Submitted:
23 July 2025
Posted:
23 July 2025
You are already at the latest version
Abstract
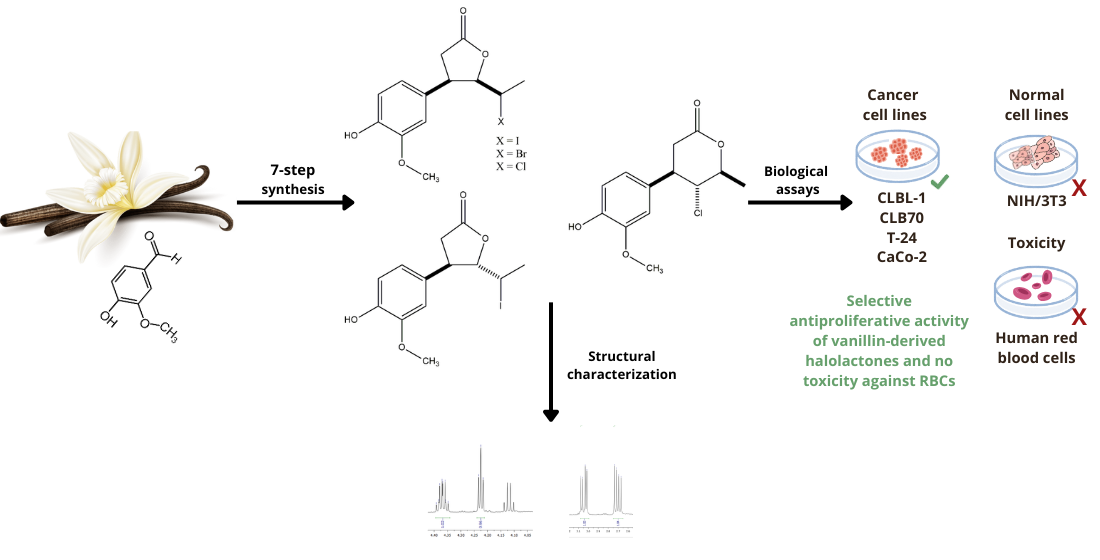
A series of novel γ-halo-δ-lactones and δ-halo-γ-lactones bearing a phenolic ring at the β-position was synthesized from the naturally occurring aromatic aldehyde vanillin. The seven-step synthetic route commenced with benzyl protection of the hydroxy group of the starting material, followed by the synthesis of a γ,δ-unsaturated carbox-ylic acid, subsequent halolactonization (iodo-, bromo-, and chlorolactonization), and concluded with selective benzyl deprotection. The structures of all synthesized com-pounds were confirmed by comprehensive spectroscopic analyses, including NMR and HRMS. The resulting halolactones were evaluated for antiproliferative activity against two canine (CLBL-1, CLB70) and two human (T-24, CaCo-2) cancer cell lines, with non-cancerous mouse embryonic fibroblasts (NIH/3T3) serving as controls. Hemolytic assays were performed to assess toxicity toward human red blood cells (RBCs). All compounds demonstrated selective inhibition of cancer cell proliferation, exhibiting no inhibitory effects on normal fibroblasts and no hemolytic toxicity. Among the tested lactones, the trans δ-iodo-γ-lactone was the most potent, particularly against CLBL-1 and T-24 cells. These findings underscore the potential of vanillin-derived halolactones as selective anticancer agents with an advantageous safety profile, owing to their lack of toxicity toward non-malignant cells.
Keywords:
vanillin
; lactones
; aromatic ring
; halolactonization
; antiproliferative activity
; hemolytic activity
1. Introduction
Lactones are intramolecular esters of hydroxy acids, found in both natural and synthetic sources. Five- and six-membered lactone rings predominate in nature due to their favorable thermodynamic stability [1]. Natural compounds containing lactone rings are produced mainly by plants [2,3,4], but also by insects [5], microorganisms [6], and marine organisms [7,8], where they function as secondary metabolites.
This structurally diverse group of compounds exhibits a wide range of biological activities, including antibacterial [9,10,11], anti-inflammatory [12,13], antifeedant [14,15,16], and antifungal properties [17,18,19]. Due to their broad spectrum of bioactivities, lactones are applied in economically important sectors such as the food industry, medicine, and agriculture. Their multifunctional nature makes them attractive candidates for the development of bioactive agents and functional materials.
However, particular attention has been paid to their anticancer properties [20,21,22,23]. Numerous studies have been conducted to demonstrate the cytotoxic activity of lactones containing an aromatic substituent against cancer or normal cell lines [24,25,26,27,28,29]. Selected β-aryl-δ-halo-γ-lactones synthesized from simple aromatic aldehydes exhibit cytotoxicity against human (e.g., Jurkat, HL-60, AGS) and canine (e.g., D17, GL-1, CLBL-1) cancer cell lines [30,31,32,33]. The mechanism of action of these compounds involves the induction of apoptosis via a mitochondrial-dependent pathway and caspase activation, as demonstrated for cuminaldehyde-derived δ-iodo-γ-lactones [34]. Enantiomeric trans β-aryl-δ-iodo-γ-lactones derived from 2,5-dimethylbenzaldehyde were shown to trigger cancer cell apoptosis by downregulating the anti-apoptotic proteins Bcl-2 and Bcl-xL. In the canine cell lines used in the study, the tested compounds also activated the receptor-mediated apoptotic pathway through fragmentation of the Bid protein, thereby enhancing their pro-apoptotic effects [35]. To gain deeper insight into their biophysical behavior and anticancer mechanisms, enantiomeric piperonal-derived trans β-aryl-δ-iodo-γ-lactones have been extensively studied for their ability to induce apoptosis and interact with biological systems, including cancer cell membranes and biomacromolecules. Membrane interaction studies revealed a reduction in membrane fluidity, correlating with a marked induction of apoptosis by these lactones [36]. Cis β-aryl-δ-iodo-γ-lactones containing phenyl or p-alkyl-substituted phenyl rings exhibited cytotoxic activity against HeLa (human cervix carcinoma) and MCF-7 (human breast adenocarcinoma) cancer cell lines, and significantly disrupted the antioxidative/oxidative balance in the NHDF (normal human dermal fibroblasts) cell line [37].
Continuing our studies on the biological activity of lactones bearing an aromatic ring, we designed and synthesized a series of novel β-arylhalolactones using vanillin (1) as the starting material. This naturally occurring aromatic aldehyde has a well-documented spectrum of biological properties, including gastro- and cardioprotective, diuretic, anti-organ toxicity, antimicrobial, anti-inflammatory, neuroprotective, and anti-infective effects, among others [38,39,40]. Numerous research groups have also reported significant anticancer activity of vanillin (1) against various human cancer cell lines, like MCF-7 (breast cancer) [41], HepG2 (hepatocellular carcinoma) [42], HT-29 (colorectal adenocarcinoma) [43], NCI-H460 (non-small cell lung carcinoma) [44], as well as A2058 (malignant melanoma) cell lines [45].
Due to the presence of various reactive functional groups, vanillin (1) is also a versatile building block for the synthesis of novel derivatives, including a wide range of heterocyclic systems such as pyrimidines, quinoxalines, imidazoles, and thiazoles, which are highly relevant in medicinal and pharmaceutical chemistry [46]. Vanillin-derived compounds have demonstrated a broad spectrum of pharmacological activities. Notably, hydrazone derivatives of vanillin (1) exhibit antimicrobial, anticancer, and enzyme inhibition properties, making them promising candidates for therapeutic applications [47]. Additionally, derivatives bearing tacrine or naphthalimido moieties have been developed as multi-target agents for Alzheimer’s disease therapy. These compounds act as antioxidants and inhibit acetylcholinesterase (AChE) as well as β-amyloid peptide aggregation, both of which are key pathological features of Alzheimer’s disease [48]. Other vanillin-based derivatives have been designed as dual inhibitors targeting both AChE and butyrylcholinesterase (BuChE), thereby enhancing their potential efficacy in neurodegenerative conditions [49]. Regarding anticancer activity, certain vanillin derivatives-such as compounds based on the SBE13 scaffold significantly outperform the parent compound by potently inducing apoptosis and reducing Plk1 kinase activity in HeLa cells, indicating strong antiproliferative effects [50]. This further emphasizes the potential of vanillin as a versatile scaffold for designing targeted anticancer therapeutics.
The presence of phenolic fragment is an advantageous factor to influence the potential of vanillin-derived halolactones as a promising bioactive compound with different therapeutic properties including cytotoxic, antioxidant, antiinflammatory which can be of high relevance in oncology. Here we would like to present the synthesis and studies on the cytotoxicity of new halolactones obtained from vanillin (1) towards different cancer cell lines. Due to the frequently observed high toxicity of anticancer agents towards healthy cells, we also decided to investigate the hemolytic properties of the synthesized halolactones towards human red blood cells (RBCs). Owing to their simple structure and crucial physiological functions, these cells are a simple and well established model to evaluate general hemolytic potential, as erythrocyte membranes are highly sensitive to chemical induced damage and serve as a convenient indicator of membrane disruptive properties of bioactive compounds.
2. Results and Discussion
2.1. Synthesis of Vanillin-Derived Halolactones
The presence of a reactive phenolic group in the benzene ring of vanillin (1) dictated our synthetic strategy, which included the benzyl protection of vanillin (1), the synthesis of the key intermediate γ,δ-unsaturated carboxylic acid 6, and subsequent halolactonization reactions, followed by a final deprotection step to obtain the target lactones. The use of benzyl protection was essential to prevent side reactions involving the phenolic OH group during the multistep synthesis.
2.1.1. Synthesis of α,β-Unsaturated Carboxylic Acid 6
The five-step synthesis of γ,δ-unsaturated carboxylic acid 6 is shown on Scheme 1.
Benzylation of vanillin (1) was carried out using an ethanolic solution of benzyl bromide in the presence of potassium carbonate. Benzylvanillin 2 was obtained in an 88% yield after crystallization from ethanol. The presence of the benzyl group was confirmed by the 1H NMR spectrum, which showed multiplets in the range of 7.32–7.47 ppm corresponding to the aromatic protons, and a distinctive singlet at 5.21 ppm attributed to the benzylic protons of the methylene group. These characteristic signals provided clear evidence of successful benzyl protection, an essential step for subsequent synthetic transformations.
In the next step, benzylvanillin 2 was converted to the α,β-unsaturated ketone 3 in high yield (84%) via Claisen–Schmidt condensation with acetone under alkaline conditions. The coupling constant between the olefinic protons observed in the 1H NMR spectrum (J = 16.2 Hz) confirmed the E-configuration of the C3–C4 double bond.
Reduction of ketone (3) by sodium borohydride afforded exclusively the corresponding allylic alcohol 4. Spectroscopic data confirmed the selective reduction of the carbonyl group, indicated by a multiplet at 4.47 ppm from H-2 and a singlet corresponding to the hydroxyl group at 1.64 ppm, as well as a strong band at 3384 cm⁻¹ attributed to O–H stretching vibrations. The retention of the E-configured double bond was confirmed by the coupling constant between the H-3 and H-4 protons (J = 15.8 Hz).
Alcohol 4 was subjected to the Johnson–Claisen rearrangement using triethyl orthoacetate in the presence of a catalytic amount of propionic acid at 138 °C. Consistently with the mechanism of Johnson - Claisen rearrangement, E configuration of double bond in γ,δ-unsaturated ester 5 was retained, as indicated by the coupling constant (J = 15.3 Hz) between olefinic protons H-4 and H-5. Ester 5, after two-step purification (first by gravity chromatography and then by flash chromatography) was hydrolyzed in a 10% ethanolic NaOH solution under reflux to afford the γ,δ-unsaturated carboxylic acid 6.
2.1.2. Halolactonizations of γ,δ-unsaturated Carboxylic Acid 6
Acid 6 was subjected to halolactonization reactions, leading to the formation of iodo-, bromo-, and chlorolactones as illustrated on Scheme 2.
The iodolactonization reaction was carried out using iodine in potassium iodide in a biphasic system Et2O/NaHCO₃. After reaction, three products were successfully isolated and purified by flash chromatography.
The major isolated products were two γ-lactones obtained as a result of 5-exo cyclization. Spectroscopic analysis (1H NMR) revealed significant differences in the chemical shifts and multiplicities of the H-4, H-5, and H-6 protons, enabling confirmation of the relative orientation of substituents at C-4 and C-5 of the γ-lactone ring. For example, in the case of the cis isomer 7a, the signal from the H-6 proton was significantly shifted upfield (3.47 ppm) compared to its chemical shift in the trans isomer 7b (4.36 ppm), which resulted from the location of H-6 within the shielding cone of the aromatic ring. The shape of the multiplet (doublet of quartets) and the coupling constant between H-6 and H-5 (J = 10.9 Hz) indicated an antiperiplanar orientation of the C5–H5 and C6–H6 bonds. For the trans δ-iodo-γ-lactone 7b, a characteristic triplet at 4.23 ppm from H-5 (J = 5.3 Hz) was observed instead of the doublet of doublets at 4.78 ppm found in the spectrum of the cis isomer.
In the case of the trans δ-iodo-γ-lactone, additional minor signals were observed in the 1H and 13C NMR spectra, resulting from a conformational equilibrium between two conformers: a predominant syn conformer in which the C–I and C–O bonds adopt a “gauche” conformation, and a minor anti conformer where these bonds are arranged in an antiperiplanar orientation. This phenomenon was previously reported and elucidated based on crystallographic analysis by Gładkowski et al. (2013) for analogs of lactone 7b, namely trans δ-iodo-γ-lactones bearing an unsubstituted phenyl ring at the β-position [31].
In the case of the minor isolated product of 5-endo iodolactonization, γ-iodo-δ-lactone 7c, the most informative signal was the triplet from the H-5 proton at 4.00 ppm. The coupling constant (J = 10.6 Hz) between the H-4 and H-6 protons observed in the 1H NMR spectrum unequivocally indicated a trans orientation of the iodine at C-5 relative to both the benzene ring at C-4 and the methyl group at C-6.
Bromo- and chlorolactonization of acid 6 were carried out using N-bromosuccinimide (NBS) and N-chlorosuccinimide (NCS), respectively, in anhydrous tetrahydrofuran with a catalytic amount of acetic acid. Both cis δ-bromo-γ-lactone 8a and cis δ-chloro-γ-lactone 9a, as well as their corresponding six-membered analogues 8b, 9b, were isolated from the product mixtures. However, attempts to isolate the pure stereoisomers of the trans γ-lactones were unsuccessful.
1H NMR data showed a high degree of resemblance in the shapes and coupling constant values of the signals from protons H-4, H-5, and H-6 compared to the corresponding signals observed in the spectra of the analogous iodolactones 7a and 7c. Among the halogens, the greatest deshielding effect for protons H-5 and H-6 was observed for iodine, followed by bromine and chlorine, which is consistent with literature data for analogous β-aryl-δ-halo-γ-lactones and β-aryl-γ-halo-δ-lactones [31].
2.1.3. Benzyl Deprotection of Halolactones 7a-c, 8a,b, 9a,b
The final stage of the synthesis involved the removal of the benzyl protecting group. To identify a selective and efficient method, various deprotection strategies were tested using cis δ-iodo-γ-lactone 7a as a model substrate. The first approach, catalytic hydrogenolysis over Pd/C, was chosen due to its general reliability and operational simplicity in benzyl group removal [51]. However, in this case, the reaction led to a mixture of more polar products than the starting iodolactone 7a. In search of greater selectivity, 7a was treated with 33% HBr in acetic acid [52], but the reaction yielded a complex mixture, and GC and NMR analysis indicated isomerization of 7a to its trans isomer 7b. The resulting products were inseparable by standard purification techniques. Similarly, treatment with concentrated HCl under reflux [53] resulted in a mixture of highly polar products. The use of aqueous NaBrO₃ and Na₂S₂O₃ solutions [54] also produced a mixture of more and less polar compounds relative to 7a, that were difficult to separate. Ultimately, the only effective deprotection method involved the use of anhydrous FeCl₃ in dry dichloromethane at room temperature under a nitrogen atmosphere [55]. This approach enabled successful removal of the benzyl group, though the initial isolated yield of the final lactone 10a after flash chromatography was only 10%, likely due to complexation of the phenolic product with Fe³⁺ ions. To improve the yield, during the work-up procedure reaction mixture was washed with 1% phosphoric(V) acid, following the protocol described by Giri et al. (2020) for FeCl₃-mediated deprotection of Boc-protected amino acids and peptides [56]. This modification increased the isolated yield of 10a to 28%, significantly enhancing the efficiency of the deprotection process.
Following this procedure, successful debenzylation of cis δ-halo-γ-lactones 7a-9a and trans δ-iodo-γ-lactone 7b was carried out, affording the corresponding deprotected lactones 10a-12a and 10b, respectively (Scheme 3). The removal of the benzyl group was confirmed by the disappearance of the aromatic proton signals in the range of 7.31–7.47 ppm and the singlet at 5.14–5.15 ppm corresponding to the benzylic methylene protons in the 1H NMR spectra. In place of these, a new singlet appeared at approximately 5.61–5.62 ppm, along with a broad band in the IR spectrum in the range of 3333–3422 cm⁻¹, corresponding to O–H stretching vibrations confirming the presence of a free hydroxy group. Moreover, the successful deprotection was additionally evidenced by the expected molecular masses of the target compounds determined by HRMS analysis. Together, these results provide strong evidence for the complete and selective removal of the benzyl protecting group under the optimized reaction conditions.
An analogous procedure was applied for the deprotection of γ-halo-δ-lactones 7c, 8b, and 9b. Among these, only in the case of γ-chloro-δ-lactone 9b was a successful deprotection achieved, leading to the isolation of the expected deprotected δ-lactone 12b as the sole product. Interestingly, NMR analysis of the product obtained from the deprotection of γ-iodo-δ-lactone 7c clearly confirmed the structure of trans δ-iodo-γ-lactone 10b, suggesting the occurrence of an intramolecular rearrangement through a translactonization process (Scheme 4). In contrast, for the bromo analogue 8b, TLC and GC analyses revealed the slow formation of two inseparable products, indicating a more complex and less selective reaction course.
The proposed mechanism of the translactonization process, which occurs during FeCl₃-mediated debenzylation of γ-iodo-δ-lactone 7c, is illustrated in Scheme 5.
The rearrangement appears to proceed in a single step via intramolecular nucleophilic substitution, facilitated by FeCl₃. Acting as a Lewis acid, FeCl₃ promotes the transposition of the iodine atom from the C-5 to the C-6 position, accompanied by a simultaneous nucleophilic attack of the lactone oxygen on the C-5 carbon, resulting in the formation of a five-membered ring. The exclusive formation of the trans isomer strongly suggests that the substitution follows an SN2-type mechanism, in which the oxygen nucleophile approaches from the side opposite to the departing iodine atom.
Translactonization converting δ-lactones into γ-lactones has been previously reported in the literature, both under chemical and microbiological halolactonization conditions. Kamizela et al. (2019) observed the unexpected formation of δ-hydroxy-γ-lactones among the products of iodolactonization of 3-methyl-5-arylpent-4-enoic acids using I₂/KI in a biphasic NaHCO₃/Et₂O aqueous system. This phenomenon was attributed to the rearrangement of initially formed γ-iodo-δ-lactones. In that case, the reaction was proposed to proceed via an SN1-like mechanism, as evidenced by the formation of both cis and trans isomers of the δ-hydroxy-γ-lactones. The process involved a dehalogenation-lactonization sequence with two simultaneous nucleophilic substitutions: replacement of the iodine atom at C-5 by the lactone oxygen, and nucleophilic attack of water at C-6 [24]. Interestingly, in one of our previous studies, a similar rearrangement was observed during the biotransformation of a chalcone-derived γ-bromo-δ-lactone in the culture of Penicillium frequentans AM 359. In contrast to the chemically induced rearrangement, the microbial process proceeded via an SN2 mechanism, which led exclusively to the formation of a single trans isomer of the hydroxylactone [57].
The translactonization of γ-halo-δ-lactones reported previously typically involved the participation of water, leading to the formation of δ-hydroxy-γ-lactones. In contrast, the FeCl₃-mediated debenzylation of γ-iodo-δ-lactone 7c was carried out under strictly anhydrous conditions, which favored an intramolecular SN2-type reaction and resulted in the selective formation of the trans δ-iodo-γ-lactone. To the best of our knowledge, this type of rearrangement has not been previously reported and may be attributed to the higher thermodynamic stability of five-membered lactone rings compared to six-membered ones. With regard to the relative reactivity of halogens in the presence of FeCl₃, the order can be classified as I > Br > Cl. This trend corresponds to the bond strength between the carbon and halogen atoms, iodine forms the weakest bond, rendering γ-iodo-δ-lactones less stable and more prone to rearrangement. Consequently, no translactonization was observed for γ-chloro-δ-lactone 9b, and the process occurred only very slowly for its bromo analogue 8b. Even after 2 h of reaction, TLC analysis revealed the presence of two distinct products, corresponding to the unrearranged δ-lactone and the newly formed γ-lactone.
2.2. Antiproliferative Activity
To evaluate their antiproliferative potential, synthesized vanillin-derived halolactones 10a,b, 11a and 12a,b as well starting vanillin (1) were subjected to in vitro MTT assay against panel of selected cancer cell lines: two lines representing hematopoietic canine cancers: canine B-cell lymphoma cell line (CLBL-1) and chronic B-cell leukemia (CLB70) and two human cancer lines: bladder carcinoma (T-24) and colorectal adenocarcinoma (CaCo-2). Normal cell line of mouse embryonic fibroblasts (NIH/3T3) was also used in these studies. The results, expressed as IC50 values are shown in Table 1.
All tested compounds exhibited measurable antiproliferative activity against the canine cancer cell lines, whereas only three compounds showed notable effects against the human cancer cell lines (T-24 or CaCo-2). This observation suggests a higher susceptibility of hematopoietic canine cancer lines compared to the relatively greater resistance displayed by the human cancer cells towards the tested compounds.
Among the tested compounds, the highest antiproliferative activity was observed for trans γ-iodo-δ-lactone 10b against the CLBL-1 cell line, with an IC₅₀ value of 46.26±4.07 µg/mL, and for vanillin (1) against the T-24 cell line (IC₅₀ = 48.46±13.16 µg/mL). Moderate activity was also noted for cis γ-bromo-δ-lactone 11a against CLBL-1 and for compound 10b against T-24. In all other cases, the tested compounds exhibited lower activity, with IC₅₀ values exceeding 70 µg/mL. Importantly, none of the compounds showed cytotoxic effects toward the normal NIH/3T3 fibroblast cell line, indicating a degree of selectivity toward cancer cells.
Comparing the antiproliferative activity of vanillin (1) with its halolactone derivatives against the CLBL-1 cell line, the trans γ-iodo-δ-lactone 10b and cis γ-bromo-δ-lactone 11a exhibited notably higher activity. In assays with CaCo-2 cells, vanillin (1) was inactive, whereas cis γ-bromo-δ-lactone 11a, cis γ-chloro-δ-lactone 12a, and γ-chloro-δ-lactone 12b demonstrated moderate activity. Conversely, vanillin (1) proved to be the most active compound against the T-24 cell line. For the CLB70 line, the antiproliferative activities of vanillin (1) and all tested vanillin-derived lactones were comparable.
When evaluating the impact of the halogen substituent on the biological activity of the lactones, iodolactones 10a and 10b demonstrated superior activity against the T-24 bladder carcinoma line compared to their bromo- and chlorolactone counterparts (11a, 12a and 12b). Conversely, for the CaCo-2 colorectal adenocarcinoma line, the trend was reversed, with bromo- and chlorolactones exhibiting relatively higher efficacy than the iodolactones.
Comparing the antiproliferative activity of cis isomers of γ-lactones against hematopoietic cell lines, the highest activity was observed for bromolactone 11a against the CLBL-1 cell line. In contrast, the activity of all tested cis δ-halo-γ-lactones (10a, 11a, and 12a) against the CLB70 cell line was comparable. These results clearly indicate that the influence of the halogen substituent on the antiproliferative activity of lactones is strongly dependent on the specific cancer cell line. Similar observations were reported in our previous studies. For instance, when studying a series of racemic halolactones bearing phenyl, p-methylphenyl, and p-isopropylphenyl substituents at the β-position of the lactone ring, bromolactones exhibited higher antiproliferative activity compared to their iodo- and chloro- analogues against the Jurkat cell line (human T-cell leukemia) [31]. In another study, β-aryl-δ-iodo-γ-lactones containing a 2’,5’-dimethylphenyl substituent showed greater activity against the CLBL-1 and D17 (canine osteosarcoma) cell lines, but lower activity against GL-1 (B-cell leukemia) and Jurkat cell lines when compared to their bromo analogs [33].
Considering the effect of the spatial structure of iodolactones on their antiproliferative properties, it is evident that trans δ-iodo-γ-lactone 10b exhibited considerably higher activity than its cis isomer 10a against the CLBL-1 and T-24 cell lines, whereas the activities of both isomers against the CLB-70 and CaCo-2 lines were comparable. A higher antiproliferative activity of trans isomers of β-aryl-δ-iodo-γ-lactones, bearing 2,5-dimethylphenyl and 1,3-benzodioxole substituents, compared to their cis isomers was also reported for analogs tested against Jurkat, GL-1, CLBL-1, and D17 cell lines [32].
In summary, among all tested halolactones, the most promising antiproliferative properties were exhibited by trans δ-iodo-γ-lactone 10b. It showed the highest activity toward the CLBL-1 and T-24 cell lines, and comparable activity against the CLB-70 cell line relative to the other tested halolactones. It is also worth mentioning that structurally related compounds, such as δ-aryl-γ-halo-δ-lactones synthesized from aryl bromides and 3-methylcrotonaldehyde [25], or trans-crotonaldehyde [24], exhibited cytotoxic effects not only toward some cancer cell lines but also against the L929 mouse fibroblast normal cell line. In contrast, in the present study, all tested vanillin-derived halolactones displayed potent and selective antiproliferative activity against cancer cells without affecting the normal NIH/3T3 fibroblast cell line. This favorable selectivity profile makes this group of lactones, particularly lactone 10b, a promising candidate for further development as a safe anticancer agents.
2.3. Cytotoxicity Toward Red Blood Cells (RBCs)
The next stage of the study focused on determining the toxicity of the tested lactones and vanillin (1) toward human red blood cells (RBCs) across a wide concentration range (0 to 200 μM) and at different incubation times (2 h, 24 h, and 72 h).
The results of these analyses are presented as the percentage of hemolysis relative to the control sample without the addition of the tested compound and the sample containing only the solvent - DMSO, allowing a precise assessment of the compounds' impact. Detailed results are shown in Figure 1 and Table 2.
The results showed that incubation of red blood cells with vanillin (1) and the tested lactones 10a,b, 11a, 12a,b for 2 h showed hemolysis between 6.65±0.14% and 7.81±0.18%, regardless of the concentration, with hemolysis levels for DMSO and control sample 6.69±0.59% and 6.63±0.28%, respectively.
A slight increase in the percentage of hemolysis was observed after 24 h of incubation, with values ranging from 7.30±0.19% to 8.99±0.69%, especially for 10a and 11a at the highest concentrations. However, that the level of hemolysis for the control and with DMSO was similar to each other, with values of 7.65±0.44% and 7.92±0.07%, respectively.
After 72 h of incubation, hemolysis increased slightly in all samples to above 9%. It was also observed that for three lactones - 10b, 11a and 12b at the highest concentration, the level of hemolysis was slightly higher compared to the other compounds and the control and amounted to 11.00±0.90%, 10.34±1.20% and 10.52±0.75%, respectively. The results obtained indicate a slight hemolytic effect associated with the prolonged incubation and very high concentration (200 μM) of the tested compounds. Nevertheless, the percentage of hemolysis increased by less than 2% compared to the control and DMSO.
The results of the study demonstrate that over a wide range of concentrations (exceeding those used in cell line studies), both vanillin and all tested vanillin-derived halolactones did not exhibit a significant increase in hemolysis compared to the control and DMSO after 2 and 24 h of exposure. Although prolonged exposure to high concentrations caused minor hemolytic effects for some halolactones, the values remained only slightly elevated relative to the control and DMSO, suggesting that the tested compounds should be considered non-toxic to erythrocytes. It should also be emphasized that hemolysis values for both DMSO and the control remained at the same level, confirming that the solvent itself had no impact on erythrocyte integrity.
The hemolytic activity profile of the tested lactones observed in this study is strongly supported by previous investigations of structurally related compounds, which likewise demonstrated no-toxic effects on erythrocyte membranes. Włoch et al. (2020) examined racemic β-aryl-δ-iodo-γ-lactones with various aromatic substituents and showed that bromolactone analogues, bearing either a methyl group or no substituent on the benzene ring, did not induce hemolysis in erythrocytes even after 48 h of incubation [37].
In another study, Włoch et al. (2025) also evaluated the toxicity of enantiomeric piperonal-derived trans β-aryl-δ-iodo-γ-lactones toward human red blood cells (RBCs) across a wide range of concentrations and various incubation times [36]. In this case as well, the results demonstrated a lack of toxicity at the tested concentrations and extended incubation periods. However, it should be noted that in both cited studies the concentration range examined (up to 100 µM) was significantly lower than in the present work. Furthermore, the incubation time in our study was longer (up to 72 h) compared to those previous reports (up to 48 h).
3. Materials and Methods
2.3. Chemicals
Vanillin (99% purity), benzyl bromide (98% purity), anhydrous methylene chloride (≥99.8% purity), anhydrous iron(III) chloride (98% purity), N-bromosuccinimide (NBS) (95% purity), N-chlorosuccinimide (NCS) (98% purity), sodium borohydride (≥96% purity), anhydrous tetrahydrofuran (THF) (≥99% purity), triethyl orthoacetate (97% purity), were purchased from Merck (Darmstadt, Germany). Other common reagents and solvents were purchased from Chempur (Piekary Śląskie, Poland or Idalia Radom, Poland).
3.2. Analysis and Purification
The reaction progress was monitored by Thin Layer Chromatography (TLC) using 0.2 mm aluminum plates coated with silica gel 60 F254 (Merck, Darmstadt, Germany). Chromatograms were visualized by spraying the plates with a 1% solution of Ce(SO4)2 and 2% H3[P(Mo3O10)4] in 10% H2SO4, followed by heating the plates at 120-200 °C.
Gas Chromatography (GC) analysis was conducted using an Agilent Technologies 6890N (Santa Clara, CA, USA) instrument equipped with an autosampler, split injection (50:1), and an FID detector, employing a DB5-HT column (Agilent, Santa Clara, CA, USA), polyimide-coated fused silica tubing, 30 m x 0.25 mm x 0.10 μm with hydrogen as the carrier gas. For analysis of lactones 10a,b, 11a, 12a,b, the following temperature program was used: injector and detector (FID) temperatures 300 °C, the column temperature was programmed from 100 °C to 200 °C at a rate of 20 °C/min, followed by a ramp from 200 °C to 300 °C at 30 °C/min, with a hold at 300 °C for 8 minutes. For intermediate product analysis 1-5, the initial column temperature was 100 °C, followed by a temperature increase from 100 °C to 200 °C at a rate of 20 °C/min, then from 200 °C to 300 °C at a rate of 30 °C/min, and finally a 1-minute hold at 300 °C.
Nuclear Magnetic Resonance (NMR) spectra, including 1H NMR, 13C NMR, 13C DEPT 135, COSY, HMQC and HMBC were recorded using either a Jeol 400 MHz Year Hold Magnet spectrometer (Jeol Ltd., Tokyo, Japan) or NMR Avance III HD 600 MHz spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany). They are available in the Supplementary Materials (Figures S1–S102). Samples were dissolved in CDCl3 (≥ 99%) or CD3OD (≥ 99%) and chemical shifts were referenced to the residual solvent signal (δH = 7.26, δC = 77.00 or δH = 3.31, δC = 49.00).
Infrared (IR) spectra were acquired using a Nicolet iS10 FTIR Spectrometer (Thermo Fisher Scientific™, Waltham, MA, USA), equipped with a monolithic diamond ATR crystal attachment. All recorded IR spectra are available in the Supplementary Materials (Figures S103-S119).
High Resolution Mass Spectra (HRMS) were recorded on either a Bruker Daltonics ESI-Q-TOF maXis impact mass spectrometer (Bruker, Billerica, MA, USA) or a Waters Xevo G2 mass spectrometer (Waters, Milford, MA, USA), with positive or negative electrospray ionization (ESI) techniques.
Flash chromatography was conducted using the puriFlash® SX520 Plus system (Interchim, Montluçon, France), which includes a gradient pump, UV detector, and fraction collector. Samples were dry loaded on a pre-column (puriFlash®) and the compounds were separated on puriFlash® SIHP F0012 or F0040 30 µm columns by gradient elution with hexane/ethyl acetate mixtures (flow rate: 26 mL/min; pressure: 15 mbar).
The melting points (uncorrected) were determined on the Boetius apparatus (Nagema, Dresden, Germany).
3.3. Preparation of Benzylvanillin (2)
Vanillin (1) (5.00 g, 33.86 mmol), potassium carbonate (5.26 g, 38.06 mmol), and benzyl bromide (4.90 mL, 41.27 mmol) were dissolved in ethanol (150 mL) and stirred at room temperature for 48 h. The reaction mixture was then filtered to remove solid impurities, and the solvent was evaporated under reduced pressure. The resulting residue was treated with 30 mL of 5% aqueous NaOH and the product was extracted with methylene chloride (3 × 30 mL). The combined organic layers were dried over anhydrous MgSO₄, filtered, and the solvent was removed under reduced pressure. The crude product was dissolved in hot ethanol and recrystallized by cooling at –20 °C for 72 h. The resulting crystals were collected by vacuum filtration using a Büchner funnel, washed with cold ethanol, and dried to afford pure benzylvanillin (2), which was characterized by the following physical and spectral data:
Yield 7.01 g (88%); light yellow crystals; mp 58-60 °C (lit. 61-63 °C) [58]; Rf = 0.45 (hexane/ethyl acetate 3:1, v/v); 1H NMR (400 MHz, CD3OD): δ 3.90 (s, 3H, -OCH3), 5.21 (s, 2H, -OCH2Ph), 7.17 (d, J = 8.2 Hz, 1H, H-5’), 7.32 (m, 1H, H-4’’), 7.35 – 7.40 (m, 2H, H-3’’, H-5’’), 7.43 – 7.47 (m, 3H, H-2’, H-2’’ and H-6’’), 7.49 (dd, J = 8.2 and 1.9 Hz, 1H, H-6’), 9.80 (s, 1H, -CHO); 13C NMR (100 MHz, CD3OD): δ 56.44 (-OCH3), 71.82 (-OCH2Ph), 111.00 (C-2), 113.91 (C-5), 127.49 (C-6), 128.69 (C-2’,C-6’), 129.17 (C-4’), 129.59 (C-3’,C-5’), 131.71 (C-1), 137.84 (C-1’), 151.50 (C-3), 155.33 (C-4), 192.94 (-CHO); IR (ATR): νmax = 2840, 1676, 1584, 1506, 1262, 1134, 748 cm-1; HRMS (ESI): m/z calcd for C15H14O3 [M+Na]+: 265.0835; found: 265.0837.
3.4. Preparation of Ketone 3 via Claisen–Schmidt Condensation
A solution of benzylvanillin (2) (7.01 g, 28.96 mmol) in acetone (230 mL) was stirred in a water bath. A 10% aqueous NaOH solution (20 mL) was added dropwise to the reaction mixture. The reaction was stirred at room temperature for 24 h, after which the mixture was acidified to pH = 2 using 1 M HCl. The resulting product was extracted with methylene chloride (3 × 40 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO₄, and filtered. The solvent was removed under reduced pressure, and the crude product was purified by flash chromatography (gradient elution from hexane to hexane/ethyl acetate 7:1, v/v) to afford pure (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3), which was characterized by the following physical and spectral data:
Yield 6.86 g (84%); yellow crystals; mp 91-93 °C (lit. 92-93 °C) [59]; Rf = 0.21 (hexane/ethyl acetate 4:1, v/v); 1H NMR (400 MHz, CDCl3): δ 2.36 (s, 3H, CH3-1), 3.92 (s, 3H, -OCH3), 5.19 (s, 2H, -OCH2Ph), 6.59 (d, J = 16.2 Hz, 1H, H-3), 6.88 (d, J = 8.3 Hz, 1H, H-5’), 7.05 (dd, J = 8.3 Hz and 2.0 Hz, 1H, H-6’), 7.09 (d, J = 2.0 Hz, 1H, H-2’), 7.31 (m, 1H, H-4’’), 7.35-7.40 (m, 2H, H-3’’, H-5’’), 7.41-7.45 (m, 2H, H-2’’, H-6’’), 7.44 (d, J = 16.2 Hz, H-4); 13C NMR (100 MHz, CDCl3): δ 27.31 (C-1), 55.95 (-OCH3), 70.78 (-OCH2Ph), 110.12 (C-2’), 113.34 (C-5’), 122.74 (C-6’), 125.31 (C-3), 127.16 (C-2’’,C-6’’), 127.61 (C-1’), 128.01 (C-4’’), 128.61 (C-3’’, C-5’’), 136.44 (C-1’’), 143.46 (C-4), 149.76 (C-3’), 150.43 (C-4’), 198.35 (C-2); IR (ATR): νmax = 1661, 1637, 1514, 1255, 1142, 974, 839, 806, 744, 697 cm-1; HRMS (ESI): m/z calcd for C18H18O3 [M+Na]+: 305.1148; found: 305.1155.
3.5. Preparation of Allylic Alcohol 4
Ketone 3 (6.86 g, 24.32 mmol) was dissolved in methanol (200 mL), and the solution was cooled in an ice bath. An aqueous solution of NaBH₄ (2.05 g in 12 mL H₂O) was added dropwise to the stirred reaction mixture. The reaction was stirred for 1 h in the ice bath and then for 23 h at room temperature. After this time, hot water (150 mL) was added, and the product was extracted with methylene chloride (3 × 30 mL). The organic layer was washed with brine, dried over anhydrous MgSO₄, and filtered. The solvent was removed under reduced pressure to afford pure (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4), which was characterized by the following physical and spectral data:
Yield 5.67 g (82%); white solid; mp 55-57 °C; Rf = 0.25 (hexane/acetone 3:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.37 (d, J = 6.4 Hz, 3H, CH3-1), 1.64 (s, 1H, -OH), 3.91 (s, 3H, -OCH3), 4.47 (m, 1H, H-2), 5.16 (s, 2H, -OCH2Ph), 6.13 (dd, J = 15.8 Hz and 6.6 Hz, 1H, H-3), 6.48 (d, J = 15.8 Hz, 1H, H-4), 6.82 (d, J = 8.2 Hz, 1H, H-5’), 6.85 (dd, J = 8.2 and 1.6 Hz, 1H, H-6’), 6.96 (d, J = 1.6 Hz, 1H, H-2’), 7.30 (m, 1H, H-4’’), 7.33-7.39 (m, 2H, H-3’’, H-5’’), 7.40-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 23.44 (C-1), 55.91 (OCH3), 69.02 (C-2), 70.92 (-OCH2Ph), 109.25 (C-2’), 113.80 (C-5’), 119.52 (C-6’), 127.19 (C-2’’, C-6’’), 127.81 (C-4’’), 128.52 (C-3’’, C-5’’), 129.19 (C-4), 130.15 (C-1’), 131.75 (C-3), 136.99 (C-1’’), 147.91 (C-4’), 149.62 (C-3’); IR (ATR): νmax = 3384, 1513, 1264, 1139, 969, 798, 858, 744, 699 cm-1; HRMS (ESI): m/z calcd for C18H20O3 [M+Na]+: 307.1305; found: 307.1317.
3.6. Preparation of Ester 5 by Johnson-Claisen Rearrangement
A mixture of alcohol 4 (5.67 g, 19.94 mmol), triethyl orthoacetate (230 mL, 1.27 mol), and a few drops of propionic acid was heated at 138 °C for 6 h with continuous distillation of ethanol. Upon completion of the reaction (monitored by TLC), the mixture was subjected to silica gel column chromatography to remove excess triethyl orthoacetate (eluent: hexane/acetone 10:1, v/v). The crude product was further purified by flash chromatography (gradient elution from hexane to hexane/ethyl acetate 19:1, v/v) to afford pure (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5), which was characterized by the following physical and spectral data:
Yield 3.32 (47%); light yellow oily liquid; Rf = 0.64 (hexane/ethyl acetate (3:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.17 (t, J = 7.1 Hz, 3H, -OCH2CH3), 1.65 (d, J = 6.0 Hz, 3H, CH3-6), 2.62 (dd, J = 14.8 and 7.5 Hz, 1H, one of CH2-2), 2.66 (dd, J = 14.8 and 8.0 Hz, 1H, one of CH2-2), 3.73 (m, 1H, H-3), 3.88 (s, 3H, -OCH3), 4.04-4.10 (two q, J = 7.1 Hz, 2H, -OCH2CH3), 5.12 (s, 2H, -OCH2Ph), 5.48 (dq, J = 15.3 and 6.0 Hz, 1H, H-5), 5.57 (ddq, J = 15.3, 7.1 and 1.1 Hz, 1H, H-4), 6.68 (dd, J = 8.2 and 2.0 Hz 1H, H-6’), 6.75 (d, J = 2.0 Hz, 1H, H-2’), 6.81 (d, J = 8.2 Hz, H-5’), 7.29 (m, 1H, H-4’’), 7.32-7.39 (m, 2H, H-3’’, H-5’’), 7.40-7.45 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 14.19 (-OCH2CH3), 17.89 (C-6), 41.18 (C-2), 44.57 (C-3), 55.98 (-OCH3), 60.26 (-OCH2CH3), 71.07 (-OCH2Ph), 111.38 (C-2’), 114.06 (C-5’), 119.12 (C-6’), 125.42 (C-5), 127.22 (C-2’’, C-6’’), 127.73 (C-4’’), 128.47 (C-3’’, C-5’’), 133.17 (C-4), 136.61 (C-1’), 137.32 (C-1’’), 146.75 (C-4’), 149.53 (C-3’), 172.03 (C-1); IR (ATR): νmax = 1731, 1511, 1259, 1137, 1025, 967, 852, 802, 734, 696 cm-1; HRMS (ESI): m/z calcd for C22H26O4 [M+Na]+: 377.1723; found: 377.1740.
3.7. Preparation of Acid 6
Ester 5 (3.32 g, 9.37 mmol) was heated under reflux for 5 h in a 10% ethanol solution of NaOH (230 mL). After evaporating the solvent under reduced pressure, the residue was diluted with water, and organic impurities were removed by extraction with diethyl ether (3 × 30 mL). The aqueous layer was acidified with 1M HCl, and the product was extracted with methylene chloride (3 × 30 ml). The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent was evaporated in vacuo to afford pure (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6) with the following physical and spectral data:
Yield 2.51 g (82%); white solid; mp 95-98 °C; Rf = 0.05 (hexane/ethyl acetate 3:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.66 (d, J = 5.8 Hz, 3H, CH3-6), 2.68 (dd, J = 15.6 and 7.4 Hz, 1H, one of CH2-2), 2.72 (dd, J = 15.6 and 8.0 Hz, 1H, one of CH2-2), 3.72 (m, 1H, H-3), 3.87 (s, 3H, -OCH3), 5.12 (s, 2H, -OCH2Ph), 5.50 (dq, J = 15.4 and 5.8 Hz, 1H, H-5), 5.57 (ddq, J = 15.4, 6.8 and 1.2 Hz, 1H, H-4), 6.68 (dd, J = 8.2 and 2.0 Hz 1H, H-6’), 6.74 (d, J = 2.0 Hz, 1H, H-2’), 6.82 (d, J = 8.2 Hz, H-5’), 7.29 (m, 1H, H-4’’), 7.33-7.39 (m, 2H, H-3’’, H-5’’), 7.40-7.45 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 17.91 (C-6), 40.67 (C-2), 44.06 (C-3), 55.98 (-OCH3), 71.05 (-OCH2Ph), 111.36 (C-2’), 114.02 (C-5’), 119.06 (C-6’), 125.68 (C-5), 127.23 (C-2’’, C-6’’), 127.76 (C-4’’), 128.49 (C-3’’, C-5’’), 132.88 (C-4), 136.25 (C-1’), 137.26 (C-1’’), 146.86 (C-4’), 149.54 (C-3’), 177.57 (C-1); IR (ATR): νmax = 2500-3500, 1702, 1518, 1236, 1142, 965, 855, 791, 744, 696 cm-1; HRMS (ESI): m/z calcd for C20H22O4 [M+Na]+: 349.1410; found: 349.1413.
3.8. Preparation of Iodolactones 7a-c
A solution of acid 6 (2.69 g, 7.70 mmol) in diethyl ether (80 mL) and saturated NaHCO₃ solution (80 mL) was stirred for 1 h at room temperature. Then, a solution of I₂ (12.70 g, 50.01 mmol) and KI (3.97 g, 23.92 mmol) in water (20 mL) was added dropwise to the stirred mixture until a stable brown color was observed. After 19 h, the reaction mixture was diluted with diethyl ether and washed with aqueous Na₂S₂O₃. The aqueous phase was shaked with diethyl ether (3 × 30 mL), and the combined organic layers were washed with saturated NaHCO₃, brine, then dried over anhydrous MgSO₄. After evaporation of the solvent under reduced pressure, the mixture of iodolactones was separated by flash chromatography (gradient elution from hexane to hexane/ethyl acetate 5:1, v/v). Physicochemical and spectral data of the isolated iodolactones 7a–c are given below:
3.8.1. Cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a)
Yield 1.81 g (52%); white solid; mp 116-118 °C; Rf = 0.27 (hexane/ethyl acetate 4:1, v/v); 1H NMR (400 MHz, CDCl3): δ 2.01 (d, J = 6.8 Hz, 3H, CH3-7), 2.70 (d, J = 17.5 Hz, 1H, one of CH2-3), 3.10 (dd, J = 17.5 and 8.4 Hz, 1H, one of CH2-3), 3.47 (dq, J = 10.9 and 6.8 Hz, 1H, H-6), 3.84 (dd, J = 8.4 and 5.0 Hz, 1H, H-4), 3.89 (s, 3H, -OCH3), 4.78 (dd, J = 10.9 and 5.0 Hz, 1H, H-5), 5.14 (s, 2H, -OCH2Ph), 6.71 (dd, J = 8.3 and 2.1 Hz, 1H, H-6’), 6.80-6.86 (m, 2H, H-2’, H-5’), 7.31 (m, 1H, H-4’’), 7.34-7.40 (m, 2H, H-3’’, H-5’’), 7.41-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 23.70 (C-6), 25.53 (C-7), 38.83 (C-3), 44.54 (C-4), 56.04 (-OCH3), 70.98 (-OCH2Ph), 87.87 (C-5), 112.80 (C-2’), 113.87 (C-5’), 120.26 (C-6’), 127.31 (C-2’’, C-6’’), 127.93 (C-4’’), 128.57 (C-3’’, C-5’’), 130.05 (C-1’), 136.86 (C-1’’), 147.81 (C-4’), 149.31 (C-3’), 176.64 (C-2); IR (ATR): νmax = 1789, 1518, 1239, 1148, 1012, 954, 857, 751, 700, 538 cm-1; HRMS (ESI): m/z calcd for C20H21IO4 [M+Na]+ 475.0377; found: 475.0390.
3.8.2. Trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b)
Yield 0.87 g (25%); white solid; mp 80-82 °C; Rf = 0.16 (hexane/ethyl acetate 4:1, v/v); 1H NMR (400 MHz, CDCl3): major signals from syn conformer: δ 1.86 (d, J = 7.1 Hz, 3H, CH3-7), 2.64 (dd, J = 18.4 and 6.6 Hz, one of CH2-3), 3.12 (dd, J = 18.4 and 10.0 Hz, 1H, one of CH2-3), 3.56 (ddd, J = 10.0, 6.6 and 5.3 Hz, 1H, H-4), 3.89 (s, 3H, -OCH3), 4.23 (t, J = 5.3 Hz, 1H, H-5), 4.36 (qd, J = 7.1 and 5.3 Hz, 1H, H-6), 5.14 (s, 2H, -OCH2Ph), 6.73 (dd, J = 8.2 and 2.1 Hz, 1H, H-6’), 6.75 (d, J = 2.1 Hz, 1H, H-2’), 6.85 (d, J = 8.2 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.34-7.40 (m, 2H, H-3’’, H-5’’), 7.41-7.45 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 23.44 (C-7), 28.45 (C-6), 37.67 (C-3), 45.17 (C-4), 56.14 (-OCH3), 71.04 (-OCH2Ph), 89.56 (C-5), 110.54 (C-2’), 114.35 (C-5’), 119.06 (C-6’), 127.23 (C-2’’, C-6’’), 127.93 (C-4’’), 128.57 (C-3’’, C-5’’), 134.31 (C-1’), 136.85 (C-1’’), 147.69 (C-4’), 150.15 (C-3’), 174.88 (C-2); minor signals from anti conformer: 1H NMR: δ 1.97 (d, J = 7.1 Hz, 3H, CH3-7), 2.76 (dd, J = 18.1 and 9.5 Hz, one of CH2-3), 3.07 (dd, J = 18.1 Hz and 9.5 Hz, 1H, one of CH2-3), 3.46 (td, J = 9.5 and 7.1 Hz, 1H, H-4), 3.73 (dd, J = 7.1 and 2.4 Hz, 1H, H-5), 3.90 (s, 3H, -OCH3), 5.30 (s, 2H, -OCH2Ph), 6.85 (d, J = 8.1 Hz, 1H, H-5’); 13C NMR: δ 25.42 (C-7), 29.02 (C-6), 37.04 (C-3), 47.41 (C-4), 89.27 (C-5), 110.70 (C-2’), 119.26 (C-6’), 132.07 (C-1’), 174.80 (C-2); IR (ATR): νmax = 1786, 1517, 1454, 1254, 1142, 1030, 970, 848, 804, 746, 697, 644, 536 cm-1; HRMS (ESI): m/z calcd for C20H21IO4 [M+Na]+: 475.0377; found: 475.0378.
3.8.3. 4-r-(4'-Benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c)
Yield 0.38 g (11%); white solid; mp 109-112 °C; Rf = 0.15 (hexane/ethyl acetate 4:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.74 (d, J = 6.2 Hz, 3H, CH3-7), 2.65 (dd, J = 17.6 and 10.6 Hz, 1H, one of CH2-3), 2.98 (dd, J = 17.6 and 6.6 Hz, 1H, one of CH2-3), 3.43 (td, J = 10.6 and 6.6 Hz, 1H, H-4), 3.90 (s, 3H, -OCH3), 4.00 (t, J = 10.6 Hz, H-5), 4.75 (dq, J = 10.6 and 6.2 Hz, 1H, H-6), 5.15 (s, 2H, -OCH2Ph), 6.65 (dd, J = 8.0 and 2.2 Hz, 1H, H-6’), 6.67 (d, J = 2.2 Hz, 1H, H-2’), 6.87 (d, J = 8.0 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.35-7.40 (m, 2H, H-3’’, H-5’’), 7.42-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 22.30 (C-7), 34.90 (C-5), 37.72 (C-3), 48.22 (C-4), 56.10 (-OCH3), 70.99 (-OCH2Ph), 81.00 (C-6), 110.46 (C-2’), 113.99 (C-5’), 118.90 (C-6’), 127.27 (C-2’’, C-6’’), 127.90 (C-4’’), 128.56 (C-3’’, C-5’’), 135.42 (C-1’), 136.90 (C-1’’), 147.79 (C-4’), 149.84 (C-3’), 169.73 (C-2); IR (ATR): νmax = 1714, 1518, 1377, 1261, 1141, 1029, 971, 851, 810, 740, 699, 574 cm-1; HRMS (ESI): m/z calcd for C20H21IO4 [M+Na]+: 475.0377; found: 475.0383.
3.9. Preparation of Bromolactones 8a,b
A solution of acid 6 (2.51 g, 7.70 mmol) and N-bromosuccinimide (1.89 g, 10.62 mmol) in THF (80 mL), with a few drops of acetic acid, was stirred at room temperature for 24 h. The mixture was then diluted with diethyl ether (40 mL) and washed with saturated NaHCO₃ solution and brine. The organic layer was dried over anhydrous MgSO₄. After solvent evaporation under reduced pressure, the product mixture was purified by flash chromatography (gradient elution from hexane to hexane/ethyl acetate 5:1, v/v). Two bromolactones, 8a and 8b, were isolated and characterized by the following data:
3.9.1. Cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a)
Yield 1.12 g (35%); white solid; mp 125-128 °C; Rf = 0.39 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.76 (d, J = 6.6 Hz, 3H, CH3-7), 2.71 (d, J = 17.5 Hz, 1H, one of CH2-3), 3.09 (dd, J = 17.5 and 8.4 Hz, 1H, one of CH2-3), 3.45 (dq, J = 10.4 and 6.6 Hz, 1H, H-6), 3.81 (dd, J = 8.4 and 5.1 Hz, 1H, H-4), 3.87 (s, 3H, -OCH3), 4.64 (dd, J = 10.4 and 5.1 Hz, 1H, H-5), 5.14 (s, 2H, -OCH2Ph), 6.68 (dd, J = 8.3 and 2.1 Hz, 1H, H-6’), 6.79 (d, J = 2.1 Hz, 1H, H-2’), 6.84 (d, J = 8.3 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.34-7.40 (m, 2H, H-3’’, H-5’’), 7.41-7.47 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 23.26 (C-7), 38.29 (C-3), 43.64 (C-4), 45.54 (C-6), 56.02 (-OCH3), 70.97 (-OCH2Ph), 86.71 (C-5), 112.58 (C-2’), 113.83 (C-5’), 120.10 (C-6’), 127.30 (C-2’’, C-6’’), 127.93 (C-4’’), 128.57 (C-3’’, C-5’’), 130.19 (C-1’), 136.87 (C-1’’), 147.82 (C-4’), 149.34 (C-3’), 176.42 (C-2); IR (ATR): νmax = 178, 1518, 1254, 1146, 1015, 966, 780, 747, 699 cm-1; HRMS (ESI): m/z calcd for C20H21BrO4 [M+Na]+: 427.0515; found: 427.0522.
3.9.2. 4-r-(4'-Benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b)
Yield 0.26 g (8%); white solid; mp 132-134 °C; Rf = 0.33 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.65 (d, J = 6.2 Hz, 3H, CH3-7), 2.70 (dd, J = 17.6 and 9.4 Hz, 1H, one of CH2-3), 3.05 (dd, J = 17.6 and 6.9 Hz, 1H, one of CH2-3), 3.40 (td, J = 9.4 and 6.9 Hz, 1H, H-4), 3.90 (s, 3H, -OCH3), 3.91 (t, J = 9.4 Hz, H-5), 4.63 (dq, J = 9.4 and 6.2 Hz, 1H, H-6), 5.14 (s, 2H, -OCH2Ph), 6.68 (dd, J = 8.1 and 2.1 Hz, 1H, H-6’), 6.71 (d, J = 2.1 Hz, 1H, H-2’), 6.87 (d, J = 8.1 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.34-7.40 (m, 2H, H-3’’, H-5’’), 7.41-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 20.62 (C-7), 37.64 (C-3), 46.78 (C-4), 54.51 (C-5), 56.10 (-OCH3), 71.00 (-OCH2Ph), 79.59 (C-6), 110.66 (C-2’), 114.05 (C-5’), 119.00 (C-6’), 127.25 (C-2’’, C-6’’), 127.90 (C-4’’), 128.57 (C-3’’, C-5’’), 133.91 (C-1’), 136.93 (C-1’’), 147.81 (C-4’), 149.88 (C-3’), 169.45 (C-2); IR (ATR): νmax = 1721, 1517, 1375, 1225, 1141, 1001, 972, 804, 758, 704 cm-1; HRMS (ESI): m/z calcd for C20H21BrO4 [M+Na]+: 427.0515; found: 427.0517.
3.10. Preparation of Chlorolactones 9a,b
A solution of acid 6 (2.51 g, 7.70 mmol) and N-chlorosuccinimide (2.32 g, 17.37 mmol) in THF (80 mL), with a few drops of acetic acid, was stirred at room temperature for 24 h. The work-up procedure and isolation described in Section 3.9 were applied to isolate chlorolactones 9a and 9b, which were characterized by the following physical and spectral data:
3.10.1. Cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a)
Yield 0.28 g (10%); white solid; mp 81-84 °C; Rf = 0.37 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.56 (d, J = 6.5 Hz, 3H, CH3-7), 2.71 (d, J = 17.6 and 0.8 Hz, 1H, one of CH2-3), 3.07 (dd, J = 17.6 and 8.4 Hz, 1H, one of CH2-3), 3.43 (dq, J = 10.1 and 6.5 Hz, 1H, H-6), 3.80 (dd, J = 8.4 and 5.2 Hz, 1H, H-4), 3.87 (s, 3H, -OCH3), 4.49 (dd, J = 10.1 and 5.2 Hz, 1H, H-5), 5.14 (s, 2H, -OCH2Ph), 6.66 (dd, J = 8.3 and 2.2 Hz, 1H, H-6’), 6.76 (d, J = 2.2 Hz, 1H, H-2’), 6.84 (d, J = 8.3 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.35-7.40 (m, 2H, H-3’’, H-5’’), 7.42-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 22.38 (C-7), 37.94 (C-3), 43.13 (C-4), 53.93 (C-6), 56.02 (-OCH3), 70.97 (-OCH2Ph), 86.58 (C-5), 112.42 (C-2’), 113.82 (C-5’), 120.03 (C-6’), 127.29 (C-2’’, C-6’’), 127.92 (C-4’’), 128.57 (C-3’’, C-5’’), 130.30 (C-1’), 136.87 (C-1’’), 147.81 (C-4’), 149.37 (C-3’), 176.34 (C-2); IR (ATR): νmax = 1789, 1516, 1454, 1238, 1144, 1019, 973, 780, 745, 698, 680 cm-1; HRMS (ESI): m/z calcd for C20H21O4Cl [M+H]+: 361.1207; found: 361.1206.
3.10.2. 4-r-(4'-Benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b)
Yield 0.5 g (18%); white solid; mp 93-96 °C; Rf = 0.24 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (400 MHz, CDCl3): δ 1.60 (d, J = 6.2 Hz, 3H, CH3-7), 2.72 (dd, J = 17.7 and 9.5 Hz, 1H, one of CH2-3), 3.06 (dd, J = 17.7 and 7.0 Hz, 1H, one of CH2-3), 3.29 (td, J = 9.5 and 7.0 Hz, 1H, H-4), 3.83 (t, J = 10.0 Hz, H-5), 3.90 (s, 3H, -OCH3), 4.50 (dq, J = 10.0 and 6.2 Hz, 1H, H-6), 5.14 (s, 2H, -OCH2Ph), 6.70 (dd, J = 8.1 and 2.2 Hz, 1H, H-6’), 6.72 (d, J = 2.2 Hz, 1H, H-2’), 6.87 (d, J = 8.1 Hz, 1H, H-5’), 7.31 (m, 1H, H-4’’), 7.35-7.40 (m, 2H, H-3’’, H-5’’), 7.42-7.46 (m, 2H, H-2’’, H-6’’); 13C NMR (100 MHz, CDCl3): δ 19.66 (C-7), 37.21 (C-3), 46.31 (C-4), 56.09 (-OCH3), 62.47 (C-5), 71.00 (-OCH2Ph), 79.35 (C-6), 110.78 (C-2’), 114.08 (C-5’), 119.10 (C-6’), 127.24 (C-2’’, C-6’’), 127.90 (C-4’’), 128.57 (C-3’’, C-5’’), 133.13 (C-1’), 136.93 (C-1’’), 147.82 (C-4’), 149.89 (C-3’), 169.33 (C-2); IR (ATR): νmax = 1732, 1511, 1258, 1219, 1139, 1016, 973, 796, 738, 696, 642 cm-1; HRMS (ESI): m/z calcd for C20H21O4Cl [M+H]+: 361.1207; found: 361.1205.
3.11. General Procedure for Benzyl Deprotection of Halolactones 7a-c, 8a,b and 9a,b
Lactone 7-9 (1 equiv) was placed in a two-necked flask equipped with a nitrogen-filled balloon and dissolved in 10 mL of anhydrous methylene chloride. The flask was placed on a magnetic stirrer, and after complete dissolution of the substrate, anhydrous FeCl3 (3 equiv) was added. The mixture was stirred at room temperature. After complete reaction of the substrate (30 min), the crude reaction mixture was filtered under reduced pressure through a Schott funnel, transferred to a separatory funnel, and washed with 1% orthophosphoric acid solution (3 × 10 mL). After phase separation, the aqueous layer was further washed with methylene chloride (3 × 15 mL). All combined organic layers were neutralized with saturated NaCl solution and dried over anhydrous MgSO4. After filtration, the solvent was evaporated under reduced pressure using a rotary evaporator. The concentrated crude product was purified by flash chromatography. Gradient elution from hexane to hexane/ethyl acetate 9:2 (v/v) was used for lactones 10a, 10b, 12a, and 12b, while for lactone 11a a gradient from hexane to hexane/ethyl acetate 3:1 (v/v) was applied. Reaction yields and the physical and spectral data of the deprotected halolactones are as follows:
3.11.1. Cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a)
Obtained from lactone 7a (1.81 g, 4 mmol) in yield 0.41 g (28%); white solid; mp 56-59 °C; Rf = 0.24 (hexane/ethyl acetate 3:1, v/v), 1H NMR (400 MHz, CDCl3): δ 2.01 (d, J = 6.8 Hz, 3H, CH3-7), 2.70 (d, J = 17.5 Hz, 1H, one of CH2-3), 3.11 (dd, J = 17.5 and 8.4 Hz, 1H, one of CH2-3), 3.48 (dq, J = 10.9 and 6.8 Hz, 1H, H-6), 3.83 (dd, J = 8.4 and 5.0 Hz, 1H, H-4), 3.89 (s, 3H, -OCH3), 4.78 (dd, J = 10.9 and 5.0 Hz, 1H, H-5), 5.62 (s, 1H, -OH), 6.74 (dd, J = 8.1 and 2.0 Hz, 1H, H-6’), 6.78 (d, J = 2.0 Hz, 1H, H-2’), 6.86 (d, J = 8.1 Hz, 1H, H-5’); 13C NMR (100 MHz, CDCl3): δ 23.72 (C-6), 25.53 (C-7), 38.91 (C-3), 44.66 (C-4), 56.01 (-OCH3), 87.93 (C-5), 111.45 (C-2’), 114.64 (C-5’), 121.20 (C-6’), 128.94 (C-1’), 145.33 (C-4’), 146.34 (C-3’), 176.67 (C-2); IR (ATR): νmax = 3367, 1762, 1518, 1178, 1129, 1033, 966, 846, 597, 527 cm-1; HRMS (ESI): m/z calcd for C13H15O4I [M+H]+: 363.0093; found: 363.0094.
3.11.2. Trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b)
Obtained from lactone 7b (0.87 g, 2.40 mmol) in yield 0.11 g (16%) and from lactone 7c (0.38 g, 1.05 mmol) in yield 0.05 g (18%); white solid; mp 39-41 °C; Rf = 0.16 (hexane/ethyl acetate 3:1, v/v); 1H NMR (600 MHz, CDCl3): δ 1.87 (d, J = 7.1 Hz, 3H, CH3-7), 2.64 (dd, J = 18.4 and 6.6 Hz, one of CH2-3), 3.13 (dd, J = 18.4 and 10.1 Hz, 1H, one of CH2-3), 3.56 (ddd, J = 10.1, 6.6 and 5.3 Hz, 1H, H-4), 3.90 (s, 3H, -OCH3), 4.23 (t, J = 5.3 Hz, 1H, H-5), 4.37 (qd, J = 7.1 and 5.3 Hz, 1H, H-6), 5.61 (s, 1H, -OH), 6.71 (d, J = 2.1 Hz, 1H, H-2’), 6.76 (dd, J = 8.1 and 2.1 Hz, 1H, H-6’), 6.89 (d, J = 8.1 Hz, 1H, H-5’); 13C NMR (150 MHz, CDCl3): δ 23.48 (C-7), 28.38 (C-6), 37.81 (C-3), 45.26 (C-4), 56.05 (-OCH3), 89.70 (C-5), 109.25 (C-2’), 115.00 (C-5’), 119.91 (C-6’), 133.26 (C-1’), 145.24 (C-4’), 147.07 (C-3’), 174.89 (C-2); IR (ATR): νmax = 3406, 1770, 1516, 1269, 1148, 1026, 969, 816, 757, 596 cm-1; HRMS (ESI): m/z calcd for C13H15O4I [M+H]+: 363.0093; found: 363.0094.
3.11.3. Cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a)
Obtained from lactone 8a (1.12 g, 3.55 mmol) in yield 0.24 g (28%); white solid; mp 43-46 °C; Rf = 0.27 (hexane/chloroform/methanol 8:2:1, v/v); Rt = 6.84 min; 1H NMR (600 MHz, CDCl3): δ 1.76 (d, J = 6.6 Hz, 3H, CH3-7), 2.71 (d, J = 17.3 Hz, 1H, one of CH2-3), 3.10 (dd, J = 17.3 and 8.5 Hz, 1H, one of CH2-3), 3.46 (dq, J = 10.4 and 6.6 Hz, 1H, H-6), 3.81 (dd, J = 8.5 and 5.1 Hz, 1H, H-4), 3.88 (s, 3H, -OCH3), 4.64 (dd, J = 10.4 and 5.1 Hz, 1H, H-5), 5.62 (s, 1H, -OH), 6.71 (dd, J = 8.1 and 2.1 Hz, 1H, H-6’), 6.73 (d, J = 2.1 Hz, 1H, H-2’), 6.87 (d, J = 8.1 Hz, 1H, H-5’); 13C NMR (150 MHz, CDCl3): δ 23.26 (C-7), 38.36 (C-3), 43.74 (C-4), 45.56 (C-6), 55.97 (-OCH3), 86.76 (C-5), 111.23 (C-2’), 114.59 (C-5’), 121.00 (C-6’), 129.08 (C-1’), 145.32 (C-4’), 146.34 (C-3’), 176.47 (C-2); IR (ATR): νmax = 3333, 1737, 1519, 1132, 967, 777, 703, 624 cm-1; HRMS (ESI): m/z calcd for C13H15O4Br [M+H]+: 315,0232; found: 315.0230.
3.11.4. Cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a)
Obtained from lactone 9a (0.28 g, 0.78 mmol) in yield 0.06 g (28%); white solid; mp 86-89 °C; Rf = 0.13 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (600 MHz, CDCl3): δ 1.57 (d, J = 6.5 Hz, 3H, CH3-7), 2.72 (d, J = 17.6 Hz, 1H, one of CH2-3), 3.08 (dd, J = 17.6 and 8.5 Hz, 1H, one of CH2-3), 3.45 (dq, J = 10.1 and 6.5 Hz, 1H, H-6), 3.80 (dd, J = 8.5 and 5.2 Hz, 1H, H-4), 3.88 (s, 3H, -OCH3), 4.50 (dd, J = 10.1 and 5.2 Hz, 1H, H-5), 5.61 (s, 1H, -OH), 6.69-6.70 (two m, 2H, H-2’ and H-6’), 6.88 (d, J = 8.4 Hz, 1H, H-5’); 13C NMR (150 MHz, CDCl3): δ 22.38 (C-7), 38.02 (C-3), 43.30 (C-4), 53.96 (C-6), 55.97 (-OCH3), 86.65 (C-5), 111.10 (C-6’), 114.60 (C-5’), 120.96 (C-2’), 129.21 (C-1’), 145.35 (C-4’), 146.41 (C-3’), 176.33 (C-2); IR (ATR): νmax = 3422, 1763, 1517, 1266, 1188, 1128, 1018, 974, 838, 785, 632, 600 cm-1; HRMS (ESI): m/z calcd for C13H15O4Cl [M-H]-: 269.0581; found: 269.0573.
3.11.5. 5-t-Chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b)
Obtained from lactone 9b (0.50 g, 1.39 mmol) in yield 0.07 g (19%); white solid; mp 52-55 °C; Rf = 0.06 (hexane/chloroform/methanol 8:2:1, v/v); 1H NMR (600 MHz, CDCl3): δ 1.60 (d, J = 6.2 Hz, 3H, CH3-7), 2.73 (dd, J = 17.8 and 10 Hz, 1H, one of CH2-3), 3.06 (dd, J = 17.8 and 7.0 Hz, 1H, one of CH2-3), 3.27 (td, J = 10.0 and 7.0 Hz, 1H, H-4), 3.83 (t, J = 10.0 Hz, H-5), 3.90 (s, 3H, -OCH3), 4.50 (dq, J = 10.0 and 6.2 Hz, 1H, H-6), 5.62 (s, 1H, -OH), 6.68 (d, J = 2.0 Hz, 1H, H-2’), 6.72 (dd, J = 8.1 and 2.0 Hz, 1H, H-6’), 6.91 (d, J = 8.1 Hz, 1H, H-5’); 13C NMR (150 MHz, CDCl3): δ 19.76 (C-7), 37.33 (C-3), 46.44 (C-4), 55.97 (-OCH3), 62.64 (C-5), 79.37 (C-6), 109.66 (C-2’), 115.04(C-5’), 119.84 (C-6’), 132.08 (C-1’), 145.30 (C-4’), 146.83 (C-3’), 169.30 (C-2); IR (ATR): νmax = 3394, 1728, 1518, 1215, 1054, 1030, 975, 735, 643, 578 cm-1; HRMS (ESI): m/z calcd for C13H15O4Cl [M+H]+: 271.0737; found: 271.0740.
3.12. Antiproliferative Activity
3.12.1. Chemicals for Biological Tests
The RPMI-1640 (Roswell Park Memorial Institute), McCoy’s 5A, Eagle’s Minimum Essential Medium (EMEM), Dulbecco’s Modified Eagle’s Medium High Glucose (DMEM HG), L-glutamine (L-Glu), DMSO (biological grade), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich® (Steinheim, Germany). The Fetal Bovine Serum (FBS) and advanced RPMI medium were purchased from Gibco/ThermoFisher (Waltham, MA, USA). Antibiotics penicillin-streptomycin (PS) was purchased from Corning (NY, USA). The T-75 cell culture flasks (EasYFlask Nunclon Delta Surface) and 96-well plates were purchased from NUNC (Kamstrupvej, Denmark).
3.12.2. Cell Lines and Cell Cultures
The CLB70 cell line (canine chronic B-cell lymphocytic leukemia) was established at the Department of Pharmacology and Toxicology, Wrocław University of Environmental and Life Sciences [60]. The CLBL-1 cell line (canine B-cell lymphoma) was kindly provided by Dr. Barbara C. Rütgen from the Institute of Immunology, Department of Pathobiology, University of Veterinary Medicine, Vienna [61]. The T-24 (human bladder carcinoma), Caco-2 (human colorectal adenocarcinoma), and NIH/3T3 (mouse embryonic fibroblasts) cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA).
Cell cultures were maintained in their respective growth media, with fetal bovine serum (FBS) concentrations ranging from 10% to 20%, depending on the cell line. Penicillin-streptomycin (PS) and L-glutamine (L-Glu) were consistently added at 1% each to all media. CLB70 cells were cultured in RPMI ADVANCE medium supplemented with 20% FBS, while CLBL-1 cells were maintained in RPMI-1640 medium with 20% FBS. T-24 cells were cultured in McCoy’s 5A medium with 10% FBS, and Caco-2 cells in Eagle's Minimum Essential Medium (EMEM) with 20% FBS. NIH/3T3 cells were maintained in high-glucose DMEM (DMEM HG) with 10% FBS. All cell cultures were grown in T-75 flasks under a humidified atmosphere at 37°C and 5% CO₂ and passaged every 3 days to maintain optimal cell density (70–80% confluence).
3.12.3. MTT Assay
Stock solutions used in the experiments were freshly prepared for each trial by dissolving the tested compounds in biological-grade DMSO. These stock solutions were then diluted with the culture medium appropriate for each specific cell line to achieve the desired final concentrations, which ranged from 3.125 to 100 µM. The final DMSO concentration in each well was maintained below 1%, a level generally considered non-toxic to cells [62]. The MTT assay was employed to evaluate the antiproliferative and cytotoxic effects of the compounds on cancer cell lines, following a previously described protocol with minor modifications [31]. Cells were seeded in 96-well plates at the following densities: 1.5 × 10⁴ cells/mL (NIH/3T3), 2 × 10⁴ cells/mL (T-24, Caco-2), and 3 × 10⁵ cells/mL (CLB70, CLBL-1). Each plate included control wells with untreated cells and wells with vehicle controls (culture medium specific to each cell line).
After 72 h of treatment with the tested compounds, 10 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL) was added to each well. The cells were subsequently incubated with MTT for an additional 3 h to allow the formation of formazan crystals. Following this, 40 µL of lysis buffer (composed of 46 g sodium dodecyl sulfate, 150 mL dimethylformamide, and 183.2 mL Milli-Q water) was added to each well to dissolve the formazan. After a 24 h incubation at room temperature, the optical density (OD) of the resulting formazan product was measured using a spectrophotometric microplate reader (Spark, Tecan, Männedorf, Switzerland) at 570 nm. To correct for background absorbance and minimize potential measurement artifacts, a reference wavelength of 630 nm, at which formazan exhibits negligible absorbance, was used.
The results were determined from three independent experiments (four wells each) and presented as mean IC50 value±SD from at least four independent experiments, each performed in quadruplicate. IC50 values (the concentration of the compound required to inhibit 50% of cell growth) were calculated from dose-response curves.
3.13. Cytototoxicity Toward Red Blood Cells (RBCs)
To evaluate the hemolytic potential and cytotoxicity of the tested compounds, a quantitative hemolysis assay was conducted using human red blood cells (RBCs), following the method described by Pruchnik et al. (2018), with minor modifications [63].
RBCs were obtained from healthy donors through the Blood Donation Center in Wrocław. In accordance with Polish law, the use of erythrocytes from blood centers for experimental purposes does not require approval from an ethics committee. The collected RBCs were gently resuspended and washed three times with cold isotonic phosphate-buffered saline (PBS, pH 7.4) to remove plasma and cellular debris.
All tested compounds were dissolved in biological-grade DMSO. A series of working solutions was prepared by diluting the stock solutions to final concentrations of 6.25, 12.50, 25, 50, 100, and 200 μM. The DMSO content in all experimental and control samples did not exceed 1% (v/v). Control samples contained no tested compounds, while vehicle control samples contained DMSO at the same concentration as the treated samples. Positive control samples (A₁₀₀%) were prepared by inducing complete hemolysis with Milli-Q water. All experiments were performed in triplicate, with three independent replicates for each condition.
The erythrocyte suspension was adjusted to a final hematocrit of 1.2% in all test tubes, with a total sample volume of 1 mL. Samples were incubated at 37 °C under static conditions for 2, 24, and h to assess both short- and long-term cytotoxic effects.
Following incubation, all samples were centrifuged at 5000 rpm for 3 minutes at room temperature. The supernatants were carefully transferred to a 96-well plate, and the absorbance of free hemoglobin was measured at 540 nm using a UV-Vis microplate spectrophotometer (EPOCH, BioTek, Santa Clara, CA, USA). The degree of in vitro hemolysis was calculated based on the amount of released hemoglobin relative to that in the completely hemolyzed positive control sample, according to the following formula:
where H [%] is the percentage of erythrocyte hemolysis, As is absorbance of hemoglobin in supernatant at 540 nm, and A100% refers to the absorbance of hemoglobin in the supernatant following complete hemolysis of the erythrocytes.
4. Conclusions
This study presents the design, synthesis, and biological evaluation of five novel vanillin-derived γ-halo-δ-lactones and δ-halo-γ-lactones bearing a phenolic substituent at the β-position, obtained via a seven-step synthetic pathway. The compounds demonstrated potent and selective antiproliferative activity against both canine (CLBL-1, CLB70) and human (T-24, Caco-2) cancer cell lines, while exhibiting no cytotoxic effects on normal mouse fibroblasts (NIH/3T3). Hemolysis assays further confirmed their safety profile, indicating no toxicity toward human red blood cells (RBCs) and preservation of erythrocyte membrane integrity. Among the tested compounds, trans δ-iodo-γ-lactone 10b showed the most promising biological activity. In addition to their antiproliferative properties, all halolactone derivatives will be subjected to further studies to evaluate their potential antioxidant and anti-inflammatory activities, as their structural features suggest broader, multi-target biological relevance.
Future research will aim first to elucidate the molecular basis of the selective antiproliferative activity exhibited by compound 10b, including a detailed investigation of its anticancer mechanism of action. Understanding these fundamental aspects will be critical to rationally guide further optimization and application. Subsequently, efforts will focus on the design and synthesis of phospholipid-based liposomal nanocarriers incorporating compound 10b to enhance its targeted delivery and therapeutic efficacy against cancer cells.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: 1H NMR spectrum of benzylvanillin (2); Figure S2: 13C NMR spectrum of benzylvanillin (2); Figure S3: DEPT 135 NMR spectrum of benzylvanillin (2); Figure S4: COSY spectrum of benzylvanillin (2); Figure S5: HMQC spectrum of benzylvanillin (2); Figure S6: HMBC spectrum of benzylvanillin (2); Figure S7: 1H NMR spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S8: 13C NMR spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S9: DEPT 135 NMR spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S10: COSY spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S11: HMQC spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S12: HMBC spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S13: 1H NMR spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S14: 13C NMR spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S15: DEPT 135 NMR spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S16: COSY spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S17: HMQC spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S18: HMBC spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S19: 1H NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S20: 13C NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S21: DEPT 135 NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S22: COSY spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S23: HMQC spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S24: HMBC spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S25: 1H NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S26: 13C NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S27: DEPT 135 NMR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S28: COSY spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S29: HMQC spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S30: HMBC spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S31: 1H NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S32: 13C NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S33: DEPT NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S34: COSY spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S35: HMQC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S36: HMBC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S37: 1H NMR spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S38: 13C NMR spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S39: DEPT 135 NMR spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S40: COSY spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S41: HMQC spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S42: HMBC spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S43: 1H NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S44: 13C NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S45: DEPT 135 NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S46: COSY spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S47: HMQC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S48: HMBC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S49: 1H NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S50: 13C NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S51: DEPT 135 NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S52: COSY spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S53: HMQC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S54: HMBC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S55: 1H NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S56: 13C NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S57: DEPT 135 NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S58: COSY spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S59: HMQC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S60: HMBC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S61: 1H NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S62: 13C NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S63: DEPT 135 NMR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S64: COSY spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S65: HMQC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S66: HMBC spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S67: 1H NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S68: 13CNMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S69: DEPT 135 NMR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S70: COSY spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S71: HMQC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S72: HMBC spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S73: 1H NMR spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S74: 13C NMR spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S75: DEPT 135 NMR spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S76: COSY spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S77: HMQC spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S78: HMBC spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S79: 1H NMR spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S80: 13C NMR spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S81: DEPT 135 NMR spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S82: COSY spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S83: HMQC spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S84: HMBC spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S85: 1H NMR spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S86: 13C NMR spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S87: DEPT 135 NMR spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S88: COSY spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S89: HMQC spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S90: HMBC spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S91: 1H NMR spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S92: 13C NMR spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S93: DEPT 135 NMR spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S94: COSY spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S95: HMQC spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S96: HMBC spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S97: 1H NMR spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S98: 13C NMR spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S99: DEPT 135 NMR spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S100: COSY spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S101: HMQC spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S102: HMBC spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b); Figure S103: IR spectrum of benzylvanillin (2); Figure S104: IR spectrum of (E)-4-(4'-(benzyloxy-3'-methoxyphenyl)but-3-en-2-one (3); Figure S105: IR spectrum of (E)-4-(4'-benzyloxy-3'-methoxyphenyl)but-3-en-2-ol (4); Figure S106: IR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoate (5); Figure S107: IR spectrum of (E)-3-(4'-benzyloxy-3'-methoxyphenyl)hex-4-enoic acid (6); Figure S108: IR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7a); Figure S109: IR spectrum of trans-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (7b); Figure S110: IR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-iodo-6-c-methyltetrahydropyran-2-one (7c); Figure S111: IR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-bromoethyl)dihydrofuran-2-one (8a); Figure S112: IR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-bromo-6-c-methyltetrahydropyran-2-one (8b); Figure S113: IR spectrum of cis-4-(4'-benzyloxy-3'-methoxyphenyl)-5-(1-chloroethyl)dihydrofuran-2-one (9a); Figure S114: IR spectrum of 4-r-(4'-benzyloxy-3'-methoxyphenyl)-5-t-chloro-6-c-methyltetrahydropyran-2-one (9b); Figure S115: IR spectrum of cis-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10a); Figure S116: IR spectrum of trans-4-(4'-hydroxy-3'-methoxyphenyl)-5-(1-iodoethyl)dihydrofuran-2-one (10b); Figure S117: IR spectrum of cis-5-(1-bromoethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (11a); Figure S118: IR spectrum of cis-5-(1-chloroethyl)-4-(4'-hydroxy-3'-methoxyphenyl)dihydrofuran-2-one (12a); Figure S119: IR spectrum of 5-t-chloro-4-r-(4'-hydroxy-3'-methoxyphenyl)-6-c-methyltetrahydropyran-2-one (12b);
Author Contributions
Conceptualization, A.D. W.G. and H.P.; methodology, W.G. H.P. and A.P.; writing-original draft, A.D.; writing-review and editing, W.G. H.P., A.W.; visualization, A.D. A.W. and J.S-Z., investigation: A.D. J.S-Z. A.W. E.D. and G.M.; supervision, W.G. and H.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was founded by the Wrocław University of Environmental and Life Sciences (Poland) as the Ph.D. research program number N020/0003/23 „Bon doktoranta SD UPWr”, from the subsidy increased by the minister responsible for higher education and science for the period 2020-2026 in the amount of 2% of the subsidy referred to Art. 387 (3) of the Act of 20 July 2018 – Law on Higher Education and Science, obtained in 2019. The article is part of a PhD dissertation titled “Chemoenzymatic synthesis and determination of biological activity of lactones, vanillin derivatives and increasing the efficiency of their delivery to cancer cells”, prepared during Doctoral School at the Wrocław University of Environmental and Life Sciences. The APC is financed by Wrocław University of Environmental and Life Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
During the preparation of this manuscript, the authors used ChatGPT (free version, OpenAI) for the purposes of language and grammar correction.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sartori, S.K.; Diaz, M.A.N.; Diaz-Muñoz, G. Lactones: Classification, Synthesis, Biological Activities, and Industrial Applications. Tetrahedron 2021, 84, 132001. [Google Scholar] [CrossRef]
- Fortuna, A.M.; de Riscala, E.C.; Catalan, C.A.N.; Gedris, T.E.; Herz, W. Sesquiterpene Lactones from Centaurea Tweediei. Biochem Syst Ecol 2001, 29, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Sokovic, M.; Ciric, A.; Glamoclija, J.; Skaltsa, H. Biological Activities of Sesquiterpene Lactones Isolated from the Genus Centaurea L. (Asteraceae). Curr Pharm Des 2017, 23, 2767–2786. [Google Scholar] [CrossRef]
- Kumar, P.; Wallis, M.; Zhou, X.; Li, F.; Holland, D.C.; Reddell, P.; Münch, G.; Raju, R. Triplinones A-H: Anti-Inflammatory Arylalkenyl α,β-Unsaturated-δ-Lactones Isolated from the Leaves of Australian Rainforest Plant Cryptocarya Triplinervis (Lauraceae). J Nat Prod 2024, 87, 1817–1825. [Google Scholar] [CrossRef]
- Lohray, B.B.; Venkateswarlu, S. Intramolecular SN2 Ring Opening of a Cyclic Sulfate: Synthesis of Erythro-(−)-6-Acetoxy-5-Hexadecanolide—a Major Component of Mosquito Oviposition Attractant Pheromone. Tetrahedron: Asymmeto’ 1997, 8, 633–638. [Google Scholar] [CrossRef]
- McGlacken, G.P.; Fairlamb, I.J.S. 2-Pyrone Natural Products and Mimetics: Isolation, Characterisation and Biological Activity. Nat Prod Rep 2005, 22, 369–385. [Google Scholar] [CrossRef]
- Zhang, L.; An, R.; Wang, J.; Sun, N.; Zhang, S.; Hu, J.; Kuai, J. Exploring Novel Bioactive Compounds from Marine Microbes. Curr Opin Microbiol 2005, 8, 276–281. [Google Scholar] [CrossRef]
- Karthikeyan, A.; Joseph, A.; Nair, B.G. Promising Bioactive Compounds from the Marine Environment and Their Potential Effects on Various Diseases. Journal of Genetic Engineering and Biotechnology 2022, 20, 1–38. [Google Scholar] [CrossRef]
- Lee, K.-H.; Ibuka, T.; Wu, R.-Y.; Geissman, T.A. Structure–Antimicrobial Activity Relationships among the Sesquiterpene Lactones and Related Compounds. Phytochemistry 1977, 16, 1177–1181. [Google Scholar] [CrossRef]
- Kudumela, R.G.; Mazimba, O.; Masoko, P. Isolation and Characterisation of Sesquiterpene Lactones from Schkuhria Pinnata and Their Antibacterial and Anti-Inflammatory Activities. South African Journal of Botany 2019, 126, 340–344. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Gawdzik, B.; Trzepizur, D.; Szymczak, M.; Skiba, G.; Raj, S.; Kramkowski, K.; Lizut, R.; Ostaszewski, R. δ-Lactones—A New Class of Compounds That Are Toxic to E. Coli K12 and R2–R4 Strains. Materials 2021, 14, 2956. [Google Scholar] [CrossRef]
- Koszelewski, D.; Borys, F.; Brodzka, A.; Ostaszewski, R. Synthesis of Enantiomerically Pure 5,6-Dihydropyran-2-Ones via Chemoenzymatic Sequential DKR-RCM Reaction. European J Org Chem 2019, 2019, 1653–1658. [Google Scholar] [CrossRef]
- Paço, A.; Brás, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-Inflammatory and Immunoregulatory Action of Sesquiterpene Lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef]
- Dancewicz, K.; Szumny, A.; Wawrzeńczyk, C.; Gabryś, B. Repellent and Antifeedant Activities of Citral-Derived Lactones against the Peach Potato Aphid. Int J Mol Sci 2020, 21, 8029. [Google Scholar] [CrossRef] [PubMed]
- Fraga, B.M.; Díaz, C.E.; Bailén, M.; González-Coloma, A. Sesquiterpene Lactones from Artemisia Absinthium. Biotransformation and Rearrangement of the Insect Antifeedant 3α-Hydroxypelenolide. Plants 2021, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, M.; Dams, I.; Wawrzeńczyk, C. Feeding Deterrent Activity of Terpenoid Lactones with the p-Menthane System Against the Colorado Potato Beetle (Coleoptera: Chrysomelidae). Environ Entomol 2005, 34, 1433–1440. [Google Scholar] [CrossRef]
- Silva, L.C.; Tauhata, S.B.F.; Baeza, L.C.; Oliveira, C.M.A.; Kato, L.; Borges, C.L.; Soares, C.M.A.; Pereira, M. Argentilactone Molecular Targets in Paracoccidioides Brasiliensis Identified by Chemoproteomics. Antimicrob Agents Chemother 2018, 62, e00737–18. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Sharma, P.; Bhardwaj, U.; Kaur, R. Dehydrocostus Lactone: A Comprehensive Review on Its Isolation, Chemical Transformations, and Pharmacological Potential. Discover Chemistry 2025, 2, 131. [Google Scholar] [CrossRef]
- Zhi, X.Y.; Zhang, Y.; Li, Y.F.; Liu, Y.; Niu, W.P.; Li, Y.; Zhang, C.R.; Cao, H.; Hao, X.J.; Yang, C. Discovery of Natural Sesquiterpene Lactone 1-O-Acetylbritannilactone Analogues Bearing Oxadiazole, Triazole, or Imidazole Scaffolds for the Development of New Fungicidal Candidates. J Agric Food Chem 2023, 71, 11680–11691. [Google Scholar] [CrossRef]
- Rasul, A.; Parveen, S.; Ma, T. Costunolide: A Novel Anti-Cancer Sesquiterpene Lactone. Bangladesh J Pharmacol 2012, 7, 6–13. [Google Scholar] [CrossRef]
- Kim, D.Y.; Choi, B.Y. Costunolide—A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential. Int J Mol Sci 2019, 20, 2926. [Google Scholar] [CrossRef]
- Wzorek, A.; Kwiatkowska, M.; Hodorowicz, M.; Kalwat, K.; Arabski, M.; Płoszaj, P.; Gonciarz, W.; Omelaniuk, A.; Gmiter, D.; Kaca, W.; et al. Syntheses, Structures and Biological Activities of New Pyridinyl Lactones. J Mol Struct 2025, 1319. [Google Scholar] [CrossRef]
- Kim, Y.; Sengupta, S.; Sim, T. Natural and Synthetic Lactones Possessing Antitumor Activities. Int J Mol Sci 2021, 22, 1052. [Google Scholar] [CrossRef]
- Kamizela, A.; Gawdzik, B.; Urbaniak, M.; Lechowicz, Ł.; Białońska, A.; Kutniewska, S.E.; Gonciarz, W.; Chmiela, M. New γ-Halo-δ-Lactones and δ-Hydroxy-γ-Lactones with Strong Cytotoxic Activity. Molecules 2019, 24, 1875. [Google Scholar] [CrossRef] [PubMed]
- Kamizela, A.; Gawdzik, B.; Urbaniak, M.; Lechowicz, Ł.; Białońska, A.; Gonciarz, W.; Chmiela, M. Synthesis, Characterization, Cytotoxicity, and Antibacterial Properties of Trans-γ-Halo-δ-Lactones. ChemistryOpen 2018, 7, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Mereyala, H.B.; Joe, M. Cytotoxic Activity of Styryl Lactones and Their Derivatives. Curr Med Chem Anticancer Agents 2001, 1, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, A.; Koszuk, J.F.; Modranka, J.; Różalski, M.; Krajewska, U.; Janecka, A.; Studzian, K.; Janecki, T. Synthesis and Cytotoxic Activity of γ-Aryl Substituted α-Alkylidene-γ-Lactones and α-Alkylidene-γ-Lactams. Bioorg Med Chem 2008, 16, 4872–4882. [Google Scholar] [CrossRef]
- Park, S.; Kim, S.; Shin, D. Arylnaphthalene Lactones: Structures and Pharmacological Potentials. Phytochemistry Reviews 2021, 20, 1033–1054. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Thi Tran, L.T.; Truong Tan, T.; Pham, P.T.V.; Nguyen, L.T.K.; Nguyen, H.T.; Ho, D.V.; Tran, M.H. Isolation, Structural Elucidation, and Cytotoxic Activity Investigation of Novel Styryl-Lactone Derivatives from Goniothalamus Elegans: In Vitro and in Silico Studies. RSC Adv 2023, 13, 17587–17594. [Google Scholar] [CrossRef]
- Wzorek, A.; Gawdzik, B.; GŁadkowski, W.; Urbaniak, M.; Barańska, A.; Malińska, M.; Woźniak, K.; Kempinska, K.; Wietrzyk, J. Synthesis, Characterization and Antiproliferative Activity of β-Aryl-δ-Iodo-γ-Lactones. J Mol Struct 2013, 1047, 160–168. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Siepka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Białońska, A.; Poradowski, D.; Drynda, A.; Urbaniak, M. Synthesis and Anticancer Activity of Novel Halolactones with β-Aryl Substituents from Simple Aromatic Aldehydes. Tetrahedron 2013, 69, 10414–10423. [Google Scholar] [CrossRef]
- Gładkowski, W.; Skrobiszewski, A.; Mazur, M.; Gliszczyńska, A.; Czarnecka, M.; Pawlak, A.; Obmińska-Mrukowicz, B.; Maciejewska, G.; Białońska, A. Chiral δ-Iodo-γ-Lactones Derived from Cuminaldehyde, 2,5-Dimethylbenzaldehyde and Piperonal: Chemoenzymatic Synthesis and Antiproliferative Activity. Tetrahedron Asymmetry 2016, 27, 227–237. [Google Scholar] [CrossRef]
- Gładkowski, W.; Włoch, A.; Pawlak, A.; Sysak, A.; Białońska, A.; Mazur, M.; Mituła, P.; Maciejewska, G.; Obmińska-Mrukowicz, B.; Kleszczyńska, H. Preparation of Enantiomeric β-(2’,5’-Dimethylphenyl) Bromolactones, Their Antiproliferative Activity and Effect on Biological Membranes. Molecules 2018, 23, 3035. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, A.; Gładkowski, W.; Mazur, M.; Henklewska, M.; Obmińska-Mrukowicz, B.; Rapak, A. Optically Active Stereoisomers of 5-(1-Iodoethyl)-4-(4′-Isopropylphenyl)Dihydrofuran-2-One: The Effect of the Configuration of Stereocenters on Apoptosis Induction in Canine Cancer Cell Lines. Chem Biol Interact 2017, 261, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, A.; Gładkowski, W.; Kutkowska, J.; Mazur, M.; Obmińska-Mrukowicz, B.; Rapak, A. Enantiomeric Trans β-Aryl-δ-Iodo-γ-Lactones Derived from 2,5-Dimethylbenzaldehyde Induce Apoptosis in Canine Lymphoma Cell Lines by Downregulation of Anti-Apoptotic Bcl-2 Family Members Bcl-xL and Bcl-2. Bioorg Med Chem Lett 2018, 28, 1171–1177. [Google Scholar] [CrossRef]
- Włoch, A.; Sengupta, P.; Szulc, N.; Kral, T.; Pawlak, A.; Henklewska, M.; Pruchnik, H.; Sykora, J.; Hof, M.; Gładkowski, W. Biophysical and Molecular Interactions of Enantiomeric Piperonal-Derived Trans β-Aryl-δ-Iodo-γ-Lactones with Cancer Cell Membranes, Protein and DNA: Implications for Anticancer Activity. Int J Biol Macromol 2025, 303, 140476. [Google Scholar] [CrossRef]
- Włoch, A.; Stygar, D.; Bahri, F.; Bażanów, B.; Kuropka, P.; Chełmecka, E.; Pruchnik, H.; Gładkowski, W. Antiproliferative, Antimicrobial and Antiviral Activity of β-Aryl-δ-Iodo-γ-Lactones, Their Effect on Cellular Oxidative Stress Markers and Biological Membranes. Biomolecules 2020, 10, 1594. [Google Scholar] [CrossRef]
- Olatunde, A.; Mohammed, A.; Ibrahim, M.A.; Tajuddeen, N.; Shuaibu, M.N. Vanillin: A Food Additive with Multiple Biological Activities. European Journal of Medicinal Chemistry Reports 2022, 5. [Google Scholar] [CrossRef]
- Kafali, M.; Finos, M.A.; Tsoupras, A. Vanillin and Its Derivatives: A Critical Review of Their Anti-Inflammatory, Anti-Infective, Wound-Healing, Neuroprotective, and Anti-Cancer Health-Promoting Benefits. Nutraceuticals 2024, 4, 522–561. [Google Scholar] [CrossRef]
- Arya, S.S.; Rookes, J.E.; Cahill, D.M.; Lenka, S.K. Vanillin: A Review on the Therapeutic Prospects of a Popular Flavouring Molecule. Advances in Traditional Medicine 2021, 21, 415–431. [Google Scholar] [CrossRef]
- Yousuf, M.; Shamsi, A.; Queen, A.; Shahbaaz, M.; Khan, P.; Hussain, A.; Alajmi, M.F.; Rizwanul Haque, Q.M.; Imtaiyaz Hassan, M. Targeting Cyclin-Dependent Kinase 6 by Vanillin Inhibits Proliferation of Breast and Lung Cancer Cells: Combined Computational and Biochemical Studies. J Cell Biochem 2021, 122, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.A.; Wu, S.L.; Lo, H.Y.; Hsiang, C.Y.; Ho, T.Y. Vanillin Inhibits Matrix Metalloproteinase-9 Expression through down-Regulation of Nuclear Factor-ΚB Signaling Pathway in Human Hepatocellular Carcinoma Cells. Mol Pharmacol 2009, 75, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Yazan, L.S.; Ismail, N.; Ismail, M. Apoptosis and Cell Cycle Arrest of Human Colorectal Cancer Cell Line HT-29 Induced by Vanillin. Cancer Epidemiol 2009, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Srinual, S.; Chanvorachote, P.; Pongrakhananon, V. Suppression of Cancer Stem-like Phenotypes in NCI-H460 Lung Cancer Cells by Vanillin through an Akt-Dependent Pathway. Int J Oncol 2017, 50, 1341–1351. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, Y.M.; Oh, T.I.; Kim, B.M.; Lim, B.O.; Lim, J.H. Vanillin Suppresses Cell Motility by Inhibiting STAT3-Mediated HIF-1α MRNA Expression in Malignant Melanoma Cells. Int J Mol Sci 2017, 18, 532. [Google Scholar] [CrossRef]
- Gendron, D. Vanillin: A Promising Biosourced Building Block for the Preparation of Various Heterocycles.
- Chem 2022, 10, 949355. [CrossRef]
- Yıldırım, M.; Ünver, H.; Necip, A.; Çimentepe, M. Design, Synthesis, and Biological Evaluation of Novel Vanillin-Derived Hydrazone Compounds with Antimicrobial, Anticancer, and Enzyme Inhibition Activities, along with Molecular Structure and Drug-Likeness Assessment. Biochem Biophys Res Commun 2025, 775, 152173. [Google Scholar] [CrossRef]
- Scipioni, M.; Kay, G.; Megson, I.L.; Kong Thoo Lin, P. Synthesis of Novel Vanillin Derivatives: Novel Multi-Targeted Scaffold Ligands against Alzheimer’s Disease. Medchemcomm 2019, 10, 764–777. [Google Scholar] [CrossRef]
- Li, Z.H.; Liu, H.M.; Fan, Z.Y.; Pang, W.; Cheng, L.P. Design, Synthesis and Evaluation of Vanillin Derivatives as Dual-Target Inhibitors for the Treatment of Alzheimer’s Disease. Bioorg Med Chem 2025, 129, 118296. [Google Scholar] [CrossRef]
- Carrasco-Gomez, R.; Keppner-Witter, S.; Hieke, M.; Lange, L.; Schneider, G.; Schubert-Zsilavecz, M.; Proschak, E.; Spänkuch, B. Vanillin-Derived Antiproliferative Compounds Influence Plk1 Activity. Bioorg Med Chem Lett 2014, 24, 5063–5069. [Google Scholar] [CrossRef]
- Li, Y.; Manickam, G.; Ghoshal, A.; Subramaniam, P. More Efficient Palladium Catalyst for Hydrogenolysis of Benzyl Groups. Synth Commun 2006, 36, 925–928. [Google Scholar] [CrossRef]
- Lanthier, C.; Payan, H.; Liparulo, I.; Hatat, B.; Lecoutey, C.; Since, M.; Davis, A.; Bergamini, C.; Claeysen, S.; Dallemagne, P.; et al. Novel Multi Target-Directed Ligands Targeting 5-HT4 Receptors with in Cellulo Antioxidant Properties as Promising Leads in Alzheimer’s Disease. Eur J Med Chem 2019, 182, 111596. [Google Scholar] [CrossRef]
- Gadhiya, S.; Madapa, S.; Kurtzman, T.; Alberts, I.L.; Ramsey, S.; Pillarsetty, N.K.; Kalidindi, T.; Harding, W.W. Tetrahydroprotoberberine Alkaloids with Dopamine and σ Receptor Affinity. Bioorg Med Chem 2016, 24, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, M.; Barone, G.; Guariniello, L.; Iadonisi, A. Facile Cleavage of Carbohydrate Benzyl Ethers and Benzylidene Acetals Using the NaBrO3/Na2S2O4 Reagent under Two-Phase Conditions. Tetrahedron Lett 1999, 40, 8439–8441. [Google Scholar] [CrossRef]
- Rodebaugh, R.; Debenham, J.S.; Fraser-Reid, B. Debenzylation of Complex Oligosaccharides Using Ferric Chloride. Tetrahedron Lett 1996, 37, 5477–5478. [Google Scholar] [CrossRef]
- Giri, R.S.; Roy, S.; Dolai, G.; Manne, S.R.; Mandal, B. FeCl3-Mediated Boc Deprotection: Mild Facile Boc-Chemistry in Solution and on Resin. ChemistrySelect 2020, 5, 2050–2056. [Google Scholar] [CrossRef]
- Gładkowski, W.; Siepka, M.; Żarowska, B.; Białońska, A.; Gawdzik, B.; Urbaniak, M.; Wawrzeńczyk, C. Chalcone-Derived Lactones: Synthesis, Whole-Cell Biotransformation, and Evaluation of Their Antibacterial and Antifungal Activity. Molecules 2023, 28, 3800. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Resch, V.; Wallner, S.; Lienhart, W.D.; Sattler, J.H.; Resch, J.; MacHeroux, P.; Kroutil, W. Biocatalytic Organic Synthesis of Optically Pure (S)-Scoulerine and Berbine and Benzylisoquinoline Alkaloids. Journal of Organic Chemistry 2011, 76, 6703–6714. [Google Scholar] [CrossRef]
- Krishnamurty, H.G.; Ghosh, S. Synthesis of Dihydrocurcumin. Indian J Chem 1986, 25B, 411–412. [Google Scholar]
- Pawlak, A.; Ziolo, E.; Kutkowska, J.; Blazejczyk, A.; Wietrzyk, J.; Krupa, A.; Hildebrand, W.; Dziegiel, P.; Dzimira, S.; Obminska-Mrukowicz, B.; et al. A Novel Canine B-Cell Leukaemia Cell Line. Establishment, Characterisation and Sensitivity to Chemotherapeutics. Vet Comp Oncol 2017, 15, 1218–1231. [Google Scholar] [CrossRef]
- Rütgen, B.C.; Hammer, S.E.; Gerner, W.; Christian, M.; de Arespacochaga, A.G.; Willmann, M.; Kleiter, M.; Schwendenwein, I.; Saalmüller, A. Establishment and Characterization of a Novel Canine B-Cell Line Derived from a Spontaneously Occurring Diffuse Large Cell Lymphoma. Leuk Res 2010, 34, 932–938. [Google Scholar] [CrossRef]
- Villarroel, A.; Duff, A.; Hu, T. DMSO Inhibits Human Cancer Cells and Downregulates the Expression of Cdk2 and Cyclin A. The FASEB Journal 2020, 34, 03158–1. [Google Scholar] [CrossRef]
- Pruchnik, H.; Włoch, A.; Bonarska-Kujawa, D.; Kleszczyńska, H. An In Vitro Study of the Effect of Cytotoxic Triorganotin Dimethylaminophenylazobenzoate Complexes on Red Blood Cells. Journal of Membrane Biology 2018, 251, 735–745. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of unsaturated γ,δ-carboxylic acid intermediate 6 from vanillin (1). Reagents and conditions: (A) BnBr, K2CO3, EtOH, r.t., 48 h (B) acetone, 10% NaOH, r.t., 24 h (C) MeOH, NaBH4, ice bath 1h, then r.t., 23 h (D) triethyl orthoacetate, propionic acid, 138 °C, 6h (E) 10% NaOH, EtOH, reflux, 5 h.
Scheme 1.
Synthesis of unsaturated γ,δ-carboxylic acid intermediate 6 from vanillin (1). Reagents and conditions: (A) BnBr, K2CO3, EtOH, r.t., 48 h (B) acetone, 10% NaOH, r.t., 24 h (C) MeOH, NaBH4, ice bath 1h, then r.t., 23 h (D) triethyl orthoacetate, propionic acid, 138 °C, 6h (E) 10% NaOH, EtOH, reflux, 5 h.
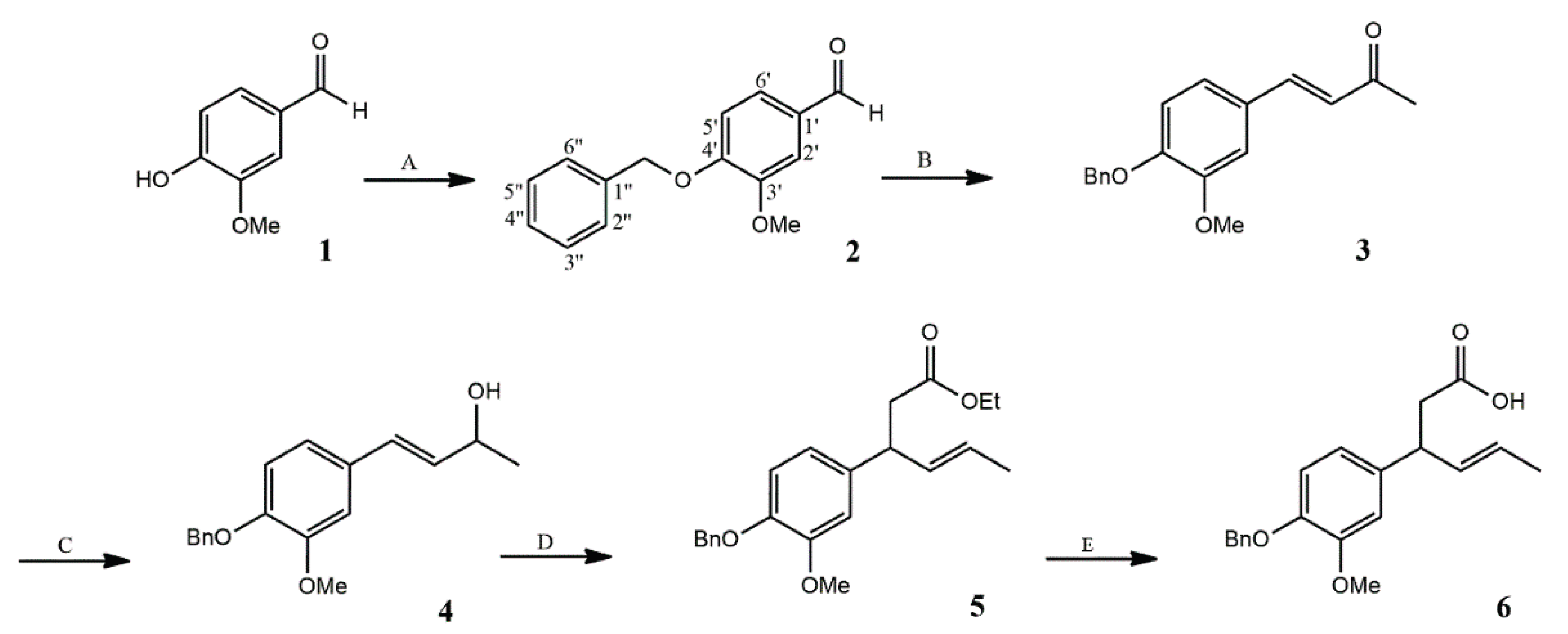
Scheme 2.
Halolactonization of acid 6. Reagents and conditions: (A) I2 in KI, Et2O, NaHCO3, r.t., 20 h (B) NBS, acetic acid, THF, r.t., 24 h (C) NCS, acetic acid, THF, r.t., 24 h.
Scheme 2.
Halolactonization of acid 6. Reagents and conditions: (A) I2 in KI, Et2O, NaHCO3, r.t., 20 h (B) NBS, acetic acid, THF, r.t., 24 h (C) NCS, acetic acid, THF, r.t., 24 h.
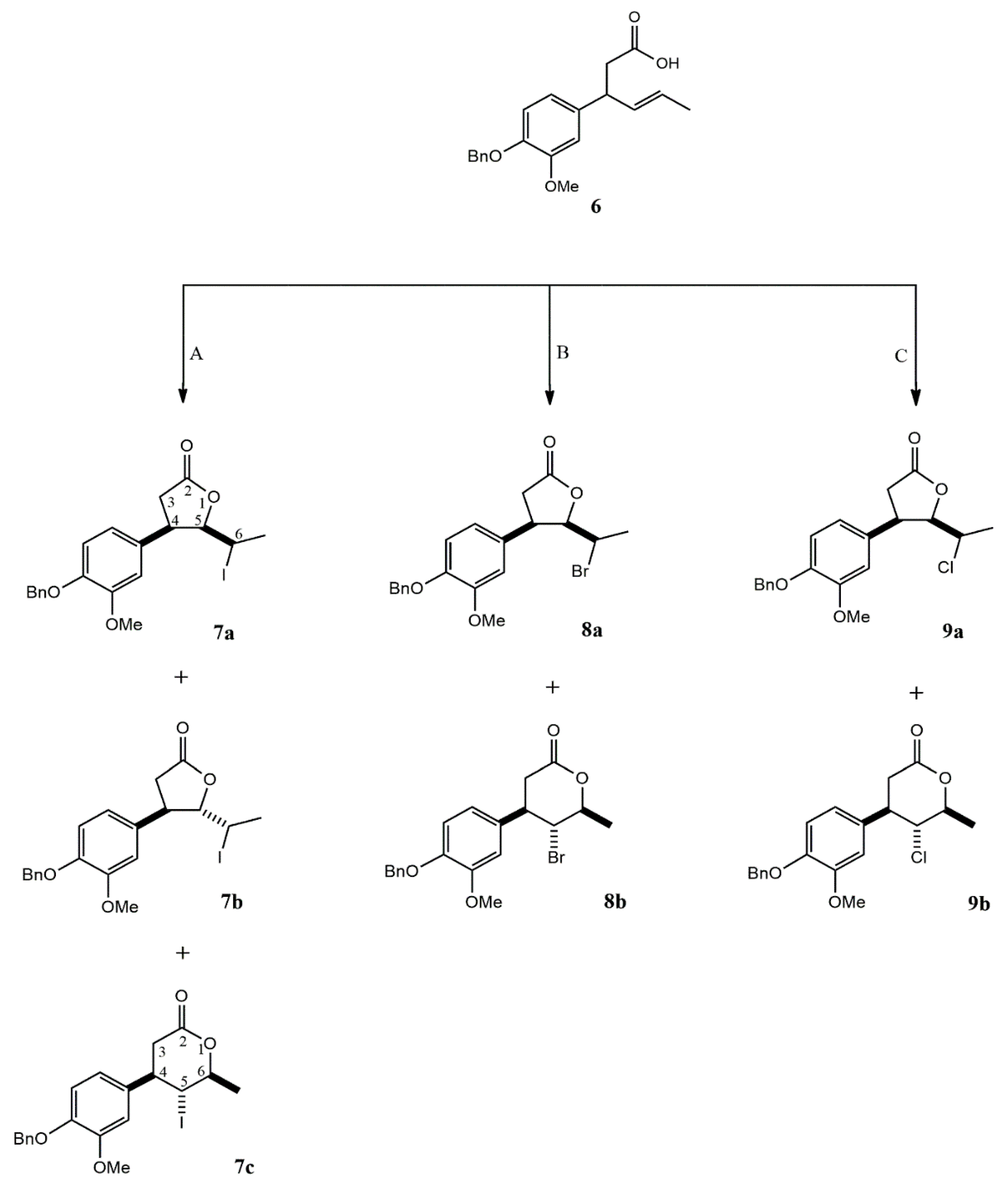
Scheme 3.
Debenzylation of cis δ-halo-γ-lactones 7a-9a and trans δ-iodo-γ-lactone 7b.
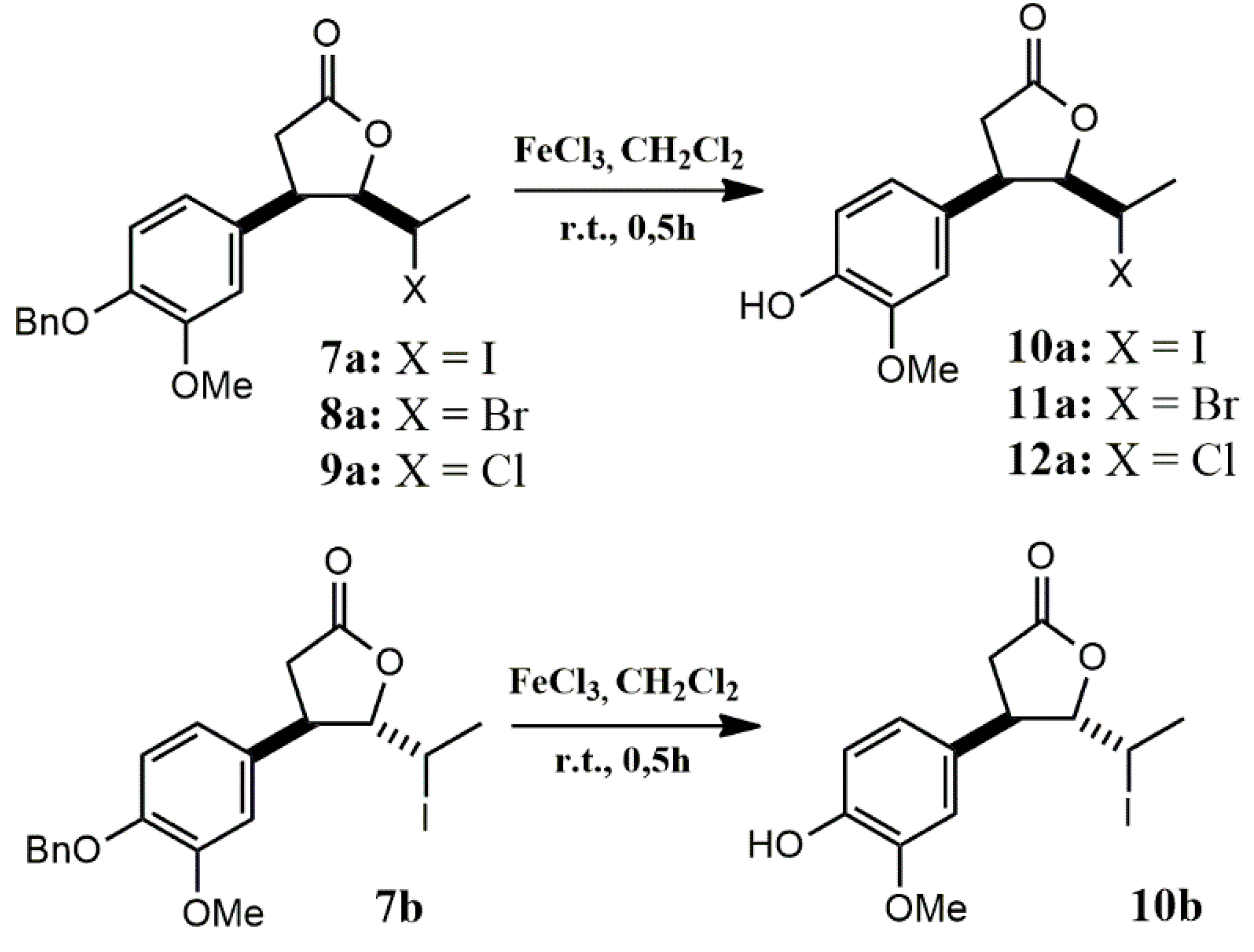
Scheme 4.
Debenzylation of γ-halo-δ-lactones 7c and 9b.
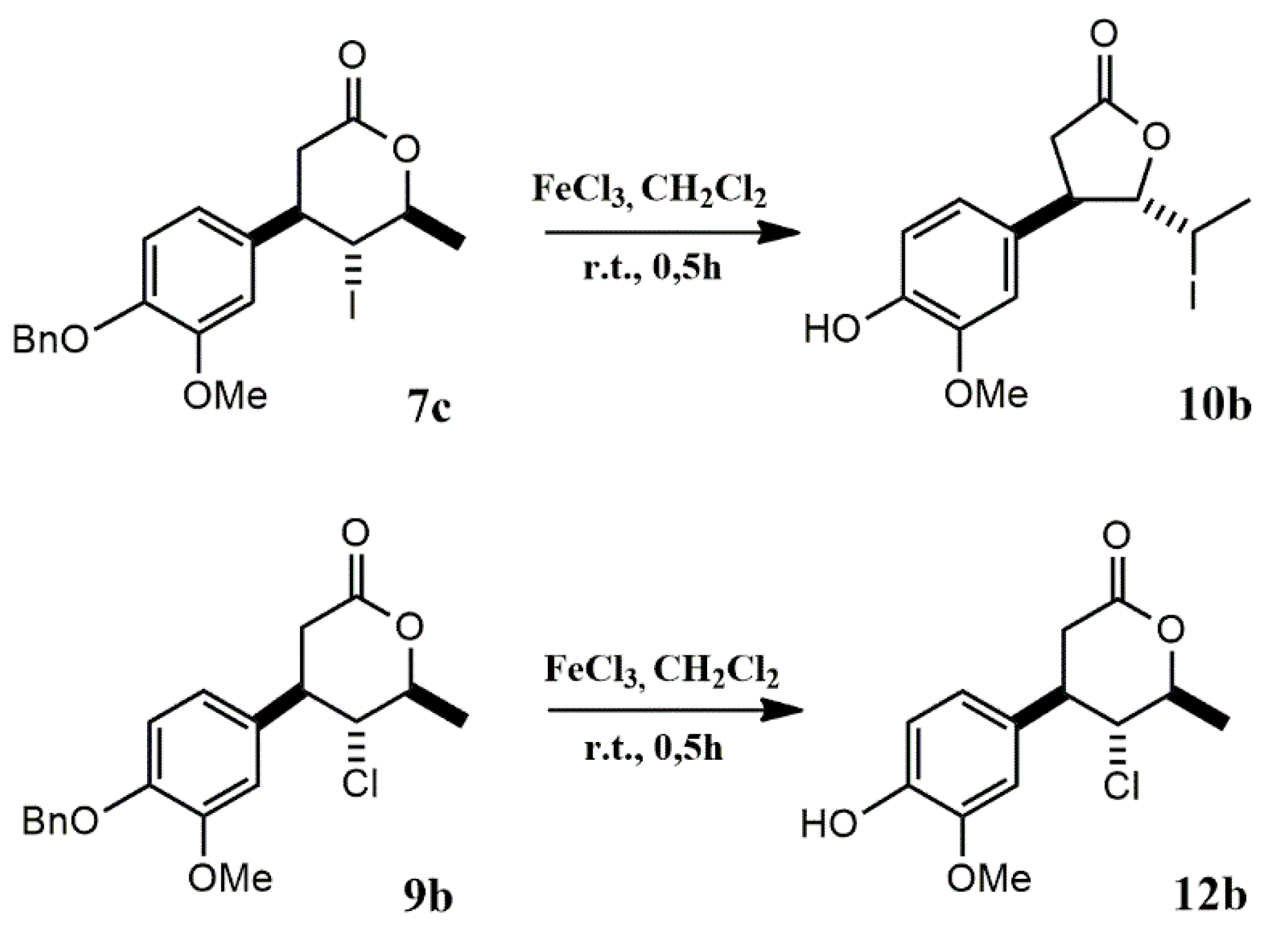
Scheme 5.
Proposed translactonization mechanism leading to the formation of trans δ-iodo-γ-lactone 10b during FeCl3-mediated debenzylation of γ-iodo-δ-lactone 7c.
Scheme 5.
Proposed translactonization mechanism leading to the formation of trans δ-iodo-γ-lactone 10b during FeCl3-mediated debenzylation of γ-iodo-δ-lactone 7c.
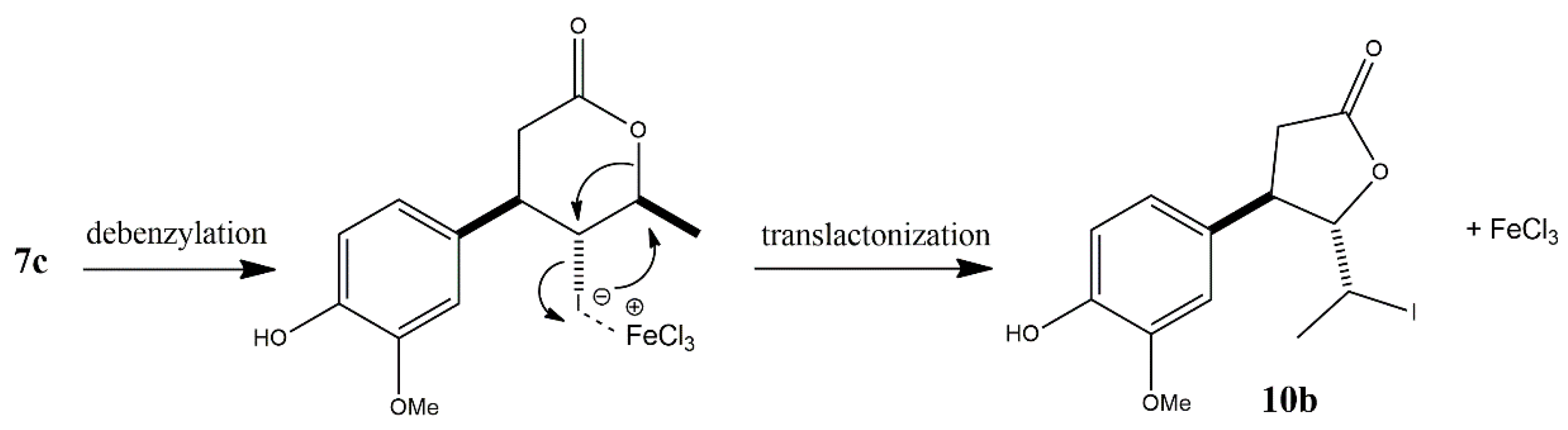
Figure 1.
Percentage of hemolysis after 2 h in the presence of the compounds. Error bars represent mean ±SD (n=3).
Figure 1.
Percentage of hemolysis after 2 h in the presence of the compounds. Error bars represent mean ±SD (n=3).
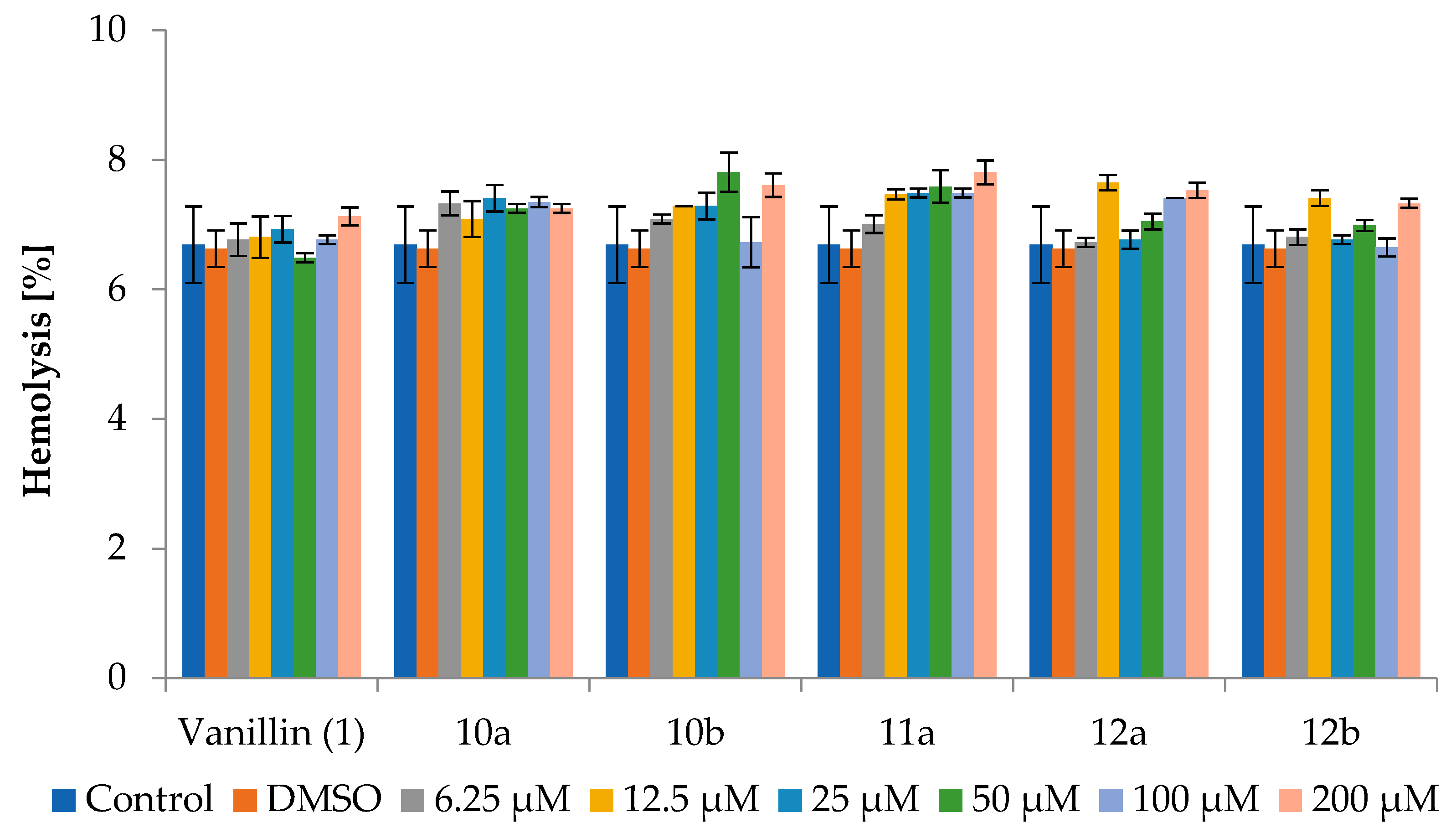

Table 1.
Antyproliferative activity of vanillin (1) and vanillin-derived halolactones (10a,b, 11a, 12a,b) toward CLBL-1, CLB70, T-24, CaCo-2 and NIH/3T3 cell lines after 72 h of exposure.
Table 1.
Antyproliferative activity of vanillin (1) and vanillin-derived halolactones (10a,b, 11a, 12a,b) toward CLBL-1, CLB70, T-24, CaCo-2 and NIH/3T3 cell lines after 72 h of exposure.
| Compound | IC50 [µg/mL] | ||||
|---|---|---|---|---|---|
| CLBL-1 | CLB70 | T-24 | CaCo-2 | NIH/3T3 | |
| 1 | 72.44±9.371 | 72.79±2.28 | 48.46±13.16 | >100 | >100 |
| 10a | 73.55±8.82 | 69.65±13.69 | 98.06±10.42 | >100 | >100 |
| 10b | 46.26±4.07 | 71.48±7.47 | 63.42±6.16 | >100 | >100 |
| 11a | 63.18±1.60 | 76.17±5.30 | >100 | 76.40±9.99 | >100 |
| 12a | 76.67±11.47 | 75.21±6.25 | >100 | 88.05±4.37 | >100 |
| 12b | 85.72±15.72 | 71.17±11.64 | >100 | 72.54±19.76 | >100 |
1 Data represent mean values ± SD from at least four independent experiments.
Table 2.
Percentage of hemolysis after 24 h and 72 h in the presence of of vanillin (1) and vanillin-derived halolactones (10a,b, 11a, 12a,b).
Table 2.
Percentage of hemolysis after 24 h and 72 h in the presence of of vanillin (1) and vanillin-derived halolactones (10a,b, 11a, 12a,b).
| 24 h | ||||||||
|---|---|---|---|---|---|---|---|---|
| [µM] | Vanillin (1) | 10a | 10b | 11a | 12a | 12b | DMSO | Control |
| 6.25 | 8.05±0.151 | 7.70±0.07 | 7.61±0.13 | 8.50±0.15 | 8.10±0.0 | 7.34±0.06 | 7.92±0.07 | 7.65±0.44 |
| 12.50 | 7.47±0.15 | 7.83±0.51 | 7.87±0.07 | 8.10±0,15 | 7.52±0.32 | 7.47±0.06 | ||
| 25 | 7.25±0.13 | 7.95±0.13 | 7.74±0.48 | 7.78±0.33 | 7.43±0.48 | 7.83±0.06 | ||
| 50 | 7.16±0.60 | 7.30±0.19 | 8.45±0.07 | 7.83±0.07 | 7.78±0.13 | 7.96±0.80 | ||
| 100 | 7.87±0.26 | 7.70±0.51 | 7.92±0.15 | 8.05±0.07 | 8.41±0.07 | 7.87±0.06 | ||
| 200 | 8.01±0.13 | 8.41±0.07 | 8.50±0.19 | 8.99±0.69 | 7.70±0.26 | 8.01±0.06 | ||
| 72 h | ||||||||
| 6.25 | 9.10±0.15 | 9.22±0.15 | 9.65±0.15 | 9.09±0.09 | 9.06±0.46 | 8.77±0.15 | 9.58±0.79 | 9.26±0.93 |
| 12.50 | 9.21±0.31 | 9.38±0.09 | 8.94±0.57 | 9.25±0.38 | 8.98±0.00 | 9.04±0.69 | ||
| 25 | 9.21±0.15 | 9.43±0.30 | 9.17±0.15 | 8.99±0.01 | 9.74±0.53 | 9.59±0.52 | ||
| 50 | 9.54±0.40 | 8.86±0.17 | 9.75±0.40 | 9.87±0.01 | 9.03±0.09 | 9.08±0.09 | ||
| 100 | 9.42±0.23 | 9.26±0.38 | 9.66±0.65 | 9.16±0.09 | 8.71±0.17 | 9.15±0.09 | ||
| 200 | 10.22±0.15 | 9.92±0.35 | 11.00±0.90 | 10.34±1.20 | 9.67±0.80 | 10.52±0.75 | ||
1 Data represent mean values ± SD from at least three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



