Submitted:
22 July 2025
Posted:
24 July 2025
Read the latest preprint version here
Abstract
Many different theories have been proposed to explain ageing, but none has yet achieved eithertheoretical consensus on its nature or sufficient empirical support to convince proponents ofcompeting theories to abandon their own in favour of a more predictive alternative. In thispaper, I present a testable proposal that may or may not achieve such a milestone oncerigorously evaluated. Most current approaches to ageing focus on identifying factors that affectits rate by observing their impact on lifespan. While this yields useful information, it does notprovide Popperian knowledge about what ageing is—only about what modifies it. Manyprevailing perspectives remain rooted in outdated paradigms such as damage accumulation orare shaped by teleologically biased reasoning that seeks purpose behind the ageing process. Incontrast, this paper offers a non-teleological explanation, grounded in a conception of life as afundamentally information-based phenomenon. From this view, ageing is understood as apurely information-processing problem. Specifically, I propose that ageing, properly speaking,emerged when eukaryotic cells began transmitting to their descendants two semi-independentcodes of biological information: the genetic code and the epigenetic code. This is not merely atheoretical concern for academic debate—our lack of a fundamental understanding of whatageing is, and how it emerged during evolution, continues to hinder the development ofinnovative biomedical strategies that could lead to more effective treatments. The frameworkdescribed here is amenable to experimental testing and thus holds the potential to generatescientifically valid Popperian knowledge—knowledge about the nature of ageing itself, notmerely about factors that influence its rate.
Keywords:
ageing theory
; epigenetic inheritance
; biological information theory
; non-genetic inheritance
1. Introduction
Ageing has long been a central enigma in biology. The prevailing view frames it as a multifactorial process. In recent years, a broad consensus has emerged among ageing researchers to study it within the framework known as the Hallmarks of Ageing [1,2,3]. This integrative model seeks to address the apparent complexity of ageing by incorporating diverse insights into a cohesive conceptual structure. The Hallmarks of Ageing concept is primarily a mechanistic framework describing nine interlinked biological processes (e.g., genomic instability, telomere attrition, mitochondrial dysfunction) that drive ageing. It draws upon both damage-based and programmatic views—along with evolutionary concepts—to explain how these hallmarks arise and persist. However, it does not explicitly unify all formal evolutionary theories of ageing, such as antagonistic pleiotropy or mutation accumulation. Rather, it acknowledges that these evolutionary theories provide the why (i.e., why organisms do not repair damage indefinitely), while the Hallmarks framework addresses the how—the molecular and cellular processes that fail with age.
Although the Hallmarks of Ageing paradigm does not inherently prioritise one hallmark over another, the historical development of ageing research has led to proposals that do exactly that. Telomere attrition, mitochondrial dysfunction, DNA damage, deregulated nutrient sensing, cellular senescence, protein damage, and others have all been proposed as primary drivers of ageing [4]. Among the various models proposed, I wish to focus on one that suggests ageing results from the loss of epigenetic information [5,6,7]. This idea has gained significant attention in recent years, particularly following its popularisation by David Sinclair and colleagues under the term “The Information Theory of Ageing.” However, I would like to clarify that this conceptual framework—ageing as a phenomenon driven by the loss of epigenetic information—was developed independently by myself and first made publicly available in a preprint in 2017 [7], prior to the earliest explicit mention of the theory by Sinclair [6]. In this paper, I present a further step in the description of the proposed framework: the Double Code Hypothesis of Ageing. This model posits that biological information is transmitted intergenerationally through both the genome and the epigenome. Ageing arises when mitotically dividing cells lose access to youthful epigenetic information that is only selectively restored during meiosis. This theory not only provides a mechanistic basis for ageing and rejuvenation, but also unifies observations on epigenetic inheritance, disease prevalence trends, and the limitations of current damage- and program-based theories.
A central argument of the model proposed in this paper is that one of the fundamental assumptions underpinning the current framework of biological knowledge is flawed: namely, the view that DNA is the sole or primary source of intergenerational transmission of biological information. This paper presents a refined and focused articulation of ideas originally developed in a broader theoretical work. The full model—including supporting data on population-wide trends, disease prevalence, and broader biomedical and societal implications—has been published as a preprint on Zenodo [8]. Here, I concentrate on formalising the central theoretical framework—the Double Code Hypothesis of Ageing—and its implications for understanding the mechanisms of ageing and rejuvenation. Ideas beyond The Double Code Hypothesis of Ageing included in [8] preprint will be made available elsewhere by this author.
2. The Double-Code Hypothesis of Ageing: Ageing as a Consequence of the Intergenerational Inheritance of a Dual Code of Information—The Genome and the Epigenome.
Modern biology largely accepts the view that the genome is the primary, if not exclusive, carrier of intergenerational information. This paper challenges that assumption. I argue that the biological code transmitted from parents to offspring comprises both genetic and epigenetic information. It is this dual mode of transmission that gives rise to the phenomenon we recognise as ageing. This model has profound implications: it redefines ageing not as a mere consequence of deterioration, but as an outcome of how life is inherited.
I propose that ageing is simply a side consequence of the way epigenetic information is transmitted across generations. Ageing succeeded as an evolutionary mechanism, enabling the emergence of more complex organisms while mitigating the increased risk of external death associated with the complex tasks that such organisms can perform [9]. Thus, ageing is an unpleasant process at the individual level, but essential at the population level to ensure the preservation of accumulated biological information [10]. To properly understand ageing, one must address this simple question: what is inherited intergenerationally, and how?
2.1. What Is Inherited Intergenerationally, and How?
Since Watson and Crick proposed the structure of DNA in 1953 [11], explaining how genetic information could be replicated and transmitted across generations [12], DNA has been regarded as the primary bearer of intergenerational information. This perspective suggested that epigenetic information was largely reset with each generation and derived solely from DNA sequences. The discovery of two waves of epigenetic reprogramming during gametic and embryonic stages initially reinforced this view [13,14,15,16,17,18,19].
However, recent research suggests that certain epigenetic marks can evade reprogramming and be inherited. Multiple studies have investigated the phenomenon of Transgenerational Epigenetic Inheritance (TEI) in various model organisms, including mammals [20], plants [21], invertebrates [22], and fission yeast [23]. A recent study demonstrated that methylation marks artificially placed at ectopic loci in mice (normally unmethylated in wild-type individuals) were transmitted to offspring, despite the well-documented waves of epigenetic reprogramming. The ectopic epigenetic marks were undetectable in the primordial germ cells (PGCs)—specialised cells that serve as embryonic precursors to gametes—or in the gametes themselves (sperm and oocytes). Similar findings were observed post-fertilisation and implantation, with the methylation marks absent in the blastocyst stage but detectable in the epiblast cells [24]. These observations suggest that, although reprogramming may erase certain marks during one of the known waves, the memory of these epigenetic marks can still be transmitted to subsequent generations. This is likely due to crosstalk and reinforcement mechanisms between different epigenetic marks [24]. This provides a mechanistic explanation for how, even in mammals—where the physiological significance of TEI remains a topic of debate [20,24]—epigenetic information can be transmitted across generations.
What, then, is the true physiological significance of TEI? The primary issue I find with most studies on TEI is that they are typically approached from a Lamarckian or ‘purpose-driven’ perspective. The flaw lies in this interpretation which is teleologically biased. In this paper, I propose a non-Lamarckian framework for understanding how epigenetic traits are transmitted across generations. This framework also explains how ageing emerged during evolution as a consequence of the uncoupling of genetic and epigenetic inheritance [9,25]. The epigenetic code is inherited in a manner that is semi-independent of the genetic code. The physiological significance of TEI extends far beyond the simplistic Lamarckian idea of trait acquisition.
I propose that germline cells possess an epigenetic repair mechanism that corrects accumulated epimutations in an organism [7]. This repaired epigenetic information is then transmitted to the offspring, who exhibit a youthful phenotype. However, neither the somatic cells of the parents nor those of the offspring can access this repair mechanism again. We are only afforded one opportunity for repair, as only germline cells have this capability. Consequently, somatic cells accumulate genetic and epigenetic defects over time, ultimately leading to cell death, primarily due to the loss of proper epigenetic information.
In certain scenarios, most likely due to saturability issues, the cellular machinery becomes unable to repair all the epigenetic errors accumulated over previous generations and present in the germline (see Figure 1D). By contrast, when no such saturability issues are present (see Figure 1C), most epigenetic defects are corrected. The concept of saturability during the proposed meiotic epigenetic repair mechanism is part of a newly proposed framework that I have termed Epigenetic Degeneration [7], further developed in [8], and to be expanded upon in a forthcoming dedicated paper. The consequence of this process is the progressive accumulation of epigenetic defects across generations.
If the continuity of life depend on recovering lost information, as the above words imply, and previously proposed [7,9], how ‘immortal’ organisms achieve extremely long lifespans? Simply, by always maintaining constant access to epigenome repair and not restricting it just to a group of cells (as ageing organisms do). By establishing a semi-separated inheritance of the genome and the epigenome, eukaryotic cells effectively invented ageing as we properly understand it. For a discussion on ageing in non-eukaryotic organisms (see bellow in section 5). In summary, for eukaryotic cells ageing is a mitotic issue. Meiotic divisions have the ability to bypass ageing.
To understand in greater detail why information management leads to ageing, it is first necessary to discuss what constitutes a living organism. This discussion is provided in the next section.
2.2. What Is Life?
At its core, life is information—but why? What exactly is life? Living beings can be defined as structures formed using accumulated biological information, containing the instructions necessary for actively interacting with the surrounding environment to ensure survival (maintaining their own structures) and self-replication (creating new ‘instances’ with the same or a similar biological code). They achieve this while preserving the accumulated information to sustain these tasks. Life ends for an organism when it can no longer sustain its own structures (see Figure 2D). However, the death of an individual does not signify the end of its species. As long as other individuals of the same species retain the accumulated information, this form of life continues to progress. For a species, extinction occurs when all its individuals have died, resulting in the irremediable loss of its accumulated information forever.
On 11 December 1867, James Clerk Maxwell proposed a thought experiment in a letter to a fellow physicist and mathematician. He imagined a hypothetical being capable of sorting gas molecules by speed. This being, later known as Maxwell’s demon, could control a small door between two chambers, allowing fast-moving molecules to pass one way and slow-moving molecules the other, thereby creating a temperature difference. This appeared to violate the second law of thermodynamics by reducing entropy without expending energy. In the 20th century, physicists such as Leo Szilard, Claude Shannon, and Rolf Landauer clarified that the demon’s sorting depends on information. The processes of acquiring, storing, and processing this information require energy, counteracting the apparent entropy decrease. Landauer’s principle states that erasing information, such as the demon’s memory, generates entropy, thereby preserving the second law of thermodynamics [26,27,28]. Maxwell’s demon is not a real physical entity, but a thought experiment that illustrates the connection between thermodynamics and information theory. Modern understanding reconciles Maxwell’s demon with the second law of thermodynamics. While the demon decreases entropy locally, the overall entropy of the universe still increases when accounting for the energy costs of information processing.
Living beings can be described as highly organised structures composed of numerous molecular mechanisms that operate similarly to Maxwell’s demons [29]. By utilising biological information, these mechanisms are able to locally decrease entropy and drive processes that would not spontaneously occur without this informational guidance. This local decrease in entropy is achieved at the expense of energy, which organisms obtain from their environment, ensuring that the second law of thermodynamics remains upheld on a universal scale. An organism can cease to be a living entity (die) through two primary mechanisms: 1) external causes of death, involving the destruction of structures built using biological information (e.g., trauma or ectopically originated diseases); or 2) internal causes of death, associated with the loss or degradation of biological information which disrupts the organism’s ability to maintain its ordered state (the ageing process).
Ageing, like life itself, should be understood as a purely information-based phenomenon.
2.3. Why Does Ageing Exist?
From an organism’s point of view, ageing does not make sense at all. If that is the case, why did ageing persist—or even succeed—during evolution? First, we must acknowledge that evolutionary success is relative. Consider Thermus aquaticus, a bacterium that thrives in environments where a human would not survive more than ten minutes [30]. Would you call such an organism a failure—or would you be the failure in a planetary environment with a mean temperature of 70 °C?
Moreover, at the population and evolutionary levels, life as we know it would be inconceivable without ageing. First, ‘non-ageing organisms’ can only succeed if they have a low chance of dying from external causes—this imply that they are not highly complex and are not exposed to a complex lifestyle [9]. According to this proposal, such organisms can continuously reconstruct their epigenome from their genome, thereby avoiding ageing—that is, avoiding internal causes of death. Second, most ageing organisms, through a programmed developmental process that favours epigenetic information loss (see below in section 5), eliminate older ‘instances’ from the population while producing new, young ‘instances’ with full functional capacity. This is achieved by controlling which information somatic cell lines (the older ‘instances’) and the germline (the source of new ‘instances’) have access to. This proposal posits that only the germline has access to the information needed to rebuild the epigenome from the genome (see Figure 1B). This mechanism not only supports a more complex lifestyle (by reducing the risk of extinction due to all individuals dying from external causes) but also accelerates the evolutionary process of complex organisms by orders of magnitude. The constant production of new, complex ‘instances’ creates opportunities for novelties to emerge in a world with limited natural resources and external risks associated with complex lifestyles. By inventing meiosis, eukaryotic cells introduced the ability to mix biological information through crossover—preventing the accumulation of genetic errors—and enabled the rebuilding of epigenetic information exclusively in new ‘instances.’ This avoided the accumulation of epigenetic errors, effectively establishing ageing as a mechanism uniquely suited for this purpose (see the section 2.4 below). At evolutionary scales, the invention of meiosis (incorporating both crossover and ageing) is analogous to the transition from horse-drawn vehicles to rocket engines in human transportation systems, enabling the achievement of more complex tasks (e.g., escaping Earth’s gravitational pull).
To explain why ageing prevailed in evolution in plain terms: if one assumes that, in nature, events occur randomly, then any complex organism that does not age would still eventually die from external causes—unless its chances of dying from such causes were extremely low. But this presents an oxymoron. A complex organism cannot have very low chances of dying from external causes; it can only have relatively lower probabilities compared to other organisms (as is the case in longer-lived species; see below in section 4.1). Therefore, even if ‘complex immortal’ organisms did arise during evolution—and it is likely that some incipient forms did—they most likely succumbed to external causes of death and thus did not survive to the present day. In non-teleological terms: evolution did not deliberately programme a loss of epigenetic information; rather, lineages that experienced such a loss were able to tolerate a level of complexity that would otherwise have been evolutionarily unsustainable [9]. In teleological terms, ageing is the price we pay for being complex [7]. Put even more simply: being complex and being ‘immortal’ don’t go together. (see below in section 3.3)
The views expressed in this section revisit Weismann’s ideas on ageing [10], which are no longer the dominant perspective in contemporary ageing research. All this ideas are discussed in more detail in section 3 below.
2.4. Interplay Between Genome and Epigenome: The Ratchet Mechanisms
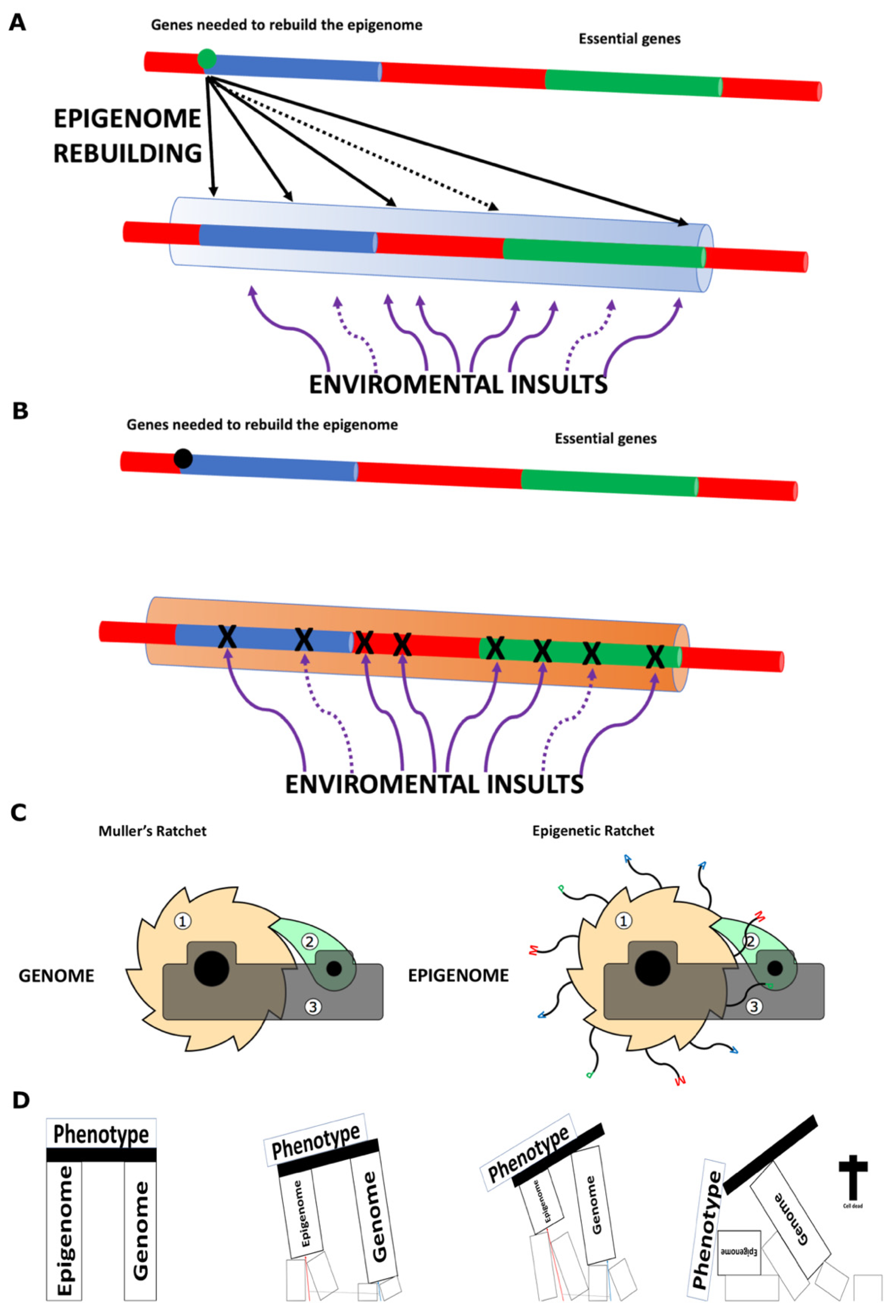
Organisms accumulate both genetic and epigenetic mistakes during ageing. While epigenetic mistakes are reversible, genetic mistakes are not. How might this impact ‘non-ageing organisms? A clue can be found in a study on Arabidopsis thaliana, which revealed a relationship between mutation bias and epigenetic features. The study showed that de novo mutations occur less frequently in coding regions, particularly in essential genes, due to the protective role of the epigenome in these regions [31]. This finding suggests a scenario where a dual protection mechanism may be at play: genome sequences can be used to repair epigenetic defects, while epigenetic regions, in turn, ‘protect’ critical genomic areas from accumulating mutations (see Figure 2A). Such a mechanism could enable ‘non-ageing organisms’ to preserve biological information—both genomic and epigenomic—for extended periods, thereby achieving remarkably long lifespans. In contrast, ageing organisms gradually lose epigenetic features, potentially rendering these regions more susceptible to mutagenic compounds. This results in the accumulation of not only epigenetic defects but also genetic defects (see Figure 2B), thereby compounding the overall burden of biological information loss.
In practical terms, this creates a dual ratchet mechanism acting during meiosis: the well-known genetic Muller’s ratchet and an epigenetic ratchet, which essentially is the ageing process [7] (see Figure 2C). The genetic ratchet prevents the excessive accumulation of genetic defects in populations of complex organisms through genetic crossover. Genetic crossovers avoid linkage of deleterious mutations in a chromosome and creates variability which is later shaped by natural selection resulting in the survival of the ones that have a functional genetic information. The epigenetic ratchet (the ageing process) serves a similar purpose but for epigenetic defects. The rejuvenation process, in which the information necessary to restore a functional epigenome is read (see Figure 1B), established, and transferred to new generations (new ‘instances’) via the epigenome, is also followed by natural selection, resulting in the survival of those with functional epigenetic information. In both cases, slightly deleterious mutations or epimutations can accumulate in asexually dividing cells. If this scenario persists over several generations, the likelihood of this evolutionary line becoming extinct increases significantly.
Ageing is the accumulation of deleterious epimutations and mutations within an organism. The soma, consisting of asexually dividing cells, is subjected to both genetic and epigenetic ratchets, whereas germline cells, through meiosis, possess the mechanisms to avoid these accumulations. Thus, ageing is fundamentally a mitotic issue. Germline cells bypass ageing while producing new ‘instances’ to preserve functional genetic and epigenetic information at the population level. Only new ‘instances’ with improved phenotypes—derived from their own genetic and epigenetic codes—can establish successful evolutionary lines and avoid extinction. At the individual level, an organism remains alive as long as its biological structures—encoded in its genetic and epigenetic codes—can withstand environmental challenges (see Figure 2D). If not killed by external factors, an organism will eventually succumb to ageing (an internal cause of death) due to accumulating defects in the epigenome and genome. Epigenetic defects accumulate faster than genetic ones [7]. Once the protective epigenetic shields are compromised, genomic errors accelerate (see Figure 2B). Ultimately, death from ageing is primarily caused by the accumulation of epigenetic defects [7].
Figure 2.
Shields and Ratchets. A. Epigenome rebuilding illustrates how, during meiosis, the correct epigenetic information required to produce the young phenotype is restored to generate a youthful phenotype. Epigenetic factors are proposed to act as shields, protecting genomic regions from mutations [31]. B. As ageing progresses and epigenetic defects accumulate (or epigenetic factors are lost), the shielding effect diminishes, leading to an increased rate of genetic mutations. C. Schematic representation of Muller’s genetic ratchet (left) and the proposed epigenetic ratchet-like mechanism (right). and B. Genetic and epigenetic information are plotted as in Figure 1. D. Schematic representation of how genomic and epigenomic information are indispensable for maintaining phenotypic functionality. The combined action of the epigenetic ratchet (the ageing process, red arrow) and genetic Muller’s ratchet (blue arrow) in non-meiotic cells compromises the phenotypic functionality of living organisms and ultimately leads to cell death. C and D. Extracted from Marsellach 2017 [7].
Figure 2.
Shields and Ratchets. A. Epigenome rebuilding illustrates how, during meiosis, the correct epigenetic information required to produce the young phenotype is restored to generate a youthful phenotype. Epigenetic factors are proposed to act as shields, protecting genomic regions from mutations [31]. B. As ageing progresses and epigenetic defects accumulate (or epigenetic factors are lost), the shielding effect diminishes, leading to an increased rate of genetic mutations. C. Schematic representation of Muller’s genetic ratchet (left) and the proposed epigenetic ratchet-like mechanism (right). and B. Genetic and epigenetic information are plotted as in Figure 1. D. Schematic representation of how genomic and epigenomic information are indispensable for maintaining phenotypic functionality. The combined action of the epigenetic ratchet (the ageing process, red arrow) and genetic Muller’s ratchet (blue arrow) in non-meiotic cells compromises the phenotypic functionality of living organisms and ultimately leads to cell death. C and D. Extracted from Marsellach 2017 [7].
3. What Distinguishes the Proposed Description of Ageing in This Work from Current Ageing Studies?
Historically, the scientific approach to ageing has been primarily shaped by two key factors: 1) theoretical considerations about the meaning and evolutionary origins of ageing (theories of ageing); and 2) experimental methods aimed at studying how ageing organisms deteriorate, with a focus that initially centred on factors affecting lifespan and more recently expanded to include healthspan. In recent years, these approaches have increasingly emphasised rejuvenation strategies, particularly inspired by the discovery and application of Yamanaka factors [32]. The evolution of experimental methods in the field has been significantly driven by groundbreaking advancements.
It should be noted that, despite the relatively large number of research papers dedicated to studying ageing, the very nature of ageing itself remains the elephant in the room—a fundamental question that appears to be deliberately avoided by much of the ageing research community and for which there is clearly no minimal consensus [7,33,34,35].
Most of the theories of ageing that have been proposed can be broadly classified into three categories:
- Evolutionary Theories of Ageing: These theories apply principles of natural selection and evolutionary trade-offs to explain why ageing exists at all, rather than being eliminated by evolution. Three classical theories are: Mutation Accumulation Theory, proposed by Peter Medawar [36], Antagonistic Pleiotropy Theory, proposed by George C. Williams [37] and Disposable Soma Theory, proposed by Thomas Kirkwood [38].
- Damage (Stochastic) Theories of Ageing: These propose that ageing results from random but cumulative damage to cells, tissues, and molecules, eventually overwhelming the body’s repair systems over time.
- Programmatic Theories of Ageing: These suggest that ageing is (at least partially) driven by genetic or regulatory programs, as if ageing is an extension or byproduct of developmental processes. This perspective is largely supported by the observation that there are conserved mechanisms, such as dietary restriction [39], as well as mutations or treatments that affect ageing.
The majority of these theories are reviewed in the following recent publications [4,40,41,42]. No single theory has yet fully explained ageing in a manner accepted by the entire scientific community. Most researchers consider ageing to be a multifaceted process involving genetic, environmental, and stochastic (random) components. As explained above, to address this complexity, integrative frameworks such as the Hallmarks of Ageing have been developed, aiming to incorporate multiple insights into a cohesive and comprehensive model [1,2,3].
In this section, I first review the Evolutionary Theories of Ageing, which—as previously explained—focus on why we age. In the following section, I discuss the Damage Theories of Ageing and the Programmatic Theories of Ageing together, as both are primarily concerned with how we age. I then examine how trade-offs have been addressed in ageing research, and conclude by emphasising the importance of considering life and ageing as phenomena fundamentally grounded in information theory.
3.1. Evolutionary Theories of Ageing
As previously explained, the basic assumption underlying this paper is that both the genome and the epigenome cooperate in the intergenerational transfer of biological information. Consequently, when analysing the evolutionary theories of ageing, I offer a key critique: most (if not all) of these theories are affected by a foundational flaw rooted in a core pillar of modern biological thinking—namely, the assumption that intergenerational information is transmitted exclusively, or nearly exclusively, through the genome. As a result, these theories attribute all evolutionary responsibility to the genetic code alone. In other words, DNA is considered the sole substrate upon which natural selection acts to shape evolution. I contend that this narrow assumption is a major reason why the ageing process remains poorly understood and fundamentally unresolved.
I propose that the phenotype—the target of natural selection—is determined entirely by the combined information from both the genome and the epigenome at any given moment (see Figure 2D). According to the ageing framework presented in this paper, ageing is primarily a process of epigenetic information loss, which can also result in genetic information loss, as illustrated in Figure 2B and discussed in the main text. This proposal implies that natural selection is continuously at play, acting as the engine that sustain life, with a prominent but often underestimated role played by negative selection. I propose that we remain alive as long as we possess the necessary biological information to sustain ourselves (see Figure 2D, Marsellach 2017 [7], and Marsellach 2021 [9]). This implies that natural selection acts more strongly—specifically through negative selection—against aged individuals who are less fit due to accumulated defects (lost information) and that even death from old age can ultimately be attributed to the action of negative selection. Life is the absence of negative selection on information-based organisms. This view contrasts with many ageing theories that argue natural selection declines in intensity with chronological age [40]. Such claims are based on a distinction between a reproductive period (when fitness is critical for passing genes to the next generation) and a post-reproductive period. However, very few wild animal species exhibit a significant post-reproductive lifespan comparable to that of human females (i.e., a well-defined menopausal period followed by many years of life). Due to predation, disease, and other ecological pressures, most wild animals simply do not live long enough to experience a prolonged post-reproductive stage, even if their physiology might allow it. Remarkably, the idea of a decline in natural selection with chronological age has gained popularity despite lacking a clear molecular mechanism to support it. This perspective can only be understood within a narrow view of natural selection—unfortunately, the prevailing one—that focuses primarily on its role at the population level (i.e., passing genes to subsequent generations) while largely overlooking the significant and continuous role of negative selection acting within individuals themselves.
The Epigenetic Degeneration paradigm that I have developed (see Marsellach, 2017 [7]; Marsellach, 2025 [8]; and a forthcoming dedicated paper based on Marsellach, 2025) proposes the existence of a saturable epigenetic repair mechanism that operates during meiosis [7]. This implies that the greater the number of epigenetic defects present at the onset of meiosis, the more defects will be transmitted to the next generation. This proposal provides a clear mechanistic explanation for why mating with a more epigenetically degenerated individual (e.g., an aged organism, according to the ageing framework advanced in this paper) would result, on average, in offspring with lower fitness. This is a testable prediction.
Indeed, the relationship between parental age and offspring health or fitness is a topic of active research across multiple species, including humans. However, no clear consensus has been reached, as contrasting results have been reported in model organisms. For instance, some studies in Drosophila suggest that older parental age can be beneficial [43], whereas studies in C. elegans indicate that offspring of young mothers may exhibit impairments compared to those of older mothers [44]. Conversely, other research demonstrates that successive generations of older parents in both species result in clear detriments [45]. In some cases, contradictory results have even been reported within the same study [46]. In humans, the bulk of evidence points to a correlation between advanced parental age (both maternal and paternal) and an increased risk of certain health or developmental issues in offspring. Although more systematic studies are necessary, these observations align well with the framework proposed in this paper for several reasons. First, in studies covering only one generation (or a few), the inheritance of a given character is expected to occur in a random manner. This can be explained by the Schrödinger’s cat-like inheritance described earlier by this author [9,25]. In a scenario involving a saturable epigenetic repair mechanism during meiosis [7], some defects are corrected while others persist, creating variability in the inheritance process. Second, while the set of studies is limited, the key insight is that several rounds of either beneficial [7] or detrimental [45] epigenetic inheritance may be required to clearly observe effects. The randomness inherent in single-generation observations may only reveal trends at the population level [8] or after multiple rounds of meiosis. Iterative corrective processes during meiosis should, over time, increase the likelihood of correcting the chosen character of interest, even if occasional failures occur due to the saturability issue. Conversely, repeated instances of saturability in meiotic epigenetic correction are expected to progressively increase the number of epigenetic defects present in a population or individual over time. Third, some studies might have simply misinterpreted their findings. For example, Aguilar et al. (2023) [43] claim that offspring of older parental flies are fitter because they weigh more than those of younger parents. However, it remains unclear whether they actually selected for fitter organisms or merely—albeit unintentionally—for obese flies.
The fact that natural selection acts on the phenotype—which, as proposed here, is governed by the combination of genotype and epigenotype (see Figure 2D)—relieves some of the excessive emphasis placed solely on the genotype. This genotype-centric view has strongly influenced modern theories of ageing by suggesting that selection acts on individuals primarily through its effects on individual genes, thereby downplaying the importance of preserving the full spectrum of biological information at the group level. While it is undeniable that selection operates at the level of individuals, this work proposes that phenotypes are shaped not only by the genomic sequences inherited from one’s parents, but also by the epigenomic information transmitted across generations.
Epigenetics has existed since life forms began to be governed by DNA, following the RNA world. DNA alone is meaningless unless it is read, and its stored information is used appropriately. Epigenetic mechanisms ensure that this information is interpreted correctly, allowing for cellular differentiation, the emergence of complex multicellular life, and, as proposed in this paper as well as in previous works of mine, the evolutionary development of ageing [7,9,25]. By controlling the accessibility of information in somatic and germline cells, epigenetics has provided a critical evolutionary advantage, helping overcome external causes of death associated with complex lifestyles and enabling the emergence of more sophisticated life forms [9]. This idea reinstates the ageing advantage for group selection. Eukaryotic cells and the organisms formed from them have an evolutionary advantage over other organisms because, thanks to epigenetic mechanisms, they are able to build multicellular organisms (as differentiated multicellularity could not exist without epigenetics), and, thanks to the ageing process (which also relies on epigenetics), multicellular organisms can speed up their evolution even though they have lost the ability of unicellular organisms to easily divide through binary fission (see below in section 5).
Moreover, ageing not only accelerates the evolutionary process by increasing the frequency of new individuals, thereby enhancing variability, but it also serves as a mechanism to eliminate older individuals while ensuring the continuity of species information preservation [10]. The allocation of fitter biological mechanisms exclusively to young individuals provides a clear and precise explanation for why natural selection operates differently as an organism or cell ages. Functional epigenetic information is concentrated in younger individuals, benefiting their descendants more effectively compared to offspring produced by older individuals.
For all the reasons outlined above, I propose a return to a Weismannian perspective on the evolutionary necessity of ageing—as a mechanism that facilitates the survival of the group [10]. While August Weismann originally framed ageing as an adaptive process that eliminates older individuals to make room for the young, the model presented in this paper offers a modern reinterpretation. It suggests that ageing arises from the intergenerational transmission of dual biological codes—genetic and epigenetic—and that the gradual accumulation of uncorrected epigenetic defects across generations (under conditions of saturable repair) may serve an implicit group-level function. By limiting the long-term propagation of increasingly defective epigenomes, this process could contribute to maintaining the overall integrity and adaptability of the population.
3.2. Damage Theories of Ageing and Programmatic Theories of Ageing
Historically, ageing was first approached through damage theories of ageing, with the concept of ‘wear and tear’ as the cause of damage accumulation. Technological advances during the Industrial Revolution inspired comparisons between living organisms and mechanical devices (e.g., steam engines, machinery), but due to the lack of knowledge about detailed biological mechanisms—such as the structure of DNA or protein metabolism— ‘wear and tear’ was more a metaphor than a rigorous hypothesis. Over time, it evolved into modern damage theories, where ‘wear and tear’ was redefined in terms of mutation accumulation, oxidative stress, DNA breaks, protein misfolding, and other molecular disruptions, providing a more precise scientific foundation for the original notion of bodily breakdown over time.
The discovery that the rate of ageing could be modulated by single mutations [47] led to the rise of programmatic theories of ageing, with more and more pathways being identified whose alteration could influence the ageing process. This, in turn, spurred the emergence of a whole new subfield within the ageing research community, often referred to as ‘The New Biology of Ageing’ [48,49]. This research effort focuses on identifying pathways and modifications that affect lifespan, with the aim of discovering ‘pieces of a puzzle’ that altogether contribute to ageing. These findings suggest that ageing might not be solely a random accumulation of damage but could, at least in part, be genetically regulated, challenging the traditional view of ageing as a purely stochastic process driven by damage accumulation. Proponents of programmatic theories of ageing argue that if ageing were entirely stochastic, it would be far more difficult to extend lifespan in a systematic, gene- or treatment-dependent manner [50].
Several key genetic pathways have been extensively studied for their roles in regulating ageing, with some of the most important including: the insulin/IGF-1 signalling pathway, which has been shown to extend lifespan through both genetic and pharmacological interventions [47,51]; the mTOR (Mechanistic Target of Rapamycin) pathway, which is central to nutrient sensing and longevity, with inhibitors like rapamycin demonstrating significant lifespan-extension effects [52,53]; the sirtuins pathway, particularly SIRT1, which regulates stress resistance and metabolism, playing a key role in ageing and longevity [54,55]; FOXO transcription factors, which contribute to stress resistance and lifespan extension across multiple species [56,57]; the AMP-activated protein kinase (AMPK) pathway, a master regulator of energy homeostasis, which influences longevity by modulating metabolic and stress responses [58,59]; the growth hormone/IGF-1 axis, which in mammals plays a complex role, with reduced signalling often correlating with lifespan extension; and the mitochondrial pathways, which impact ageing by regulating mitochondrial dynamics, biogenesis, and reactive oxygen species (ROS) production [60].
Ultimately, damage-based and programmatic theories of ageing converged into the previously mentioned unified framework known as the Hallmarks of Ageing paradigm [1,2,3], which established a shared conceptual structure for studying ageing as a multifactorial process. Many of the pathways discussed above were incorporated into the hallmark of deregulated nutrient sensing. According to this paradigm, ageing is driven by the intricate interplay of all the hallmarks, reinforcing the view that it is a highly complex and interconnected biological phenomenon.
This proposal challenges the prevailing perspective by arguing that, among all currently recognised hallmarks of ageing, only one is truly fundamental: the epigenome [7]. The widespread assumption—shared by both damage-based and programmatic theories—that all hallmarks are merely interconnected “pieces of a puzzle” has led the ageing field into a conceptual dead-end, where it largely remains today. As stated earlier, the primary reason for this impasse is the foundational flaw addressed in this paper: the failure to recognise epigenetic intergenerational inheritance as a core biological mechanism.
In summary, the proposal defended in this paper outlines a model suggesting that ageing, properly speaking, originated alongside the emergence of meiosis in eukaryotic cells. It posits that ageing is linked to how biological information is managed during cellular division. Cells undergoing mitosis are thought to have limited access to certain information necessary for maintaining youthfulness, whereas meiotic cells retain this access. As a result, mitotically dividing cells undergo ageing, while meiotic cells can reset or avoid it. It is worth noting that multicellular organisms, although they reproduce sexually as a species, are fundamentally composed of mitotically dividing cells [7]. This model thus provides an explanation for why organisms age, despite reproducing through a process that—at least temporarily—escapes ageing.
This model helps to resolve the dilemma identified by Sinclair and colleagues as the central mystery underlying the model that they propose, the “Information Theory of Ageing”: the location of the “backup copy” of functional epigenetic information and how it is re-established during rejuvenation [5]. According to the framework proposed in this paper, new generations receive their “backup copy” of youthful epigenetic information through the epigenome inherited from their parents. The conceptual flaw outlined in this paper—namely, the neglect of epigenetic intergenerational inheritance—prevents Sinclair and colleagues from arriving at this explanation.
This proposal arose from serendipitous observations in fission yeast, which led me to formulate a theoretical model in which the loss of epigenetic information is not just a correlate of ageing, but its fundamental cause. While Sinclair’s work has been instrumental in both conceptualizing and in demonstrating that epigenetic marks can be manipulated to induce partial rejuvenation [61,62,63], his model focuses more on the somatic loss and reprogramming of epigenetic states. In contrast, this hypothesis frames ageing as a consequence the transmission of information across generations tightly coupled to the mechanism of inheritance itself. In this view, ageing and transgenerational epigenetic inheritance are two sides of the same coin. My approach provides a mechanistic and evolutionary answer to their central mystery. The epigenetic intergenerational transmission is, in effect, the source of the rejuvenated epigenetic state Sinclair’s model postulates. Thus, while both models agree on the central role of epigenetic information loss in ageing, they diverge in their mechanistic premises. My framework views the dual inheritance of genome and epigenome as the root cause of both ageing and rejuvenation, whereas Sinclair’s model remains centred on the reprogrammability of somatic epigenomes and assumes the existence of an undefined epigenetic archive. The distinction has both conceptual and experimental implications.
The conceptual ideas defended in this paper were already implicitly present in my previous publications [7,9,25], which were openly shared and actively promoted by this author to relevant individuals who might have had an interest. Ironically, I arrived at the model proposed in this paper by serendipitously identifying a potential solution to the central question of Sinclair’s framework—possibly well before that question had even been formally articulated by their group [7].
The model presented in this paper not only challenges Sinclair’s framework but also has profound—and potentially uncomfortable—implications for proponents of damage-based and programmatic theories of ageing. It suggests that much of the work many researchers have actively pursued may, in fact, address only indirect aspects of ageing. According to this proposal, such research primarily identifies mechanisms that modulate the rate of ageing, rather than contributing to the emergence of the phenomenon itself.
First, the model I propose defines ageing without invoking damage as one of its primary causes. In this framework, damage is viewed as a consequence of ageing rather than its origin. The only form of damage that truly matters is the loss of biological information—primarily epigenetic, though also genetic. As this information is progressively lost, the efficiency of damage repair pathways declines, leading to the accumulation of molecular damage commonly observed in aged organisms. This reframes damage accumulation as a downstream effect of ageing, rather than a direct cause.
Second, I propose that the pathways traditionally understood to regulate the rate of ageing actually control the speed at which the developmental epigenetic program advances—ultimately culminating in death by ageing, as defined in this paper. According to this model, death results from the progressive loss of biological information, primarily epigenetic. Metaphorically, these pathways determine the speed of the epigenetic ratchet (see Figure 2C), governing how quickly an organism progresses toward functional decline and eventual death.
The ‘Hallmarks of Ageing’ paradigm posits that ageing arises from a synergistic interaction among multiple core processes or hallmarks, rather than being caused by any single factor. However, I must argue that this is not true. I fully acknowledge that such a statement may be met with resistance, particularly from researchers who have dedicated their careers to working within this framework. Fortunately, some researchers are beginning to embrace the necessity of being open to the possibility of being proven wrong [64]. Ultimately, experiments will provide the answers. The framework I have developed offers a means to test the model I propose, and to get answers, in the Popperian sense, about the nature of ageing itself (see Marsellach 2017 [7] and see section 6 below), and not just on the factors that affects its rate as has been the main focus that, according to this proposal, lead ageing research to a dead-end.
3.3. Trade-Offs in Ageing
Most ageing theories involve trade-offs between short-term reproductive or survival advantages and long-term maintenance and longevity (especially evolutionary based, but not exclusively). A few examples: Mutation Accumulation Theory argues that harmful mutations acting late in life are less strongly selected against, thus accumulating in the gene pool and contributing to ageing (late-acting deleterious effects vs. early-life reproduction) [36]. Antagonistic Pleiotropy considers that a single gene may have beneficial effects for early-life reproduction or survival but harmful effects later in life (increased reproductive success early on vs. late-life decline) [37]. Disposable Soma Theory posits that organisms allocate a finite pool of resources between reproduction and somatic maintenance (investing heavily in reproduction vs. allocating more to repair) [38]. Some Programmatic Ageing Theories suggest that ageing might be at least partially regulated or ‘programmed’ (genes that optimize early fitness vs. long-term survival). Finally, Damage Theories of Ageing hold that ageing results from accumulated damage (e.g., oxidative stress, DNA breaks, protein misfolding) exceeding repair capacity (short-term function vs. long-term maintenance), and so on.
Contrary to those traditional trade-offs, I propose that the trade-off central to understanding why ageing exists is: a non-ageing, non-complex lifestyle versus an ageing, complex lifestyle [9]. This perspective requires approaching the trade-offs scenario from a non-teleological standpoint and conceiving both life and ageing as information-based phenomena.
We humans often interpret the functioning of many aspects of the universe from a teleological perspective, likely because this mirrors how we approach tasks: analysing, planning, and executing them with purpose. However, nature does not operate in this way. Teleological language is, though, deeply embedded in our daily lives and has not been entirely excluded from scientific reasoning. Indeed, it has been suggested that contemporary biological research is still influenced by teleological thinking [65]. On the other hand, some argue that the negative effects of humans’ innate tendency toward teleological thinking have been largely overcome in science [66]. While it can be claimed that besides the fact that the concept of purpose remains a natural part of everyday discourse in biology, it has largely fallen out of favour as a foundational element of scientific explanation since the works of Newton and Darwin [67,68]. Newton’s mechanistic worldview and Darwin’s theory of evolution—grounded in chance and natural selection—eliminated the need for purpose as a scientific explanation [69]. In my view, however, alongside the flaw in how biological information is transferred intergenerationally, as proposed in this paper, the ‘teleological obstacle’ [65] further hinders a deeper understanding of how nature works, and therefore to the nature of ageing. One piece of evidence for this is the tendency of many of today’s approaches to TEI to adopt a teleological (Lamarckian) perspective, much like when Lamarck proposed his theory of evolution [70,71]. The ‘teleological obstacle’ hinders our ability to fully embrace randomness as an essential force shaping our existence. I argue that ageing is a direct consequence of the randomness that drives and underpins our lives [7,25]. This randomness leads to the fundamental trade-off behind ageing: the choice between being simple and amortal or complex and mortal.
3.4. Information-Based Conception of Life and Ageing
The model defended in this paper aligns well with recent advances in understanding biology as a purely information-based science. In recent years, a theoretical framework using information theory has been proposed to address the fundamental question, ‘What exactly makes something an individual?’ [72]. This framework suggests that an individual can be defined as an informationally coherent entity—where its constituent parts ‘hang together’ in a specific way with respect to information flow. To extend this paradigm to ageing, I propose two key ideas:
- 1)
- Biological information is always composed of both the genome and the epigenome, meaning that a cells or individual’s phenotype is determined in ‘real time’ by the specific genomic and epigenomic code it carries (see Figure 2D).
- 2)
- To describe the intergenerational transfer of information between individuals (parents and offspring), I suggest borrowing the concept of ‘instantiation’ from the Object Oriented Programming Paradigm (OOP) in computer science, a concept I have been employing throughout this paper.
In this framework, a specific organism can be conceptualized as an entity instantiated using a particular biological code (composed of the inherited genomic and epigenomic information). For example, monozygotic twins can be described as ‘two distinct objects’ instantiated from a single ‘class’ that contains the shared initial biological code (the zygote). When twins are produced, resources are ‘allocated’ to create two individuals instead of one, following a division at the embryonic stage. In contrast, when no twins are produced, resources are ‘allocated’ to produce a single individual from the biological code contained in the ‘class’. See Box 1 for clarification of these terms.
Box 1. A conceptual analogy between object-oriented programming and biology: the genome–epigenome system acts as a class blueprint, with each organism as an instantiation — highlighting how identical twins emerge from a shared but independently instantiated biological code.

From this perspective, ageing in an organism can be viewed as the evolution of its genetic and epigenetic code—primarily the epigenetic code—over the course of the organism’s life as the ‘program’ is run. Each individual ‘object’ (organism) undergoes its own unique evolution, influenced by environmental inputs that impact both its genetic and epigenetic code.
I propose that the majority of environmental effects act primarily through their influence on the epigenetic code. In a given ‘object’ (individual), these epigenetic modifications accumulate over time and are not repaired. The epigenetic code is only repaired during the process of producing new ‘instances’ (offspring), ensuring that the next generation starts with a functional epigenome. This mechanism allows for the intergenerational transfer of epigenetic information while maintaining the continuity of life through rejuvenation in successive generations [9].
Moreover, I like others, propose in this paper to model ageing processes using concepts borrowed from software-related problems [6,73]. However, this paper presents five significant departures from previous perspectives:
- Analogy of Biological Codes: The most appropriate analogy for describing the two biological codes of information (genome and epigenome) is not the digital-analogue pair suggested by Sinclair and LaPlante [6], but rather the OOP paradigm. In the digital-analogue comparison, both systems convey the same message through different formats. In contrast, the OOP analogy better captures the interplay between the genome and epigenome. OOP introduces additional coding capabilities to the epigenetic code (the derived code) that the genome (the original code) does not inherently possess. Although the genome encodes the instructions for constructing the epigenome, once built, the epigenome functions as a semi-independent code. While not a perfect analogy, the OOP paradigm is the closest conceptual framework for understanding the genome-epigenome relationship.
- A further notable difference between the proposal defended in this paper and that of Sinclair and colleagues is the causative relationship between DNA mutations and epimutations. According to Sinclair and colleagues, DNA mutations drive the emergence of epimutations [5,61,74], whereas the framework proposed here posits the reverse: that epimutations can lead to DNA mutations (see Figure 2A and Figure 2B). While it is likely that both factors contribute to the overall defect burden, my view strongly supports epimutations as the primary driver, ultimately leading to the progressive accumulation of DNA mutations over time. A recent study has shown the correlation between mutations and DNA methylation epimutations in cancer samples, though it has not yet determined which is the primary causal agent [75]. I propose that we consider the dual protection mechanism hypothesized to exist between the genome and the epigenome, as suggested by the study conducted by Monroe et al. (2022) [31] (see Figure 2A and Figure 2B).
- Perspective on Ageing: Ageing is not a design flaw. While it is often perceived as a flaw when analysed from an individual human perspective [73], this perception arises from our recognition of ageing’s negative implications for individuals. From a teleological viewpoint, ageing appears flawed because humans would never intentionally design a process with such detrimental consequences. However, ageing did not arise from intentional design—it emerged randomly, guided by Weismannian logic. While ageing is detrimental to individuals, it is essential for species maintenance and evolution, acting as a trade-off that supports the perpetuation of biological information across generations.
- The Instantiation Mechanisms: Building on the flaw proposed in today’s biological knowledge framework and inspired by the OOP paradigm analogy, this paper proposes that new ‘instances’ (offspring) are created using both genetic and epigenetic information inherited from parental cells or individuals. Once formed, the somatic cells of a new individual can modify the epigenome to produce differentiated cells but cannot restore the ‘young epigenome.’ This capability is exclusive to germline cells, which create new ‘instances’ that do not contribute to the parent’s body. Some epigenetic information in somatic cells exists in its current state because it was inherited from progenitors and cannot be reconstructed from the individual’s genome. This is not due to a lack of necessary information in the genome but rather to the restricted access somatic cells have to this information. Consequently, the epigenetic code of an individual functions as a partially independent code from the genome.
- Phenotype as a Dynamic Outcome: The phenotype of a given cell or organism is determined by the ‘real-time’ biological information contained in its genome and epigenome. For multicellular organisms, a developmental program runs alongside the ageing program from the moment of ‘instantiation.’ Many ageing-associated diseases arise from the intrinsic loss of information—primarily epigenetic but also genetic—which ultimately leads to the organism’s death when it no longer retains the necessary information to sustain life.
Ultimately, all these reasonings leads to a concise and clear definition of life as the state in which negative selection mechanisms—whether external (such as predation or injury) or internal (such as ageing)—are absent in information-based organisms.
4. The Epigenetics of Ageing
As I propose that epigenetics is the only relevant factor in explaining ageing, I summarise what has already been achieved in the field of epigenetics and its relationship with ageing in this separate section. This distinction underscores the pivotal role I attribute to epigenetics, distinguishing it from other mechanisms traditionally implicated in the ageing process. By focusing solely on the interplay between epigenetics and ageing, this section aims to provide a targeted overview of the current state of research, highlighting how it aligns with or diverges from the framework proposed in this paper.
The relationship between ageing and epigenetics is not a novel concept that has emerged recently. As early as the 1970s and 1980s, studies began linking ageing to epigenetic features [76,77,78,79]. Observations of DNA methylation changes in ageing tissues led scientists to coin the term ‘epigenetic drift,’ referring to the gradual, stochastic alterations in epigenetic marks over time [80,81,82]. Soon after, other epigenetic marks beyond DNA methylation were also linked to ageing, including histone modifications and chromatin remodelling [55,74,83,84,85,86]. Over time, the connection between epigenetics and ageing has become increasingly evident, solidifying their intertwined roles in understanding the ageing process. Today, epigenetics is widely recognised as one of the key hallmarks of ageing [1,2,3].
In 2013, a revolution occurred in recognising the relationship between epigenetics and ageing. That year, Steve Horvath published a seminal paper introducing a multi-tissue ‘epigenetic clock’—a predictive model based on DNA methylation levels at specific CpG sites across the genome [87]. This clock was groundbreaking due to its ability to estimate chronological age with remarkable accuracy across a wide variety of human tissues (over 50 types), including blood, brain, liver, and more. Its precision surpassed that of telomere length, which had been the most accurate biomarker of ageing to date. Interestingly, Horvath faced initial resistance when trying to publish this work, with some reviewers dismissing it as ‘too good to be true’ [88]. Around the same time this revolution began, at the end of 2013 and the beginning of 2014, I started gathering serendipitously obtained data that led me to consider a model of ageing based exclusively on epigenetic parameters. I encountered many surprising and unexpected results while dissecting tetrads from fission yeast, particularly during self-crosses, which indirectly pointed to epigenetic phenomena occurring in my experiments which lead me to understand that the value of a self-cross seen in wild fission yeast strain was a biomarker of their age (see Marsellach (2017) [7] and sections 6 below). Anyway, following Horvath’s landmark study, additional epigenetic clocks were developed, such as Hannum’s clock [89], PhenoAge [90], and GrimAge [91]. Each of these clocks was designed for specific populations or endpoints, further advancing our understanding of ageing through epigenetic markers. For a recent review on ageing clocks, see Teschendorff and Horvath (2025) [92].
These epigenetic clocks were soon shown to be associated with all-cause mortality [93,94]. Additionally, other studies demonstrated a relationship between DNA methylation patterns (not specifically related to epigenetic clocks) and all-cause mortality [95,96,97]. These findings further solidified the connection between epigenetic modifications and fundamental aspects of ageing and health outcomes, emphasizing the critical role of DNA methylation in lifespan prediction and mortality risk. This was consistent with the serendipitous data I obtained, which pointed towards lethality in old age being a consequence of epigenetic information loss [7]. Clocks were also developed for other species, accelerating their use as biomarkers to study ageing and its modifications [98,99,100,101,102,103,104,105,106,107,108,109]. Moreover, this concept began to be used to investigate whether diseases [110,111,112,113,114,115,116,117,118,119,120], lifestyle changes (e.g., diet, exercise) [121,122,123,124], pharmacological agents (e.g., metformin, rapamycin) [125,126,127,128,129,130,131,132,133], or experimental therapies (e.g., Yamanaka-factor-based partial reprogramming or chemical reprogramming) could alter the pace of these epigenetic clocks [62,134,135,136,137,138].
With the information-based conception of life and ageing framework described in the sections above in mind, the mechanisms behind Yamanaka-based approaches become clearer. The four Yamanaka factors appear to ectopically reprogram somatic cells, effectively resetting their epigenome without altering their genetic code [32]. Beyond altering cell type, reprogrammed cells exhibit rejuvenated characteristics compared to their original state [87,139,140,141]. This reprogramming enables somatic cells to restart the developmental program from scratch, mimicking the epigenetic state of an embryonic stem cell necessary to initiate a new ‘instance.’ In contrast, unmodified somatic cells cannot naturally perform this reset as they lack the ability to restore a ‘young epigenome.’ However, this process likely occurs naturally in germline cells, as evidenced by earlier experiments by Gurdon, Wilmut, and colleagues, where somatic nuclei exposed to oocyte cytoplasm were successfully reprogrammed [142,143]. This similarity reinforces the idea that germline cells inherently possess mechanisms akin to those artificially induced by Yamanaka factors. Notably, Yamanaka factors were also shown to be applicable in vivo [144,145,146], though their continuous expression in adult animals often led to tumorigenesis, making them unsuitable for direct rejuvenation purposes.
To address the challenges posed by continuous Yamanaka factor expression, partial reprogramming strategies emerged, focusing on the transient expression of these factors to ameliorate ageing phenotypes without inducing tumorigenesis. The first success in this area, though limited to a mouse model of progeria, was achieved by Ocampo et al. in 2016 [138]. Since then, numerous studies have built on this foundation (for a review, see Cipriano et al., (2023) [147]). While these approaches have demonstrated the ability to alleviate ageing phenotypes and extend lifespan, they fall short of providing an eternal ‘fountain of youth.’ Even when applied periodically within a limited timeframe, they fail to prevent death from old age, achieving instead a lifespan extension of approximately 25% [148]. This limited efficacy raises an important question: why does complete and indefinite rejuvenation remain unattainable? To address this, examining the characteristics of long-lived species in nature and understanding how they achieve exceptional lifespans may offer valuable insights.
4.1. Epigenetics of Long-Lived Organisms
When examining longevity across the tree of life in wild populations, researchers have focused on two distinct groups: organisms often referred to as ‘immortal’ (e.g., jellyfish like Turritopsis dohrnii and hydra) and those with exceptionally long lifespans compared to closely related species (e.g., naked mole-rats vs. other rodents). First, it is important to clarify that, in strict scientific terms, the term ‘immortal’ should be avoided, as it originates from mythology and lacks relevance in the real world. Even the longest-lived animal will die if its biological structures are destroyed. A more accurate term would be ‘amortal’ or ‘ageless,’ denoting an absence of—or extremely minimal—age-related changes over time. In contrast, mortal animals exhibit either rapid or gradual, but progressive, time-dependent functional decline, which increases their vulnerability to disease and ultimately leads to death—a process we call ageing. Among the first group (the amortal organisms), the vast majority are relatively simple animals with non-complex lifestyles that minimize their exposure to external causes of death. In this scenario, their biology may have evolved to allow continuous access to the information required to maintain a ‘young’ (functional) epigenome. This capability enables them to remain fully functional indefinitely, avoiding the accumulation of epimutations in their epigenome—since it is constantly being rebuilt—and mutations in their DNA, protected by the epigenome’s regulatory shield (see Figure 2A). These organisms are capable, individually, of circumventing both genetic and epigenetic ratchets. In contrast, mortal animals evolved with a restriction on the ability of somatic cells to fully restore their epigenome. This limitation leads to a progressive accumulation of epimutations and mutations within an individual, ultimately causing its death (see Figure 2D)—a phenomenon we recognise as death from old age. This accumulation of epimutations and mutations is also the underlying cause of age-related diseases. Mortal organisms are individually subjected to both genetic and epigenetic ratchets. They mitigate the negative effects of these ratchets by passing their biological information to successive generations, thereby creating new functional ‘instances’ with an, ideally, fully restored and functional biological code. This process ensures the continuity of life despite the progressive deterioration experienced by individual organisms [9].
If this is how it works, why then can’t complex organisms like the naked mole-rat become truly amortal, as simpler organisms like hydra do? To address this, I propose the ‘turning an ocean liner in a river’ metaphor: ‘Imagine you have a massive cruise ship (an ocean liner) that typically sails in open seas. Now, you need to turn that enormous ship around within the tight confines of a narrow river. Because of the ship’s size and the river’s constraints, even the smallest directional adjustments become slow, challenging, and require significant effort and planning.’ Similarly, complex organisms require a developmental process to exist—an intricate set of biological instructions needed to build an entire organism from a single cell, the zygote. The developmental and ageing processes (the latter essential for the Weismannian conservation of the species beyond the human dream of immortality) appear to have become intrinsically linked during evolution (see below in section 5). This intertwining means that, for complex organisms, reversing or halting the developmental process is as challenging as turning an ocean liner in a river. The intricacy and scale of biological systems in complex organisms make achieving amortality nearly impossible due to the inherent limitations imposed by their developmental complexity. Does this mean achieving true amortality in complex organisms is impossible? Simply: we do not know. We have only just begun to experiment with ectopic reprogramming approaches to reverse ageing [147]. For instance, it has been demonstrated that using Yamanaka factors can be beneficial for certain tissues but detrimental to others [135]. In summary, while partial reprogramming offers clear benefits for some ageing-related phenotypes, its expression across an entire organism has shown detrimental or incomplete effects, failing to fully ‘turn the ocean liner in the river’ [148]. Yamanaka factors appear to restore the epigenetic code necessary to start a new ‘instance’ from scratch, even this might not suffice, as problems have been observed in offspring derived from iPSCs. While some iPSC-derived animals are viable, fertile, and produce healthy offspring, others exhibit reduced lifespans, immune dysfunction, or hidden phenotypes, indicating that being ‘normal’ at birth does not guarantee lifelong health [149,150,151,152,153,154,155]. Metaphorically, the challenge ahead is akin to dismantling the ocean liner within the river and reassembling it in reverse orientation—without letting it sink during the process.
It is worth noting that in complex organisms with exceptionally long lifespans compared to closely related species, longevity is often associated with effective protection against external causes of death (e.g., turtles) or low natural predation (e.g., sharks and whales). Similarly, aquatic and aerial species can achieve longer lifespans that terrestrial ones, because they are less likely to die from falls or other such mechanical accidents [156,157,158,159]. This observation aligns with a well-established principle in evolutionary biology: lower external mortality often correlates with longer lifespans. From the perspective of the framework proposed in this manuscript, organisms with low exposure to external threats can extend their lifespans by modulating ageing pathways, which ultimately regulate the pace of the epigenetic ratchet. This ability allows them to maintain their species’ biological information, avoid extinction, and achieve longer lifespans. On the other hand, in organisms with high predation rates, individuals with slow-ageing traits are unlikely to persist, as they would succumb to predation before passing their biological information to new ‘instances.’ In such cases, short lifespans coupled with rapid reproduction ensure the continual generation of new ‘instances,’ preserving the species’ biological information and preventing extinction [9]. In some species, such as social insects, these longevity differences manifest within individuals of the same species. In ants, for example, castes are epigenetically regulated, and research has shown that ageing pathways directly influence the pace of ageing within different castes [160,161]. The relationship between the pace of a biomarker of ageing—such as epigenetic clocks—and the rate of ageing is well established and supported by robust empirical data [103,162,163,164,165,166,167,168,169]. A general pattern observed across species is that DNA methylation change rates negatively scale with maximum lifespan—reflecting, in terms of the model proposed in this paper, a slowing down of the epigenetic ratchet.
Within the context of the hallmarks of ageing paradigm—which attributes ageing to the interplay of multiple independent pathways—the rise of epigenetic clocks based on a few DNA methylation marks across the genome has catalysed the development of various other types of ageing clocks. These include AI-based clocks [170], metabolomics-based clocks [171,172,173,174], microbiome-based clocks [175], mutational clocks [176], proteomics-based clocks [172,177,178,179,180], and transcriptomics-based clocks [181,182,183,184,185,186], among others integrating multiple data inputs. From the perspective of the framework proposed in this manuscript, these clocks are secondary as they measure downstream effects or correlates of the primary driver of ageing: epigenetic phenomena. Efforts should therefore focus on characterising diverse epigenetic marks, building on existing studies [187,188]. DNA methylation clocks, which have been widely adopted, are among the most validated across diverse cohorts and tissues, often showing strong correlations (>0.90) with chronological age. Although the precise mechanisms linking methylation to ageing remain elusive, DNA methylation is directly tied to gene regulation, with numerous studies implicating it in ageing pathways (potentially as an endpoint of these pathways).
It is notable that many organisms lacking DNA methylation pathways also age, highlighting the need for new approaches to detect epigenetic biomarkers of ageing in such species. My early work with self-crosses in fission yeast indirectly underscored the potential of epigenetic markers. Observing spore survival values in these experiments, I hypothesised that they were biomarkers of ageing. I noted a decrease in spore survival after environmental insults (e.g., cold shock), as well as a correlation between spore survival and pre-meiotic cell survival. The observed cell death exhibited ploidy-dependent and sequence-independent patterns, suggesting epigenetic causation through the accumulation of epigenetic defects [7]. Remarkably, similar to how DNA methylation clocks predict mortality, these phenomena appeared to be epigenetically driven. Furthermore, spore survival seemed to be repaired during meiosis [7], paralleling findings by Unal et al. in Saccharomyces cerevisiae [189], where gametogenesis resets lifespan, and mimicking the ectopic effects seen with Yamanaka factors [87].
5. Random vs. Programmed Ageing Processes: Information Maintenance in Unicellular vs. Multicellular Organisms
The study of ageing in unicellular organisms—including prokaryotes, naturally unicellular eukaryotes, and derived unicellular eukaryotes like yeasts—primarily revolves around the concept of damage accumulation, framed within the ‘Hallmarks of Ageing’ paradigm [1,2,3]. These studies generally assume that ageing results from the accumulation of cellular damage. Bacterial ageing was initially considered paradoxical due to their single-cell nature, lack of a defined lifecycle, and the potential for rejuvenation through damage dilution during cell division. However, studies have demonstrated ageing-like behaviours in bacteria, sparking debates about whether asymmetry or stressors are prerequisites for ageing to manifest [190,191,192,193,194,195]. While bacterial ageing does not strictly parallel eukaryotic ageing, Asymmetric Damage Segregation (ADS) is now recognised as a central mechanism in bacterial ageing [196,197,198]. Yeasts, particularly budding yeast and, more recently, fission yeast, serve as widely used model organisms for ageing research. Most studies done about ADS use yeasts [199,200,201,202,203,204] as model organism. Two types of ageing are studied in yeasts: replicative ageing, defined as the number of daughter cells a mother cell can produce before senescence, and chronological ageing, which refers to the survival of non-dividing, quiescent cells [205]. Yeasts are also extensively employed in exploring pharmacological and genetic interventions that affect lifespan [206,207,208,209,210]. Naturally unicellular eukaryotes like Paramecium, Tetrahymena, Chlamydomonas, and Dictyostelium exhibit ageing-like phenomena despite their theoretical immortality under ideal conditions. Although fewer in number, studies on these organisms similarly focus on damage accumulation as the primary driver of ageing [211,212,213,214].
In summary, research on unicellular ageing highlights the central role of damage accumulation while exploring diverse mechanisms, from ADS in replicative systems to stress resistance in quiescent states.
As stated earlier, the model I defend in this paper defines ageing without relying on damage as one of its causes. According to this model, the biological code that a newborn inherits should contain the information necessary to produce flawless biological structures—structures that either do not accumulate damage or are optimally equipped to handle the damage produced during normal biological processes. Damage accumulates during ageing because the information required for flawless functioning is progressively lost. This loss of information drives ageing and represents the evolutionary origin of the ageing process.
Unicellular organisms, most—if not all—can divide through binary fission or mitosis-related mechanisms, sometimes producing millions of copies of themselves, as observed in certain bacteria and yeast under laboratory conditions. In natural environments, this ability is one of their most effective strategies for propagating and preserving their accumulated biological information under favourable conditions. In harsher environments, many unicellular organisms have developed specialised survival strategies, such as spore formation. Bacterial endospores, formed by species like Bacillus and Clostridium, are highly resistant structures designed solely for survival. In contrast, yeast spores, which are formed after meiosis, serve both as survival adaptations and reproductive units, capable of germinating into new cells when conditions improve. These spores are specialised cell types that can endure extreme environmental conditions, ensuring the organism’s long-term persistence.
In all multicellular organisms, there exists the concept of somatic and germline cells. However, while many multicellular organisms exhibit a clear distinction between somatic and germline cells, this is not universal. In some organisms, the lines between somatic and germline cells are blurred, and the distinction may be less rigid or absent. According to the model presented in this paper, germline cells are ‘amortal’ compared to somatic cells, not because of how they handle damage, but because of how they manage and preserve biological information. Although some ADS mechanisms have been identified, they likely represent part of the normal biological processes that maintain flawless structures. So far, there is no conclusive evidence to date that damage removal is the primary reason for the youthfulness of a newborn individual. The framework I developed offers an opportunity to test whether information management alone is sufficient to explain the youthfulness of newborns (see Marsellach (2017) [7] and section 6).
Unicellular organisms lack the defining developmental processes observed in most multicellular organisms, where a single cell grows, divides, and differentiates into specialised structures and tissues in an organised sequence. While developmental processes offer evident advantages—allowing the formation of more complex biological structures—they eliminate the simplicity of creating a new organism through mechanisms like binary fission or mitosis. This imposes limitations on the ease with which multicellular organisms can preserve their accumulated biological information. Moreover, the increased likelihood of external death associated with a complex lifestyle further underscores the necessity of a mechanism like ageing to prevent extinction and ensure the preservation of biological information [9]. Once a developmental program and the need for a mechanism like ageing align for survival, it becomes plausible for both processes to intertwine. Thus, it is reasonable to describe ageing in multicellular organisms as a programmatic process. In unicellular organisms, where developmental programs are unnecessary, ageing is more likely a stochastic (random) process rather than a programmed one. In derived unicellular eukaryotes like yeasts, a dual scenario may exist, with ageing exhibiting characteristics of both a programmatic and a random process.
In summary, I propose that the multiple reasons why ageing succeeded during evolution are all closely tied to the challenge of maintaining biological information—particularly in relation to the levels of complexity that can be sustained by such information. The existence of ageing allowed for the following:
- The continuous replacement of older individuals with ‘young’, flawless newborns, thereby reducing the risk of extinction associated with the increasing vulnerability to external causes of death that accompany greater behavioural and structural complexity.
- The ability of complex organisms—which inherently require a developmental process—to maintain a stable balance between the creation and removal of individuals, without relying on simpler reproductive strategies such as binary fission or spore formation, which are used by less complex organisms that do not undergo proper ageing.
- An acceleration in the emergence of evolutionary novelties, by reinforcing the need for population renewal. This mechanism helped overcome the limits imposed by external threats on organismal complexity. As a result, increasingly complex organisms were able to evolve over time—organisms that would likely not have arisen in the absence of an ageing process.
6. An Empirical Proposal to Test the Nature of Ageing in a Popperian Framework—Beyond Identifying Factors that Influence Its Pace
The framework I developed—based on serendipitously obtained data [7]—allows us to do precisely what the title of this section suggests: address the fundamental nature of ageing itself. To my surprise, much of current ageing research avoids engaging with this question directly. Many studies proceed with the uncritical assumption that if a factor modifies the pace of ageing, or is observed to accumulate in aged individuals, it must be causally involved in producing the ageing phenomenon. Ageing is frequently labelled as inherently complex, but this complexity is often asserted rather than explained—without any rigorous justification for why that should be the case.
The serendipitous observations that led to the development of this framework suggest the following:
- Fission yeast self-cross survival is a biomarker of ageing, where ageing is defined, as described in this paper, as the number of epigenetic defects accumulated in a given clone [7].
- The lower the self-cross survival value of a clone, the greater the inferred impact of accumulated epigenetic defects on the survival of its mitotically derived progeny. This reflects the influence of stochastic variation during the epigenetic copying process in mitotic divisions.
- Different clones may show different self-cross survival values due to the accumulation of distinct epigenetic defects at different epiloci.
- During meiosis, a saturable “epigenetic repair mechanism” operates, which may or may not improve fitness at any given epilocus. From the perspective of a single epilocus, the outcome is random-like due to this saturability. Improvements in fitness can be detected as increased self-cross spore survival.
- An individual cell dies when the real-time biological information it carries is no longer sufficient to sustain life (see Figure 2D).
- A clonal lineage will cease to exist when all of its mitotically derived progeny reach the same informational threshold described in point 5, i.e., when all cells from a given colony lose viability due to excessive epigenetic damage (Figure 2D).
- This creates a scenario where lethality due to old age is attributable to the epigenetic state of a clonal group of cells—that is, a population-level property rather than a purely individual one.
This last point offers a clear experimental test for the hypothesis that loss of epigenetic information is the causal mechanism of ageing. If the assumptions just exposed are correct, then any intervention known to modify the pace of ageing should also influence the dynamics of self-cross spore survival. As described, this metric provides an a priori estimation of the number and/or severity of accumulated epigenetic defects, which would eventually produce the death of all of the individuals due to the accumulation in all of them of epigenetic defects (see step 6). Self-cross spore survival thus becomes a proxy for estimating the probability that individual cells in a clonal line will die. In practice, it provides an early predictor of how a culture will behave under a fixed environmental conditions.
I observed this behaviour only circumstantially in ageing-focused experiments, but I repeatedly noted it during experiments involving cold shock-like external stress. Such stress may destabilise the epigenetic machinery, leading to reduced accuracy in the copying of epigenetic information in the subsequent generation. This, in turn, increases the burden of accumulated epigenetic defects. In other words, repeated cold shocks appear to accelerate the ageing process—at least in yeast—by exacerbating the accumulation of epigenetic errors.
This observation resonates with a well-known phenomenon in yeast laboratories: when old glycerol stocks stored at –80 °C perform poorly—producing only a few colonies—researchers restore performance simply by creating a new stock. This occurs because a hidden process of epigenetic selection takes place during culture renewal, favouring the fitter cells. In such cases, environmental conditions are not held constant, as fresh media are used. What actually happens is that when a new culture is ‘instantiated’ from an old one, a form of epigenetic selection is triggered: the fitter cells within the loop used to inoculate the new culture disproportionately contribute to the next population, effectively purging epigenetically unfit individuals (i.e., “old individual instances”). This process explains why organisms like bacteria or yeast appear “immortal” under standard laboratory conditions.
This phenomenon may also have a parallel in human biology. Individuals conceived through Assisted Reproductive Technologies (ART) have been reported to exhibit an increased risk of ageing-related diseases [215]. This could potentially be explained by the mechanisms described above. During ART procedures, freezing and thawing of gametes or embryos is common, and—as previously discussed—such processes may increase the burden of epigenetic defects, potentially accelerating the ageing process from the earliest stages of development. This is a scenario that warrants careful scientific investigation, as it may carry uncomfortable implications for individuals conceived via ART. It is ethically inconsistent to prohibit experimental studies on human samples while simultaneously turning a blind eye to potentially harmful outcomes when such risks arise from personal or societal desires.
The empirical observation of a consistent correlation between spore survival and the viability of mitotically dividing cells (i.e., cells that have never undergone meiosis), as observed at least once by serendipity [7], does not in itself prove the epigenetic nature of the phenomenon. However, I also serendipitously observed a case that supports such an interpretation. While working simultaneously with haploid and diploid versions of fission yeast strains that shared nearly identical genetic backgrounds (differing only in the selectable markers used for their construction), I noticed that most haploid and diploid strains responded similarly to cold shock stress. Yet one strain—JB953—exhibited a striking divergence: the haploids developed a defect much faster than the diploids under the same conditions. This observation, alongside several others, led me to hypothesise that the defects I was observing were epigenetic in nature. Cases in which haploids and diploids behaved similarly could be explained by dominant epigenetic defects. In contrast, the JB953 case suggests the presence of a recessive epigenetic defect—one in which both epialleles must accumulate alterations for the phenotype to manifest. Such a scenario, however, is only plausible if epigenetic inheritance is taken into account [7].
While more observations of this kind would be valuable, they will not be sufficient to confirm the model. Ultimately, specific epigenetic defects must be mapped, identified, and reproducibly introduced into multiple independent strains in order to gain robust experimental support. Fortunately, the experimental malleability of yeast may make this possible. For example, transposon-based approaches such as Hermes insertion mapping [216,217] could be used to detect ectopic linkage between genetic markers and phenotypic differences in clonal strains that show no sequence variation upon whole-genome sequencing. Once such genomic regions are identified, downstream analyses such as quantitative proteomics [218] or other high-throughput assays could be applied to pinpoint the molecular basis of phenotypic divergence in genetically identical but phenotypically discordant clonal populations.
Overall, the ideas and experimental approaches discussed throughout this paper make it possible to subject the hypothetical model proposed herein to proper Popperian testing—a standard not consistently achieved by many ageing hypotheses presented to date.
Author Contributions
Conceptualization, XM. Writing, review, and editing: XM, with the collaboration of Large Language Models (LLMs) developed by OpenAI, which supported the refinement of the manuscript’s final form.
References
- G. Kroemer, A. B. Maier, A. M. Cuervo, V. N. Gladyshev, L. Ferrucci, V. Gorbunova, B. K. Kennedy, T. A. Rando, A. Seluanov, F. Sierra, E. Verdin, C. López-Otín, From geroscience to precision geromedicine: Understanding and managing aging. Cell 188, 2043–2062 (2025). [CrossRef]
- C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, G. Kroemer, Hallmarks of aging: An expanding universe. Cell 186, 243–278 (2023). [CrossRef]
- C. López-Otín, M. A. Blasco, L. Partridge, M. Serrano, G. Kroemer, The Hallmarks of Aging Cell 153, 1194–1217 (2013).
- J. P. de Magalhães, An overview of contemporary theories of ageing. Nat Cell Biol (2025).
- Y. R. Lu, X. Tian, D. A. Sinclair, The Information Theory of Aging Nature Aging 3, 1486–1499 (2023).
- D. A. Sinclair, M. D. LaPlante, Lifespan: Why We Age—and Why We Don’t Have To (Atria Books, 2019).
- X. Marsellach, A non-genetic meiotic repair program inferred from spore survival values in fission yeast wild isolates: a clue for an epigenetic ratchet-like model of ageing bioRxiv (2017).
- X. Marsellach, Ageing is not just ageing Zenodo (2025).
- X. Marsellach, The Principle of Continuous Biological Information Flow as the Fundamental Foundation for the Biological Sciences. Implications for Ageing Research Preprints (2021).
- A. Weismann, Essays upon heredity and kindred biological problems. (Clarendon Press, Oxford, 1889). [CrossRef]
- J. D. WATSON, F. H. CRICK, Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953). [CrossRef]
- J. D. WATSON, F. H. CRICK, Genetical implications of the structure of deoxyribonucleic acid. Nature 171, 964–967 (1953). [CrossRef]
- P. W. S. Hill, H. G. Leitch, C. E. Requena, Z. Sun, R. Amouroux, M. Roman-Trufero, M. Borkowska, J. Terragni, R. Vaisvila, S. Linnett, H. Bagci, G. Dharmalingham, V. Haberle, B. Lenhard, Y. Zheng, S. Pradhan, P. Hajkova, Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 555, 392–396 (2018). [CrossRef]
- J. A. Hackett, M. A. Surani, DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond B Biol Sci 368, 20110328 (2013). [CrossRef]
- M. Cowley, R. J. Oakey, Resetting for the next generation. Mol Cell 48, 819–821 (2012).
- H. Kobayashi, T. Sakurai, M. Imai, N. Takahashi, A. Fukuda, O. Yayoi, S. Sato, K. Nakabayashi, K. Hata, Y. Sotomaru, Y. Suzuki, T. Kono, Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8, e1002440 (2012). [CrossRef]
- Z. D. Smith, M. M. Chan, T. S. Mikkelsen, H. Gu, A. Gnirke, A. Regev, A. Meissner, A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484, 339–344 (2012). [CrossRef]
- R. Hirasawa, H. Chiba, M. Kaneda, S. Tajima, E. Li, R. Jaenisch, H. Sasaki, Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 22, 1607–1616 (2008). [CrossRef]
- E. Li, Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3, 662–673 (2002). [CrossRef]
- A. Bird, Transgenerational epigenetic inheritance: a critical perspective Frontiers in Epigenetics and Epigenomics 2, 0 (2024).
- S. Cao, Z. J. Chen, Transgenerational epigenetic inheritance during plant evolution and breeding. Trends Plant Sci 29, 1203–1223 (2024). [CrossRef]
- K. Moelling, Epigenetics and transgenerational inheritance. J Physiol 602, 2537–2545 (2024). [CrossRef]
- L. Duempelmann, M. Skribbe, M. Bühler, Small RNAs in the Transgenerational Inheritance of Epigenetic Information. Trends Genet 36, 203–214 (2020). [CrossRef]
- Y. Takahashi, M. Morales Valencia, Y. Yu, Y. Ouchi, K. Takahashi, M. N. Shokhirev, K. Lande, A. E. Williams, C. Fresia, M. Kurita, T. Hishida, K. Shojima, F. Hatanaka, E. Nuñez-Delicado, C. R. Esteban, J. C. Izpisua Belmonte, Transgenerational inheritance of acquired epigenetic signatures at CpG islands in mice. Cell 186, 715–731.e19 (2023). [CrossRef]
- X. Marsellach, A non-Lamarckian model for the inheritance of the epigenetically-coded phenotypic characteristics: a new paradigm for Genetics, Genomics and, above all, Ageing studies bioRxiv (2018).
- R. Landauer, Irreversibility and heat generation in the computing process IBM journal of research and development 5, 183–191 (1961).
- C. E. Shannon, A mathematical theory of communication The Bell System Technical Journal 27, 379–423 (1948).
- L. Szilard, über die Entropieverminderung in einem thermodynamischen System bei Eingriffen intelligenter Wesen Zeitschrift für Physik 53, 840–856 (1929).
- E. Mizraji, The biological Maxwell’s demons: exploring ideas about the information processing in biological systems. Theory Biosci 140, 307–318 (2021). [CrossRef]
- T. D. Brock, H. Freeze, Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile Journal of bacteriology 98, 289–297 (1969).
- J. G. Monroe, T. Srikant, P. Carbonell-Bejerano, C. Becker, M. Lensink, M. Exposito-Alonso, M. Klein, J. Hildebrandt, M. Neumann, D. Kliebenstein, M. L. Weng, E. Imbert, J. Ågren, M. T. Rutter, C. B. Fenster, D. Weigel, Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 602, 101–105 (2022). [CrossRef]
- K. Takahashi, S. Yamanaka, Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors Cell 126, 663–676 (2006). [CrossRef]
- V. N. Gladyshev, B. Anderson, H. Barlit, B. Barré, S. Beck, B. Behrouz, D. W. Belsky, A. Chaix, M. Chamoli, B. H. Chen, K. Cheng, J. Chuprin, G. A. Churchill, A. Cipriano, A. Colville, J. Deelen, Y. Deigin, K. K. Edmonds, B. W. English, R. Fang, M. Florea, I. M. Gershteyn, D. Gill, L. H. Goetz, V. Gorbunova, P. T. Griffin, S. Horvath, M. Borch Jensen, X. Jin, S. Jovanovska, K. M. Kajderowicz, T. Kasahara, C. Kerepesi, S. Kulkarni, V. M. Labunskyy, M. E. Levine, S. Libert, J. Y. Lu, Y. R. Lu, R. E. Marioni, B. M. McCoy, W. Mitchell, M. Moqri, F. Nasirian, P. Niimi, H. S.-H. Oh, B. Okundaye, A. A. Parkhitko, L. Peshkin, M. Petljak, J. R. Poganik, G. Pridham, D. E. L. Promislow, W. Prusisz, M. Quiniou, K. Raj, D. Richard, J. L. Ricon, J. Rutledge, M. Scheibye-Knudsen, N. J. Schork, A. Seluanov, M. Shadpour, A. V. Shindyapina, S. R. Shuken, S. Sivakumar, T. Stoeger, A. Sugiura, N. R. Sutton, A. Suvorov, A. E. Tarkhov, E. C. Teeling, A. Trapp, A. Tyshkovskiy, M. Unfried, C. K. Ward-Caviness, S. H. Yim, K. Ying, J. Yunes, B. Zhang, A. Zhavoronkov, Disagreement on foundational principles of biological aging. PNAS Nexus 3, pgae499 (2024). [CrossRef]
- J. R. Poganik, V. N. Gladyshev, We need to shift the focus of aging research to aging itself. Proc Natl Acad Sci U S A 120, e2307449120 (2023). [CrossRef]
- A. A. Cohen, B. K. Kennedy, U. Anglas, A. M. Bronikowski, J. Deelen, F. Dufour, G. Ferbeyre, L. Ferrucci, C. Franceschi, D. Frasca, B. Friguet, P. Gaudreau, V. N. Gladyshev, E. S. Gonos, V. Gorbunova, P. Gut, M. Ivanchenko, V. Legault, J.-F. Lemaître, T. Liontis, G.-H. Liu, M. Liu, A. B. Maier, O. T. Nóbrega, M. G. M. Olde Rikkert, G. Pawelec, S. Rheault, A. M. Senior, A. Simm, S. Soo, A. Traa, S. Ukraintseva, Q. Vanhaelen, J. M. Van Raamsdonk, J. M. Witkowski, A. I. Yashin, R. Ziman, T. Fülöp, Lack of consensus on an aging biology paradigm? A global survey reveals an agreement to disagree, and the need for an interdisciplinary framework. Mech Ageing Dev 191, 111316 (2020). [CrossRef]
- P. B. Medawar, An unsolved problem of biology (1952).
- G. C. Williams, PLEIOTROPY, NATURAL SELECTION, AND THE EVOLUTION OF SENESCENCE Evolution 11, 398–411 (1957).
- T. B. Kirkwood, Evolution of ageing. Nature 270, 301–304 (1977).
- C. M. McCay, M. F. Crowell, L. A. Maynard, The effect of retarded growth upon the length of life span and upon the ultimate body size: one figure The journal of Nutrition 10, 63–79 (1935).
- M. T. Mc Auley, The evolution of ageing: classic theories and emerging ideas. Biogerontology 26, 6 (2024). [CrossRef]
- R. Pamplona, M. Jové, J. Gómez, G. Barja, Programmed versus non-programmed evolution of aging. What is the evidence? Exp Gerontol 175, 112162 (2023). [CrossRef]
- V. N. Gladyshev, S. B. Kritchevsky, S. G. Clarke, A. M. Cuervo, O. Fiehn, J. P. de Magalhães, T. Mau, M. Maes, R. Moritz, L. J. Niedernhofer, E. Van Schaftingen, G. J. Tranah, K. Walsh, Y. Yura, B. Zhang, S. R. Cummings, Molecular Damage in Aging. Nat Aging 1, 1096–1106 (2021). [CrossRef]
- P. Aguilar, B. Dag, P. Carazo, Z. Sultanova, Sex-specific paternal age effects on offspring quality in Drosophila melanogaster. J Evol Biol 36, 720–729 (2023). [CrossRef]
- M. F. Perez, M. Francesconi, C. Hidalgo-Carcedo, B. Lehner, Maternal age generates phenotypic variation in Caenorhabditis elegans. Nature 552, 106–109 (2017). [CrossRef]
- C. Lenzi, A. Piat, P. Schlich, J. Ducau, J.-C. Bregliano, H. Aguilaniu, A. Laurençon, Parental age effect on the longevity and healthspan in Drosophila melanogaster and Caenorhabditis elegans Aging (Albany NY) 15, 11720 (2023). [CrossRef]
- K. Sanghvi, T. Pizzari, I. Sepil, What does not kill you makes you stronger? Effects of paternal age at conception on fathers and sons. Evolution 78, 1619–1632 (2024). [CrossRef]
- C. Kenyon, J. Chang, E. Gensch, A. Rudner, R. Tabtiang, A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 (1993). [CrossRef]
- L. Partridge, J. Thornton, G. Bates, The new science of ageing. Philos Trans R Soc Lond B Biol Sci 366, 6–8 (2011).
- L. Partridge, The new biology of ageing. Philos Trans R Soc Lond B Biol Sci 365, 147–154 (2010).
- T. C. Goldsmith, On the programmed/non-programmed aging controversy. Biochemistry (Mosc) 77, 729–732 (2012). [CrossRef]
- D. J. Clancy, D. Gems, L. G. Harshman, S. Oldham, H. Stocker, E. Hafen, S. J. Leevers, L. Partridge, Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292, 104–106 (2001).
- D. E. Harrison, R. Strong, Z. D. Sharp, J. F. Nelson, C. M. Astle, K. Flurkey, N. L. Nadon, J. E. Wilkinson, K. Frenkel, C. S. Carter, M. Pahor, M. A. Javors, E. Fernandez, R. A. Miller, Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009). [CrossRef]
- M. Kaeberlein, R. W. Powers, K. K. Steffen, E. A. Westman, D. Hu, N. Dang, E. O. Kerr, K. T. Kirkland, S. Fields, B. K. Kennedy, Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196 (2005). [CrossRef]
- M. Kaeberlein, M. McVey, L. Guarente, The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13, 2570–2580 (1999). [CrossRef]
- D. A. Sinclair, L. Guarente, Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 91, 1033–1042 (1997). [CrossRef]
- K. Lin, J. B. Dorman, A. Rodan, C. Kenyon, daf-16 : An HNF-3/forkhead Family Member That Can Function to Double the Life-Span of Caenorhabditis elegans Science 278, 1319–1322 (1997).
- M. E. Giannakou, M. Goss, L. Partridge, Role of dFOXO in lifespan extension by dietary restriction in Drosophila melanogaster: not required, but its activity modulates the response. Aging Cell 7, 187–198 (2008).
- J. Apfeld, G. O’Connor, T. McDonagh, P. S. DiStefano, R. Curtis, The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18, 3004–3009 (2004). [CrossRef]
- W. Mair, I. Morantte, A. P. C. Rodrigues, G. Manning, M. Montminy, R. J. Shaw, A. Dillin, Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 470, 404–408 (2011). [CrossRef]
- A. Dillin, A. L. Hsu, N. Arantes-Oliveira, J. Lehrer-Graiwer, H. Hsin, A. G. Fraser, R. S. Kamath, J. Ahringer, C. Kenyon, Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401 (2002). [CrossRef]
- J. H. Yang, M. Hayano, P. T. Griffin, J. A. Amorim, M. S. Bonkowski, J. K. Apostolides, E. L. Salfati, M. Blanchette, E. M. Munding, M. Bhakta, Y. C. Chew, W. Guo, X. Yang, S. Maybury-Lewis, X. Tian, J. M. Ross, G. Coppotelli, M. V. Meer, R. Rogers-Hammond, D. L. Vera, Y. R. Lu, J. W. Pippin, M. L. Creswell, Z. Dou, C. Xu, S. J. Mitchell, A. Das, B. L. O’Connell, S. Thakur, A. E. Kane, Q. Su, Y. Mohri, E. K. Nishimura, L. Schaevitz, N. Garg, A. M. Balta, M. A. Rego, M. Gregory-Ksander, T. C. Jakobs, L. Zhong, H. Wakimoto, J. El Andari, D. Grimm, R. Mostoslavsky, A. J. Wagers, K. Tsubota, S. J. Bonasera, C. M. Palmeira, J. G. Seidman, C. E. Seidman, N. S. Wolf, J. A. Kreiling, J. M. Sedivy, G. F. Murphy, R. E. Green, B. A. Garcia, S. L. Berger, P. Oberdoerffer, S. J. Shankland, V. N. Gladyshev, B. R. Ksander, A. R. Pfenning, L. A. Rajman, D. A. Sinclair, Loss of epigenetic information as a cause of mammalian aging. Cell S0092–8674(22)01570 (2023).
- Y. Lu, B. Brommer, X. Tian, A. Krishnan, M. Meer, C. Wang, D. L. Vera, Q. Zeng, D. Yu, M. S. Bonkowski, J. H. Yang, S. Zhou, E. M. Hoffmann, M. M. Karg, M. B. Schultz, A. E. Kane, N. Davidsohn, E. Korobkina, K. Chwalek, L. A. Rajman, G. M. Church, K. Hochedlinger, V. N. Gladyshev, S. Horvath, M. E. Levine, M. S. Gregory-Ksander, B. R. Ksander, Z. He, D. A. Sinclair, Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129 (2020). [CrossRef]
- D. A. Sinclair, P. Oberdoerffer, The ageing epigenome: damaged beyond repair Ageing Res Rev 8, 189–198 (2009).
- M. Fossel, ‘We need to get comfortable with being wrong about aging’. (available at https://longevity.technology/news/we-need-to-get-comfortable-with-being-wrong-about-aging/).
- M. G. Ribeiro, A. L. Larentis, L. A. Caldas, T. C. Garcia, L. L. Terra, M. H. Herbst, R. V. Almeida, On the debate about teleology in biology: the notion of “teleological obstacle”. Hist Cienc Saude Manguinhos 22, 1321–1333 (2015). [CrossRef]
- E. Takemura, EL DEBATE FILOSÓFICO DE LA TELEOLOGÍA EN LA EXPLICACIÓN BIOLÓGICA:¿ UN MAL NECESARIO? Journal of Teleological Science (2021).
- C. Darwin, On the origin of species (D. Appleton and Co., New York : 1871).
- I. Newton, Philosophiae Naturalis Principia Mathematica (Jussu Societatis Regiae, London, 1687).
- P. Siregar, N. Julen, P. Hufnagl, G. Mutter, A general framework dedicated to computational morphogenesis Part II - Knowledge representation and architecture. Biosystems 173, 314–334 (2018). [CrossRef]
- J.-B. P. A. D. M. Lamarck, Histoire naturelle des animaux sans vertèbres (Verdière, Paris, 1815).
- J.-B. P. A. D. M. Lamarck, Philosophie zoologique, ou Exposition des considérations relatives à l’histoire naturelle des animaux (Dentu & L’Auteur, Paris, 1809).
- D. Krakauer, N. Bertschinger, E. Olbrich, J. C. Flack, N. Ay, The information theory of individuality. Theory Biosci 139, 209–223 (2020).
- J. P. de Magalhães, Ageing as a software design flaw. Genome Biol 24, 51 (2023).
- P. Oberdoerffer, S. Michan, M. McVay, R. Mostoslavsky, J. Vann, S. K. Park, A. Hartlerode, J. Stegmuller, A. Hafner, P. Loerch, S. M. Wright, K. D. Mills, A. Bonni, B. A. Yankner, R. Scully, T. A. Prolla, F. W. Alt, D. A. Sinclair, SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008). [CrossRef]
- Z. Koch, A. Li, D. S. Evans, S. Cummings, T. Ideker, Somatic mutation as an explanation for epigenetic aging. Nat Aging (2025). [CrossRef]
- V. L. Wilson, R. A. Smith, S. Ma, R. G. Cutler, Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem 262, 9948–9951 (1987). [CrossRef]
- K. A. Wareham, M. F. Lyon, P. H. Glenister, E. D. Williams, Age related reactivation of an X-linked gene. Nature 327, 725–727 (1987). [CrossRef]
- V. L. Wilson, P. A. Jones, DNA methylation decreases in aging but not in immortal cells. Science 220, 1055–1057 (1983). [CrossRef]
- B. F. Vanyushin, L. E. Nemirovsky, V. V. Klimenko, V. K. Vasiliev, A. N. Belozersky, The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia 19, 138–152 (1973).
- M. F. Fraga, E. Ballestar, M. F. Paz, S. Ropero, F. Setien, M. L. Ballestar, D. Heine-Suñer, J. C. Cigudosa, M. Urioste, J. Benitez, M. Boix-Chornet, A. Sanchez-Aguilera, C. Ling, E. Carlsson, P. Poulsen, A. Vaag, Z. Stephan, T. D. Spector, Y. Z. Wu, C. Plass, M. Esteller, Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102, 10604–10609 (2005). [CrossRef]
- K. M. Kim, D. Shibata, Tracing ancestry with methylation patterns: most crypts appear distantly related in normal adult human colon. BMC Gastroenterol 4, 8 (2004). [CrossRef]
- C. A. Cooney, Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging 57, 261–273 (1993).
- W. Dang, K. K. Steffen, R. Perry, J. A. Dorsey, F. B. Johnson, A. Shilatifard, M. Kaeberlein, B. K. Kennedy, S. L. Berger, Histone H4 lysine 16 acetylation regulates cellular lifespan Nature 459, 802–807 (2009). [CrossRef]
- B. Sarg, E. Koutzamani, W. Helliger, I. Rundquist, H. H. Lindner, Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem 277, 39195–39201 (2002). [CrossRef]
- H. A. Tissenbaum, L. Guarente, Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410, 227–230 (2001). [CrossRef]
- B. K. Kennedy, M. Gotta, D. A. Sinclair, K. Mills, D. S. McNabb, M. Murthy, S. M. Pak, T. Laroche, S. M. Gasser, L. Guarente, Redistribution of Silencing Proteins From Telomeres to the Nucleolus is Associated With Extension of Life Span in S. Cerevisiae. Cell 89, 381–391 (1997). [CrossRef]
- S. Horvath, DNA methylation age of human tissues and cell types. Genome Biol 14, R115 (2013).
- W. W. Gibbs, Biomarkers and ageing: The clock-watcher. Nature 508, 168–170 (2014). [CrossRef]
- G. Hannum, J. Guinney, L. Zhao, L. Zhang, G. Hughes, S. Sadda, B. Klotzle, M. Bibikova, J. B. Fan, Y. Gao, R. Deconde, M. Chen, I. Rajapakse, S. Friend, T. Ideker, K. Zhang, Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49, 359–367 (2013). [CrossRef]
- M. E. Levine, A. T. Lu, A. Quach, B. H. Chen, T. L. Assimes, S. Bandinelli, L. Hou, A. A. Baccarelli, J. D. Stewart, Y. Li, E. A. Whitsel, J. G. Wilson, A. P. Reiner, A. Aviv, K. Lohman, Y. Liu, L. Ferrucci, S. Horvath, An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591 (2018). [CrossRef]
- A. T. Lu, A. Quach, J. G. Wilson, A. P. Reiner, A. Aviv, K. Raj, L. Hou, A. A. Baccarelli, Y. Li, J. D. Stewart, E. A. Whitsel, T. L. Assimes, L. Ferrucci, S. Horvath, DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 11, 303–327 (2019). [CrossRef]
- A. E. Teschendorff, S. Horvath, Epigenetic ageing clocks: statistical methods and emerging computational challenges. Nat Rev Genet (2025). [CrossRef]
- C. McCrory, G. Fiorito, B. Hernandez, S. Polidoro, A. M. O’Halloran, A. Hever, C. Ni Cheallaigh, A. T. Lu, S. Horvath, P. Vineis, R. A. Kenny, GrimAge Outperforms Other Epigenetic Clocks in the Prediction of Age-Related Clinical Phenotypes and All-Cause Mortality. J Gerontol A Biol Sci Med Sci 76, 741–749 (2021). [CrossRef]
- R. E. Marioni, S. Shah, A. F. McRae, B. H. Chen, E. Colicino, S. E. Harris, J. Gibson, A. K. Henders, P. Redmond, S. R. Cox, A. Pattie, J. Corley, L. Murphy, N. G. Martin, G. W. Montgomery, A. P. Feinberg, M. D. Fallin, M. L. Multhaup, A. E. Jaffe, R. Joehanes, J. Schwartz, A. C. Just, K. L. Lunetta, J. M. Murabito, J. M. Starr, S. Horvath, A. A. Baccarelli, D. Levy, P. M. Visscher, N. R. Wray, I. J. Deary, DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 16, 25 (2015). [CrossRef]
- Y. Zhang, R. Wilson, J. Heiss, L. P. Breitling, K. U. Saum, B. Schöttker, B. Holleczek, M. Waldenberger, A. Peters, H. Brenner, DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun 8, 14617 (2017). [CrossRef]
- J. Ma, C. M. Rebholz, K. V. E. Braun, L. M. Reynolds, S. Aslibekyan, R. Xia, N. G. Biligowda, T. Huan, C. Liu, M. M. Mendelson, R. Joehanes, E. A. Hu, M. Z. Vitolins, A. C. Wood, K. Lohman, C. Ochoa-Rosales, J. van Meurs, A. Uitterlinden, Y. Liu, M. A. Elhadad, M. Heier, M. Waldenberger, A. Peters, E. Colicino, E. A. Whitsel, A. Baldassari, S. A. Gharib, N. Sotoodehnia, J. A. Brody, C. M. Sitlani, T. Tanaka, W. D. Hill, J. Corley, I. J. Deary, Y. Zhang, B. Schöttker, H. Brenner, M. E. Walker, S. Ye, S. Nguyen, J. Pankow, E. W. Demerath, Y. Zheng, L. Hou, L. Liang, A. H. Lichtenstein, F. B. Hu, M. Fornage, T. Voortman, D. Levy, Whole Blood DNA Methylation Signatures of Diet Are Associated With Cardiovascular Disease Risk Factors and All-Cause Mortality. Circ Genom Precis Med 13, e002766 (2020). [CrossRef]
- J. B. Lund, S. Li, J. Baumbach, A. M. Svane, J. Hjelmborg, L. Christiansen, K. Christensen, P. Redmond, R. E. Marioni, I. J. Deary, Q. Tan, DNA methylome profiling of all-cause mortality in comparison with age-associated methylation patterns. Clin Epigenetics 11, 23 (2019). [CrossRef]
- Y. Haluza, J. A. Zoller, A. T. Lu, H. E. Walters, M. Lachnit, R. Lowe, A. Haghani, R. T. Brooke, N. Park, M. H. Yun, S. Horvath, Axolotl epigenetic clocks offer insights into the nature of negligible senescence bioRxiv 0 (2024).
- A. T. Lu, Z. Fei, A. Haghani, T. R. Robeck, J. A. Zoller, C. Z. Li, R. Lowe, Q. Yan, J. Zhang, H. Vu, J. Ablaeva, V. A. Acosta-Rodriguez, D. M. Adams, J. Almunia, A. Aloysius, R. Ardehali, A. Arneson, C. S. Baker, G. Banks, K. Belov, N. C. Bennett, P. Black, D. T. Blumstein, E. K. Bors, C. E. Breeze, R. T. Brooke, J. L. Brown, G. G. Carter, A. Caulton, J. M. Cavin, L. Chakrabarti, I. Chatzistamou, H. Chen, K. Cheng, P. Chiavellini, O. W. Choi, S. M. Clarke, L. N. Cooper, M. L. Cossette, J. Day, J. DeYoung, S. DiRocco, C. Dold, E. E. Ehmke, C. K. Emmons, S. Emmrich, E. Erbay, C. Erlacher-Reid, C. G. Faulkes, S. H. Ferguson, C. J. Finno, J. E. Flower, J. M. Gaillard, E. Garde, L. Gerber, V. N. Gladyshev, V. Gorbunova, R. G. Goya, M. J. Grant, C. B. Green, E. N. Hales, M. B. Hanson, D. W. Hart, M. Haulena, K. Herrick, A. N. Hogan, C. J. Hogg, T. A. Hore, T. Huang, J. C. Izpisua Belmonte, A. J. Jasinska, G. Jones, E. Jourdain, O. Kashpur, H. Katcher, E. Katsumata, V. Kaza, H. Kiaris, M. S. Kobor, P. Kordowitzki, W. R. Koski, M. Krützen, S. B. Kwon, B. Larison, S. G. Lee, M. Lehmann, J. F. Lemaitre, A. J. Levine, C. Li, X. Li, A. R. Lim, D. T. S. Lin, D. M. Lindemann, T. J. Little, N. Macoretta, D. Maddox, C. O. Matkin, J. A. Mattison, M. McClure, J. Mergl, J. J. Meudt, G. A. Montano, K. Mozhui, J. Munshi-South, A. Naderi, M. Nagy, P. Narayan, P. W. Nathanielsz, N. B. Nguyen, C. Niehrs, J. K. O’Brien, P. O’Tierney Ginn, D. T. Odom, A. G. Ophir, S. Osborn, E. A. Ostrander, K. M. Parsons, K. C. Paul, M. Pellegrini, K. J. Peters, A. B. Pedersen, J. L. Petersen, D. W. Pietersen, G. M. Pinho, J. Plassais, J. R. Poganik, N. A. Prado, P. Reddy, B. Rey, B. R. Ritz, J. Robbins, M. Rodriguez, J. Russell, E. Rydkina, L. L. Sailer, A. B. Salmon, A. Sanghavi, K. M. Schachtschneider, D. Schmitt, T. Schmitt, L. Schomacher, L. B. Schook, K. E. Sears, A. W. Seifert, A. Seluanov, A. B. A. Shafer, D. Shanmuganayagam, A. V. Shindyapina, M. Simmons, K. Singh, I. Sinha, J. Slone, R. G. Snell, E. Soltanmaohammadi, M. L. Spangler, M. C. Spriggs, L. Staggs, N. Stedman, K. J. Steinman, D. T. Stewart, V. J. Sugrue, B. Szladovits, J. S. Takahashi, M. Takasugi, E. C. Teeling, M. J. Thompson, B. Van Bonn, S. C. Vernes, D. Villar, H. V. Vinters, M. C. Wallingford, N. Wang, R. K. Wayne, G. S. Wilkinson, C. K. Williams, R. W. Williams, X. W. Yang, M. Yao, B. G. Young, B. Zhang, Z. Zhang, P. Zhao, Y. Zhao, W. Zhou, J. Zimmermann, J. Ernst, K. Raj, S. Horvath, Universal DNA methylation age across mammalian tissues. Nat Aging (2023). [CrossRef]
- J. A. Zoller, E. Parasyraki, A. T. Lu, A. Haghani, C. Niehrs, S. Horvath, DNA methylation clocks for clawed frogs reveal evolutionary conservation of epigenetic aging. Geroscience (2023). [CrossRef]
- K. J. Peters, L. Gerber, L. Scheu, R. Cicciarella, J. A. Zoller, Z. Fei, S. Horvath, S. J. Allen, S. L. King, R. C. Connor, L. A. Rollins, M. Krützen, An epigenetic DNA methylation clock for age estimates in Indo-Pacific bottlenose dolphins (Tursiops aduncus). Evol Appl 16, 126–133 (2023). [CrossRef]
- S. Horvath, A. Haghani, J. A. Zoller, A. Naderi, E. Soltanmohammadi, E. Farmaki, V. Kaza, I. Chatzistamou, H. Kiaris, Methylation studies in Peromyscus: aging, altitude adaptation, and monogamy. Geroscience 44, 447–461 (2022). [CrossRef]
- S. Horvath, A. Haghani, N. Macoretta, J. Ablaeva, J. A. Zoller, C. Z. Li, J. Zhang, M. Takasugi, Y. Zhao, E. Rydkina, Z. Zhang, S. Emmrich, K. Raj, A. Seluanov, C. G. Faulkes, V. Gorbunova, DNA methylation clocks tick in naked mole rats but queens age more slowly than nonbreeders. Nat Aging 2, 46–59 (2022). [CrossRef]
- S. Horvath, A. Haghani, S. Peng, E. N. Hales, J. A. Zoller, K. Raj, B. Larison, T. R. Robeck, J. L. Petersen, R. R. Bellone, C. J. Finno, DNA methylation aging and transcriptomic studies in horses. Nat Commun 13, 40 (2022). [CrossRef]
- S. Horvath, J. A. Zoller, A. Haghani, A. J. Jasinska, K. Raj, C. E. Breeze, J. Ernst, K. L. Vaughan, J. A. Mattison, Epigenetic clock and methylation studies in the rhesus macaque. Geroscience 43, 2441–2453 (2021). [CrossRef]
- A. Caulton, K. G. Dodds, K. M. McRae, C. Couldrey, S. Horvath, S. M. Clarke, Development of Epigenetic Clocks for Key Ruminant Species. Genes (Basel) 13, 96 (2021). [CrossRef]
- R. Lowe, A. F. Danson, V. K. Rakyan, S. Yildizoglu, F. Saldmann, M. Viltard, G. Friedlander, C. G. Faulkes, DNA methylation clocks as a predictor for ageing and age estimation in naked mole-rats, Heterocephalus glaber. Aging (Albany NY) 12, 4394–4406 (2020). [CrossRef]
- M. J. Thompson, K. Chwiałkowska, L. Rubbi, A. J. Lusis, R. C. Davis, A. Srivastava, R. Korstanje, G. A. Churchill, S. Horvath, M. Pellegrini, A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854 (2018). [CrossRef]
- M. J. Thompson, B. vonHoldt, S. Horvath, M. Pellegrini, An epigenetic aging clock for dogs and wolves. Aging 9, 1055–1068 (2017). [CrossRef]
- M. E. Sehl, W. Guo, C. Farrell, N. Marino, J. E. Henry, A. M. Storniolo, J. Papp, J. J. Li, S. Horvath, M. Pellegrini, P. A. Ganz, Systematic dissection of epigenetic age acceleration in normal breast tissue reveals its link to estrogen signaling and cancer risk bioRxiv 0 (2024).
- S. Haefliger, O. Chervova, C. Davies, C. Loh, R. Tirabosco, F. Amary, N. Pillay, S. Horvath, S. Beck, A. M. Flanagan, I. Lyskjær, Epigenetic age acceleration is a distinctive trait of epithelioid sarcoma with potential therapeutic implications. Geroscience 46, 5203–5209 (2024). [CrossRef]
- K. L. Thrush, D. A. Bennett, C. Gaiteri, S. Horvath, C. H. V. Dyck, A. T. Higgins-Chen, M. E. Levine, Aging the brain: multi-region methylation principal component based clock in the context of Alzheimer’s disease. Aging (Albany NY) 14, 5641–5668 (2022). [CrossRef]
- P. R. Matías-García, C. K. Ward-Caviness, L. M. Raffield, X. Gao, Y. Zhang, R. Wilson, X. Gào, J. Nano, A. Bostom, E. Colicino, A. Correa, B. Coull, C. Eaton, L. Hou, A. C. Just, S. Kunze, L. Lange, E. Lange, X. Lin, S. Liu, J. C. Nwanaji-Enwerem, A. Reiner, J. Shen, B. Schöttker, P. Vokonas, Y. Zheng, B. Young, J. Schwartz, S. Horvath, A. Lu, E. A. Whitsel, W. Koenig, J. Adamski, J. Winkelmann, H. Brenner, A. A. Baccarelli, C. Gieger, A. Peters, N. Franceschini, M. Waldenberger, DNAm-based signatures of accelerated aging and mortality in blood are associated with low renal function. Clin Epigenetics 13, 121 (2021). [CrossRef]
- D. Nachun, A. T. Lu, A. G. Bick, P. Natarajan, J. Weinstock, M. D. Szeto, S. Kathiresan, G. Abecasis, K. D. Taylor, X. Guo, R. Tracy, P. Durda, Y. Liu, C. Johnson, S. S. Rich, D. Van Den Berg, C. Laurie, T. Blackwell, G. J. Papanicolaou, A. Correa, L. M. Raffield, A. D. Johnson, J. Murabito, J. E. Manson, P. Desai, C. Kooperberg, T. L. Assimes, D. Levy, J. I. Rotter, A. P. Reiner, E. A. Whitsel, J. G. Wilson, S. Horvath, S. Jaiswal, T.-O. F. P. M. T. O. P. M. C. NHLBI, Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 20, e13366 (2021). [CrossRef]
- K. C. Paul, A. M. Binder, S. Horvath, C. Kusters, Q. Yan, I. D. Rosario, Y. Yu, J. Bronstein, B. Ritz, Accelerated hematopoietic mitotic aging measured by DNA methylation, blood cell lineage, and Parkinson’s disease. BMC Genomics 22, 696 (2021). [CrossRef]
- S. Horvath, J. Oshima, G. M. Martin, A. T. Lu, A. Quach, H. Cohen, S. Felton, M. Matsuyama, D. Lowe, S. Kabacik, J. G. Wilson, A. P. Reiner, A. Maierhofer, J. Flunkert, A. Aviv, L. Hou, A. A. Baccarelli, Y. Li, J. D. Stewart, E. A. Whitsel, L. Ferrucci, S. Matsuyama, K. Raj, Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 10, 1758–1775 (2018). [CrossRef]
- C. R. Gale, R. E. Marioni, S. E. Harris, J. M. Starr, I. J. Deary, DNA methylation and the epigenetic clock in relation to physical frailty in older people: the Lothian Birth Cohort 1936. Clin Epigenetics 10, 101 (2018). [CrossRef]
- G. R. Fries, I. E. Bauer, G. Scaini, M. J. Wu, I. F. Kazimi, S. S. Valvassori, G. Zunta-Soares, C. Walss-Bass, J. C. Soares, J. Quevedo, Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Transl Psychiatry 7, 1283 (2017). [CrossRef]
- R. E. Marioni, S. Shah, A. F. McRae, S. J. Ritchie, G. Muniz-Terrera, S. E. Harris, J. Gibson, P. Redmond, S. R. Cox, A. Pattie, J. Corley, A. Taylor, L. Murphy, J. M. Starr, S. Horvath, P. M. Visscher, N. R. Wray, I. J. Deary, The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol 44, 1388–1396 (2015). [CrossRef]
- S. Horvath, A. J. Levine, HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J Infect Dis 212, 1563–1573 (2015). [CrossRef]
- F. Galkin, O. Kovalchuk, D. Koldasbayeva, A. Zhavoronkov, E. Bischof, Stress, diet, exercise: Common environmental factors and their impact on epigenetic age. Ageing Res Rev 88, 101956 (2023). [CrossRef]
- K. N. Fitzgerald, R. Hodges, D. Hanes, E. Stack, D. Cheishvili, M. Szyf, J. Henkel, M. W. Twedt, D. Giannopoulou, J. Herdell, S. Logan, R. Bradley, Potential reversal of epigenetic age using a diet and lifestyle intervention: a pilot randomized clinical trial. Aging (Albany NY) 13, 9419–9432 (2021). [CrossRef]
- G. Fiorito, S. Caini, D. Palli, B. Bendinelli, C. Saieva, I. Ermini, V. Valentini, M. Assedi, P. Rizzolo, D. Ambrogetti, L. Ottini, G. Masala, DNA methylation-based biomarkers of aging were slowed down in a two-year diet and physical activity intervention trial: the DAMA study. Aging Cell 20, e13439 (2021). [CrossRef]
- Quach, M. E. Levine, T. Tanaka, A. T. Lu, B. H. Chen, L. Ferrucci, B. Ritz, S. Bandinelli, M. L. Neuhouser, J. M. Beasley, L. Snetselaar, R. B. Wallace, P. S. Tsao, D. Absher, T. L. Assimes, J. D. Stewart, Y. Li, L. Hou, A. A. Baccarelli, E. A. Whitsel, S. Horvath, Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY) 9, 419–446 (2017). [CrossRef]
- P. Chiavellini, M. Lehmann, M. D. Gallardo, M. Canatelli Mallat, D. C. Pasquini, J. A. Zoller, J. Gordevicius, M. Girard, E. Lacunza, C. B. Herenu, S. Horvath, R. G. Goya, Young Plasma Rejuvenates Blood Dna Methylation Profile, Extends Mean Lifespan And Improves Physical Appearance In Old Rats. J Gerontol A Biol Sci Med Sci glae071 (2024).
- S. Horvath, K. Singh, K. Raj, S. I. Khairnar, A. Sanghavi, A. Shrivastava, J. A. Zoller, C. Z. Li, C. B. Herenu, M. Canatelli-Mallat, M. Lehmann, S. Habazin, M. Novokmet, F. Vučković, L. C. Solberg Woods, A. G. Martinez, T. Wang, P. Chiavellini, A. J. Levine, H. Chen, R. T. Brooke, J. Gordevicius, G. Lauc, R. G. Goya, H. L. Katcher, Reversal of biological age in multiple rat organs by young porcine plasma fraction. Geroscience 46, 367–394 (2024). [CrossRef]
- Y. Yang, X. Lu, N. Liu, S. Ma, H. Zhang, Z. Zhang, K. Yang, M. Jiang, Z. Zheng, Y. Qiao, Q. Hu, Y. Huang, Y. Zhang, M. Xiong, L. Liu, X. Jiang, P. Reddy, X. Dong, F. Xu, Q. Wang, Q. Zhao, J. Lei, S. Sun, Y. Jing, J. Li, Y. Cai, Y. Fan, K. Yan, Y. Jing, A. Haghani, M. Xing, X. Zhang, G. Zhu, W. Song, S. Horvath, C. Rodriguez Esteban, M. Song, S. Wang, G. Zhao, W. Li, J. C. Izpisua Belmonte, J. Qu, W. Zhang, G.-H. Liu, Metformin decelerates aging clock in male monkeys. Cell S0092–8674(24)00914 (2024). [CrossRef]
- J. Sanz-Ros, N. Romero-García, C. Mas-Bargues, D. Monleón, J. Gordevicius, R. T. Brooke, M. Dromant, A. Díaz, A. Derevyanko, A. Guío-Carrión, A. Román-Domínguez, M. Inglés, M. A. Blasco, S. Horvath, J. Viña, C. Borrás, Small extracellular vesicles from young adipose-derived stem cells prevent frailty, improve health span, and decrease epigenetic age in old mice. Sci Adv 8, eabq2226 (2022). [CrossRef]
- A. V. Shindyapina, Y. Cho, A. Kaya, A. Tyshkovskiy, J. P. Castro, A. Deik, J. Gordevicius, J. R. Poganik, C. B. Clish, S. Horvath, L. Peshkin, V. N. Gladyshev, Rapamycin treatment during development extends life span and health span of male mice and Daphnia magna. Sci Adv 8, eabo5482 (2022). [CrossRef]
- S. Horvath, J. A. Zoller, A. Haghani, A. T. Lu, K. Raj, A. J. Jasinska, J. A. Mattison, A. B. Salmon, DNA methylation age analysis of rapamycin in common marmosets. Geroscience 43, 2413–2425 (2021). [CrossRef]
- G. M. Fahy, R. T. Brooke, J. P. Watson, Z. Good, S. S. Vasanawala, H. Maecker, M. D. Leipold, D. T. S. Lin, M. S. Kobor, S. Horvath, Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 18, e13028 (2019). [CrossRef]
- S. Horvath, A. T. Lu, H. Cohen, K. Raj, Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging (Albany NY) 11, 3238–3249 (2019). [CrossRef]
- T. Wang, B. Tsui, J. F. Kreisberg, N. A. Robertson, A. M. Gross, M. K. Yu, H. Carter, H. M. Brown-Borg, P. D. Adams, T. Ideker, Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18, 57 (2017). [CrossRef]
- S. Horvath, E. Lacunza, M. C. Mallat, E. L. Portiansky, M. D. Gallardo, R. T. Brooke, P. Chiavellini, D. C. Pasquini, M. Girard, M. Lehmann, Q. Yan, A. T. Lu, A. Haghani, J. Gordevicius, M. Abba, R. G. Goya, Cognitive rejuvenation in old rats by hippocampal OSKM gene therapy. Geroscience (2024). [CrossRef]
- A. Parras, A. Vílchez-Acosta, G. Desdín-Micó, S. Picó, C. Mrabti, E. Montenegro-Borbolla, C. Y. Maroun, A. Haghani, R. Brooke, M. Del Carmen Maza, C. Rechsteiner, F. Battiston, C. Branchina, K. Perez, S. Horvath, C. Bertelli, C. Sempoux, A. Ocampo, In vivo reprogramming leads to premature death linked to hepatic and intestinal failure. Nat Aging (2023). [CrossRef]
- J. H. Yang, C. A. Petty, T. Dixon-McDougall, M. V. Lopez, A. Tyshkovskiy, S. Maybury-Lewis, X. Tian, N. Ibrahim, Z. Chen, P. T. Griffin, M. Arnold, J. Li, O. A. Martinez, A. Behn, R. Rogers-Hammond, S. Angeli, V. N. Gladyshev, D. A. Sinclair, Chemically induced reprogramming to reverse cellular aging. Aging (Albany NY) 15, 5966–5989 (2023). [CrossRef]
- T. J. Sarkar, M. Quarta, S. Mukherjee, A. Colville, P. Paine, L. Doan, C. M. Tran, C. R. Chu, S. Horvath, L. S. Qi, N. Bhutani, T. A. Rando, V. Sebastiano, Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat Commun 11, 1545 (2020). [CrossRef]
- A. Ocampo, P. Reddy, P. Martínez-Redondo, A. Platero-Luengo, F. Hatanaka, T. Hishida, M. Li, D. Lam, M. Kurita, E. Beyret, T. Araoka, E. Vazquez-Ferrer, D. Donoso, J. L. Roman, J. Xu, C. Rodriguez Esteban, G. Nuñez, E. Nuñez Delicado, J. M. Campistol, I. Guillen, P. Guillen, J. C. Izpisua Belmonte, In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719–1733.e12 (2016). [CrossRef]
- L. Lapasset, O. Milhavet, A. Prieur, E. Besnard, A. Babled, N. Aït-Hamou, J. Leschik, F. Pellestor, J. M. Ramirez, J. De Vos, S. Lehmann, J. M. Lemaitre, Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev 25, 2248–2253 (2011). [CrossRef]
- R. M. Marion, K. Strati, H. Li, A. Tejera, S. Schoeftner, S. Ortega, M. Serrano, M. A. Blasco, Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141–154 (2009). [CrossRef]
- S. T. Suhr, E. A. Chang, R. M. Rodriguez, K. Wang, P. J. Ross, Z. Beyhan, S. Murthy, J. B. Cibelli, Telomere dynamics in human cells reprogrammed to pluripotency. PLoS One 4, e8124 (2009). [CrossRef]
- J. B. GURDON, Adult frogs derived from the nuclei of single somatic cells. Dev Biol 4, 256–273 (1962). [CrossRef]
- I. Wilmut, A. E. Schnieke, J. McWhir, A. J. Kind, K. H. Campbell, Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810–813 (1997). [CrossRef]
- R. M. Marión, I. López de Silanes, L. Mosteiro, B. Gamache, M. Abad, C. Guerra, D. Megías, M. Serrano, M. A. Blasco, Common Telomere Changes during In Vivo Reprogramming and Early Stages of Tumorigenesis. Stem Cell Reports 8, 460–475 (2017). [CrossRef]
- K. Ohnishi, K. Semi, T. Yamamoto, M. Shimizu, A. Tanaka, K. Mitsunaga, K. Okita, K. Osafune, Y. Arioka, T. Maeda, H. Soejima, H. Moriwaki, S. Yamanaka, K. Woltjen, Y. Yamada, Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677 (2014). [CrossRef]
- M. Abad, L. Mosteiro, C. Pantoja, M. Cañamero, T. Rayon, I. Ors, O. Graña, D. Megías, O. Domínguez, D. Martínez, M. Manzanares, S. Ortega, M. Serrano, Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 502, 340–345 (2013). [CrossRef]
- A. Cipriano, M. Moqri, S. Y. Maybury-Lewis, R. Rogers-Hammond, T. A. de Jong, A. Parker, S. Rasouli, H. R. Schöler, D. A. Sinclair, V. Sebastiano, Mechanisms, pathways and strategies for rejuvenation through epigenetic reprogramming Nature Aging (2023).
- R. Klausner, You Can Live Longer! (available at https://www.youtube.com/watch?v=Elt4xGalQu4&t=1s).
- K. Hochedlinger, R. Jaenisch, Induced Pluripotency and Epigenetic Reprogramming. Cold Spring Harb Perspect Biol 7, a019448 (2015). [CrossRef]
- T. Zhao, Z.-N. Zhang, Z. Rong, Y. Xu, Immunogenicity of induced pluripotent stem cells. Nature 474, 212–215 (2011).
- R. Lister, M. Pelizzola, Y. S. Kida, R. D. Hawkins, J. R. Nery, G. Hon, J. Antosiewicz-Bourget, R. O’Malley, R. Castanon, S. Klugman, M. Downes, R. Yu, R. Stewart, B. Ren, J. A. Thomson, R. M. Evans, J. R. Ecker, Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73 (2011). [CrossRef]
- K. Kim, A. Doi, B. Wen, K. Ng, R. Zhao, P. Cahan, J. Kim, M. J. Aryee, H. Ji, L. I. R. Ehrlich, A. Yabuuchi, A. Takeuchi, K. C. Cunniff, H. Hongguang, S. McKinney-Freeman, O. Naveiras, T. J. Yoon, R. A. Irizarry, N. Jung, J. Seita, J. Hanna, P. Murakami, R. Jaenisch, R. Weissleder, S. H. Orkin, I. L. Weissman, A. P. Feinberg, G. Q. Daley, Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290 (2010). [CrossRef]
- J. M. Polo, S. Liu, M. E. Figueroa, W. Kulalert, S. Eminli, K. Y. Tan, E. Apostolou, M. Stadtfeld, Y. Li, T. Shioda, S. Natesan, A. J. Wagers, A. Melnick, T. Evans, K. Hochedlinger, Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28, 848–855 (2010). [CrossRef]
- K. Miura, Y. Okada, T. Aoi, A. Okada, K. Takahashi, K. Okita, M. Nakagawa, M. Koyanagi, K. Tanabe, M. Ohnuki, D. Ogawa, E. Ikeda, H. Okano, S. Yamanaka, Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol 27, 743–745 (2009). [CrossRef]
- M. J. Boland, J. L. Hazen, K. L. Nazor, A. R. Rodriguez, W. Gifford, G. Martin, S. Kupriyanov, K. K. Baldwin, Adult mice generated from induced pluripotent stem cells. Nature 461, 91–94 (2009). [CrossRef]
- Y. Voituron, M. de Fraipont, J. Issartel, O. Guillaume, J. Clobert, Extreme lifespan of the human fish (Proteus anguinus): a challenge for ageing mechanisms. Biol Lett 7, 105–107 (2011). [CrossRef]
- E. Beccari, P. Capdevila, R. Salguero-Gómez, C. P. Carmona, Worldwide diversity in mammalian life histories: Environmental realms and evolutionary adaptations Ecology Letters 27, e14445 (2024). [CrossRef]
- D. Sol, A. Prego, L. Olivé, M. Genovart, D. Oro, A. Hernández-Matías, Adaptations to marine environments and the evolution of slow-paced life histories in endotherms. Nat Commun 16, 4265 (2025). [CrossRef]
- E. Pennisi, Greenland shark may live 400 years, smashing longevity record Science Magazine 11, (2016).
- K. M. Glastad, J. Roessler, J. Gospocic, R. Bonasio, S. L. Berger, Long ant life span is maintained by a unique heat shock factor. Genes Dev 37, 398–417 (2023). [CrossRef]
- H. Yan, C. Opachaloemphan, F. Carmona-Aldana, G. Mancini, J. Mlejnek, N. Descostes, B. Sieriebriennikov, A. Leibholz, X. Zhou, L. Ding, M. Traficante, C. Desplan, D. Reinberg, Insulin signaling in the long-lived reproductive caste of ants. Science 377, 1092–1099 (2022). [CrossRef]
- J. A. Zoller, A. T. Lu, A. Haghani, S. Horvath, T. Robeck, Enhancing epigenetic aging clocks in cetaceans: accurate age estimations in small endangered delphinids, killer whales, pilot whales, belugas, humpbacks, and bowhead whales. Sci Rep 15, 4048 (2025). [CrossRef]
- A. Haller, J. Risse, B. Sepers, K. van Oers, Independent avian epigenetic clocks for aging and development bioRxiv 0 (2024).
- C. Z. Li, A. Haghani, Q. Yan, A. T. Lu, J. Zhang, Z. Fei, J. Ernst, X. W. Yang, V. N. Gladyshev, T. R. Robeck, A. S. Chavez, J. A. Cook, J. L. Dunnum, K. Raj, A. Seluanov, V. Gorbunova, S. Horvath, Epigenetic predictors of species maximum life span and other life-history traits in mammals. Sci Adv 10, eadm7273 (2024). [CrossRef]
- E. M. Bertucci-Richter, B. B. Parrott, The rate of epigenetic drift scales with maximum lifespan across mammals. Nat Commun 14, 7731 (2023). https. [CrossRef]
- K. M. Parsons, A. Haghani, J. A. Zoller, A. T. Lu, Z. Fei, S. H. Ferguson, E. Garde, M. B. Hanson, C. K. Emmons, C. O. Matkin, B. G. Young, W. R. Koski, S. Horvath, DNA methylation-based biomarkers for ageing long-lived cetaceans. Mol Ecol Resour (2023).
- S. J. C. Crofts, E. Latorre-Crespo, T. Chandra, DNA methylation rates scale with maximum lifespan across mammals. Nat Aging (2023). [CrossRef]
- N. A. Prado, J. L. Brown, J. A. Zoller, A. Haghani, M. Yao, L. R. Bagryanova, M. G. Campana, J. E Maldonado, K. Raj, D. Schmitt, T. R. Robeck, S. Horvath, Epigenetic clock and methylation studies in elephants. Aging Cell 20, e13414 (2021). [CrossRef]
- G. S. Wilkinson, D. M. Adams, A. Haghani, A. T. Lu, J. Zoller, C. E. Breeze, B. D. Arnold, H. C. Ball, G. G. Carter, L. N. Cooper, D. K. N. Dechmann, P. Devanna, N. J. Fasel, A. V. Galazyuk, L. Günther, E. Hurme, G. Jones, M. Knörnschild, E. Z. Lattenkamp, C. Z. Li, F. Mayer, J. A. Reinhardt, R. A. Medellin, M. Nagy, B. Pope, M. L. Power, R. D. Ransome, E. C. Teeling, S. C. Vernes, D. Zamora-Mejías, J. Zhang, P. A. Faure, L. J. Greville, L. G. Herrera M, J. J. Flores-Martínez, S. Horvath, DNA methylation predicts age and provides insight into exceptional longevity of bats. Nat Commun 12, 1615 (2021). [CrossRef]
- A. Zhavoronkov, P. Mamoshina, Q. Vanhaelen, M. Scheibye-Knudsen, A. Moskalev, A. Aliper, Artificial intelligence for aging and longevity research: Recent advances and perspectives. Ageing Res Rev 49, 49–66 (2019). [CrossRef]
- J. Mutz, R. Iniesta, C. M. Lewis, Metabolomic age (MileAge) predicts health and life span: A comparison of multiple machine learning algorithms. Sci Adv 10, eadp3743 (2024). [CrossRef]
- E. Hantikainen, C. X. Weichenberger, N. Dordevic, V. V. Hernandes, L. Foco, M. Gögele, R. Melotti, C. Pattaro, M. Ralser, F. Amari, V. Farztdinov, M. Mülleder, P. P. Pramstaller, J. Rainer, F. S. Domingues, Identifying Metabolomic and Proteomic Biomarkers for Age-Related Morbidity in a Population-Based Cohort - the Cooperative Health Research in South Tyrol (CHRIS) study medRxiv 0 (2024).
- A. A. Cohen, L. Ferrucci, T. Fülöp, D. Gravel, N. Hao, A. Kriete, M. E. Levine, L. A. Lipsitz, M. G. M. Olde Rikkert, A. Rutenberg, N. Stroustrup, R. Varadhan, A complex systems approach to aging biology Nature Aging 2, 580–591 (2022).
- C. Menni, G. Kastenmüller, A. K. Petersen, J. T. Bell, M. Psatha, P.-C. Tsai, C. Gieger, H. Schulz, I. Erte, S. John, M. J. Brosnan, S. G. Wilson, L. Tsaprouni, E. M. Lim, B. Stuckey, P. Deloukas, R. Mohney, K. Suhre, T. D. Spector, A. M. Valdes, Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol 42, 1111–1119 (2013). [CrossRef]
- F. Galkin, P. Mamoshina, A. Aliper, E. Putin, V. Moskalev, V. N. Gladyshev, A. Zhavoronkov, Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 23, 101199 (2020). [CrossRef]
- L. B. Alexandrov, P. H. Jones, D. C. Wedge, J. E. Sale, P. J. Campbell, S. Nik-Zainal, M. R. Stratton, Clock-like mutational processes in human somatic cells. Nat Genet 47, 1402–1407 (2015). [CrossRef]
- M. A. Argentieri, S. Xiao, D. Bennett, L. Winchester, A. J. Nevado-Holgado, U. Ghose, A. Albukhari, P. Yao, M. Mazidi, J. Lv, I. Millwood, H. Fry, R. S. Rodosthenous, J. Partanen, Z. Zheng, M. Kurki, M. J. Daly, A. Palotie, C. J. Adams, L. Li, R. Clarke, N. Amin, Z. Chen, C. M. van Duijn, Proteomic aging clock predicts mortality and risk of common age-related diseases in diverse populations. Nat Med 30, 2450–2460 (2024). [CrossRef]
- C.-L. Kuo, Z. Chen, P. Liu, L. C. Pilling, J. L. Atkins, R. H. Fortinsky, G. A. Kuchel, B. S. Diniz, Proteomic aging clock (PAC) predicts age-related outcomes in middle-aged and older adults. Aging Cell 23, e14195 (2024). [CrossRef]
- L. Coenen, B. Lehallier, H. E. de Vries, J. Middeldorp, Markers of aging: Unsupervised integrated analyses of the human plasma proteome. Front Aging 4, 1112109 (2023). [CrossRef]
- B. Lehallier, M. N. Shokhirev, T. Wyss-Coray, A. A. Johnson, Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. Aging Cell e13256 (2020). [CrossRef]
- A. Tyshkovskiy, D. Kholdina, K. Ying, M. Davitadze, A. Molière, Y. Tongu, T. Kasahara, L. M. Kats, A. Vladimirova, A. Moldakozhayev, H. Liu, B. Zhang, U. Khasanova, M. Moqri, J. M. Van Raamsdonk, D. E. Harrison, R. Strong, T. Abe, S. E. Dmitriev, V. N. Gladyshev, Transcriptomic Hallmarks of Mortality Reveal Universal and Specific Mechanisms of Aging, Chronic Disease, and Rejuvenation bioRxiv 1 (2024).
- E. D. Sun, O. Y. Zhou, M. Hauptschein, N. Rappoport, L. Xu, P. Navarro Negredo, L. Liu, T. A. Rando, J. Zou, A. Brunet, Spatial transcriptomic clocks reveal cell proximity effects in brain ageing. Nature (2024). [CrossRef]
- S. Jung, J. Arcos Hodar, A. Del Sol, Measuring biological age using a functionally interpretable multi-tissue RNA clock. Aging Cell 22, e13799 (2023). [CrossRef]
- A. Tyshkovskiy, S. Ma, A. V. Shindyapina, S. Tikhonov, S. G. Lee, P. Bozaykut, J. P. Castro, A. Seluanov, N. J. Schork, V. Gorbunova, S. E. Dmitriev, R. A. Miller, V. N. Gladyshev, Distinct longevity mechanisms across and within species and their association with aging. Cell 186, 2929–2949.e20 (2023). [CrossRef]
- M. T. Buckley, E. D. Sun, B. M. George, L. Liu, N. Schaum, L. Xu, J. M. Reyes, M. A. Goodell, I. L. Weissman, T. Wyss-Coray, T. A. Rando, A. Brunet, Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain. Nat Aging 3, 121–137 (2023). [CrossRef]
- N. Holzscheck, C. Falckenhayn, J. Söhle, B. Kristof, R. Siegner, A. Werner, J. Schössow, C. Jürgens, H. Völzke, H. Wenck, M. Winnefeld, E. Grönniger, L. Kaderali, Modeling transcriptomic age using knowledge-primed artificial neural networks. NPJ Aging Mech Dis 7, 15 (2021). [CrossRef]
- L. P. de Lima Camillo, M. H. Asif, S. Horvath, E. Larschan, R. Singh, Histone mark age of human tissues and cell types. Sci Adv 11, eadk9373 (2025). [CrossRef]
- F. Morandini, C. Rechsteiner, K. Perez, V. Praz, G. Lopez Garcia, L. C. Hinte, F. von Meyenn, A. Ocampo, ATAC-clock: An aging clock based on chromatin accessibility. Geroscience 46, 1789–1806 (2024). [CrossRef]
- E. Unal, B. Kinde, A. Amon, Gametogenesis eliminates age-induced cellular damage and resets life span in yeast. Science 332, 1554–1557 (2011). [CrossRef]
- A. M. Proenca, C. U. Rang, A. Qiu, C. Shi, L. Chao, Cell aging preserves cellular immortality in the presence of lethal levels of damage. PLoS Biol 17, e3000266 (2019). [CrossRef]
- U. Łapińska, G. Glover, P. Capilla-Lasheras, A. J. Young, S. Pagliara, Bacterial ageing in the absence of external stressors. Philos Trans R Soc Lond B Biol Sci 374, 20180442 (2019). [CrossRef]
- C. U. Rang, A. Y. Peng, A. F. Poon, L. Chao, Ageing in Escherichia coli requires damage by an extrinsic agent. Microbiology (Reading) 158, 1553–1559 (2012). [CrossRef]
- E. J. Stewart, R. Madden, G. Paul, F. Taddei, Aging and death in an organism that reproduces by morphologically symmetric division. PLoS Biol 3, e45 (2005). [CrossRef]
- M. Ackermann, S. C. Stearns, U. Jenal, Senescence in a bacterium with asymmetric division. Science 300, 1920 (2003). [CrossRef]
- L. Partridge, N. H. Barton, Optimality, mutation and the evolution of ageing. Nature 362, 305–311 (1993).
- L. Chao, A model for damage load and its implications for the evolution of bacterial aging PLoS genetics 6, e1001076 (2010).
- J. Winkler, A. Seybert, L. König, S. Pruggnaller, U. Haselmann, V. Sourjik, M. Weiss, A. S. Frangakis, A. Mogk, B. Bukau, Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J 29, 910–923 (2010). [CrossRef]
- S. Vedel, H. Nunns, A. Košmrlj, S. Semsey, A. Trusina, Asymmetric Damage Segregation Constitutes an Emergent Population-Level Stress Response. Cell Syst 3, 187–198 (2016). [CrossRef]
- C. Zhou, B. D. Slaughter, J. R. Unruh, A. Eldakak, B. Rubinstein, R. Li, Motility and segregation of Hsp104-associated protein aggregates in budding yeast. Cell 147, 1186–1196 (2011). [CrossRef]
- B. Liu, L. Larsson, V. Franssens, X. Hao, S. M. Hill, V. Andersson, D. Höglund, J. Song, X. Yang, D. Öling, J. Grantham, J. Winderickx, T. Nyström, Segregation of protein aggregates involves actin and the polarity machinery. Cell 147, 959–961 (2011). [CrossRef]
- Y. M. Yamashita, Asymmetric Stem Cell Division and Germline Immortality. Annu Rev Genet 57, 181–199 (2023). [CrossRef]
- H. Y. Lee, J. C. Chao, K. Y. Cheng, J. Y. Leu, Misfolding-prone proteins are reversibly sequestered to an Hsp42-associated granule upon chronological aging. J Cell Sci 131, jcs220202 (2018). [CrossRef]
- J. Yang, M. A. McCormick, J. Zheng, Z. Xie, M. Tsuchiya, S. Tsuchiyama, H. El-Samad, Q. Ouyang, M. Kaeberlein, B. K. Kennedy, H. Li, Systematic analysis of asymmetric partitioning of yeast proteome between mother and daughter cells reveals “aging factors” and mechanism of lifespan asymmetry. Proc Natl Acad Sci U S A 112, 11977–11982 (2015). [CrossRef]
- M. Coelho, S. J. Lade, S. Alberti, T. Gross, I. M. Tolić, Fusion of protein aggregates facilitates asymmetric damage segregation. PLoS Biol 12, e1001886 (2014). [CrossRef]
- L. Legon, C. Rallis, Genome-wide screens in yeast models towards understanding chronological lifespan regulation. Brief Funct Genomics 21, 4–12 (2022). [CrossRef]
- R. Dahiya, T. Mohammad, M. F. Alajmi, M. T. Rehman, G. M. Hasan, A. Hussain, M. I. Hassan, Insights into the Conserved Regulatory Mechanisms of Human and Yeast Aging. Biomolecules 10, 882 (2020). [CrossRef]
- A. Arlia-Ciommo, A. Leonov, A. Piano, V. Svistkova, V. I. Titorenko, Cell-autonomous mechanisms of chronological aging in the yeast Saccharomyces cerevisiae. Microb Cell 1, 163–178 (2014). [CrossRef]
- M. G. Mirisola, V. D. Longo, Yeast Chronological Lifespan: Longevity Regulatory Genes and Mechanisms. Cells 11, 1714 (2022). [CrossRef]
- C. He, C. Zhou, B. K. Kennedy, The yeast replicative aging model. Biochim Biophys Acta Mol Basis Dis 1864, 2690–2696 (2018).
- E. Janssens, L. M. Veenhoff, Evidence for the hallmarks of human aging in replicatively aging yeast. Microb Cell 3, 263–274 (2016). [CrossRef]
- D. Y. Damoo, D. G. Durnford, Long-term survival of Chlamydomonas reinhardtii during conditional senescence. Arch Microbiol 203, 5333–5344 (2021). [CrossRef]
- P. Sharma, P. Jain, A. Shrivastava, S. Saran, “Can Autophagy Stop the Clock: Unravelling the Mystery in Dictyostelium discoideum” Eds. 2020), pp. 235–258.
- I.-T. Lin, J.-L. Chao, M.-C. Yao, An essential role for the DNA breakage-repair protein Ku80 in programmed DNA rearrangements in Tetrahymena thermophila. Mol Biol Cell 23, 2213–2225 (2012). [CrossRef]
- G. E. Holmes, N. R. Holmes, Accumulation of DNA damages in aging Paramecium tetraurelia. Mol Gen Genet 204, 108–114 (1986). [CrossRef]
- M. Chen, L. K. Heilbronn, The health outcomes of human offspring conceived by assisted reproductive technologies (ART). J Dev Orig Health Dis 8, 388–402 (2017). [CrossRef]
- L. Grech, D. C. Jeffares, C. Y. Sadée, M. Rodríguez-López, D. A. Bitton, M. Hoti, C. Biagosch, D. Aravani, M. Speekenbrink, C. J. R. Illingworth, P. H. Schiffer, A. L. Pidoux, P. Tong, V. A. Tallada, R. Allshire, H. L. Levin, J. Bähler, Fitness Landscape of the Fission Yeast Genome. Mol Biol Evol 36, 1612–1623 (2019). [CrossRef]
- J. M. Park, A. G. Evertts, H. L. Levin, The Hermes transposon of Musca domestica and its use as a mutagen of Schizosaccharomyces pombe. Methods 49, 243–247 (2009). [CrossRef]
- J. Gunaratne, A. Schmidt, A. Quandt, S. P. Neo, O. S. Saraç, T. Gracia, S. Loguercio, E. Ahrné, R. L. Xia, K. H. Tan, C. Lössner, J. Bähler, A. Beyer, W. Blackstock, R. Aebersold, Extensive mass spectrometry-based analysis of the fission yeast proteome: the Schizosaccharomyces pombe PeptideAtlas. Mol Cell Proteomics 12, 1741–1751 (2013).
Figure 1.
Intergenerational Flow of Epigenetic Information. A. Epigenetic information is represented by a blue cylinder atop the DNA (red cylinder). Arrows indicate the flow of functional information derived from the genome (red) and the epigenome (blue). Adapted from Marsellach, 2021 [9]. B. Silenced loci in somatic cells are represented as closed black dots, while the same loci, expressed in germline cells, are shown in green. Red lines indicate how gene expression re-establishes youthful epigenetic information, effectively repairing epimutations present in the previous generation (orange cylinders). C. When no saturability issues occur during meiotic epigenetic repair, all defects are corrected, and the resulting organism is free of epigenetic defects. D. Persistent saturability problems during meiotic epigenetic repair, occurring over successive generations, lead to the progressive accumulation of epigenetic defects within the population.
Figure 1.
Intergenerational Flow of Epigenetic Information. A. Epigenetic information is represented by a blue cylinder atop the DNA (red cylinder). Arrows indicate the flow of functional information derived from the genome (red) and the epigenome (blue). Adapted from Marsellach, 2021 [9]. B. Silenced loci in somatic cells are represented as closed black dots, while the same loci, expressed in germline cells, are shown in green. Red lines indicate how gene expression re-establishes youthful epigenetic information, effectively repairing epimutations present in the previous generation (orange cylinders). C. When no saturability issues occur during meiotic epigenetic repair, all defects are corrected, and the resulting organism is free of epigenetic defects. D. Persistent saturability problems during meiotic epigenetic repair, occurring over successive generations, lead to the progressive accumulation of epigenetic defects within the population.
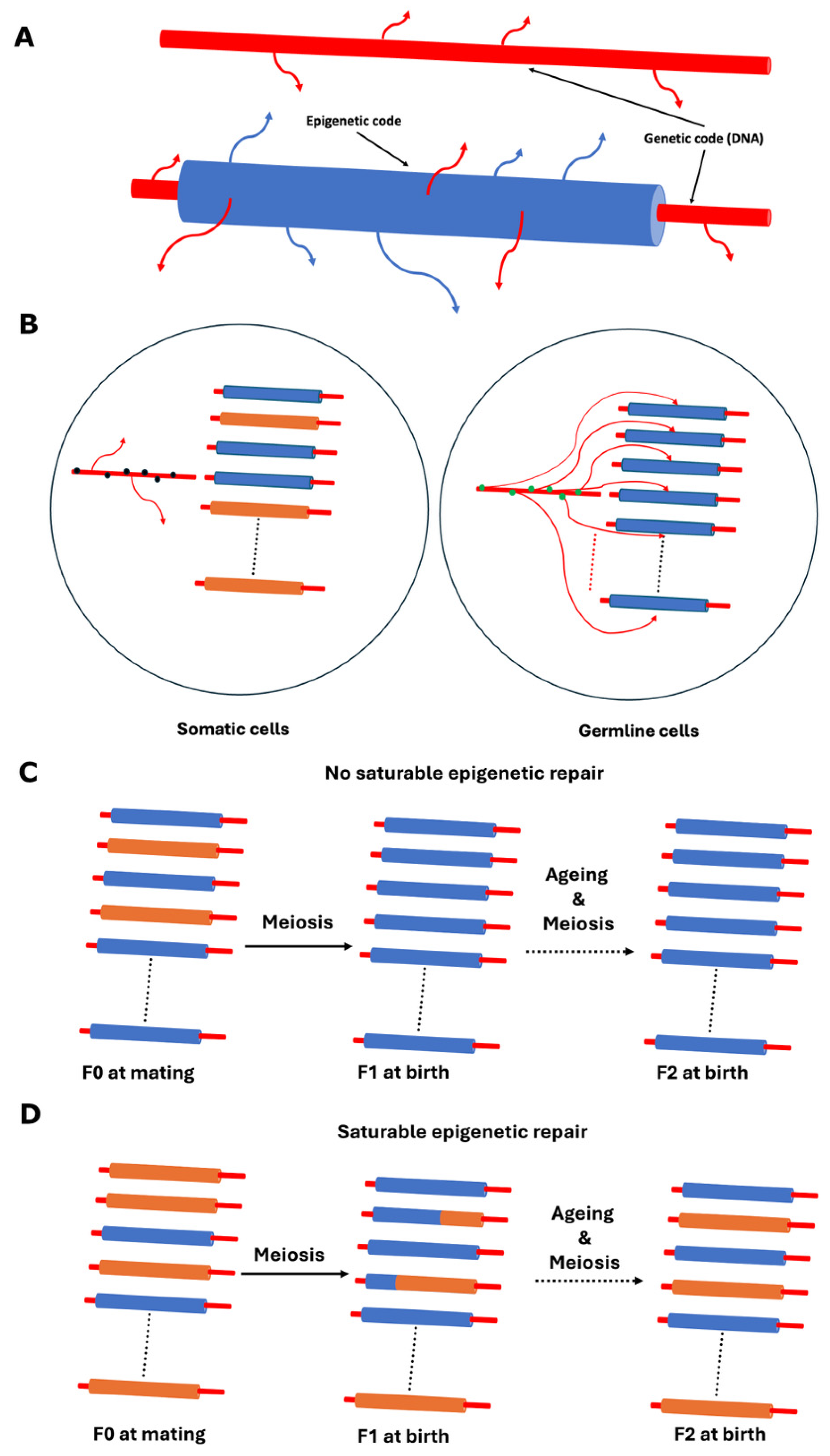
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



