvironment Immune Cell Profiling
The TIMER platform (https://cistrome.shinyapps.io/timer/) serves as a dedicated computational resource for deconvoluting immune cell infiltration landscapes within tumor microenvironments. Leveraging TCGA RNA-Seq datasets, it quantifies abundance profiles of six major immune lineages. We employed this analytical framework to delineate TNFRSF12A-mediated modulation of immune infiltration dynamics in TNBC.
2.8. Single-Cell Data Analysis
The TISCH2 database (http://tisch.comp-genomics.org) curates single-cell transcriptomic data encompassing 6,297,320 cells derived from 190 distinct datasets. Leveraging this resource, we deciphered the transcriptomic landscape across individual cells within breast carcinoma tissues. This analytical approach enabled comprehensive mapping of TNFRSF12A expression profiles among diverse cellular subtypes and subpopulations, thereby elucidating its spatial distribution patterns and functional relevance within the tumor ecosystem.
2.9. Drug Sensitivity Analyses
GSCA (https://guolab.wchscu.cn/GSCA/) represents an integrative computational platform for comprehensive oncogenomic investigations, consolidating functionalities spanning single-gene profiling, multi-gene signature analysis, immune infiltration quantification, mutational landscape assessment, and pharmacological responsiveness evaluation. This repository encompasses data from TCGA and Genomics of Drug Sensitivity in Cancer (GDSC) across 33 malignancy types. Utilizing its drug sensitivity module, we systematically assessed correlations between TNFRSF12A expression levels and chemotherapeutic responsiveness.
2.10. Molecular Docking Simulation of TNFRSF12A and Docetaxe
The PubChem database can be utilized to identify the three-dimensional structure of docetaxel, while the tertiary structure of TNFRSF12A was acquired from PDB. Molecular preprocessing of both ligand (docetaxel) and receptor (TNFRSF12A), including binding site specification, was performed using AutoDockTools (v1.5.7). Subsequent molecular docking simulations employed AutoDock Vina (v1.2.3) utilizing its integrated Iterated Local Search global optimization algorithm to identify optimal binding conformations. Resultant docking complexes underwent structural analysis and visualization primarily through PyMOL molecular graphics software.
2.11. The Culture of Triple-Negative Breast Cancer Cell Lines
MDA-MB-231 and HCC1806 (the American Type Culture Collection, ATCC, USA) were utilized. e cultured MDA-MB-231 cells. We used high-glucose DMEM medium. This medium contained 10% FBS and 1% penicillin-streptomycin. while HCC1806 cells were grown in RPMI 1640 containing identical concentrations of FBS and antibiotic/antimycotic. All cultures were maintained at 37°C in a humidified 5% CO₂ incubator, with routine testing confirming mycoplasma-, bacterial-, and fungal-free status.
2.12. Cell Transfection
All small interfering RNAs (siRNAs) utilized in this investigation were synthesized by RiboBio (Guangzhou, China). We seeded cells in logarithmic growth phase into 6-well plates. Upon reaching 70%–90% confluence, transfection was conducted using Lipofectamine 3000® (Invitrogen), with all procedures adhering strictly to manufacturer protocols. Nucleotide sequences of experimental siRNAs are detailed in Table 1.
2.13. Real-Time Fluorescent Quantitative Polymerase Chain Reaction
The RNA was extracted using the RNA Isolater reagent (Vazyme, China). The extracted RNA was converted into cDNA using reverse transcription reagent (TransGen Biotech, China). Then, the cDNA was analyzed by quantitative polymerase chain reaction (qPCR) according to a standardized protocol. We used the 2^(−ΔΔ) Ct method to calculate the expression levels of the target genes. The primer sequences listed in Table 2 are primers designed for the gene in this study.
2.14. In Vitro T-Cell Cytotoxicity Assay
Post-transfection, tumor cells subsequently cultivated in a 12-well plate. (1 × 10⁶ cells) and incubated for 24 h under standard conditions. Activated peripheral blood mononuclear cells (PBMCs) were then introduced at a 1:5 effector-to-target ratio (tumor cells: PBMCs) for 48-hour co-culture. After 48 hours of coculture, discard the old culture medium. Then, fix the cells at room temperature with 4% paraformaldehyde for 30 minutes. Finally, stain with 0.1% crystal violet to assess tumor cell death.
2.15. Tube Formation Assay
HUVEC cells were cultured into a 48-well plate (1.6 × 10⁴ cells per well) and cultured using conditioned medium collected under different treatment conditions. After an 8-hour incubation period under standard conditions (37°C, 5% CO₂), images of endothelial tubulogenesis were acquired using light microscopy. The resulting tubular networks were quantitatively assessed utilizing the Angiogenesis Analyzer toolset within ImageJ software.
2.16. CCK-8 Cytotoxicity Assay
At 24 hours post-transfection, cells were plated in 96-well plates (5×10³ cells/well) with triplicate wells per group and cultured for 24 hours at 37°C. Docetaxel was then administered at graded concentrations. After a 48-h drug exposure, 10 μL of CCK-8 reagent was added per well, with incubation proceeding for 2 h. Absorbance (OD₄₅₀) was measured using a microplate reader. Cell viability. IC₅₀ was derived from dose-response relationships.
2.17. Statistical Analysis
The GraphPad Prism 9.0 software was utilized to execute all statistical analyses in this study. Unpaired Student’s t-tests were employed for intergroup comparisons and assessments against control groups. For qPCR analyses of patient-derived samples, paired Student’s t-tests were applied. All experiments comprised ≥3 independent biological replicates. p ≤ 0.05 is considered to be statistically significant.
3. Results and Discussion
3.1. Identify TNFRSF12A for Subsequent Studies Based on Bioinformatics Analysis
Publicly available breast cancer gene expression datasets were acquired from the GEO database: GSE69194 (153 breast cancer and 11 normal samples), GSE38959 (30 breast cancer and 13 normal samples), GSE45827 (130 breast cancer and 11 normal samples), and GSE42568 (104 breast cancer and 17 normal samples). Differential gene expression analysis was conducted on each dataset using limma R package. We selected differentially expressed genes (DEGs) based on the following criteria: log2 fold change (log2FC) ≥ 1.0 and P-value ≤ 0.05. Concurrently, a compendium of immune-related genes (IRGs) was sourced from the ImmPort database. Intersection analysis via Venn diagram identified 40 overlapping genes common to both the DEG sets and the IRG list. Prognostic analysis of these 40 candidate genes revealed TNFRSF12A as possessing significant prognostic value, designating it as the primary target for subsequent mechanistic investigation (Figure 1).
TNFRSF12A (officially designated Fn14), a fibroblast growth factor-responsive immediate-early gene product, functions as a constitutively active type I transmembrane receptor within the tumor necrosis factor receptor superfamily (TNFRSF) [20]. Current evidence substantiates that TNFRSF12A upregulation promotes bone metastasis in prostate cancer [21] and non-small cell lung carcinoma (NSCLC) [22]. In cutaneous squamous cell carcinoma, TNFRSF12A activation drives neoplastic proliferation [23], while cholangiocarcinoma studies demonstrate its pro-tumorigenic role through tumor microenvironment (TME) remodeling [24]. Collectively, TNFRSF12A propels pan-cancer progression by orchestrating proliferation, metastasis, and TME reprogramming. Therapeutically, leveraging TNFRSF12A’s tumor-selective membrane localization, Li et al. [25] engineered TNFRSF12A×CD3 bispecific T-cell engagers (BiTEs) and TNFRSF12A-targeted CAR-T, both exhibiting potent oncosuppressive efficacy in preclinical models—highlighting substantial translational potential. Consequently, systematic elucidation of TNFRSF12A’s molecular mechanisms and therapeutic vulnerabilities in TNBC will streamline its clinical translation pipeline.
3.2. Clinical Validation Confirms Marked TNFRSF12A Upregulation in TNBC Correlating Significantly with Adverse Patient Outcomes
Public omics database analyses revealed TNBC-specific TNFRSF12A overexpression (Figure 2A-C). To further characterize its expression pattern, qRT-PCR profiling demonstrated significantly elevated TNFRSF12A levels in 19 TNBC specimens compared to matched adjacent normal tissues (p = 0.04) (Figure 2D). Further validation through the KM-Plotter prognostic platform (https://kmplot.com/analysis/ ) established that TNFRSF12A overexpression portends poorer TNBC prognosis (Figure 2E), evidenced by: Shortened overall survival (OS: HR = 1.89, (1.28–2.81); p = 0.0012); Reduced recurrence-free survival (RFS: HR = 1.47,(1.17–1.84); p = 0.00077); Worsened distant metastasis-free survival (DMFS: HR = 1.86,(1.35–2.57); p = 0.00011). These findings position TNFRSF12A as a promising therapeutic target with clinical translatability in TNBC, underscoring the imperative to dissect its molecular mechanisms for improving patient prognosis.
3.3. TNFRSF12A Promotes Angiogenesis in TNBC
To delineate the tumor-promoting mechanisms of TNFRSF12A in TNBC, This study used the STRING database to identify proteins that interact with TNFRSF12A. Then, it performed a functional enrichment analysis on these potential interacting proteins. Results demonstrated significant enrichment of these interacting proteins in tumor necrosis factor-mediated signaling pathways, signal transduction cascades, and fibroblast growth factor receptor (FGFR) signaling pathways (Figure 3A). This implicates TNFRSF12A in cooperatively orchestrating TNBC progression through inflammatory microenvironment modulation and growth factor signaling. These findings align with established literature: Aberrant activation of TNF signaling drives tumor cell survival and metastasis via NF-κB-mediated transcriptional reprogramming [26,27], while FGFR signaling underpins the aggressive phenotype of TNBC [28,29]. Expression-correlated genes of TNFRSF12A were retrieved from UALCAN platform and subjected to functional enrichment analyses via the DAVID. The top ten enriched terms are presented below: Biological Processes: Preferential enrichment in RNA polymerase II-regulated transcription, positive regulation of cell migration, inflammatory response, angiogenesis, positive regulation of apoptosis, response to hypoxia, and cell-cell adhesion. Cellular Components: Significant clustering in extracellular regions including exosomes, extracellular matrix, and plasma membrane. Molecular Functions: Dominant enrichment in metal ion binding, calcium ion binding, protein binding, and RNA polymerase II binding. KEGG Pathways: Primary enrichment in ECM-receptor interaction and TNF signaling pathway. Convergence of biological processes—particularly positive regulation of cell migration, angiogenesis, and hypoxia response—suggests involvement in metastatic dissemination and stromal remodeling in TNBC. Critically, exosome and extracellular matrix enrichment in cellular components implies TNFRSF12A may facilitate tumor-stromal crosstalk through exosome-mediated intercellular communication or ECM modification, a mechanism extensively implicated in TNBC distant metastasis. These findings posit TNFRSF12A as a potential orchestrator of pro-angiogenic and pro-metastatic signaling via exosomes. Further mechanistic dissection and validation could position TNFRSF12A as an actionable therapeutic target for TNBC intervention.
3.4. TNFRSF12A Knockdown Attenuates Angiogenic Capacity
Functional enrichment analyses implicated TNFRSF12A in angiogenic regulation. To validate this hypothesis, we conducted in vitro tubulogenesis assays using HUVECs. Initial validation of siRNA-mediated knockdown efficiency in MDA-MB-231 and HCC1806 cell lines confirmed potent silencing by siRNA-1 and siRNA-2 (Figure 4A, B), which were subsequently employed in functional studies. Tubulogenesis assays demonstrated markedly attenuated vascular network formation upon TNFRSF12A knockdown (Figure 4C, D). These findings collectively substantiate TNFRSF12A’s role in modulating tumor-associated angiogenesis.
3.5. TNFRSF12A Exerts an Inhibitory Impact on Anti-Tumor Immunity by Hindering Immune Cell Infiltration into Tumors and Suppressing T-Cell Cytotoxic Activity
Tumor immune microenvironment (TIME) constitutes a dynamic ecosystem of immune/stromal cells, cytokines, and metabolites [30], driving oncogenesis through the immunoediting cascade (elimination-equilibrium-escape) [31,32]. Prior studies indicate TNFRSF12A targeting enhances T-cell activation/proliferation in gastric cancer [33]. To elucidate TNFRSF12A’s immunomodulatory role, we first analyzed its correlation with immune infiltration via TIMER (https://cistrome.shinyapps.io/timer/), revealing significant inverse correlations with B-cell (Cor = -0.164, p= 6.80e-02) and CD8⁺ T-cell infiltration (Cor = -0.165, p = 6.74e-02), alongside variable impacts on other populations:CD4⁺ T cells: Cor = -0.071, p = 4.36e-01; Macrophages: Cor = 0.071, P = 4.33e-01; Neutrophils: Cor = -0.022, p = 8.23e-01; Dendritic cells: Cor = -0.005, p = 9.57e-01(Figure 5A, B). In vitro T-cell cytotoxicity assays demonstrated significantly enhanced tumor cell killing upon TNFRSF12A silencing (Figure 5C, D), corroborating bioinformatic data and indicating TNFRSF12A suppresses cytotoxic T-lymphocyte recruitment/effector functions. Mechanistically, TNFRSF12A likely promotes an immune-excluded phenotype—trapping lymphocytes in peri-tumoral stroma rather than infiltrating tumor cores. This spatial constraint impairs immunosurveillance, representing a key immunoediting escape mechanism. Future studies should validate these mechanisms in vivo and delineate downstream lymphocyte-trafficking signals. Our findings provide compelling rationale for targeting TNFRSF12A to reverse immune exclusion, potentially synergizing with checkpoint blockade in “cold” TNBC tumors to improve patient outcomes.
3.6. CAF-Specific TNFRSF12A Elevation Correlates with Increased Stromal Abundance
The tumor microenvironment (TME)—a complex ecosystem comprising malignant cells, immune populations, stromal constituents, and extracellular matrix—plays a pivotal role in tumorigenesis, metastasis, and therapeutic resistance [34,35]. Advancements in single-cell genomics now enable unprecedented resolution for deconvoluting TME cellular architecture, state transitions, and molecular networks [36]. To delineate TNFRSF12A’s functional role within the TME, we leveraged the TISCH2 database (http://tisch.comp-genomics.org) to map its expression landscape across cellular subtypes in breast carcinoma. Our analysis revealed predominant TNFRSF12A enrichment in cancer-associated fibroblasts (CAFs) (Figure 6A, B). Subsequent analyses revealed a direct relationship with increased CAF abundance in TNBC. (MCPcounter algorithm:ρ = 0.316, p = 2.15 × 10⁻⁵; TIDE algorithm: ρ = 0.329, p = 9.12 × 10⁻⁶) (Figure 6C, D). Moreover, correlational analysis demonstrated pervasive positive associations between TNFRSF12A and established CAF marker genes(GPR77(cor=-0.189,p=2.52e-10);MME(Cor=0.221,p=1.41e-12);CD74(Cor=0.127,p=2,52e-05); MCAM(cor=0.195,p=6.36e-11); CAV1(Cor=0.081,p=7.36-03); S100A4(Cor=0.294,p=2.26e-23); FAP(Cor=0.304,p=6.21e-25); PDGFRB(Cor=0.18,p=1.85e-09); PDPN(Cor=0.364,p=7.28e-36); CD70(Cor=0.223,p=7.53e-14)) (Figure 6E). Cancer-associated fibroblasts (CAFs) constitute pivotal stromal components within the tumor microenvironment (TME) that drive malignant progression [37]. Expression of their heterogeneity markers (e.g., FAP [38], PDGFRB [39], S100A4 [40,41]) correlates significantly with breast cancer invasiveness and adverse prognosis. Our study identified CAF-specific TNFRSF12A overexpression, aligning with prior observations: in hepatocellular carcinoma, elevated TNFRSF12A similarly associates with CAF enrichment and Treg infiltration, accelerating tumor aggressiveness via apoptotic and invasive pathway dysregulation [42,43]. The pervasive positive correlation between TNFRSF12A and key CAF markers—particularly its strong association with FAP (ρ = 0.304) and PDPN (p = 0.364)—implies its involvement in TME remodeling through dual mechanisms: FAP⁺ CAFs secrete matrix metalloproteinases (MMPs) that degrade extracellular matrix [38], facilitating peritumoral dissemination; PDPN⁺ CAFs exclude T cells via CXCL12-mediated chemorepulsion [44,45], establishing immune-privileged niches. Notably, TNFRSF12A-PDPN co-expression (ρ = 0.364) suggests synergistic suppression of anti-tumor immunity. Consequently, future investigations should delineate TNFRSF12A’s role in CAF subset differentiation and validate its therapeutic targetability or prognostic utility, providing critical rationale for novel TNBC treatment strategies.
3.7. Elevated TNFRSF12A Expression Promotes Sensitivity to Docetaxel Treatment
The triple-negative phenotype (lacking ER, PR, and HER2) drives therapeutic limitations, elevated resistance risk, and inferior survival outcomes [21,22]. This study focuses on TNFRSF12A, which is highly expressed in TNBC, investigating its potential role in regulating tumor drug resistance. Drug resistance analysis utilizing the GSCA database integrated with the GDSC drug database revealed that elevated TNFRSF12A expression levels correlated with increased sensitivity to docetaxel (Figure 7A). To elucidate the underlying molecular mechanism, molecular docking analysis (Figure 7B-D) identified a potential interaction between TNFRSF12A and docetaxel (binding energy: -6.7 kcal/mol). The docking model indicates that residues 10Ala, 11Pro, 28Lys, and 30Met of TNFRSF12A form hydrophobic interactions with docetaxel, while residues 17Ser, 20Ser, 28Lys, and 6Ser form hydrogen bonds. Furthermore, in vitro CCK-8 assay results confirmed that knockdown of TNFRSF12A led to increased IC50 values for docetaxel in both MDA-MB-231 and HCC1806 cells (Figure 7E). Based on these findings, our study suggests that TNFRSF12A may serve as a biomarker for predicting docetaxel efficacy, and its binding sites (e.g., Lys28, Ser17) may represent novel targets for developing synergistic agents. Consequently, combining TNFRSF12A-targeted therapy with docetaxel chemotherapy represents a potential strategy for treating TNBC patients. The key advantage of such combination therapy lies in generating synergistic effects through drugs with complementary mechanisms. During tumor treatment, secondary genetic mutations or bypass pathway activation often led to monotherapy failure; combination therapy significantly delays disease progression by concurrently inhibiting multiple resistance pathways. However, realizing this combination approach requires further elucidation of the precise mechanism of action of TNFRSF12A in TNBC.
4. Conclusions
This study systematically elucidated the pro-tumor mechanism and therapeutic value of TNFRSF12A in triple-negative breast cancer (TNBC) by integrating multi-omics analysis with experimental validation. Using public databases, the study identified TNFRSF12A overexpression in TNBC tissues, which was confirmed in clinical TNBC patient samples. Additionally, Prognostic analysis indicated that high TNFRSF12A expression is significantly linked to poorer overall survival (OS, HR = 1.89), recurrence-free survival (RFS, HR = 1.47), and distant metastasis-free survival (DMFS, HR = 1.86). These findings support the potential of TNFRSF12A as an independent prognostic marker for TNBC. Subsequent functional enrichment analysis indicated that TNFRSF12A was markedly enriched in positive regulation of cell migration, angiogenesis, and hypoxia response, thereby suggesting its potential involvement in the metastatic spread and microenvironment remodeling of TNBC. In vitro experiments demonstrated that silencing TNFRSF12A significantly inhibited the tube-forming ability of HUVEC cells (p < 0.01), corroborating the activation of the angiogenesis pathway suggested by functional enrichment analysis. Bioinformatics analysis revealed a negative correlation between TNFRSF12A and CD8+ T cell (r = -0.165) and B cell (r = -0.164) infiltration. In vitro experiments demonstrated that silencing TNFRSF12A enhances T cell killing efficiency (p < 0.05). The fifth point indicates that it weakens antitumor immunity by inducing an “immune rejection” phenotype. A single-cell transcriptomics analysis (TISCH2 database) revealed that TNFRSF12A is highly expressed in cancer-associated fibroblasts (CAFs) and positively correlated with CAF markers (FAP: ρ= 0.304; PDPN: ρ= 0.364). Quantitative analysis confirmed parallel upregulation of PDPN and CAF infiltration markers in TNBC biopsies. This correlation was statistically significant. The results of this study suggest that the progression of TNBC is promoted by CAF-mediated microenvironmental remodeling. A recent study revealed that high TNFRSF12A expression enhances docetaxel sensitivity. Moreover, molecular docking revealed that the drug directly binds to the protein via key residues (10ALA/28LYS, etc.) with a binding energy of -6.7 kcal/mol. Gene silencing experiments confirmed that TNFRSF12A deficiency increases the docetaxel IC50 value in MDA-MB-231/HCC1806 cells, supporting its role as a chemotherapy sensitization target. Consequently, a synergistic treatment regimen targeting TNFRSF12A in combination with docetaxel is proposed to overcome TNBC resistance by reversing immune suppression and enhancing chemotherapy response. This study is pioneering in its exploration of the multifaceted role of TNFRSF12A in TNBC, delineating its function as a “diagnostic marker-microenvironment regulatory factor-chemotherapy sensitization target.” The findings of this study lay the groundwork for the development of combination therapies that capitalize on the potential of TNFRSF12A inhibitors.
Author Contributions
Conceptualization, C.J; methodology, C.J.; software, Z.W.Z.; validation, C.J.; formal analysis, C.J.; investigation, C.J.; resources, M.W.; data curation, C.J.; writing—original draft preparation, C.J.; writing—review and editing, G.S. and G.Y.; visualization, Z.W.Z.; supervision, M.W.; project administration, M.W.; funding acquisition, M.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Research Ethics Committee of Central South University.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All datasets analyzed in this study are available for download from the T ImmPort database (https://immport.org/) and the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
Acknowledgments
The study was financially supported by project grants from the National Natural Science Foundation of China (No. 82303841 and No. 81572900), and also by the Natural Science Foundation of Hunan Province (No. 2025JJ40071 and No. 2023JJ40800), and the Central South University Innovation-Driven Research Program (No. 2025ZZTS0946). Top-Notch Innovation Base of Basic Medicine, Central South University provides strong support.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Abbreviations | Full name |
| TNBC | Triple-negative breast cancer |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| OS | Overall survival |
| RFS | Recurrence-Free Survival |
| DMFS | Distant Metastasis Free Survival |
| HR | Hazard Ratio |
| CAFs | Cancer-associated fibroblasts |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ECM | Extracellular matrix |
| HUVECs | Human Umbilical Vein Endothelial Cells |
| DEGs | Differentially expressed genes |
| PPI | Protein-Protein Interaction Networks |
| BP | Biological process |
| MF | Molecular function |
| CC | Cellular component |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| Cor | correlation coefficient |
References
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024; 74: 229-63. [CrossRef]
- Tong L, Yu X, Wang S, Chen L, Wu Y. Research Progress on Molecular Subtyping and Modern Treatment of Triple-Negative Breast Cancer. Breast Cancer (Dove Med Press). 2023; 15: 647-58. [CrossRef]
- Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019; 9: 176-98. [CrossRef]
- Obidiro O, Battogtokh G, Akala EO. Triple Negative Breast Cancer Treatment Options and Limitations: Future Outlook. Pharmaceutics. 2023; 15. [CrossRef]
- Xiong N, Wu H, Yu Z. Advancements and challenges in triple-negative breast cancer: a comprehensive review of therapeutic and diagnostic strategies. Front Oncol. 2024; 14: 1405491. [CrossRef]
- Zagami P, Carey LA. Triple negative breast cancer: Pitfalls and progress. NPJ Breast Cancer. 2022; 8: 95. [CrossRef]
- Rong L, Li N, Zhang Z. Emerging therapies for glioblastoma: current state and future directions. J Exp Clin Cancer Res. 2022; 41: 142. [CrossRef]
- Keenan TE, Tolaney SM. Role of Immunotherapy in Triple-Negative Breast Cancer. J Natl Compr Canc Netw. 2020; 18: 479-89. [CrossRef]
- Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC Med. 2019; 17: 90. [CrossRef]
- Lan J, Chen L, Li Z, Liu L, Zeng R, He Y, et al. Multifunctional Biomimetic Liposomes with Improved Tumor-Targeting for TNBC Treatment by Combination of Chemotherapy, Antiangiogenesis and Immunotherapy. Adv Healthc Mater. 2024; 13: e2400046. [CrossRef]
- Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho JW, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell. 2022; 185: 1694-708.e19. [CrossRef]
- Ruiz BI, Lowman XH, Yang Y, Fan Q, Wang T, Wu H, et al. Alpha-Ketoglutarate Regulates Tnfrsf12a/Fn14 Expression via Histone Modification and Prevents Cancer-Induced Cachexia. Genes (Basel). 2023; 14. [CrossRef]
- Xu RD, Feng F, Yu XS, Liu ZD, Lao LF. miR-149-5p inhibits cell growth by regulating TWEAK/Fn14/PI3K/AKT pathway and predicts favorable survival in human osteosarcoma. Int J Immunopathol Pharmacol. 2018; 32: 2058738418786656. [CrossRef]
- Xu Q, Fan G, Shao S. Role of TNFRSF12A in cell proliferation, apoptosis, and proinflammatory cytokine expression by regulating the MAPK and NF-κB pathways in thyroid cancer cells. Cytokine. 2025; 186: 156841. [CrossRef]
- Liu T, Pu J, Theil S, Liu Y, Jiang L, Liu H, et al. Potential role of TNFRSF12A in linking glioblastoma and alzheimer’s disease via shared tumour suppressor pathways. Sci Rep. 2025; 15: 21535. [CrossRef]
- Wang C, Zhao Y, Chen Y, Shi Y, Yang Z, Wu W, et al. The Oncogenic Role of TNFRSF12A in Colorectal Cancer and Pan-Cancer Bioinformatics Analysis. Cancer Res Treat. 2025; 57: 212-28. [CrossRef]
- Wu ZH, Niu X, Wu GH, Cheng Q. Decreased expression of TNFRSF12A in thyroid gland cancer predicts poor prognosis: A study based on TCGA data. Medicine (Baltimore). 2020; 99: e21882. [CrossRef]
- Guo L, Chen Q, Xu M, Huang J, Ye H. Communication between alveolar macrophages and fibroblasts via the TNFSF12-TNFRSF12A pathway promotes pulmonary fibrosis in severe COVID-19 patients. J Transl Med. 2024; 22: 698. [CrossRef]
- Svobodová B, Löfdahl A, Nybom A, Wigén J, Hirdman G, Olm F, et al. Overlapping Systemic Proteins in COVID-19 and Lung Fibrosis Associated with Tissue Remodeling and Inflammation. Biomedicines. 2024; 12. [CrossRef]
- Brown SA, Richards CM, Hanscom HN, Feng SL, Winkles JA. The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J. 2003; 371: 395-403. [CrossRef]
- Yin J, Liu YN, Tillman H, Barrett B, Hewitt S, Ylaya K, et al. AR-regulated TWEAK-FN14 pathway promotes prostate cancer bone metastasis. Cancer Res. 2014; 74: 4306-17. [CrossRef]
- Cheng E, Whitsett TG, Tran NL, Winkles JA. The TWEAK Receptor Fn14 Is an Src-Inducible Protein and a Positive Regulator of Src-Driven Cell Invasion. Mol Cancer Res. 2015; 13: 575-83. [CrossRef]
- Hu G, Liang L, Liu Y, Liu J, Tan X, Xu M, et al. TWEAK/Fn14 Interaction Confers Aggressive Properties to Cutaneous Squamous Cell Carcinoma. J Invest Dermatol. 2019; 139: 796-806. [CrossRef]
- Dwyer BJ, Jarman EJ, Gogoi-Tiwari J, Ferreira-Gonzalez S, Boulter L, Guest RV, et al. TWEAK/Fn14 signalling promotes cholangiocarcinoma niche formation and progression. J Hepatol. 2021; 74: 860-72. [CrossRef]
- Li G, Zhang Z, Cai L, Tang X, Huang J, Yu L, et al. Fn14-targeted BiTE and CAR-T cells demonstrate potent preclinical activity against glioblastoma. Oncoimmunology. 2021; 10: 1983306. [CrossRef]
- Zhu C, Zhang L, Liu Z, Li C, Bai Y. TWEAK/Fn14 interaction induces proliferation and migration in human airway smooth muscle cells via activating the NF-κB pathway. J Cell Biochem. 2018; 119: 3528-36. [CrossRef]
- Liu JY, Jiang L, He T, Liu JJ, Fan JY, Xu XH, et al. NETO2 promotes invasion and metastasis of gastric cancer cells via activation of PI3K/Akt/NF-κB/Snail axis and predicts outcome of the patients. Cell Death Dis. 2019; 10: 162. [CrossRef]
- Morales-Guadarrama G, Méndez-Pérez EA, García-Quiroz J, Avila E, Ibarra-Sánchez MJ, Esparza-López J, et al. The Inhibition of the FGFR/PI3K/Akt Axis by AZD4547 Disrupts the Proangiogenic Microenvironment and Vasculogenic Mimicry Arising from the Interplay between Endothelial and Triple-Negative Breast Cancer Cells. Int J Mol Sci. 2023; 24. [CrossRef]
- Wu Y, Yi Z, Li J, Wei Y, Feng R, Liu J, et al. FGFR blockade boosts T cell infiltration into triple-negative breast cancer by regulating cancer-associated fibroblasts. Theranostics. 2022; 12: 4564-80. [CrossRef]
- Lv B, Wang Y, Ma D, Cheng W, Liu J, Yong T, et al. Immunotherapy: Reshape the Tumor Immune Microenvironment. Front Immunol. 2022; 13: 844142. [CrossRef]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002; 3: 991-8. [CrossRef]
- Roerden M, Spranger S. Cancer immune evasion, immunoediting and intratumour heterogeneity. Nat Rev Immunol. 2025; 25: 353-69. [CrossRef]
- Xia L, Jiang L, Chen Y, Zhang G, Chen L. ThPOK transcriptionally inactivates TNFRSF12A to increase the proliferation of T cells with the involvement of the NF-kB pathway. Cytokine. 2021; 148: 155658. [CrossRef]
- Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021; 221: 107753. [CrossRef]
- Mottini C, Auciello FR, Manni I, Pilarsky C, Caputo D, Caracciolo G, et al. The cross-talk between the macro and micro-environment in precursor lesions of pancreatic cancer leads to new and promising circulating biomarkers. J Exp Clin Cancer Res. 2024; 43: 198. [CrossRef]
- Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020; 52: 1428-42. [CrossRef]
- Raaijmakers K, Adema GJ, Bussink J, Ansems M. Cancer-associated fibroblasts, tumor and radiotherapy: interactions in the tumor micro-environment. J Exp Clin Cancer Res. 2024; 43: 323. [CrossRef]
- Mori N, Jin J, Krishnamachary B, Mironchik Y, Wildes F, Vesuna F, et al. Functional roles of FAP-α in metabolism, migration and invasion of human cancer cells. Front Oncol. 2023; 13: 1068405. [CrossRef]
- Zhang L, Wang K, Zhang J, Qian X, Zhang X, Wang Y, et al. PDGFR-β/Cav1-induced autophagy via mTOR/FIP200/ATG13 activation in cancer-associated fibroblasts promotes the malignant progression of breast cancer. J Transl Med. 2025; 23: 784. [CrossRef]
- Liu Y, Geng YH, Yang H, Yang H, Zhou YT, Zhang HQ, et al. Extracellular ATP drives breast cancer cell migration and metastasis via S100A4 production by cancer cells and fibroblasts. Cancer Lett. 2018; 430: 1-10. [CrossRef]
- Qu S, Wu J, Bao Q, Yao B, Duan R, Chen X, et al. Osterix promotes the migration and angiogenesis of breast cancer by upregulation of S100A4 expression. J Cell Mol Med. 2019; 23: 1116-27. [CrossRef]
- Liao M, Liao J, Qu J, Shi P, Cheng Y, Pan Q, et al. Hepatic TNFRSF12A promotes bile acid-induced hepatocyte pyroptosis through NFκB/Caspase-1/GSDMD signaling in cholestasis. Cell Death Discov. 2023; 9: 26. [CrossRef]
- Wang T, Ma S, Qi X, Tang X, Cui D, Wang Z, et al. Knockdown of the differentially expressed gene TNFRSF12A inhibits hepatocellular carcinoma cell proliferation and migration in vitro. Mol Med Rep. 2017; 15: 1172-8. [CrossRef]
- Shindo K, Aishima S, Ohuchida K, Fujiwara K, Fujino M, Mizuuchi Y, et al. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol Cancer. 2013; 12: 168. [CrossRef]
- Feng C, Yu A, Wang Z, Wang K, Chen J, Wu Y, et al. A novel PDPN antagonist peptide CY12-RP2 inhibits melanoma growth via Wnt/β-catenin and modulates the immune cells. J Exp Clin Cancer Res. 2024; 43: 9. [CrossRef]
Figure 1.
Identification Workflow for Key Immune-Related Genes Impacting TNBC Prognosis. (A) Schematic of the screening methodology. (B)Intersection of DEGs across four datasets with immune-related gene sets.
Figure 1.
Identification Workflow for Key Immune-Related Genes Impacting TNBC Prognosis. (A) Schematic of the screening methodology. (B)Intersection of DEGs across four datasets with immune-related gene sets.
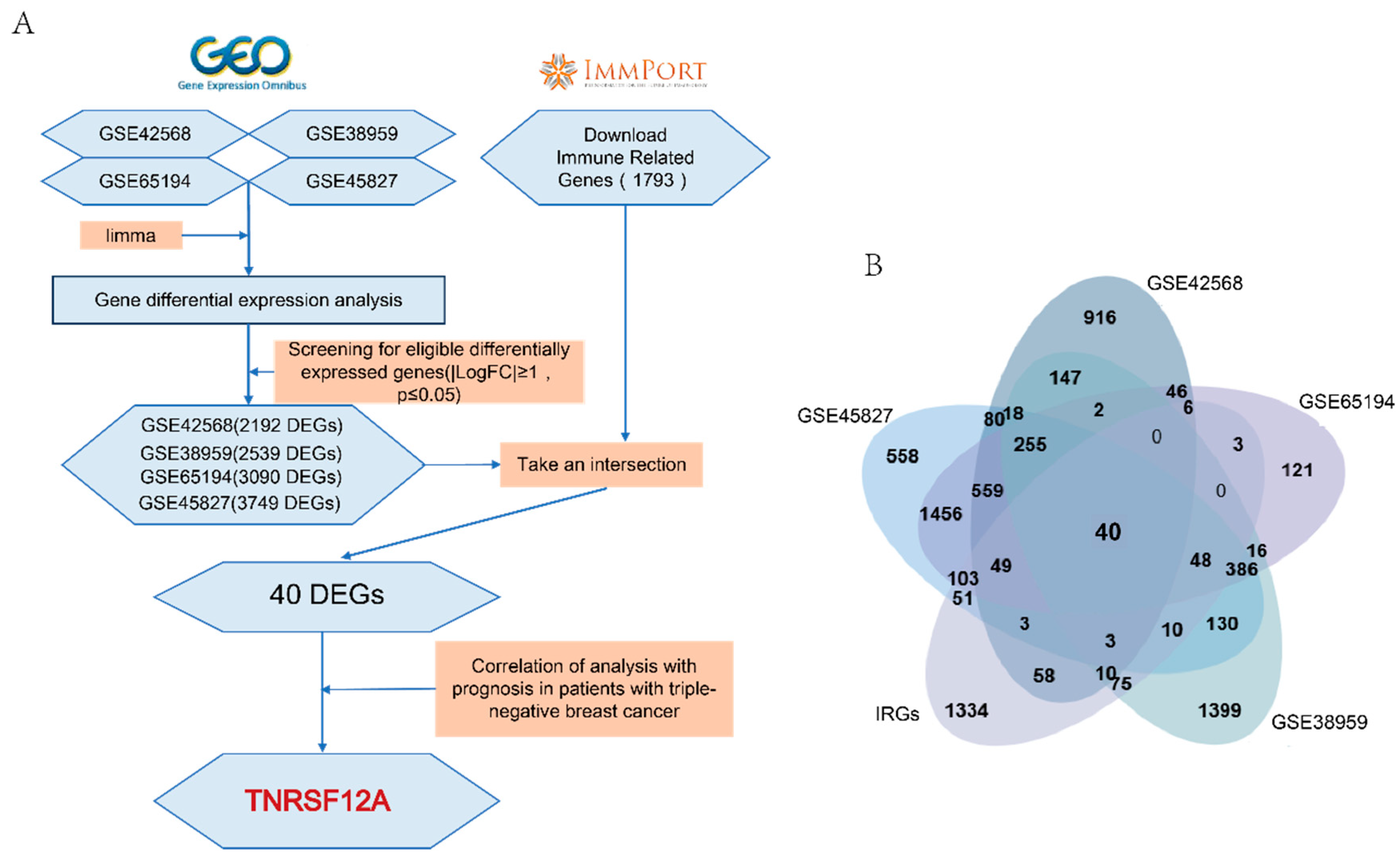
Figure 2.
Expression Profile and Prognostic Significance of TNFRSF12A in TNBC. (A) Immunohistochemical quantification of TNFRSF12A protein expression in breast carcinoma specimens. (B–C) Differential mRNA expression patterns of TNFRSF12A across breast adenocarcinoma subtypes and molecular classifications. (D) Significantly elevated TNFRSF12A transcript levels in 19 TNBC tissues versus matched adjacent non-tumor controls (p = 0.04). (E) Kaplan-Meier survival curves demonstrating adverse prognostic impact of high TNFRSF12A expression (stratified by median cut-off): Overall Survival (OS): HR = 1.89, P = 0.0012; Recurrence-Free Survival (RFS): HR = 1.47, P = 0.00077; Distant Metastasis-Free Survival (DMFS): HR = 1.86, P = 0.00011.
Figure 2.
Expression Profile and Prognostic Significance of TNFRSF12A in TNBC. (A) Immunohistochemical quantification of TNFRSF12A protein expression in breast carcinoma specimens. (B–C) Differential mRNA expression patterns of TNFRSF12A across breast adenocarcinoma subtypes and molecular classifications. (D) Significantly elevated TNFRSF12A transcript levels in 19 TNBC tissues versus matched adjacent non-tumor controls (p = 0.04). (E) Kaplan-Meier survival curves demonstrating adverse prognostic impact of high TNFRSF12A expression (stratified by median cut-off): Overall Survival (OS): HR = 1.89, P = 0.0012; Recurrence-Free Survival (RFS): HR = 1.47, P = 0.00077; Distant Metastasis-Free Survival (DMFS): HR = 1.86, P = 0.00011.
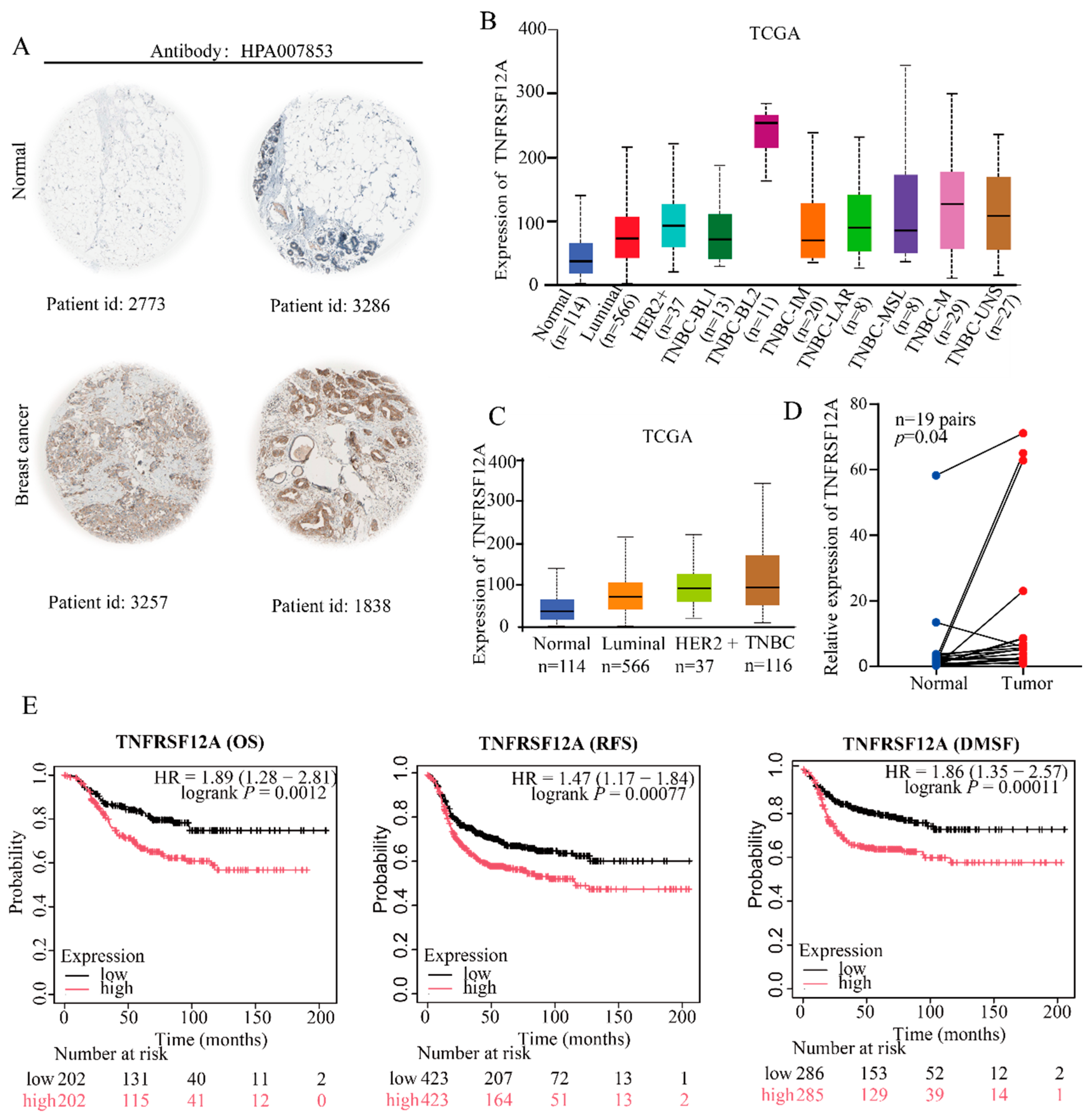
Figure 3.
Functional Enrichment Profiling of TNFRSF12A-Associated Pathways. (A) PPI network of putative TNFRSF12A-binding partners. (B) GO enrichment analysis of potential TNFRSF12A interactors. (C–F) Functional annotation clustering of TNFRSF12A expression-correlated genes.
Figure 3.
Functional Enrichment Profiling of TNFRSF12A-Associated Pathways. (A) PPI network of putative TNFRSF12A-binding partners. (B) GO enrichment analysis of potential TNFRSF12A interactors. (C–F) Functional annotation clustering of TNFRSF12A expression-correlated genes.
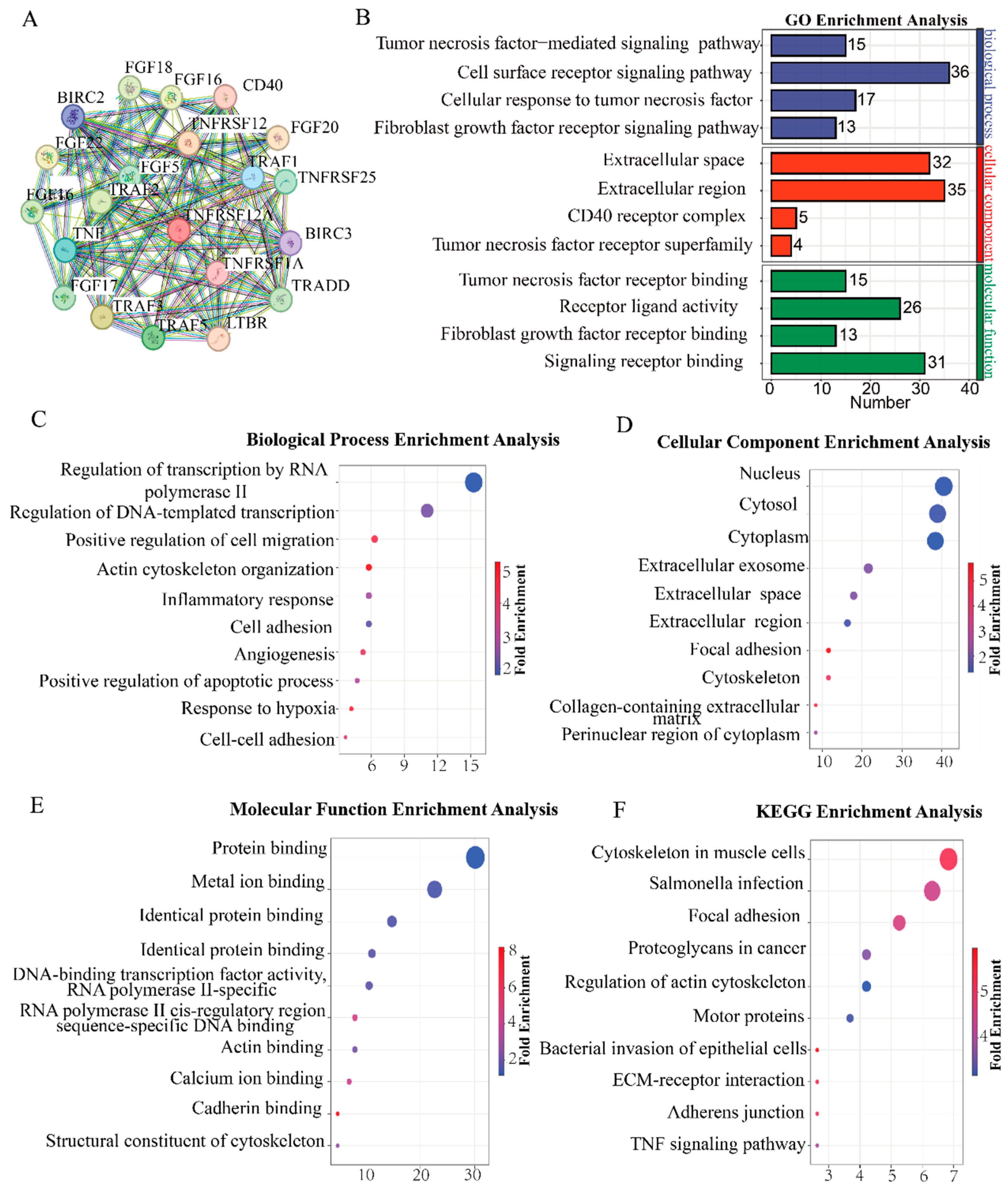
Figure 4.
In Vitro Angiogenesis Assay. (A–B) Validation of siRNA-mediated knockdown efficiency in MDA-MB-231 and HCC1806 cell lines. (C) Representative images of HUVEC tubulogenesis. (D) Statistical quantification of total tubular length. (E) Statistical quantification of branching points. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
Figure 4.
In Vitro Angiogenesis Assay. (A–B) Validation of siRNA-mediated knockdown efficiency in MDA-MB-231 and HCC1806 cell lines. (C) Representative images of HUVEC tubulogenesis. (D) Statistical quantification of total tubular length. (E) Statistical quantification of branching points. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****).
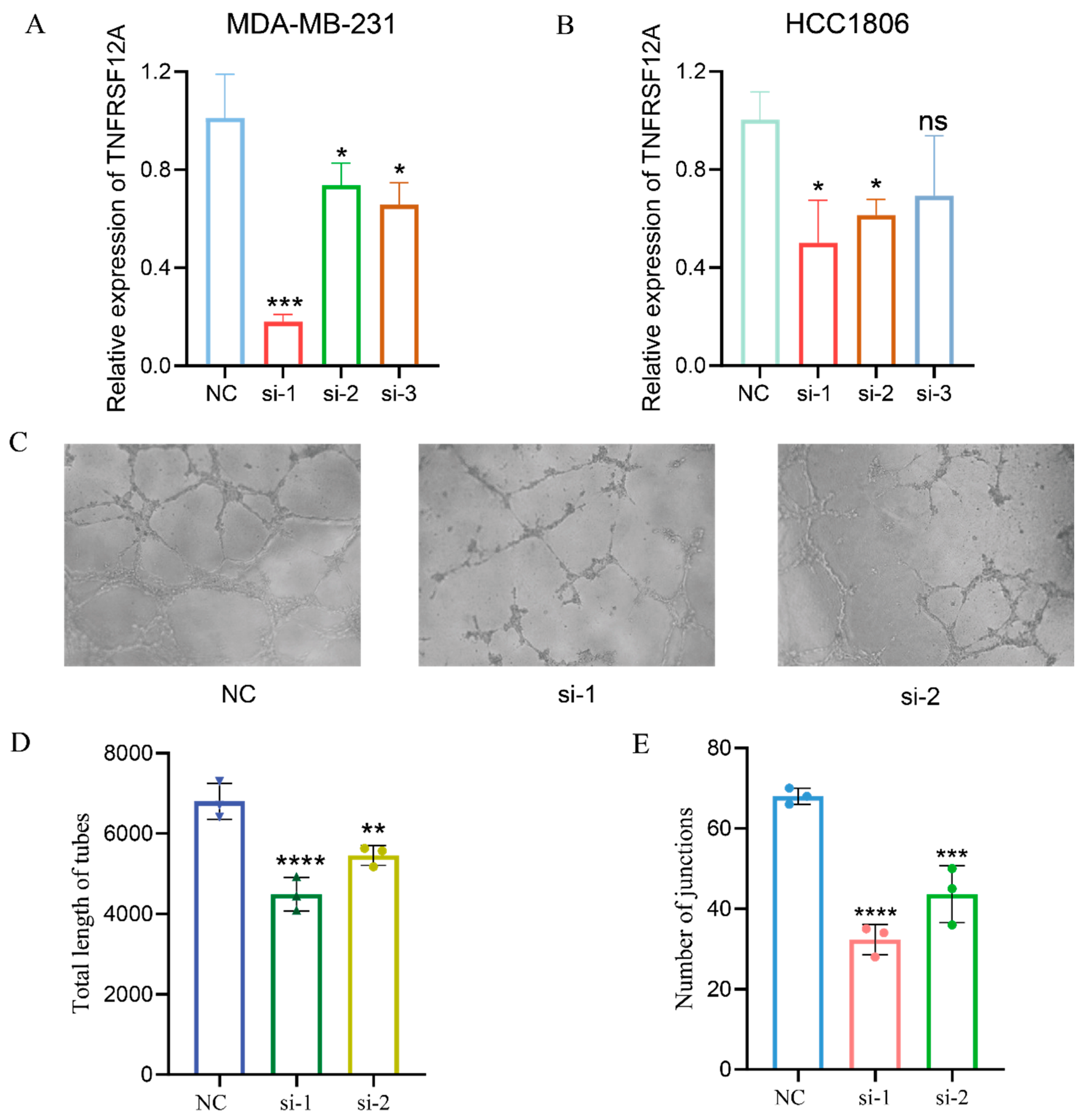
Figure 5.
Modulation of the tumor immune landscape by TNFRSF12A in TNBC. (A) Correlational profiling of TNFRSF12A expression with infiltration levels of B cell (Cor = -0.164, p = 6.80e-02), CD8⁺ T cells (Cor = -0.165, p = 6.74e-02), and CD4⁺ T cells (Cor = -0.071, p = 4.36e-01).(B) Correlation analysis of TNFRSF12A expression with macrophage (Cor = 0.071, p = 4.33e-01), neutrophil (Cor = -0.022, p = 8.23e-01), and dendritic cell infiltration (Cor = -0.005, p = 9.57e-01). (C) Representative data from in vitro T-cell cytotoxicity assays. (D) Statistical quantification of T-cell-mediated tumor cell killing. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
Figure 5.
Modulation of the tumor immune landscape by TNFRSF12A in TNBC. (A) Correlational profiling of TNFRSF12A expression with infiltration levels of B cell (Cor = -0.164, p = 6.80e-02), CD8⁺ T cells (Cor = -0.165, p = 6.74e-02), and CD4⁺ T cells (Cor = -0.071, p = 4.36e-01).(B) Correlation analysis of TNFRSF12A expression with macrophage (Cor = 0.071, p = 4.33e-01), neutrophil (Cor = -0.022, p = 8.23e-01), and dendritic cell infiltration (Cor = -0.005, p = 9.57e-01). (C) Representative data from in vitro T-cell cytotoxicity assays. (D) Statistical quantification of T-cell-mediated tumor cell killing. p < 0.05 (*), p < 0.01 (**), p < 0.001 (***).
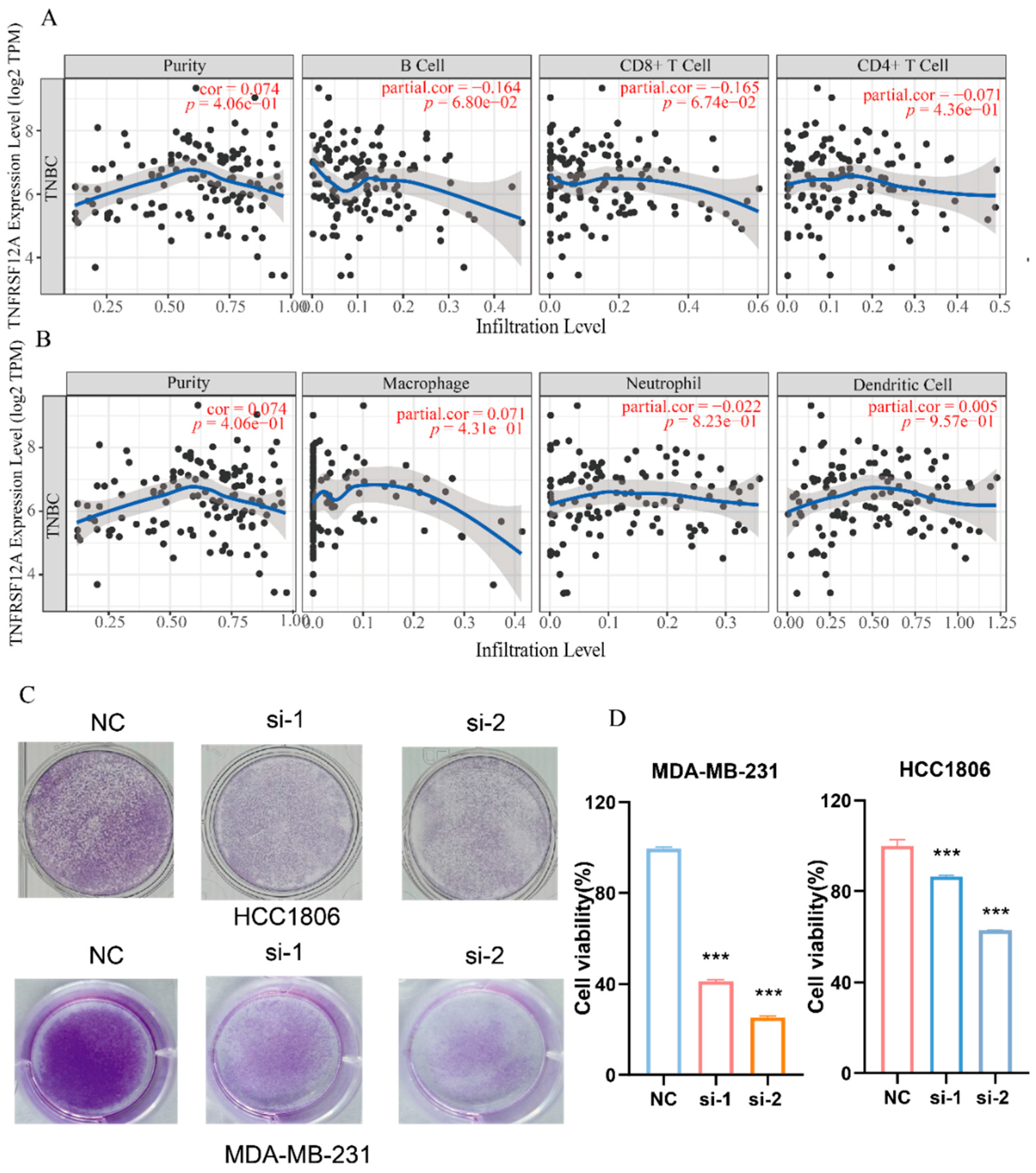
Figure 6.
Single-cell expression landscape of TNFRSF12A and its correlation with fibroblast infiltration. (A) Expression signatures with single-cell granularity of TNFRSF12A in the BRCA-EMTAB11077 dataset. (B) Expression signatures with single-cell granularity of TNFRSF12A in the BRCA-GSE161529 dataset. (C) Correlation analysis between TNFRSF12A expression levels and fibroblast infiltration levels (MCPcounter algorithm:ρ = 0.316, P = 2.15 × 10⁻⁵; TIDE algorithm: ρ = 0.329, P = 9.12 × 10⁻⁶). (D) Correlation analysis between TNFRSF12A and fibroblast signature genes (GPR77(Cor=-0.189,p=2.52e-10; MME(Cor=0.221,p=1.41e-12; CD74(Cor=0.127. p=2,52e-05); MCAM(Cor=0.195,p=6.36e-11); CAV1(Cor=0.081,p=7.36-03); S100A4(Cor=0.294,p=2.26e-23); FAP(Cor=0.304,p=6.21e-25); PDGFRB(Cor=0.18,p=1.85e-09); PDPN(Cor=0.364,p=7.28e-36); CD70 (Cor=0.223,p=7.53e-14).
Figure 6.
Single-cell expression landscape of TNFRSF12A and its correlation with fibroblast infiltration. (A) Expression signatures with single-cell granularity of TNFRSF12A in the BRCA-EMTAB11077 dataset. (B) Expression signatures with single-cell granularity of TNFRSF12A in the BRCA-GSE161529 dataset. (C) Correlation analysis between TNFRSF12A expression levels and fibroblast infiltration levels (MCPcounter algorithm:ρ = 0.316, P = 2.15 × 10⁻⁵; TIDE algorithm: ρ = 0.329, P = 9.12 × 10⁻⁶). (D) Correlation analysis between TNFRSF12A and fibroblast signature genes (GPR77(Cor=-0.189,p=2.52e-10; MME(Cor=0.221,p=1.41e-12; CD74(Cor=0.127. p=2,52e-05); MCAM(Cor=0.195,p=6.36e-11); CAV1(Cor=0.081,p=7.36-03); S100A4(Cor=0.294,p=2.26e-23); FAP(Cor=0.304,p=6.21e-25); PDGFRB(Cor=0.18,p=1.85e-09); PDPN(Cor=0.364,p=7.28e-36); CD70 (Cor=0.223,p=7.53e-14).
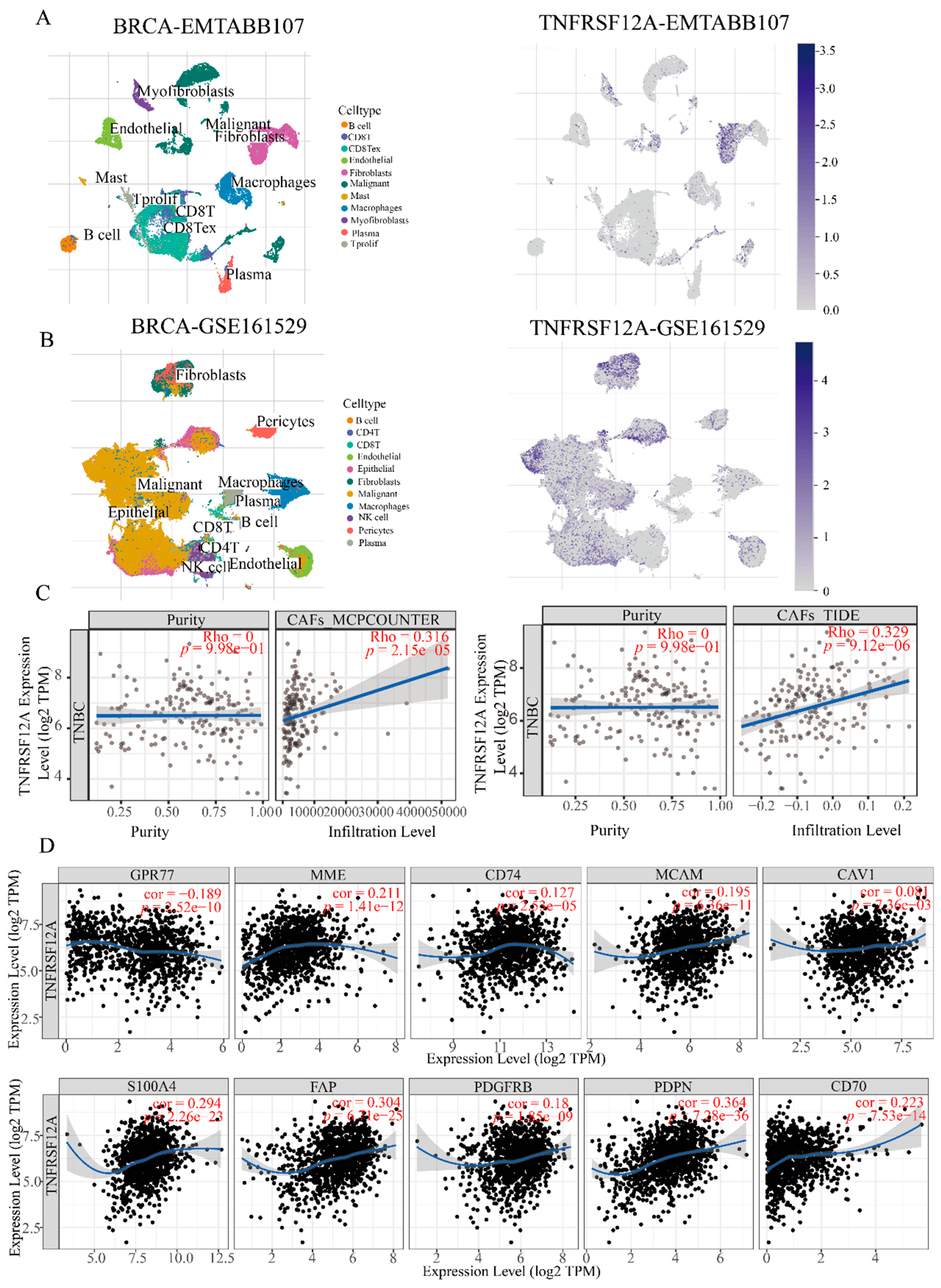
Figure 7.
Drug resistance analysis of TNFRSF12A. (A) Correlation analysis between TNFRSF12A expression levels and therapeutic drug resistance. (B) Three-dimensional structural representations of TNFRSF12A and the drug docetaxel. (C) Three-dimensional molecular docking model depicting the interaction between TNFRSF12A and docetaxel. (D) Two-dimensional interaction diagram of the molecular docking results between TNFRSF12A and docetaxel. (E) Determination of IC50 values for docetaxel in TNBC cell lines treated with varying concentrations of the drug, measured absorbance at 450 nm to assess cell viability.
Figure 7.
Drug resistance analysis of TNFRSF12A. (A) Correlation analysis between TNFRSF12A expression levels and therapeutic drug resistance. (B) Three-dimensional structural representations of TNFRSF12A and the drug docetaxel. (C) Three-dimensional molecular docking model depicting the interaction between TNFRSF12A and docetaxel. (D) Two-dimensional interaction diagram of the molecular docking results between TNFRSF12A and docetaxel. (E) Determination of IC50 values for docetaxel in TNBC cell lines treated with varying concentrations of the drug, measured absorbance at 450 nm to assess cell viability.

Table 1.
Sequence of TNFRSF12A siRNA.
| siRNA name | Sequence |
| siRNA-1 | 5’-AGAGAGAAGTTCACCACC -3’ |
| SiRNA-2 | 5’- CACTCATCATTCATTCATC-3’ |
Table 2.
Primer sequence for qPCR.
| Gene name | Forward | Reverse |
| TNFRSF12A | GACCTGGACAAGTGCAT | GGTGGTGAACTTCTCTCTC |
| U6 | CTCGCTTCGGCAGCACA | AACGCTTCACGAATTTGCGT |
| β-actin | CTCTTCCAGCCTTCCTTCCT | AGCACTGTGTTGGCGTACAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



