
Submitted:
28 July 2025
Posted:
29 July 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Background: The mutation of a new virus is key to its ability to adapt, as it allows the virus to change and survive in new environments. We try to capture the full extent of SARS-CoV-2 diversity in Asturias. Methods: Samples from nasopharyngeal swabs belonging to SARS-CoV-2 infected patients were characterized by the whole genome sequencing. Results: A total of 4,001 sequences were analysed between 2020 and July 2024. During this time, 5302 mutations were found (157 occurred in more than 5% of the viral population). Between 2022 and 2024, 88 new non-synonymous mutations occurred in over 5% of the viral population, of which 31 are currently maintained and were fixed 250±46 (93-620) days after their appearance. Besides, 55 new circulating variants (NCVs) belonging to 41 pangolin lineages were detected. Of these NCVs, 24 were originated throughout the world, and the other 31 in Asturias (10 were only detected in the region, 8 also in Spain, and 13 around the world).Conclusions: New variants circulating around the world continually appear in our region, leading to a constant rotation of lineages. Although most of the changes have little or no impact on the virus's properties, the continued emergence of new variants warrant continued genomic surveillance.
Keywords:
SARS-CoV-2
; new variants
; surveillance
; phylogenetic analysis
1. Introduction
Since the start of the pandemic, the rate of mutation of the SARS-Cov-2 has been decreasing [1]. However, the number of SARS-Cov-2 infections continues to rise and outbreaks still occur despite the majority of the population being vaccinated. Thankfully, the current COVID-19 fatality rate is lower than it was during the early stages of the pandemic [1]. This is because the causative agent of COVID-19, SARS-CoV-2, is constantly evolving as it spreads from person to person, with new sub-lineages emerging all the time. Therefore, the SARS-CoV-2 genome should be sequenced to identify viral strains and investigate their local and worldwide dissemination. In addition, the comprehensive sequencing of the entire viral genome associated with infection is a valuable tool in elucidating outbreak dynamics [2,3]. Our group started sequencing the whole genome of the SARS-Cov-2 samples, to identify the genotypes of the virus circulating in our area and to analyze genomic diversity, the types of mutations, and the emergence of new variants of SARS-CoV-2 by June 2022 [4,5,6].This study monitored and tracked the SARS-CoV-2 epidemic in Asturias during the post-Covid period (July 2022 to July 2024).
2. Materials and Methods
2.1. Sample Collection
From July 2022 to July 2024, 2268 (1733 recorded from March 2020 to July 2022) samples from nasopharyngeal swabs belonging to SARS-CoV2 infected patients were characterized by the whole genome sequencing (WGS) method and were uploaded to GISAID Data on age, sex, date and pangolin lineage were collected (Supplementary Table S1).
2.2. WGS
Selected SARS-CoV-2 positive samples were sequenced by using the Ion AmpliSeq SARS-CoV-2 research panel (Thermo Fisher Scientific, Waltham, MA, USA) following the instructions set out in the manufacturer’s user guide. Libraries were prepared on the Ion Chef system as described in the user’s guide. Amplified samples were then sequenced, using Ion 540 chips with the Ion S5 system following the instructions set out in the manufacturer’s user guide (Alessandrini et al. 2020).
The obtained sequences were uploaded to GISAID database (https://www.gisaid.org/)
2.3. Classification/Characterization
The SARS-CoV-2 genomes were aligned using MAFFT (https://mafft.cbrc.jp/alignment/software/) and then manually curated using MEGA 7 (https://www.megasoftware.net/). The best-fit nucleotide substitution model GTR+I was identified according to the Akaike information criterion using jModel-Test v2.1.10. (https://github.com/ddarriba/jmodeltest2). Phylogenetic trees were reconstructed by ML with FastTree (http://www.microbesonline.org/fasttree/ ) for large trees or IQ-TREE (http://www.iqtree.org/). Bootstrap values were estimated using the SH test and ultrafast bootstrap. The Wuhan-Hu-1 reference genome (MN908947.3) was used as an outgroup.
Each of the coding regions (ORF1a, RDPD, ORF1ab, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N, ORF10) was extracted separately from the alignments and nucleotides in the coding regions were converted to their corresponding encoded amino acid residues (SeaView https://doua.prabi.fr/software/seaview). SNPs were retrieved from the aligned regions in line with the reference genome. Non-synonymous mutations with a frequency of more than 5 % (number of strains with a specific mutation/total number of strains) were considered majority in the population and were used in subsequent analyses. A mutation is considered to have become fixed in the population when it reaches a frequency of 95% (number of strains with a specific mutation/total number of strains in a month) and remains until today.
Dated phylogeny was reconstructed using Bayesian inference through a Markov chain Monte Carlo (MCMC) framework implemented in BEAST v1.10 (https://beast.community/). The days until the date of the most recent sequence were used as the sampling date. An uncorrelated relaxed clock model was employed to estimate the time to a most recent common ancestor (TMRCA). Bayesian Skyline analysis was used to infer how the population size is expected to change over time. MCMC chains were run for 100 million steps with sampling every 10,000 steps from the previous distribution. Convergence was evaluated by calculating the effective sample sizes of the parameters using Tracer v1.7.1 (https://beast.community/tracer). Trees were summarized as maximum clade credibility trees using TreeAnnotator v1.8.4 after discarding the first 10% as burn-in, and then visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
A phylogeographic analysis was performed in the BEAST program considering geographic locations as discrete states in a Bayesian statistical framework. An asymmetric substitution model and an uncorrelated relaxed molecular clock were applied to the Bayesian Stochastic Search Variable selection (BSSVS) method to identify the number of non-zero transitions (migrations) rates between states.
Using unusual SNPs in the circulating lineages in Asturias and monophyletic clades with more than five sequences, possible new lineages to study were defined. Lineage specific mutations were obtained from analysis of WGS genomes against the Wuhan-Hu-1 reference genome (MN908947.3) and filtered for substitutions. The pattern of specific mutations of possible new lineages was searched for in sequences obtained from GISAID in order to analyze their distribution around the world.
Diversity (D = 1 − ∑f2) of pangolin lineages was analyzed, a measure of variability that takes into account the frequencies (f) of all types.
3. Results
3.1. Mutations
A total of 4,001 sequences were analysed in Asturias (northern Spain) between 2020 and July 2024. During this time, 5302 mutations were found in 9769 viral aminoacid of genome. Of these, 157 (3% of mutations, and 1.6% of total genome) occurred in more than 5% of the viral population. In the same period, 158 (1.6% of the genome) mutations were found in Spain, and 116 (1.2%) were found worldwide. (Supplementary Table S2, Table 1).
The estimated relative mutation rates for the codon were 1.34 for the third position, 0.81 for the second position and 0.85 for the first position.
The estimated mean rate was 9.20 x10-4 (8.07-10.00) replacements per site, per year in the 2022/24, compared to 7.92 x10-4 (7.30–8.54) replacements per site, per year in the 2020/22.
Between 2022 and 2024, 88 new non-synonymous mutations (0.9% of the genome) occurred in over 5% of the viral population, of which 31 are currently maintained and were fixed 250±46 (93-620) days after their appearance. Between March 2020 and July 2022, 105 (1.1% of the genome) major mutations were identified, of which 51 were fixed after 344±94 (0-1209) days (Supplementary Table S3, Table 2, Figure 1).
In contrast to Asturias, as of June 2024, 50 mutations have been established worldwide: 29 in the gene S, 8 in ORF1a, 4 in the N gene, 2 in the M gene, 3 in ORF1ab, and 1 in RDPD, ORF3a, the E gene and ORF6 (Supplementary Table S2).
The P323L mutation (RDPD gene) and the D614G mutation (S gene) appeared in over 5% of the viral population by day 0. The following mutations were delayed two months: M gene-A63T, M gene-Q19E, ORF1a gene_P3395H, ORF1ab gene_I643V, S gene_N679K, S gene_N764K, S gene_N969K, S gene_Q954H, and S gene_S373P.
Of the 82 mutations fixed, 41 occurred between February and June 2022, and 41 (10 of which appeared in first period, N_Q229K, ORF1a_A2710T, ORF1a_T4175I, ORF6_D61L, S_A264D, S_E484A, S_G446S, S_L216F, S_R21T, S_S939F) between January and June 2024 (Figure 2).
3.2. Lineages
From June 2022 to July 2024, 313 pangolin lineages were identified in Asturias, of which BA.5.1 (6.1%), DV.7.1 (5.8%) and BQ.1.1 (5.5%) accounted for over 5% of cases (see Supplementary File S4, Figure 3). In this period, diversity reached a maximum of 0.980. During the first period (June 2020–22), diversity was 0.929 and 111 lineages were identified.
From 2020 to 2024, 55 new circulating variants (NCVs) belonging to 41 pangolin lineages were detected (Supplementary Table S5). The distribution of the 91 mutations gained from these NCVs were 21 in NSP3 (ORF1a gene), 17 in S, 9 in ORF3a, 8 in NSP2 and 6 in NSP14 (Supplementary Table S5, Table 3).
Of these 55 NCVs, 24 were circulating throughout the world, two of them (BF.5+S_D80E+S_A701V+NSP1_G94C and JN.1.16.1+NSP3_S1428L+NSP5_K90R+NSP6_L37F+S_F59S) mainly in Asturias, during 59±16 (6-169) days; only variant XBB.1.5.8+NSP14_V328F in March 2023 belong to the same transmission clade in the region (Figure 4) . The other 31 originated in Asturias: 10 of them were only detected in the region during 92±20 (31-129) days, 8 also in Spain during 107±31 (43-154) days, and 13 around the world during 103±20 (44-184) days (Table 4).
Figure 3C shows the variants circulating in the region, both imported and indigenous, at the time.
3.3. Variants of Interest
Of the 82 non-synonymous mutations fixed in Asturias by 2024, 17 were used to define a variant of interest (Table 5).
Figure 5 show the number of fixed mutations found in each variant of interest in Asturias and the rest of the world (Supplementary File S4).
4. Discussion
Viruses constantly change through mutation and some changes allow the virus to spread more easily or make it resistant to treatments or vaccines. As the virus spreads, it may change and become harder to stop. The mutation rate may not be as high as one might think for the effects to be significant; the highest levels of adaptability occur at intermediate mutation rates, regardless of how rapidly the environment fluctuates [7]. Effective surveillance requires the sequencing of enough sequence data from representative populations to detect new variants and monitor trends in circulating variants. In 2022, the Spanish Ministry of Health took the lead in adapting the general strategy followed until now, going on to monitor the impact of the disease on vulnerable people, hospitalizations and deaths, and monitor changes that may generate a modification in the favorable trends that are being observed at this time[8]. Consensus genomic sequences of SARS-CoV-2 variants represent the genomes most frequently found in the clinical samples of patients, which have been widely used to monitor the global spread of the virus[9,10]. However, these genomes represent the most frequently observed viral genomes and do not capture the full extent of their diversity.
Currently, there are growing signs that adaptive evolution of SARS-CoV-2 has stalled, and purifying selection is the dominant evolutionary force acting on non-synonymous mutations in the Omicron lineage[11]. Because mutations at the third nucleotide of a codon often leads to no change in the amino acid sequence, the most frequent change occurs in the third position of the codon [12]. However, despite the fact that SARS-CoV-2 is an RNA virus with a high mutation rate, changes that are maintained only occur in 1% of the viral genome. As you might expect, these changes occur mostly in the S protein, although ORF1a gene mutations, protein involved in regulating the host response, are also notable [13,14].
In Asturias, we observed 157 mutations out of 5% of the viral population. This is a similar proportion to that in Spain and slightly higher if all the viral strains circulating in the world are taken into account where 116 mutations were observed. This is because the number of viral strains is greater, making it more difficult to obtain that 5% viral population for a given mutation.
If we compare the latter period with the start of the pandemic, it is striking that the estimated mutation rate is higher in the post-pandemic era. However, of the non-synonymous mutations that occurred, 31 were fixed in the post-pandemic period, compared to 51 in the first period. This is because, once the initial changes are in place, subsequent changes are more difficult to establish, especially if the first mutations have evolutionary advantages.
In a previous study and as reflected by other authors [3,5,15], mutations can emerge, giving rise to new variants, as the number of infected people increases. This second study provides additional information: it appears that the mutations are fixed within 18 months of the onset period, as illustrated in Figure 2. It will be interesting to see if the pattern of mutation fixation is maintained in 2026 and the number of established mutations continues to decrease due to purifying selection.
High vaccination rates and the universal use of masks were considered important factors in slowing the spread of the virus that prevented many variants of the virus from appearing. However, the diversity of the virus remains, being greater than during the pandemic. In post-pandemic era, until 313 lineages were identified in Asturias, but only four represent more than 5% of viral population. Furthermore, the gain of 91 mutations led to the classification of 55 new circulating variants according to the criteria for designation of a new Pango lineage (necessary but not sufficient).
As mentioned above, the most frequent mutations are in S and ORF1a genes. Of the four structural proteins, only the S protein (17 mutations,), which serves as the primary antigen targeted by the host immune response, accumulates changes in the new variants. In general, mutations in viral proteins, particularly in the S protein, can strongly impact viral infectivity, virulence and immunogenicity and therefore require continuous monitoring. The RBD, particularly the RBM, is the core part of the S protein that directly binds to ACE2 in host cells. Some mutations in the RBM (3 mutations,) can induce significant changes in SARS-CoV-2 phenotypes. Genetic mutations at the V445 influencing membrane fusion and entry into diverse target cells, mutations at the site F456 often resulted in resistance to class A antibodies and mutations at the F486 site escape the epitopes of class B antibodies [16,17].P1263L showed a significant increase in fusión [18] P1263Q. Most mutations that generate new variants occur in accessory proteins: ORF7a (3 mutations) can suppress the IFN-I response by inhibiting STAT2 and ORF8 (2 mutations,) downregulates the presentation of viral antigens via the class I major histocompatibility complex [19]; ORF10 (1 mutation) has exhibited the ability to suppress the IFN-I signaling pathway by interacting with mitochondrial antiviral signaling protein [19,20]; ORF3a (9 mutations) modifies crucial cellular processes such as apoptosis and autophagy [19,21]; Both ORF3a and ORF7a have been reported to induce the expression of inflammatory cytokines through the activation of NF-κB signaling [21]; and Nsp2 (8 mutations) is involved in disruption of signalling in host cells [22]. More surprising are the abundant mutations in NSP3 (21 mutations), which is involved in promoting RNA replication and transcription and cleavage of proteins involved in the host innate immune immunity [23,24].
The COVID-19 pandemic has shown a tendency to generate variants in specific geographic areas and cross-border transmission patterns [25,26,27,28]. Tourism, transportation routes, and global migration patterns have facilitated the introduction and dissemination of the virus across different regions [29]. The increase in incidence may be accompanied by the appearance of new variants originating in the region. New variants circulating around the world continually appear in our region, this makes them the ones that are detected for the longest time as they are more difficult to control, although none of them generate large outbreaks. In fact, the NCVs from Asturias are the majority. But over time, the absence of new local variants causes a rebound with imported variants. None of the predominant lineages in the epidemic remain for long, and only two are actively transmitted in the region. This indicates a constant turnover of lineages, each of which predominates for a couple of months at most and without causing any serious problems.
Although, most changes have little to no impact on the properties of the virus, this high mutation rate and the continuous appearance of NCVs require genomic surveillance, since some change can lead a more aggressive variant that can spread and predominate to the rest of the world. Therefore, lineages may retain mutations that became dominant in the global pandemic over time, which may have positive effects on the fitness of the virus and facilitate the emergence of new variants. The WHO has established a dedicated group to monitor the evolution of the virus. This group has been operational since June 2020, tracking SARS-CoV-2 variants [30].
The Omicron variant has taken hold worldwide and is currently the predominant. Increased infectivity and immune evasion and a substantially larger number of mutations compared to previous variants of concern (VOCs) characterize the Omicron variant [31]. Mutations originate from the previous variants, within the RBD of Omicron K417N and N501Y (Beta) are largely responsible for the failure of these monoclonal antibodies to neutralize Omicron S [32].Some of these mutations increased affinity for ACE2 (T478K -Delta-, N501Y), while others decreased ACE2 affinity (K417N) [31]. Other mutations fixed from previous variants of interest are: RDPD_ P323L (Alpha), required for polymerase activity and predicted to diminish antiviral drugs efficacy and S_D614G (Alpha) that increases infectivity [33,34]; S_H655Y from gamma, this variant may have reduced neutralization by monoclonal antibody therapies, convalescent sera, and post-vaccination sera [35], enhance viral growth and the S protein cleavage by furin [36] and governs entry through endosomes, as suggested by the significant increase in viral infectivity [37]. The N protein mutations, including P13L (Lambda), R203K and G204R/K (Alpha), may assist with increasing the transmission of the SARS-CoV-2 but also be associated with reduced severity of disease and, therefore, lower mortality rates when compared to individuals with the wild-type [38,39,40] P13L itself has been identified epidemiologically as the most important driver of fitness in N-protein [41] The mutation S_E484K present in beta has reappeared today, it escapes the neutralizing effect of several monoclonal antibodies, convalescent plasma, and post-vaccine sera [42]. The mutation ORF1a_T3255I (nsp4_ T492I) present in Lambda, increases the replication capacity and infectiveness of the virus and improves its ability to evade host immune responses [43].
The global SARS-CoV-2 pandemic is decreasing but the populations affected by the virus continue to appear. Geo-epidemiological investigations shed light on the transmission dynamics of the COVID-19 pandemic, elucidating how the virus spreads within and between communities, which serves to identify and contain chains of transmission [44,45]. Fortunately, all newly detected circulating variants appear to be extinct, but further examination of the spatial patterns and transmission dynamics of COVID-19 at various scales, from local communities to global populations, is necessary to implement localized lockdown measures to contain the spread, the day a really dangerous variant is generated
5. Conclusions
Although the global pandemic of SARS-CoV-2 is fading, populations affected by the virus continue to increase, and a continued focus on it is necessary. Continuous efforts to understand and adapt to the evolving viral landscape will be crucial in controlling the impact of COVID-19 on a global scale.
This study facilitates tracing the origins and sources of circulating SARSCoV-2 variants and the identification and comparative analysis of emerging variations within Asturias. It is possible that low-frequency lineages, together with circulating VOCs, may result in the next lineage that can further increase transmissibility, infectivity, and escape vaccine-induced or natural host immunity.
Mutations that increase SARS-CoV-2 transmission and may be associated with less disease severity are those that are becoming established in the virus population.
In addition to direct intervention measures, sustained monitoring of SARS-CoV-2 plays a crucial role in controlling variants. Continuous surveillance facilitates the early identification of variants that undergo substantial changes in their adaptability, which allows rapid adjustments to preventive measures and will contribute to strengthening preparedness for the next pandemic.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Asturian sequences uploaded to GISAID database until July 2024; Table S2: Mutations occurred in >5% of the viral population sequenced. amutations fixed in the world (>95% population); Table S3: Number of mutations fixed occurring in each gene in the Asturias strains during Covid (March 2020 to June 2022) and post-Covid period (July 2022 to July 2024);. Table S4: Number of pangolin lineages detected in Asturias from June 2022 to July 2024;. Table S5: New circulating variants from 2020 to 2024 in Asturias.
Author Contributions
Conceptualization, J.M.G.-A., Z.P.M., S.R.-A., J.G.d.O., M.E.Á.-A., C.O.V., M.R.P. and S.M.G.; Data curation, J.M.G.-A., Z.P.M., S.R.-A., J.G.d.O., M.E.Á.-A., C.O.V., M.R.P. and S.M.G.; Formal analysis J.M.G.-A., Z.P.M., S.R.-A., J.G.d.O., M.E.Á.-A., C.O.V., M.R.P. and S.M.G.; Writing—original draft J.M.G.-A., Z.P.M., S.R.-A., J.G.d.O., M.E.Á.-A., C.O.V., M.R.P. and S.M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
Data availability in GISAID(https:www.gisaid.org/).
Ethical Statement: This study was conducted in accordance with the Declaration of Helsinki, as revised in 2013. Approval from a Research Ethics Committee was not required, as Organic Law 3/2018, of 5 December, on the Data Protection and Guarantee of Digital Rights provides, with respect to the processing of health data, that the health authorities and public institutions with public health monitoring powers, may carry out scientific research without the data subject’s consent in situations of exceptional relevance and seriousness for public health.
Acknowledgments
European Commission/Carlos III Health Institute (Ministry of Science and Innovation) HaDEA RELECOV 2.0 EU4H-2022-DGA-MS-IBA-01-02 and the Ministry of Science, Business, Training and Employment of the Principality of Asturias (GRUPIN-24-IDE/2024/000719)
Conflicts of Interest
The authors declare no conflicts of interest
References
- Zhou, C.-M.; Qin, X.-R.; Yan, L.-N.; Jiang, Y.; Yu, X.-J. Global trends in COVID-19. Infect. Med. 2022, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Lino, A.; Cardoso, M.A.; Martins-Lopes, P.; Gonçalves, H.M.R. Omicron—The new SARS-CoV-2 challenge? Rev. Med. Virol. 2022, 32, e2358. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alba JM, Rojo-Alba S, Perez-Martinez Z, Boga JA, Alvarez-Arguelles ME, Gomez J, Herrero P, Costales I, Alba LM, Martin-Rodriguez G, Campo R, Castelló-Abietar C, Sandoval M, Abreu-Salinas F, Coto E, Rodriguez M, Rubianes P, Sanchez ML, Vazquez F, Antuña L, Álvarez V, Melón García S. Monitoring and tracking the spread of SARS-CoV-2 in Asturias, Spain. Access Microbiol. 2023 Sep 27;5(9):000573.v4. [CrossRef] [PubMed]
- Alba JMG, Pérez-Martínez Z, Boga JA, Rojo-Alba S, de Oña JG, Alvarez-Argüelles ME, Rodríguez GM, Gonzalez IC, González IH, Coto E, García SM. Emergence of New SARS-CoV2 Omicron Variants after the Change of Surveillance and Control Strategy. Microorganisms. 2022 Sep 30;10(10):1954. [CrossRef] [PubMed]
- Ministerio de Sanidad. Salud Publica. Available online: https://www.sanidad.gob.es/areas/alertasEmergenciasSanitarias/alertasActuales/nCov/variantesSARS-COV-2/home.htm (accessed on 1 July 2024 ).
- Peck, K.M. Laurin A.S. Complexities of viral mutation rates. J. Virol. 2018;92(14):1031–1037. [CrossRef]
- Ministerio de Sanidad. Salud Publica. Available online https://www.sanidad.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Nueva_estrategia_vigilancia_y_control.pdf.
- Colson, P. Bader, W., Fantini, J., Dudouet, P., Levasseur, A., Pontarotti, P., et al. (2023). From viral democratic genomes to viral wild bunch of quasispecies. J. Med. Virol. 95:e29209. [CrossRef]
- Messali, S. Rondina, A., Giovanetti, M., Bonfanti, C., Ciccozzi, M., Caruso, A., et al. (2023). Traceability of SARS-CoV-2 transmission through quasispecies analysis. J. Med. Virol. 95:e28848. [CrossRef]
- Liu, Y. Is SARS-CoV-2 facing constraints in its adaptive evolution? Biomol Biomed. 2025 Jun 9. [CrossRef]
- Bloom JD, Neher RA. Fitness effects of mutations to SARS-CoV-2 proteins. Virus Evol. 2023 Sep18;9(2):vead055. Erratum in: Virus Evol. 2024 Mar 26;10(1):veae026. [CrossRef] [PubMed]
- Maiti, AK. Progressive Evolutionary Dynamics of Gene-Specific ? Led to the Emergence of Novel SARS -CoV-2 Strains Having Super-Infectivity and Virulence with Vaccine Neutralization. Int J Mol Sci.2024 Jun 7;25(12):6306. [CrossRef] [PubMed]
- Emam M, Oweda M, Antunes A et al. Positive selection as a key player for SARS-CoV-2 pathogenicity: Insights into ORF1ab, S and E genes. Virus Res. 2021 Sep;302:198472. [CrossRef] [PubMed]
- Cocherie T, Zafilaza K, Leducq V et al. Epidemiology and Characteristics of SARS-CoV-2 Variants of Concern: The Impacts of the Spike Mutations. Microorganisms. 2022 Dec 22;11(1):30. [CrossRef] [PubMed]
- Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, Huang W, Li Q, Wang P, An R, Wang J, Wang Y, Niu X, Yang S, Liang H, Sun H, Li T, Yu Y, Cui Q, Liu S, Yang X, Du S, Zhang Z, Hao X, Shao F, Jin R, Wang X, Xiao J, Wang Y, Xie XS. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature. 2022 Feb;602(7898):657-663. [CrossRef] [PubMed]
- Yang S, Yu Y, Jian F, Song W, Yisimayi A, Chen X, Xu Y, Wang P, Wang J, Yu L, Niu X, Wang J, Xiao T, An R, Wang Y, Gu Q, Shao F, Jin R, Shen Z, Wang Y, Cao Y. Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86. Lancet Infect Dis. 2023 Nov;23(11):e457-e459. [CrossRef] [PubMed]
- Barrett CT, Neal HE, Edmonds K, Moncman CL, Thompson R, Branttie JM, Boggs KB, Wu CY, Leung DW, Dutch RE. Effect of clinical isolate or cleavage site mutations in the SARS-CoV-2 spike protein on protein stability, cleavage, and cell-cell fusion. J Biol Chem. 2021 Jul;297(1):100902. [CrossRef] [PubMed]
- Zandi M, Shafaati M, Kalantar-Neyestanaki D, Pourghadamyari H, Fani M, Soltani S, Kaleji H, Abbasi S. The role of SARS-CoV-2 accessory proteins in immune evasion. Biomed Pharmacother. 2022 Dec;156:113889. [CrossRef] [PubMed]
- Li X, Hou P, Ma W, Wang X, Wang H, Yu Z, Chang H, Wang T, Jin S, Wang X, Wang W, Zhao Y, Zhao Y, Xu C, Ma X, Gao Y, He H. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol Immunol. 2022 Jan;19(1):67-78. [CrossRef] [PubMed]
- Wong, L. R. and Perlman, S. (2022). Immune dysregulation and immunopathology induced by SARS-CoV -2 and related coronaviruses—are we our own worst enemy? Nat. Rev. Immunol. [CrossRef]
- Cornillez-Ty CT, Liao L, Yates JR 3rd, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol. 2009 Oct;83(19):10314-8. [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Moustaqil M, Ollivier E, Chiu HP, Van Tol S, Rudolffi-Soto P, Stevens C, Bhumkar A, Hunter DJB, Freiberg AN, Jacques D, Lee B, Sierecki E, Gambin Y. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg Microbes Infect. 2021 Dec;10(1):178-195. [CrossRef] [PubMed]
- Gohari K, Kazemnejad A, Sheidaei A, Hajari S. Clustering of countries according to the COVID-19incidence and mortality rates. BMC Public Health. (2022)22:632. [CrossRef]
- Darques R, Trottier J, Gaudin R, Ait-Mouheb N. Clustering and mapping the first COVID-19 outbreak in France. BMC Public Health. (2022)22:1279. [CrossRef]
- Arora P, Mrig S, Goldust Y, Kroumpouzos G, Karadağ AS, Rudnicka L, Galadari H, Szepietowski JC, Di Lernia V, Goren A, Kassir M, Goldust M. New Coronavirus (SARS-CoV-2) Crossing Borders Beyond Cities, Nations, and Continents: Impact of International Travel. Balkan Med J. 2021 Jul;38(4):205-211. [CrossRef] [PubMed]
- Xiang L,Ma S, Yu L,WangW, Yin Z.Modeling the global dynamic contagion of COVID-19. Front Public Health. (2022) 9:809987. [CrossRef]
- Findlater A, Bogoch II. Human mobility and the global spread of infectious diseases: a focus on air travel. Trends Parasitol. (2018) 34:772–83. [CrossRef]
- www.who.
- Parsons RJ, Acharya P. Evolution of the SARS-CoV-2 Omicron spike. Cell Rep. 2023 Dec 26;42(12):113444. [CrossRef] [PubMed]
- Syed AM, Ciling A, Khalid MM, Sreekumar B, Chen PY, Kumar GR, Silva I, Milbes B, Kojima N, Hess V, Shacreaw M, Lopez L, Brobeck M, Turner F, Spraggon L, Taha TY, Tabata T, Chen IP, Ott M, Doudna JA. Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. medRxiv [Preprint]. 2022 Jan 2:2021.12.20.21268048. Update in: Proc Natl Acad Sci U S A. 2022 Aug 2;119(31):e2200592119. [CrossRef] [PubMed]
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI; Sheffield COVID-19 Genomics Group; McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020 Aug 20;182(4):812-827.e19. [CrossRef] [PubMed]
- Hartley PD, Tillett RL, AuCoin DP, Sevinsky JR, Xu Y, Gorzalski A, Pandori M, Buttery E, Hansen H, Picker MA, Rossetto CC, Verma SC. Genomic surveillance of Nevada patients revealed prevalence of unique SARS-CoV-2 variants bearing mutations in the RdRp gene. J Genet Genomics. 2021 Jan 20;48(1):40-51. [CrossRef] [PubMed]
- Wang P, Casner RG, Nair MS, Wang M, Yu J, Cerutti G, Liu L, Kwong PD, Huang Y, Shapiro L, Ho DD. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021 ;29(5):747-751.e4. 12 May. [CrossRef] [PubMed]
- Minami S, Kotaki T, Sakai Y, Okamura S, Torii S, Ono C, Motooka D, Hamajima R, Nouda R, Nurdin JA, Yamasaki M, Kanai Y, Ebina H, Maeda Y, Okamoto T, Tachibana T, Matsuura Y, Kobayashi T. Vero cell-adapted SARS-CoV-2 strain shows increased viral growth through furin-mediated efficient spike cleavage. Microbiol Spectr. 2024 Apr 2;12(4):e0285923. [CrossRef] [PubMed]
- Qu P, Evans JP, Kurhade C, Zeng C, Zheng YM, Xu K, Shi PY, Xie X, Liu SL. Determinants and Mechanisms of the Low Fusogenicity and High Dependence on Endosomal Entry of Omicron Subvariants. mBio. 2023 Feb 28;14(1):e0317622. [CrossRef] [PubMed]
- Subramoney K, Mtileni N, Giandhari J, Naidoo Y, Ramphal Y, Pillay S, Ramphal U, Maharaj A, Tshiabuila D, Tegally H, Wilkinson E, de Oliveira T, Fielding BC, Treurnicht FK. Molecular Epidemiology of SARS-CoV-2 during Five COVID-19 Waves and the Significance of Low-Frequency Lineages. Viruses. 2023 ;15(5):1194. 18 May; Erratum in: Viruses. 2023 Jul 04;15(7):1502. [CrossRef] [PubMed]
- Wu H, Xing N, Meng K, Fu B, Xue W, Dong P, Tang W, Xiao Y, Liu G, Luo H, Zhu W, Lin X, Meng G, Zhu Z. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe. 2021 Dec 8;29(12):1788-1801.e6. [CrossRef] [PubMed]
- Alsuwairi FA, Alsaleh AN, Alsanea MS, Al-Qahtani AA, Obeid D, Almaghrabi RS, Alahideb BM, AlAbdulkareem MA, Mutabagani MS, Althawadi SI, Altamimi SA, Alshukairi AN, Alhamlan FS. Association of SARS-CoV-2 Nucleocapsid Protein Mutations with Patient Demographic and Clinical Characteristics during the Delta and Omicron Waves. Microorganisms. 2023 ;11(5):1288. 15 May. [CrossRef] [PubMed]
- Nguyen A, Zhao H, Myagmarsuren D, Srinivasan S, Wu D, Chen J, Piszczek G, Schuck P. Modulation of biophysical properties of nucleocapsid protein in the mutant spectrum of SARS-CoV-2. Elife. 2024 Jun 28;13:RP94836. [CrossRef] [PubMed]
- Yang WT, Huang WH, Liao TL, Hsiao TH, Chuang HN, Liu PY. SARS-CoV-2 E484K Mutation Narrative Review: Epidemiology, Immune Escape, Clinical Implications, and Future Considerations. Infect Drug Resist. 2022 Feb 3;15:373-385. [CrossRef] [PubMed]
- Lin X, Sha Z, Trimpert J, Kunec D, Jiang C, Xiong Y, Xu B, Zhu Z, Xue W, Wu H. The NSP4 T492I mutation increases SARS-CoV-2 infectivity by altering non-structural protein cleavage. Cell Host Microbe. 2023 Jul 12;31(7):1170-1184.e7. [CrossRef] [PubMed]
- Fang Y, Nie Y, PennyM. Transmission dynamics of the COVID-19 outbreak and effectiveness of government interventions: a data-driven analysis. J Med Virol. (2020) 92:645–59. [CrossRef]
- Brand SPC, Ojal J, Aziza R, Were V, Okiro EA, Kombe IK, Mburu C, Ogero M, Agweyu A, Warimwe GM, Nyagwange J, Karanja H, Gitonga JN, Mugo D, Uyoga S, Adetifa IMO, Scott JAG, Otieno E, Murunga N, Otiende M, Ochola-Oyier LI, Agoti CN, Githinji G, Kasera K, Amoth P, Mwangangi M, Aman R, Ng'ang'a W, Tsofa B, Bejon P, Keeling MJ, Nokes DJ, Barasa E. COVID-19 transmission dynamics underlying epidemic waves in Kenya. Science. 2021 Nov 19;374(6570):989-994. [CrossRef] [PubMed]
- Principio del formulario.
Figure 1.
Rate of main mutations circulating in Asturias that have been fixed ( ) and lost (
) and lost ( ) in the population in two period analyzed.
) in the population in two period analyzed.
) and lost () in the population in two period analyzed.
Figure 1.
Rate of main mutations circulating in Asturias that have been fixed () and lost () in the population in two period analyzed.
) and lost () in the population in two period analyzed.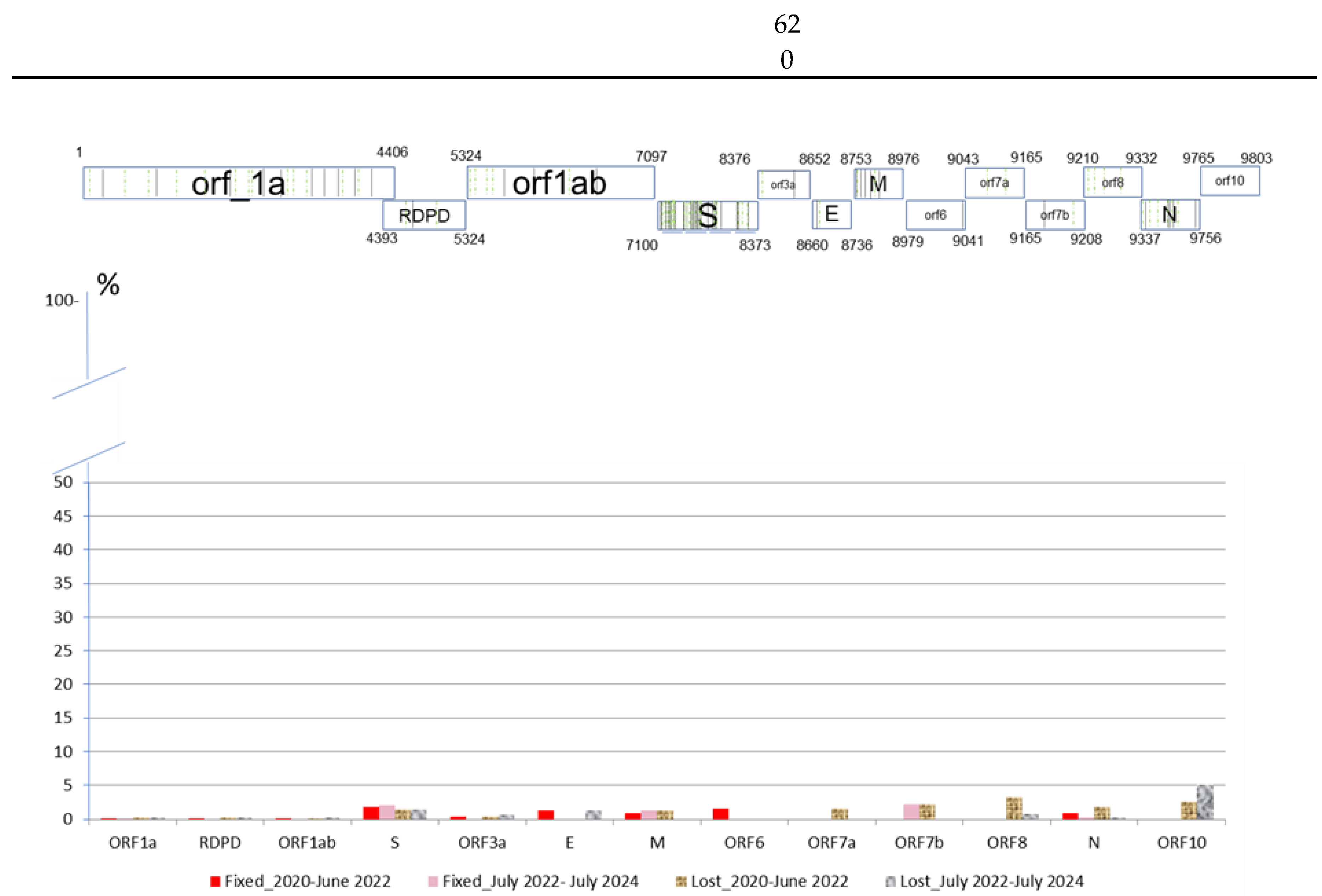
Figure 2.
Number of major SARS-COV2 mutations that occurred in the Asturian population, date of appearance and fixation.
Figure 2.
Number of major SARS-COV2 mutations that occurred in the Asturian population, date of appearance and fixation.
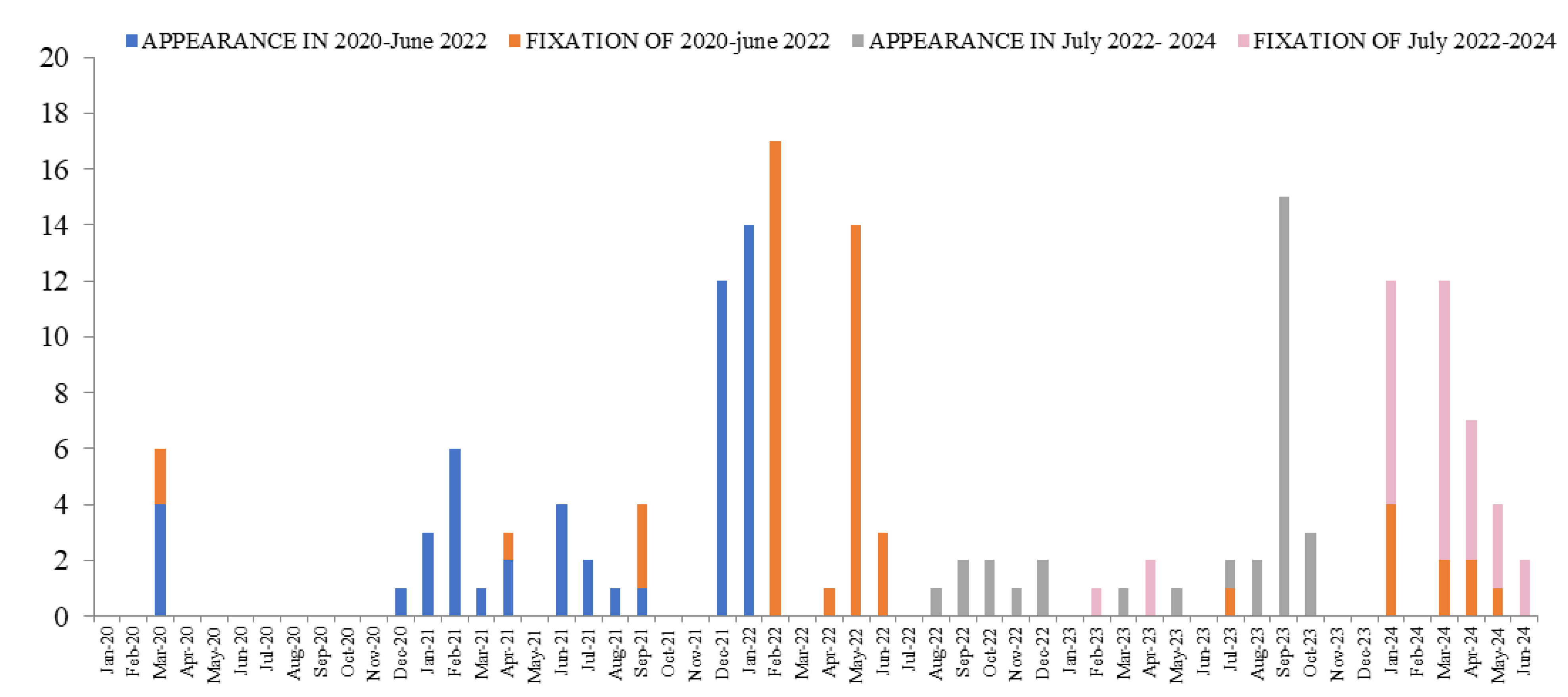
Figure 3.
Number of pangolin lineages detected in Asturias (A). Percentage of pangolin lineages mostly detected over time (>1%) (B).Number of new variants in circulation and incidence of SARS-Cov2 (C).
Figure 3.
Number of pangolin lineages detected in Asturias (A). Percentage of pangolin lineages mostly detected over time (>1%) (B).Number of new variants in circulation and incidence of SARS-Cov2 (C).
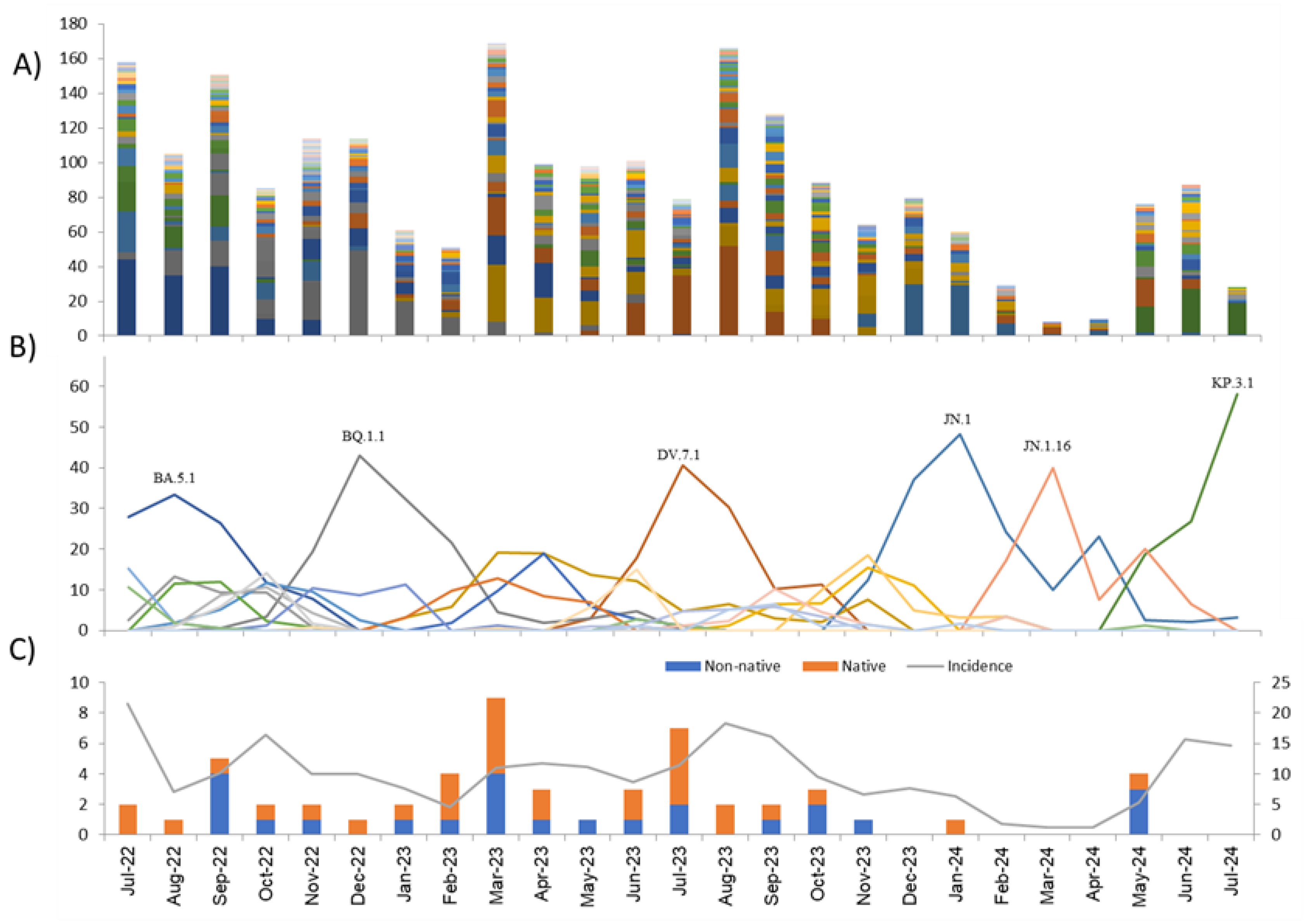
Figure 4.
Dated phylogeny of variant XBB.1.5.8+NSP14_V328F, the days until the date of the most recent sequence were used as the sampling date.
Figure 4.
Dated phylogeny of variant XBB.1.5.8+NSP14_V328F, the days until the date of the most recent sequence were used as the sampling date.
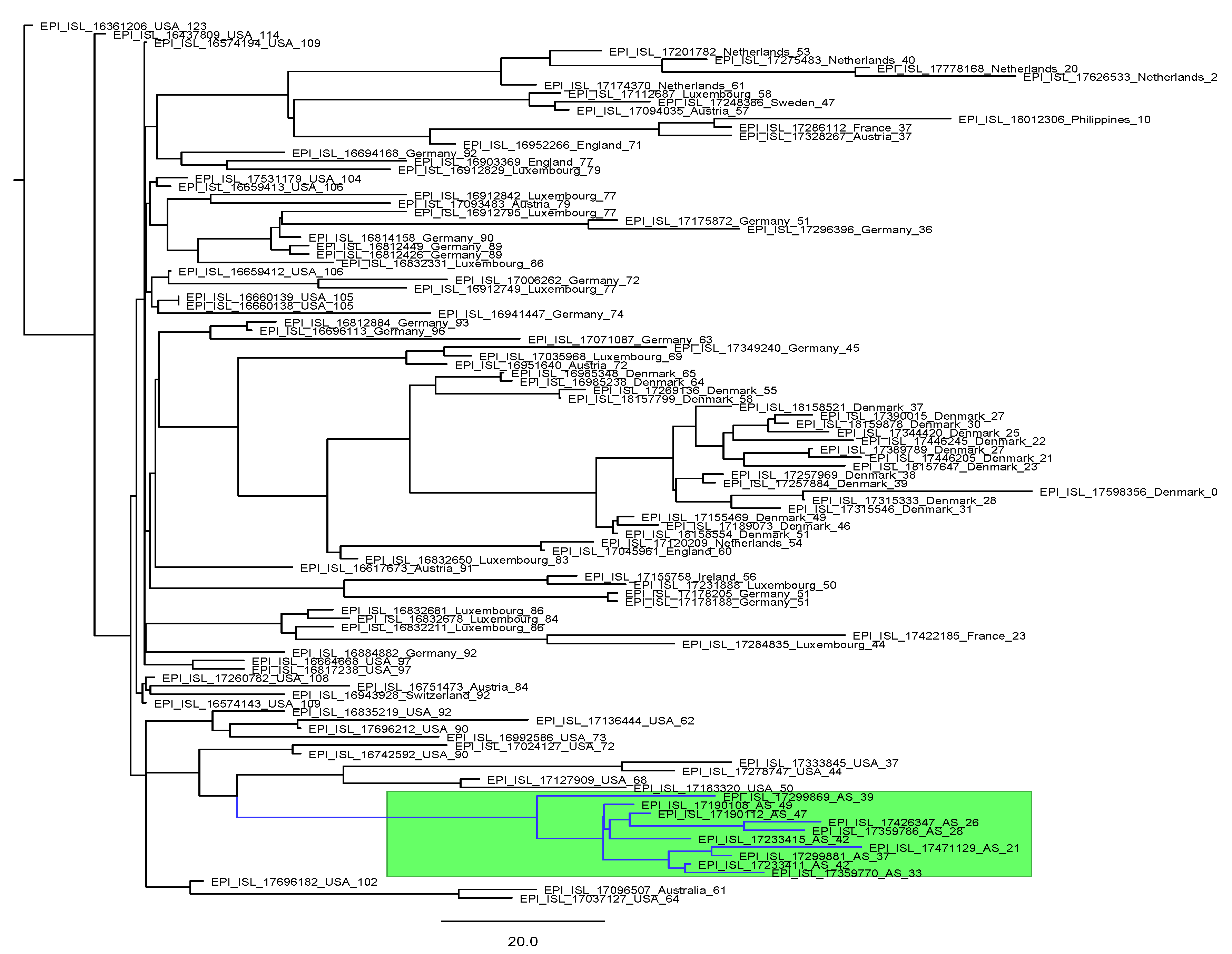
Figure 5.
Number of major mutations per gene currently fixed and the variants of interest in which they were initially found. a: In Asturias and in the world, b: in the world but not in Asturias, c: in Asturias but not in the world.
Figure 5.
Number of major mutations per gene currently fixed and the variants of interest in which they were initially found. a: In Asturias and in the world, b: in the world but not in Asturias, c: in Asturias but not in the world.
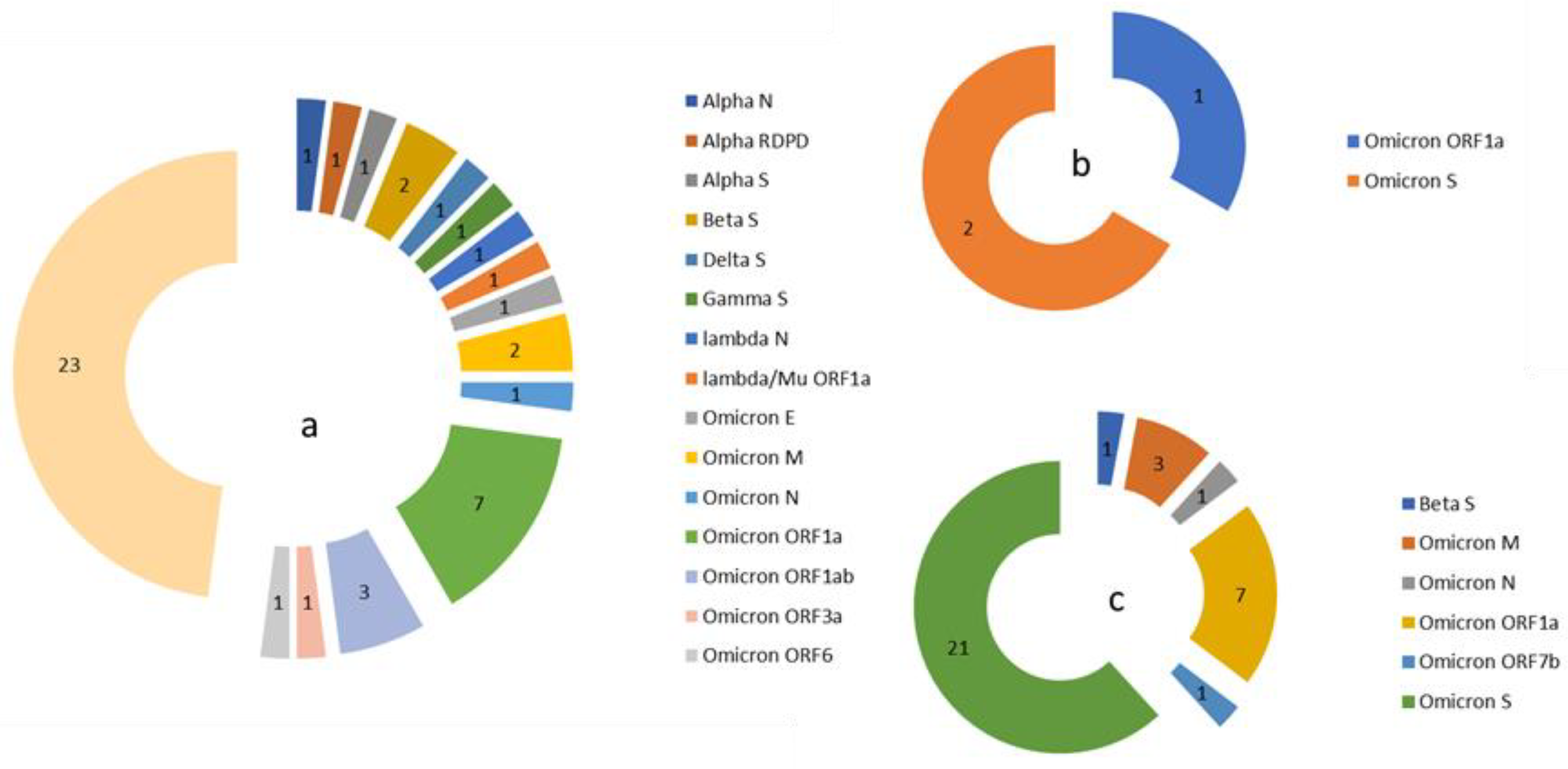

Table 1.
Number of mutations occurred in >5% of the viral population sequenced in GISAID in each gene between 2020 and July 2024.
Table 1.
Number of mutations occurred in >5% of the viral population sequenced in GISAID in each gene between 2020 and July 2024.
| ORF1a | RDPD | ORF1ab | S | ORF3a | E | M | ORF6 | ORF7a | ORF7b | ORF8 | N | ORF10 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GISAID | 21 | 2 | 6 | 59 | 2 | 2 | 5 | 1 | 2 | 1 | 4 | 11 | 116 | |
| SPAIN | 29 | 4 | 7 | 82 | 2 | 2 | 8 | 1 | 2 | 2 | 4 | 14 | 1 | 158 |
| ASTURIAS | 31 | 3 | 8 | 82 | 2 | 2 | 7 | 1 | 2 | 2 | 4 | 12 | 1 | 157 |
Table 2.
Number of mutations fixed occurring in each gene in the Asturias strains during Covid (March 2020 to June 2022) and post-Covid period (July 2022 to July 2024). Time is the average number of days it took to become fixed with its confidence interval.
Table 2.
Number of mutations fixed occurring in each gene in the Asturias strains during Covid (March 2020 to June 2022) and post-Covid period (July 2022 to July 2024). Time is the average number of days it took to become fixed with its confidence interval.
| Mutations | ORF1a | RDPD | ORF1ab | S | ORF3a | E | M | ORF6 | ORF7a | ORF7b | ORF8 | N | ORF10 | Total | |
| 2020-22 | Occurred | 21 | 3 | 5 | 47 | 2 | 1 | 5 | 1 | 2 | 1 | 4 | 12 | 1 | 105 |
| Fixed | 8 | 1 | 3 | 29 | 1 | 1 | 2 | 1 | 5 | 51 | |||||
|
Days or Mean± CI |
62/124(x4)/217/775 930 | 0 | 62 124 124 |
358±132 (0-1209) |
496 | 372 | 62 62 |
868 | 124/310 403/713961 |
344±94 (0-1209) |
|||||
| 2022-24 | Ocsurred | 20 | 3 | 6 | 47 | 2 | 1 | 3 | 1 | 1 | 2 | 2 | 88 | ||
| Fixed | 6 | 21 | 3 | 1 | 31 | ||||||||||
|
Days or Mean± CI |
93/124(x2) 217(x3) | 257±53 (124-527) |
248 278 620 |
217 | 250±46 (93-620) |
||||||||||
Table 3.
Mutation gained by new circulting variants in Asturias.
| ORF_1a | RDPD | ORF_1ab | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | NSP6 | NSP8 | NSP9 | NSP10 | NSP12 | NSP13 | NSP14 | NSP15 | NSP16 | S | ORF3a | E | ORF7a | ORF8 | N | ORF10 |
| G94C | A357S | D112N | A128V | K90R | F235L | P10S | G38V | T115I | A4774V | V157L | A320T | D36G | K160R | A67V | D155Y | T9V | L5F | D119Y | P151S | I27T |
| P6S | E574D | D1764G | V13I | V35L | L37F | T21I | I4563M | I15T | P205S | P215L | A701V | D27H | Q94L | P38S | ||||||
| V121A | G265S | E119K | M5021V | N71S | A845S | L140F | T28I | |||||||||||||
| G285S | E387D | S4621N | P203L | D253G | M260K | |||||||||||||||
| L24F | I541V | Q22H | D80E | Q185H | ||||||||||||||||
| N254S | M988L | V328F | F456L | Q57H | ||||||||||||||||
| S591I | N1322S | F486V | S171L | |||||||||||||||||
| T103I | N1680K | F59S | T270I | |||||||||||||||||
| P153L | G252V | V273L | ||||||||||||||||||
| Q167R | L249F | |||||||||||||||||||
| R1297G | N354K | |||||||||||||||||||
| S126L | P1263Q | |||||||||||||||||||
| S1428L | Q675H | |||||||||||||||||||
| S454G | T547I | |||||||||||||||||||
| T1203I | T572I | |||||||||||||||||||
| T424N | V1264L | |||||||||||||||||||
| T720I | V445P | |||||||||||||||||||
| T970M | ||||||||||||||||||||
| V1385I | ||||||||||||||||||||
| V1673I | ||||||||||||||||||||
| Y1535H | ||||||||||||||||||||
Table 4.
Asturian new circulating variants between 2020 and 2024.
| NVC | N(%Total identified) | First detection date | Last detection date | Time of detection (days) |
|---|---|---|---|---|
| Frst detected in the world | ||||
| BA.5.2.1 + NSP3_S454G | 6 (43) | 17/09/2022 | 22/10/2022 | 35 |
| BA.5.2.6 + NSP3_M988L | 7 (3) | 22/09/2022 | 06/11/2022 | 45 |
| BF.5 + S_D80E + S_A701V + NSP1_G94C | 8 (62) | 22/09/2022 | 08/11/2022 | 47 |
| BA.5.1 + NSP12_M5021V | 5 (7) | 24/09/2022 | 16/11/2022 | 53 |
| BF.7 + NSP14_P203L | 13 (29) | 19/10/2022 | 07/12/2022 | 49 |
| BQ.1.1.18 + S_T547I | 5 (28) | 16/11/2022 | 11/01/2023 | 56 |
| XBB.1.5 + NSP3_T1203I | 12 (6) | 24/01/2023 | 01/05/2023 | 97 |
| FL.5 + NSP14_Q22H + NSP3_P153L | 6 (2) | 15/02/2023 | 24/05/2023 | 98 |
| XBB.1.5.8 + NSP14_V328F | 10 (10) | 01/03/2023 | 29/03/2023 | 28 |
| BQ.1.18 + NSP14_I15T | 20 (17) | 04/03/2023 | 29/06/2023 | 117 |
| XBB.1.5 + NSP2_S591I + NSP3_N1322S | 12 (1) | 04/03/2023 | 20/08/2023 | 169 |
| XBB.1.5 + NSP5_V35L | 6 (1) | 09/03/2023 | 07/07/2023 | 120 |
| XBB.1.5.71 + NSP1_P6S + ORF3a_Q57H + NSP12_I4563M | 5 (4) | 28/04/2023 | 13/06/2023 | 46 |
| XBB.1.5.71 + NSP1_P6S + NSP12_I4563M | 6 (2) | 12/05/2023 | 03/06/2023 | 22 |
| EG.5.1 + ORF3a_D27H | 5 (1) | 20/06/2023 | 24/08/2023 | 65 |
| XBB.1.16.11 + NSP2_N254S | 6 (1) | 20/07/2023 | 16/10/2023 | 88 |
| DV.7.1 + NSP3_E119K | 6 (18) | 26/07/2023 | 08/09/2023 | 44 |
| EG.5.1.3 + NSP3_I541V | 5 (42) | 05/09/2023 | 14/09/2023 | 9 |
| JD.1.1 + ORF3a_M260K | 5 (2) | 13/10/2023 | 28/11/2023 | 46 |
| JG.3 + ORF3a_S171L | 7 (14) | 27/10/2023 | 04/12/2023 | 38 |
| JN.1.31 + S_T572I | 6 (23) | 29/11/2023 | 13/03/2024 | 105 |
| JN.1.16 + S_A67V + S_L249F + S_V445P | 6 (10) | 07/05/2024 | 26/05/2024 | 19 |
| JN.1.32 + S_F456L | 7 (1) | 12/05/2024 | 06/06/2024 | 25 |
| JN.1.16.1 + NSP3_S1428L + NSP5_K90R + NSP6_L37F + S_F59S | 5 (83) | 13/05/2024 | 19/05/2024 | 6 |
| First detected in Asturias | ||||
| BA.4.6 + NSP2_G265S + NSP3_D112N + ORF7a_Q94L | 7 (100)** | 10/06/2022 | 17/10/2022 | 129 |
| CH.1.1.28 + NSP16_K160R | 14 (100)** | 22/12/2022 | 22/03/2023 | 90 |
| XBB.1.5 + NSP3_T970M + NSP12_A4774V | 6 (100)** | 27/01/2023 | 13/04/2023 | 76 |
| XBB.1.5.77 + S_G252V + NSP2_E574D + ORF7a_L5F + ORF7a_T28I | 6 (100)** | 14/02/2023 | 07/04/2023 | 52 |
| XBB.1.5.1 + NSP15_D36G + NSP3_S126LL + NSP1_V121A + ORF8_P38S | 7 (100)** | 03/04/2023 | 04/05/2023 | 31 |
| XBB.2.3 + E_T9V + NSP3_N1680K + NSP3_Y1535H + ORF10_I27T | 5 (100)** | 16/04/2023 | 11/08/2023 | 117 |
| EG.1.4 + NSP12_S4621N + S_A845S | 6 (100)** | 09/05/2023 | 03/08/2023 | 86 |
| DV.7.1 + NSP3_R1297G + ORF3a_Q185H | 6 (100)** | 21/05/2023 | 31/08/2023 | 102 |
| JG.3 + NSP3_V1385I | 8 (100)** | 04/10/2023 | 22/01/2024 | 110 |
| JN.1.16.2 + S_A67V + S_V445P + S_L249F | 8 (100)** | 24/01/2024 | 25/05/2024 | 122 |
| BQ.1.1.15 + NSP3_T1203I | 6 (55)* | 26/07/2022 | 27/12/2022 | 154 |
| BQ.1.1.66 + NSP3_E387D + NSP9_G38V + NSP10_T115II | 10 (83)* | 23/11/2022 | 15/03/2023 | 112 |
| XBB.1.5.37 + NSP9_T21I | 7 (78)* | 15/12/2022 | 23/04/2023 | 129 |
| XBB.2.3.13 + NSP3_Q167R | 6 (60)* | 28/02/2023 | 31/07/2023 | 153 |
| XBB.2.3.13 + NSP2_A357S + NSP6_F235L + S_A701V | 13 (93)* | 12/03/2023 | 24/04/2023 | 43 |
| XBB.1.5.71 + NSP15_P205S + NSP2_L24F | 5 (83)* | 10/05/2023 | 27/09/2023 | 140 |
| BA.4.1 + NSP13_V157L | 7 (88)* | 09/06/2022 | 15/08/2022 | 67 |
| BE.1 + ORF3a_T270I + ORF3a_V273L | 6 (86)* | 10/07/2022 | 08/09/2022 | 60 |
| BF.7 + NSP4_A128V + S_Q675H + S_P1263Q | 10 (19) | 29/06/2022 | 01/12/2022 | 155 |
| CK.2.1.1 + S_V1264L + ORF8_D119Y | 5 (63) | 03/10/2022 | 21/12/2022 | 79 |
| EL.1 + NSP2_T103I + NSP3_T720I | 35 (90) | 16/11/2022 | 19/05/2023 | 184 |
| EF.1.2 + S_D253G + NSP4_V13I + ORF3a_D155Y | 10 (67) | 20/12/2022 | 16/03/2023 | 86 |
| XBB.2.3.13 + NSP2_A357S | 12 (16) | 05/02/2023 | 09/06/2023 | 124 |
| EL.1 + NSP14_A320T + NSP2_G265S + NSP3_T970M | 6 (75) | 14/02/2023 | 19/05/2023 | 94 |
| DV.7.1 + NSP14_N71S | 8 (57) | 28/04/2023 | 02/09/2023 | 127 |
| XBB.1.5 + NSP3_V1673I + ORF3a_L140F | 13 (81) | 08/06/2023 | 19/09/2023 | 103 |
| EG.6.1 + NSP3_S1428L | 11 (79) | 16/06/2023 | 02/10/2023 | 108 |
| DV.7.1 + NSP8_P10S + NSP3_D1764G | 5 (16) | 23/06/2023 | 10/09/2023 | 79 |
| EG.5.1.5 + S_N354K + N_P151S | 12 (46) | 13/07/2023 | 26/08/2023 | 44 |
| HV.1 + NSP14_P203L | 6 (29) | 05/08/2023 | 13/10/2023 | 69 |
| JN.1 + S_F486V | 6 (75) | 29/10/2023 | 25/01/2024 | 88 |
**detected only in Asturias; *detected also in Spain.
Table 5.
Mutations fixed in Asturias used to define the genetic characteristics of SARS-CoV-2 variants of interest by the WHO.
Table 5.
Mutations fixed in Asturias used to define the genetic characteristics of SARS-CoV-2 variants of interest by the WHO.
| Variant Mutation |
Alpha | Beta | Gamma | Delta | Zeta | Eta | Theta | Iota | Kappa | Lambda | Mu | Epsilon | Omicron |
| E_T9I | X | X | X | X | |||||||||
| M_A63T | X | ||||||||||||
| N_G204R | X | X | X | X | X | X | |||||||
| N_P13L | X | X | |||||||||||
| N_R203K | X | X | X | X | X | X | |||||||
| ORF1a_T3255I | X | X | X | ||||||||||
| RDPD_P323L | X | X | X | X | X | X | X | X | X | X | X | X | X |
| S_D614G | X | X | X | X | X | X | X | X | X | X | X | X | X |
| S_E484K | X | X | X | X | X | X | |||||||
| S_H655Y | X | X | |||||||||||
| S_K417N | X | ||||||||||||
| S_N501Y | X | X | X | X | |||||||||
| S_N679K | X | ||||||||||||
| S_P681R | X | X | |||||||||||
| S_Q954H | X | ||||||||||||
| S_S373P | X | ||||||||||||
| S_T478K | X | X |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



