
Submitted:
08 July 2025
Posted:
09 July 2025
You are already at the latest version
Abstract
Autism spectrum disorders (ASD) is a complex, heterogenous, and prevalent neurodevelopmental disorder characterized by core symptoms, including social communication deficits, restrictive interests and repetitive behaviors. Although environmental factors contribute to the etiology of ASD, the disorder has a strong genetic basis. Previous studies have focused on the cerebral cortex, hippocampus, and associated brain regions to uncover the underpinnings of ASD. However, dysfunction of the cerebellum has emerged as one of the most consistent associates of ASD. Although traditionally thought to function solely in motor control, more recent studies have established that projections from the cerebellum make mono- and polysynaptic connections to a variety of non-motor areas including cerebral cortex, hypothalamus, hippocampus, and is involved in a range of cognitive, sensory, and behavioral functions. Whereas the cellular and molecular mechanisms underlying ASD are far from understood, information gained from genetic studies in humans and animal models of ASD have identified a large number of genes and several signaling pathways the dysfunction of which may contribute to the disorder. In this review, we summarize recent evidence on the key role that the cerebellum plays in the development of ASD and then focus on genetic variations that cause ASD, focusing, to the extent possible, on the cerebellum. We have divided the ASD-associated gene in two subgroups – those that have been identified through a candidate gene approach with knowledge of their function in the cerebellum and their relationship to ASD subsequently confirmed in experimental models, and those identified through unbiased genetic analyses of individuals with ASD, many of which have not yet been characterized extensively and/or not studied in animal models.
Keywords:
autism
; cerebellum
; Purkinje cells
; genes
; signaling pathways
; synaptic dysfunction
ASD is a heterogeneous and behaviorally-defined group of conditions sharing three core phenotypic manifestations - impairment in social communication and interaction, repetitive patterns of behavior, and restricted interests, which typically are displayed early during childhood (Volpe, 2009, 2021; Lord et al., 2020). Although not widely appreciated, about 90% of individuals with ASD also have difficulties in sensory processing (Levy et al., 2009; Marco et al., 2011; Robertson and Baron-Cohen, 2017; Nimbley et al., 2022) and about ~30% display intellectual disability (Baio et al., 2018). About 15% of young children with ASD have macrocephaly, as a result of accelerated brain growth (Lainhart et al., 2006; Sacco et al., 2015). In contrast to macrocephaly in children, brain size is generally reduced in adults with ASD (Courchesne et al., 2011). Based on current estimates from the CDC’s Autism and Developmental Disabilities Monitoring (ADDM), about 1 in 36 children in the U.S. are diagnosed with ASD (Shaw et al., 2025). Worldwide, the prevalence of ASD has quadrupled over the past three decades. Although much of this increase has been attributed to increased awareness of ASD, increased screening, and better diagnostic tools, a substantial increase in the development of ASD cannot be ruled out.
Based on clinical criteria, ASD is classified as “syndromic” or “non-syndromic”. Whereas individuals with non-syndromic ASD, also referred to as “idiopathic ASD”, exhibit the core symptoms that define the disorder, syndromic ASD is clinically highly heterogenous and includes other phenotypic abnormalities, such as epilepsy, speech impairment, intellectual disability, and/or dysmorphic features (Sztainberg and Zoghbi, 2016; Weuring et al., 2021). In most cases, syndromic ASD is monogenetic, with the gene mutation causing another neurodevelopmental disorder the manifestations of which are accompanied by ASD symptoms. Such disorders include Fragile X syndrome, Down syndrome, Asperger’s syndrome, Rett syndrome, and Tuberous Sclerosis Complex (TSC) (Moss and Howlin, 2009). It is noteworthy that the affected genes within this category identified so far account have disparate biological functions and account for only 1-2% of ASD cases. In contrast, non-syndromic ASD is polygenic with many genes each making minor contributions, and that act in combination with prenatal or perinatal environmental factors (Sztainberg and Zoghbi, 2016; Weuring et al., 2021). While contribution of environmental factors in the pathogenesis of idiopathic ASD is well-accepted, input from genetic factors is much higher and consequently more attention has been placed on identifying the latter. It is noteworthy, however, that there is no known genetic cause for about 80% of individuals with ASD.
The vast majority of effort into understanding the neurobiological underpinnings of ASD has focused on the cerebral cortex and to a lesser extent, the amygdala and hippocampus. Critically, however, a large and growing body of evidence indicates that atypicality in the development and functioning of the cerebellum is more commonly associated with ASD than other brain regions (Fatemi et al., 2012; Becker and Stoodley, 2013; D’Mello and Stoodley, 2015a; D’Mello et al., 2016; Stoodley et al., 2017; Kelly et al., 2020). In this review we summarize findings that support a key role for the cerebellum in ASD pathogenesis. In particular, we describe many of the genes and signaling pathways the dysfunction of which have been implicated in ASD focusing more on those that are expressed and function in the cerebellum.
Cerebellar Structure and Function
The cerebellum is comprised of three lobes (anterior, posterior, and flocculonodular) which are further subdivided into ten bilateral lobules (Roostaei et al., 2014; D’Angelo, 2018). The outer portion, the cerebellar cortex, is composed of three layers – the internal granule layer (IGL) containing billions of granule neurons, the single-cell Purkinje cell (PC) layer, and a cell sparse molecular layer (ML) in which inhibitory basket and stellate interneurons are located and where synapses involving cell types and granule and PCs exist (Figure 1). The PCs are flanked by Bergman glial (BG) cells, a specialized type of glial cell present only in the cerebellum, which play a crucial role in the development of the cerebellum and the functioning of PCs through adulthood. Coming into the cerebellar cortex are mossy fibers from brainstem nuclei and the spinal cord, which synapse with provide excitatory input to granule neurons (Figure 1). PCs receive excitatory input from the granule neuron parallel fibers in the molecular layer and from the climbing fibers of the inferior olive. The parallel fibers of granule cell also stimulate basket and stellate interneurons within the molecular layer and Golgi cells, the cell bodies of which reside within the IGL. PCs represent the sole output from the cerebellum receiving inhibitory modulation primarily from stellate and basket cells in the molecular layer (see Figure 1). PCs, which are GABAergic, connect to the deep cerebellar nuclei – the main output nuclei of the cerebellum (Roostaei et al., 2014; D’Angelo, 2018). Unlike neurons within the layers of the cerebral cortex, cerebellar cytoarchitecture within the three layers and ten lobules has long been thought to be relatively uniform and simple. However, recent findings document considerable functional heterogeneity within the same neuronal types across the cerebellum, and with complex region- and cell population-specific transcriptomic and splicing programs underlying its development and functioning (Farini et al., 2020, 2021).
Historically considered a motor structure, it is now established that the cerebellum makes functional and anatomical connections to several association areas of the cerebral cortex including the prefrontal cortex and several whole-brain networks that are important for cognition (D’Mello and Stoodley, 2015a; Frosch et al., 2022). Indeed, like the cerebral cortex, the cerebellum is involved in a variety of cognitive and behavioral functions, including speech and language, emotions, learning and memory, mentalizing, decision making, and reward and social behavior (Dickson et al., 2017; Carta et al., 2019; Chabrol et al., 2019; Deverett et al., 2019; Schmahmann, 2019; Schmahmann et al., 2019; Chen et al., 2022b; Olson et al., 2023; Jobson et al., 2024). Additionally, and through converging connections to sensorimotor cortices, thalamus, and basal ganglia the cerebellum is involved in sensory-motor processing that fine-tunes motor actions (Glickstein and Doron, 2008; Strick et al., 2009; Sivalingam and Pandian, 2024).
A large body of evidence indicates that defective formation, maintenance, and plasticity of synapses plays a central role in ASD and other neurodevelopmental disorders (Bourgeron, 2015; Parenti et al., 2020). Synaptic dysfunction affects neuronal circuitry through a number of effects, chief among which is the disruption of the interplay between excitatory and inhibitory neurotransmission, widely referred to as excitatory/inhibitory (E/I) balance (Gao and Penzes, 2015; Sohal and Rubenstein, 2019). Excitatory (primarily glutaminergic) and inhibitory (primarily GABAergic) synapses are morphologically distinct, contain different protein components, and display different localization.
Cerebellum in Human Studies of Autism
Although several regions of the brain show atypicalities in studies of autism, the cerebellum is strikingly the most consistent site of abnormality. Postmortem studies of autism find reduced PCs in the posterior cerebellum, a region most associated with cognitive processing (Stoodley and Schmahmann, 2010; Van Overwalle et al., 2022; Ciricugno et al., 2024). Structural neuroimaging studies in humans also report differences in the cerebellum. Posterior cerebellar regions show reduced gray matter and individual differences in cerebellar grey matter volume are associated with increased symptom severity in core ASD diagnostic criteria (social interaction, language, and repetitive behaviors (Zhao et al., 2022; Christensen et al., 2024). Reduced cerebellar grey matter has also been associated with language delays in autistic children – one of the first signs of ASD which spurs parents to seek out diagnoses for their child. Finally, cerebellar differences in autistic children and adults have also been documented by functional neuroimaging studies. Posterior cerebellar regions (Crus I/II) implicated in cognition show atypical patterns of connectivity with the cerebral cortex, including regions critical for speech and language, the prefrontal cortex, and other association areas important for social interaction, executive function, and communication (Verly et al., 2014; Khan et al., 2015; Alho et al., 2023). Atypical functional connections between the cerebral cortex and sensorimotor cerebellar regions (in the anterior lobe and lobule VIII) have also been associated with sensory over-responsivity, a core diagnostic criterion in autism (Cakar et al., 2024).
Crucially, differences in the cerebellum may be an early indicator of long-term outcomes in ASD. For instance, altered cerebro-cerebellar connections in 9-month old infants at risk for autism were associated with greater probability of social communication difficulties years later (Okada et al., 2022). This suggests that dysfunction in cerebro-cerebellar circuits may be useful predictors of social communication disruptions in ASD children even before these behaviors are exhibited.
Cerebellar Development in ASD
In humans, the cerebellum starts developing from about 4 weeks of gestational age extending into second year of life, with the third trimester of gestation representing a critical period during which there are dynamic changes (ten Donkelaar et al., 2003; Leto et al., 2016).
Cerebellar lesions, damage resulting from tumor-resection, and genetically-caused neurodevelopmental disorders that disrupt cerebellar development, particularly during the critical period, are associated with ASD (Becker and Stoodley, 2013; D’Mello and Stoodley, 2015b; Stoodley, 2016). Reduction in cerebellar volume is a common feature both in individuals with ASD and in various mouse genetic models of the disorder (Courchesne, 1991; Ellegood and Crawley, 2015; McKinney et al., 2022). Specific cerebellar subregions have been identified that are associated with ASD, such as right Crus 1 (RCrus1), which is located in the right posterolateral portion of the cerebellar cortex, and involved in higher-order social and language processing (Stoodley et al., 2017). Compelling evidence that functional connections between the cerebellum and cortex are disrupted in ASD has been described (D’Mello and Stoodley, 2015b). The cerebellum makes direct connections with the ventral tegmental area (VTA), a brain area critical for the control of social behaviors and reward perception, which in turn makes dopaminergic connections with the prefrontal cortex (PFC) (Carta et al., 2019). Optogenetic inhibition of cerebellum-VTA projections in mice abolishes social preference demonstrating the importance of this circuit for normal social behavior. Stimulation of the dentate nucleus (DN) of the cerebellum results in the release of dopamine in the medial prefrontal cortex (mPFC)(Rogers et al., 2013) possibly via the VTA. Altered connectivity between the RCrus1 region of the cerebellum and the PFC has been described in children with ASD and in ASD mice (Stoodley et al., 2017). Together, these findings suggests that reduced or delayed information processing between the cerebellum and regions within the cortex, as well as other brain regions, may manifest as the cognitive, language, motor, and social interactions displayed in ASD. Studies using various genetic models have confirmed the association of cerebellar dysfunction in ASD-like behavior, including sensory defects (C et al., 2014; Kloth et al., 2015; Fernández et al., 2021; Hanzel et al., 2024). It should be noted that while cerebellar underdevelopment or lesions cause cerebellar dysfunction, there are other mechanisms by which dysfunction can occur without obvious changes in cerebellar morphology or volume. For example, exposure to a number of toxins early in life, including mercury, lead, valproic acid, and alcohol (Sanders et al., 2009; Minami et al., 2010; Kern et al., 2012; Kumar et al., 2013; Stoodley and Tsai, 2021; Guerra et al., 2023; Mitoma et al., 2024), as well as prenatal stressors and infections (Wang et al., 2014; Beversdorf et al., 2018; Zawadzka et al., 2021), affect the functioning of cerebellar cells and circuitry without noticeable macro-morphological alterations.
Of particular significance to ASD pathogenesis are cerebellar PCS, the elaborate dendritic branching of which is the most complex among CNS neurons (Hampson and Blatt, 2015). The dendrites of PCs are rich in spines which change in form, number, and function in response to stimuli (Tanaka, 2009; Nishiyama, 2014; Fernández Santoro et al., 2024). Post-mortem examinations of individuals with ASD consistently find a reduction in the number, size, and dendritic arborization of PCs regardless of age or sex (Bauman and Kemper, 1985; Courchesne, 1991; Bailey et al., 1998; Fatemi et al., 2002a; Whitney et al., 2009; Fatemi et al., 2012; Skefos et al., 2014; Hampson and Blatt, 2015). In addition to PC numbers, cerebellar weight and size is often reduced in the ASD brain (Courchesne, 1991; Wang et al., 2018b; Ma and Kwan, 2022). Chimeric mice generated using the Lurcher mutant (which displays total loss of PCs) and normal mice, display repetitive behavior and increased activity, consistent with a causal a role for decreased PC numbers in ASD-related symptoms (Martin et al., 2010). Similarly, genetically-induced dysfunction of PCs or their outputs to the deep cerebellar nuclei in mice induces ASD-like behavior, including social interaction deficits, and repetitive and stereotyped behavior (Xing et al., 2018; Yang et al., 2022; Cai et al., 2024). Vice versa, commonly utilized genetic mouse models of ASD that are not constructed by targeting PCs, display loss of PC and cerebellar dysfunction (Jaber, 2023). In addition to reduced PC numbers, disorganization of cerebellar nuclei and a reduction in white matter volume and integrity in the cerebellum has also been described in mouse models of ASD (Cheung et al., 2009; Hatten, 2020; Yeh et al., 2022). In contrast to these changes, neither the density of basket and stellate cells or the number synaptic connections to PCs by these cell types is reduced prior to the loss of PCs (Whitney et al., 2009).
Midgestational exposure of rodents to valproic acid (VPA) recapitulates several of the neurochemical and behavioral features of ASD and is a commonly used non-genetic model of the disorder (Nicolini and Fahnestock, 2018). Similar to its effect in rodents, exposure to VPA, a drug used to treat epilepsy, during pregnancy increases the risk of ASD (DiLiberti et al., 1984; Clayton-Smith and Donnai, 1995; Nicolai et al., 2008; Bromley et al., 2013) strengthening the physiological validity of the experimental model. In VPA-exposed mice, the reduction in PC numbers is unequal across the cerebellar cortex, with only some lobes displaying it (Wang et al., 2018b; Ma and Kwan, 2022). Chemogenetic inhibition of PCs in RCrus1 of normal mice generates deficits in social interaction, whereas optogenetic stimulation of this circuit has been described to reduce these symptoms in a mouse genetic model of ASD (Stoodley et al., 2017). This identifies RCrus1 as being one area in which loss of PC could have significant effect on ASD development. Like RCrus1, the cerebellar posterior vermis connects to the mPFC (Kelly et al., 2020). Stimulation of PCs in the posterior vermis, but not some other cerebellar regions, alleviates repetitive behaviors. This finding suggests that PCs in different parts of the cerebellum control different aspects of ASD (Kelly et al., 2020).
Multiple studies have described gene expression changes in PCs of humans with ASD (Fatemi et al., 2002b; Yip et al., 2007; Brandenburg et al., 2022). Among the genes that are downregulated is GAD67, which is the major GAD isoform in PCs and in the brain. Given that GAD is the enzyme that catalyzes conversion of glutamate to GABA, a significant reduction of GAD67 expression would lead to reduced GABA production in PCs (Gonzalo-Ruiz et al., 1990; Yamamoto et al., 1992; Middleton and Strick, 2001). Thus, along with reduced PC numbers, the reduced GAD expression would substantially decrease GABAergic neurotransmission to the deep cerebellar nuclei from the cerebellum to the thalamus and cortical regions with functional consequences. Reduced GAD expression has also been described in the mouse VPA model along with along with manifestation of ASD-like behaviors, including anxiety, sleep disturbances, and social interaction deficits (Cusmano and Mong, 2014; Olexová et al., 2016; Win-Shwe et al., 2018; Ma and Kwan, 2022).
An argument by Baizer against a role for the cerebellar dysfunction in ASD has also been presented (Baizer, 2024). Central to this argument is the observation that cerebellar damage does not consistently cause ASD in children. While this is the case, it is undisputable that cerebellar damage increases the risk of developing ASD and is, in fact, regarded as one of the highest risk factors for ASD (d’Oleire Uquillas et al., 2025). Given the highly diverse and heterogenous clinical manifestations and etiology of ASD, it is unlikely that cerebellar damage and dysfunction is the sole contributor to ASD diagnosis. Dysfunction of non-cerebellar brain regions may account for a subset of ASD cases with symptoms that incompletely overlap with cerebellar ASD. Additionally, non-cerebellar areas likely make greater contributions to the systemic symptoms often displayed in ASD, such as circadian rhythm disruption, immune system dysfunction, and gut microbiota alterations (Fattorusso et al., 2019; Hughes et al., 2023; Yurdakul et al., 2023). Finally, even though cerebellar damage greatly increases the risk for ASD, contribution of other genetic and environmental factors may be necessary.
It has also been pointed out that cerebellar damage in adults does not cause the same types of behavioral alterations that ASD children with early-life cerebellar damage exhibit (Baizer, 2024). This is not surprising, however, because early-life damage would have broad impact the development of the cerebellum, its cytoarchitecture, and circuitry within it as well as from it to other brain regions. Being a period of great neuroplasticity, some of the damage resulting from prenatal damage would be alleviated by compensatory mechanisms explaining the milder, but broader spectrum of symptoms exhibited by prenatal damage compared with the primarily motor deficits observed in adults.
Another argument made, specifically discounting the role of PCs is that ASD is not observed in all conditions in which there is reduced PC numbers (Baizer, 2024). It deserves mention, however, that reduction in PCs could be due to decreased production during cerebellar development or resulting from increased loss degeneration after these cells are produced in correct numbers. Existing information points to increased death of PCs in ASD, rather than a reduction in their production (Gerhant et al., 2013). In many mutant mouse lines, including those displaying ASD features, PC reduction results from early postnatal degeneration (Dusart et al., 2006; Wang and Morgan, 2007; Armstrong et al., 2011; Cendelin, 2014) which could impact refinement of neural connectivity in the cerebellum, which would impact functioning of other brain regions that are connected by the cerebellum. In adults, reduction of PC numbers could only result of a loss of pre-formed neurons. Recent findings that subregions within the cerebellar cortex and nuclei have distinct functions (King et al., 2019; Kebschull et al., 2020) indicating that the location of PC reduction within the cerebellum would also impact the consequences. Consistent with this, it is known that in different neurodevelopmental disorders, different cerebellar lobules are affected (Daskalakis et al., 2005; Becker and Stoodley, 2013; Hampson and Blatt, 2015; Wang et al., 2020). Additionally, GWAS analyses of different subregions of the cerebellar hemispheres and vermis have described heritable genetic variability across the different anatomical regions of the cerebellum (Chambers et al., 2022; Carrión-Castillo and Boeckx, 2024). Whether the loss of PCs is accompanied by loss of the associated granule neurons or interneurons could also influence outcome with regard to clinical outcome. Although convincing evidence supports involvement of the cerebellum in ASD, this does not exclude involvement of non-cerebellar areas also, particularly with relation to the systemic symptoms often displayed in ASD, such as circadian rhythm disruption, immune system dysfunction, and gut microbiota alterations (Fattorusso et al., 2019; Hughes et al., 2023; Yurdakul et al., 2023).
Strong correlation between circadian rhythm disorders and ASD (as well as other neurodevelopmental disorders) has been documented (Abdul et al., 2022; Yurdakul et al., 2023). In humans, and other mammals, the circadian timing system is composed of a central clock in the suprachiasmatic nuclei (SCN) of the hypothalamus and a number of secondary clocks (also referred to as oscillators) in the brain and peripheral organs. The peripheral clocks are synchronized by the central clock through neuronal activity and humoral signals (Dibner et al., 2010; Schibler et al., 2015). Circadian rhythms both in the central and peripheral clocks are generated by auto-regulatory feedback loops controlled by the CLOCK and BMAL1 proteins, which regulate the transcription of a large number of genes in different organs and cell types to generate oscillations (Dibner et al., 2010; Schibler et al., 2015). Dysfunction of BMAL1 and some of the clock-controlled genes affect synaptic function and are associated with ASD susceptibility (Geoffray et al., 2016). The cerebellum is one of the many regions outside the central clock that participates in the regulation of circadian rhythm function of the brain (Mendoza et al., 2010; Rath et al., 2012; Bering et al., 2021). The cerebellar oscillator has been proposed to reside in PCs (Mendoza et al., 2010; Mordel et al., 2013). Genetic or environmental factors that cause the dysfunction or degeneration of PCs could therefore disrupt the working of the central clock resulting in neurodevelopmental abnormalities, including ASD.
Although it is widely assumed that the underpinnings of ASD lie in neuronal dysfunction, a growing number of studies indicate contributions from abnormalities in astrocytes and microglia. Compelling evidence from both patients and mouse models indicates alterations in the numbers, morphology, and functioning of glial cell types in ASD (Petrelli et al., 2016, 2016; Scuderi and Verkhratsky, 2020; Gzielo and Nikiforuk, 2021; Cantando et al., 2024). By regulating synaptic pruning, microglia play an essential role in the establishment of functional of neuronal circuitry (Paolicelli et al., 2011; Schafer et al., 2012). Dysfunction in microglial function resulting in reduced or excessive synaptic pruning disrupts the E/I balance in neuronal circuits affecting brain function and behavior (Zhan et al., 2014; Koyama and Ikegaya, 2015). Astrocytes play critical roles in the regulation of a variety of neurodevelopmental processes, including neuronal migration, axon guidance, dendritic morphology, neurotransmitter uptake, and neuroinflammation (Linnerbauer et al., 2020; Andrade-Talavera et al., 2023; Valles et al., 2023). It is now well-documented that astrocytes regulate synaptic development, maturation, and function, and form tripartite synapses with neurons (Farhy-Tselnicker and Allen, 2018; Park and Chung, 2023). Some of the neurodevelopmental functions of astrocytes involves interaction with microglia (Petrelli et al., 2016; Sun et al., 2023). Studies of postmortem ASD patients have described increased expression of both astrocyte and microglial markers in the PFC (Edmonson et al., 2014). In the cerebellum however, only an increase in astrocyte markers is found in the ASD along with reduced expression of neuronal markers (Edmonson et al., 2014). Of particular significance to ASD-associated cerebellar dysfunction are BGs (Bellamy, 2006; Ben Haim and Rowitch, 2017; Chrobak and Soltys, 2017). BG play a crucial role in multiple aspects of cerebellar development including neuronal migration, maturation, synapse formation, and regulation of neuronal activity (Yamada et al., 2000; Lordkipanidze and Dunaevsky, 2005; Wang et al., 2012; H et al., 2013; Chrobak and Soltys, 2017). With regard to PCs, BGs regulate the growth and shaping of the dendrites of adjacent PCs (Lordkipanidze and Dunaevsky, 2005; Bellamy, 2006). In the mature cerebellum, BG processes cover PC synapses, and through their ability to regulate the membrane potential, control the activity of PCs (Wang et al., 2012; Chrobak and Soltys, 2017). Some evidence suggests that BG regulate the survival of PCs through regulation of glutamate homeostasis (Chrobak and Soltys, 2017). Thus, dysfunction of BG could cause the degeneration and loss of PCs in ASD. Neuroinflammation, oxidative stress and endoplasmic stress in the cerebellum (and other brain regions) are other features described in ASD (Vargas et al., 2005; Sajdel-Sulkowska et al., 2009, 2011; Fatemi et al., 2012; Dong et al., 2018; Matta et al., 2019) and that can result from dysfunction of BGs and other types of glial cells.
The Genetic Basis of ASD
ASD is highly heritable - the concordance rate for monozygotic (MZ) twins has been reported to be as high as 90%, and about 31% for dizygotic (DZ) twins (Rosenberg et al., 2009; Lichtenstein et al., 2010; Ho et al., 2022; Vorstman et al., 2022). Indeed, ASD is the most heritable of all psychiatric disorders. Consequently, much of the research on the molecular underpinnings of ASD has focused largely on genetic factors. While genetic contribution is indisputable, the less than absolute concordance in twin studies indicates that other pre-, peri-, and postnatal environmental factors protect or are necessary for full manifestation of ASD. While the nature of the environmental factors remains poorly understood, maternal lifestyle, pregnancy-related factors including viral infections, birth complications, and parental age have been implicated (Modabbernia et al., 2017; Bai et al., 2019, 2019; Cheroni et al., 2020; Masini et al., 2020; Andrade, 2024).
Consistent with the clinical heterogeneity, different types of inherited or spontaneous genetic mutations, including point-mutations, chromosomal rearrangements, and copy number variants, can cause or contribute to ASD (Chahrour et al., 2016; Masi et al., 2017; Havdahl et al., 2021; Vashisth and Chahrour, 2023). Another complication in identifying genes associated with ASD is that it can occur with other behavioral disorders as schizophrenia, bipolar disorder, ADHD, anxiety disorders, epilepsy, obsessive compulsive disorder, and learning or communication disorders. Results of genetic studies indicate that the genetic risk for ASD can have causal effects on one of aforementioned disorders and vice versa (Salenius et al., 2024).
Although the genetic variations that produce the core symptoms of ASD are far from clear, significant progress has been made over the past decade. Various transcriptomic analyses both at the level of brain regions and subregions and from single cells in these locations have been conducted (Harrison et al., 2015; Qian et al., 2018; Jin et al., 2020; Morabito et al., 2023; Wamsley et al., 2024). Much of this information that has come from the analyses of the cortex, and particularly the PFC. Encouragingly, studies using patients and mouse models have identified a number of common genes the altered expression of which is associated with ASD. But how the heterogenous etiologies and genetic variation in ASD converge on to these common and broadly shared transcriptional changes is currently unclear. In comparison with the cortex, considerably less attention has been placed on ASD-associated genetic variation in the cerebellum although based on existing information, a majority of the genes that have been found cause or increase risk of ASD genes in the cortex and other brain regions are co-expressed in the cerebellum (van der Heijden et al., 2021; Guerra et al., 2023). Despite the finding of location-dependent functional differences within the cerebellum, analyses conducted so far using UK Biobank GWAS summary data have not identified specific subregions of the cerebellum displaying significant genetic correlations with ASD at the whole genome-level (Chambers et al., 2022; Carrión-Castillo and Boeckx, 2024). The lack of genetic correlation may be explained by the canceling out of genetic changes occurring in opposing directions or limited sample size.
In this review, we describe genetic variations that cause or increase the risk of syndromic and/or non-syndromic ASD, focusing, to the extent possible, on the cerebellum. Emphasis has been placed on studies conducted more recently. For this review, we have divided the genes associated with ASD in to two subgroups – those that have been identified primarily through a candidate gene approach and their relationship to ASD then confirmed in experimental models, and those identified through unbiased genetic analyses of individuals with ASD, many of which have not yet been characterized extensively and/or not studied in animal models.
- I.
- Candidate Gene Approach
The candidate gene approach involves identification based on clinical studies, or experimental evidence derived from the analyses of pathophysiological animal models exhibiting ASD-like behavior. Based on the analyses of such candidate genes, primarily using rodent models, a diverse set of cellular abnormalities have been implicated in ASD. Most common among these are abnormal synaptic development, maintenance, functioning, and plasticity (Zoghbi, 2003; Garber, 2007; Nagappan-Chettiar et al., 2023). Not surprising therefore, most of the genes that cause or increase risk of ASD encode for synaptic proteins. We subdivide this section into genes that function primarily at the synapse, and those that have other functions including the development of the cerebellum. Although numerous genes have been implicated in both these categories, focus has been restricted on those that have been implicated in multiple studies conducted by different laboratories.
- Protein Regulating Synaptic Function and Neurotransmission
SHANK3: The SHANK3 (SH3 and multiple ankyrin repeat domains protein) gene is part of a family of three genes, SHANK 1 – 3, which encode postsynaptic multi-domain scaffolding proteins at glutamatergic synapses in the CNS (Monteiro and Feng, 2017). As scaffolding proteins, SHANK proteins interact with a large number of postsynaptic proteins and these associations are critical for synapse formation, function, and plasticity (Monteiro and Feng, 2017). In humans and rodents, all three SHANK proteins are expressed in several brain regions and cell types (Monteiro and Feng, 2017). Within the human and mouse cerebellum, SHANK2 displays highest expression in the cerebellum (Wan et al., 2021; Woelfle et al., 2023). While SHANK1 and 2 are expressed by PCs whereas SHANK3 is expressed in granule neurons (Wan et al., 2021; Woelfle et al., 2023). The patterns of intracellular localization also differ - SHANK3 mRNA, is localized in the soma of cerebellar cells, whereas SHANK1 and 2 mRNAs are in dendrites (Böckers et al., 2004).
Variations in all three of the SHANK genes are associated with ASD, which totally account for ~2% of ASD cases (Leblond et al., 2014). Based on mRNA expression and localization analysis, SHANK1 and SHANK2 mRNAs are expressed widely early in the brain, particularly after birth (Böckers et al., 2004). In contrast, SHANK3 is expressed selectively inn the cerebellum and thalamus where expression increases after birth and through adulthood (Böckers et al., 2004). Within the cerebellum, SHANK 1 and SHANK2 mRNA localize selectively to parallel fibers of granule cells. The localization of SHANK mRNAs to dendrites in the cerebellum suggests roles in synaptic activity-induced alterations through local translation.
Most research of the involvement of SHANKS in ASD has been on SHANK3 (Peça et al., 2011; Uchino and Waga, 2013; Mashayekhi et al., 2021). SHANK3 gene haploinsufficiency is the major cause of Phelan-McDermid syndrome (PMDS), a syndromic form of ASD (Phelan et al., 1993; Mitz et al., 2024). Other loss-of-function mutations of SHANK3, which include large deletions and insertions, point mutations, and splicing mutations, have been linked to non-syndromic ASDs (Durand et al., 2007; Moessner et al., 2007; Boccuto et al., 2013; Leblond et al., 2014). SHANK3-deficient mice and rats display disrupted E/I balance, and alterations in dendritic and spine morphology in multiple brain regions, including the cerebellum. Furthermore, the mutant animals display ASD-like behaviors including repetitive grooming and impaired social interaction (Peça et al., 2011; Durand et al., 2012; J et al., 2015; Jacot-Descombes et al., 2020). Loss of SHANK3 function also produces ASD-like behavior in other species, including zebrafish (Kareklas et al., 2023), dogs (Tian et al., 2023), and macaques (Zhou et al., 2019). Results of a recent study described deregulated glutaminergic receptors at granule neuron – mossy fiber synapses in SHANK3 mutant mice, which correlated with behavioral alterations (Kshetri et al., 2024).
Several studies have documented ASD-linked mutations and polymorphisms in the SHANK1 gene (Sato et al., 2012b; Gong and Wang, 2015; Qiu et al., 2019). ASD core symptoms are recapitulated in SHANK1 knockout mice (Sungur et al., 2014, 2016, 2018). Knock-in mice expressing the R882H mutation, a common SHANK1 mutation in humans with ASD, also display core symptoms of ASD indicating that the mutation impairs function. The development of symptoms is associated with structural changes in the frontal cortex, cerebellum, and hippocampus and a reduction of mGluR1-IP3R1-calcium signaling in these brain regions (Qin et al., 2022). Impaired mGluR1 signaling is also displayed in knock-in carrying another common ASD-associated mutation, SHANK1-P1812L (Qin et al., 2023). Interestingly, while mice hemizygous for the mutant allele display core ASD symptoms, homozygous mutant mice displayed long-tern memory impairment but not ASD behaviors (Qin et al., 2023).
Loss-of-function mutations in the SHANK2 gene are a particularly penetrant cause of ASD with cerebellum-regulated motor impairment (Leblond et al., 2014). PC-specific deletion of SHANK2 in mice results in their dysfunction and in an ASD-like phenotype (Peter et al., 2016). In mouse models of both idiopathic and syndromic ASD, GABA-A receptor density is reduced in the cerebellum which disrupts E/I balance (Nardi et al., 2023). Interestingly GABA-A receptor density is elevated in the ASD hippocampus in SHANK2-deficient mice suggesting brain-region-specific perturbations. Together, these and other studies indicate that dysfunction of both excitatory and inhibitory neurotransmission contribute to ASD.
Neuroligins (NLGNs): One of the many proteins that interact with SHANK proteins in the postsynaptic terminal are the neuroligins (NLGNs), a family of four transmembrane cell adhesion proteins, (NLGN1-4), that regulate synapse formation, organization, and function (Nguyen et al., 2020). Besides associating with postsynaptic proteins though their intracellular domains, the extracellular domain of NLGNs bind to presynaptic transmembrane neurexin proteins forming bridges across synapses bridges. Whereas the other NLGNs act at both excitatory and inhibitory synapses, NLGN2 acts exclusively at inhibitory synapses (Ali et al., 2020). Mutations in all for NLGN-encoding genes display a high penetrance linkage to ASD (Chen et al., 2014). The mutations are generally de novo and occurring in the germline (Chen et al., 2014). Among the NLGNs, genes for NLGN-3 and NLGN-4 are on the X-chromosome (Nguyen et al., 2020).
Each NLGN plays an essential role in synapse maintenance as RNAi-mediated knockdown of each of them results in extensive loss of both excitatory and inhibitory synapses (Chih et al., 2005a). Analyses of the localization of the four NLGN proteins has revealed that NLGN1 is specifically present in excitatory neurons, NLGN2 in inhibitory neurons, and NLGN4 in glycinergic neurons (Chih et al., 2005b; Varoqueaux et al., 2006; Craig and Kang, 2007; Bolliger et al., 2008). In contrast, NLGN3 is expressed in both excitatory and inhibitory neurons. While the intracellular region of the NLGNs associate with postsynaptic proteins, including SHANK proteins, the extracellular region interacts with neurexins (NXNs), which are transmembrane presynaptic proteins. The NLGN-NXN interaction is necessary for synapse formation and maintenance (Reissner et al., 2008; Südhof, 2008a; Koehnke et al., 2010). Within the postsynaptic cell, NLGNs also play a key role in localizing neurotransmitter receptors and channels (Ozaki, 2001). Different NLGN mRNAs can be co-expressed in the same neurons where they are localized to distinct types of synapses.
All four NLGNs are expressed in the cerebellum. Selective deletion of NLGNs in cerebellar PCs either singly or in combination reveals that NLGNs have both shared and distinct roles in regulating synaptic transmission (Zhang et al., 2015a; Zhang and Südhof, 2016; Qin et al., 2024). Ablation of all NLGNs in PCs results in the reduction of climbing fiber synapses (Zhang et al., 2015a). NLGNs also localize within cerebellar astrocytes with about 40% of the total NLGN expressed in BG cells, the predominant astrocytes in the cerebellum.
In the cerebellum, NLGN1 localizes mostly to synapses between parallel fibers, processes of interneurons in the ML, and synapses of mossy fibers and granule cell dendrites where is colocalizes with PSD-95, which marks excitatory postsynaptic sites. In contrast, localization of NLGN1 is low in PC dendrites and soma. NLGN2 localization in the cerebellum is restricted to inhibitory synapses and plays a critical role in regulating climbing fiber synapse numbers (Zhang et al., 2015b). NLGN3 is localized predominantly in the cell body of BG cells with much lower presence in the processes (Qin et al., 2024). Little is known about the localization pattern or function of NLGN4 is the cerebellum.
NLGN1 and ASD-NLGN2 knockout mice display ASD-like behaviors (Blundell et al., 2009, 2010). In the Fragile-X mouse model of ASD, the expression of NLGN1 is decreased in the brain (Dahlhaus and El-Husseini, 2010). Normalizing NLGN1 expression alleviates impairment of social behavior in mutant mice, but not the deficit in learning and memory suggesting that the reduced level of NLGN1 is a key contributor to at least some ASD-like behavioral impairment (Dahlhaus and El-Husseini, 2010).
In the context of ASD, most work has been done on NGLN-3. In the cerebellum, NLGN-3 is expressed both in PCs and granule neurons. Knock-in mice with an ASD-associated mutation in NLGN-3, R451C, a mutation first identified in individuals with ASD (Jamain et al., 2003; Yan et al., 2005), display increased cortical inhibitory synaptic strength along with impaired social interaction (Ellegood et al., 2011; Etherton et al., 2011; Ellegood and Crawley, 2015; Speed et al., 2015). Another study that compared the effect of the R451C mutation in different brain regions described increased excitatory synaptic transmission in the hippocampus but increased inhibitory transmission in the somatosensory cortex indicating that the same mutation induces region-specific changes in synaptic function (Etherton et al., 2011). In the cerebellum of NLGN-3-R451C mice, the expression of NLGN3 is reduced by ~90% suggesting that the mutation renders NLGN3 unstable (Lai et al., 2021). Furthermore, the normal postnatal pruning of supernumerary climbing factor synapses with PCS is impaired, leading to abnormal E/I balance in PCs (Lai et al., 2021). Abnormal synapse elimination is thought to contribute to ASD and other neurodevelopmental disorders (Watanabe and Kano, 2024). In contrast to mice with the R451C mutation, NLGN-3 global knockout mice don’t display an ASD phenotype suggesting possibly a gain-of-function effect of the R451C mutation (Tabuchi et al., 2007). NLGN-3 is also expressed in cerebellar BG cells although its absence in these cells in mice does not affect morphology, synaptic number, or synaptic function (Qin et al., 2024). However, BG-specific NLGN-3 deletion does alter gene expression in other cerebellar cell types, including PCs, granule neurons, and oligodendrocytes, as well as in the cortex (Qin et al., 2024). The contribution of BG-initiated changes in gene expression in the cerebellum and cortex to ASD associated with NLGN3 deficiency remains to be clarified. Somewhat puzzlingly, NLGN-3-R451C mutation in mice of another genetic background than the one used in the aforementioned studies, display minor behavioral issues but not ASD-like behaviors (Jaramillo et al., 2018) indicating that ASD-like behavioral outcome caused by the R451C mutation is dependent on genetic factors..
Aberrant cerebellar-cortical communication is likely to be an important contributor to ASD (D’Mello and Stoodley, 2015a; Stoodley, 2016; Stoodley et al., 2017). In addition to alterations within the cerebellum, inhibitory output from the deep cerebellar nuclei to the thalamus, midbrain and brainstem is reduced in ASD-NLGN-3 mice leading to impaired social interaction (Cai et al., 2024). Of these different pathways, chemogenetic inhibition of the pathway connecting to the zona incerta (ZI), a subthalamic region, rescues social impairment in ASD-NLGN3 mice (Cai et al., 2024). Specific deletion of NLGN3 in cerebellar astrocytes, which normally express it highly, results in modest transcriptional changes among multiple cell types, but not in synapse number, synaptic transmission, or morphology of the astrocytes (Qin et al., 2024). In contrast, deletion of NLGN2 in astrocytes, albeit of the cortex, affects morphology and synaptic transmission in addition to gene expression changes (Stogsdill et al., 2017). The consequences of NLGN2 in cerebellar astrocytes or BG cells has not been studied.
Mutations in the X-linked NLGN4 gene represent one of the most common monogenic causes associated with ASD with more than 50 mutations identified in humans (Veenstra-VanderWeele and Cook, 2004; Kumar and Christian, 2009; Lord et al., 2020). NLGN4 is widely expressed in the brain localizing primarily to inhibitory glycinergic synapses where it is required for the organization and functioning of these synapses (Jamain et al., 2008; Hoon et al., 2011; Zhang et al., 2018). Localization to excitatory synapses has also been described (Marro et al., 2019; Cast et al., 2021). Mutations of NLGN4 could thus contribute to ASD by affecting synaptic transmission and impairment of E/I balance. Mice lacking NLGN4 display impaired social interaction, repetitive behavior, and other ASD behaviors (Jamain et al., 2008; El-Kordi et al., 2013). Knock-in mice heterozygous for an ASD-associated NGNL4 mutation, P89L, also display abnormal social behavior suggesting that the mutation reduces function. The P89L mutation affects the cellular localization of NLGN4 and impairs dendritic spine formation (Nakanishi et al., 2017). Another ASD-associated genetic alteration in NLGN4 is missense mutation at an arginine conserved in all four NLGN members, R704C. In NLGN4 the R470C mutation elevates AMPA-receptor-mediated synaptic responses. However, when the same mutation is created in NLGN3, it enhances AMPA-receptor internalization resulting in reduced postsynaptic AMPA-receptor at the synapse (Chanda et al., 2016). Therefore, while both NLGN3 and NLGN4 affect the AMPA-receptor they have different effects on it and consequently on AMPA-receptor-mediated synaptic function. In contrast, another ASD-associated mutation, R87W, blocks the transport of NLGN4 to the cell surface thus blocking its actions on synaptic formation and function (Zhang et al., 2009). Similarly, an ASD-linked R101Q mutation impairs maturation of NLGN4 resulting in its retention in the ER and Golgi reducing its transport to the membrane (Cast et al., 2021).
Classical neurexins (NRXNs): NRXNs consist of a superfamily of presynaptic cell adhesion proteins that are subdivided into two groups - classical NRXNs encoded by three genes (NXRN1-3) and a large number of related proteins belonging to the contactin-associated protein (CASPR) subfamily. The NXRN 1 – 3 genes produce a multitude of transcripts through alternative splicing and promoter usage and that are expressed in a cell-specific manner (Missler et al., 1998; Reissner et al., 2013). Besides interacting with NLGNs, NRXNs associate with a variety of postsynaptic proteins (Craig and Kang, 2007; Südhof, 2008b; Reissner et al., 2013). Through these interactions, NRXNs regulate a number of synaptic properties, including assembly of the synapse, the organization of the presynaptic release machinery, and in the regulation of E/I balance (Khoja et al., 2023). Mutations in all three NRXNs are linked to ASD (Vaags et al., 2012; Khoja et al., 2023; Cooper et al., 2024). Interestingly, the three NRXNs display distinct patterns of expression in the developing human cortex (Harkin et al., 2017) suggesting that their dysfunction contributes to ASD through distinct mechanisms.
In the cerebellum, NRXNs connect to postsynaptic proteins through interaction with cerebellins (CLBNs), a family of four secreted adaptor proteins (Uemura et al., 2010). Association of NRXNs with CLBNs, particularly CLBN1, is instrumental for the development of cerebellar parallel-fiber synapses (Uemura et al., 2010).
Genetic variants in all three NRXN genes are associated with ASD (Gauthier et al., 2011; Reissner et al., 2013; Wang et al., 2018a). In the context of ASD, most attention has been placed on NRXN1 (Kasem et al., 2018; Gerik-Celebi et al., 2024; Shan et al., 2024). Mice lacking NRXN1 display electrophysiological abnormalities and exhibit ASD-like behavior (Etherton et al., 2009; Grayton et al., 2013; Armstrong et al., 2020) indicating a loss-of-function mechanism in ASD. Similarly, NRXN2 knockout mice display synaptic abnormalities and ASD-like behavior (Born et al., 2015; Khoja et al., 2023). Analysis of NXN3 knockout mouse lines reveal that NLGN3 has distinct roles in different brain regions and on excitatory versus inhibitory synapses (Aoto et al., 2015).
CNTNAP2: CNTNAP2 is the gene that encodes contactin-associated protein 2 (CASPR2), a transmembrane protein belonging to the CASPR subfamily of the NXRN super-family. CASPR2 is widely expressed in the CNS (Peñagarikano and Geschwind, 2012; Rodenas-Cuadrado et al., 2014). Besides functioning as a cell adhesion molecule, CASPR2 is involved in localizing K+ channels at Nodes of Ranvier, regulating myelination, and in cell-cell interactions (Poliak et al., 1999; Rodenas-Cuadrado et al., 2014). CNTNAP2 mutations (Arking et al., 2008; Bakkaloglu et al., 2008; Nascimento et al., 2016) and expression-reducing variants of the CNTNAP2 promoter region (Chiocchetti et al., 2015) are associated with ASD in humans. CNTNAP2-deficient mice also display synaptic dysfunction and ASD-like behaviors (Peñagarikano et al., 2011) supporting a loss-of-function mechanism.
CNTNAP2 is also widely expressed in the cerebellum (Gordon et al., 2016). Individuals with ASD-related mutations in the CNTNAP2 gene display reduced gray matter volume in the cerebellum (Tan et al., 2010). Similarly, mice lacking CNTNAP2 display reduced cerebellar volume (Crawley, 2007). CASPR2 is abundant at cerebellar synapses and CNTNAP2 knockout mice display disrupted dendrite development of PCs and impaired motor coordination (Argent et al., 2020). Another study described altered morphology of PCs in cerebellar Crus I/II region of mutant mice and impaired electrical responses to somatosensory stimulation, indicating abnormal sensory processing by the cerebellum (Fernández et al., 2021). CNTPNAP-deficient mice have been reported to develop cerebellar heterotopias (as do a subset of ASD patients) although one study has attributed this to the genetic background of C57BL/6 mice, which develop spontaneous cortical and cerebellar heterotopias (Otazu et al., 2021).
Much more is known about the role of CNTNAP2 in the cortex. During cortical development, CNTNAP2 is highly expressed in early-born excitatory cortical neurons. Loss of CNTNAP2 disrupts the excitatory neuron differentiation as well as overall neural circuit assembly in the developing cortex (Anderson et al., 2012; St George-Hyslop et al., 2023). Other functions of CNTNAP2 in the cortex include the regulation of neuronal migration, glutamate receptor organization, and morphology of dendritic arborization and synaptic spines (Anderson et al., 2012; Varea et al., 2015).
Cadherins are a large family of about 100 cell adhesion proteins, which in the nervous system are involved in the formation of neural circuitry, organization of synapses, and regulation of synaptic plasticity (Obst-Pernberg and Redies, 1999; Huntley et al., 2002; Basu et al., 2015; de Agustín-Durán et al., 2021). Besides mediating cell-cell contact, cadherins also regulate intracellular signaling (Redies et al., 2011, 2012; Basu et al., 2015). The large family of cadherin proteins is subdivided into multiple sub-families, including Type I cadherins, which are widely distributed, Type II cadherins, which that are expressed in specific brain regions and subcellular compartments, protocadherins, and atypical cadherins (Yagi and Takeichi, 2000). In the cerebellum, many cadherins are expressed at the earliest stage of development and where their function is required for proper cerebellar development (Redies et al., 2011). One function of the cadherins in the developing cerebellum is to regulate migration of PCs to parasaggital regions and ensure proper connectivity (Arndt et al., 1998; Luo et al., 2004; Redies et al., 2011). Unbiased genetic studies have identified mutations in several cadherins in neurodevelopmental disorders, including ASD (Betancur et al., 2009; Hussman et al., 2011; Lin et al., 2016). Indeed, copy number variations of multiple Type-II cadherins have been linked to ASD (Costa et al., 2022). A study focusing on two ASD-associated Type II cadherins, CDH9 and CDH11 within the cerebellum, described high expression in non-overlapping populations of PCs during early development but a decrease in as cerebellar development proceeded (Wang et al., 2019). In rodents, CDH11 expression is largely restricted to dorsal lobules of the vermis and the lateral hemisphere area equivalent to the Crus I and Crus II areas in the human cerebellum (Wang et al., 2019). The high expression CDH11 expression correlates with the expression of the calcium-binding protein, calbindin, and is associated with a delayed maturation of PCs (Wang et al., 2019). Together, these results suggest that the two ASD-linked cadherins function to regulate the development of the cerebellum and the circuitry within it. Although elevated expression affects PC development, deletion of a portion of the CDH11 locus has been described in idiopathic ASD (Crepel et al., 2014). Similarly, organoids generated from ASD patient-derived iPSCs display reduced CDH11 expression (Frei et al., 2021b). Another study using cultured CDH11-deficient hippocampal neurons described increased expression of postsynaptic density (PSD)-95 and NLFG1 that accompanied changes in dendritic morphology and electrical properties (Frei et al., 2021b). The somewhat counterintuitive elevation in PSD-95 and NLGN1 expression was suggested to represent a compensatory effect. In sum, both increase and decrease of CDH11 function could contribute to defective maturation and function of neurons that contribute to ASD. In addition of CDH9 and CDH11. Mutations in CDH13, a cadherin expressed specifically within Golgi cells of the cerebellum, has been linked to ASD (Chapman et al., 2011; Costa et al., 2022). Mice lacking CDH13 in Golgi cells exhibit reduced display deficits in cognitive performance and social behavior, but not in motor function (Tantra et al., 2018). Mice lacking CDH13 display deficits in adaptation to early-life stress (Kiser et al., 2019).
Besides Type-1 and Type-II cadherins, some members of the atypical cadherin subfamily have been linked to ASD. Among these is FAT1 (FAT atypical cadherin-1), a protein that is expressed highest in the postnatal cerebellum, where it is localized to granule neurons and Golgi cells in the IGL and inhibitory interneurons in the molecular layer (Neale et al., 2012; Cukier et al., 2014; Frei et al., 2021b). The expression of FAT1 is reduced in iPSC-derived neural cells from individuals with ASD (Frei et al., 2021a). Another, atypical cadherin Celsr3, which is necessary for proper brain development (Goffinet and Tissir, 2017; Boucherie et al., 2018), is expressed highly in PCs. Mice with elective deletion of Celsr3 in PCs display some ASD-like abnormalities, including reduced dendritic arborization and synaptic plasticity, and motor impairment (Zhou et al., 2021).
CUB and sushi multiple domains 3 (CSMD3): Mutations in CSDM3, a member of a family of three proteins (CSDM1-3), have been linked to ASD (Shimizu et al., 2003; Floris et al., 2008). CSMD3 is a large oligomeric transmembrane protein, expressed primarily in the fetal and adult brain (Shimizu et al., 2003). In the postnatal mouse hippocampus, CSDM3 localizes to apical dendrites (Mizukami et al., 2016). Elevating CSDM3 expression promotes dendritic branching in cultured hippocampal neurons, an activity that requires its extracellular domain suggesting that interaction with another protein is involved (Mizukami et al., 2016). In fact, CSDM3 has been proposed to act as a coreceptor with some other as yet unidentified protein (Mizukami et al., 2016). Mice lacking CSMD3 display core ASD symptoms and motor deficits which has been attributed to cerebellar dysfunction (Xi et al., 2023). Specifically, CSMD3-deficient mice display abnormal PC morphology in Right Crus I / Crus II lobules along with E/I imbalance in PC synapses within these cerebellar lobules. Besides its effects in the cerebellum, CSMD3 deficiency impairs neurogenesis and synaptogenesis in the cortex perturbing functional neural networks (Song et al., 2022).
- B.
- Proteins Involved in Cerebellar Development and Other Cellular Functions:
Engrailed-2 (En2-): En-2 is a homeobox transcription factor that is expressed at high levels in the cerebellum and hindbrain and is necessary for pattern formation and connectivity of the cerebellum during development. En-2 knockout mice display reduced cerebellar size, altered foliation of lobes, reduced number of PCs, and abnormal mossy fibers (Joyner et al., 1991; Mw et al., 1996; Kuemerle et al., 1997; Sudarov and Joyner, 2007; Cheng et al., 2010). The number of astrocytes and microglia are increased in the cerebellum of En-2 knockout mice (Lazzarini et al., 2024). More recent studies describe roles of En2 in regulating neuronal differentiation, diversity and survival of cerebellar neurons (Krishnamurthy et al., 2024). In mice expressing En-2 beyond the time that it is normally downregulated results in a delay in the onset of PC differentiation suggesting that En-2 controls the timing of PC differentiation (Jankowski et al., 2004).
Genetic studies conducted by multiple groups have identified En-2 as an ASD susceptibility gene (Gharani et al., 2004; Benayed et al., 2005; Yang et al., 2008; Sen et al., 2010). Susceptibility in humans is conferred by intronic SNPs that increase transcription of the En-2 gene (Benayed et al., 2009; Choi et al., 2014). Additionally, elevation of En-2 gene transcription in the ASD cerebellum results from increased activating histone H3K27 trimethylation (James et al., 2013) or by reduced repressive binding of MeCP2 (Methyl CpG-binding protein) to methylated regions in the 5’ region of the En-2 gene promoter (James et al., 2014). In view of the finding that En-2 negatively regulates PC differentiation (Jankowski et al., 2004), the increased En-2 expression in the ASD cerebellum could interfere with normal PC maturation and function, thereby resulting in cerebellar dysfunction. Surprising in view of the elevated En-2 expression humans with ASD, mice lacking En-2 also display ASD-like neurochemical alterations and behavioral phenotypes, including cognitive impairment and deficits in social interaction (Cheh et al., 2006; Brielmaier et al., 2012). Transcriptomic analysis of the cerebellum of mice lacking En-2 reveal increased expression of genes regulating immune function with reduced levels of pro-inflammatory molecules and chemokines further establishing relationship between immune dysfunction and cerebellar deficits in ASD and a role for EN-2 in this imbalance (Pangrazzi et al., 2022).
Tuberous sclerosis complex (TSC) proteins: Loss-of-function mutations of the TSC1 or TSC2 gene in humans causes a multiorgan disorder called TSC, which is characterized by non-malignant tumors in the brain (and several other organs) , white matter abnormalities, intellectual disability, and epilepsy (Boer et al., 2008; Orlova and Crino, 2010). A majority of TSC patients also display ASD behavior (McDonald et al., 2017; Dickinson et al., 2019; Samanta, 2020) with TSC2 mutations causing more severe symptoms than TSC1 mutations (Kashii et al., 2023). Mice in which TSC1 is specifically deleted in PCs exhibit core ASD features, including social interaction deficits, repetitive behavior and vocalizations, and cognitive deficits (Tsai et al., 2012). Similarly, mice lacking TSC2 display progressive loss of PCs, cerebellar dysfunction, and ASD behavior (Reith et al., 2013). Interestingly, TSC2-deficient mice that are seizure- and lesion-free, do not display cognitive impairment (Goorden et al., 2007) supporting the possibility suggested by some, that cognitive deficits in individuals with ASD might be caused or potentiated by seizure-induced damage (Capal and Jeste, 2024).
TSC1 and TSC2 negatively regulate mammalian target of rapamycin (mTOR), which forms two distinct types of protein complexes, mTOR complex-1 (mTORC1) and mTORC2 that differ in composition, intracellular localization, regulation, and substrates (Wullschleger et al., 2006; Szwed et al., 2021; Battaglioni et al., 2022). With regard to differences in composition, TORC1 contains the scaffolding protein Raptor, which is essential for its activity, whereas TORC2 contains Rictor, a protein required for its activity (Figure 2). Additionally, while mTORC1 is substantially more sensitive to rapamycin than mTORC2. A major function of TORC1 is enhancing of protein synthesis, with other functions including negatively-regulating autophagy, while the major function of TORC2 activates kinases that regulate cell survival and metabolism, including lipogenesis and glucose transport (Gaubitz et al., 2016; Xie et al., 2018).
In neurons, mTORC1 regulates dendritic morphology and synaptic density (Tavazoie et al., 2005; Bowling and Klann, 2014). Abnormal increase of mTORC1 activity (because of reduced TSC1/TSC2 inhibition) along with abnormal dendritic morphology has been described in patients with ASD with the severity of symptoms correlating with the level of increase (Bowling and Klann, 2014; Winden et al., 2018; Rosina et al., 2019). In mice, increased mTORC1 signaling leads to extra-numerary striatal-cortical excitatory synapses. The surplus synaptic connection in TSC2-deficient mice has been attributed to reduced autophagy and synaptic pruning resulting from overactivity of mTORC1 (Tang et al., 2014; Pagani et al., 2021). In a TSC2-deficient rat model of ASD, the cerebellar vermis shows enlarged white matter, a thickened molecular layer, and demyelination of the central tract of the vermis (Kútna et al., 2022). Furthermore, and as observed in mutant mice and ASD patients, ASD-like rats display reduced PC numbers and increased numbers of astrocytes and microglia. However, BG numbers and morphology is unchanged. Pharmacological inhibition of mTORC1 stimulates autophagy, reduces the synaptic hyperactivity, and ameliorates ASD behaviors in mice and restores normal neuronal connectivity and activity in iPSC-derived neurons from humans with TSC2 mutations (Sato et al., 2012a; Tang et al., 2014; Alsaqati et al., 2020; Pagani et al., 2021). Based on such results, mTORC1 has been considered as a target for the treatment of ASD and clinical trials are underway to test the efficacy to mTORC1 inhibitors (Sato, 2016; Winden et al., 2018).
Knocking out TORC1 activity specifically in PCs through deletion of the Raptor gene results in social interaction impairment and progressive loss of PCs (Angliker et al., 2015). In contrast, PC-specific deletion of Rictor, which eliminates mTORC2 activity, results in motor coordination and the gait alterations and disruption of climbing fiber synapses (Angliker et al., 2015). These results demonstrate that both mTORC1 or mTORC2 play important roles in cerebellar development and that deletion of either produces non-overlapping ASD-like phenotypes. Furthermore, these results show that either loss or gain of mTOR activity produces ASD-like pathologies pointing to the importance of careful regulation of mTOR activity for proper cerebellar development.
An important downstream target of mTORC1 is eIF4E, a key component of a multiprotein complex required for translation of capped mRNA (Thoreen et al., 2012; Zhang et al., 2021). Within the complex, binding of eIF4E to the mRNA cap leads to the recruitment of the ribosomal subunits for translation to initiate. The formation of the eIF4E complex depends on the phosphorylation of eIF4E-BP2, a binding protein that interacts with eIF4E inhibiting it. Phosphorylation of eIF4E-BP2 by mTORC1 causes its disassociation from eIF4E resulting in eIF4E-mediated complex formation and mRNA translation resulting in increased protein synthesis (Figure 2). Thus, mTORC1 promotes cap-depended protein synthesis by reducing the inhibitory action of eiF4E.
Several lines of evidence point to elevated eIF4E function as a key contributor of ASD. Deletion of the gene encoding 4E-BP2 or overexpression of eIF4E in mice increases protein synthesis in the brain and leads to ASD-like behaviors (Banko et al., 2007; Gkogkas et al., 2013a; Santini et al., 2013). 4E-BP2-lacking mice also display a disruption of the E/I balance in the hippocampus (Banko et al., 2005; Gkogkas et al., 2013b). Surprisingly, cell type-specific deletion of 4E-BP2 in GABAergic interneurons of mice produces ASD-like behaviors, whereas deletion in forebrain glutaminergic excitatory neurons or astrocytes do not (Wiebe et al., 2019). Mice in which 4E-BP2 is specifically deleted in PCs display a reduced number of PCs, aberration in neuronal activity, and impaired motor and spatial learning (Hooshmandi et al., 2021). However, these mice do not display social interaction or repetitive behavior suggesting that in the cerebellum, ASD-like impairment of social and repetitive behavior is controlled by a mechanism that is different from the control of motor and spatial memory (Hooshmandi et al., 2021).
Among the mRNAs that display increased translation in the hippocampus of 4EBP2 knockout mice is NLGN1-4 (Gkogkas et al., 2013a). Knockdown of NLGN1 but not NLGN2 restores E/I balance, restores normal protein levels, and reverses ASD-like behavior in the 4E-BP2 knockout mice (Gkogkas et al., 2013a). As observed in 4E-BP2 knockout mice, pharmacological inhibition of NLGN1 or its knockdown reverses ASD-like behaviors in Tsc2-deficient mice without a reduction in mTORC1 activity (Chalkiadaki et al., 2023), confirming that hyperactivated mTORC1-mediated ASD-like phenotype results from eIF4E-induced NLGN1 synthesis. Interestingly, recent evidence suggests that the ASD-associated deregulation of eIf4E function occurs in microglia (Xu et al., 2020). Overexpression of eIf4E in microglia, but not neurons or astrocytes, leads to ASD-like behaviors (Xu et al., 2020). Interestingly, although microglial eIF4E overexpression elevates translation in both sexes, dysfunction of microglia occurs only in male mice (Xu et al., 2020).
Phosphatase and tensin homolog (PTEN): PTEN is a tumor suppressor that is involved in the control of the cell cycle, apoptosis, and cell migration, acting as a lipid phosphates to by negatively regulate the PI-3 kinase-Akt and mTOR pathways (Worby and Dixon, 2014). PTEN is required for the normal development of the cerebellum, regulating cerebellar cytoarchitecture and migration of neurons and glial cells (Marino et al., 2002). Not unexpectedly, germline PTEN mutations in humans results in structural and functional abnormalities in the cerebellum (Gambini et al., 2024). Astroglial deletion of PTEN results in disorganization of cytoarchitecture predominantly in the cerebellum and hippocampus (Wen et al., 2013).
Initial findings of reduced PTEN levels in ASD patients with macrocephaly (Frazier et al., 2015) led to genetic analyses that revealed ASD-associated germ-line mutations in the PTEN gene (Butler et al., 2005; Buxbaum et al., 2007; Herman et al., 2007; Rademacher and Eickholt, 2019; Yehia et al., 2020). Germ-line PTEN mutations in humans with ASD are associated with extreme macrocephaly (Butler et al., 2005; Greer and Wynshaw-Boris, 2006). While only responsible for a relatively small proportion of ASD cases, most individuals with loss-of-function PTEN mutations and macrocephaly present with ASD (those that do are designated as ASD-PTEN). PTEN-deficiency in the brain results in hypertrophy of the soma and abnormal migration, increased dendritic elaboration, dentritic overgrowth, and higher spine density, which together result in neuronal hyperexcitability (Kwon et al., 2001, 2006; Luikart et al., 2011; Haws et al., 2014; Williams et al., 2015; Getz et al., 2016, 2022; Skelton et al., 2020). Mice in which PTEN is selectively deleted in PCs display structural and functional PC abnormalities and exhibit ASD-like behavior(Cupolillo et al., 2016). As described above, PTEN inhibits Akt activity, which inhibits TSC1/TSC2, which in inhibit mTORC1/mTORC2. Not unexpectedly therefore, mTORC1 activity is elevated in ASD-PTEN patients and in PTEN-deficient mice and chronic administration of rapamycin (which also inhibits mTORC2 upon prolonged exposure) ameliorates ASD-like behavior and brain abnormalities in PTEN-deficient mice (Kwon et al., 2006; Zhou et al., 2009; Haws et al., 2014; Getz et al., 2016).
It is noteworthy that although much of the effects of PTEN are mediated through its actions at the plasma membrane, where is inhibits PI-3 kinase-Akt and mTOR signaling, PTEN also localizes to the nucleus. Selective reduction of nuclear PTEN in mice results in macrocephaly at birth, reduced neuronal soma size in the cortex, cerebellum, and hippocampus, and enhanced seizure susceptibility (Igarashi et al., 2018). The contribution of nuclear PTEN to ASD remains uninvestigated.
Brain and Muscle ARNT-Like 1 (Bmal1): ASD is often associated with circadian clock perturbations, which result in sleep disturbances (Johnson and Zarrinnegar, 2024; Sidhu et al., 2024). Based on a number of studies indicate that 50 – 80% of developing children with ASD have sleep disturbances (Geoffray et al., 2016; Pinato et al., 2019; Sidhu et al., 2024). In the VPA-induced ASD mouse model, circadian behavior and expression of circadian clock-regulatory genes is altered (Ferraro et al., 2021). A key protein in the regulation of the circadian clock is BMALl, transcription factor which heteromerizes with the CLOCK transcription factor to activate transcription of the PER and CRY genes. Once produced PER and CRY form a complex that inhibits the transcriptional actions of BMAL1-CLOCK resulting in oscillatory transcriptional- feedback loops (Takahashi et al., 2008). Both BMAL1 null- and -haplosufficient mice develop core behavioral deficits of ASD including social impairments, repetitive behaviors, and learning disabilities (Singla et al., 2022; Liu et al., 2023). BMAL1-deficient mice also display aberrant cell density and immature morphology of dendritic spines. Moreover, the mutant mice display abnormal cell density in the cerebellum along with immature morphology, and electrophysiological properties of PCs (Liu et al., 2023). Deletion of Bmal1 only in PCs recapitulates the ASD-like phenotype, displayed by BMAL1-null mice (Liu et al., 2023) underscoring the importance of PC dysfunction in ASD.
Besides being a critical component of circadian rhythm regulatory machinery, BMAL1 has other functions including the regulation of mRNA translation (Guerrero-Morín and Santillán, 2020), a process that is under the control of TORC1 activity (as described above). Interestingly, mTORC1 and eIF4E activity, deregulation of which is associated with ASD, is increased in the BMAL1-knockout cerebellum (Liu et al., 2023). Additionally, the transcriptional profile in the cerebellum is altered with changes in the expression of several ASD-associated genes. Pharmacological inhibition of mTORC1 signaling reverses the ASD-like behavior in BMAL1-deficient mice, indicating that the impairments resulting from BMAL1 deletion are mediated by hyperactivation of mTORC1 (Liu et al., 2023).
While BMAL1 deficiency leads to an hyperactivation of mTORC1, mTORC1 prevents the degradation of BMAL1 through its phosphorylation by S6 kinase, an effector target of mTORC1. Consequently, in BMAL1 protein expression is increased in Tsc2-deficient cells and mice, an experimental model of ASD (Lipton et al., 2017). Treatment with an mTOCR1 inhibitor reverses the increased BMAL1 expression whereas reducing the level of BMAL in mice normalizes the circadian phenotype in Tsc2-deficient mice. These findings indicate that the ASD-promoting effect of BMAL1 deficiency results from mTORC1 hyperactivation (Lipton et al., 2017).
It is noteworthy that besides regulating the circadian clock and protein translation, BMAL1 regulates mitochondrial fission and mitophagy (Li et al., 2020), autophagy (McKee et al., 2023), neuroinflammation (Liu et al., 2020), synaptic pruning (Iweka et al., 2023), and myelination (Rojo et al., 2023), all processes that are dysregulated in ASD. Therefore, BMAL1-deficiency can contribute to ASD by TORC1 dependent and independent mechanisms.
Retinoic acid-related Orphan Receptor alpha (RORa): (Sayad et al., 2017; Ribeiro and Sherrard, 2023). RORa is best known as a nuclear receptor that is widely-expressed in the brain and that is activated through binding by retinoic acid (RA), a powerful signaling molecule produced through the oxidation of retinal (vitamin A) (Sayad et al., 2017; Ribeiro and Sherrard, 2023). RA plays a variety of critical roles during CNS development, including the regulation of stem cell production, neuronal differentiation, and morphogenesis of brain structures (Maden and Holder, 1991, 1992). RA also regulates synaptic plasticity, E/I balance, and other important processes in the adult brain (Moramarco and McCaffery, 2024).
Nuclear RA-bound RORa exerts its biological effects by binding to genomic DNA at ROR response elements (RORE) located within the promoters of a large number of genes (Maden, 2007). More importantly, RORα binds to the promoters of over 400 genes that have been implicated in ASD (Hu et al., 2009, 2015; Xu et al., 2012; Ribeiro and Sherrard, 2023). In humans, mutations in the RORa gene can cause either ASD or cerebellar ataxia depending on whether the mutation produces loss-of-function or a toxic gain-of-function, respectively (Guissart et al., 2018).
RORα plays a key role at different stages of cerebellar development and through adulthood (Ribeiro and Sherrard, 2023). In the cerebellum, RORα is most highly expressed in PCs and is required for the development and survival of these neurons (Boukhtouche et al., 2006b, 2006a; Chen et al., 2013; Takeo et al., 2015). Mice lacking RORα display defective interactions between PCs and climbing fibers, atrophy of the soma and dendrites of PCs followed by their death resulting in cerebellar dysfunction (Doulazmi et al., 2001; Jarvis et al., 2002; Chen et al., 2013). In the prenatal VPA-exposure rat model of autism, reduced PC numbers and arborization is accompanied by reduced level of RA and decreased expression of RORα. Administration of RA to the VPA-exposed rats normalizes expression of RORα, PC number and dendritic arborization, and improves motor function (Yuan et al., 2023). Although exactly how reduced function of RORα contributes to ASD is not known, given the large number of target genes with varied functions, it is possible that deregulation cab contributes to several of the diverse symptoms in ASD. One target gene that could be particularly important is RA-induced 1 (RAI1), a protein that is expressed at high levels in the cerebellum and in PCs, and that is involved in cognitive and motor function (Bi et al., 2007; Fragoso et al., 2015). RA administration stimulates expression of RAI1, while decreased function of RAI1 has been described to contribute to multiple neurodevelopmental disorders (Cao et al., 2014; Fragoso et al., 2015; Chang et al., 2022).
Besides neurons, RORα is expressed in astrocytes where is acts to suppress inflammation and promote neuronal survival (Journiac et al., 2009). It also expressed in microglia, a cell type that changes morphology during sleep (Nakanishi et al., 2021). Reduced RORα in microglia function leads to reduced BMAL1 expression and disrupts the microglial clock system, which contributes to sleep disruption but also deficits in social interaction and cognitive impairment (Nakanishi et al., 2021). Microglial RORα also suppresses neuroinflammation (Nakanishi et al., 2021), an alteration linked to ASD.
Neuregulins: NRGs are a family of four major signaling proteins (NRG1-4) characterized by an EGF-like domain through which they interact with the Erb4 receptor tyrosine kinase (Lemke, 1996; Esper et al., 2006; Ou et al., 2021). NRGs play key roles during neurodevelopment and are involved in the functioning of the brain in adulthood (Esper et al., 2006; Ledonne and Mercuri, 2019). The expression patterns of NRG1-3 show differences during development and within the brain. For example, in the postnatal rodent brain, NRG1 is expressed highest at P0 whereas NGN2 expression increases through development (Longart et al., 2004). The best studied of the NRGs is under complex control with multiple transcription initiation sites and splice variants as well as posttranslational modifications (Brown et al., 2004; Steinthorsdottir et al., 2004; Talmage and Role, 2004). NRG1 is expressed in several brain regions and cell types, including cerebellar PCs (Ding et al., 2023), whereas NRG3, the most highly expressed NGN in the brain, is expressed mostly in the cortex, hippocampus, and cerebellum (Rahman et al., 2019). Within the cerebellum, NGN3 is expressed in PCs and granule neurons (Rahman et al., 2019). NRG2 is much more restricted than NRG1 and NRG3, with expression restricted to the cerebellum, hippocampus and olfactory bulb (Busfield et al., 1997; Longart et al., 2004). NRGs have been associated with schizophrenia (Rico and Marín, 2011), but emerging evidence suggest that these NRGs may contribute to ASD development also. Indeed, ASD-associated genetic variations have been reported for both NRG1 and NRG2 in humans (Yoo et al., 2015; Chien et al., 2024). NRG1 levels are significantly higher in serum from ASD patients and associate with behavioral impairment (Abbasy et al., 2018; Esnafoglu, 2018). In mice exposed to VPA prenatally, NRG1 level is elevated and NRG1/Erb4 signaling increased. This is the result of increased Notch1/Hes signaling (Deng et al., 2024). Pharmacological inhibition of Notch1 signaling reduces NRG1 expression and signaling, and ameliorates ASD-like behavior (Deng et al., 2024). It is noteworthy that besides neurons, NRGs are expressed in astrocytes, and microglia in the brain (Ikawa et al., 2017; Deng et al., 2024). Indeed, the increased expression of NRG1 expression in the mouse model of VPA-induced as well as genetic models of ASD occurs mostly in microglia associated with the neuroinflammatory function of microglia (Ikawa et al., 2017; Deng et al., 2024).
Although NRG2 has no active kinase domain, it interacts with Erb4 and through this interaction regulates internalizing of NMDA receptor reducing its activity in interneurons (Vullhorst et al., 2015). However in a negative feedback loop mechanism, NMDA receptor activation promotes shedding of NRG2 from dendritic membranes (Vullhorst et al., 2015, 2023; Vullhorst and Buonanno, 2019). Expression of NRG2 is reduced in ASD individuals (Chien et al., 2024).
UBE3A: UBE3A is a protein which functions both as an E3 ubiquitin ligase and a transcriptional regulator for steroid hormone receptors that functions to regulates synaptic function and plasticity in the brain. It is expressed widely in the brain, with high expression in the cerebellum where it is expressed in PCs and Golgi cells (Albrecht et al., 1997; Gustin et al., 2010; Bruinsma et al., 2015). In healthy individuals, the region encompassing the UBE3A gene is silenced by imprinting in the paternal copy of the chromosome resulting in only the maternal copy of the gene being expressed. Mutations or imprinting errors that disrupt expression of the maternal allele results in reduction or complete loss of UBE3A gene expression, causing Angelman syndrome (AS), a disorder characterized by ASD features (Vatsa and Jana, 2018; Roy et al., 2023). In AS mice, maternal allele imprinting caused marked reduction of UBE3A in the cerebellum and hippocampus, whereas loss in other brain regions was low to moderate (Albrecht et al., 1997; Fang et al., 1999). Within the cerebellum of AS mice, a substantial loss of UBE3A occurs in a subset of GABAergic Golgi interneurons in the granule cell layer (Dindot et al., 2008; Bruinsma et al., 2015). Duplication, triplication, or gain-of-function mutations in the UBE3A gene are also associated with ASD behavioral features indicating that the expression and activity of UBE3A must be tightly regulated during brain development. The proteins degraded through the activity E3 ligase activity of UBE3A remain largely unidentified.
Elevated UBE3A affects dendritic maturation and spine morphology, abnormalities that have been attributed to its action in astrocytes (Dindot et al., 2008; Gardner et al., 2024). AS mice also display disrupted electrical communication between cerebellar PCs and the cortex resulting in abnormal E-I balance (Cheron et al., 2014). UBE3A-haplosufficient mice display only mild impairment of cerebellar function with deficits in learning deficits but not in locomotor function (Bruinsma et al., 2015). Consistent with loss of expression in GABAergic interneurons, AS mice display a reduction in the tonic inhibition of granule neurons (Bruinsma et al., 2015). In cerebellar PCs, UBE3A-deficiency results in enhanced autophagy through the activation of AMPK and inhibition of mTORC1 (Hao et al., 2023). Deregulated E-I balance and autophagy have both been implicated in ASD pathogenesis (Kim et al., 2017; Angrand et al., 2022).
Oxytocin (OXT): Best known for its role in uterine contractions during labor and in lactation, OXT, a nine-amino-acid neuropeptide regulates social behaviors such as including social attachment, social cognition, anxiety, aggression, and immune functioning (Jones et al., 2017; Carter et al., 2020; Froemke and Young, 2021). These actions of OXT are mediated through binding to its G-protein coupled receptor. A meta-analysis examining the linkage of OXT with ASD identified multiple SNPs (LoParo and Waldman, 2015). Other studies have identified genetic variations in the OXT receptor that are associated with ASD (Harrison et al., 2015; LoParo and Waldman, 2015; Watanabe et al., 2017; Al-Ali et al., 2022). In mice, mutation of the genes expressing OXT or its receptor results in altered circuit connectivity and deficits in social interaction and memory (Ferguson et al., 2000; Winslow and Insel, 2002; Choe et al., 2022). OXT expression is altered by alterations in the functioning of other ASD-linked proteins. For example, OXT level is reduced in Cntnap2, SHANK3, and NLGN3-deficient mice (Hörnberg et al., 2020; Reichova et al., 2020; Choe et al., 2022). Administration of oxytocin, activation of endogenous OXT release via chemogenetic or by pharmacological strategies alleviates social behavior deficits in both mutant Cntnap2 and SHANK3 mice (Peñagarikano et al., 2015; Choe et al., 2022; Szabó et al., 2024). On the other hand, administration of pharmacological activators of NLGN3 signaling in NLGN3-knockout mice restores OXT levels while also alleviating ASD-like behavior (Hörnberg et al., 2020). Assuming that the four proteins are part of a common signaling pathway, this puts OXT upstream of Cntnap2 and SHANK3 and downstream of NLGN3.
Although the cerebellum expresses the OXT receptor and it is known that OXT modulates circuitry between the cerebellum and the striatum (Zhao et al., 2019), the functions of OXT in the cerebellum is not clear. Within the cerebellum, the OXT receptor is expressed in PCs of lobule Crus I, dysfunction of which is implicated in ASD. However, blockage of OXT receptor in PCs of lobule Crus I in wild-type mice does not induce autistic-like social, stereotypic, cognitive, and anxiety-like behaviors. Similarly, activation of the OXTCN in this brain area does not alter the electrophysiological properties of Crus I PCs nor alleviate symptoms in ASD mice (Shen et al., 2023). Although this may suggest that OXT action in PCs of Crus1 are not involved in ASD, it is possible that it contributes to but is not sufficient for ASD development. A recent study described that cerebellar damage, which has been associated with ASD, upregulates OXT receptor expression in BG cells much more so than occurs in PCs (Inutsuka et al., 2024).
Chromodomain Helicase DNA-binding protein-2 and -8 ( CHD2 and CHD8): CHD proteins are a family of chromatin remodeling proteins that have been linked to neurodevelopmental disorders (Weissberg and Elliott, 2021; Muhammad et al., 2023). Among the CHD proteins, CHD2 and CHD8 are linked to ASD (Kasah et al., 2018). Although expressed in the cerebellum, both CHD2 and CHD8 are expressed more highly in the hippocampus and cortex. While function of these proteins is unclear, existing information suggests that they promote proliferation of neural progenitor cells. Deletion of CHD8 specifically in proliferating cerebellar granule progenitor cells resulting in reduced proliferation and differentiation of these cells and ectopic localization of PCs with the mice displaying cerebellar hypoplasia, ataxia, and psychiatric symptoms (Kawamura et al., 2021; Chen et al., 2022a). Within granule neuron precursor cells, CHD8 regulates the expressed of many genes, including genes linked to ASD (Kawamura et al., 2021). Although CHD8 mutant mice display motor coordination defects, other cognitive or behavioral ASD-associated defects are not observed (Kawamura et al., 2021).
Astrotactin 2 (ASTN2): ASTN2 is a transmembrane glycoprotein that displays highest expression in the cerebellum, with low levels in the cortex and hippocampus (Wilson et al., 2010; Behesti et al., 2018). Within the cerebellum, ASTN2 is expressed in PCs and granule neurons regulating synaptic activity and trafficking of receptors and other synaptic proteins (Wilson et al., 2010; Behesti et al., 2018). Recent research has identified ASTN2 mutations, including CNVs, that cause or increase risk for ASD and other neurodevelopmental disorders (Lionel et al., 2014; Hanzel et al., 2024). ASTN2 knock have altered cerebellar circuitry abnormal dendritic morphology in Crus1 and posterior vermis, disrupted E/I balance and display ASD-like behaviors (Hanzel et al., 2024) indicating that the effect of ASD-linked mutations of ASTN2 is due to loss-of-function. Mice with PC-specific deletion of ASTN2 also display ASD-like behavior underscoring the importance of cerebellar dysfunction in ASD (Hanzel et al., 2024).
- C.
- Whole Genome Analyses
Identification of disease modifying genes by GWAS and whole-exome sequencing are not reliant on an a priori hypothesis for the development of the disorder. While these unbiases analyses have been productive for many genetically-influenced disorders, a fundamental challenge in understanding the genetic mechanisms underlying ASD is the heterogeneity of the disorder. An estimated 1,100 genes are implicated in ASD (De Rubeis et al., 2014), of which close to 200 have been identified with high-confidence carrying rare de novo and transmitted genetic variants. Further complicating the understanding of the genetic basis of ASD is that over 90% of the high-confidence genes are expressed during early development and most of these are associated with other neurodevelopmental disorders (Deciphering Developmental Disorders Study, 2017; Courchesne et al., 2019; Kaplanis et al., 2020; Nóbrega et al., 2024). In children, ASD often presents alongside comorbidities including attention-deficit-hyperactivity disorder (ADHD), intellectual deficiency (ID), and epilepsy (McPartland and Volkmar, 2012; Genovese and Butler, 2023). In adults, symptoms of ASD are frequently observed in individuals with schizophrenia, bipolar disorder, and other disorders with neurodevelopmental underpinnings (McPartland and Volkmar, 2012; Genovese and Butler, 2023). In this review, we focus on peer-revied publications limited to the past 5 years. While finding the significance of the genes identified in these large-scare studies to cerebellar function has been attempted, information is limited for a majority of the genes described below.
A study of patients with either ASD or ADHD and that used Genomic Structural Equation Modeling (SEM) to GWAS summary statistics combined with transcriptome-wide analysis, identified 83 unique genes with expression changes associated with ASD but not other closely-linked neurodevelopmental disorders, many of which were novel (Schaffer et al., 2024). Interestingly, many of these ASD-specific genes overlapped with gene sets implicated in bacterial skin disease and erythema. This is consistent with the previously described association between ASD and immune system dysfunction, particularly involving the skin (Zerbo et al., 2015; Muratori et al., 2017; Jameson et al., 2022; Man et al., 2023). One limitation of the study is that study only included individuals of European ancestry.
A recent study in which the genetic variation of 91 circulating inflammatory factors was analyzed from genome wide association studies (GWAS) database of over 18,000 individuals of European ancestry with ASD suggested positive association with ASD of multiple immune system-associated genes, including those of CD244, CD5, FLT3LG, sulfotransferase (SULT) 1A1, and TNFSF10, while levels of IL-7, IL2Rβ, and IL-2 were negatively associated (Long et al., 2024). SULT1A is expressed in both neurons and glia in specific regions of the brain with high expression in the cerebellum (Salman et al., 2009).
A meta-analyses of a GWAS database of 18,000 individuals with ASD (but without other neurodevelopmental disorders), reported the identification of five loci and many common genetic variants that contributing to ASD alone. These genes included CADPS, KCNN2, KMT2E, MACROD2, NEGR1, and PTBP2 (Grove et al., 2019). Of these SNPs in the PTBP2, CADPS, KCCN2 and KMT2E gene have previously been linked to ASD by GWAS, and as described below, in the case of CADPS, by clinical and experimental research. Interestingly, CADPS and NEGR1 were identified in another recent genetic analysis aimed at identifying genes that are co-linked with ASD and another neurodevelopmental disorder(s) (Salenius et al., 2024). In this study both CAPDS and NEGR1 were also associated with epilepsy. Furthermore, NEGR1, which encodes a neuronal growth regulator, displayed statistically significant genetic association with ASD only in females.
Several studies have described aberrant splicing and genetic variations of the CADPS2 (calcium-dependent activator protein for secretion 2) gene (which is closely related to CADPS) causes ASD (Sadakata et al., 2013b, 2013a; Bonora et al., 2014). Additionally, duplication, deletion, and translocation within the CADPS2 gene locus have been linked to ASD (Okamoto et al., 2011; Aristidou et al., 2017; Pavone et al., 2019). Mice lacking CADPS2 also display ASD-like behavior (Sadakata et al., 2013a). CADPS2 is most highly expressed in granule neurons of the developing cerebellum where it stimulates release of BDNF and NT3 from parallel fiber terminals. CADPS2 knockout mice exhibit ASD-like symptoms along with impaired release of BDNF and NT3 leading to , delayed cerebellar development along with death of granule neurons and stunted PC branching (Sadakata and Furuichi, 2009). CADPS2 is also critically involved in the normal release of oxytocin, a social interaction modulatory protein, from the pituitary (Fujima et al., 2021). Although CADPS2 levels are high in the hypothalamus and pituitary, its release from the pituitary of CADPS2 knockout mice (Fujima et al., 2021).
KCNN2 (potassium voltage-gated channel subfamily C member 2) is the gene that encodes SK2, a member of the small-conductance Ca2+ -activated potassium channel family widely expressed in the brain and localizing to synaptic terminals (Rahman et al., 2023). By affecting neuronal excitability and signaling, SK2 regulates memory and learning, and in synaptic plasticity (Sun et al., 2015). The level of SK2 levels at synaptic terminals is regulated by UBE3A, which inhibits the recycling of endocytosed SK2 back to the synaptic membrane (Sun et al., 2015, 2020b). As described above, increased expression of UBE3A increases risk of ASD whereas deficiency causes Angelman syndrome (Sun et al., 2020a, 2022). SK2 regulates PC development, and following cerebellar development, SK regulates activity-dependent dendritic plasticity of PCs (Ohtsuki et al., 2012a, 2012b; Grasselli et al., 2016, 2020). Humans with KCNN2 gene mutations display ataxia, impairment in language development, and intellectual disabilities (Rahman et al., 2023).
KMT2E (Lysine methyltransferase 2E) is a N-methyltransferase that negatively regulates gene transcription by methylation of histone 3 (Zhang et al., 2017). De novo haplosufficient mutations and variants in the KMT2E gene are linked to a variety of neurodevelopmental disorders, including ASD, often accompanied by macrocephaly (Pais et al., 1993; O’Donnell-Luria et al., 2019; Li et al., 2023). Non-functional variants resulting from truncating mutations or defective splicing in KMT2E produces cerebellar hypoplasia and ASD symptoms (O’Donnell-Luria et al., 2019; Li et al., 2021; Abreu et al., 2022). Prenatal alcohol exposure in mice increases expression of KMT2E in the developing postnatal cerebellum which disrupts proliferation and neuronal differentiation (Schaffner et al., 2020).
PTBP2 (Polypyrimidine tract-binding protein 2) is a neuronal splicing factor that plays a crucial role in brain development by regulating neurogenesis through the control of differentiation and maturation of neurons (Licatalosi et al., 2012; Zhu et al., 2023). Alterations in splicing has been reported in the ASD brain, although the extent to which PTBP2 dysfunction contributes to it. While studies linking PTBP2 dysfunction in the cerebellum to ASD are limited, the protein's critical role in neuronal RNA splicing suggests that deregulation of its function could contribute to abnormal neurogenesis in the cerebellum.
Other genes and loci recently linked to ASD by analysis of summary statistics of previously reported GWAS data are XRN2, NKX2-4, PLK1S1, CRHR1-IT1, C8orf74 and LOC644172 (Alonso-Gonzalez et al., 2019). Multiple genetic studies have identified the MACROD2 gene as being linked to ASD (Jones et al., 2014; Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium, 2017; Bacchelli et al., 2020, p. 1616; Alnak et al., 2021). MACROD2 is best known for its role in suppressing colorectal and other cancers (Jin and Burkard, 2018). While MACROD2 expression is not altered in the ASD brain, the expression of RPS10P2-AS1, a long-coding RNA that is transcribed from a region close to the MACROD2 gene on Chromosome 5, is increased 7-fold in postmortem cortex of ASD patients and regulates development of human neural progenitor cells (DeWitt et al., 2016b, 2016a; Bilinovich et al., 2019). It has been suggested that the GWAS linkage to ASD is likely connected to RPS10P2-AS1 rather than the coding MACROD2 gene (Bilinovich et al., 2019).
A large-scale exome sequencing study combining data from about 36,000 individuals, including about 12,000 with ASD, identified 102 ASD risk genes most of which represented de novo variants (Satterstrom et al., 2020). Four risk genes (SLC6A1, DEAF1, KCNQ3, SCN1A) that were identified in which mutations confer a gain-of-function effect (Satterstrom et al., 2020). Gain-of-function variants in the KCNQ3 gene, which encodes a K+ channel, has previously been reported to cause ASD (Sands et al., 2019). Mutations in SCN1A, which encodes the alpha 1 subunit of the sodium channel, causes or is associated with several epilepsy disorders. Of the 102 risk genes, 12 genes were located in regions impacted by copy number variation (CNV). Another gene that has been identified in other studies, KMT2E, was also identified as a rick gene in this study. Based on genetic, cognitive, and behavioral criteria used in the analyses, the authors could separate 53 genes as conferring risk predominantly for ASD, whereas the other 49 had broader relevance connecting to both ASD and other neurodevelopmental disorders (NDDs). Most of the 102 risk genes were found to affect neuronal communication or gene expression regulation (GER). Expression analyses in the normal human cortex indicated that the risk genes participated in mid-to-late fetal development and with most being expressed in maturing and mature neurons that were both excitatory and inhibitory (Satterstrom et al., 2020). Analysis of the GER genes suggested involvement in the regulation of E/I balance.
Another parent-offspring trio whole-exome sequencing (WES) study conducted in a group of 60 individuals with both ID and ASD, identified variants in previously identified 8 ID/ASD-linked (SYNGAP1, SMAD6, PACS1, SHANK3, KMT2A, KCNQ2, ACTB, and POGZ) (Bruno et al., 2021). Other studies have identified SYNGAP as an ASD risk gene (Agarwal et al., 2019; Wright et al., 2024a, 2024b). SYNGAP1 protein (synaptic Ras GTPase-activating protein-1) is present at high amounts in the postsynaptic cells of excitatory synapses (Chen et al., 1998; Kim et al., 1998). Interestingly, although most studied in the context of synaptic function, SYNGAP1 also regulated neurogenesis (Birtele et al., 2023). Additionally, 4 novel candidate ASD/ID genes with de novo truncating variants (MBP, PCDHA1, PCDH15, PDPR) were identified (Bruno et al., 2021). The pyruvate dehydrogenase phosphatase regulatory subunit (PDPR) is variably expressed in the brain, with the highest levels in the corpus callosum and in the cerebellum.
Most whole-genome studies have focused on de novo variants identified from parent offspring trios. An integrated model composed of a two-stage analysis of de novo and inherited genetic variants, which provides yields greater power to identify risk genes (He et al., 2013), was used to study about 43,000 ASD cases, including ~35,000 new cases identified 60 genes including five new moderate-risk genes (NAV3, ITSN1, MARK2, SCAF1 and HNRNPUL2). These five genes represent rare loss-of-function variants with modest effect size, not stronglyassociated with ID compared with five of the most common highly-penetrant genes (CHD8, SCN2A, ADNP, FOXP1 and SHANK3) (Varghese et al., 2017; Zhou et al., 2022). Finding related to SHANK 3 and CHD8 in ASD with are described above. Activity-dependent neuroprotective protein (ADNP) is most highly expressed in the cerebellum and hippocampus and best known for its neuroprotective activity (Gozes et al., 1999). In the ASD brain, ADNP dysfunction caused disrupts DNA methylation and mitochondrial gene expression in the cerebellum (D’Incal et al., 2024).
The SCN2A gene encodes the Na(v)1.2 channel protein that is expressed highly and selectively in the cerebellum in rodents and non-human primates (Gould and Kim, 2021). SCN2A variations in humans causes seizures, ataxia, and cerebellar atrophy (Murray et al., 2023). Clinical exome sequencing has identified specific loss of function variations in SNCA, which carries one of the highest risk for ASD of any gene in humans (Satterstrom et al., 2020; Fu et al., 2022). In mice, ASD-associated SCNA variants disrupt granule neuron to PC communication and impairs neuronal plasticity in the cerebellum (Wang et al., 2024).
Given the importance of inherited genetic factors, an approach that was used recently to better understand the ASD architecture is the identification of moderate effect size (MES) genes in ASD (Caballero et al., 2024). MES genes do not qualify as risk genes by themselves but contribute to ASD when paired with specific other MES genes. A first step of the study identified inherited variants in two distinct genes overrepresented in offspring with ASD withnon-ASD offspring potentially inheriting either, but not both variants. Analysis of about 11,000 families led to the identification of 97 MES genes forming 50 significant gene pairs (Caballero et al., 2024). One pair of genes identified as ASD-associated was SNAP25 (Synaptosome Associated Protein 25) and SLPI (Secretory Leukocyte Peptidase Inhibitor). A study using whole genome sequencing of 32 Chinese ASD trios found de novo mutations, inherited variants, copy number variants (CNVs) and genomic structural variants (Wu et al., 2018). This study identified 87 potentially risk genes from 4832 genes containing variants (Wu et al., 2018). Among these were microcephaly-related genes and genes involved in centrosomal function and chromatin remodeling
A recent transcriptomic analysis of postmortem cerebellar PCs isolated by laser capture and pooled from postmortem ASD tissue identified about 430 differentially expressed genes, which were involved in regulating neurodevelopmental processes and connectivity, organization of the ECM, calcium response, and immune function (Brandenburg et al., 2022). Despite the compelling results of this and other genetic studies utilizing both ASD patients and mice, two recent studies analyzing the UK Biobank summary data failed to identify specific subregions of the cerebellum displaying significant genetic correlations with ASD at the whole genome-level, suggesting smaller pleiotropic cerebellar subregions than were analyzed and/or cancelling out of opposing genetic correlations across the genome.
Conclusion
Much progress has been made in our understanding of ADS since the time it was referred to as “infantile psychosis” and blamed on a cold and emotionally distant “refrigerator mothers” (Kanner and Lesser, 1958; Tonge et al., 1994). Despite this, our understanding of the molecular and cellular underpinnings of ASD remains limited. The phenotypic and genetic heterogeneity of ASD and its frequent co-occurrence in other neurodevelopmental and psychiatric disorders has made it challenging to identify specific genes that are causally involved in the development of the disorder. As our review describes, however, compelling evidence from human genetic analyses and experimental studies conducted in rodents point to a key role for cerebellar dysfunction in the development and progression of ASD. Although most attention has been placed on PCs, accumulating evidence suggests that dysfunction of granule neurons, other neuronal types, and glial cells, as well as disruption in the normal interactions between PCs and these other cell types, contribute to ASD. Cerebellar dysfunction impacts the functioning of other brain regions connected to by the cerebellum. Besides synaptic dysfunction, the identification and analyses ASD-linked genes had indicated that deregulation of neurogenesis as a common cellular feature of ASD. Recent studies have suggested important roles for alterations in gut microbiota (Fattorusso et al., 2019; Chernikova et al., 2021), innate immune system dysfunction (Robinson-Agramonte et al., 2022; Hughes et al., 2023) and maternal inflammation (Han et al., 2021; Kwon et al., 2022; Hughes et al., 2023) in the development of ASD. How these factors interface with dysfunction of the brain, and specifically the cerebellum, to cause ASD remains to be resolved.
Author Contributions
The manuscript was written by SRD.
Funding
Salary support of the author was provided by the Louisiana State University Shreveport and the Manipal Institutes of Higher Education.
Acknowledgments
The author wishes to acknowledge Anila D’Mello for her careful review of the manuscript and insightful suggestions.
Conflicts of Interest
The author declares no conflict of interest.
References
- Abbasy, S.; Shahraki, F.; Haghighatfard, A.; Qazvini, M.G.; Rafiei, S.T.; Noshadirad, E.; et al. Neuregulin1 types mRNA level changes in autism spectrum disorder, and is associated with deficit in executive functions. EBioMedicine 2018, 37, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Abdul, F.; Sreenivas, N.; Kommu, J.V.S.; Banerjee, M.; Berk, M.; Maes, M.; et al. Disruption of circadian rhythm and risk of autism spectrum disorder: role of immune-inflammatory, oxidative stress, metabolic and neurotransmitter pathways. Rev Neurosci 2022, 33, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Abreu, N.J.; Siemon, A.E.; Baylis, A.L.; Kirschner, R.E.; Pfau, R.B.; Ho, M.-L.; et al. Novel truncating variant in KMT2E associated with cerebellar hypoplasia and velopharyngeal dysfunction. Clin Case Rep 2022, 10, e05277. [Google Scholar] [CrossRef]
- Agarwal, M.; Johnston, M.V.; Stafstrom, C.E. SYNGAP1 mutations: Clinical, genetic, and pathophysiological features. Int J Dev Neurosci 2019, 78, 65–76. [Google Scholar] [CrossRef]
- Al-Ali, Z.; Yasseen, A.A.; Al-Dujailli, A.; Al-Karaqully, A.J.; McAllister, K.A.; Jumaah, A.S. The oxytocin receptor gene polymorphism rs2268491 and serum oxytocin alterations are indicative of autism spectrum disorder: A case-control paediatric study in Iraq with personalized medicine implications. PLoS One 2022, 17, e0265217. [Google Scholar] [CrossRef]
- Albrecht, U.; Sutcliffe, J.S.; Cattanach, B.M.; Beechey, C.V.; Armstrong, D.; Eichele, G.; et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet 1997, 17, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Alho, J.; Samuelsson, J.G.; Khan, S.; Mamashli, F.; Bharadwaj, H.; Losh, A.; et al. Both stronger and weaker cerebro-cerebellar functional connectivity patterns during processing of spoken sentences in autism spectrum disorder. Hum Brain Mapp 2023, 44, 5810–5827. [Google Scholar] [CrossRef]
- Ali, H.; Marth, L.; Krueger-Burg, D. Neuroligin-2 as a central organizer of inhibitory synapses in health and disease. Sci Signal 2020, 13, eabd8379. [Google Scholar] [CrossRef]
- Alnak, A.; Kuşcu Özücer, İ.; Okay Çağlayan, A.; Coşkun, M. Peripheral Expression of MACROD2 Gene Is Reduced Among a Sample of Turkish Children with Autism Spectrum Disorder. Psychiatry Clin Psychopharmacol 2021, 31, 261–268. [Google Scholar] [CrossRef]
- Alonso-Gonzalez, A.; Calaza, M.; Rodriguez-Fontenla, C.; Carracedo, A. Novel Gene-Based Analysis of ASD GWAS: Insight Into the Biological Role of Associated Genes. Front Genet 2019, 10, 733. [Google Scholar] [CrossRef]
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol Autism 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Galfin, T.; Xu, W.; Aoto, J.; Malenka, R.C.; Südhof, T.C. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc Natl Acad Sci U S A 2012, 109, 18120–18125. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. Maternal Cannabis Use in Pregnancy and Autism Spectrum Disorder or Attention-Deficit/Hyperactivity Disorder in Offspring. J Clin Psychiatry 2024, 86, 24f15717. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Pérez-Rodríguez, M.; Prius-Mengual, J.; Rodríguez-Moreno, A. Neuronal and astrocyte determinants of critical periods of plasticity. Trends Neurosci 2023, 46, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Angliker, N.; Burri, M.; Zaichuk, M.; Fritschy, J.-M.; Rüegg, M.A. mTORC1 and mTORC2 have largely distinct functions in Purkinje cells. Eur J Neurosci 2015, 42, 2595–2612. [Google Scholar] [CrossRef]
- Angrand, L.; Masson, J.-D.; Rubio-Casillas, A.; Nosten-Bertrand, M.; Crépeaux, G. Inflammation and Autophagy: A Convergent Point between Autism Spectrum Disorder (ASD)-Related Genetic and Environmental Factors: Focus on Aluminum Adjuvants. Toxics 2022, 10, 518. [Google Scholar] [CrossRef]
- Aoto, J.; Földy, C.; Ilcus, S.M.C.; Tabuchi, K.; Südhof, T.C. Distinct circuit-dependent functions of presynaptic neurexin-3 at GABAergic and glutamatergic synapses. Nat Neurosci 2015, 18, 997–1007. [Google Scholar] [CrossRef]
- Argent, L.; Winter, F.; Prickett, I.; Carrasquero-Ordaz, M.; Olsen, A.L.; Kramer, H.; et al. Caspr2 interacts with type 1 inositol 1,4,5-trisphosphate receptor in the developing cerebellum and regulates Purkinje cell morphology. J Biol Chem 2020, 295, 12716–12726. [Google Scholar] [CrossRef]
- Aristidou, C.; Koufaris, C.; Theodosiou, A.; Bak, M.; Mehrjouy, M.M.; Behjati, F.; et al. Accurate Breakpoint Mapping in Apparently Balanced Translocation Families with Discordant Phenotypes Using Whole Genome Mate-Pair Sequencing. PLoS One 2017, 12, e0169935. [Google Scholar] [CrossRef]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 2008, 82, 160–164. [Google Scholar] [CrossRef]
- Armstrong, C.L.; Duffin, C.A.; McFarland, R.; Vogel, M.W. Mechanisms of compartmental purkinje cell death and survival in the lurcher mutant mouse. Cerebellum 2011, 10, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.C.; Caruso, A.; Servadio, M.; Andreae, L.C.; Trezza, V.; Scattoni, M.L.; et al. Assessing the developmental trajectory of mouse models of neurodevelopmental disorders: Social and communication deficits in mice with Neurexin 1α deletion. Genes Brain Behav 2020, 19, e12630. [Google Scholar] [CrossRef]
- Arndt, K.; Nakagawa, S.; Takeichi, M.; Redies, C. Cadherin-Defined Segments and Parasagittal Cell Ribbons in the Developing Chicken Cerebellum. Mol Cell Neurosci 1998, 10, 211–228. [Google Scholar] [CrossRef]
- Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism 2017, 8, 21. [CrossRef]
- Bacchelli, E.; Cameli, C.; Viggiano, M.; Igliozzi, R.; Mancini, A.; Tancredi, R.; et al. An integrated analysis of rare CNV and exome variation in Autism Spectrum Disorder using the Infinium PsychArray. Sci Rep 2020, 10, 3198. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; et al. A clinicopathological study of autism. Brain 1998, 121 Pt 5, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years - Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Baizer, J.S. Neuroanatomy of autism: what is the role of the cerebellum? Cereb Cortex 2024, 34, 94–103. [Google Scholar] [CrossRef]
- Bakkaloglu, B.; O’Roak, B.J.; Louvi, A.; Gupta, A.R.; Abelson, J.F.; Morgan, T.M.; et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet 2008, 82, 165–173. [Google Scholar] [CrossRef]
- Banko, J.L.; Merhav, M.; Stern, E.; Sonenberg, N.; Rosenblum, K.; Klann, E. Behavioral alterations in mice lacking the translation repressor 4E-BP2. Neurobiol Learn Mem 2007, 87, 248–256. [Google Scholar] [CrossRef]
- Banko, J.L.; Poulin, F.; Hou, L.; DeMaria, C.T.; Sonenberg, N.; Klann, E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci 2005, 25, 9581–9590. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Taylor, M.R.; Williams, M.E. The classic cadherins in synaptic specificity. Cell Adh Migr 2015, 9, 193–201. [Google Scholar] [CrossRef]
- Battaglioni, S.; Benjamin, D.; Wälchli, M.; Maier, T.; Hall, M.N. mTOR substrate phosphorylation in growth control. Cell 2022, 185, 1814–1836. [Google Scholar] [CrossRef]
- Bauman, M.; Kemper, T.L. Histoanatomic observations of the brain in early infantile autism. Neurology 1985, 35, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.B.E.; Stoodley, C.J. Autism spectrum disorder and the cerebellum. Int Rev Neurobiol 2013, 113, 1–34. [Google Scholar] [CrossRef]
- Behesti, H.; Fore, T.R.; Wu, P.; Horn, Z.; Leppert, M.; Hull, C.; et al. ASTN2 modulates synaptic strength by trafficking and degradation of surface proteins. Proc Natl Acad Sci U S A 2018, 115, E9717–E9726. [Google Scholar] [CrossRef]
- Bellamy, T.C. Interactions between Purkinje neurones and Bergmann glia. Cerebellum 2006, 5, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Benayed, R.; Choi, J.; Matteson, P.G.; Gharani, N.; Kamdar, S.; Brzustowicz, L.M.; et al. Autism-associated haplotype affects the regulation of the homeobox gene, ENGRAILED 2. Biol Psychiatry 2009, 66, 911–917. [Google Scholar] [CrossRef]
- Benayed, R.; Gharani, N.; Rossman, I.; Mancuso, V.; Lazar, G.; Kamdar, S.; et al. Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am J Hum Genet 2005, 77, 851–868. [Google Scholar] [CrossRef] [PubMed]
- Bering, T.; Hertz, H.; Rath, M.F. The Circadian Oscillator of the Cerebellum: Triiodothyronine Regulates Clock Gene Expression in Granule Cells in vitro and in the Cerebellum of Neonatal Rats in vivo. Front. Physiol. 2021, 12, 706433. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Sakurai, T.; Buxbaum, J.D. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci 2009, 32, 402–412. [Google Scholar] [CrossRef]
- Beversdorf, D.Q.; Stevens, H.E.; Jones, K.L. Prenatal Stress, Maternal Immune Dysregulation, and Their Association With Autism Spectrum Disorders. Curr Psychiatry Rep 2018, 20, 76. [Google Scholar] [CrossRef]
- Bi, W.; Yan, J.; Shi, X.; Yuva-Paylor, L.A.; Antalffy, B.A.; Goldman, A.; et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum Mol Genet 2007, 16, 1802–1813. [Google Scholar] [CrossRef]
- Bilinovich, S.M.; Lewis, K.; Grepo, N.; Campbell, D.B. The Long Noncoding RNA RPS10P2-AS1 Is Implicated in Autism Spectrum Disorder Risk and Modulates Gene Expression in Human Neuronal Progenitor Cells. Front. Genet. 2019, 10, 970. [Google Scholar] [CrossRef]
- Birtele, M.; Del Dosso, A.; Xu, T.; Nguyen, T.; Wilkinson, B.; Hosseini, N.; et al. Non-synaptic function of the autism spectrum disorder-associated gene SYNGAP1 in cortical neurogenesis. Nat Neurosci 2023, 26, 2090–2103. [Google Scholar] [CrossRef]
- Blundell, J.; Blaiss, C.A.; Etherton, M.R.; Espinosa, F.; Tabuchi, K.; Walz, C.; et al. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci 2010, 30, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.; Tabuchi, K.; Bolliger, M.F.; Blaiss, C.A.; Brose, N.; Liu, X.; et al. Increased anxiety-like behavior in mice lacking the inhibitory synapse cell adhesion molecule neuroligin 2. Genes Brain Behav 2009, 8, 114–126. [Google Scholar] [CrossRef]
- Boccuto, L.; Lauri, M.; Sarasua, S.M.; Skinner, C.D.; Buccella, D.; Dwivedi, A.; et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet 2013, 21, 310–316. [Google Scholar] [CrossRef]
- Böckers, T.M.; Segger-Junius, M.; Iglauer, P.; Bockmann, J.; Gundelfinger, E.D.; Kreutz, M.R.; et al. Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3’ untranslated region of Shank1 mRNA. Mol Cell Neurosci 2004, 26, 182–190. [Google Scholar] [CrossRef]
- Boer, K.; Troost, D.; Jansen, F.; Nellist, M.; van den Ouweland, A.M.W.; Geurts, J.J.G.; et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 2008, 28, 577–590. [Google Scholar] [CrossRef]
- Bolliger, M.F.; Pei, J.; Maxeiner, S.; Boucard, A.A.; Grishin, N.V.; Südhof, T.C. Unusually rapid evolution of Neuroligin-4 in mice. Proc Natl Acad Sci U S A 2008, 105, 6421–6426. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Graziano, C.; Minopoli, F.; Bacchelli, E.; Magini, P.; Diquigiovanni, C.; et al. Maternally inherited genetic variants of CADPS2 are present in autism spectrum disorders and intellectual disability patients. EMBO Mol Med 2014, 6, 795–809. [Google Scholar] [CrossRef]
- Born, G.; Grayton, H.M.; Langhorst, H.; Dudanova, I.; Rohlmann, A.; Woodward, B.W.; et al. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front Synaptic Neurosci 2015, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Boucherie, C.; Boutin, C.; Jossin, Y.; Schakman, O.; Goffinet, A.M.; Ris, L.; et al. Neural progenitor fate decision defects, cortical hypoplasia and behavioral impairment in Celsr1-deficient mice. Mol Psychiatry 2018, 23, 723–734. [Google Scholar] [CrossRef]
- Boukhtouche, F.; Doulazmi, M.; Frederic, F.; Dusart, I.; Brugg, B.; Mariani, J. RORalpha, a pivotal nuclear receptor for Purkinje neuron survival and differentiation: from development to ageing. Cerebellum 2006, 5, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Boukhtouche, F.; Janmaat, S.; Vodjdani, G.; Gautheron, V.; Mallet, J.; Dusart, I.; et al. Retinoid-related orphan receptor alpha controls the early steps of Purkinje cell dendritic differentiation. J Neurosci 2006, 26, 1531–1538. [Google Scholar] [CrossRef]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015, 16, 551–563. [Google Scholar] [CrossRef]
- Bowling, H.; Klann, E. Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron 2014, 83, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, C.; Griswold, A.J.; Van Booven, D.J.; Kilander, M.B.C.; Frei, J.A.; Nestor, M.W.; et al. Transcriptomic analysis of isolated and pooled human postmortem cerebellar Purkinje cells in autism spectrum disorders. Front Genet 2022, 13, 944837. [Google Scholar] [CrossRef] [PubMed]
- Brielmaier, J.; Matteson, P.G.; Silverman, J.L.; Senerth, J.M.; Kelly, S.; Genestine, M.; et al. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One 2012, 7, e40914. [Google Scholar] [CrossRef] [PubMed]
- Bromley, R.L.; Mawer, G.E.; Briggs, M.; Cheyne, C.; Clayton-Smith, J.; García-Fiñana, M.; et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry 2013, 84, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.J.; Lin, B.; Holguin, B. Expression of neuregulin 1, a member of the epidermal growth factor family, is expressed as multiple splice variants in the adult human cornea. Invest Ophthalmol Vis Sci 2004, 45, 3021–3029. [Google Scholar] [CrossRef]
- Bruinsma, C.F.; Schonewille, M.; Gao, Z.; Aronica, E.M.A.; Judson, M.C.; Philpot, B.D.; et al. Dissociation of locomotor and cerebellar deficits in a murine Angelman syndrome model. J Clin Invest 2015, 125, 4305–4315. [Google Scholar] [CrossRef]
- Bruno, L.P.; Doddato, G.; Valentino, F.; Baldassarri, M.; Tita, R.; Fallerini, C.; et al. New Candidates for Autism/Intellectual Disability Identified by Whole-Exome Sequencing. Int J Mol Sci 2021, 22, 13439. [Google Scholar] [CrossRef]
- Busfield, S.J.; Michnick, D.A.; Chickering, T.W.; Revett, T.L.; Ma, J.; Woolf, E.A.; et al. Characterization of a neuregulin-related gene, Don-1, that is highly expressed in restricted regions of the cerebellum and hippocampus. Mol Cell Biol 1997, 17, 4007–4014. [Google Scholar] [CrossRef]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.-P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005, 42, 318–321. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Cai, G.; Chaste, P.; Nygren, G.; Goldsmith, J.; Reichert, J.; et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 2007, 144B, 484–491. [Google Scholar] [CrossRef]
- C, P.; Ad, K.; G, G.; Hk, T.; H, N.; K, H.; et al. Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nature communications 2014, 5, 5586. [Google Scholar] [CrossRef]
- Caballero, M.; Satterstrom, F.K.; Buxbaum, J.D.; Mahjani, B. Identification of moderate effect size genes in autism spectrum disorder through a novel gene pairing approach. medRxiv 2024. [Google Scholar] [CrossRef]
- Cai, X.-Y.; Wang, X.-T.; Guo, J.-W.; Xu, F.-X.; Ma, K.-Y.; Wang, Z.-X.; et al. Aberrant outputs of cerebellar nuclei and targeted rescue of social deficits in an autism mouse model. Protein Cell 2024, 15, pwae040. [Google Scholar] [CrossRef]
- Cakar, M.E.; Okada, N.J.; Cummings, K.K.; Jung, J.; Bookheimer, S.Y.; Dapretto, M.; et al. Functional connectivity of the sensorimotor cerebellum in autism: associations with sensory over-responsivity. Front. Psychiatry 2024, 15, 1337921. [Google Scholar] [CrossRef] [PubMed]
- Cantando, I.; Centofanti, C.; D’Alessandro, G.; Limatola, C.; Bezzi, P. Metabolic dynamics in astrocytes and microglia during post-natal development and their implications for autism spectrum disorders. Front Cell Neurosci 2024, 18, 1354259. [Google Scholar] [CrossRef]
- Cao, L.; Molina, J.; Abad, C.; Carmona-Mora, P.; Cárdenas Oyarzo, A.; Young, J.I.; et al. Correct developmental expression level of Rai1 in forebrain neurons is required for control of body weight, activity levels and learning and memory. Hum Mol Genet 2014, 23, 1771–1782. [Google Scholar] [CrossRef]
- Capal, J.K.; Jeste, S.S. Autism and Epilepsy. Pediatr Clin North Am 2024, 71, 241–252. [Google Scholar] [CrossRef]
- Carrión-Castillo, A.; Boeckx, C. Insights into the genetic architecture of cerebellar lobules derived from the UK Biobank. Sci Rep 2024, 14, 9488. [Google Scholar] [CrossRef]
- Carta, I.; Chen, C.H.; Schott, A.L.; Dorizan, S.; Khodakhah, K. Cerebellar modulation of the reward circuitry and social behavior. Science 2019, 363, eaav0581. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Kenkel, W.M.; MacLean, E.L.; Wilson, S.R.; Perkeybile, A.M.; Yee, J.R.; et al. Is Oxytocin “Nature’s Medicine”? Pharmacol Rev 2020, 72, 829–861. [Google Scholar] [CrossRef]
- Cast, T.P.; Boesch, D.J.; Smyth, K.; Shaw, A.E.; Ghebrial, M.; Chanda, S. An Autism-Associated Mutation Impairs Neuroligin-4 Glycosylation and Enhances Excitatory Synaptic Transmission in Human Neurons. J Neurosci 2021, 41, 392–407. [Google Scholar] [CrossRef]
- Cendelin, J. From mice to men: lessons from mutant ataxic mice. Cerebellum Ataxias 2014, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Chabrol, F.P.; Blot, A.; Mrsic-Flogel, T.D. Cerebellar Contribution to Preparatory Activity in Motor Neocortex. Neuron 2019, 103, 506–519.e4. [Google Scholar] [CrossRef]
- Chahrour, M.; O’Roak, B.J.; Santini, E.; Samaco, R.C.; Kleiman, R.J.; Manzini, M.C. Current Perspectives in Autism Spectrum Disorder: From Genes to Therapy. J Neurosci 2016, 36, 11402–11410. [Google Scholar] [CrossRef]
- Chalkiadaki, K.; Statoulla, E.; Zafeiri, M.; Haji, N.; Lacaille, J.-C.; Powell, C.M.; et al. Reversal of memory and autism-related phenotypes in Tsc2+/- mice via inhibition of Nlgn1. Front Cell Dev Biol 2023, 11, 1205112. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.; Escott-Price, V.; Legge, S.; Baker, E.; Singh, K.D.; Walters, J.T.R.; et al. Genetic common variants associated with cerebellar volume and their overlap with mental disorders: a study on 33,265 individuals from the UK-Biobank. Mol Psychiatry 2022, 27, 2282–2290. [Google Scholar] [CrossRef]
- Chanda, S.; Aoto, J.; Lee, S.-J.; Wernig, M.; Südhof, T.C. Pathogenic mechanism of an autism-associated neuroligin mutation involves altered AMPA-receptor trafficking. Mol Psychiatry 2016, 21, 169–177. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Kowalczyk, M.; Fogerson, P.M.; Lee, Y.-J.; Haque, M.; Adams, E.L.; et al. Loss of Rai1 enhances hippocampal excitability and epileptogenesis in mouse models of Smith-Magenis syndrome. Proc Natl Acad Sci U S A 2022, 119, e2210122119. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.H.; Estes, A.; Munson, J.; Bernier, R.; Webb, S.J.; Rothstein, J.H.; et al. Genome-scan for IQ discrepancy in autism: evidence for loci on chromosomes 10 and 16. Hum Genet 2011, 129, 59–70. [Google Scholar] [CrossRef]
- Cheh, M.A.; Millonig, J.H.; Roselli, L.M.; Ming, X.; Jacobsen, E.; Kamdar, S.; et al. En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res 2006, 1116, 166–176. [Google Scholar] [CrossRef]
- Chen, H.J.; Rojas-Soto, M.; Oguni, A.; Kennedy, M.B. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 1998, 20, 895–904. [Google Scholar] [CrossRef]
- Chen, J.; Yu, S.; Fu, Y.; Li, X. Synaptic proteins and receptors defects in autism spectrum disorders. Front Cell Neurosci 2014, 8, 276. [Google Scholar] [CrossRef]
- Chen, X.; Chen, T.; Dong, C.; Chen, H.; Dong, X.; Yang, L.; et al. Deletion of CHD8 in cerebellar granule neuron progenitors leads to severe cerebellar hypoplasia, ataxia, and psychiatric behavior in mice. J Genet Genomics 2022, 49, 859–869. [Google Scholar] [CrossRef]
- Chen, X.; Du, Y.; Broussard, G.J.; Kislin, M.; Yuede, C.M.; Zhang, S.; et al. Transcriptomic mapping uncovers Purkinje neuron plasticity driving learning. Nature 2022, 605, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.R.; Heck, N.; Lohof, A.M.; Rochefort, C.; Morel, M.-P.; Wehrlé, R.; et al. Mature Purkinje cells require the retinoic acid-related orphan receptor-α (RORα) to maintain climbing fiber mono-innervation and other adult characteristics. J Neurosci 2013, 33, 9546–9562. [Google Scholar] [CrossRef]
- Cheng, Y.; Sudarov, A.; Szulc, K.U.; Sgaier, S.K.; Stephen, D.; Turnbull, D.H.; et al. The Engrailed homeobox genes determine the different foliation patterns in the vermis and hemispheres of the mammalian cerebellum. Development 2010, 137, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, M.A.; Flores, G.D.; Kilroy, E.; Labus, J.S.; Mayer, E.A.; Aziz-Zadeh, L. The Brain-Gut-Microbiome System: Pathways and Implications for Autism Spectrum Disorder. Nutrients 2021, 13, 4497. [Google Scholar] [CrossRef] [PubMed]
- Cheron, G.; Márquez-Ruiz, J.; Kishino, T.; Dan, B. Disruption of the LTD dialogue between the cerebellum and the cortex in Angelman syndrome model: a timing hypothesis. Front Syst Neurosci 2014, 8, 221. [Google Scholar] [CrossRef]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Cheung, C.; Chua, S.E.; Cheung, V.; Khong, P.L.; Tai, K.S.; Wong, T.K.W.; et al. White matter fractional anisotrophy differences and correlates of diagnostic symptoms in autism. J Child Psychol Psychiatry 2009, 50, 1102–1112. [Google Scholar] [CrossRef]
- Chien, W.-H.; Chen, C.-H.; Cheng, M.-C.; Wu, Y.-Y.; Gau, S.S.-F. Neuregulin 2 Is a Candidate Gene for Autism Spectrum Disorder. International Journal of Molecular Sciences 2024, 25, 5547. [Google Scholar] [CrossRef]
- Chih, B.; Engelman, H.; Scheiffele, P. Control of excitatory and inhibitory synapse formation by neuroligins. Science 2005, 307, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Chih, B.; Engelman, H.; Scheiffele, P. Control of excitatory and inhibitory synapse formation by neuroligins. Science 2005, 307, 1324–1328. [Google Scholar] [CrossRef]
- Chiocchetti, A.G.; Kopp, M.; Waltes, R.; Haslinger, D.; Duketis, E.; Jarczok, T.A.; et al. Variants of the CNTNAP2 5’ promoter as risk factors for autism spectrum disorders: a genetic and functional approach. Mol Psychiatry 2015, 20, 839–849. [Google Scholar] [CrossRef]
- Choe, K.Y.; Bethlehem, R.A.I.; Safrin, M.; Dong, H.; Salman, E.; Li, Y.; et al. Oxytocin normalizes altered circuit connectivity for social rescue of the Cntnap2 knockout mouse. Neuron 2022, 110, 795–808.e6. [Google Scholar] [CrossRef]
- Choi, J.; Ababon, M.R.; Soliman, M.; Lin, Y.; Brzustowicz, L.M.; Matteson, P.G.; et al. Autism associated gene, engrailed2, and flanking gene levels are altered in post-mortem cerebellum. PLoS One 2014, 9, e87208. [Google Scholar] [CrossRef]
- Christensen, Z.P.; Freedman, E.G.; Foxe, J.J. Autism is associated with in vivo changes in gray matter neurite architecture. Autism Res 2024, 17, 2261–2277. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, A.A.; Soltys, Z. Bergmann Glia, Long-Term Depression, and Autism Spectrum Disorder. Mol Neurobiol 2017, 54, 1156–1166. [Google Scholar] [CrossRef]
- Ciricugno, A.; Ferrari, C.; Battelli, L.; Cattaneo, Z. A chronometric study of the posterior cerebellum’s function in emotional processing. Curr Biol 2024, 34, 1844–1852.e3. [Google Scholar] [CrossRef] [PubMed]
- Clayton-Smith, J.; Donnai, D. Fetal valproate syndrome. J Med Genet 1995, 32, 724–727. [Google Scholar] [CrossRef]
- Cooper, J.N.; Mittal, J.; Sangadi, A.; Klassen, D.L.; King, A.M.; Zalta, M.; et al. Landscape of NRXN1 Gene Variants in Phenotypic Manifestations of Autism Spectrum Disorder: A Systematic Review. J Clin Med 2024, 13, 2067. [Google Scholar] [CrossRef]
- Costa, C.I.S.; da Silva Montenegro, E.M.; Zarrei, M.; de Sá Moreira, E.; Silva, I.M.W.; de Oliveira Scliar, M.; et al. Copy number variations in a Brazilian cohort with autism spectrum disorders highlight the contribution of cell adhesion genes. Clin Genet 2022, 101, 134–141. [Google Scholar] [CrossRef]
- Courchesne, E. Neuroanatomic imaging in autism. Pediatrics 1991, 87, 781–790. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; et al. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef]
- Courchesne, E.; Pramparo, T.; Gazestani, V.H.; Lombardo, M.V.; Pierce, K.; Lewis, N.E. The ASD Living Biology: from cell proliferation to clinical phenotype. Mol Psychiatry 2019, 24, 88–107. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.M.; Kang, Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol 2007m 17, 43–52. [CrossRef]
- Crawley, J.N. Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 2007, 17, 448–459. [Google Scholar] [CrossRef]
- Crepel, A.; De Wolf, V.; Brison, N.; Ceulemans, B.; Walleghem, D.; Peuteman, G.; et al. Association of CDH11 with non-syndromic ASD. Am J Med Genet B Neuropsychiatr Genet 2014, 165B, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism 2014, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Cupolillo, D.; Hoxha, E.; Faralli, A.; De Luca, A.; Rossi, F.; Tempia, F.; et al. Autistic-Like Traits and Cerebellar Dysfunction in Purkinje Cell PTEN Knock-Out Mice. Neuropsychopharmacology 2016, 41, 1457–1466. [Google Scholar] [CrossRef]
- Cusmano, D.M.; Mong, J.A. In utero exposure to valproic acid changes sleep in juvenile rats: a model for sleep disturbances in autism. Sleep 2014, 37, 1489–1499. [Google Scholar] [CrossRef]
- Dahlhaus, R.; El-Husseini, A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav Brain Res 2010, 208, 96–105. [Google Scholar] [CrossRef]
- D’Angelo, E. Physiology of the cerebellum. Handb Clin Neurol 2018, 154, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Daskalakis, Z.J.; Christensen, B.K.; Fitzgerald, P.B.; Fountain, S.I.; Chen, R. Reduced cerebellar inhibition in schizophrenia: a preliminary study. Am J Psychiatry 2005, 162, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- de Agustín-Durán, D.; Mateos-White, I.; Fabra-Beser, J.; Gil-Sanz, C. Stick around: Cell-Cell Adhesion Molecules during Neocortical Development. Cells 2021, 10, 118. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [CrossRef]
- Deng, Y.; Ma, L.; Du, Z.; Ma, H.; Xia, Y.; Ping, L.; et al. The Notch1/Hes1 pathway regulates Neuregulin 1/ErbB4 and participates in microglial activation in rats with VPA-induced autism. Prog Neuropsychopharmacol Biol Psychiatry 2024, 131, 110947. [Google Scholar] [CrossRef]
- Deverett, B.; Kislin, M.; Tank, D.W.; Wang, S.S.-H. Cerebellar disruption impairs working memory during evidence accumulation. Nat Commun 2019, 10, 3128. [Google Scholar] [CrossRef]
- DeWitt, J.J.; Grepo, N.; Wilkinson, B.; Evgrafov, O.V.; Knowles, J.A.; Campbell, D.B. Impact of the Autism-Associated Long Noncoding RNA MSNP1AS on Neuronal Architecture and Gene Expression in Human Neural Progenitor Cells. Genes (Basel) 2016, 7, 76. [Google Scholar] [CrossRef]
- DeWitt, J.J.; Hecht, P.M.; Grepo, N.; Wilkinson, B.; Evgrafov, O.V.; Morris, K.V.; et al. Transcriptional Gene Silencing of the Autism-Associated Long Noncoding RNA MSNP1AS in Human Neural Progenitor Cells. Dev Neurosci 2016, 38, 375–383. [Google Scholar] [CrossRef]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.; Varcin, K.J.; Sahin, M.; Nelson, C.A.; Jeste, S.S. Early patterns of functional brain development associated with autism spectrum disorder in tuberous sclerosis complex. Autism Res 2019, 12, 1758–1773. [Google Scholar] [CrossRef] [PubMed]
- Dickson, P.E.; Cairns, J.; Goldowitz, D.; Mittleman, G. Cerebellar contribution to higher and lower order rule learning and cognitive flexibility in mice. Neuroscience 2017, 345, 99–109. [Google Scholar] [CrossRef] [PubMed]
- DiLiberti, J.H.; Farndon, P.A.; Dennis, N.R.; Curry, C.J. The fetal valproate syndrome. Am J Med Genet 1984, 19, 473–481. [Google Scholar] [CrossRef]
- D’Incal, C.; Van Dijck, A.; Ibrahim, J.; De Man, K.; Bastini, L.; Konings, A.; et al. ADNP dysregulates methylation and mitochondrial gene expression in the cerebellum of a Helsmoortel-Van der Aa syndrome autopsy case. Acta Neuropathol Commun 2024, 12, 62. [Google Scholar] [CrossRef]
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet 2008, 17, 111–118. [Google Scholar] [CrossRef]
- Ding, C.-Y.; Ding, Y.-T.; Ji, H.; Wang, Y.-Y.; Zhang, X.; Yin, D.-M. Genetic labeling reveals spatial and cellular expression pattern of neuregulin 1 in mouse brain. Cell Biosci 2023, 13, 79. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Moore, D.M.; Crocetti, D.; Mostofsky, S.H.; Stoodley, C.J. Cerebellar gray matter differentiates children with early language delay in autism. Autism Res 2016, 9, 1191–1204. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci 2015, 9, 408. [Google Scholar] [CrossRef]
- D’Mello, A.M.; Stoodley, C.J. Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci 2015, 9, 408. [Google Scholar] [CrossRef]
- d’Oleire Uquillas, F.; Sefik, E.; Li, B.; Trotter, M.A.; Steele, K.A.; Seidlitz, J.; et al. Multimodal evidence for cerebellar influence on cortical development in autism: structural growth amidst functional disruption. Mol Psychiatry 2025, 30, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Zielke, H.R.; Yeh, D.; Yang, P. Cellular stress and apoptosis contribute to the pathogenesis of autism spectrum disorder. Autism Res 2018, 11, 1076–1090. [Google Scholar] [CrossRef]
- Doulazmi, M.; Frédéric, F.; Capone, F.; Becker-André, M.; Delhaye-Bouchaud, N.; Mariani, J. A comparative study of Purkinje cells in two RORalpha gene mutant mice: staggerer and RORalpha(-/-). Brain Res Dev Brain Res 2001, 127, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Perroy, J.; Loll, F.; Perrais, D.; Fagni, L.; Bourgeron, T.; et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 2012, 17, 71–84. [Google Scholar] [CrossRef]
- Dusart, I.; Guenet, J.L.; Sotelo, C. Purkinje cell death: differences between developmental cell death and neurodegenerative death in mutant mice. Cerebellum 2006, 5, 163–173. [Google Scholar] [CrossRef]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol Autism 2014, 5, 3. [Google Scholar] [CrossRef]
- El-Kordi, A.; Winkler, D.; Hammerschmidt, K.; Kästner, A.; Krueger, D.; Ronnenberg, A.; et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav Brain Res 2013, 251, 41–49. [Google Scholar] [CrossRef]
- Ellegood, J.; Crawley, J.N. Behavioral and Neuroanatomical Phenotypes in Mouse Models of Autism. Neurotherapeutics 2015, 12, 521–533. [Google Scholar] [CrossRef]
- Ellegood, J.; Lerch, J.P.; Henkelman, R.M. Brain abnormalities in a Neuroligin3 R451C knockin mouse model associated with autism. Autism Res 2011, 4, 368–376. [Google Scholar] [CrossRef]
- Esnafoglu, E. Levels of peripheral Neuregulin 1 are increased in non-medicated autism spectrum disorder patients. J Clin Neurosci 2018, 57, 43–45. [Google Scholar] [CrossRef]
- Esper, R.M.; Pankonin, M.S.; Loeb, J.A. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev 2006, 51, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.; Földy, C.; Sharma, M.; Tabuchi, K.; Liu, X.; Shamloo, M.; et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci U S A 2011, 108, 13764–13769. [Google Scholar] [CrossRef] [PubMed]
- Etherton, M.R.; Blaiss, C.A.; Powell, C.M.; Südhof, T.C. Mouse neurexin-1alpha deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A 2009, 106, 17998–18003. [Google Scholar] [CrossRef]
- Fang, P.; Lev-Lehman, E.; Tsai, T.F.; Matsuura, T.; Benton, C.S.; Sutcliffe, J.S.; et al. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 1999, 8, 129–135. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Farini, D.; Cesari, E.; Weatheritt, R.J.; La Sala, G.; Naro, C.; Pagliarini, V.; et al. A Dynamic Splicing Program Ensures Proper Synaptic Connections in the Developing Cerebellum. Cell Rep 2020, 31, 107703. [Google Scholar] [CrossRef]
- Farini, D.; Marazziti, D.; Geloso, M.C.; Sette, C. Transcriptome programs involved in the development and structure of the cerebellum. Cell Mol Life Sci 2021, 78, 6431–6451. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Halt, A.R.; Realmuto, G.; Earle, J.; Kist, D.A.; Thuras, P.; et al. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol Neurobiol 2002, 22, 171–175. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.C.; Realmuto, G.R. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Fattorusso, A.; Di Genova, L.; Dell’Isola, G.B.; Mencaroni, E.; Esposito, S. Autism Spectrum Disorders and the Gut Microbiota. Nutrients 2019, 11, 521. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.N.; Young, L.J.; Hearn, E.F.; Matzuk, M.M.; Insel, T.R.; Winslow, J.T. Social amnesia in mice lacking the oxytocin gene. Nat Genet 2000, 25, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Sánchez-León, C.A.; Llorente, J.; Sierra-Arregui, T.; Knafo, S.; Márquez-Ruiz, J.; et al. Altered Cerebellar Response to Somatosensory Stimuli in the Cntnap2 Mouse Model of Autism. eNeuro 2021, 8, ENEURO. [Google Scholar] [CrossRef] [PubMed]
- Fernández Santoro, E.M.; Karim, A.; Warnaar, P.; De Zeeuw, C.I.; Badura, A.; Negrello, M. Purkinje cell models: past, present and future. Front Comput Neurosci 2024, 18, 1426653. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, S.; de Zavalia, N.; Belforte, N.; Amir, S. In utero Exposure to Valproic-Acid Alters Circadian Organisation and Clock-Gene Expression: Implications for Autism Spectrum Disorders. Front Behav Neurosci 2021, 15, 711549. [Google Scholar] [CrossRef]
- Floris, C.; Rassu, S.; Boccone, L.; Gasperini, D.; Cao, A.; Crisponi, L. Two patients with balanced translocations and autistic disorder: CSMD3 as a candidate gene for autism found in their common 8q23 breakpoint area. Eur J Hum Genet 2008, 16, 696–704. [Google Scholar] [CrossRef]
- Fragoso, Y.D.; Stoney, P.N.; Shearer, K.D.; Sementilli, A.; Nanescu, S.E.; Sementilli, P.; et al. Expression in the human brain of retinoic acid induced 1, a protein associated with neurobehavioural disorders. Brain Struct Funct 2015, 220, 1195–1203. [Google Scholar] [CrossRef]
- Frazier, T.W.; Embacher, R.; Tilot, A.K.; Koenig, K.; Mester, J.; Eng, C. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry 2015, 20, 1132–1138. [Google Scholar] [CrossRef]
- Frei, J.A.; Brandenburg, C.; Nestor, J.E.; Hodzic, D.M.; Plachez, C.; McNeill, H.; et al. Postnatal expression profiles of atypical cadherin FAT1 suggest its role in autism. Biol Open 2021, 10, bio056457. [Google Scholar] [CrossRef]
- Frei, J.A.; Niescier, R.F.; Bridi, M.S.; Durens, M.; Nestor, J.E.; Kilander, M.B.C.; et al. Regulation of Neural Circuit Development by Cadherin-11 Provides Implications for Autism. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Froemke, R.C.; Young, L.J. Oxytocin, Neural Plasticity, and Social Behavior. Annu Rev Neurosci 2021, 44, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Frosch, I.R.; Mittal, V.A.; D’Mello, A.M. Cerebellar Contributions to Social Cognition in ASD: A Predictive Processing Framework. Front Integr Neurosci 2022, 16, 810425. [Google Scholar] [CrossRef]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Fujima, S.; Yamaga, R.; Minami, H.; Mizuno, S.; Shinoda, Y.; Sadakata, T.; et al. CAPS2 Deficiency Impairs the Release of the Social Peptide Oxytocin, as Well as Oxytocin-Associated Social Behavior. J Neurosci 2021, 41, 4524–4535. [Google Scholar] [CrossRef]
- Gambini, D.; Ferrero, S.; Bulfamante, G.; Pisani, L.; Corbo, M.; Kuhn, E. Cerebellar phenotypes in germline PTEN mutation carriers. Neuropathol Appl Neurobiol 2024, 50, e12970. [Google Scholar] [CrossRef]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med 2015, 15, 146–167. [Google Scholar] [CrossRef]
- Garber, K. Neuroscience. Autism’s cause may reside in abnormalities at the synapse. Science 2007, 317, 190–191. [Google Scholar] [CrossRef]
- Gardner, Z.; Holbrook, O.; Tian, Y.; Odamah, K.; Man, H.-Y. The role of glia in the dysregulation of neuronal spinogenesis in Ube3a-dependent ASD. Exp Neurol 2024, 376, 114756. [Google Scholar] [CrossRef]
- Gaubitz, C.; Prouteau, M.; Kusmider, B.; Loewith, R. TORC2 Structure and Function. Trends Biochem Sci 2016, 41, 532–545. [Google Scholar] [CrossRef]
- Gauthier, J.; Siddiqui, T.J.; Huashan, P.; Yokomaku, D.; Hamdan, F.F.; Champagne, N.; et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet 2011, 130, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes (Basel) 2023, 14, 677. [Google Scholar] [CrossRef] [PubMed]
- Geoffray, M.-M.; Nicolas, A.; Speranza, M.; Georgieff, N. Are circadian rhythms new pathways to understand Autism Spectrum Disorder? J Physiol Paris 2016, 110, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Gerhant, A.; Olajossy, M.; Olajossy-Hilkesberger, L. [Neuroanatomical, genetic and neurochemical aspects of infantile autism]. Psychiatr Pol 2013, 47, 1101–1111. [Google Scholar]
- Gerik-Celebi, H.B.; Bolat, H.; Unsel-Bolat, G. Rare heterozygous genetic variants of NRXN and NLGN gene families involved in synaptic function and their association with neurodevelopmental disorders. Dev Neurobiol 2024, 84, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Getz, S.A.; DeSpenza, T.; Li, M.; Luikart, B.W. Rapamycin prevents, but does not reverse, aberrant migration in Pten knockout neurons. Neurobiol Dis 2016, 93, 12–20. [Google Scholar] [CrossRef]
- Getz, S.A.; Tariq, K.; Marchand, D.H.; Dickson, C.R.; Howe Vi, J.R.; Skelton, P.D.; et al. PTEN Regulates Dendritic Arborization by Decreasing Microtubule Polymerization Rate. J Neurosci 2022, 42, 1945–1957. [Google Scholar] [CrossRef]
- Gharani, N.; Benayed, R.; Mancuso, V.; Brzustowicz, L.M.; Millonig, J.H. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry 2004, 9, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef]
- Glickstein, M.; Doron, K. Cerebellum: Connections and Functions. Cerebellum 2008, 7, 589–594. [Google Scholar] [CrossRef]
- Goffinet, A.M.; Tissir, F. Seven pass Cadherins CELSR1-3. Semin Cell Dev Biol 2017, 69, 102–110. [Google Scholar] [CrossRef]
- Gong, X.; Wang, H. SHANK1 and autism spectrum disorders. Sci China Life Sci 2015, 58, 985–990. [Google Scholar] [CrossRef]
- Gonzalo-Ruiz, A.; Leichnetz, G.R.; Hardy, S.G. Projections of the medial cerebellar nucleus to oculomotor-related midbrain areas in the rat: an anterograde and retrograde HRP study. J Comp Neurol 1990, 296, 427–436. [Google Scholar] [CrossRef]
- Goorden, S.M.I.; van Woerden, G.M.; van der Weerd, L.; Cheadle, J.P.; Elgersma, Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol 2007, 62, 648–655. [Google Scholar] [CrossRef]
- Gordon, A.; Salomon, D.; Barak, N.; Pen, Y.; Tsoory, M.; Kimchi, T.; et al. Expression of Cntnap2 (Caspr2) in multiple levels of sensory systems. Mol Cell Neurosci 2016, 70, 42–53. [Google Scholar] [CrossRef]
- Gould, E.; Kim, J.H. SCN2A contributes to oligodendroglia excitability and development in the mammalian brain. Cell Rep 2021, 36, 109653. [Google Scholar] [CrossRef]
- Gozes, I.; Bassan, M.; Zamostiano, R.; Pinhasov, A.; Davidson, A.; Giladi, E.; et al. A novel signaling molecule for neuropeptide action: activity-dependent neuroprotective protein. Ann N Y Acad Sci 1999, 897, 125–135. [Google Scholar] [CrossRef]
- Grasselli, G.; Boele, H.-J.; Titley, H.K.; Bradford, N.; van Beers, L.; Jay, L.; et al. SK2 channels in cerebellar Purkinje cells contribute to excitability modulation in motor-learning-specific memory traces. PLoS Biol 2020, 18, e3000596. [Google Scholar] [CrossRef]
- Grasselli, G.; He, Q.; Wan, V.; Adelman, J.P.; Ohtsuki, G.; Hansel, C. Activity-Dependent Plasticity of Spike Pauses in Cerebellar Purkinje Cells. Cell Rep 2016, 14, 2546–2553. [Google Scholar] [CrossRef]
- Grayton, H.M.; Missler, M.; Collier, D.A.; Fernandes, C. Altered social behaviours in neurexin 1α knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS One 2013, 8, e67114. [Google Scholar] [CrossRef] [PubMed]
- Greer, J.M.; Wynshaw-Boris, A. Pten and the brain: sizing up social interaction. Neuron 2006, 50, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019, 51, 431–444. [Google Scholar] [CrossRef]
- Guerra, M.; Medici, V.; Weatheritt, R.; Corvino, V.; Palacios, D.; Geloso, M.C.; et al. Fetal exposure to valproic acid dysregulates the expression of autism-linked genes in the developing cerebellum. Transl Psychiatry 2023, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Morín, J.G.; Santillán, M. Crosstalk dynamics between the circadian clock and the mTORC1 pathway. J Theor Biol 2020, 501, 110360. [Google Scholar] [CrossRef]
- Guissart, C.; Latypova, X.; Rollier, P.; Khan, T.N.; Stamberger, H.; McWalter, K.; et al. Dual Molecular Effects of Dominant RORA Mutations Cause Two Variants of Syndromic Intellectual Disability with Either Autism or Cerebellar Ataxia. Am J Hum Genet 2018, 102, 744–759. [Google Scholar] [CrossRef]
- Gustin, R.M.; Bichell, T.J.; Bubser, M.; Daily, J.; Filonova, I.; Mrelashvili, D.; et al. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis 2010, 39, 283–291. [Google Scholar] [CrossRef]
- Gzielo, K.; Nikiforuk, A. Astroglia in Autism Spectrum Disorder. Int J Mol Sci 2021, 22, 11544. [Google Scholar] [CrossRef] [PubMed]
- H, X.; Y, Y.; X, T.; M, Z.; F, L.; P, X.; et al. Bergmann glia function in granule cell migration during cerebellum development. Molecular neurobiology 2013, 47. [Google Scholar] [CrossRef]
- Hampson, D.R.; Blatt, G.J. Autism spectrum disorders and neuropathology of the cerebellum. Front Neurosci 2015, 9, 420. [Google Scholar] [CrossRef]
- Han, V.X.; Patel, S.; Jones, H.F.; Dale, R.C. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol 2021, 17, 564–579. [Google Scholar] [CrossRef]
- Hanzel, M.; Fernando, K.; Maloney, S.E.; Horn, Z.; Gong, S.; Mätlik, K.; et al. Mice lacking Astn2 have ASD-like behaviors and altered cerebellar circuit properties. Proc Natl Acad Sci U S A 2024, 121, e2405901121. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Sun, J.; Zhong, L.; Baudry, M.; Bi, X. UBE3A deficiency-induced autophagy is associated with activation of AMPK-ULK1 and p53 pathways. Exp Neurol 2023, 363, 114358. [Google Scholar] [CrossRef]
- Harkin, L.F.; Lindsay, S.J.; Xu, Y.; Alzu’bi, A.; Ferrara, A.; Gullon, E.A.; et al. Neurexins 1-3 Each Have a Distinct Pattern of Expression in the Early Developing Human Cerebral Cortex. Cereb Cortex 2017, 27, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.J.; Gamsiz, E.D.; Berkowitz, I.C.; Nagpal, S.; Jerskey, B.A. Genetic variation in the oxytocin receptor gene is associated with a social phenotype in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet 2015, 168, 720–729. [Google Scholar] [CrossRef]
- Hatten, M.E. Adding cognitive connections to the cerebellum. Science 2020, 370, 1411–1412. [Google Scholar] [CrossRef] [PubMed]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol Med 2021, 51, 2260–2273. [Google Scholar] [CrossRef]
- Haws, M.E.; Jaramillo, T.C.; Espinosa, F.; Widman, A.J.; Stuber, G.D.; Sparta, D.R.; et al. PTEN knockdown alters dendritic spine/protrusion morphology, not density. J Comp Neurol 2014, 522, 1171–1190. [Google Scholar] [CrossRef]
- He, X.; Sanders, S.J.; Liu, L.; De Rubeis, S.; Lim, E.T.; Sutcliffe, J.S.; et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 2013, 9, e1003671. [Google Scholar] [CrossRef]
- Herman, G.E.; Butter, E.; Enrile, B.; Pastore, M.; Prior, T.W.; Sommer, A. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am J Med Genet A 2007, 143A, 589–593. [Google Scholar] [CrossRef]
- Ho, A.; Towheed, A.; Luong, S.; Zucker, S.; Fethke, E. Clinical Discordance in Monozygotic Twins With Autism Spectrum Disorder. Cureus 2022, 14, e24813. [Google Scholar] [CrossRef] [PubMed]
- Hoon, M.; Soykan, T.; Falkenburger, B.; Hammer, M.; Patrizi, A.; Schmidt, K.-F.; et al. Neuroligin-4 is localized to glycinergic postsynapses and regulates inhibition in the retina. Proc Natl Acad Sci U S A 2011, 108, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Hooshmandi, M.; Truong, V.T.; Fields, E.; Thomas, R.E.; Wong, C.; Sharma, V.; et al. 4E-BP2-dependent translation in cerebellar Purkinje cells controls spatial memory but not autism-like behaviors. Cell Rep 2021, 35, 109036. [Google Scholar] [CrossRef]
- Hörnberg, H.; Pérez-Garci, E.; Schreiner, D.; Hatstatt-Burklé, L.; Magara, F.; Baudouin, S.; et al. Rescue of oxytocin response and social behaviour in a mouse model of autism. Nature 2020, 584, 252–256. [Google Scholar] [CrossRef]
- Hu, V.W.; Sarachana, T.; Kim, K.S.; Nguyen, A.; Kulkarni, S.; Steinberg, M.E.; et al. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res 2009, 2, 78–97. [Google Scholar] [CrossRef]
- Hu, V.W.; Sarachana, T.; Sherrard, R.M.; Kocher, K.M. Investigation of sex differences in the expression of RORA and its transcriptional targets in the brain as a potential contributor to the sex bias in autism. Mol Autism 2015, 6, 7. [Google Scholar] [CrossRef]
- Hughes, H.K.; R J Moreno, null, and Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain Behav Immun 2023, 108, 245–254. [CrossRef] [PubMed]
- Huntley, G.W.; Gil, O.; Bozdagi, O. The cadherin family of cell adhesion molecules: multiple roles in synaptic plasticity. Neuroscientist 2002, 8, 221–233. [Google Scholar] [CrossRef]
- Hussman, J.P.; Chung, R.-H.; Griswold, A.J.; Jaworski, J.M.; Salyakina, D.; Ma, D.; et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Molecular Autism 2011, 2, 1. [Google Scholar] [CrossRef]
- Igarashi, A.; Itoh, K.; Yamada, T.; Adachi, Y.; Kato, T.; Murata, D.; et al. Nuclear PTEN deficiency causes microcephaly with decreased neuronal soma size and increased seizure susceptibility. J Biol Chem 2018, 293, 9292–9300. [Google Scholar] [CrossRef]
- Ikawa, D.; Makinodan, M.; Iwata, K.; Ohgidani, M.; Kato, T.A.; Yamashita, Y.; et al. Microglia-derived neuregulin expression in psychiatric disorders. Brain Behav Immun 2017, 61, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Inutsuka, A.; Hattori, A.; Yoshida, M.; Takayanagi, Y.; Onaka, T. Cerebellar damage with inflammation upregulates oxytocin receptor expression in Bergmann Glia. Molecular Brain 2024, 17, 41. [Google Scholar] [CrossRef]
- Iweka, C.A.; Seigneur, E.; Hernandez, A.L.; Paredes, S.H.; Cabrera, M.; Blacher, E.; et al. Myeloid deficiency of the intrinsic clock protein BMAL1 accelerates cognitive aging by disrupting microglial synaptic pruning. J Neuroinflammation 2023, 20, 48. [Google Scholar] [CrossRef]
- J, L.; C, C.; S, H.; D, L.; Dy, K.; H, K.; et al. Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Frontiers in cellular neuroscience 2015, 9. [Google Scholar] [CrossRef]
- Jaber, M. Genetic and environmental mouse models of autism reproduce the spectrum of the disease. J Neural Transm (Vienna) 2023, 130, 425–432. [Google Scholar] [CrossRef]
- Jacot-Descombes, S.; Keshav, N.U.; Dickstein, D.L.; Wicinski, B.; Janssen, W.G.M.; Hiester, L.L.; et al. Altered synaptic ultrastructure in the prefrontal cortex of Shank3-deficient rats. Mol Autism 2020, 11, 89. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Radyushkin, K.; Hammerschmidt, K.; Granon, S.; Boretius, S.; Varoqueaux, F.; et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A 2008, 105, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry 2013, 3, e232. [Google Scholar] [CrossRef]
- James, S.J.; Shpyleva, S.; Melnyk, S.; Pavliv, O.; Pogribny, I.P. Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl Psychiatry 2014, 4, e460. [Google Scholar] [CrossRef]
- Jameson, C.; Boulton, K.A.; Silove, N.; Guastella, A.J. Eczema and related atopic diseases are associated with increased symptom severity in children with autism spectrum disorder. Transl Psychiatry 2022, 12, 415. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Holst, M.I.; Liebig, C.; Oberdick, J.; Baader, S.L. Engrailed-2 negatively regulates the onset of perinatal Purkinje cell differentiation. J Comp Neurol 2004, 472, 87–99. [Google Scholar] [CrossRef]
- Jaramillo, T.C.; Escamilla, C.O.; Liu, S.; Peca, L.; Birnbaum, S.G.; Powell, C.M. Genetic background effects in Neuroligin-3 mutant mice: Minimal behavioral abnormalities on C57 background. Autism Res 2018, 11, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.I.; Staels, B.; Brugg, B.; Lemaigre-Dubreuil, Y.; Tedgui, A.; Mariani, J. Age-related phenotypes in the staggerer mouse expand the RORα nuclear receptor’s role beyond the cerebellum. Molecular and Cellular Endocrinology 2002, 186, 1–5. [Google Scholar] [CrossRef]
- Jin, N.; Burkard, M.E. MACROD2, an Original Cause of CIN? Cancer Discovery 2018, 8, 921–923. [Google Scholar] [CrossRef]
- Jin, X.; Simmons, S.K.; Guo, A.; Shetty, A.S.; Ko, M.; Nguyen, L.; et al. In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 2020, 370, eaaz6063. [Google Scholar] [CrossRef]
- Jobson, K.R.; Hoffman, L.J.; Metoki, A.; Popal, H.; Dick, A.S.; Reilly, J.; et al. Language and the Cerebellum: Structural Connectivity to the Eloquent Brain. Neurobiol Lang (Camb) 2024, 5, 652–675. [Google Scholar] [CrossRef]
- Johnson, K.P.; Zarrinnegar, P. Autism Spectrum Disorder and Sleep. Psychiatr Clin North Am 2024, 47, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Barrera, I.; Brothers, S.; Ring, R.; Wahlestedt, C. Oxytocin and social functioning. Dialogues Clin Neurosci 2017, 19, 193–201. [Google Scholar] [CrossRef]
- Jones, R.M.; Cadby, G.; Blangero, J.; Abraham, L.J.; Whitehouse, A.J.O.; Moses, E.K. MACROD2 gene associated with autistic-like traits in a general population sample. Psychiatr Genet 2014, 24, 241–248. [Google Scholar] [CrossRef]
- Journiac, N.; Jolly, S.; Jarvis, C.; Gautheron, V.; Rogard, M.; Trembleau, A.; et al. The nuclear receptor ROR(alpha) exerts a bi-directional regulation of IL-6 in resting and reactive astrocytes. Proc Natl Acad Sci U S A 2009, 106, 21365–21370. [Google Scholar] [CrossRef]
- Joyner, A.L.; Herrup, K.; Auerbach, B.A.; Davis, C.A.; Rossant, J. Subtle cerebellar phenotype in mice homozygous for a targeted deletion of the En-2 homeobox. Science 1991, 251, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L.; Lesser, L.I. Early infantile autism. Pediatr Clin North Am 1958, 5, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Kaplanis, J.; Samocha, K.E.; Wiel, L.; Zhang, Z.; Arvai, K.J.; Eberhardt, R.Y.; et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020, 586, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Kareklas, K.; Teles, M.C.; Dreosti, E.; Oliveira, R.F. Autism-associated gene shank3 is necessary for social contagion in zebrafish. Mol Autism 2023, 14, 23. [Google Scholar] [CrossRef]
- Kasah, S.; Oddy, C.; Basson, M.A. Autism-linked CHD gene expression patterns during development predict multi-organ disease phenotypes. J Anat 2018, 233, 755–769. [Google Scholar] [CrossRef]
- Kasem, E.; Kurihara, T.; Tabuchi, K. Neurexins and neuropsychiatric disorders. Neurosci Res 2018, 127, 53–60. [Google Scholar] [CrossRef]
- Kashii, H.; Kasai, S.; Sato, A.; Hagino, Y.; Nishito, Y.; Kobayashi, T.; et al. Tsc2 mutation rather than Tsc1 mutation dominantly causes a social deficit in a mouse model of tuberous sclerosis complex. Hum Genomics 2023, 17, 4. [Google Scholar] [CrossRef]
- Kawamura, A.; Katayama, Y.; Kakegawa, W.; Ino, D.; Nishiyama, M.; Yuzaki, M.; et al. The autism-associated protein CHD8 is required for cerebellar development and motor function. Cell Rep 2021, 35, 108932. [Google Scholar] [CrossRef]
- Kebschull, J.M.; Richman, E.B.; Ringach, N.; Friedmann, D.; Albarran, E.; Kolluru, S.S.; et al. Cerebellar nuclei evolved by repeatedly duplicating a conserved cell-type set. Science 2020, 370, eabd5059. [Google Scholar] [CrossRef]
- Kelly, E.; Meng, F.; Fujita, H.; Morgado, F.; Kazemi, Y.; Rice, L.C.; et al. Regulation of autism-relevant behaviors by cerebellar-prefrontal cortical circuits. Nat Neurosci 2020, 23, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Audhya, T.; King, P.G.; Sykes, L.K.; Geier, M.R. Evidence of parallels between mercury intoxication and the brain pathology in autism. Acta Neurobiol Exp (Wars) 2012, 72, 113–153. [Google Scholar] [CrossRef]
- Khan, A.J.; Nair, A.; Keown, C.L.; Datko, M.C.; Lincoln, A.J.; Müller, R.-A. Cerebro-cerebellar Resting-State Functional Connectivity in Children and Adolescents with Autism Spectrum Disorder. Biol Psychiatry 2015, 78, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Khoja, S.; Haile, M.T.; Chen, L.Y. Advances in neurexin studies and the emerging role of neurexin-2 in autism spectrum disorder. Front Mol Neurosci 2023, 16, 1125087. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liao, D.; Lau, L.F.; Huganir, R.L. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 1998, 20, 683–691. [Google Scholar] [CrossRef]
- King, M.; Hernandez-Castillo, C.R.; Poldrack, R.A.; Ivry, R.B.; Diedrichsen, J. Functional boundaries in the human cerebellum revealed by a multi-domain task battery. Nat Neurosci 2019, 22, 1371–1378. [Google Scholar] [CrossRef]
- Kiser, D.P.; Popp, S.; Schmitt-Böhrer, A.G.; Strekalova, T.; van den Hove, D.L.; Lesch, K.-P.; et al. Early-life stress impairs developmental programming in Cadherin 13 (CDH13)-deficient mice. Prog Neuropsychopharmacol Biol Psychiatry 2019, 89, 158–168. [Google Scholar] [CrossRef]
- Kloth, A.D.; Badura, A.; Li, A.; Cherskov, A.; Connolly, S.G.; Giovannucci, A.; et al. Cerebellar associative sensory learning defects in five mouse autism models. Elife 2015, 4, e06085. [Google Scholar] [CrossRef]
- Koehnke, J.; Katsamba, P.S.; Ahlsen, G.; Bahna, F.; Vendome, J.; Honig, B.; et al. Splice form dependence of beta-neurexin/neuroligin binding interactions. Neuron 2010, 67, 61–74. [Google Scholar] [CrossRef]
- Koyama, R.; Ikegaya, Y. Microglia in the pathogenesis of autism spectrum disorders. Neurosci Res 2015, 100, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Lee, A.S.; Bayin, N.S.; Stephen, D.N.; Nasef, O.; Lao, Z.; et al. Engrailed transcription factors direct excitatory cerebellar neuron diversity and survival. Development 2024, 151, dev202502. [Google Scholar] [CrossRef]
- Kshetri, R.; Beavers, J.O.; Hyde, R.; Ewa, R.; Schwertman, A.; Porcayo, S.; et al. Behavioral decline in Shank3Δex4-22 mice during early adulthood parallels cerebellar granule cell glutamatergic synaptic changes. Mol Autism 2024, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- Kuemerle, B.; Zanjani, H.; Joyner, A.; Herrup, K. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci 1997, 17, 7881–7889. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; LaVoie, H.A.; DiPette, D.J.; Singh, U.S. Ethanol neurotoxicity in the developing cerebellum: underlying mechanisms and implications. Brain Sci 2013, 3, 941–963. [Google Scholar] [CrossRef]
- Kumar, R.A.; Christian, S.L. Genetics of autism spectrum disorders. Curr Neurol Neurosci Rep 2009, 9, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Kútna, V.; O’Leary, V.B.; Hoschl, C.; Ovsepian, S.V. Cerebellar demyelination and neurodegeneration associated with mTORC1 hyperactivity may contribute to the developmental onset of autism-like neurobehavioral phenotype in a rat model. Autism Res 2022, 15, 791–805. [Google Scholar] [CrossRef]
- Kwon, C.H.; Zhu, X.; Zhang, J.; Knoop, L.L.; Tharp, R.; Smeyne, R.J.; et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 2001, 29, 404–411. [Google Scholar] [CrossRef]
- Kwon, C.-H.; Luikart, B.W.; Powell, C.M.; Zhou, J.; Matheny, S.A.; Zhang, W.; et al. Pten regulates neuronal arborization and social interaction in mice. Neuron 2006, 50, 377–388. [Google Scholar] [CrossRef]
- Kwon, H.-K.; Choi, G.B.; Huh, J.R. Maternal inflammation and its ramifications on fetal neurodevelopment. Trends Immunol 2022, 43, 230–244. [Google Scholar] [CrossRef]
- Lai, E.S.K.; Nakayama, H.; Miyazaki, T.; Nakazawa, T.; Tabuchi, K.; Hashimoto, K.; et al. An Autism-Associated Neuroligin-3 Mutation Affects Developmental Synapse Elimination in the Cerebellum. Front Neural Circuits 2021, 15, 676891. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, J.E.; Bigler, E.D.; Bocian, M.; Coon, H.; Dinh, E.; Dawson, G.; et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A 2006, 140, 2257–2274. [Google Scholar] [CrossRef]
- Lazzarini, G.; Gatta, A.; Miragliotta, V.; Vaglini, F.; Viaggi, C.; Pirone, A. Glial cells are affected more than interneurons by the loss of Engrailed 2 gene in the mouse cerebellum. J Anat 2024, 244, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet 2014, 10, e1004580. [Google Scholar] [CrossRef] [PubMed]
- Ledonne, A.; Mercuri, N.B. On the Modulatory Roles of Neuregulins/ErbB Signaling on Synaptic Plasticity. Int J Mol Sci 2019, 21, 275. [Google Scholar] [CrossRef]
- Lemke, G. Neuregulins in development. Mol Cell Neurosci 1996, 7, 247–262. [Google Scholar] [CrossRef]
- Leto, K.; Arancillo, M.; Becker, E.B.E.; Buffo, A.; Chiang, C.; Ding, B.; et al. Consensus Paper: Cerebellar Development. Cerebellum 2016, 15, 789–828. [Google Scholar] [CrossRef]
- Levy, S.E.; Mandell, D.S.; Schultz, R.T. Autism. Lancet 2009, 374, 1627–1638. [Google Scholar] [CrossRef]
- Li, E.; Li, X.; Huang, J.; Xu, C.; Liang, Q.; Ren, K.; et al. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 2020, 11, 661–679. [Google Scholar] [CrossRef]
- Li, Y.; Fan, L.; Luo, R.; Yang, Z.; Yuan, M.; Zhang, J.; et al. Case Report: De novo Variants of KMT2E Cause O’Donnell-Luria-Rodan Syndrome: Additional Cases and Literature Review. Front Pediatr 2021, 9, 641841. [Google Scholar] [CrossRef]
- Li, Y.-J.; Li, C.-Y.; Li, C.-Y.; Hu, D.-X.; Xv, Z.-B.; Zhang, S.-H.; et al. KMT2E Haploinsufficiency Manifests Autism-Like Behaviors and Amygdala Neuronal Development Dysfunction in Mice. Mol Neurobiol 2023, 60, 1609–1625. [Google Scholar] [CrossRef]
- Licatalosi, D.D.; Yano, M.; Fak, J.J.; Mele, A.; Grabinski, S.E.; Zhang, C.; et al. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 2012, 26, 1626–1642. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Carlström, E.; Råstam, M.; Gillberg, C.; Anckarsäter, H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry 2010, 167, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Frei, J.A.; Kilander, M.B.C.; Shen, W.; Blatt, G.J. A Subset of Autism-Associated Genes Regulate the Structural Stability of Neurons. Front Cell Neurosci 2016, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Lionel, A.C.; Tammimies, K.; Vaags, A.K.; Rosenfeld, J.A.; Ahn, J.W.; Merico, D.; et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum Mol Genet 2014, 23, 2752–2768. [Google Scholar] [CrossRef]
- Lipton, J.O.; Boyle, L.M.; Yuan, E.D.; Hochstrasser, K.J.; Chifamba, F.F.; Nathan, A.; et al. Aberrant Proteostasis of BMAL1 Underlies Circadian Abnormalities in a Paradigmatic mTOR-opathy. Cell Rep 2017, 20, 868–880. [Google Scholar] [CrossRef]
- Liu, D.; Nanclares, C.; Simbriger, K.; Fang, K.; Lorsung, E.; Le, N.; et al. Autistic-like behavior and cerebellar dysfunction in Bmal1 mutant mice ameliorated by mTORC1 inhibition. Mol Psychiatry 2023, 28, 3727–3738. [Google Scholar] [CrossRef]
- Liu, W.-W.; Wei, S.-Z.; Huang, G.-D.; Liu, L.-B.; Gu, C.; Shen, Y.; et al. BMAL1 regulation of microglia-mediated neuroinflammation in MPTP-induced Parkinson’s disease mouse model. FASEB J 2020, 34, 6570–6581. [Google Scholar] [CrossRef]
- Long, J.; Dang, H.; Su, W.; Moneruzzaman, M.; Zhang, H. Interactions between circulating inflammatory factors and autism spectrum disorder: a bidirectional Mendelian randomization study in European population. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef]
- Longart, M.; Liu, Y.; Karavanova, I.; Buonanno, A. Neuregulin-2 is developmentally regulated and targeted to dendrites of central neurons. J Comp Neurol 2004, 472, 156–172. [Google Scholar] [CrossRef]
- LoParo, D.; Waldman, I.D. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: a meta-analysis. Mol Psychiatry 2015, 20, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; et al. Autism spectrum disorder. Nat Rev Dis Primers 2020, 6, 5. [Google Scholar] [CrossRef]
- Lordkipanidze, T.; Dunaevsky, A. Purkinje cell dendrites grow in alignment with Bergmann glia. Glia 2005, 51, 229–234. [Google Scholar] [CrossRef]
- Luikart, B.W.; Schnell, E.; Washburn, E.K.; Bensen, A.L.; Tovar, K.R.; Westbrook, G.L. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci 2011, 31, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Treubert-Zimmermann, U.; Redies, C. Cadherins guide migrating Purkinje cells to specific parasagittal domains during cerebellar development. Mol Cell Neurosci 2004, 25, 138–152. [Google Scholar] [CrossRef]
- Ma, S.Y.; Kwan, K.M. Size anomaly and alteration of GABAergic enzymes expressions in cerebellum of a valproic acid mouse model of autism. Behav Brain Res 2022, 428, 113896. [Google Scholar] [CrossRef] [PubMed]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Maden, M.; Holder, N. The involvement of retinoic acid in the development of the vertebrate central nervous system. Dev Suppl 1991, Suppl 2, 87–94. [Google Scholar] [CrossRef]
- Maden, M.; Holder, N. Retinoic acid and development of the central nervous system. Bioessays 1992, 14, 431–438. [Google Scholar] [CrossRef]
- Man, M.-Q.; Yang, S.; Mauro, T.M.; Zhang, G.; Zhu, T. Link between the skin and autism spectrum disorder. Front Psychiatry 2023, 14, 1265472. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.J.; Hinkley, L.B.N.; Hill, S.S.; Nagarajan, S.S. Sensory processing in autism: a review of neurophysiologic findings. Pediatr Res 2011, 69, 48R–54R. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Krimpenfort, P.; Leung, C.; van der Korput, H.A.G.M.; Trapman, J.; Camenisch, I.; et al. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 2002, 129, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.G.; Chanda, S.; Yang, N.; Janas, J.A.; Valperga, G.; Trotter, J.; et al. Neuroligin-4 Regulates Excitatory Synaptic Transmission in Human Neurons. Neuron 2019, 103, 617–626.e6. [Google Scholar] [CrossRef]
- Martin, L.A.; Goldowitz, D.; Mittleman, G. Repetitive behavior and increased activity in mice with Purkinje cell loss: a model for understanding the role of cerebellar pathology in autism. Eur J Neurosci 2010, 31, 544–555. [Google Scholar] [CrossRef]
- Mashayekhi, F.; Mizban, N.; Bidabadi, E.; Salehi, Z. The association of SHANK3 gene polymorphism and autism. Minerva Pediatr (Torino) 2021, 73, 251–255. [Google Scholar] [CrossRef]
- Masi, A.; DeMayo, M.M.; Glozier, N.; Guastella, A.J. An Overview of Autism Spectrum Disorder, Heterogeneity and Treatment Options. Neurosci Bull 2017, 33, 183–193. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; et al. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int J Mol Sci 2020, 21, 8290. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav Immun 2019, 79, 75–90. [Google Scholar] [CrossRef]
- McDonald, N.M.; Varcin, K.J.; Bhatt, R.; Wu, J.Y.; Sahin, M.; Nelson, C.A.; et al. Early autism symptoms in infants with tuberous sclerosis complex. Autism Res 2017, 10, 1981–1990. [Google Scholar] [CrossRef]
- McKee, C.A.; Polino, A.J.; King, M.W.; Musiek, E.S. Circadian clock protein BMAL1 broadly influences autophagy and endolysosomal function in astrocytes. Proc Natl Acad Sci U S A 2023, 120, e2220551120. [Google Scholar] [CrossRef]
- McKinney, W.S.; Kelly, S.E.; Unruh, K.E.; Shafer, R.L.; Sweeney, J.A.; Styner, M.; et al. Cerebellar Volumes and Sensorimotor Behavior in Autism Spectrum Disorder. Front Integr Neurosci 2022, 16, 821109. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.; Volkmar, F.R. Autism and related disorders. Handb Clin Neurol 2012, 106, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Pévet, P.; Felder-Schmittbuhl, M.-P.; Bailly, Y.; Challet, E. The cerebellum harbors a circadian oscillator involved in food anticipation. J Neurosci 2010, 30, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.A.; Strick, P.L. Cerebellar projections to the prefrontal cortex of the primate. J Neurosci 2001, 21, 700–712. [Google Scholar] [CrossRef]
- Minami, T.; Miyata, E.; Sakamoto, Y.; Yamazaki, H.; Ichida, S. Induction of metallothionein in mouse cerebellum and cerebrum with low-dose thimerosal injection. Cell Biol Toxicol 2010, 26, 143–152. [Google Scholar] [CrossRef]
- Missler, M.; Fernandez-Chacon, R.; Südhof, T.C. The making of neurexins. J Neurochem 1998, 71, 1339–1347. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Shaikh, A.G. Alcohol Toxicity in the Developing Cerebellum. Diagnostics (Basel) 2024, 14, 1415. [Google Scholar] [CrossRef]
- Mitz, A.R.; Boccuto, L.; Thurm, A. Evidence for common mechanisms of pathology between SHANK3 and other genes of Phelan-McDermid syndrome. Clin Genet 2024, 105, 459–469. [Google Scholar] [CrossRef]
- Mizukami, T.; Kohno, T.; Hattori, M. CUB and Sushi multiple domains 3 regulates dendrite development. Neurosci Res 2016, 110, 11–17. [Google Scholar] [CrossRef]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet 2007, 81, 1289–1297. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Morabito, S.; Reese, F.; Rahimzadeh, N.; Miyoshi, E.; Swarup, V. hdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell Rep Method s 2023, 3, 100498. [Google Scholar] [CrossRef]
- Moramarco, F.; McCaffery, P. Retinoic acid regulation of homoeostatic synaptic plasticity and its relationship to cognitive disorders. J Mol Endocrinol 2024, 72, e220177. [Google Scholar] [CrossRef] [PubMed]
- Mordel, J.; Karnas, D.; Pévet, P.; Isope, P.; Challet, E.; Meissl, H. The Output Signal of Purkinje Cells of the Cerebellum and Circadian Rhythmicity. PLOS ONE 2013, 8, e58457. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Howlin, P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res 2009, 53, 852–873. [Google Scholar] [CrossRef]
- Muhammad, T.; Pastore, S.F.; Good, K.; Ausió, J.; Vincent, J.B. Chromatin gatekeeper and modifier CHD proteins in development, and in autism and other neurological disorders. Psychiatr Genet 2023, 33, 213–232. [Google Scholar] [CrossRef]
- Muratori, F.; Tonacci, A.; Billeci, L.; Catalucci, T.; Igliozzi, R.; Calderoni, S.; et al. Olfactory Processing in Male Children with Autism: Atypical Odor Threshold and Identification. J Autism Dev Disord 2017, 47, 3243–3251. [Google Scholar] [CrossRef]
- Murray, M.; Martindale, J.M.; Otallah, S.I. SCN2A- Associated Episodic and Persistent Ataxia with Cerebellar Atrophy: A Case Report. Child Neurol Open 2023, 10, 2329048X231163944. [Google Scholar] [CrossRef]
- Mw, V.; Z, J.; K, M.; Al, J. The Engrailed-2 homeobox gene and patterning of spinocerebellar mossy fiber afferents. Brain research. Developmental brain research 1996, 96. [Google Scholar] [CrossRef]
- Nagappan-Chettiar, S.; Yasuda, M.; Johnson-Venkatesh, E.M.; Umemori, H. The molecular signals that regulate activity-dependent synapse refinement in the brain. Curr Opin Neurobiol 2023, 79, 102692. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Ni, J.; Nonaka, S.; Hayashi, Y. Microglial circadian clock regulation of microglial structural complexity, dendritic spine density and inflammatory response. Neurochem Int 2021, 142, 104905. [Google Scholar] [CrossRef]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; et al. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet 2017, 13, e1006940. [Google Scholar] [CrossRef]
- Nardi, L.; Chhabra, S.; Leukel, P.; Krueger-Burg, D.; Sommer, C.J.; Schmeisser, M.J. Neuroanatomical changes of ionotropic glutamatergic and GABAergic receptor densities in male mice modeling idiopathic and syndromic autism spectrum disorder. Front Psychiatry 2023, 14, 1199097. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, P.P.; Bossolani-Martins, A.L.; Rosan, D.B.A.; Mattos, L.C.; Brandão-Mattos, C.; Fett-Conte, A.C. Single nucleotide polymorphisms in the CNTNAP2 gene in Brazilian patients with autistic spectrum disorder. Genet Mol Res 2016, 15. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Lehr, A.W.; Roche, K.W. Neuroligins and Neurodevelopmental Disorders: X-Linked Genetics. Front Synaptic Neurosci 2020, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, J.; Vles, J.S.H.; Aldenkamp, A.P. Neurodevelopmental delay in children exposed to antiepileptic drugs in utero: a critical review directed at structural study-bias. J Neurol Sci 2008, 271, 1–14. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp Neurol 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Nimbley, E.; Golds, L.; Sharpe, H.; Gillespie-Smith, K.; Duffy, F. Sensory processing and eating behaviours in autism: A systematic review. Eur Eat Disord Rev 2022, 30, 538–559. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H. Learning-induced structural plasticity in the cerebellum. Int Rev Neurobiol 2014, 117, 1–19. [Google Scholar] [CrossRef]
- Nóbrega, I. de S.; Teles E Silva, A.L.; Yokota-Moreno, B.Y.; Sertié, A.L. The Importance of Large-Scale Genomic Studies to Unravel Genetic Risk Factors for Autism. Int J Mol Sci 2024, 25, 5816. [Google Scholar] [CrossRef] [PubMed]
- Obst-Pernberg, K.; Redies, C. Cadherins and synaptic specificity. J Neurosci Res 1999, 58, 130–138. [Google Scholar] [CrossRef]
- O’Donnell-Luria, A.H.; Pais, L.S.; Faundes, V.; Wood, J.C.; Sveden, A.; Luria, V.; et al. Heterozygous Variants in KMT2E Cause a Spectrum of Neurodevelopmental Disorders and Epilepsy. The American Journal of Human Genetics 2019, 104, 1210–1222. [Google Scholar] [CrossRef]
- Ohtsuki, G.; Piochon, C.; Adelman, J.P.; Hansel, C. SK2 channel modulation contributes to compartment-specific dendritic plasticity in cerebellar Purkinje cells. Neuron 2012, 75, 108–120. [Google Scholar] [CrossRef]
- Ohtsuki, G.; Piochon, C.; Adelman, J.P.; Hansel, C. SK2 channel modulation contributes to compartment-specific dendritic plasticity in cerebellar Purkinje cells. Neuron 2012, 75, 108–120. [Google Scholar] [CrossRef]
- Okada, N.J.; Liu, J.; Tsang, T.; Nosco, E.; McDonald, N.M.; Cummings, K.K.; et al. Atypical cerebellar functional connectivity at 9 months of age predicts delayed socio-communicative profiles in infants at high and low risk for autism. J Child Psychol Psychiatry 2022, 63, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Hatsukawa, Y.; Shimojima, K.; Yamamoto, T. Submicroscopic deletion in 7q31 encompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder and PHPV. Am J Med Genet A 2011, 155A, 1568–1573. [Google Scholar] [CrossRef]
- Olexová, L.; Štefánik, P.; Kršková, L. Increased anxiety-like behaviour and altered GABAergic system in the amygdala and cerebellum of VPA rats - An animal model of autism. Neurosci Lett 2016, 629, 9–14. [Google Scholar] [CrossRef]
- Olson, I.R.; Hoffman, L.J.; Jobson, K.R.; Popal, H.S.; Wang, Y. Little brain, little minds: The big role of the cerebellum in social development. Dev Cogn Neurosci 2023, 60, 101238. [Google Scholar] [CrossRef]
- Orlova, K.A.; Crino, P.B. The tuberous sclerosis complex. Ann N Y Acad Sci 2010, 1184, 87–105. [Google Scholar] [CrossRef]
- Otazu, G.H.; Li, Y.; Lodato, Z.; Elnasher, A.; Keever, K.M.; Li, Y.; et al. Neurodevelopmental malformations of the cerebellum and neocortex in the Shank3 and Cntnap2 mouse models of autism. Neurosci Lett 2021, 765, 136257. [Google Scholar] [CrossRef]
- Ou, G.-Y.; Lin, W.-W.; Zhao, W.-J. Neuregulins in Neurodegenerative Diseases. Front Aging Neurosci 2021, 13, 662474. [Google Scholar] [CrossRef]
- Ozaki, M. Neuregulins and the shaping of synapses. Neuroscientist 2001, 7, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Pagani, M.; Barsotti, N.; Bertero, A.; Trakoshis, S.; Ulysse, L.; Locarno, A.; et al. mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat Commun 2021, 12, 6084. [Google Scholar] [CrossRef] [PubMed]
- Pais, L.; Rodan, L.; O’Donnell-Luria, A. KMT2E-Related Neurodevelopmental Disorder. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., et al., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK602945/ (accessed on 26 July 2024).
- Pangrazzi, L.; Genovesi, S.; Balasco, L.; Cerilli, E.; Robol, C.; Zunino, G.; et al. Immune dysfunction in the cerebellum of mice lacking the autism candidate gene Engrailed 2. J Neuroimmunol 2022, 367, 577870. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci 2020, 43, 608–621. [Google Scholar] [CrossRef]
- Park, J.; Chung, W.-S. Astrocyte-dependent circuit remodeling by synapse phagocytosis. Curr Opin Neurobiol 2023, 81, 102732. [Google Scholar] [CrossRef]
- Pavone, P.; Corsello, G.; Marino, S.D.; Ruggieri, M.; Falsaperla, R. 7q31.32 partial duplication: First report of a child with dysmorphism, autistic spectrum disorder, moderate intellectual disability and, epilepsy. Literature review. Epilepsy Res 2019, 158, 106223. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011, 147, 235–246. [Google Scholar] [CrossRef]
- Peñagarikano, O.; Geschwind, D.H. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med 2012, 18, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Peñagarikano, O.; Lázaro, M.T.; Lu, X.-H.; Gordon, A.; Dong, H.; Lam, H.A.; et al. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med 2015, 7, 271ra8. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Ten Brinke, M.M.; Stedehouder, J.; Reinelt, C.M.; Wu, B.; Zhou, H.; et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat Commun 2016, 7, 12627. [Google Scholar] [CrossRef]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and Microglia and Their Potential Link with Autism Spectrum Disorders. Front Cell Neurosci 2016, 10, 21. [Google Scholar] [CrossRef]
- Phelan, K.; Rogers, R.C.; Boccuto, L. Phelan-McDermid Syndrome-SHANK3 Related. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1198/ (accessed on 10 January 2025).
- Pinato, L.; Galina Spilla, C.S.; Markus, R.P.; da Silveira Cruz-Machado, S. Dysregulation of Circadian Rhythms in Autism Spectrum Disorders. Curr Pharm Des 2019, 25, 4379–4393. [Google Scholar] [CrossRef]
- Poliak, S.; Gollan, L.; Martinez, R.; Custer, A.; Einheber, S.; Salzer, J.L.; et al. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999, 24, 1037–1047. [Google Scholar] [CrossRef]
- Qian, Z.; Zhang, R.; Zhou, J.; Sun, S.; Di, Y.; Ren, W.; et al. RNA-Seq data on prefrontal cortex in valproic acid model of autism and control rats. Data Brief 2018, 18, 787–789. [Google Scholar] [CrossRef]
- Qin, L.; Liu, Z.; Guo, S.; Han, Y.; Wang, X.; Ren, W.; et al. Astrocytic Neuroligin-3 influences gene expression and social behavior, but is dispensable for synapse number. Mol Psychiatry. 2024. [CrossRef] [PubMed]
- Qin, Y.; Du, Y.; Chen, L.; Liu, Y.; Xu, W.; Liu, Y.; et al. A recurrent SHANK1 mutation implicated in autism spectrum disorder causes autistic-like core behaviors in mice via downregulation of mGluR1-IP3R1-calcium signaling. Mol Psychiatry 2022, 27, 2985–2998. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, X.-Y.; Liu, Y.; Ma, Z.; Tao, S.; Li, Y.; et al. Downregulation of mGluR1-mediated signaling underlying autistic-like core symptoms in Shank1 P1812L-knock-in mice. Transl Psychiatry 2023, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, Y.; Bai, Y.; Shi, J.; Cui, H.; Gu, Y.; et al. SHANK1 polymorphisms and SNP-SNP interactions among SHANK family: A possible cue for recognition to autism spectrum disorder in infant age. Autism Res 2019, 12, 375–383. [Google Scholar] [CrossRef]
- Rademacher, S.; Eickholt, B.J. PTEN in Autism and Neurodevelopmental Disorders. Cold Spring Harb Perspect Med 2019, 9, a036780. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Weber, J.; Labin, E.; Lai, C.; Prieto, A.L. Developmental expression of Neuregulin-3 in the rat central nervous system. J Comp Neurol 2019, 527, 797–817. [Google Scholar] [CrossRef]
- Rahman, M.A.; Orfali, R.; Dave, N.; Lam, E.; Naguib, N.; Nam, Y.-W.; et al. KCa 2.2 (KCNN2): A physiologically and therapeutically important potassium channel. J Neurosci Res 2023, 101, 1699–1710. [Google Scholar] [CrossRef]
- Rath, M.F.; Rohde, K.; Møller, M. Circadian oscillations of molecular clock components in the cerebellar cortex of the rat. Chronobiol Int 2012, 29, 1289–1299. [Google Scholar] [CrossRef]
- Redies, C.; Hertel, N.; Hübner, C.A. Cadherins and neuropsychiatric disorders. Brain Res 2012, 1470, 130–144. [Google Scholar] [CrossRef]
- Redies, C.; Neudert, F.; Lin, J. Cadherins in cerebellar development: translation of embryonic patterning into mature functional compartmentalization. Cerebellum 2011, 10, 393–408. [Google Scholar] [CrossRef]
- Reichova, A.; Bacova, Z.; Bukatova, S.; Kokavcova, M.; Meliskova, V.; Frimmel, K.; et al. Abnormal neuronal morphology and altered synaptic proteins are restored by oxytocin in autism-related SHANK3 deficient model. Mol Cell Endocrinol 2020, 518, 110924. [Google Scholar] [CrossRef] [PubMed]
- Reissner, C.; Klose, M.; Fairless, R.; Missler, M. Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A 2008, 105, 15124–15129. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol 2013, 14, 213. [Google Scholar] [CrossRef]
- Reith, R.M.; McKenna, J.; Wu, H.; Hashmi, S.S.; Cho, S.-H.; Dash, P.K.; et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiology of Disease 2013, 51, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Sherrard, R.M. Cerebellum and neurodevelopmental disorders: RORα is a unifying force. Front Cell Neurosci 2023, 17, 1108339. [Google Scholar] [CrossRef]
- Robertson, C.E.; Baron-Cohen, S. Sensory perception in autism. Nat Rev Neurosci 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Robinson-Agramonte, M. de L. A.; Noris García, E.; Fraga Guerra, J.; Vega Hurtado, Y.; Antonucci, N.; Semprún-Hernández, N.; et al. Immune Dysregulation in Autism Spectrum Disorder: What Do We Know about It? Int J Mol Sci 2022, 23, 3033. [Google Scholar] [CrossRef] [PubMed]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet 2014, 22, 171–178. [Google Scholar] [CrossRef]
- Rogers, T.D.; Dickson, P.E.; McKimm, E.; Heck, D.H.; Goldowitz, D.; Blaha, C.D.; et al. Reorganization of circuits underlying cerebellar modulation of prefrontal cortical dopamine in mouse models of autism spectrum disorder. Cerebellum 2013, 12, 547–556. [Google Scholar] [CrossRef]
- Rojo, D.; Dal Cengio, L.; Badner, A.; Kim, S.; Sakai, N.; Greene, J.; et al. BMAL1 loss in oligodendroglia contributes to abnormal myelination and sleep. Neuron 2023, 111, 3604–3618.e11. [Google Scholar] [CrossRef]
- Roostaei, T.; Nazeri, A.; Sahraian, M.A.; Minagar, A. The Human Cerebellum: A Review of Physiologic Neuroanatomy. Neurologic Clinics 2014, 32, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Law, J.K.; Yenokyan, G.; McGready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med 2009, 163, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Rosina, E.; Battan, B.; Siracusano, M.; Di Criscio, L.; Hollis, F.; Pacini, L.; et al. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl Psychiatry 2019, 9, 50. [Google Scholar] [CrossRef]
- Roy, B.; Amemasor, E.; Hussain, S.; Castro, K. UBE3A: The Role in Autism Spectrum Disorders (ASDs) and a Potential Candidate for Biomarker Studies and Designing Therapeutic Strategies. Diseases 2023, 12, 7. [Google Scholar] [CrossRef]
- Sacco, R.; Gabriele, S.; Persico, A.M. Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Res 2015, 234, 239–251. [Google Scholar] [CrossRef]
- Sadakata, T.; Furuichi, T. Developmentally regulated Ca2+-dependent activator protein for secretion 2 (CAPS2) is involved in BDNF secretion and is associated with autism susceptibility. Cerebellum 2009, 8, 312–322. [Google Scholar] [CrossRef]
- Sadakata, T.; Shinoda, Y.; Oka, M.; Sekine, Y.; Furuichi, T. Autistic-like behavioral phenotypes in a mouse model with copy number variation of the CAPS2/CADPS2 gene. FEBS Lett 2013, 587, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Sadakata, T.; Shinoda, Y.; Sato, A.; Iguchi, H.; Ishii, C.; Matsuo, M.; et al. Mouse models of mutations and variations in autism spectrum disorder-associated genes: mice expressing Caps2/Cadps2 copy number and alternative splicing variants. Int J Environ Res Public Health 2013, 10, 6335–6353. [Google Scholar] [CrossRef]
- Sajdel-Sulkowska, E.M.; Xu, M.; Koibuchi, N. Increase in cerebellar neurotrophin-3 and oxidative stress markers in autism. Cerebellum 2009, 8, 366–372. [Google Scholar] [CrossRef]
- Sajdel-Sulkowska, E.M.; Xu, M.; McGinnis, W.; Koibuchi, N. Brain region-specific changes in oxidative stress and neurotrophin levels in autism spectrum disorders (ASD). Cerebellum 2011, 10, 43–48. [Google Scholar] [CrossRef]
- Salenius, K.; Väljä, N.; Thusberg, S.; Iris, F.; Ladd-Acosta, C.; Roos, C.; et al. Exploring autism spectrum disorder and co-occurring trait associations to elucidate multivariate genetic mechanisms and insights. BMC Psychiatry 2024, 24, 934. [Google Scholar] [CrossRef]
- Salman, E.D.; Kadlubar, S.A.; Falany, C.N. Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain. Drug Metab Dispos 2009, 37, 706–709. [Google Scholar] [CrossRef]
- Samanta, D. An Updated Review of Tuberous Sclerosis Complex-Associated Autism Spectrum Disorder. Pediatr Neurol 2020, 109, 4–11. [Google Scholar] [CrossRef]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic effects and biomarkers of lead exposure: a review. Rev Environ Health 2009, 24, 15–45. [Google Scholar] [CrossRef] [PubMed]
- Sands, T.T.; Miceli, F.; Lesca, G.; Beck, A.E.; Sadleir, L.G.; Arrington, D.K.; et al. Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann Neurol 2019, 86, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Huynh, T.N.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Sato, A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol Disord Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 2012, 3, 1292. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am J Hum Genet 2012, 90, 879–887. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Sayad, A.; Noroozi, R.; Omrani, M.D.; Taheri, M.; Ghafouri-Fard, S. Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab Brain Dis 2017, 32, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Schaffer, L.S.; Breunig, S.; Lawrence, J.M.; Foote, I.F.; Grotzinger, A.D. Characterizing Genetic Pathways Unique to Autism Spectrum Disorder at Multiple Levels of Biological Analysis. medRxiv 2024. [Google Scholar] [CrossRef]
- Schaffner, S.L.; Lussier, A.A.; Baker, J.A.; Goldowitz, D.; Hamre, K.M.; Kobor, M.S. Neonatal Alcohol Exposure in Mice Induces Select Differentiation- and Apoptosis-Related Chromatin Changes Both Independent of and Dependent on Sex. Front Genet 2020, 11, 35. [Google Scholar] [CrossRef]
- Schibler, U.; Gotic, I.; Saini, C.; Gos, P.; Curie, T.; Emmenegger, Y.; et al. Clock-Talk: Interactions between Central and Peripheral Circadian Oscillators in Mammals. Cold Spring Harb Symp Quant Biol 2015, 80, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D. The cerebellum and cognition. Neurosci Lett 2019, 688, 62–75. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Guell, X.; Stoodley, C.J.; Halko, M.A. The Theory and Neuroscience of Cerebellar Cognition. Annu Rev Neurosci 2019, 42, 337–364. [Google Scholar] [CrossRef]
- Scuderi, C.; Verkhratsky, A. The role of neuroglia in autism spectrum disorders. Prog Mol Biol Transl Sci 2020, 173, 301–330. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Singh, A.S.; Sinha, S.; Chatterjee, A.; Ahmed, S.; Ghosh, S.; et al. Family-based studies indicate association of Engrailed 2 gene with autism in an Indian population. Genes Brain Behav 2010, 9, 248–255. [Google Scholar] [CrossRef]
- Shan, D.; Song, Y.; Zhang, Y.; Ho, C.W.; Xia, W.; Li, Z.; et al. Neurexin dysfunction in neurodevelopmental and neuropsychiatric disorders: a PRIMSA-based systematic review through iPSC and animal models. Front. Behav. Neurosci. 2024, 18. [Google Scholar] [CrossRef]
- Shaw, K.A.; Williams, S.; Patrick, M.E.; Valencia-Prado, M.; Durkin, M.S.; Howerton, E.M.; et al. Prevalence and Early Identification of Autism Spectrum Disorder Among Children Aged 4 and 8 Years - Autism and Developmental Disabilities Monitoring Network, 16 Sites, United States, 2022. MMWR Surveill Summ 2025, 74, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-P.; Li, W.; Pei, L.-Z.; Yin, J.; Xie, S.-T.; Li, H.-Z.; et al. Oxytocin Receptor in Cerebellar Purkinje Cells Does Not Engage in Autism-Related Behaviors. Cerebellum 2023, 22, 888–904. [Google Scholar] [CrossRef]
- Shimizu, A.; Asakawa, S.; Sasaki, T.; Yamazaki, S.; Yamagata, H.; Kudoh, J.; et al. A novel giant gene CSMD3 encoding a protein with CUB and sushi multiple domains: a candidate gene for benign adult familial myoclonic epilepsy on human chromosome 8q23.3-q24.1. Biochem Biophys Res Commun 2003, 309, 143–154. [Google Scholar] [CrossRef]
- Sidhu, N.; Wong, Z.; Bennett, A.E.; Souders, M.C. Sleep Problems in Autism Spectrum Disorder. Pediatr Clin North Am 2024, 71, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Mishra, A.; Lin, H.; Lorsung, E.; Le, N.; Tin, S.; et al. Haploinsufficiency of a Circadian Clock Gene Bmal1 (Arntl or Mop3) Causes Brain-Wide mTOR Hyperactivation and Autism-like Behavioral Phenotypes in Mice. Int J Mol Sci 2022, 23, 6317. [Google Scholar] [CrossRef]
- Sivalingam, A.M.; Pandian, A. Cerebellar Roles in Motor and Social Functions and Implications for ASD. Cerebellum 2024. [CrossRef]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; et al. Regional alterations in purkinje cell density in patients with autism. PLoS One 2014, 9, e81255. [Google Scholar] [CrossRef] [PubMed]
- Skelton, P.D.; Poquerusse, J.; Salinaro, J.R.; Li, M.; Luikart, B.W. Activity-dependent dendritic elaboration requires Pten. Neurobiol Dis 2020, 134, 104703. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Song, W.; Li, Q.; Wang, T.; Li, Y.; Fan, T.; Zhang, J.; et al. Putative complement control protein CSMD3 dysfunction impairs synaptogenesis and induces neurodevelopmental disorders. Brain Behav Immun 2022, 102, 237–250. [Google Scholar] [CrossRef]
- Speed, H.E.; Masiulis, I.; Gibson, J.R.; Powell, C.M. Increased Cortical Inhibition in Autism-Linked Neuroligin-3R451C Mice Is Due in Part to Loss of Endocannabinoid Signaling. PLoS One 2015, 10, e0140638. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, F.; Haneklaus, M.; Kivisild, T.; Livesey, F.J. Loss of CNTNAP2 Alters Human Cortical Excitatory Neuron Differentiation and Neural Network Development. Biol Psychiatry 2023, 94, 780–791. [Google Scholar] [CrossRef]
- Steinthorsdottir, V.; Stefansson, H.; Ghosh, S.; Birgisdottir, B.; Bjornsdottir, S.; Fasquel, A.C.; et al. Multiple novel transcription initiation sites for NRG1. Gene 2004, 342, 97–105. [Google Scholar] [CrossRef]
- Stogsdill, J.A.; Ramirez, J.; Liu, D.; Kim, Y.H.; Baldwin, K.T.; Enustun, E.; et al. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 2017, 551, 192–197. [Google Scholar] [CrossRef]
- Stoodley, C.J. The Cerebellum and Neurodevelopmental Disorders. Cerebellum 2016, 15, 34–37. [Google Scholar] [CrossRef]
- Stoodley, C.J.; D’Mello, A.M.; Ellegood, J.; Jakkamsetti, V.; Liu, P.; Nebel, M.B.; et al. Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci. 2017, 20, 1744–1751. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Schmahmann, J.D. Evidence for topographic organization in the cerebellum of motor control versus cognitive and affective processing. Cortex 2010, 46, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J.; Tsai, P.T. Adaptive Prediction for Social Contexts: The Cerebellar Contribution to Typical and Atypical Social Behaviors. Annu Rev Neurosci 2021, 44, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Strick, P.L.; Dum, R.P.; Fiez, J.A. Cerebellum and nonmotor function. Annu Rev Neurosci 2009, 32, 413–434. [Google Scholar] [CrossRef]
- Sudarov, A.; Joyner, A.L. Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev 2007, 2, 26. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Baudry, M.; Bi, X. SK2 channel regulation of neuronal excitability, synaptic transmission, and brain rhythmic activity in health and diseases. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118834. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Y.; Hao, X.; Baudry, M.; Bi, X. Lack of UBE3A-Mediated Regulation of Synaptic SK2 Channels Contributes to Learning and Memory Impairment in the Female Mouse Model of Angelman Syndrome. Neural Plast 2022, 2022, 3923384. [Google Scholar] [CrossRef]
- Sun, J.; Liu, Y.; Zhu, G.; Cato, C.; Hao, X.; Qian, L.; et al. PKA and Ube3a regulate SK2 channel trafficking to promote synaptic plasticity in hippocampus: Implications for Angelman Syndrome. Sci Rep 2020, 10, 9824. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, G.; Liu, Y.; Standley, S.; Ji, A.; Tunuguntla, R.; et al. UBE3A Regulates Synaptic Plasticity and Learning and Memory by Controlling SK2 Channel Endocytosis. Cell Rep 2015, 12, 449–461. [Google Scholar] [CrossRef]
- Sun, M.; You, H.; Hu, X.; Luo, Y.; Zhang, Z.; Song, Y.; et al. Microglia-Astrocyte Interaction in Neural Development and Neural Pathogenesis. Cells 2023, 12, 1942. [Google Scholar] [CrossRef]
- Sungur, A.Ö.; Schwarting, R.K.W.; Wöhr, M. Early communication deficits in the Shank1 knockout mouse model for autism spectrum disorder: Developmental aspects and effects of social context. Autism Res 2016, 9, 696–709. [Google Scholar] [CrossRef]
- Sungur, A.Ö.; Schwarting, R.K.W.; Wöhr, M. Behavioral phenotypes and neurobiological mechanisms in the Shank1 mouse model for autism spectrum disorder: A translational perspective. Behav Brain Res 2018, 352, 46–61. [Google Scholar] [CrossRef]
- Sungur, A.Ö.; Vörckel, K.J.; Schwarting, R.K.W.; Wöhr, M. Repetitive behaviors in the Shank1 knockout mouse model for autism spectrum disorder: developmental aspects and effects of social context. J Neurosci Methods 2014, 234, 92–100. [Google Scholar] [CrossRef]
- Szabó, J.; Mlynár, M.; Feješ, A.; Renczés, E.; Borbélyová, V.; Ostatníková, D.; et al. Intranasal oxytocin in a genetic animal model of autism. Mol Psychiatry 2024, 29, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci 2016, 19, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Takahashi, J.S.; Hong, H.-K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 2008, 9, 764–775. [Google Scholar] [CrossRef]
- Takeo, Y.H.; Kakegawa, W.; Miura, E.; Yuzaki, M. RORα Regulates Multiple Aspects of Dendrite Development in Cerebellar Purkinje Cells In Vivo. J Neurosci 2015, 35, 12518–12534. [Google Scholar] [CrossRef] [PubMed]
- Talmage, D.A.; Role, L.W. Multiple personalities of neuregulin gene family members. J Comp Neurol 2004, 472, 134–139. [Google Scholar] [CrossRef]
- Tan, G.C.Y.; Doke, T.F.; Ashburner, J.; Wood, N.W.; Frackowiak, R.S.J. Normal variation in fronto-occipital circuitry and cerebellar structure with an autism-associated polymorphism of CNTNAP2. Neuroimage 2010, 53, 1030–1042. [Google Scholar] [CrossRef]
- Tanaka, M. Dendrite formation of cerebellar Purkinje cells. Neurochem Res 2009, 34, 2078–2088. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Tantra, M.; Guo, L.; Kim, J.; Zainolabidin, N.; Eulenburg, V.; Augustine, G.J.; et al. Conditional deletion of Cadherin 13 perturbs Golgi cells and disrupts social and cognitive behaviors. Genes Brain Behav 2018, 17, e12466. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alvarez, V.A.; Ridenour, D.A.; Kwiatkowski, D.J.; Sabatini, B.L. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 2005, 8, 1727–1734. [Google Scholar] [CrossRef]
- ten Donkelaar, H.J.; Lammens, M.; Wesseling, P.; Thijssen, H.O.M.; Renier, W.O. Development and developmental disorders of the human cerebellum. J Neurol 2003, 250, 1025–1036. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Tian, R.; Li, Y.; Zhao, H.; Lyu, W.; Zhao, J.; Wang, X.; et al. Modeling SHANK3-associated autism spectrum disorder in Beagle dogs via CRISPR/Cas9 gene editing. Mol Psychiatry 2023, 28, 3739–3750. [Google Scholar] [CrossRef]
- Tonge, B.J.; Dissanayake, C.; Brereton, A.V. Autism: fifty years on from Kanner. J Paediatr Child Health 1994, 30, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Waga, C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev 2013, 35, 106–110. [Google Scholar] [CrossRef]
- Uemura, T.; Lee, S.-J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; et al. Trans-Synaptic Interaction of GluRδ2 and Neurexin through Cbln1 Mediates Synapse Formation in the Cerebellum. Cell 2010, 141, 1068–1079. [Google Scholar] [CrossRef]
- Vaags, A.K.; Lionel, A.C.; Sato, D.; Goodenberger, M.; Stein, Q.P.; Curran, S.; et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet 2012, 90, 133–141. [Google Scholar] [CrossRef]
- Valles, S.L.; Singh, S.K.; Campos-Campos, J.; Colmena, C.; Campo-Palacio, I.; Alvarez-Gamez, K.; et al. Functions of Astrocytes under Normal Conditions and after a Brain Disease. Int J Mol Sci 2023, 24, 8434. [Google Scholar] [CrossRef]
- van der Heijden, M.E.; Gill, J.S.; Sillitoe, R.V. Abnormal Cerebellar Development in Autism Spectrum Disorders. Dev Neurosci 2021, 43, 181–190. [Google Scholar] [CrossRef]
- Van Overwalle, F.; Pu, M.; Ma, Q.; Li, M.; Haihambo, N.; Baetens, K.; et al. The Involvement of the Posterior Cerebellum in Reconstructing and Predicting Social Action Sequences. Cerebellum 2022, 21, 733–741. [Google Scholar] [CrossRef]
- Varea, O.; Martin-de-Saavedra, M.D.; Kopeikina, K.J.; Schürmann, B.; Fleming, H.J.; Fawcett-Patel, J.M.; et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc Natl Acad Sci U S A 2015, 112, 6176–6181. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; et al. Neuroligins determine synapse maturation and function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef]
- Vashisth, S.; Chahrour, M.H. Genomic strategies to untangle the etiology of autism: A primer. Autism Res 2023, 16, 31–39. [Google Scholar] [CrossRef]
- Vatsa, N.; Jana, N.R. UBE3A and Its Link With Autism. Front Mol Neurosci 2018, 11, 448. [Google Scholar] [CrossRef]
- Veenstra-VanderWeele, J.; Cook, E.H. Molecular genetics of autism spectrum disorder. Mol Psychiatry 2004, 9, 819–832. [Google Scholar] [CrossRef]
- Verly, M.; Verhoeven, J.; Zink, I.; Mantini, D.; Peeters, R.; Deprez, S.; et al. Altered functional connectivity of the language network in ASD: role of classical language areas and cerebellum. Neuroimage Clin 2014, 4, 374–382. [Google Scholar] [CrossRef]
- Volpe, J.J. Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J Child Neurol 2009, 24, 1085–1104. [Google Scholar] [CrossRef]
- Volpe, J.J. Commentary - Cerebellar underdevelopment in the very preterm infant: Important and underestimated source of cognitive deficits. J Neonatal Perinatal Med 2021, 14, 451–456. [Google Scholar] [CrossRef]
- Vorstman, J.A.S.; Freitag, C.M.; Persico, A.M. From Genes to Therapy in Autism Spectrum Disorder. Genes (Basel) 2022, 13, 1377. [Google Scholar] [CrossRef]
- Vullhorst, D.; Bloom, M.S.; Akella, N.; Buonanno, A. ER-PM Junctions on GABAergic Interneurons Are Organized by Neuregulin 2/VAP Interactions and Regulated by NMDA Receptors. Int J Mol Sci 2023, 24, 2908. [Google Scholar] [CrossRef]
- Vullhorst, D.; Buonanno, A. NMDA Receptors Regulate Neuregulin 2 Binding to ER-PM Junctions and Ectodomain Release by ADAM10 [corrected]. Mol Neurobiol 2019, 56, 8345–8363. [Google Scholar] [CrossRef]
- Vullhorst, D.; Mitchell, R.M.; Keating, C.; Roychowdhury, S.; Karavanova, I.; Tao-Cheng, J.-H.; et al. A negative feedback loop controls NMDA receptor function in cortical interneurons via neuregulin 2/ErbB4 signalling. Nat Commun 2015, 6, 7222. [Google Scholar] [CrossRef]
- Wamsley, B.; Bicks, L.; Cheng, Y.; Kawaguchi, R.; Quintero, D.; Margolis, M.; et al. Molecular cascades and cell type-specific signatures in ASD revealed by single-cell genomics. Science 2024, 384, eadh2602. [Google Scholar] [CrossRef]
- Wan, L.; Ai, J.-Q.; Yang, C.; Jiang, J.; Zhang, Q.-L.; Luo, Z.-H.; et al. Expression of the Excitatory Postsynaptic Scaffolding Protein, Shank3, in Human Brain: Effect of Age and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Wang, C.; Derderian, K.D.; Hamada, E.; Zhou, X.; Nelson, A.D.; Kyoung, H.; et al. Impaired cerebellar plasticity hypersensitizes sensory reflexes in SCN2A-associated ASD. Neuron 2024, 112, 1444–1455.e5. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Y.-H.; Wang, Y.; Blatt, G.; Yuan, X.-B. Segregated expressions of autism risk genes Cdh11 and Cdh9 in autism-relevant regions of developing cerebellum. Mol Brain 2019, 12, 40. [Google Scholar] [CrossRef]
- Wang, F.; Xu, Q.; Wang, W.; Takano, T.; Nedergaard, M. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc Natl Acad Sci U S A 2012, 109, 7911–7916. [Google Scholar] [CrossRef]
- Wang, J.; Gong, J.; Li, L.; Chen, Y.; Liu, L.; Gu, H.; et al. Neurexin gene family variants as risk factors for autism spectrum disorder. Autism Res 2018, 11, 37–43. [Google Scholar] [CrossRef]
- Wang, R.; Tan, J.; Guo, J.; Zheng, Y.; Han, Q.; So, K.-F.; et al. Aberrant Development and Synaptic Transmission of Cerebellar Cortex in a VPA Induced Mouse Autism Model. Front Cell Neurosci 2018, 12, 500. [Google Scholar] [CrossRef]
- Wang, S.S.-H.; Kloth, A.D.; Badura, A. The cerebellum, sensitive periods, and autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef]
- Wang, T.; Morgan, J.I. The Purkinje cell degeneration (pcd) mouse: an unexpected molecular link between neuronal degeneration and regeneration. Brain Res 2007, 1140, 26–40. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Q.; Zuo, C.; Zhao, L.; Hao, L. Longitudinal Changes of Cerebellar Thickness in Autism Spectrum Disorder. Neurosci Lett 2020, 728, 134949. [Google Scholar] [CrossRef]
- Watanabe, T.; Kano, M. Molecular and cellular mechanisms of developmental synapse elimination in the cerebellum: Involvement of autism spectrum disorder-related genes. Proc Jpn Acad Ser B Phys Biol Sci 2024, 100, 508–523. [Google Scholar] [CrossRef]
- Watanabe, T.; Otowa, T.; Abe, O.; Kuwabara, H.; Aoki, Y.; Natsubori, T.; et al. Oxytocin receptor gene variations predict neural and behavioral response to oxytocin in autism. Soc Cogn Affect Neurosci 2017, 12, 496–506. [Google Scholar] [CrossRef]
- Weissberg, O.; Elliott, E. The Mechanisms of CHD8 in Neurodevelopment and Autism Spectrum Disorders. Genes (Basel) 2021, 12, 1133. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Choudhury, G.R.; He, R.; Yang, T.; Liu, R.; et al. Astroglial PTEN Loss Disrupts Neuronal Lamination by Dysregulating Radial Glia-guided Neuronal Migration. Aging Dis 2013, 4, 113–126. [Google Scholar]
- Weuring, W.; Geerligs, J.; Koeleman, B.P.C. Gene Therapies for Monogenic Autism Spectrum Disorders. Genes 2021, 12, 1667. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of cerebellar basket and stellate cells in autism: evidence for a late developmental loss of Purkinje cells. J Neurosci Res 2009, 87, 2245–2254. [Google Scholar] [CrossRef]
- Wiebe, S.; Nagpal, A.; Truong, V.T.; Park, J.; Skalecka, A.; He, A.J.; et al. Inhibitory interneurons mediate autism-associated behaviors via 4E-BP2. Proc Natl Acad Sci U S A 2019, 116, 18060–18067. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; DeSpenza, T.; Li, M.; Gulledge, A.T.; Luikart, B.W. Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci 2015, 35, 943–959. [Google Scholar] [CrossRef]
- Wilson, P.M.; Fryer, R.H.; Fang, Y.; Hatten, M.E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J Neurosci 2010, 30, 8529–8540. [Google Scholar] [CrossRef]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu Rev Neurosci 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Win-Shwe, T.-T.; Nway, N.C.; Imai, M.; Lwin, T.-T.; Mar, O.; Watanabe, H. Social behavior, neuroimmune markers and glutamic acid decarboxylase levels in a rat model of valproic acid-induced autism. J Toxicol Sci 2018, 43, 631–643. [Google Scholar] [CrossRef]
- Winslow, J.T.; Insel, T.R. The social deficits of the oxytocin knockout mouse. Neuropeptides 2002, 36, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Woelfle, S.; Pedro, M.T.; Wagner, J.; Schön, M.; Boeckers, T.M. Expression profiles of the autism-related SHANK proteins in the human brain. BMC Biology 2023, 21, 254. [Google Scholar] [CrossRef]
- Worby, C.A.; Dixon, J.E. PTEN. Annu Rev Biochem 2014, 83, 641–669. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Kenny, A.; Eley, S.; McKechanie, A.G.; Stanfield, A.C. Visual social attention in SYNGAP1-related intellectual disability. Autism Res 2024, 17, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Kenny, A.; Mizen, L.A.M.; McKechanie, A.G.; Stanfield, A.C. The Behavioral Profile of SYNGAP1-Related Intellectual Disability. Am J Intellect Dev Disabil 2024, 129, 199–214. [Google Scholar] [CrossRef]
- Wu, J.; Yu, P.; Jin, X.; Xu, X.; Li, J.; Li, Z.; et al. Genomic landscapes of Chinese sporadic autism spectrum disorders revealed by whole-genome sequencing. J Genet Genomics 2018, 45, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Xi, K.; Cai, S.-Q.; Yan, H.-F.; Tian, Y.; Cai, J.; Yang, X.-M.; et al. CSMD3 Deficiency Leads to Motor Impairments and Autism-Like Behaviors via Dysfunction of Cerebellar Purkinje Cells in Mice. J Neurosci 2023, 43, 3949–3969. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. Who does TORC2 talk to? Biochem J 2018, 475, 1721–1738. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Sapuan, A.; Dineen, R.A.; Auer, D.P. Life span pigmentation changes of the substantia nigra detected by neuromelanin-sensitive MRI. Mov. Disord. 2018, 33, 1792–1799. [Google Scholar] [CrossRef]
- Xu, L.-M.; Li, J.-R.; Huang, Y.; Zhao, M.; Tang, X.; Wei, L. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res 2012, 40, D1016–1022. [Google Scholar] [CrossRef]
- Xu, Z.-X.; Kim, G.H.; Tan, J.-W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat Commun 2020, 11, 1797. [Google Scholar] [CrossRef]
- Yagi, T.; Takeichi, M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev 2000, 14, 1169–1180. [Google Scholar] [CrossRef]
- Yamada, K.; Fukaya, M.; Shibata, T.; Kurihara, H.; Tanaka, K.; Inoue, Y.; et al. Dynamic transformation of Bergmann glial fibers proceeds in correlation with dendritic outgrowth and synapse formation of cerebellar Purkinje cells. J Comp Neurol 2000, 418, 106–120. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoshida, K.; Yoshikawa, H.; Kishimoto, Y.; Oka, H. The medial dorsal nucleus is one of the thalamic relays of the cerebellocerebral responses to the frontal association cortex in the monkey: horseradish peroxidase and fluorescent dye double staining study. Brain Res 1992, 579, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef]
- Yang, P.; Lung, F.-W.; Jong, Y.-J.; Hsieh, H.-Y.; Liang, C.-L.; Juo, S.-H. H. Association of the homeobox transcription factor gene ENGRAILED 2 with autistic disorder in Chinese children. Neuropsychobiology 2008, 57, 3–8. [Google Scholar] [CrossRef]
- Yang, X.; Yin, H.; Wang, X.; Sun, Y.; Bian, X.; Zhang, G.; et al. Social Deficits and Cerebellar Degeneration in Purkinje Cell Scn8a Knockout Mice. Front Mol Neurosci 2022, 15, 822129. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Tseng, R.-Y.; Ni, H.-C.; Cocchi, L.; Chang, J.-C.; Hsu, M.-Y.; et al. White matter microstructural and morphometric alterations in autism: implications for intellectual capabilities. Mol Autism 2022, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu Rev Med 2020, 71, 103–116. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J.-J.; Blatt, G.J. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol 2007, 113, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Woo, R.-S.; Cho, S.-C.; Kim, B.-N.; Kim, J.-W.; Shin, M.-S.; et al. Genetic association analyses of neuregulin 1 gene polymorphism with endopheontype for sociality of Korean autism spectrum disorders family. Psychiatry Res 2015, 227, 366–368. [Google Scholar] [CrossRef]
- Yuan, B.; Luo, L.; Hu, C.; Lin, F.; Yang, T.; Chen, J.; et al. Retinoic acid supplementation ameliorates motor incoordination via RARα-CBLN2 in the cerebellum of a prenatal valproic acid-exposed rat autism model. Neurosci Lett 2023, 809, 137316. [Google Scholar] [CrossRef] [PubMed]
- Yurdakul, E.; Barlas, Y.; Ulgen, K.O. Circadian clock crosstalks with autism. Brain Behav 2023, 13, e3273. [Google Scholar] [CrossRef]
- Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int J Mol Sci 2021, 22, 11516. [Google Scholar] [CrossRef] [PubMed]
- Zerbo, O.; Leong, A.; Barcellos, L.; Bernal, P.; Fireman, B.; Croen, L.A. Immune mediated conditions in autism spectrum disorders. Brain Behav Immun 2015, 46, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014, 17, 400–406. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.-J.; Gokce, O.; et al. Neuroligins Sculpt Cerebellar Purkinje-Cell Circuits by Differential Control of Distinct Classes of Synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.-J.; Gokce, O.; et al. Neuroligins Sculpt Cerebellar Purkinje-Cell Circuits by Differential Control of Distinct Classes of Synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef]
- Zhang, B.; Gokce, O.; Hale, W.D.; Brose, N.; Südhof, T.C. Autism-associated neuroligin-4 mutation selectively impairs glycinergic synaptic transmission in mouse brainstem synapses. J Exp Med 2018, 215, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Südhof, T.C. Neuroligins Are Selectively Essential for NMDAR Signaling in Cerebellar Stellate Interneurons. J Neurosci 2016, 36, 9070–9083. [Google Scholar] [CrossRef]
- Zhang, C.; Milunsky, J.M.; Newton, S.; Ko, J.; Zhao, G.; Maher, T.A.; et al. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci 2009, 29, 10843–10854. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, X.; Hou, Q.; Hu, Z.; Wang, Y.; Wang, Z. Regulation of mTORC1 by amino acids in mammalian cells: A general picture of recent advances. Anim Nutr 2021, 7, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Novera, W.; Zhang, Y.; Deng, L.-W. MLL5 (KMT2E): structure, function, and clinical relevance. Cell Mol Life Sci 2017, 74, 2333–2344. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, S.; Cao, Y.; Cheng, P.; Lin, Y.; Sun, Z.; et al. Abnormalities of Gray Matter Volume and Its Correlation with Clinical Symptoms in Adolescents with High-Functioning Autism Spectrum Disorder. Neuropsychiatr Dis Treat 2022, 18, 717–730. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, X.; Geng, Y.; Zhao, W.; Zhou, F.; Wang, J.; et al. Oxytocin differentially modulates specific dorsal and ventral striatal functional connections with frontal and cerebellar regions. Neuroimage 2019, 184, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.-H.; Zhang, W.; Sinton, C.; et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 2009, 29, 1773–1783. [Google Scholar] [CrossRef]
- Zhou, Q.; Qin, J.; Liang, Y.; Zhang, W.; He, S.; Tissir, F.; et al. Celsr3 is required for Purkinje cell maturation and regulates cerebellar postsynaptic plasticity. iScience 2021, 24, 102812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Feliciano, P.; Shu, C.; Wang, T.; Astrovskaya, I.; Hall, J.B.; et al. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet 2022, 54, 1305–1319. [Google Scholar] [CrossRef]
- Zhou, Y.; Sharma, J.; Ke, Q.; Landman, R.; Yuan, J.; Chen, H.; et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature 2019, 570, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Fisher, E.; Li, L.; Zhong, P.; Yan, Z.; Feng, J. PTBP2 attenuation facilitates fibroblast to neuron conversion by promoting alternative splicing of neuronal genes. Stem Cell Reports 2023, 18, 2268–2282. [Google Scholar] [CrossRef]
- Zoghbi, H.Y. Postnatal neurodevelopmental disorders: meeting at the synapse? Science 2003, 302, 826–830. [Google Scholar] [CrossRef]
Figure 1.
Neuronal circuity in the cerebellum. The mature cerebellar cortex consists of the internal granule cell layer (IGL), which contains the cell bodies of granule neurons and Golgi cells, the Purkinje cell layer (PCL), which contains the cell bodies of a single monolayer of PCs, and the molecular layer (ML), which contains the dendrites of the PC and granule neurons (parallel fibers), and the basket and stellate cells, two types of GABAergic interneurons. Granule neurons receive excitatory input from the mossy fibers whereas Purkinje cells are stimulated by neurons of the inferior olivary neurons. The sole output from the cerebellum is from the PC and through the neurons of the deep cerebellar nuclei. Bergman glia cells, which localize close to PCs, are not shown.
Figure 1.
Neuronal circuity in the cerebellum. The mature cerebellar cortex consists of the internal granule cell layer (IGL), which contains the cell bodies of granule neurons and Golgi cells, the Purkinje cell layer (PCL), which contains the cell bodies of a single monolayer of PCs, and the molecular layer (ML), which contains the dendrites of the PC and granule neurons (parallel fibers), and the basket and stellate cells, two types of GABAergic interneurons. Granule neurons receive excitatory input from the mossy fibers whereas Purkinje cells are stimulated by neurons of the inferior olivary neurons. The sole output from the cerebellum is from the PC and through the neurons of the deep cerebellar nuclei. Bergman glia cells, which localize close to PCs, are not shown.
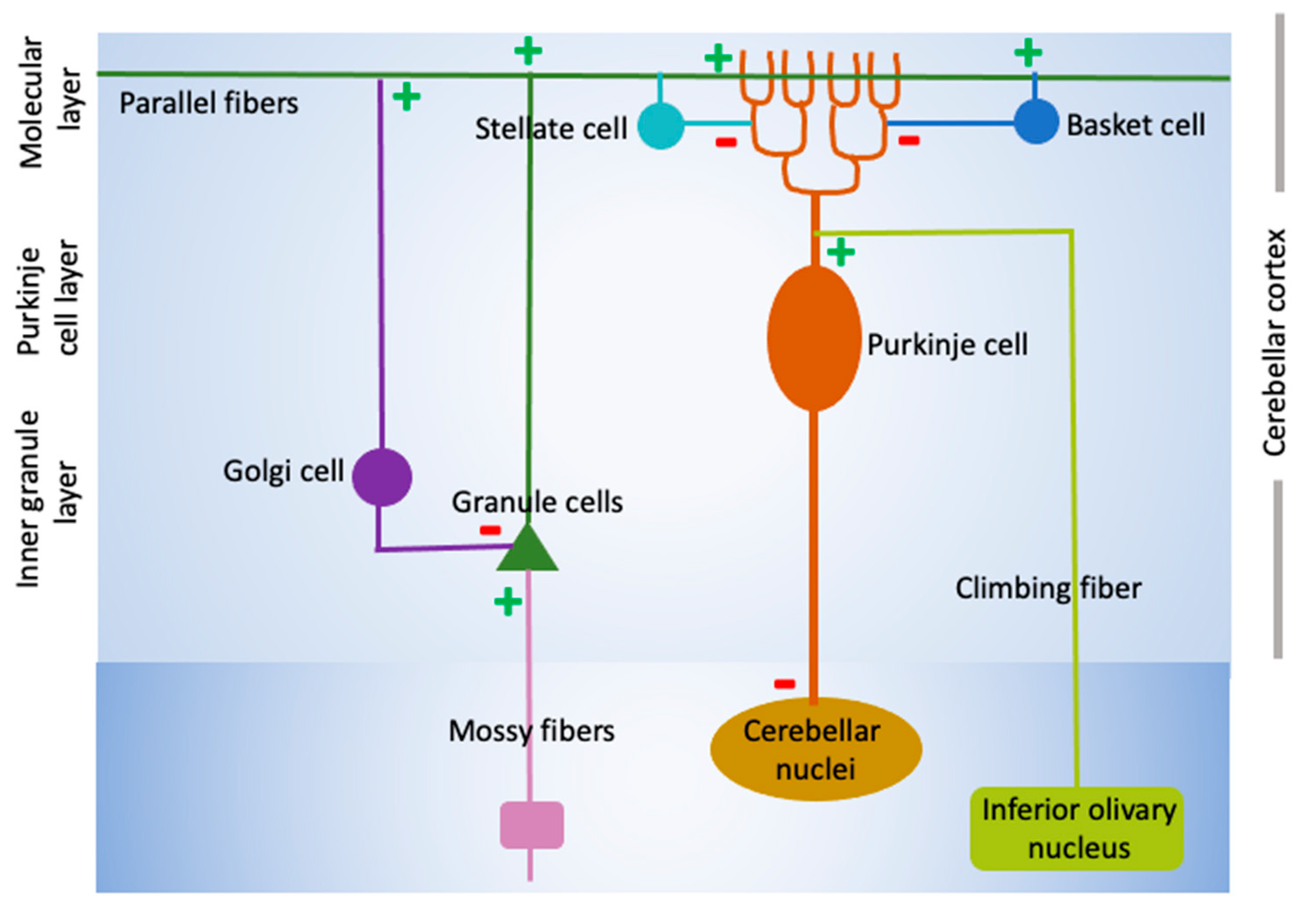
Figure 2.
The Akt-mTORC signaling pathway. Through phosphorylation of membrane-associated phosphoinositides, activation of the lipid kinase PI-3 kinase following the binding of growth and neurotrophic factors to their receptors leads the activation of AKT, a cell survival-promoting protein kinase. AKT inhibits the TSC1/TSC2 protein complex which inhibits mTOR. mTOR, along with Raptor and Rictor, is part of multiprotein complexes, TORC1 and TORC2, respectively. Through phosphorylation,n mTORC1 inhibits 4E-BP, which normally binds the translation initiation factor eIF4E inhibiting it. Phosphorylation by 4E-BP2 causes its disassociation of 4E-BP2 from eIF4E promoting ribosome recruitment and protein translation. AKT-mTORC1 signaling can be inhibited by PTEN, a lipid phosphatase by reversing PI-3K-mediated phosphorylation of phosphoinositides.
Figure 2.
The Akt-mTORC signaling pathway. Through phosphorylation of membrane-associated phosphoinositides, activation of the lipid kinase PI-3 kinase following the binding of growth and neurotrophic factors to their receptors leads the activation of AKT, a cell survival-promoting protein kinase. AKT inhibits the TSC1/TSC2 protein complex which inhibits mTOR. mTOR, along with Raptor and Rictor, is part of multiprotein complexes, TORC1 and TORC2, respectively. Through phosphorylation,n mTORC1 inhibits 4E-BP, which normally binds the translation initiation factor eIF4E inhibiting it. Phosphorylation by 4E-BP2 causes its disassociation of 4E-BP2 from eIF4E promoting ribosome recruitment and protein translation. AKT-mTORC1 signaling can be inhibited by PTEN, a lipid phosphatase by reversing PI-3K-mediated phosphorylation of phosphoinositides.
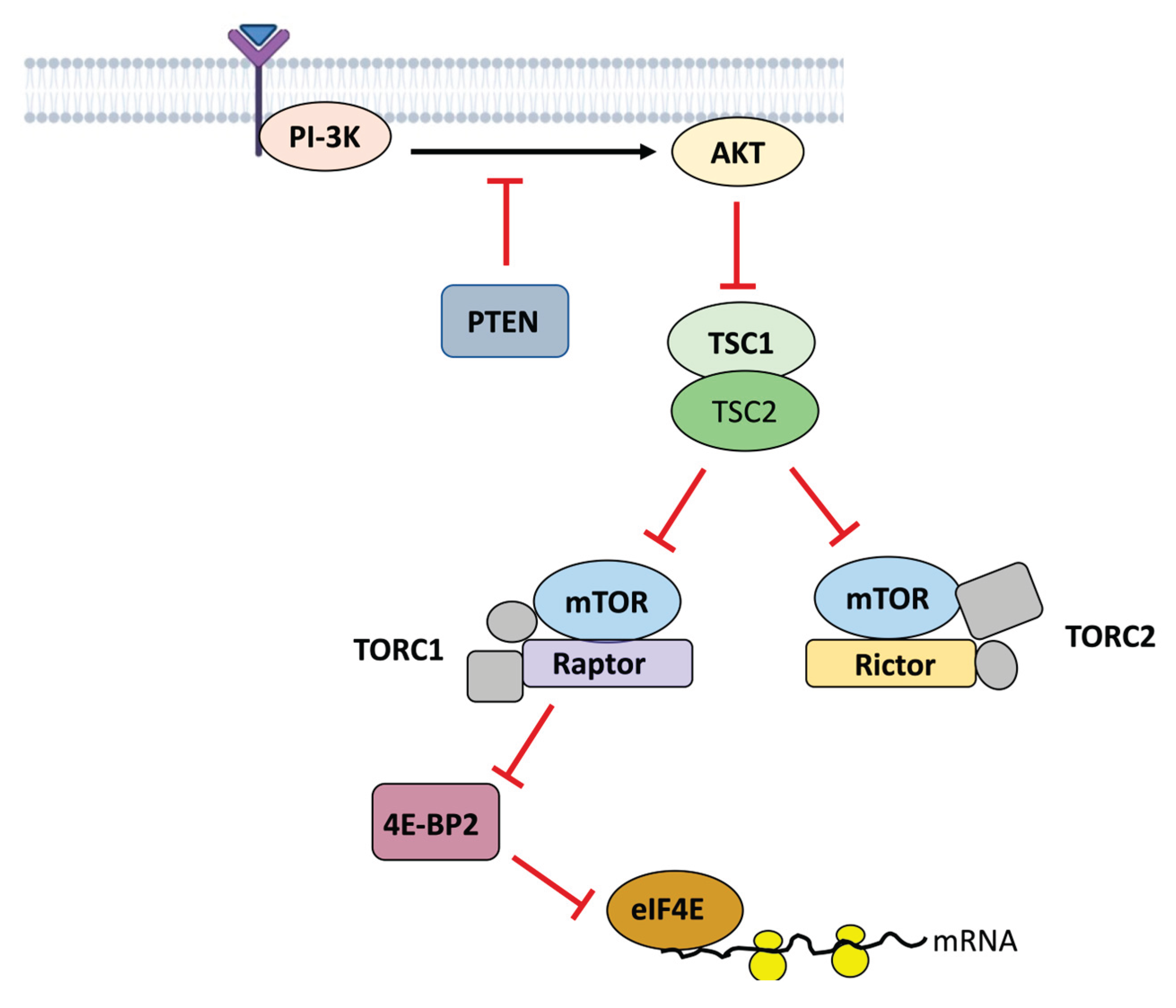
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



